Note: Descriptions are shown in the official language in which they were submitted.
' D-20,012 2 1 6 2 7 4 0
-- 1 --
OXYGEN-SELECTIVE SORBENTS
Background of the Invention
Field of the Invention - The invention relates to
the recovery of oxygen from gas mixtures. More
particularly, it relates to oxygen-selective sorbents
useful for separating or purifying oxygen-containing
gas mixtures.
Description of the Prior Art - For many years, air
has been separated by cryogenic distillation, for which
operating temperatures are set by the vapor-liquid
equilibrium of the liquefied mixtures. Air may also be
separated by sorption processes that operate at
temperatures that are set by the gas-solid equilibrium
of the sorbents. Cryogenic separation plants are
capital intensive, especially for production rates
below several hundred tons per day. More recently,
adsorption processes based on nitrogen-selective
zeolite adsorbents have been used at such production
rates, as well as much lower rates. Considerable
progress has been made in reducing product costs for
such noncryogenic pressure swing adsorption (PSA)-type
processes. However, such nitrogen-selective sorbents
are required to handle most of the feed air passed to
adsorption processes, and the available selectivities
of the nitrogen-selective sorbents impose process
restrictions and limit feed separation.
Oxygen-selective sorbents can constitute effective
materials for sorption processes that do not require
cryogenic temperatures. Such oxygen-selective
materials can reduce the size of sorbent inventory
required and make possible process cycle
simplifications. Oxygen-selective sorbents are
D-20,012 216~74D
-- 2
especially appropriate for sorption processes that
produce nitrogen. Such sorbents, with good sorption
capabilities and high selectivities for oxygen, can
reduce the cost of noncryogenic nitrogen production
significantly. Oxygen-selective sorbents can lead to
lower sorbent inventory, simpler processes with less
equipment, and higher purity nitrogen with less power
consumption.
It will be appreciated that oxygen-selective
sorbents with good sorption capacities and high
selectivities can provide attractive alternatives to
post-purification techniques for low purity nitrogen
and crude argon, presently accomplished by
chemisorption on a finely divided metal, such as
palladium, or by cryogenic distillation. The
chemisorption technique has an added cost factor
associated with the requirement for hydrogen to
regenerate the chemisorbent. The cryogenic
distillation approach requires the use of a large,
costly distillation column to remove the oxygen
impurity in the post-purification treatment.
Two different classes of oxygen-selective sorbents
are known in the art for oxygen separation or removal
purposes, said sorbents differing in the mechanism of
separation. "Rate-selective" adsorbents discriminate
molecules being sorbed based on critical dimension, so
that smaller molecules, such as oxygen, adsorb and
desorb faster than larger ones, such as nitrogen.
Several types of materials exhibit rate selectivity,
but oxygen will always be selectively adsorbed in
preference to nitrogen. "Equilibrium-selective"
sorbents discriminate molecules interacting with them
based on equilibrium affinity, leading to either
D-20,012 2162~ 4~
nitrogen or oxygen selectivity. Nitrogen selectivity
is observed, for example, for specific physical
adsorption on zeolites, such as 5A or 13X molecular
sieve material, while oxygen selectivity is observed
with mild chemical reactions on numerous cobalt
complexes.
With respect to rate-type oxygen-selective
sorbents, carbon molecular sieves have been used to
produce nitrogen commercially in PSA-type processes
since the early 1980's. These adsorbents are amorphous
carbons having sharper pore size distributions than
most carbons activated for gas adsorption. The
preparation and characterization of carbon molecular
sieves, as well as processes for their use, are well
known and described in the art.
Certain hexacyano compounds, such as Ce[Fe(CN6)],
exhibit rate selectivity for oxygen over nitrogen, and
their utility for separating gas mixtures containing
oxygen has likewise been described in the art. These
hexacyano compounds are crystalline solids that are
similar to zeolites in some structural characteristics
and adsorptive properties, but the compositions and
chemical structures nevertheless differ from those of
said zeolites.
Some small-pore zeolites, such as 4A material,
exhibit rate-selectivity for oxygen over nitrogen at
cycle times shorter than the usual, that is, in seconds
rather than minutes. Zeolites with larger pores, on
the other hand, e.g. sodium mordenite LP, can be
modified chemically to yield rate-selective adsorbents.
With respect to equilibrium-type oxygen-selective
sorbents, the reversible reactions of oxygen with
condensed materials may be classified into two groups,
D-20,012 21627 ~~
namely those in which the O=O double bond is broken,
and those in which this bond remains intact. Oxygen
reaction with oxidic materials are typical of the first
group. Examples of such reactions are known with solid
oxides and with molten nitrates. Temperatures hundreds
of degrees above ambient are required to achieve
practical reaction rates in such processing, so that
energy recovery is essential, and applications of this
equilibrium-type reaction are favored in high
temperature processes, e.g. in steel making.
Numerous transition element complexes (TEC's) are
known to react reversibly at or below ambient
temperatures without breaking the O=O double bond. The
use of TEC's to selectively remove oxygen from its
mixtures with other gases has been disclosed for
solutions of TEC's, for TEC solids or slurries of said
solids, for TEC's supported physically on solid
supports, for TEC's incorporated in zeolites and for
TEC's bound chemically to physical supports. Each of
these approaches for the use of TEC's have been beset
by one or more of the following problems: (1)
insufficient oxygen capacity, (2) slow reaction rates,
and (3) decreasing reactivity with time. None of such
TEC systems has yet been employed in commercially
acceptable embodiments for air separation or oxygen
removal from gas stream applications.
The principal disadvantage of rate-selective
oxygen sorbents is the pronounced decrease in
selectivity encountered over time as a consequence of
the operable separation mechanism. Since short
processing cycles must be employed with rate-selective
sorbents, restrictions are necessarily imposed on the
processing cycles that can be employed, limiting the
~ D-20,012
2 1 6~7 4~
use thereof to higher power requirement cycles than
those of equilibrium cycles. For a given adsorbent
material, higher selectivities can sometimes be
obtained, but at a cost of slower adsorption rates.
The disadvantage of equilibrium-selective oxygen
sorbents relate to the temperatures at which they are
used and the manner in which they are deployed. Oxygen
sorbents of the oxide type require elevated
temperatures sufficiently high to obtain practical
operating rates without too much loss of adsorptive
capacity. The processing cycles employed must be able
to cope with the high reaction enthalpies that pertain,
and with the side reactions initiated by the high
temperatures employed. The availability of
equilibrium-type oxygen-selective sorbents that operate
near or below ambient temperatures would be desirable
in the art, so that such problems associated with high
temperature operations could be avoided.
Among the TEC-based products referred to above,
those that are used in the liquid phase are found to
have a greater potential for deactivation in use than
TEC's deployed in the solid phase. In the liquid
phase, a given TEC can be attacked and oxidized by a
different oxygenated TEC, owing to the mobility of the
TEC's. In solutions, the solvent may also deactivate
the TEC in several ways. In addition, the use of a
solvent imposes further restrictions. Thus, the
solvent must have a low vapor pressure, must be safe to
use in high oxygen concentrations, and yet must not be
too viscous for the desired use.
In light of such factors, solid TEC-based sorbents
are of genuine interest in the art. Early attempts to
use solid TEC-based sorbents have, however, shown
. D-20,012
216~7~
-- 6 --
relatively poor performance for a variety of reasons
related to the chemistry of TEC's and the
characteristics of solid state reactions. Such solid
TEC's are expensive sorbents and, in order to maximize
their adsorptive capacity, they have sometimes been
used without support. However, reactive diffusion in
the pure TEC crystals is very slow at their optimum
operating temperatures. For crystalline, unsupported
TEC's, the critical dimensions of crystals obtained by
solvent removal are on the order of micrometers to
millimeters. Although the surface layers can react
readily, only very small fractions of TEC's occupy said
surface layers. As a result, reaction times of many
hours are required in order to utilize more than 50% of
the TEC's employed in a given application.
In some cases, the TEC's have been deposited
physically on common support materials, such as
diatomaceous earth, alumina and silica gel. Such
efforts have met with little success, because the TEC's
have not been properly deployed. Crystallization on
low surface area materials, for example on catalyst
supports having surface areas of about 50 m2/g, usually
produces crystal sizes such as those described above
with respect to crystalline, unsupported TEC's. Even
if very thin layers are obtained on such low surface
area support materials, the adsorptive capacities of
the resulting composites are too low for practical
commercial operation. On the other hand, physical
deposition on high surface area materials, such as
adsorbents and the like having surface areas on the
order of about 500 m2/g, commonly leads to pore
blockage. Since most of the surface area in such high
surface area supports is in micropores, typically less
D-20,012
2~4~
than 50~, low adsorptive capacities will also result
when such high surface area supports are employed.
Special techniques are required in order to take
advantage of the large internal surface of such high
surface area supports. The physical deposition
techniques heretofore employed have not been well
suited for this task.
In order to disperse the TEC's on the support,
efforts have been made to incorporate them in the
adsorption cavities of zeolites. In type X zeolites,
for example, one TEC per cavity would correspond to a
maximum of on the order of 0.5 to 0.6 mmol/g, if all of
the TEC's were active. For most TEC's, this is
unattainable due to accessibility problems. First, the
size of most TEC's relative to the "window" diameters
of the zeolites makes it difficult to transport or
assemble the TEC's in the zeolite crystal interior.
Second, even if all of the cavities were to be
occupied, transport of oxygen to those in the interior
of the zeolites would be very slow, if it could occur
at all.
Another method considered for dispersing TEC's on
a solid support is to attach them chemically to
specific groups in a polymer chain. The attachment is
normally performed by contacting a solution of the TEC
with either a dissolved or solvent swollen polymer.
With the polymers used so far, poor capacities and/or
low reaction rates have been observed. Two types of
problems have been identified that have yet to be
overcome. First, the high concentrations of attached
TEC's needed for practical capacities lead to use of
crystalline polymers in which diffusion is very slow.
Second, the polymer environment not only retards access
D-20,012 21627 4~
of the feed gas mixture to the bound TEC, but can also
prevent the TEC from reacting with the feed gas mixture
by blocking the oxygen binding site, either physically
or chemically.
It will be appreciated, therefore, that further
improvements in the art are needed to enable adsorption
processes to satisfy the requirements of the art. In
particular, further improvements are desirable with
respect to transitional element complexes in order to
enhance the use thereof as oxygen-selective sorbents,
especially solid TEC's in supported form.
It is an object of the invention, therefore, to
provide improved oxygen-selective sorbents.
It is another object of the invention to provide
improved TEC oxygen-selective sorbents.
