Note: Descriptions are shown in the official language in which they were submitted.
21~1789
WO 94/26731 PCT/CA94/00264
- 1 -
TITLE OF THE INVENTION
2-SUBSTITUTED-3,4-DL~RYLTHIOPHENE DERIVATIVES AS
IN~BITORS OF CYCLOOXYGENASE
5 BACKGROUND OF THE INVENTION
This invention relates to compounds and ph~rm~ceutical
compositions for the treatment of cyclooxygenase mediated diseases and
methods of treatment thereof.
Non-steroidal, antiinfl~mm~tory drugs exert most of their
antiinfl~mm~tory, analgesic and antipyretic activity and inhibit
hormone-induced uterine contractions and certain types of cancer
growth through inhibition of prostaglandin G/H synthase, also known as
cyclooxygenase. Up until recently, only one form of cyclooxygenase
had been characterized, this corresponding to cyclooxygenase-1 or the
15 constitutive enzyme, as originally identified in bovine seminal vesicles.
Recently the gene for an inducible form of cyclooxygenase
(cyclooxygenase-2) has been cloned, sequenced and characterized from
chicken, murine and human sources. This enzyme is distinct from the
cyclooxygenase-1 which has now also been cloned, sequenced and
20 characterized from sheep, murine and hllm~n sources. The second form
of cyclooxygenase, cyclooxygenase-2, is rapidly and readily inducible
by a number of agents including mitogens, endotoxin, hormones,
cytokines and growth factors. As prostaglandins have physiological and
pathological roles, we have concluded that the constitutive enzyme,
25 cyclooxygenase-1, is responsible, in large part, for endogenous basal
release of prost~gl~nclinc and hence is important in their physiological
functions such as the m~intenance of gastrointestinal integrity and renal
blood flow. In contrast, we have concluded that the inducible form,
cyclooxygenase-2, is mainly responsible for the pathological effects of
30 pros~gl~n~lin~ where rapid induction of the enzyme would occur in
response to such agents as infl~mm~tory agents, hormones, growth
factors, and cytokines. Thus, a selective inhibitor of cyclooxygenase-2
will have similar antiinfl~mm~tory, antipyretic and analgesic properties
to a conventional non-steroidal ~ntiinfl~mm~tory drug (NSAID), and in
21~178~
WO 94/26731 PCT/CA94/00264
addition would inhibit hormone-induced uterine contractions and have
potential anti-cancer effects, but will have a ~limini~hed ability to induce
some of the mech~ni~m-based side effects. In particular, such a
compound should have a reduced potential for gastrointestinal toxicity,
a reduced potential for renal side effects, a reduced effect on bleeding
times and possibly a lessened ability to induce asthma attacks in aspirin-
sensitive asthmatic subjects.
SUMMARY OF THE INVENTION
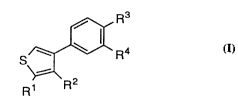
The invention encompasses compounds of Formula I
S~ R4
R1 R2
which are useful in the treatment of cyclooxygenase mediated diseases,
20 in particular cyclooxygenase-2 mediated diseases.
The invention also encomp~ses ph~ eutical
compositions for inhibiting cyclooxygenase and for treating
cyclooxygenase mediated diseases as disclosed herein comprising a
ph~ ceutically acceptable carrier and a non-toxic therapeutically
25 effective amount of compound of Formula I as described herein.
The invention also encompasses me~ods of inhibiting
cyclooxygenase and treating cyclooxygenase mediated diseases
comprising:
~lmini~tration to a patient in need of such tre~tmtont of a non-toxic
30 therapeutically effective amount of a compound of Formula I as
disclosed herein.
DETAILED DESCRIPTION OF THE ~VENTION
The invention encompasses compounds of Formula I
~ WO 94/26731 2 1 ~ 1 7 ~ !~ PCT/CA94/00264
- 3 -
S~ R4
R1 R2
and pharmaceutically acceptable salts thereof wherein:
o R1 is selected from the group consisting of
(a) hydrogen,
(b) halo, including fluoro, chloro, bromo and iodo,
(c) CN,
(d) N02,
(e) CF3, and
(f) Cl 6aLkYl;
R2 is selected from the group consisting of
(a) C3 6alkyl,
(b) mono or di substituted phenyl wherein the substitutents are
selected from the group consisting of
(1) hydrogen,
(2) halo as defined above,
(3) C1 6alkoxy,
(4) C1 6alkylthio,
(S) CN,
(6) CF3,
(7) C1 6aLkyl, and
(8) N3,
(c) mono or di substituted heteroaryl wherein heteroaryl is
(1) a monocyclic aromatic ring of ~ atoms, cont~ining
one hetero atom which is S, O or N and optionally 1,
2, or 3 additional hetero atoms which are N, or
(2) a monocyclic aromatic ring of 6 atoms, con~ining 1,
2, 3, or 4 hetero atoms which are N, and
2161789
WO 94/26731 PCTICA94/00264 ~
wherein the sub~ uLel-ts on the heteroaryl are selected
from the group consisting of
(1 ) hydrogen,
(2) halo as defined above,
(3) C1 6aLkoxy,
(4) Cl 6alkylthio,
(5) CN,
(6) CF3,
(7) C1 6a1~cyl,
o (8) N3,
R3 is selected from the group consisting of
(1 ) -S(0)2CH3,
(2) -S(O)(NH)CH3,
(3) -S(O)NH2, and
4) -S(0)2N~2;
R4 is selected from the group consisting of
(1) hydrogen,
(2) halo as defined above,
(3) carboxy, and
(4) CF3.
