Note: Descriptions are shown in the official language in which they were submitted.
WO 95108541 217 ~ J ~ ~ pCT/US94/10794
-1-
CONFORMATIONALLY LOCKED NUCLEOSIDE ANALOGUES
FIELD OF THE INVENTION
This invention relates to nucleoside analogues and
methods for their synthesis. More specifically, it relates to
nucleoside analogues containing a cyclopropane-fused
carbocyclic ring.
BACKGROUND OF THE INVENTION
Nucleoside analogues, such as those species lacking the
3' hydroxyl group or both the 2' and 3' hydroxyl groups, of
the naturally-occurring nucleosides can act as chain
terminators of the DNA into which they are incorporated.
Intense effort has focused on the design and use of these
compounds as inhibitors of viral replication (Van Roey et al . ,
(1990) Ann. NY Acad. Sci., 616: 29). Although the
conformation of the sugar moiety in these analogues is
believed to play a critical role in modulating biological
activity, including the anti-HIV 1 activity mediated by
derivatives such as 3'-azido-3'-deoxythymidine (AZT) and
dideoxyinosine (ddI), the main problem encountered in
attempting to correlate a specific type of sugar conformation
with the biological activity of nucleoside analogues is that
the sugar ring is quite flexible and its conformation in
solution can differ markedly from its conformation in the
solid state (Jagannadh, et al., (1991) Biochem. Biophys. Res.
Commun., 179: 386; Plavec et al., (1992) Biochem. Biophys.
Methods, 25: 253.). Thus, any structure-function analysis
based solely on solid state conformational parameters would be
inaccurate unless it was previously determined that both
solution and so~id-state conformations were the same.
Some [3.1.OJ-fused 2',3'-modified cyclopropane-fused
dideoxynucleosides (Figure lA; Wu and Chattopadhyaya, (1990)
Tetrahedron Lett., 46: 2587; Okabe and Sun, (1989) Tetrahedron
Lett., 30: 2203; Beard et al., (1990) Carbohyd. Res., 205: 87;
Codington et al., (1962) J. Org. Chem., 27: 163) appear quite
rigid and their altered sugar moiety shows the same
conformational preference in solution as in the solid state.
However, the conformation of the furanose ring in these
WO 95/08541 PCT/US94/10794
_2_
compounds is well outside the typical range of the Northern
(N) or Southern (S) geometry conformations that are
characteristic of nucleosides (Koole et al., (1991) J. Org.
Chem., 56: 6884). A different type of [3.1.0) fusion, an
epoxide ring between carbons 4' and 6', is found in the
naturally-occurring carbocyclic nucleoside analogue neplanocin
C (Kinoshita et al., (1985) Nucleosides & Nucleotides, 4:
661), which allows this compound to adopt a rigid N-geometry.
In solution there is a dynamic equilibrium between N and
S type furanose conformers (Taylor et al. , (1990) An ti viral
Chem. Chemother., 1: 163-173). The conformations of
nucleosides and their analogues can be described by the
geometry of the glycosyl link (syn or anti), the rotation
about the exocyclic C4'-C5' bond and the puckering of the
sugar ring leading to formation of the twist and envelope
conformations. Two conformations are preferred for ribose
ring puckering: C3'-endo (N) and C2'-endo (S). The endo and
exo refer to displacement of the atom above or below the plane
of the ribose ring, respectively. The torsion angles X [C2-
N1-C1'-04' (pyrimidines) or C4-N9-C1'-04' (purines)l and y
(C3'-C4'-C5'-05') describe, respectively, the orientations of
the base and the 5'-hydroxyl group relative to the ribose
ring.
In ribonucleosides and 2'-deoxyribonucleosides, two types
of sugar puckering are generally energetically preferred,
namely the C3'-endo (N) and the C2'-endo (S) conformations.
In DNA duplexes, a 2'-endo (S) conformation of the repeating
nucleoside unit confers upon the double helix a B
conformation, whereas the 3'-endo (N) conformation induces an
A-conformation double helix. The A and B forms of DNA differ
in the number of base pairs per turn, the amount of rotation
per base pair, the vertical rise per base pair and the helical
diameter. In addition, in stretches of DNA containing
alternating purines and pyrimidines, a left-handed helix
called Z-DNA may form.
Since DNA in solution may exist in several different
conformations, the present invention provides a means of
CA 02172534 2004-07-16
. _ . . . , ~fls ~"~l y
I~ ~
n f ff~ ff3 ffff !! !!f.!
f f r , : f ! f ! f f ! t
p ~ r r ' S ! f ! ! f !~ O ! ! ! ~,
locking DNfA iFj.to a SpeG~ifc ~co~forrlatio.r ~ This can be useful
r r : r,. !~. ,.,. ~r r !! !!! .
in elucidating the structural requirements influencing DNA
protein, DNA-DNA and DNA-RNA interactions and the development
of valuable therapeutics able to specifically block these
interactions.
SUMMARY OF THE INVENTION
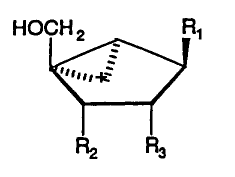
The present invention provides conformationelly locked
compounds having the formula
' HOCH 2 R~
...."*
i
wherein R1 is adenine, guanine, cytosine, thymine, uracil or
a derivative thereof and RZ and R3 are independently H or OH.
In a preferred embodiment, the compounds are locked in the
Northern configuration. In various embodiments of the present
invention either Rz=R3=H, RZ=OH and R3=H or Ra=R3=OH. According
to a preferred embodimgnt of the invention, there are provided
carbocyclic-4',6'-cyclopropane-fused-2',3'-derivatives of
dideoxypurines, dideoxypyrimidines, ~ deoxypurines,
deoxypyrimidines, purine ribonucleosides, and pyrimidine
ribonucleosides. The invention also includes the
corresponding conformationally locked nucleot~iides . According
to another aspect of the invention there are provided
oligonucleotides comprising the aforementioned compounds
wherein RZ=OH and R3=H or R2=R3=OH and oligonucleotides
consisting essentially of the aforementioned compounds wherein
RZ=OH and R3=H or R2=R3=OH .
According to another aspect of the invention there is
provided a process for preparing the adenosine and guanosine
species of the above compounds comprising providing a
cyclopropane-fused cyclopentane compound,. condensing the
cyclopropane-fused cyclopentane compound with a 6-halopurir~e
or a 2-amino-6-halopurine to form a condensation product
wherein said purine derivative is linked to said cyclopentane
AMENDED S~-IEET
I P EA/E P
CA 02172534 2004-07-16
:~
-4-
' ' ~ f r r r r : r . r r r r r r
r . r r . ~ (' f f ( ~ r f
' compound t'~roLgh a Clf' ~~9 rbc~nd; r aizd ,tH~~~ displacing the halo
r r r r r r
group of the 2=amino=6 ~ nalopuf irf~' with az ami.no group to form
a guanosine analogue. In a particular embodiment of the
process, the cyclopropane-fused cyclopentane compound is a
dihydroxy cyclopropanated allylic alcohol, and the
condensation product is a purine ribonucleoside analogue.
According to another aspect of the invention, there is
provided a process for preparing conformationally locked
pyrimidine nucleoside analogues of the invention, comprising
the steps of providing a cyclopropane-fused cyclopentane
compound, and condensing the cyclopentane compound with an N3-
protected thymine or an N3-protected uracil to form a
condensation product wherein the thymine or uracil derivative
is linked to the cyclopentane compound through a C1'-N1 bond.
A corresponding cytidine analogue is formed by the further
steps of protecting the hydroxyl groups or a uracil analogue
of the invention, prepared as described above, treating that
analogue to form a triazole intermediate at C4 of uracil, and
displacing the triazole group by aminolysis to form the
cytidine analogue. In-;a particular embodiment of this aspect
of the invention, the cyclopropane-fused cyclopentane compound
is a protected dihydroxy-cyclopropanated allylic alcohol, and
the product comprises a pyrimidine ribonucleoside analogue.
Deoxynucleoside analogues of the conformationally locked
nucleoside analogues of the invention are.prepared by a
radically-induced deoxygenation of the C-2' hydroxyl function
of the corresponding ribose nucleoside analogue.
The present invention also provides the compound
3 0 RCI-tz0
~~~''OH
3 5 Me3C0
wherein R is selected from the group consisting of alkyl,!
40 aryl, alkylaryl and aroyl.
AMENDED S'~icET
IPEA/EP
21725x4
WO 95/08541 PCT/US94/10794
-5-
St.i 11 another embodiment of the invention is a method for
preparing the above compound, comprising the steps of:
(a) providing
R ~CH2o
~~OH
OX
(b) reacting the compound of step (a) with trimethyl
aluminum to form tent-butyloxy and hydroxy substituents
at the 4' and 5' positions, respectively;
(c) protecting the hydroxy group at the 1' position
by silylating the compound of step (b) with a bulky
group;
(d) forming an O-(methylthio)thiocarbonyloxy group
at the 5' position;
(e) removing said O-(methylthio) thiocarbonyloxy
group;
(f) removing said silyl group from the 1' position
to form an allylic alcohol; and
(g) cyclopropanating said allylic alcohol to form a
carbocyclic alcohol.