It is a further object of the invention to provide
improved solid TEC sorbents deployed on supports.
With these and other objects in mind, the
invention is hereinafter described in detail, the novel
features thereof being pointed out in the appended
claims.
SUMMARY OF THE INVENTION
TEC's are deployed in the solid phase, and are
immobilized and spaced so as to avoid undesired
deactivating reactions that occur with TEC's in the
liquid phase. The TEC's are supported and provide for
ready access of feed gas mixtures thereto at the high
TEC concentrations needed for practical oxygen
adsorption capabilities.
D-20,012 2 l 627 4
BRIEF DESCRIPTION OF THE DRAWINGS
The invention is hereinafter described in detail
with reference to the accompanying drawings in which:
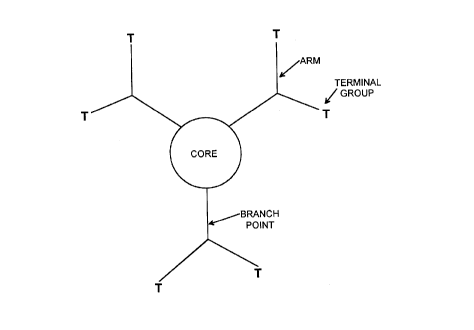
Fig. 1 is a schematic representation of a general
amplifying linking agent, illustrating a trifunctional
core, with each of the three branching into two units;
Fig. 2 is a schematic representation of the
molecular structure of DPy6 and DIm6 linking agents;
Fig. 3 is a schematic representation of the
orientation of an amplifying linking agent with respect
to the surface of a supporting substrate;
Fig. 4 is a representation of the synthesis of a
TEC modified silica surface;
Fig. 5 is a schematic representation of TEC
structures, with Fig. 5(a) being a tetradentate ligand
with exogenous axial base, and Fig. 5(b) being a
pentadentate Ligand;
Fig. 6 is a schematic representation of the
introduction of linking agent and TEC onto the surface
of a substrate, with Fig 6(a) representing simultaneous
loading and Fig. 6(b) representing sequential loading.
DETAILED DESCRIPTION OF THE INVENTION
The objects of the invention are accomplished by
the deployment of solid TEC's on supports in order to
(a) fix large numbers of TEC's in space to provide high
loadings and storage of selectively adsorbed oxygen,
(b) obtain faster rates of adsorption and release of
oxygen by maintaining accessibility and utilization of
the individual TEC's, and (c) increase the useful
lifetime of the sorbents by reducing the probabilities
of deactivating reactions or unfavorable physical
processes. The practice of the invention has two
D-20,012 2 162~ 4~
-- 10 --
principal advantages over early methods for deploying
TEC's. First, the invention yields TEC distributions
that are more uniformly spaced, at distances that
correspond to high oxygen adsorption capacities, but
with low probabilities for bimolecular interactions.
Second, the invention provides ready access of the feed
gas mixture to the TEC's, even at the high TEC
concentrations needed for practical oxygen capacities.
These advantages derive from the chemical structure of
the linking agents employed. The chemical interactions
with the surfaces of these linking agents is simpler,
so that it is easier to attach them to the pore walls
inside an oxide or to the chains of a microporous
polymer. In addition, the linking agents provide local
control of TEC concentration and spacing. As a result,
the TEC's are deployed so as to achieve superior
performance as oxygen-selective sorbents, i.e., ones
that react reversibly with oxygen, but not with
nitrogen, argon or carbon dioxide.
High surface area supports for the TEC's may be
achieved with either microporous particles with concave
porosity, or very small particles with convex surfaces.
The TEC's are distributed in either of two modes.
First, the TEC's can be distributed essentially as
monolayers attached to either concave or convex
surfaces by novel linking agents. Second, the TEC's
can be deployed as thin multilayers on convex surfaces
either as amorphous or crystalline solids or as polymer
attached TEC's, i.e. PATEC's. Two key novel features
of the invention are (1) the application of
polyfunctional molecules, including oligomers, as
agents to link TEC's to support surfaces in monolayer
coatings, and (2) the use of very small particles, i.e.
D-20,012 216~ -
~ -- 11 --
less than lOOA, as sorbent supports for either
monolayers or thin multilayers of TEC's.
Practical sorbent materials are determined by the
performance criteria that relate to, but are
independent of, the PSA processing cycles that might be
employed with the sorbents. Of primary interest is the
equilibrium capacity for oxygen. Acceptable values
typically fall in the loading range of 0.3 to 0.6 mmol
~2 per g sorbent, the storage range of 0.3 to 0.8 mmol
~2 per cm3 sorbent particle, or the storage range of
0.2 to 0.5 mmol ~2 per cm3 of a bed of particles.
Higher values are desirable, but these values are
acceptable, especially if the sorption rates are fast.
Ranges of values are indicated because of differences
in the shapes of equilibrium isotherms and differences
in material densities. For these capacities to be
useful, oxygen sorption rates of at least 0.3 mmol ~2
per g sorbent per minute are desired. To obtain these
rates, the pressure driving forces can be on the order
of 1 atm. The selectivity of the sorbents for oxygen
can be expressed in various ways, depending on the
application involved. For air separation, an air
separation factor can be calculated for synthetic air
from pure-gas data, either at equilibrium or at some
cycle time, in the case of rate-selective sorbents.
Desirable separation factor values for loading fall in
the range of 10 to 20, while, for bed storage, in the
range of 3 to 5.
The equilibrium-type supported TEC's of the
invention are more desirable than rate-selective
sorbents for two reasons. First, the selectivities can
be much higher and are not time-dependent. This allows
greater flexibility in process design, since the
D-20,012 21627 4
- 12 -
sorbent is not required to operate at short cycle
times. The equilibrium selectivity arises from the
fact that the TEC reacts reversibly with oxygen, but
not with nitrogen or argon, so that there is a strong
thermodynamic driving force that underlies the
selectivity. Rate selectivity, on the other hand,
depends on differences in the rates of sorption, and
the kinetic driving forces can even be in the opposite
direction of the thermodynamic driving forces. As soon
as the less selectively adsorbed component catches up
with the more selectively adsorbed component, the rate
selectivity is lost.
Second, higher limiting oxygen capacities are
possible below about 1,000 torr oxygen partial
pressures in the practice of the invention. The shapes
of the loading vs. pressure isotherms for the TEC's of
the invention can be quite sharp at low pressures, in
contrast to the equilibrium isotherms of known
rate-selective materials. The higher limiting oxygen
capacities are the result of stronger interactions
between the sorbent and oxygen in the case of
equilibrium selectivity. This is reflected in much
more negative enthalpies of oxygen sorption for the
mild chemical reactions of the TEC's than those
observed for physical adsorption on rate-selective
materials. Such TEC-based sorbents are much better
suited for the removal of very low concentrations of
oxygen in the purification of nitrogen or argon than
are the rate-selective sorbents of the art.
The oxygen-selective sorbents of the invention are
also more desirable than oxide-type, equilibrium-
selective sorbents because the sorbents of the
invention are not required to operate at high
D-20,012 2162~ 4~
- 13 -
temperatures, such as above 300~C. In addition, the
oxygen-selective sorbents of the invention possess less
negative heats of reaction. The advantage of operating
at ambient temperatures leads to lower capital costs,
because special materials of construction and heat
exchange equipment for higher temperature operations
are not required. The large difference in operating
temperatures arises from the differing mechanisms of
oxygen reactions. Oxide-type equilibrium-selective
sorbents require that the O=O double bond be broken in
order to react to add O-atoms and/or O~ions, and this
condition necessarily requires high temperatures for
appreciable sorption rates. With the use of TEC
sorbents, oxygen can react as a molecule, without
breaking a double bond as with oxide-type sorbents.
This is a much milder reaction, which is evidenced by a
more positive heat of reaction, i.e. -10 to -20,
compared to -18 to -55 kcal/mol oxygen.
The use of TEC's in the solid phase, as in the
invention, is advantageous over the use of TEC's in the
liquid phase. First, deactivating reactions, such as
bimolecular oxidations, that occur readily in solutions
owing to the mobility of the TEC's is avoided. In the
practice of the invention, the TEC's are immobilized
and spaced so as to avoid such bimolecular reactions.
Second, by not using a solvent as in liquid phase
TEC's, conflicting compromises that must be made in
selecting a solvent are avoided. When using liquid
phase TEC sorbents, the solvent employed must balance
high solubility, low vapor pressure, low viscosity, and
must be safe to use at relatively high oxygen
concentrations.
D-20,012
~ Z 7 4 0 - 14 -
Compared to unsupported solid TEC's, the supported
solid TEC's of the invention are advantageous in that
the supported TEC's can be dispersed in very thin
layers, resulting in better utilization of the TEC's
and faster sorption rates. For the TEC's of the
invention, the intrinsic reaction rates at the TEC site
are very fast, based on the results of laser flash
photolysis spectroscopy and of reactions in solution.
For such TEC's in the solid phase, the sorption rates
are controlled by the diffusion to the reaction site,
whether it is the phase boundary, as in TEC crystals,
or a dispersed site, as in polymer-attached TEC's, i.e.
PATEC's. The sorption rates in crystalline TEC's are
proportional to the ratio [D/(d) 2], where "D" is the
diffusion coefficient and "d" is the smallest dimension
of the crystal. For example, at constant D, the
sorption rate per unit area of phase boundary in a 500A
crystal layer will be 1600 times that in a 2 micrometer
crystal. Depending on the value of D, and the time
allowed for reaction, some of the TEC's inside a 2
micrometer crystal may never be utilized at all. The
fraction reacted at time "t" for a planar reaction
boundary is proportional to [D*t/(d2)]%. For equal
values of "t" and "D", the volume fraction reacted in
the 500A layer is 40 times that in the 2 micrometer
crystal.
The term "supported TEC's" will be understood to
include dispersed configurations, such as monolayers on
adsorbent supports or multilayers within polymeric
supports, as well as solid (amorphous or crystalline)
layers. For the dispersed configurations, the TEC's
are attached to the support through chemical bonds.
Examples are (1) coupling agents on SiO2 gel, and (2)
D-20,012 ~ 74
the organic nitrogen bases of the copolymers of octyl
methacrylate and either vinyl pyridine or vinyl
imidazole. In contrast to earlier attempts to couple
TEC's to adsorbent supports, the preparation methods of
the invention provide novel, superior chemical linkages
to adsorbent supports that lead to high coverages and,
therefore, practical oxygen capacities.
The use of polymer-attached TEC's, i.e. PATEC's,
can be an effective way to disperse the TEC's,
especially when using TEC's whose crystals are densely
packed and have very low diffusion coefficients.