For purposes of this specification aLkyl is defined to include
linear, branched, and cyclic structures, with Cl 6alkyl including
including methyl, ethyl, propyl, 2-propyl, s- and t-butyl, butyl, pentyl,
hexyl, 1,1-dimethylethyl, cyclopropyl, cyclobutyl, cyclopentyl and
cyclohexyl. Sirnilarly, Cl 6aLkoxY is inten-led to include alkoxy groups
of from 1 to 6 carbon atoms of a straight, branched, or cyclic
configuration. Examples of lower aLkoxy groups include methoxy,
O ethoxy, propoxy, isopropoxy, cyclopropyloxy, cyclohexyloxy, and the
3 like. Likewise, C1 6alkylthio is intended to include alkylthio groups of
from 1 to 6 carbon atoms of a straight, branched or cyclic
configuration. Examples of lower alkylthio groups include methylthio,
propylthio, isopropylthio, cycloheptylthio, etc. By way of illustration,
the propylthio group signifies -SCH2CH2CH3.
~WO 94/26731 2 1 6 ~ 7 8 9 PCT/CA94/00264
- 5 -
In one genus the invention encompasses compounds of
Formula I
~R3
S~ R4
R1 R2
and pharmaceutically acceptable salts thereof wherein:
R4 is hydrogen, and
R2 is selected from the group consisting of
(a) C3 6alkyl,
(b) mono or di substituted phenyl, and
(c) mono or di substituted heteroaryl wherein heteroaryl is
selected from the group consisting of
(1) furanyl,
(2) diazinyl, triazinyl, tetrazinyl,
(3) imidazolyl,
(4) isoxazolyl,
(S) isothiazolyl,
(6) oxadiazolyl,
(7) oxazolyl,
(8) pyrazolyl,
(9) pyrrolyl,
(10) th~ 7olyl~
(11 ) thiazolyl,
(12) thienyl,
3 (13) triazolyl,
(14) pyridyl, and
(15) tetrazolyl.
Exemplifying the invention are the compounds of Table 1
including:
3 -(4-fluorophenyl)-4-(4-(methylsulfonyl)phenyl)thiophene,
V~O 94/26731 , ~ PCT/CA94/00264 ~
2-Nitro-3 -(4-fluorophenyl)-4-((4 -methylsulfonyl)phenyl)thiophene,
2-Bromo-3 -(4-fluorophenyl)-4-((4-methylsulfonyl)phenyl)thiophene,
and
3 -(4-Fluorophenyl)-4-(4-sulfamoylphenyl)thiophene.
In a second embo-lim~nt, the invention encompasses
ph~rm~ceutical compositions for inhibiting cyclooxygenase and for
treating cyclooxygenase mediated diseases as disclosed herein
comprising a ph~ ceutically acceptable carrier and a non-toxic
therapeutically effective amount of compound of Formula I as described
above.
Within this embodiment the invention encompasses
ph~ eutical compositions for inhibiting cyclooxygenase-2 and for
treating cyclooxygenase-2 mediated diseases as disclosed herein
comprising a pharmaceutically acceptable carrier and a non-toxic
therapeutically effective amount of compound of Formula I as described
above.
In a third emborliment, the invention encompasses a method
of inhibiting cyclooxygenase and treating cyclooxygenase mediated
diseases as disclosed herein comprising:
~dmini~tration to a patient in need of such treatment of a non-toxic
therapeutically effective amount of a compound of Formula I as
disclosed herein.
Within this embodiment the invention encompasses a
method of inhibiting cyclooxygenase-2 and treating cyclooxygenase-2
mediated diseases as disclosed herein comprising:
~lmini~tration to a patient in need of such treatment of a non-toxic
therapeutically effective amount of a compound of Formula I as
disclosed herein.
The pharmaceutical compositions of the present invention
comprise a compound of Formula I as an active ingredient or a
ph~ ceutically acceptable salt, thereof, and may also contain a
pharmaceutically acceptable carrier and optionally other therapeutic
ingredients. The term "pharmaceutically acceptable salts" refers to salts
prepared from ph~ ceutically acceptable non-toxic bases including
.
~WO 94/26731 2 16 17 ~ ~ PCT/CA94/00264
inorganic bases and organic bases. Salts derived from inorganic bases
include aluminum, ammonium, calcium, coRer, ferric, ferrous,
lithium, m~gnesium, manganic salts, manganous, potassium, sodium,
zinc, and the like. Particularly preferred are the ammonium, calcium,
5 m~ nesium, potassium, and sodium salts. Salts derived from
ph~ ceutically acceptable organic non-toxic bases include salts of
primary, secondary, and tertiary amines, substituted amines including
naturally occurring substituted amines, cyclic amines, and basic ion
exchange resins, such as algi~ le, betaine, caffeine, choline, N,N-
diberl~ylethylenerli~mine, diethyl~mine, 2-diethylaminoethanol, 2-
dimethylaminoethanol, ethano!~mine, ethylene(li~mine, N-ethyl-
morpholine, N-ethylpiperidine, glucamine, glucosamine, histidine,
hydrabamine, isopropyl~mine, lysine, methylglucamine, morpholine,
piperazine, piperidine, polyamine resins, procaine, purines,
15 theobromine, triethyl~mine, trimethylamine, tripropyl~mine,
trometh~mine, and the like.
It will be understood that in the discussion of methods of
treatment which follows, references to the compounds of Formula I are
meant to also include the pharmaceutically acceptable salts.
