Note: Descriptions are shown in the official language in which they were submitted.
~11 1 57q
The present invention relates to piperidine
derivatives, a process for their preparation and their
application in therapeutics.
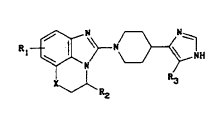
The present invention provides a compound of
formula (I)
R~ N~,~, H (I)
X`J` R3
in which
X represents an oxygen atom or a methylene group,
Rl represents a chlorine or fluorine atom or a methyl,
methoxy or amino group, and
R2 and R3 independently represent a hydrogen atom or a
methyl group, in the form of the free base or an
addition salt with a pharmaceutically acceptable acid.
The preferred compounds according to the
invention are those in which,
X represents an oxygen atom and
Rl is at position 8 on the phenyl ring.
The particularly preferred compounds of
formula ~I) are (S)-8-fluoro-4-methyl-2-t4-(5-methyl-
lH-imidazol-4-yl)piperidin-1-yl]-
4,5-dihydroimidazotl,5,4-de]tl,4]benzoxazine and (S)-
8-fluoro-4-methyl-2-t4-(lH-imidazol-4-yl)piperidin-1-
yl]-4~5-dihydroimidazo[l~5~4-de]tl,4]benzoxazine.
Some compounds of formula (I) contain an asymmetric
~ 71~79
asymmetric carbon atom. They can therefore exist in the
form of enantiomers. These enantiomers, pure or in the
form of mixtures, including racemic mixtures, form part
of the invention.
The mesomeric forms of the compounds of formula (I)
also form part of the invention.
The compounds of formula (I) can be prepared
by reaction of a compound of formula (II), in which X,
Rl and R2 are as defined in formula (I) and Y represents
a halogen atom, in particular a chlorine atom, with a
compound of formula ~III), in which R3 is as defined in
formula (I), according to Scheme 1.
Scheme 1
(~1) (111) (1)
The starting compounds are commercially
available or described in the literature or can be
prepared according to methods which are described
therein or which are known to the person skilled in the
art.
Thus, the compound of formula (III) in which
R3 is a hydrogen atom is described in Arch. Pharmaz.,
306 (12), 934-942 (1973).
The compound of formula (III) in which R3 is a methyl
group can be prepared according to the process
2 1 7 1 579
described in EP-A-0,507,650.
The compounds of formula (II), in which Y is a chlorine
atom and X, Rl and R2 are as defined in formula (I), can
be prepared by the reduction of a compound of formula
(IV)
N02
~ % ~ (IV)
which is then reacted with urea to give a compound of
formula (V)
Rl~ >=O (V)
,~,1~
Rz
which is then treated with phosphoryl chloride.
The compounds of formula (IV) can be prepared
by reaction of a compound of formula (VI)
Rl ~ Nx ~ Rz (VI)
with bis(1,1-dimethylethyl) dicarbonate,
nitration of a compound of formula (VII)
2171579
coo~- ~u
R~ ~ Nx ~ Rz (VII)
and hydrolysis of a compound of formula ~VIII)
t~2 COO~ U
~N~ (VIII)
~ XJ
Some compounds of formula ~IV) can also be
prepared from compounds of formula (IX)
NO
1 2 H
~ x ~ (IX)
in which X and R2 are as defined in formula (I).
In particular, compounds of formula (IVa)
NOz H
Cl ~ x ~ (IVa)
can be prepared by chlorination of the compounds of
formula (IX).
The preparation of the compound of formula
(IX), in which R2 is a methyl group and X an oxygen
atom, is described in EP-A-0,646,583.
The compound of formula (IX) in which R2 is a
hydrogen atom and X an oxygen atom can be prepared by
217157~
An analogous process.
Compounds of formula ~IVb)
N02
~ ~ R2 (IVb)
in which R2 is as defined in formula (I)
can also be prepared by nitration of a compound of
formula ~X)
~N~R2 (X)
The compounds of formula (X) are typically
prepared by cyclization of 2-bromo-5-fluorophenol with
a compound of formula (XI)
HO ~ COO~u (X~
R2
The following Examples illustrate the
preparation of compounds according to the present
invention. Microanalyses and NMR spectra have confirmed
the structures of the products obtained. The (y:x)
ratios correspond to the (base:acid) ratio. The numbers
between brackets refer to the table given later which
21 7 157~
illustrates the chemical structures and the physical
properties of some compounds according to the
invention.
Example 1 (Compound No. 1)
~S)-8-chloro-4-methyl-2-[4-(5-methyl-lH-imidazol-
4-yl)piperidin-1-yl]-4,5-dihydroimidazo-
[1,5,4-de]~1,4]benzoxazine
1.1. (S)-7-chloro-3-methyl-5-nitro-3,4-dihydro-2H-
1,4-benzoxazine
5.82 g (0.030 mol) of (S)-3-methyl-5-nitro-
3,4-dihydro-2H-1,4-benzoxazine are dissolved in 135 ml
of acetic acid at 45C. 5.16 g (0.038 mol) of
_-chlorosuccinimide are rapidly added and the reaction
mixture is stirred for 4 hours at 50C. It is then
poured into water and extracted 3 times with diethyl
ether. The organic phases are combined, washed with
water and then with an aqueous sodium hydroxide
solution and dried. The solvent is evaporated to
dryness.
