Note: Descriptions are shown in the official language in which they were submitted.
2~66~21
'~O 95I01980 PCT/IB94/00156
-1_
BICYCLIC TETRAHYDRO PYRAZOLOPYRIDINES
Background of the Invention
This invention relates to a series of bicyclic tetrahydro pyrazolopyridines
which
are selective inhibitors of phosphodiesterase (PDE) type IV or the production
of tumor
necrosis factor (hereinafter TNF) and as such are useful in the treatment of
asthma,
arthritis, bronchitis, chronic obstructive airways disease, psoriasis,
allergic rhinitis,
dermatitis and other inflammatory diseases; and AIDS, septic shock and other
diseases
involving the production of TNF.
This invention also relates to a method of using such compounds in the
treatment of the above diseases in mammals, especially humans and to
pharmaceutical
compositions useful therefor.
Since the recognition that cyclic AMP is an intracellular second messenger
(E.W. Sutherland, and T. W. Rall, Pharmacol. Rev., 1960, 12, 265), inhibition
of the
phosphodiesterases has been a target for modulation and, accordingly,
therapeutic
intervention in a range of disease processes. More recently, distinct classes
of PDE
have been recognized (J.A. Beavo and D. H. Reifsnyder, TIPS, 1990, 11, 150),
and their
selective inhibition has led to improved drug therapy (C.D. Nicholson, R. A.
Challiss and
M. Shahid, TIPS, 1991, 12, 19). More particularly, it has been recognized that
inhibition
of PDE type IV can lead to inhibition of inflammatory mediator release (M.W.
Verghese
et al., J. Mol. Cell Cardiol., 1989) 12 (Suppl. II), S 61 ) and airway smooth
muscle
relaxation (T. J. Torphy in Directions for New Anti-Asthma Druas, eds S. R.
O'Donnell
and C. G. A. Persson, 1988, 37, Birkhauser-Verlag). Thus, compounds that
inhibit PDE
type IV, but which have poor activity against other PDE types, would inhibit
the release
of inflammatory mediators and relax airway smooth muscle without causing
cardiovascular effects or antiplatelet effects.
TNF is recognized to be involved in many infectious and auto-immune diseases
(W. Friers) FEBS Letters, 1991, 285, 199). Furthermore, it has been shown that
TNF is
the prime mediator of the inflammatory response seen in sepsis and septic
shock (C.E.
Spooner et al., Clinical Immunology and Immunopatholo4y, 1992, 62, S11 ).
21667 21
- 2 -
Summary of the Invention
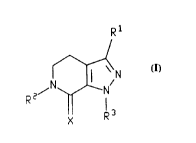
The present invention relates to compounds of the
formula:
I
and the pharmaceutically acceptable salts thereof;
wherein
R1 is hydrogen, (C1-C~)alkyl, (C2-C3)alkenyl,
(C3-C5)cycloalkyl or methylene (C3-C5)cycloalkyl (i.e.,
(C3-C5)cycloalkyl-methyl), wherein each alkyl or alkenyl group
may be optionally substituted with up to two (C1-C2)alkyl or
trifluoromethyl groups or up to three halogens;
X is oxygen or two hydrogen atoms;
R2 and R3 are each independently selected from the group
consisting of hydrogen, (C1-C14)alkyl, (C3-C~)cycloalkyl,
cyclopropylmethyl, (C1-C14)alkoxy, (C2-C~)alkenyl, 2-indanyl,
(C4-C~)heterocyclic group containing oxygen, sulphur, S02 or
NR5 wherein R5 is hydrogen or (C1-C4)alkyl, or a group of the
formula:
(R4)a II
(wherein a is an integer from 1 to 5; b and c are 0 or 1; R4
is hydrogen, hydroxyl, (C1-C5)alkyl, (C2-C5)alkenyl,
(C1-C5)alkoxy, (C3-C6)cycloalkoxy, halogen, trifluoromethyl,
64680-865
,y~~
- 3 - 21 667 21
C02R6, CONR6R~, NR6R~, N02 or S02NR6R~ wherein R6 and R~ are
each independently hydrogen or (C1-C4)alkyl; wherein Z is
oxygen, sulphur, S02 or NR8 wherein R8 is hydrogen or
(C1-C4)alkyl; and Y is (C1-C5)alkylene or (C2-C6)alkenylene
each optionally substituted with up to two (C1-C~)alkyl or
(C3-C~)cycloalkyl groups; or a group of the formula:
R9
W
wherein p is an integer from 1 to 3, W is oxo or hydroxyl, R9
is (C1-C3)alkyl; wherein each of the alkyl, alkenyl,
cycloalkyl, alkoxy or heterocyclic group may be optionally
substituted with one to fourteen, preferably one to five,
substituents selected from the group consisting of
(C1-C2)alkyl, trifluoromethyl and halogen,
with the proviso that:
(1) when R1 is ethyl and R2 is 4-methylphenyl, then R3
cannot be hydrogen, methyl, phenyl, 4-fluorophenyl or
2-pyridyl,
(2) when R2 is 4-methylphenyl and R3 is 4-fluorophenyl,
then R1 cannot be phenyl, methyl or n-propyl,
(3) when R1 is ethyl and R2 is phenyl, then R3 cannot be
4-chlorophenyl, 4-fluorophenyl or 4-methylphenyl,
(4) when Rl is ethyl and R2 is 4-methoxyphenyl, then R3
cannot be 4-fluorophenyl, and
(5) one of R2 or R3 must be other than hydrogen,
64680-865
21 667 2 1
- 4 -
(C1-C14)alkyl, (C2-C~)alkenyl, (C3-C~)cycloalkyl,
cyclopropylmethyl, pyridyl, (C1-C2)alkylpyridyl or a group of
the formula:
~4)a II
wherein b is one and c is zero or b and c are each zero, a is
zero or one and R4 is (C1-C5)alkyl, (C1-C5)alkoxy, halo,
trifluoromethyl, or di(C1-C4)alkylamino.
In one embodiment, the invention relates to a
compound of formula I wherein R1 is (C1-C3)alkyl and R2 and R3
are each independently selected from the group consisting of
(C3-C~)cycloalkyl, (C4-C~)heterocyclic group containing S02 or
a group of the formula:
~4)a
ITI
wherein a is an integer from 1 to 5 and R4 is hydrogen,
hydroxyl, (C1-C5)alkyl, (C1-C5)alkoxy or halogen. An example
of the (C4-C~)heterocyclic group containing S02 is
3-sulfolanyl.
In another embodiment, the invention relates to a
compound of formula I wherein R1 is ethyl or isopropyl; R2 is
phenyl, 2-methylphenyl, 3-methylphenyl, 2-methoxyphenyl,
3-methoxyphenyl or 3-trifluoromethylphenyl and R3 is
cyclobutyl, cyclopentyl, cyclohexyl, 3-sulfolanyl,
64680-865
- 4a - 21 6 6 7 2 1
4-fluorophenyl or 3,4-dichlorophenyl.
The present invention further relates to a
pharmaceutical composition for the inhibition of
phosphodiesterase (PDE) type IV and the production of tumor
necrosis factor (TNF) comprising a pharmaceutically effective
amount of a compound according to formula I and the
pharmaceutically acceptable salts thereof, and a
pharmaceutically acceptable carrier.
The present invention further relates to a use of a
compound according to formula I and the pharmaceutically
acceptable salts thereof for the inhibition of
phosphodiesterase (PDE) type IV and the production of tumor
necrosis factor (TNF).
The present invention further relates to a use of a
compound of the formula I and the pharmaceutically acceptable
salts thereof for treating an inflammatory condition in
mammals.
The present invention further relates to a
pharmaceutical composition for the treatment of asthma,
arthritis, bronchitis, chronic obstructive airways disease,
psoriasis, allergic rhinitis, dermatitis and other
inflaa~atory diseases, AIDS, septic shock and other diseases
involving the production of TNF comprising a pharmaceutically
effective amount of a compound according to formula I and the
pharmaceutically acceptable salts thereof together with a
pharmaceutically acceptable carrier.
