Note: Descriptions are shown in the official language in which they were submitted.
~ W095/0~ 2 16 8 0 9 ~ PCT~4/02354
BENZOXAZINONES AND BENZ~THIAZINON~S ENDOWED WITH
TEERAPEUTIC A~lvll~
The present invention relates to the therapeutic
use of 2,3-dihydro-4H-1,3-benzoxazin- and -benzo-
thiazin-4-ones.
2,3-Dihydro-4H-1,3-benzoxazin-4-ones unsubstituted
on the nitrogen atom were described by B.W. Horrom et
al., J. Org. Chem., 72, 721 (1950) as endowed with
analgesic activity. Other 2,3-dihydro-4H-1,3-
benzoxazin-4-ones were disclosed by R.B. Gammil, J.
Org. Chem., 46, 3340 (1981).
10N-substituted derivatives of said heterocycle were
described by J. Finkelstein et al., J. Med. Chem., 11,
1038 (1968) as antinflammatory agents. Also, 6-amino
derivatives showing antinflammatory activity were
disclosed by F. Fontanini et al., Riv. Farmacol. Ter.,
154(1), 119 (1973) (Chem. Abs. 73745n, Vol. 79, page. 40,
1073).
The publication of the patent application EP-A-0
490 183 (in the nam~ of the applicant) shows 2,3-
dihydro-4H-1,3-benzoxazin- and -benzothiazin-4-ones
having a -ONO2 group: these compounds are endowed with
a good cardiovascular activity, particularly against
angina.
The publication of the patent application EP-A-0
359 627 describes benzoxazin-4-ones useful as agents
inducing bradycardia, anti-ischemiscs and calcium-
antagonists.
It has been now surprisingly found that 2,3-
dihydro-4H-1,3-benzoxazin- and -benzothiazin-4-ones,
WO9~/0~ ~ PCT~4/02354~
some of them being already known as intermediates for
the synthesis of compounds claimed in patent
applications EP-A-0 490 183 and EP-A-0 359 627, are
therapeutically active, particularly in the
cardiovascular field.
Therefore, the present invention relates to
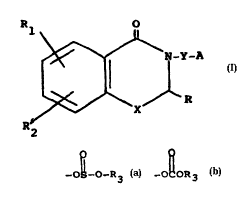
compounds of the general formula (I)
0
~ N-Y-A
¦ (I)
~--X
R2
wherein X is an oxygen or a sulphur atom;
Y represents methylene, ethylene, (C3 6)alkylene
optionally branched, or cyclopentylene, cyclohexylene
or cycloheptylene optionally substituted with
(Cl 4)alkyl;
A is hydroxy; (Cl 6)alkoxy, formyloxy; (C2 6)acyloxy;
mercapto; (Cl 6)alkyl-mercapto; mesyloxy; (C2 6)alkyl-
sulfonyloxy; tosyloxy; phenyl-sulfonyloxy optionally
substituted
~ Q
with (C2 6)alkyl; -O~-O-R3 or -O~OR3 wherein R3 is a
(Cl 6)alkyl group, methylene, ethylene or (C3 6)alky-
lene ~substituted with a 2,3-dihydro-4H-1,3-benzo-
xazin-4-one-N-yl residue;
R represents hydrogen, (Cl 6)alkyl or phenyl;
Rl and R2 are independently hydrogen, halogen,
(Cl 6)alkoxy, trifluoromethyl, and (Cl 6)alkyl;
and the pharmaceutically acceptable acid or base salts
~W095/0~ 21 6 8 0 9 ~ PCT~4/02354
. i ,... .
thereof, useful as therapeutically active substances.
; Specifically, the invention relates to the use of
the compounds of formula (I) as agents useful in the
cardiovascular field.
As intended hereinbelow, the alkyl groups
essentially identify methyl, ethyl, propyl, i-propyl,
butyl, 2-methyl-propyl, n-pentyl, 3-methylbutyl, i-
pentyl, n-hexyl and the like, while the alkoxy groups
are preferably selected from the group consisting of
methoxy, ethoxy, propoxy, i-propoxy, butoxy, 2-
methylbutoxy and t-butoxy. As linear or branched (C3
6)alkylene group it is intended, e.g., 2-
methylethylene, 1,3-propylene, 1,4-butylene, 2-
ethylethylene, 3-methylpropylene, 1,5-pentylene, 2-
ethylpropylene, 2-methylbutylene, 1,6-hexylene, 1-
ethyl-l-methylpropylene, 3-methylpentylene and the
like. As (C2 6)acyloxy it is intended, e.g., acetyloxy,
propionyloxy, butyryloxy, hexanoyloxy, oxalyloxy,
malonyloxy, succinyloxy.
The compounds of the present invention are
prepared according to procedures known to the skilled
in the art. For example, the compounds of formula (I)
wherein X, Y, R, Rl and R2 are as defined above, A is
hydroxy, (Cl 6)alkoxy, formyloxy or (C2 6)acyloxy, may
be obtained according to what taught by the publication
of the Patent application EP-A-0 490 183, by reacting a
salicylamide or thiosalicylamide of formula (II)
W095/0~ ~9~ PCT~ ~4/02354
~/J~\'NH--Y--XR4
~ XH (II)
wherein Rl, R2, X and Y are as defined above, and R4 is
hydrogen, (Cl_6)alkyl or (Cl_6)acyl, with an aldehyde
of formula (III)
R-CH0 (III)
wherein R is as above, or a derivative or a precursor
thereof. The condensation generally occurs in an acidic
environment, for example in a system constituted by a
strong mineral acid and acetic acid, thereby obtaining
compounds of formula (I) wherein R4 is acetyl, or by
molecular sieves in the presence of sulfonic acids such
as p-toluenesulfonic acid, methanesulfonic acid, ~ and
~-naphthalenesulfonic acids, phosphoric acids, esters
and analogues thereof. The condensation is carried out
in the presence of an organic solvent, preferably an
inert organic solvent such as ethyl acetate,
acetonitrile, benzene, nitrobenzene or chlorobenzene,
halogenated aliphatic hydrocarbons such as methylene
chloride, chloroform, 1,2-dichloroethane or 1,1,2-
trichloroethylene, cyclohexane, tetrahydrofuran,tetrahydropyran, dimethylformamide, dimethylacetamide.
The reaction temperature may vary within quite wide
limits withou~ prejudice for the course of the
reaction. The preferred range of temperature is
comprised between about -10C and the reflux
temperature of the reaction mixture, and the reaction
~W095/~ 21 68 ~99 PCT~ ~4/02354
is completed in a period of time ranging between about
2 and about 30 hours.
The molar quantities of the reagents of formula
(II) and (III) are not critical for the good course of
the cyclization, and such reagents may be used in the
widest stoichiometric ratios. When compounds of formula
(I) wherein R is hydrogen or methyl are desired,
precursors of the compound of formula (III) such as
paraformaldehyde and the paraldehyde are preferably
employed.