6.7 g of product are obtained.
Melting point : 95C
1.2. (S)-8-chloro-4-methyl-4,5-dihydroimidazo-
[1,5,4-de][1,4]benzoxazin-2(lH)-one
A suspension between 8.9 g (0.039 mol) of
(S)-7-chloro-3-methyl-5-nitro-3,4-dihydro-2H-1,4-
benzoxazine in 250 ml of ethanol and a catalytic amount
2~ 7 1 579
of Raney nickel are introduced into a Parr apparatus.
Hydrogenation is carried out for 2 hours at room
temperature under a pressure of 70 kPa (10 psi). The
catalyst is then filtered off and washed with ethanol,
the filtrate is recovered and the solvent is evaporated
to dryness.
3.4 g ~0.056 mol) of urea are ~dded to the residue thus
obtained and heating is carried out with an oil bath at
170-180C for 1 hour. The solid obtained is then taken
up in a mixture of water and diethyl ether (50:50), and
the precipitate formed is filtered off, washed with
diethyl ether and then with water and dried under
vacuum over phosphorous pentoxide. 6.3 g of product are
obtained, which product is purified by chromatography
on a silica gel column with a mixture of
dichloromethane and methanol (97:3).
5.2 g of product are obtained.
1.3 (S)-2,8-dichloro-4-methyl-
4,5-dihydroimidazo[1,5,4-de][1,4]benzoxazine
100 ml of phosphoryl chloride are poured onto
5.2 g (0.023 mol) of (S)-8-chloro-4-methyl-
4,5-dihydroimidazo[1,5,4-de][1,4]benzoxazin-2(lH)-one.
The mixture is heated at reflux for 2 hours, the
solvent is then evaporated to dryness and the residue
is taken up in ice-cold water ~nd then in a
concentrated aqueous ammonia solution. Extraction is
then carried out with diethyl ether, the organic phases
2 1 7 1 579
are combined and dried and the solvent is evaporated to
dryness. 4.5 g of residue are obtained, which residue
is purified by chromatography on a silica gel column
with a mixture of hexane and ethyl acetate (97:3).
4.1 g of product ~re obtained in the form of an oil.
1.4. ~S)-8-chloro-4-methyl-2-[4-(5-methyl-lH-imidazol-
4-yl)piperidin-l-yl]-4~5-dihydroimidazo-
t1,5,4-de][1,4]benzoxazine
A mixture of 1.35 g (0.0082 mol) of
4-(5-methyl-lH-imidazol-4-yl)piperidine, 1 g
(0.0041 mol) of (S)-2,8-dichloro-4-methyl-
4,5-dihydroimidazotl,5,4-de]tl~4]benzoxazine and 4.5 ml
of isoamyl alcohol is heated at 120C for 12 hours with
stirring. The solvent is then evaporated to dryness and
the residue is purified by chromatography on a silica
gel column with a mixture of dichloromethane, methanol
and aqueous ammonia (95:5:0.5).
After recrystallization from acetone, 0.7 g of product
is obtained in the base form.
Melting point : 208C
[~]DO = -23.8 (c = 0.01, methanol)
Example 2 (Compound No. 2)
(S)-8-chloro-2-[4-(lH-imidazol-4-yl)piperidin-1-yl]-
4-methyl-4,5-dihydroimidazotl,5,4-de]tl,4]tbenzoxazine
There is obtained, from a mixture of (S)-
2,8-dichloro-4-methyl-4,5-dihydroimidazo-
21 7 i 57~
g
[l~5~4-de]tl~4]benzoxazine and 4-(lH-imidazol-4-
yl)piperidine, treated under the conditions of Example
1.4, after recrystallization from acetone, 0.6 g of
product in the base form.
S Melting point : 215C
[cr~D = -3-7 (c = O.01, methanol)
Example 3 (Compound No. 5)
8-fluoro-2-t4-(lH-imidazol-4-yl)piperidin-1-yl]-
4-methyl-5,6-dihydro-4H-imidazot4,5,1-ij]quinoline
3.1. 1,1-dimethylethyl 6-fluoro-2-methyl-
1,2,3,4-tetrahydroquinoline-1-carboxylate
A mixture of 16.5 g ~0.1 mol) of 6-fluoro-
2-methyl-1,2,3,4-tetrahydroquinoline and 32 g (0.146
mol) of bis(l,l-dimethylethyl) dicarbonate in 100 ml of
15 tetrahydrofuran is heated at 60C for 3 days. The
solvent is then evaporated to dryness and the remaining
dicarbonate is driven off using a vane pump. The
residue is purified by chromatography on silica gel
with a mixture of hexane and ethyl acetate (so:10).
