Note: Descriptions are shown in the official language in which they were submitted.
21688~
- WO 95/04277 - PCT/US94/07780
A METHOD FOR PREPARING AND SELECTING
PHARMACEUTICALLY USEFUL NON~ E COMPOUNDS
FROM A STRUCTURALLY DIVERSE UNIVERSAL LIBRARY
CROSS-REFERENCE TO RELATED APPLICATIONS
This application is a co..~ on-in-part application of co-pending U.S. Patent
Application Serial No. 08/101,074, filed August 3, 1993.
TECHNICAL FIELD
The invention relates to a method for pl~,~dl Ulg and selecting low molecular weight
non-peptide collll)uullds having desired ph~ `c..~ 1 or other biological utility. More
particularly, the invention is a method for prc~alulg a sllul;lulally diverse library of low
molecular weight coulpoullds and then selecting from t_e library those colllpùunds having
the desired ph~ ologic activity.
BACKGROUND INFORMATION
A key step in pl~C~dlillg and selPcting ph~. Ill~rclll ;r~lly or other biologically useful
compounds is itlPntifir~tion of sLlu;~uldlly-unique lead co~ uullds. In 1990 it was
e~ ed that nearly one-third of the $231 Million average cost for making a new
thc.a~culic compound available for widespread public use was spent in idc.lliryillg and
opl;..,i,;.,g a lead chPmir~l structure. TraAition~lly and ~;ullclllly mass sc.eerullg of large
numbers of collll,ùunds and mixtures of colll~uunds has been and is the most sl~rcessful
method for id~"lliryillg chPn ir~l leads. Recent availability of robotic, rapid, high
throughput biological screens is beghll~illg to make possible err,ciclll scl~ g of hull~eds
of thousands of colllpoullds per year.
Most screenillg libraries consist of a hi~lo~ical collection of compounds sy~
in the course of ph~,."~l c~ir~ sealch, natural products, and, more lccenlly, peptide
libraries. Each of these libraries has limit~tions. Historical phs.. ~rcu~ l collections
of synthPsi7Pd collll)uullds contain a limited number of diverse structures which Icpres~
only a small fraction of total structural diversity possibilities. ~ imit~tions of natural
products libraries include the structural complexity of the leads i~entifiP~ and the rliffirulty
of reducing these leads to useful ph~,."~cc~ll;r~l agents. Peptide libraries are limited to
peptides or peptide mimics; to date conversion of peptide chPmir~l leads into
SUBSTITUTE SHEET (RULE 26)
WO 95/04277 ~16 8 8 8 o PCT/US94/07780
phz....~relltir~lly useful, orally active, non-peptide drug c~ntli~tPs in the absence of a
small molecule çhPmir~l lead has been met with limited success.
Some of the peptide and peptide mimic libraries lcf.,llcd to above were pll ~al~d
using combi~tolial ch~ y. The ch~llPnge facing medicinal Ch~ is to translate thesuccess using colllbinatolial el.. .~ y to prepare peptide and peptide-like compounds into
technology suitable for effiriPntly yr~yaling large libraries of low molecular weight
non-peptide colllyùullds~ Solid phase c~ l.y for pl~,~aling low molecular weightcol,lyounds is desirable to effect such a translation. The following lcfelcllces are
examples of the types of solid phase ch....i~l.y methods that may be useful in low
molecular weight colllpoulld colllbi~lolial ch ..;~I . y.
In 1974, F. Camps _ ah (Annales De Quimica 70, 848) rc~ollcd solid phase
~yll~L~i.is of four related bPn7o~ yilRs~ More lccen~ly Bunin and Ellman (J. Am.Chem. Soc. (1992) 114, 10997) and S. H. DeWitt et ah (PNAS (1993) 90, 6909) also~-,pol~ed pl~al~lion of a small number of benzodiazeyi~les using solid phase ch~lli~ll.y.
Two tetra~ecPnP-1-ol acetates also have been yl~ ,d on solid ~Uyyul~ (C. C. Leznoff
et ah, Can. J. Chem. (1977) 55, 1143). Additionally, solid phase ~ylllllesis of
4,4'-stilbcnccall,aldehyde has been l~p~Jllcd (J. Y. Wong et ah, Angew. Chem. Int. Ed.
(1974) 13, 666).
The following lcr~,,e.lces are examples of biphenyl and Lliyh~llyl colllyoullds that
have been yl~yal~d by well known synthetic organic h~ iral mPthn-ic. A. A. P~chcu
_ ah recently reported that certain biphenyl acylsulfon~mi~les and biphenyl
sulfonylc~,b~ s are orally active antagonists of the angiotensin II lccep~o~ (Me~iirin~l
Chrrni.ctry Abstract #80 (1993) ACS Meeting-Chicago). Other recently reported
angiotensin II antagonists include several imi~l~7Oyyli~h~e and tetrazole-substituted
biphenyl compounds (E. M. Naylor _ ah, Medicinal t~h~ Abstract #76 (1993) ACS
Meeting-Chicago) and a series ûf carbon-tethered biphenyl pyrrole colllyoullds (J. M.
Hamby et ~ Me~iirin~l Chemistry Abstract #72 (1993) ACS Meeting-Chicago). Another
rec~ ly rcyol~ed substitnte~ biphenyl angiotensin II receptor antagonist inr~ les an
exocyclic niLlogen link (A. S. Tasker _ al., Mcdicinal Chemistry Abstract #338 (1993)
ACS Meeting - Chicago). Others recently have reported that certain
ortho-biphenylphenols are leukotriene antagonists (M. J. Sofia _ ah, Medicinal Chcllli~
Abstract #5 (1993) ACS Meeting-Chicago).
SUBSTITUTE SHEET (RULE 26)
W O 95/04277 216 ~ 8 8 ~ ~CTrUS94/07780
Plc~-dlion of various other ~.~b~ d biphenyls has been reported. An example
of the many lcre,~ ces describing methoxy substituted biphenyls is M. G. Banwell et ah
which desclibes certain trimethoxy and Icllall~ OXy biphtllyls that have tubulin binding
p~ope,Lies (CA118(19):191308u (1992)). Another such lerclcnce desclibes synthesis of
several methoxy and ethoxy-substitllt~d bi~hcllyls for use in a peroxidase in~lir~or system
for basic media (CA118(1):341 la (1992)). 2,4' ,5-Trimethoxy~-biphenylcarboxylic acid
has been reported to have e~Llogel ic activity (CA54: 19584c (1959)).
Sylllllesis of 2,2',5,5'-(klla~lu~,yl-1-1-oxy)bil,he lyl has been reported without
indication ûf its use (CA116(11): 105745p (1991)). Similarly,
2,2',6,6'-tetrabenzyloxybenzyl has been reported (CA110(21):192346b (1988)) and
2,2',3,3'-LeLl~llcLlloxymethylbiphenyl (CA97(11):91847y (1982)) have been I~Glt~d
without a suggested utility. Plcpalali~n of several l~ L~ and tetr~ bslil~lrd
biphenyls and lel~he,l~ls has been reported (CA118(21):212566u (1993)).
ala~ion of various sub~ d bis-[2,3-dihydl~y~h~"lyl]..-~ Ih~nFS has been
reported without an in~1ie~tion of their utility (Marsh _ ah, Ind. Eng. Chem. (1949) 41,
2176). TnrlvrlPd in this lcf~,."lce is a desr, ;l-ti-)n of syllLlRsis of
2,3-dihydroxyphenyl-3',4'-dih,~droxy~he,lyl...~ l-F. P~ lionof~.h~ dl)is~he~
collll,uullds having SO2, S, CMe2, or O lllo.e.t;es b~ n the rings has been l~,~oltcd for
use in making sc,lli~e,llledble COlll~OSiLc mlmhT~nPS for liquid separation
(CA109(18):151003y (1986)).
Thus, there remains a need for mPthorls to effiriPntly prepare large libraries of low
molecular weight non-peptide collll)oullds and to select from such liblaries colllpou~ds
having desired ~h~ c.llir~l utility.
SUMMARY OF THE INVENTION
The ~vlese~lLly invented method for plc~alil1g and selP~ g low molecular weight
non-peptide collllJuullds having desired ph~rm~reutir~l or other biological utility inrhl.1Ps
a system for rapidly generating large rationally ~esign~od libraries of structurally diverse
small molecule collll,ounds to explore mnl~ ~alllcL~r space that overcomes many of the
disadvantages associated with using ~;ull~lllly available libraries as a basis for ide,lLiryhlg
and selecting new phz....~r~"l;rzl agents. The disclosed invention makes possible
pl~alaLion of libraries of low molecular weight organic rh. ."ir~l colll~(~ullds which have
SUBSTITUTE SH EET (RULE 26)
W O 95/04277 216 X 8 8 6 1~CTrUS94/07780
diverse ch~mirAI ~LlucLulcs that are known and can be controlled. Additionally, other
characteristics of the collll)oullds that are illl~OlL~lll for phArmAreutirAI utility, such as
solubility, can be controlled. Most illlpolL~Lly, however, because the colllpollllds
pl-,~al~d using this invention are low molecular weight non-peptide colll~,ounds they are
e~l,ccled to be useful in a much broader sl~e~;Llulll of thl,lalJ~,uLic applications than peptides
which generally can only be ~minictered by injection or inhAlAtion.
DETAILED DESCRIPTION OF THE INVENTION
The pl~sellLly invented method for ~lcpalillg and selP -~ g low molecular weightcol,l~,vullds having desired ph5~ r~ ;r~l or other biologic utility inr~ s a multiple
combil~tolial a~ploach to prepare sLlu~Lulally diverse libraries which contain biologically
useful collll,oullds. Combinàt~lial rh~ y takes advantage of the nature of the
interaction bc~ n biological ligates such as antibodies, lece~tol~, e~llles, ion ch~ f 1~,
and llans~ lion factors, and their ligands such as qntigen~, hollllol~es, nculuLlAll~
and ph~ eu~ ;ral agents. It generally is agreed that ligate/ligand affinity and illLe.aclion
results from binding or hlL~,laclion b~L~ at least three fi---- ~ 1 groups or c~- ..,ir~l
fu~ ;on~liti~s on the ligand and complt ..~ ly sites on the ligate. Strong illl~la~;Lions
b.,lwccn ligates and ligands are d~pf-~ upon the ~lu~cllies and three rlim~n~io
spacial oli.,~lL~Lion of the functional groups or ch~ Al functionalities on the îigqn~ls
High affinity specific ligands for a given ligate have fu.-~-l;on~l groups that: (1) bind
tightly to the binding sites on the ligate and (2) are positioned to bring the functional
groups into close proAillliLy with the ligate binding sites in the biological milieu where the
hlL~laclions occur.
Colll~ullds pl~pal~,d using the invented method have molecular weights of
b.,lwccn about 200 and 1000 daltons, preferably btLween about 300 and 600 daltons, and
contain two colllpollent parts: (1) scaffold moieties and (2) at least three functional groups.
As used herein a scaffold" is a molecule onto which functional groups can be attqrhrd
in a manner that when two or more scaffold moieties are ~hrhrd results in the desired
spacial Ol ;~ l ;OI~ of the functional groups. Scaffold moieties preferably are se!rctPd such
that they can be ~ d from available materials by known chPmirAl reactions and
readily allow for qttqrhmrnt of desired functional groups and/or other scaffold moieties
in a variety of positions on the molecule. In this specification and claims, as in~lirqted
SUBSTITUTE SHEET (RULE 26)
~0 9S/04277 ;~ l ~i 8 8 8 6 PcTtuss4/07780
by the context, "scaffold" may also refer to two or more att~rhP~l scaffold moieties.
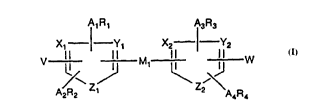
Suitable scaffolds are coll,poullds of the following formula:
X. Y, X~, Y2
V I I M ~ W
Zl Z2
wh~eill:
M, and M2 in~1~pen~ ntly are a bond or CRR', CRR'CRR', CR=CR', or C_C
wh~ R and R' i~ F~.~ .lly are H or C,~alkyl;
X" Yl and Z, are any ~cce~ .le cOlllbi~ ioll of CH, CHCH, O, S, N, and NH
provided that at least one is CH or CHCH and not more than one is CHCH;
X2, Y2, and Z2 are any acces~ihle co",bina~io,l of CH, CHCH, O, S, N, and NH
provided that at least one is CH or CHCH and not more than one is CHCH;
WisHor
X, Y3
M2 1 ~¦
X3, Y3 and Z3 are any ~rces~ihle co",bi,lalion of CH. CHCH, O, S, N, and NH
provided that at least one is CH or CHCH and not more than one is CHCH; and
V is H, C,~alkyl, halo, (C0,alkyl)OH, (CO,alkyl)SH, or (CO,alkyl)NRR, or
(C0~alkyl)CO2R wlRr~ each R in~ nr~ ntly iS H or C,~alkyl.
Useful fimrtion~l groups include the side chains of the 19 naturally oc~..;..g
L-amino acids and the side chains of nucleotides found in nature. Additionally,
non-naturally oc~;ulling mimics of these groups are useful. Pref~lled colllp~w~ds of the
invention which are pl~pal~d by combining ~lel~l~cd scaffold moieties with l"efell,,d
functional groups are shown in Formula I below:
SUBSTITUTE SHEET (RULE 26)
WO 95/04277 PCT/US94/07780
,!
-- 6
AlRl A~R3
X. I--Yl X~ I--Y2
V I I I M 1 1 I W
~Z,J ~zJ
A2R2 A4R4
Formula I
Wll~
X" Y, and Zl are any ~rcescihle colllbil,ation of CH, CHCH, O, S, N, and NH
provided that at least one is CH or CHCH and not more than one is CHCH;
X2, Y2, and Z2 are any ~rcec.ci~ le combination of CH, CHCH, O, S, N, and NH
provided that at least one is CH or CHCH and not more than one is CHCH;
WisHor
A5R~
X: I Y3
M2 I JJ
~`Z3
A6R6
X3, Y3 and Z3 are any ~ce~;ble colllbil~ion of CH, CHCH, O, S, N, and NH
provided that at least one is CH or CHCH and not more than one is CHCH;
M, and M2 independently are a bond or CRUR~5, CR,,,R,,5CRUR~5, CR~,=CR~, or
C _C;
V is H, C,.5alkyl, halo, (C0JaL~cyl)OH, (C0,alkyl)SH, (CO,alkyl)NR22R23, or
(co~alkyl)co2R7b;
A" A2, A3, A4, A5, and A6 independently are absent or present as O, S, NR60; or
CO.5alkylC(O)NR2,, provided that at least three are present;
R" R2, R3, R~, R5 and Rb independently are H, C~balkylCOR,5 C,JalkylR,bR,7,
C,JalkylOR2l except methoxymethyl, C,JaLkylNR25R2b, C~balkylNR~OC(NR~,)NRt2R~3.
C,Jalkylindole, or CO~5alkyl-D;
D is any one or multiple fused saturated or ul~dLul.lled five or six membered cyclic
hydlocdll,on or h~ ocyclic ring system CO~ g one or more O, N, or S atoms that
SUBSTITUTE SHEET (RULE 26)
WO 95/04277 i~ 8 ~ PCT/US94/07780
are ~ b~ ed or sl~bstinuted by any arceccihle combination of 1 to 4 s~1bstin~ontc
selected from C,~alkyl, NR7R~, OR9, SR,o, or COR", halogen, CF3;
R7, Rt R9, R~o~ R,9, R20, R2" Rn~ R23, R60, Rto~ Rt" R~2, Rt3, Rt4 and Rt5 in~pen~ntly
are H or C,~alkyl;
R,2, R,3, R," R,6, R,7, R,t, R24, R25, R26, and R76 in(l~ lly are H, C,6aL~yl,phenyl, or suh~ ed phenyl;
R" is OR,2 or NR,3R,4;
R,, is OR,~ or NR,9R20; or
any pharmaceutically useful salt thereof except
2,2',5,5'-(tcllaylOyylll-l-Oxy)biyh~llyl and salts t_ereof and co.. l.o~,ulc wl,~,.c u at least
three of A" A2, A3, A4, A5, and A5 are oxygen and at least three of R" R2, R3, Rl, R5. and
R6 are hydrogen, methyl, ethyl, or phenyl and salts thereof.
