Note: Descriptions are shown in the official language in which they were submitted.
WO 95/12398 2 1 7 4 2 3 4 PCT/US94/12516
TITLE OF THE INVENTION
7-SUBSTITUTED-4-AZA-STEROID DERIVATIVES AS
5-ALPHA- REDUCTASE INHIBITORS
This application is a continll~tion-in-part of pending
International application number PCT/US93/04643, filed on
May 14, 1993, which in turn was a contiml~tion-in-part of U.S.
application serial no. 886,572, (Merck case no. 18670) filed
May 20, 1992, and now abandoned.
F~ELD OF THE ~NVENTION
The present invention provides novel compounds, novel
compositions, methods of their use and methods of their manufacture,
where such compounds are generally ph~rm~cologically useful as agents
in therapies whose mech~ni~m of action rely on the inhibition of Soc-
reductase, and more particularly, the dual inhibition of the 5a-reductase 1
and 5a-reductase 2 isozymes.
BACKGROUND OF THE INVENTION
2 0 Certain undesirable physiological manifestations, such as
acne vulgaris, seborrhea, female hir--tism, androgenic alopecia which
includes female and male pattern baldness, and benign prostatic
hyperplasia, are the result of hyperandrogenic stim~ tion caused by
an excessive accllmlll~tion of testosterone ("T") or similar androgenic
hormones in the metabolic system. Early attempts to provide a
chemotherapeutic agent to counter the undesirable results of
hyperandrogenicity resulted in the discovery of several steroidal
antiandrogens having undesirable hormonal activities of their own.
The estrogens, for example, not only counteract the effect of the
androgens but have a femini7in~ effect as well. Non-steroidal
antiandrogens have also been developed, for example, 4'-nitro-3'-
trifluoromethyl-isobutyranilide. See Neri, et al., Endocrinol. 1972, 91
(2). However, these products, though devoid of hormonal effects,
compete with all natural androgens for receptor sites, and hence have
a tendency to femini7e a male host or the male fetus of a female host
W0 9S/12398 ' ~ 7' S 2 1 7 4 2 3 4 PCT/US94/12~;16
and/or ini*~te feed-back effects which would cause hyperstimlllation
of the testes.
The principal mediator of androgenic activity in some target
organs, e.g. the prostate, is 5a-dihydrotestosterone ("DHT"), formed
5 locally in the target organ by the action of testosterone-Sa-reductase (or
simply Sa-reductase). Inhibitors of 5a-reductase will serve to prevent or
lessen symptoms of hyperandrogenic stim~ tion in these organs. See
especially United States Patent Nos. 4,377,584, issued March 22, 1983,
and 4,760,071, issued July 26, 1988, both assigned to Merck & Co., Inc.
It is now known that a second Sa-reductase isozyme exists, which
interacts with skin tissues, especially in scalp tissues. See, e.g., G. Harris,
et al., Proc. Natl. Acad. Sci. USA, Vol. 89, pp. 10787-10791 (Nov. 1992).
The isozyme that principally interacts in skin tissues is conventionally
desi~n~te~l as 5a-reductase 1 (or Sa-reductase type 1), while the isozyme
15 that principally interacts within the prostatic tissues is designated as Sa-
reductase 2 (or Sa-reductase type 2).
In ~e treatment of hyperandrogenic disease conditions, e.g.
benign prostatic hyperplasia (BPH) and/or the prevention and treatment
of prostatic cancer, and the treatment of prostatitis, it would be desirable
20 to have one drug entity which is active against bo~ isozymes to
significantly inhibit dihydrotesterone production. It would also be
desirable to have one drug entity that is active as a dual inhibitor of both
isozymes for the treatment of conditions of the skin and scalp, e.g. acne
vulgaris, seborrhea, female hirsutism, and androgenic alopecia.
Additionally, such a dual inhibitor of Sa-reductase 1 and 2 could be used
in combination with a Sa-reductase 1 inhibitor or wi~ a 5a-reductase 2
inhibitor, e.g. finasteride (PROSCAR(g)), for combin~tion therapy in the
tre$ment of hyperandrogenic conditions. The dual isozyme inhibitor
could also be used in combination with a potassium channel opener, e.g.
3 minoxidil, for the treatment of male ~allelll baldness. The present
invention addresses this by providing compounds ~at are active as dual
Sa-reductase 1 and 2 inhibitors.
WO 95/12398 PCT/US94/12S16
~ 2 1 7 4 2 3 4
~UMMARY OF THE INVENTION
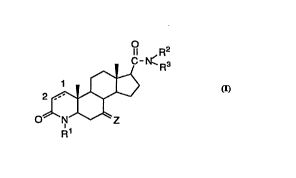
The novel compounds of the present invention are those of
structural Formula I:
O
Il R2
C- N~ 3
O~N~Z
R1
or a ph~rm~ceutically acceptable salt or ester thereof, and are dual
inhibitors of 5a-reductase isozymes 1 and 2. The compounds of Formula
I are useful in the oral, systemic, parenteral or topical treatment of acne
vulgaris, androgenic alopecia including female and male pattern baldness,
2 o benign prostatic hyperplasia, female hirsutism, prostatitis, and the
prevention and treatment of prostatic carcinoma.
Therefore it is an object of this invention to provide
compounds that have sufficient activity in the inhibition of both 5a-
reductase isozymes. It is an additional object of this invention to provide
2 5 methods of using the compounds of Formula I for the treatment and/or
prevention of hyperandrogenic conditions such as BPH, female hirsutism,
male pattern baldness, acne vulgaris, androgenic alopecia, and prostatic
cancer, as well as prostatitis. It is a further object of this invention to
provide ph~ ceutical compositions for the compounds of Formula I.
3 Another object of this invention is to provide compounds of Formula I in
combination with a Sa-reductase 2 inhibitor, such as finasteride, or a 5a-
reductase 1 inhibitor, or a pot~sillm ch~nnel opener, such as minoxidil,
wherein such combinations would be useful in one or more of the above-
mentioned methods of treatment or ph~rm~-~eutical compositions.
