Note: Descriptions are shown in the official language in which they were submitted.
2~2~3
WO 94/26725 PCT/I;R94/00533
1
NOUV~UX D~RIV~ DU T~upAN~ T~T`uR PRT'PARATION F~'T T.T`~ COMPOSITIONS
PHARMA~T~`UTIOUT'~ OUI T.T~'~ CONTIT~`NNT'NT
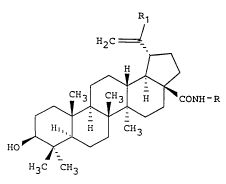
La présente invention concerne des dérivés du lupane, de formule
générale :
H2C
CH3 ~ CONH-R
(I)
. Hl, H CH3
HO
H3C CH3
leurs sels, leur préparation et les compositions pharmaceutiques qui
les contiennent.
Dans la ~Pm~n~ de brevet japonais J 01 143 832 ont été décrits des
dérivés de la bétuline de formule générale :
H 2 C =l"
`H2--R2
R1 3
H3C CH3
dans laquelle R1 et R2 sont hydroxy ou acyloxy et R3 est notamment
méthyle. Ces dérivés sont utiles dans le ~o~-ne des anticancéreux.
Il a été maintenant trouvé que les dérivés du lupane de formule géné-
rale ~I) dans laquelle :
R représente un radical de formule générale :
W O 94/26725 PCT~R94/00533
2~627~3 2
N
(CH2)n - X - (CH2)m - Y (C)p ~ / ll (II)
R R N
X est une liaison ou représente un radical carbamoyle, N-méthyl
carbamoyle, aminocarbonyle ou N-méthylaminocarbonyle,
Y est une liaison ou représente un radical méta ou para phénylène,
R et R identiques ou différents sont des atomes d'hydrogène ou des
radicaux méthyle (étant entendu que tous les motifs -CRR ne sont
pas nécessairement identiques entre eux) et,
n est un nombre entier de 6 à 12, m est un nombre entier de 0 à 3 et
p est un nombre entier de 0 a 2 étant entendu que m + n + p est com-
pris entre 6 et 14, et
R1 est un radical méthyle, hydroxyméthyle ou un radical -CH2OR' ou
-CH2SR' pour lequel R' est alcoyle ou hydroxyalcoyle,
ainsi que leurs sels pharmaceutiquement acceptables, manifestent un
effet cytoprotecteur de cellules infectées par un virus HIV (Human
Immunodeficiency Virus) ainsi qu'une activité inhibitrice de la
production de transcriptase inverse d'un virus HIV.
Dans la formule générale ci-dessus, il est entendu que les radicaux
alcoyle sont droits ou ramifiés et contiennent 1 à 4 atomes de car-
bone.
Dans les cas où R et R sont différents, les produits de formule
générale (I) présentent des formes stéréoisomères, il est également
entendu que les formes stéréoisomeres ainsi que leurs mélanges en-
trent aussi dans le cadre de la présente invention.
Selon l'invention, les dérivés du lupane de formule générale (I) peu-
vent également être préparés par action d'un azoture sur un nitrile
de formule générale :
j~) 94/26725 ~16 2 ~ ~ 3 PCT/FR94/00533
R1
H2C I
~ ~ ~ CoNH-(cH2\n-X-(CH2)m-Y- (C)p-CN
¦ H~, H CH3 R R
HO ~
H3C CH3 (III)
dans laquelle R1 R, R, X, Y, n, m, et p sont définis comme
précé~^m~^nt.
L'azoture est avantageusement choisi parmi l'azoture de tri
n.butylétain, l'azoture de triméthylsilyle ou un azoture alcalin.
Lorsque l'on utilise l'azoture de tri n.butylétain la réaction
s'effectue dans un solvant organique tel que le diméthoxy-1,2 éthane,
l'éthoxy-2 éthanol ou un mélange de tels solvants, a la température
de reflux du mélange réactionnel. Lorsque l'on fait agir l'azoture de
triméthylsilyle, on opère en présence de trichlorure d'aluminium,
dans un solvant chloré comme par exemple le dichlorométhane, à une
température comprise entre 10 et 40C.
Selon l'invention le dérivé du lupane de formule générale (I) pour
lequel X (dans le radical R) représente un radical carbamoyle ou
aminocarbonyle éventuellement N-méthylé et les autres radicaux sont
définis comme précé~^mm~nt, peut être également obtenu à partir d'un
dérivé du lupane de formule générale :
R1
H2C I
CH3 ~ CONH-R (IV)
¦ Hl, H CH3
HO
H3C CH3
W O 94/26725 PCT~R94/00533
~1627~3
dans laquelle R1 est défini comme pr~cé~ -rt et R" représente un
radical de formule générale :
(CH2)n - COOH (IVa)
ou
(CH2)n-NH-R"' (IVb)
dans lesquelles n est défini comme précé~f ~rt et R"' est un atome
d'hydroqène ou un radical méthyle, par action respectivement d'une
amine ou d'un acide de formules générales :
N ~ N
R"'-HN - (CH2)m - Y /C)P ~ N ~N (V a)
R R H
ou
N - N
HO-CO - (CH2)m - Y (C)p ~ ~ N (V b)
R R H
dans lesquelles R, R, m et p sont définis comme précé~Pmm~nt et
R"' est défini comme ci-dessus.
Il est entendu que les radicaux hydroxy du dérivé du lupane de for-
mule générale (IV) sont de préférence protégés par des radicaux
compatibles qui peuvent être mis en place et fli mi n~S sans toucher au
reste de la molécule. A titre d'exemple les groupements protecteurs
peuvent être choisis parmi les radicaux décrits par T.W. GREENE,
Protective Groups in Organic Synthesis, J. WILEY-Interscience
Publication (1991) ou par Mc OMIE, Protective Groups in Organic
Chemistry, Plenum Press (1973). On choisira des radicaux pouvant être
~limin~5 en milieu neutre, basique ou acide.
Notamment, les radicaux hydroxy peuvent être protégés ~ l'état d'es-
ter (formyloxy, acétoxy, i.butoxy, trichloracétyloxy, phénoxyacétyl-
oxy, benzyloxy)i dans ce cas, l'~liminAtion s'effectue par hydrolyse
en milieu basique, notamment en présence de soude à une température
comprise entre 10 et 50C. La protection peut également être effec-
tuée ~ l'état de cétone, sous forme de carbonate par un radical
wo 94,26725 2 I 6 ~ 7 0 ~ PCT~R94/00533
-COORa dans lequel Ra est un radical alcoyle ou benzyle éventuelle-
ment substitués ou encore par un radical trialcoylsilyle.
La réaction s'effectue selon les méthodes habituelles de condensation
d'une amine sur un acide. Notamment, on opère en présence d'un accep-
teur d'acide tel qu'une base organique azotée (trialcoylamine, pyri-
dine, N-méthyl-morpholine, diaza-1,8 bicyclo[5.4.0]11n~cène-7, diaza-
1,5 bicyclo[4.3.0] nonène-5 par exemple) dans un solvant organique
comme un solvant chloré (dichlorométhane, dichloréthane, chloroforme
par exemple) ou un amide (diméthylformamide par exemple). Il est éga-
lement possible d'opérer en présence d'un agent de condensation telqu'un carbodiimide (dicyclohexylcarbodiimide, chlorhydrate de 1-(3-
diméthylaminopropyl)-3-éthylcarbodiimide par exemple) et éventuelle-
ment en présence d' Ul catalyseur tel que le N-hydroxybenzotriazole ou
le N-hydroxysuccinimide, à une température comprise entre -20 et
40C.
La condensation de l'amine de formule générale (Va) s'effectue de
préférence à partir du chlorhydrate de l'amine.
Le nitrile de formule générale (III) peut être préparé par
deshydratation de l'amide correspondant, par toute méthode connue qui
n'altère pas le reste de la molécule.
On opère notamment au moyen d'anhydride trifluoracétique dans un
éther (tétrahydrofuranne par exemple), à une température comprise
entre 10 et 66C
L'amide correspondant au nitrile de formule générale (III) peut être
lui-même obtenu à partir de l'acide selon les méthodes connues et/ou
citées ci-après dans les exemples.
L'acide correspondant peut être préparé par action d'un ~tno~cide de
formule générale :
H2N - (cH2)n-x-(cH2)m-y-~c~p-cooH (VI)
- R R
W O 94/26725 PCT~R94/00533
~lS2703 6
dans laquelle R, R, X, Y, n, m, et p sont définis comme précédem-
ment et dont la fonction acide est pr~AlAhl~ -nt protégée, sur le
chlorure de l'acide de formule générale :
R1
H2
CH3 ~ COOH (VII)
~ Hl, H CH3
HO
H3C CH3
dans laquelle Rl est défini comme précé~ t et dont les radicaux
hydroxy sont préalablement protégés, suivie de l'~1imin~tion du/des
radicaux protecteurs.
La réaction de l'amine sur le chlorure de l'acide lupèn-20(29)oïque-
28 de formule générale (VII) s'effectue selon les méthodes habi-
tuelles qui n'altèrent pas le reste de la molécule. On opère notam-
ment en présence d'une base organique azotée telle qu'une trialcoyla-
mine (triéthylamine par exemple) dans un solvant organique tel qu'un
solvant chloré (chloroforme, dichloro-1,2 éthane, dichlorométhane) ou
dans le tétrahydrofurane ou dans un mélange de ces solvants, à une
température comprise entre 15 et 30C.
Il est entendu que le/les radicaux hydroxy sont de préférence
préalablement protégés. La protection et la libération des radicaux
protecteurs s'effectuent comme décrit préc~ rt.
A titre d'exemple, les radicaux protecteurs d'acide peuvent être
choisis parmi les radicaux alcoyle (méthyle, éthyle, t.butyle), al-
coyle substitué (trichloréthyle, haloéthyle, p.toluènesulfonyl-
éthyle, benzyle, benzyle substitué par un radical nitro, benzhydryle,
triphénylméthyle, benzyloxyméthyle...), alcoyloxyalcoyle (méthoxymé-
thyle), tétrahydropyranyle ou triméthylsilyle. La mise en place et
l'él~minAtion de ces radicaux s'effectue selon les méthodes
habituelles citées précedemment.
~o 94,26725 ~ ~ 6 ~ PCT~R94/00533
Lorsque le radical X (dans R) est un radical carbamoyle ou
inoc~rbonyle éventuellement N-méthylé, le nitrile de formule géné-
rale (III) peut aussi être préparé par condensation d'une amine ou
d'un acide de formule générale :
R"'-NH - (CH2)m~Y- (C~p - CN tVIIa)
R R
ou
HO-CO - (CH2)m-Y- (C)p - CN (VIIb)
R R
dans lesquelles R"', R, R, Y, m, et p sont définis comme précédem-
ment, sur un dérivé du lupane de formule générale (IVa) ou (IVb).
La réaction s'effectue dans des conditions identiques à celles de la
préparation d'un dérivé du lupane de formule générale (I) ~ partir
d'une amine ou d'un acide de formule générale (Va) ou (Vb) et d'un
dérivé du lupane de formule générale (IV).
L'acide dérivé du lupane, de formule générale (IVa) peut être préparé
par analogie avec la réaction de l'Amino~cide de formule générale
(VI) sur le chlorure de l'acide dérivé du lupane de formule générale
(VII), par action d'un aminoacide de form,ule générale :
H2N~(CH2)n~CH (VIII)
dans laquelle n est défini comme précé~ ~nt et dont la fonction
acide est préalablement protégée.
L'amine dérivée du lupane, de formule générale (IVb), pour laquelle
R''' est un atome d'hydrogène, peut être préparée a partir d'un
dérivé du lupane de formule générale (VII), par action d'une ~i~ine
de formule générale :
H2N-(cH2)n-NH2 (IX)
dans laquelle n est défini comme préc~e cnt~
Les dérivés (IVb) pour lesquels R''' est un radical méthyle peuvent
être obtenus par N-méthylation selon la méthode décrite par
W O 94/26725 21~ 2 7 ~ 3 PCT~Rg4/00533
A. KATRITSKY, J. Chem. Soc. Perkin Trans. I, 2539 (1987), à partir du
produit de formule générale (IVb) pour lequel R''' est un atome
d'hydrogène.
Les produits de formule générale (Va) ou (Vb) peuvent être préparés
selon ou par analogie avec les méthodes citées ou décrites ci-après
dans les exemples.
Les dérivés du lupane de formule générale (IV) ou (VII) pour lesquels
Rl est un radical hydroxyméthyle peuvent être préparés par action de
l'acétate d'argent sur le dérivé bromé correspondant pour lequel R1
est bromométhyle et pour lequel, le radical hydroxy est préalablement
protégé. La réaction s'effectue avantageusement dans un solvant orga-
nique comme par exemple le toluène, à une température comprise entre
20C et la température de reflux du mélange réactionnel. Le dérivé
bromé de départ peut être obtenu par action d'un agent de bromation
comme par exemple le tribromure de tétrabutyl~mm~r--um ou le N-
bromosuccinimide sur le dérivé correspondant de l'acide lup-20(29)-
èn-28-oïque. La réaction s'effectue dans un solvant chloré comme le
chlorure de méthylène, le chloroforme, ou le tétrachlorure de carbone
à une température voisine de 25C.
Les dérivés du lupane de formule générale (VII) pour lesquels R1 est
un radical -CH2OR' ou -CH2SR' peuvent être préparés à partir du dé-
rivé du lupane pour lequel R1 est bromométhyle, par action de
l'alcoolate ou du thiolate correspondant.
Le chlorure d'acide du dérivé du lupane de formule générale (VII)
peut être préparé selon les méthodes connu~s A titre d'exemple, on
opère par action du chlorure d'oxalyle ou du chlorure de thionyle,
dans un solvant chloré comme le chloroforme, le dichloroéthane ou le
dichlorométhane
Les nouveaux dérivés du lupane de formule générale (I) peuvent être
purifiés le cas échéant par des méthodes physiques telles que la
cristallisation ou la chromatographie.
Lorsque le dérivé du lupane de formule générale (I) présente des
formes stéréoisomères, la préparation s'effectue au moyen de d~rivés
O 94/26725 2 1 ~ 2 ~ ~ ~ PCT~R94/00533
chiraux de formule générale (Va), (Vb), (VI), (VIIa) ou (VIIb). Il
est entendu que lorsque l'on veut obtenir un intermédiaire chiral, la
séparation s'effectue selon les méthodes habituelles qui n'affectent
pas le reste de la molécule, telles que par exemple la chromatogra-
phie sur phase chirale, ou par conversion du l~]~nge racémique endiastéréoisomères et séparation par cristallisation ou chromatogra-
phie.
Les produits selon l'invention peuvent être transformés en sels mé-
talliques avec les bases fortes selon les méthodes connues en soi.
Ces sels peuvent être obtenus par action d'une base métallique dans
un solvant approprié. Le sel formé précipite après concentration
éventuelle de la solution ; il est séparé par filtration.
Comme exemples de sels pharmaceutiquement acceptables peuvent être
cités les sels avec les métaux alcalins (sodium, potassium, lithium).
Les nouveaux dérivés du lupane selon la présente invention sont
particulièrement utiles pour la prophylaxie et le traitement du SIDA
(syndrome d'immunodéficience acquise) et de syndromes associés [ARC
(AIDS related complex)]. Par prophylaxie nous sous-entendons le
traitement des sujets qui ont été exposés aux virus HIVs, en particu-
lier les séropositifs asymptomatiques ,qui présentent le risque dedévelopper la maladie dans les mois ou les années à venir après la
primoinfection.