BRIEF DESCRIPTION OF THE DRAWINGS
Figure 1 shows the 4',6'-cyclopropane-fused carbocyclic
dideoxynucleoside analogues according to the present
invention: adenosine (5), guanosine (6), thymidine (7),
uracil (8) and cytidine '(9). ,
Figure 2 shows the conformation of compound 12 as deduced
by 1H-NMR. The double arrows represent reciprocal positive
enhancements in a 1-D Nuclear Overhaeuser Effect (NOE)
difference experiment.
Figure 3 shows a cytopa~hic effect assay using HIV 1-
infected ATH8 cells. The drug concentration is indicated on
the x-axis and the number of viable cells x 10,000 is
indicated on the y-axis. 80A10 corresponds to compound (5)
and ddI is dideoxyinosine. The open bars indicate uninfected
cells treated with the drug. The closed bars indicate
infecte~= yells treated with the drug.
WO 95/08541 PCT/US94/10794
6
DETAILED DESCRIPTION OF THE INVENTION
The compounds of the present invention represent the
first examples of carbocyclic dideoxynucleosides that in
solution exist locked in a defined N-geometry (C3'-endo)
conformation typical of conventional nucleosides. These
analogues exhibit increased stability due to the cyclopropane-
fused group and the substitution of carbon for oxygen in the
ribose ring. The dideoxyadenosine analogue exhibits anti-HIV
activity in vi tro.
As used herein, the terms oligonucleoside and
oligonucleoside refer to two or more contiguous nucleosides or
nucleotides in 3'-5' phosphodiester linkage. In a chemical
synthesis, the term "protection" refers to the addition of a
chemical substituent prior to a reaction in order to prevent
the group to which the substituent is attached from reacting.
"Deprotection" refers to the removal of the protecting group.
This invention includes 4',6'-cyclopropane-fused
carbocyclic dideoxynucleosides, 2'-deoxynucleosides and
ribonucleosides as well as oligonucleotides derived from these
analogues. These oligonucleotides may be synthesized
exclusively from the nucleoside analogues of the present
invention. In addition, oligonucleotides derived from one or
more of the nucleoside analogues in combination with the
naturally-occurring nucleosides are also within the scope of
the present invention. These compounds comprise the five
naturally-occurring nitrogenous bases adenine, guanine,
cytosine, thymine and uracil. The present invention also
includes methods of synthesis of these dideoxynucleoside
analogues as well as the corresponding 2'-deoxyribo-
nucleosides and ribonucleosides.
The synthetic scheme for preparing the adenosine,
guanosine, cytidine, thymidine and uridine dideoxynucleoside
analogues of the present invention is described in the
following example. These analogues contain the naturally-
occurring nitrogenous bases found in DNA and RNA in either N9
(purines) or N1 (pyrimidines) linkage to the cyclopropane-
fused carbocyclic ribose ring. The synthesis and utilization
?17L~34
WO 95/08541 PCT/US94/10794
_7_
of cyclopropane-fused carbocyclic nucleoside analogues
containing modified bases by virtue of substitution of the
purine and pyrimidine rings is also within the scope of the
present invention.
The salient features of the nucleoside analogue
synthetic scheme that is exemplified include a "hydroxyl-
directed" cyclopropanation of a 5'-OH-protected allylic
alcohols 10 and 29 via a samarium (2+) carbenoid intermediate
(Molander and Etter, (1987) J. Org. Chem. 52: 3942; Molander
and Harring, (1989) J. Org. Chem., 54: 3525) to generate
4',6'-cyclopropane-substituted carbocyclic alcohols 11 and 30
(Examples 1,2,21). Thi chemistry can be carried out
analogously using 2',3',5'-OH protected carbocyclic compounds.
The 2'-deoxynucleoside analogs are prepared according to
the same synthetic schemes as the dideoxynucleoside analogs,
with the exception that the carbocyclic alcohol contains a
tert-butyl group at the 4 position (corresponding to the 3'
position in the corresponding deoxynucleoside) which protects
the OH group during the reaction steps. Since this tert
butyl-substituted carbocyclic alcohol 30 is a chiral material,
no optical resolution of the synthesis products is required to
separate out the active isomer from a racemic mixture. The
synthesis of the tert-butyl carbocyclic alcohol is briefly
described below, and in detail in Examples 16-22.
(1S,4R,5S)-3-[(benzyloxy)methyl)-4,5-O-isopropylidene-2-
cyclopenten-1-of (24) is prepared from the 1-carbonyl (23)
according to the procedure of Marquez et al., (J.~ Org. Chem.,
53:5709, 1988). Although a benzyloxy group was present at
position 3, the use of any 3-aryloxy, alkyloxy, alkylaryloxy
or aroyloxy group for the subsequent synthetic steps is within
the scope of the present invention. Compound 24 is then
reacted with trimethylaluminum to form Compound 25 containing
tert-butyloxy and hydroxy substituents at the 4 and 5
positions, respectively. The tert-butyl group effectively
protects the 4-hydroxy group in all subsequent synthetic
steps. Compound 25 is then silylated to F~~tect the hydroxy
group at the 1 position, forming compound 2~, . It is important
WO 95/08541 PCT/US94/10794
_8_
~, J
2.~ 1 , ,
that the silylating agent be a large, bulky chemical group in
order to effectively repel the tert-butyl group, thus allowing
the next synthetic step to proceed efficiently. Compound 26
is reacted with methyl iodide and carbon disulfide to form a
(methylthio) carbonyloxy group at the 5 position (Compound
27). This bulky group is able to fit between the silyl and
tert-butyl group due to repulsive forces acting between the
bulky groups. The entire O-(methylthio) carbonyloxy group is
then removed with tri-n-butyltin hydride to form compound 28.
The silyl group is then removed with tetrabutylammonium
fluoride to form the allylic alcohol 29. The allylic alcohol
is then cyclopropanated as described above to form the
cyclopropanated carbocyclic alcohol 30 containing a tert-butyl
substituent at the 4 position which protects the hydroxy group
at this position.
The purine nucleoside analogues are then prepared as
described in Examples 3-10. Both carbocyclic alcohols 11 and
30 are used as starting materials for purine synthesis. The
carbocyclic alcohol 11 is reacted with a 6-halo-substituted
purine ring in a Mitsunobu-type condensation (Mitsunobu,
(1980) Synthesis 1,; Jenny et al., (1991) Tetrahedron Lett.
32: 7029; Jenny et al., (1992) Helv. Chim. Acta. 75: 1944) to
form halogenated intermediates 13 (Example 5) and 14 (Example
6). Although 6-chloropurine and 2-amino-6-chloropurine were
used, it is envisioned that other substitutions at the 6-
position could be incorporated into the synthetic scheme to
generate modified purine analogues. Furthermore, the use of
other halogen-substituted purines is also contemplated. The
substituent chlorine group is then displaced by ammonia to
generate the 5'-OH protected adenosine 16 (Example 7) or
displaced by benzylate to generate the 5'-OH and 6-OH
protected guanosine 17 (Example 8) derivatives which are then
deblocked to generate the adenosine and guanosine derivatives
5 (Example 9) and 6 (Example 10).
The main features of the pyrimidine analogue synthetic
scheme are described in Examples 11-15. This scheme includes
condensation of N-3 protected uracil or N-3 protected thymine
l 7?534
WO 95/08541 PCTIUS94/10794
-9-
with carbocyclic alcohols 11 or 30 to generate the protected
Mitsunobu condensation products 18 (Example 11) and 19
(Example 12). The use of substituted N-3 protected bases is
also within the scope of the present invention. Compounds 18
and 19 are then deprotected to generate the thymidine analogue
7 (Example 13) and the uracil analogue 8 (Example 14). The
5' -OH group of compound 8 is then protected with an acetyl
group to generate intermediate 21 (Example 15) and the
resulting compound is derivatized with a triazole group at C4
of the pyrimidine ring, although the use of a number of groups
for this derivatization is contemplated. The triazole group is
then displaced by ammonia to generate the cytidine derivative
9 (Example 15).
Synthesis of Conformationallv Locked Ribonucleosides
The ribonucleoside species of the invention are prepared
by replacing the carbocyclic allylic alcohol of Examples 3, 4,
9, and 10 with a the equivalent isopropylidene-protected
carbocyclic allylic alcohol formed from D-ribose according to
the procedure set forth in Marquez, V. et al. (Compound 8a in
J. Org. Chem. (1988), 53: 5709-5714). This compound is then
cyclopropanated as described in Example 1. The remainder of
the synthetic steps are the same commencing with the Mitsunobu
coupling reaction as described in Examples 3, 4, 9, and 10.
One of ordinary skill in the art will appreciate that
there are a number of alternative ways in which to perform
certain synthetic steps of the present invention. using well
known reactions.
