Note: Descriptions are shown in the official language in which they were submitted.
WO95/1402~ 21 74 65~ PCT/IJS94/13483
- 1 -
TITLE OF THE INVENTION
PLPERIDINYLCAMPHORSULFONYL OXYTOCIN ANTAGONISTS
FIELD OF THE INVENTION
This application is a cnntin~ tinn-in-part of U.S. Seriai No.
08/153,521, filed November 16, 1993, the contents of which are hereby
incorporated by reference, which is a continll ~rinn-in-part of U.S. Serial
No. 07/954,596, filed September 30, 1992, now abandoned, which is a
cul,lillu~ion of U.S. Serial No. 07/759,254, filed September 13, 1991,
now abandoned, which is a cnn~in~ ion-in-part of U.S. Serial No.
07/612,344, filed November 13, 1990, now ~h~n~lnnP~I the contents of
all previous app]ications are hereby incorporated by reference.
The present imvention provides novel compounds, novel
compositions, methods of their use and methods of their m~nllf~chlre7
such compounds generally pharmacologically useful as agents in
obstetric and gynecologic therapy. The aforementioned phammacologic
activities are useful in the treatment of m~mm~l~ More specifically, the
compounds of the present invention can be used in the treatment of
preterm labor, stopping labor preparatory to Caesarean delivery, and in
the treatment of dysmenorrhea. At the present time, there is a need in
the area of obstetric and gynecologic therapy for such agents.
BACKGROUND OF THE INVENTION
In the field of obstetrics, one of the most important
problems is the management of preterm labor. A significant number of
the pregnancies progressing past 20 weeks of gestation experience
p~Clll~Ult~ labor and delivery, which is a leading cause of neonatal
morbidity and mortality. Despite major advances in neonatal care,
retention of the fetus in utero is preferred in most instances.
Tocolytic (uterine-relaxing) agents that are currently in
use include ~2-adrenergic agonists, m~nPsillm sulfate and ethanol.
Ritodrine, the leading ~2-adrenergic agonist, causes a number of
cardiovascular and metabolic side effects in the mother, including
tachycardia, increased renin secretion, hyperglycemia (and reactive
.... , . . . , .. .. . . ... . _ _ _ _ _ _ _ _ _
WO 95/14025 PCr~US94113483
21 7~650
- 2 --
hypoglycemia in the infant). Other ~2-adrenergic agonists, including
terbutaline and albuterol have side effects similar to those of ritodrine.
nf~Sillm sulfate at plasma concentrations above the therapeutic range
of 4 to 8 mg/dL can cause inhibition of cardiac conduction and
5 neuromuscular transmission, ~ dloly depression and cardiac arrest,
thus malcing this agent unsuitable when renal function is impaired.
Ethanol is as effect;ve as ritodrine in preventing pl~lllalulc labor, but it
does not produce a corresponding reduction in the incidence of fetal
d~ y distress that administration of ritodnne does.
It has been proposed that a selective oxytocin antagonist
would be the ideal tl~colytic agent. In the last few years, evidence has
~rcllm~ tPd to strongly suggest that the hormone oxytocin may be a
physiological initiat~r of labor in several m~mm~ n species including
humans. Oxytocin is believed to exert this effect in part by directly
5 contracting the uterine myometrium and in part by ~nh~nrin~ the
synthesis and release of contractile pr )st~ n-lins from the uterine
endometrium/decid~a. These pr st~ n~lins may, in addition, be
illll,ollallL in the cer~/ical ripening process. By these ml~l h~nicms, the
process of labor (term and preterm) is initiated by a heightened
20 sensitivity of the ut~rus to oxytocin, resulting in part as a result of a
well-documented increase in the number of oxytocin receptors in this
tissue. This "up-regulation" of oxytocin receptors and enhanced uterine
sensitivity appears to be due to trophic effects of rising plasma levels of
estrogen towards term. By blocking oxytocin, one would block both the
25 direct (contractile) and indirect (enhanced prostaglandin synthesis)
effects of oxytocin on the uterus. A selective oxytocin blocker, or
antagonist, would lilcely be more efficacious for treating preterm labor
than current regimens. In addition, since oxytocin at term has major
effects only on the uterus, such an oxytocin antagonizing compound
30 would be expected t~ have few, if any, side effects.
The compounds of the present invention are also useful in
the treatment of dysmenorrhea. Thi~ condition is characteri~ed by
cyclic pain associated with menses during ovulatory cycles. The pain i.s
thought to result from uterine contractions and ischemia, probably
WO 95/14025 2 1 7 4 6 5 0 Pcr~Sg4/l3483
- 3 -
mediated by the effect of prost~ nrlins produced in the secretory
endometrium. By blocking both the direct and indirect effects of
oxytocin on the uterus, a selective oxytocin antagonist can be more
efficacious for treating dy~ eno~lllea than current regimens.
An additional use for the present invention is for the
stoppage of labor ~ ,al~lluly to Caesarean delivery. Certain
spiroindanylpiperidines and spiroind~llyl~ilJelidines are known (U.S.
Patents 3,654,287 and 3,666,764), however, they are reported to be
useful as ~n~sth!~ti~ agents which is quite distinct from the utility of the
o present invention.
It is, therefore, a purpose of this invention to provide
sllbst:lnces which more effectively antagonize the function of oxytocin in
disease states in animals, preferably m~mm~l~, especially in humans. It
is another purpose of this invention to prepare novel compounds which
more selectively inhibit oxytocin. It is still another purpose of this
invention to provide a method of antagonizing the functions of oxytocin
in disease states in m~mm:~lc It is also a purpose of this invention to
develop a method of preventing or treating oxytocin-related disorders
of preterm labor and dy~lllenullllea by antagonizing oxytocin.
It has now been found that compounds of the present
invention are antagonists of oxytocin and bind to the oxytocin receptor.
When the oxytocin receptor is bound by the compounds of the present
invention, oxytocin is antagonized by being blocked from its receptor
and thus being unable to exert its biologic or pharmacologic effects.
25 These compounds are useful in the treatment and prevention of
oxytocin-related disorders of animals, preferably m~mm~ls and
especially humans. These disorders are primarily preterm labor and
dysmenorrhea. The compounds would also find usefulness for stoppage
of labor preparatory to Caesarean delivery. Additionally, such
30 compounds are useful in inducing cu-l~ldc~plion in m:~mm~l~ inasmuch
a~ oxytocin antagonists have now been shown to inhibit the release of
oxytocin-stimulated llltPini~in~ hormone (LH) by anterior pituitary
cells.
WO 95/140Z5 PCI/US94/13483
2 1 7~
- 4 -
Compounds of the present invention are also inhibitors of
vasopressin and can bind to the vasopressin receptor. These compounds
are useful in inducillg vasodilation, treating hypertension, inducing
diuresis and inhibiting platelet ~ tin~fion.
Additionally, it has now been found that the compounds of
the present inventioll are balanced oxytocin and vasopressin antagonists
useful for treating preterm labor and dysmenorrhea,
SU~IARY OF TH~ NTION
0 The compounds and their pharrn~( e--fic~lly acceptable salt.s
and esters of the present invention are those of the general structural
formula I
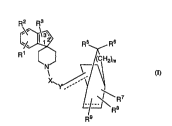
~2 R3
~\~ Rs ,R6
~ R Y
L~ R8
R9
wherem
X is
o
C
3 o CH2.
P02H,
P02Z where Z is Cl lo alkyl,
so2~
S02NH or
-
WO 95/14025 ~ PCrlUS94/13483
- 5 -
o
C - NH .
Y is absent or is
CH or
CH2;
m is an integer of from zero to one;
o n is an integer of from zero to five;
p is an integer of from zero to five;
q is an integer of from zero to five;
Rl and R2 are each independently
hydrogen,
halogen,
hydroxy,
Cl -10 alkyl,
Cl lo alkoxy or
trifluoromethyl;
R3 is
hydrogen,
halogen,
hydroxy or
oxo with the proviso that when R3 is oxo then the 2,3 bond
is saturated;
R4is
3 0 hydrOgell,
phenyl, or
Cl lo alkyl,
R5 and R6 are in-l~pçn~ ntly
hy ~roge .,
WO 95/14025 Pcr/uss4ll34s3
.
- - 21 1~6
- 6 -
C1 1o alkyl or
Cl 1o hydroxyalkyl; or
RS and R6 together are
oxo or
n~nbstitllted or substituted C3-6 cycloalkyl, where said
substituents are
hydroxy,
Cl lo alkyl,
CI -10 hydroxyalkyl,
Cl lo alkoxy or
Cl-3 alkoxyalkoxyalkoxyalkyl;
R7and R8 are independently
hydrogen or
hydroxyl;
R9 is
C7 1 0 alkoxy,
Cl -10 alkoxycarbonyl,
Cl l o alkoxycarbonylalkylarninocarbonyl,
cyano,
--O--P-OH
OH
Cl lo alkyl substituted phosphonate,
--O-S- OH
Cl lo alkyl substituted sulfonate,
--(CH2)n --N--R~2
W0 95/14025 2 1 7 4 ~ ~ ~ PCrlUss4/l34s3
R11
--(CH2)p --N- CO--Rl~
--(CH2)q--CO-NH--R14 or
llnsllbstihl~d or substituted C1 l0 alkyl where said
lr ~ ~ l is
Rl; or
R7 and R8 are, together with the carbons to which they are attached,
o joined to form a 5-membered hetercyclic ring containing 2 hetero atoms
where said hetero atoms are N and O;
R10 is
hydroxyl,
carboxyl,
C1 loalkoxy,
C1 1o alkoxycarbonyl,
R1S or
cyano;
Rll iS
hydrogen,
C 1- l o alkyl,
Cl lo carboxyalkyl or
C1 1o alkoxycarbonylalkyl;
R12is
hydrogen,
Cl lo alkylsulfonyl,
Cl_10 alkarylsulfonyl,
C1-10 aralkylsulfonyl,
Cl 1o alkoxyarylsulfonyl,
aminosulfonyl,
Cl lo alkylaminosulfonyl,
Cl lo dialkylaminosulfonyl,
WO 95114025 PCTIIJS94113483
21 74650
- 8 -
llncllhstihlt~d or sllh~stihltpd C4 15 cycloalkylalkyl, bicycloalkyl or
tricycloalkyl where said substituent is
oxo or
sulfonyl sllh.s~ih~tPd by uul~ub~Lilul~d or s~lhstih-t~d C7 ls
cycloalkyl, bicycloalkyl or tricycloalkyl where said
,lr~ are
oxo,
o~ime or
hydroxy;
R13 is
amino,
Cl lo alkylarnino,
Cl lo alkyl which is llnsllhstihltPd or mono- or di-substituted by
6,
C2 l0 alkeny~ which is unsubstituted or substituted by Rl6,
phenyl substituted by R 17,
lln~lhctitlltPd or sllh~tihlt~d C3-8 cycloalkyl where said
sllhstitllPnt.~ al-e
Cl lo ~Ikyl or
carboxy,
N riic--hstitlltPd by Cl lo alkyl,
llnsllhstihltPd or suhstih-tPd C7 l5 bicycloalkyl or tricycloalkyl
where said s--l-~tit-~Pnt i.s
carbox~,
Cl lo alkoxy or
R18;
Rl4 i.s
Cl-lO alkyl,
Cl lo aminoalkyl,
Cl lo alkoxycarbonylalkyl, or
C l l o carboxyalkyl,
WO 95/14025 2 1 7 4 6 5 0 PCT/IJS94/13483
_ 9 _
R15 is
lm~lhstihltl-d or SllhstitlltPd 5 membered heterocyclic rings
containirlg I hetero atom where said hetero atom is N and said
substituents are
0X0,
amino,
Cl 1o alkylamino,
Cl 1o carboxyalkylcarbonylamino,
Cl lo dicarboxyalkylamino or
o Cl lo alkyloxycarbonylalkylamino;
R16 is
llr~cllhstihlt~d or s~lbstih~t~d C4-8 cycloalkyl where said
substituents are
hydroxy or
carboxy,
C10 15 bi- or tricycloalkyl,
halogen,
hydroxy,
carboxy,
oxo,
oxime,
Cl lo aikylthio,
Cl lo alkylsulfunyl,
Cl_10 alkylsulfonyl,
Cl lo alkoxycarbonyl,
R20,
R21,
amino,
aminocarbonyl,
Cl lo dialkylaminocarbonyl,
Cl lo alkylamino,
Cl lo dialkyiamino,
Cl lo alkylcarbamate,
WO 95/14025 PCrlUS94113483
.
~7~5
- 10 -
Cl lo alkylcarbonate,
C1 loalkylureide,
Cl lo aralkyl.;a-l,all.dl~,
lm~llh~titlltl~d or sl~hstihlt~d aryloxy where said substituents
are
amino,
C1 1o alkyl or
Cl lo aminoalkyl,
Cl lo aralkoxy,
n~llhStihlt~d or ,~llhstihlf~d Cl 1o alkaryloxy where said
sllhstihl.ont is
Cl lo ~Ikylcarbamate,
N di~ub~Lilu~d by Cl lo alkyl and Cl lo carboxyalkyl or
N tri-substituted by two Cl lo alkyls and by Cl lo
alkoxycarbonylalkyl with the proviso that a trifluoroacetic
acid counterion be present;
Rl7 is
amino,
2 halogen,
Cl_lo alkyl,
Cl loalkoxy,
nitro,
phenylcarbon~l or
un~ub~LiLul~d ~r ~l-bctihlt~d 5 membered heterocyclic rings
c-nt~inin~ I hetero atom where said hetero atom is O and
wherein said ~ub~Li~u~ is
oxo;
R 18 is ull~ul~liLul~d or ~lhstihll~d heterocyclic rings selected from
azetidinyl,
pyrrolidinyl,
pyrrolyl,
piperidinyl,
;
WID 9~114025 2 1 7 4 6 5 0 PCrlUS94/13483
- 11 -
piperizinyl,
pyridinyl,
pyrimidinyl,
tetrahydrofuranyl,
furanyl7
dioxolanyl,
thienyl,
1, 3-thiazolidinyl or
tetrahydrooxazolyl; where said s~lhstit-~PnS~ are one or more of
1 o oxo,
hydroxy,
carboxy,
amino,
Cl lo carboxyalkyl,
Cl_10 alkyl,
Cl 1oalkoxy,
C1 -10 aralkoxy,
C1 1oalkaryloxy,
C1 1o alkoxycarbonyl,
Cl-10 alkoxycarbollylamino~
CI-10 alkoxycarbonylalkyl,
Cl lo aralkoxycarbonyl or
s-lh~titl-tPd or lln~llhstitlltPd phenyl where said sub~Lilu~
are
2~ Cl_5 alkyl,
carboxy or
halogen;
R20 is lln~lhstitlltPd or substituted heterocyclic rings selected from
azetidinyl
pyrrolidinyl,
pyrrolyl,
tetrahydroimidazolyl,
irnidazolyl,
WO 9S114025 PCI'IUS94/13483
2~ 7~65~
- 12 -
tetrazolyl,
piperidinyl,
pyridinyl,
hexah~dloa~ yl,
thienYI~
1, 3-thiazolidinyl or
tetrahydrothi~lzinyl; where said sllhsfitll~ntc are one or
more of
C1-10 alkyl,
o Cl-10 aralkyl.