Les produits selon l'invention, qui sont inhibiteurs de l'effet
cytopathogène du HIV et inhibiteurs de la production de transcriptase
inverse en culture cellulaire à des concentrations dépourvues d'effet
cytotoxique ou cytostatique, sont particulièrement intéressants.
Il a été également démontré que les produits selon l'invention sont
actifs pour le traitement des affections dans lesquelles
interviennent des virus de la famille des herpès.
La famille des herpès est en effet à l'origine de nombreuses
affections dont certaines peuvent être très graves. Elle comprend
l'Herpès simplex, le varicella-zoster, le cytomegalovirus et le virus
d'Epstein-Barr. L'Herpès simplex peut varier de formes bénignes comme
W O 94/26725 PCT~R94/00533 ~
2~9~7~3 ~o
l'herpès labial aux formes plus sérieuses comme l'herpès génital et
peut même être responsable d'encéphalites mettant la vie du patient
en danger. Le varicella zoster est le virus responsable de la
varicelle et du zona, il peut également être à l'origine d'affections
plus graves parmi lesquelles des encéphalites. Les infections à
cytomégalovirus sont en général asymptomatiques chez les sujets
sains, mais peuvent devenir la cause d'infections ophtalmiques graves
(retinites) pouvant conduire à la cécité, de morbidité et de
mortalité chez des sujets immunodéprimés (m~ es atteints du SIDA ou
de toute autre immunodéficience, par exemple après transplantation
d'organes ou après chimiothérapie anticancéreuse). Le cytomégalovirus
est également responsable de manifestations cliniques sévères pour le
foetus ou le nouveau-né dans le cas d'une primo-infection pendant la
grossesse ou lors de la transfusion de sang séropositif à un nouveau-
né séronégatif.
Les activités ont été mises en évidence dans les tests suivants :
Activité vis-à-vis de l'effet cytopatho~ène du vin ~ HIV
Les produits en poudre ont été mis en solution à raison de 2 mg de
produit par 2 ml (environ 4 x 10 M) dans du diméthylformamide (DMF)
pour obtenir une solution stock de produit à 100 % de DMF. Le test
est réalisé sur la lignée lymphoh1~toïde CEM 4. Dans une microplaque
de 96 puits on dépose 25 ~l/puits d'une solution de produit à tester
dans du tampon phosphate isotonique (TPI) ou de TPI seul dans le cas
des contrôles. Les produits sont étudiés à 8 concentrations, à raison
de 6 puits par concentration. On ajoute alors 125 ~l d'une suspension
de cellules C~M (8 x 10 cellules par ml) dans le milieu RPMI
contenant 10 % de sérum de veau foetal, 100 UI/ml de pénicilline, 100
~g/ml de streptomycine et 2 ~moles/ml de glutamine et les
microplaques sont incubées une heure à 37C, sous une atmosphère
contenant 5 % de gaz carbonique. Pour chaque concentration, l'essai
est partagé en deux parties : une partie (3 puits) sur cellules
infectées, pour la détermination de l'activité antivirale et l'autre
partie (3 puits) sur cellules non infectées, pour déterminer la
cytotoxicité des produits. On infecte alors la première série avec
HIV-1 (100 ~l par puits d'une suspension de virus LAV-1-BRU contenant
~ 0 94/2672~ 21 6 ~ ~ ~ 3 PCT~R94/00533
1 1
200-300 TCID50) tandis que l'autre s~rie reçoit 100 ~l de milieu RPMI
tel que défini préc~m~nt. Au bout de 5 jours d'incubation, 100 ~l
de cellules sont prélevés pour mesurer la viabilité cellulaire
[déterminée selon une modification de la technique décrite par R.
Pauwels et coll., J. Virol. Meth., ~Q, 309-321 (1988)]. On ajoute à
ce prélèvement 10 ~l d'une solution contenant 7 mg de M~T [bromure de
3-(4,5-diméthyl 2-thiazolyl)-2,5-diphényltétrazolium] par ml de
tampon phosphate isotonique. Après 3 heures d'incubation à 37C le
surnageant est enlevé. Le MTT est converti en un sel de formazan
(bleu) uniquement à l'intérieur des cellules vivantes. On ajoute
alors 100 ~l d'isopropanol (contenant 0,04 mole/l d'acide
chlorhydrique) et les microplaques sont agitées jusqu'à la
solubilisation du bleu de formazan. L'absorbance à 540 nm est lue
avec un lecteur automatique de réactions ELISA en microplaques. Cette
absorbance est proportionnelle à la quantité de cellules vivantes.
Le taux de protection (en ~) d'un produit donné est déterminé à par-
tir des densités optiques (DO) par la formule :
DO~cell. traitées et inf.)-DO(cell. non traitées et inf.)
DO(cell. traitées et non inf.)-DO(cell. non traitées et inf.)
Le cas échéant la concentration inhibitrice 50 % est déterminée.
Les résultats démontrent que pour des concentrations de produit testé
comprises entre 0,01 et 10 ~g/ml, on obtient une ~ nution signifi-
cative de l'effet cytopathogène.
Le taux de protection procuré par les produits selon l'invention est
25 compris entre 20 et 100 %
La concentration inhibitrice 50 ~ des produits selon l'invention est
comprise entre 0,05 et 50 ~g/ml lorsqu'elle peut etre déterminée.
D~ter~in~tion ~ la multiplication virale par ~os~e ~e la tran~criD-
tase inverse du virns HIV
L'activité de la transcriptase inverse est mesurée dans 10 ~l de
surnageant de culture A l'aide du kit de dosage poly r(A) reverse
transcriptase [3H]-SPA enzyme assay (RPQN 0100, Amersham, GB) selon
W O 94/26725 2 ~ 6 ~ q 0 3 12 PCT~R94/00533
la procédure préconisée par le fabricant. Le temps de réaction est de
6 heures et la radioactivité comptée à l'aide d'un compteur microbeta
tTOPCOUNT ~ PACKARD].
On observe des inhibitions de la production de transcriptase inverse
associée au virus, comprises entre 20 et 100 % aux concentrations
testées (0,01 à 10 ~g/ml).
Action sur les virus de la fAmille des her~ès :
L'action des dérivés de formule qénérale (I) sur les virus de la
famille des herpès a été mise en évidence dans la technique décrite
par NEYTS et coll., Virology, 179, 41-50 (1990).
La technique employée consiste dans la mesure de l'effet
cytopathogène du virus et de sa protection par utilisation des
produits de formule générale (I). L'activité antivirale est appréciée
par la mesure de la CE50 (concentration nécessaire pour inhiber 50
de l'effet cytopathogène induit par le virus).
Les résultats obtenus dans cette technique sont donnés ci-après :
L'activité sur cytomégalovirus a été étudiée sur les souches Davis et
DER 648 (souche résistante au Ganciclovir) :
, Souche Davis DER 648
N de l exemple
CE50 (~g/ml) CE50 (~g/ml)
1 0,86 0,57
0,8 0,76
13 sup. 2 sup.0,5
14 sup. 2 2
Tableau I
L'activité sur varicella zoster a été étudiée sur la souche
résistante thymidine-kinase négative (TK-) YS/R :
O 94/26725 ~ 1 6 2 7 0 ~ PCT~R94/00533
13
N de l'exemple Souche YS/R
CE50 (~g~ml)
1 sup. 2
13 sup. 2
14 4
Tableau II
Les exemples suivants donnés à titre non limitatif illustrent la pré-
sente invention.
~mD le 1
A une solution de 0,5 g d'acide N-(3~-acétoxylup-20(29)-èn-28-oyl)-8-
aminooctanoïque dans 90 cm de dichlorométhane, on a~oute 125 mg de
chlorhydrate de 5-~min~thyl-tétrazole, 141 mg d'hydrate de
1-hydroxybenzotriazole, 380 mq de chlorhydrate de 1-(3-diméthylamino-
propyl)-3-éthylcarbodiimide et 0,48 cm de triethy1~in~. La solution
est agitée pendant 24 heures à 20C puis le mélange réactionnel est
lavé 5 fois avec 100 cm d'eau distillée. La phase organique est en-
suite séchée sur du sulfate de magnesium et evaporee à sec sous pres-
sion réduite (2,7 kPa) à une temperature voisine de 40C. Le residu
obtenu (600 mg) est chromatographié sur une colonne de silice (0,02-
15 0,045 mm) éluée d'abord avec 2 litres d'un ~ nge de cycloh~Y~ne et
d'acétate d'éthyle (30-70 en volumes), puis avec 2 litres d'un mé-
lange de méthanol et d'acétate d'éthyle (30-70 en volumes) ; les
fractions contenant le produit sont réunies et concentrées sous
pression réduite (2,7 kPa) à une température voisine de 40C. Le
résidu est repris avec 10 cm d'éther éthylique, filtré, lavé 2 fois
avec 5 cm d'éther éthylique et séché sous pression réduite (13,5 Pa)
a 20C. On obtient ainsi 290 mg de 5-{N'-[N-(3~-acétoxylup-20(29)-~n-
W O 94/26725 ~ 7 ~3 PCT~Rg4/00533
28-oyl)-8-aminooctanoyl]-Am;n~m~thyl}-tétrazole sous la forme d'une
poudre hlAnch~ (Rf = 0,26 ; chromatographie sur couche mince de
silice ; éluant : chloroforme-méthanol-A~m~n-Aque à 20 % (12-3-0,5 en
volumes)).
Une solution de 1,07 g de 5-{N'-[N-(3~-acétoxylup-20(29)-èn-28-oyl)-
8-aminooctanoyl]-~minométhyl}-tétrazole obtenu comme décrit ci-des-
sus, 5,7 cm de soude 5N, 18,4 cm de tétrahydrofurane et 36,9 cm3 de
méthanol est agitée 20 heures à une température voisine de 20C. Le
mélange est refroidi à 5C, puis acidifié avec de l'acide chlorhy-
drique 5N, et dilué avec 100 cm d'eau distillée. Après 1 heure sous
agitation à 5C, le solide obtenu est séparé par filtration, lavé 5
fois avec 25 cm d'eau distillée et séché sous pression réduite
(15,5 Pa) à une température voisine de 20C. On obtient ainsi 880 mg
de 5-{N'-[N-(3~-hydroxylup-20(29)-èn-28-oyl)-8-aminooctanoyl]-amino-
méthyl}-tétrazole, sous la forme d'un solide blanc fondant vers
154C
L'acide N-(3~-acétoxylup-20(29)-èn-28-oyl)-8-aminooctanoïque peut
être préparé de la manière suivante :
Après avoir chauffé au reflux pendant 3 heures un mélange de 330 mg
d'acide 8-aminooctanoïque et 410 mg de chlorotriméthylsilane dans
50 cm de dichlorométhane, la solution est refroidie vers 20C et on
ajoute 1 g de chlorure de 3~-acétoxylup-20(29)-èn-28-oyle (préparé à
partir d'acide 3~-acétoxylup-20(29)-èn-28-oïque) puis 0,81 cm3 de
triéthylamine. L'agitation est maintenue pendant 15 heures a une tem-
pérature voisine de 20C et le solvant est évaporé à sec sous pres-
sion réduite (2,7 kPa) à une température voisine de 35C. Le résidu
est chromatographié sur une colonne de silice (0,02-0,045 mm) éluée
avec un mélange de cyclohexane et d'acétate d'éthyle (60-40 en vo-
lumes). Les fractions contenant le produit sont rass~ hl~ et
concentrées à sec sous pression réduite (2,7 kPa) à une température
voisine de 35C. On obtient ainsi 1,1 g d'acide N-(3~-acétoxylup-
20(29)-èn-28-oyl)-8-aminooctanoïque sous la forme d'un meringue
hlAnche (Rf - 0,4 ; chromatographie sur couche mince de silice ;
éluant : cyclohexane, acétate d'éthyle (60-40 en volumes)).
O 94/26725 ~ 1 S 2 ~ ~ 3 PCT~R94/00~33
Le chlorhydrate de 5-~min~m~thyl-tétrazole peut être préparé selon la
méthode décrite dans la ~m~n~ de brevet DE 3 626 130.
F~ mnle 2
A une solution de 640 mg d'acide N-(3~-acétoxylup-20(29)-èn-28-oyl)-
8-aminooctanoïque dans 50 cm de chloroforme, on ajoute 134 mg de
5-amino-lH-tétrazole, 168 mg d'hydrate de 1-hydroxybenzotriazole,
50 cm3 de diméthylformamide, 692 mg de chlorhydrate de 1-(3-diméthy-
laminopropyl)-3-éthylcarbodiimide, et 0,70 cm de triéthylamine en
solution dans 5 cm de chloroforme. La solution est agitée pendant 2
10 jours à 20C puis le mélange réactionnel est lavé 3 fois avec 50 cm3
d'acide chlorhydrique lN, puis 5 fois avec 100 cm d'eau distillée.
La phase organlque est séchée sur du sulfate de magnésium et évaporée
à sec sous pression réduite (2,7 kPa) à une température voisine de
40C. Le résidu obtenu (730 mg) est chromatographié sur une colonne
15 de silice (0,02-0,045 mm) éluée avec 800 cm d'acétate d'éthyle puis
avec 800 cm d'un mélange de dichlorométhane et de méthanol (95-5 en
volumes). Les fractions contenant le produit sont réunies et concen-
trées sous pression réduite (2,7 kPa) à une température voisine de
40C. Le résidu est repris dans 15 cm d'oxyde d'isopropyle, filtré,
20 séché sous pression réduite (13,5 Pa) à 20C. On obtient ainsi 265 mg
de 5-{N'-[N-(3~-acétoxylup-20(29)-èn-28-oyl)-8-aminooctanoyl]-amino}-
tétrazole sous la forme d'une poudre beige (Rf 0,40 ; chromatogra-
phie sur couche mince de silice ; éluant : chloroforme-méthanol-ammo-
niaque à 20 % (12-3-0,5 en volumes)).
25 Une solution de 260 mg de 5-{N'-[N-(3~-acétoxylup-20(29)-èn-28-oyl)-
8-aminooctanoyl]-amino}-tétrazole, 0,92 cm3 d'hydroxyde de li-
thium 1N, 2,5 cm3 de tétrahydrofurane et 5 cm de méthanol est agitée
20 heures à une température voisine de 20C. Le mélange est acidifié
avec de l'acide chlorhydrique lN, et dilué avec 80 cm d'eau distil-
lée. On extrait ensuite le mélange par 100 cm d'acétate d'éthyle
puis 2 fois par 25 cm d'acétate d'éthyle. Les phases organiques sont
rassemblées, lavées 4 fois par 40 cm d'eau distillée, séchées sur du
sulfate de magnésium et évaporées ~ sec sous pression réduite
(2,7 kPa) à une température voisine de 45C. Le résidu obtenu
35 (170 mg) est chromatographié sur une colonne de silice (0,02-
W O 94/26725 PCT~R94/00533
~27~3 16
0,045 mm) eluée avec un melange de chloroforme, methanol et
A -ni~que à 20 % t12-3-0,5 en volumes). Les fractions contenant le
produit sont reunies et concentrées sous pression réduite (2,7 kPa) à
une temperature voisine de 40C. Le residu est repris par 5 cm
d'oxyde d'isopropyle, filtre, lave 2 fois par 1 cm d'oxyde
d'isopropyle et séche sous pression réduite (15,5 Pa) à 20C. On
obtient ainsi 80 mg de 5-{N'-[N-(3~-hydroxylup-20(29)-en-28-oyl)-8-
aminooctanoyl]-amino}-tétrazole, sous la forme d'un solide blanc
fondant vers 165-170C.