As disclosed elsewhere in this specification in further
detail, these diseases include pain, fever and infl~mm~tion of a variety
of conditions including rheumatic fever, symptoms associated with
influenza or other viral infections, common cold, low back and neck
pain, dysmenorrhea, headache, toothache, sprains and strains, myositis,
25 neuralgia, synovitis, ar~ritis, including rheumatoid ~llllilis
degenerative joint diseases (osteoarthritis), gout and ankylosing
spondylitis, bursitis, burns, injuries.
Compounds of Formula I are useful for the relief of pain,
fever and infl~mm~tion of a variety of conditions including rheumatic
fever, symptoms associated with influenza or other viral infections,
common cold, low back and neck pain, dysmenorrhea, headache,
toothache, sprains and strains, myositis, neuralgia, synovitis, arthritis,
including rheumatoid arthritis degenerative joint diseases
(osteoarthritis), gout and ankylosing spondylitis, bursitis, burns,
2161789
WO 94/26731 f~ ~ - PCT/CA94/00264
- 8 -
injuries, following surgical and dental procedures. In addition, such
compounds may inhibit cellular neoplastic transformations and metastic
tumor gro~vth and hence can be used in the treatment of cancer.
Compounds of Formula I will also inhibit prostanoid-induced smooth
5 muscle contraction by preventing the synthesis of contractile prostanoids
and hence may be of use in the treatment of dysmenorrhea, premature
labor and asthma and Alzheimers disease.
By virtue of their high cyclooxygenase-2 (COX-2) activity
and/or their specificity for cyclooxygenase-2 over cyclooxygenase-1
(COX-1), compounds of Formula I will prove useful as alternatives to
conventional non-steroidal anti-infl~mm~tory drugs (NSAID'S)
particularly where such non-steroidal anti-infl~mm~tory drugs may be
contra-indicated such as in patients with peptic ulcers, gastritis, regional
enteritis, ulcerative colitis, diverticulitis or with a recurrent history of
15 gastrointestinal lesions; GI bleeding, coagulation disorders including
anemia such as hypoprothrombinemia, haemophilia or other bleeding
problems; kidney disease; those prior to surgery or taking
anticoagulants .
Similarly, compounds of Formula I, will be useful as a
partial or complete substitute for conventional NSAID'S in preparations
wherein they are presently co-~(lmini~tered with other agents or
ingredients. Thus in further aspects, the invention encompasses
pharmaceutical compositions for treating cyclooxygenase-2 mediated
diseases as defined above comprising a non-toxic therapeutically
25 effective amount of compound of Formula I as defined above and one
or more ingredients such as another pain reliever including
acetaminophen or phenacetin; a potentiator including caffeine; an H2-
antagonist, all~minllm or m~gnesium hydroxide, simethicone, a
decongestant including phenylephrine, phenylpropanol~mine,
pseudophedrine, oxymetazoline, ephinephrine, naphazoline,
xylometazoline, propylhexedrine, or levo-desoxyephedrine; an
~ntitllssive including codeine, hydrocodone, caramiphen,
carbetapentane, or dextramethorphan; a diuretic; a sedating or non-
~ WO 94/26731 21817 8 ~ PCT/CA94/00264
sedating ~ntihi,st~mine. In addition the invention encompasses a methodof treating cyclooxygenase mediated diseases comprising
~dmini~tration to a patient in need of such treatment a non-toxic
therapeutically effective amount of compound of Formula I, optionally
5 co-administered with one or more of such ingredients as listed
immediately above.
Compounds of the present invention are inhibitors of
cyclooxygenase-2 and are thereby useful in the treatment of
cyclooxygenase-2 mediated diseases as enumerated above. This activit~
is illustrated by their ability to selectively inhibit cyclooxygenase-2 over
cyclooxygenase-l. Accordingly, in one assay, the ability of the
compounds of this invention to treat cyclooxygenase mediated diseases
can be demonstrated by measuring the amount of prostaglandin E2
(PGE2) synthesized in the presence of arachidonic acid,
5 cyclooxygenase-l or cyclooxygenase-2 and a compound of Formula I.
The ICS0 values represent the concentration of inhibitor required to
return PGE2 synthesis to 50% of that obtained as compared to the
uninhibited control. Illustrating this aspect, we have found that
Compounds 1 through 25 are more than 100 times more effective in
inhibiting COX-2 than they are at inhibiting COX-1. In addition they
all have a COX-2 IC50 of 1 nM to 1 ,uM. By way of comparison,
Ibuprofen has an ICS0 for COX-2 of 1 ~lM, and Indomethacin has an
ICS0 for COX-2 of approximately 100 nM.
For the treatment of any of these cyclooxygenase mediated
25 diseases compounds of Formula I may be ~-lmini.ctered orally, topically,
parenterally, by inh~l~tion spray or rectally in dosage unit formulations
cont~ining conventional non-toxic pharmaceutically acceptable carriers,
adjuvants and vehicles. The term parenteral as used herein includes
subcutaneous injections, intravenous, intramuscular, intrasternal
3 injection or infusion techniques. In addition to the treatment of warm-
blooded ~nim~l~ such as mice, rats, horses, cattle, sheep, dogs, cats, etc.,
the compounds of the invention are effective in the tre~tment of
hllm~n.~.
2161~8~
WO 94/26731 PCT/CA94/00264
- 10-
As indicated above, phàImaceutical compositions for
treating cyclooxygenase-2 mediated diseases as defined may optionally
include one or more ingredients as listed above.