Cl lo aralkoxy,
Cl lo alkaryl,
amino,
C1 1o alkylarnino,
Cl -10 dialkylarnino,
oxo,
oxime,
fused phenyl,
C1 1o alkoxycarbonyl,
C1 1o ;llkylcarbonate,
C1 lo alkylureide,
Cl lo alkyl~,dll)al.laL~, or
llncllhs~itllt~-l or substituted ~-membered heterocyclic
rings ha~ing I hetero atom where said hetero atom is
N and said s~lhstitl~nt is one or more of
oxo or
fused phenyl;
R21 iS
unsub~tituted or s~hstit--~d phenyl where said substituent~ are
halogen,
C1 1o alkyl,
C I 10 carboxyalkyl,
Cl lo alkoxy,
r
WO9S/14025 2~ 74~ PCI/US94113483
- 13 -
5- or 6-membered heterocyclic rings having l or 2 hetero
atoms where said hetero atoms are N or S,
hydro~y,
carbo~y or
--O-S--OH
o
In one embodiment of the instant invention are compounds
the formula
R2 R3
~,
X~y~
R9 R
~vherein
Xis
o
C,
CH2,
Po2H7
PO2Z ~vhere Z is C1 l0 alkyl, or
SO2;
Rl and R2 are each indPp~n~l~n~ly
hydrogen,
halogen or
Cl lo alkyl;
WO 95/14025 PC~IUS94/13483
21 7~65G
- 14 -
R4 is
hydrogen or
phenyl;
RS and R6 are independently
hydrogen or
C1 1o alkyl; or
RS and R6 together are
oxo or
n~-hstihlt~d or ~llhctihltt~d C3-6 cycloalkyl, where said
sllhstit-l.onts are
hydroxy,
CI-10 hydroxyalkyl or
C1-3 al~oxyalkoxyalkoxyalkyl;
R9 is
C7-1o alkoxy,
C1 1o alkoxycarbonyl,
C1 1o alkoxycarbonylalkylarninocarbonyl,
--O--P-OH
C1 1o alkyl substituted OH
ll
o
phosphonate,
Cl lo alkyl substituted sulfonate,
R11
--(CH2)n --N--R12
R11
--(CH2)p --1\1- CO--Rl3
llnsllh~titllt~d or substituted Cl lo alkyl where said substituent i.s
WO 95/14025 2 1 7 ~ ~ 5 ~ PCT/IJS94113483
- 15 -
R10; or
R7 and R8 are, together with the carbons to which they are attached,
joined to form a 5-membered heterocyclic ring containing 2 hetero
5 atoms where said hetero atoms are N and O;
R12 iS
hydrogen,
Cl loalkylsulfonyl,
o C1 1oalkarylsulfonyl,
Cl lo alkoxyarylsulfonyl,
~min--sulfonyl,
C1 1odialkylaminosulfonyl,
ns~lh~titlltPd or sllbstit~lt~ d C3-15 cycloalkylalkyl where said
1 5 .s. .~ iS
oxo or
sulfonyl sl~hstihlt~d by IIII~III).,Iill~l~d or substituted C3 15
cycloalkyl where said ~ ll is
oxo
R 14 is
Cl lo alkyl,
Cl lo arninoalkyl or
Cl loalkoxycarbonylalkyl;
R16 iS
lln~llhctihltt~d or ~llhstitllt~d C5-6 cycloaLkyl where said s~lhstitll~nt
is
hydroxy,
C10-15 tricycloalkyl7
halogen,
hydroxy,
carboxy,
oxo,
WO 9~/1402~ PCTIUS94113483
2~ 74650
- - 16 -
Cl loalkyltlhio,
C1 lo alkylsulfonyl,
C1 lo alkoxycarbonyl,
R20,
R21,
amino,
Cl -10 alkylamino,
C1 1o dialkylamino,
Cl lo alkylc~lb~ulla~
Cl loaralkyl~all,~"là~,
unsubstituted or ~ub~Li~ul~d aryloxy where said sllh~tit~ nt~
are
arnino,
Cl 1o alkyl or
C1 1oaminoalkyl,
C1 1o aralkoxy,
lln~llhstitlltPd or sllhstitllt~d C1 l0 alkaryloxy where said
l l r l l l is
Cl lo alkyl ,alballlat~,
N ,li~ d by Cl lo alkyl and Cl lo carboxyalkyl; or
N tri-s--hstitllt~d by two Cl-lo alkyls and by Cl-lO
alkoxycarborlylalkyl with the proviso that a counterion be
present from tlhe group c--n~i~tin~ of Cl ~ halogenated
carboxylic acids;
Rl~ is llncllh~titllt~d or s--h~tin-t~d II~Lelu~j~lic rings selected from
azetidinyl,
pyrrolidinyl,
pyrrolyl,
plpendmyl,
piperizinyl,
pyridinyl,
pyrimidinyl,
tetrahydrofuranyl,
WO 95/14025 2 ~ 7 ~ 6 5 0 Pcr/usg4/l3483
.
- 17 -
furanyl,
dioxolanyl,
thienyl,
1, 3-thiazolidinyl or
tetrahydrooxazolyl, where said substituents are one or more of
oxo,
hydroxy,
carboxy,
Cl lo carboxyalkyl,
o C1 loalkyl,
C1 1o alkoxy,
Cl lo aralkoxy,
C1 1oalkoxycarbonyl,
Cl lo alkoxycarbonylalkyl or
C1 1o aralkoxycarbonyl;
R20 is l~n~llhstinl~Pd or substituted heterocyclic rings selected from
hexahydroazepinyl,
pyrrolidinyl,
pyrrolyl,
tetrahydroimidazolyl,
imidazolyl,
tetrazolyl,
piperidinyl,
pyridinyl,
azetidinyl,
thienyl,
1, 3-thiazolidinyl or
tetrahydrothiazinyl; where said ~llb~titl-~nt~ are one or more of
Cl-lO alkyl,
Cl lo aralkyl,
Cl lo aralkoxy,
arnino,
~.~
WO 9S/14~nS PCrlUS94113483
- 18 -
fused phenyl,
Cl 1o alkoxycarbonyl,
Cl lo alkylcarbamate, or
lm~llh~titlltt-d or s~lhstitlltPd S-membered heterocyclic rings
having I hetero atom where said hetero atom is N and said
Ut;lll iS
oxo or
fused phenyl;
10 R21 iS
unsubstituted or substituted phenyl ~vhere said substituent~ are
Cl-10 alkyl,
Cl la carboxyalkyl,
Cl lo alkoxy,
S- or 6-membered heterocyclic rings having I or 2 hetero
atoms where said hetero atoms are N or S,
hydroxy,
carboxy or
o
--O-S--OH
a
In a class are the compounds selected from
WO95/14025 21 74 650 PC!rluss4ll34s3
.
- 19 -
CH3 ,CH3
N : ~
o ~CH3
HN ~H3
So CH3
[~ CH3 ,CH3
:~o N
2s ~H
HN~So CH3
In another class are the compounds selected from
WO 9511402!i PCrrllS94/13483
2 1~65~ --
- 20 -
~2~?oH o O NHz
N ~ ;~
~\ or
~OH
S2 1l
H--~- ?
H
Illustral:ive of the invention is a pharmaceutical
composition, Cu~ g a compound of formula I in a
pharmacologically effective amount for antagonizing the binding of
oxytocin to its receptor site, and a ph~-l-la~u~ically acceptable carrier.
Illustrations of the present invention include methods of
antagonizing the binding of oxytocin to its receptor site, preventing
preterrn labor, treating dysmenorrhea, and stopping labor preparator~
to cesarean delivery, in a mammal in need thereof, comprising the ~tep
of ~.l,,,i,,;~lP, jllg to the mammal a pharmacologically effective amount
of a compound of formula I.
Exemplifying the invention are method~ of antagonizing
vasopressin from binding to it~ receptor site, inducing vasodilation,
treating hypertension, inducing diuresi~. and inhibiting platelet
:~g~ tin~tion, in a mammal in need thereof comprising the step of
wo 95/14025 2 1 7 4 ~ ~ ~ PC~rllJS94113483
- 21 -
rl illg to the mammal a pharmacologically effective amount of a
compound of formula 1.
An example of the invention is a method of increasing
fertility and embryonic survival in a farm animal comprising
5 adl~ elillg to the farm animal a pharmacologically effective amount
of a compound of the present invention.
Further illustrating the invention is a method for
improving survival of a farm animal neonate CUIIIIJI i~illg controlling
timing of parturition to effect delivery of the neonate during daylight
hours by adlllilli~l~lillg to a farm animal which is expected to deliver
the neonate within 24 hours a pharmacologically effective amount of a
compound of the present invention.
Another illustration of the invention is a method of
controlling the timing of estrus in a farm animal, comprising
15 ~-lminist~ring to the farm animal a pharmacologically effective amount
of a compound of the present invention.
Also included in the instant invention is a compound of the
formula n
2 5
HN
n `loT ~ ~
H
and the pharm~relltir~lly acceptable salt.s thereof.
An embodiment of the invention is a ph~rm~elltical
composition, comprising the compound of formula n in a
. .
WO 95/1402~ PCTIUS94/13483
21 7~55
- 22 -
pharmacologically effective amount for antagonizing the binding of
oxytocin to its receptor site, and a pharm~cel-tir ~lly acceptable carrier.
Illustrative of the invention are methods of antagonizing the
binding of oxytocin to its receptor site, preventing preterm labor,
5 treating dysmrnnrrhp~ and stopping labor preparatory to cesarean
delivery, in a mammal in need thereof, comprising the step of
ad.~ li..g to the mammal a pharmacologically effective amount of a
compound of formula II.
Also ~nmp~ed in the inst~nt invention are methods of
treating preterm labor and d~ lellolll-ea in a mammal in need thereof,
c~,lll~,lisillg ~ rl;ll~ to the mammal a pharmacologically effective
amount of a compoumd which binds to a human oxytocin receptor with a
binding affinity which is no more than ten-fold higher or ten-fold lower
than the binding affinity with which the compound binds to a human
15 arginine-vasopressill-vla (AVP-Vla) receptor.
More particularly illustrating the the invention are the
methods of treating preterm labor and dysmenorrhea, wherein the
compound binds to the human oxytocin receptor with a binding affinity
which is no more than five-fold higher or five-fold lower than the
20 binding affinity with which the compound binds to the human AVP-VIa
receptor.
Exemplifying the invention are the methods of treating
preterrn labor and dysmenorrhea, wherein the compound is selected
from
~2 -- OH o NH2
NH J~
or
WO 95/14025 2 1 7 4 6 5 ~ PCI/US94/13483
- 23 -
~2
Salts and esters encompassed within the term
"ph:lrm~relltir~lly acceptable salts and esters" refer to non-toxic salts of
the compounds of this invention which are generally prepared by
reacting the free base with a suitable organic or inorganic acid.
15 Rep.~se.llalive salts and esters include the following:
Acetate, Ben7~n~s~1fonate, Benzoate, Bicarbonate,
Bisulfate, Bitartrate, Borate, Bromide, Calcium Edetate, Camsylate,
Carbonate, Chloride, Clavulanate, Citrate, Dihydrochloride, Edetate,
Edisylate, Estolate, Esylate, Fumarate, Gluceptate, Gluconate,
20 Glutamate, Glycollylarsanilate, Hexyl~so~ a~e, Hydrabamine,
Hydrobromide, Hydrochloride, Hydroxynaphthoate, lodide, Isothionate,
Lactate, Lactobionate, Laurate, Malate, Maleate ,Mandelate, Mesylate,
Methylbromide, Methylnitrate, Methylsulfate, Mucate, Napsylate,
Nitrate, N-methylgl~ min~ ammonium salt, Oleate, Oxalate, Pamoate
25 (Embonate), Palmitate, Pantothenate, Phosphate/diphosphate,
Polygalacturonate, Salicylate, Stearate, Sulfate, Subacetate, Succinate,
Tannate, Tartrate, Teoclate ,Tosylate, Triethiodide and Valerate.
The term "pharmacologically effective amount" shall mean
that amount of a drug or pharmaceutical agent that will elicit the
30 biological or medical response of a tissue, system, animal or human that
is being sought by a researcher or clinician.
The term "alkyl" shall mean straight or branched chain
alkanes of one to ten total carbon atoms, or any number within this
range.
WO 95/14025 PCT/US94/13483
2~ 7465~ --
- ~4 -
The term "lower alkyl" shall mean straight or branched
chain alkanes of orle to five total carbon atoms, or any number within
this range. ~
The term "alkenyl" shall mean straight or branched chain
alkenes with one o'r more degrees of unsaturation at any position on the
chain, of two to ten total carbon atoms, or any number within this
range.
The term "alkynyl" shall mean straight or branched chain
alkynes with one or more degrees of u~lsi~lu~aLion at any position on the
chain, of two to terl total carbon atoms, or any number within this
range.
The term "aryl" shall mean phenyl, naphthyl or fluorenyl.
The term "cycloalkyl" shall mean cyclic rings of alkanes of
three to eight total carbon atoms.
The term "trihaloalkylsulfonyloxo" shall mean the
sllhstihl~nt
o
O
Whenever the term "alkyl" or "aryl" or either of their
prefix roots appear in a name of a ~ ctihl~nt (e.g. aralkoxyaryloxy), it
shall be interpreted as including those limitations given above for
"alkyl" or "aryl." Designated numbers of carbon atoms (e g Cl-10)
shall refer independently to the number of carbon atoms in an alkyl or
cyclic alkyl moiety or to the alkyl portion of a larger substituent in
which alkyl appears as it.s prefix root.
The terrn "oxo" shall refer to the substituent =0.
The term "halogen" shall include iodine, bromine, chlorine
and fluorine.
The term "preterm labor" shall mean expulsion from the
uterus of a viable il~fant before the normal end of gestation, or more
particularly, onset of labor with effacement and dilation of the cervix
before the 37th weel~ of gestation. It may or may not be associated with
vaginal bleeding or rupture of the membranes.
-
WO 95/14025 2 1 7 4 6 ~ ~ PC~/IJS94113483
- 25 -
The term "dysmenorrhea" shall mean painful menstruation.
The term "Caesarean delivery" shall mean incision through
the abdominal and uterine walls for delivery of a fetus.
As used herein, the definition of each expression when it
5 occurs more than once in any structure, can be independent of its
definition elsewhere in the same structure.
The term "substituted" shall be deemed to include multiple
degrees of substitution by a named ~ub~ u~
Where multiple substituent moieties are disclosed or
claimed, the sl-hstihlt~d compound can be independently sllhtitlltl~d by
one or more of the disclosed or claimed substituent moieties, singly or
plurally .
The compounds of the present invention, may have
asymmetric centers and occur as racemates, racemic mixtures and as
15 individual diastereomers, or enantiomers with all isomeric forms being
included in the present invention. Therefore, where a compound is
chiral, the separate enantiomers, sllhst~nti~lly free of the other, are
included within the scope of the invention; further included are all
mixtures of the two enantiomers. Also included within the scope of the
20 invention are polymorphs and hydrates of the compounds of the instant
invention.
Oxytocin (OT) and arginine vasopressin (AVP) are
structurally related peptide hormones of the pituitary gland having
distinct biological functions. OT is released from the pituitary in
25 response to stimuli related to parturition (e.g., labor, suckling of the
neonate). Circulating OT then stimulates uterine activity to promote
labor and delivery and contracts mammary gland myoepithelium to
elicit milk-letdown postpartum. AVP, on the other hand, is secreted
into the bloodstream by di~lu~ es in hemostasis such as reduced
30 blood pressure or blood volume and/or increased plasma osmolality.