This invention further relates to a use of a
compound according to formula I and the pharmaceutically
64680-865
2166721
- 4b -
acceptable salts thereof for treating or preventing a
condition selected from the group consisting of asthma,
arthritis, bronchitis, chronic obstructive airways disease,
psoriasis, allergic rhinitis, dermatitis and other
inflaaanatory diseases, AIDS, septic shock and other diseases
involving the production of TNF.
Specific preferred compounds disclosed in this
specification include both within and outside the scope
claimed in this application, as follows:
3-ethyl-1-(4-methoxyphenyl)-6-phenyl-7-oxo-4,5,6,7-
tetrahydro-1H-pyrazolo[3,4-c]pyridine;
3-ethyl-1-cyclopentyl-6-phenyl-7-oxo-4,5,6,7-
tetrahydro-1H-pyrazolo[3,4-c]pyridine;
3-ethyl-1-(3,4-dichlorophenyl)-6-(3-methoxyphenyl)-
7-oxo-4,5,6,7-tetrahydro-1H-pyrazolo[3,4-c]pyridine;
3-ethyl-1-cyclopentyl-6-(3-methoxyphenyl)-7-oxo-
4,5,6,7-tetrahydro-1H-pyrazolo[3,4-c]pyridine;
3-ethyl-1-(4-fluorophenyl)-6-(2-methoxyphenyl)-7-
oxo-4,5,6,7-tetrahydro-1H-pyrazolo[3,4-c]pyridine;
3-ethyl-1-cyclopentyl-6-(3-methylphenyl)-7-oxo
4,5,6,7-tetrahydro-1H-pyrazolo[3,4-c]pyridine;
3-ethyl-1-cyclopentyl-6-(3-trifluoromethylphenyl)-7-
oxo-4,5,6,7-tetrahydro-1H-pyrazolo[3,4-c]pyridine;
3-ethyl-1-cyclohexyl-6-(3-methoxyphenyl)-7-oxo-
4,5,6,7-tetrahydro-1H-pyrazolo[3,4-c]pyridine;
64680-865
''O 95I01980 ~ ~ ~ PCT/1B94/00156
-5-
3-isopropyl-1-cyclopentyl-6-(3-methoxyphenyl)-7-oxo-4,5,6,7-tetrahydro-1 H-
pyrazolo[3,4-c]pyridine;
3-ethyl-1-cyclobutyl-6-(3-methoxyphenyl)-7-oxo-4,5,6,7-tetrahydro-1 H-
pyrazolo[3,4-c]pyridine;
3-ethyl-1-cyclopentyl-6-phenyl-4,5,6,7-tetrahydro-1 H-pyrazolo[3,4-c]pyridine;
3-ethyl-1-cyclopentyl-6-(2-methylphenyl)-7-oxo-4,5,6,7-tetrahydro-1 H-
pyrazolo[3,4-
c]pyridine;
3-ethyl-1-(3-sulfolanyl)-6-(3-methylphenyl)-7-oxo-4,5,6,7-tetrahydro-1 H-
pyrazolo [3,4-c] pyridine;
3-ethyl-1-(3-sulfolanyl)-6-(3-methoxyphenyl)-7-oxo-4,5,6,7-tetrahydro-1 H-
pyrazolo[3,4-c]pyridine;
3-ethyl-1-cyclobutyl-6-(3-methylphenyl)-7-oxo-4,5,6,7-tetrahydro-1 H-
pyrazolo[3,4-
c]pyridine;
3-ethyl-1-(3-sulfolanyl)-6-(3-trifluoromethylphenyl)-7-oxo-4,5,6,7-tetrahydro-
1 H-
pyrazolo[3,4-c]pyridine;
3-ethyl-1-cyclobutyl-6-(3-trifluoromethylphenyl)-7-oxo-4,5,6,7-tetrahydro-1 H-
pyrazolo[3,4-c]pyridine;
3-ethyl-1-cyclobutyl-6-(2-methylphenyl)-7-oxo-4,5,6,7-tetrahydro-1 H-
pyrazolo[3,4-
c]pyridine.
Detailed Description of the Invention
The term "halogen", as used herein, unless otherwise indicated, includes
chloro,
fluoro and bromo.
Unless indicated otherwise, the alkyl, alkoxy and alkenyl groups referred to
herein may be straight chained or if comprising three or more carbons may be
straight
chained, branched, cyclic or a combination of cyclic and branched or straight
chained
moieties.
The "inflammatory diseases" which can be treated according to this invention
include, but are not limited to asthma, chronic obstructive pulmonary disease,
bronchitis and arthritis.
R' , R2 and R3, as used herein, unless otherwise indicated, are as defined
above
with reference to formula I.
The following reaction schemes illustrate, but are not limiting to the
preparation
of the compounds of the present invention.
21b6721
WO 95/01980 PCT/IB94/00156
SCHEnE i
0 0
NH ly ~NRZ 2 I R 1
R2~N OH
Iv v 0
VI
13
0
wRi
R~~N OCH3
0
VII
SCHEnE 2
0
Ri
i
~R
~ N I 0 R 5 ~.. N I N.~J
R R~
0 0 R3
VIII IX
i
Ri
~N~N.N
R
R3
X
2 i 66l'2
- 'NO 95l01980 PCT/IB94/00156
_7_
In Reaction 1 of Scheme 1, the 2-pyrrolidinone compound of formula IV is
converted to the corresponding N-(aryl)-2-pyrrolidone compound V wherein
"aryl" is a
group of the formula II by reacting IV with an aryl halide neat in the
presence of copper
power and potassium carbonate. Suitable aryl halides include 1-iodo- or 1-
bromo- 4-
methoxybenzene, 3-methoxybenzene, 2-methoxybenzene, 3-methylbenzene, 4-
methylbenzene) 2-methylbenzene, 3-trifluoromethylbenzene) 2-
trifluoromethylbenzene)
3,4-dimethoxybenzene or 3-cyclopentoxy-4-methoxybenzene. The reaction
temperature
will generally be in the range of about 110 ~ C to about 170 ~ C, preferably
about 150 ~ C,
for a time period of about 14 hours to about 22 hours, preferably about 18
hours, under
inert reaction conditions.
In Reaction 2 of Scheme 1, R' halide is added to a suspension of magnesium
in an anhydrous aprotic solvent. The reaction mixture is heated to reflux
until all the
magnesium is consumed and thereafter cooled to atemperature between about -
15~C
to about 15 ~ C, preferably about 0 ~ C. The N-(aryl)-2-pyrrolidone compound
of formula
V is then added and the reaction mixture is warmed to room temperature while
being
stirred for a time period between about 1.5 hours to about 2.5 hours,
preferably about
2 hours. Suitable alkyl halides include bromomethane, bromoethane or
bromopropane. The preferred anhydrous aprotic solvent is anhydrous ether. Upon
completion of the reaction, the desired intermediate may be isolated in a
conventional
manner, e.g., by first washing the combined organics with water and brine,
then drying
over sodium sulfate, filtering and concentrating under reduced pressure to
afford a
readily-recoverable precipitate in the form of a white solid.
The above precipitate is converted to the corresponding 1 (2,5,6-
tetrahydropyridine compound of formula VI by dispersing the precipitate in a
mixture
of a non-polar aprotic solvent and base. Upon vigorous stirring, ethyl oxalyl
chloride
is added and the reaction mixture is heated to reflux for a time period
between about
1.5 hours to about 4.5 hours, preferably about 3.0 hours. The preferred non-
polar
aprotic solvent is benzene and the preferred base is sodium hydroxide. The
solvents
are removed and the resulting residue is treated with a solution of sodium
alkoxide in
ethanol. After heating at reflux for a time period between about 1 hour and
about 3
hours, preferably about 1.5 hours, the mixture is concentrated under reduced
pressure
and acidified to pH=3 with hydrochloric acid.