The dideoxynucleoside analogue compounds of the present
invention are incapable of forming oligonucleotides, DNA or
RNA due to the absence of a 3' hydroxyl group necessary for
chain elongation; however the ribonucleoside and
deoxyribonucleoside species, having the 3'-hydroxyl present,
can react to form the polynucleotides in the manner of tr-e
naturally occurring nucleoside species.
Once these 2'-deoxyribonucleoside and ribonucleoside
analogues are synthesized, and phosphoramidites of the base
WO 95/08541 PCT/US94/10794
J~ i
-10-
analogues, which will react like conventional
phosphoramidites, are prepared, one of ordinary skill will be
able to synthesize oligodeoxyribonucleotides and
oligoribonucleotides using readily available automated
S synthesizers by methods well known in the art (Gait, M.J. ed.,
Oligonucleotide Synthesis: A Practical Approach, Washington,
D.C.: IRL Press, 1984). In addition, the preparation of the
corresponding nucleotides containing attached phosphate groups
using well known chemical methods is also within the scope of
the present invention.
These 2'-deoxyribonucleoside and ribonucleoside analogues
are expected to adopt a rigid N-geometry conformation in
solution. The 2'-deoxyribonucleoside and ribonucleoside
analogues are useful for the same purposes as are conventional
nucleosides. They can be used, for example, to synthesize
nucleic acid coding sequences, regulatory sequences known to
interact with DNA binding proteins, and to synthesize probes
for hybridization analysis. These oligonucleotide analogues
can also be inserted into expression vectors from which
functional sequences will be transcribed. Each analogue is
thus useful in forming functional DNA or RNA oligonucleotide
analogues. DNA and RNA oligonucleotides containing the 2'-
deoxynuceoside and ribonucleoside analogs will exhibit
increased stability and will be useful as antisense molecules
for inhibiting mRNA translation for a gene of interest.
Furthermore, the nucleoside analogues can be used as a
scientific tool to synthesize and lock DNA into the A-
conformation thus allowing subsequent discoveries.
The synthesis of 2'-deoxyribonucleoside and
ribonucleoside analogues will allow the preparation of
molecules which will interact more strongly with proteins and
enzymes requiring Northern conformers for substrates. Since
the conformation of a particular nucleic acid is an
equilibrium conformation, the overall interaction of DNA and
RNA binding proteins with DNA and RNA, respectively, is the
sum of the interactions with the various conformations. DNA
and RNA with nucleotide units locked in the N-geometry
217254
WO 95/08541 PCT/US94/10794
-11-
conformation will be useful in elucidating the interactions
between nucleic acids and nucleic acid binding proteins. This
will lead .to novel therapeutics which can inhibit specific
DNA-protein interactions thus inhibiting the synthesis of
deleterious gene products and/or increasing the synthesis of
beneficial gene products. It is also envisioned that other
types of cyclopropane-fused nucleosides prepared using this
technology will generate compounds locked in the opposite (S)
configuration by simply changir:g the relative position of the
base and the hydroxymethyl groups.
Compared to conventional oligonucleotides,
oligonucleotides synthesized using the nucleoside analogues of
the present invention will have a significantly smaller
entropy change upon duplex formation due to their rigidity.
This will result in a more favorable negative free energy
change upon hybridization relative to conventional DNA
fragments. Using this new methodology, by combining natural
nucleosides with the analogues of the present invention, DNA
fragments can be constructed to create a specific "bend" in
t'~e double helix which may lead to either increased or
decreased binding to proteins and/or nucleic acids.
The foregoing examples are intended to be illustrative
rather than limiting with the full scope of the invention
being defined by the following appended claims.
All chemical reagents were obtained from commercial
sources. Silica gel column chromatography was performed on
silica gel 60, 230-400 mesh (Merck). Analytical thin layer
chromatography (TLC) was performed on Analtech Uniplates
silica gel GF. Proton and 13C-NMR spectra were recorded in a
Brucker AC-250 instrument at 250 and 62.9 MHz, respectively.
NMR conformational studies were performed in a Brucker AMX-500
instrument at 500 MHz. Chemical shifts are expressed as b
values with reference to tetramethylsilane. Positive-ion fast
bombardment (FAB) mass spectra wer obtained on a VG 7070E
mass spectrometer equipped with a FAB ion source. The sample
was dissolved in a glycerol matrix, and ionization was
effected by a beam of xenon atoms. Elemental analyses were
WO 95/08541 PCT/US94/10794
21 1 ~ 5_~ T
-12-
performed by Atlantic Microlab, Inc., Norcross, GA. W
spectra were recorded in a Shimadzu Model UV-2101PC
spectrophotometer. These values are listed after each
synthetic step described below.
A convergent approach was used to incorporate the purine
and pyrimidine bases onto the carbocyclic moiety in one step.
Mitsunobu-type condensations with the appropriate bases were
performed using the common intermediate carbocyclic alcohol,
(+/-) -5- [ (benzyloxy) methyl] -2-hydroxy-cis-bicyclo [3 .1. 0] hexane
(11, Schemes 1 and 2).
The present invention is described below in detail using
the following examples, but the methods described are broadly
applicable for the preparation of all of the nucleoside
analogues described herein and are not limited to the examples
given below.
EXAMPLE 1
Svnthesis of the Intermediate Carbocyclic Alcohol
1-Benzvloxvmethvl-cis-bicyclof3 1 Olhexane-4-of 11
Samarium metal (5.04 g, 33.6 mmol) was added to a dry
round-bottomed flask which was simultaneously flushed with
argon and flamed dry. Anhydrous tetrahydrofuran (THF; 50 ml)
was then added followed by a solution of mercuric chloride
(HgCl2; 0.88 g, 3.2 mmol) in 10 ml THF. The mixture was
stirred for 10 min followed by addition of allylic alcohol 10
(1.52 g, 7.45 mmol). The reaction mixture was cooled to -78'C
followed by addition of chloroiodomethane (CH2IC1; 2.32 ml, 32
mmol) and the mixture was stirred at -78°C overnight. The
mixture was allowed to warm to room temperature and stirred
for one hour. The reaction was quenched with 300 ml saturated
Na2C03 and extracted several times with methylene chloride.
The combined organic layers were washed with brine, dried over
MgS04, filtered and evaporated yielding pure compound 11 as a
colorless oil in nearly theoretical yield. The product was
used in the next step without further purification.
FAB MS (m/z, %) - 201 ({MH-H20}+,9);91 (100); 71 (98).
1H-NMR (CDC13) b - 0.47 (dd, J = 5.2, 8.0 Hz, 1H, H-6 exo) ;
~1 ~~~~:~4
WO 95/08541 PCT/US94/10794
-13-
0.85 (t, J = 5.2 Hz, 1H, H-6 enct~) ; 1.40 (m, 1H, H-5) ; 3.42
(s, 2H, H-7); 4.50 (s, 2H, PhCH2-); 4.55 (m, 1H, H-4); 7.25-
7.40 (m, 5H, aromatic protons). 13C-NMR (CDC13) b - 8.93 (C-
6); 27.17 (C-5); 27.51 (C-3); 28.25 (C-1); 29.64 (C-2); 72.39
(C-7); 73.60 (C-4); 74.42 (C-8); 127.36 (C-3' & C-4'); 128.17
(C-2' ) 138.35 (C-1' ) .
EXAMPLE 2
4-Acetoxv-1-benzyloxy-cis-bicyclo-f3 1 O1-hexane 12
A solution of compound 11 (109 mg, 0.5 mmol) in 3 ml
anhydrous pyridine was treated with 2 ml acetic anhydride and
the mixture was stirred overnight at room temperature. The
solvent was evaporated and the residue purified by flash
chromatography eluting with hexane-ethyl acetate (4:1) to
yield mg of the stable acetate derivative 12.
Results from the NOE difference 1H-NMR spectra for the
acetate 12 (Figure 2) agreed well with the disposition of the
bicyclic system as inferred by the mode of cyclopropanation.
1H-NMR (CDCL3) b=0.54 (dd, J = 5.4, 8. 0 Hz, 1H, H-6 exo) ;
0.85 (t, J = 5.4 Hz, 1H, H-6 endo); 1.53 (m, 1H, H-5); 2.02
(s, 3H, COCH3); 3.43 (s, 2H, H-7); 4.50 (s, 2H, PhCH2-); 5.30
(dt, J = 4.7, 8.2 H, 1H, H-4); 7.30 (m, 5H, aromatic protons).