AVP acts to correct these imh~ n(~ec by enhancing peripheral va.scular
resistance and by promoting water reabsorption by the kidney. These
effects of AVP are mediated by distinct receptors of the vascular
smooth muscle (AVP-Vla receptor~) and kidney (Avp-v2 receptors),
WO 9~i/14025 PCT/US94/13483
2 ~
- 26 -
respectively. AVP Vla receptors are also located in uterine smooth
muscle to cause contraction and on platelets to mediate aggregation. By
contrast, the contra~tile responses of the uterus and mammary glands to
OT appear to be transduced by a separate, single receptor subtype. A
5 third, relatively obscure vas~ ,OOill receptor subtype, AVP-Vlb, has
been identified in tlle pituitary gland where it mediates the stimulatory
effects of AVP on adrenocorticotrophic hormone (AC~I) release. The
human OT, Avp-vla and Avp-v2 receptors have recently been cloned
and expressed.
The ability of the compounds of the present invention to
antagonize oxytoc~ makes these compounds useful as pharmacologic
agents for m~mm~l~, especially for humans, for the treatment and
prevention of disorders wherein oxytocin may be involved. Examples
of such disorders irnclude preterm labor and dysmenorrhea. These
15 compounds may also find usefulness for stoppage of labor ~l~paldtOIy
to Cesarean deliver~.
The oxytocin antagonist compounds of the present
invention are also Lseful for improving reproductive efficiency in farm
animals. In certain farm animals (e.g., sheep, cattle, swine and goats),
20 the beginning of the estrous cycle is typically marked by behavioral
estrus when the female animal accepts the male for mating. Ovulation
of the ovarian follicle occurs shortly after onset of estrus and cells in
the follicle give rise to the corpus luteum. The cells that form the
corpus luteum produce progesterone and they also produce oxytocin.
25 The secretion of oxytocin from the corpus luteum and/or pituitary acts
on the uterine endollletliulll to stimulate the secretion of pro.~t~ n-1inc
(in particular PGF) which, in turn, causes the regression of the corpus
luteum of the ovary. PGF is, therefore, the luteolytic hormone. In the
cycling animal (i.e., where mating and fertilization have not occurred),
30 destruction of the corpus luteum removes the source of progesterone
which is key to the preparation of the uterus for pregnancy. The
presence of a viable conceptus (i.e., the embryo and its associated
membranes) is necessary to prevent the luteolytic process. In fact, the
first key signal that the conceptus must produce is the one to prevent
WO95/14025 2 l 746~ PCT/US94113483
- 27 -
regression of the corpus luteum (i.e., the maternal recognition of
pregnancy signal). Thus, in the animal where mating and fertilization
have occurred, the conceptus secretes a factor that antagonizes the action
of oxytocm to induce luteolysis. This results in ~ rl~AIloe of a
S functioning corpus luteum and the continued secretion of progesterone
which is obligatory to the initiation of pregnancy.
Adl.lilli~LIdLion of an oxytocin antagonist of the present
invention at this critical period after fertilization (i.e., just prior to or
during the period of maternal recognition of pregnancy) supplements
o the natural signal from the conceptus (i.e., maternal recognition of
pregnancy) to prolong corpus luteal function. The result is to increase
pregnancy rates by enhancing the chances of impregnation through a
reduction in embryonic loss. Thus, to improve fertility in a farm
animal, a mated animal, for example, a mated ewe, IS treated with an
oxytocin antagonist compound beginning on between day 10 to day 15
after onset of estrus. The oxytocin antagonist compound is ad~ L~Icd
to the mated animal for a period of one day to three weeks, preferably
one week to three weeks, most preferably one week to two weeks.
The compounds of the present invention are also useful in
farm animals for controlling the timing of parturition so that delivery
of neonates occurs during the daytime. Approximately 80% of
livestock are delivered at night and up to 5 to 10% of newborns die
because the deliveries are not monitored properly. An oxytocin
antagonist compound of the present invention A~ lt;d to the
mother on the evening before expected delivery delays parblrition so
that the delivery occurs during the daylight hours. By delaying the
timing of parturition, proper monitoring of the delivery and the
neonates is ensured, resulting in increased survival rates of the
newborns.
ln addition, the oxytocin antagonists of the instant invention
can also be used to control the timing of estrus in a cycling farm animal
by preventing luteal regression. An oxytocin antagonist compound of
the instant invention is administered to a cycling farrn animal prior to
expected e~strus to prevent regression of the corpus luteum. Daily
= = = ~ , ,
WO 95114025 PCT/USg4/13483
21 74~5~ ~
- 28 -
administration of tlle compound retards estrus until administration of
the compound ceases. Preferably, the oxytocin antagonist compound is
t~ l,;d at lea~t I day prior to expected estrus. By delaying estrus
in a group of farm animals, a farmer can synchronize estrus among the
5 group to provide time and cost savings in farm management.
The compounds of the present invention also bind to the
vasopressin receptors and are therefore useful as vasopressin
antagonists. Vasopressin antagonists are useful in the treatment or
prevention of disease states involving vas~ disorders, including
o their use as diuretics and their use in congestive heart failure.
It has llow been found that compounds of the present
invention are balanced oxytocin and vasopressin antagonists useful for
treating preterm labor and dysmenorrhea. The terms "balanced
oxytocin and vasopressin antagonist(s)," "balanced oxytocin/vasopressin
15 compound(s)," and "balanced oxytocin/arginme-vasopressin (AVP)-VIa
compound(s)," as used herein, are defined as a compound which has a
ten-fold or less sep;lration between the binding affinity of the compound
for a human oxytocin receptor and the binding affinity of the compound
for a human AVP-`Vla receptor. Preferably, there is a five-fold or less
20 separation between the binding affinities of the compound to the
oxytocin and AVP-Vla receptors. It is believed that a balanced
oxytocin/vasopressin compound will provide a more effective treatment
for preterm labor a~d dysmenorrhea. The fact that oxytocin receptors
are up-regulated in the uterine myometrium during labor and before
25 and during menstruation (AVP-Vla receptors are also present but are
not up-regulated), t~he observation that the uterus responds to both
oxytocin and AVP and that the sensitivity of the uterus to the contractile
effects of oxytocin is highest during labor and menstruation, and the
fact that oxytocin/A.VP-stim~ t- d synthesis of contractile pros~ n-lin~;
30 occurs during labor and ~ lu~ion indicates that a balanced
oxytocin/vasopressin antagonist compound would provide effective
treatment for both preterm labor and dysmenorrhea. In addition, it
appears that the vasoconstrictor activity of AVP (AVP-Vla response)
contributes to uterine ischemia and pain. Thus, balanced
WO95/14025 2 ~ 7 ~ PCT/US94/13483
- 29 -
oxytocin/v~sul,lc~ compounds, such as the balanced
oxytocin/vasopressin compounds disclosed herein, provide a novel
approach fûr the treatment of preterm labor and dysmenorrhea.
The compounds of the present invention can be
5 ~11minist~red in such oral dosage forms as tablets, capsules (each
including timed release and sustdined release formulations), pills,
powders, granules, elixers, tinctures, suspensions, syrups and emulsions.
Likewise, they may also be ~mini~t~red in imtravenous (both bolus and
irlfusion), intraperitoneal, subcutaneous or intramuscular form, all using
forms well known to those of ordinary skill in the pharmaceutical arts.
An effective but non-toxic amount of the compound desired can be
employed as a tocolytic agent.
The dosage regimen utili~ing the compoumds of the present
invention is selected in accordance with a variety of factors including
15 type, species, age, weight, sex and medical condition of the patient; the
severity of the condition to be treated; the route of administration; the
renal and hepatic function of the patient; and the particular compound
or salt thereof employed. An ordinarily skilled physician or
v~le~illdli~l can readily d~L~ li"e and prescribe the effective amount of
20 the drug required to prevent, counter or arrest the progress of the
condition.
Oral dosages of the present invention, when used for the
indicated effects, will range between about 0.3-6.0 gm/day orally.
Intravenously, the most preferred doses will range from 0.1 to about 10
25 mg/minute during a constant rate infusion. Advantdgeously, compounds
of the present invention may be administered in a single daily dose, or
the total daily dosage may be administered in divided doses of two,
three or four times daily. Furthermore, preferred compounds for the
present invention can be administered in intranasal forrn via topical use
30 of suitable intranasal vehicles, or via transdermal routes, using those
forms of transdermal skin patches well known to those of ordinary skill
in that art. To be ~ll",illi.~ d in the form of a transdermal delivery
system, the dosage administration will, of course, be continuous rather
than intermittant throughout the dosage regimen.
. . _ ~
WO 9~/14025 PCrNS94/13483
~ 14~
- 3() -
In the ]nethods of the present invention, the compounds
herein described in detail can forrn the active ingredient, and are
typically ~rlminist~led in admixture with suitable pharmaceutical
diluents, excipients or carriers (collectively referred to herein as
5 "carrier" materials) suitably selected with respect to the intended form
of administration, tllat is, oral tablets, capsules, elixirs, syrups and the
like, and consistl~nt with conventional pharm~e-lticAI practices.
For instance, for oral adl.lil.i~L-~llion in the form of a tablet
or capsule, the acti~e drug component can be combined with an oral,
non-toxic pharm~el-tir~lly acceptable inert carrier such as ethanol,
glycerol, water and the like. Moreover, when desired or necessary,
suitable binders, lubricants, disintegrating agents and coloring agents
can also be incorporated into the mixture. Suitable binders include
starch, gelatin, natural sugars such as glucose or beta-lactose, corn
5 sweeteners, natural and synthetic gums such as acacia, tragacanth or
sodium alginate, carboxymethylcellulose, polyethylene glycol, waxes
and the like. Lubricants used in these dosage forms include sodium
oleate, sodium stearate, m~n( sillm stearate, sodium benzoate, sodium
acetate, sodium chloride and the like. Disintegrators include, without
20 limit:~tinn, starch, methyl cellulose, agar, bentonite, zanthan gum and
the like.
The compounds of the present invention can also be
adl..i--i~ d in the IForm of liposome delivery systems, such as small
unilamellar vesicles, large unilamellar vesicles and m~ m~llar
25 vesicles. Liposome3 can be formed from a variety of phospholipids,
such as cholesterol, stearylamine or phosphatidylcholines.
Compounds of the present invention may also be delivered
by the use of monoclonal antibodies as individual carriers to which tlle
compound molecules are coupled. The compounds of the present
3~ invention may al.so be coupled with soluble polymers as targetable drug
carriers. Such polymers can include polyvinylpyrrolidone, pyran
copolymer~ polyhydroxypropylmethacrylamidephenol,
polyhydroxyethylasl~artamidephenol, or polyethyleneoxidepolylysine
substituted with palmitoyl residue.~. Furthermore, the compounds of the
WO 95/~4025 2 1 7 4 6 ~ G PC170S94/13483
- 31 -
present invention may be coupled to a class of biodegradable polymers
useful in achieving controlled release of a drug, for example, polylactic
acid, polepsilon caprolactone, polyhydroxy butyric acid,
polyorthoesters, polyacetals, polydihydropyrans, polycyanoacrylates and
cross-linked or a llL,llil,alllic block copolymers of hydrogels.
The present invention is also directed to combinations of
the compounds of formula I with one or more agents useful in the
treatment of oxytocin related disorders such as preterm labor,
dysmenorrhea and stopping labor prior to cesarean delivery. For
example, the compounds of the instant invention may be effectively
administered in combination with effective amounts of other agents
used in the treatment of preterm labor, such as antenatal steroids
(e.g., ~irxi1~,rlllAcolle). Preferred combinations are ~imlllt~n~ous or
alternating tlca~lllC.Il~, of an oxytocin receptor antagonist of the
present invention and an antenatal steroid. These combinations have
beneficial effects on the neonate by both decreasing uterine activity
to prolong gestation and increasimg fetal maturation. In accordance
with the method of the present invention, the individual components
of the combination can be A(lmini~tered separately at different times
during the course of therapy or concurrently in divided or single
combimation forms. The instant invention is therefore to be
understood as embracing all such regimes of ~imllltAn~ous or
alternating treatment and the term "A~lminictrring" is to be
interpreted accordingly. It will be understood that the scope of
combinations of the compounds of this invention with other agents
useful for treating oxytocin related conditions includes in principle
any combination with any ph~rmArelltirAl composition useful for
treating preterm labor, dysmenorrhea or stopping labor prior to
cesarean delivery.
The compounds of the instant invention can be prepared
readily accordin~ to the following reaction schemes and Examples or
modifications the,-eof using readily available starting materials, reagents
and conventional synthesis procedures. In these reactions, it is also
possible to make use of variants which are themselves known to those of
... . . .. .. .. _ _ _ _ _ _
WO 95114025 PCrNS94/13483
2~ 74~
- 32 -
ordinary skill in th] s art, but are not mentioned in greater detail.
Reference can also be made to U.S. Patent No. 5,091,387, issued
February 25, 1992, the entire disclosure of which is incorporated by
reference. Additiollal reference can also be made to the method of
Matier, et al., J. Org. Chem., Vol. 36, No. 5, 650-654 (1971), the entire
disclosure of which is incorporated by reference, on elaboration of
indenes and their 2-oxo derivatives to ~,uilului~elidine analogs. Also
irlcorporated by reference is a variant of this procedure described in
Matier, et al., J. Org. Chem., Vol. 36, No. 5, 650-654 (1971).
0 The most preferred compounds of the invention are any or
all of those specifically set forth in these Examples. These compounds
are not, however, ta be construed as forming the only genus that is
considered as the invention, and any combination of the compounds or
their moieties may itself form a genus. The following examples further
5 illustrate details for the preparation of the compounds of the present
invention. Those sk.illed in the art will readily understand that known
variations of the cnrl~iitic)nc and processes of the following preparative
procedures can be used to prepare these compounds. All ~tillll)eldLUl~S
are degrees Celsius lmless noted otherwise.