WO 95I01980 6 PCT/IB94/00156
_g_
In Reaction 3 of Scheme 1, the compound of formula VI is converted to the
corresponding 3-methoxy-1,2,5,6-tetrahydropyridine compound VII by heating to
reflux
a reaction mixture of VI and 3-methyl-1-p-tolyltriazene in an aprotic solvent.
The
preferred aprotic solvent is 1,2-dichloroethane. The time period for the
reaction is
between about 30 minutes to about 120 minutes, preferably about 45 minutes.
In Reaction 1 of Scheme 2, the 1,2,5,6-tetrahydropyridine compound of formula
VIII, wherein R5 is hydrogen or methyl, is converted to the corresponding
4,5,6,7-
tetrahydro-7-oxo-1 H-pyrazolo[3,4-c]pyridine compound IX by reacting VIII with
a
hydrazine of the formula R3HNNHz. Both derivatives of the compound of formula
VIII,
3-hydroxy and 3-methoxy, may be used as starting materials under one of three
different sets of reaction conditions.
Under one set of reaction conditions, the 1,2,5,6-tetrahydropyridine compound
of formula VIII is converted to the corresponding compound of formula IX by
reacting
VIII with a hydrazine hydrochloride and sodium alkoxide in an anhydrous polar
erotic
solvent. The preferred sodium alkoxide is sodium methoxide and the preferred
anhydrous polar erotic solvent is anhydrous ethanol. The reaction mixture is
heated
to reflux for a time period between about 9 hours to about 15 hours,
preferably about
12 hours.
Under a second set of reaction conditions, the 1,2,5,6-tetrahydro-pyridine
compound VIII is converted to the corresponding compound of formula IX by
reacting
VIII with hydrazinobenzoic acid in an anhydrous polar erotic solvent,
preferably ethanol.
The reaction mixture is heated to reflux for a time period between about 16
hours to
about 24 hours, preferably about 20 hours. The compound IX so formed may be
further reacted to give the corresponding 1-(4-benzamide)-7-oxo-4,5,6,7-
tetrahydro-1 H-
pyrazolo(3,4-c]pyridine compound by reacting IX with sodium methoxide in a
polar
erotic solvent, preferably methanol, for a time period between about 15
minutes to
about 45 minutes, preferably 30 minutes. The polar erotic solvent is removed
under
reduced pressure, the solid residue is suspended in a non-polar aprotic
solvent,
perferably benzene, and thereafter) the non-polar solvent is removed under
reduced
pressure. The resulting dry solid is suspended in cold ether and treated with
oxalyl
chloride and N,N-dimethylformamide and allowed to stir for a time period
between
about 30 minutes .to about 90 minutes, preferably 60 minutes. The solvent is
then
removed and the crude residue is dissolved in dry tetrahydrofuran. The
resulting
VO 95/01980 PCT/IB94/00156
2166721
-g_
solution is added dropwise to stirred ammonium hydroxide at a temperature
between
about -10~C to about 10~C, preferably 0~C.
Under a third set of reaction conditions, the 1,2,5,6-tetrahydropyridine
compound of formula VIII is converted to the corresponding compound of formula
IX
by reacting VIII with a hydrazine hydrochloride in a polar erotic solvent,
preferably
methanol. The reaction mixture is heated to a temperature between about 70~C
to
about 110 ~ C, preferably about 90 ~ C, under a gentle stream of nitrogen
until all of the
solvent is removed. The neat mixture is then heated to a temperature between
about
120 ~ C to about 180 ~ C, preferably about 150 ~ C, for a time period between
about 30
minutes to about 90 minutes, preferably 60 minutes.
The compounds so formed of formula IX may be converted to the
corresponding 6-Rz-4,5,6,7-tetrahydro-7-oxo-1 H-pyrazolo [3,4-c]pyridine
compound,
wherein R2 is other than the group of formula II, by reacting a solution of IX
in a polar
aprotic solvent, preferably acetonitrile, with a solution of ammonium cerium
(IV) nitrate
in water at a temperature between about -15~C to about 15~C, preferably about
0~C,
for a time period between about 20 minutes to about 50 minutes, preferably
about 35
minutes. Upon completion of the reaction, the mixture is diluted with water
and
extracted with ethyl acetate. The combined organics are then washed with
saturated
sodium bicarbonate followed by sodium sulfite. The compound so formed in a
polar
aprotic solvent, preferably tetrahydrofuran, is treated with sodium hydride,
heated to
reflux and stirred for a time period between about 30 minutes to about 60
minutes,
preferably 45 minutes. The reaction mixture is cooled to a temperature between
about
20 ~ C to about 30 ~ C, preferably about 25 ~ C, and an alkyl halide of
formula RZ halide,
wherein R~ is as defined with reference to formula I other than a group of
formula II, is
added. The reaction mixture is stirred and heated to reflux for a time period
between
about 12 hours to about 20 hours, preferably 16 hours.
In Reaction 2 of Scheme 2, the 2-oxo-4,5,6,7-tetrahydro-1 H-pyrazolo[3,4-
c]pyridine compound IX is converted to the corresponding compound of formula X
by
reacting IX with a reducing agent, preferably lithium aluminum hydride, in a
non-polar
aprotic solvent, preferably ether. The reaction is stirred for a time period
between about
12 hours to about 20 hours, preferably 16 hours. Water and base) preferably
sodium
hydroxide, is then added and the reaction mixture is stirred for a time period
between
WO 95/01980 ~ PCT/IB94/00156
-10-
about 1.5 hours to about 2.5 hours, preferably 2 hours, and filtered. The
filtrate is
concentrated to a white solid.
The ability of the compounds or the pharmaceutically acceptable salts thereof
to inhibit phosphodiesterase IV (PDEQ) and, consequently, demonstrate their
effectiveness for treating inflammatory diseases is shown by the following in
vitro assay.
BIOLOGICAL ASSAY
(Human lung PDE,~)
Thirty to forty grams of human lung tissue is placed in 50 ml of pH 7.4
Tris/phenylmethylsulfonyl fluoride (PMSF)/sucrose buffer and homogenized using
a
Tekmar Tissumizer~ (Tekmar Co., 7143 Kemper Road, Cincinnati, Ohio 45249) at
full
speed for 30 seconds. The homogenate is centrifuged at 48,000 x g for 70
minutes at
4~C. The supernatant is filtered twice through a 0.22,um filter and applied to
a Mono-D
FPLC column (Pharmacia LKB Biotechnology, 800 Centennial Avenue, Piscataway,
New
Jersey 08854) pre-equilibrated with pH 7.4 Tris/PMSF buffer. A flow rate of 1
ml/minute
is used to apply the sample to the column, followed by a 2 ml/minute flow rate
for
subsequent washing and elution. Sample is eluted using an increasing, step-
wise NaCI
gradient in the pH 7.4 Tris/PMSF buffer. Eight ml fractions are collected.
Fractions are
assayed for specific PDE,~ activity, determined by [3H]CAMP hydrolysis and the
ability
of a known PDE,~ inhibitor (e.g. rolipram) to inhibit that hydrolysis.
Appropriate
fractions are pooled, diluted with ethylene glycol (2 ml ethylene glycol/5 ml
of enzyme
prep) and stored at -20~C until use.
Compounds are dissolved in DMSO at a concentration of 10 mM and diluted
1:25 in water (400 ,uM compound, 4% DMSO). Further serial dilutions are made
in 4~~
DMSO to achieve desired concentrations. Final DMSO concentration in assay tube
is
1 %. In duplicate the following are added, in order, to a 12 x 75 mm glass
tube (all
concentrations are given as final concentrations in assay tube).
i) 25 ,ul compound or DMSO (1 %, for control and blank)
ii) 25 NI pH 7.5 Tris buffer
iii) [3H]cAMP (1 ,uM)
iv) 25 NI PDE", enzyme (for blank, enzyme is preincubated in boiling water
for 5 minutes)
The reaction tubes are shaken and placed in a water bath (37~C) for 20
minutes, at which time the reaction is stopped by placing the tubes in a
boiling water
WO 95I01980 ~ b ~ PCTIIB94/00156
-11-
bath for 4 minutes. Washing buffer (0.5 ml, 0.1 M 4-(2-hydroxyethyl)-1-
piperazine-
ethanesulfonic acid (HEPES)/0.1 M NaCI, pH 8.5) is added to each tube on an
ice bath.