13C-NMR (CDC13) b - 9. 99 (C-6) ; 21.12 (COCH3) ; 24.49 (C-5) ;
26.36 (C-3); 27.05 (C-C-2); 28.50 (C-1); 72.52 (C-7); 74.19
(C-8); 76.77 (C-4); 127.41 (C-3' & C-4'); 128.23 (C-2');
138.32 (C-1' ) ; 171.23 (CO) . Anal . Calculated for C16H2oO3 = C
73.82 H 7.74. Found C 73.71, H 7.79.
WO 95108541 PCT/US94/10794
-14-
Synthesis of Purine Dideoxynucleoside Analogues
Scheme 1
Bn0 Bn0 C-CI=purine
CH21C1 - or
OH S~ ~ , ~ y ,,%R ~_ NH., _ 6_ CI-purine
HgCl2 ° Ph3 P /DEAD
THF (- 78 C) THF, rt
(100%) ~11, R = H (?8%-38io)
1?, R =Ac
X
~N ~ w
h
Bn0 N N/ Y Bn0 N
~~iit + ~~iit I
13, X = CI, Y = H
14, X = CI, Y = NH2
NH3/MeOH 65°C (54%)
OR
PHCH,,ONo, PhCH,,OH, rt (77%)
X ~ X
N ~ N
Bn0 N N~ Y HO N N~ 1
~yt ~yt
BC13 , CH ~ CI,,
-78°C (55%-86%)
16, X = NHS , Y = H 5. X = NH_~ , Y - H
17, X = OBn, Y - NH" ~~. X= 0H. 1 - NH.,
SUBSTITUTE SHEET (RULE 26)
2 i 7 % '~ 3 'f~
WO 95/08541 PCT/US94/10794
-15-
EXAMPLE 3
5'-Benzvloxv-4' 6'-c~rclopropyl-2' 3'-dideoxy-6
chloroadenosine 13
To a suspension of 6-chloropurine (148 mg, 0.96 mmol) in
3 ml anhydrous THF was added diethylazodicarboxylate (DEAD;
206 mg, 0.96 mmol) and the resulting mixture was stirred
vigorously for 10 min. Alcohol 11 was then added (218 mg, 1
mmol in 5 ml THF) and the reaction was stirred overnight at
room temperature. The solvent was evaporated and the residue
was adsorbed on silica gel and purified by column
chromatography. The products eluted with hexane: ethyl acetate
(3:2) resulting in 75 mg (21% yield) of pure compound 13 as a
white solid and 24 mg of 7-N derivative 15 as a pale yellow
oil.
Compound 13, m.p. - 118-119°C. FAB MS (m/z, o) - 357
(12), 355 (MH+, 35), 247 (11), 155 (34) 91 (100). 1H-NMR
(CDC13)b --- 0.76 (m, 2H, H-7'); 1.56 (dd, J = 4.3, 8.2 Hz,
1H, H-6'); 1.65-2.00 (m, 3H, H-2' & H-3' a); 2.25 (m, 1H, H-
3'(3); 3.29 (d, J = 9.9 Hz, 1H, H-5a'); 3.95 (d, J = 9.9 Hz,
1H, H-5b' ) ; 4.63 (s, 2H, PhH~-) ; 5.22 (d. J = 5.5 Hz, H-1' ) ;
7.36 (m, 5H, aromatic protons); 8.74 (s, 1H, H-8); 9.00 (s,
1H, H-2). 13C-NMR (CDCl3b = 12.30 (C-7'); 26.22 (C-6'); 26.28
(C-2'); 30.26 (C-3' & 4'); 56.94 (C-1'); 72.86 (C-5'); 73.20
(PhCHz-); 127.49 (C-4"); 127.64 (C-3"); 128.52 (C-2"); 131.74
(C-5); 137.95 (C-1"); 144.78 (C-8); 150.65 (C-4); 151.30 (C-
6 ) ; 151. 51 ( C-2 ) . Anal . Calculated for C1gH190N4C1 = C 64 . 38 ,
H 5.41, N 15.82, C1 9.87; Found C 64.43, H 5.47, N 15.82, C1
9.80.
EXAMPLE 4
Carbocyclic-5'-benzyl-4',6'-c5rclopro~wl-2',3'-dideoxy-6-
chloroguanine 14
To a suspension of triphenylphosphine (Ph3P; 2.672 g,
10.24 mmol) and 2-amino-6-chloropurine in 60 ml anhydrous THF
was added DEAD ( 1 . 76 ml , 11 . 26 mmol ) under an argon atmosphere
and the resulting yellow mixture was stirred for 10 minutes at
WO 95/08541 PCT/US94/10794
~~ 7~~c~ jt;.
-16-
room temperature. A solution of compound 11 (670 mg, 3.07
mmol) in 5 ml THF was added and the mixture was stirred for 18
hours at room temperature . The solvent was evaporated and the
residue absorbed on silica gel and purified by column
chromatography eluting with hexane-ethyl acetate to obtain 435
mg (38o yield) of compound 14 as a white solid.
m.p. - 135-137°C. FAB MS (m/z, %) - 372 (15); 370 (MH+,
42) ; 172 (14) ; 170 (43) ; 91 (100) . 1H-NMR (CDCl,b = 0.69 (m,
2H, H-7'); 1.50 (dd, J = 3.9, 8.4 Hz, 2H, H-5'); 3.30 (d, J =
9.9 Hz, 1H, H5a'); 3.88 (d, J = 9.9 Hz, 1H, H5b'); 4.60 (AB q.
J = 14.8 Hz, 2H, PhCHz-); 4.95 (d, J = 5.3 Hz, 1H, H-1'); 5.05
(s, 2H, -NH2); 7.35 (m. 5H, aromatic protons); 8.56 (s. 1H, H-
8) . 13C-NMR (CDC13) b - 12.21 (C-7' ) ; 26.29 (C-6' ) ; 26.46 (C-
2'); 30.19 (C-3')*; 30.26 (C-4')*; 56.26 (C-1'); 73.00 (C-5');
73.17 (PhCH2-); 125.54 (C-5); 127.65 (C-4"); 127.75 (C-3");
128.51 (C-2"); 138.09 (C-1"); 141.84 (C-8); 150.99 (C-6);
153.29 (C-2); 158.76 (C-4. Anal. Calculated for
C19H2oON5C1.2/3Hz0 - C 59.76, H 5.63, N 18.34, C1 9.28. Found
C 59.56, H 5.46, N 18.47, C1 9. 53.
EXAMPLE 5
Carbocyclic-5'-benzyloxy-4' 6'-cyclopro~ yl-2' 3'
dideoxvadenosine 16
Compound 13 (215 mg) was treated with 5 ml of a saturated
solution of ammonia in methanol in a sealed tube and stirred
at 70°C overnight. The mixture was cooled to room temperature
and the solvent was evaporated. The residue was purified by
column chromatography on silica gel and eluted with CHC13
isopropanol (9:1) yielding 109 mg (54% yield) of pure compound
16 as a white solid.
m.p. - 170°C. 1H-NMR (CD30D)b = 0.76 (m, 2H, H-7'); 1.59
(t, J = 6 Hz, 1H, H-6' ) ; 1.65-2. 00 (m. 3H, H-2' & H-3' a) ;
2.20 (m, 1H, H-3'~3); 3.40 (d, J = 10.0 Hz, 1H, H5a'); 3.97 (d,
J = 10.0 Hz, 1H, H5b'); 4.57 (s, 2H, PhCH2-); 5.02 (d, J = 5.7
Hz, 1H, H-1'); 7.2-7.4 (m, 5H, aromatic protons); 8.18 (s, 1H,
H-8); 8.60 (s, 1H, H-2). 13C-NMR (CD30D)b - 12.72 (C-7');
i 7534
WO 95/08541 PCT/US94/10794
-17-
27.27 (C-6'); 27.54 (C-2'); 31.12 (C-3'); 31.36 (C-4'); 57.96
(C-1'); 74.23 (C-5'); 74.80 (PhCHz-); 120.06 (C-5); 128.64 (C-
4"); 128.70 (C-3"); 129.50 (C-2"); 139.80 (C-1"); 141.08 (C-
8); 149.99 (C-4); 153.50 (C-2); 157.27 (C-6). Anal.
Calculated for Cl9H~lONS - C 68.04, H 6.31, N 20.88; Found C
67.86, H 6.34, N 20.80.
EXAMPLE 6
Carbocvclic-5'-benzyl-4' 6'-cyclopropyl-2' 3'-dideoxy-6
benzylQUanidine 17
Anhydrous benzyl alcohol (PhCH20H) was treated with 100
mg sodium and the suspension was vigorously stirred under an
argon atmosphere until no sodium metal remained. Compound 14
was treated with 1.5 ml sodium benzylate and stirred for 10
min. The reaction was quenched with 25 ml water followed by
the addition of 30 ml methylene chloride. The organic layer
was washed with water until pH=7, dried with MgS04 and
evaporated. The residue was purified by flash chromatography
eluting with hexane-ethyl acetate (1:1) to yield 161 mg (77%
yield) of pure compound 17 as a white solid.