The colllyvullds of Formula I col.~ a ulli~ al library of collly()ullds that
i n~lllrl~s ph A I I I IA~ l I ;f ~1 ly useful coluyoullds .
As used in Formula I and clsc~ .e in this sye~;r.r-';o~ and the claimc,
"C,~ yalkyl" is a straight chain or ~lallched, saluldLcd or ~ salulatcd aLkyl group CO1II~A;~
x to y carbon atoms wh~lcln x and y are hll~gcl~ and "halo" in~lurlPs bromo, chloro,
fluoro, and iodo, and ~ i--h ~ phenyl" iS a phenyl group ~ k~ by any arcescihle
combination of halo, CF3, OH, C,~,aL~cyl, C,~aL~coxy, COOH, COOC,~alkyl, NRR', or
CONRR' wh~lcill R and R' ;iul. ~)f ~ y are H or ,~aL~cyl.
In more plercllcd compounds of the invention X, to X3, Y, to Y3, and Z, to Z3 are
select~d so that one or more of the ring systems is pyrrole, furan, thiophene, lJylidi~
pyra_ole, pyrimidine, or isoxazole with phenyl being most plcf~llcd. Also in more
plcr~ d collll,oullds of the invention D is one of the following ring systems sllh~ d
as described above: pyrrole, furan, imirlA7ole, thiophene, ~ylidine, pyræole, pyrimidine,
pyridæine, or isoxæole with phenyl being most plcr~ ,d.
More plc;fe,,cd colll~oullds of the invention are shown in the following FormulaII:
SUBSTITUTE SH EET (RULE 26)
w o gs/04277 2 1 6 8 8 8 6 T~r~US94/07780
-- 8
A7(C,.~2lkYI)R30
Ag(C1 4alkYI)R32
V1 ~ ~ M1
A8(C 1 .calkyl)R3~
A1O(c1 4alCyl)R33
Formula II
wll~,.cul:
M, is a bond or CH2, CH2CH2 CH=CH, or C_C;
Vl is H, CH3, OH, or CH20H;
A7, At, A9, and A~o j".1~p~nrlPntly are absent or present as O provided that three
are O; and
R30, R3,, R32, and R33 in(1~pen~ nfly are OH, NH2, CO2H, phenyl, snb~ d
phenyl, CONH2, NR3OC(NRt,)NRt2Rt3, C,~alkyl, imirl~7ole, or indole wl~e.e;ll R80 to Rt3 are
H or C,JaLkyl.
ph~ r~Jl;r~lly useful salts of the above co~ )ùullds include, for eY~mrl~,
so~ m, pot~Cci~lm~ trialkyl ~ o..;~.., c~lri--m~ zinc, lifhillm~ m 3g~ .., alu...i..~
~ieth~n-lamine, ethyl~nf~ r-~ megulmine, acetate, maleate, Çulllalate, lactate, oxalate,
I..rlh~h~.-lfonate, ~Ih~nP~ fonate, be~ f~ lfonate, tartrate, citrate, hydlochloride,
hydrobromide, sulfate, pho~hdlc~ and nitrate. Other ph~rmi~rentir~lly useful salts are
readily appal~ to skilled ,.,-~.lir;"~l ch~
Some of the colll~uullds inrll-ded in Formula I can exist in more than one chiral
form and thus exhibit stereoisolllc.i~lll. Formula I inrhlfles all purified stereoisomers and
racemic lllib~Lul~s of the colll~uunds within its scope.
In one aspect of the invention a p~ step in pl~ali,lg and selecting
compounds having desired ph~rm~reutir~l or other biologic utility is prepdldtion of a
ullivcl~al library. As stated above mr~liein~l chrrnict~ and ph~ rologists generally agree
that interactions b~Lwæn biological ligates and ligands require that the ligand contain at
least three functional groups in a spacial olicllLdLion that is comple~ .y to the binding
sites on the ligate. It also is known that the ~ re between the binding sites on ligates
SUBSTITUTE SHEET (RULE 26)
~Vo 95/04277 ~ 3 8 8 ~ PCT/US94/07780
is de~ .I.;..~d by the col~llllation of the ligate as it exists in its native enviroll,llcllL and
that effective ligands are those that have functional groups positioned to be compl~ . y
to such col~""aLion. Rec~lls~ ligates are three rlimPn.cional in their natural setting, for
any selected intramolecular ~ e bel~n binding sites an eccenti~lly infinite number
of possible specific positions for the binding sites exist. Thus there similarly is a very
large llulllber of possible rull~liol~al group positions on the ligands that errccLively interact
with particular ligates. As used in this sl,ecir~r~;on and claims a ul~ al library is a
collection of related small molecular weight COlll~OUlldS that with respect to spacial
oli~llL~tion of functional groups errccLivcly samples a large se~ of the possible
specific positions within a selçc~d ~ re and a sub-ulli~ al library is a universal
library that is l~g~,tcd to a particular biological ligate.
P~ dldLion of bradykinin antagonists provides an c~ le of the general
approach to ~leci~..;..g a sub-universal library. Bradykinin is a naturally-oc~ g
tide (Arg-Pro-Pro-Gly-Phe-Ser-Pro-Phe-Arg) that is formed e..Ly-.-~l;r~lly in
the blood and extra^el~ r fluids after injury.
At least two distinct lccclJLdr types, Bl and B2 appear to exist. ~lthol-gh
activation of B2 fecc~tul~ appears to ~ the most lele~,dlll biological actions of
kinins, both B1 and B2 lcce~Lol~ could be illl~lL~L in developing Lh~,la~euLic sLla~gies.
Bradykinin is a major pain producing s~ e that excites and sen~ s sensory nervesfollowing trauma, burns, injury and infection. Peptide bradykinin antagonists block
bradykinin-in~ ced pain in animal models suggcsLing that a bradykinin antagonist would
be crrecLivc for the Ll~ of a variety of painful disorders. Bradykinin has also been
found in plasma rY~ t~s taken from the scalp of mi~.d.. ,, and has been shown to cause
severe vascular head painupon hlLIa\~lWUS injection s~lggc j~;..g that bradykinin antagonists
would be useful for the Ll~ - .1 of hr~ eh~. Bradykinin is a potent v~co~ tor of most
pclilJhclal arteries and also causes neurogenic ;"ll~."...~tinn by the peripheral release of
subst~nre P, neurokinin A, and CGRP from sensory nerve fibers. Bradykinin has also
been found in fluid from a,LhliLic joints. These results suggest that bradykinin antagonists
might have an illl~OlL~lL role as ~nriinfl~.. ~uly agents. Bradykinin has been proposed
to play a role in the pathogenesis of asthma as well.
While an orally-active bradykinin antagonist is likely to be of ;.---..rll~e thel~euLic
benefit, the potent bradykinin agonists and antagonists reported to date have been peptide
SUBSTITUTE SHEET (RULE 26)
WO 95/04277 Z 16 8 8 8 S PCT/US94/07780
- 10 -
delivaLives similar in size to bradykinin (which like bradykinin are e~-e~ to be rapidly
degraded in body fluids).
Peptide analogs of bradykinin have shown that in general, repl~ ..f It of Pro 7
with D-Phe or COl~ Qn~lly-col~L~ailled analogues as well âS repl~rf,~m~nt of Phe 5 and
8 with Lllie~lalanine or Cfj~O~ n~lly-col~Llailled phenyl analogues affords Cfj~ eLi~ ive
and seleeLi~,~, antagonists of bradykinin. The C t. .~..;..~l algh~i~e is crucial for l~c~plol
activity. It appears that the N-tf~nnin~l amino group is not n~c~Cc_~,y for activity since it
can be acylâted or removed without ~;g. ;ri~ Oss of activity. Bl selective antagoni~L~
are obtained by making the des-9 Arg analogues.
As an eY~mrl~, D-Arg-Hyp3-Thi5-D-Tic~-Oic~-bradykinin is a specific, potent, andlong-lasting bradykinin ~nt~gonict being developed by Hoechst (Hoe-140) for allergic
rhinitis and asthma.
FU1L11~L111O1C~ Kyle et al. have h~collJula~cd unndluldl amino acids in the
C LC~ IC of blLIdykinill which introduce 13-turn stability and conclude that a B-turn in
the four C-t~rmin~l amino acid residues might be a plc,~ui~i~e for high lcc~lJt()l affinity
(D. J. Kyle _ ah, J. Med. Chem. (1991) 34 (3): 1230-33).
Using the illfo..---~ion d~s~ e~ above it is possible to design a sub-uni~ àl
library that is likely to possess bladyl~illill antagonist activity. The 13-turn likely at the
C-t~rrnin~l portion of bradykinin sll~gest~ that the peptide antagonists are not fully
çxtrnrlPd at the lcccp~O~ and likely occupy a ~ nre of 10 -18A. This is an ideal size
to be mimirlrPd by a bi~l hellyl scaffold and the size, shape, and group vdlidlions are
explored by IJlC~dlillg a large library of collll,ounds guided, or limited, by previously
~ Jolled SAR studies on bradykinin leceplor antagonist,s. This app~uach can be carried
over to B, l~,C.,~tOl~ by leaving out the al~i~ulC mimic on the A-ring. Using the
previously desclibcd SAR data on bradykinin peptide antagonists the following compounds
of Formula III are e~c.,~d to include bradykinin antagonists:
CH~ j M~ F
Formula III E
B' F'
SUBSTITUTE SHEET (RULE 26)
W O 95/04277 ~16 8 8 8 6 T~CTAUS94/07780
- 11 -
wL~.eill:
M, is a bond or CH2, CH2CH2 CH=CH, or CeC;
B and B' are H, O(CH2)nNR~C(NR~,)NR~2R6,, or O(CH2)n.NR~3R~, whelc~ R,"
R~2. R43, R" and R6, inAepenAPntly are H or C, 3aLlcyl, n and n' are 2 or 3; provided one of
BandB' isH;
E is R
o (CH2)~
/ ~CH)n'
wherein X is CH, N, NH, O, or S; n is 1-3; and n' is 1 or 2;
F, F', and F" are H, O(CH2)nNR,5C(NRU~ 7R62, or O(CH2)n.NR~ 9 wll. lci~ 5,
R~6, R", R~ R~, and R62 ;,~Af~ Af~ntly are H or C,.3aLkyl, and n and n' are 2 or 3;
provided two of F, F', and F" are H;
G and G' are H, O(CH2)nOR~, or
Rsl
O (CH2)n~
~ (CH)n'
X'
wh~leil~ X' is CH, N, NH, O, or S;
R50 is H or C, 3alkyl;
R5, is H, C, 3aLkyl, halogen, OH, or OC,.3aL~cyl; n is 1-3; and n' is 1 or 2; provided
oneofGandG' isH.
In plefell~d co,ll~ou"ds:
B or B' is OCH2CH2NHC(NH)NH2;
E is
OCH
or
SUBSTITUTE SHEET (RULE 26)
WO 95/04277 ~ 8~ 8 6 PCT/US94/07780
OC~I2CH2~3
F or F" are OCH2CH2NHC(NH)NH2; and
G and G' are H.
SyllLhcsis of the following l lef,,lled cullllx)und is inr.ln~le~ in the Examples below:
~H
/( 2)2\~NH ~CH
CH3~ ~ ~ I~'H
/(CH2)2\ 1 NH2
(1-methyl-2,5'-diethoxy~ -3'-o~y~ylbilJh.,~yl)
These coulpuu,,ds are tested for bladykillill antagonist activity using a high-volume
bi- ch~ iral binding assay such as is leÇ~e.~ced in the examples below. For potential use
in rapid mass sc~eel~ng~ a rat B2 leceptor has been cloned by Jarnagin _ ah (PNAS
(1991) 88, 7724). It appears to be a 7-Ll,.~ ...hrane domain G-protein coupled l.,cc~lor
with a molecular weight of 42 kD and 366 amino acids. FulLh~ ore, a human B2
recc~ol was cloned by Hess et ah (RiochPTn Biophys. Res. Comm. (1992) 184, 260) and
has a molecular weight of 41.1 kD and 364 amino acids with 81~ seqmPnre homology to
the rat B2 ~c~Lor. The binding assays are followed by e~ ion of the co~oulldin ~ in vitro smooth muscle ~c~,~aLion. Punctional activity is acseccecl by eY~---i--i-.g
in vitro PI lul"ovel. In vivo models include bradykinin paw plei,~uie in rats, both IP and
PO.
Most of the rPTn~ining seven L ~ bldlle G-protein coupled receptors (GPCR)
are viable c~nrii~i~tps for the approach described herein. Such lcceptol~ include, but are
not limited to, CCK, angiotensin, bolllbesill, bradykinin, endothelin, lltulo~ Lide Y,
ntulote~ , opiod, sol..alo~l;,l;.-, tachykil~,n (NK" NK2, NK3), thromboxane A2, and
SUBSTITUTE SHEET (RULE 26)
W O 95/04277 ~ 1 6 8 8 8 S ~CTrUS94/07780
- 13
vasop,es~ . The angiotensin-2 re~,eptor might be of particular interest as a test case in
light of the l.,cel.lly reported activity of a number of functionalized bisy~llyl molecules.
The ligands for many of the GPCRs range from small---~PAi~-- sized 01~ ~U1iCS tosmall-m~ m peptides (4-35 amino acids). Most of tnese ligands are e~l~e~led to occupy
a 10-30 cubic A volume making them ideal c~n~ Ps for the libraries desc,ibed herein.
An i~ easing number of modeling and mllt~gen~,sic studies are not only in~ir~ting the
app,~,iaLc approximate size but are also giving specific il~lll.aLion on pOl ~ L residues
of the ecepLor that interact with the ligand. This h~ll-laLion can be readily applied to
the design of ,~ccplor specific ~ubuni~ al libraries.
Some c~ylcs of l~c~.lLly available illrO....~ .. inrll-des the TXA2r~c~lor
(Yamamoto _ ak, J. Med. Chem. (1993) 36, 820-25). These wol~ls propose the TXA2
binding site and suggest specific residues of the rcc~,~tor that are ilLIyOlLallL for ligand
binding, inrl~ Ser-201, Arg-295, and Trp-258. Groups that are compli...r ~ y to
these residues would be built into the sub-universal library.
The NK 1-3 ~CC-,~tOl~ have been cloned and e~ylcssed and mllt~tion~l studies areongoillg which suggest the binding site for NK, antagonists is likely to be around the
junction of ç~trarel~ r loop 2 and the top of TMV and TMVI. Furthermore, the
i(~entifir~tion of non-peptide leads for the NK-1 l~,ceptor suggests some groups that allow
initial selection of groups for a sub-universal library (Watling, TIPS (1993) 14, 81). It
is believed that NK, antagonists will be useful for treating pain, i-~ ion~ arthritis,
and asthma.
ir.r~lion of residues for design of a som~tost~tin sub-universal library is
guided by the work of ~i~cl.",~nl~ et ak (J. Amer. Chem. Soc. (1992) 114, 9217).~ alion of compounds that interact with ion rh~n~.~lc provides another example
of d~signing a sub-ul~i~ al library. Ion ch~lm~lc are pl~,leills which span cell membranes
providing ya~hways for the flow of ions such as chloride or pot~Ccium These channel
yloleills are involved in many cellular functions such as nerve cign~ling, muscle
contraction and hormone secretion. Over the past several years there has been anexplosive growth in the number of cloned and e~yl~;,sed ion ch~nn~lc, as well as in
discoveries which link channels to disease. Moreover, now that it is clear that there are
many subtypes of ion ch~nn~olc dirr.,l.,n-ially di~llibul~d throughout the body, the
possibilities for selective ~lgeLing of specific ch~nn~lc in specific tissues are unlimit~d.