Compounds of formula (I) wherein X, Y, R, Rl and
R2 are as above, and A is hydroxy, may be used as
reagents for preparing other compounds of formula (I).
~ or example, when compounds of (I) wherein X, Y,
lS R, Rl and R2 are as above, and A represents formyloxy,
(C2 6)acyloxy, mesyloxy, (C2 6)alkylsulfonyloxy,
tosyloxy, phenyl-sulfonyloxy optionally substituted
with (C2 6)alkyl,
C
-OS-O-R3 or -O OR3 wherein R3 is a (Cl 6)alkyl group,
methylene, ethylene or (C3 6)alkylene R~substituted by
a 2,3-dihydro-4H-1,3-benzoxazin-4-one-N-yl residue are
desired, a compound of formula (I) wherein A is hydroxy
is treated with a suitable carboxylic or sulfonic acid
activated in form of anhydride, halide or imidazolide,
used in excess, preferably in the presence of an
organic base such as pyridine. Suitable solvents for
such synthesis are, e.g., chloroform or methylene
chloride, while the reaction temperature ranges between
about -10C and the room temperature, and the reaction
time is of about 1-20 hours. In the case of
wo gs/o~ 9 PCT~4/~354~
-OS-O-R3 or -O~OR3, at the end of the reaction a sui- -
table amount of the R30H desired alcohol is added.
Also, the compounds of formula (I) wherein A is
hydroxy may yield compounds of formula (I) wherein A is
mercapto or (Cl 6)alkyl-mercapto, by an intermediate of
formula (IV)
0
~'-Y-Halo
,~ I
(IV)
~/\ ~ /~
R2
wherein X, Y, R, Rl and R2 are as above, and Halo is an
halogen atom, preferably chlorine. Such intermediate is
obtained by treating the OH group with halogenating
agents such as, e.g., thionyl chloride, sulfuryl
chloride, phosphorous trichloride, phosphorous
pentachloride, phosphorous oxytrichloride, phosphorous
tribromide, sulfuryl bromide and the like. Said
halogenation reaction occurs in an organic solvent,
preferably in an inert organic solvent selected from
the ones employed for the synthesis of the compound of
formula (I) described above, at a temperature ranging
between the room temperature and the reflux temperature
of the reaction mixture. The compound of formula (IV)
is converted into a compound of formula (I) wherein A
is mercapto, by reaction with thiourea in alcoholic
solution, at the reflux temperature of the reaction
mixture, for about 5-12 hours, and subsequent
hydrolysis with strong organic bases such as an alkali
W095/0~ ; PCT~4/02354
2~ ~8~93
metal hydroxide, at the reflux temperature for about 2-
~ 10 hours. Subsequently, the compound of formula (I)
wherein A is mercapto may be converted into a compound
of formula (I) wherein A is (Cl 6)alkyl-mercapto, by
treatment with the suitable acyl halide in the presence
of an organic base such as diazabicycloundecene,
according to what taught by Patai, 2nd part, pages 721-
735.
The compounds of formula (I) wherein A is hydroxy
may also provide compounds of formula (I) wherein A is
(Cl 6)alkoxy, according to the so-called Williamson's
reaction carried out with the suitable acyl halide.
In general, compounds of formula (I) may be
converted into other compounds of formula (I) by means
of suitable procedures for modifying the substituents
Rl and R2, wholly familiar to the skilled in the art.
In any case, the conversion of an Rl or R2 group into
another Rl or R2 group having a ~eAning comprised by
formula (I) may occur following conventional procedures
familiar to the skilled in the art. These procedures
are in the scope of the invention together with the
obvious modifications of the just disclosed methods for
preparing the compounds of the invention.
HereinbeloW preparation examples for some of the
compounds of the invention are provided. Unless
otherwise indicated the H-NMR spectra were carried out
in dimethylsulfoxide (DMS0).
~XAMPLE 1
2,3-Dihydro-3-(2'-hydroxyethyl)-4H-1,3-benzoxazin-4-one
A) N-(2'-hydroxyethyl)salicylamide was prepared as
described in Aust. J. Chem., 25, 1797 (1972).
W095/0~ ~99 PCT~ ~4/02354
B) 5.5 g (0.03 mole) of the compound under A) and
0.57 g (0.003 mole) of p-toluenesulfonic acid were
dissolved in 100 ml of benzene, and 2.7 g of
paraformaldehyde were added to the resulting
mixture. The reaction mixture was heated to 90C
for 3 hours, distilling off the formed water and,
after cooling to room temperature, washed with
water. The organic phase was recovered and, after
evaporation of the solvent, there were obtained
4.7 g of a residue which was dissolved in 150 ml
of tetrahydrofuran and 14 ml of lN HCl. The
mixture was refluxed for 8 hours, cooled to room
temperature and extracted with ethyl acetate. The
organic phase was dried and evaporated under
vacuum, thus yielding 3.76 g of the title product.
m.p. 59-61C (methylene chloride~acetone 1:9 vlv).
H-NMR (200 MHz): 7.82 (lH, dd); 7.54 (lH, dt); 7.18
(lH, t); 7.07 (lH, d); 5.34 (2H, s); 3.57 (4H, s).
EXAMPLE 2
3-(2'-Acetoxyethyl)-2,3-dihydro-4H-~1,3-benzoxazin-4-one
A solution of 10 g (0.05 mole) of the compound of
Example 1 in 200 ml of chloroform and 4.18 ml (0.05
mole) of pyridine was added with 19.6 ml (0.207 mole)
of acetic anhydride. The solution was left at room
temperature for 12 hours, then washed with water, and
the organic phase was dried over sodium sulfate and
evaporated to dryness. The crude obtained was purified
by flash chromatography (eluent: hexane/ethyl acetate
6:4), thus yielding 2.4 g of the title product.
m.p. 67C (ethyl acetate/hexane).
lH-NMR (200 MHz): 7.82 (lH, dd); 7.55 (lH, dt); 7.15
_ W095lO~ i ; PCT~4/02354
21~68o9~
(2H, m); 5.35 (2H, s); 4.17 (2H, t); 3.79 (2H, t); 2.00
- (3H, s).
; EXAMPLE 3
3-(2'-Acetoxyethyl)-2,3-dihydro-6-methyl-4H-1,3-
benzoxazin-4-one
A) 8.5 g of methyl 5-methyl-salicylate (J. Chem.
Soc., 661, 1961) in 3.7 ml of 2-aminoethanol were
heated to 170C for 3 hours. After cooling to room
temperature the reaction mixture was taken up in
ethyl acetate, washed with 5~ hydrochloric acid
and dried over anhydrous sodium sulfate. There
were obtained 9 g of ~-(2'-hydro:~yethyl)-5-methyl-
salicylamide.
m.p. 73-75C (hexan~)
B) A solution ~f 8 g ~.041 mole) of the compound
under ~ 0~ ml of chloroform and 11 ml of
gla~i~l acetic acid was added with 4 g of
~ar2formal~ehyde. The mixture was cooled to 0C
3n~ ~ded with 10 g of gaseous hydrochloric acid
in 30 minutes, then the resulting solution was
stirr~d at room temperature for 24 hours. The oily
layer was eliminated and the chloroform phase was
washed with water and dried over sodium sulfate.