~mnle 3
A une solutlon de 100 mg d'acide N-(3~,30-diacétoxylup-20(29)-èn-28-
oyl)-8-aminooctanoïque dans 50 cm de dichlorométhane, on ajoute
22 mg de chlorhydrate de 5-~min~ thyl-tétrazole, 25 mg d'hydrate de
1-hydroxybenzotriazole, 70 mg de chlorhydrate de 1-(3-diméthylamino-
propyl)-3-éthylcarbodiimide et 0,09 cm de triéthylamine. La solution
est agitee pendant 24 heures à 20C puis le mélange réactionnel est
lavé successivement 3 fois avec 50 cm d'eau distillee, 2 fois avec
25 cm d'acide chlorhydrique 1N, puis 3 fois avec 50 cm d'eau
distillee . La phase organique est ensuite sechee sur du sulfate de
magnesium et evaporee à sec sous pression reduite (2,7 kPa) à une
temperature voisine de 40C. Le residu obtenu (110 mg) est chromato-
graphie sur une colonne de silice (0,02-0,045 mm) eluée avec un mé-
lange de chloroforme, methanol et Ammnni~que a 20 % (12-3-0,5 en vo-
lumes). Les fractions contenant le produit sont reunies et concen-
trees sous pression reduite (2,7 kPa) à une temperature voisine de40C. On obtient ainsi 65 mg de 5-{N'-[N-(3~,30-diacetoxylup-20(29)-
èn-28-oyl)-8-aminooctanoyl]-Amin~thyl}-tetrazole sous la forme
d'une meringue blanche (Rf = 0,40 ; chromatographie sur couche mince
de silice ; eluant : chloroforme-méthanol-~ ;~que à 20 % (12-3-0,5
en volumes)).
Une solution de 160 mg de 5-{N'-[N-(3~,30-diacétoxylup-20(29)-èn-28-
oyl)-8-aminooctanoyl~-~min~m~thyl}-tetrazole, 1,3 cm de soude 5N,
6 cm de tetrahydrofurane et 3 cm de methanol est agitee 20 heures à
une temperature voisine de 20C. Le melange est refroidi à 5C, puis
acidifie avec de l'acide chlorhydrique 5N, et dilué avec 30 cm3 d'eau
~ 0 94/2672~ 21 6 2 7 0 3 PCT~R94/00533
17
distillée Après 1 heure sous agitation à 5C, on ajoute 150 cm
d'eau distillée et le mélange est extrait 5 fois par 50 cm d'un mé-
lange de dichlorométhane et de méthanol (9-1 en volumes~. Les phases
organiques sont rassemblées, séchées sur du sulfate de maqnésium et
évaporées à sec sous pression réduite (2,7 kPa) à une température
voisine de 40C Le résidu obtenu (140 mg) est chromatographié sur
une colonne de silice (0,02-0,045mm) éluée avec un ~el~nqe de chloro-
forme, méthanol et ~m~Q~i~que à 20 % (12-3-0,5 en volumes). Les frac-
tions contenant le produit sont réunies et concentrées sous pression
réduite (2,7 kPa) à une température voisine de 40C. Le résidu est
repris par 2 cm d'acétonitrile, filtré et séché sous pression ré-
duite (15,5 Pa) à 20C. On obtient ainsi 48 mg de 5-{N'-[N-(3~,30-di-
hydroxylup-20(29)-èn-28-oyl)-8-aminooctanoyl]-aminométhyl}-tétrazole,
sous la forme d'un solide blanc fondant vers 145-150C
15 L'acide N-(3~,30-diacétoxylup-20(29)-èn-28-oyl)-8-aminooctanoique est
synthétisé par analogie avec l'acide N-(3~-acétoxylup-20(29)-èn-28-
oyl)-8-aminooctanoïque à partir de chlorure de 3~,30-diacétoxylup-
20(29)-èn-28-oyle et de 8-aminooctanoate de méthyle (R = 95 %; Rf =
0,34 ; chromatographie sur couche mince de silice ; éluant : cyclo-
hexane-acétate d'éthyle (50-50 en volumes)).
Le chlorure de 3~,30-diacétoxylup-20(29)-èn-28-oyle est préparé de la
manière suivante :
A une solution de 560 mg d'acide 3~,30-diacétoxylup-20(29)-èn-28-
oïque dans 25 cm de dichlorométhane, on ajoute 0,22 cm de chlorure
d'oxalyle. Après 15 heures d'agitation à une température voisine de
20C, le solvant est évaporé sous pression réduite (22 kPa) et à une
température voisine de 40C On obtient 600 mg d'une meringue hlAn~he
qui est utilisée sans autre purification.
L'acide 3~,30-diacétoxylup-20(29)-èn-28-oïque est obtenu de la ma-
30 nière suivante :
Une suspension de 1,8 g d'acide 3~-acétoxy-30-bromolup-20(29)-èn-28-
oïque et 800 mg d'acétate d'argent dans 50 cm de toluène est agitée
48 heures à une température voisine de 20C puis filtrée. La solution
obtenue est ensuite concentrée à sec sous pression réduite (2 kPa).
-
-
W O 94l26725 PCT~R94/00~33 ~
~2~ ~3 ` 18
Le résidu est mis en solution dans 50 cm3 d'acétate d'éthyle. La
phase organique est décantée et lavée par 60 cm au total d'eau
distillée, séchée sur du sulfate de magnésium anhydre, puis concen-
trée sous pression réduite (2,7 kPa) à une température vosine de
40C. Le solide blanc obtenu (1,7 g) est chromatographié sur une co-
lonne de 1,7 cm de diamètre et contenant 35 g de silice (0,02-
0,045 mm) éluée avec un mélange de cycloh~n~ et d'acétate d'éthyle
80-20 ten volumes), en rec~ nt des fractions de 15 cm . Les 4
premières fractions obtenues sont ~l imin~, les 10 suivantes sont
concentrées à sec sous pression réduite (13,5 Pa) a une température
voisine de 40C. On obtient ainsi 1,1 g d'acide 3~,30-diacétoxylup-
20(29)-èn-28-oique sous forme d'une meringue blanche suffi~Amment
- pure pour les transformations ultérieures.
L'acide 3~-acétoxy-30-bromolup-20(29)-en-28-oïque est obtenu de la
manière suivante :
Une solution de 2,5 g d'acide 3~-acétoxy-lup-20(29)-èn-28-oïque et
2,41 g de tribromure de tétrabutyl~mm~nium dans 50 cm3 de chloroforme
est agitée 5 jours à une température voisine de 25C. Le mélange ré-
actionnel est lavé successivement 2 fois par 25 cm d'eau distillée,
2 fois par 25 cm d'une solution de thiosulfate de sodium 0,lN et par
2 fois 25 cm d'eau distillée puis séché sur du sulfate de magnésium
anhydre. La phase organique est concentrée a sec sous pression
réduite (2,7 kPa) et a une température voisine de 40C. On obtient un
solide pâteux jaune (4 g) qui est chromatographié sur une colonne de
3,2 cm de diametre contenant 100 g de silice (0,02-0,045 mm) et éluée
avec un mélange de cyclol-~x~n~ et d'acétate d'éthyle 85-15 (en
volumes) en recueillant des fractions de 20 cm . Les 5 premières
fractions obtenues sont ~l; mi n~, les 10 suivantes sont évaporées
sous pression réduite (13,5 Pa) à une température voisine de 40C. On
obtient ainsi 2,7 g d'acide 3~-acétoxy-30-bromolup-20(29)-~n-28-oïque
sous forme d'un solide blanc fondant a une température voisine de
l 90C
L'acide 3~-acétoxylup-20(29)-en-28-oïque peut être préparé selon
BRUCXNER, KOVACS et KOCZKA, J. Chem. Soc., 948 (1948).
_~YO 94/26725 PCT~R94/00533
`~ 2~6~0~
,9
~m~ le 4
A une solution de 400 mg d'acide N-[3~-acétoxylup-30-(2'-aCétoxyé-
thylthio)-20(29)-èn-28-oyl]-8-aminooctanoique, 90 mg de chlorhydrate
de 5-aminométhyltétrazole, 100 mg d'hydrate de 1-hydroxybenzotria-
zole, 250 mg de chlorhydrate de 1-(3-diméthylaminopropyl)-3-éthyl-
carbodiimide dans 100 cm de dichlorométhane, est ajouté en environ
10 minutes, 0,32 cm de triéthylamine. La solution est agitée pendant
15 heures à une température voisine de 20C, additionnée de 50 cm
d'eau distillée puis acidifiée a pH voisin de 1 par 15 cm d'une so-
lution aqueuse d'acide chlorhydrique N. La phase organique est décan-
tée, la phase aqueuse est extraite par 3 fois 50 cm de dichlorom~-
thane. Les phases organiques réunies sont ensuite s~chées sur du sul-
fate de magnésium et évaporées a sec sous pression réduite (2,7 kPa)
à une température voisine de 40C. Le résidu obtenu (400 mg) est
15 chromatographié sur colonne de silice (0,02-0,045 mm), éluée avec un
mélange de dichlorométhane et de méthanol (90-10 en volumes). Les
fractions contenarlt le produit sont réunies et concentrés sous pres-
sion réduite (2,7 kPa) à une température voisine de 40C. On obtient
ainsi 300 mg de 5-{N'-[N-(3~-acétoxylup-30-(2'-acétoxyéthylthio)-
20 20(29)-en-28-oyl)-8-aminooctanoyl]aminométhyl}-tétrazole sous la
forme d'une meringue blanche (Rf = 0,40 ; chromatographie sur couche
mince de silice ; éluant : chloroforme-méthanol-~mmoniAque à 20 %
(12-3-0,5 en volumes)).
Une solution de 300 mg de 5-{N'-[N-(3~-acétoxylup-30-(2'-acétoxyé-
25 thylthio)-20(29)-en-28-oyl)-8-aminooctanoyl]aminométhyl}-tétrazole,
2,2 cm de soude N, 10 cm de t~trahydrofurane et 20 cm de méthanol
est agitée 48 heures à une température voisine de 20C. Le r' 1 An~e
est acidifié à pH 1 par addition de 4 cm d'une solution aqueuse
d'acide chlorhydrique 4N puis diluée par 70 cm d'eau distillée.
Apres 2 heures sous agitation à une température voisine de 20C, le
solide est filtré et lavé 5 fois par 10 cm d'eau distillée. Le so-
lide est dissout dans 20 cm d'éthanol. La solution est filtrée et le
- filtrat est évaporé sous pression réduite (2,7 kPa) à une température
voisine de 40C. Le résidu obtenu (300 mg) est chromatographié sur
35 colonne de silice (0,02-0,045 mm), éluée avec un mélange de chlo-
roforme, méthanol et ~mmQni~que à 20 % (24-6-1 en volumes). Les frac-
W O 94/26725 PCT~R94/00533 ~
~6~7 ~ 20
tions contenant le produit sont reunies et concentrees sous pression
réduite (2,7 kPa) a une temperature voisine de 40C. Le solide obtenu
(150 mg) est traité par 5 cm d'oxyde d'isopropyle, le solide est
filtré, lavé 2 fois avec 1 cm d'oxyde d'isopropyle puis séché sur
hydroxyde de potassium. On obtient ainsi 100 mg de 5-{N'-[N-(3~-
hydroxylup-30-(2'-hydroxyéthylthio)-20(29)-èn-28-oyl)-8-aminoocta-
noyl]-A~tnom~thyl}-tetrazole sous la forme d'un solide blanc fondant
vers 140C.
L'acide N-[3~-acetoxylup-30-(2'-acetoxyethylthio)-20(29)-èn-28-oyl]-
8-aminooctanoïque peut être obtenu de la manière suivante :
A une solution de 500 mg d'acide N-[3~-acetoxylup-30-(2'-hydroxyé-
thylthio)-20(29)-èn-28-oyl]-8-aminooctanoïque et 600 mg d'acétate de
sodium dans 35 cm d'anhydride acétique est porté à une température
proche du reflux durant environ 3 heures. Le mélange réactionnel est
réfroidi à 20C puis concentré sous pression réduite (2,7 kPa) à une
température voisine de 40C. Le résidu est traité par un mélange de
15 cm de pyridine et 15 cm d'eau et agite durant 12 heures à une
temperature voisine de 20C. Le melange réactionnel est concentré à
sec sous pression réduite (2,7 kPa) a une temperature voisine de
50C. Le residu est traite par 50 cm d'eau distillee et 10 cm d'une
solution aqueuse d'acide chlorhydriqu,e N. La phase organique est
décantée, la phase aqueuse est extraite par 3 fois 40 cm3 de
dichloromethane. Les phases organiques réunies sont lavées 3 fois
avec 20 cm d'eau distillée, séchees sur sulfate de magnésium puis
évaporée à sec sous pression reduite (2,7 kPa) a une température voi-
sine de 40C. Le résidu obtenu (500 mg) est recristallisé dans 5 cm3
d'un mélange éthanol-eau (75-25 en volumes). Le solide obtenu est
filtré, puis lavé 2 fois par 1 cm d'un mélange éthanol-eau (75-25 en
volumes) et séché sous pression réduite (13,5 Pa) sur pentoxyde de
phosphore On obtient ainsi 400 mg d'acide N-[3~-acétoxylup-30-(2'-
acétoxyéthylthio)-20(29)-èn-28-oyl]-8-aminooctanoïque sous la forme
d'une meringue blanche (Rf = 0,25 ; chromato~raphie sur couche mince
de silice ; éluant : chloroforme-méthanol-Amm~n;~que à 20 % (24-6-1
en volumes)).
~ O 94/26725 ~ ~ ~ 2 7 ~ 3 PCT~Rg4/00533
21
L'acide N-[3~-acétoxylup-30-(2'-hydroxyéthylthio)-20~29)-èn-28-oyl~ -
8-aminooctanoïque peut être obtenu de la manière suivante :
A une solution de 1,3 q d'acide N-[3~-acétoxylup-30-bromo-20(29)-èn-
28-oyl~-8-aminooctanoïque dans 10 cm d'éthanol, on ajoute une solu-
tion de 0,60 q de 2-hydroxyéthanethiolate de sodium dans 35 cm3
d'éthanol. On laisse agiter le mélanqe réactionnel 48 heures à une
température voisine de 20C. On ajoute qoutte à goutte en 10 minutes
environ, 5 cm d'une solution aqueuse d'acide chlorhydrique N puis
dilue par 350 cm d'eau. Le mélange réactionnel est extrait 3 fois
par 50 cm de dichlorométhane. Les phases organiques réunies sont
lavée 3 fois par 30 cm d'eau distillée puis séchées sur du sulfate
de maqnésium et évaporées à sec sous pression réduite (2,7 kPa) à une
température volslne de 40C L'huile obtenue (1,1 q) est chromatoqra-
phié sur colonne de silice (0,02-0,045 mm), éluée avec un mélange de
dichlorométhane et de méthanol (98-2 en volumes). Les fractions
contenant le produit sont réunies et concentrés sous pression réduite
(2,7 kPa) à une température voisine de 40C. On obtient ainsi 500 mg
d'acide N-[3~-acétoxylup-30-(2'-hydroxyéthylthio)-20(29)-èn-28-oyl]-
8-aminooctanoïque sous la forme d'une meringue blanche (Rf 0,40 ;
chromatographie sur couche mince de silice ; éluant : chloroforme-
m~thanol-ammoniaque à 20 % (12-3-0,5 en volumes)~.