SUBSTITUTE SHEET (RULE 26)
W O 95/04277 216 8 8 8 6 I~CT~US94/07780
This selective L~ tLillg will reduce ullwdnlcd drug-related side effects and toxicities. The
plasma lllelllbl~le locali7~tion of ion ch~nnPIc el;...;.-~tes the need for complex delivery
systems required for drugs dire~;led at intr~rel~ r or intr~nl~rlP~r targets.
POLassiu~l rh~nnPIc can be divided into at least 6 major classes, and 15 subclasses,
each with its own distinct biophysical and ph~rm~rological identity. Agents w_ich
m~lll~te specific pot~ccillm (~h~nnPIc in specific tissues are exrectPd to target select
disease states without altering normal Ç ....-~iOI~c. Potassium rh~nnPl.c are largely
~,.,~ollsible for m~;,.l~..~nre functions like establisning the llle.llbl~le potcnlial in
t~P~ cells, or in switching on, or off, a cell's elPrtrir~l activity. Thus, these
rh~nnPIc in part control the cell's cd~a~ y for ~ uus Ll,...~ ;on, muscle contraction
and seclelion. Due to their integral roles in almost all normal signal pn~ce~,~illg, agents
w_ich mo-h-l~te potassium ch~nnPls are likely to be useful for treating con~litions such as
het~PS and mncc~ r sclerosis, cardiac ~lhy l~ias and vascular hy~laeli~ity.
Various types of ligand-activated and voltage-a.;~ atéd ion ch~nnplc have now been
cloned and functionally e~lcssed. SequPnre colll~alisons and llydlopallly analyses have
revealed a great deal of ~Lluclulal homology among these ch~..n~lc. Each channel seqllpnre
is composed of a lc~e~l;..g motif of l~ .llhlane S~ ;..g ~lom~inc which co,llbille in
various ways to form ch~nnPlc (For a recent review of the field, see Andersen and
Koeppe, II, Physiological Reviews (1992) Vol. 72).
Site-direc~cd mutagenesis has allowed le;,eal~;he,~ to probe the pfilllaly structure
of ion channel ploleills for critical amino acid residues involved in the binding sites of
drug molecules. These studies will allow for the development of agents L~lg.,ted for
specific channel ~ub~y~es and binding sites. To date, several classes of ion ch~nn~olc,
inrlllrling pol;-~;ll ll and chloride, have received intensive ch~aclcl~LLion leading to a
basis on which to conci~lpr structure-based drug design.
Toxins, such as those from scorpion venoms, have proven useful in defining
poL~llLial drug illL~laclion sites on ion ch~nnPIc as well as ~efining physiological roles for
channels. These peptide toxins are 36-38 residues long, contain three lic--lfi~P bridges,
and share strong sequ~pnre similarity among isoforms, block both voltage-gated and
Ca-activated K rh~nnplc with nanomolar affinity. Within this group of toxins, there are
specific subtypes which bind to specific subtypes of pot~c.cinm ch~nnPlc. Ele~;uo~Lalic
interactions between charybdotoxin (CTX), a specific peptide pore blocker of K rh~nnPlc
SUBSTITUTE SH EET (RULE 26)
W O 95/04277 ~ ~ 6 8 8 8 ~ I~CT~US94/07780
- 15 -
and a Ca-activated K channel have been e~ ively investi~q-t~d Charybdotoxin has eight
po~ilively charged residues (four Iysines, three al~illilles, and one I~ ). Ele~Ll-)slaLic
forces are known to favor CTX binding to the negatively charged mouths of K chqnnPIc.
However, only repIq~PmPnt of Arg25, Lys27, or Lys34 with a Gln residue sLIo~ly
decreased the affinity of the toxin for the chq-nn~l These three residues are located close
to one another on one side of the CTX moIPc~llP and make direct contact with the channel
mouth. On the oppo~ile side are five charged residues whose nPlltrqli7qtion show little
effect. Thc,~fole the posilively charged groups on CTX plolllole toxin channel illt~laclion
in two ways; by weak through space cle~LIu~Lic infll)em~s and by direct and ;~ q~e
contact with the channel on one side of the toxin molecule. The solution ~u~;lulc; of CTX
has been lecelllly ~ fd (Bontems _ ah, Biorh- ..;~ (1992) 31, 7756) and it has
been shown that Arg25 and Lys34 are located within loA of Lys27 and each is crucial
for high affinity binding of CTX. The l~ceptol site in the channel's mouth must be wide
(>22A) and flat to accommo~lqte the CTX molecule. The wide mouth must narrow
abruptly into an ion-selc~ r. pore in order to provide a sele~ ., K binding site with
which Lys27 illl~ln~l~ (Miller and Park, Bior~ y (1992) 31, 749, and Neuron (1992)
2, 307). These studies reveal a molecular surface of CTX which makes direct contact
with the extra~elllllqr mouth of the K channel and a single CTX molecule physically
occlud~s the K conducliol- p~ w~ly by binding to a l~c~l,lor located in the
eYt~rn~lly-facing mouth of the channel protein.
Using the il~llllation described above, a sub-ulli~ al library ~;.~d to K
ch~nnPIc which mimics the three illl~)Ullillll binding ~,i.lues both ele~;llonically (three
positive charges) and spatially (6-18A total separation) is deci~Pd. Such a library is
eYpec~d to identify non-peptide CTX mimics with IhCl~)~:UliC po~n~ial. The collll,oullds
of Formula IV l,,~l.s~illl a sub-ulli~ al library targeted to ~,ol-c~ .. ch~nn~olc.
J' M
CH
I=\ ~\
J a Q'
Formula IV
SUBSTITUTE SHEET (RULE 26)
Wo 95l04277 PCT/US94/07780
wL~,eh~ G
Ml is a bond or CH2, CH2CH2, CH=CH, or CsC;
J, J', and M intlfpe~-lfntly are O(CH2)nNR50C(NR,,)NR52R55 or O(CH2)n.NR53R"
whcleill R50, R5" R52, R53, Rs~ and R65 ;~ p,n~lfL,~Ily are H or Cl3alkyl, and n and n'
in~lf~f~ie~nly are 2-3;
Q and Q' are H or O(C,~alkyl)T Wl~.~ T is C,.6alkyl, CO2R55, OR,6, or
X7~U
~d
(CH2)n
wll.,.f .~l:
X7 is CH, N, NH, S, or O;
n"' is 1 or 2;
U is H, C,~alkyl, halogen, CF3, or OR57; and
R55, R56, and R57 i~Af~ Af-~lly are H or Cl,alkyl; ~ vided that Q or Q' is H.
The p~sc;~llly ill~ ted multiple colllbi~olial method for p~palillg and sf~lf ctir~
small molecular weight colllpounds having pk~ ;ral utility or other biologic utility
is used to err;~ lly prepare ulliv~ al li'Gl~if s. As used herein a multiple combi~ o.;~l
method is a method for ~le~iu g culllp~ullds that uses nvo or more scaffold molecules
each callying Ç~ ;on~l groups that have been ~ hf~d in a colll'G;.~tori~l fachion
Generally, co...l.ou~.~iC colllpli..hlg two scaffold llloit:lies are used for ligates of about 12
to 20 A and colll~oullds having three scaffold llloitlies yield ligands for ligates of about
20 to 35 A.
The power of the invented multiple combi~lolial method is ~IfL~..ol.~ led by the,.---..h~-, of colll~loullds that can be ~ ed quickly and e rl;ni,~ .lly. For example, using
two scaffold molecules each co~ .;..g two of twenty possible funrtio~l groups a~l~nged
in four dirre-~-ll Ol;~ I;onc yields more than 1,000,000 compounds. Using the same
pal~ll~tel~ with a third scaffold molecule allows for plep~lion of a universal library
cont~ining more than 1,000,000,000 compounds. The compounds of Formula I are an
example of a Ulli~ .al Iibra~y of compounds that are prepared according to the invention.
In another aspect the invention is used to prepare large qn~ntitif,s of a desired
target colll~oulld rather than small ~.-...u..l~i of multiple compounds as is the case in
SUBSTITUTE SHEET (RULE 26)
WO 951042M ~ 8 ~ ~ PCr/US94/07780
pl-,palulg u~ elsdl or sub-universal libraries. Preferably when ~l~p~ulg universal or
sub-universal libraries multiple compounds are ~lc~a~d by sim-llt~nPously con~ cting
dirrclcllL ch- l-ir~l reactions in multiple reaction vessels. Preferably, reactions are
contl~ctr~l sim~lt~n~ously in about 25 reaction vessels, more preferably in about 100
reaction vessels, and most preferably in ~d~d 96 well plates. To prepare large
qll~ntitirs of a se1Pcttocl cc,llll)~und the same reaction is carried out simlllt~n~o~lcly in
multiple reaction vessels. For example, 2',4,5'-trimetho~yl,il.hcllyl 4'-carboxylic acid,
a colll~ound known to exhibit e~lloge~ic activity, (CA54:19584c (1959)) is prepared
according to the invention as described in the Examples below.
The compounds and libraries of the invention preferably are pl~,~cd according
to ScllPm~ I below. In Srll~m~ I the ~lefellcd method of ~yl~lllrc;~ g the colllpoullds on
a solid support is depicted. The libraries and colll~uullds of the invention, however, also
can be ~lepd~cd using solution phase ch~
[This space intentionally left blank]
SUBSTITUTE SH EET (RULE 26)
WO 95/04277 ` 8~ PCT/US94/07780
- 18 -
Scheme I
OP OP
( ~ Linker + AG ~X' ~ (~ Linker~X'
(1) (2)
OP
1. Deprotect OFG
2. Introduce Functional ~ r,l~ 2
Group ~ ( SS )--Linker~ (~) )--X'
3. Repeat if Desired ~ / (4)
(3)
OFG ~ OP
OFG2 I 1. Deprotect
~ r~ r~ 2. Introduce Functional
( SS ~--Linker ( (J~--M~ Group
~' \ \~\ 3. Repeat if Desired
OP'
(5)
OFG1 OFG3 1. Modify Ml if needed
~1~ OFG2 ~1~ 2. Rernove Protecting
(~Linker~M1~ Groups
(6)
OFG1 OFG3
V~FG2 {~
OFG4
(~
SUBSTITUTE SHEET (RULE 26)
W O 95/04277 ~ 1 ~ 8 8 8 6 1~CTrUS94/07780
- 19 -
Scheme I demo~LIa~cs the invented method of pl~,paling uni~e~àl libraries of
com~vullds. Accol~ing to this scheme functional groups are attArhP~ to a first scaffold
moiety to yield a colll~u~lld CvlllpliSiug a scaffold and one or two f~lnrtionAl groups
(Compound 3). Thel.,arL~ r a second scaffold molecule (CompuLIlld 4) is added followed
by addition of fi~nrtir,nAI group(s) to the second scaffold moiety to yield Compvu~d 6
which can have 3 or 4 fi~nrtionAl groups. Collly~ul~ds of Formula I wh~ c-ll M, is a bond
then are l le~ ,d by cleaving Col~vulld 6 from the solid support. Co~ -v~ s of the
invention wherein M, is CH2, CH2CH2, CH=CH, or CeC are p~~ .,d as described in
Example 4.
In Scheme I "SS" is a solid support mAteriAI such as the clv~sli.lLrd polystyrene
resin known as the Merrifield resin (R. S. Merrifield, J. Am. Chem. Soc. (1963) 85,
2149). Al~elllaLivcly, any other suitable polymeric resin or other support material such
as, for example, silica, glass, cotton, and cPIllllose is used. Also in Scheme I "AGr is
any suitable group for ~tt~k".. .-1 to the lin.~er such as, for example, OH, NH2, COOH,
CH2Br, CHO, CH2CI, CH2SH, SH, V and M, are the same as in Formula I.
The linker group shown in Scheme I is any group that can be reacted with the first
scaffold (CollllJoulld 1) to attach it to a solid support, is stable to the reaction CQI~fl;l;O
nFcesSA~y to complete the ~yrlLLesis, and is easily clca~ablc upon co...l~ ;on of the
synthesis. Suitable lin.~ers are, for example, an OH, NH2, halogen, SH, or COOH group.
An olefin group also is used as a linker. In such case, for example, AG in Collll~vund
1 is CHO and it is AttAr~Pd to the solid support using a Wittig-like reaction. When an
olefin group is used the final product is cleaved from the linker by l~eAI...~ with ozone
or other known mPth~-rlc. A sulfide or oxygen bond is another suitable linker. When a
sulfide or oxygen bond is the desired linker AG in Colllpuund 1 is CH2 halogen,
preferably Br, and the bond between the solid support and Colllpound 1 is formed by
reaction b~lwccn the AG on Collll.oulld 1 and an SH or OH group on the solid support.
Upon completion of the ~ylllllesis a sulfide or oxygen bond linker is cleaved by, for
example, llc~l..-- -l with hydrogenolysis, Raney~ nickel or dissolving metal reductions.
P and P' in Scheme I are protecting groups for aronlalic hydroxy groups. P and
P' can be the same or different to allow for selective deplotec,lion. Choice of P and P'
also is inflllPnretl by compAtihility with the rllr~ to be used in the rPmAinrlPr of the
synthesis. Plcfcllcd protecting groups are C(O)CH3 and Ph-CO whelcin "Ph" is phenyl.
SUBSTITUTE SHEET (RULE 26)
WO 95/04277 216 8 8 8 t~ PCT/US94/07780
- 20 -
Del)~ok.;lion of a C(O)CH3 is p~,.Çol~cd by llca~ -l with an amine according to known
ploce.lul.,s and de~l~Lcclion of a Ph-CO group is accomplished by tlc~ .l with anucleophile such as methoxide using known con~liti-nc and ~locedules.
In Scheme I X' and Y' are groups that allow for ~tt~ hmPnt of the scaffold ringsand illLl~-lucLion of the a~lo~liate M, group. A plcfell d method for joining the rings
is the method of Stille (J. Am. Chem. Soc. (1987) ~2., 5478-5486) wh.,rcill X' and Y'
are an Ol~dllO~ group and a halogen or triflate, lci,~ecli~.,ly. The following is an
example of using this method:
~G (OT~OP ~Op
When col~ ,ullds having more than two scaffold llloieties are desired the
ploce-lule of Scheme I is mof~ifiPd by l~ g the steps needed to add one or more
qrltlitionql scaffolds before cleaving from the solid support. Also, the general plocedurc
shown in SrhPmP I is used when scaffolds other than phenyl rings are used. Thus, any
of the colll~oui~ds inr~ ed in Formula I can be pl~palcd using SrhPmP I m~rlifiPd as may
be l~c~ to a~cc mmo~l?t~ dirL,~ scaffold moieties. Any such nPce~
morlifir~tion~ are appalC;lll to those skilled in the organic chPmirql synthetic arts.