After evaporating the solvent, the resulting crude
wzs purified on a silica gel column by eluting
with methylene chloride/acetone (85:15 v/v) thus
yielding 6 g of the title product.
m.p. 53-55C (hexane).
l~-NMR (80 MHz): 7.61 (lH, d); 7.30 (lH, dd); 6.92 (lH,
d); 5.30 (2H, s); 4.15 (2H, t); 3.71 (2H, t); 2.02 (3H,
s ) .
W095/0~ ~ PCT~ ~4/0235~
~63~
EgAMPLE 4
2,3-Dihydro-3-(2'-hydroxyethyl)-6-methyl-4H-1,3-
benzoxazin-4-one
A solution of 6 g (0.024 mole) of the compound of
Example 3, in 120 ml of methanol was added with 1.4 g
(0.013 mole) of sodium carbonate, and the resulting
mixture was left at room temperature for 12 hours.
After evaporating the solvent, the resulting crude was
taken up in methylene chloride, and the resulting
organic phase was washed with water and dried over
sodium sulfate. After evaporating methylene chloride,
there were obtained 4.2 g of the title product.
m.p. 59-61C (diethyl ether)
lH-NMR (80 MHz): 7.60 (lH, d); 7.30 (lH, dd); 6.90 (lH,
d); 5.30 (2H, s); 4.83 (lH, t); 3.56 (4H, m); 2.30 (3H,
5).
EXAMPLE S
3-(2'-Acetoxyethyl)-7-chloro-2,3-dihydro-4H-1,3-
benzoxazin-4-one
A) 4-Chloro-N-(2'-hydroxyethyl)-salicylamide was
prepared following the procedure of Example 3,A)
starting from 20 g of methyl 4-chlorosalicylate
(Chem. Abs. 81, 3624q~ and 8 ml of 2-aminoethanol.
Yield: 11 g m.p. 95-97C (chloroform).
B) From 11 g (0.051 mole) of the compound under A)
and 4.5 g of paraformaldehyde, following the
procedure of Example 3,~), there were obtained 9 g
of the title product.
m.p. 92-94C (hexane).
lH-NMR (80 MHz): 7.82 (lH, d); 7.30T7.o8 (2H, m); 5.35
(2H, s); 4.15 (2H, t); 3.70 (2H, t); 2.00 (3H, s).
095/0~ ~ 09 PCT~4/02~54
EXAMPLE 6
7-Chloro-2,3-dihydro-3-(2'-hydroxyethyl)-4H-1,3-
benzoxazin-4-one
From 8 g (0.03 mole) of the compound of Example 5
and following the procedure of Example 4, there were
obtained 6.1 g of the title product.
m.p. 104-106C (hexane).
H-NMR (80 MHz): 7.83 (lH, d); 7.30~7.10 (2H, m); 5.36
(2H, s); 4.86 (lH, t); 3.60 (4H, m).
~XAMPL~ 7
3-(2'-Acetoxyethyl)-2,3-dihydro-7-methyl-4H-1,3-
benzoxazin-4-one
A~ N-(2'-hydroxyethyl)-4-methylsalicylamide was
prepared according to the procedure of Example
3,A) starting from 20 g of methyl 4-
methylsalicylate (Chem. Abs. 64, 6568d) and 9 ml
of 2-aminoethanol. Yield: 16.7 g m.p. 78-80C
(hexane).
B) From 16 g (0.081 mole) of the compound under A)
and 4.5 g of paraformaldehyde, and following the
procedure of Example 3,B), there were obtained 13
g of the title product as an oil.
lH-NMR (200 MHz): 7.59 (lH, d); 6.98 (lH, d); 6.91 (lH,
s); 5.19 (2H, s); 4.18 (2H, t); 3.68 (2H, t); 2.01 (3H,
s).
EXAMPLE 8
2,3-Dihydro-3-(2'-hydroxyethyl)-7-methyl-4H-1,3-
benzoxazin-4-one
From 12 g of the compound of Example 7 and
following the procedure of Example 4, there were
obtained 9 g of the title product as an oil.
W095/0~ 2~ 9 PCT~4/02354
12
H-NMR (80 MHz): 7.73 (lH, d); 6.96s6.63 (2H, m); 5.20
(2H, s); 3.96T3.56 (4H, m); 2.36 (3H, s).
~XAMPLE 9
3-(2'-Acetoxyethyl)-2,3-dihydro-2-methyl-4H-1,3-
benzoxazin-4-one
Starting from 18.4 g (0.101 mole) of the compound
of Example l,A) and 8.13 ml (0.061 mole) of
paraldehyde, and following the procedure of Example
3,B), there were obtained 6.7 g of the title product as
an oil.
H-NMR (200 MHz): 7.74 (lH, dd); 7.52 (lH, dt); 7.17
(lH, t); 7.02 (lH, d); 5.70 (lH, q); 4.22 (2H, t);
4.63T3.83 (lH, m); 3.S6T3.23 (lH, m); 2.05 (3H, s);
1.56 (3H, d).
EXAMPL~ 10
2,3-Dihydro-3-(2'-hydroxyethyl)-2-methyl-4H-1,3-
benzoxazin-4-one
From 6 g (0.029 mole) of the compound of Example 9
and operating as described in Example 4, there were
obtained 3.8 g of the title product as an oil.
H-NMR (200 MHz): 7.74 (lH, dd); 7.52 (lH, dt); 7.17
(lH, t); 7.01 (lH, d); 5.50 (lH, q); 3.90T3.57 (4H, m);
1.52 (3H, d).
EXAMPLE 11
3-(2'-Acetoxyethyl)-2,3-dihydro-2,7-dimethyl-4H-1,3-
benzoxazin-4-one
From 6 g (0.03 mole) of the compound of Example
7,A) and 2.4 ml (0.018 mole) of paraldehyde and
operating as described in Example 3,B), there were
obtained 2.5 g of the title product as an oil.
lH-NMR (200 MHz): 7.63 (d, lH); 6.92 (d, lH); 6.88 (s,
W095/0~ PCT~ ~4/02354
lH); 5.66 (q, lH); 4.28 (t, 2H); 4.08~3.78 (m, lH);
3.52T3.22 (m, lH); 2.35 (s, 3H); 2.02 (s, 3H); 1.52 (s,
3H)
EXAMPLE 12
2,3-Dihydro-2,7-dimethyl-3-(2'-hydroxyethyl)-4H-1,3-
benzoxazin-4-one
From 2.3 g (0.009 mole) of the compound of Example
11, following the procedure of Example 4, there were
obtained 2.1 g of the title product as an oil.
lH-NMR (80 MHz): 7.63 (lH,d); 7.OOT6.76 (2H,m); 5.66
(lH,q); 4.86 (lH, t); 3.70T3.43 (4H, m); 2.36 (3H, s);
1.50 (3H, d).