m.p. - 171°C. FAB MS (m/z, %) - 442 (MH+, 49); 242 (34);
91 (100). 'H-NMR (CDC138 = 0.66 (m, 2H, H-7'); 1.49 (dd, J =
3.8 8.4 Hz, 1H, H-6'); 3.36 (d, J = 9.9 Hz, 1H, H-5a'); 3.82
(d, J = 9. 9 Hz, 1H, H-5b' ) ; 4.58 (AB q. J = 14. 0 Hz, 2H, PhCH~-
at 0-5' ) ; 4.84 (s, 2H, -NH2) ; 4. 93 (d, J = 5.2 Hz, 1 H, H-
1'); 5.58 (AB q, J = 14.8 Hz, 2 H, PhCH2- at C-6~; 7.26-7.54
(m. lOH, aromatic protons); 8.24 (s. 1H, H-8). 13C-NMR
(CDC13)b = 12.16 (C-7'); 26.47 (C-2'); 26.70 (C-6'); 30.26 (C-
3' & C-4'); 55.78 (C-1'); 67.91 (PhCH2- at C-6); 73.09 (C-5);
73.23 (PhCH2- at 0-5'); 115.69 (c-5); 127.56, 127.63 (C-4");
127.45, 128.18 (C-3"); 128.30, 128.45 (C-2"), 136.61 (C-8);
138.21, 138.63 (C-1"); 153.65 (C-6); 158.90 (C-2); 1'60.90 (C-
4 ) . Anal . Calculated for C26HZ,ONzNS - C 70 . 73 , H 6 . 16 , N
15.86. Found C 70.72, 6.21, N 15.83.
WO 95108541 PCT/US94/10794
,)~_ ~a
' -18-
EXAMPLE 7
Carbocyclic-4',6'-cyclo~ropvl-2',3'-dideoxyadenosine 5
Palladium on 10% charcoal (300 mg) was purged with argon
for 15 min followed by addition of 50 mg compound 16 dissolved
in 5 ml methanol. The resulting mixture was treated with 1 g
ammonium formate and refluxed for 3 hours. The mixture was
allowed to cool to room temperature, filtered and the solvent
was evaporated. The residue was purified using a reverse
phase column eluted with water to yield 12 mg (33o yield) of
pure compound 5 as a pale yellow solid.
m.p. - 251°C (d) . W (MeOH) ~",aX 260.7 (E 15200) . FAB MS
(m/z, o) - 338 ({MH+glycerine}+, 12), 246(MH+, 100), 136 (84),
500 MHz 'H-NMR (DMSO-db) b - 0.66 (m, 2H, H-7' ) ; 1.48 (dd, J
- 3.9, 8.3 Hz, 1H, H-6'); 1.58 (dd, J = 8.2, 14.3 Hz, 1H, H-
3'a); 1.67 (dd, J = 8.0, 12.5 Hz, 1H, H-2'/3); 1.84 (m, 1H, H-
2' a) ; 2 . 07 (dt, J = 8. 0, 12 . 0 Hz, 1H, H-3' (3) ; 3 . 37 (dd, J =
11.4 Hz, J = 5.1 Hz, 1 H, H-5a'); 3.86 (dd, J = 11.4 Hz, J =
5.1 Hz, 1 H, H-5b'); 4.90 (d, J = 6.0 Hz, 1H, H-1'); 4.99 (t,
J = 5.2 Hz, 1H, -OH); 7.17 (s, 2H, -NHS); 8.11 (s, 1H, H-B);
8.37 (s, 1H, H-2). 13C-NMR (CD30D-D~O)b = 12.51 (C-7'); 26.88
(C-6'); 27.11 (C-2'); 30.95 (C-3'); 32.99 (C-4'); 58.34 (C-
1'); 66.21 (C-5'); 119.84 (C-5); 141.05 (C-8); 149.57 (C-4);
153.25 (C-2) ; 156.84 (C-6) . Anal. Calculated for C12H150N5 = C
58.76, H 6.16, N 28.55; Found C 58.63, H 6.19, N 28.52.
EXAMPLE 8
Carbocvclic-4',6'-cycloprogvl-2',3'-dideoxyauGnosine 6
A solution of compound 17 (154 mg, 0.35 mmol) in 35 ml
anhydrous methylene chloride was cooled at -78°C under an
argon atmosphere, treated with 3.0 ml 1.0 M boron trichloride
(BC13) and stirred for 6 hours at -78°C. The solvent was
evaporated and the residue dissolved in 30 ml CH2Clz. The
organic layer was washed with saturated NaHC03 (3 x 30 ml) and
water (2 x 30 ml), dried with MgS04 and evaporated. The
residue was purified using a reverse phase column eluted with
water to yield 50 mg (55 o yield) of pure compound 6 as a white
solid.
WO 95/08541 1 > ~ ~ PCT/LTS94/10'794
-19-
m.p.>300°C. W (MeOH) ~",aX254.4 (e 10500). FAB MS (m/z,
%) - 354 ({MH+glycerine}+, 14), 262(MH+, 100), 152 (66). 1H-
NMR (DMSO-d6) b - 0.60 (m, 1H, H-7' exo) ; 0.84 (m, 1H, H-7'
endo); 1.40 (dd, J = 3.7, 8.1 Hz, H-6'); 3.81 (dd, J = 5.2,
10.4 Hz, 1H, H-Sa'); 4.13 (dd, J = 5.2, 10.4 Hz, 1H, H-5b');
4.64 (d, J = 5.8 Hz, 1H, H-1); 4.95 (t, J = 5.2 Hz, 1H, -OH);
6.60 (s, 2H, -NH2); 7.94 (s, 1H, H-8); 10.66 (s, 1H, H-1).
mC_NMR (DMSO-d6) b = 11.03 (C-7'); 25.50 (C-2'); 25.87 (C-6');
29.73 (C-4'); 31.89 (C-3'); 55.31 (C-1'); 63.80 (C-5'); 116.52
(C-5); 135.19 (C-8); 150.44 (C-6); 153.53 (C-2);56.79 (C-4).
Anal. Calculated for C1zH150zN5.1/6 H20 = C 54.54,
H 5.85, N 26.50; Found C 54.5II.
The pyrimidine derivatives were also synthesized under
Mitsunobu conditions in comparable yields using protected N-3-
benzoyl thymine or N-3-benzoyl uracil (Cruickshank et al.,
(1984) s.Tetrahedron Lett., 681) as described in the following
examples 9-13.
WO 95/08541 PCT/US94110'794
rl ~ ~ .% ~ ~ 4T _20_
L.-
Synthesis of Pyrimidine Dideoxynucleoside Analogues
Scheme ''
Me
0
R~ ~~
N-3-BzUr ~ NRz
or N ~ NR,,
11 N-3-BzThy
Ph3 P/DEAD Bn0 N 0 Bn0
- 78° C (38%) _ = 0
~''~+ + ~''~+
18, R~ = CHI , RL = Bz 20, R" = Bz
19, R ~ = H, R,, = Bz
1. NH40H/MeOH 2. BC13
CH2 C12 ,- 78 C
(46-87%)
0 0
R~
NH ~ NH
HO N \0 Ac 0/pyr Ac0 N \0
= rt ( 100%) __
~iit ~ ~iit
POC13
1,2,4- triazoleF ? 1
7, R~ = CH3
Et3 N, MeCN
g, R1= H N~ _
NH,, N
N~
w
\ N NH40H \NH
dloxane
rt
HO N 0 NHS, MeOH Ac0 N
= rt -
~''t (46%) ~ '' ~+
SUBSTITUTE SHEET (RULE 26)
WO 95108541 ~ ~ PCT/US94/10794
-21-
EXAMPLE 9
Carbocvclic-5'-benzoyloxy-4' 6'-cyclopropvl-2' 3'-dideoxy
3-benzo~rlth~midine 18
To a solution of triphenylp'~osphine (Ph3P; 1.340 g, 5.10
mmol) in 16 ml anhydrous THF was added DEAD (860 ~l, 5.0
mmol). The mixture was stirred for 30 min at 0°C then cooled
to 45°C. To this suspension was added a solution of 3-N
benzoylthymine (N-3-BzThy; 920 mg, 4 mmol) and carbocyclic
alcohol 11 (460 mg, 2.10 mmol) in 16 ml THF over 45 min and
the mixture was stirred overnight at 45°C. The mixture was
warmed to room temperature and the solvent was evaporated.
The residue was purified by flash chromatography, eluting with
hexane-ethyl acetate (7:3) yielding a mixture of N-alkylation
and O-alkylation products. This mixture was repurified by
silica gel column chromatography, eluting with CH~C12-ether
(97.5:2.5) to yield 330 mg (36% yield) of the desired N-
alkylation product 18 as a white solid and 400 mg (44% yield
of the undesired O-alkylation product 20 as an oil. In this
solvent system, compound 18 eluted faster than compound 20.
m.p. - 182-184°C. FAB MS (m/z, %) - 431 (MH+, 30), 323
(6), 231 (13), 105 (100), 91.(79). 1H-NMR (CDC13)b = 0.58 (m,
1H, H-7' exo); 0.71 (m, 1H, H-7' endo); 1.32 (dd, J = 3.7, 8.8
Hz, 1H, H-6'); 1.55 (d, J = 0.8 Hz, 3H, Me at C-5); 1.60-1.90
(m, 3H, H-2' & H-3'a); 2.25 (m, 1H, H-3'(3); 3.26 (d, J = 9.9
Hz, 1H, H-5a'); 4.05 (d, J = 9.9 Hz, 1H, H-5b'); 4.57 (AB q,
J = 16.5Hz, 2H, PhCHZ-); 4.98 (d, J = 5.8 Hz, 1H,'H-1'); 7.35
(m, 5H, benzylic protons) ; 7.50 (t, J = 7.4 Hz, 2H, H-2' ") ;
7 . ~2 (t, J = 7 .4 Hz, 1H, H-4 "' ) ; 7. 91 (d, J = 7.4 Hz, 2H, H-
2"') ; 8.00 (d. j = 0.8 Hz, 1H, H-6) . 13C-NMR (CDC13) 6 = 11.77
(Me at C-5); 12.18 (C-7'); 25.71 (C-2'); 26.31 (C-6'); 30.34
(C-3'); 31.02 (C-4'); 57.48 (C-1'); 73.53 (C-5')*; 73.65
(PhCH2-)*; 110.25 (C-5); 127.99 (C-2 "'); 128.53 (C-3", C-4" &
C-3'"); 129.00 (C-2"); 130.37 (C-4 "'); 134.73 (C-1'"); _. 7.90
(C-6); 138.19 (C-1"); 149.95 (C-2); 162.90 (C-4); 169.46
(C=O) . Anal. Calculated for Cz6HzsO4Nz.1/10 CH2Clz = C
71.41, H 6.02, N 6.38. Found C 72.46, H 5.98, N 6.39.