3~
-
WO 951140~5 2 1 7 4 ~ 5 0 Pcr/uss4ll34s3
- 33 -
Abbreviations used in the Examples are as follows:
BOP = bell~,u~lid/ul-l-yloxytris(dimethylamino)phosphonium
hexafluorophosphate
5 DCM = dichloromethane
DIEA = diisu~-u~lethylamine
DMF = dimethylformamide
EtOAc = ethyl acetate
EtOH = ethanol
EDC = l-ethyl-3-(3-dimethylaminopropyl)carbodiimide
FAB MS = fast atom bombardment mass spectroscopy
HOBT = 1-hydroxybenzotriazole
HPLC = high pressure liquid chromatography
MeOH = methanol
15 NMR = nuclear magnetic resonance
THF= tetrahydrofuran
TLC = thin layer chromatography
WO 95/14025 PCI/US94/13483
21 7465~
- 34 -
lEXAMPLE 1
`~O2C~
H NH2
clcocH2CH=cH2
~5O2C~
H NHCOCH2CH=CH2
WO 9511402~ PCTIUS94/13483
21 7~5~
- 35 -
EXAMPLE 2
5 ~
SO2C~
H NH2
ClSO2N(CH3)3
H N
so2CH2
H NHSO2N(CH3)2
WO 95/14025 PCIIUS94/13483
~46~ --
- 36 -
EXAMPLE 3
~O2C' ~
H NH2
ClCO2CH2CH3
20 5O2CH~
2s H NHCO2CH2CH3
PCTIUS94113483
W095/14025 2 1 7 4 ~ 5 ~
- 37 -
EXAMPLE 4
~02C~
H NH2
CICO~
:~o C~ ~
so2CH2 NHC
H
PCT/U594/13483
wo 95/~402s
2 1 ~
- 38 -
FXAMPLE 5
~O~C~
H NH2
CICO~
N
2 o 52C~
H NHCO~N
WO 95/14025 PCIIUS94113483
21 74S~i~
- 39 -
EXAMPLE 6
~02C~/
H NH2
CO2H ,~
Step 1
CO2CI
Step 2
2s ~ ~ A,
SO2CH2 /~
H NHCO~ ¦
PC~IU594/13483
wo 95/141)25
2~ 7~
- 40 -
FXAMPLE 8
H NH2
HO2CC H2--N ,N
2~ ~ `r'
SO2CH2 HCOCH2--N' ,N
\=N
, I--II~Y 217465C PCT~US 94/13483
~1 Rec'd PCT/PTO 2 4 ~T~995
- 41 -
REACTIONS~HFMF`S PART2
~02C~
H NH2
CICO
EXAMPLE 9
`~O2C~
H NHCO/~
ll,lFND~D S~
rr~Y
21 74~5G ~ q 4~r 1 7 ~
~1 Re~'d ~CTIPTO 24 OCT19~5
-41A -
~02CH~
H NH2
HNOC"
H~
H o
EXAMPLE 10
So~CH~$
H 'N- C
951 4025 PCTIUS94/13483
wo 1 21 74650
- 42 -
REACTION SCHEMES. PART 3
.o2cH2~
H NH2
CICOCH2CH2CO2CH2CH3
EXAMPLE 7
N ~
so2CH2
CH3CH202CCH2CH2CONH H
I R~ Y 2 1 7 ~GT a ~ I 1 3 ~ 8
2 4 aGT
- 43 -
REACTION SCHEMES, PART 3 cont'd
SO2CH~
H NH2
HO~O
to ~N~
EXAMPLE 1 1
SO2C~ ,,~OCH
H NHCO Nl
co2c(CH3)3
EXAMPLE 12
S02C~> ~c~OH
H NHCO N
C02C(CH 3)3
~MENDED SllEEr
,~Y 21 74~PCT~US 9~/13483
" ~ .' 2~ GCT1995
-44 -
REACTIQN S~HFMF~ PART 3 (~ONTD
EXAMPLE 12
~O2C~
H NHCO N
co2c(CH3)3
HCI
EXAMPLE 1
~'
S02CH 2 ,
H NHCO N
H HCI
~, ;,F~
WO 9~/1402!; PCT/US94113483
21 7465'J
- 45 -
REACTION SCHEMES. PART 3 CONT'D
H N
so2CH2
H NH2
HOOCCH2
H
EXAMPLE 14
so2CH2
H NHCOCH
N
H
~VO 9~11402~i PCT/US94/13483
2 ~ 7 ~b~O
- 46 -
REACTION SCHEMES PART 4
5 ~ ~
so2CH2
H NH
o 2
EXAMPLE 15
N
SO2CH2
H NHCOCH2~
0 NHCO2C(CH3)3
W11)95/14025 2 ~ l'CT/US94/13483
.
-47 -
REACTION SCHEMES. PART 4 CONT'D
EXAMPLE 16
5 ~`J~
SO2CH2
H NHCOCH2~
o NH2 HCI
WO 95/14025 PCT/US94113483
21 7465~ --
- 48 -
REACTION SCHEMES~ PART ~
@~ ~ HOOCCHCH2~NH
~ NHCO2C(CH3)3
so2C~I2 y,
H NH2
(, N=\
H NHCOCHCH2~,NH
NHCO2C(cH3)3
EXAMPLE 17
~2C~, N
H NHCO IHCH2~,NH
NH2HCI
WO g5/14025 PCI'IUS94/13483
2~ 74~150
- 49 -
REACTION SCHE~F.`~. PART 5 CONT'D
~, NHCO2C(CH3)3
'~ HOOClHCH2~qCH
H NH2
EXAMPLE 18
~,~
SO2CH2 HCOfHCH2~
NHCO2C(CH3)3
2s EXAMPLE 19
H ~HcocHcH
H2HCI
=-- ~
WO 95/14025 PCT/~IS94~13483
2 1 ~ U
- 50 -
]~EACTION SCHl~MF~. PART 6
[~ HOOCCH2--N~)
~ ~ NHC02C(CH3)3
s02CH2 ,y"
H NH2
LXAMPLE 20
SO2CH2 y.
H NHCOCH2--N
~--<
NHco2c(cH3)3
EXAMPLE 21
SO2CH2
H NHCOCH2--N~
N H2HCL
WO 9~/14025 2 1 7 4 6 5 ~ PCT/US94/13483
- 51 -
REACTION SCHEMES. PART 7
(CICH2CH2)2NH HCI + [(CH3)3COC0]20
Et3N
(ClCH2CH2)2NCO2-t-Bu
~ + (ClCH2CH2)2NCO2-t-Bu
LiN[Si(CH3)312
N~o t Bu
HCI, EtOAc
WO 95/14û25 PCT/US94/13483
~ 1746~
- 52 -
~F~CTION SCHEMES. PART 7 CONT'D
5 [~ + r~
H HCI O
Et3N
0
52 ~
O
-
WC 95114025 ~ 1 74 ~ PCTIUS94113483
- ~3 -
REACTION SCHEMES. PART 7 CONT'D
H2NOH HCI
pyridine
1 0
N-OH
Ni(R), H2
' 3 i~
NH2
3c Endo Isomer
WO 95/14~25 PCT/US94/13483
21 74650
- ~4 -
RE~ACTION SCHEMES. PART 7 CONTD
HO2C
H
EDC, HBT, NEt3, DMF
lo 2.
HCI
~2 ~H
HN~ N~,
O N~
H HCI
III~Y 217465~ PCT/US 94/1~483
51 Re~'d P~P~O 24 ~rl9~S
- 55 -
REACTION SC~ . PART X
H HCI
CISO\.
N Et3
N
SO2~
Benzene
AMENDED SltE~T
WO 95/14025 PCT/US94/13483
~7~
- ~6 -
RF.~CTION SCHEMES. PART ~ CONT'D
N H~
+ TRACE OF EXO ISOMER
1. BH3 THF
H202, OH
2. pyridinium
chlorochromate,
CH2CI2
2
EXAMPLE 3 1
~V095/14025 21 74650 PCT/US94/13483
- 57 -
REACTION SCHEMES. PART 8 CONrD
- 5 ~J~
SO2
Anthracene
Toluene
I2 - L_ ~r ~
EXAMPLE 32
WO 9~/14025 PCT/US94/13483
2 1 ~ 4 ~
~F~CTION SCHEMES~ PART 8 CONT'D
'~ 1~>
so2,o
~ Q
~INI~ L~
EXAMPLE 33
WO 95114025 2 1 7 4 6 5 0 PCT/US94/13483
_ 59 _
REACTION SCHEMES. PART ~ CONT'D
so2,~
2 s FXAMPLE 34
' ,A ~
7465
21
.
PCT/US 9 4/13483
p ~ ~ 2 4 ~G ~ ,~S
- 60 -
REA(3TIONSCHFI~F~ PART9
N
SO2~
V--OH
Toluene
SO2
CICH 20CH2CH 20CH3
D1 EA
[~ ~f ~ CH3
-->
EXAMPLE 35
~MENDEI) SHEET
WO 95114025 2 1 7 4 6 5 0 PCINS94/13483
- 61 -
REACTION SCHEMES~ PART 10
SO2 OH
CH2NH2
1. Methyliminodiacetic acid
anhydride, toluene
THF
2. CH31, DMF
7\~oH
SO2 CH2NH2
~0
2 5 <
c02CH3
EXAMPLE 36
WO 9~/14025 -- PCT/US94113483
2~74650
RE~CTIQN SCHEMES. PART 11
Nl 2
~ ~o;~~
EXAMPLE 37
~CT/US 94/13483
61 R~c'd PCTIPTO 24 OCT199S
- 63 -
RF~CT~ON ScHFl\/lF~ PART 11 CQN~D
¢~
N
, \
Zn - HOAc
N
E~AMpl F 3
~MENDED ~H~
Wo 95114025 PCI~/US94/13483
21 7~650
- 64 -
EXAMPLE A
~02CH2~
o '"'NH
Endo-(1 S)- I '(((2-amino-7,7-dimethylbicyclo(2.2. 1 )-hept- I -yl)-
melllyl)-sulfonyl)~piro(lH-indan-l .4'-piperidine)
Di-t-butyl dicarbonate (31g, 0.14 mole available from
Aldrich) and bis(2-chloroethyl)amine hydrochloride (21.6g, 0.12 mole
5 Aldrich) were combined in CH2C12 (250 ml) stirred at ambient
ldLul~ and tr~ated with triethylamine (12.8 g, 0.127 mole) added
dropwise over 15 minutes. After I hour, another 1.5 ml of
triethylamine was added. After a total of 2.5 hours, the mixture was
poured onto a silica gel column packed with CH2C12:hexane
20 (1:1), and eluted ~ith CH2C12. The combined product fractions were
evaporated to dryness in vacuo to give N,N-bis(2-chloroethyl)-t-butyl-
carbamate.
To a solution of indene (10.3 g, 89 mmole) in dry
tetrahydrofuran (TMF, 18 ml) cooled in an ice bath and maintained
25 under a nitrogen blanket was added lithium bis(trimethylsilyl)amide
(Aldrich, 177 ml of a l.OM ~olution in THF; 177 mmole) over 15
minutes. The mixl:Ure was stirred in the cold for 30 minutes, then
added over 15 minutes to a solution of N,N-bis(2-chloroethyl)-t-
butylcarbamate (2~1.2 g, 88 mmole) stirred in an ice bath. The mixture
30 was ~tirred for 2 h~urs in the cold and for 30 minutes at ambient
temperature under nitrogen, then evaporated in vacuo to a foam.
CH2C12 was added and the resulting mixture poured onto a silica gel
column packed with 40% hexane in CH2C12. The column
~, ?,, ~
~0 9S/1402~i PCT/~JS94/13483
21 74650
- 65 -
was eluted with 40~o hexane m CH2cl2 followed by CH2C12, and the
product fractions were evaporated to dryness in vacuo to provide l'-(t-
butyloxycarbonyl)-spiro(indene- 1 ,4'-piperidine).
- 1'-(t-Butyloxycarbonyl)spiro(indene-1,4'-piperidine) (16 g,
5 56 mmole) in ethyl acetate (250 ml) was stirred in an ice bath and
saturated with HCl(g) for 30 minutes. The mixture was evaporated
to dryness. Ethyl acetate was added and removed in vacuo three times,
and the residue was triturated with diethyl ether and filtered to provide
spiro(lH-indene-1,4'-piperidine) hydrochloride. The free base was
obtained by slurrying the hydrochloride in aqueous sodium bicarbonate
solution and extracting with CH2cl2. The organic layer was separated,
dried over sodium sulfate, filtered, and evaporated to dryness in vacuo
to provide spiro(lH-indene-1,4'piperidine.
Spiro(lH-indene-1,4'piperidine) (308 mg, 1.66 mmol) and
15 (+)-lo-~a~ hol~ulfonyl chloride (418 mg, 1.66 mmol) were combined
in CH2C12 and treated with triethylamine (0.23 ml). The mixture was
stirred at ambient ~ ldlUI~ for 15 minutes, then poured onto a silica
gel column and eluted with 1:1 CH2cl2:hexane. The product fractions
were combined and evaporated to dryness in vacuo to provide (IS)-I'-
2 (((7,7-dimethyl-2-oxobicicylo-(2.2. 1 ) hept- I -yl)-methyl)sulfonyl)spiro-
(lH-indene-1.4'-piperidine) as a solid which was recrystallized from
petroleum ether and dried ovemight in vacuo at ambient l~ ,elalul~.
( 1 S)- 1 '-(((7,7-dimethyl-2-oxobicyclo(2.2. 1 )hept- 1-
yl)methyl)sulfonyl)spiro(lH-indene-1,4'-piperidine) (30 g, 0.075 mole)
25 irl pyridine (500 mL) was heated in an oil bath to 70C (internal).
Hydroxylamine hydrochloride (30 g) was added in three portions over
ca. 20 minutes. After 2 hours, an additional 10 g of hydroxylamine
hydrochloride was added (over 10 minutes). At 30, 40, and 50 minutes
additional elap~ed time, further 3 g lots of hydroxyl-amine
30 hydrochloride were added. After another 30 minutes, the mixture was
poured into water (2 L) and extracted 3 times with ethyl acetate (300
mL portions). The organic layers were combined, washed with I N HCI
(600 mL total), dried over sodium sulfate, filtered, and evaporated to
dryness in vacuo. EtOH (abs; ca. 250 mL) was added to the resulting
WO 95/14025 PCrJUS94113483
.
~7
- 66 -
thick syrup and the solution allowed to stand at ambient temperature
overnight. The m~xture was filtered aLLd the fLltrate boiled down to ca.
80 mL. After standing, the mixture was again filtered and boiled down
to ca. 20 mL. After a third filtration, the filtered solids were combined
to give (lS)-1'-(((7,7-dimethyl-2-oximinobicyclo(2.2.1)hept-1-yl)-
methyl) sulfonyl)spiro(lH-indene-1,4'-piperidine) (28 g).
Fresh~y prepared, activated Raney Nickel catalyst (ca. 30 g)
in water was allowed to settle and the water decanted. Abs. ethalLol
(300 mL) was added, and the mixture swirled and again allowed to
settle. The solven~ was decanted. lwo more wash-decant cycles with
150 mL of ethanol were similarly carried out. (lS)-1'-(((7,7-dimethyl-
2-oximinobicyclo(2.2.1)hept-1-yl)methyl)sulfonyl)-spiro(lH-indene-
1,4'-piperidine) (30 g) was stirred in a mixture of abs. ethanol (450
mL) and 2-methoxyethanol (900 mL), nitrogen was bubbled through the
5 suspension/solutioll, and the Raney Nickel catalyst was added. The
mixture was hydrogenated under 50 psi overlLight. TLC (9:1
CH2C12MeOH, silica gel) showed the reaction to be complete. The
catalyst was remo~led by filtration, and the filtrate evaporated to dryness
in ~Ç~Q. The crlLde solid (27 g) was divided into 7 g batches, and each
20 batch was dissolved in methylene chloride (ca. 200 mL) and flash
c~Lromatographed on silica (700 g in a 100 mm column, packed and
eluted with 8% (v~v) methanol ilL methylene chloride), taking 200 mL
fractions. The exo isomer of the title amine was obtained in fractions
ca. 5-7, and the desired endo isomer in fractions ca. 8-16. TLC was on
25 silica, eluted with 8% methanol-methylene chloride, phosphomolybdic
acid stain. The combined product fractions were evaporated to dryness
to provide the title compound (4.5 g from each 7 g lot, ca. 18 g total) a.s
a colorless solid.
r
WO 95/14025 PCI/~JS94/13483
21 7465~
- 67 -
EXAMPLE I
- 5 0
N
s02CH2 \~ H
0 HN~
o
Endo-(lS)-1'-(((2-(but-3ene-1-ylamino)7,7-dimethyl-bicyclo(2.2.1)-
hept-l -vl)-methyl)sulfonyl)~piro(lH-indan-1 .4'-piperidine)
To a solution of endo-lS-1'-(((2-amino-7,7-dimethyl-
bicyclo(2.2. 1 .)-hept- I -yl)-methyl)-sulfonyl)spiro(l H-indan- 1,4'-
piperidine) (50 mg, 0.000125 m) in methylene chloride (3 ml) in an
atmosphere of nitrogen was added crotonyl chloride (13.1 mg, 14 ml,
0.000125 M) via a syringe. After two minutes, triethylamine (50 ml)
20 was added to make the solution basic (ph=9). After I hour, the reaction
mixture was concentrated and the oil was chromatographed on a silica
"flash" column using 40% ethyl acetate in hexane as solvent. Fraction~
27-40 contained a fluorescent spot. These were concentrated and ether-
hexane was added and collc~ aled to yield the title compound
25 as a white foam.