EXAHPLE 13
3-(2~-Acetoxyethyl)-6-chloro-2,3-dihydro-4H-1,3-
benzoxazin-4-one
A) 5-Chloro-N-(2'-hydroxyethyl)-salicylamide was
prepared following the procedure of Example 3,A)
starting from 19 g of methyl 5-chlorosalicylate
(Arch. Pharm. 296(10), 714, 1963) and 7.5 ml of 2-
aminoethanol. Yield: 13.8 g
m.p. 100-102C (hexane).
B) From 13 g (0.06 mole) of the compound under A) and
4.5 g of paraformaldehyde, following the method of
Example 3,B), there were obtained 11 g of the
title product.
m.p. 93-95C (hexane).
1H_NMR (80 MHz): 7.73 (lH, d); 7.53 (lH, dd); 7.10 (lH,
d); 5.35 (2H, s); 4.16 (2H, t); 3.71 (2H, t); 2.02 (3H,
s ) .
EXAMPLE 14
6-Chloro-2,3-dihydro-3-(2'-hydroxyethyl)-4H-1,3-
W095/~ 6~ PCT~ ~4/0235
14
benzoxazin-4-one
From 10 g (0.037 mole) of the compound of Example
13 and operating as described in Example 4, there were
obtained 7 g of the title product.
m.p. 88-90C (diethyl ether).
lH-NMR (80 MHz): 7.73 (lH, d); 7.53 (lH, dd); 7.10 (lH,
d); 5.36 (2H, s); 3.56 (4H, m).
EXAMPLE 15
N-(5'-Acetoxypentyl)-2,3-dihydro-4H-1,3-benzoxazin-4-
one
A) N-(5'-hydroxypentyl)salicylamide was prepared
following the procedure of Example 3,A) starting
from 17.6 g of methyl salicylate and 8.5 ml of 5-
aminopentanol. Yield: 11 g. The compound was an
oil used as such in the subsequent step.
B) From 11.4 g (0.051 mole) of the compound under A)
and 4.5 g of paraformaldehyde and following the
procedure of Example 3,B), there were obtained 9 g
of the title product as an o}l.
lH-N~R (200 MHz): 7.7g (lH, dd); 7.51 (lH, dt); 7.14
(lH, t); 7.04 (lH, t); 5.29 (2H, s); 4.03 (2H, t); 3.44
(2H, t); 2.04 (3H, s!; 1.6OT1.19 (6H, m).
EXAMPLE 16
2,3-Dihydro-3-(5'-hydroxypentyl)-4H-1,3-benzoxazin-4-
o
From 8.5 g (0.031 mole) of the compound of Example
15 and operating as described in Example 4, there were
obtained 5.3 g of the title product as an oil.
lH-NMR (200 MHz): 7.79 (lH, dd); 7.51 (lH, dt);
7.14T7.04 (2H, m); 5.29 (2H, s); 3.44 (4H, m);
1.6OT1.19 (6H, m).
095/0~ ~1 ~ PCT~ ~4/02354
EXAMPLE 17
2,3-Dihydro-3-(2'-hydroxyethyl)-7-methoxy-4H-1,3-
benzoxazin-4-one
A) N-(2'-hydroxyethyl)-4-methoxy-salicylamide was
prepared following the procedure described in
Example 3,A), starting from 16.9 g of methyl 4-
methoxysalicylate (J. Org. Chem., 23, 756, 1958)
and 7 ml of 2-aminoethanol.
Yield: 9.5 g.
10 m.p. 92-94C (hexane).
B) 9 g (0.042 mole) of the compound under A) and 0.81
g (0.004 mole) of p-toluenesulfonic acid were
dissolved in 130 ml of benzene, and the resulting
solution was added with a 4 A molecular sieve and
1.55 g of paraformaldehyde. The mixture was
refluxed for 2 hours and, after cooling to room
temperature, added with 300 ml of ethyl acetate.
The molecular sieve was filtered off, the solution
was washed with water and the organic phase was
recovered and dried over sodium sulfate. After
evaporation of the solvent there were obtained
10.3 g of a residue which was purified by a silica
gel column by eluting with ethyl acetate/hexane _
8:2 (v/v). Yield: 2.1 g of the title product as an
oil.
H-NMR (200 MHz): 7.83 (lH, d); 6.63 (lH, dd); 6.42
(lH, d); 5.36 (2H, s); 4.88 (lH, t); 3.62~3.47 (4H, m);
3.81 (3H, s).
EXAMPLE 18
2,3-Dihydro-N-(2-hydroxyethyl)-4H-1,3-benzothiazin_4_
o
wo gs/o~ ~9~ PCT~ ~4/0235 ~
A) A solution of 5 g (0.03 mole) of methyl
thiosalicylate (Synthesis, 59, 1974) in 4 ml (0.06
mole) of ethanolamine was heated to 140C while
distilling off the methanol. After 2 hours, the
solution was poured into water and extracted with
ethyl acetate. The organic phase was dried and
evaporated under vacuum, thus yielding 4.2 g (0.01
mole) of bis-[2-(2-hydroxyethyl)carboxamidophe-
nyl]disulfide which were dissolved as such in 32
ml of ethanol. The solution was heated to 65C and
dropwise added with 0.4 g (0.01 mole) of sodium
borohydride in 21 ml of ethanol. The solution
temperature was kept at 65C for 1 hour, then
brought to room temperature. The residue obtained
by evaporation of the solvent was chromatographed
on a silica gel column by eluting with ethyl
acetate. There were obtained 1.4 g of N-(2-
hydroxyethyl)-2-mercaptobenzamide as an oil which
was used as such in the subsequent step.
B) A solution of 3.5 g (0.017 mole) of the compound
under A) in 50 ml of dioxane was added with 1.59 g
(0.~53 mole) of paraformaldehyde. The solution was
cooled to 0C and saturated with gaseous
hydrochloric acid, then brought to room
temperature and stirred for 3 days. After dilution
with water, the ~.ixture was extracted with ethyl
acetate. The organic phase was dried over sodium
sulfate and evaporated under vacuum, and the
resulting residue was chromatographed through a
silica gel column [eluent: ethyl acetate/hexane
8:2 (v/v)]. There were obtained 1.5 g of the title
~ 2~1 r6 ~ o 9 ~ PCT~4/02354
product as an oil.
lH-NMR (80 MHz): 7.93.-7.80 (lH, m); 7.50.7.13 (3H, m);
; 4.83 (2H, s); 3.63 (4H, m).