WO 9S/12398 PCTIUS94/12S16
~S~ 1 74234
-4 -
DETAILED DESCRIPTION OF THE lNVENTION
The novel compounds of this invention have the structural
Formula I:
1I R2
C- N~ 3
2~/
O~N~Z
R1
or a ph~ eutically acceptable salt or ester thereof, wherein:
the Cl-C2 bond deei~n~te~ ~ " represents a single or double
bond;
R1 is selected from:
1) -H,
2) -CH3 and
3) -CH2CH3;
Z is selected from:
1) oxo,
2) a-H and a ~-substituent selected from:
a) Cl 4 aLkyl,
b) C2 4 alkenyl,
c) -CH2 COOH,
d) -OH,
3 e) -COOH,
f~ -COO (Cl 4 alkyl),
g) -oCoNR4R5 wherein
R4 and R5 are independently selected from
i) -H,
ii) -Cl 4 alkyl,
WO 95/12398 PCT/US94/12516
~ 21 74234
iii) phenyl and
iv) benzyl;
or R4 and RS taken together with the nitrogen to
which they are attached represent a 5-6 membered
saturated heterocycle optionally cont~ining one other
heteroatom selected from -O-, -NH- and -S-;
h) Cl 4 alkoxy,
i) C3-6 cycloalkoxy,
j) -oC(o)R7~ wherein R7 is C1 6aL~yl or phenyl,
k) halo,
1) halo-Cl 2 alkyl,
m) -CF3, and
n) C3-6 cycloalkyl;
3) =CHR6;
4) spirocyclopropane either unsubstituted or substituted with
R6;
R2 is selected from -H and C1 6 alkyl, either unsubstituted or substituted
with one or more of aryl, heteroaryl, -COOH or -OH;
R3 is -C1 6 alkyl substituted with one or more of aryl, heteroaryl,
-COOH, -OH or di-aryl amino; and
R6 is selected from -H and Cl 4 alkyl.
Combinations of substituents and/or variables are
permissible only if such combinations result in stable compounds.
In one embodiment of the in~t~nt invention are
compounds of Formula I wherein R 1 is hydrogen or methyl; R2 is
hydrogen, methyl, ethyl or propyl; Z is an alpha hydrogen and a beta
substituent selected from C14 alkyl, C24 alkenyl, C1 4 alkoxy, halo-
Cl 4 aLkyl, -CF3, and C3-6 cycloalkyl; and R3 is Cl 4 aLkyl di-
3 substituted with phenyl, wherein each phenyl ring is independentlyunsubstituted or subsLiLu~ed.
In one class of this embodiment are compounds of Formula I
that have structural Formula II:
WO 95/12398 PCT/US94/12516
t,
2 1 74234
_ C-~CH~
2~
O N Z
R
II
or a ph~ ceutically acceptable salt thereof wherein
R1 is hydrogen or methyl; R2 is hydrogen or methyl, and Z is alpha
hydrogen and beta methyl.
Some examples of compounds within this class are:
N~diphenylme~yl)-3-oxo4-aza-7,~-methyl-Sa-androstane- 1 7~-
carbox~mide;
N~diphenylmethyl)-3-oxo-4-aza-7~-methyl-Sa-androst- 1 -ene- 1 7,B-
carboxamide;
N~diphenylmethyl)-3-oxo4-aza-4,7~-dimethyl-Sa-androstane- 17~-
carboxamide; and
N-(methyl),N~diphenylmethyl)-3-oxo4-aza-4,7~-dimethyl-Sa-
androstane-17~-carbox~mi~le.
As used herein "aLkyl" is intended to include both
branched- and straight-chain saturated aliphatic hydrocarbon groups
having the specified number of carbon atoms, e.g., methyl (Me), ethyl
(Et), propyl, butyl, iso-propyl (i-Pr), iso-butyl (i-Bu), sec-butyl (s-Bu),
tert-butyl (t-Bu). "ALkyloxy" (or "alkoxy") represents an aLkyl group
30 having the indicated number of carbon atoms ~ ched through an
oxygen bridge, e.g., methoxy, ethoxy, propyloxy, iso-propoxy, n-
butoxy, iso-butoxy, sec-butoxy, t-butoxy and the like. "ALkenyl" is
intended to include hydrocarbon groups having ~e speci~led number
of carbon atoms of either a straight or branched con~iguration with
WO 95/12398 PCT/US94/12516
~ ~ S i~ 7423 4
one or more carbon-carbon double bonds which may occur in any
stable point along the chain, such as ethenyl, allyl, l-propen-l-yl, 1-
propen-2-yl, l-buten-l-yl, 1-buten-2-yl, pentenyl, and the like.
Included in this invention are all E, Z diasteriomers.
The term "C3-C6 cycloalkyl" as used herein is meant to
include, e.g., cyclopropyl, cyclobutyl, cyclopentyl, and cyclohexyl.
The term C3-C6 cycloalkoxy" as used herein is meant to include a
cycloalkyl group ~ ched through an oxygen bridge, e.g., cyclo-
propyloxy, cyclobutyloxy, cyclopentyloxy, and cyclohexyloxy.
Representative examples of Z are where the a-substituent
(dashed lines) is hydrogen and the b-substituent (wedge) is e.g.
methyl, ethyl, propyl, allyl, carboxymethyl, hydroxy, methoxy,
ethoxy, cyclopropyloxy, cyclopentyloxy, acetoxy, fluoro, chloro,
bromo, trifluorome~yl, trichloromethyl, fluoromethyl, chloromethyl,
carboxy, N,N-dimethylcarbamoyl, hydroxymethyl, methoxymethyl,
and the like. Representative examples where Z is an alkenyl
substituent, =CH-R6, include, e.g. =CH2, =CH-CH3, =CH-CH2CH3,
and the like.
Representative examples where Z is spirocyclopropane
include:
2s \\/ R1~ includes: \~ H,
\~ CH3 \~ CH2CH3,
stereoisomers thereof and the like.
WO 9S/12398 PCT/US94/12516
S 2 1 7 4 2 3 4
Unless otherwise indicated the 17-position substituent is
assumed to be in the beta configuration.
As used herein ~e term "aryl" is intended to include
phenyl or 1- or 2-naph~yl, either unsubstituted or mono- or di-
substituted. The term "halo" is intended to include chloro, fluoro,
bromo and iodo.
The phrase "R4 and RS taken together with the nitrogen
to which they are ~ ched represent a S- to 6-membered saturated
heterocycle optionally cont~inin~ one other heteroatom selected from
N, -O-, -NH- and -S-" is intended to include ring groups such as N-
piperidinyl, N-morpholinyl, N-piperazinyl, N-(4-methyl)piperazinyl,
N-thiomorpholinyl, N-pyrrolidinyl, N-imidazolidinyl and the like.
The term "heteroaryl" is intended to include the
following either unsubstituted or mono- or di-substituted: pyridyl,
furyl, pyrryl, thienyl, iso~iazolyl, imicl~7olyl, ben_imidazolyl,
tetrazolyl, pyrazinyl, pyrimidyl, quinolyl, isoquinolyl, benzofuryl,
isobenzofuryl, benzothienyl, pyrazolyl, indolyl, isoindolyl, purinyl,
carbazolyl, isoxazolyl, thiazolyl, oxazolyl, ben7tlli~7olyl, and
benzoxazolyl. The heteroaryl ring may be attached wi~in structural
Formula I or substituted at any heteroatom (N, O or S) or carbon atom
in the ring which results in the creation of a stable structure.