As used in Scheme I FG" is a functional group which may be the same or
different at dirr."c,l~ positions on the compounds. Suitable functional groups are the R,
through R6 groups as defined in Formula I above. Although Scheme I shows pl.,p~dlion
of colllpoullds having two scaffold moieties and four functional groups such compounds
having three r!.nr~;onal groups are ple~cd by using a scaffold having one functional
group in place of Colll~c und 1 or Coll~oulld 4. Also, Co~ oullds 1 and 4 provide for
al~ h...f-.l of functional groups through an oxygen. By suitable replqrPmPnt of these
colll~oul1ds a sulfur atom, a niL~ogci1 atom, or an N-aLkylamide group can be used in place
of one or more of the oxygens. Plocedùlcs for introducing functional groups onto the
scaffolds are inrl~lded in the examples below.
SchPmP II is a mo(1ific~tion of the Scheme I procedure that is used to prepare
compounds wh~,cill the functional group is ~ltt~ hPd to the scaffold moiety using a
SUBSTITUTE SH EET (RULE 26)
W O 95/04277 Z 1~6 8 ~ 8 ~ P~rAUS94/07780
- 2 1 -
(CH2)n.ClO)NR' and n' is O and R' is H or C,~alkyl. In SrhrmP II AG, X', Y', and FG
have the same mF~ g~ as in SrhlomP I.
Scheme II
CN CN CN
(~L;nker . AG~X _ (~Linker~cXN'
~8) (9)
CO2H
Hydrolyze (~Linker~ X HN(CH3)FG
(1 o) CO2H
C(O)N(CH3)FG CN
rl~CN C(O)N(CH3)FG 1
Linker~ X Y~ 1~ CN CN
C(O)N(CH3)FG , (~Linker{ÇI>~M1~
(1 1) l
C(O)N(CH3)FG2
(12)
C(O)N(CH3)FG 1
C, (O)N(CH3)FG3
1. Hydrolyze ~ r ~ rl~
2. Introduce ~Linker~ ~M~
Groups C(O)N(CH3)FG2 C(O)N(CH3)FG4
(1 3)
According to Scheme II a scaffold molecule having two cyano groups ~ rhrd
(Co~ uulld 8) first is ~n~rhPd to a solid support via a linker and then is hydrolyæd to
yield free carboxylic acid groups (Colll~oulld 10). Then, functional groups are ~ hPd
by I with HN(CH3)FG to yield a scaffold with two functional groups (Colllpuulld
11). Next a second scaffold moiety with two cyano groups is a~ rhrd as desclil,ed in
Scheme I followed by addition of functional groups to yield Colll~oulld 13. Compounds
to be included in the libraries of the invention then are ~lcparcd by adjusting the M, group
SUBSTITUTE SHEET (RULE 26)
2l68886
WO 951W~M PCT/US94/07780
as needed. del)lolccl,llg and cleaving Colllpoulld 13 from the solid support as described
in Scheme I.
Scheme III is a variation of Scheme II wh~.ein the scaffold moiety substit~~ent~ are
plolecled prior to addition of the functional groups. In Scheme III X' Y' P, P, and FG
have the same mP.~n;t~g~ as in Scheme I.
Scheme III
C(O)N(CH3)P 1 Deprotect
~ r~ . Functional C(O)N(CH3)
(~Linker~y~x Groups ,l~
( ) ( H3)P 3. Repeat 1 and (~Linker~ X'
(14) 2 i~ Desired
C(O)N(CH3)FG2
(15J
C(O)N(cH3)P
Y' ~
C(O)N(CH3)FG 1
C(O)N(cH3)P ¦ C(O)N(CH3)P 1 Deprotect
(16) ~ ~Linker~ --C~) Groups
C(O)N(CH3)P
C(O)N(CH3)FG2 3. Repeat 1 and
C(O)N(CH3)FG 1
C(O)N(CH3)FG3
--M 1{~)
C(o)N(cH3)FG2 C(O)N(CH3)FG4
(~8)
According to Scheme III Compound 14 is pl~al~d by adding HN(CH3)P or
HN(CH3)P' to the COOH functionalities of Colllpoulld 10 from Scheme II. Compound15 then is pl~ ed by depl.,tc-;~ing dirr~cll~ially if desired and introducing functional
groups onto Compound 14. Compound 16 then is added to Compound 15 using the
p~ucedu~e for ~ rhing scaffold moieties described in Scheme I to yield Compound 17.
SUBSTtTUTE SHEt T (RULE 26)
WO 95/04277 21~ 8 g 8 ~ PCTIUS94/07780
Colll~uulld 18 next is pl~alud by delJlut~ g, dirf~len~ially if desired, and intro~ucin~
functional groups onto Colllpoulld 17. CoLI~uullds included in the invented libraries are
pl~l,a,cd by adjusting the M, group as needed, dep~lceLillg and cleaving Compound 18
from the solid support as des~ ed in Scheme I.
Scheme IV desclibes an ~ t~ method of producing compounds whelcul the
filnrti~n~l groups are linked to the scaffold moieties via a C(O)N(CHl) residue. In
Scheme IV X', Y', P, P', and FG have the same ...~-n;.~ as in .~h~m~ I.
Scheme IV
COOR' 1. Deprotect
rl~ 2. Introduce C(O)N(CH3)FG1
(~ Linker~((~))--XFunctional ~ ~1~
COOR 3. Repeat if Unker~ X
(19) Desired C(O)N(CH3)FG2
(20)
COOR'
I
y, ~O~ C(O)N(CH3)FG1 1. Deprotect
~i~ COOR' 2. Introduce
COOR~ ~ r ~ rl~ Functional
Linker~O~M~ Groups
COOR~ 3. Repeatif
C(O)N(CH3)FG2 Desired
(22)
C(O~N(CH3)FG1
C(O)N(CH3)FG3
} {(~
C(O)N(CH3)FG2 C(O)N(CH3)FG4
(23)
The starting cûlllpoulld in Scheme IV (Colllpound 19) is ~ .al~;d by s~u~dard
prûcedures. Colllpoullds inrl~ ed in the invented libraries are p~alcd by cleaving
Compound 23 from the solid support.
SUBSTITUTE SH EET (RULE 26)
WO 9S/04277 i~ :1 6 8 8 8 6 PCT/US94/07780
- 24 -
,~lt. . .~ rely~ two or more scaffolds are i."l,~ ently d~,~ivali~cd with one or two
functional groups, then are coll,billed in a convergent approach. In a p.~ d method of
this app,oach, two scaffolds are i~ el~Pntly attvq~hPd through a sepd,dle linker to a
se~ e solid support material. The linkers and solid ~uyl~ulL~ can be the same ordirr~.e,l-. The scaffolds can have handles for introducing side chains that are optionally
pro~clcd or dirr~ - d as desrribe(l herein. After the al~rk",l -~l of one or twofunctional groups to each scaffold, one de.ivalizcd scaffold can be cleaved from its solid
support, then re~ rhPd to the other scaffold through an a~pro~,iale coupling reaction.
After any ~l~litir~n-ql desired or needed ~yllLl~elic Lld,~Çc"lllaLions (e.g., side chain
p~l~ u~ g group removal), the fimrtion~li7Pd scaffold(s) is cleaved from the ,c...
solid support to give cûlll~uunds of the h~ cd libraries.
When colll~ûullds having more than two scaffolds are desired, a third scaffold can
be in~Ppenti~Pntly funrtion~li7Pd then coupled in the desired manner to one or both of the
other scaffolds qtt~hP(l to a solid support or a co",bi"aLion of the two ~LlaLcgies can be
employed wL~.~ two scaffolds are ~ fd tog~Lhcl on a solid support in the manner
described in the SchPmPs herein (a linear approach), then a third filnrtionqli7Pd scaffold
derived from a s~ le solid support is ~ rhPd In any case two or more scaffolds can
be sepalalcly Ç~ io~li7P~ in a parallel, ~im--l~ fashion.
The ~i~rlos~P~ invention inrll~es the following Formula V cul~ uul~ds which are
useful as i-~ t~s in ~Ic~alhlg the ill~el,Led libraries and CCIll~u~lS:
Al R'l A3R~3
X. I Yl X,--I--Y2
V~ I ~ M 1 1 ~I W
A2R'2 A4R~4
Formula V
wll~i~cill:
W is H or A R'
s s
X --Y3
M2 1
~Z3
A6R~6
SUBSTITUTE SH EET (RULE 26)
WO 95/W277 ~16 ~ ~ 8 ~ PCT/US94J07780
- 25 -
R'" R'2, R'3, R'~, R'5, R'6 are a plolecLillg group or R" R2, R3, R4, R5, and R6 as
defined in Formula I, provided that at least one of R', to R'6 is a prote~;Lh,g group;
V' is V as defined in Formula I or a bond to a solid support; and
the 1~ g variables are as defined in Formula I.
As used in Formula V, a proLc~;Lillg group is any of the well known plùLecLiLIg
groups that is suitable in view of the synthetic con~iitionc used. P~efe.lcd protc.;LiL,g
groups are C(O)CH3 and Ph-CO.
Pl~ aldLion of lib,d,ies of Formula I compounds is the first step in the ill~e.lL~d
method of pl~aling and selçcting coL.,poullds having phz....~r~ ;r~l or other biologic
utility. After the libraries are p~,pdl` d they are tested in a wide variety of in vitro and
in vivo assays that are predictive of biologic activity and generally involve cont~rting the
cGLIlJuunds with biological targets of interest and dæ~----;--;ng the ~Ll~ Lh of the
interaction ~ ,L~ ,ll the coLul)uullds and the biological target. Such assays are well known
and include, without limh~tion, enzyme inhibition assays, such as protein kinase C and
angiotensin coL~_-ling enzyme"~,c~lor binding assays, such as Se~uLOn~l~ and excitatory
amino acids, ion channel blocking, such as c~lrillm, pot~csil~m and chloride, and
Lldnscli~Lioll factor il~ al~Lion. Generally, any activity identifi~d in vitro is cc nfi....Pd by
evaluation in a suitable animal model if such is available and pr~ .;Livc of human
ph~....~r~ ir~l activity. The examples below include assays that are useful to select
collll,uullds of the invention that have ~h-....~ræ..~ir~l utility.
The col,l~ounds of Formula I that are useful as ph~rm~re~tir~l agents can be
inco,~oldted into convenient dosage unit forms such as caps~ os, tablets, or injectable
ple~dLions. pl.~.".~c,ll;r~l carriers which can be employed include, among others,
syrup, peanut oil, olive oil, and water. Similarly, the carrier or diluent may include any
time delay material, such as glyceryl l,lono~e~ldte or glyc~.yl distearate, alone or with
a wax. The amount of solid carrier will vary widely but, preferably, will be from about
25 mg to about 1 g per dosage unit. If a liquid carrier is used, the L,l~p~lldLion will be in
the fûrm of a syrup, emulsion, soft gelatin capsule, sterile injectable liquid such as an
ampule, or an ~q~leollc or non-aqueous Sl~ -.`;Oll.
ph~rm~reutir~l plel~dLions are made following conventional techniques of a
ph~rTn~reutir~l chPrnict involving mixing, gr~mll~ting, and COlll,ul~;,S~g, when -rcess~y,
SUBSTITUTE SHEET (RULE 26)
wo g5/04277 2 1 6 ~: 8 8 6 PCT/US94/07780
- 26 -
for tablet forms, or mixing, filling, and dissolving the iL~l~die.lLs, as ayplupliaLc~ to give
the desired oral or yal~lllclal end products.
Doses of the ph~ tuPutir~lly useful compounds of the invention will be an
effective amount, that is, an amount l-Pce~cA-y to produce the desired effect without
producing ullLuw~d toxicity selectPd from the range of 0.1-1000 mg/kg of active
couuyouud, preferably 10-100 mg/kg. The selPctPd dose is a~l.nini~t~ .~d to a patient in
need of Lle~ -1 from 1-5 times per day, orally, rectally, by bolus injection, or by
infusion.
EXAMPLES
The following examples illustrate but do not limit the scope of the invention
disclosed in this .cpecifi~tion.
EXAMPLE 1
General P~oce.lules for Formula I Co.l.,Joul.ds Wherein M, is a Bond
Il~t.o(l~ of the phenyl ring onto the solid support: Merrifield resin (5g;
con~;,.;..g 1.42 mmol Cl/g) and the ayyloylialcly sllb~ d stannylated benzyl alcohol
(for general pluce-lules useful in the yll~p~ation of stannylated benzyl alcohols see:
Wynberg and Meijer, Org. React. (1982) 65, 1; Hodgson _ ~ J. Amer. Chem. Soc.
(1929) 1639, and Aziaian, J. Orgmetal. Chem. (1981) 215, 49) (25 mmol) in 60 mL of
dry pyridine was stirred at room ~e~ aLulc for 48h. The polymer was filtered in air,
washed with 4 x 25 mL pyridine, 25 mL ether, washed in a Soxhlet c~L~a~;101 for 8 h with
ether and dried in vacuo.
Removal of the acetate group and illlr~ r of the r....~
group-cQ~ .t side chain: The resin bound material was placed in acetone and 2N
ammonium hydroxide was added and the solution left at room tel.lpclal~lrc for 24 h
(Haslam et ah, J. Chem. Soc. (1964) 2137). The resin was filtered, washed and subjected
to the following general alkylation scheme (Venuti et ah, J. Med. Chem. (1988) 31,
2132). The resin-bound material (8.3 mmol) was placed in a ~ ure of 100mL CHCI3,
50mL MeOH and anhydrous powdered pot~csi-lm carbonate (5.0g, 36.18 mmol) was
added (18-crown-6 can be added if solubility is a problem). The reaction was heated at
SUBSTITUTE SH EET (RULE 26)
W O 95/04277 P~rrUS94/07780
~16888G `
- 27 -
S~C for 15 min, then side chain bromide (9.24 mmol) was added and the Illi~lUlC
refluxed for 4h. After filtration, the residue was washed.
~ Iio..of3~ lo~y-5 ~tu~ henoltriflate: Phloroglucinol(16.2g,0.1
mol) was dissolved/,usl,ellded in H20. The pH was adjusted to 8 with 10% NaOH.
Bromobel-,f -~ (300 mL) was added and NaOH (60 mL) and benzoyl chloride (18 rnL)were added via ~ùppillg funnel tog~ el over 30 min. The reaction was stirred for an
additional 1 hr, filtered, washed with 150 mL bromob~e.~ and then twice with 500 mL
warm water until the filtrate was pH 7. The water wdSll~ngS were back e~ ,d withethyl acetate (2 x 50 mL), dried and ev~olatcd. The co.llbi. cd solids were dried in
vacuo and le,~ lli7P~ from 90mL ethyl acetate and 80 mL hexane to afford 7.7g (33 %
un~7~ cd) of desired product. This material (21.8g, 94.6 mmol) was dissolved in
CH2Clt (220 mL) and triethylamine (66 mL, 0.473 mol) and acetic a~y&ide (26.78 mL,
0.283 mol) was added. The reaction was cooled in an ice bat'n and DMAP (2.31 g)
added. The l~ ul~e was stirred o~ li,hL and allowed to warm to room ~ ,f-~ e
during this period. The reaction was diluted with ethyl acetate, washed witn sat NH4CI,
sat NaCI, back e~ctPd with ethyl acetate, dried, and evapul~lcd. The m~tPri~l
rem~ining was purified by quick pass through silica gel; eluting with hexane/ethyl acetate
4: 1. Obtained 29.59 g of desired product (99 % yield). This l~t~Prj~l (1.0 g, 3.18 mmol)
was dissolved in bc~lLcne (8 mL)/ethanol (10 mL) under argon. Pot~ m hydroxide (178
mg, 3.18 mmol) in ethanol (4 mL) was added dlu~v~i~e over 10 min. The reaction was
stirred for 10 min, diluted with ether and washed with 0.5M H2SO4, sat NaHCO3, sat
NaCI, back extracted with ether, dried and evaporated. The material was purified by
flash chromatography (hexane/ethyl acetate 4:1). Obtained 0.91g, 82% yield of desired
product. This material (5.86g, 21.5 mmol) was dissolved in CH2CI2 and cooled in ice.