EXAMPLE 19
2,3-Dihydro-trans-3-(2'-hydroxycyclohexyl)-4H-1,3-
benzoxaz~in-4-one
A) Trans-N-(2'-hydroxycyclohexyl)salicylamide was
prepared following the procedure of Example 3,A)
from 16 g of methyl salicylate and 12 g of trans-
2-amino-cyclohexanol. Yield: 9.1 g
m.p.: 91-96C (hexane).
B) The title product was prepared according to the
procedure of Example 17,B), starting from 8.1 g
(0.0319 mole) of the compound under A). Yield: l.S
g.
m.p.: 87-89C (hexane).
lH-NMR (80 MHz): 7.90 (lH, dd); 7.33 (lH, dt);
7.03.6.70 (2H, m); 3.56 (2H, m); 2.10.1.15 (8H, m).
EXAMPLE 20
2,3-Dihydro-3-(1'-methyl-2'-hydroxyethyl)-4H-1,3-
benzoxazin-4-one
A) N-(l'-methyl-2'-hydroxyethyl)salicylamide was
prepared following the procedure of Example 3,A)
starting from 18 g (0.118 mole) of salicylic acid
methyl ester and 18.8 ml of 2-amino-1-propanol.
- Yield: 19 g. The compound was an oil which was
used as such in the subse~uent step.
B) Following the procedure of Example l,B) and
starting from 19 g (0.973 mole) of the compound
under A), there were obtained 7.76 g of the title
product as an oil.
W095/0~ PCT~ ~4/02354~h
'i~,6~
18
H-NMR (200 MHz): 7.82 (lH, dd); 7.52 (lH, dt); 7.17
tlH, t); 7.05 (lH, d); 5.30 (2H, m); 4.50 (lH, m); 2.50
(2H, m); 1.20 (3H, d).
EXAMPL~ 21
2,3-Dihydro-N-(2'-hydroxyethyl)-6-methoxy-4H-1,3-
benzoxazin-4-one
A) N-(2'-hydroxyethyl)-5-methoxysalicylamide was
prepared following the procedure of Example 3,A)
starting from 25 g (0.137 mole) of 5-methoxy-
salicylic acid methyl ester and 9.94 ml of 2-
aminoethanol. Yield: 25 g. The compound was used
as such in the subsequent step.
B) The title product was prepared as described in
Example l,B) starting from 20 g (0.094 mole) of
the compound under A). Yield: 17.2 g as an oil.
H-NMR (80 MHz): 7.30~6.90 (3H, m); 5.30 (2H, s); 3.80
(3H, s); 3.60 (4H, s).
~XAMPLE 22
2,3-Dihydro-N-(2'-hydroxyethyl)-7-trifluoromethyl-4H-
1,3-benzoxazin-4-one
A) N-(2'-hydroxyethyl)-4-trifluoromethyl-salicylamide
was prepared following the procedure of Example
3,A) starting from 26 g (0.118 mole) of 4-
trifluoromethyl-salicylic acid methyl ester [J.
Am. Chem. Soc., 76, 1051-4 (1954)] and 100 ml of
2-aminoethanol. Yield: 24 g. The compound obtained
was used as such in the subsequent step.
B) The title product was prepared as described in
Example l,B) starting from 20 g (0.08 mole) of the
compound under A). Yield: 13 g.
m.p.: 63-65 C (hexane).
~ 095/0~ PCT~4/02354
,~, 2168~99
lH-NMR (80 MHz): 7.92 (lH, d); 7.40 (lH, d); 7.36 (lH,
s); 5.36 (2H, s); 3.53 (4H, s).
EXAMPLE 23
N-(2'-Acetoxyethyl)-2,3-dihydro-7-trifluoromethyl-4H-
1,3-benzoxazin-4-one
Following the procedure of Example 2 and starting
from 5.1 g (0.019 mole) of the compound of Example 22,
there were obtained 4.7 g of the title product.
lH-NMR (200 MHz): 8.00 (lH, d); 7.50 (lH, d); 7.48 (lH,
s); 5.43 (2H, s); 4.17 (2H, t); 3.72 (2H, t); 2.00 (3H,
s) .
EXAMPLE 24
2,3-Dihydro-3-(2'-hydroxyethyl)-8-methyl-4H-1,3-
benzoxazin-4-one
15 A) N-(2'-hydroxyethyl)-3-methyl-salicylamide was
prepared following the procedure of Example 3,A)
starting from 24.2 g (0.145 mole) of 3-methyl-
salicylic acid methyl ester and 100 ml of 2-
aminoethanol. Yield: 28.9 g. The compound was used
as such in the subsequent step.
B) The title product was prepared as described in
Example l,B) starting from 28.3 g (0.145 mole) of
the compound under A). Yield: 15 g as an oil.
lH-NMR (80 MHz): 7.59 (lH, d); 7.34 (lH, d); 7.00 (lH,
t); 5.33 (2H, s); 3.58 (4H, m); 2.23 (3H, s).
EXAMPL~ 25
2,3-Dihydro-3-(2~-hydroxyethyl)-5-methyl-4H-1,3-
benzoxazin-4-one
A) N-(2'-hydroxyethyl)-6-methyl-salicylamide was
prepared following the procedure of Example 3,A)
starting from 18.8 g (0.113 mole) of 6-methyl-
W095/0~ - PCT~ ~4/0235 ~
?,'~6~9~
salicylic acid methyl ester and 78 ml of 2-
aminoethanol. Yield: 19.3 g.
m.p.: 134-135C (hexane).
B) The titl~ product was prepared as described in
Example 1,B) starting from 13.6 g (0.069 mole) of
the compound under A). Yield: 6.6 g as an oil.
lH-NMR (80 MHz): 7.30 (lH, t); 6.86 (2H, m); 5.23 (2H,
s); 3.56 (4H, m); 2.63 (3H, s).
~XANPLE 26
2,3-Dihydro-N-(3'-hydroxypropyl)-4H-1,3-benzoxazin-4-
o
A) N-(3'-hydroxypropyl)salicylamide was prepared
following the procedure of Example 3,A) starting
from 20 g (0.131 mole) of salicylic acid methyl
ester and 13.2 ml of 3-aminopropanol. Yield: 24 g
as an oil which was used as such in the subsequent
step.
B) ~he title product was prepared as described in
Example l,B) starting from 17 g (0.087 mole) of
the compound under A). There were obtained 7.75 g
of the title product as an oil.
H-NMR (80 MHz): 7.76 (lH, dd); 7.50 (lH, dt); 7.10
(2H, m); 5.33 (2H, s); 3.52 (4H, m); 1.73 (2H, m).