The contents of each tube are applied to an Affi-Gel 601 column (Biorad
Laboratories,
P.O. Box 1229, 85A Marcus Drive, Melville, New York 11747) (boronate affinity
gel, 1
ml bed volume) previously equilibrated with washing buffer. [3H)cAMP is washed
with
2 x 6 ml washing buffer, and [3H]5'AMP is then eluted with 4 ml of 0.25M
acetic acid.
After vortexing, 1 ml of the elution is added to 3 ml scintillation fluid in a
suitable vial,
vortexed and counted for [3H].
inhibition = 1 - avera a cpm test compound) - average cpm Lblank)
average cpm (control) - average cpm (blank)
ICSO is defined as that concentration of compound which inhibits 50% of
specific
hydrolysis of [3H]CAMP to [3H]5'AMP.
(TNF)
The ability of the compounds or the pharmaceutically acceptable salts thereof
to inhibit the production of TNF and, consequently, demonstrate their
effectiveness for
treating diseases involving the production of TNF is shown by the following in
vitro
assay:
Peripheral blood (100 mls) from human volunteers is collected in
ethylenediaminetetraacetic acid (EDTA). Mononuclear cells are isolated by
Ficoll/Hypaque and washed three times in incomplete HESS. Cells are
resuspended
in a final concentration of 1 x 1 O6 cells per ml in pre-warmed RPMI
(containing 5% FCS,
glutamine, pen/step and nystatin). Monocytes are plated as 1 x 106 cells in
1.0 ml in
24-well plates. The cells are incubated at 37~C (5% carbon dioxide) and
allowed to
adhere to the plates for 2 hours, after which time non-adherent cells are
removed by
gentle washing. Test compounds (10N1) are then added to the cells at 3-4
concentrations each and incubated for 1 hour. LPS (10N1) is added to
appropriate
wells. Plates are incubated overnight (18 hrs) at 37~C. At the end of the
incubation
period TNF was analyzed by a sandwich ELISA (R&D Quantikine Kit). ICSo
determinations are made for each compound based on linear regression analysis.
Pharmaceutically-acceptable acid addition salts of the compounds of this
invention include, but are not limited to, those formed with HCI, HBr, HN03,
HZS04,
H3P04, CH3S03H, p-CH3C6HQS03H, CH3COZH, gluconic acid, tartaric acid, malefic
acid
and succinic acid. Pharmaceutically-acceptable cationic salts of the compounds
of this
WO 95/01980 2 ~ ~; 6 7 21 PCT/IB94/00156
-12-
invention of formula I wherein R4 is COZRs and R6 is hydrogen include, but are
not
limited to, those of sodium, potassium, calcium, magnesium) ammonium, N,N'-
dibenzylethylenediamine, N-methylglucamine (meglumine), ethanolamine and
diethanolamine.
For administration to humans in the curative or prophylactic treatment of
inflammatory diseases, oral dosages of the compounds of formula I and the
pharmaceutically acceptable salts thereof (hereinafter also referred to as the
active
compounds of the present invention) are generally in the range of from 0.1-100
mg
daily for an average adult patient (70 kg). Thus for a typical adult patient,
individual
tablets or capsules contain from 0.1 to 50 mg of active compound, in a
suitable
pharmaceutically acceptable vehicle or carrier. Dosages for intravenous
administration
are typically within the range of 0.1 to 10 mg per single dose as required.
For
intranasal or inhaler administration, the dosage is generally formulated as a
0.1 to 1 ~~
(w/v) solution. In practice the physician will determine the actual dosage
which will be
most suitable for an individual patient and it will vary with the age, weight
and response
of the particular patient. The above dosages are exemplary of the average case
but
there can, of course, be individual instances where higher or lower dosage
ranges are
merited, and all such dosages are within the scope of this invention.
For administration to humans for the inhibition of TNF, a variety of
conventional
routes may be used including orally, parenterally and topically. In general,
the active
compound will be administered orally or parenterally at dosages between about
0.1 and
mg/kg body weight of the subject to be treated per day, preferably from about
0.3
to 5 mg/kg. However, some variation in dosage will necessarily occur depending
on
the condition of the subject being treated. The person responsible for
administration
25 will, in any event, determine the appropriate dose for the individual
subject.
For human use, the active compounds of the present invention can be
administered alone, but will generally be administered in an admixture with a
pharmaceutical diluent or carrier selected with regard to the intended route
of
administration and standard pharmaceutical practice. For example, they may be
administered orally in the form of tablets containing such excipients as
starch or
lactose, or in capsules or ovales either alone or in admixture with
excipients, or in the
form of elixirs or suspensions containing flavoring or coloring agents. They
may be
injected parenterally; for example, intravenously, intramuscularly or
subcutaneously.
~VO 95/01980 ~ ~ ~ PCT/IB94/00156
-13-
For parenteral administration, they are best used in the form of a sterile
aqueous
solution which may contain other substances; for example, enough salts or
glucose to
make the solution isotonic.
Thus in a further aspect the invention provides pharmaceutical compositions
comprising a compound of the formula I and the pharmaceutically acceptable
salts
thereof together with a pharmaceutically acceptable diluent or carrier.
The present invention is illustrated by the following examples, but it is not
limited
to the details thereof.
Example 1
3-Ethyl-1-~4-methoxyehenyl)-6-phenyl-7-oxo-4,5,6,7-tetrah~dro-1 H-pyrazolof3,4-
c ridine
A mixture of 3-hydroxy-2-oxo-1-phenyl-4-propionyl-1,2,5,6-tetrahydro-pyridine
(1.0
g, 4.1 mmole), 4-methoxyphenylhydrazine hydrochloride (0.8 g, 4.6 mmole) and
sodium
methoxide (0.11 grams, 2 mmole) in 35 ml anhydrous ethanol (distilled from Mg)
was
heated at reflux. After 12 hours, the solvent was removed by rotory
evaporation under
reduced pressure, and the crude residue was chromatographed on a 4x20 cm
silica
column using 1:1 ether/hexane as eluent to give 345 mg of the title compound
as a red
oil that crystallized upon standing at room temperature. The desired 1-(4-
methoxyphenyl) regioisomer is less polar than the 2-(4-methoxyphenyl)
byproduct.
M.P. 43-45~C, IR (chloroform) lactam C=O, 1665 cm-'; 'H NMR (300 MHz, CDC13) d
1.32(t,J=7.6Hz,3H),2.74(q,J=7.6Hz,2H),2.96(t,J=6.6Hz,2H),3.79(s,3H),
4.10 (t,J = 6.6 Hz, 2H), 6.89 (d,J = 9.0 Hz, 2H), 7.22-7.39 (m, 5H), 7.45 (d,J
= 9.0 Hz,
2H); Anal. calcd. for C2, Hz, N302: C, 72.60; H, 6.09; N, 12.09. Found; C,
72.48; H,
6.08; N, 11.66; MS m/z (M+) 347.
Examples 2-15
Reaction of the appropriate hydrazine hydrochloride with the requisite 4-
alkanoyl-3-hydroxy-2-oxo-1,2,5,6-tetrahydropyridine, analogous to the
procedure of
Example 1, affords the following compounds.