The substituents on the aryl and heteroaryl groups named
above are independently selected from: C1 6 alkyl, C2 6 aLkenyl,
phenyl, halo such as chloro, bromo, iodo and fluoro, trifluoromethyl,
cyano, carboxy, C1 6 a~yloxycarbonyl, hydroxy, C1 6 aLkoxy,
C1 6 alkylcarbonyloxy, Cl-6 alkylthio, Cl-6 alkylsulfinyl, C1-6
alkylsulfonyl, Cl-6 alkylcarbonyl, amino, Cl 6 alkylarnino, di(cl-6
alkyl)amino, C1 6 alkylcarbonylamino, Cl 6 alkyloxycarbonylamino,
C1 6 alkylsulfonyl-amino and C1 6 aLkyl~minosulfonyl.
3 Whenever the te~ns "aLkyl", "alkenyl", "alkyloxy (or
aLI~oxy)'', "aryl" or "heteroaryl", or one of their prefix roots, appear in
a name of a substituent in FoImula I, (e.g. aralkoxyaryloxy) they shall
have the same definitions as those described above for "alkyl",
"aLkenyl", "aLkyloxy (or alkoxy)", "aryl" and "heteroaryl",
WO 95/12398 PCT/US94/12516
2 1 7 4 2 3 ~
respectively. Designated numbers of carbon atoms (e.g. C1 1o) shall
refer independently to the number of carbon atoms in an alkyl or
alkenyl moiety or to the alkyl or alkenyl portion of a larger substituent
in which alkyl or alkenyl appears as its prefix root.
Also included within the scope of this invention are
ph~rm~ceutically acceptable salts of the compounds of Formula I,
where a basic or acidic group is present on the structure. When an
acidic substituent is present, i.e. -COOH, there can be formed the
ammonium, sodium, potassium, calcium salt, and the like, for use as
the dosage form. Where a basic group is present, i.e. amino or a basic
heteroaryl radical such as, e.g., 4-pyridyl, an acidic salt, i.e.
hydrochloride, hydrobromide, acetate, pamoate, and the like, can be
used as the dosage form.
Also, in the case of the -COOH group being present,
ph~ çeutically acceptable esters can be employed, e.g. acetate,
maleate, pivaloyloxymethyl, and the like, and those esters known in
the art for modifying solubility or hydrolysis characteristics for use as
sustained release or prodrug forrn~ tions.
Representative salts include the following salts:
20 acetate, lactobionate, benzenesulfonate, laurate, benzoate, m~l~te,
bicarbonate, maleate, bisulfate, m~n~lelate, bild~ dte, mesylate, borate,
methylbromide, bromide, methylnitrate, calcium edetate, methylsulfate,
camsylate, mllc~te, carbonate, napsylate, chloride, nitrate, clav~ n~te, N-
methylgluc~mine, citrate, ammonium salt, dihydrochloride, oleate,
25 edetate, oxalate, edisylate, pamoate (embonate), estolate, p~lmit~te,
esylate, pantothenate, fulllardte, phosphate/diphosphate, gluceptate,
polygalacturonate, gluconate, salicylate, glllt~m~t.o, stearate,
glycollylars~nil~te, sulfate, hexylresorcinate, subacetate, hydrabamine,
succinate, hydrobromide, t~nn~te, hydrochloride, tartrate,
30 hydroxynaphthoate, teoclate, Iodide, tosylate, isothionate, triethiodide,
lactate, and valerate.
In addlition, some of the compounds of the instant
invention may form solvates with water or common organic solvents.
Such solvates are encompassed within the scope of this invention.
WO 95/12398 PCT/IJS94/12516
'` h ~ ~ t~
- 21 74234
- 10-
The compounds of the present invention have
asymmetric centers and may occur as racemates, racemic mixtures and
as individual enantiomers or diastereomers, with all isomeric forms
being included in the present invention as well as mi~ res thereof.
Fur~ermore, some of the crystalline forms for compounds of the
present invention may exist as polymorphs and as such are intended to
be included in the present invention.
The term "therapeutically effective amolmt" shall mean that
amount of a drug or pharmaceutical agent that will elicit the biological or
medical response of a tissue, system, ~nim~l or l~llm~n that is being
sought by a researcher, veterinarian, medical doctor or other clinician.
The present invention has the objective of providing
methods of treating the hyperandrogenic conditions of androgenic
alopecia including male pattern baldness, acne vulgaris, seborrhea,
and female hirsutism by oral, systemic, parenteral or topical
~lmini~tration of the novel compounds of Formula I either alone or in
combination with a Sa-reductase 2 inhibitor andlor a Sa-reductase 1
inhibitor and/or a potassium channel opener. The term "treating
androgenic alopecia" is intended to include the arresting and/or
reversing of androgenic alopecia, and the promotion of hair grow~.
I'he present invention has the further objective of providing methods
of treating benign prostatic hyperplasia, prostatitis, and treating and/or
preventing prostatic carcinoma by oral, systemic or parenteral
~lmini~tration of the novel compounds of Formula I either alone or in
combination with a Sa-reductase 2 inhibitor and/or in combination
with a Sa-reductase 1 inhibitor.
The present invention also has ~e objective of providing
suitable topical, oral, systemic and parenteral ph~ reutical
forrmll~tions for use in the novel methods of treatment of the present
invention. The compositions cont~inin~; the present compounds as ~lLe
active ingredient for use in ~e trea~nent of the above-noted
conditions can be ~lmini~tered in a wide variety of ~erapeutic dosage
forms in conventional vehicles for systemic ~lmini~tration. For
example, the compounds can be ~lminictered in such oral dosage
WO 95/12398 . PCT/US94/12516
~ 2 1 7 4 2 3 4
- 11 -
forms as tablets, capsules (each including timed release and sustained
release formulations), pills, powders, granules, elixirs, tinctures,
solutions, suspensions, syrups and emulsions, or by injection.
Likewise, they may also be ~tlmini~tered in intravenous (both bolus
and infusion), intraperitoneal, subcutaneous, topical with or without
occlusion, or illtl~lluscular form, all using forms well known to those
of ordinary skill in the ph~ ceutical arts. An effective but non-toxic
amount of the compound desired can be employed as an
antiandrogenic agent.
o The daily dosage of the products may be varied over a
wide range from 0.01 to 1,000 mg per adult hllm~n/per day. For oral
~(1mini~tration, the compositions are preferably provided in the form
of scored or unscored tablets collt~ illg 0.01, O.OS, 0.1, 0.5, 1.0, 2.5,
S.0, 10.0, lS.0, 25.0, and S0.0 milligrams of the active ingredient for
the symptomatic adjustment of the dosage to the patient to be treated.