EXAMPLE 27
2,3-Dihydro-N-(2'-hydroxy-1'-methyl-1'-propyl)-4H-1,3-
benzoxazin-4-one
A) 31.24 g (0.146 mole) of salicylic acid methyl
ester and 13 g (0.146 mole) of 3-amino-2-butanol
was heated to 80C for 4 hours then cooled to room
temperature. The crude obtained was washed with
hexane and purified by silica gel column
~ 095/0~PCT~4/OZ354
~ 21 6~09~
chromatography [eluent: chloroform /acetone 10/1
- v/v]. There were obtained 12 g of N-(l'-methyl-2'-
hydroxy-l'-propyl)salicylamide as an oil which was
used as such in the subsequent step.
B~ The compound was prepared as described in Example
l,B) starting from 11 g (0.052 mole) of the
compound under A). There were obtained 8.13 g of
the title product as an oil.
1H_NMR (80 MHz): 7.76 (lH, dd); 7.46 (lH, dt); 7.10
10(2H, m~; 5.33 (2H, s); 4.30 (lH, m); 3.76 (lH, m); 1.30
(6H, dd).
EXAMPLE 28
2,3-Dihydro-3-(2'-hydroxy-1'-propyl)-4H-1,3-benzoxazin-
4-one
A) N-(2'-hydroxy-1'-propyl)salicylamide was prepared
following the procedure of Example 3,A) starting
from 18 g (0.118 mole) of salicylic acid methyl
ester and 18.3 ml of 1-amino-2-propanol. Yield: 24
g as an oil which was used as such in the
subsequent step.
B) The title product was prepared as described in
Example l,B) starting from 20 g (0.102 mole) of
the compound under A). There were obtained 18 g as
an oil.
lH-NMR (200 MHz): 7.83 (lH, dd); 7.50 (lH, dt);
7.33T7.00 (2H, m); 5.40 (2H, s); 4~OOT3.1O t3H, m);
1.10 (3H, d).
EXAMPLE 29
2,3-Dihydro-3-(2'-hydroxyethyl)-7-isopropoxy-4H-1,3-
benzoxazin-4-one
A) N-(2'-hydroxyethyl)-4-isopropoxy-salicylamide was
W095/0~ PCT~ ~4/023~
6~Q9~
prepared following the procedure of Example 3,A)
starting from 25.3 g (0.166 mole) of 4-isopropoxy-
salicylic acid methyl ester [obtained by treating
4-isopropoxy-salicylic acid prepared according to
Synthesis 758-760 (1984), with methanol and
sulfuric acid~ and 100 ml of 2-aminoethanol.
Yield: 22 g. The compound was used as such in the
subsequent step.
B) The title product was prepared as described in
Example l,B) starting from 5 g (0.028 mole) of the
compound under A). There were obtained 3.1 g as an
oil.
H-NMR (200 MHz): 7.70 (lH, d); 6.69 (lH, dd); 6.58
(lH, d); 5.31 (2H, s); 4.88 (lH, bs); 4.71 (lH, m);
3.53 (4H, mt; 1.29 (6H, d).
EXAMPLE 30
2,3-Dihydro-3-(2'-mercaptoethyl)-4H-1,3-benzoxazin-4-
one
A) A solution of 9 g (0.046 mole) of the compound of
Example 1 in 70 ml of chloroform was added with
3.54 ml (0.048 mole) of methylene chloride. The
solution was heated to 70C for 3 hours, then
brought again to room temperature, washed with a
solution of 5O~ sodium hydrogen carbonate, then
with water. The organic phase was dried over
sodium sulfate and evaporated. There were obtained
9.3 g of 2,3-dihydro-3-(2'-chloroethyl)-4H-1,3-
benzoxazin-4-one.
B) A solution of 15 g (0.071 mole) of the compound
under A) in 500 ml of ethanol was added with 16.1
g (0.2 mole) of thiourea. The mixture was refluxed
095/0~ 21 68 0~g PCT~ ~4/02354
,
23
for 8 hours, then brought to room temperature,
treated with 42 ml of 10% NaOH and refluxed again
~or 5 hours. The solution was evaporated to
dryness, the crude was taken up in water and
extracted with chloroform. The organic phase was
dried over sodium sulfate and evaporated. The
crude was purified by flash-chromatography
(eluent: chloroform), and there were obtained 2.3
g of the title product.
m.p.: 44-46C (chloroform)
H-NMR (200 MHz): 7.82 (lH, dd); 7.54 (lH, dt); 7.17
(lH, t); 7.07 (lH, d); 5.38 (2H, s); 3.63 (2H, t); 2.72
(2H, q); 2.46 (lH, t).
~XAMPLE 31
2,3-Dihydro-3-(2'-methoxyethyl)-4H-1,3-benzoxazin-4-one
A) A solution of 10 g of methyl salicylate in 15 ml
of acetonitrile was added with 5.5 ml of 2-
methoxyethylamine, and then refluxed for 24 hours.
At the end of the heating, the solution was
evaporated to dryness under vacuum, taken up in
ethyl acetate and washed with lN hydrochloric acid
and then with water. The organic phase was dried
over sodium sulfate and concentrated to small
volume. The formed solid was filtered and dried
under vacuum thus yielding 9 g of N-(2'-
methoxyethyl)salicylamide.
B) Following the procedure of Example l,B) and
starting from 9 g (0.046 mole) of the compound
under A), there were obtained 8.3 g of the title
product.
1H-NMR (200 MHz, DMSO) 7.82 (lH, dd); 7.55 (lH, dt);
W095/0~ ~99 PCT~ ~4/0235
24
7.17 (lH, t); 7.07 (lH, d); 5.32 (2H, s); 3.67 (2H, m);
3.51 (2H, m); 3.29 (3H, s).
EXAMPLE 32
2,3-Dihydro-3-(2'-tosyloxyethyl)-4H-1,3-benzoxazin-4-
o
A solution of 3 g (0.015 mole) of the compound of
Example 1 in 15 ml of chloroform was added with 3.7 ml
(0.046 mole) of pyridine and 8.88 g (0.046 mole) of
tosyl chloride. After 2 hours the reaction mixture was
washed with lN HCl. The organic phase was dried over
sodium sulfate and evaporated, and the resulting crude
was purified by flash-chromatography (eluent: methylene
chloridelethyl acetate 98:2) thus yielding 3.34 g of
the title product.
m.p.: 122-124C (methylene chloride)
1H-NMR (200 MHz~: 7~77T7.71 (3H, m); 7.55 (lH, dt);
7.33 (2H, ~.~; 7.17 (lH, t); 7.06 (lH, d); 5.20 (2H, s);
4.23 (2H, t); 3.72 (2H, t); 2.32 (3H, s).