Ex.#R' Rz R3 M.p.C Mass Spectra Mass Spectra
or or
Analysis (ealcd.)Analysis (found)
%C, %H, %N %C, %H) %N
2 ethyl phenyl methyl 80-83 70.56, 6.71, 70.61, 6.77,
16.46 15.51
3 ethyl phenyl ten-butyl120-121?2.70, 7.79, 72.50, 7.96,
14.13 14.16
WO 95/01980 PCT/IB94/00156
-14-
4 ethyl 4-methoxy4-methoxy42-45 70.01, 6.14, 70.05, 6.07,
11.13 11.00
phenyl phenyl
ethyl 4-fluorotert-butyl92-94 (M') 315.1747HRMS (M')
phenyl 315.1741
6 ethyl phenyl 3,4- (oil) [M' ] 386.26 MS m/z [M+]
386
dichloro-
phenyl
7 ethyl 4-fluoro4-methoxy129-130'69.03, 5.52, 68.75, 5.37,
11.50 11.43
phenyl phenyl
5 8 methylphenyl 4-fluoro139-140[M'] 321.3 MS m/z [M')
322
phenyl
9 ethyl phenyl cyclopent73-75 (M+) 309.1841HRMS (M+)
yl 309.1823
methylphenyl 4-methoxy167-168(M+) 333.1477HRMS (M+)
phenyl 333.1477
11 ethyl phenyl 5-phenyl(oil) (M+) 388.2389HRMS (M+)
pentyl 388.239S
12 methyl4-methoxy4-fluoro140-14268.36, 5.16, 67.92, 5.03,
11.96 11.72
phenyl phenyl
10 13 methyl4-methoxy3-fluoro133-13868.36, 5.16, 68.04, S.04,
11.96 11.7S
phenyl phenyl
14 ethyl 4-methoxy3,4- 50-60 60.59, 4.60, 60.34, 4.56,
10.09 9.86
phenyl dichloro-
phenyl
ethyl 3-methoxymethyl (oil) [M' ] 285.3S MS m/z [M'
] 286
phenyl
Recrystallizing solvents: aisopropyl ether. b5% Ethyl acetate in petroleum
ether.
15 Example 16
3-Ethyl-1-(4-phenylcarboxylic acid)-6-phenyl-7-oxo-4,5,6,7-tetrahydro-1 H-
pyrazolo f3,4-clp~rridine
A mixture of 3-hydroxy-2-oxo-1-phenyl-4-propionyl-1,2,5,6-tetrahydro-pyridine
(1.0
grams, 4.08 mmole), 4-hydrazinobenzoic acid (0.68 grams, 4.49 mmole) and 30 ml
of
anhydrous ethanol was heated at reflux. After 20 hours, the mixture was
concentrated
by rotory evaporation under reduced pressure, and the solid residue was
suspended
in a mixture of ethyl acetate (500 ml) and pH 4 buffer (200 ml). The organic
layer was
separated (leaving behind most of the 2-(4-phenylcarboxylic acid) byproduct),
washed
with brine, dried over sodium sulfate, filtered and concentrated under reduced
pressure.
WO 95/01980 216 6 7 21 PCT/1B94/00156
-15-
Recrystallization from methanol gives 0.64 grams of the title compound as an
orange
solid. M.P. 261-263~C, 'H NMR (300 MHz, DMSO-d6j, d 1.23 (t,J = 7.6 Hz, 3H),
2.68
(q,J - 7.6 Hz, 2H), 2.94 (t,J = 6.5 Hz, 2H), 4.05 (t,J = 6.5 Hz, 2H), 7.20-
7.41 (m) 5H),
7.65 (d,J = 8.6 Hz) 2H), 7.96 (d,J = 8.6 Hz) 2H)) 13.05 (s) 1 H); MS m/z (M+)
362.
Example 17
1-(4-Benzamide)-3-ethyl-6-(4-methoxyphenyll-7-oxo-4.5.6,7-tetrahydro-1 H-
prrazolof3,4-clpyridine
To a stirred solution of sodium methoxide in methanol (prepared from 6.6 mg
Na) is added 3-ethyl-6-(4-methoxyphenyl)-1-(4-phenylcarboxylic acid)-7-oxo-
4,5,6,7-
tetrahydro-1 H-pyrazolo[3,4-cjpyridine (96 mg, 0.25 mmole). After 30 minutes,
methanol
was removed under reduced pressure, the solid residue was suspended in
benzene,
and the benzene was removed under reduced pressure. The resulting dry solid
was
suspended in cold ether (ice-bath) and treated with oxalyl chloride (31 NI,
0.35 mmole)
and anhydrous N,N-dimethylformamide (1 drop). After stirring for 1 hour the
valatiles
are removed under reduced pressure, and the crude residue was dissolved in dry
tetrahydrofuran. The resulting solution was added dropwise to briskly stirred
ammonium hydroxide at 0 ~ C. After warming to ambient temperature over 2 hours
the
reaction mixture was concentrated under reduced pressure until a yellow solid
begins
to precipitate. At this time the mixture was diluted with water to
approximately 100 ml
and filtered, and the precipitate was washed with water to give 81 mg of the
title
compound. Decomposition point 243-245 ~ C; ' H NMR (DMSO-ds) 1.24 (t,J = 7.6
Hz,
3H), 2.68 (q,J = 7.6 Hz, 2H), 2.93 (t,J = 6.5 Hz, 2H), 3.75 (s, 3H), 3.99 (t,J
= 6.5 Hz,
2H), 6.94 (d,J = 9.1 Hz, 2H), 7.27 (d,J = 9.0 Hz, 2H), 7.43 (s, 1 H), 7.59
(d,J = 8.5 Hz,
2H), 7.90 (d,J = 8.6 Hz, 2H), 8.04 (s, 1 H); Anal. calcd. for CZZHZZN403: C,
67.68; H,
5.68; N, 14.35. Found: C, 67.19; H) 5.31; N, 13.55. HRMS calcd. for CZZHzzNa03
[M+]
391.1770. Found 391.1781.
The starting 3-ethyl-6-(4-methoxyphenyl)-1-(4-phenylcarboxylic acid)-7-oxo-
4,5,6,7-tetrahydro-1 H-pyrazolo [3,4-cj pyridine was prepared using the
appropriate
reagents according to the procedure of example 16.
WO 95/01980 2 ~ 6 6 7 21 PCT/IB94/00156
-16-
Example 18
1-(3 4-dichloropheny~-3-ethy~3-methoxyphenyl)-7-oxo-4.5.6,7-tetrahydro-1 H-
pyrazolo f 3,4-cl pyridine
A stirred mixture of 3-methoxy-1-(3-methoxyphenyl)-2-oxo-4-propionyl-1,2,5,6-
tetrahydro-pyridine (0.49 grams,1.7 mmole), 3,4-dichlorophenylhydrazine
hydrochloride
(0.40 grams, 1.87 mmole) and sodium methoxide (46 mg, 0.85 mmole) in anhydrous
ethanol was heated to reflux. After 16 hours, the mixture was concentrated
under
reduced pressure and chromatographed on a silica gel column using 1:4 ethyl
acetate/hexane as eluent to give a white solid. Recrystallization from ether
gave 0.46
grams of white needles. M.P. 97-99~C,'H NMR (250 MHz, CDCI3) 1.31 (t,J = 7.5
Hz,
3H), 2.73 (q,J = 7.6 Hz. 2H), 2.96 (t,J = 6.6 Hz, 2H), 3.79 (s, 3H), 4.09 (t,J
= 6.6 Hz,
2H), 6.78-6.91 (m, 3H), 7.29-7.49 (m, 3H), 7.73 (d,J = 1.8 Hz, 1 H); MS m/z
[M+] 416.
Examples 19-42
Reaction of the appropriate hydrazine hydrochloride with the requisite 4-
alkanoyl-3-methoxy-2-oxo-1,2,5,6-tetrahydropyridine, analogous to the
procedure of
Example 18, affords the following compounds.