The ph~rm~celltical compositions cont~ining the active
5 ingredient may be in a form suitable for oral use, for example, as
tablets, troches, lozenges, aqueous or oily suspensions, dispersible
powders or granules, emulsions, hard or soft capsules, or syrups or
elixirs. Compositions intended for oral use may be prepared according
to any method known to the art for the manufacture of ph~rm~ceutical
compositions and such compositions may contain one or more agents
selected from the group consisting of sweetening agents, flavoring
agents, coloring agents and preserving agents in order to provide
ph~rTn~ceutically elegant and palatable preparations. Tablets contain the
active ingredient in ~-lmixt~lre with non-toxic pharmaceutically
acceptable excipients which are suitable for the m~nuf~rture of tablets.
These excipients may be for example, inert diluents, such as calcium
carbonate, sodium carbonate, lactose, calcium phosphate or sodium
phosphate; gran~ ting and disintegrating agents, for example, corn
starch, or alginic acid; binding agents, for example starch, gelatin or
acacia, and lubricating agents, for example m~gnesium stearate, stearic
acid or talc. The tablets may be uncoated or they may be coated by
known techniques to delay ~ integration and absorption in the
gastrointestinal tract and thereby provide a sustained action over a
longer period. For example, a time delay material such as glyceryl
5 monostearate or glyceryl distearate may be employed. They may also
be coated by the techniques described in the U.S. Patents 4,256,108;
4,166,452; and 4,265,874 to form osmotic ~erapeutic tablets for
control release.
Forrn~ tions for oral use may also be presented as hard
30 gelatin capsules wherein the active ingredient is mixed with an ~nert
solid diluent, for example, calcium carbonate, calcium phosphate or
kaolin, or as soft gelatin capsules wherein the active ingredient is mixed
with water or an oil medium, for example peanut oil, liquid paraffin, or
olive oil.
~ ~=
~WO 94126731 21 G 1 7 8 ~ PCT/CA94100264
- 11 -
Aqueous suspensions contain the active materials in
admixture with excipients suitable for the m~nllf~cture of aqueous
suspensions. Such excipients are suspending agents, for example sodium
carboxymethylcellulose, methylcellulose, hydroxypropylmethyl-
5 cellulose, sodium ~l~in~te, polyvinyl-pyrrolidone, gum tragacanth and
gum acacia; dispersing or wetting agents may be a naturally-occurring
phosphatide, for example lecithin, or condensation products of an
aLkylene oxide with fatty acids, for example polyoxyethylene stearate,
or condensation products of ethylene oxide with long chain aliphatic
alcohols, for example heptadecaethyleneoxycetanol, or condensation
products of ethylene oxide with partial esters derived from fatty acids
and a hexitol such as polyoxyethylene sorbitol monooleate, or
condensation products of ethylene oxide with partial esters derived from
fatty acids and hexitol anhydrides, for example polyethylene sorbitan
monooleate. The aqueous suspensions may also contain one or more
preservatives, for example ethyl, or n-propyl, p-hydroxybenzoate, one
or more coloring agents, one or more flavoring agents, and one or
more sweetening agents, such as sucrose or saccharin.
Oily suspensions may be form~ ted by suspending the
20 active ingredient in a vegetable oil, for example arachis oil, olive oil,
sesame oil or coconut oil, or in a mineral oil such as liquid paraffin.
The oily suspensions may contain a thickening agent, for example
beeswax, hard paraffin or cetyl alcohol. Sweetening agents such as
those set forth above, and flavoring agents may be added to provide a
palatable oral preparation. These compositions may be preserved by the
addition of an anti-oxidant such as ascorbic acid.
Dispersible powders and granules suitable for preparation
of an aqueous suspension by the addition of water provide the active
ingredient in admixture with a dispersing or wetting agent, suspen~1ing
agent and one or more preservatives. Suitable dispersing or wetting
agents and suspending agents are exemplified by those already
mentioned above. Additional excipients, for example sweetening,
flavoring and coloring agents, may also be present.
21~1~89
WO 94/26731 PCT/CA94/00264 ~
The pharmaceutical compositions of the invention may also
be in the form of oil-in-water emulsions. The oily phase may be a
vegetable oil, for example olive oil or arachis oil, or a mineral oil, for
example liquid paraffin or mixtures of ~ese. Suitable emulsifying
5 agents may be naturally-occurring gums, for example gum acacia or
gum tr~c~nth, naturally-occurring phosphatides, for example soy
bean, lecithin, and esters or partial esters derived from fatty acids and
hexitol anhydrides, for example sorbitan monooleate, and contlenc~tion
products of the said partial esters with ethylene oxide, for example
polyoxyethylene sorbitan monooleate. The emulsions may also contain
sweetening and flavoring agents.
Syrups and elixirs may be form~ te-l with sweetening
agents, for example glycerol, propylene glycol, sorbitol or sucrose.
Such formulations may also contain a demulcent, a preservative and
flavoring and coloring agents. The ph~rm~ceutical compositions may
be in the form of a sterile injectable aqueous or oleagenous suspension.
This suspension may be formulated according to the known art using
those suitable dispersing or wetting agents and suspending agents which
have been mentioned above. The sterile injectable preparation may also
be a sterile injectable solution or suspension in a non-toxic parenterally-
acceptable diluent or solvent, for example as a solution in 1,3-butane
diol. Among the acceptable vehicles and solvents that may be employed
are water, Ringer's solution and isotonic sodium chloride solution. In
addition, sterile, fixed oils are conventionally employed as a solvent or
suspending merlillm. For this purpose any bland fixed oil may be
employed including synthetic mono- or diglycerides. In addition, fatty
acids such as oleic acid find use in the preparation of injectables.