L'acide N-[3~-acétoxylup-30-bromo-20(29)-èn-28-oyl]-8-aminooctanoïque
peut être obtenu de la manière suivante :
A un mélange de 640 mg d'acide N-[3~-acétoxylup-20(29)-èn-28-oyl]-8-
aminooctanoïque et de 30 cm de tétrachlorure de carbone, ajouter en
une seule fois 350 mg de N-bromosuccinimide. La suspension est agitée
12 heures à une température voisine de 20C. Le solide est filtré,
lavé par 3 fols 10 cm de tétrachlorure de carbone. Le filtrat est
évaporé sous pression réduite (2,7 kPa) à une température voisine de
30 40C. On obtient 800 mg d'acide N-[3~-acétoxylup-30-bromo-20(29)-èn-
28-oyl]-8-aminooctanoïque sous la forme d'une meringue hl~n~he (Rf -
0,25 ; chromatographie sur couche mince de silice ; éluant : chlo-
roforme-méthanol-ammoniaque à 20 ~ (24-6-1 en volumes)).
L'acide N-[3~-acétoxylup-30-bromo-20(29)-èn-28-oyl]-8-aminooctanoïque
est préparé comme décrit dans l'exemple 1.
W O 94/26725 PCT~R94/00533
3 22
L'hydroxyéthanethiolate de sodium est préparé par action de l'éthy-
late de sodium sur le 2-hydroxyéthanthiol en solution dans l'éthanol.
Le chlorure de 3~-acétoxylup-20(29)-èn-28-oyle est préparé d'après J.
PROVITA et A. VYSTRCIL, Collect. Czech. Chem. Commun. 41, 1200
(1976).
~mn le 5
A une solution de 1,9 g d'acide N-[[3~-acétoxylup-20(29)-an-28-oyl]-
8-aminooctanoïque dans 55 cm de dichlorométhane, on ajoute 2,2 cm3
de chlorure de thionyle. La solution est agitée pendant 24 heures a
20C puis le mélange réactionnel est concentré à sec sous pression
réduite (2,7 kPa) à une température voisine de 35C. Le résidu obtenu
est mis en solution dans 25 cm de dichlorométhane et la solution est
à nouveau évaporée à sec sous pression réduite (2,7 kPa) a une tempé-
rature voisine de 35C. Le solide obtenu (1,97 g) et 485 mg de 5-(4-
aminophényltétrazole) sont mis en solution dans 60 cm de dichloromé-
thane et 1,41 cm de triéthylamine sont ajoutés goutte à goutte. La
solution obtenue est agitée 24 heures a une température voisine de
20C. La solution obtenue est concentrée sous pression réduite
(2,7 kPa) à une température voisine de 35C. Au solide obtenu est
ajouté 50 cm de dichlorométhane et 25 cm d'une solution d'acide
chlorhydrique N La phase organique est décantée, lavée 2 fois avec
10 cm d'une solution d'acide chlorhydrique N puis 2 fois avec 10 cm
d'eau distillée La phase organique est sechée sur sulfate de sodium
puis concentrée à sec sous pression réduite (2,7 kPa) à une
température voisine de 35C. On obtient ainsi 2,0 g d'un solide qui
est chromatographié sur colonne de silice (0,02-0,045 mm) éluée, avec
un mélange de méthanol et d'acétate d'éthyle (10-90 en volumes), les
fractions contenant le produit sont réunies et concentrées sous
pression réduite (2,7 kPa) à une température voisine de 40C. Le
30 résidu (280 mg) est chromatographié sur colonne de silise (0,02-
0,045 mm) éluée avec un mélange de chloroforme, de méthanol et
d'Ammon;Aque (24-6-1 en volumes), les fractions contenant le produit
sont réunies et concentrées sous pression réduite (2,7 kPa) a une
température voisine de 35C. On obtient ainsi 310 mg de 5-~4-{N-[N'-
35 (3~-acétoxylup-20(29)-èn-28-oyl)-8-aminooctanoyl]-amino}-phényl]-té-
~ 0 94/26725 21 ~ 2 7 ~ 3 PCT~R94/00533
23
trazole sous la forme d'une poudre blanche (Rf 0,32 ; chromatogra-
phie sur couche mince de silice ; éluant : chloroforme-méthanol-am-
moniaque ~ 20 ~ (24-6-1 en volumes)).
Une solution de 0,31 q de 5-[4-{N-[N'(3~-acétoxylup-20(29)-èn-28-
oyl)-8-aminoctyll-amino}-phényl]-tétrazole, 1 cm de soude 4N, 3 cm3
de tétrahydrofurane et 1,5 cm de méthanol est agitée 26 heures à une
température voisine de 20C. Le mélange est évaporé sous pression
réduite (2,7 kPa) à une température voisine de 35C, puis dilué avec
cm d'eau distillée et acidifié avec 1,2 cm d'une solution
d'acide chlorhydrique 4N. Après 1 heure d'agitation, à une tempéra-
ture voisine de 20C, le solide est séparé par filtration, lavé 6
fois avec 5 cm d'eau distillée et séché sous pression réduite
(15,5 Pa) à une température voisine de 30C. On obtient ainsi 250 mg
de 5-[4-{N-[N'-(3~-hydroxylup-20(29)-èn-28-oyl)-8-aminooctanoyl]-
amino}-phényl]-tétrazole, sous la forme d'un solide blanc fondant
vers 200C.
Le 5-(4-aminophényle)tétrazole peut être préparé d'après J.M. McMANUS
et R. HERBST, J Org. Clem , 24, 1044 (1959).
Ex~mole 6
A une solution de 1,9 g d'acide N-(3~-acétoxylup-20(29)-èn-28-oyl)-8-
aminooctanoïque dans 55 cm de dichlorométhane, on ajoute 2,2 cm3 de
chlorure de thionyle. La solution est agitée pendant 24 heures à 20C
puis le mélange réactionnel est concentré à sec sous pression réduite
(2,7 kPa) à une température voisine de 35C. Le résidu obtenu est mis
en solution dans 25 cm de dichlorométhane et la solution est à
nouveau concentrée a sec sous pression réduite (2,7 kPa) à une tempé-
rature voisine de 35C. Le solide obtenu (1,97 g) et 485 mg de 5-(3-
aminophényltétrazole) sont mis en solution dans 60 cm de dichloromé-
thane et 1,41 cm de triéthylamine sont ajoutés goutte à goutte. La
solution obtenue est agitée 24 heures à une température voisine de
20C. La solution obtenue est concentrée sous pression réduite
(2,7 kPa) à une température voisine de 35C. Au solide obtenu est
ajouté 50 cm de dichlorométhane et 25 cm d'une solution d'acide
chlorhydrique N. La phase organique est décantée, lavée 2 fois avec
10 cm d'une solution d'acide chlorhydrique N puis 2 fois avec 10 cm
W O 94/26725 ~ PCT~R94/00533 ~
7~ ~ 24
d'eau distillée. La phase organique est sechée sur sulfate de sodium
puis concentrée à sec sous pression réduite (2,7 kPa) ~ une
température voisine de 35C. On obtient ainsi 2,2 g d'un solide qui
est chromatographié sur colonne de silice (0,02-0,045 mm) éluée, avec
un mélange de chloroforme, de méthanol et d'ammoniaque (24-6-1 en
volumes), les fractions contenant le produit sont réunies et
concentrées sous pression réduite (2,7 kPa) à une température voisine
de 35C. On obtient ainsi 840 mg d'un solide qui est à nouveau
chromatographié sur colonne de silice (0,02-0,045 mm) éluée, en
premier lieu avec de l'acétate d'éthyle puis avec un mélange de
chloroforme, de méthanol et d'Ammoni~que (24-6-1 en volumes), les
fractions contenant le produit sont réunies et concentrées sous
pression réduite (2,7 kPa) à une température voisine de 35C. On
obtient ainsi 390 mg de 5-[3-{N-[N'-(3~-acétoxylup-20(29)-èn-28-oyl)-
8-aminooctanoyl]-amino}-phényl]-tétrazole sous la forme d'une poudre
blanche (Rf = 0,36 ; chromatographie sur couche mince de silice ;
éluant : chloroforme-méthanol-A~ ;aque à 20 ~ (24-6-1 en volumes)).
Une solution de 380 mg de 5-[3-{N-[N'-(3~-acétoxylup-20(29)-èn-28-
oyl)-8-aminooctalloyll-amino}-phényl]-tétrazole, 1,2 cm de soude 4N,
4 cm de tétrahydrofurane et 2 cm de méthanol est agitée 22 heures à
une température voislne de 20C. Le mélange est évaporé sous pression
réduite (2,7 kPa) à une température voisine de 35C, puis dilué avec
25 cm d'eau distillée et acidifié avec 1,4 cm d'une solution
d'acide chlorhydrique 4N. Après 1 heure d'agitation, à une tempéra-
ture voisine de 20C, le solide est séparé par filtration, lavé 6
fois avec 5 cm d'eau distillée et séché sous pression réduite
(15,5 Pa) à une température voisine de 30C. On obtient ainsi 270 mg
de 5-[3-{N-[N'-(3~-hydroxylup-20(29)-èn-28-oyl)-8-aminooctanoyl]-
amino}-phényl]-tétrazole, sous la forme d'un solide blanc fondant
vers 170C.
Le 5-(3-aminophényle)tétrazole peut être préparé d'après J.M. McMANUS
et R. HERBST, J. Org. Chem., 24, 1044 (1959).
~n le 7
670 mg de N-(3~-acétoxylup-20(29)-èn-28-oyl)-11-aminonn~c~n~nitrile
et 1 g d'a~oture de tributylétain sont mis en solution dans 5 cm3 de
~ 0 94/2672~ 2 1~ 2~ o~ PCT~R94/00533
1~2-diméthoxyéthane~ Le mélange est chauffé au reflux pendant
48 heures. On rajoute 0,5 g d'azoture de tributylétain et le mélange
est à nouveau chauffé au reflux pendant 48 heures. Le mélanqe réac-
tionnel est dilué par 50 cm d'eau distillée puis extrait avec
100 cm d'acétate d'éthyle. La phase organique est ensuite lavée avec
75 cm d'eau distillée, séchée sur du sulfate de magnésium et évapo-
rée à sec sous pression réduite ~2,7 kPa) à une température voisine
de 40C. Le résidu obtenu ~2,2 g~ est chromatographié sur une colonne
de silice ~0,02-0,045 mm) éluée avec un mélange de chloroforme, mé-
thanol et ammoniaque à 20 % ~12-3-0,5 en volumes). Les fractions
contenant le produit sont réunies et concentrées sous pression ré-
duite (2,7 kPa) à une température voisine de 40C. On obtient ainsi
700 mg de 5-~N-(3~-acétoxylup-20(29)-èn-28-oyl)-10-aminodécyl]-té-
trazole sous la forme d'une meringue blanche (Rf - 0,32 ; chromato-
graphie sur couche mirlce de silice ; éluant : chloroforme-méthanol-
Amm~niAque à 20 % (12-3-0,5 en volumes)).
Une solution de 290 mg de 5-[N-(3~-acétoxylup-20~29)-èn-28-oyl)-10-
aminodécyl]-tétrazole, 0,82 cm de soude 5N, 3 cm de tétrahydrofu-
rane et 6 cm3 de méthanol est agitée 20 heures à une température voi-
sine de 20C. Le mélange est acidifié avec de l'acide chlorhydrique5N, et dilué avec 40 cm d'eau distillée. Le 51 An~e est extrait par
50 cm , puis 2 fois 25 cm , d'acétate d'~thyle. Les phases organiques
sont rassemblées, lavées par 100 cm d'eau distillée, séchées sur du
sulfate de magnésium et évaporées à sec sous pression réduite
(2,7 kPa) à une température voisine de 40C. Le résidu obtenu
(280 mg) est chromatographié sur une colonne de silice (0,02-
0,045 mm) éluée avec un mélange de chloroforme, méthanol et
A --lAqUe à 20 % (12-3-0,5 en volumes). Les fractions contenant le
produit sont réunies et concentrées sous pression réduite (2,7 kPa) à
une température voisine de 40C. Le résidu est repris par 30 cm3
d'oxyde d'isopropyle, filtré, lavé avec 10 cm d'oxyde d'isopropyle
et séché sous pression réduite (15,5 Pa) à 20C. On obtient ainsi
185 mg ,de 5-[N-(3~-hydroxylup-20(29)-èn-28-oyl)-10-aminodécyl]-
tétrazole, sous la forme d'un solide blanc fondant vers 200C.
L'azoture de tributylétain peut être préparé d'après H.R. KRICHELDORF
et E. LEPPERT, Synthesis, 329 (1976).
WO 94/26725 ~ PCT/FR94/00533 ~
7 ~ 3 26
Le N-(313--acétoxylup-20(29)-èn-28-oyl)-11-amino~ln~l~CcAnF~nitrile peut
être obtenu de la façon suivante:
A une solution de 2,5 g de N-(3¦3-acétoxylup-20 (29)-èn-28-oyl)-11-ami-
noundécanamide dans 300 cm de tétrahydrofurane sec, on ajoute goutte
5 à goutte 1,92 cm d'anhydride trifluoroacétique. La solution est agi-
tée 15 heures à température ambiante puis 1,9 cm supplémentaires
d'anhydride trifluoroacétique sont ajoutés et la solution est agitée
48 heures à température ambiante. La solution est neutralisée par
addition d'une solution saturée de bicarbonate de sodium puis
10 concentrée sous pression réduite (2,7 kPa). Le résidu est repris par
100 cm d'eau distillée et le mélange est extrait 2 fois avec 150 cm
d'acétate d'éthyle. Les phases organiques sont rassemblées, lavées 3
fois avec 100 cm d'eau distillée, séchées sur du sulfate de magné-
sium et évaporées A sec sous pression réduite (2,7 kPa) à une tempé-
15 rature voisine de 40C. Le résidu obtenu (2,5 g) est chromatographiésur une colonne de silice (0,02-0,045 mm) éluée avec du dichloromé-
thane. Les fractions contenant le produit sont réunies et concentrées
sous pression réduite (2,7 kPa) à une température voisine de 40C. On
obtient ainsi 1,6 g de N-(313-acétoxylup-20(29) -en-28-oyl)-11-
20 amino--n~lécAn~nitrile sous la forme d'une meringue blAn~ he ~Rf -- 0,6;
chromatographie sur couche mince de silice ; éluant : cycl~h~cAne
acétate d'éthyle (60-40 en volumes)) .
Le N-(3~3--acétoxylup-20(29)-èn-28-oyl)-11-aminonn~l~c~n~Tnide peut être
obtenu de la manière suivante:
25 A une solution de 2,6 g d'acide N-[313-acétoxylup-20(29)-èn-28-oyl]-
11-aminonndécAnoïque dans 240 cm de dichlorométhane, on ajoute
3,5 cm d'Amm~njAque à 20 %, 580 mg d'hydrate de 1 -hy-
droxybenzotriazole, et 1,45 g de chlorhydrate de 1- (3-
diméthylaminopropyl)-3-éthylcarbodiimide. La solution est agitée pen-
30 dant 2 jours à 20C puis la phase organique est décantée, lavée suc-
cessivement avec 50 cm d'eau distillée, 3 fois 25 cm d'acide chlor-
hydrique lN, 2 fois 40 cm d'eau distillée, puis séchée sur du sul-
fate de magnésium, filtrée et évaporée à sec sous pression réduite
(2,7 kPa) à une température voisine de 40C. On obtient ainsi 2,7 g
35 de N-(3~3-acétoxylup-20 (29)-èn-28-oyl)-11-aminol~nll~cAnA~;de sous la
~o 94/26725 ~ 3 PCT~R94/00533
27
forme d'une meringue couleur crème (Rf ! 0,15 ; chromatographie sur
couche mince de silice ; éluant : cyclohPxAne-acétate d'éthyle (30-70
en volumes)).