Ex.#R' RZ R3 M.p.C Mass Spectra Mass Spectra
or or
Analysis (calcd.}Analysis (found)
,
%C, %H, %N %C, %H, %N
19 methyl4-methoxy3-4- 143-144'59.71, 4.26, 56.13, 4.02,
10.45 9.65
phenyl dichloro-
phenyl
20 ethyl 3-methoxycyclo- 64-65 [M' ] 340.2025HRMS [M+]
phenyl pentyl 340.2046
21 ethyl 4-methoxycyclo- 96-98 70.77, 7.42, 70.44, 7.68,
12.38 11.69
phenyl pentyl
22 methyl4-methoxycyclo- 121-12270.13, 7.12, 69.48, 7.10,
12.91 12.70
phenyl pentyl
23 iso- phenyl 3,4- oil [M'] 400.0983HRMS [M']
propyl dichloro 400.0966
phenyl
24 ethyl 3,4-dimeth-cyclo- 107-108[M'] 369.46 MS m/z [M']
369
oxyphenylpentyl
25 ethyl 3,4-dimeth-3,4- 190-19159.20, 4.74, 59.41, 4.46,
9.41 9.71
oxyphenyldichloro-
phenyl
PCT/IB94J00156
vo 95/01980 216 6 7 21
-17-
Ex.# R' R2 R3 M.p.C Mass Spectra Mass Spectra
or or
Analysis (calcd.)Analysis (found)
%C, %H, %N %C, %H, %N
26 iso- 4-methoxy3,4- 145-147'61.40, 4.92, 61.29, 4.81,
9.76 9.53
propylphenyl dichloro
phenyl
27 propyl4-methoxycyclo- 102-103'71.36, 7.70, 70.98, 7.66,
11.89 11.73
phenyl pentyl
28 iso- 3-methoxy3,4- 126-12T61.40, 4.92, 61.55, 5.10,
9.76 9.97
propylphenyl dichloro-
phenyl
29 ethyl 4-methoxy-3,4- 54-56 62.40, 5.44, 62.15, 5.S0,
8.40 7.97
3-cyclo-dichloro-
pentoxy-phenyl
phenyl
30 ethyl 4-methoxy-cyclo- 88-89 [M' ] 423.S5 MS m/z [M'
) 423
3-cyclo-pentyl
pentoxy-
phenyl
31 ethyl 3-methoxy4-fluoro-139-140'69.03, 5.79, 69.05, 5.42,
11.50 11.57
phenyl phenyl
32 ethyl 2-methoxycyclo- 119-12070.77, 7.42, 70.63, 7.16,
12.38 12.01
phenyl pentyl
33 ethyl 2-methoxy4-fluoro-103-104'[M' ] 365.41 MS m/z [M'
) 366
phenyl phenyl
34 ethyl 3-methylcyclo- oil 74.27, 7.79, 74.54, 7.89,
12.99 12.63
phenyl pentyl
35 ethyl 3-methyl4-fluoro-oil 72.19,5.77, 72.06, 5.55,
12.02 11.52
phenyl phenyl
36 ethyl 3-trifluoro-cyclo- oil 63.65, 5.87, 63.9S, 5.73,
11.13 10.97
methyl- pentyl
phenyl
37 ethyl 3-trifluoro-4-fluoro-139-140'62.53) 4.25, 62.60, 4.08,
10.42 10.41
methyl- phenyl
phenyl
38 ethyl 4-methyl-cyclo- 93-94 74.27, 7.79, 74.10, 7.52,
12.99 12.59
phenyl pentyl
39 ethyl 2-methyl-4-fluoro-141-142b72.19, S.77, 72.36) 5.52)
12.03 12.09
phenyl phenyl
40 ethyl 2-methyl-cyclo- 130-131MW 323.44 MS m/z 323
phenyl pentyl
WO 95I01980 PCT/IB94100156
21 ~6~2~
-18-
Ex.# R' Rz R3 M.p.C Mass Spectra Mass Spectra
or or
Analysis (calcd.)Analysis (found)
%C, %H, %N %C, %H, %N
41 ethyl 2-trifluoro-4-fluoro-48-50 MW 403.38 MS m/z 404
methyl- phenyl
phenyl
42 ethyl 3-methyl-3-sulfo-oil MW 373.47 MS m/z 374
phenyl lanyl
Recrystallizing solvents: B 5% Ethyl acetate/petroleum ether. b Isopropyl
ether.
' Ethyl acetate/hexane. dEthyl ether. e5% Ethyl acetate/pentane. 'Pentane.
Example 43
1-Cvclohexvl-3-ethyl-6-(3-methoxvphenvl)-7-oxo-4.5.6.7-tetrahvdro-1 H-
pyrazolo(3,4-clpyridine
A solution of 3-methoxy-1-(3-methoxyphenyl)-2-oxo-4-propionyl-1,2,5,6-pyridine
(0.80 grams, 2.8 mmole) and cyclohexylhydrazine hydrochloride (0.54 grams, 3.6
mmole) in methanol (15 ml) was warmed to 90~C under a gentle stream of
nitrogen
until all of the solvent was removed. The neat mixture was then heated to
approximately 150~C under nitrogen for 1 hour. After cooling to room
temperature, the
mixture was dissolved in ether and washed with 1 N hydrochloric acid followed
by brine,
dried over sodium sulfate, filtered and concentrated under reduced pressure.
Chromatography on silica gel using 1:1 ethyl acetate/hexane as eluent gives
0.47
grams of the title compound as a yellow oil. ' H NMR (250 MHz, CDCI3) 1.20-
1.52 (m,
6H, including t at 1.23, J = 7.6 Hz, 3H), 1.64-1.74 (m, 1 H), 1.80-2.06 (m,
6H), 2.67 (q,J
= 7.6 Hz, 2H), 2.87 (t,J = 6.7 Hz, 2H), 3.82 (s, 3H), 3.97 (t,J = 6.7 Hz, 2H),
5.13 (tt, J
= 4.3 and 11.3 Hz, 1 H), 6.79-6.93 (m, 3H), 7.31 (t,J = 8.1 Hz) 1 H); HRMS
calculated
for C2,H2,N30z[M+]: 353.2103. Found: 353.2094.
Examples 44-57
Reaction of the appropriate hydrazine hydrochloride with the requisite 4
alkanoyl-3-methoxy-2-oxo-1,2,5.6-tetrahydropyridine, analogous to the
procedure of
Example 43, affords the following compounds.
Ex.# R' Rz R3 M.p.C Mass Spectra Mass Spectra
or or
Analysis (calcd.)Analysis (found)
%C, %H, %N %C, %H, %N
44 iso- 4-methoxycyclo- 102-103'[M'] 354 MS [M'] 354
propylphenyl pentyl
p VO 95/01980 ~ PCT/IB94100156
-19-
Ex.# R' R2 R3 M.p.C Mass Spectra Mass Spectra
or or
Analysis (calcd.)Analysis (found)
%C, %H, %N %C, %H, %N
45 iso- 3-methoxycyclo- 99-10071.36, 7.70, 71.10, 7.56,
11.89 11.73
propylphenyl pentyl
46 ethyl 3-methoxycyclobutyl73-74b70.13, 7.12, 70.10, 7.22,
12.91 12.93
phenyl
47 ethyl phenyl methylene60-62'73.19, 7.17, 73.34, 7.08,
14.23 13.95
cyclo-
propyl
48 ethyl 3-methoxymethyleneoil [M' ] 326 MS [M' ] 326
phenyl cyclo-
propyl
49 ethyl 4-methoxy-phenyl 156-157b72.60, 6.09, 72.35, 5.91,
12.10 12.02
phenyl
50 ethyl 3-methoxy-3-sulfo-oil 58.59, 5.95, 58.46, 6.03,
10.79 9.82
phenyl lanyl
51 ethyl 3-methoxy-4-trifluoro-124-12563.61, 4.85, 63.40, 4.51,
10.12 10.09
phenyl methyl-
phenyl
52 ethyl 3-methyl-cyclobutyloil 73.75, 7.49, 73.22, 7.56,
13.S8 13.03
phenyl
53 ethyl 3-trifluoro-3-sulfo-oil MW 427.44 MS m/z 428
methyl- lanyl
phenyl
54 ethyl 3-trifluoro-cyclobutyloil 62.80, 5.55, 63.01, 5.54,
i 1.56 i 1.19
methyl-
phenyl
55 ethyl phenyl 2-indanyl155-156'77.28, 6.49, 77.35, 6.48,
11.76 11.08
56 ethyl 2-methyl-cyclobutyl100-102MW 309.41 MS m/z 310
phenyl
57 ethyl 3-methoxy-2-indanyl60-62'MW 387.48 MS m/z 388,
389,
phenyl 390
Recrystallization solvents: ~Ethyl acetate/pentane. bEthyl ether/pentane.