20 24.5 g of product are obtained in the form of an oil.
3.2 1,1-dimethylethyl 6-fluoro-2-methyl-8-nitro-
1,2,3,4-tetrahydroquinoline-1-carboxylate
A solution of 18.4 g (0.0697 mol) of
l,l-dimethylethyl 6-fluoro-2-methyl-
1,2,3,4-tetrahydroguinoline-1-carboxylate and 13.8 ml
217i~7~
of anhydrous N,N,N',N'-tetramethylethylenediamine in
350 ml of Anhydrous diethyl ether is cooled to -78C.
64.2 ml of a 1.3N solution of sec-butyl iodide in a
mixture of cyclohexane and pentane are then added over
30 minutes. The reaction mixture is stirred for 1 hour
at -78C and then 12.3 g (0.103 mol) of isobutyl
nitrate are added. The reaction mixture is stirred for
1.5 hours at -70C and is then neutralized by being
poured into ice-cold water. Extraction is carried out
with diethyl ether, the organic phases are combined,
washed with water and dried and the solvent is then
evaporated to dryness. The residue is purified by
chromatography on a silica gel column, elution being
carried out with a mixture of hexane and ethyl acetate
~90:10).
8.1 g of product are obtained.
3.3. 6-fluoro-2-methyl-8-nitro-
1,2,3,4-tetrahydroguinoline
A mixture of 8.6 g (0.0261 mol) of
1,1-dimethylethyl 6-fluoro-2-methyl-8-nitro-
1,2,3,4-tetrahydroquinoline-1-carboxylate, 30 ml of
concentrated hydrochloric acid, 20 ml of toluene and
30 ml of water is heated at the reflux temperature for
one day. The mixture is then allowed to return to room
temperature, separation is carried out by settling and
extraction is carried out with toluene. The organic
phases are combined, washed with water And dried and
2171579
the solvent is then evaporated to dryness.
5.6 g of product are obtained.
Melting point: 56C
3.4. 8-fluoro-4-ethyl-5,6-dihydro-4H-imidazo-
t4,5,1-i~]quinol-2(lH)-one
A suspension of 5.5 g (0.026 mol) of
6-fluoro-2-methyl-8-nitro-1,2,3,4-tetrahydroquinoline
in 150 ml of ethanol and a catalytic amount of Raney
nickel are introduced into a Parr apparatus and
hydrogenation is then carried out for 1 hour at room
temperature under a pressure of 0.21 MPa (30 p5i). The
catalyst is then filtered off and washed with ethanol,
the filtrate is recovered and the solvent i8 evaporated
to dryness. The residue is added to 2.6 g (0.043 mol)
of urea and heating is carried out with an oil bath at
175C for 1.5 hours. The solid obtained is taken up in
a mixture of water and diethyl ether (50:50) and the
precipitate formed is then filtered off, washed with
diethyl ether and with water and dried under vacuum
over phosphorus pentoxide.
4.2 g of product are obtained.
3.5. 2-chloro-8-fluoro-4-methyl-5,6-dihydro-4H-
imidazo[4,5,1-i~]guinoline
100 ml of phosphoryl chloride are poured onto
4.2 g (0.02 mol) of 8-fluoro-4-methyl-5,6-dihydro-4H-
imidazot4,5,1-i ]quinol-2(lH)-one and the mixture is
21 7 1 51~
then heated at reflux for 2 hours. The solvent is then
evaporated to dryness and the residue is taken up in
ice-cold water And then in a concentrated Aqueous
ammonia solution. Extraction is carried out with
diethyl ether, the organic phases are combined and
dried and the solvent is then evaporated to dryness.
The residue is purified by chromatography on a silica
gel column, elution being carried out with a mixture of
hexane and ethyl acetate (70:30).
2.4 g of product are obtained.
Melting point : 80C
3.6. 8-fluoro-2-~4-(lH-imidazol-4-yl)piperidin-1-yl]-
4-methyl-5,6-dihydro-4H-imidazot4,5,1-ij]guinoline
A mixture of 0.94 g ~0.00625 mol) of 4-(lH-
imidazol-4-yl)piperidine, 0.7 g (0.0031 mol) of
2-chloro-8-fluoro-4-methyl-5,6-dihydro-4H-imidazo-
t4,5,1-i~]quinoline and 3.5 ml of isoamyl alcohol is
heated at 120C for 18 hours with stirring. The solvent
is then evaporated to dryness and the residue is
purified by chromatography on a silica gel column with
a mixture of dichloromethane, methanol and aqueous
ammonia (95:5:0.5).
After recrystallization from acetone, 0.85 g of product
is obtained in the base form.