Triethylamine (4.5 mL, 1.5 eq) followed by triflic anhydride (3.98 mL, 23.6 mmol) was
added dropwise. To this final ll~ UlC was added DMAP (130 mg, 0.05 eq). After 5 min
stirring the reaction was diluted with 500 mL ethyl acetate, washed with 0.5M H2SO4 (200
mL), sat NaHCO3 (200 mL), sat NaCI, and back extracted with ethyl acetate. Dried,
filtered, and evaporated. Purified via flash chlulllatugraphy (W. C. Still et ah, J. Org.
Chem. (1978) 43, 2923) (hexane/ethyl acetate; 19:1) to afford 6.22 g, 72~ yield of the
desired triflate.
SUBSTITUTE SH EET (RULE 26)
WO 95/04277 216 8 8 8 6 - PCT/US94/07780
- 28 -
Palladium-Catalyzed Cross ~~ Of Aryl Triflates with O~ -o,~ anes:
General l,r~ce-lulc (Saa et ah, J. Org. Chem. (1992) 57, 678 and ibid (1993) 58, 1963).
The phenol triflate (0.5 mmol), anhy-llous LiCI (0.171g, 4.2 mmol),
Lli~he lylpho~hille (0.079g, 0.30 mmol), and PdCI2(PPh3) (0.037g, 0.06 mmol) is
s~lspPnAPd in DMF (4.5mL). The resin-bound o~ n~ n~ was added and a crystal
of inhibitor (2~6-di-tert-butyl-4-ln~ h~nol) was added, and the llli~lUlc was then heated
under an inert a~ .hf .c of argon at 120nC for 2-8h. The resin was filtered and washed.
Removal of acetate group (a), i~tlu~ n of side chain (b), removal of
group (c), ulln~ of side chain (d):
Steps a, b, and d are carried out as described above. Removal of the bf n7o~lc is carried
out as ~csc~il e~ by Bell (Tet. Lett. (1986) 27, 2263). The resin-bound material (0.u~v5
mol) was placed in toluene (20 mL) and n-butylamine (3.65 g, 0.05 mol) was added. The
mixture was stirred at room t~,l~elalulc for 3 h followed by filtration and washing of the
resin.
Removal of all plo~r~ groups: The resin-bound m~tori~l was placed in
CH2CI2 (10 mL) and trifluv~vacctic acid (0.5 mL) added. The ",i~-lu,c was stirred at
room le.~ .alu,e for one hour then the resin filtered and washed.
Removal of the final product from the resin: p~ Aillm (II) acetate (3 mol eq)
was dissolved in warm (40nC) DMF (about 10 mL DMF/g of resin) and the resin added.
After allowing 15 min for resin swelling and catalyst diffusion the reaction is shaken with
hydrogen at a~ Os~h~,licplc~s~ulc. The resin turned black and hydrogen uptake co~ d
for 24 h. The catalyst and resin were removed by filtration to afford the final plo-lucl in
DMF (Sc~l~tt~r et ah, Tet. Lett. (1977) ~ ).
~ ,udtion of Heteroaromatic Systems: The co".,sl,ol,dillg compounds where
the phenyl ring is replaced with a hel~,.u~yclic ring can be ~ylllh~ Pd by the same general
procedures as described above with slight modification of the coupling step as described
below. Each morlifir~tion has strong precedent in the liL~la~ul~. As such, the systems
co-.~ ing all h~,t~,rocyclic rings or a col..bil~Lion of phenyl and h~te.u~yclic rings can be
readily p-.~ d. In general~ the same type of rh~ y that is used to couple two (or
more) phenyl rings can be utilized to prepare the mixed, or pure, he~.ucyclic systems.
For example, Godard _ ah (Tetrah~oAron(l992)48~4l23) have demon~llat~d the
phenyl-pyridyl and pyridyl-pyridyl coupling reactions in good to exrP~Iler~t yields using
SUBSTITUTE SHEET (RULE 26)
Wo 95/04277 ~ 1 6 8 ~ 8` ~ PCT/US94/07780
- 29 -
either the Suzuki-type or Stille-type couplings utilized in the biph~llyl construction,
FulLh~llllore, these authors carry out the reaction on O-aLkyl sl1bs~ Pd substrates in
analogy with our proposed usage.
The following general ~rocedu,c illustrates the coupling of a phenyl ring with apyridine under Suzuki-type conditions: To the phenylboronic acid on resin (3.96 mmol
of boronic acid) in toluene (20 mL) was added a solution of the iodopyridine (3.3 mmol),
p~ m tetraki~(Lli~hellyl)lJho~h~ o (0.115 g, 0.1 mmol), sodiurn calllvnat~ (3 mL aq
2M solution) in 1.7 mL ethyl alcohol and 20 mL toluene. The l~a~;lion was refluxed
under argon for 12 h. The desired products were obtained after filtration and washing of
the resin bound material.
The following procedure illustrates the coupling of pylidille rings with pyridine-like
systems using the Stille l)locedule (taken from Godard et ah): To the
(2-quinolyl)llilll~,Lhyl~ nP (2.4 mmol based on the sl;~n~r) in Aioy~n~ (25 mL) was
added a solution of 2-~yli-lylL~iflate (2 mmol) and LiCI (0.254 g, 6 mmol) in AioY~n~ (25
mL). p~ lm tetrakis(Llil,Lllyl)phosphin~ (69 mg, 3%) was added and the ~ lule
refluxed for 12-72 hr. Filtration and washillg afforded the resin-bound product.Terashima _ ah (Hek_lo~;ycles (1985) 23, 2375) have described the couplin~ of
nitrogen-co-,~ g hct~,lucy~;les with phenyl, pyridyl, thienyl, and furanyl colll~u~ ds
utili7in~ the reaction of a borane with a bromide under p~ illm catalysis. The reaction
occurs when the groups are a~ .h.~A in the 2 and 3-posilions of either ring system and
Ll-~,lefore appears general in nature. The yields are moderate to good.
Finally, a review ~A~t~iling the scope of co~lpling ~t~loc~clic rings together can
be found by Kalinin (Russ. Chem. Rev. (1991) 60, 339).
EXAMPLE 2
Plc~,al.lLion of l-Methvl-2.5'-dietho~y~uanidino-3'-o,~vl,~ lbiphenyl
tion of il~t~,"r.~
~ (Tr.n~lL~ 2-~etu~ l alcohol (A): Plocedul~s used in tne
pl~ ion of stannylated benzyl alcohols: Wynberg and Meijer, Org. React. (1982) 65,
l; Hodgson et ah, J. Amer. Chem. Soc. (1929) 1639, and Aziaian, J. Orgmetal. Chem.
(1981) 215, 49.
SUBSTITUTE SHEET (RULE 26)
W O 95/04277 ~16 8 8 8 ~ I~CTrUS94/07780
- 30 -
(2-N-PMC-guanidino)-(1-methanesulfonyl)eth~rol (B): Ethanolarnine (10.Og,
0.163 mol) was dissolved in CH2CI2 (250 mL) and imitl~701e (24.41g, 0.358 mol) was
added. The reaction was cooled to OoC and TBDMS (27.14g, 0.18 mol) was added. The
mixture was stirred at OnC for two hours then room tcn~eld~u~c for an additional two
hours. Ethyl acetate (500mL) was added and the mixture washed with O.5M H2SO4
(400mL), sat'd NaHCO3 (400mL) and sat'd NaCI (400mL), dried, e~d~GldLtd and the
resulting material (12.0g, 42% yield) used as is. Form~mi~inPs~llfonic acid (1.Og, 8.05
mmol; Tet. Lett. (1988) 29, 3183) and the above material (1.41g, 8.05 mmol) weredissolved in dry ~ A~ I (lOmL) and stirred for 2h at room ~ .alule. The solvent
was removed in vacuo and the product dissolved in acetone (27mL), water (7mL) and
NaOH (lOmL, 3.2M) added. The reaction was cooled to O~C and PMCCI (3.66g, Raylo
ChP~nir~lc, Alberta, Canada) was added in acetone (8mL). After stirring for lh at OnC
the reaction was diluted with ethyl acetate, washed one time each with 25mL sat'd NH,CI,
water, and sat'd NaCl, dried and ~va~olated. The product was purified by flash
chromatography (silica, hexane/ethyl acetate 1:1) to afford 1.71g (46%) of desired
product.
The product (0.57g, 1.23 mmol) was dissolved in THF (lOmL), cooled to O~C and
tetrabutyl~.. o-~ .. noride (371mg, 1.42 mmol) added. After 30 min the reaction was
worked up by diluting with ethyl acetate, wa~ g one time each with 25mL sat'd NH~CI,
water, and sat'd NaCl, dried and evaporated. The product was purified by flash
cl~ullla~ography (silica, CH2Cl2/m~oth~n~ l; 19:1) to afford 0.43g (94%) of desired product.
This material (64mg, 0.186 mmol) was dissolved in CH2CI2 (2mL), cooled to O C and
DMAP added (2.2mg). ~elh~,~P~--Ifonyl chloride (35.6mg, 0.204 mmol) was added and
reaction was complete after 20 min. Eval)uldlion of the llli~Luie was followed by
purification (silica, CH2Cl2) to afford 95mg (92% yield) of desired product.
3-BenzyloA~-5-acetoAy~ enoltriflate (C): Phloroglucinol (16.2g, 0.1 mol) was
dissolved/sl-spen-iPd in H20. The pH was adjusted to 8 with 10% NaOH. Bromobel.7ene
(300 mL) was added and NaOH (60 mL) and benzoyl chloride (18 mL) were added via
.llu~illg funnel together over 30 min. The reaction was stirred for an additional 1 hr,
filtered, washed with 150 mL bromobel~e"e and then twice with 500 mL warm water
until the filtrate was pH 7. The water washings were back extracted with ethyl acetate
(2 x 50 mL), dried and evaporated. The combined solids were dried in vacuo and
SUBSTITUTE SHEET (RULE 26)
W O 95/04277 ~ 1 S ~ 8 8 G~ ~ P~CTrUS94/07780
le~ 7P~l from 90mL ethyl acetate and 80 mL hexane to afford 7.7g (33 %
u~ ed) of desired product. This material (21.8g, 94.6 mmol) was dissolved inCH2Cl2 (220 mL) and triethylamine (66 mL, 0.473 mol) and acetic al~d.ide (26.78 mL,
0.283 mol) was added. The reaction was cooled in an ice bath and DMAP (2.31 g)
added. The mixture was stirred ove.lligllL and allowed to warm to room tclllpc~alulc
during this period. The reaction was diluted with ethyl acetate, washed with sat NH~Cl,
sat NaCl, back extracted with ethyl acetate, dried, and e~dyoldt~ d. The material
rem~ining was purified by quick pass through silica gel; eluting with hexanelethyl acetate
4: 1. Obtained 29.6 g of desired product (99% yield). This material (1.0 g, 3.18 mmol)
was dissolved in b~ Lelle (8 mL)/ethanol (10 mL) under argon. Pul ~c~ .. hydroxide (178
mg, 3.18 mmol) in ethanol (4 mL) was added dLuywi~e over 10 min. The reaction was
stirred for 10 min, diluted with ether and washed with 0.5M H2SO" sat NaHCO3, sat
NaCl, back extracted with ether, dried and e~ayolaLed. The material was purified by
flash c~malography (hexanelethyl acetate 4:1). Obtained 0.91g, 82% yield of desired
product. This material (5.86g, 21.5 mmol) was dissolved in CH2Cl2 and cooled in ice.
Triethylamine (4.5 mL, 1.5 eq) followed by triflic a~ d.ide (3.98 mL, 23.6 mmol) was
added dropwise. To this final llli~Lul~ was added DMAP (130 mg, 0.05 eq). After S min
stirring the reaction was diluted with 500 rnL ethyl acetate, washed with 0.5M H2SO~ (200
mL), sat NaHCO3 (200 mL), sat NaCl, and back çlrtracted with ethyl acetate. Dried,
filtered~ and e~apo,dted. Purified via flash chromatography (hexanelethyl acetate; 19:1)
to afford 6.22 g, 72% yield of the desired triflate.
N-t-Boc-1-hro--~o~ yl~lc (D): Goel and Beylin, (Org. Prep. Proc. Int. (1987)
9, 78).
Y~paldtion of Final ~oducl:
Illkoil~- I ;nn of the phenyl ring (A) onto the solid support: Merrifield resin (Sg;
cont~ining 1.42 mmol Cl/g) and stannylated benzyl alcohol (A, 25 mmol) in 60 mL of dry
pyridine was stirred at room l~llly~dLult for 48h. The polymer was filtered in air,
washed with 4 x 25 mL pyridine, 25 mL ether, washed in a Soxhlet extractor for 8 h with
ether and dried in vacuo.
Removal of the acetate group and illllo~lu~ n of the f m~innql
group-co-~ g side chain (B): The resin bound material was placed in acetone and
SUBSTITUTE SHEET (RULE 26)
~1~888~ ,
WO 95/04277 PCT/US94/07780
2N ~mmonillm hydroxide was added and the solution left at room ~ulpe.atule for 24 h
(Haslam et ak, J. Chem. Soc. (1964) 2137). The resin was filtered, washed. The
resin-bound material (8.3 mmol) was placed in a llliAlulc of 100mL CHCl3, 50mL MeOH
and anhy-lruus powdeled potassium carbonate (5.0g, 36.18 mmol) was added (18-crown-6
can be added if solubility is a plobleul). The reaction was heated at 50 C for 15 min,
then side chain mesylate (B, 9.24 mrnol) was added and the llliA~UlC; refluxed for 4h.
After filtration, the residue was washed.
Palladium-Calal,~L~ Cross Col~pling.c of Aryl Triflate (C) with Resin-Bound
A: The phenoltriflate (0.5 mmol), anllydluus LiCl (0.171g, 4.2 mmol),
triphenylphns~h~ (0.079g, 0.30 mmol), and PdC12(PPh3) (0.037g, 0.06 mmol) is
s~cr~PnrlPd in DMF (4.5mL). The resin-bound org~n~ was added and a crystal
of inhibitor (2,6-di-tert-butyl-4-1llcLllyl,uhenol) was added, and the llliALul~ was then heated
under an inert atmnsphPre of argon at 120nC for 2-8h. The resin was filtered and washed.
Removal of acetate group (step a), introduction of side chain (B) (step b),
r~."u~al of he-- n~'e group (c), ulln~ I;n-- of side chain (D)(step d):
Steps a, b, and d are carried out as described above with step d using side chain bromide
(D). Removal of the be~.,o~ç is carried out as descliLed by Bell (Tet. Lett. (1986) 27,
2263). The resin-bound material (0.005 mol) was placed in toluene (20 mL) and
n-butylamine (3.65 g, 0.05 mol) was added. The llliA~Ul~; was stirred at room telll~ldLulc
for 3 h followed by filtration and washing of the resin.
Removal of all prote~tin~ ~;lUU~IS The resin-bound material was placed in
CH2Cl2 (10 mL) and trifluurûac~lic acid (0.5 mL) added. The lniAIul~ was stirred at room
~,n~eldLure for one hour then resin filtered and washed.
Removal of the final ~l o~lu~l from the resin: p~ lm (II) acetate (3 mol eq)
was dissolved in warTn (40OC) DMF (about 10 mL DMF/g of resin) and the resin added.
After allowing 15 min for resin swelling and catalyst diffusion the reaction is shaken with
hydrogen at atmo~h~,lic plC~Ul~. The resin turned black and hydrogen uptake continued
for 24 h. The catalyst and resin were removed by filtration to afford the final product in
DMF (Srhl~tter et al., Tet. Lett. (1977) 2851).