NMR (CDC13) was c~llsi~ lt with structure.
HPLC~ 210-97 .181% 254-99.3 86%
Mass Spectra: Calculated, m/e-470.679; Found, m/e=471.3
30 Analysis c~ t~d for C27H3gN2O3S
C, 68.90; H, 8.14; N, 5.95
Found: C, 68.99; H, 8.19; N, 5.7
wo 95/14025 PCr~sg4/l3483
2~ 7465~
- 68 -
EXAMPLE 2
5 0
2--
SO ~, .
- H CH3
- I
HN `SO' N `CH
Endo-( 1 S)- I '-(((2-(dimethylaminosulfonylamino)-7 ,7-dimethyl-
bicyclo(2.2. 1 )-hept- I -yl)-methyl)-sulfonyl)spiro( I H-indan- 1,4-
idine)
In an oven-dried flasl; (50 ml) under nitrogen was
dissolved endo- I S- I '-(((2-amino-7,7-dimethylbicyclo(2.2. 1 .)-hept- I -yl)-
methyl)-sulfonyl)-spiro(lH-indan-1,4'-piperidine)(50 mg, 0.000125 M)
in methylene chlor;de (3 ml). Dimethylaminosulfonyl chloride (17.9
mg, 0.000125 M) was added, and after two minutes, triethylamine (50
ml) was added. After one hour, the reaction mixture was concentrated
under reduced pressure (20 mm). A silica "flash" column using
methylene chloride(9)-methanol(l) as solvents was used to purify the
title compound as a tan solid after adding and concentrating with
hexane.
NMR (CDC13) was con~ l with structure.
HPLC: 210-96.4Z% 254-100%
Mass Spectra: Calculated, 509.736; Found, 510.3
Analysis calculated for C2sH3gN3O4S2
C, 5951; H, 7.93; N, 8.04
Found: C, 59.58; H, 7.71; N, 7.87
wo 9~/14025 PCrN594113483
21 74~5~
- 69 -
EXAMPLE 3
5
2--
SO ~.
HN\I~O~/
Endo-( 1 S)- I '-(((2-(ethoxycarbonylamino)-7,7-dimethyl-bicyclo-
(2.2.1 )-hept- I -vl)-methYI)-sulfon~d)spiro( 1 H-indan- 1 .4-piperidine)
~ an oven-dried flask (50 ml) under nitrogen was
dissolved endo- 1 S- I '-(((2-amino-7 ,7-dimethylbicyclo(2.2. 1 .)-hept- I -yl)-methyl)-sulfonyl)-spiro(lH-indan-1,4'-piperidine)(S0 mg, 0.000125 M)
in methylene chloride (3 ml). Ethyl chloluru~ a~ (13.6 mg, 12 ml,
0.000125 M) was âdded via syringe. After two minutes, triethylamirle
(50 ml) was added to make the solution basic (pH=9). After 30 minutes,
the reaction mixture was concentrated under reduced pressure (20 mm).
The oil was chromatographed on a silica "flash" column with 40% ethyl
acetate in hexane as eluent. Fractions 12-20 contained the product.
These fractions were concentrated under reduced pressure (20 mm).
Hexane-ether was added to the oil and removed under reduced pressure
to yield 17 mg of the title compound as a white powder.
NMR (CDC13) was consi~ with structure.
HPLC 210-99.~17% 254-100%
Mass Spectra: Calculated 474.668, Found 475.3
Analysis c:~lrlll~t~d for C26H3gN2O4S
C, 65.79; H, 8.07; N, 5.90
Found: C, 6G.05; H, ~.22; N, 5.72
WO 9~/14025 PCrllJS94/13483
~1 74~5~
- 70 -
EX~MPLE 4
~`32-~
o H~ ~
Endo-(lS)-I '-(((2-~2-thiophenecarbonylamino)-7,7-dimethylbicyclo-
(2.2.1)-hept-1 -vl)-~nethyl)-sulfonyl)spiro(lH-indan-l .4-piperidine)
In an oven-dried flask (50 ml) under nitrogen was
dissolved endo- I S - I '-(((2-amino-7,7-dimethylbicyclo(2.2. 1 .)-hept- I -yl)-methyl)-sulfonyl)-spiro(lH-indan-1,4'-piperidine)(50 mg, 0.000125 m)
in methylene chloride (3 ml). The 2-thiophene carbonyl chloride (18
20 mg, 13.4 ml, 0.000125 m) was added via syringe. Triethylamine (50
ml) was added to ni~ake the mixture basic. The reaction mixture was
concentrated to oil and chromatographed using a silica "flash" column
with 40% ethyl acetate in hexane as eluent. Fractions 12-20 contained
the product. Fractions 11-18 contained the desired product and these
were corl~cntr~t~d to an oil. Ether-hexane was added and removed to
yield 27 mg of the title compound as a white amorphous powder.
HPLC 210-98.873% 254-97/931%
Mass Spectra: Cal~ulated, 512.738; Found, 513.3
Analysis calculated for C28H36N203S2 0.72 hexane
C,63.~7; H,7-11; N,5-33
Found: C, 63~9; H, 6.7~; N, 5.08
W~ 95/14025 PCINS94/13483
21 7465a
- 71 -
FXAMPLE 5
~l2
HN~
Endo-(lS)-I '-(((2-(isonicotinylamino)-7,7-dimethyl-bicyclo-
(2.2.1 )-hept- 1 -yl)-methyl)-sulfonyl)spiro( I H-indan- 1 ~4-piperidine)
In an oven-dried flask (50 ml) under nitrogen was
dissolved endo- 1 S- I '-(((2-amino-7,7-dimethylbicyclo(2.2. 1 .)-hept- I -yl)-
methyl)-sulfonyl)-spiro( l H-indan- 1 ,4'-piperidine)(50 mg, 0.000 125 m)
in methylene chloride (3 ml). The isonicotinoyl chloride dihydro-
chloride (22.38 mg, 0.000125 m) was added. After 2 minutes,
triethylamine (50 ml) was added so the pH equaled 9. The solvent was
removed under reduced pressure and the product was purified by a
"flash" silica column using methylene chloride (9)-methanol (1) as
solvents. The product was contained in fractions 6-10 which were
cnnl çntr~t~d. The oil was treated with hexane-ether which was
removed under reduced pressure. This gave 18 mg of the title
compound as a white foam.
NMR (CDC13) was CollSi~ with structure.
HPLC 210-95.84% 254-98.04%
3 Mass Spectra~ lc~ tPrl 507.7; Found, 508.3
Analysis calculated for C2gH37N3O3S
C, 68.61; H, 7.35; N, 8.28
Found: C, 6~.46; H, 6.99; N, 7.90
WO 9~14025 PCT/US94/13483
2l 7~
- 72 -
EXAMPLE 6
~O~c5i~
lo HN
o
Endo-( 1 S)- I '-(((2-(noradamantyl- 1 -carbonylamino)-7,7-dimethyl-
bicyclo(2.2. 1 )-hept- I -yl)-methyl)-sulfonyl)spiro( I H-indan- 1,4-
piperidine)
Step 1: Noradamantyl-l-carbonyl chloridç
To a solution of noradarnantylcarboxylate (20.8 mg,
0.000125 M) in melhylene chloride (4 ml) in a round bottom flask (50
ml) under an atmos]?here of nitrogen at 0C was added oxalyl chloride
(120 ml) via syringe. Dimethylformamide (7 ml) was added and the
solution was stirrçd at 0C for 15 minutes. The reaction mixture was
allowed to warm to room ~ ul~ and stir for 45 minutes. The
solvent was removed under reduced pressure (20 mm). Twice
?"1~1iti~n:~1 methylene chloride was added and removed under reduced
pressure. The acid chloride of the title compound was kept under high
vacuum (0.05 mm) for one hour before using in the next step.
Step 2: Endo-( I S)- I '-(((2-(noradamantyl- 1 -carbonylamino)-7,
3 7-dime~hylbicyclo(2.2. 1 )-hept- I -yl)-methyl)sulfonyl)-
~piro( I H-indan- I ~4'-piperidine)
Adamalltyl-l-carbonyl chloride was dissolved in methylene
chloride (4 ml) and endo-lS-1'-(((2- amino-7,7-dimethylbicyclo(2.2.1)-
hept- I -yl)-methyl)-sulfonyl)spiro( I H-indan- I ,4'-piperidine) (50 mg,
0.000125 m) was added. After two minutes, triethylamine ( 100 ml) wa~s
WO 95114025 PCTIUS94/13483
2~ 7465G
- 73 -
introduced via syringe. The solvent was removed under reduced
pressure and the oil was chromatographed on silica with 40% ethyl
acetate in hexane as elutant. The product fractions were collected and
concentrated to an oil, which became an amorphous white solid upon
5 addition and removal of ether-hexane. This gave 10 mg of the title
compourld.
NMR (CDC13) was COII~ ell~ with structure.
HPLC 210-98.427% 254-100%
o Mass Spectra: Calculated, m/e 550.81; Found, m/e 551.3
Analysis calculated for C33H46N2O3S
C, 71.96; H, 8.42; N, 5.09
Found: C, 71.57; H, 8.71; N, 4.77
EXAMPLE 7
SO2CH2 0
H NHJ~O
O
Exo-(lS)-1'-(((2-(ethyl succinoylamino)-7,7-dirnethylbicyclo(2.2.1)-
hept-l -Yl)-methvl)-.sulfonyl)s~iro(lH-indan-l .4'-pireridine)
To a solution of exo-(lS)-1'-(((2-(amino)-7,7-
dimethylbicyclo(2.2.1)-hept-1-yl)methyl)-sulfonyl)spiro(lH-indan-1,4'-
30 piperidine) (50 mg, 0.000125 M) in methylene chloride (3 ml) under ablanket of nitrogen was added ethyl succinoyl chloride (20.6 mg,
0.000125 M). After two minutes, triethylamine (50 ml) was added with
a syringe. The pH of the solution was 9. After 15 minutes, a new spot
wa~s observed on the tlc (silica~0% ethyl acetate in hexane). The
reaction mixture was concentrated and the oil was chromatographed on
WO 9!i/14025 PCIIUS94/13483
.
21 7~650
- 74 -
a silica "flash" column with 40% ethyl acetate as solvent. Fractions 24-
48 contained the product. These fractions were collected and
concentrated. Eth~r-hexane was added and removed to yield 21.3 mg of
the title compound as a white powder.
NMR (CDC13) was consistent with structure.
HPLC 210-99.190% 254-96.914%
Mass Spectra: Calculated mle, 530.732; Found m/e, 531.5
Analysis calculated for C2gH42N2OsS 0.25 H2O
C, 65.08; H, 8.00; N, 5.23
Found: C, 65.16; H, 8.03; N, 5.09
EXAMPLE 8
'2C~ H N =N
HN ~ ~N
o
Endo-( 1 S)- I '-(((2-(tetrazole- 1 -acetylamino)-7 ,7-dimethylbicyclo-(2.2.1)-hept-1-vl)-meth~ ulfonyl)-spiro(lH-indan-l.4-piperidine)
In an oven-dried flask (50 ml) under nitrogen was
dissolved endo-1$-1'-(((2-amino-7,7-dimethylbicyclo(2.2.1.)-hept-1-yl)-
methyl)-sulfonyl)-spiro(lH-indan-1.4'-piperidine)(50 mg, 0.000125 M).
tetrazole-l-acetic acid (16 mg, 0.000125 M), and benzotriazol-l-
yloxytris(dimethylamino)phosphonium-hexafluorophosphate (Sequalog)
(55 mg, 0.000125 M) in acetonitrile (3 ml). Diisopropylethylamine (50
ml) was added to make the solution basic. After tlc (silica-methylene
chloride(9)-methallol(l)) showed a new spot, the reaction was
concentrated to an oil. The oil was dissolved in methylene chloride-
-
Wl)95/14025 PCrlUS94113483
21 74650
- 75 -
ether (1:3) and the solution was washed with water, sodium bicarbonate
(sat., aqueous), water, 10% potassium bisulfate, and brine. After
drying with sodium sulfate, the solution was filtered and concentrated to
oil, which was chromatographed on a silica "flash" column with 10%
5 methanol in methylene chloride. Fractions 5-10 contained the product
and were collected and concentrated. Ether-hexane was added and
removed to yield the title compound as a white powder.
NMR (CDC13) was consistent with structure.
HPLC: 210-98% 254-100%
Mass Spectra: Calculated, 513.679; Found, 513.3
Analysis c~ ted for C26H36N6O3S 0-25 H2O
C, 60.37; H, 7.11; N, 16.25
Found: C, 60.28; H, 6.96; N, 16.18
EX~MPLE 9
20 ~ ~
SO2CH2 H
HN~o
o
Endo-( 1 S)- I '-(((2-(2-furoylamino)-7 ,7-dimethylbicyclo-(2.2. 1)-
hept- I -yl)-methyl)sulfonyl)spiro( I H-indan- I .4-piperidine)
In an oven-dried flask (50 ml) under nitrogen was
dissolved endo- 1 S- I '-(((2-arnino-7,7-dimethylbicyclo(2.2. 1 .)-hept- I -yl)-
methyl)-sulfonyl)-spiro(lH-indan-1,4'-piperidine)(50 mg, 0.000125 M)
in methylene chloride (3 ml) and 2-furoyl chloride (16.3 mg, 0.000125
M) was added. After two minutes, triethylamine (150 ml) was added so
the pH equaled 9. The solvent wa.s removed under pressure and the
,, _ _ , . _ . , . _ . . . ... . . . . . ..
WO 95114025 PtCTlUS94/13483
.
21 74
- 76 -
product was purified by a silica "flash" column using 40% ethyl acetate
in hexane as solvents. The product was contained in fractions 6-10
which were concentrated. The oil was treated with hexane-ether which
was removed under reduced pressure.
NMR (CDC13) was consistent with structure.
HPLC: 210-99.341% 254-99.576%
Mass Spectra~ It~ (l, 496.674; Found, 497.3
Analysis calculated for C2gH36N2O4S
o C,67.71;H,7.31;N,5.64
Found: C, 67.~1; H, 7.39; N, 5.46
EXAMPLE 10
~o2cH2 7 \ /,
HN
o
Endo-(lS)-1'-(((2-(S-(1)-2-oxo-pyrrolidin-5-yl-carbonyl)amino)-7,7-
dimethylbicyclo(2.2.1)-hept-1-yl)-methyl)-sulfonyl)spiro(lH-indan-
I ~4-piperidine)
In an oven-dried flask (50 ml) under nitrogen was
dissolved endo- I S- I '-(((2-amino-7,7-dimethylbicyclo(2.2. 1 .)-hept- I -yl)-
methyl)-sulfonyl)-spiro(lH-indan-1,4'-piperidine)(50 mg, 0.000125 M),
S-(1)-2-oxo-pyrrolidine-5-carboxylic acid (16.1 mg, O.OOOI'25 M), and
benzotriazol- I -yloxytris(dimethyl-amino)phosphonium-hexafluoro-
phosphate (Sequalo~) (55 mg, 0.000125 M) in acetonitrile (3 ml).