Melting point : 175-177C
2 1 71 579
Example 4 (Compound No. 6)
8-fluoro-4-methyl-2-[4-(5-methyl-lH-imidazol-
4-yl)piperidin-1-yl]-5,6-dihydro-4H-imidazo-
[4,5,1-i ]quinoline
There is obtained, from 0.7 g (0.0031 mol) of
2-chloro-8-fluoro-4-methyl-5,6-dihydro-4H-
imidazo[4,5,1-i~quinoline and 1 g (0.00623 mol) of
4-(5-methyl-lH-imidazol-4-yl)piperidine, treated under
the conditions of Example 3.6, after recrystallization
from acetone, 0.7 g of product in the base form.
Melting point : 255C
Example 5 (Compound No. 8)
(S)-4-methyl-2-[4-(5-methyl-1-imidazol-4-yl)piperidin-
1-yl]-4,5-dihydroimidazo[1,5,4-de][1,4]benzoxazin-
8-amine
5.1. (S)-5,7-dinitro-3-methyl-3,4-dihydro-2H-
1,4-benzoxazine
7.7 g (0.04 mol) of (S)-3-methyl-5-nitro-
3,4-dihydro-2H-1,4-benzoxazine are dissolved in 176 ml
of acetic acid at 60C. 2.8 ml (0.044 mol) of nitric
acid are added dropwise, the reaction mixture is
stirred for 45 minutes at 600C and then 176 ml of water
are added. The reaction mixture is cooled to 0C and
the precipitate formed is filtered off, washed with
water and dried under vacuum.
8.13 g of product are obtained.
2 1 7 1 579
14
Nelting point : 170C
5.2. ~S)-3-methyl-7-nitro-3,4-dihydro-2H-
1,4-benzoxazin-5-amine
7.63 g (0.032 mol) of (S)-5,7-dinitro-
3-methyl-3,4-dihydro-2H-1,4-benzoxazine are heated to
50C and a solution of 30.52 g (0.391 mol) of sodium
sulphide and of 10.70 g of sodium hydrogencarbonate
dissolved in 46 ml of water is added dropwise.
This reaction mixture is slowly brought to the reflux
temperature and then, after stirring for 30 minutes, a
quantity of water is poured in. The reaction mixture is
allowed to cool and is extracted three times with ethyl
ether. The organic phases are combined, washed with
water and dried and the solvent is then evaporated to
dryness.
5.98 g of product are obtained.
5.3. (S~-4-methyl-8-nitro-4,5-dihydroimidazo-
t1,5,4-de]tl,4]benzoxazin-2(lH)-one
3.43 g (0.057 mol) of urea are added to
5.98 g (0.029 mol) of (S)-3-methyl-7-nitro-3,4-dihydro-
2H-1,4-benzoxazin-5-amine and the mixture is heated for
2 hours at 175C. It is taken up in boiling water and
the precipitate is filtered off and washed with water.
The filtrate is extracted with dichloromethane and the
organic extracts are washed with water, dried and
evaporated under vacuum. The residue is purified by
2171~79
chromatography on a silica gel column, elution being
carried out with a dichloromethane:methanol (98:2)
mixture.
3.86 g of product are obtained.
5.4. ~S)-2-chloro-4-methyl-8-nitro-
4,5-dihydroimidazotl,5,4-de]tl,4]benzoxazine
3.86 g of (0.021 mol) of (S)-4-methyl-
8-nitro-4,5-dihydroimidazot1,5,4-de][l,4]benzoxazin-
2(lH)-one are heated at the reflux temperature for 3
hours in the presence of 76 ml of phosphoryl chloride.
The solvent is evaporated to dryness and the residue is
taken up in ice. The reaction mixture is basified with
agueous ammonia and the precipitate formed is filtered
off, rinsed with water and pulled dry. It is dried in a
vacuum oven. The residue is purified by chromatography
on a silica gel column, elution being carried out with
a dichloromethane:methanol ~99:1) mixture.
3.6 g of product are obtained.
5.5. (S)-4-methyl-8-nitro-2-[4-(5-methyl-lH-imidazol-
4-yl)piperidin-1-yl]-4,5-dihydroimidazo-
[1,5,4-de]tl,4]benzoxazine
A mixture of 1.87 g (0.00788 mol) of
4-(5-methyl-lH-imidazol-4-yl)piperidine, 1 g (0.00394
mol) of (S)-2-chloro-4-methyl-8-nitro-
4,5-dihydroimidazotl,5,4-de][1,4]benzoxazine and 5 ml
of isoamyl alcohol is heated at 120C for 5 hours with
2 i 7 1 57~
16
stirring. The solvent is evaporated to dryness and the
residue is purified by chromatography on a silica gel
column, elution being carried out with a mixture of
dichloromethane, methanol and aqueous ammonia
(94:6:0.6).
After recrystallization from ethanol, 1.3 g of product
are obtained in the base form.