WO 95!08541 PCT/US94/10'794
~~ L.. L
_.
L ..' -22-
l_ ,
EXAMPLE 10
Carbocvclic-5'-benzyloxv-4' 6'-cyclopropyl-2',3'-dideoxy-3
benzoyluridine 19
To a solution of Ph3P (857 mg, 3.26 mmol) in 10 ml
anhydrous THF was added DEAD (0.500 ml, 3.2 mmol). The
mixture was stirred at 0°C for 30 min and cooled to -78°C. A
suspension of 3-N-benzoyluracil (N-3-BzUr; 550 mg, 2.56 mmol)
and carbocyclic alcohol 11 (280 mg, 1.27 mmol) in 25 ml THF
was added to the reaction mixture over 10 min. The mixture
was stirred overnight at 50°C, allowed to warm to room
temperature and the solvent evaporated. The residue was
purified by column chromatograph(silica gel) using CH2C12-ether
(98:2) as eluent to yield 150 mg (28o yield) of pure compound
19 as a colorless oil.
FAB MS (m/z, %) - 417 (MH+, 48) , 309 (9) , 217 (9) , 105
(100) , 91 (76) . 1H-NMR (CDC13) b - 0.61 (dd, J = 3.5, 5.7 Hz
1 H, H-7' exo); 0.74 (dd, J = 5.7, 8.5 Hz 1H, H-7' endo); 1.31
(dd, J = 3.5, 8.5 Hz, 1H, H-6'); 1.60-1.90 (m, 3H, H-2' & H-
3' a) ; 2 . 20 (m, 1H, H-3' (3) ; 3 . 28 (d, J = 9 . 9 Hz, 1H, H-5a' ) ;
4.07 (d, J = 9.9 Hz, 1H, H-5b'); 4.51 (AB q, J = 16.5Hz, 2H,
PhCH~-); 4.95 (d, J = 5.4 Hz, 1H, H-1'); 5.42 (d, J = 8.1 Hz,
1H, H-5 ; 7.30-7.40 (m, 5H, benzylic protons); 7.47 (t, J =
7.4 Hz, 2H, H-3'"); 7.62 (dt, J = 7.4, 1.0 Hz, 1H, H-4'");
7.92 (dt, J = 7.4, 1.0 Hz, 1H, H-2"') ; 8.26 (d. j - 8.1 Hz,
1H, H-6). 13C-NMR (CDC13)b = 12.25 (C-7'); 25.29 (C-2'); 26.18
(C-6'); 30.22 (C-3'); 30.98 (C-4'); 57.83 (C-1'); 73.28 (C-
5')*; 73.59 (PhCHz-)*; 101.35 (C-5); 127.45 (C-4"); 127.88 (C-
3)*; 128.44 (C-2")*; 128.98 (C-2 "')*; 130.30 (C-3 "'); 131.55
(C-4 "'); 134.80 (C-1"'); 137.95 (C-1"); 142.45 (C-6); 149.88
(C-2); 162.14 (C-4); 169.14 (CO). Anal. Calculated for
CZSH290qNz = C 72 . 10 , H 5 . 81, N 6 . 73 . Found C 71 . 84 , H 5 . 8 9 , N
6.59.
WO 95/08541 ~ ~ ~ ~ ~ J ~ PCT/US94/10794
-23-
EXAMPLE 11
Carbocvclic-4' 6'-cyclo_propyl-2' 3'-dideoxythymidine 7
Compound 18 (150 mg, 0.35 mmol) was suspended in 100 ml
methanol. Concentrated ammonia (4 ml) was then added and the
mixture was stirred at room temperature for 16 hours. The
solvent was evaporated and the residue was dissolved in 30 ml
CH2C12. The organic layer was washed with saturated NaHC03 (3
x 30 ml) and water (2 x 30 ml), dried with MgS04 and
evaporated. The residue was purified by flash chromatography
(silica gel) using CH2Clz-isopropanol (97:3) as eluent to
obtain 105 mg (92% yield) of the N-3 deblocked intermediate as
a white solid.
A solution of the N-3 deblocked intermediate (85 mg in 20
ml anhydrous CH2C12) cooled to -78°C under argon was treated
with BC13 (1.0 M in hexane, 1.80 ml) and stirred for 6 hours
at -78°C. Methanol (4.0 ml) was added at the same temperature
and the mixture was allowed to warm to room temperature. The
solvent was evaporatEd and 4 ml methanol was again added
followed by evaporation of the solvent. This procedure was
repeated 6 times. The residue was purified by C-18 reverse
phase chromatography eluting with water to yield 28 mg (46%
yyeld) of pure compound 7 as a white so~id.
m.p. - 205-207°C. 1H-NMR (CDC13)b - 0.57 (dd, J = 4.0,
5.6 Hz, 1H, H-7' exo) ; 0.68 (dd, J = 5.6, 7.8 Hz, 1H, H--7'
endo); 1.27 (dd, J = 4.0, 7.7 Hz, 1H, H-6'); 1.54 (d, J = 1.0
Hz, 3 H, Me at C-6); 1.65-1.90 (m, 3H, H-2' & H-3'~); 2.20 (m,
1H, H-3'(3); 3.34 (d, J = 9.9 Hz, 1H, H-5a'); 4.01 (d, J = 9.9
Hz, 1H, H-5b'); 4.55 (AB q. J = 16.5 Hz, 2H, PhCH2-); 4.98 (d,
J = 6.1 Hz, 1H, H-1'); 7.34 (m, 5H, aromatic protons), 7.88
(d, J = 1.0 Hz, 1H, H-6); 8.15 (s, 1H, NH). 13C-NMR 9CDC13)b
- 11.78 (Me at C-5); 12.14 (C-7'); 25.78 (C-2'); 26.34 (C-6');
30.25 (C-3'); 30.96 (C-4'); 57.16 (C-1'); 73.47 (C-5')*; 73.63
(PhCH~-)*; 110.22 (C-5); 127.92 (C-4"); 128.50 (C-3"); 128.56
(C-2"); 137.94 (C-6); 138.39 (C-1"); 151.01 (C-2). Anal.
Calculated for C19Hz203N2 - C 69.92, H 6.79, N 8.58. Found C
69.78, H 6.85, N 8.53.
WO 95/08541 PCT/US94/10794
L~ ~ L~~~'~ -24-
,I
EXAMPLE 12
Carbocyclic-4',6'-cyclopropyl-2',3'-dideoxyuridine 8
A solution of compound 19 (120 mg, 0.29 mmol) in 60 ml
methanol was treated with concentrated ammonia and the mixture
was stirred for 16 hours at room temperature. The solvent was
evaporated and the residue was purified by flash
chromatography eluting with CH2C12-isopropanol ( 97 : 3 ) to obtain
77 mg (86% yield) of the N-3 deblocked intermediate as a white
solid.
A solution of this N-3 deblocked intermediate (67 mg,
0.21 mmol) in 16 ml anhydrous CH2C12 was cooled at -78°C under
argon was treated with 1.50 ml 1.0 M BC13 in hexane and
stirred for 6 hours at -78°C. The reaction was quenched as
described for compound 7. The residue was purified by flash
chromatography using CHZC12-isopropanol (9:1) as solvent to
yield 41 mg (87% yield) of pure compound 8 as a white solid.
m.p. - 154-156°C. 1H-NMR (CDC13)b - 0.59 (dd, J = 3.8,
5.7 Hz, 1H, H-7' exo) ; 0.72 (dd, J = 5.7, 8.7 Hz, 1H, H-7'
endo); 1.26 (dd, J = 3.8, 8.7 Hz, 1H, H-6'); 1.55-1.90 (m, 3H,
H-2' & H-3'a); 2.15 (m, 1H, H-3',Q); 3.27 (d, J = 9.9 Hz, 1H,
H-5a'); 4.40 (d, J = 9.9 Hz, 1H, H-5b');4.52 q. J = 16.4 Hz,
2H, PhCH2-); 5.00 (d, J = 5.8 Hz, 1H, H-1'); 5.37 (dd, J =
1.4, 8.0 Hz, 1H, H-5); 8.11 (d, J = 8.0 Hz, 1H, H-6); 9.90 (d,
J = 1 .4 Hz, 1H, -NH) ; 13C-NMR (CDC13) b - 12.19 (C-7' ) ; 25.42
(C-2'); 26.24 (C-6'); 30.16 (C-3'); 30.92 (C-4'); 57.35 (C-
1'); 73.25 (C-5')*; 73.62 (PhCH2-)*; 101.59 (C-5); 127.39 (C-
4"); 127.83 (C-3"); 128.42 (C-2"); 137.96 (C-1"); 142.58 (C-
6 ) ; 151 . 21 (C-2 ) ; 163 . 74 (C-4 ) . Anal . Calculated for C18H2o03N2
- c 69.21, h 6.45, n 8.97. Found C 69.13, H 6.44, N 9.02.