An effective amount of the drug is ordinarily supplied at a dosage
level of from about 0.0002 mglkg to about S0 mg/kg of body weight
per day. The range is more particularly from about 0.001 mg/kg to 7
mg/kg of body weight per day.
Advantageously, compounds of the present invention
may be ~lmini~tered in a single daily dose, or the total daily dosage
may be ~lmini~tered in divided doses of two, three or four times
daily. Furthermore, compounds for the present invention can be
~lmini~tered in i~tranasal form via topical use of suitable intranasal
vehicles, or via transdermal routes, using those forms of transdermal
skin patches well known to those of ordinary skill in that art. To be
~lmini~tered in the form of a transdermal delivery system, the dosage
~lmini~tration will, of course, be continuous rather than intermittent
throughout the dosage regimen.
For the tre~tment of androgenic alopecia including male
pattern baldness, acne vulgaris, seborrhea, and female hirsutism, the
compounds of the present invention may be ~timini~tered in a
pharmaceutical composition comprising the active compound in
combination with a pharmaceutically acceptable carrier adapted for
WO 95/12398 PCT/US94/12S16
~c~ 21 74234 e
topical ~lmini~tration. Topical ph~rm~ceutical compositions may be,
e.g., in the form of a solution, cream, ointment, gel, lotion, shampoo
or aerosol formulation adapted for application to the skin. These
topical phalmaceutical compositions cont~inin~ the compounds of the
present invention ordinarily include about 0.001% to 15% by weight
of the active compound in ~ lix~ with a ph~ ceutically
acceptable vehicle.
For the treatment of acne vulgaris, androgenic alopecia
including male pattern baldness, seborrhea, female hirsutism, benign
prostatic hyperplasia, prostatitis and the prevention and/or tre~tment
of prostatic cancer, the compounds of the instant invention can be
combined with a therapeutically effective amount of a 5a-reductase 2
inhibitor, such as finasteride, or a Soc-reductase 1 inhibitor, such as
4,7~-dimethyl-4-aza-Sa-cholestan-3-one, in a single oral, systemic, or
15 parenteralph~ ceuticaldosageform~ tion. Alternatively,a
combined therapy can be employed wherein the compound of
Formula I and the 5a-reductase 1 or 2 inhibitor are ~(lmini~tered in
separate oral, systemic, or parenteral dosage form~ tions. Also, for
the skin and scalp related disorders of acne vulgaris, androgenic
2 alopecia including male pattern baldness, seborrhea, and female
hirsllti~m, the compounds of the instant invention and a Sa-reductase
1 or 2 inhibitor can be formlll~ted for topical ~lmini~tration. For
example, a compound of Formula I and finasteride can be
~lminictered in a single oral or topical dosage form~ tion, or each
25 active agent can be ~lmini~tered in a separate dosage formlll~tion,
e.g., in separate oral dosage forrnlll~tions, or an oral dosage
fonnlll~tion of finasteride in combination with a topical dosage
folm~ ion of a compound of Formula I. See, e.g., U.S. Patent No.'s
4,377,584 and 4,760,071 which describe dosages and formulations for
3 Sa-reductase inhibitors.
Fur~e~nore, ~rlmini~tration of a compound of the
present invention in combination with a ~erapeutically effective
amount of a potassium channel opener, such as ~ninoxidil, crom~k~lin,
pinacidil, a compound selected from ~e classes of S-triazine, thiane-
WO 95/12398 PCT/US94/12516
~ ` 2 ~ 7 4 2 3 4
1-oxide, benzopyran, and pyridinopyran derivatives or a
ph~rm~ceutically acceptable salt thereof, may be used for the
treatment of androgenic alopecia including male pattern baldness.
The active agents can be ~tlmini~tered in a single topical dosage
form~ tion, or each active agent can be ~lmini~tered in a separate
dosage formulation, e.g., in separate topical dosage form~ tions, or
an oral dosage formulation of a compound of Formula I in
combination with a topical dosage formlll~tion of, e.g., minoxidil.
See, e.g., U.S. Patent No.'s 4,596,812, 4,139,619 and WO 92/02225,
published 20 February 1992, for dosages and formulations of calcium
channel openers.
For combination treatment with more than one active
agent, where the active agents are in separate dosage form~ tions, the
active agents can be ~lmini~tered concomit~ntly, or they each can be
~lmini~tered at separately staggered times.
The dosage regimen l-tili7ing the compounds of the
present invention is selected in accordance with a variety of factors
including type, species, age, weight, sex and medical condition of the
patient; the severity of the condition to be treated; the route of
~lmini~tration; the renal and hepatic function of the patient; and the
particular compound thereof employed. A physician or veterinarian of
ordinary skill can readily dete~ e and prescribe the effective
amount of the drug required to prevent, counter or arrest the progress
of the condition. Optimal precision in achieving concentration of drug
within the range that yields efficacy without toxicity requires a
regimen based on the kinetics of the drug's availability to target sites.
This involves a consideration of the distribution, equilibrium, and
elimin~tion of a drug.
ln the methods of the present invention, the compounds
herein described in detail can form the active ingredient, and are
typically ~tlmini~tered in a&~ lule with suitable ph~ ceutical
diluents, excipients or carriers (collectively referred to herein as
"carrier" materials) suitably selected with respect to the intended form
WO 95/12398 PCT/US94/12S16
~ i S 2 1 7 4 2 3 4
- 14-
Of ~(lmini~tration, that is, oral tablets, capsules, elixirs, syrups and the
like, and consistent with conventional ph~ ceutical practices.
For instance, for oral ~lmini~tration in the form of a
tablet or capsule, the active drug component can be combined with an
oral, non-toxic pharmaceutically acceptable inert carrier such as
ethanol, glycerol, water and the like. Moreover, when desired or
necessary, suitable binders, lubricants, disintegrating agents and
coloring agents can also be incorporated into the Illix(~ . Suitable
binders include, without limit~tion, starch, gelatin, natural sugars such
as glucose or beta-lactose, corn sweeteners, natural and synthetic
gums such as acacia, trag~c~nth or sodium alginate, carboxymethyl-
cellulose, polyethylene glycol, waxes and ~e like. Lubricants used in
these dosage forms include, without limit~tion, sodium oleate, sodium
stearate, m~gnPsium stearate, sodium benzoate, sodium acetate,
sodium chloride and the like. Disintegrators include, without
limit~tion, starch, methyl cellulose, agar, beutol~i~, x~nth~n gum and
the like.
The liquid forms in suitably flavored suspenclin or
dispersing agents such as ~e synthetic and natural gums, for example,
trag~c.~nth, acacia, methyl-cellulose and the like. Other dispersing agents
which may be employed include glycerin and the like. For parenteral
~lmini~tration, sterile suspensions arld solutions are desired. Isotonic
preparations which generally contain suitable preservatives are employed
when intravenous ~(lmini~tration is desired.