However, prior attempts to employ PATEC's have all
utilized copolymers prepared by radical chain
polymerization, which leads to problems analogous to
those encountered with TEC's in liquid form. For
example, the incorporation of the axial bases, which
are needed for TEC's, into the polymer chain leads to a
distribution of these groups that is more nearly random
than equally spaced. Clusters of two or three
closely-spaced TEC's will be relatively common. As a
result, bimolecular oxidations and blocking reactions
that occur in liquids can also occur in prior art
PATEC's.
As indicated above, TEC's can be distributed
essentially as monolayers attached to either concave or
convex surfaces by novel linking agents, or as thin
multilayers on convex surfaces either as amorphous or
crystalline solids, or as PATEC's. The monolayer
coatings of TEC's are linked to the support surfaces by
polyfunctional linking agents. Very small particles
are used as sorbent supports for either monolayers or
thin multilayers. For the attaching of TEC's to a
support surface through the use of polyfunctional
D-20,012 ~162
- 16 -
linking agents, the most effective means is through the
use of an axial base linking agent. For this mode of
TEC attachment, the best configuration of TEC's in a
supported monolayer is: (1) a dense side-by-side array
of TEC's, (2) situated with the axis defined by the
oxygen-metal-axial base bonds normal to the surface of
the support layer, so that (3) the oxygen binding sites
face away from the support surface. This is
accomplished by providing each TEC with a linking agent
molecule having an axial base group at one end and one
or more groups at the other for bonding to the support
surface. Examples of such linking agents are
derivatives of silane "coupling agents", alkoxysilane
derivatives with a single substituent containing the
axial base group. It has been found that the three
ideal conditions listed above are difficult to achieve
with these reagents, because these molecules tend to
react with each other as much as with the support
surface. This problem is even more difficult inside
the pores of microporous support particles.
It has been found that certain amplifying linking
agents provide sorption performance superior to that
obtained with only one axial base functionality. In
this regard, amplifying linking agents are defined
herein as chemical species wherein the number of TEC's
associated with each linking agent is greater than the
number of surface interaction sites. Typically, the
amplifying linking agent provides two or more Lewis
base sites, such as pyridine or imidazole derivatives,
capable of binding TEC's to promote reversible
oxygenation. In addition, there must be one or more
sites capable of interacting with the surface of a
support to attach and orient the linking agent and
D-20,012 21627 4
TEC's with respect to the local support surface.
Examples of such amplifying linking agents are
derivatives of starburst dendrimers and derivatives of
arborols.
Linking agents are required for the preparation of
properly spaced and oriented TEC monolayers. Up until
the present time, coupling agents have been attached by
covalent bonds to support surfaces, usually by
condensation reactions that release product alcohols.
One monofunctional linking agent must be reacted with
the support for every TEC attached. The amplifying
linking agents of the invention rely on milder
interactions that do not release product molecules.
These interactions include acid-base interactions
between a Lewis base site on the linking agent and an
acidic surface site, such as an OH group. For a
polyfunctional linking agent such as the DIm6
dendrimer, 3-6 TEC's can be attached for each dendrimer
attached to the surface. The advantages of the
polyfunctional linking agents used in the practice of
the invention are: (1) greater ease of attachment to
support surfaces, (2) more efficient TEC attachment to
linking agents, (3) better control of TEC spacing to
the surface, and (4) more efficient control of
supported TEC reactivity.
Very small particle sizes are required for TEC
monolayers on non-porous supports. For example, to
obtain an oxygen loading of 0.3 mmol/g with a TEC
monolayer, the surface area of the support must be
300m2/g for a TEC molecular "footprint" of 167(A)2. To
obtain a surface area of 300m2/g with dense, spherical
particles, the required diameters are: 200A for
l.Og/cm3, 100~ for 2.Og/cm3, and 50A for 4g/cm3. As a
D-20,012 21627 40
_
- 18 -
result, the particle diameters must be equal to, or
less than 100A for common, dense support materials,
such as alumina, carbon and silica. Linking agents for
attaching TEC's of various types can be used, but the
polyfunctional agents described above are preferred for
the reasons indicated.
Very small supports are desirable for TEC
multilayer coatings, such as PATEC's or neat solid
TEC's. The advantage of small particles is that
thinner multilayers can be used to obtain the same
loadings of TEC's on the support. For constant
densities of TEC and support, the TEC loading varies
only with the ratio of the diameters of coated and
uncoated particles. Consequently, at a fixed TEC
loading, the coating thickness varies as the particle
size, so that the smaller the particle, the thinner the
coating. As noted above, thin layers are advantageous
in achieving higher utilization of the reactants for a
given adsorption time cycle. Multilayer coatings may
be applied by various methods, such as deposition of
solid TEC's or PATEC's from solutions in which the
small support particles are dispersed.
The invention is further described with respect to
various elements of the overall oxygen-selective
sorbents of particular commercial interest.
A. DEPLOYMENT OF O~-SELECTIVE SITES (FUNCTION)
Each TEC is a potential site for the reversible
binding Of ~2 if: (1) it is reactive, (2) it is
accessible within the cycle time allowed, and (3) it is
one of those that can react under the thermodynamic
constraints for the applied conditions. The limiting
D-20,012 216~7 4~
-- 19 --
number Of ~2 molecules per unit weight that can be
bound by a given unsupported TEC is given by the
expression:
L1im (mmol ~2 / g TEC) = 1000 / (MWTEC) /
where MWTEC is the molecular weight in grams of the TEC
(including the axial base). For a supported TEC, the
limiting number is given by:
LmaX (mmol ~2 j g sorbent)
( 10 O O WTEC / MWTEC ) / ( WTEC + W5UP ) ~
where WTEC and WsUp are the weights of the TEC and
support, respectively. The actual amount of ~2 bound
at equilibrium, Leq(O2), by a supported TEC at a given
temperature and ~2 pressure can be approximated by the
Langmuir Isotherm to be:
Leq (~2) (mmol/g) = fr * LmaX * Lang
Keq * P (~2)
where Langfrac
+ Keq * P (~2)
and P (O2) is the ~2 partial pressure and Keq (in units
of inverse pressure) is the affinity coefficient for
given sorbent and temperature.
D-20, 012 216274~
-- 20 --
Here, the fraction of TEC ' s that are reactive, fr, is
taken into account. Keq is the equilibrium coefficient
for the reversible oxygenation reaction:
TEC + ~2 ~ [ TEC ~ ~2 ] ~
If a sorbent, initially at equilibrium, is
displaced from equilibrium by a pressure step,
~ P (~2) Pfinal (~2) Pinitial (~2) ~
the time dependence of the loading, Ltime (O2), in units
of mmol ~2 per unit weight sorbent per unit time, can
be expressed as:
Ltime(~2) = [fa * Leq(O2) ] * [ratefrac(T, ~p, L, . . .) ] .
Here fa is the fraction of reactive TEC ' s that are
accessible, and ratefraC is the fractional rate of
unhindered approach to the new Leq (~2) that corresponds
to Pfinal. The fractional rate depends not only on the
applied conditions as shown, but also on the
coefficients kon and koff for the intrinsic ~2 reactions.
For convenience, the diffusional effects have been
assigned to fa, which will depend on the materials
properties of the supported TEC, such as thickness and
porosity .
Although such analysis pertains to sorption per
unit weight, it can be converted to sorption per unit
volume in two stages . The storage ( loading per unit
volume ) for an individual piece of the supported
sorbent can be obtained from the appropriate loading
values by multiplying by the piece density, Ppiece~ in
units of g/cm3~ for example. The storage for a bed of
such individual pieces must account not only for the
D-20,012 - 21 - ~16274~
storage on the pieces, but also for the storage in the
interparticle voids. Thus, the bulk density, PbUlk~ and
the gas densities must also be known.
The subject invention is directed particularly to
maximize Ltime (~2) ~ even at the expense of lower TEC
loadings or storage. From the preceding equations,
following the expression is obtained:
Ltime ( ~2 ) = fr~fa*Llim~wTEc*Langfrac~ratefrac
WTEC + W5UP
To a first approximation, the terms fr, fa, WTECI and WsUp
depend on deployment, while the others depend primarily
on the identity of the TEC. The invention deliberately
uses finite or increased values of WsUp in order to
obtain greater increase in the value of the utilization
(the product [fr ~ Fa])~ so that a net increase in
Ltime (~2) is obtained. To maximize fr, we deploy the
TEC's on solid supports in order to avoid the
deactivating reactions in solutions. To maximize fa,
the TEC's are attached either in monolayers or in thin
multilayers. To maximize WTEC for a given value of WsUp,
high number density forms of these coatings are
employed.
The particulars of the invention relate to this
strategy as follows. Novel classes of amplifying
linking agents for producing TEC monolayers on support
surfaces are disclosed and claimed herein. To achieve
practical ~2 loadings and storage, particle diameters <
loOA are used for dense TEC supports, whether for
monolayers or thin multilayers.
D-20,012 21627~
- 22 -
B. LINKING AGENTS
1. Function
The function of the linking agent is to attach the
TEC's to the support in a monolayer in such a way as to
maximize the number of reactive TEC's. The linking
agent can be a separate species, part of the TEC, or
part of the support particle, as in the case of an
organic polymer supports. The linking agent can be
attached to the support by means of various
interactions, including covalent bonds and acid-base
interactions. TEC's may be attached to the linking
agents in several ways. A particularly efficient way
to attach TEC's to a substrate is through the axial
base. Attachment through other groups on the TEC is
also possible, e.g., to the tetradentate ligand.
However, another molecule must either be the axial base
or provide it, and the ~2 binding site must be
accessible. Previously, others have used
monofunctional linking agents, i.e., those with one TEC
attached per linking agent. In the practice of the
invention, amplifying linking agents are those with
more than one TEC attached per linking agent.
The role of the linking agent is evident in the
relationship between the support particle surface area
and the maximum ~2 loading of the attached TEC's. For
a monofunctional linking agent with a cross-sectional
area less than that of the TEC, the "footprint" area,
A~EC/ ~f the attached TEC will determine the minimum
spacing and the maximum number of TEC's in a monolayer
on a given surface. The minimum surface area of a
D-20,012 2162~ 4Q
- 23 -
support, As~ required for a TEC monolayer to provide a
given LmaX is:
As(m /g) = [LmaX * NAVO * A~EC (A/molecule)] *10-23,
where NAVO is Avogadro's Number. For a TEC with a
footprint of 167 A2, a minimum surface area of 300m2/g
is required to provide a value of LmaX = 0.3 mmol/g.