EXAMPLE 13
Carbocyclic-4',6'-cyclopropyl-2',3'-dideoxycytidine 9
Compound 8 (120 mg, 0.54 mmol) was dissolved in 3 ml
anhydrous pyridine, treated with 2 ml acetic anhydride and
stirred overnight. Evaporation of the solvent produced the
5'-monoacetyl derivative 21 in almost theoretical yield. This
WO 95108541 /~ i l ~ ~ 3 l'~ PCT/US94/10794
-25-
compound was used in the next step without further
purification.
Triethylamine (Et3N; 540 ~C1, 3.88 mmol) was added to a
mixture of 1,2,4-triazole (280 mg, 4.05 mmol), phosphorous
oxychloride (F '13; 81 ul, 0.867 mmol) and anhydrous
acetonitrile (2.3 ml) under argon. Compound 21 (100 mg, 0.45
mmol) in 2.0 ml acetonitrile was then added and the reaction
mixture was stirred at room temperature for 24 hours. Et3N
( 375 ~.1, 2 . 67 mmol ) and water ( 97 ~.1 ) were then added, the
mixture was stirred for 10 min and the solvent was evaporated.
The residue was partitioned between CH2C12 (50 ml) and
saturated NaHC03 (50 ml). The organic layer was removed and
the aqueous layer was extracted with CHzCl2 (2 x 50 ml). The
combined organic layers were dried with MgS04 and the solvent
evaporated to obtain compound 22 which was used in the next
step without purification.
The residue was dissolved in 10 mo dioxane, treated with
1.6 ml aqueous ammonia and stirred overnight. The solvent was
evaporated and the residue was treated with methanolic ammonia
(saturated at -70°C and stirred for 20 hours at room
temperature. The solvent was evaporated and the residue
purified by preparative TLC using CHZC12-isopropanol-Et3N
(70:30:1) as eluent to obtain 31 mg (37% yield) of pure
compound 9 as a white solid.
m.p. - 222-224°C. W (MeOH) a",ax 276.5 (E 8400) . FAB MS
(m/z, %) - 314 ((MH + glycerine}+,9); 222 (MH+, 90); 152 (7);
112 (100. 1H-NMR (DMSOds)b - 0.50 (dd, J = 3.7, 5.1 Hz, 1H,
H-7' exo); 0.60 (dd, J = 5.1, 8.6 Hz, 1H, H-7' endo); 1.15
(dd, J = 3.7, 8.6 Hz, 1H, H-6'); 3.32 (dd, J = 5.0, 11.4 Hz,
1H, H-5a'); 3.78 (dd, J = 5.0, 11.4 Hz, 1H, H-5b'); 4.77 (d,
J = 6.3 Hz, 1H, H-1'); 4.89 (t, J = 5.0 Hz, 1H, -OH); 5.67 (d,
J = 7.3 Hz, 1H, H-5); 7.08 (broad, 2H, -NH2); 7.86 (d, J = 7.3
Hz, 1H, H-6). 13C-NMR (DMSOd6)b = 11.07 (C-7'); 24.85 (C-2');
25.75 (C-6'); 29.70 (C-3'), 32.51 (C-4'); 57.11 (C-1'); 63.78
(C-5'); 92.99 (C-5); 142.72 (C-6); 154.88 (C-2); 164.59 (C-
4 ) . d Anal . Calculated for C11H1s03N2 . 1/3Hz0 - C 58 . 14 , H 6 . 95 ,
- CA 02172534 2004-07-16
. ~ -26- ~
. r.- rr rr , f~ ffr r
~r f-r- f r r rr r
N 18.49. Found C SS.fø~~H 6-.8~,rrlvrl~.~~.~ ~fff
r f . f r r
r f . , r f C
f f EX~pL~ r14 f ~ f ,. /~
Conformational analysis of dideoxynucleoside analocTs
With the exception of signals corresponding to the
individual aglycons, the 1H NMR spectra of compounds 5-9. were
nearly identical and no apparent changes in the coupling
constants were observed between 25°C and 80°C. This indicated
that these compounds had a highly similar rigid conformation
in solution. Using compound 5 as a prototype, the
pseudoanomeric signal appeared as a doublet with a coupling
constant of 6.0 Hz centered at b=4.90. To understand the
multiplicity of this signal, models of N- and S-conformers of
5 were constructed using the QUANTA program version 3.2.4
using CHARMrri version 21 with the standard parameter set . The
. 15 structures were minimized by systematic conformational search
and the implicated torsion angles were measured for both
conformers. For the N-conformer the values were: H6'-C6'-
C1'-H1' (-86.1°), H1'-C1'-C2'-H2'(3 (91.3°) and H1'-C1'-C2'-
H2'a 9 (-23.9°) . .
Although the Karplus equation, defined as an empirically-
derived correlation between the coupling constant J for the
H/H interaction in a H-C1-C2-H system and the dihedral angle
(6) formed between the planes that contain H-C1-C2 and C1-C2-
H; amy not apply perfectly due to the distortion produced by
the fused cyclopropane ring, the torsion angle values measured
for the N-conformer suggest that two of the three coupling
constants should be very close to zero. Conversely, none of
the same torsion angles measured for the S-conformer
approached 90° (-134.7°, 175.7° and 60.9°). Thus,
the torsion
angles measured for the N-geometry agree better with the
multiplicity observed for the pseudoanomeric signal of 5
(doublet, J=6 Hz), in the 1H NMR spectrum. These torsion
angles are also similar to those measured from the crystal
structure of neplanocin C, suggesting that structures 5-9 have
equivalent N-geometries in solution. The N-geometry in
AllfiENGED Sv~IEET
IPEA/EP
4'~
WO 95108541 ~ ~ ~ ~ ~ f~. PCT/US94110794
-27-
compounds 5-9 can only be achieved if the bicyclo [3 . 1 . 0] hexane
system exists as a pseudoboat, since a pseudochair
conformation would correspond to the S-geometry. A search for
compounds containing unrestricted bicyclo [3 .1 . 0] hexanes in the
Cambridge Structural Data Base (Allen, et al., (1979) Acta.
Crystallogr., B35, 2331) revealed that in the seven examples
found the pseudoboat was the only form of puckering observed.
EXAMPLE 15
Evaluation of anti-HIV activity of dideoxynucleosides
Compounds 5-9 were evaluated against HIV-1 in
immortalized OKT4' T-cells (ATH8 cells) by the cytopathic
effect assay (Mitsuya and Broder, (1986) Proc. Natl. Acad.
Sci. U.S.A., 83: 1911). Compound 5, the adenosine derivative,
was the only analogue which exhibited anti-HIV activity. A
dose-dependent increase in the number of viable cells was
observed from 5-50 ~.M, although the drug itself exhibited some
toxicity in this range (Figure 3). Since compound 5 is a
racemic mixture, it is anticipated that separation of the two
enantiomers will result in one responsible for toxicity and
one responsible for the antiviral effect.
The synthetic approach for the 2' tent-butyl-substituted
carbocyclic alcohol 30 is shown in Scheme 3 and described in
Examples 16-21 below.
WO 95/08541 PCT/US94/10794
21-12c34 -28-
n
U ~O
h
2
O
Z p O
IIIO =
III p
N II O
I I I O U ~ ~ N I S
M ai D II10
O O ~ ~ n z
s aH ~ O
z
U a E r o ~
U d Z
N n
Q ~ I U U
Q U ., Z
2
+r U U p
ci d Z
III p
N
IIIO
I I I O ~ I I I ~ O ~ Z I!
II IO N ~ 1111~
II10 O ~ II10
U U
O
O c a °' N- 0
U N tn Z y~ 2
~ m c _
U N
L ~ ~ m Q N v
O~
+r D7 U O ~ U
Z U ~ n = = o
t~ U
O O In r'
v
(n 1110
Z
O v7
N G C~ O
II10 I
C
III p
O N
Z IIIO 11 (~~I
U U 10
L n U
d O ~ n
N ~ O a~
N
U
t
d L
a
SUBSTITUTE SHEET (RULE 26)
WO 95/08541 L GJ ~ ~ ~ PCT/US94/10794
-29-
Example 16
(1S,4R,5S)-3-((Benzyloxy)methvl-4 5-O-isopropylidene-2-
cyclopenten-1-of 24
Compound 24 was prepared from compound 23 according to
the procedure of Marquez et al., J. Org. Chem., 53:5709
(1988) .