Diisopropyl-ethylamine (50 ml) was added to make the solution basic.
After tlc(silica-methylene chloride(9)-methanol(l)) showed a new SpOt.
WO 95114025 PCrlUS94113483
21 74650
- 77 -
the reaction was concentrated to an oil. The oil was dissolved in
methylene chloride-ether(1:3) and the solution was washed with water,
sodium bicarbonate (sat., aqueous), water, 10% potassium bisulfate, and
brine. After drying with sodium sulfate, the solution was filtered and
5 concentrated to an oil. Ether-hexane was added and removed under
reduced pressure to give the title compound as a white solid.
NMR (CDC13) was consistent with structure.
HPLC: 210-93.102% 254-92.292%
0 Mass Spectra: Calculated, 513.705; Found, 514.3
EXAMPLE l I
\~
~0
o
Endo-(lS)-I '-(((2-(L-N-(tert.-butoxycarbonyl)-4-benzyl-oxy-
2s prolinoylamino)-7,7-dimethylbicyclo(2.2. 1 )-hept- I -yl)-methyl-
.sulfonyl)~spiro( I H-indan- l ~4-piperidine)
In an oven-dried flask (50 ml) under nitrogen was
dissolved endo-lS-1'-(((2-amino-7,7-dimethylbicyclo(2.2.1.)-hept-1-yl)-
methyl)-sulfonyl)spiro( I H-indan- I ,4'-piperidine)(50 mg, 0.000125 M),
30 L-N(tert.-butoxycarbonyl)-4-benzyloxy-proline (40.2 mg, 0.000125
M), and benzotriazol-l-yloxytris(dimethylamino)phosphonium-
hexafluorophosphate sequalog (55 mg, 0.000125 M) in acetonitrile (3
ml). Diisopropylethylamine (50 ml) wa~ added to make the solution
basic. After tlc(silica-methylene chloride(9)-methanol(1)) showed a
new spot, the reaction was concentrated to an oil. The oil was dissolved
wo 951140~ PCr/US94/13483
~i ~4~5~
- 78 -
in methylene chloride-ether(l :3) and the solution was washed with
water, sodium bicarbonate (sat., aqueous), water, 10% potassium
bisulfate, and brin~. After drying with sodium sulfate, the solution was
filtered and concentrated to an oil which was purified by "flash" silica
5 chromatography with 40% ethyl acetate in hexane as solvent. Fractions
15-24 contained the product and they were collected and concentrated.
Ether-hexane treatrnent provided a white solid which was the title
compound. A 10 ~lg sample was used for characte~ization and testing
with the remainder being used in s~lbceq ll~nt steps.
NMR (CDC13) was consistent with structure.
HPLC: 210-99.OQ3% 254-100%
Mass Spectra: Cal'd, 705.965; Found, 706
Analysis calculated for C40HssN306S 0.25 H20
C, 67.62; H, 7.87; N, 5.91
Found: C, 67.56; H, 8.1 1; N, 5.59
EXAMPLE 12
~O2C~
HN N~
Endo-( I S)- I '-(((2-(L-N(tert . -butoxycarbonyl)-4-hydroxy-
prolinoylamino)-7,7-dimethylbicyclo(2.2. 1 )-hept- I -yl)methyl-
~u If onyl)~piro( I H-indan- I .1 -piperidine)
A mix~ure of Endo-(lS)-1'-(((2-(L-N(tert.-butoxy-
carbonyl)-4-benzyloxy-prolinoylamino)-7,7-dimethyl)bicyclo(2.2. 1 )-
WO 95/14025 PCT/US94/13483
21 7465~
- 79 -
hept- 1 -yl)-methyl)-sulfonyl)spiro( 1 H-indan- 1 ,4-piperidine), ethanol (5
ml), and Pd(OH)2 (60 mg) was hydrogenated on the Parr apparatus at
50 psi for 18 hours. I'he catalyst was removed by filtration and the
solvent was removed under reduced pressure to yield the title compound
5 as a white solid.
NMR (CDC13) was cu,lsi~ with structure.
HPLC: 210-93.5% 254-(7.381%
Mass Spectra: ('~lr~ t~-1, 615 840; Found, 616
o Analysis calculated for C33H49N3O6S 1.5 H2O
C, 61.65; H, 8.15; N, 6.53
Found: C, 61.46; H, 7.88; N, 6.90
EXAMPLE 13
SO2CH2 H ~>
HN~NH
o
Endo-( I S)- I '-(((2-(L-4-hydroxyprolinoylamino)-7,7-dimethyl-
bicyclo(2.2. 1 )-hept- I -yl)-methyl)-sulfonyl)spiro( I H-indan- 1,4-
piperidine)hydrochloride
- A mixture of Endo-(lS)-1'-(((2-(L-N(tert.-butoxy-
carbonyl)-~-hydroxyprolinoylamino)-7,7-dimethyl)bicyclo('2.2. 1 )-hept-
I-yl)-methyl)-sulfonyl)spiro(lH-indan-1,4-piperidine), was dissolved in
ethyl acetate (5 ml) and cooled to -5C. This was placed under a
nitrogen atmosphere and hydrogen chloride gas was bubbled in for 10
minutes. The solvent was removed under reduced pressure. Ether was
WO 95114025 PC'rlUS94/13483
21 74651~
- 80 -
added and removed under reduced pressure to give 15.48 mg of the title
compound as a white solid.
NMR (CDC13) was consistent with structure.
HPLC: 210-93.50% 254-93.812%
Mass Spectra: Calculated, 515.721 (free base); ~ound, 516.4
Analysis calculated for C28H41N34S HCI 0-25 H2O
C, 60.41; H, 7.69; N, 7.54
Found: C, 60.08; H, 7.73; N, 7.35
EXAMPLE 14
A~
so2CH2 H
N
H
Endo-( 1 S)- I '-(((2-(3-indolylacetylamino)7 ,7-dirnethyl-bicyclo-
(2.2.1)-hept-1-yllmethyl-sulfonyl~spiro(lH-indan-1.4-piperidine)
In an oven-dried flask (50 ml) under nitrogen was
dissolved endo- I S - I '-(((2-amino-7,7-dimethylbicyclo(2.2. 1 .)-hept - I -yl)-
methyl)-sulfonyl)spiro(lH-indan-1,4'-piperidine)(50 mg, 0.000125 M),
3-indolyl acetic acid (21.9 mg, 0.000125 M), and benzotriazol-l-
yloxytris(dimethylamino)phosphonium-hexafluorophosphate (Sequalog)
(55 mg, 0.0001'25 ~)in acetonitrile (3 ml). Diisopropylethyl~mine (50
ml) was added to mlake the solution basic. After tlc(silica-methylene
chloride(9)-methanol(l)) showed a new spot, the reaction was
concentrated to an oil. The oil was dissolved in methylene chloride-
ether (1:3) and the solution was washed with water, sodium bicarbonate
(sat., aqueou.s)~ water, 10% potassium bisulfate~ and brine. After
WO 9~/14025 2 1 7 4 ~ ~ ~ PCT/IJS94/13483
- 81 -
drying with sodium sulfate, the solution was filtered and concentrated to
an oil which was purified by "flash" silica chromatography with 10%
methanol in methylene chloride as solvent. Fractions 12-19 contained
the product and they were collected and concentrated. Ether-hexane
5 treatment provided a white solid which was the title compound.
MR (CDC13) was consistent with structure.
HPLC: 210-99.829% 254-92.699%
Mass Spectra: Calculated, 559.777; Found, 560.3
o Analysis calculated for C33H41N3O3S 0.5 H2O
C, 69.69; H, 7.44; N, 7.51
Found: C, 69.70; H, 7.57; N, 7.20
EXAMPLE 15
~02CH, 7~
O ~ H
Endo-(lS)-I '-(((2-(S-3-(tert.-butoxycarbonyl)amino)-2-oxo-1-pyrro-
lidineacetylamino)7 ,7-dimethylbicyclo-(2.2. 1 )hept- I -yl)-methyl-
sulfonyl)~piro( I H-indan- I .4-piperidine)
In an oven-dried flask (50 ml) under nitrogen was
dissolved endo-lS-1'-(((2-amino-7,7-dimethylbicyclo(2.2.1.)-hept-1-yl)-
methyl)-sulfonyl)spiro(lH-indan-1,4'-piperidine)(50 mg, 0.000125 M),
S-3-((tert.-butoxycarbonyl)amino)-2-oxo-1-pyrrolidine acetic acid (32.2
mg, 0.000125 M), and l,el~uL-iazol-l-yloxytris (dimethylamino)-
phosphoniumhexafluoro-phosphate (Sequalog) (55 mg, 0.000125 M) in
acetonitrile (3 ml). Diisopropylethylamine (50 ml) wa~ added to make
WO 95/14025 PCT/US94/13483
21 7~0
- 82 -
the solution basic. After tlc(silica-methylene chloride(9)-methanol(l))
showed a new spo~, the reaction was concentrated to an oil. The oil was
dissolved in methylene chloride-ether(1:3) and the solution was washed
with water, sodium bicarbonate (sat., aqueous), water, 10% potassium
5 bisulfate, and brine. After drying with sodium sulfate, the solu-
tion was filtered and ~ C~ lal~d to an oil which was purified by
"flash" silica chrornatography with 10% methanol in methylene chloride
as solvent. Fractions 15-28 cnnt~inPd the product and they were
collected and concentrated. Ether-hexane treatrnent provided a white
o solid which was the title compound.
NMR (CDC13) was consistent with structure.
HPLC: 210-94.005% 254-96.33%
Mass Spectra: ('~ tPd m/e, 642.859; Found m/e, 643.4
Analysis c:~ t~dforC34HsoN4O6S"0.1hexane
C, 63.79; H, 7.95; N, 8.59
Found: C, 63.~6; H, 8.32; N, 8.29
EXAMPLE 16
~DO2CH~ O
3C
WO 9~/1402~ PCr/US94113483
21 746513
- 83 -
Endo-(lS)-1'-(((2-(S-3-amino)-2-oxo-1-pyrrolidine-acetylamino)7,
7-dimethylbicyclo(2.2. 1 )-hept- l -yl)-methyl)-sulfonyl)spiro( I H-indan-
1 ,4-piperidine)hydrochloride
Endo-lS-1 '-(((2-(S-3-(tert.-butoxycarbonyl)amino)-2-oxo-
1 -pyrrolidineacetylamino-)-7,7-dimethyl-bicyclo(2.2. 1 .)hept- 1 -yl)-
methyl)-sulfonyl)spiro(lH-indan-1,4'-piperidine) was dissolved in ethyl
acetate (5 ml) and cooled to -5C. This was placed under a nitrogen
atmosphere and hydrogen chloride gas was bubbled in for 10 minutes.
The solvent was removed under reduced pressure. Ether was added and
o removed under reduced pressure to give 23.7 mg of the title compound
as a white solid.
NMR (CDC13) was ~.",~ "l with structure.
HPLC: 210-96.562% 254-95.493%
Mass Spectra: Calculated for free base m/e, 542; Found m/e, 543.3
Analysis calculated for C2gH42N4O4S " H2O " HCI
C, 58.32; H, 7.59; N, 9.38
Found: C, 58.14; H, 7.97; N, 9.21
EXAMPLE 17
so2CH2
HN ~ NH
0
Step 1: Endo-( I S)- I '-(((2-(L-N-tertbutoxycarbonyl-histidinoyl -
amino)-7 ,7-dimethylbicyclo(2.2. 1 )-hept- I -yl)-methyl-
~ulfonvl)spiro( I H-indan- I ,4-piperidine)
WO 95/14025 PCI/US94/13483
~ i 14~
- 84 -
In an oven dried flask (50 ml) under nitrogen were
dissolved endo-lS-1'-(((2-amino-7,7-dimethylbicyclo(2.2.1.)hept-1-yl)-
methyl)-sulfonyl)spiro(lH-indan-1,4'-piperidine) (50 mg, 0.000125 M),
L-N-tert, butoxyc~rbonyl histidine (29.9 mg, 0.000125 M), and f
benzotriazol-l-yloxytris(dimethylamino)phosphonium-hexafluoro-
phosphate (Sequalog) (55 mg, 0.000125 M) in acetonitrile (3 ml).
Diisopropylethylamine (50 ml) was added to make the solution basic.
After tlc(silica-me~ylene chloride(9)-methanol(l)) showed a new spot,
the reaction was concentrated to an oil. The oil was dissolved in
methylene chloride-ether (1:3) and the solution was washed with water,
sodium bicarbonate (sat., aqueous), water, 10% potassium bisulfate, and
brine. After drying with sodium sulfate, the solution was filtered and
concentrated to an oil, which was purified by "flash" silica
chromatography with 10% methanol in methylene chloride as solvent.
Fractions 15-28 contained the product and they were collected and
c~ncrntratf~-l Ether-hexane treatment provided a white solid which as
the title cornpound.
NMR (CD30D) was consistent with structure.
HPLC: 210-97.822% 254-100%
Mass Spectra: Calculated m/e, 639.859; Found m/e, 640.3
Step 2: Endo-( 1 S)- I '-(((2-(L-N-histidinoylamino)-7,7-dimethyl-
bicyclo(2.2. 1 )-hept- I -yl)-methyl-sulfonyl)spiro( I H-
indan- I .4-piperidine~
Endo- I S- 1 '-(((2-(L-N-tert.butoxycarbonyl-histidinoyl -
amino)-7,7-dimethylbicyclo(2.2.1 .)-hept-l -yl)-methyl)-sulfonyl)-
spiro(lH-indan-1,4'-piperidine) (50 mg, 0.000125 M) was dissolved in
ethyl acetate (5 ml) and cooled to -5C. This was placed under a
30 nitrogen atmosphe~e and hydrogen chloride gas was bubbled in for 10
minutes. The solvent was removed under reduced pressure. Ether was
added and nemoved und~r reduced pressure to give 27.5 mg of the title
compound as a white solid.
WO 95/14025 PCT/US94/13483
21 74650
- 85 -
NMR (CD30D) was consistent with structure.
HPLC. 210-98.652% 254-100%
Mass Spectra: Calculated for free base m/e, 539.7; Found m/e, 540.3
Arlalysis calculated for C29H4lN5o3s 2HC1 2H2
C, 53.69; H, 7.30; N, 10.79
Found: C, 53.58; H, 7.45; N, 11.00
EXAMPLE 18
SO2CH2 H
~ H~O~,
o
Endo-( l S)- I '-(((2-(D-N-(tert.butoxycarbonyl)-im-benzylhistidino-
ylamino)-7,7-dimethylbicyclo(2.2. 1 )-hept- I -yl)-methyl)sulfonyl)-
spiro( I H-indan- I .4'-piperidine)
In an oven-dried flask (50 ml) under nitrogen were
dissolved endo-lS-I '-(((2-amino-7,7-dimethylbicyclo(2.2.1 .)-hept-l -yl)-
methyl)-sulfonyl)spiro(lH-indan-1,4'-piperidine) (50 mg, 0.000125 M),
D-N-tert. butoxycarbonylim-benzyl-histidine (43 mg, 0.000125 M), and
benzotriazol- I -yloxytris(dimethylamino)pho~ i " " ~hf~xafluoro
phosphate (Sequalog) (55 mg, 0.000125 M) in acetonitrile (3 ml).