Melting point : 125C
5.6. (S)-4-methyl-2-[4-(5-methyl-lH-imidazol-
4-yl)piperidin-1-yl]-4,5-dihydroimidazo-
[1,5,4-de][1,4]benzoxazin-8-amine
A suspension of 1.1 g (0.00287 mol) of (S)-
4-methyl-8-nitro-2-[4-(5-methyl-lH-imidazol-
4-yl)piperidin-1-yl]-4,5-dihydroimidazo-
[1,5,4-de][1,4]benzoxazine in 50 ml of ethanol and a
catalytic amount of platinum oxide are introduced into
a Parr flask and hydrogenation is then carried out for
1 hour at room temperature under a pressure of 0.28 MPa
(40 psi). The catalyst is filtered off and washed with
ethanol and with ethyl acetate, the filtrate is
recovered and the solvent is evaporated to dryness.
0.8 g of product is obtained, which product
crystallizes in the base form.
Melting point : 245C
[~] 20 = -23.4 (c = 0.01, methanol).
2171579
17
Example 6 (Compound No. 7)
(S)-2-[4-(lH-imidazol-4-yl)piperidin-1-yl]-4-methyl-
4,5-dihydroimidazo[1,5,4-de][1,4]benzoxazin-8-amine
There are obtained, from a mixture of (S)-
2-chloro-4-methyl-8-nitro-4,5-dihydroimidazo-
[1,5,4-de]tl,4]benzoxazine and 4-(lH-imidazol-4-
yl)piperidine, treated under the conditions of Example
5.5, after chromatography on a silica gel column,
elution being carried out with a mixture of
dichloromethane, methanol and aqueous ammonia
(94:6:0.6), 1.3 g of product, which product is treated
under the conditions of Example 5.6.
0.7 g of product is obtained in the base form.
Nelting point : 235-238C
[~] 20 = _ 1. 4 (c = 0.01, methanol)
Example 7 (Compound No. 14)
(S)-8-fluoro-4-methyl-2-[4-(5-methyl-lH-imidazol-
4-yl)piperidin-1-yl]-4,5-dihydroimidazo-
[l~5~4-de][1,4]benzoxazine
7.1. 1,1-dimethylethyl (S)-[2-(2-bromo-
5-fluorophenoxy)-1-methylethyl]carbamate
130 g (0.74 mol) of 1,1-dimethylethyl (S)-
(2-hydroxy-1-methylethyl)carbamate and 191 g ~0.728
mol) of triphenylphosphine are placed in a 4-litre
three-necked, round-bottomed flask containing 2.3
litres of toluene. The mixture is cooled with an ice
2171~7~
18
bath and 115 ml (0.726 mol) of diethyl azodicarboxylate
are added dropwise. The reaction mixture is left
stirring for 1 hour, 100 g (0.52 mol) of 2-bromo-5-
fluorophenol are added dropwise, the reaction mixture
is stirred for 2 hours at 0C, the temperature of the
reaction mixture is allowed to return to room
temperature and the reaction mixture is left stirring
overnight. The precipitate obtained is then filtered
off ~nd the filtrate is washed with lN sodium hydroxide
solution and water, dried and evaporated. The residue
obtained is purified by chromatography on a silica gel
column, elution being carried out with a mixture of
dichloromethane and heptane (70:30).
170 g of product are obtained.
[~]20 = _ 45.6O (c = 0.01, dichloromethane)
7.2. ~S)-1-(2-bromo-5-fluorophenoxy)propan-2-amine
170 g (0.48 mol) of 1,1-dimethylethyl (S)-
t2-(2-bromo-5-fluorophenoxy)-1-methylethyl]carbamate
dissolved in 520 ml of water and 260 ml of 12N
hydrochloric acid are heated at reflux for 2 hours.
500 ml of ice-cold water are poured in and extraction
is carried out once with 600 ml of toluene and once
with 400 ml of ether. The aqueous phase is cooled in an
ice-cold water bath and is basified with lON sodium
hydroxide solution. Extraction is carried out three
times with 500 ml of ether and the organic phases are
combined, washed twice with water and once with a
2`171~79
19
saturated aqueous sodium chloride solution, dried and
evaporated under reduced pressure.
118 g of product are obtained in the form of an oil.
[C~]D0 = ~ 2.8 (c = 0.01, dichloromethAne)
7.3. (S)-7-fluoro-3-methyl-3,4-dihydro-2H-
1,4-benzoxazine
72 g (0.768 mol) of sodium tert-butoxide are
placed in 1 litre of toluene in a 4-litre three-necked
flask under an inert atmosphere. 5 g of
tetrakis(triphenylphosphine)palladium and then a
solution of 120 g (0.48 mol) of (S)-1-(2-bromo-
5-fluorophenoxy)propan-2-amine in 250 ml of toluene are
~dded. The reaction mixture is heated to 110C and is
left stirring for 2.5 hours. 3 g of catalyst are again
added and, after stirring for 5.5 hours at 110C, the
reaction mixture is allowed to cool and 1 litre of
water is added. The reaction mixture is stirred, is
allowed to separate by settling and the aqueous layer
is extracted with toluene. The organic phases are
combined, washed with water and dried and the solvent
is evaporated under reduced pressure. Purification is
carried out by chromatography on a silica gel column,
elution being carried out with dichloromethane.