The compounds of forrnula (I) may also be ~lministered in
30 the form of suppositories for rectal ~tlmini.ctration of the drug. These
compositions can be prepared by mixing ~e drug with a suitable non-
irritating excipient which is solid at ordinary temperatures but liquid at
the rectal temperature and will therefore melt in the rectum to release
the drug. Such materials are cocoa butter and polyethylene glycols.
~ WO 94/26731 2 1 6 1 7 8 !~ PCT/CA94/00264
- 13 -
For topical use, creams, ointments, jellies, solutions or
suspensions, etc., cont~ining the compounds of Formula (I) are
employed. (For purposes of this application, topical application shall
include mouth washes and gargles.)
Dosage levels of the order of from about 0.01 mg to about
140 mg per kilogram of body weight per day are useful in the treatment
of the above-indicated conditions, or alternatively about 0.5 mg to about
7 gms per patient per day. For example, infl~mm~tion may be
effectively treated by the a~lministration of from about 0.01 to 50 mg of
the compound per kilogram of body weight per day, or alternatively
about 0.5 mg to about 3.5 gms per patient per day.
The amount of active ingredient that may be combined with
the carrier materials to produce a single dosage form will vary
depending upon the host treated and the particular mode of
minictration. For example, a form~ tion intended for the oral
~lminictration of hllm~nc may contain from 0.5 mg to 5 gm of active
agent compounded with an appropriate and convenient amount of
carrier material which may vary from about 5 to about 95 percent of
the total composition. Dosage unit forms will generally contain between
from about 1 mg to about 500 mg of an active ingredient, typically 25
mg, 50 mg, 100 mg, 200 mg, 300 mg, 400 mg, 500 mg, 600 mg, 800
mg, or 1000 mg.
It will be understood, however, that the specific dose level
for any particular patient will depend upon a variety of factors
including the activity of the specific compound employed, the age, body
weight, general health, sex, diet, time of ~lminictration, route of
~lminictration, rate of excretion, drug combination and the severity of
the particular disease undergoing therapy.
The compounds of the present invention are conviently
prepared using the procedures described below.
Method A
The compounds of the present invention can be prepared by
the general method described by J. Nakayama et al., Tetrahedron Lett.,
2 ~
WO 94/26731 PCT/CA94/00264 ~
~ - 14-
26 (16), 1983-1984 (1985). Accordingly, intermediate III, obtained
from the base catalyzed coupIing of an a-mercaptoacetophenone and an
a-haloacetophenone, is treated with tit~nium tetrachloride and zinc at
low temperature in an inert solvent such as tetrahydrofuan to give an
5 intermediate 3,4-dihydroxythiolane IV. This compound can then be
dehydrated by heating it in a solvent such as toluene in the presence of
an acid such as p-toluenesulfonic acid to yield II. This compound can
be oxidized with a reagent such as the magnesium salt of mono-
peroxyphtalic acid (MMPP) or m-chloroperoxybenzoic acid (mCPBA)
to yield I.
METHOD A
~SMe 2 base
HS~R4 + X~R
O O
O O ,~SMe
MeS ~ ~ R2 TiCI4/Zn S/~J~R4
111 R1 OH
IV
2s H+ S~ SMe ~SRO42Me
heat R2 [ R2
R1 Rl
ll 1 a (R3 = SO2Me)
~WO 94/26731 21 G 17 ~ 9 PCT/CA94/00264
Method B
If I contains a substituent R1 which can be introduced by an
aromatic electrophilic substitution, this can be carried out either on I or
II. Accordingly, I can be treated with a halogenating agent such as
5 bromine in a solvent such as glacial acetic acid to give the desired 2-
bromothiophene (R1 = Br). When it is desired to have a nitrogen
substituent at this position, for example R1 = N02, a cold mixhlre of I
in a solvent such as acetic anhydride is treated with nitric acid to
introduce the nitro group at the desired position.
METHOD B
la (R~ = H) R ~SO2Me
R1 Rl
IC- NO2
2161~89
WO 94/26731 PCT/CA94/00264 ~
- 16 -
Method C
When R3 is a sulfonarnide group such as R3 = S02NH2,
this substituent can be introduced by treating V with a base such as n-
BuLi at low temperature and quenching ~e anion with sulfur dioxide to
5 give a lithium arylsulfinate intermediate. This intermediate can then be
converted to an arylsulfonyl chloride which will react with NH3 to
provide Id. This general procedure has been described by T. Hameda
and O. Yonemitsu, Synthesis, 852 (1986).
METHOD C
1S ~R4 2)S2 ~SO2NH2
R2 3) SO2CI2 R2
R1 4) NH3 R1
V Id (R3=So2NH2)
Table I illustrates compounds of Formula I, which are
representative of the present invention. Representative biological data
and the assays utilized to generate the data is provided immediately
thereafter.