Diisopropyl-ethylamine (50 ml) was added to make the solution basic.
After tlc(silica-methylene chloride(9)-methanol(1)) showed a new spot,
the reaction was concentrated to an oil. The oil was dissolved in
methylene chloride-ether (1:3) and the solution was washed with water,
sodium bicarbonate (sat., aqueous), water, 10% potassium bisulfate, and
brine. After drying with sodium sulfate, the solution was filtered and
WO 9~;114025 PCINS94/13483
~7 7~5~
- 86 -
concentrated. Eth~r-hexane provided a white solid which was the title
compound.
NMR (CDC13) was consistent with structure.
HPLC: 210-99551%
Mass Spectra: Calculated, m/e 730.Q62; Found, m/e 730.6
Analysis calcul~lted for C41HssNsOsS: 0.65 H20 0.4 hexane
C,67.15;H,8.04;N,9.02
Found: C, 67.18; H, 7.73; N, 9.02
EXAMPLE 19
HN NH2
Endo-(lS)-I '-(((2-(D-im-benzylhistidinoylamino)-7,7-dimethyl-
bicyclo-(2.2. 1 )-hept- I -yl)-methyl)-sulfonyl)spiro( I H-indan- 1,4-
25 piperidine)-hydrochloride
Endo-~ I S)- I '-(((2-(D-N-(tert.butoxycarbonyl)-im-
ben~ylhistidinoylamino)-7,7-dimethylbicyclo(2.2.1 .)-hept- I -yl)-
methyl)-sulfonyl)sl~iro(lH-indan-1.4'-piperidine) was dissolved in ethyl
acetate (5 ml) and cooled to -5C. This was placed under a nitrogen
30 atmosphere and hydrogen chloride gas was bubbled in for 10 minute.s.
The solvent was removed under reduced pressure. Ether was added ~nd
removed under reduced pressure to give the title compound as a white
solid.
NMR (CDC13) Wd.S consistent with structure.
-
WO95/1402S 21 7 465~ PCr/uss41l34s3
- 87 -
HPLC: 210-100%
Mass Spectra: (~ 1 m/e, 629.879; Found m/e, 630
Analysis calculated for C36H47NsO3S HCI 0.1
ethyl acetate 1.4 H2O
C, 59.33; H, 7.20; N, 9.51
Pound: C, 59.36; H, 7.03; N, 9.50
EXAMPLE 20
SO2CH2 H
HN
O N~
O
Endo-(lS)-I '-(((2-(S-3-tert.butoxycarbonylamino-2-oxo-1-
ylamino)-7,7-dimethylbicyclo(2.2. 1 )-hept- I -y 1 )-methyl-
sulfonyl)~piror I H-indan- I ,4-piperidine)
In an oven-dried flask (50 ml) under nitrogen was
2 5 dissolved endo- 1 S- I '-(((2-amino-7,7-dimethylbicyclo(2.2. 1 .)-hept- 1 -yl)-
methyl)-sulfonyl)spiro( I H-indan- I ,4'-piperidine)(50 mg, 0.000125 M),
S-3-tert.butoxycarbonylamino-2-oxo-1-azepine acetic acid (32.8 mg),
and benzotria~ol-l-yloxytris (dimethylamino)phosphonium-hexafluoro-
phosphate sequalog (55 mg, 0.000125 M) in acetonitrile (3 ml).
30 Diisopropylethylamine (50 ml) was added to make the solution basic.
After tlc(silica-methylene chloride(9)-methanol(l)) showed a new spot,
the reaction was concentrated to an oil. The oil was dissolved in
methylene chloride-ether(1.3) and the solution was washed with water,
sodium bicarbonate (sat., aqueous), water, 10% potassium bisulfate, and
, .. . . . , _ . .
WO 95/14025 PCrlUS94/13483
2 ~ i7 ~
- 88 -
brine. After drying with sodium sulfate, the solution was filtered and
concentrated to an oil, which was purified by "flash" silica
chromatography with 10% methanol in methy~ene chloride as solvent.
The fractions which contained the product were collected and
cnnc~n~ p~ Ether-hexane treatment provided a white solid which was
the title compound. A 10 mg sample was saved for characterization and
testing.
NMR (CDC13) was Co~ L~ with structure.
o HPLC 210-100%
Mass Spectra calculated m/e 670.918 found m/e 671
Analysis calcul~ted for C36Hs4N4O6S
C, 64.44; H, 8.11; N, 8.35
Found: C, 64.12; H, 8.23; N, 8.03
lS
EXAMPLE 21
20 ~ ~
SO2CH2 . H ~NH2
\~ N~j
o
Endo-(1 S)- I '-(((2-(S-3-amino-2-oxo- 1 -azepine acetyl-amino)-7,7-
dimethylbicyclo(2.2. 1 )-hept- I -yl-methyl)-sulfonyl)spiro( 1 H-indan-
30 1.4'-piperidine) hvdrochloride
Endo( I S)- I '-(((2-(S-3-tert-butoxycarbonyl-amino-2-oxo- 1-
azepine acetylamino)-7,7-dimethylbicylo-(2.2. 1 .)-hept- I -yl)-methyl)-
sulfonyl)spiro(lH-indan-1,4'-piperidine) was dissolved in ethyl acetate
(~ ml) and cooled to 5C. This was placed under a nitro~en atmosphere
and hydrogen chloride gas wa~ bubbled in for 10 minutes. The solvent
WO 95/14025 PCTNS94/13483
21 7465û
.
- 89 -
was removed under reduced pressure. Ether was added and removed
under reduced pressure to give the title compound as a white solid.
NMR(DCDl3) was Collsi~ t with structure.
HPLC 210-100%
Mass Spectra: Calculated m/e, 570.802; Found m/e, 571
Analysis calculated for C3 lH46N4O4S " HCI 1.10
H2O " 0.40 ethyl acetate
C, 59.11; H 7.97, N, 8.46
Found: C, 59.09; H 8.00, N, ~.45
EXAMPLE 22
1 5
SO2CH2 H
HN ~6~`~
O NH
Endo-(lS)-1'-(((2-(4-imidazoleacetylarnino)-7,7-dirnethylbicyclo-
25 (2.2.1)-hept-1-yl)-methyl)-sulfonyl)spiro(lH-indane-1,4'-piperidine)-
hydrocloride
N~N-bis(2-chloroethyl)-t-butyIcarbamate
Di-t-butyldicarbonate (62 g, 0.28 mole, available from
30 Aldrich) and bis(2-chloroethyl)amine hydrochloride (55 g, 0.21 mole
,available from Aldrich) were stirred together in methylene chloride
(400 mL). Triethylamine (42 mL, 0.3 mole) was added dropwise but
briskly to the stirred suspen.sion. After 10 minutes, additional
triethylamine (ca. 5-6 mL) was added to adjust the pH of the mixture (a~s
determined by spotting a sarnple on E. Merck pH 5-10 colorpHast sticks,
WO 9511402~ PC.rll~S94/13483
2~ ,74~
- 90 -
moistened with water) to 9-9.5. The mixture was stirred for 1 hour at
ambient tc~ Jclalu,~7 then filtered. The filtrate was divided iJn half, and
each half (ca. 300 mL) was flash chromatographed on a separate 15",
2"diameter silica column packed with 3:2 (v:v) methylene
5 chlori~lf h~Y:ln~ and eluted with 9:1 methylene chloride:hexane. 100
mL fractions were taken, and the product was obtained in fractions ca.
6-12. Assay was b~ TLC on silica gel plates, eluted with methylene
chloride and visualized with phosphomolybdic acid stain (Rf ca. 0.75).
Evaporation of the combined product fractions from both columns in
vacuo provided the title compound (70 g) as a colorless oil. The oil was
twice dissolved in dry THF and evaporated in vaGuo to remove
methylene chloride.
I '-(t-Butyloxycarb~nyl)spiro( I H-indene- 1 ,4'-piperidine)
To a solution of indene (36.2 g, 310 mmole) in dry
tetrahydrofuran (TH~F, 40 mL) cooled in an ice bath and mAinlAin~d
under a nitrogen blanket was added (dropped funnel) lithium
bis(trimethylsilyl)-amide (620 mL of a 1.0 M solution in THF; 620
mmole, available from Aldrich) over 30 minutes. The mixture was
20 stirred in the cold for 30 minutes, then Llall~rcll~,d by cannula over 20
minutes to a solution of N,N-bis(2-chloroethyl)-t-butylcarbamate (70 g,
290 mmole) in TH[F (40 mL), stirred in an ice bath. The mixture was
stirred for 2 hours iln the cold and for 30 minutes at ambient
temperature under nitrogen, then evaporated in vacuo to a foam.
25 CH2C12 (400 mL) was added and the resulting mixture divided in half.
Each half was poured onto a silica gel column (15", 2" id) packed and
eluted with 1:1 hexane:CH2C12 (2 L) followed by 1:4 hexalne: CH2cl2
The product fractiolls (fractions 3-9 of 200 mL fractions) were
evaporated to drynes~ in vacuo to provide l'-(t-butyloxycarbonyl)spiro
30 (indene-1,4'piperidille)~ 90 g total, as a crude yellow solid. The solid
was taken up in boiling hexane (400 mL), cooled, and the crystallized
solid filtered. The filtrate was repeatedly boiled down to 1/2 its
volume, cooled and filtered to obtain successive crops, providing a total
WO 95114025 PCT/US94/13483
2 ~ 7~6~
- 91 -
of 54 g of pure product. The residue was rechromatographed to
provide another 6.7 g (60.7 g total).
Spiro(l H-indene- I ~4'-piperidine) hvdrochloride
1'-(t-Butyloxycarbonyl)spiro(indene-1,4'-piperidine) (60.7
g) in ethyl acetate (700 mL) was stirred in an ice bath and saturated
with HCI (g) for 30 minutes, keeping the internal L~ eldlul~ < 12C.
The mixture was stirred in the cold an additional 30 mimutes, then
evaporated to dryness. Ethyl acetate was added and removed in vacuo
o three times, and the residue was triturated with diethyl ether and
filtered to provide spiro(lH-indene-1,4'-piperidine) hydrochloride.
( 1 S)- 1'-(((7 ,7-dimethyl-2-oxobicyclo(2.2. 1 )hept- 1 -)methyl)-sulfonyl)-
spiro( I H-indene- 1 ~4'-piperidine)
Spiro(lH-indene-1,4'piperidine) hydrochloride (45.4 g, 0.2
mole) and (+)-10-~,a l,~hol~ulfonyl chloride (62.5 g, 0.25 mole,
available from Aldrich) were combined in CH2C12 (700 mL) and
treated with triethylamine (68.5 mL, 0.5 mole). Additional
triethylamine was added as needed to adjust the pH of the mixture to 9-
9 5 (moistened E. Merck colorpHast sticks). The mixture was stirred at
ambient I~ Jtla~ul~ for 1 hour, then poured onto a silica gel column
(10", 2" id) packed with CH2CL2 and eluted with 1:1 Et2o:cH2cl2.
The product fractions were combined and evaporated to dryness in
vacuo to provide the title compound as a solid which was recrystallized
from petrolium ether and dried 6 hours in vacuo at arnbient
lalul~: (m.p. 146-147C).
TLC: Rf=0.44 silica gel (CH2C12).
NMR: Consistent with structure.
HPLC: >99.7% pure.
MS: Molecular ion at m/e - 399
Analysis calculated for C23H2gNO3S
C, 69.14; H, 7.32; N, 3.51
Found: C, 6~.97; H, 7.2; N, 3.38
WO 95/14025 PCT/US94/13483
2~ 7465~ --
.
- 92 -
( I S)- 1 '-(((7,7-dime~hyl-2-oximinobicyclo(2.2. 1 )hept- I -yl)-methyl)-
sulfonyl)spiro( I H-indene- I .4'-piperidine)
(lS)-1'-(((7,7-dimethyl-2-oxobicyclo(2.2.1)hept-1-
yl)methyl)sulfonyl)spiro(lH-indene-1,4'piperidine) (30 g, 0.075 mole)
5 in pyridine (500 mL) was heated in an oil bath to 70C (irlternal).
Hydroxylamine hyclrochloride (30 g) was added in three portions over
ca. 20 minutes. Af~er 2 hours, an ~l(1itian~l 10 g of hydroxylamine
hydrochloride was ~Idded (over 10 minutes). At 30, 40 and 50 minutes
~ltlition~l elapsed time, further 3 g lots of hydroxyl-
amine hydrochloride were added. After another 30 minutes, the
mixture was poured into water (2 L) and extracted 3X with ethyl acetate
(300 mL portions). The organic layers were combined, washed with
IN HCI (600 mL tatal), dried over sodium sulfate, filtered, and
evaporated to dryness in vacuo. EtOH ~abs; ca. 25Q mL) was added to
15 the resulting thick syrup and the solution allowed to stand at ambient
t~ dlUI~ overnight. The mixture was filtered and the filtrate boiled
down to ca. 80 mL. After standing, the mixture was again filtered and
boiled down to ca. 20 mL. After a third filtration, the filtered solids
were combined to give the title compound (28 g).
Endo-(lS)-1'(((2-amino-7,7-dimethylbicyclo(2.2.1)hept-1 -yl)-
methyl)sulfonyl)spiro( I H-indane- I .4'-piperidine)
Freshly prepared, activated Raney Nickel catalyst (ca. 30 g)
in water was allowed to settle and the water decanted. Abs. ethanol
25 (300 mL) was added, and the mixture swirled and again allowed to
settle. The solvent ~as decanted. Two more wash-decant cycle~ with
150 mL of ethanol were similarly carried out. (lS)-1'(((7,7-dimethyl-
2-oximinobicyclo(2.2. 1 )hept- I -yl)methyl)sulfonyl)-spiro( I H-indene-
1,4'-piperidine) (30 g) was ~tirred in a mixture of abs. ethanol (450
30 mL) and 2-methoxyethanol (900 mL), nitrogen was bubbled through the
suspension/~olution, and the Raney Nickel catalyst was added. The
mixture was hydrogenated under 50 psi overnight- TLC (9:1 CH2cl2:-
MeOH. silica gel) showed the reaction to be complete. The catalyst was
removed by filtration, and the flltrate evaporated to dryness in vacuo.
.
WO 95/14025 PCrlUS94113483
~2~ 746~0
- 93 -
The crude solid (27 g) was divided into 7 g batches, and each batch wasdissolved in methylene chloride (ca. 200 mL) and flash chromato-
graphed on silica (700 g in a 100 mm column, packed and eluted with
- 8% (v/v) methanol in methylene chloride), taking 200 mL fractions.
5 The exo isomer of the title amine was obtained in fractions ca. 5-7, and
the desired endo isomer in fractions ca. 8-16. TLC was on silica, eluted
with 8% methanol/methylene chloride, phosphomolybdic acid stain.
The combined product fracbons were evaporated to dryness to provide
the title compound (4.5 g from each 7 g lot, ca 18 g total) as a colorless
solid.
Endo-(l S)- I '(((2-(4-irnidazoleacetylamino)-7,7-dimethylbicyclo-
(2.2.1)-hept-1-yl)methyl)sulfonyl)spiro(lH-indane-1 ,4'-piperidine
hydrochloride
4.56 g (11.3 mmols) of Endo-(lS)-1'(((2-amino-7,7-
dimethylbicyclo-(2.2.1)hept-1-yl)methyl)sulfonyl)spiro(lH-indane-1 ,4'-
piperidine) was dissolved in 50 mL DMF and the solution treated with
2.30 g (14.1 mmols) of 4-imidazole acetic acid, 1.9 g (14.1 mmols) of
l-hydroxyl,~l,,ulliazule hydrate (HBT), and 2.7 g (14.1 mmols) of 1-
20 ethyl-3-(3-dimethylall-il-op-~yl) carbodiimide HCI (EDC). The
pH of the suspension was adjusted to 9.5 with 4.65 mL (33.4 mmols) of
triethylamine and the reaction mixture stirred at 25C for 18 hours.