SUBSTITUTE SHEET (RULE 26)
wo 95~w277 ~ 88~ PCT/US94/07780
EXAMPLE 3
Pn,~Jalation of 2'~4.5'-Trimethoxybiphenyl-4'-carboxvlic Acid
~"~.~tio.. of i~ ,...r li-~,y
~ (Tri~ lh~l~l~llyl)-2,5-~1;r-rPtc~ylJ~.~oicacid (A): This co"l~uul1d is pl~;pal~d
from 4-bromo-2,5-~;~rrtol~yl~oic acid whose plelJalalion is described in Kamil et ah,
Pak. J. Sci. Ind. Res. (1971) 14, 59, by conversion of the bromo to the coll~ulld~g
Lfi~ethyl~ following the general p~uce.lul~ of Aziaian (J. Orgmetal. Chem. (1981)
215, 49).
~ MelhoAyphenol triflate (B): 4-Metho~y~henol (Aldrich, 1.24g, 10.0 mmol)
was dissolved in CH2CI2 and cooled in ice. Triethylamine (2.1 mL, 1.5 eq) followed by
triflic anhydride (0.98 mL, 11.0 mmol) was added dlu~,~ise. To this final l~ u~e was
added DMAP (60 mg, 0.05 eq). After 5 min stirring the reaction was diluted with 50 mL
ethyl acetate, washed with 0.5M H2SO, (20 mL), sat NaHCO3 (20 mL), sat NaCl, and back
e~raete~l with ethyl acetate. Dried, filtered, and e~àpoldt~d. Purified via flash
cl~omatography (hexane/ethyl acetate; 19:1) to afford 2.38 g (93%) yield of the desired
triflate.
~ ation of the Final ~ udu.l;
Illtrû~ of the phenyl ring (A) onto the solid support: Merrifield resin (Sg;co..~ g 1.42 mmol Cl/g) and stannylated benzoic acid (A, 25 mmol) were reacted as
described by Merrifield (J. Amer. Chem. Soc. (1963) 85, 2149). The polymer was filtered
in air, washed with 4 x 25 mL yylidille~ 25 mL ether, washed in a Soxhlet e~ acLor for
8 h with ether and dried in vacuo.
Removal of the acetate group and illlrollu~lion of the methyl ~. Ull~s: The resin
bound material was placed in acetone and 2N ~",non,u,n hydroxide was added and the
solution left at room temperature for 24 h (Haslam et ah J. Chem. Soc. (1964) 2137).
The resin was filtered, washed. The resin-bound material (8.3 mmol) was exhaustively
methylated with excess dimethylsulfate (5.23 g, 41.5 mmol; Org Synth. Coll. (1941) Vol.
I, 58). After filtration, the residue was washed.
p~ m-catalyzed Cross Couplings of Aryl Triflate (B) with Resin-Bound
A: The phenoltriflate (0.5 mmol), al~,ydrùus LiCl (0.171g, 4.2 mmol),
triphenylphosphine (0.079g, 0.30 mmol), and PdC12(PPh3) (0.037g, 0.06 mmol) is
SUBSTITUTE SHEET (RULE 26)
wo 95/04277 ~ ~ 6 8 8 8 6 PCT/US94/07780
sllsp~Pd in DMF (4.5mL) . The resin-bound ol~,~nnsL;1nn~nP was added and a crystal
of inhibitor (2,6-di-tert-butyl~-methylphenol) was added, and the mixture was then heated
under an inert ~tm-)srhP~e of argon at 120~C for 2-8h. The resin was filtered and washed.
Removal of the final product from the resin: The s~nd~ Merrifi~ltl procedure
was employed (Merrifield, J. Amer. Chem. Soc. (1963) 85, 2149). The catalyst and resin
were removed by filtration to afford the final product.
EXAMPLE 4
General P~ocedu,es for P~wal~lion of Formula I
Collll)oullds Wherein M, is CH.. CH.CH.. CH =CH. or C e C
SchPmPs V and Va and the plucedul~,s which follow describe procedures for
making the title colllpoullds.
[This space int~-lti-)n~lly left blank]
SUBSTITUTE SHEET (RULE 26)
WO 95/04277 ~ 1 6 9 8 ~ PCT/US94/07780
- 35 -
Scheme V
OAc OAc
(~ SH + Bt~ Br -- (~ 5~ 3~ Bt
3~ ) 03Z OBZ
(32) (33)
1. RNH2 AcO
2. Br~CH2~n FGlP 5~ l0~2)nFGlP ç~ H (35)
4. B-(CH2)nFG2P (34) ( 2)n Ph3P, Pd(ll~(OAC)2
O(CH2)nFG ~P OAc 1. RNH2
~ ~,3 2. Br(CH2~n FG3P
O(CH2~nFG2? 03Z 3. R'NH2
(36) 4. Br(CH2~FG~P
(CH2)nFGlP iO(CH2)nFG3? O(CH2)nFGl
(~ 5~ 31--; ~ ~ ~ 2 hv CH3~ o(CH2)nFG3
O(CH2)nFG2? O(CH2)nFG'P O(CH2)nFG2 (CH2)nFG'~
(37) (38)
H2
l Lindlar ca:alyst
(C ~ so(cH2)nFG3p 1 H~ ~
O(CH2)nFG2P O(CH2)nFG'P O(CH2)nFG2 o(CH2)nFG4
¦ H2, Pd-C
f(CH2)nFGlp o(cH2)nFG3p (CH2)nFGl o(CH2)nFG3
(~ 5~1~ 1 H~ CH ~13~,~
O(CH2)nFG2? 0(CH2)nFG~p O(CH2)nFG2 o(CH2)nFG4
(41) (~2)
SUBSTiiTUTE SHEET (RULE 26)
W O 95/04277 ~16888 6 }~CTrUS94/07780
- 36 -
Scheme Va
OAc
SH + Br~ ~ SnMe3 --_ OAc
OBz
(53)
(54)
1. RNH2 Br
2. Br(CH2~ FGlP O(CH2)nFGlP r
3. R NH2 ~ S~ 3--SnMe3 BzOJ~ OAc (56)
4. 8r(CH2~FG 2p
O(CH2)nFG2P 'Pd-
(55)
O(CH2)~F OAc 1 RNH2
3. R'NH2
O(CH2)nFG2P 4. Br(CH2~FG~P
(57)
O(CH2)nFG 'P
O(CH2)nFG3P
(CH2)nFG~P
O(CH2)nFG2P
(58) IO~C~ G~ O(CH2) FG3
O(CH2)nFG2 O(,CH2)nFG~
(59)
SUBSTITUTE SHEET (RULE 26)
WO 9~/04277 216 8 8 8 ~ PCT/US94/07780
- 37 -
~dion of resin, 31.
2-Methoxy methyl phenylacetate was prepa.~,d by leflw~"~g 2-methoxy
phenylacetate (10.2 g, 61.3 mmole), 70 mL of a,lhydluLls "\~.hA~ol and 1.5 mL ofconc."llldted sulfuric acid for 17 hours. The solvent was removed and the oil was
dissolved in 100 mL of diethyl ether, washed with saturated NaHCO3, dried, filtered and
cva~old~d to give 9.26 grams (83%) of 2-methoxymethyl phenylacetate. This m~t~ori~l
(10.0 g, 55.0 mmole) dissolved in 6 mL of tetrachlo,oe~.ane was added over a period of
25 min-ltf~s ( making sure that the tellli)cldlulc of the reaction ~ lurce did not exceed
50nC) to AlCI3 (15 g, 112 mmole) in 50 mL of tetrachloroethane to which was added
2-~rullloplul)iollyl chloride (5.7 mL, 56.5 mmole) and the llliAIUl~ heated at 45nC for 20
mimlt~S The .~aclion was allowed to stir at 50nC for 5 hours then at room t~ ,ldLu~e
for 10 hours, poured onto 150 mL ice and 0.5 mL of conce,.l.dled HCl was added. The
llli~lUlC was e~Lldcled with CH2Cl2 and the organic layer washed with 10% NaOH, and
H20, dried, filtered and e~,~o,dtcd to give a dark purple-red oil which was purified by
flash chlu,,,atography (SiO2, first with 50% hexane-CH2CI2 then with CH2CI2) to afford
11.6 grams (66% yield) of methyl [3-(2-~u--,opn,pionyl)-6-metho~yl,h~ l]acetate as a
thick oil: Rf= 0.44 (SiO2, CH2CI2). This material (11.4 g, 36.3 mmole) was dissolved in
70 mL of acetone and 15 mL of conc~,llLIatcd HCI, 20 mL of H.O were added and the
r~slllting solution refluxed for 6 hours. The volatiles were removed to give an oil/water
which was then dissolved in 100 mL of CH2CI2. The mixture was extracted with
150 mL of saLu,dted NaHCO3. The aqueous was then removed and acidified with
co"ce--L-dted HCI to a pH = 1. The ~queous llli~ lC was then quickly extracted with 100
mL of CH2Cl2. The organic layer was then dried, filtered, and evdpoldled to give 6.0
g (64%) of as a white solid: Rf = 0.6 (SiO2, 10% m~th~nol-CH2Cl2). To the sodium salt
of 2-methyl-2-plol,dlkLhiol (0.9 g, 8.82 mmole) was added sodium hydride (0.25g, 11.3
mmole) and 15 mL of anhydrous tetrahydruruldn and mixture cooled to 0nC. To th
mi~lul~ was added the above 3-[(2-chlo,ol"upionyl)-6-methoxyphenyl]acetate (1.5g, 5.84
mmole) dissolved in 15 mL of anhydrous THF over a period of 10 mimltf C. After
addition, the reaction Illib~lul~ was stirred, at room l~ eldlul~ under nitrogen for 18
hours, the volatile components removed and dissolved in 80 mL of H2O and the aqueous
washed with 100 mL of diethyl ether. The aqueous layer was then acidified with 1 mL
of conce,lL~dted HCI (pH=1) and extracted with ether. The organic layers were
SUBSTITUTE SHEET (RULE 26)
WO 95lW277 - - PCT/US94/07780
21588~6
- 38 -
combined, dried, filtered, and evaporated to give 1.75 grams of the t-butyl thioether
product which was used without any further purification. To 1.75 g (5.64 mmole) of this
material was added 2 mL of DMF, 4 mL of concellllated acetic acid, and 1 mL of H20.
To the solution was then added 2-llillobel~7~ ~f ~.~lfenyl chloride (1.6 g, 8.44 mrnole) then
stirred for 24 hours. The volatile COlll~)OllC~ were removed under reduced pres~ to
give an oil/water mixture. To the mixture was then added 15 mL of H2O, cooled torl~illg, and then lyophilized overnight. After lyophili7~tion, the l~.n~ ;n~ solid was
taken up in CH2Cl2 and purified by flash chromatography (SiO2, first with CH2Cl2, then
with 50% diethyl et_er-CH2CI2 and then with 10% mPth~nr)l-CH2Cl2) to isolate a yellow
oil which cryst~lli7Pd upon 5li~i,rli~g to give 1.41 grams (59%) of a yellow crystalline
solid: Rf- 0.37(SiO2,10%~ h~ l-CH2CI2). CouplingofthismaterialtoTentaGel~
resin was ~compli.~hP-cl by placing TentaGel~ (3.0g, 0.87 mmole of amine), 50 mLCH2CL2 and 1 mL DIEA (6.46 mmole) in a peptide syl~ ;.is vessel and the llli~lUlc
shaken for 5 ...;..~lles followed by washing with CH2Cl2. To this was added 20 mL CH2Cl2
followedby3-[2-~(2-nil,o~h~ l)dithio]~lu~iollyl]-6-metho~yl~h~ e~ fromabove
(0.9 g, 2.2 mmole) dissolved in 30 mL of CH2Cl2 and llPi~lul~ shaken for 30 seconds. To
the lllib~lUle was added (0.3 mL, 2.1 m_ole) diiso~lop~lcabo~;i-ni~le (DIC) and llli~lUl~
shaken for 7 hours, filtered, washed with CH2Cl2, ~ l, and CH2CI2. The resin was
then placed under pump vacuum for several hours to give 3.2 grams of the final resin
material as a yellow solid. The amount of r~ lfitie on the resin was dt~ ed by am~ifiP~ Ellman ~I,e~;llupholûlll~llic assay at 490 nm (0.18 mmole of lliC~Ifi-l~P/g of resin).
I~tr~duclion of the mono- or di-oxygen substituted Ib;~ l bromides 32,
or mono- or di-oxygen ~ le~ yl~ l benzyl bromides 53, onto the resin
to give 33 or 54: The mono- or di-oxygen ~.lh~ r~ bromobenzyl bromides, 32 can be
pl~pal~d by mPthorlc well known to those skilled in the art . For an example see Example
S below. The llilneLllylstannyl group can be introduced if needed to afford 53, by reacting
the ppnnltim~te ;-.l~ .",P~ to bromobellze,1e 32, the tolyl coll.~.~und, (8.78 mmol) with
Pd(PPh3)4 (71 mg, 0.061 mmol) in toluene (8.8 mL) to which was added hP~mPthylditin
(5 g, 15.26 mmol). The reaction was heated to 120OC for l.5h which after work-up and
purification afforded the desired product. Conversion to the benzylbromide occurs with
NBS under standard co~litiQn~.
SUBSTITUTE SHEET (RULE 26)
W O 95/04277 ~ 3 8 ~ ~CTrUS94/07780
- 39 -
To the above resin, 31 (2.3 g, 0.43 mmole) was added 150 mL DMF,
"lc..;d~l()eth~nnl (0.25 mL, 3.5 mmole) and diiso~ )ylethylamine (0.4 mL, 2.3 mmole)
and the lllL~Ullc shaken for 2-3 ..~ s, filtered, and the process repeated two more times
using the same qu~ntiti~ of BME and DIEA. The resin was then washed five times with
DMF, three times with mPth~nnl, four times with CH2Cl2 and then three times with DMF.
To the resin was then added 32 or 53 (1.21 mmole) dissolved in lS mL DMF and DIEA
(0.5 mL, 2.87 mmole) added and the mixture shaken for 6.5 hours, filtered, and washed
five times with DMF, three times with mPth~nnl, and six times with CH2CI2. The resin
was then dried under pump vacuum to give 33 or 54.
Removal of acetate group, illt~v.l~- I c~ of side chain, removal of l~ c-~e
group, and i~ vJ~ of side chain to give 34 or 55: The resin bound material
(46.0g, 8.3 mmol) was placed in acetone (300 mL) and excess 2N ~mmonillm hydroxide
was added and the solution left at room t~ c.alule for 24 h (Haslam et ah, J. Chem.
Soc., 2137 (1964)). The resin was filtered, washed, and subjected to the following
general alkylation scheme (Venuti _ ah, J. Med. Chem. 31, 2132 (1988)):
The ~ Uil~d bromides are either collllllcl.;ially available as needed, or as thebromide having side chains which require plotecl;vl~ with s~d~d acid labile plotccLillg
groups. ~ ivcly the alcohols are available which require co~c.~ion to the
cGlle;,~ondillg bromides or mesylates by .... ll,n 15 known to those skilled in the art. In
several cases the materials were ~ alcd by a several step synthetic sequenre as dcsclibcd
in the specific exarnple below.
The resin-bound material (46.0g, 8.3 mmol) was placed in a llliALLne of 300mL
CHCl3, 150mL MeOH and anhydruus powdered polas~iulll carl,ol~le (5.0g, 36.18 mmol)
was added (18-crown-6 can be added if solubility is a problem). The reaction was heated
at S~C for 15 min, then side chain bromide (9.24 mmol) was added and the llliA~UlC
refluxed for 4h. After filtration, the residue was washed.