L'acide N-(3~-acétoxylup-20(29)-èn-28-oyl)-11-aminonn~écAnoïque est
synthétisé par analogie avec l'acide N-(3~-acétoxylup-20(29)-èn-28-
oyl)-8-aminooctanoïque a partir de chlorure de 3~-acétoxylup-20(29)-
èn-28-oyle et de 11-aminoundécanoate de méthyle (R = 38 % ; Rf
0,57 ; chromatographie sur couche mince de silice ; éluant
dichlorométhane-méthanol (90-10 en volumes)).
~m~le 8
1 g de N-(3~-acétoxylup-20(29)-èn-28-oyl)-8-aminooctanenitrile et
1,6 g d'azoture de tributylétain sont mis en solution dans 25 cm de
1,2-diméthoxyéthane. Le mélange est chauffé au reflux pendant
24 heures. On ajoute 35 cm de 2-éthoxyéthanol, distille le 1,2-dimé-
thoxyéthane et le mélanqe est à nouveau chauffé au reflux pendant48 heures. Le mélange réactionnel est refroidi à température
ambiante, versé dans une solution de 100 cm d'acide chlorhydrique
lN. Après 15 mlnutes d'agitation, le mélange est extrait avec 3 fois
50 cm d'acétate d'éthyle. Les phases organiques sont rassemblées,
lavées avec 7 fois 30 cm d'eau distillée, séchées sur du sulfate de
magnésium et concentrées à sec sous pression réduite (2,7 kPa) à une
température voisine de 40C. Le résidu obtenu (2,7 g) est
chromatographié sur une colonne de silice (0,02-0,045 mm) éluée avec
un mélange de chloroforme, méthanol et A~oniAque à 20 % (12-3-0,5 en
volumes). Les fractions contenant le produit sont réunies et
concentrées sous pression réduite (2,7 kPa) à une température vo;ci n~
de 40C. Le résidu obtenu est lavé avec 5 fois 75 cm de pentane,
filtré, séché sous pression réduite (2,7 kPa) à 40C. On obtient
ainsi 920 mg de 5-[N-(3~-acétoxylup-20~29)-èn-28-oyl)-7-~ inoheptyl]
r 30 tétrazole sous la forme d'une poudre blanc crème (Rf = 0,28
chromatographie sur couche mince de silice ; éluant : chloroforme-
méthanol-ammoniaque à 20 ~ (12-3-0,5 en volumes)).
Une solution de 900 mg de 5-[N-(3~-acétoxylup-20(29)-èn-28-oyl)-7-
Aminoheptyl]-tétrazole, 2,7 cm de soude 5N, 10 cm de tétrahydrofu-
35 rane et 20 cm de méthanol est agitée 15 heures a une température
W O 94/26725 PCT~R94/00533
2~27~3
28
voisine de 20C. Le mélange est acidifié avec de l'acide chlorhy-
drique 1N, et dilué avec 175 cm d'eau distillée. Le mélange est ex-
trait par 3 fois 100 cm d'acétate d'éthyle. Les phases organiques
sont rAcse~hlées, lavées par 250 cm d'eau distillée, séchées sur du
sulfate de magnésium et concentrées à sec sous pression réduite
(2,7 kPa) à une température voisine de 40C. Le résidu obtenu
~865 mg) est chromatographié sur une colonne de silice (0,02-
0,045 mm) éluée avec un mélange de chloroforme, méthanol et
~ que à 20 % (12-3-0,5 en volumes). Les fractions contenant le
produit sont réunies et concentrées sous pression réduite (2,7 kPa) à
une température voisine de 40C. Le résidu est repris par 10 cm3
d'oxyde d'isopropyle, filtré, lavé avec 6 cm d'oxyde d'isopropyle et
- séché sous pression réduite (15,5 Pa) à 20C. La poudre obtenue (510
mg) est à nouveau chromatographiée sur une colonne de silice (0,02-
0,045 mm) éluée avec un mélange de dichlorométhane et de méthanol
(95-5 en volumes) Les fractions contenant le produit sont réunies et
concentrées sous pression réduite (2,7 kPa) à une température voisine
de 40C. Le résidu est repris par 5 cm d'oxyde d'isopropyle, filtré,
lavé avec 2 cm3 d'oxyde d'isopropyle et séché sous pression réduite
20 (15,5 Pa) à 20C. On obtient ainsi 224 mg de 5-[N-(3~-hydroxylup-
20(29)-èn-28-oyl)-7-~minoheptyl]-tétrazole, sous la forme d'un solide
blanc fondant vers 150-155C.
Le N-(3~-acétoxylup-20(29)-èn-28-oyl)-8-aminooctanenitrile peut être
obtenu de la facon suivante :
25 A une solution de 2,4 g de N-(3~-acétoxylup-20(29)-èn-28-oyl)-8-
aminooctanamide dans 200 cm de tétrahydrofurane sec, on ajoute
goutte à goutte 1,97 cm d'anhydride trifluoroacétique. La solution
est agitée 15 heures à température ambiante puis 5,0 cm
supplémentaires d'anhydride trifluoroacétique sont ajoutés et la
solution est agitée 48 heures à température ambiante. La solution est
neutralisée par addition d'une solution saturée de bicarbonate de
sodium, puis diluée par 100 cm d'eau distillée. Le tétrahydrofurane
est évaporé sous pression réduite (2,7 kPa) à 40C. Le melange est
ensuite repris par 200 cm3 d'acétate d'éthyle. La phase aqueuse est
extraite par 50 cm d'acétate d'éthyle. Les phases organiques sont
rassemblées, lavées 2 fois avec 100 cm d'eau distillée, séchées sur
~ O 94/2672~ 216 2 7 0 ~ PCT~R94/00533
29
du sulfate de magnésium, filtrées et évaporées à sec sous pression
réduite ~2,7 kPa) à une température voisine de 40C. On obtient ainsi
2,2 g de N-(3~-acétoxylup-20t29)-èn-28-oyl)-8-aminooctanenitrile sous
la forme d'une meringue blanc crème (Rf - 0,23 ; chromatoqraphie sur
couche mince de silice ; éluant : cycloh~An~-acétate d'éthyle (80-20
en volumes)).
Le N-(3~-acétoxylup-20(29)-èn-28-oyl)-8-aminooctanamide peut être
obtenu de la manière suivante :
A une solution de 2,4 g d'acide N-(3~-acétoxylup-20(29)-èn-28-oyl)-8-
10 aminooctanoïque dans 240 cm de dichlorométhane, on ajoute 3,8 cm3
d'Armo~iAque à 20 %, 580 mg d'hydrate de 1-hydroxybenzotriazole, et
1,45 g de chlorhydrate de 1-(3-diméthylaminopropyl)-3-éthylcarbodii-
mide. La solution est agitée 24 heures à température A~hi~nte puis la
phase organique est décantée, lavée avec 5 ~ois 250 cm d'eau distil-
lée, séchée sur du sulfate de magnésium, filtrée et évaporée à sec
sous pression réduite (2,7 kPa) à une température voisine de 40C. On
obtient ainsi 2,4 g de N-(3~-acétoxylup-20(29)-èn-28-oyl)-8-aminooc-
tanamide sous la forme d'une meringue hlAnche (Rf 0,1 ; chromato-
graphie sur couche mince de silice ; éluant : cycloh~An~-acétate
d'~thyle (50-50 en volumes)).
ExemPle 9
1,3 g de N-(3~-acétoxylup-20(29)-èn-28-oyl)-12-aminodo~cAn~nitrile
et 1,27 g d'azoture de tributylétain sont mis en solution dans 50 cm
de 2-éthoxyéthanol. Le mélange est chauffé au reflux pendant
25 48 heures. Le mélange réactionnel est versé dans 300 cm3 d'eau dis-
tillée glacée. Après 5 minutes d'agitation, le r~lAnqe est acidifié
avec une solution d'acide chlorhydrique 2N puis extrait avec 3 fois
80 cm de dichlorométhane. Les phases organiques sont réunies, lavées
jusqu'à neutralité par de l'eau distillée, séchées sur du sulfate de
magnésium et évaporées à sec sous pression réduite (2,7 kPa) à une
température voisine de 40C. Le résidu obtenu (1,4 q) est chromato-
graphié sur une colonne de silice (0,02-0,045 mm) éluée avec un mé-
lange de chloroforme, méthanol et A ~ Aque à 20 % (12-3-0,5 en vo-
lumes). Les fractions contenant le produit sont réunies et concen-
trées sous pression réduite (2,7 kPa) à une température voisine de
W O 94/26725 PCT~R94/00533 _
2 ~ 3
40C. On obtient ainsi 1,2 g de 5-[N-(3~-acétoxylup-20(29)-èn-28-
oyl)-11-aminoundécyl]-tétrazole sous la forme d'une meringue blanche
(Rf - 0,45 ; chromatographie sur couche mince de silice ; éluant :
chloroforme-méthanol-~ ~ni~que a 20 % (12-3-0,5 en volumes)).
5 Une solution de 1,2 q de 5-[N-(3~-acétoxylup-20(29)-èn-28-oyl)-11-
aminoundécyl~-tétrazole, 3,3 cm de soude 5N, 12 cm de tétrahydrofu-
rane et 24 cm de méthanol est agitée 20 heures à une température
voisine de 20C. Le mélange est acidifié avec de l'acide chlorhy-
drique 5N, et dilué par 150 cm d'eau distillée. Le mélange est ex-
trait par 3 fois 60 cm de dichlorométhane. Les phases organiques
sont rassemblées, lavées jusqu'à neutralité par de l'eau distillée,
séchées sur du sulfate de magnésium et évaporées à sec sous pression
réduite (2,7 kPa) à une température voisine de 40C. Le résidu obtenu
(1,1 g) est chromatographié sur une colonne de silice (0,02-0,045 mm)
éluée avec un mélange de dichlorométhane, méthanol et ~rmoniAque a
20 % (12-3-0,5 en volumes). Les fractions contenant le produit sont
réunies et concentrées sous pression réduite (2,7 kPa) à une tempéra-
ture voisine de 40C. Le résidu est repris par 15 cm d'éther éthy-
lique, filtré, lavé avec 12 cm d'éther éthylique et séché sous pres-
20 sion réduite (15,5 Pa) à 20C. On obtient ainsi 340 mg de 5-[N-(3~-
hydroxylup-20(29)-èn-28-oyl)-11-aminoundécyl]-tétrazole, sous la
forme d'un solide blanc fondant vers 200-205C.
Le N-(3~-acétoxylup-20(29)-èn-28-oyl)-12-aminodo~P~AnPnitrile peut
être obtenu de la façon suivante :
25 A une solution de 1,5 g de N-(3~-acétoxylup-20(29)-èn-28-oyl)-12-ami-
nodod~cAn~mlde dans 150 cm de tétrahydrofurane sec, on a~oute goutte
à goutte 1,9 cm d'anhydride trifluoroacétique. La solution est agi-
t~e 60 heures à température ambiante, puis diluée par 30 cm3 d'eau
distillée. Le tétrahydrofurane est évaporé ~ sec sous pression ré-
30 duite (2,7 kPa) à 40C. Le mélange est repris par 100 cm3 d'eau dis-
tillée, neutralisé par addition d'une solution saturée de bicarbonate
de sodium puis extrait par 3 fois 50 cm3 de dichlorométhane. Les
phases organiques sont rassemblées, lavées par 3 fois 40 cm3 d'eau
distillée, séchées sur du sulfate de magnésium et évaporées à sec
sous pression réduite (2,7 kPa) à une température voisine de 40C. On
O 94/26725 ~ ~ ~ 2 7 ~ 3 PCT~R94/00533
31
obtient ainsi 1,36 g de N- (3~-acétoxylup-20(29)-èn-28-oyl)-12-ami-
nodo~5c~ne~itrile sous la forme d'une meringue hl~nche (Rf ~ 0,35 ;
chromatographie sur couche mince de silice ; éluant : cycloh~YAne-
acétate d'éthyle (80-20 en volumes)).
Le N-(3~-acétoxylup-2o(29)-èn-28-oyl~-12-aminodo~c~n~mlde peut être
obtenu de la façon suivante :
A une solution de 1,5 g d'acide N-(3~-acétoxylup-20(29)-èn-28-oyl)-
12-aminododécanoïque dans 250 cm de dichlorométhane, on ajoute
2,15 cm3 d'ammoniaque ~ 20 %, 330 mg d'hydrate de 1-hydroxybenzo-
triazole, et 826 mg de chlorhydrate de 1-(3-diméthylaminopropyl)-3-
éthylcarbodiimide. La solution est agitée 15 heures à température
ambiante puis la phase organique est décantée, lavée suçcessivement
avec 5 fois 80 cm d'eau distillée, 3 fois 80 cm d'acide chlorhy-
drique lN, 5 fois 80 cm d'eau distillée, puis séchée sur du sulfate
15 de magnésium, filtrée et concentrée à sec sous pression réduite
(2,7 kPa) à une température voisine de 40C. On obtient ainsi 1,5 g
de N-(3~-acétoxylup-20(29)-èn-28-oyl)-12-aminododécanamide sous la
forme d'une meringue couleur crème (Rf = 0,47 ; chromatographie sur
couche mince de silice ; éluant : dichlorométhane-méthanol (90-10 en
20 volumes))
~'acide N-(3~-acétoxylup-20(29)-èn-28-oyl)-12-aminodod~écanoïque est
synthétisé par analogie avec l'acide N-(3~-acétoxylup-20(29)-èn-28-
oyl)-8-aminooctanoïque à partir de chlorure de 3~-acétoxylup-20(29)-
èn-28-oyle et de 12-aminododécanoate de méthyle (R = 75 %; Rf =
25 0,45 ; chromatographie sur couche mince de silice ; éluant
dichlorométhane-méthanol (90-10 en volumes)).
~X~mDle 1 0
490 mg de N'- [N- (3~-acétoxylup-20(29)-èn-28-oyl)-8-aminooctanoyl]-3-
r aminopropanenitrile et 700 mg d'azoture de tributylétain sont mis en
30 solution dans 10 cm de 1,2-diméthoxyéthane. Le mélange est chauffé
- au reflux pendant 6 jours. Le mélange réactionnel est refroidi à tem-
pérature ambiante, dilué avec 100 cm de dichlorométhane et acidifié
avec 25 cm d'acide chlorhydrique 2N. Après 5 minutes d'agitation, la
phase organique est décantée, lavée par 5 fois 40 cm3 d'eau
W O 94/26725 PCT~R94/00533 ~
2~ ~7~3 32
distillée, sechée sur du sulfate de magnésium, filtrée et concentrée
à sec sous pression réduite (2,7 kPa) à une température voisine de
40C. Le résidu obtenu est lavé par 5 fois 10 cm de pentane, chroma-
tographié sur une colonne de silice ~0,02-0,045 mm) éluée avec un
mélange d'acétate d'éthyle et de méthanol (80-20 en volumes). Les
fractions contenant le produit sont réunies et concentrées sous pres-
sion réduite (2,7 kPa) a une température voisine de 40C. On obtient
ainsi 435 mg de 5-{N'-[N-(3~-acétoxylup-20(29)-en-28-oyl)-8-aminooc-
tanoyl]aminoéthyl}-tétrazole sous la forme d'une meringue hlAnrh~,
(Rf 0,39 ; chromatographie sur couche mince de silice ; éluant :
chloroforme-méthanol-Ammo~i~que à 20 % (12-3-0,5 en volumes)).