EXAMPLE 33
2,3-Dihydro-3-(2'-mesyloxyethyl)-4H-1,3-benzoxazin-4-
one
A solution of 5 g (0.026 mole) of the compound of
Example 1, in 80 ml of chloroform and 5.8 ml (0.072
mole) of pyridine, cooled to 0C, was added with 5.6 ml
(0.072 mole) of mesyl chloride dissolved in 20 ml of
chloroform. After 6 hours the solution was washed with
1N HCl, water, 5~ sodium hydrogen carbonate and finally
with water. The organic phase, dried over sodium
sulfate, was evaporated under vacuum. There were
obtained 2.7 g of the title product.
m.p.: 76-78C (methylene chloride)
~ ~ PCT~4/02354
~ 6~099
H-NMR (200 MHz): 7.83 (lH, dd); 7.55 (lH, dt); 7.18
(lH, t); 7.09 (lH, d); 5.36 (2H, d); 4.39 (2H, t); 3.83
(2H, d); 3.22 (3H, s).
EX~MPLE 34
Methyl 2-(2',3'-~ihydro-4'-oxo-1',3'-benzoxazin-3l-
yl)ethylsulfite
A solution of 10 g (0.05 mole) of the compound of
Example 1 in 200 ml of chloroform, cooled to -15C was
added with 3.77 ml (0.051 mole) of methylene chloride.
After 1 hou~, 2.1 ml of methanol were added. The
reaction mixture was brought to room temperature and,
after S hours, was evaporated. The resulting crude was
purified by flash chromatography (eluent: hexane/ethyl
acetate 1:1). There were obtained 4 g of the title
product as an oil.
H-NMR (200 MHz): 7.82 (lH, dd); 7.54 (lH, dt); 7.17
(lH, t); 7.07 (lH, d); 5.35 (2H, s); 4.14 (2H, m); 3.77
(2H, m); 3.59 (3H, s).
EXAMæLE 35
Bis-2'-(2,3-dihydro-4-oxo-1,3-benzoxazin-3-
yl)ethylcarbonate
A solution of 4 g (0.02 mole) of the compound of
Example 1 in 200 ml of methylene chloride was added
with 120 mg (0.002 mole) of sodium methylate and 1.92 g
(0.012 mole) of carbonyldiimidazole. The solution was
left at room temperature for 23 hours, then washed with
water. The organic phase was dried over sodium sulfate
and evaporated to dryness. The crude was purified by
flash-chromatography (eluent: hexane/ethyl acetate 6:4)
thus yielding 2.5 g of the title product.
m.p.: 74-75C (hexane/ethyl acetate 1:1)
W095/0~ PCT~ ~410235~
Q9~
26
H-NMR (200 MHz): 7.82 (2H, dd); 7.54 (2H, dt); 7.18
(2H, t); 7.07 (2H, d); 5.30 (4H, s); 4.25 (4H, t); 3.70
(4H, t~.
EXAMPL~ 36
2,3-Dihydro-3-(2'-ethoxyethyl)-4H-1,3-benzoxazin-4-one
Operating as in Example 31, from 12 g of 2-
ethoxyethylamine, there were obtained 1.4 g of the
title product.
lH-NMR (200 MHz): 7.83 (lH, dd); 7.52 (lH, dt); 7.15
(lH, t); 7.05 (lH, d); 5.32 (2H, s); 3.65 (2H, m); 3.54
(2H, m); 3.45 (2H, q); 1.10 (3H, t).
~XAMPLE 37
Isopropyl 2-(2',3'-dihydro-4'-oxo-1l,3l-benzoxazin-3~-
yl)ethylsulfite
The title product was prepared as described in
Example 34 starting from 10 g (0.051 mole) of the
compound of Example 1, 3.78 ml (0.051 mole) of thionyl
chloride and 398 ml (5.17 moles) of isopropyl alcohol.
There were obtained 1.2 g of the title product as an
oil.
H-NMR (200 MHz): 7.82 (lH, dd); 7.55 (lH, dt); 7.17
(lH, t); 7.08 (lH, d); 5.35 (2H, s); 4.71 (lH, m); 4.13
(2H, m); 3.78 (2H, m); 1.24 (3H, d); 1.21 (3H, d).
EXAMPLE 38
6,8-Dichloro-2,3-dihydro-3-(2'-hydroxyethyl)-4H-1,3-
benzoxazin-4-on~
A) 3,5-Dichloro-N-(2~-hydroxyethyl)-salicylamide was
prepared following the procedure of Example 3,A)
starting from 20.9 g (0.094 mole) of methyl 3,5-
dichloro-salicylate and 6.85 ml (0.113 mole) of 2-
aminoethanol. Yield: 14 g.
~ 095/0~ PCT~4/02354
' ' ' . . .
27 2168og9
m.p. 137-139C (hexane)
B) From 14 g (0.055 mole) of the compound under A)
and 3.07 g of paraformaldehyde, and operating as
described in example 3,B), there were obtained 2.7
g of the title product.
m.p. 98-100C (hexane)
lH-NMR (200 MHz): 7.88 (lH, d); 7.72 (lH, d); 5.47 (2H,
s); 4.91 (lH, t); 3.57 (4H, m).
EXaMPLE 39
2,3-Dihydro-4,5-dimethoxy-3-(2'-hydroxyethyl)-4H-1,3-
benzoxazin-4-one
A) 4,5-Dimethoxy-N-(2'-hydroxyethyl)-salicylamide was
prepared by reacting 23 g (0.108 mole) of 4,5-
dimethoxy-salicylic acid methyl ester (obtained as
described in Synthesis, 758, 1984) with 100 ml of
2-aminoethanol at 170C for 3 hours. After cooling
to room temperature, the mixture was taken up in
ethyl acetate, washed with 5% aqueous
hydrochloric acid and dried over sodium sulfate.
Yield: 18 g. The compound was used as such in the
subsequent step.
B) The title product was prepared according to the
procedure described in Example 17,B), starting
from 15 g (0.062 mole) of the compound under A).
Yield: 13 g.
lH-NMR (200 MHz~: 7.21 (lH, s); 6.67 (lH, s); 5.27 (2H,
s); 4.84 (lH, 7); 3.81 (3H, s); 3.76 (3H, s); 5.52 (4H,
m).
EXAMPLE 40
2,3-Dihydro-6-fluoro-3-(2'-hydroxyethyl)-4H-1,3-
benzoxazin-4-one
wo gs/o~ ~ 9 PCT~ ~4/0235 ~
A) 5-Fluoro-N-(2'-hydroxyethyl)-salicylamide was
prepared as described in Example 3 A), starting
from 4.9 g (0.03 mole) of 5-fluoro-salicylic acid
methyl ester and 2.08 ml (0.034 mole) of 2-
aminoethanol. Yield: 5.29 g.
b) The title product was prepared according to theprocedure described in Example 17 B), starting
from 5.2 g (0.026 mole) of the compound under A)
and 2.37 g of paraformaldehyde. Yield: 3.1 g.
m.p.- 128-130C (hexane)
H-NMR (200 MHz): 7.52 (lH, dd); 7.42 (lH, dt); 7.15
(lH, dd); 5.32 (2H, s); 4.90 (lH, t); 3.57 (4H, m).