Removal of the ben70~te is carried out as described by Bell (Tet. Lett., 27, 2263
(1986)). The resin-bound material (46.0g, 8.3 mmol) was placed in toluene (300 mL) and
n-butylamine (3.65 g, 50 mmol) was added. The mixture was stirred at room tc~ clalulc
for 3 h followed by filtration and washing of the resin.
For introduction of the second functional group, the resin-bound material (46.0g,
8.3 mmol) was placed in a mixture of 300mL CHC13, 150mL MeOH and al~hydrous
SUBSTITUTE SHEET (RULE 26)
W O 95104277 ~ 1 6 8 8 8 6 PCTrUS94/07780
- 40 -
powdered pot~sillm callJonàte (5.0g, 36.18 mmol) was added (18-crown-6 can be added
if solubility is a problem). The reaction was heated at 50OC for 15 min, then side chain
bromide (9.24 mmol) was added and the mixture refluxed for 4h. After filtration, the
residue was washed.
EXAMPLE 4a
Plc~)alaLion of Formula I Coll"~ounds Wherein M, is C--C
Preparation of the substituted IJh~ c~ylelle 3~ and C~ iOI~ of
34 to 36: The substitl~tecl phenylacetylene 35 was ~lcpalcd from the coll~s~onding
iodobel~l~ collll)oulld by the general pl~,cedulc described by Lau et ah descflbed below
(J. Org. Chem., 1981, 46, 2280).
The dir~.~lllially ~loLccL~d dihydlv~LyiodobenJ~.-r was ~lc~ d from the
~lim~thoxyaniline (Aldrich) by didzoLi~aLion and iodine h~ luclion followed by
demethylation of the methoxy groups all under sL~ldald co~ n~. Dirr~lcll~ial
protection was accomplished from the ioc~oE-h~nnl (16.0 mmol) which was dissolved in
CH2CI2 (30mL). Triethylamine (11.15mL, 80 mmol), acetic anhydride (4.55 mL, 48
mmol) and DMAP (390 mg, 3.2 mmol) were added and the reaction stirred for 16 h. The
reaction was e ~a~Olat~d to dryness and this m~ l (12.8 mmol) was dissolved in a mix
of ethanol (32 mL) and b~ (16 mL). POIassi~lll, hydroxide (0.72g, 12.8 mmol) was
dissolved in 8 mL ethanol and added over 30 min. After 30 min the reaction was diluted
with ether, washed, dried, and e~à~ola~d. The monoacet~t~ was dissolved in CH2CI2
(50 mL) and triethylamine (3.45 mL, 24.8 mmol), DMAP (0.3g, 2.5 mmol) and benzoyl
chloride (1.8 mL, 15.5 mmol) were added. The reaction was complete in 10 min then
diluted with CH2Cl2, washed, and dried to afford the monoacetoxy
monobenzoyloxyiodob~clle. This material (3 mmol) was placed together with
TMS-acetylene (0.47g, 4.8 mmol), Pd(II)acetate (10 mg), and triphenylphosphine (20 mg)
in dry triethylamine (5 mL). The mixture was heated at reflux for 4 h, cooled, the solid
removed by filtration, the filtrate concc.lllatcd, mixed with sodium bicarbonate (20 mL),
extracted with CH2Cl2 dried, and collcellLIa~cd to afford the TMS-phenylacetylene. This
material (2 mmol) was co,,~clkd to the free acetylene by dissolving in THF (8 mL) and
adding tetrabutyl ammonium fluoride (3 mL of lM in THF) and stirring for 3h at room
SUBSTITUTE SHEET (RULE 26)
WO 95/W277 ~ 1 ~ B~86 PCT/US94/07780
- 41 -
le,l~p~.aLu,e. After ~ldald workup the desired substituted phenylacetylene 35 was
obtained.
The resin bound bromobcnzcl~ 34 (16.5g, 3 mmol) was sll~pPn-lPd in DMF (30
mL) and triethylamine (6 mL) added along with the acetylene 35 (8 mmol), Pd(II)acetate
(20 mg), and ~ hcl~ylphosphine (40 mg) in dry triethylamine (5 mL). The mixture was
heated at reflux for 4 h, cooled, filtered, and washed in the ~ldnddld fashion to afford 36.
EXAMPLE 4b
Pl~paldlion of Formula I Compounds Wherein M, is CH~
~ ioll of benzyll)ro~ e 56 and con~;.io~. of 5S to 57: The ~-~bs~
benzylbromide 56 was prepared from the corresponding
monoacetoxy-monobel~yloxytoluenebyreactionwithN-b,ù.no~,~r~ Punder~ da,d
conditions. The mo~oacetoxy-monobenzyloxy toluene was in turn prepared from the
co,,.,s~ùllding dihydroxytoluene by the same protection scheme used to prepare the
phenylacetylene 35, above.
For the conversion of 55 to 57, the general pl~cedwl; of Milstein and Stille (JACS
(1979) 101, 4992) was employed. The resin-bound 55 (5.5g, 1 mmol; p,e~d as
described above) was s,l~l.e~ded in lOmL hF ~ hyll~ho~h. ~ . To this was added
benzylchlorobis(L~i~hc"yl-FhosphinP)-pq~ lm (II) (0.05 mmol) and the benzylbromide
56 (5 mmol). The reaction was heated to 65~C for lOh, cooled, filtered and washed in
the usual fashion.
Completion of 38, 40, 42, 59.
Removal of acetate group, introA-~ti~n of side chain, rc~,.o~al of ~r~YOnl~
group, and i~ltrOJIJ l;-~n of side chain to afford 37 or 58: The resin bound material
(4.6g 0.83 mmol) was placed in acetone (20 mL) and excess 2N ammonium hydroxide
was added and the solution left at room l~l"~c.ature for 24 h (Haslam et ah, J. Chem.
Soc., 2137 (1964)). The resin was filtered, washed, and subjected to the following
general alkylation scheme (Venuti et ah, J. Med. Chem. 31, 2132 (1988)):
The required bromides are either col"",clcially available or as the bromide having
side chains which require protection with ~da,-l acid labile l~uLecling groups.
~ltern~tively the alcohols are available which require conversion to the corresponding
SUBSTITUTE SH EET (RULE 26)
W O 95/04277 ~16 8 8 8 ~ P~CTrUS94/07780
- 42
bromides or mesylates by mlotho-1c known to those skilled in the art. In several cases the
bromides or mesylates were plepalcd via several step synthetic procedures such as that
described in the specific example below.
The resin-bound material (4.6g, 0.83 mmol) was placed in a llli~lule of 20mL
CHCl3, 10mL MeOH and ar~dluus powdered potassium carbonate (3.6 mmol) was added
(18-crown-6 can be added if solubility is a problem). The reaction was heated at 50nC for
15 min, then side chain bromide (0.92 mmol) was added and the llPL~lUlC refluxed for 4h.
After filtration, the residue was washed.
Removal of the b~ '7o~r~ is carried out as described by Bell (Tet. Lett., 27, 2263
(1986)). The resin-bound material (4.6g, 0.83 mmol) was placed in toluene (lOmL) and
n-butylamine (5.0 mmol) was added. The mi~ulc was stirred at room k~ elaLulc for3 h followed by filtration and washing of the resin.
For ulLlùduLlion of the second functional group, the resin-bound material (4.6g,0.83 mmol) was placed in a mixture of 20mL CHCI3, 10mL MeOH and anil~uu~
powdered pot~ccil~m call,onate (3.6 mmol) was added (18-crown-6 can be added if
solubility is a l~ro~ ). The Icaclioll was heated at 50nC for 15 min, then side chain
bromide (0.92 mmol) was added and the llli~lUl~ l~llu~ed for 4h. After filtration, the
residue was washed.
12e~ of the ace ~le.le 37 to the oleffn 39: This selective re~lction of the
acetylene to the coll~i~ondillg olefin is accomplished with Lindlar catalyst plcp~d as
described in Org. Syn. Coll., Vol. V, 880.
To the resin-bound 37 (5.5g, 1 mmol) in 10 mL hexane was added 10mg of
Lindlar catalyst and 50mL of quinoline. The reaction vessel is evacuated and placed
under a slight positive pl~ iUlC of hydrogen gas for 3 hours. filtered, and washed to
afford 39.
l2P~ rtion of thé olefin 39 to the ell.yl~..c allalo~.-e 41: To the resin-bound 39
(5.5g, 1 mmol) in 10 rnL ethyl acetate was placed 50mg of Pd(OAc)2 and the reaction was
~ubjcclcd to a pO~ilivc pl~ ule of hydrogen gas for 6 h. The mixture was filtered and
washed in the s~dar~ fashion to obtain 41.
Remo~al of all ~ ot~l;..g groups from 37, 39, 41, and 58: The resin-bound
material (11.0g, 2mmol) was placed in CH2CI (100 mL) and triflouloact~ic acid (0.5 mL)
~ SUBSTITUTE SHEET (RULE 26)
W O 95/04277 ~ 1 ~ g 8 8 6 }~CTrUS94/07780
- 43
added. The ~ ulc was stirred at room te~ ulc for one hour then the resin filtered
and washed.
Removal of the final product from the resin to give 38, 40, 42, and S9: The
resin (3.0 g, 0.6 mmol ) s~cpen~1ed in 25 mL of ace~ollillile. The stirred l~ lulc was
irradiated under nil~ogell atmosphere using a Rayonet photorh~ iral reactor (Con~
of sixteen black light pho~l,hor bulbs having a m~rim--m wavcle~ lte~ily at 350 nm)
for 4 hours. After irra~ ion, the mixture was filtered to afford the desired products 38,
40, 42 and 59 in solution.
EXAMPLE S
Plepdl~lion of l-r4-Methyl-3-etho~Y~ ;..v~henyll-2
r3'-etho~Yb~ l-S'-etho,~y~ ni~ o~ "llethane (70)
and (4-Methvl-3-etho,~,Y~u~..;-i;.~. ph~.lyl)-(3'-etho~cybcl~l-
5'-ethu~y~ ;-.o~)h~.,yl),.,.,~ (87)
SrllPmP VI, SellrmP VIa, and the proce-lu.es that follow describe pl~p~lion of
the title COlllp~ unds.
[This space intentionally left blank]
SUBSTITUTE SHEET (RULE 26)
~16888~
W O 95/04277 }~CTAUS94/07780
- 44
Scheme ~
OAc OAc
(~ SH + 8r~_b--Br -- (~ S b_ 8r
(31) (60) (61)
H~H
1. NH,OH O~ CH2)2N NHPMC
NH (~ S~ E~ BzO~GL 163)
2. MsO(CH2k-NH NHPMC ~62)
Ph3P, Pd(ll)(OAC)2
NH
0~2NH NH?t~C OAc 1 NH,OH ~
S~} ~ NH
OBz 3. BuNH2 Jl~
(6~) ~. M~o(cH2)2NH NH?MC
CH2)2NH~ NHPMC O(CI12)2~ H2)2NH~ NH2 (CHz)
(~S~--~ NH 2 hv CH3~8 ~P
(65) O(CH2)2~ NH?MC (r,6) O(CH2)2NH ~ NH2
NH
H2
Lindhr catalyst
(~ s$~ ~ h'H 2 hv ~ ~(CH2)2~3
(67) O CH NHJ~ (6~) O(CH2kNHy 2
( 2~2 NHPMC NH
, Pd-C
NtH O(cH2)2NHJl` NH2 O(CH2)2~
O(CH2)2NH~ NH~O(CH2)2~ ~. _ CH3~--~ tNH
(~ S~~~ 2. hv O(CH2)2~H NH2
(69) o(CH2)2~H~ NHPI.~C
SUBSTITUTE SHEET (RULE 26)
Wo 95/04277 ~ 1 6 8 8 8~ PCTrus94l07780
- 45 -
Scheme VIa
OAc
(~ SH + Br~ SnMe3 ~ 3 SnMe3
(31 ) (81 ) (82)
NH
1. NH~OH O(CH2)2N~ NHPMC r Br
NH (~ S~SnMe3 ~ (84)
2. MsO(CH2)2NHJ~ NHPMC BzO OAc
(83)
Benzykhl~ rub,s(~ hcnyl-
phosphine)palladium(ll)
HJ~H
o~CH2)2~' NH?MC
~OAc 2 Br(CH
(85) OEz 3 BUNHC2H )
H~H
O~CH2)2~'' NHPMC
~ o(CH2)Z~
(~ 5~~~ NH
(86) O(CH2~2N~ NbPMC
0~2NH NH2 O(CH )~
CH3~--~ HlH
~87) O(CH2)2N NH2
SUBSTITUTE SH EET (RULE 26)
WO 95/W277 ~ 16 ~ 8 8 6 PCT/US94/07780
- 46 -
Introduct;on of the 2-acetoxy-4-bromobenzylbromide, 60 or
2~ to~y-4lr,~ lLyl~lannylbenzylbromide, 81 onto resin 31 to give 61 or 82: Resin31 is plc~al~d as described in Example 3 above.
The starting materials 60 and 81 are plcpâled from commercially available
materials by re~tionc well known to those skilled in the art of organic ~yllLllcsis. For
example, 2-acetoxy4-bromobenzyl bromide is pl~alcd from 2-methyl-5-nitroaniline
(Aldrich) by ~ 7Oti~ ion under ~dald con~litions. The diazo coln~oulld is thermolyzed
in acetic acid for 1 h at 80^C as des-;lil~cd in Chem. Lett., 1991, 459 to afford the
coll._sponding acetoxy co,lll).,und. The bromo group is introduced via nitro re(l~ctio~,
~ 7c~ti,~lion, and bromide displ~ P .I all under standard conditions and finally, the
desired product is obtained by benzylic ~lu...i~ Q~ with N-brnmn~rc;..;...irie under
standard con-iition~.
The COllc~ul~ g Llilllclllylstannyl derivative 81 is ple~ d from the p~mlltim~t~interm.odi~tP above, 2-acetoxy-4-l)lulllotoluene. The Llilllclllyl stannyl group is introduced
by placing the bromotoluene (2.0g, 8.78 mmol) in a dry flask with Pd(PPh3), (71 mg,
0.061 mrnol). Toluene (8.8 mL) was added then h~.... lh~rlditin (5 g, 15.26 mmol) was
added via syringe. The reaction was heated to 120~C for 1.5h. The reaction was cooled,
filtered through Celite, and ~apOlal~,d. The residue was dissolved in ether and washed
with 3 x 50rnL of 50% KF. The organic layers were dried (NaSO,) and evaporated which
after ~ulificalion afforded the desired product (77% yield). Finally, the benzylic bromide
is ill~lo~ ced by reaction with NBS under standard conditions.
The resin, 31 (2.3 g, 0.43 mmol) was s~ Pd in 45 mL of anhydrous DMF.
15 mL DMF, b-lllel~,alJluell~lol (0.25 mL, 3.5 mmole) and diisoplu~ylethylamine (0.4
mL, 2.3 mmole) were added and the mixture shaken for 2-3 mimlt~s, filtered and the
process repeated two more times using the same q~-~ntiti~s of BME and DIEA. The resin
was then washed five times with DMF, three times with mPth~nnl, four times with CH2Cl2
and then three times with DMF. To the resin was then added 60 (0.42g, 1.21 mmole)
dissolved in 15 mL DMF and DIEA (0.5 mL, 2.87 mmole) added and the l~ cLul~ shaken
for 6.5 hours, filtered and washed five times with DMF, three times with m~th~nnl and
six times with CH2Cl2. The resin was then dried under pump vacuum to give 2.2 grams
of 61. The same procedure is used for the conversion of 81 to 82.