Topical preparations co~ g the active drug component
can be ~lmixed wi~ a variety of carrier materials well known in the art,
such as, e.g., alcohols, aloe vera gel, ~ ntoin, glycerine, vitamin A and E
oils, mineral oil, PPG2 myristyl propionate, and the like, to form, e.g.,
alcoholic solutions, topical cleansers, cleansing creams, skin gels, skin
3 0 lotions, and shampoos in cream or gel form~ tions. See, e.g., EP O 285
382.
The compounds o~ the present invention can also be
lmini~tered in the form of liposome delivery systems, such as small
mil~mellar vesicles-, large lmil~mellar vesicles and m~lltil~m~llar
WO 9S/12398 PCT/US94/12516
~ 2 1 7423~
vesicles. Liposomes can be formed from a variety of phospholipids,
such as cholesterol, stearyl~mine or phosphatidylcholines.
Compounds of the present invention may also be
delivered by the use of monoclonal antibodies as individual carriers to
5 which the compound molecules are coupled. The compounds of the
present invention may also be coupled with soluble polymers as
targetable drug carriers. Such polymers can include polyvinyl-
pyrrolidone, pyran copolymer, polyhydroxypropylmethacryl-
amidephenol, polyhydroxy-ethylaspartamidephenol, or polyethyl-
eneoxidepolylysine sub~LiLu~ed with palmitoyl residues. Furthermore,
the compounds of the present invention may be coupled to a class of
biodegradable polymers useful in achieving controlled release of a
drug, for example, polylactic acid, polyepsilon caprolactone,
polyhydroxy butyric acid, polyorthoesters, polyacetals, polydihydro-
pyrans, polycyanoacrylates and cross-linked or amphipathic block
copolymers of hydrogels.
The compounds of the present invention can be prepared
readily according to the following P.x~mples or modifications thereof
using readily available starting materials, reagents and conventional
20 synthesis procedures. In these reactions, it is also possible to make
use of variants which are themselves known to those of ordinary skill
in this art, but are not mentioned in greater detail. The examples are
not intended to be limit~tions on the scope of the instant invention in
any way, and they should not be so construed. Furthermore, the
compounds described in the following examples are not to be
construed as forming the only genus that is considered as the
invention, and any combination of the compounds or their moieties
may itself form a genus. Those skilled in the art will readily
understand that known variations of the conditions and processes of
3 the following preparative procedures can be used to prepare these
compounds. All temperatures are in degrees Celsius unless noted
otherwise.
WO 9S/12398 , PCT/US94/12S16
7 ~ ; 2 1 74234
- 16-
~XAMPLE 1
S~
N ~NH2
Dioxane/Reflux
o H
O N~
O N
N-Diphenylmethyl-3-oxo-4-aza-7,B-methyl-5a-androstane- 17,B-
carboxamide
To a solution of S-2'-pyridyl-3-oxo-4-aza-7~-methyl-5a-
25 androstane-17,~-thiocarboxylate (600 mg, 1.406 mmol) in dioxane (20
ml) was added amino diphenylme~ane (512 mg, 2.79 mmol). After
stirring the reaction ~ Lule for overnight at 80, the reaction mixture
was concentrated and partitioned between ethyl acetate and water. The
organic layer was washed with brine, dried and concentrated. The
3 0 residue was purified by preparatat*e TLC (thin layer chromatography) to
afford pure product; mp 261-263. Mass spec. (MS) M+ calculated, 498;
observed m/e 499 (m+1) (FAB); lH NMR (CDCl3, 400 MHz, Key
Peaks) ~(ppm) 0.666 (s), 0.8358 (s), 0.9975 (d, J=5.9 Hz), 3.051 (dd,
J=3.9 Hz, J=1 1.85 Hz).
WO 95/12398 PCT/US94/12516
~ . 2 t 7 4 2 3 4
EXAMPLE 2
H
Oq~N ~J~
NaH/DMF/Mel
o O N
H
CH3
O~N
O N
CH3
N-Diphenylmethyl-N-methyl-3 -oxo-4-aza-4,7~-dimethyl-Sa-androstane-
17,B-carboxamide
To a solution of N-diphenylmethyl-3-oxo-4-aza-7,13-methyl-
25 Sa-androstane-17~-carboxamide (70 mg, 0.14 mmol) in 1 ml of N,N-
dimethylform~mide (DMF) was added sodium hydride (40 mg, 50%
suspension). After sti~ing the reaction mixll-re for 15 min., methyl
iodide (200 ,ul) was added. The reaction -~;xll~c was allowed to stir for
overnight at room temperalule, quenched with water and extracted with
30 ethyl acetate. The organic layer was washed with water, brine, dried,
concentrated and purified by prep. TLC to give pure product. Mass spec.
(MS) M+ calculated, 526; observed m/e 527 (m+1) (FAB); lH NMR
(CDCl3, 400 MHz, Key Peaks) ~(ppm) 0.814 (s), 0.840 (s), 1.056 (d,
J=6.06 Hz), 3.02 (dd, J=3.22 Hz, J=12.45 Hz).
WO 9S/12398 PCT/US94/12516
2 t 7423~ --
- 18-
EXAMPLE 3
0 N ~
NaH/THF/Mel
o O N
H
H
Oq~N~
O N
CH3
N-Diphenylmethyl-3-oxo-4-aza-4,7,13-dimethyl-Sa-androstane-17~-
carboxamide
To a solution of N-diphenylmethyl-3-oxo-4-aza-7,B-methyl-
Sa-androstan-17~-carboxamide (70 mg, .14 mmol) in 1 ml of
tetrahydrofuran (I~IF) was added sodium hydride (40 mg, 50%
suspension). After stirring ~e reaction mixhlre for lS min., methyl
iodide (200 ~1) was added. The reaction mi~ture was allowed to stir for
30 overnight at room tempe,dlul~, quenched with water and extracted with
ethyl acetate. The organic layer was washed wi~ water, brine, dried,
concentrated and purified by prep. TLC to g*e pure product; mp 1 18-
120. Mass spec. (MS) M+ calculated, 512; observed m/e 513 (m+1)
(FAB); lH NMR (CDCl3, 400 MHz, Key Peaks) ~(ppm) 0.659 (s), 0.815
(s), 1.025 (d, J=6. 1 Hz), 3.01 (dd, J=3.38 Hz, J=12.73 Hz).