The values of ATEC change with the structure and
composition of the TEC. These values can be estimated
from models of the known structure. For example, the
value for Co(TpivPP)/Im is about 225A /molecule, for
attachment through the axial base.
The actual surface area values will be greater,
and the molecular weights and dimensions of both TEC
and linking agent must be considered. The larger and
heavier the linking agent per TEC, the higher the
surface area required for a given Lm3X. For very small
particles, the mass and volume of the coated particle
are significantly different from the uncoated, so that
the molecular properties of the TEC and linking agent
affect both ~ and LmaX. From a geometrical point of
view, small, rod-shaped, monofunctional linking agents
would be superior to relatively large, bulkier
amplifying ones. From a preparative point of view,
however, the linking agents of the invention are much
simpler to attach, and they provide local control of
spacing that the monofunctional ones do not.
2. Types
Two categories of linking agents are identified as
components of TEC based sorbents. These are linear
(non-amplifying) linking agents, and amplifying linking
D-20,012
216~740
- 24 -
agents. The term non-amplifying ("linear") linking
agents indicates linking agents where the number of
sites available to TEC's is less than or equal to the
number of interaction sites or regions with the
support. The nature of the interaction sites in the
linking agent for the TEC and the support can be
similar or different, and the interactions can be
either covalent or non-covalent.
Non-amplifying linking agents are particularly
suited to supports where the number of interaction
sites per unit area is high. A convenient
configuration occurs when the linking agent provides a
Lewis base group to serve as an axial donor to a
transition metal center to give an oxygen-selective
TEC. The modification of surfaces by commercial silane
coupling agents, X3SiRY, has been described in the art
where X is a group for attachment to "mineral
surfaces", Y is an organic functional group, and R is a
hydrocarbon group. This example represents
non-amplifying covalent attachment at multiple sites,
and examples of coupling agents include
(EtO)3SiCH2CH2CH2NH2 and (MeO)3CH2CH2CH2NHCH2CH2NH2.
Similar approaches have been used for the
immobilization of "homogeneous" organometallic complex
catalysts.
Alternative examples of linear linking agents
which provide Lewis base groups to give oxygen-
selective TEC sites include substituted heterocycles
where the functionality is selected for interaction
with surface groups on a porous support or on small
particles either covalently (using known synthetic
organic transformations) or non-covalently. Examples
of the former category include the condensation of
D-20,012
2162~40
- 25 -
substituted pyridine carboxylic acid esters with a
porous solid support containing primary or secondary
amine functionalities. An example of the latter class
is provided by the interaction of an aminoalkylpyridine
or an aminoalkylimidazole with an acidic support. This
particular example relies on a difference in pKa
between the functional groups present. The linking
agent can also be selected where the functional groups
used for interaction with the surface and the
transition metal center are the same. Examples of this
class include diazabicyclo[2.2.2]octane and
hexamethylenetetramine. The Lewis base groups serve as
axial ligands giving rise to oxygen-selective TEC'S.
These cage structures orient the Lewis base with
respect to the surface due to steric considerations.
Surface amplifying linking agents are defined as
linking agents where the number of TEC's associated
with each linking agent is greater than the number of
surface interaction sites. Polydisperse systems
containing multiple Lewis base sites can serve as
surface amplifiers with an acidic substrate and
examples include poly(ethyleneimine) and
poly(ethyleneglycol). In these examples, the Lewis
base is involved in surface attachment and axial
donation to provide oxygen-selective sites. However,
transport into porous supports is expected to be slow,
and it will be difficult to control the amount of
donors àvailable to transition metal centers and the
orientation with respect to the surface. Therefore,
alternative configurations are preferred. Monodisperse
surface amplifying linking agents have been prepared
and a general form is shown schematically in Figure 1.
These linking agents contain one or more branching
D-20,012
2162~40
- 26 -
points and steric effects serve to orient the Lewis
base groups away from the surface. Lewis base groups
suitable for coordination to a transition metal chelate
to give an oxygen-selective sorbent include oxygen,
nitrogen, or sulfur heterocycles, and ethers, amines,
or thioethers. Examples include substituted pyridines
and substituted imidazoles.
The invention comprises the use of new surface
amplifying linking agents for ANY oxygen-selective
sorbent and with ANY TEC and/or substrate. Also
claimed are compositions based on non-amplifying
(linear) linking agents where the linking agent
provides a Lewis base donor for axial coordination to a
transition metal chelate to give an oxygen-selective
TEC, but excluding the use of silane coupling agents
with mineral surfaces.
Examples of amplifying linking agents bearing
Lewis base groups are provided by modified dendrimers,
particularly the low generation examples. The general
structures and syntheses of dendrimers has been
reviewed recently in the art. Modifications to
dendrimer structures to provide terminal Lewis base
groups suitable for interaction with TEC's are
accomplished using synthetic transformations used in
dendrimer growth but with alternative reagents. The
amplifying linking agents DIm6 and DPy6, as shown in
Figure 2, have been prepared using general methods
described in the art for synthesis of PAMAM dendrimers
but using tris(aminoethyl)amine as a core, methyl
acrylate for branching, and an aminoalkyl-substituted
heterocycle to provide terminal groups. Related
structures are possible where the core and the
D-20,012
'- 216274~
- 27 -
composition of the terminal unit (heterocycle, linking
arm) are varied.
In general terms, the structure and composition of
the amplifying linking agent is highly versatile.
Variations can conveniently be made to one or more of
the following features: the core (including
functionality present and multiplicity); the
multiplicity of branching points; the nature of linking
arms; the functionality present and reaction types; and
the structure and substitution patterns of terminal
groups.
The TEC units can be either incorporated as part
of the amplifying linking agent (covalently attached to
the ligand periphery) or the linking agent can provide
Lewis base donors which interact with transition metal
chelates in an axial manner to give oxygen-selective
TEC'S. In the latter category, the Lewis base groups
are typically nitrogen, sulfur, or oxygen heterocycles,
amines, ethers or thioethers.
The linking agent also serves to interact with the
substrate surface in a manner such that the terminal
groups project away from the surface, as shown in
Figure 3. This can be accomplished using either
covalent or non-covalent interactions between the
support surface and functionality associated with
either the core, branching groups, or linking arms.
These points are illustrated for DIm6 with a silica gel
support and a transition metal chelate. The DIm6
molecule has a central core which contains strongly
basic tertiary amine groups which combine with an
acidic surface, leaving the N-substituted imidazole
termini to project away from the surface and combine
with the transition metal centers to form the oxygen-
D-20,012 216~7 4~
- 28 -
selective TEC'S. The presence of a monolayer coating
of DIm6 onto the interior surfaces of SiO2 supported by
uptake studies of DIm6, from solution onto silica gel.
The availability of imidazole groups for coordination
to transition metal sites is indicated indirectly by
sorption studies after the TEC coating is formed.
Advantages of amplifying linking agents over
linear (non-amplifying) linking agents is that they can
be utilized with support surfaces which possess only a
small population of interaction sites per unit area.
Steric effects favor the projection of terminal groups
away from the surface so that TEC orientation is
accomplished.
A synthetic method using coupling agents for
covalent attachment of non-amplifying linking agents is
shown in Figure 4 based on methods described by Basolo,
et al J. Amer. Chem. Soc. 97, 5125 (1975). The oxygen
sorption capacities of materials prepared in this
manner are inferior relative to related materials using
DIm6. In addition, the use of acid-base interactions
is simple and involves no byproducts. However, it
should be noted that covalent attachment of amplifying
linking agents could be utilized where the amplifier is
either grown from the surface or a preformed amplifier
is attached to a surface.
C. TRANSITION ELEMENT COMPLEXES
The function of the TEC is to provide a site for
the reversible, selective binding of oxygen. The
selectivity of the TEC for oxygen over other gases such
as N2 and Ar gives rise to the 02-selectivity of the
TEC-based sorbents of this invention. The desired
D-20,012 216274~
- 29 -
performance of a given sorbent will rely on matching
the structure and composition of the TEC and its
associated axial base with the ~2 concentrations of the
gas mixtures to be separated.
The definition of transition element complexes
(TEC's) is limited to those transition element
complexes which have reversible and selective
interactions with molecular oxygen. Figure 5 shows
schematic representations of TEC structures and their
components. Three components are essential for a TEC
to function as an 02-selective compound: (1) a
transition metal ion located centrally in the complex,
(2) a polydentate ligand which chelates the transition
metal ion, and (3) an axial base which is bound to the
transition metal ion. The axial base may be exogenous,
as shown in Figure 5(a), or endogenous, i.e., part of
the ligand structure, as shown in Figure 5(b).
Cyanometallate salts such as lithium
pentacyanocobaltate solvates are not included since the
ligands do not chelate (simultaneously bind using
different sites to the same metal ion). Polymeric
chelating ligands are included.
With regard to the TEC's of this invention, we
claim appropriate combinations of the following
components: 1) metal ions: Co(II), Fe(II), Ni(II),
Mn(II), Ru(II), Ru(III), Cu(IJ,and Rh(III), 2) ligands:
porphyrins, Schiff bases, polyamines, polyoxoamides,
oximes and their derivatives, and cyclidenes, and 3)
axial bases: pyridine, imidazole, and their
derivatives. Preferred embodiments include appropriate
combinations of the following: 1) cobalt(II) as the
metal ion, 2) picket-fence porphyrin and related
porphyrin dianions as ligands, low molecular weight
D-20,012
2l6274Q
- 30 -
ligands such as malen and related Schiff bases, and
tetraazaannulene ligands, and 3) axial bases of
N-substituted imidazoles and 3- and/or 4-substituted
pyridines as axial bases.
D. SUBSTRATES
The function of the substrate is to provide a
solid support on which to deploy TEC's (with or without
linking agent), to distribute said TEC's, and to serve
as heat sinks for the adsorption processes. The
substrate should be inert with respect to TEC's in both
oxygenated and non-oxygenated forms. Where necessary,
a linking agent can be used to ensure that these
criteria are met. The substrate should be available in
a suitable form for coating with monolayers or thin
multilayers of TEC's (and linking agents if necessary)
to provide practical oxygen storage capacities. In
addition, the substrate surface should be suitable for
coating with linking agent and/or TEC units.
Two alternative substrate configurations for
coating are considered. These are small particles or
clusters of particles (convex surface) and porous
materials (concave surface). In each case, the surface
area available for coating is of particular interest.
In order to have a surface area greater than 300m2/g,
for a pore volume of 0.9 cm3/git is necessary to have a
pore diameter less than 130A. For a pore volume of 0.5
cm3/g, a pore diameter less than 70~ is required.
It should be noted that additional features should
accompany the surface area requirement (2300 m2/g).