~XAMPLE 41
2,3-~ihydro-6-ethyl-3-(2~-hydroxyethyl)-4H-1,3-
benzoxazin-4-one
A) 5-Ethyl-N-(2'-hydroxyethyl)-salicylamide was
prepared as described in Example 3 A), starting
from 26.8 g (0.148 mole) of 5-ethyl-salicylic acid
methyl ester (prepared as described in Synthesis,
758, 1984) and 10.7 ml (0.178 mole) of 2-
aminoethanol. Yield: 29.8 g.
B) The title product was prepared according to the
procedure described in Example 17 B), starting
from 25 g (0.119 mole) of the compound under A)
and 10.7 g of paraformaldehyde. Yield: 11.9 g.
H-NMR (200 MHz): 7.62 (lH, d); 7.37 (lH, dd); 6.97
(lH, d); 5.32 (2H, s); 4.87 (lH, t); 3.57 (4H, m); 2.62
(2H, q); 1.20 (3H, t).
EXAMPLE 42
2,3-Dihydro-6,7-dimethyl-3-(2'-hydroxyethyl)-4H-1,3-
benzoxazin-4-one
~ 095/0~ - PCT~4/02354
21 68~
A) 4,5-Dimethyl-N-(2'-hydroxyethyl)-salicylamide was
~ prepared as described in Example 3,A), starting
; from 33.6 g (0.186 mole) of 4,5-dimethyl-salicylic
acid methyl ester (prepared as in Synthesis, 758,
1984) and 13.5 ml (0.223 mole) of 2-aminoethanol.
Yield: 34.4 g.
B) The title product was prepared according to the
procedure described in Example 17,B), starting
from 30.5 g (0.146 mole) of the compound under A)
and 13.14 g of paraformaldehyde. Yield: 17.7 g.
m.p.= 89-91C (hexane)
lH-NMR (200 MHz): 7.55 (lH, s); 6.85 (lH, s); 5.27 (2H,
s); 4.87 (lH, t); 3.55 (4H, m); 2.25 (3H, s); 2.20 (3H,
s) .
EXAMPLE 43
6-Bromo-2,3-dihydro-3-(2'-hydroxyethyl)-4H-1,3-
benzoxazin-4-one
A) 5-Bromo-N-(2'-hydroxyethyl)-salicylamide was
prepared as described in Example 3,A), starting
from 5.2 g (0.022 mole) of 5-bromo-salicylic acid
methyl ester and 1.63 ml (0.027 mole) of 2-
aminoethanol. Yield: 5.53 g.
B) The title product was prepared according to the
procedure described in Example 17,B), starting
from 5.4 g (0.021 mole) of the compound under A)
and 1.87 g of paraformaldehyde. Yield: 1.84 g.
m.p.= 78-81C (hexane)
lH-NMR (200 MHz): 7.87 (lH, d); 7.70 (lH, d); 7.07 (lH,
d); 5.37 (2H, s); 4.90 (lH, t); 3.57 (4H, m).
As already said above, the compounds of the
invention possess cardiovascular activity. In
W095/0~ PCT~ ~4/023
particular they showed a remarkable antianginal and
antiischemic activity in the lab animal.
These favourable biological properties are, in
general, accompanied with a negligible hypotensive
effect. Thus the compounds of the invention can be
considered as potential drugs with a specific
antianginal activity.
The in vivo antianginal activity was determined on
anaesthetized Sprague Dawley rats (average weight
350-400 g), according to the method described by M.
Leitold et al., Arzneim. Forsch. 36, 1454, 1986. The
test was carried out by intravenously administering the
animals with 1 U.I/kg, equal to 3 mg/kg of Arg-
vasopressin which induce a coronary spasm reproducible
and electrocardiographically detectable by an increase
of the T-wave. The animals were then intravenously
treated with four increasing doses of compounds
representative of the invention to measure their ED50,
i.e. the dose yielding a 50% of inhibition of the
increasing of the T-wave. The compounds of the
invention showed to have ED50 ranging between about 1
and about 300 ygJKg. Specifically, the compounds of
Examples 1, 2 and 34 showed an ED50 of. respectively,
87, 215 and 10.5 yg/Kg. Also, some compounds were
tested as above, but orally administered. For example,
the compound of Example 1 showed an ED50 of 0.23 mg/Kg.
The antianginal activity of the claimed compounds
was also tested by the metacholine-induced angina test
described by Sakai K. et al., Pharmacol. Met., 5, 325-
336, 1981. The percentage of inhibition of the ST-wave
increase induced by metacholine (0.8 ug/kg i.v.) was
095/040~ 21 6809~ PCT~4/02354
measured after intravenous administration of the
- compounds of the invention. The results are set forth
in the following Table 1.
TABLE 1
____________________
Example % of Inhibition at 3 ~glkg
1 min. 10 min. 30 min.
_________________________________________
4 60.0 45.0
6 57.1 51.0 28.6
8 31.1
14 71.4 54.7 73.8
17 45.1 39.2
18 37.2 28.0
15 21 25.0 38.6 34.1
22 29.3 22.0 34.1
30.2 48.8 37.2
31 30.0 20.4
38 40.6
20 39 23.0 52.3
_________________________________________
Following the same method the ED50 of the compound
of Example 1 was determined to be 73.5 ~g/kg.
The test above was repeated by orally
administering 0.3 mg/kg of the claimed compounds. The
- results are set forth in the following Table 2.
-
W095/0~ ~ 9~ PCT~410235
TABLE 2
__________________________________________ .
Example % of Inhibition
30 minutes 120 minutes
__ _______________
4 52.6
14 71.2 73.1
34 46.8 21.8
21 28
------______________________
The same test showed that the compounds of the
invention have an ED50 per os ranging between about 100
and about 0.01 mg/kg.
The favourable biological properties above are
also accompanied by a low toxicity. The LD50 values,
calculated according to the method of Lichtfield and
Wilcoxon, J. Pharm. Expt. Ther. 96, 99, 1949, are in
fact higher than 500 mg/kg i.p. in mouse and 800 mg/kg
p.o. in rat.
Object of the present invention is also the use of
the claimed compounds as antianginal agents and agents
useful in the treatment of ischemic cardiopathies, in
connection with all the industrial aspects and
applications of said use, comprising their
incorporation into pharmaceutical compositions.
Examples of these compositions are tablets, sugar-
coated and film-coated tablets, syrups and phials,
these latter being suitable for both the oral and the
intramuscular or intravenous administration. They
contain the active principle alone or in combination
with common pharmaceutically acceptable carriers and
216809c~ PCT/EP94/02354
excipients.
The dosages of active principle used in the
; antianginal therapy or to treat ischemic cardiopathies
may vary within wide limits depending on the specific
S compound employed, and are chosen to provide the
patient with un effective therapeutic protection for.
For example, ~nit doses of from about 0.01 to about 1
mg may be administered from 1 to 4 times a day
depending on the patient's necessity (prophylaxis,
therapy, emergency).