21.5 g of product are obtained.
[tr]20 = - 4.8 (c = 0.01, methanol)
21 7 1 579
7.4. (S)-4-acetyl-7-fluoro-3-methyl-3,4-dihydro-2H-
1,4-benzoxazine
A solution of 21.5 g (0.128 mol) of (S)-
7-fluoro-3-methyl-3,4-dihydro-2H-1,4-benzoxazine in
60 ml of pyridine is cooled with an ice bath, 15.1 g
(0.15 mol) of acetic anhydride are added and the
mixture is allowed to return to room temperature And is
stirred for 48 hours. It is poured onto ice-cold water
and extracted twice with ether. The organic phases are
combined, washed successively with water, with dilute
hydrochloric acid, with water and with brine and are
then dried and the solvent is evaporated under reduced
pressure.
24.2 g of product are obtained.
t~] 20 = t 102.9 (c = o.01, dichloromethane)
7.5. ~S)-7-fluoro-3-methyl-5-nitro-3,4-dihydro-2H-
1,4-benzoxazine
24 g (0.114 mol) of (S)-4-acetyl-7-fluoro-
3-methyl-3,4-dihydro-2H-1,4-benzoxazine are dissolved
in 350 ml of acetic acid. 8.4 ml (0.134 mol) of nitric
acid are added dropwise and the reaction mixture is
left stirring for 4 hours at approximately 100C. The
acetic acid is evaporated under reduced pressure,
100 ml of toluene and 500 ml of 3N sodium hydroxide
solution are added and the reaction mixture is left
stirring at the reflux temperature for 3 hours. The
reaction mixture is separated by settling and the
21 21 71579
agueous layer is extracted with toluene. The organic
phases are combined, washed with water, dried and
evaporated under reduced pressure. The residue obtained
is purified by chromatography on a silica gel column,
elution being carried out with a mixture of
dichloromethane and heptane (50:50). Two products are
separated.
3.8 g of (S)-7-fluoro-3-methyl-5-nitro-3,4-dihydro-2H-
1,4-benzoxazine are obtained
Melting point : 116C
t~20 = - 65 (c = 0.016, dichloromethane)
and 15 g of its regioisomer, (S)-7-fluoro-3-methyl-
6-nitro-3,4-dihydro-2_-1,4-benzoxazine are obtained
Melting point : 132C
[~] 20 = _79.7 (c = 0.01, dichloromethane)
7.6. (S)-7-fluoro-3-methyl-3,4-dihydro-2H-
1,4-benzoxazin-5-amine
A solution of 3.45 g (0.0162 mol) of ~S)-
7-fluoro-3-methyl-5-nitro-3,4-dihydro-2H-1,4-
benzoxazine in 90 ml of ethanol and a catalytic amountof platinum oxide are introduced into a Parr flask.
Hydrogenation is carried out for 1 hour at room
temperature under a pressure of 0.28 MPa (40 psi). The
catalyst is filtered off and washed with ethanol, the
filtrate is recovered and the solvent is evaporated to
dryness. 2.9 g of crystalline product are obtained.
Melting point : 72C
2 1 7 1~79
[~]D0 = _ 70.30 (c = 0.01, dichloromethane)
7.7. (8)-8-fluoro-4-methyl-4,5-dihydroimidazo-
tl,5,4-de][1,4]benzoxazin-2(lH)-one
2.9 g (0.0162 mol) of (S)-7-fluoro-3-methyl-
3,4-dihydro-2H-1,4-benzoxazin-5-amine are heated at
175C with an oil bath for 1.5 hours in the presence of
1.8 g (0.03 mol) of urea. The residue is taken up in a
water:ether (50:50) mixture and is triturated in order
to obtain a precipitate which is filtered off, washed
with water and with ether and then dried.
3.1 g of product are obtained.
Melting point : 175C
t~ 20 = + 33.9o (c = o.o1, dimethylformamide)
7.8. (S)-2-chloro-8-fluoro-4-methyl-
4,5-dihydroimidazo[1,5,4-de]t1,4]benzoxazine
3.1 g (0.0148 mol) of tS)-8-fluoro-4-methyl-
4,5-dihydroimidazo[1,5,4-de][1,4]benzoxazin-2(lH)-one
are heated at the reflux temperature for 3 hours in
70 ml of phosphoryl chloride. The solvent is evaporated
to dryness, the residue is taken up in ice-cold water
and the pH of the mixture is adjusted to 8 with aqueous
ammonia. Extraction is carried out twice with ether,
the organic phases are combined, washed with water and
dried and the solvent is evaporated to dryness.