For example, the dimensions of linking agents and TEC's
restricts the internal coating of porous substrates to
thin layers. In addition, it is necessary to provide
D-20,012
- ~16~7 4~
- 31 -
sufficient accessible pore volume for the transport of
linking agent and TEC to interior regions of the
substrate.
Calculations based on a TEC footprint of 167(~) 2
have indicated that surface areas 2300m2/g are required
for practical materials. To achieve a surface area
2300m2/g for dense spherical small particle substrates
it is necessary to have a particle diameter below 200A
(particle density 1.0 g/cm3) or 100A (particle density
2.0 g/cm3). For common, dense supports such as SiO2
and Al2O3, particle diameters below 100~ are required.
Small dense particles are suitable for coating with
either monolayers or thin multilayers of TEC (linking
agent or a Lewis base containing species can be a
component of the TEC system). Notice that clusters of
small particles are included as useful substrates.
The use of substrates for TEC coatings employing
linking agents requires that the linking agent and
substrate interact. Similar interactions can be
obtained for any porous or non-porous small particle
substrate incorporating acidic surface groups (Lewis or
Bronsted). In general, the choice of linking agents
will be dependent on substrate surface groups. For
example, a linking agent bearing acidic groups could be
employed for a substrate containing basic groups.
Other interactions can also be exploited including
H-bonding, electrostatic, and covalent bonding. Porous
substrates or dense small particle substrates can be
used where the surface contains Lewis base groups
suitable for interaction with transition metal sites to
provide oxygen-selective TEC coatings (i.e. no linking
agent).
The composition of the substrate for both porous
and dense small particle categories includes inorganic
~ D-20,012
. . _,.,
- 32 -
(mineral) supports such as: single oxides (SiO2, Al203,
TiO2); mixed oxides (Al203-SiO2, glasses, clays);
carbons, modified carbons, and carbon foams. Also
included as substrates are porous polymeric
compositions such as crosslinked polymers and
copolymers, macroreticular re~sins, phase separated
polymers (e.g N~FI()~ FLE~lIO ~ , and "microporous"
polymers (such as PTMSP). Porous condensation polymers
such as polyamides and polyimides are also included.
These polymer categories can be used with or without
linking agent. Porous polymers and copolymers
containing potential axial donors (e.g.
polyvinylpyridine type or polyvinylimidazole type) or
groups which are easily modified to provide potential
axial donors are included.
All substrates are claimed from the categories
above which fit the minimum surface area requirement
(2300m2/g) or that provide an effective surface area
2300m2/g when combined with a linking agent.
Substrates should be inert~with respect to TEC (using
linking agent if necessary) and be coatable with a
suitable linking agent and/or TEC. Substrates are
preferred that have a high solid density, low cost, and
are available in various forms or sizes. Examples
which meet this preferred criteria include SiO2, Al203,
TiO2 porous glasses, clays and pillared clays, and
carbon adsorbents.
Depending on the substrate, a variety of
configurations may be available which could be used as
adsorbents. Examples of configurations are listed
below with examples of substrates shown in parentheses:
microporous particles (SiO2, Al203); dense small
particlesi beads (porous glass, clays); fibers
(adsorbent carbons); sheets ("microporous" polymers
D-20,012 ~16~:74~
, ~
- 33 -
such as PTMSP); and composites (ceramic monoliths,
pillared clays).
E. PREPARATION OF O~SELECTIVE SORBENTS
1. Generic Methods
It should be noted that coating of substrates with
TEC's can actually be accomplished with or without the
intervention of a linking agent. Examples where the
linking agent is not required includes the ion-exchange
of cationic TEC's with ion exchangeable units on the
substrate surface. In addition, combinations of
transition metal chelates with Lewis bases which do not
function as linking agents can be used to give oxygen-
selective sorbents where the coating is amorphous
(non-oriented) or crystalline. The use of linking
agents, however, is generally preferred such that TEC
orientation is accomplished relative to the substrate
surface.
Fig. 6 illustrates two alternative methods that
have been employed for the introduction of TEC's
incorporating linking agents onto substrates. A
simultaneous method is shown in Fig. 6(a), and
sequential method, is illustrated in Figure 6(b).
These methods are useful for coating porous substrates
and non-porous small particle substrates with TEC's
including Lewis base/TEC combinations. Pretreatment of
the substrate may be necessary. For example, silica
gel substrates were dried under vacuum at 100~C for
several days prior to use. Silica small particles were
dried at 100~C/0.02 torr until no change in pressure
was observed for the drying system over several hours.
D-20,012 ~16~7 40
- 34 -
Other substrates should be dried or activated to remove
adsorbed materials using methods appropriate to the
substrate and the adsorbate. The coating procedures
are performed under dry, anaerobic conditions using
anhydrous solvents. Solvents are selected based on TEC
solubility and compatibility with TEC and other
components. For sequential methods (see below), it is
desirable to use solvents where the linking agent shows
a substantially lower solubility than the TEC.
The simultaneous coating method involves the
formation of a slurry of the substrate with a solution
containing the TEC ~or TEC precursor) and either a
linking agent or an additional component that can serve
as an axial base. Solvent removal is used to deposit a
layer of the oxygen-selective TEC (and linking agent if
present) onto the support surface. It is sometimes
convenient to preform a solid containing linking agent
and TEC by concentration of a mixed solution. This
mixed solid can be combined with the substrate using a
solvent in a subsequent step. This method is not
distinguished from a method where the linking agent,
TEC, and substrate are mixed directly in an appropriate
solvent.
The sequential coating method is used to deposit a
monolayer of linking agent onto the support surface in
an initial step. This method is preferred when there
is significant interaction strength between the linking
agent and the substrate. The linking agent is coated
onto the substrate by adsorption from a solution
containing an excess of the linking agent, then the
coated support is collected by filtration and dried.
Introduction of the TEC component is performed by
addition of the coated substrate to a solution
D-20,012 21~2~4Q
- 35 -
containing TEC under conditions where extraction of the
linking agent from the surface is minimized (typically
ambient temperature for several days), then the solvent
is allowed to evaporate slowly. Alternative ways to
collect the particles are applicable and these include
filtration and centrifugation. In both sequential and
simultaneous methods, the oxygen-selective TEC-coated
substrate is dried under vacuum at temperatures from 25
to 100~C prior to use as sorbents.
We have found that materials prepared using
amplifying linking agents by simultaneous and
sequential loading methods do not show identical
properties. ~or example, the equilibrium oxygen uptake
(expressed in mmol/g) is higher for current materials
based on simultaneous methods, whereas the oxygen
uptake and release rates tend to be faster for
sequentially loaded samples. This is thought to
reflect differences in composition (TEC content and
location). It is felt that samples prepared by
simultaneous methods with a SiO2 substrate contain
oxygen-selective TEC units as internal or external
multilayers and that these are responsible for slower
uptake and release rates compared to sequentially
prepared samples.
2. Examples of Specific Embodiments
a) General Materials and Procedures
All reagents were obtained from Aldrich unless
otherwise noted and were purified by standard methods
if necessary. Solvents were purchased as anhydrous
reagents in Sure/seal vessels. The preparation of
meso-tetra (a, a, a, a-o-pivalamidophenyl)
D-20,012
~-- 2162~Q
- 36 -
-porphyrinatocobalt(II) (abbreviated as Co(TpivPP)) was
based on procedures reported by Collman. All synthetic
procedures excluding preparation of amplifying linking
agents were performed under inert atmosphere
conditions.
Silica gel used for dispersal in the examples
shown was a 60A grade with 130-270 mesh and a BET
surface area of 500m2/g and a pore volume of 0.75
cm3/g. The silica gel was dried in a vacuum oven at
100~C for 16h then transferred to a glove box. Asahi
80A porous glass was cleaned using hydrogen peroxide at
90~C, rinsed with water, then dried under nitrogen
purge at 80~C , followed by heating under vacuum to
140~C. Small particle SiO2 xerogels were prepared by
desolvation of a colloidal sol of 50~ SiO2 small
particles (pH = 9.0; concentration = ~O.lM SiO2). SiO2
small particles were prepared by grinding the xerogels
to a fine powder, followed by drying at 100~C/0.02 torr
until no change in pressure was observed for the drying
system over several hours.
DIm6, was prepared in two steps by the reaction of
tris(2-aminoethyl)amine in methanol with six
equivalents of methyl acetate in methanol followed by
six equivalents of N-(2-aminoethylimidazole) in
methanol using the general procedures described by the
art for the preparation of the so-called PAMAM
dendrimers. The product was dried under vacuum with
temperature maintained below 50~C. An infrared
spectrum of the product recorded as a thin film
contains signals at 1740, 1665, 1565, and 1515 cm~1.
Impregnation of SiO2 with DIm6 was performed for
sequential loading procedures and is illustrated here
for DIm6/SiO2. Predried silica gel (0.4995g) was
~ D-20,012
21627 4~
- 37 -
treated with DIm6 (0.235g) in chloroform (lOml) under
an inert atmosphere at room temperature. The system
was stirred for 24h then the solid was collected by
filtration. The DIm6 content of the solid lies in the
region 16-18 wt. % and extraction of the physically
adsorbed DIm6, with CHCl3 is not favorable. A similar
procedure was used for the preparation of DPy6/SiO2 to
give solid containing 20 wt. % DPy6.
b) Examples of Amplifying Linking Agents
Example 1: Co(Tpivpp)/DIm6
A solution of DIm6 (0.0228 g) in methanol (5 ml)
was added to Co(TpivPP) (0.1050 g), then methanol was
added to give a solution volume of 10 ml. After
stirring for 3h, the solution was filtered to remove
excess Co(TpivPP). The filtrate was allowed to
concentrate by slow evaporation and was then dried
under vacuum to give a brown solid, mass 0.0858g.
Assuming that the DIm6 remains in the methanol and that
only Co(TpivPP) is removed by filtration, the
composition of the solid corresponds to 76 wt. %
C~ ( TpiVPP ) .
Example 2: Co(Tpivpp)/DIm6
Co(TpivPP) (0.1225 g) in chloroform (5 ml) was
added to a solution of DIm6 (0.0467 g) in chloroform (5
ml). The solution was stirred for lh, then the
solution was concentrated to dryness under vacuum.
Based on the amounts of reagents used, the solid
contains 72 wt. ~ Co(TpivPP) (0.678 mmol/g).
D-20,012 -
2162~Q
- 38 -
Example 3: Co(TpivPP)/DIm6/SiO2, simultaneous
Chloroform (0.5 ml) was added to a mixture of
DIm6/Co(TpivPP) (47.8 mg, composition 76 wt. % Co(TpivPP))
and silica gel (5.6 mg) then the solvent was allowed to
evaporate slowly overnight to give a purple solid. The
sample was dried under vacuum prior to sorption
studies. The sample composition corresponds to 10 wt.