'Isopropyl ether/pentane. dEthyl/acetate/petroleum ether. ~Ethyl acetate.
'Ethyl
acetate/hexane.
WO 95/01980 ~ ~ ~ PCT/IB94/00156
-20-
Example 58
3-Ethyl-6-(4-fluorophenyl -~1-(4-methoxVphenyl)-4,5.6,7-tetrahydro-1 H-
p~rrazolo(3,4-clp riy dine
To a stirred solution of 3-Ethyl-6-(4-fluorophenyl)-1-(4-methoxyphenyl)-7-oxo-
4,5,6,7-tetrahydro-1 H-pyrazolo[3,4-c]pyridine (0.3 grams, 0.82 mmole) in 50
ml ether
was added lithium aluminum hydride (33 mg, 0.86 mmole). After stirring for 16
hours
water (0.5 ml) was added followed by 3N sodium hydroxide (1 ml). After
stirring for 2
hours the white precipitate was filtered through celite and the filtrate is
concentrated
under reduced pressure. Chromatography on a silica gel column using 1:3 ethyl
acetate/hexane as eluent gives 0.12 grams of the title compound as a pale
yellow
paste. ' H NMR (250 MHz, CDCI3) 1.28 (t,J = 7.6 Hz, 3H), 2.66 (q,J = 7.6 Hz,
2H), 2.71
(t,J = 5.7 Hz, 2H), 3.49 (t,J = 5.7 Hz, 2H), 3.84 (s, 3H), 4.23 (s, 2H), 6.84-
6.99 (m, 6H),
7.36 (d,J = 9.0 Hz, 2H); MS m/z [M+] 352.
Examples 59-63
Reaction of the appropriate 7-oxo-2,5,6,7-tetrahydro-1 H-pyrazolo[3,4-
c]pyridine
with lithium aluminum hydride, analogous to the procedure of Example 58,
affords the
following compounds.
Ex.#R' Rz R3 M.p.C Mol. WeightMass Spectra
(M'] (found)
59 ethyl 4-methoxy-3-cyclopentyloil 409.57 409
cyclopentoxy
phenyl
60 ethyl phenyl 3,4-dichloro-oil 372.30 371.373
phenyl
61 ethyl phenyl cyclopentyloil 295.43 296
62 ethyl 3-methoxy cyclobutyloil 311.43 312
phenyl
63 ethyl 3-methoxy cyclohexyloil 339.48 340
phenyl .
Example 64
1-cyclopentyl-3-ethyl-7-oxo-4,5,6.7-tetrahydro-1 H-pyrazolo(3,4-cl~rridine
A stirred solution of 1-cyclopentyl-3-ethyl-6-(4-methoxyphenyl)-7-oxo-4,5,6,7-
tetrahydro-1 H-pyrazolo(3,4-c]pyridine (2.58 grams, 7.60 mmoles) in
acetonitrile (90 ml)
at 0~C is treated with a solution of ceric ammonium nitrate (12.5 grams, 22.8
mmoles)
~''VO 95I01980 216 6 7 21 PCT/IB94/00156
-21-
in water (110 ml). After stirring for 35 minutes the mixture is diluted with
water (550 ml)
and extracted with ethyl acetate (100 ml x 4). The combined organics are
washed with
50~~ saturated sodium bicarbonate (250 ml) followed by 10% sodium sulfite
until the
aqueous wash becomes pale yellow. The organic layer is then washed further
with
saturated bicarbonate and brine, and treated with decolorizing charcoal. After
stirring
for 30 minutes the mixture is dried over sodium sulfate, filtered through
celite and
concentrated under reduced pressure. The brown residue is recrystallized from
ether
to give .814 grams of a tan solid. M. P. 143-145 ~ C; MS (M/Z) 234; ' H NMR
(250 MHz,
CDCl3) 1.21 (t, J = 7.6 Hz, 3H), 1.62-2.13 (m, 8H), 2.62 (q, J = 7.6 Hz, 2H))
2.73 (t, J
= 6.8 Hz, 2H), 3.51 (dt, J = 2.7 and 6.8 Hz, 2H), 5.47 (s, 1 H), 5.61 (pentet,
J = 7.7 Hz,
1 H).
Example 65
1-c5rclopentyl-3-ethyl-6-cycloprop I~yl-7-oxo-4.5.6,7-tetrahydro-1 H-
pyrazolo(3,4-clpyridine
A solution of 1-cyclopentyl-3-ethyl-7-oxo-4,5,6,7-tetrahydro-1H-pyrazolo[3,4-
c]pyridine (0.21 grams, 0.92 mmoies) in THF (5 ml) is treated with 60% sodium
hydride
in mineral oil (40 mg, 1.01 mmoles). After stirring at reflux over 45 minutes
the reaction
mixture is cooled to 25 ~ C and (bromomethyl) cyclopropane (0.31 grams, 2.29
mmoles)
is added. The mixture is stirred at reflux for 16 hours and then cooled to
25~C before
concentrating under reduced pressure. Chromatography on silica gel eluting
with 1:1
ethyl acetate/hexane gives 0.19 grams of the title compound as a colorless
oil. MS m/z
[M+] 288; ' H NMR (300 MHz, CDC13) 0.26-0.31 (m, 2H), 0.50-0.56 (m, 2H), 0.85-
1.06
(m, 1 H), 1.20 (t, J = 7.6 Hz, 3H), 1.62-2.08 (m, 8H), 2.61 (q, J = 7.6 Hz,
2H), 2.74 (t,
J=6.8Hz,2H),3.39(d,J=6.9Hz,2H),3.63(t,J=6.8Hz,2H),5.67(pentet,J=
7.8 Hz, 1 H).
Preparation 1
4-isobutyryl-3-methoxy-1-phenyl-2-oxo-1,2.5,6-tetrahydropyridine
A stirred solution of freshly distilled diisopropylamine (0.16 ml, 2.21 mmole)
in
anhydrous tetrahydrofuran (4 ml) was cooled to 0 ~ C and treated with 2.5 M n-
butyl
lithium (0.85 ml, 2.11 mmole). After 15 minutes the mixture was cooled to -
78~C and
a pre-cooled solution of 4-propionyl-3-methoxy-1-phenyl-2-oxo-1,2,5,6-
tetrahydropyridine (0.52 grams, 2.0 mmole) in tetrahydrofuran (4 ml) was added
dropwise via cannula. After approximately 20 minutes methyl iodide (0.20 ml,
3.0
WO 95/01980 PCT/IB94/00156
2166721
-22-
mmole) was added to the bright orange-red solution and the mixture was allowed
to
come to room temperature over 2.5 hours. The reaction mixture is poured into
saturated aqueous ammonium chloride and the organic layer is washed with
brine,
dried over sodium sulfate, filtered and concentrated under reduced pressure.
Chromatography on a silica gel column using 1:4 ethyl acetate/hexane as eluent
gives
0.12 grams of the title compound as a yellow oil and 0.1 grams of recovered
starting
material. ' H NMR (250 MHz, CDCI3) 1.15 (d, 6H), 2.72 (t, 2H), 3.47 (heptet, 1
H), 3.82
(t, 2H), 3.97 (s, 3H), 7.21-7.45 (m, 5H); MS m/z [M+] 274.
Preparations 2-3
Reaction of the appropriate 3-methoxy-2-oxo-4-propionyl-1,2,5,6-
tetrahydropyridine with lithium diisopropylamine and methyl iodide, analogous
to the
procedure of preparation 1, affords the following compounds of formula VII.