Example 17
(1S,4R.5S)-3-f(Benzyloxy)methyll-4-tert-butyloxy-5-hydroxy-2-
cyclopenten-1-of 25
A solution of 24 (0.61 g, 2.20 mmol) was stirred in
anhydrous CHZC12 (25 ml) at -78°C and treated with a solution
of trimethylaluminum in toluene (2 M, 7.8 ml, 15.6 mmol).
After the addition, the reaction was allowed to reach room
temperature and stirring was continued for 18 hours. The
reaction mixture was cooled again to -78°C and quenched with
an aqueous saturated solution of NH4C1 (10 ml). Since this is
a very exothermic process, the addition of NH4C1 was done
slowly. The mixture was filtered after reaching room
temperature and the solid was washed with CHC13. The filtrate
was extracted with CHC13 (3 x 50 ml) and the combined organic
extract was washed with water (50 ml), dried (NazS04), and
concentrated under vacuum. The crude product was purified by
flash column chromatography over silica gel with a 0-50%
gradient of ethyl acetate in hexane as eluant to give 0.349 g
(54%) of compound 25 as a thick oil. Analysis calculated for
C1,H24O4: C, 69.83; H, 8.27. Found: C, 69.57; H; 8.27.
Example 18
(1S,4R.5S)-1-(tert-Butyldimethylsilyloxy)-3-f(Benzovloxy)
methvll-4-tert-butyloxy-5-hydroxy-2-cyclopentene 26
A solution of 25 (8.04 g, 27.5 mmol) and imidazole (7.05
g, 103.55 mmol) in anhydrous DMF (80 ml) was treated with
tert-butyldimethylsilyl chloride (6.70 g, 44.45 mmol). The
mixture was stirred at room temperature under a blanket of
argon for 40 min and quenched by the addition of water (100
ml). The reaction mixture was extracted with ethyl acetate (3
x 100 ml), and the combined organic extract was washed with
WO 95/08541 PCTIUS94/10794
-30- 21 ?534
brine (2 x 100 ml) and dried over Na2S0q. The solvent was
evaporated and the product purified by flash column
chromatography over silica gel to give 9.77 g (87.4x) of pure
26 as an oil . Analysis calculated for C23H38O4S1 ~ 0 . 5 H~O: C,
66.46; H, 9.46. Found: C, 66.41; H, 9.31.
Examgle 19
(1S,4R,5S)-1-(tert-Butyldimethylsilyloxv)-3-[(Benzyloxy)
methyl]-4-tert-butyloxy-5-[(methylthio)thiocarbon~loxy-2-
cyclopentene 27
A solution of 26 (9.77 g), 24.02 mmol) in anhydrous THF
(100 ml) was treated with carbon disulfide (10.2 ml, 168.8
mmol). The mixture was stirred at 0°C for 5 min, and NaH (80%
suspension in oil, 2.2 g, 73.3 mmol) was added in portions.
The mixture was stirred at room temperature for 30 min.
Methyl iodide (19.5 ml, 313.2 mmol) was added, and after
further stirring for 30 min, the reaction mixture was cooled
to 0°C, and excess NaH was destroyed by the slow addition of
water (very exothermic process). The organic layer was
separated and the aqueous extract was dried (Na2S09) and
concentrated under vacuum. The crude product was purified by
flash column chromatography over silica gel using a 0-50
gradient of ethyl acetate in hexane to give 9.83 g (82.4%) of
pure 27 as an oil . Analysis calculated for C25HqoO4S2Si ~ 0 . 25
HBO: C, 59.90; H, 8.10; S, 12.77. Found: C, 59.84; H, 8.10;
S, 12.72.
Example 20
(1S,4R)-1-(tert-Butyldimethylsilyloxy)-3-[(Benzyloxy)methyl]-
4-tert-butyl-2-cyclopentene 28
A solution of 27 (9.82 g, 19.76 mmol) and
azobis(isobutyronitrile) (AIBN, 2.04 g, 12.42 mmol) in
anhydrous toluene (100 ml) under a blanket of argon was heated
to ca. 50°C and treated slowly with tri-n-butyltin hydride (22
ml, 81.8 mmol). After the addition was complete, the mixture
was heated (oil bath temp. 120°C) for 1.5 hours and cooled to
room temperature. The solvent was evaporated and the crude
product was purified by flash column chromatography over
~~7~~~q.
WO 95/08541 PCT/US94I10794
-31-
silica gel with a gradient of 0-5% ethyl acetate in hexane to
give compound 28 (5.94 g, 77%) as an oil. Analysis calculated
for C23H3e03Si ~ 0. 5H20: C, 69.12; H, 9. 83 . Found: C, 69.21; H,
9.71.
Example 21
(1S,4R) -3- f (Benzylox~r)methyll -4-tert-butylox~,r-2-cyclopenten-1-
ol 29
A solution of 28 (4.82 g, 12.36 mmol) in anhydrous THF
(80 ml) was treated with a solution of tetrabutylammonium
fluoride in THF (1 M, 51 ml) and the resulting mixture was
stirred at room temperature overnight. The solvent was
evaporated and the residue treated with water and extracted
with ethyl acetate (3 x 100 ml). The combined organic extract
was washed with brine (2 x 100 ml) and dried (Na2S09) . The
solvent was removed under reduced pressure and the crude
product purified by flash column chromatography over silica
gel using a gradient of 50-66% ethyl acetate in hexane to give
compound 29 (3.152 g, 92%) as a clear oil. Analysis
calculated for C1,H240a ~ 0 . 75H20: C, 70 . 43 ; H, 8 . 86 . Found: C,
70.62; H, 8.54.
Example 22
(1R,2S,4R,5S)-1-f(Benzovloxy)methyll-2-tert-but~loxy-4-
hydroxybi cyc 1 o f 3 .1. 0 J hexane 3 0
Samarium metal (4.40 g, 29.3 mmol) was placed in a flask
and dried with a flame under a stream of argon. Anhydrous THF
(30 ml) and a solution of mercuric chloride (0.76~g, 2.8 mmol)
in 10 ml THF were added and the mixture was stirred for 10 min
prior to the addition of a solution of compound 29 (1.80 g,
6.50 mmol) in THF (30 ml). The reaction mixture was cooled to
-78°C and treated with chloroiodomethane (2.20 ml, 30 mmol).
The resulting mixture was stirred continuously starting at -
78°rr and allowing the temperature to reach room temperature
during the course of the night. The following day, the
reaction was quenched with a saturated solution of potassium
carbonate (200 ml) and extracted with methylene chloride (3 x
100 ml). The combined organic extract was washed with brine
WO 95/08541 PCT/US94110794
L -32-
(100 ml), dried (Na2S04), filtered and evaporated to give
nearly pure compound 30 quantitatively as a colorless oil.
This product was used in the condensation steps described in
the purine and pyrimidine synthetic steps described above.
Compounds 31 and 32 corresponding to the thymidine and
adenosine analogs, respectively, were synthesized using
compound 30 as the starting material following reaction
Schemes 2 and 1, respectively, starting with the step
following cyclopropanation.
(1R 2S 4S 5S)-1-Hydroxymethyl-4-(5-methyl-2,4(1H,3H)-
dioxopyrimidin-1-yl ) bicyclo (3 . 1 . OJ hexane 31
The thymidine analog 31 was obtained as a white
crystalline product having the following parameters: melting
point - 239-241°C; [a]D25 - +47.14; FAB MS m/z (relative
intensity) 253 (MH+, 100), 127 (b + 2H, 40). Analysis
calculated for C12H,604Nz ~ 0 . 33H20: C, 55. 81; H, 6 . 50; N, 10 . 85.
Found: C, 55.91; H, 6.51; N, 10.73.
(1R,2S,4S,5S)-1-Hydroxymethyl-4-(6-amino-9-purinyl)bicyclo
f 3 . 1 . 0 J hexane 32
The adenosine analog 32 was obtained as a white solid,
melting point 259-261°C; FAB MS m/z (relative intensity) 262
(MH+, 100), 136 (b + 2H, 58).
In the final step of the purine synthetic scheme (Scheme
1), and in the thymidine/uracil synthetic step of the
pyrimidine synthetic scheme (Scheme 2), the benzyl protecting
groups are removed by cleavage with BC13. This same treatment
removes the tert-butyl group at the 2' position when compound
is used as the starting material for synthesis of the
nucleoside analogs, thus leaving a 2'-OH group.
30 The description above is illustrative and not
restrictive. Accordingly, many variations of the invention
will be apparent to one skilled in the art on review of this
disclosure, and the invention can be embodied in these various
specific forms without departing from it in spirit or
essential characteristics. For example, it should be apparent
from the foregoing that various purine and pyrimidine
WO 95/08541 l I 7 2 5 3 ~ PCT/US94/10794
-33-
analogues can be substituted in the Examples to achieve
similar results. The scope of the invention is therefore
indicated by the appended claims rather than by the foregoing
description.