% sio2.
Example 4: Co(TpivPP)/DIm6/SiO2, simultaneous
Chloroform (0.5 ml) was added to a mixture of
DIm6/Co(TpivPP) (20.9 mg, composition 76 wt. % Co(TpivPP))
and silica gel (8.7 mg) then the solvent was allowed to
evaporate slowly overnight to give a purple solid. The
sample was dried under vacuum prior to sorption
studies. The sample composition corresponds to 29 wt.
% sio2.
~xample 5: Co(TpivPP)/DIm6/porous glass,
simultaneous
Chloroform (2 ml) was added to a mixture of
DIm6/Co(TpivPP) (31.5 mg, composition 76 wt. % Co(TpivPP))
and Asahi 80A porous pieces. The sealed system was
allowed to stand for 18h, then the solvent was allowed
to evaporate slowly. The solid obtained was dried
under vacuum for 6h prior to sorption studies. The
sample composition corresponds to 30 wt. % porous
glass.
D-20,012
~16~4~
- 39 -
Example 6: Co(TpivPP)/DIm6/SiO2, sequential
Benzene (2 ml) was added to a mixture of DIm6/SiO2
(0.1718 g, 18 wt. % DIm6) and Co(TpivPP)(0.0538 g). The
system was sealed, then the contents were stirred for
24 h. The solvent was allowed to evaporate slowly to
give a purple solid. The sample was dried
under vacuum prior to sorption studies. The sample
composition corresponds to 24 wt. % Co(TpivPP).
Example 7: C~(TpivPP)/DPY6/SiO2~ sequential
Chloroform (2 ml) was added to a mixture of
DPy6/SiO2 (0.1010 g, 20 wt. % DPy6) and Co(TpivPP) (0.0538
g). The system was sealed and allowed to stand at room
temperature for 17h. The solvent was allowed to
evaporate slowly overnight to give a purple solid. The
sample was dried under vacuum for 6h prior to sorption
studies. The sample composition corresponds to 35 wt.
% Co ( TpivPP ) -
Example 8: Co(acacen)/DIm6/SiO2, sequential
Chloroform (2 ml) was added to a mixture of
DIm6/SiO2 (0.1058 g, 18 wt. % DIm6) and Co(acacen)
(0.0260 g). The system was sealed and allowed to stand
at room temperature for 2h then the solvent was allowed
to evaporate slowly overnight to give a orange solid.
This solid was dried under vacuum for 3h. The sample
composition corresponds to 20 wt. % Co(acacen).
- D-20,012 2 16 2 7 4 ~
- 40 -
Example 9: Co(TPP)/DIm6/SiO2, sequential
Chloroform (2 ml) was added to a mixture of
DIm6/SiO2(0.0319 g, 19 wt. % DIm6) and Co(TPP) (1.045
g). The system was sealed and allowed to stand for 2h
then the solvent was allowed to evaporate slowly to
give a purple solid. This solid was dried under vacuum
for 6h. The sample composition corresponds to 77 wt. %
Co(TPP).
Example 10: Co(TpivPP)/DIm6/PTMSP sequential
PTMSP (36.8 mg) and DIm6/Co(TpivPP) (54.4 mg) were
dissolved in chloroform (5 ml) then the solution was
stirred for 3 days. The viscous solution was cast onto
a teflon dish then the surface was covered with a glass
dish to slow the solvent evaporation process. After
one day the surface appeared dry. The sample was
slowly placed under vacuum to remove residual solvent.
c) Examples of Small Particles
Example 11: Co(TpivPP)/BzIm
Co(TpivPP) (0.874 g) was added to a stirred
solution of 1-benzylimidazole (0.0246 g) in chloroform
(4 ml) and the solution was allowed to evaporate slowly
over several days, with stirring, to give a dark solid.
The sample was dried under vacuum for 18.5h prior to
sorption testing. This unsupported solid (0 wt. %
SiO2) was used for comparison with SiO2supported
samples.
D-20,012
2162740
- 41 -
Example 12: Co(Tpivpp)/BzIm/sio2
l-Benzylimidazole (0.0246 g) was added to a
stirred suspension of SiO2 small particles (0.0305 g)
in chloroform (2 ml). The suspension was stirred for
several minutes, Co(TpivPP) (0.0874 g) was added, and
the solution was allowed to evaporate slowly over
several days, with stirring, to give a dark solid. The
sample was dried under vacuum for 22h prior to sorption
testing. The sample composition corresponds to 8 wt. %
sio2 .
Example 13: Co(Tpivpp)/BzIm/sio2
l-Benzylimidazole (0.0246 g) was added to a
stirred suspension of SiO2 small particles (0.0305 g)
in chloroform (2 ml). The suspension was stirred for
several minutes, Co(TpivPP) (0.0874 g) was added, the
reaction vessel was capped, and the mixture was allowed
to stir for 1.5 hr. The vessel was then uncapped and
solvent was allowed to evaporate over several weeks to
give a dark solid. The solid was ground then dried
under vacuum for 10 h prior to sorption testing. The
sample composition corresponds to 21 wt. % SiO2.
Example 14: co(malen)/Me2Im/sio2
Chloroform (15 ml) was added to a mixture of
Co(malen) (0.3254 g), Me2Im (0.2234 g), and SiO2
(0.5020 g) then the solvent was allowed to evaporate.
When the sample appeared dry, additional chloroform (10
ml) was added and slow evaporation was allowed to
occur. The solid was dried under vacuum prior to
D-20,012
21627 4o
- 42 -
sorption studies. The sample composition corresponds
to 31 wt. % Co(malen).
Example 15: Co(Tpp)/Me2Im/sio2
Chloroform (15 ml) was added to a mixture of
C~(TPP)(0.4032 g), Me2Im (0.1740g), and SiO2 (0.5098 g)
then the solvent was allowed to evaporate. When the
sample appeared dry, additional chloroform (10 ml) was
added and slow evaporation was allowed to occur. The
sample was dried under vacuum prior to sorption
studies. The sample composition corresponds to 37 wt.
% Co(TPP).
Example 16: Co(TpiVPP)/BzIm/PTMSP
PTMSP (0.10 g) was dissolved in chloroform (15 ml)
with stirring for 2h. A mixture containing Co(TpivPP)
(0.10 g) and BzIm (0.027 g) was dissolved in chloroform
(5 ml) then the two solutions were mixed. The combined
solution was cast onto a teflon dish and the sample was
covered to slow the evaporation rate. Upon drying, the
sample was slowly placed under vacuum to remove
residual volatile components. The purple membrane was
used for sorption studies.
Example 17: Co(TpivPP)/MeIm/PTMSP
PTMSP (0.30 g) was dissolved in chloroform (15 ml)
with stirring for 3h. Co(TpivPP) (0.25 g) in chloroform
(5 ml) was added to the polymer solution, then MeIm
(0.1 ml) was added by syringe. The combined solution
was cast onto a teflon dish and the sample was covered
D-20,012
216274~
- 43 -
to slow the evaporation rate. Upon drying, the sample
was slowly placed under vacuum to remove residual
volatile components. The purple membrane was used for
sorption studies.
F. GAS SEPARATION APPLICATIONS
1. General
Supported TEC sorbents which are the subject of
this invention are useful for gas separation
applications. Equilibrium oxygen-selective sorbents of
this type can be used either for bulk separation of
oxygen-nitrogen containing mixtures (e.g. air), or can
be used for purifications such as oxygen removal from
low purity nitrogen or crude argon. The materials
and/or conditions should be matched to a particular
application based on the properties of the oxygen-
selective TEC sites. For bulk separation of air, the
oxygen affinity (frequently expressed as Pso (~2) ) under
the operating conditions employed should be similar to
the oxygen partial pressure in the feed so that
significant oxygen binding occurs. The oxygen affinity
of the TEC sites is a function of the donors (ligands)
interacting with the transition metal ion in the deoxy
form (equatorial and axial), the transition metal ion
selected, structural effects in the vicinity of the
oxygen interaction site together with conditions (e.g.
temperature). The selection of each structural
component can be inferred based on data available from
solution and solid state studies either directly or by
analogy and implication.
D-20,012
216274~
- 44 -
2. Examples of Sorption Performance
Sorption data for oxygen, nitrogen, and argon were
obtained using a gravimetric method with pure gases.
Sorption experiments were performed at 27~C unless
otherwise stated.
Example 18: Co(TpivPP)/DIM6/SiO2, simultaneous, 29 wt.
% SiO2, 58 wt. % Co(TpivPP)(0.54 mmol/g)
Sorption studies revealed that this material is
oxygen-selective at equilibrium. Equilibrium and rate
data are shown in Table 1 and Table 2, respectively.
Oxygen uptake to 2000 torr is 98% complete after 5
minutes, and release from 2000 torr is 95% complete
within 30 minutes.
D-20, 012
2162~4~
-- 45 --
TABLE 1
SORPTION DATA FOR DIm6/Co ~TpiVPP) /SiO2 AT 27~C
Adsorbate Step # Pressure Loading Approach
(torr) (mmol/g) (S or D)
~2 8 125 0.108 S
~2 9 250 0.154 S
~2 10 375 0.181 S
~2 11 500 0.200 S
~2 12 750 0.226 S
~2 13 1000 0.249 S
~2 14 2000 0.301 S
~2 1 2000 0.295 S
~2 16 2000 0.317 S
N2 3 2000 0.025 S
N2 4 3779 0.042 S
N2 6 3779 0.042 D
N2 5 5000 0.057 S
Ar 18 1000 0.030 S
Ar 21 1000 0.028 D
Ar 20 1500 0.043 D
Ar 22 1500 0.041 S
Ar 19 2500 0.071 S
D-20,012
216274P
- 46 -
TABLE 2
SORPTION DATA FOR DIm6/CO(TPLVPP)/SiO2 AT 27 ~C
DATA PRESENTED FOR UPTAKE TO 2000 TORR,
AND RELEASE FROM 2000 TORR
Time ~minutes) Sorption (mmol/g) De~orption (mmol/g)
0 0.0030.317
1 0.241-0.041
2 0.2830.052
3 0.3010.060
4 0.3090.057
0.3120.055
0.3140.039
0.3170.032
0.3170.026
0.019
0.008
0.003
120 0.003
D-20,012
-
- 47 - ~16274~
Example 19: Co(TpivPP)/DIM6/SiO2, simultaneous, 29 wt.