Une solution de 600 mg de 5-{N'-~N-(3~-acétoxylup-20(29)-en-28-oyl)-
8-aminooctanoyl]aminoéthyl}-tétrazole, 1,6 cm de soude 5N, 10 cm de
tétrahydrofurane et 20 cm de méthanol est agitée 15 heures a une
température voisine de 20C. Le mélange est acidifié avec de l'acide
chlorhydrique lN, et dilué avec 120 cm d'eau distillée. Le mélange
est agité 1 heure a 20C, filtré, lavé jusqu'à neutralité par de
l'eau distillé, puis par 5 fois 5 cm de pentane, et séché sous pres-
sion réduite (15,5 Pa) a 20C La poudre obtenue (553 mg) est chroma-
tographiée sur une colonne de silice (0,02-0,045mm) éluée avec un
mélange de chloro~orme, méthanol et ammoniaque a 20 ~ (12-3-0,5 en
volumes) Les fractions contenant le produit sont réunies et concen-
trées sous pression réduite (2,7 kPa) a une température voisine de
40C. Le résidu est repris par 15 cm d'oxyde d'isopropyle, filtré,
et séché sous pression réduite (15,5 Pa) à 20C. On obtient ainsi
430 mg de 5-{N'-[N-(3~-hydroxylup-20(29)-en-28-oyl)-8-aminoocta-
noyl]aminoéthyl}-tétrazole, sous la forme d'un solide blanc fondant
vers 155-160C.
Le N'-[N-(3~-acétoxylup-20(29)-èn-28-oyl)-8-aminooctanoyl]-3-ami-
nopropanenitrile peut être obtenu de la façon suivante :
A une solution de 960 mg de N'-[N-(3~-acétoxylup-20(29)-èn-28-oyl)-8-
aminooctanoyl]-3-aminopropanamide dans 70 cm de tétrahydrofurane
sec, on ajoute goutte à goutte 0,94 cm d'anhydride trifluoroacé-
tique. La solution est agitée 15 heures à température ambiante, puis
le tétrahydrofurane est évaporé sous pression réduite (2,7 kPa) à
~ 0 94/2672~ 216 2 7 0 3 PCT~R94/00~33
33
40C. Le mélange est ensuite repris par 70 cm de dichlorométhane. La
phase organique est décantée, lavée par 3 fois 50 cm d'eau distil-
lée, séchée sur du sulfate de magnésium et évaporée à sec sous pres-
sion réduite (2,7 kPa) à une température voisine de 40C. Le résidu
obtenu (887 mg) est chromatographié sur une colonne de silice (0,02-
0,045 mm) éluée avec un mélange d'acétate d'éthyle et de cyclohexane
(70-30 en volumes). Les fractions contenant le produit sont réunies
et concentrées sous pression réduite (2,7 kPa) à une température voi-
sine de 40C. On obtient ainsi 692 mg de N'-[N-(3~-acétoxylup-20(29)-
èn-28-oyl)-8-aminooctanoyl]-3-amino propanenitrile sous la forme
d'une meringue blanche (Rf = 0,27 ; chromatographie sur couche mince
de silice ; éluant : cycloh~x~ne-acétate d'éthyle (30-70 en
~ volumes)).
Le N'-[N-(3~-acétoxylup-20~29)-èn-28-oyl)-8-aminooctanoyl]-3-ami-
nopropanamide peut être obtenu de la manière suivante :
A une solution de 967 mg d'acide N'-[N-(3~-acétoxylup-20(29)-èn-28-
oyl)-8-aminooctanoyll-3-aminopropanoïque dans 150 cm de dichloro-
méthane, on ajoute 1 cm3 d'~m~oni~que à 20 ~, 208 mg d'hydrate de
1-hydroxybenzotriazole, et 521 mg de chlorhydrate de 1-(3-
diméthylaminopropyl)-3-éthylcarbodiimide. La solution est agitée
50 heures à température ambiante puis la phase organique est
décantée, lavée avec 60 cm d'acide chlorhydrique 1N, 5 fois 50 cm3
d'eau distillée, séchée sur du sulfate de magnésium, filtrée et
évaporée à sec sous pression réduite (2,7 kPa) a une température
25 voisine de 40C. On obtient ainsi 960 mg de N'-[N-(3~-acétoxylup-
20(29)-èn-28-oyl)-8-aminooctanoyl]-3-aminopropanamide sous la forme
d'une meringue blanc crème (Rf = 0,68 ; chromatographie sur couche
mince de silice ; éluant : chloroforme-méthanol-A i~que à 20 %
(12-3-0,5 en volumes)).
30 L'acide N'-[N-(3~-acétoxylup-20(29)-èn-28-oyl)-8-aminooctanoyl]-3-
aminopropanoïque peut être obtenu de la manière suivante :
A une solution anhydre de 2,3 g de iodure de lithium et de 100 cm3 de
collidine, on ajoute 1,3 g de N-[(3~-acétoxylup-20(29)-èn-28-oyl)-8-
aminooctanoyl]-3-aminopropionate de méthyle. La solution est chauffée
au reflux pendant 3 heures, puis concentrée sous pression réduite
W O 94/26725 PCTn~R94/00533 ~
3 34
(2,7 kPa) à une température voisine de 40C. Le résidu est repris par
150 cm d'eau distillée et 150 cm d'acétate d'éthyle, puis refroidi
à 0C. La phase organique est extraite, lavée par 3 fois 50 cm
d'acide chlorhydrique lN, puis par de l'eau distillée jusqu'à neutra-
lité, séchée sur du sulfate de magnésium et évaporée à sec sous pres-
sion réduite ~2,7 kPa) à une température voisine de 40C. Le résidu
est repris par 20 cm d'acétate d'éthyle et le mélange est concentré
sous pression réduite (2,7 kPa) à une température voisine de 40C. On
obtient ainsi 1,07 g d'acide N'-[N-(3~-acétoxylup-20(29)-en-28-oyl)-
8-aminooctanoyl]-3-aminopropanoïque sous la forme d'une meringue
beige (Rf = 0,22 ; chromatographie sur couche mince de silice
éluant : chloroforme-méthanol-ammoniaque à 20 ~ (12-3-0,5)).
Le N'-[N-(3~-acétoxylup-20(29)-èn-28-oyl)-8-aminooctanoyl]-3-amino
propionate de méthyle peut être préparé de la manière suivante :
A une solution de 2 g d'acide N-(3~-acétoxylup-20(29)-èn-28-oyl)-8-
aminooctanoïque dans 260 cm de dichlorométhane, on ajoute 524 mg de
chlorhydrate d'amino-3 propionate de méthyle, 480 mg d'hydrate de
1-hydroxybenzotriazole, l,2 g de chlorhydrate de 1-(3-diméthylamino-
propyl)-3-éthylcarbodiimide, et 1,1 g de triéthylamine en solution
dans 15 cm de dichlorométhane. La solution est agitée 50 heures a
température ambiante puis la phase organique est décantée, lavée avec
6 fois 150 cm d'eau distillée, séchée sur du sulfate de magnésium,
filtrée et évaporée a sec sous pression réduite (2,7 kPa) a une tem-
pérature voisine de 40C. Le résidu obtenu (2,4 g) est chromatogra-
phié sur une colonne de silice (0,02-0,045 mm) éluée avec un mélange
d'acétate d'éthyle et de cyclohexane (50-50 en volumes). Les frac-
tions contenant le produit sont réunies et concentrées sous pression
réduite (2,7 kPa) a une température voisine de 40C. On obtient ainsi
2 g de N'-[N-(3~-acétoxylup-20(29)-en-28-oyl)-8-aminooctanoyl]-3-
amino propionate de méthyle sous la forme d'une meringue hlAnche (Rf
= 0,62 ; chromatographie sur couche mince de silice ; éluant : acé-
tate d'éthyle)
~ emn l e 11
720 mg de [N-(3~-acétoxylup-20(29)-en-28-oyl)-8-aminooctanoyl-méthyl-
amino]-acétonitrile et 1,1 g d'azoture de tributylétain sont mis en
0 94/26725 ~ 1 ~ 2 7 Q 3 PCT~R94/00533
, ,.
solution dans 15 cm de 1,2-diméthoxy~thane. Le mélange est chauffé
au reflux pendant 3 jours. Le mélange réactionnel est dilué avec 40
cm d'eau distillée et 15 cm d'acide chlorhydrique 2N, puis extrait
avec 3 fois 80 cm de dichlorométhane. La phase organique est séchée
sur du sulfate de magnésium, filtrée et évaporée à sec sous pression
réduite (2,7 kPa) a une température voisine de 40C. Le résidu obtenu
(1,8 g) est chromatographié sur une colonne de silice (0,02-0,045 mm)
éluée avec un mélange de chloroforme, méthanol et ~oni~que à 20 %
(12-3-0,5 en volumes). Les fractions contenant le produit sont
réunies et concentrées sous pression réduite (2,7 kPa) à une
température voisine de 40C. Le résidu est lavé 5 fois avec 20 cm de
pentane, puis séché sous pression réduite (15,5 Pa) à 20C. On
obtient ainsi 820 mg de 5-[N-(3~-acétoxylup-20(29)-èn-28-oyl)-8-
aminooctanoyl-~etllyl-alnillométhyl]-tétrazole sous la forme d'une
meringue blanche (Rf = 0,33 ; chromatographie sur couche mince de
silice ; éluant : chloroforme-méthanol-~m~oni~que à 20 % (12-3-0,5 en
volumes)).
A une solution de 800 mg de 5-[N-(3~-acétoxylup-20(29)-èn-28-oyl)-8-
aminooctanoyl-méthyl-A~inom~thyl]-tétrazole, 2,7 cm de soude 5N, 10
cm3 de tétrahydrofurane et 20 cm de méthanol est agitée 48 heures à
une température voisine de 20c. Le mélange est acidi~ié avec de
l'acide chlorhydrique lN, et dilué avec~100 cm d'eau distillée. Le
mélange est agité 1 heure à 20C, filtré, lavé jusqu'à neutralité par
de l'eau distillé, puis par 5 fois 5 cm de pentane, et s~ché sous
pression réduite ( 15,5 Pa) à 20C. Le produit obtenu (740 mg) est
chromatographié sur une colonne de silice (0,02-0,045 mm) éluée avec
un melange de chloroforme, méthanol et ammoniaque à 20% (12-3-0,5 en
volumes). Les fractions contenant le produit sont réunies et
concentrées sous pression réduite (2,7 kPa) à une température voisine
de 40C. Le résidu est repris par 15 cm d'oxyde d'isopropyle,
filtré, et séché sous pression réduite (15,5 Pa) à 20C. On obtient
ainsi 360 mg de 5-[N-(3~-hydroxylup-Z0(29)-èn-28-oyl)-8-
aminooctanoyl-méthyl-aminométhyl]-tétrazole, sous la forme d'un
solide blanc fondant vers 151C.
Le [N-(3~-acétoxylup-20(29)-èn-28-oyl)-8-aminooctanoyl-méthyl-amino]-
acétonitrile peut etre obtenu de la façon suivante :
W O 94/26725 PCT~R94/00533
~ 36
A une solution de 1,2 g de [N-(3~-acétoxylup-20(29)-èn-28-oyl)-8-
aminooctanoyl-méthyl-amino~-acétamide dans 70 cm de tétrahydrofurane
sec, on ajoute goutte à goutte 1 cm d'anhydride trifluoroacétique.
La solution est agitée 15 heures à température ambiante, puis
neutralisée par une solution saturée de bicarbonate de sodium. Le
tétrahydrofurane est évaporé sous pression réduite (2,7 kPa) à 40C.
Le mélange est ensuite repris par 200 cm d'eau distillée et extrait
par 3 fois 80 cm de dichlorométhane. Les phases organiques sont ré-
unies, lavées par 3 fois 50 cm d'eau distillée, séchée sur du sul-
fate de magnésium et évaporée à sec sous pression réduite (2,7 kPa) àune température voisine de 40C. Le résidu obtenu (1,0 g) est chroma-
tographié sur une colonne de silice (0,02-0,045 mm) éluée avec un
mélange d'acétate d'éthyle et de cycloh~x~ne (30-70 en volumes). Les
fractions contenant le produit sont reunies et concentrées sous pres-
sion réduite (2,7 kPa) à une température voisine de 40C. On obtientainsi 720 mg de [N-(3~-acétoxylup-20(29)-èn-28-oyl)-8-aminooctanoyl-
méthyl-amino]-acétonitrile sous la forme d'une meringue blAn~h~ (Rf -
0,32 ; chromatographie sur couche mince de silice ; éluant
cyclohexane-acétate d'éthyle (50-50 en volumes)).
Le [N-(3~-acétoxylup-20(29)-èn-28-oyl)-8-aminooctanoyl-méthyl-amino]-
acétamide peut être obtenu de la manière suivante :
A une solution de ,00 mg d'acide [N-(3~-acétoxylup-20(29)-èn-28-oyl)-
8-aminooctanoyl-métllyl-amino]-acétique dans 100 cm de di-
chlorométhane, on ajoute 0,72 cm d'A~oniAque à 20 %, 150 mg d'hy-
drate de 1-hydroxybenzotriazole, et 380 mg de chlorhydrate de 1-(3-
diméthylaminopropyl)-3-éthylcarbodiimide. La solution est agitée
15 heures à température ambiante puis la phase organique est
décantée, lavée avec 30 cm d'acide chlorhydrique 1N puis avec 5 fois
40 cm d'eau distillée, séchée sur du sulfate de magnésium, filtrée
et concentrée à sec sous pression réduite (2,7 kPa) à une température
voisine de 40C. Le résidu obtenu (630 mg) est chromatographié sur
une colonne de silice (0,02-0,045 mm) éluée avec un mélanqe de di-
chlorométhane et de méthanol (97-3 en volumes). Les fractions conte-
nant le produit sont réunies et concentrées sous pression réduite
(2,7 kPa) à une température voisine de 40C. On obtient ainsi 500 mg
de [N-(3~-acétoxylup-20(29)-èn-28-oyl)-8-aminooctanoyl-méthyl-amino]-
O 94/26725 ~ ~ ~ 2 7 ~ 3 PCT~R94/00533
37
acétamide sous la forme d'une meringue blanc crème (Rf - 0,32
chromatographie sur couche mince de silice i éluant : dichloromé-
thane-méthanol (95-5 en volumes)).