WO 95/12398 PCT/US94/12516
~ f
2 1 7~23~
- 19-
EXAMPLE 4
H,
O~N
, ~ J . / ~ DDQ/BSTFA
~ N-- Trifilic Acid/Toluene
H
H
O~N
2 0 0 N,
N-Diphenylmethyl-3-oxo-4-aza-7,13-methyl-Soc-androst- 1 -ene- 17,B-
carboxamide
2s To a solution of N-diphenylmethyl-3-oxo-4-aza-7~-methyl-
Soc-androstan-17,13-carboxamide (200 mg, .402 mmol) in toluene (10 ml)
was added bis(trimethylsilyl)trifluoroacetamide (BSTFA) (454 mg, 1.768
mmol), trifilic acid (trifluoromethanesulfonic acid)(4 ~ll) and dichloro-
30 dicyano benzoquinone (DDQ) (122 mg, 0.442 mmol). After stirring thereaction mi~lur~ overnight at room temperature, methyl acetoacetate (9.3
ml, 0.08 mmol) was added and reaction ~ Lure refluxed for 6 hrs. The
lure was cooled to 23, diluted with ethyl acetate, washed with aq.,
sodium carbonate, aq., sodium bisulfite, brine, dried and concentrated.
Crude product was purified by prep. TLC to afford pure product; mp 142-
W O 95/12398 PCTrUS94/12516
21 74234r ~
.
- 20 -
144. Mass spec. (MS) M+ calculated, 496; observed m/e 497 (m+l)
(FAB); lH NMR (CDCl3, 400 MHz, Key Peaks) ~(ppm) 0.68 (s), 0.89
(s), 1.01 (d, J=5.72 Hz), 3.32 (dd, J=3.72 Hz, J=12.36 Hz).
P,XAMPLE S
O: OH
ol ,~ ~ J~ _ ,
H
H
[S-(2-Pyridyl)-7,B-me~yl-3-oxo-4-aza-Sa-androstane-17~-
thiocarboxylate
To a solution of 7,B-methyl-3-oxo-4-aza-Sa-androstane-17,~
carboxylic acid (1.3 g, 3.9 mmol) in toluene (20 ml) was added aldrithiol
(1.7 g, 7.8 mmol) and triphenylphosphine (2.04 g, 7.8 mmol). After
stirring at room tempel~lule overnight the reaction ,~,;x~.,,e was eluted
through 200 ml of silica gel with 10% acetone in methylene chloride to
g*e crystalline product. Recryst~ tion from ethyl acetate afforded the
title compound.
EXAMPLE 6
Preparation of 7,B-methyl-3-oxo-4-aza-5a-androstane- 17,~-carboxylic
acid
As seen below in Flowsheets A and B, pregnenolone-3-
acetate P is first reduced to the alcohol II by sodium borohydride in
ethanol at -10 to 0C. The alcohol II is then protected by a dime~yl-t-
butyl silyl (TBS) group in DMF with TBS chloride and imidazole as a
WO 95/12398 PCT/US94/12516
~ . ~ . t ~ 2 ~ 7 ~ 2 3 ~
- 21 -
base at room temperature. The protected alcohol is then oxidized to the
corresponding 5-en-7-one m by treatment with hydrogen t-butyl peroxide
and cllronliulll hexacarbonyl in e.g., acetonitrile, at reflux. The 7-methyl
group can be introduced at this point by a Grignard reaction using e.g.,
5 methyl magnesium chloride in e.g., anhydrous THF at 0 to -10C to
produce the 7-methyl-7-hydroxy adduct IV. This is then oxidized with
e.g. ~ isopropoxide and cyclohexanone (Oppenauer oxidation
conditions) in refluxing toluene solvent to produce the 7-methyl-4,6-dien-
3-one V. This in turn is reduced via e.g., metal-ammonia, THF and
toluene at -78C to selectively yield the 7-beta-methyl-S-en-3-one VI. In
the next step the delta-S double bond is isomerized to the 4-ene by use of
DBU (1,8-diazabicyclo[5.4.0] undec-7-ene) in e.g. reflllxin~
tetrahydrofuran (THF) to produce the 7-methyl-4-ene-3-one, VII. The A
ring is next cleaved by treatment with e.g. pot~illm pe~n~n~n~e,
5 sodium periodate in t-butyl alcohol at 80C to produce the corresponding
seco-acid VIII.
Tre~tmtqnt of the seco-acid with ammonium acetate in
glacial acetic acid at 120C yields e.g., the 7-methyl-4-aza-pregn-5-en-3-
one IX. This in turn is selectively reduced with e.g., PtO2, to remove the
2 S-double bond to produce the Sa-hydrogen compound X. The TBS
protecting group is next removed by aqueous HF in acetonitrile at room
temperature and then oxidized by tell~ru~ylammonium perruthenate/4-
methylmorpholine N-oxide in methylene chloride at room temperature to
yield the 17-acetyl compound XI. This is treated with a sodium
25 hypol~r~llliL~/sodium hydroxide solution in dioxane at 10-15C to form
the title 17-carboxylic acid XII, mp. 311-312C. This is then used as
described above to make the 2-thiopyridyl ester and the resulting
azasteroidal amides.
The 1,2-double bond in the A ring can be introduced into
3 XII by DDQ oxidation (see procedure in U.S. Patent 5,084,574) to
produce xm, mp. 328-330C. Formation of ~e 2-thiopyridyl
intermediate XIV, analogously as described above, and reaction with
benzhydryl~mines as described above produces the corresponding
WO 95/12398 PCT/US94112516
t ~ 2 ~ 74234
- 22 -
amides. In the schemes below, "Ac" represents an acetyl group and "Ar"
represents an aryl group.
FLOWSHEET A
~ ~_OH
(~ NaBH4/EtOH ~ ~
--/ ~(1) TBSCI/DMF
AcO~--~J AcO'~ (2) Cr(CO)6/t-BuOOH
~_OTBS ~_OTBS
~~ MeMgCI/THF ~
Oxidation
AcOJ--~O HO~HCH3
111 IV
\~OTBS
(1 ) Li/NH3/THF/Toluene \~ OTBS
/ (2) CH2BrCH2Br/THF ~~~
~ (3) NH4CI ~ ~/ DBU.