~WO 94/26731 ~ 1 G 17 ~ 9 PCT/CA94/00264
- 17 -
- TABLE I
~R3
S/~--R4
~,
R1 R2
Compound R1 R2 R3 R4
H 4-fluorophenyl -S(0)2CH3 H
2 N2 -S(0)2CH3 H
3 Br " -S(0)2CH3 H
4 H " -S(0)2NH2 H
F " -s(o)2cH3 H
6 Cl " -S(0)2CH3 H
2 0 7 I .. -S(0)2CH3 H
8 CH3 cyclohexyl -s(o)2cH3 H
9 CF3 4-fluorophenyl -S(0)2CH3 H
CN ~ -S(0)2CH3 H
11 H " -S(O)(NH)CH3 H
12 H 2-pyridyl -S(0)2CH3 H
13 H 2-thienyl -S(0)2CH3 H
- 3 14 H -n-pentyl -S(0)2CH3 H
15 H 4-cyanophenyl -s(o)2cH3 H
16 H 4-fluorophenyl -S(0)2CH3 Br
17 H 4-fluorophenyl -S(0)2CH3 C02H
WO 94/26731 PCT/CA94/00264 ~
- 18 -
Whole Cell Cvclooxygenase Assays
Human osteosarcoma 143.98.2 cells were cultured in
DULBECCOS MOD~D EAGLES MEDIUM (SIGMA) cont~inin~;
3.7 g/l NaHCO3 (SIGMA), 100 ,ug/l ge~t~",icin (GIBCO), 25 mM
HEPES, pH 7.4 (SIGMA), 100 IU/ml penicillin (FLOW LABS), 100
,ug/ml streptomycin (FLOW LABS), 2 mM gl~ ";lle (FLOW LABS)
and 10% fetal bovine serum (GIBCO). Cells were rn~int~ined at 37C,
6% C02 in 150 cm2 tissue culture flasks (CORNING). For routine
subculturing, media was removed from confluent cultures of cells,
which were then incubated with 0.25% trypsin/0.1% EDTA (JR~I
BIOSCIENCES) and incubated at room temperature for approxirnately
5 minutes. The trypsin solution was then aspirated, and cells
resuspended in fresh medium and dispensed at a ratio of 1:10 or 1:20
into new flasks.
U-937 cells (ATCC CRL 1593) were cultured in 89%
RPMI-1640 (SIGMA), 10% fetal bovine serum (GIBCO), cont~ining 50
IU/ml penicillin (FLOW LABS), 50 ~g/ml streptomycin (FLOW
LABS) and 2 g/l NaHCO3 (SIGMA). Cells were m~in~ined at a density
of 0.1-2.0 x 1o6lml in 1 liter spinner flasks (CORNING) at 37C, 6%
CO2. For routine subculturing, cells were diluted in fresh medium and
transferred to fresh flasks.
Assay Protocol
For cyclooxygenase assays, osteosarcoma 143.98.2 cells
2s were cultured in 1 ml of media in 24-well multidishes (NUNCLON)
until confluent. The number of cells per assay was determined from
replicate plates prior to assays, using standard procedures. Lmmediately
prior to cyclooxygenase assays, media was aspirated from cells, and the
cells washed once with 2 ml of Hanks b~l~n~ed salts solution (~SS;
SIGMA) prewarmed to 37C. 1 ml of prewarmed HBSS was then added
per well.
Irnmediatley prior to cyclooxygenase assays, the
appropriate number of U-937 cells were removed from spinner cultures
and centrifuged at 300 x g for 10 minlltes. The supern~t~nt was
~WO 94/26731 21617 ~ 9 PCT/CA94/00264
- 19-
decanted and cells washed in 50 ml of HBSS prewarmed to 37C. Cells
were again pelleted at 300 x g for 10 ~ s and resuspended in
prewarmed HBSS to a final cell density of approximately 1.5 x 106
cells/ml. 1 ml aliquots of cell suspension were transferred to 1.5 ml
5 microcentrifuge tubes or 24-well multidishes (NUNCLON).
Following washing and resuspension of osteosarcoma 143
and U-937 cells in 1 ml of HBSS, 1 ,ul of test compounds or DMSO
vehicle were added, and samples gently mixed. All assays were
performed in triplicate. Samples were then incubated for 15 minlltes at
37C, prior to the addition of 10 ~l of peroxide-free arachidonic acid
(CAYMAN) diluted to 1 mM in HBSS. Control samples contained
ethanol vehicle instead of arachidonic acid. Samples were again gently
mixed and incubated for a further 10 minlltes at 37C. For
osteosarcoma cells, reactions were then stopped by the addition of 100
5 ~11 of lN HCl, with mixing, or by the rapid removal of media directly
from cell monolayers. For U-937 cells, reactions in multiwell dishes or
microcentrifuge tubes were stopped by the addition of 100 ~l of lN
HCl, with mixing. Samples assayed in 24-multidishes were then
transferred to microcentrifuge tubes. If necessary, samples were stored
20 at 4C prior to analysis of PGE2 levels.