L'acide [N-(3~-acétoxylup-20(29)-èn-28-oyl)-8-aminooctanoyl-méthyl-
- 5 amino]-acétique peut être obtenu de la manière suivante :
Une solution de 167 mg de sarcosine, 406 mg de chlorotriméthylsilane
et 75 cm de dichlorométhane est chauffée au reflux pendant
15 heures. Cette solution est ensuite ajoutée à une solution conte-
nant 1 g d'acide N-(3~-acétoxylup-20(29)-èn-28-oyl)-8-aminoocta-
10 noïque, 239 mg d'hydrate de 1-hydroxybenzotriazole, 600 mg de chlor-
hydrate de 1-(3-diméthylaminopropyl)-3-éthylcarbodiimide, et 75 cm3
de dichlorométhane. On ajoute ensuite 1,5 cm de triéthylamine en
solution dans 10 cm de dichlorométhane. La solution est agitée
50 heures à température ambiante puis le mélange réactionnel est lavé
15 par 70 cm d'acide chlorhydrique 2N, puis par 3 fois 80 cm d'eau
distillée, séché sur du sulfate de magnésium, filtré et concentré à
sec sous pression réduite (2,7 kPa) à une température voisine de
40C. Le résidu obtenu (2,4 q) est chromatographié sur une colonne de
silice (0,02-0,045 mm) éluée avec un mélange de dichlorométhane,
20 méthanol et ammoniaque à 20 ~ (12-3-0,5 en volumes) Les fractions
contenant le produit sont réunies et concentrées sous pression ré-
duite (2,7 kPa) à une température voisine de 40C. On obtient ainsi
712 mg d'acide [N-(3~-acétoxylup-20(29)-èn-28-oyl)-8-aminooctanoyl-
méthyl-amino]-acétique sous la forme d'une meringue hlAn~he (Rf
0,32 ; chromatographie sur couche mince de silice ; éluant
chloroforme-méthanol-AmmoniAque à 20 ~ (12-3-0,5 en volumes)).
E8~mnl e 12
A une solution de 1,4 g de N-(3~-acétoxylup-20(29)-èn-28-oyl)-8-
aminooctylcarbamoylacétonitrile dans 20 cm de dichlorométhane, on
30 ajoute 0,8 cm d'azoture de triméthylsilyle et 270 mg de trichlorure
d'aluminum. Le mélange réactionnel est agité pendant 72 heures à 20C
puis versé goutte à goutte en 30 minutes sur 50 cm d'une solution
aqueuse à 10 % d'hydrogénocarbonate de sodium. La phase organique est
décantée, la phase aqueuse est extraite par 2 fois 50 cm3 de
dichlorométhane. Les phases organiques réunies sont lavées 3 fois
-
W O 94l26725 PCT~R94/00533 ~
~2~ ~3 38
avec 50 cm d'eau distillée, séchées sur sulfate de magnésium puis
évaporée à sec sous pression réduite (2,7 kPa) a une température voi-
sine de 30C. Le résidu obtenu (1,9 g) est chromatographié sur co-
lonne de silice (0,02-0,045 mm), éluée avec un mélange de chloro-
forme, méthanol et ammoniaque à 20 % (24-6-1 en volumes). Les frac-
tions contenant le produit sont réunies et concentrées sous pression
réduite (2,7 kPa) à une température voisine de 40C. On obtient ainsi
1 g de 5-[N-(3~-acétoxylup-20(29)-èn-28-oyl)-8-aminooctylcarba-
moylméthyl]-tétrazole sous la forme d'une meringue hl~nrh~ (Rf =
0,25 ; chromatographie sur couche mince de silice ; éluant : chloro-
forme-méthanol-ammoniaque à 20 % (24-6-1 en volumes)).
Une solutlon de 1 g de 5-[N-(3~-acétoxylup-20(29)-èn-28-oyl)-8-ami-
nooctylcarbamoylméthyl~-tétrazole, 5,4 cm de soude N, 20 cm de
tétrahydrofurane et 40 cm de méthanol est agitée 72 heures à une
température voisine de 20C. Le mélange est versé dans 10 cm d'une
solution d'acide chlorhydrique N puis dilué avec 250 cm d'eau dis-
tillée. Après 1 heure sous agitation à une température voisine de
20C, le solide est filtré et lavé 4 fois par 10 cm d'eau distillée
puis dissout dans 30 cm d'éthanol. La solution est filtrée, le fil-
trat est évaporé sous pression réduite (2,7 kPa) à une température
voisine de 50C Le résidu obtenu (140 mg) est repris par 15 cm
d'oxyde d'isopropyle Le solide obtenu est fitré, lavé par 2 fois
5 cm d'oxyde d'isopropyle puis séché sous vide (13,5 Pa) à une tem-
pérature voisine de 40C. On obtient ainsi 700 mg de 5-[N-(3~-hy-
25 droxylup-20(29)-èn-28-oyl)-8-aminooctylcarbamoylméthyl]-tétrazole
sous la forme d'un solide blanc fondant vers 175C
Le N-(3~-acétoxylup-20(29)-èn-28-oyl)-8-aminooctylcarbamoylacétoni-
trile peut être obtenu de la manière suivante :
A une solution de 3,6 g de N-[3~-acétoxylup-20(29)-èn-28-oyl]-1,8-
30 ~i~minooctane~ 500 mg d'acide cyanoacétique, 1,06 g d'hydrate de
1-hydroxybenzotriazole, 2,76 g de chlorhydrate de 1-(3-dimethylamino-
propyl)-3-éthylcarbodiimide dans 75 cm de dichlorométhane, est
ajouté en environ 10 minutes, 3,6 cm de triéthylamine. La solution
est agitée pendant 15 heures à une température voisine de 20C puis
le mélange réactionnel est lavé 3 fois avec 40 cm d'eau distillée.
~ O 94/26725 21~62~ ~ ~ PCT~R94/00533
39
La phase organique est ensuite séchée sur du sulfate de magnésium et
évaporée à sec sous pression réduite (2,7 kPa) à une température voi-
sine de 40C. Le résidu obtenu (4,8 g) est chromatographié sur co-
lonne de silice (0,02-0,045 mm), éluee avec un mélange de dichloromé-
thane et de métharlol (99-1 en volumes). Les fractions contenant le
produit sont réunies et concentrés sous pression réduite (2,7 kPa) à
une température voisine de 40C. On obtient ainsi 3,7 g de N-(3~-
acétoxylup-20(29)-èn-28-oyl)-8-aminooctylcarbamoylacétonitrile sous
la forme d'une meringue jaune (Rf = 0,40 ; chromatographie sur couche
mince de silice ; éluant : chloroforme-méthanol-~mm~niaque à 20 %
(12-3-0,5 en volumes))
Le N-(3~-acéto:~ylup-20(29)-erl-28-oyl)-1,8-diaminooctane peut être
obtenu de la ma~ re suivante :
50 cm d'une solution dans le dichlorométhane de chlorure de 3~-acé-
15 toxylup-20(29)-èn-28-oyle (préparée à partir de 5 g d'acide 3~-acé-
toxylup-20(29)-èn-28-oïque) sont ajoutés goutte à goutte en environ 3
heures à une solution de 8,6 g de 1,8~ ;n~octane dans 20 cm de
dichlorométhane. Le mélange réactionnel est agité durant 2 heures à
une température voisine de 20C puis concentré à sec sous pression
réduite (2,7 kPa) à une température voisine de 40C. Le résidu obtenu
est chromatographié sur colonne de silice (0,02-0,045 mm), éluée avec
un mélange de chloroforme, méthanol et A -ni~que à 20 % (24-6-1 en
volumes). Les fractions contenant le produit sont réunies et concen-
trées sous pression réduite (2,7 kPa) à une température voisine de
25 40C. On obtient ainsi 5,9 g de N-[3~-acétoxylup-20(29)-èn-28-oyl]-
1,8-diaminooctane sous la forme d'une meringue jaune (Rf G 0~ 25 ;
chromatographie sur couche mince de silice ; éluant : chloroforme-
méthanol-ammonlaque à 20 ~ (24-6-1 en volumes)).
~n le 13
30 A une solution de 2,1 g de N-(3~-acétoxylup-20(29)-èn-28-oyl)-7-ami-
noheptycarbamoylacétonitrile dans 25 cm de dichlorométhane, on
ajoute 1,07 cm d'azoture de triméthylsilyle et 410 mg de trichlorure
d'aluminum. Le mélange réactionnel est agité pendant 72 heures à 20C
puis versé goutte à goutte en 30 minutes sur 50 cm d'une solution
aqueuse à 10 % d'hydrogénocarbonate de sodium. La phase organique est
W O 94/26725 I PCT~R94/00533 ~
211~2703 40
décantée, la phase aqueuse est extraite par 2 fois 60 cm de
dichlorométhane. Les phases organiques réunies sont lavées 3 fois
avec 50 cm d'eau distillée, séchées sur sulfate de magnésium pUi5
concentrées à sec sous pression réduite (2,7 kPa) ~ une température
voisine de 40C. Le résidu obtenu (2,5 g) est chromatographié sur co-
lonne de silice ~0,02-0,045 mm), éluée avec un mélanqe de chloro-
forme, méthanol et ~nmoni~que à 20 % (24-6-1 en volumes). Les frac-
tions contenant le produit sont réunies et concentrées sous pression
réduite (2,7 kPa) à une température voisine de 40C. On obtient ainsi
1,4 g de 5-[N-(3~-acétoxylup-20(29)-èn-28-oyl)-7-Am1nohPptylcarba-
moylméthyl]-tétrazole sous la forme d'une meringue hl~n~h~ (Rf
0,25 ; chromatographie sur couche mince de silice ; éluant: chloro-
forme-méthanol-ammoniaque à 20 % (24-6-1 en volumes)).
Une solution de l g de 5-rN-(3~-acétoxylup-20(29)-èn-28-oyl)-7-
aminoheptylcarbamoyllnétllyl]-tétrazole, 8 cm de soude N, 25 cm de
tétrahydrofurane et 50 cm de méthanol est agitée 48 heures à une
température voisine de 20C. Le mélange réactionnel est évaporé par-
tiellement sous pression réduite (2,7 kPa) a une température voisine
de 40C jusqu'à l'apparition d'un trouble. Le mélange est alors aci-
difié a un p~ compris entre 1 et 2 par addition d'une solutionaqueuse d'acide chlorhydrique N. Après 2 heures sous agitation à une
température voisine de 20C, le solide est filtré et lavé 5 fois par
5 cm d'eau distillée puis dissout dans 50 cm3 d'éthanol. La solution
est filtrée, le filtrat est evaporé sous pression réduite (2,7 kPa) à
une température voisine de 40C. Le résidu obtenu est repris par
20 cm d'oxyde d'isopropyle. Le solide obtenu est fitré, lavé par 2
fois 5 cm d'oxyde d'isopropyle puis séché sous pression réduite
(13,5 Pa) a une température voisine de 40C. Le résidu obtenu est
chromatograpllié sur colonne de silice (0,02-0,045 mm), éluée avec un
mélange de chloroforme, métllanol et ~ ni~que à 20 % (24-6-1 en
volumes) Les fractions contenant le produit sont réunies et
concentrées sous pression réduite (2,7 kPa) à une température voisine
de 40C. Le solide obtenu est mis en solution dans 20 cm3 de
chloroforme, la solution est filtrée, le filtrat est évaporé sous
pression réduite (2,7 kPa) a une température voisine de 40C. Le
résidu obtenu est repris par 20 cm d'oxyde d'isopropyle. Le solide
obtenu est fitré, lavé par 2 fois 5 cm d'oxyde d'isopropyle puis
O 94/26725 ~ 16 2 ~ ~ 3 PCT~R94/00533
41
séché sous vide (13,5 Pa) a une température voisine de 40C. On
obtient ainsi 900 mg de 5-[N-(3~3-hydroxylup-20(29)-~n-28-oyl)-7-
Aminoheptylcarbamoylméthyl]-tétrazole sous la forme d'un solide blanc
fondant vers 160C.
5 Le N-(3~3-acétoxylup-20(29)-èn-28-oyl)-7-~minohPptylcarbamoyl-acét
nitrile peut être obtenu de la manière suivante :
A une solution de 2,45 g de N-(3~-acétoxylup-20(29)-èn-28-oyl)-1,7-
~i~minoh~ptane~ 340 mg d'acide cyanoacétique, 730 g d'hydrate de
1-hydroxybenzotriazole, 1,9 g de chlorhydrate de 1-(3-diméthylamino-
propyl)-3-éthylcarbodiimide dans 120 cm de dichlorométhane, est
ajouté en environ 10 minutes, 2,5 cm de triéthylamine. La solution
est agitée pendant 15 heures à une température voisine de 20C puis
le mélange réactionnel est lavé 3 fois avec 50 cm d'eau distillée.
La phase organlque est ensuite séchée sur du sulfate de magnésium et
évaporée à sec sous pression réduite (2,7 kPa) à une température voi-
sine de 40C. Le résidu obtenu (2, 7 g) est chromatographié sur co-
lonne de silice (0,02-0,045 mm), éluée avec un mPlAnge de dichloromé-
thane et de méthanol (99-1 en volumes). Les fractions contenant le
produit sont réunies et concentrés sous pression réduite (2,7 kPa) à
20 une température voisine de 40C. On obtient ainsi 3,7 g de N-(3~-
acétoxylup-20(29)-èn-28-oyl) -7-~mi n~hpptylcarbamoylacétonitrile sous
la forme d'une meringue blanche (Rf r 0,40 ; chromatographie sur
couche mince de silice ; éluant : chloroforme-méthanol-~m~on1~que à
20 ~ (12-3-0,5 en volumes)~.
25 Le N-[3~3-acétoxylup-2o(29)-èn-28-oyl]-l~7-~iAminoheptane peut être
obtenu de la manière suivante :
1000 cm d'une solution dans le dichlorométhane de chlorure de 3~3-
acétoxylup-20(29)-èn-28-oyle ~préparée à partir de 30 g d'acide 3~-
acétoxylup-20(29)-èn-28-oïque) sont ajoutés goutte à goutte, en envi-
30 ron 10 heures, à une solution de 46,8 g de 1,7-~i~minohPptane dans
120 cm de dichlorométhane Le mélanqe réactionnel est agité durant
15 heures à une température voisine de 20C puis concentré à sec sous
pression réduite (2, 7 kPa) à une température voisine de 40C. Le ré-
sidu est repris par 2000 cm d'eau distillée. Le solide est filtré,
lavé par 3 fois 200 cm d'eau distillée, puis mis en solution dans
W O 94/26725 ~16 % 7 0 3 42 PCT~R94/00533 ~
500 cm d'éthanol. La solution est filtrée, le solide est lavé 2 fois
avec 50 cm d'éthanol et les filtrats sont évaporés à sec sous pres-
sion réduite (2,7 kPa) a une température voisine de 50C. Le résidu
obtenu est chromatographié sur colonne de silice ~0,02-0,045 mm),
éluée avec Ull mélange de chloroforme, méthanol et ~m~on;~que à 20 %
(24-6-1 en volumes). Les fractions contenant le produit sont réunies
et concentrées sous pression réduite (2,7 kPa) à une température voi-
sine de 40C. On obtient ainsi 28 g de N-(3~-acétoxylup-20(29)-èn-28-
oyl)-1,7-~ in~heptane sous la forme d'une meringue jaune (Rf =
0,25 ; chromatographie sur couche mince de silice ; éluant : chloro-
forme-méthanol-ammoniaque à 20 ~ (24-6-1 en volumes)).