V o~
~_OTBS Vl \~OTBS
, ~ KMnO4/NalO
3 o ~ ~~~ Na2CO3/t-BuOH
0~ HOOC O,
Vll Vlll
WO 9S/12398 PCT/US94/12S16
2 1 7 4 2 3 4
FLOWSHEET B
OTBS ~ OTBS
H2/ACOH/Pto2
HOOC O O N
Vlll H IX
~_ OTBS ~o
(1) Aq. HF/CH~CN ~ ~
~X~ (2) Oxidation ~ NaOBr
O ,N O,N~~ Xl
H~o
` DDQ ~
O ,N ~
H Xll O N--
H Xlll
¢~ N/CHAr
~~ ArCHR'NHR, f ~~ R'
O ,N~~ ~
H XIV O~N~~ XV
H
WO 95112398 PCT/US94rl2516
~ S h ~ ~ 2 1 7 4 2 3 4
- 24 -
Biological Assays
Preparation of Human prostatic and scalp 50c-reductases
Samples of hl-m~n tissue were pulverized using a freezer
5 mill and homogenized in 40 mM potassium phosphate, pH 6.5, 5 mM
magnesium sulfate, 25 mM potassium chloride, 1 mM phenylmethyl-
sulfonyl fluo~de, 1 mM dithiothreitol (Dl'r) cont~inin~ 0.25 M sucrose
using a Potter-Elvehjem homogenizer. A crude nuclear pellet was
prepared by centri~ugation of the homogenate at 1,500 x g for 15 min.
o The crude nuclear pellet was washed two times and resuspended in two
volumes of buffer. Glycerol was added to the resuspended pellet to a
final concentration of 20%. The enzyme suspension was frozen in
aliquots at -80C. The prostatic and scalp reductases were stable for at
least 4 months when stored under these conditions.
5a-reductase assay
The reaction ll~ix~ for the type 1 5a-reductase contained
40 mM potassium phosphate, pH 6.5, 5 mM [7-3H]-testosterone, 1 mM
dithiothreitol and 500,uM NADPH in a final volume of l OO ,ul. The
2 reaction mixture for the type 2 Sa-reductase contained 40 mM sodium
citrate, pH 5.5, 0.3 mM [7-3H]-testosterone, 1 mM di~iothreitol and 500
~M NADPH in a f;nal volume of 100 ~ul. Typically, the assay was
init;~ted by the addition of 50-100 ,ug prostatic homogenate or 75-200 ~g
scalp homogenate and incubated at 37C. After 10-50 min. the reaction
25 was quenched by extraction wi~ 250 ,ul of a mi~ e of 70%
cyclohexane: 30% ethyl acetate cont~inin~ 10 ,ug each DHT and T. The
aqueous and organic layers were separated by centrifugation at 14,000
rpm in an Eppendorf microfuge. The organic layer was subjected to
no~nal phase HPLC (10 cm Wh~ n partisil S silica column
30 equilibrated in 1 ml/min 70% cyclohexane: 30% ethyl acetate; retention
times: DHT, 6.8-7.2 min.; androstanediol,7.6-8.0 min.; T, 9.1-9.7 min).
~he HPLC system consisted of a Waters Model 680 Gradient System
equipped with a Hitachi Model 655oc autosampler, Applied Bio~yslems
Model 757 variable UV detector, and a Radiomatic Model A120
WO 95/12398 PCT/US94/12516
r ~ 2 1 7 4 2 3 4
radioactivity analyzer. The conversion of T to DHT was monitored using
the radioactivity flow detector by mixing the HPLC efflllent with one
volume of Flo Scint 1 (Radiomatic). Under the conditions described, the
production of DHT was linear for at least 25 min. The only steroids
5 observed with the hllm~n prostate and scalp preparations were T, DH'r
and androstanediol.
Inhibition studies
Compounds were dissolved in 100% ethanol. IC50 values
represent the concentration of inhibitor required to decrease enzyme
activity to 50% of the control. IC50 values were determined using a 6
point titration where the concentration of the inhibitor was varied from
0.1 to 1000 nM. Representative compounds of this invention were tested
in the above desribed assay for Sa-reductase type 1 and type 2 inhibition.
A compound ~fe~cd to herein as a Soc-reductase 2 inhibitor
is a compound that shows inhibition of the Sa-reductase 2 isozyme in the
above-described assay, having an IC50 value of about or under 100 nM.
A compound lcfelled to herein as a Sa-reductase 1 inhibitor
is a compound that shows inhibition of the Sa-reductase 1 isozyme in the
2 above-described assay, having an IC50 value of about or under 100 nM.
Human Dermal Papilla Cell Assay
The dermal papilla is a small group of cells at the base of
each hair follicle, and it is presently ~ought that these cells are stem
cells that form the basis for hair growth. These cells have been shown
to have S alpha reductase activity, and it is therefore possible to test
inhibitors of 5 alpha reductase in ~ese cell culture systems.
Isolated and cultured dermal papilla cells are prepared
according to the methods of Messenger, A.G., "The Culture of Dermal
Papilla Cells From H--man Hair Follicles", Br. J. Dermatol. 110:685-
689, 1984 and Itami, S. et.al., "Sa-Reductase Activity In Cultured
Human Dermal Papilla Cells From Beard C~m~ared With Reticular
Dermal Fibroblasts", J. Invest. Dermatol. 94:150-152, 1990. Beard
dermal papilla cells and occipital scalp hair of t~,vo different
W O 95112398 PCTrUS94112516
2 1 7 4 2 3 4
- 26 -
individuals are used ~roughout the study. All experiments are
performed at confluency after the fourth to sixth subculture.
Confluent monolayers are rinsed twice with phosphate-buffered
saline, scraped from dishes by rubber policemen, and collected into a
centrifuge tube. The cell suspensions are centrifuged at 1,500 rpm for
10 min at 4C. The pellets are resuspended in 20 mM Tris-HCl
buffer, pH 7.5, at 4C, cont~inin~ 250 mM sucrose, 1 mM MgC12, and
2 mM CaC12, by vortexing and 10 passes through a 25-gauge needle.
The crude homogenate is further homogenized by a teflon-glass
homogenizer, and is used as the cell homogenate. For the study of
subcellular loc~li7~*0n of 5a-reductase, the cell homogenate is
centrifuged at 800 x g for 10 min to yield a crude nuclear pellet. The
resultant supernatant is centrifuged at 10,000 x g for 15 min to
produce a crude mitochondrial pellet. The supern~t~nt is centrifuged
at 100,000 x g for 60 min to yield a microsomal pellet and cytosol.
Each particulate fraction is washed twice and resuspended in ~e
buffer.
A standard incubation mixture will consist of 50 nM
[3H]-testosterone, 1 mM NADPH, 100 mM sodium citrate, pH 5.5 or
100 mM Tris-HCl, pH 7.5, and 50 ml of the cell homogenate, in a
final volume of 100 ml. Each tube cont~in~ 50-100 rng of cellular
protein. Incubation is carried out at 37C for 30 min. During this
incubation, the reaction is proportional to the time. For the study of
op~ pH, citrate buffer is used at pH 4.5-6.5, and the Tris HCl
buffer at pH 7.0-9Ø The protein content is detellllilled by the method
of Lowry, et al., "Protein Measurement With The Folin Phenol
Reagent" J. Biol. Chem.. 193:265-275, 1951.