SUBSTITUTE SHEET (RULE 26)
Wo 95/04277 ~16 8 ~ 8 5 PCTIUS94/07780
- 47 -
Removal of acetate group and ill~ru~ l;nrl of side chain to give 62 and 83:
The resin bound material 61 (4.6g. 0.83 mmol) was placed in acetone (30mL) and excess
2N ~mmnnillm hydroxide was added and the solution left at room le~ dlure for 24 h
(Haslam et ah, J. Chem. Soc., 2137 (1964)). The resin was filtered, washed, and
subjected to the following general alkylation scheme of Venuti et ah (J. Med. Chem.
1988, 31, 2132).
The resin-bound m~teri~l (4.6g, 0.83 mrnol) was placed in a llli~clule of 30mL
CHCl3, 15mL MeOH and anhydrous po~delcd po!;~c~ .. c~bol~le (0.5g, 3.62 mmol)
was added. The reaction was heated at 500C for 15 min, then
(2-N-PMC-gl~ni~linn)-(l-mr~ lf~ fonyl)ethanol~ (0.92 mmol, see pl~alion below)
was added and the llli~lul~ refluxed for 4h. After filtration, the residue was washed in
the ~Landal-:l fashion. To this material in THF (10 mL) was added tetrabuLyl~ll~llonium
fluoride (2.0 mL of lM solution in THF) and the reaction stirred at room L~ clal~lle for
3h. After filtration and washing 62 was obtained. The same ~ cedul~ was used for the
conversion of 82 to 83.
Preparation of (~N-PMC-guanidino)-(l-methanesulfonyl)ethanol: F~ n~
(lO.Og, 0.163 mol) was dissolved in CH2CI2 (250 mL) and im~ 7~le (24.41g, 0.358 mol)
was added. The reaction was cooled to OnC and TBDMSCI (27.14g, 0.18 mol) was added.
The mixture was stirred at O~C for two hours then room te~ for an ~rl(lition~l two
hours. Ethyl acetate (500mL) was added and the llli~lUl~ washed with 0.5M H2SO4
(400mL), sat'd NaHCO3 (400mL) and sat'd NaCI (400mL), dried, evaporated and the
reslll~ing material (12.0g, 42% yield) used as is. Form~mi~linrsulfonic acid (l.Og, 8.05
mmol; Tet. Len., 29, 3183, (1988)) and the above material (1.41g, 8.05 mmol) were
dissolved in dry mrth~nr~l (lOmL) and stirred for 2h at room t~ ldlule. The solvent
was removed in vacuo and the product dissolved in acetone (27mL), water (7mL), and
NaOH (lOmL, 3.2M) added. The reaction was cooled to OnC and PMCCI (3.66g, Raylo
ChPInir~l~, Alberta, Canada) was added in acetone (8mL). After stirring for lh at OnC
the reaction was diluted with ethyl acetate, washed one time each with 25mL sat'd NH4CI,
water, and sat'd NaCI, dried and evaporated. The product was purified by flash
chlollldtography (silica, hexane/ethyl acetate 1:1) to afford 1.71g (46%) of desired
product.
SUBSTITUTE SHEET (RULE 26)
~168886
Wo 95/04277 PCT/US94/07780
- 48 -
The product (0.57g, 1.23 mmol) was dissolved in THF (lOmL), cooled to 0nC and
tetrabutyl~mmn~ oride (371mg, 1.42 mmol) added. ARer 30 min the reaction wasworked up by diluting with ethyl acetate, washing one time each with 25mL sat'd NH,Cl,
water, and sat'd NaCl, dried and e~apolalcd. The product was purified by flash
c}~ulllalography (silica, CH2Cl2/...~ nnl; 19:1) to afford 0.43g (94%) of desired product.
This material (64mg, 0.186 mmol) was dissolved in CH2Cl2 (2mL), cooled to 0~C and
DMAP added (2.2mg). Me~ P~Ifonyl chloride (35.6mg, 0.204 mmol) was added and
reaction was complete after 20 min. Evaporation of the mixture was followed by
;r~lion (silica, CH2CI2) to afford 95mg (92% yield) of desired product.
J~dt;(Jl- of ~ k~ t~d phch~l&c~t~ c 63 and formation of diphenyl
ac~ ,.c 64: 3,5-Dihydroxyiodobtl.2cne was p~ al~d from 3~5-tlimpthnxyaniline
(Aldrich) by diazoli~alion and iodine hlLluducLion followed by delllcLhylation of the
methoxy groups all under sLandald con~ition~. This material (3.78g 16.0 mmol) was
dissolved in CH2CI2 (30mL). Triethylamine (11.15mL, 80 mmol), acetic anhydride (4.55
mL, 48 mmol) and DMAP (390 mg, 3.2 mmol) were added and the reaction stirred for16 h. The reaction was evapolaLcd to dryness, and passed through a plug of silica gel
eluting with 4:1 hexane:ethyl acetate to afford the desired product. This material (12.8
mmol) was dissolved in a mix of ethanol (32 mL) and ~.~e.R (16 mL). PoL~ssiw
hydroxide (0.72g, 12.8 mmol) was dissolved in 8 mL ethanol and added over 30 min.
After 30 min the reaction was diluted with ether and washed with 0.5N H2SO" sat
NaHCO3, sat NaCl, dried over Na2SO, and evaporated. The product was recryst~lli7Pd
from toluene to afford 86% yield of the mono~cet~t~ which was dissolved in CH2CI2 (50
mL) and triethylamine (3.45 mL, 24.8 rnmol), DMAP (0.3g, 2.5 mmol) and benwyl
chloride (1.8 mL, 15.5 mmol) was added. The reaction was col~le~e in 10 min thendiluted with CH2Cl2 and washed with sat NH,Cl, sat NaHCO3, and sat NaCl. The solution
was dried (Na2SO,) and e\/a~olal~d. Purifir~tion was accomplished via silica
ch~ latography 19:1 hexane:ethyl acetate to afford 4.4g (95 % yield) of desired
3 -acetoxy-5 -ben_oyloxy-iodobenzel1e .
The substituted phenylacetylene 63 was ~lc;pal~d from this iodobel~elle by the
general procedure described by Lau et ah (J. Org. Chem., 1981, 46, 2280).
The iodobenzene (1.15g, 3 mmol) was placed together with TMS-acetylene (0.47g,
4.8 mmol), Pd(II)acetate (10 mg), and triphenylphosphine (20 mg) in dry triethylamine
SUBSTITUTE SHEET (RULE 26)
W O 9S/04277 ~16 ~ ~ 8 6 F~CTrUS94/07780
- 49 -
(5 mL). The ll~ ulc was heated at reflux for 4 h, cooled, the solid removed by filtration,
the filtrate concenLldled, mixed with sodium bicarbonate (20 mL), extracted with CH2CI2
dried, and conccllLlalcd to afford the TMS-phenylacetylene. This material was converted
to the free acetylene by dissolving in THF (8 mL) and adding tetrabutyl ammoniumfluori(le (3 mL of lM in THF) and stirring for 3h at room te~ alulc. After standard
workup and ch,umaLogl~hic purification the desired substituted phenylacetylene 63 was
obtained.
The resin bound bromobc.lL~ne 62 (5.5g, 1 mmol) was s~cpen~l~od in DMF (10
mL). To this ~s~cn~ion was added the acetylene 63 (0.85g, 3 mmol), Pd(II)acetate (10
mg), and triphenyll,hos~hine (15 mg) in dry lliclllrlall~uR (5 mL). The mixture was
heated at reflux for 4 h, cooled, filtered, and washed in the ~Ldnd~.l fashion to afford 64.
P~ ~ d1.io.l of benzylbron~ide 84 and ~ G~ ~;(J.. of 83 to 85: The substituted
benzylbromide 84 was ~l~,pal~d from the c~ll.,s~ondillg 3-acetoxy-5-benzyloxytoluene by
reaction with N-bromosuccinimide under standard conditions.
3-Acetoxy-5-benzyloxytoluene was in turn pl~ d from orcinûl (Aldrich) by the same
protection scheme used to prepare the phenylacetylene 63, above.
For the coll~ ion of 83 to 85, ~e general l rocedul~ of Milstein and Stille (JACS,
1979, 101, 4992) was employed. The resin-bound 83 (5.5g, 1 mmol; pl~ d as
described above) was sllcpenA~d in 10mL hr~ .yl-phosphoramide. To this was addedbenzylchlorobis(Llil)hc~l~ho~hine)-p~ m(2) (0.05 mmol) and the benzylbromide 84
(1.75g, 5 mmol). The reaction was heated to 65~C for 10h, cooled, filtered and washed
in the usual fashion to afford 85.
The following conditions apply to 64 and 85 to afford 65 and 86.
Removal of acetate group, il-l.o~ n of side chain, r~...o.~l of ~ te
group, and il-lr~lu~lion of side chain to afford 65 and 86: The resin bound material
(4.6g, 0.83 mmol) was placed in acetone (30 mL) and excess 2N ammonium hydroxidewas added and the solution left at room ~ n~ldlulc for 24 h (Haslam et al., J. Chem.
Soc., 2137 (1964)). The resin was filtered, washed and subjec~ed to the following general
alkylation scheme (Venuti et ah, J. Med. Chem. 31, 2132 (1988)):
The resin-bound material (4.6g, 0.83 mmol) was placed in a mixture of 30mL
CHCI3, 15mL MeOH and anhydrous powdered potassium carbonaLe (0.5g, 3.6 mmol) wasadded. The reaction was heated at 50nC for 15 min, then (2-bromoethyl)bel.,e.le (171mg,
SUBSTITUTE SHEET (RULE 26)
-
~168~8~
Wo 95/W277 PCT/US94/07780
- 50 -
0.92 mmol, Aldrich) was added and the mixture refluxed for 4h. After filtration, the
residue was washed.
Removal of the ben70~tP~ is carried out as described by Bell (Tet. Lett., 27, 2263
(1986)). The resin-bound material (4.6g, 0.83 mmol) was placed in toluene (30 mL) and
n-butylamine (0.37 g, 5.0 mmol) was added. The mixture was stirred at room
te~ c.~Lule for 3 h followed by filtration and washing of the resin.
For introduction of the second functional group, the resin-bound material (4.6g,0.83 mmol) was placed in a mixture of 30mL CHC13, l5mL MeOH and a~ ydr~us
powdered ~/u!;~ - Cal'l)Ol~l~c (0.5g, 3.6 mmol) was added. The reaction was heated at
50OC for 15 min, then (2-N-PMC-~ n~ n~,.lfonyl)ethanol, (0.92 mmol, seeLion above) was added and the mixture rellu~ed for 4h. After filtration, the
residue was washed in the ~.L~ldal :1 fashion.
Reduction of the ac~ c 65 to the olefin 67: This selective reduction of the
acetylene to the corresponding olefin is accomplished with Lindlar catalyst ~ J~cd as
described in Org. Syn. Coll., Vol. V, 880. To the resin-bound 65 (5.Sg, 1 mmol)
sllspen(lPd in 10 mL hexane was added 10mg of Lindlar catalyst and 50mL of quinoline.
The reaction vessel is evacuated and placed under a slight positive pl~....u~e of hydrogen
gas for 3 h, filtered, and washed to afford 67 (~ e~lrd to be exclusively the Z-olefin).
R~ of the olefin 67 to the elk~lene analogue 69: To the resin-bound 67
(5.Sg, 1 mmol) ~ ~ n~lPd in 10 rnL ethyl acetate was added 50mg of Pd(OAc)2 and the
reaction was subjected to a positive ~ .ule of hydrogen gas for 1 h. The mixture was
filtered and washed in the sL;~ldal-i fashion to obtain 69.
Removal of all prote~ting ~IV.~S; The resin-bound material (ll.lg, 2mmol) was
placed in CH2Cl2 (100 mL) and Llinuoloact;lic acid (2.0 mL) added. The mixture was
stirred at room te~ ul~ for one hour then the resin filtered and washed.
Removal of the final l"odu.l from the resin to give 66, 68, 70, and 87: The
resin bound material (3.3 g, 0.6 mmol ) was s~spe~ Pd in 50 mL of acelol~iLIile. The
stirred mixture was irradiated under nitrogen atmosphere using a Rayonet photoc7nPmir~l
reactor (consis~ g of sixteen black light phosphor bulbs having a m~ximllm wavclell~Lh
illL~usily at 350 nm) for 4 hours. After irradiation, the mixture was filtered to afford the
desired products 66, 68, 70, and 87 in solution.
SU8STITUTE SHEET (RULE 26)
WO 9~/W277 21~ ~ 8 8 ~ PCT/US94/07780
- 51 -
EXAMPLE 6
Protein Kinase C Activitv Dele~.llination
Colllp~lullds of the invention are tested for ability to inhibit protein kinase C using
rat brain as the enzyme source accol-lillgly to widely used procedures such as described
by A. C. McArdle and P. M. Conn, Methods in Enzymology (1989) 168. 287-301, and
by U. Kikkawa et ah, Biochem. Biophys. Res. Comm~n (1986), 135, 636-634.
~It~rn~tively, protein kinase C activity is i~t.. ;I-~d using l u~ ed human protein kinase
C isozymes by methods such as described in P. Basta et ah, Biochim. Biophys. Acta.
(1992) 1132. 154-160.
EXAMPLE 7
Me",blàne Receptor Affinitv Det~llllillalioQs
A. Bradvkinin Receptor
The bradykinin lece~tul affinity of conl~ou,lds L/l.,pal~,d acco,dil~g to this invention
is ~ele l..in~d by testing for ability to displace [3Hl bladykinill binding from guinea pig
ileal membrane as desclil,ed in S. G. Farmer et ~ J. pl.~ ol. Exp. Ther. (1989) 248,
677.
B. Other Receptol~
Generally applicable mPth~c for testing lcc~ or affinity of the collll,oullds of the
invention are described by H. I. Y~ lllllla et ah, Methods in N~U1UL1~ e~ Receptor
Analysis, Raven Press, 1990.
EXAMPLE 8
Measul~,~le-~L of Interaction with Tar~et Enz~mes
A. An~iotensin Con~e,Li..~ IIIC
Methods useful for ~rl~ ing the ability of compounds of the invention to inhibitangiotensin COll~ illg enzyme are disclosed by J. W. Ryan, Methods in Enzymology(1988) 164, 194-211.
SUBSTITUTE SHEET (RULE 26)
WO 9~/04277 216 ~ ~ 8 G PCT/US94/0778~
- 52 -
B. Phospholipase A.
A procedure useful to test eff1cacy of the invented collluoullds in inhibilillg
phospholipase A2 is described by J. Reynolds et ak, Methods in Enzymology (1991) 197,
3-23.
EXAMPLE 9
D~....,;.,~lion of Ion Ch~nn~ol Bindin~
Xenopus oocytes are well known as tools for studying ion ch~nnrlc and rcce~lol~.A. L. Buller and M. M. White have described mPtho~s useful to uleasule interaction
b.,~ n coulp~uuds of the invention and various ion rh~nnrlc or ll,ce~lol~. Methods in
Enzymology (1992) 207, 368-375.
EXAMPLE 10
Tlal~cliulion Factor Effect.
J. M. Gottesfeld is an example of a lcr._l~,nce desc,il~iulg a p~ce.lulc suitable for
analyzing the ability of the ill~ lcd coull)ouuds to inflnPnre L~ansc-iulion factor ÇullcLion.
Methods in Enzymology (1977) 170, 346-359.
The ~icclosllres of all ~cç~l~,nces cited in this ~yccirr~lion herein are mcGluolaL~d
in their euLuet;es by lcr~,le~.,e.
SUBSTITUTE SHEET (RULE 26)