~em~le 14
A une solutlon de 2,3 g de N-(3~-acétoxylup-20(29)-èn-28-oyl)-7-ami-
noheptylcarbamoylpropionitrile dans 25 cm3 de dichlorométhane, on
ajoute 1,30 cm d'azoture de triméthylsilyle et 450 mg de trichlorure
d'aluminium. Le mélange réactionnel est agité pendant 48 heures à
20C puis 32 heures à une température proche du reflux. On ajoute à
nouveau 1,30 cm d'azoture de triméthylsilyle et 450 mg de trichlo-
rure d'aluminium et on chauffe 24 heures à une température voisine du
reflux. Le mélange réactionnel est refroidi à 20C puis versé goutte
à goutte en 30 mlnutes sur 40 cm d'une solution aqueuse à 10 % d'hy-
drogénocarbonate de sodium. La phase organique est décantée, la phase
aqueuse est extraite par 3 fois 50 cm de dichlorométhane. Les phases
organiques réunies sont lavées 3 fois avec 50 cm d'eau distillée,
séchées sur sulfate de magnésium puis évaporées à sec sous pression
réduite ~2,7 kPa) à une température voisine de 40C. Le résidu obtenu
(2,5 g) est traité par 25 cm d'oxyde d'isopropyle, puis lavé 3 fois
par 5 cm d'oxyde d'isopropyle. Le solide obtenu (1,8 g) est mis en
solution dans 25 cm de dichlorométhane. La solution est filtrée sur
celite. Le filtrat est concentré à sec sous pression réduite
(2,7 kPa) à une température voisine de 40C. Le solide est traité par
20 cm d'oxyde d'isopropyle puis séché sous pression réduite
(2,7 kPa) à une température voisine de 40C. On obtient ainsi 1,5 g
de 5-[N-(3~-acétoxylup-20(29)-èn-28-oyl)-7-A~i noheptylcarbamoylé-
thyl]-tétraæole sous la forme d'une meringue hl~n~he (Rf 0,25 ;
WO 94/26725 ~ 1 ~ 2 7 0 3 PCT/FR94/00533
. . .
43
chromatographie sur couche mince de silice; éluant: chloroforme-
méthanol-~ ni~que à 20 % ~24-6-1 en volumes)) .
Une solution de 1 g de 5-[N-(313-acétoxylup-20(29) -èn-28-oyl) -7-
aminoheptylcarbamoyléthyl]-tétrazole, 20 cm de soude N, 25 cm de
tétrahydrofurane et 50 cm de méthanol est agitée 72 heures à une
température voisine de 20C. Le mélange dilué par 250 cm d'eau dis-
tillée et acidifié à pl~ l par addition d'une solution aqueuse d'acide
chlorhydrique N Après 1 heure sous agitation à une température voi-
sine de 20C, le solide est filtré et lavé 5 fois par 10 cm d'eau
distillée puis traité par 5 fois 40 cm dlacétate d'éthyle. Les
phases organiques réunies sont évaporées sous pression réduite
(2,7 kPa) à une température voisine de 40C. Le résidu obtenu
(700 mg) est chromatographié sur colonne de silice (0,02-0,045 mm),
éluée avec un mélange de chloroforme, méthanol et ~mmor~i aque à 20 %
(24-6-l en volumes). Les fractions contenant le produit sont réunies
et concentrées sous pression réduite (2,7 kPa) à une température
voisine de 40C. Le solide obtenu (600 mg) est traité par 15 cm
d'oxyde d'isopropyle, le solide est filtré, lavé 2 fois avec 5 cm
d'oxyde d'isopropyle puis séché sur hydroxyde de potassium. On
obtient ainsi 140 mg de 5-tN- (313-hydroxylup-20(29) -èn-28-oyl) -7-
aminoheptylcarbamoyléthyle]-tétrazole sous la forme d'un solide blanc
fondant vers 110C.
Le N-(3f~-acétoxylup-20(29)-èn-28-oyl)-7-aminoheptylcarbamoylpropio-
nitrile peut être obtenu de la manière suivante:
A une solution de 2,45 g de N-(3~3-acétoxylup-20 (29) -èn-28-oyl) -1,7-
diaminoheptane, 400 mg d'acide 2-cyanopropionique, 730 g d'hydrate de
1-hydroxybenzotriazole, 1,9 g de chlorhydrate de 1-(3-diméthylamino-
propyl)-3-éthylcarbodiimide dans 120 cm de dichlorométhane, est
ajouté en environ 10 minutes, 2,5 cm de triéthylamine. La solution
est agitée pendant 15 heures à une température voisine de 20C. Le
mélange réactionnel est lavé 3 fois avec 50 cm d'eau distillée. La
phase orqanique est ensuite séchée sur du sulfate de magnésium et
évaporée à sec sous pression réduite (2,7 kPa) à une température voi-
sine de 40C. Le résidu obtenu (3,5 g) est chromatographié sur co-
lonne de silice (0,02-0,045 mm), éluée avec un mélange de dichloromé-
W O 94/26725 , PCT~R94/00533
~1 &~7~3 44
thane et de méthanol (99-l en volumes). Les fractions contenant le
produit sont réunies et concentrés sous pression réduite (2,7 kPa) à
une température voisine de 40C. On obtient ainsi 2,3 g de N-(3~-
acétoxylup-20(29)-èn-28-oyl)-7-Aminoheptylcarbamoylpropionitrile sous
la forme d'une meringue blanche (Rf = 0,40 ; chromatographie sur
couche mince de silice ; éluant : chloroforme-méthanol-A~niAque à
20 ~ (12-3-0,5 en volumes)).
Le N-(3~-acétoxylup-2o(29)-èn-28-oyl)-1~7-~iAmin~h~ptane peut être
obtenu de la manière suivante :
1000 cm3 d'une solution dans le dichlorométhane de chlorure de 3~-
acétoxylup-20(29)-èn-28-oyle (préparée à partir de 30 g d'acide 3~-
acétoxylup-20(29)-èn-28-oïque) sont ajoutés goutte à goutte en envi-
ron 10 heures à une solution de 46,8 g de 1,7-~;~minoh~ptane dans
120 cm de dichlorométhane. Le mélange réactionnel est agité durant
15 heures à une température voisine de 20C puis concentré à sec sous
pression réduite (2,7 kPa) à une température voisine de 40C. Le ré-
sidu est repris par 2000 cm3 d'eau distillée. Le solide est filtré,
lavé par 3 fois 200 cm d'eau distillée, puis mis en solution dans
500 cm d'éthanol. La solution est filtrée, le solide est lavé 2 fois
avec 50 cm3 d'éthanol et les filtrats sont évaporés à sec sous pres-
sion réduite (2,7 kPa) à une température voisine de 50C. Le résidu
obtenu est chromatoqraphié sur colonne de silice (0,02-0,045 mm),
éluée avec un mélange de chloroforme, méthanol et ammoniaque à 20 ~
(24-6-1 en volumes) Les fractions contenant le produit sont réunies
et concentrées sous pression réduite (2,7 kPa) a une température voi-
sine de 40C. On obtient ainsi 28 g de N-(3~-acétoxylup-20(29)-èn-28-
oyl)-1,7-~iArinoheptane sous la forme d'une meringue jaune (Rf 2
0,25 ; chromatographie sur couche mince de silice ; éluant : chloro-
forme-méthanol-~ -niAque à 20 ~ (24-6-1 en volumes)).
Exemele 15
En opérant par analogie avec l'exemple 10, le 5-{N-{[3~-hydroxylup-
20(29)-èn-28-oyl]-8-aminooctanoyl}-2-aminopropyl (R,S)} tétrazole a
été préparé a partir de l'acide N-(3~-acétoxylup-20(29)-èn-28-oyl)-8-
aminooctanoyl}-2-aminobutanoïque (R,S)}. Le produit se présente sous
la forme d'une poudre blanche; pF=130C.
W O 94/26725 2 ~ 6 2 7 o 3 E~r~Rg4/00533
L'acide N-(3~-acétoxylup-20(29)-èn-28-oyl)-8-aminooctanoyl}-3-
aminobutanoïque (R,S)} peut être préparé par analogie avec
l'acide [N-(3~-acétoxylup-20(29)-èn-28-oyl)-8-~inooctanoyl}-méthyl-
amino~acétique (exemple 11). L'acide N-(3~-acétoxylup-20(29)-èn-28-
oyl)-8-aminooctanoyl}-3-aminobutanoïque (R,S)} se présente sous la
forme d'une meringue blanche (Rf=0,44 ; chromatographie sur couche
mince de silice ; éluant : dichloromethane, acétate d'éthyle et
méthanol 40-40-20 (en volumes)).
~ pmnle 16
En opérant par analogie avec l'exemple 13, le 5-{N-[3~-hydroxylup-
20(29)-èn-28-oyl]-7-aminoheptyl-(N-méthylcarbamoyl)}méthyltétrazole a
été préparé à partir de N-méthyl N-(3~-acétoxylup-20(29)-èn-28-oyl)-
1,7-~ inoheptane, pFs183C
Le N-méthyl-N'-(3~-acétoxylup-20(29)-èn-28-oyl)-1,7-di~minoheptane
peut être obtenu de la manière suivante :
A une solution de 15,2 g d'iodure de N-méthyl 2-méthylthiobenzothia-
zolium dans 300 cm3 de dichlorométhane, on ajoute une solution de
28,7 g de N-(3~-acétoxylup-20(29)-èn-28-oyl)-1,7-~;~m;noheptane dans
50 cm3 de dichlorométhane puis en approximativement 15 minutes, 6,6
cm3 de triéthylamine. La solution est agitée à une température
voisine de 20C durant 2 heures puis le mélange réactionnel est
concentré sous pression atmosphérique à une température voisine de
60C. A l'huile obtenue (35,6 g), on ajoute 150 cm3 d'iodure de
méthyle et le mélanqe réactionnel est porté durant 30 heures à une
température voisine du reflux. Le mélange réactionnel est concentré
sous pression réduite (2,7 kPa) à une température voisine de 50C. Le
résidu huileux est repris par 300 cm3 d'oxyde de diéthyle et tritur~.
On obtient ainsi 39,8 g d'un solide orangé qui est mis en solution
dans 100 cm3 de dichlorométhane. A la solution obtenue, on ajoute 4,3
cm3 de n-butylamine. Le miélange réactionnel est agité 4 heures à une
température voisine de 25C, puis dilué par 300 cm3 de
dichlorométhane et lavé par 450 cm3 d'eau distillée. La phase
organique est ensuite séchée sur du sulfate de magnésium et
concentrée à sec sous pression réduite (2,7 kPa) à une température
W O 94/26725 PCT~R94/00533
~627~3 46
voisine de 40C. Le résidu obtenu est chromatographié sur une colonne
de silice (0,02 - 0,045 mm) éluée avec un mélange de dichlorométhane
et de méthanol 19-1 (en volumes). Les fractions contenant le produit
sont réunies et concentrées sous pression réduite (2,7 kPa) ~ une
température voisine de 40C. On obtient ainsi 7,1 g (26%) de N-
méthyl-N'-(3~-acétoxylup-20(29)-èn-28-oyl)-1,7-~i~minoheptane sous la
forme d'une meringue blAnch~, pF~158C.
~m~le 17
En opérant par analogie avec l'exemple 14, on prépare le 5-{3-[N-(3~-
hydroxylup-20(29)-èn-28-oyl)-7-AminohPptylcarbamoyl]-2,2-diméthylpro-
pyl}tétrazole sous forme d'un solide blanc ; pF = 155C.
La présente invention concerne également les compositions pharmaceu-
tiques contenant au moins un produit de formule générale (I) éven-
tuellement sous forme de sel, ~ l'état pur ou sous forme d'une asso-
ciation avec un ou plusieurs diluants ou adjuvants compatibles et
pharmaceutiquement acceptables, ou avec un autre agent destiné au
traitement du SIDA, un agent antiviral, immunomndulateur ou antimi-
crobien.
La composition selon l'invention est capable de maintenir en vie les
cellules infectées par un virus HIV et donc de réduire la progression
vers le SIDA ou de diminuer sa gravité chez les sujets déja infectés
en réduisant la mortalité des cellules infectées. Les compositions
peuvent être utilisées par voie orale, parentérale ou rectale.
Les compositions peuvent être utilisées à titre curatif ou à titre
préventif chez des sujets présentant une immunodéficience et/ou in-
fectés par un virus HIV. Bien entendu, la constitution de ces compo-
sitions sera adaptée au cas particulier du tractus digestif des immu-
nodéprimés.
Comme compositions solides pour administration orale peuvent etre
utilisés des comprimés, des pilules, des gélules, des poudres ou des
granul~s. Dans ces compositions, le produit actif selon l'invention
est mélangé à un ou plusieurs diluants ou adjuvants inertes, tels que
s~h~Arose, lactose ou amidon.
W O 94/26725 2 ~ ~ 2 7 ~ 3 PCT~R94/00s33
47
Ces compositions peuvent comprendre des substances autres que les
diluants, par exemple un lubrifiant tel que le stéarate de magnésium
ou un enrobage destiné à une libération contrôlée.
Comme compositlons liquides pour administration orale, on peut
utiliser des solutions pharmaceutiquement acceptables, des suspen-
sions, des émulsions, des sirops et des élixirs contenant des
diluants inertes tels que l'eau ou l'huile de paraffine. Ces
compositions peuvent également comprendre des substances autres que
les diluants, par exemple des produits mouillants, édulcorants ou
aromatisants.
Les compositions pour administration parentérale, peuvent être des
solutions stériles ou des émulsions. Comme solvant ou véhicule, on
peut employer le propylèneqlycol, un polyéthylèneglycol, des huiles
végétales, en particulier l'huile d'olive, des esters organiques
injectables, par exemple l'oléate d'éthyle.
Ces compositions peuvent également contenir des adjuvants, en
particulier des agents mouillants, isotonisants, émulsifiants,
dispersants et stabilisants.
La stérilisation peut se faire de plusieurs façons, par exemple à
l'aide d'un filtre bactériologique, par irradiation ou par chauffage.
Elles peuvent également être préparées sous forme de compositions
solides stériles qui peuvent être dissoutes au moment de l'emploi
dans de l'eau stérile ou tout autre milieu stérile injectable.
Les compositions par administration rectale sont les suppositoires ou
les capsules rectales, qui contiennent outre le principe actif, des
excipients tels que le beurre de cacao, des glycérides semi-
synthétiques ou des polyéthylèneglycols.
En thérapeutique humaine, le médecin déterminera la posologie qu'il
estime la plus appropriée en fonction d'un traitement préventif ou
curatif, en fonction de l'âge, du poids, du degré de l'infection et
des autre facteurs propres au sujet à traiter. Généralement, les
doses sont comprises entre 10 et 100 mg/kg par voie orale pour un
adulte.
W O 94/26725 PCT~R94/00533
3 48
La présente invention concerne également les associations constituées
d'un ou plusieurs dérivés du lupane de formule générale (I), et/ou le
cas échéant leurs sels, et d'un autre principe actif connu pour son
activité anti-rétrovirus, éventuellement en présence d'excipients
pharmaceutiquement acceptables.
Les agents anti-rétrovirus pouvant être associés sont choisis parmi
des agents compatibles et inertes vis-à-vis du dérivé du lupane de
formule générale (I). A titre non limitatif ces agents sont choisis
parmi des inhibiteurs de la reverse transcriptase [zidovudine (AZT),
didanosine (DDI), didéoxycytidine (DDC), TIBO, névirapine, PMEA, D4T,
pyridones, ~-APA, HEPT... ], parmi des inhibiteurs de la protéase
[comme par exemple le RO 31-8959 ou le A 77003], ou parmi des inhibi-
teurs de la protélne tat rcomme par exemple le RO 24-7429].
L'exemple suivant illustre une composition selon l'invention.
Exempl~
On prépare selon la technique habituelle des comprimés de produit
actif ayant la composition suivante :
- 5-[N'-[N-[3~-hydroxylup-20(29)-en-28-oyl]-8-aminooctanoyl]-
Aminom~thyl]-tétrazole , 100 mg
- amidon 332 mg
- silice 120 mg
- stéarate de magnésium 12 mg