% SiO2,68 .%Co(TpivPP)(0.637 mmol/g)
Sorption studies revealed that this material is
oxygen-selective at equilibrium. Equilibrium and rate
data are shown in Table 3 and 4 respectively. Oxygen
uptake to 1000 torr is 95% complete after 15 minutes,
and release from 1000 torr is 85% complete within 60
minutes. The utilization of TEC sites is poor compared
to the sample with higher SiO2 content (Example 18).
D-20, 012 2~1 6z~40
-- 48 --
TABLE 3
SORPTION DATA FOR DIm6/Co ~Tp~VPP) /SiO2 AT 27 ~C
Adsorbate Step # Pressure Loading Approach
(torr) (mmol/g) (S or D)
~2 5 125 0.128 S
~2 6 250 0.167 S
~2 7 375 0.190 S
~2 8 500 0.204 S
~2 9 750 0.223 S
~2 3 1000 0.236 S
~2 10 1000 0 . 238 S
~2 11 1500 0.258 S
~2 12 5000 0.313 S
~2 4 0 0.000 D
~2 13 0 0.000 D
N2 1 1000 O.011 S
N2 14 3779 0.009 S
N2 2 0 0.006 D
N2 15 0 0.000 D
Ar 16 1000 0.011 S
Ar 19 1000 0.009 D
Ar 18 1500 0.012 D
Ar 20 1500 0.011 S
Ar 17 2500 0.019 S
Ar 21 0 0.000 D
D-20,012 21627 4~
- 49 -
TABLE 4
SORPTION DATA FOR DIm6/Co(Tp~vPP)/SiO2 AT 27 ~C
DATA PRESENTED FOR UPTAKE TO 1000 TORR,
AND RELEASE FROM 1000 TORR
Time (minute~)Sorption ~mmol/g) De~orption ~mmol/g)
0 0.000 0.236
1 0.096 0.099
2 0.133 0.119
3 0.153 0.119
4 0.168 0.115
0.179 0.110
0.208 0.091
0.223 0.078
0.227 0.070
0.233 0.056
0.235 0.035
0.236 0.026
120 0.020
150 0.014
180 0.013
210 0.011
D-20,012
-~ 2~6~274~
- 50 -
Example 20: Co(TpivPP)/DIm6,76 .%Co(TpivPP)(0.711
mmol/g)
Sorption studies revealed that this material is
oxygen-selective at equilibrium. Equilibrium and rate
data are shown in Table 5 and 6 respectively. Oxygen
uptake to 1000 torr is 87% complete after 30 minutes,
and release form 1000 torr is 73% complete after 120
minutes. The sample shows high oxygen loadings
compared to supported samples (see Examples 18 and 19),
but uptake and release rates are very slow. These slow
rates demonstrate the need for thin supported layers.
-
D-20, 012
2-162740
-- 51 --
TABLE 5
SORPTION DATA FOR DIm6/Co (Tp~VPP) /SiO2 AT 27 ~C
Adsorbate Step # Pressure Loading Approach
(torr) (mmol/g) (S or D)
~2 30125 0.285 S
~2 30125 0.258 S
~2 14250 0.351 S
~2 35375 0.423 S
~2 35375 0.402 S
~2 32500 0.434 S
~2 37750 0.496 S
~2 37750 0.472 S
~2 202000 0.587 S
N2 13779 0.029 S
N2 33779 0.033 D
Ar 51000 0.014 S
Ar 81000 0.015 D
Ar 91500 0.022 S
Ar 71500 0.022 D
.
D-20,012 21~7 4~
- 52 -
TABLE 6
SORPTION DATA FOR DIm6/CoTplvPP) AT 27 C ~15,749-12)
DATA PRESENTED FOR UPTAKE AND RELEASE FOR 2000 TORR
Time (minutes) Sorption (mmol/g) Desorption (mmol/g)
0 0.001 0.573
1 0.140 0.408
2 0.212 0.420
3 0.259 0.417
4 0.293 0.409
0.319 0.402
0.400 0.367
0.443 0.340
0.468 0.318
0.500 0.285
0.537 0.223
0.550 0.184
120 0.558 0.157
150 0.563
180 0.564
210 0.569
240 0.570
270 0.572
D-20,012
216274~
- 53 -
Example 21: Co(TpivPP)/DIm6/SiO2, sequential, 23.8 wt.
% Co(TpivPP) (0.293 mmol/g)
Sorption studies revealed that this material is
oxygen-selective at equilibrium. Equilibrium and rate
data are shown in Table 7 and 8, respectively. Oxygen
uptake to 1000 torr is 93% complete after 1 minute, and
release from 1000 torr is 95% complete after 2 minutes.
The observed rates are fast compared to samples
prepared using simultaneous methods (see Examples 18,
19, and 20) but equilibrium oxygen loadings are lower
(reflects lower TEC contents). The fast rates support
organization of the TEC coating into thin oriented
layers.
D-20, 012 21 62740
-- 54 --
Table 7
SORPTION DATA FOR DIm6/SiO2/Co (To~vPP) AT 27 ~C
Adsorbate Step # Pressure Loading Approach
(torr) (mmol/g) (S or D)
~2 5 125 0.072 S
~2 6 250 0.094 S
~2 7 375 0.109 S
~2 8 500 0.120 S
~2 16 500 0.126 D
~2 9 750 0.135 S
~2 1 1000 0 . 148 S
~2 10 1000 0 . 147 S
~2 15 1000 0.152 D
~2 11 1500 0.165 S
~2 12 2000 0.180 S
~2 13 3000 0.206 S
~2 14 4000 0.229 S
N2 3 1000 0.014 S
N2 18 1000 0.018 S
N2 22 1000 0.018 D
N2 19 3779 0.051 S
N2 21 3779 0.050 D
Ar 24 1000 0.019 S
Ar 27 1000 0.020 D
Ar 28 1500 0.028 S
Ar 26 1500 0.028 D
Ar 25 2500 0.044 S
~ D-20,012 2162740
_
- 55 -
Table 8
SORPTION DATA FOR DIm6/SiO2/Co(Tpi~PP) AT 27 ~C
DATA PRESENTED FOR UPTAKE FOR 1000 TORR,
AND RELEASE FROM 1000 TORR
~ime (minutes)Sorption (mmol/g)Desorption (mmol/g)
0 0.000 0.148
1 0.137 -0.005
2 0.138 0.008
3 0.140 0.008
4 0.143 0.006
0.143 0.006
0.146 0.006
0.146 0.005
0.147 0.005
0.147 0.005
0.148 0.006
72 0.006
~ D-20,012 216274~
-
- 56 -
Example 22: Co(TpivPP)/DPy6/SiO2, sequential, 34.8 wt.
% Co(TpivPP) (0.326 mmol/g)
Sorption studies revealed that this material is
oxygen-selective at equilibrium. Equilibrium and rate
data are shown in Table 9 and 10 respectively. Oxygen
uptake to 1000 torr is 93% complete after 1 minute, and
release from 1000 torr is 98% complete after 2 minutes.
The observed rates are fast compared to samples
prepared using a simultaneous methods (see Examples 18,
19, and 20) and are similar to a related sample
containing DIm6 but the oxygen binding equilibrium
constant for TEC sites is shifted to higher pressures
relative to the DIm6, material (see Example 21).
D-20,012 2162740
-- 57 --
TABLE 9
SORPTION DATA FOR DPy6/Co (T~,l"PP) /SiO2 AT 27 ~C
Adsorbate Step #Pressure Loading Approach
(torr)(mmol/g) (S or D)
N2 1 1000 0.004 S
N2 2 3779 0.013 S
N2 3 5000 0.014 S
N2 4 3779 0.010 D
N2 5 1000 0.006 D
N2 6 0 0.003 D
~2 7 250 0.045 S
~2 8 500 0.066 S
~2 9 1000 O. 092 S
~2 1 ~ 1000 0 . 093 S
~2 10 1500 0.109 S
~2 11 2000 0.124 S
~2 12 2500 0.135 S
~2 13 3000 0.144 S
~2 14 5000 0.174 S
~2 15 1000 0.096 D
~2 16 0 -0.001 D
Oz 2' 0 0.003 D
Ar 3' 1000 0.007 S
Ar 4' 2500 0.016 S
Ar 5' 1500 0.013 D
Ar 6' 1000 0.010 D
Ar 7' 1500 0.015 S
Ar 8' 0 0.006 D
D-20,012
21627'lO
- 58 -
Table 10
Oxygen Binding Kinetics for DPy6/Co(TpivPP)/SiO2 at 27 ~C
Uptake and Release for 1000 torr
Time (minutes) Sorption (mmol/g) Desorption (mmol/g)
0 0.0020.093
1 0.086-0.012
2 0.0890.002
3 0.0900.000
4 0.0920.005
0.0920.003
0.0920.003
0.0920.003
0.0920.003
0.002
0.003
D-20,012 21627~0
- 59 -
Example 23: Co(TpivPP)/BzIm/dense 50 A sio2, Small
Particles, 21 wt. % SiO2,
This example describes a superior performing
TEC-supported small particle sample. The sample is
comprised of a Co(TpivPP)/BzIm coating on 50 ~ diameter
SiO2 small particles using 21 wt. % SiO2. The sorption
behavior for this sample is characterized by high Oz
loadings, fast uptake and release rates, and high
selectivities for oxygen over nitrogen. The isotherm
data are tabulated in Table 11. Uptake ~f ~2 to 1000
torr is 97% complete after 5 minutes and ~2 release
from 1000 torr is 95% complete in 15 minutes. The ~2
loading vs. time data for this sample are tabulated in
Table 12.
~ D-20,012 2162740
, _
- 60 -
Table 11
BzIm/Co~Tp~PP) on SiO2
Loading vs. Pressure
21 wt. % SiO2
Pressure ~2 Loading N2 Loading Ar Loading
(torr) (mmol/g) (mmol/g) (mmol/g)
O O.000 0.000 0.000
100 0.188
250 0.296
500 0.383
1000 0.421 0.010 0.008
2000 0.468
3000 0.489
3779 0.005
4500 0.517
-
~ D-20,012 ~6274 0
. ~
- 61 -
Table 12
~2 Uptake and Release data to and From 1000 Torr
BzIm/Co~Tp~vPP) on SiO2
21 wt. % SiO2
Time (minutes) ~2 Loading - Uptake ~2 Loading - Release
to 1000 Torr From 1000 Torr
(mmol/g) (mmol/g)
0 0.001 0.415
1 0.355 -0.016
2 0.382 0.047
3 0.392 0.050
4 0.398 0.047
0.401 0.044
0.409 0.030
0.411 0.020
0.412 0.019
0.413 0.016
36 0.415
53 0.014
D-20,012
216274~
- 62 -
Example 24: Co(TpivPP)/BzIm/dense 50 A sio2 Small
Particles, 8 wt. % SiO2,
This example shows the advantage of using thin
layers of Co(TpivPP)/BzIm coatings on small particle
supports. In this experiment, two samples were
prepared which contained the same amounts of the two
coating components, Co(TpivPP) and BzIm, but different
amounts of SiO2 which resulted in a different thickness
of TEC coating for each sample. The first sample,
comprised of 8 wt. % SiO2, had a thick TEC coating
relative to that of the second sample, comprised of 21
wt. % SiO2 (see Example 23). The 8 wt. % SiO2 sample
(thick coating) shows a lower ~2 loadings than the 21
wt. % SiO2 sample (thin coating). Loading data for the
8 wt. % SiO2 are tabulated in Table 13. The lower
loadings observed for this sample are probably a result
of the poor accessibility of the TEC to ~2 due to
diffusion constraints imparted by the thicker coating.