Purification is carried out by chromatography on a
silica gel column, elution being carried out with a
2l7 l 57~
mixture of ethyl acetate and heptane (50:50).
1.85 g of product are obtained.
Melting point : 102C
[~]DO = _ 15.85 (c = 0.01, dichloromethane)
7.9. ~S)-8-fluoro-4-methyl-2-[4-(5-methyl-lH-imidazol-
4-yl)piperidin-1-yl]-4,5-dihydroimidazo-
[1,5,4-de][1,4]benzoxazine
A mixture of 1.47 g (0.00617 mol) of
4-(5-methyl-la-imidazol-4-yl)piperidine, 0.7 g (0.00308
mol) of (S)-2-chloro-8-fluoro-4-methyl-
4,5-dihydroimidazo[1,5,4-de][1,4]benzoxazine and 4 ml
of isoamyl alcohol is heated at 120C for 24 hours with
stirring. The solvent is evaporated to dryness and the
residue is purified by chromatography on a silica gel
column, elution being carried out with a mixture of
dichloromethane, methanol and agueous ammonia
(95/5/0.5)-
After recrystallization from ethanol, 0.6 g of product
is obtained in the base form.
Melting point: 224C
[~]20 = _ 11.0 (c = 0.991, methanol)
Example 8 (Compound No. 13)
(S)-8-fluoro-2-[4-(lH-imidazol-4-yl)piperidin-1-yl]-
4-methyl-4,5-dihydroimidazo[1,5,4-de][1,4]benzoxazine
(Z)-but-2-enedioate (1:2)
There is obtained, from a mixture of (S)-
21 71~79
2-chloro-8-fluoro-4-methyl-4,5-dihydroimidazo-
[1,5,4-de][1,4]benzoxazine and 4-~lH-imidazol-4-
yl)piperidine, treated under the conditions of Example
7.9., the product in the base form which is purified by
chromatography on a silica gel column, elution being
carried out with a mixture of dichloromethane, methanol
and aqueous ammonia (95/5/0.5).
By adding to the base two equivalents of
maleic acid in methanol, 0.26 g of maleate is obtained.
Melting point : 142C
[h]20 = + 8.13 (c = 0.504, methanol)
Rey to the table :
in the "Salt" column : the ratio between
brackets represents the ratio (salt:base); the absence
of any mention means that the product is in the base
form; "HCl" corresponds to a hydrochloride and "mal."
corresponds to a maleate
2171~79
~ o ~
.C N ~ N
t~
P~ U a~ 0 N 1
O .~
_ N N N NU~ N Ul N
~I N
,1~1
~r ~ I 1 5 ~ I I I I
Z~
f ~ ~ U~ I I U~ I U
~ZJ
Z~z~ I X ~ U~
)~x
U U U U
,~ ~
# O O O O ~ ~ O O
2 i 7 1~7~
., --, ..
~, o ,, .,
,c I I I I 1
r~ N ~ N N ~ ~ N
o~J Ir~ ~ ~ N
~, _~1 0 ~ ~ N r~
~I
U ~ U C~
P: I I I I I I I I
X O O O O O O O O -I
33
O 0, o ,1 ~, 1 n ,, 0
o
o o
U ~,
21 71~9
27
The compounds of the invention formed the
subject of pharmacological tests which demonstrAted
their advantage as therapeutically active substances.
They were in particular tested for their
inhibitory effects on the binding of t3H]-(8)-zacopride
to type 5-HT3 serotoninergic receptors of the rat
cortex, according to the method described by
N.M. Barnes et al., in J. Pharm. Pharmacol., ~0, 548-
551 ~1988).
Male Sprague-Dawley rats (OFA, Iffa credo) weighing 200
to 250 g are humanely killed and their brains are
removed. The cortex is dissected and is homogenized
using a Polytron- mill (7.20 s position) in 20 volumes
of 25 mM Tris buffer ~pH = 7.4, 22C). The homogenate
is centrifuged for 10 min at 45000 x g (in a 8Orvall
centrifuge equipped with an 8S34 rotor) and the pellet
is then resuspended in 10 volumes of Tris buffer and
incubated at 37C for 10 min with stirring. The
suspension is then diluted to 20 volumes using Tris
buffer and centrifuging is carried out under the same
conditions as above. The pellet obtained is resuspended
in 5 volumes of Tris buffer and then divided into 5 ml
aliquot fractions which are frozen at -80C. On the day
of the experiment, the preparation is defrosted to 4C
and then diluted 1.2 times using Tris-NaCl incubation
buffer (25 mM Tris, 150 mM NaCl, pH = 7.4, 22C).
The membrane suspension (100 ~1, 1 mg of proteins) is
incubated at 25C for 25 min in the presence of 0.5 nM
2 1 7 1 579
28
of [3H]-(S)-zacopride (specific activity :
75-85 Ci/mmol, Amersham, Little Chalfont, United
Kingdom) in a final volume of 500 ~l Tris-NaCl buffer,
in the absence or in the presence of the test compound.