After incubation, the reaction is stopped by adding 4
times volume of chloroform-methanol (2/l:V/V) CO~ i"g 110 mg
30 each of carrier steroids. The extracted steroids are analyzed by thin-
layer chromatography as previously described by Gomez, et al., "In
Vitro Metabolism Of Testosterone-4-14C and D-androstene-3, 17-
dione-4-14C In Human Skin" Biochem., 7:24-32,1968, and the purity
of each steroid is detellllined by the recryst~lli7~tion method. The
WO 95/12398 PCT/US94112516
t ~
2 1 74234
- 27 -
activity of Sa-reductase is expressed by the sum of dihydro-
testosterone, androstanediol and androstanedione formed. [1,2-3H]-
testosterone (55.2 Ci/mmol) is obtainable from New Fngl~n(l Nuclear
Corporation (Boston, MA) and unlabeled steroids can be purchased
from Sigma Chemical Company (St. Louis, MO). Fetal calf serum is
obtainable from Hazleton (Lenaxa, K~nc~). All other chemicals are
of reagent grade.
Fuzzy Rat Acne Model
o Adult fuzzy rats are a variety of rat that has stunted hair
growth, brown colored seborrhea covering their entire back skin and
abnormally increased sebum production after puberty that has been
demonstrataed to be due to circulating androgens. 0.1, 0.05 and
0.025% solutions of a selected Sa-reductase inhibitor of interest are
15 prepared in a vehicle of propylene glycol, isopropanol, isopropyl
myristate and water (50/30/2/18%), and is topically applied onto the
backs of adult male fuzzy rats, 0.2 ml per ~nim~l daily for 4 weeks.
Controls receive the vehicle alone and S of them are castrated. After 2
weeks seborrhea will be dose-dependently depleted and after 4 weeks
bromodeoxyuridine (BrdU, 200 mg~g) is intraperitoneally injected 2
hours before sacrifice. The skin tissues are incubated with EDTA (20
mM) in phosphate buffer, 1.5 hours at 37C. The pilo-sebaceous unit
attached to the epidermis is striped from the dermis and fixed with
form~lin for i~ o-staining of BrdU. DNA synthesis cells showing
a BrdU-positive nucleus are located in the outer ~l~ncl~ r border. The
number of S-phase cells per lobe is dete~ led with a micro-image
apparatus. Using form~lin fixed skin, frozen serial sections are
stained with 1% o~millm and the size of the lobes is measured. A
positive inhibitor of skin 5a-reductrase will induce suppression of
30 sebum production by inhibiting the rate of ~l~n(llll~r cell turnover, and
showing reduced lobular size.
The following describes an example methodology that can
be used for detection of hair growth.
WO 95/12398 PCT/US94112516
~ C S h ~ ~ ~ 21 74234
- 28 -
MACROPHOTOGRAPHY AND GLOBAL PHOTOGRAPHY
PROCEDURE FOR DETECTION OF HAl[R GROWTH
A. Macrophotographic Procedure Location: ID card
Haircount target area
Equipment: Film: Kodak-T-max 24 exposure each of same emulsion lot
number
Camera: Nikon N-6000
Lens: Nikkor 60 mm f2.8
Flashes: Nikon SB-21B Macroflash
Device: registration device
Photo~raphic Procedure:
In these clinical photographs, the only variable allowed is
the haircount. Film emulsion, li~hlin~;, f,~llillg, exposure, and
reproduction ratios are held constant.
1. The haircount area on the patient is prepared as follows: A
small (~lmm) dot tattoo is placed at the beginning of the
study at the le~ling edge of ~e bald area directly interior to
the center of the vertex bald spot, using a commercial
tattooing machine or m~ml~lly (needle and ink). An area
approximately one square inch in size, centered at the tattoo
at the le~(lin~; edge of the balding area, is clipped short
(~2mm). Cut hairs removed from the area to be
photographed, using tape. (~ompressed air andlor ethanol
wipes may also be used to facilitate removal of cut hairs.
2. Magnification: Each lens supplied has a fixed reproduction
ratio of 1:1.2.
3 Aperture: Every photograph is taken at f/22.
Film: T-Max 100 (24 exposure) is used.
3. Patient's haircount target area. Three exposures (-2/3, 0, and
+2/3 f-stop).
WO 95/12398 PCT/US94112~16
i ~ 2 1 7 4 2 3 4
- 29 -
B. Global Photographic Procedure
Locations: Color card/patient Id
Global photograph
Equipment: Film: Kodachrome KR-64 24 exposure each of same
emulsion lot number
Camera: Nikon N-6000
Lens: Nikkor 60 mm f2.8
Flashes: Nikon SB-23
Color card/patient Id
Photographic Procedure
In these clinical photographs, the only variable allowed is
the global area's appearance. Anything extraneous to the area (clothing,
r",.,;l",~, walls, etc.) is elimin~ted from the fields to be photographed.
1. Patients will have global photographs taken prior to hair
clipping with the head in a fixed position (determined by the
supplied stereotactic device). Hair on the patient's head is
positioned consistently so as to not obscure the bald area.
2. Magnification: Eachlenssuppliedhasafixedreproduction
ratio of 1:6.
Aperture: Every photograph will be taken at f/11.
Film: Kodachrome (24 exposure) is used.
3. Patient's global photographs. Three exposures at zero
compensation.
A trained technician places a transparency over the
photograph and, using a felt tip pen, places a black dot over each visible
hair. The dot map transparency is then counted using image analysis
with co,ll~uler assistance.
3 Photographs are coded with a random number corresponding
to study site, visit number and patient allocation number to insure
blin(lin~ to time. At Month 6, baseline and Month 6 photographs are
counted and data analyzed for i.lL~ n analysis. At Month 12, baseline,
WO 95/12398 PCT/US94/12516
~1 74234 ~
- 30 -
Month 6 and Month 12 photographs are counted and data analyzed for
the primary endpoint.
Methodology for detection of hair growth is also described
in Olsen, E.A. and Delong, E., J. American Academy of Dermatology,
Vol. 23, p. 470 (1990).
While the invention has been described and illustrated
with reference to certain particular embodiments thereof, ~ose skilled
in the art will appreciate that various changes, modi~lcations and
substitutions can be made therein without departing from ~e spirit
and scope of the invention. For example, effective dosages other than
the particular dosages as set forth herein above may be applicable as a
consequence of variations in the responsiveness of the m~mm~l being
treated for any of the indications for the compounds of the invention
indicated above. Likewise, the specific ph~ cological responses
15 observed may vary according to and depending upon the particular
active compound selected or whether there are present ph~rm~ceut-ical
ca~iers, as well as the type of form~ tion and mode of ~lmini~tration
employed, and such expected variations or dir~lel,ces in the results
are colltelll~lated in accordance with the objects and practices of the
present invention. It is intended, therefore, that the invention be
defined by the scope of ~e claims which follow and ~at such claîms
be intel~leled as broadly as is reasonable.