This argument is also supported by the lower rates
observed for the 8 wt. % SiO2 sample compared to those
of the 21 wt. % sample. A comparison of these two
samples (Table 14) shows the uptake ~f ~2 to 1000 torr
is only 83% complete after 5 minutes for the 8 wt. %
SiO2 sample, but 97% complete for the 21 wt. % sample.
~2 release rates from 1000 torr follow a similar trend:
the release process is only 82% complete after 20
minutes for the 8 wt. % SiO2 sample, but 95% complete
for the 21 wt. % sample. The ~2 loading vs. time data
for this sample are given in Table 15.
D-20,012 216274~
-- 63 --
Table 13
BzIm/Co (TpiVPP) on SiO2
Loading vs. Pressure
8 wt. % SiO2
Pressure~2 LoadingN2 Loading Ar Loading
(torr) (mmol/g) (mmol/g) (mmol/g)
O O.000 0.000 O.000
200 0.080 0.002
500 0.124 0.003
1000 0.162
1691 0.003 0.008
2000 0.199
3000 0.233 0.003
4500 0.245 0.003 0.019
D-20,012 216 2 7 4 ~
'_
- 64 -
Table 14
Comparison of % ~2 Uptake and Release for
BzIm/Co(TpivPP) on SiO2
8 and 21 wt. % SiO2 Samples
Time 21 wt. ~ SiO2 8 wt. % SiO2 21 wt. % SiO2 8 wt. % SiO2
(min)% ~2 % ~2 Uptake ~ ~2 % ~2 Release
Uptake Release
O O 0 100 100
3 94 76 88 68
97 83 89 71
98 89 93 76
99 95 95 82
97 85
89
91
D-20,012
2 1 ~274~
- 65 -
Table 15
~2 Uptake and Release Data to and From 1000 Torr
BzIm/Co(TpivPP) on SiO2
8 wt. % SiO2
Time (minutes)~2 Loading - Uptake~2 Loading - Release
to 1000 Torr From lO00 Torr
(mmol/g) (mmol/g)
0 0.001 0.161
1 0.098 0.042
2 0.115 0.052
3 0.123 0.052
4 0.129 0.049
0.133 0.047
0.144 0.038
0.149 0.032
0.153 0.029
0.157 0.024
49 0.161
0.017
0.015
120 0.014
150 0.014
166 0.014
D-20,012
2162740
- 66 -
Example 25: Unsupported Co(TpivPP)/BzIm
This example illustrates the benefits of using
small particle supports in improving TEC performance.
In this experiment, the sorption behavior of three
samples were compared: 1) a Co(TpivPP)/BzIm/SiO2 sample
using 21 wt. % SiO2 (see Example 23), 2) a Co(TpivPP)/
BzIm/SiO2 sample using 8 wt. % SiO2 (see Example 24),
and 3) a Co(TpivPP)/BzIm sample which was unsupported.
A comparison of the ~2 isotherms for the three samples
illustrates that the loading of Co(TpivPP) is not
improved using an 8 wt. % SiO2 support. However, a
substantial improvement in loading can be seen at the
21 wt. % SiO2 level. The sorption data for the
unsupported BzIm/Co(TpivPP) sample is given in Table 16.
Improvements in the percent utilization of Co(TpivPP)
can be achieved by using small particle supports. The
percent utilization of Co(TpivPP) increased from 24% in
the unsupported sample to 71% in the 21 wt. % SiO2
sample, as shown in Table 17. Lastly, improvements in
~2 uptake and release rates of Co(TpivPP)/BzIm can be
achieved by using small particle supports. The uptake
and release Of ~2 iS slow for the Co(TpivPP)/BzIm
sample, but increases for the 8 and 21 wt. % SiO2
samples, as shown in Table 18. The ~2 loading vs. time
data for the unsupported sample are given in Table 19.
D-20,012
21~40
- 67 -
Table 16
BzIm/Co(T~ivPP) Unsupported
Loading vs. Pressure
Pressure ~2 Loading N2 Loading Ar Loading
(torr) (mmol/g) (mmol/g) (mmol/g)
O O.000 0.000 O.000
200 0.079
S00 0.125 0,003 0.004
1000 0.164
1691 0.003 0.009
2000 0.201
3000 0.224
4500 0.247 0.004 0.017
D-20,012
~16~7 4D
- 68 -
Table 17
Percent Utilization of Co~TpivPP) in BzIm/Co~TpivPP),
Unsupported and on SiO2
SiO2 Content% Utilization of C~~TpivPP)
O % 24
8 % 27
21 % 71
D-20,012
- 69 - 21~2740
Table 18
Comparison of % ~2 Uptake and Release for
BzIm/C~(Tpi~pp)
Unsupported and on SiO2
Time 21 wt % 8 wt. % Unsup- 21 wt. % 8 wt. ~ Unsup-
(min) SiO2 % ~2 sio2% ~2 ported 5i~2 % ~2 SiO2 % ~2 ported
Uptake Uptake% ~2Uptake Release % ~2
Uptake Release
O O O 0 100 100 100
3 94 76 59 88 68 51
97 83 67 89 71 54
98 89 77 93 76 61
99 95 86 95 82 68
97 90 85 73
96 89 79
100 91 83
D-20,012
2i62740
- 70 -
Table 19
~2 Uptake and Release Data to and From 1000 Torr
BzIm/Co(TpivPP) Unsupported
Time (minutes)~2 Loading - Uptake ~2 Loading - Release
to 1000 Torr From 1000 Torr
(mmol/g) (mmol/g)
0 0.000 0.160
1 0.071 0.073
2 0.086 0.081
3 0.095 0.078
4 0.102 0.076
0.108 0.074
0.123 0.062
0.132 0.056
0.137 0.051
0.144 0.043
0.154 0.033
0.160 0.027
100 0.160
120 0.024
150 0.022
180 0.020
200 0.019
D-20,012 - 71 - 21~ ~7 ~ ~
It will be understood that various changes can be
made in the details of the invention without departing
from the scope of the invention as recited in the
appended claims.
1. Composition
The composition of the supported TEC can be varied
by substitution of the supports, i.e. composition and
configuration, the TEC; and, if used, the linking agent
or exogenous Lewis base. Criteria for selecting
supports are based primarily on surface area. The
range of TEC's includes all such materials that
interact reversibly with oxygen in either solution or
solid embodiments. It will be understood that various
changes and modifications can be made with respect to
metal ions, ligands and axial bases used in the TEC's
of the invention. Sorbents using Cr(II) or Pt(II) and
other transition metal ions that can react reversibly
with oxygen can be employed. Similarly, axial bases,
such as oxygen-sulfur-containing hetrocycles, ethers,
thioethers or amines can be used in addition to those
previously mentioned.
The range of TEC's includes TEC's based on
chelating ligands such as pentadentate or tetradentate
systems. Under the application conditions for the
supported TEC, the transition element site should exist
in a five coordinate state in the absence of oxygen.
For TEC's based on tetradentate ligands, this is
conveniently accomplished by coordination in an axial
sense using a Lewis base which can be derived from
either the support surface, the linking agent, or can
be part of an additional component.
D-20,012
- 2l~;2l14~)
- 72 -
2. Preparation
The preparation of TEC coated substrates has been
performed by a number of methods including solvent
evaporation of particle slurries and casting methods
for materials based on porous, soluble polymers. For
non-soluble substrates, other isolation procedures are
applicable including filtration and centrifugation. In
addition, column-based loading methods should be
included where a solution containing the oxygen-
selective coating material is allowed to pass through a
bed containing the substrate. Other coating methods
should also be included such as chemical vapor
deposition ~for TEC and/or other components).
3. Conditions and Applications
As previously noted, oxygen-selective sorbents can
be applied to either bulk separation or purification
processes. The conditions which are employed for these
processes is an important variable. Since the basis of
the selectivity is an equilibrium process with negative
enthalpy, operation at different temperatures can be
used to match performance characteristics to a
particular application. In addition, the sorption and
release of oxygen will respond to changes in either
pressure (or partial pressure) or temperature. In
addition to operating conditions, structural variations
can be used to give desirable oxygen affinities
(Pso (~2) ) ~ oxygenation enthalpies (~H (~2) ) ~ and
uptake/release rates including changes in the nature of
the TEC and the axial donor (Lewis base).
The supported TEC ' s based on small particle
substrates can be used in a number of ways. The
application of TEC' s supported on small particles can
be extended to include assemblies attached to heat
D-20,012
- 216274o
- 73 -
exchanger surfaces or compacted into porous polymers.
In addition, assemblies of TEC coated small particles
can be applied to membrane gas separation processes.
Possible configurations include TEC coated small
particles embedded into dense or microporous polymeric
matrices. Microporous membranes based on TEC coated
small particles are expected to show high oxygen
permeation selectivities due to selective surface
diffusion. The permeation selectivity will be
dependent on a number of factors including the free
pore diameter (a function of mean particle size and
distribution), the spacing of TEC sites, the surface
diffusion coefficient of oxygen, and the oxygen
interaction characteristics of the TEC coating (site
uptake and release rates, energetics).
To this point, the use of supported TEC's has been
described to take advantage of an oxygen selectivity.
However, the use of alternative TEC's can provide the
basis for alternative small molecule separations. The
minimum performance criteria and methods to achieve
them remain the same, but the TEC component should be
substituted for examples appropriate to the desired
separation in solution or solid state embodiments.
The invention provides highly desirable oxygen-
selective sorbents incorporating supported TEC's in the
solid phase capable of enhanced utilization and
sorption rates. Thus, the sorbents of the invention
are highly advantageous for practical commercial
operations.