Prep# Rz m.p. C M.W. Mass Spectra
[M+]
2 4-methoxyphenyloil 303.36 304
3 3-methoxyphenyloil 303.36 304
Preparation 4
3-Methoxy-1-(4-methylphenyl)-2-oxo-4-propionyl-1,2,5,6-tetrahydropyridine
A solution of 3-hydroxy-1-(4-methylphenyl)-2-oxo-4-propionyl-1,2,5,6-
tetrahydropyridine (5.9 grams, 23 mmole) and 3-methyl-1-p-tolyltriazine (5.1
grams, 34
mmole) in 1,2-dichloroethane was heated to reflux for 45 minutes. The mixture
was
allowed to cool to room temperature and was poured into water and acidified
with 6N
hydrochloric acid. The aqueous layer was extracted 3 times with methylene
chloride,
and the combined organics are washed with 1 N hydrochloric acid followed by
water
and brine, dried over magnesium sulfate, filtered and concentrated under
reduced
pressure. The resulting quantitative brown oil was clean by thin layer
chromatography
and ' H NMR and was used without purification. ' H NMR (300 MHz, CDCI3) 1.12
(t,J =
7.2 Hz, 3H), 2.34 (s, 3H), 2.71 (t,J = 6.7 Hz, 2H), 2.93 (q,J = 7.2 Hz, 2H),
3.77 (t,J =
6.8 Hz, 2H), 3.94 (s, 3H), 7.20 (s, 4H); MS [M+] 273.
Preparations 5-14
WO 95/01980 2 ~ b 6 7 21 PCT/IB94/00156
-23-
Reaction of the appropriate 3-hydroxy-1-aryl-2-oxo-4-alkanoyl-1,2,5,6-
tetrahydropyridine with 3-methyl-1-p-tolyltriazine, analogous to the procedure
of
Preparation 4, affords the following compounds of formula VI.
Prep# R' Rz m.p. M.W. Mass
C Spectra
IM+l
5 ethyl phenyl oil 259.31 260
6 methyl 4-methoxyphenyl oil 275.30 275
7 ethyl 4-methoxyphenyl 81-82 289.33 289
8 n-propyl 4-methoxyphenyl oil 303.36 303
9 ethyl 3-methoxyphenyl 59-60 289.33 289,
290
10 ethyl 2-methoxyphenyl oil 289.33 289
11 ethyl 3,4-dimethoxyphenyloil 319.26 319
12 ethyl 3-cyclopentoxy-4-oil 373.45 373
methoxyphenyl
13 ethyl 3-methylphenyl oil 273.33 273
14 ethyl 3-trifluoromethylphenyloil 327.30 327
Preparation 15 -
3-Hvdroxv-1-(3-methvlphenvl)-2-oxo-4-propionvl-1.2.5.6-tetrahvdropvridine
To a stirred suspension of magnesium turnings (1.9 grams, 79 mmole) in 30 ml
of anhydrous ether was added dropwise bromoethane (5.9 ml, 79 mmole). A mild
reflux was initiated after approximately 1 ml was added. After all of the
magnesium was
consumed, the reaction mixture was cooled to 0~C and N-(3-methylphenyl)-2-
pyrrolidone (8.7 grams, 50 mmole) was added at once. After warming to room
temperature and stirring for 2 hours the reaction mixture was poured over ice
and
extracted with ethyl acetate. The combined organics are washed with water and
brine,
dried over sodium sulfate, filtered and concentrated under reduced pressure to
afford
8.8 grams of a white solid.
The above solid is dispersed in a mixture of 40 ml benzene and 86 ml 1 N
sodium hydroxide, and with vigorous mechanical stirring ethyl oxalyl chloride
(7.2 ml,
64 mmole) was added. After stirring at reflux over 1.5 hours the layers are
separated
and the aqueous layer was extracted with ethyl acetate. The combined organics
are
PCT/IB94/00156
WO 95I01980
-24-
washed with water and brine, dried over magnesium sulfate, filtered and
concentrated
under reduced pressure to give an amber oil. GCMS [M+] 305.
The above intermediate was dissolved in 20 ml anhydrous ethanol and treated
with a solution of sodium methoxide in methanol (prepared from the careful
addition of
sodium (1.0 grams) to 10 ml anhydrous methanol). After being stirred at reflux
over 1.5
hours, the mixture was concentrated under reduced pressure and 100 ml of water
was
added. The mixture was acidified to pH 3 with 6N hydrochloric acid and the
dull yellow
precipitate was filtered and washed with water. Recrystallization from 75 ml
isopropyl
ether affords 6.8 grams of pale yellow crystals. M.P. 115-116~; 'H NMR (300
MHz,
CDC13) 1.16 (t,J = 7.2 Hz, 3H), 2.37 (s, 3H), 2.74-2.82 (m, 4H), 3.85 (t,J =
6.8 Hz, 2H),
7.08-7.14 (m) 3H), 7.30 (t,J = 7.7 Hz, 1 H); MS m/z (M+] 259.
Preparations 16-29
Reaction of the appropriate 2-pyrrolidinone with the requisite alkylmagnesium
bromide, followed by treatment with ethyl oxalyl chloride and base, analogous
to that
reported in Preparation 15, affords the following compounds of formula VI.
Prep# R' RZ m.p. M.W. Mass
C Spectra
(M+]
16 methyl phenyl oil 231.25 231
17 ethyl phenyl 140-142 245.28 245
18 ethyl 4-fluorophenyl 133-135 263.27 263
19 methyl 4-methoxyphenyl oil 261.28 262
20 ethyl 4-methoxyphenyl 121-122 275.30 276
21 n-propyl 4-methoxyphenyl 125-126 289.33 289
22 ethyl 3-methoxyphenyl 129-130 275.30 275
23 ethyl 2-methoxyphenyl 119-120 275.30 275
24 ethyl 4-methylphenyl 110-112 259.30 260
25 ethyl 2-methylphenyl oil 259.30 259
26 ethyl 3-trifluoromethylphenyl117-118 313.28 313
27 ethyl 2-trifluoromethylphenyloil 313.28 313
28 ethyl 3,4-dimethoxyphenyl179-180 305.33 306
''VO 95I01980 ~ b ~ ~ PCT/IB94/00156
-25-
Prep#R' Rz m.p. M.W. Mass
C
Spectra
[M+]
29 ethyl 3-cyclopentoxy-4-133-134 359.42 360
methoxyphenyl
Preparation 30
N-(2-MethoxVphenyl?-2-pVrrolidone
A mixture of 2-pyrrolidone (15.0 grams, 176 mmole), 2-iodoanisole (7.6 ml, 59
mmole), copper powder (7.5 grams, 117 mmole) and potassium carbonate (8.1
grams,
59 mmole) are stirred under nitrogen at 150~C. After 18 hours, the reaction
mixture
was filtered through a 6x15 cm pad of silica gel eluting with 1:1 ethyl
acetate/hexane
to give a pale yellow oil. The unreacted reagents are removed by vacuum
distillation
(0.6 mm, 80-100~C) Veaving 9.2 grams of the title compound as a honey-like
oil. 'H
NMR (300 MHz, CDCI3) 2.20 (pentet, 2H), 2.55 (t, 2H), 3.75 (t, 2H), 3.82 (s,
3H), 6.93-
7.02 (m, 2H), 7.25-7.30 (m, 2H); MS m/z [M+] 191.
Preparations 31-39
Reactions of the appropriate iodo- or bromobenzene with 2-pyrrolidinone,
analogous to that reported in Preparation 30, affords the following compounds
of
formula V.
Prep# R M.W. Mass Spectra
[M+]
31 4-methoxyphenyl 191.22 191
32 3-methoxyphenyl 191.22 191
33 3-methylphenyl 175.23 175
34 4-methylphenyl 175.23 175
35 2-methylphenyl 175.23 175
36 3-trifluoromethylphenyl229.20 229
37 2-trifluoromethylphenyl229.20 229
38 3,4-dimethoxyphenyl221.26 221
39 3-cyclopentoxy-4-275.35 275
methoxyphenyl