Incubation is halted by filtration using ~hatman GF/B
filters pretreated with polyethyleneimine (0.1%). Each
reaction tube is prediluted with 4 ml of Tris-NaCl
buffer and then rinsed 3 times with 4.5 ml of Tris-NaCl
buffer.
The filters are cut up beforehand before drying in an
oven (120C, 5 min). The radioactivity retained on the
filters is measured by liquid scintigraphy. The non-
specific binding is determined in the presence of 10 ~M
of MDL 72222.
For each concentration of study compound, the
percentage of inhibition of the specific binding of
[3H]-(S)-zacopride and then the concentration of the
compound which inhibits 50% of the specific binding of
[3H]-(S)-zacopride (IC50) are determined.
The IC50 values of the compounds of the invention lie
between 0.5 nM and 1 ~M.
The compounds of the invention were also
studied for their affinity with respect to 5-HT4
receptors in the striatum of guinea pigs according to
the method described by Grossman et al. in Br. J.
Pharmacol., 109, 618-624 (1993).
Guinea pigs ~Hartley, Charles River) weighing 300 to
400 g are humanely killed and their brains are removed.
217157~
The striata are excised and are frozen at -80C. On the
day of the experiment, the tissue is defrosted to +4C
in 33 volumes of 50 mM Hepes-NaOH buffer (pH 7.4 at
20C) ~nd is homogenized using a Polytron~ mill. The
homogenate is centrifuged for 10 min at 48,000 x g, the
pellet is recovered, i8 resuspended and is
recentrifuged under the same conditions. The final
pellet is suspended in Hepes-NaOH buffer l30 mg of
fresh tissue/ml). This membrane suspension is used as
iS.
100 ~l of the membrane suspension are incubated at 0C
for 120 minutes, in the presence of 0.1 nM of
[~]GR113808 (specific activity 80-85 Ci/mmol), in a
final volume of 1 ml of Hepes-NaOH buffer (50 mM, pH =
7.4), in the absence or in the presence of the compound
under test. Incubation is halted by filtration through
Whatman GF/BR filters pretreated with 0.1%
polyethyleneimine, each tube is rinsed with 4 ml of
buffer at 0C and filtered again. The radioactivity
retained on the filters is measured by liquid
scintigraphy. The non-specific binding is determined in
the presence of 30 ~M serotonin.
The specific binding represents 90% of the total
radioactivity recovered on the filter.
For each concentration of study compound, the
percentage of inhibition of the specific binding of
[3H]GR118808 and then the concentration of the tested
compound which inhibits 50% of the specific binding
2 1 7 1 579
(IC~) are determined.
The IC~ values of the compounds of the invention lie
between 0.02 and 2 ~M.
The results of the biological tests show that
the compounds of the invention are antagonists of 5-HT3
And/or 5-HT4 serotoninergic receptors.
They may hence be used for the treatment and prevention
of disorders in which 5-HT3 and 5-HT4 receptors are
involved, such as nausea and vomiting, for example
following antitumour treatment or the administration of
an anaesthetic; disorders of the central nervous system
such as schizophrenia, mania, anxiety and depression;
disorders of cognition such as senile dementia or
Alzheimer's presenile dementia; dyskinesia, pain,
migraine and headache; disorders associated with
alcohol or drug dependence or withdrawal; disorders of
gastrointestinal function such as dyspepsia, peptic
ulcer, heartburn, flatulence; disorders of the
cardiovascular system and respiratory disorders.
They may also be used for the treatment and prevention
of disorders such as diarrhoea, irritable colon,
oesophageal reflux, intestinal motor disorders,
disorders of intestinal secretion, cystic fibrosis of
the pancreas, carcinoid syndrome and incontinence.
The present invention also provides a
pharmaceutical composition which comprises a compound
of formula (I) and an excipient, which may be in any
form suitable for oral or parenteral administration,
2l71579
31
such as a tablet, dragée, capsule, including a hard
gelatin capsule, or a suspension or solution to be
swallowed or injected, and in doses that enable 0.005
to 5 mg/kg to be administered 1 to 4 times a day.
The present invention al~o provides a compound of
formula (I) for use in a method of treatment of the
human or animal body.
The present invention further provides the use of
a compound of formula (I) in the manufacture of a
medicament for use in the treatment or prevention of a
disorder in which 5-HT3 and/or 5-HT4 receptors are
involved.
There is also disclosed a method of treatment of a
subject suffering from a disorder in which 5-HT3 and/or
5-HT4 receptors are involved which comprises
administering to that subject an effective amount of a
compound of formula (I).
The present invention also provides a composition
for treating or preventing a disorder in which 5-HT3
and/or 5-HT4 receptors are involved comprising a
compound of formula (I) and a pharmaceutically
acceptable adjuvant.