Ouantitation of PGE~ Concentrations
Osteosarcoma 143.98.2 and U-937 samples were
neutralized by the addition of 100 ,ul of lN NaOH. Samples were then
25 mixed by vortexing, and PGE2 levels measured using a PGE2 radio-
immllnoassay (NEW ENGLAND NUCLEAR-DUPONT) according to
the manufacturers instructions. This procedure was automated using a
BIOMEK 1000 (BECKMAN). Levels of PGE2 were calculated from
the standard curve determined using BECKMAN IMMUNO~l l
30 EL~/RLA analysis software.
2161~89
WO 94/26731 PCT/CA94/0026~ ~
- 20 -~ ~
.~
Results
TABLE 2
Compound Drug Concen- % Of Inhibition
tration nM Whole Cells Mic-osomes
COX2 COX 1 COX2 COX 1
(osteosar- (U937) (osteosar- (U937)
coma) coma)
38
100 87 -2 98 15
2 10 27 0 20 5
100 95 0 45
3 10 62 0 36 3
100 84 0 81 -5
4 10 69 8 54 -5
100 94 21 63 -8
EXAMPLES
The invention will now be illustrated by the following non-
limiting examples. Unless stated otherwise: (i) all operations were
carried out at room or ambient temperature, that is, at a temperature in
the range 18-25C; (ii) evaporation of solvent was carried out using a
25 rotary evaporator under reduced pressure (600-4000 pascals: 4.5-30
me. Hg) with a bath temperature of up to 60C; (iii) the course of
reactions was followed by ~in layer chromatography (TLC) and
reaction times are given for illustration only; (iv) melting points are
uncorrected and `d' indicates decomposition; the melting points given
30 are those obtained for the materials prepared as described;
polymorphism may result in isolation of materials with different
melting points in soee preparations; (v) the structure and purity of all
final products were assured by at least one of the following techniques:
TLC, mass spectrometry, nuclear m~gnetic resonance (NMR)
spectrometry or microanalytical data; (vi) yields are given for
~ WO 94/26731 21 6 17 8 9 PCT/CA94/00264
- 21 -
- illustration only; (vii) when given, NMR data is in the form of delta (~)
values for major diagnostic protons, given in parts per million (ppm)
relative to tetramethylsilane (TMS) as internal standard, determined at
300 MHz or 400 MHz using the indicated solvent; conventional
5 abbreviations used for signal shape are: s. singlet; d. doublet; t. triplet;
m. multiplet; br. broad; etc.: in addition "Ar" signifies an aromatic
signal; (viii) chemical symbols have their usual me~nin~;~; the following
abbreviations have also been used v (volume), w (weight), b.p. (boiling
point), m.p. (melting point), L (liter(s)), mL (milliliters), g (gram(s)),
mg (milligrams(s)), mol (moles), mmol (millimoles), eq (equivalent(s)).
Further, unless otherwise stated the following abbreviations have the
indicated m~nin~:
DMF = N,N-dimethylformamide
DMSO = dimethyl sulfoxide
Et3N = triethyl~mine
MMMP = m~p;nesium monoperoxyphth~l~te
Ph = phenyl
r.t. = room temperature
2 THF = tetrahydrofuran
TLC = thin layer chromatography
Alkyl group abbreviations
Me = methyl
Et = ethyl
n-Pr = normal propyl
i-Pr = isopropyl
n-Bu = normal butyl
i-Bu = isobutyl
s-Bu = secondary butyl
t-Bu = tertiary butyl
Optical Isomers - Diastereomers - Geometric Isomers
Wa~9~;t2~731 ~ PCT/CA94/00264 ~
- 22 -
Some of the compounds described herein contain one or
more asymmetric centers and may thus give rise to diastereomers and
optical isomers. The present invention is meant to comprehend such
possible diastereomers as well as their racemic and resolved,
5 enantiomerically pure forms and ph~ ceutically acceptable salts
thereof.
Some of the compounds described herein contain olefinic
double bonds, and unless specified otherwise, are meant to include both
E and Z geometric isomers.
EXAMPLE 1 (Compound 1)
3-(4-Fluorophenyl)-4-(4-(methylsulfonyl)phenvl)thiophene
5 Step 1: 4'-(Methylthio)-2-chloroacetophenone
To a -5C solution of thioanisole (26.4 g) and chloroacetyl
chloride (27 g) in dichloromethane (600 mL) was added AIC13 (33.2 g)
portion-wise. The mixture was allowed to warm up to 25C and was
stirred for 16 h. It was poured over ice water and stirred for 1.~ h.
20 The mi~ul~e was extracted with CH2C12 (2 x 600 mL) and the
combined extracts were washed with brine and dried with MgSO4. The
solvent was removed in vacuo and the residue swished in 1:25 ethyl
acetate:hexanes. The solid was filtered and dried to yield 18 g of the
title compound.
25 lH NMR (CD3COCD3): ~ 2.55 (3H, s), 4.95 (2H, s), 7.35-8.0 (4H,
m).
Step 2: 4'-(Methylthio)-2-(acetylthio)acetophenone
To a 0C suspension of 4'-(methyl~io)-2-chloro-
30 acetophenone (18 g) from Step 1 in THF (20 mL) and DMF (180 mL)was added potassium thioacetate (11.6 g). The mixture was warmed to
25C and stirred 1.5 h. It was poured over ice/dilute NaHCO3. The
mixture was extracted with ethyl acetate:ether (2 x 200 mL) and the
combined extracts were washed with H20, brine and dried with
MgSO4. After removal of the solvents in vacuo, the residue was
¦~wo 94/26731 2 ~ G 17 8 9 PCT/CA94/00264
- 23 -
- swished in 1:25 ethyl acetate-hex~nes and the solid was filtered and
dried to yield 18 g of the title compound.
lH NMR (CD3COCD3): ~ 2.35 (3H, s), 2.55 (3H, s), 4.45 (2H, s),
7.35-7.95 (4H, m)-
Step 3: 4'-(Methylthio)phenacyl-4-fluorophenacyl sulfide
To a -5C solution of 4'-(methylthio)-2-(acetylthio)-
acetophenone (18 g) from Step 2 in THF (50 mL) and DMF (100 mL)
was added hydrazine (2.6 mL). After 0.5 h Cs2C03 (26 g) and 4-
fluorophenacylchloride (21.6 g) were added and the mixtllre was
poured over ice/dilute HCl. It was extracted with ethyl acetate (2 x 200
mL) and the combined extracts were washed with H20, brine and dried
with MgS04. After removal of the solvents the residue was purified by
chromatography to yield the title compound (12.76 g).
5 lH NMR (CD3COCD3): ~ 2.55 (3H, s), 4.05 (2H, s), 4.1 (2H, s), 7.2-
8.2 (8H, m).
Step 4: 3-(4-Fluorophenyl)-4-((4-methylthio)phenyl)thiophene
To a -35C solution of 4'-(methylthio)phenacyl-4-
20 fluorophenacyl sulfide (8.8 g) from Step 3 in THF (225 mL) was added
TiCl4 (23.71 g) dropwise and the mix~lre was stirred for 0.5 h. Zinc
powder (16.4 g) was added portionwise with vigourous stirring and the
dark green suspension was stirred for 1 h at -35C. The mixture was
transferred via a c~m-l~ to ice cold 1 M tartaric acid (1000 mL) and the
2s mixture was stirred for 1 h. It was extracted with ethyl acetate (3 x 200
mL) and the combined extracts were washed with brine and dried with
MgS04. After removal of the solvents the residue was dissolved in
toluene (50 mL) cont~inin~ p-TsOH (50 mg) and the mixture was
refluxed for 3 h. The solvent was removed and the residue purified by
30 chromatography to afford the title compound.
lH NMR (CD3COCD3): ~ 2.45 (3H, s), 7.0-7.5 (lOH, m).
21 ~1789
WO 94/26731 PCT/CA94/00264
- 24 -
Step 5: 3-(4-Fluorophenyl)-4-(4-(methylsulfonyl)phenyl)thiophene
To a 0C suspension of 3-(4-fluorophenyl)-4-((4-
methylthio)phenyl)thiophene (3 g) fr~om Step 4 in MeOH and CH2cl2
(50 mL) was added portionwise MMPP (6.8 g) and the mixtl-re was
allowed to react for 3 h while warming to 25C. It was diluted with
CH2C12 (50 mL) and filtered through celite. The filtrate was
concentrated to dryness and purified by chromatography to yield the
title compound (3.16 g).
lH NMR (CD3COCD3): ~ 3.05 (3H, s), 6.95-7.9 (lOH, m).
EXAMPLE 2 (Compound 2)
2-Nitro-3 -(4-fluorophenyl)-4-((4-methylsulfonyl)phenyl)thiophene
To a 0C suspension of 3-(4-fluorophenyl)-4-((4-
methylsulfonyl)phenyl) thiophene (1.32 g) from Example 1 in
nitromethane (10 mL) and acetic anhydride (10 mL) was added 70%
aqueous HN03 (320 ,uL). The cold bath was removed and the mixture
was stirred 2 h at 25C. It was then poured over ice H20 and extracted
with ethyl acetate (3 x 25 mL). The combined extracts were washed
with brine, dried with MgS04 and the solvents were removed in vacuo.
The residue was purified by chromatography to afford the title
compound (858 mg).
Analysis calculated for Cl7Hl2FNo4s2:
C, 54.10; H, 3.21; N, 3.71.
2s Found: C, 53.77; H, 3.39; N, 3.62.
EXAMPLE 3 (Compound 3)
2-Bromo-3-(4-fluorophenyl)-4-(f4-methylsulfonyl)phenyl)thiophene
To a 0C solution of 3-(4-fluorophenyl)-4-((4-methyl-
sulfonyl)phenyl)thiophene (332 mg) from Example 1 in CH2Cl2 (2 mL)
and acetic acid (2 mL) was added a 1 M solution of Br2 in CCl4 (1.1
mL) and the mixtllre was reacted for 2 h at 0C. The mi~ re was then
concentrated to dryness and the residue purified by chromatography to
afford the title compound (203 mg).
WO 94/26731 PCT/CA94/00264
2161789
- 25 -
- Analysis calculated for cl7Hl2BrFs2o2:
C, 49.65; H, 2.94.
Found: C, 49.83; H, 2.89.
EXAMPLE 4 (Compound 4)
3 -(4-Fluorophenyl)-4-(4-sulfamoylphenyl)thiophene
Step 1: 3-(4-Fluorophenyl)-4-(4-bromophenyl)thiophene
Following the procedures of Example 1, Steps 2-7, but
replacing 4'-(methyl~io)-2-chloroacetophenone by 4-bromophenyl
bromide in Step 2, there was obtained the title compound.
1H NMR (CD3COCD3): o 7.0-7.3 (4H, m), 7.4-7.6 (6H, m)
5 Step 2: 3-(4-Fluorophenyl)-4-(4-sulfamoylphenyl)thiophene
To a -78C solution of 3-(4-fluorophenyl)-4-(4-
bromophenyl)thiophene (999 mg) in tetrahydrofuran (10 mL) was
added 2.27 M n-BuLi (1.45 mL) and the mixtllre was stirred for 0.5 h
at -78C. It was then transferred dropwise into a -78C solution of
20 sulfur dioxide (10 mL) and tetrahydrofuran (10 r~T ) and the mi~ture
was allowed to warm slowly to 25C. The solvents were removed in
vacuo and the solid obtained was suspended in hexanes (15 mL) and the
suspension was cooled to 0C. A 1 M dichloromethane solution of
sulfuryl chloride (3 mL) was then added dropwise and the mixh-re was
2S stirred for 0.5 h at 25C. It was cooled again at 0C and filtered. The
solid obtained was taken into tetrahydrofuran and the mixture was
cooled to 0C before NH3 was bubbled in for a few minlltes. Removal
of the solvents followed by purification on silica gel yielded the title
compound (350 mg).
1H NMR (CD3COCD3): ~ 6.5-6.6 (2H, broad), 7.0-7.4 (6 H, m), 7.55
(l H, d), 7.65 (lH, d), 7.75-7.8 (2H, d).