DMF was removed in vacuo and the crude purplish residue
treated with water and extracted with EtOAc (3X). The organics were
2s combined, washed with H2O (IX), brine (iX), dried over Na2SO4,
filtered and stripped to dryness in vacuo. Flash chromatography of the
crude product on silica gel (114/10/1 of CH2cl2lMeoHlconc. NH40H)
gave 5.29 g (92%) of desired product as a white foam.
5.17 g (10.1 mmols) of this product was dissolved in 100
30 mL EtOAc. While stirring vigorously, a solution of HCI (g) in EtOAc
was added dropwise until precipatation ceased. The slightly gurnmy
mixture was stirred 15 minutes at 25C, then evaporated to dryness in
vacuo. The residue was restripped 3X from EtOAc, then 3X from
WO g511402~ PCr/US94/13483
21 74650
- 94 -
Et2O. The white solid was scraped from the walls of the flask,
t~iturated with Et2O and 5.3 g of hydrochloride salt collected.
M.P.: 93-167C (slow fo~m)
5 HPLC: 99.4%
PMR: (~nn~i~t~nt with structure, plus 0.30 ethyl acetate, 0.05 ether
and H2O.
M.S.: M+H + 511 ~FAB)
Analysis calculated for C2gH3gN4O3S HCI 0.30 C4Hgo2 0.05
C4H1oO " 0.4 H2O. (M.W. 584.48)
C, 60 41; H, 7.36; N, g.59
Found: C, 60.38; H, 7.46; N, 9.33
~XAMPLE 23
SO2CH2~_
Br~(~
\/
I -[[6-bromo- 1 ,7-dimethyl -2-oxobicyclo[2.2. 1 ]hept-7-yl)methyl] -
sulfonyll-spirol IH-indene-l .4'-piperidinel
Dissol~ed 100 mg of (+)-3-Bromocamphor-sulfonic acid
30 ~mmnnillm salt (.304 mM) in 15 mL of DLM. Added 5 eq of SOC12
(FW=I 18.97; d=1.631; 1.52 mM; 120 mL). The mixture was allowed
to react overnight, l~hen concentrated to obtain 92.7 mg of the crude
sulfonyl chloride. The 92.7 mg of sulfonyl chloride was dissolved in 25
mL of dry THF. T~ this was added 1.1 eq of indene salt (68.59 mg)
WO 95/14025 PCI`/US94/13483
2l~74650
and 2 eq of TFA, which was allowed to react at room temperature for
- three hours. The product was concentrated and washed with HCI, H2O
and brine. Flask chromatography in 20% EtOAc/petroleum ether.
HPLC 97.7% at 13.52 min.
EXAMPLE 24
SO2
2~'
20 (IR-syn)-1'-[[(1,7-dimethyl-2-oxobicyclo[2.2.1]hept-7-yl)methyl)-
sulfonyll -spiror I H-indene- I .4'-piperidinel
To a solution of the product of Example 23 (107 mg, 0.300
mmol) in glacial acetic acid (10 mL) was added zinc dust (24 mg, 0.367
mmol). The temperature was then increased to reflux. After 30
25 mmutes, the mixture was cooled to room temperature, filtered, then
concentrated under reduced pressure. Purification by flash
chromatography (35% ethyl acetate in petroleum ether as eluent)
afforded P~0 mg of the product as a white amorphous foam.
NMR (300 MHz, CDC13): Consistent with structure.
HPLC (Vydac C18 column, gradient from 95/5 to 0/100 H20/CH3CN
with 0.1% TFA, 15 min. gradient, flow rate = 1.5 mL/min): purity =
98%, r.t. = 12.67 min.
FABMS: [M+l]+] at400.91
Analysis calculated for C23H2gNO3S
i
wo ssrl402s PcrNss4ll3483
.
2~ i 4~5
- 96 -
C, 69.13; H, 7.32; N, 3.51
Found: C, 69.25; H, 7.30; N, 3.48
E~XAMPLE 25
`so2c~-~\D//o
o,3~-CF3
o
(IS)-1'(((2-(trifluoromethyl)sulfonyl)oxy)2,3-ene-7,7-dimethyl-
bicyclo(2.2. 1 )hept- I -yl)methyl)sulfonyl)-spiro(l H-indene- 1,4'-
piperidine)
To a solution of (IS)-1(((7,7-dimethyl-2-bicyclo(2.2.1)-
hept-l-yl)methyl)sulfonyl)spiro(lH-indene-1,4'-piperidine) (2g, 5.0
20 mmol) and 2,6-di-tert-butyl-4-methylpyridine (1.5 g, 7.5 mmol) in
CH2C12 (25 ml) was added triflic anhydride (1.3 ml, 7.5 mmol) and the
mixture was stirrea at room L~ e-a~u-~ for 30 minutes. The mixture
was then diluted with CH2C12 (30 ml) and filtered. The filtrate was
then washed with 5% HCI (2x50 ml), saturated NaHCO3 (2x50 ml), and
25 brine, dried over Na2SO4, and evaporated.
The crude triflate was purified by flash chromatography
eluting with 20% et~lylacetate in hexanes to yield 1.7 g of the title
product as a white foam (64%).
3 0 HPLC RT = 12.75 min.
NMR (CDC13) in agreement with title compound.
FAB MS: 532 (M+l)
Analysis calculat~d for C42H2gNS2OsF3
N, 2.63. C, 54.22, H, 2.63
Found: N, 2.37: C, 54.~2; H, 5.34
WO 95114025 PCTIUS94113483
2 1 7 ~ 6 ~ 0
- 97 -
EXAMPLE 26
5 1~
N\ /~1
~0
H3CO \
OC H3
( 1 S)- I '-(((2-(dimethylphosphonyl)oxy)2,3-ene-7 ,7-dimethyl-
yc1O(2.2.1)hept-1-yl)sulfonyl)spiro(lH-indene-1.4'-pipendine)
A mixture of the triflate product of Example 25 (50 mg,
0.097 mrnol), dimethyl phosphite (13 ml, 0.14 mmol), triethylamine (61
ml, 0.44 mmol), and tetrakis(triphenylphosphine)r~ lm (5 mg,
0.005 mmol) in DMF (3 ml) was stirred under an atmosphere of argon
20 for I hour. The reaction was then diluted with CHC13 (25 ml). The
chloroform was washed with 5% HCI (2x25 ml) and brine, dried over
Na2SO4 and evaporated to dryness. Purification via flash chroma-
tography (25% ethylacetate in hexanes) afforded 40 mg of the title
compound as a white solid (~6%).
HPLC Rl` = 10.31 min.
NMR(CDC13) in agreement with title compound
FAB MS: 492 (M+l)
Analysis calculated for C2sH34NSOsP 0.5 H2O
~o N, 2.80; C, 59.99; H, 7.05
Found: N, 2.71; C, 59.90; H, 7.49
wo 95/14025 PCr/US94113483
21 ,~G~i~
- 98 -
FXAMPLE 27
so2cH2~?
N ~ O/~
S~
[l-[[[[exo-2-hydroxy-777-dimethyl-l-[spiro[lH-indene-l74-piperidin]-
I -yl-sulphonyl)met 1yl]bicyclo]2.2. 1 ]hept-2-yl]methyl]amino]carbonyl]-
3-(methvlthio)propyll-carbamic acid-l.l-dimethylethyl ester
To a stirred solution of (IS)-I'-(((exo-2-hydroxy-endo-2-
:~min~m!~thyl-7,7-dimethylbicyclo(2.2.1)hept-1-yl)methyl)sulfonyl)-
spiro(lH-indene-1,4'-piperidine) (600 mg, 1.39 mmole) in 5 ml of
dry, degassed N,N-dimethylformamide was added Boc-L-methionine
(381 mg, 1.53 mrnole) and 743 mg (1.68 mmole) of benzotriazol-l-
yloxy tris(dimethylamino)phosphonium hexafluorophosphate (BOP) at
room temperature. The resulting reaction mixture was protected from
moisture and the p~ was adjusted to 8-9 with diusopropylethylamine.
After one hour, all volatile components were removed under reduced
pressure and the residue was dissolved in 250 ml of ethyl acetate. This
601ution was washed in succession with 10% aqueous citric acid, 50%
sodium bicarbonate solution, and brine. The organic phase was dried
(sodium sulfate) ani concentrated. Chromatography of the crude
reaction product on silica gel (1:1 ethyl acetate-hexane elution) afforded
the title compound as an amorphous solid: m.p. 82-92C.
HPLC: >92% pure at 214 nM
NMR: Consistent ~vith structure and verified presence of solvent
WO 95/14025 PCr/lJS94/13483
21 74~
99
FAB MS: 670 (M+ + thiogIycerol)
Analysis calculated for C34Hs1N3O6S2 O.5CHC13
C, 57.42; H, 7.19; N, 5.82
Found: C, 57.58; H, 7.50; N, 5.83
EXAMPLE 28
~r~
- OH o
H~S~
'
2-amino-N-[[exo-2-hydroxy-7,7-dimethyl- 1 -[(spiro[lH-indene-l ,4'-
piperidin] I 'ylsulfonyl)methyl]bicyclo]2.2. 1 ]hept-2-yl]methyl] -4-
methylthio)-butanamide
A c-~ntinl~o~l~ stream of dry HCI gas was passed for S
minutes into an ice cold solution of ethyl acetate (2 ml) containing 20
mg of the product of Example 27. After 30 minutes at 0C, the reaction
mixture was cu~ ai~d and the residue was chromatographed on two
0.25 mm precoated silica gel plates (chloroform-methanol-ammonium
hydroxide, 93:7:0.7 v/v elution). The title compound was obtained as a
solid (10.7 mg): m.p. 78-80"C.
TLC: Rf=0.28 (CHC13-CH30H-NH40H, 95:5:0.5)
NMR: Consistent with structure and verifies presence of solve
FABMS: 562(M++H)
Analysis calculated for C2gH43N3O4S2âO.75CHCI-~
C, 54.86; H, 6.77; N, ~.45
Found: C, 54.52; H, 6.90, N, 6.3Z
WO 95114025 PCT/US94/13483
~ 7 ~
- 100-
EXAMPLE 29
s
SO2
0 ~OH~ ~
5-(1,3-dihydro-1,3-dioxo-2H-isoindol-2-yl)-N-[~2-hydroxy-7,7-
dimethy- I -[(spiro[ I H-indene- I ,4'-piperidin] - I '-ylsulfonyl)methyl] -
bicyclo[2.2.1]hept-2-yl]-methyl]dihydro-4-oxo-2H-1,3-Thiazine-
3(4H)-~et~m j~l~
To a slirred solution of (lS)-I'-(((exo-2-hydroxy-2-
20 aminomethyl-7,7-di~ llyl~icyclo(2.2.1)hept-1-yl)-sulfonyl)spiro(lH-
indene-1,4'-piperidine) (67 mg, 0.16 mmole) in 1.5 ml of dry, degassed
N~N-dilll~lllylru~ dlllide was added 4-oxo-~-phthalyl-1,3-thiazine-acetic
acid (62 mg, 0.192 mmole) and 88 mg (0.20 mmole) of l~ell~ullia~
yloxytris(dimethylamino)-phosphonium hexafluorophosphate (BOP) at
2s room l~ ,eldlul~;. The resulting reaction mixture was protected from
moisture and the pH was adjusted to 8-9 with diisopropylethylamine.
After 24 hours all volatile components were removed under reduced
pressure and the residue was suspended in 5 ml of toluene.
Cnn~ntr~tion of this suspension afforded the crude product which wa.
30 chromatographed on silica gel (96:4:0.4 CHC13-CH30H-NH40H) to
give the title compcund (109 mg): m.p. 134-137 C.
NMR: Consistent vvith structure and verifies presence of solvent;
HPLC: >94% pure at 214 nM;
FAB MS: 733 (M+~H)
WO 95/14025 PCTNS94113483
21 7465~
- 101 -
Analysis calculated for C3gH44N4O7S21.1CHC13
C, 54.34; H, 5.26; N, 6.48
Found: C, 54.12; H, 5.22; N, 6.42
EXAMPLE 30
H ~H2
5-arnino-N-[[1 -[[spiro[lH-indene-l ,4'-piperidin]-1-yl)-sulfonyl]-
methyl]-exo-2-hydroxy-7,7-dimethylbicyclo-[ 1.1. I ]hept-2-yl]-
methylldihvdro-4-oxo-2H- I .3-Thiazine-3(4H)-acetamide
The product of Example 29 (60 mg) was dissolved in 3 ml
of methanol and treated with 50 ml (1.59 mmole) of 95% hydrazine at
23C. After 24 hours the solvent was removed under vacuum and the
residue was suspended in toluene. Rotoevaporation of this suspension
gave the crude product, which was chromatographed on four 0.5 mm
procoated silica gel plates (chloroform-methanol-ammonium hydroxide,
92:8:0.~ v/v elution). The title compound was obtained as an
amorphous solid (27 mg): m.p. 103-106C.
NMR: Consistent with structure and verifies presence of solvent
HPLC: >97% pure at 214 nM;
FAB MS: 605 (M++H)
Analysis calculated for C30H44N4OsS2 0.6CHC13
C, 54.33; H, 6.64; N, 8.28
Found: C, 54.12; H, 6.60; N, 8.20
WO 9!i11402~ PCT/US94/13483
21,7~
- 102-
E~XAMPLE 31
5 g3
N ~/--
\SO2 ~
1 '-[(5-oxospiro[bicyclo[2.2.1]heptane-7,1 '-cyclopro-pan]-2-yl)-
sulfonyll-spirorlH[-indane-l ~4'-piperidinel
Spiro( I H-indane- I ,4'-piperidine)hydrochloride(3 g, 13 .S
mmole) was dissolved in 30 ml of methylene chloride. The resulting
solution was cooled to 0C and treated with 3.76 ml of triethylamine.
This was followed by the dropwise addition of chloroethyl-
sulfonylchloride (1.57 ml, 15 mmole). The reaction mixture was
allowed to warm to room temperature over one hour and was filtered.
The filtrate wa~ washed with water~ sodium bicarbonate solution, and
brine. The dried organic washings were ~ullct;llllal~d and the residue
was column chromatographed on silica gel (98:2 methylene chloride-
ether elution) to give 1.88 g of l'-(vinylsulfonyl)spiro(lH-indane-
1,4'piperidine)(m.p. 114-115C).
I '-(Vinylsulfonyl)spiro( I H-indane- 1 74'piperidine) ( 185 mg,
0.67 mmole) was combined with 5.1 g (55 mmole) of ~ ulle~diene in
4 ml of benzene arld heâted to reflux for 48 hours. The solvent and
excess reagent were removed under reduced pressure and the residual
ûil was chrûmatûglâphed on ~ilica gel (4:1 hexâne-ethyl âcetate elution)
to give 216 mg of a solid which was recrystallized from methanol to
give white needles (m.p. 176-177C). Of this material, 110.7 mg was
dissolved in 5 ml of dry THF under nitrogen. This solution was cooled
to 0C and treated with 0.1 ml of borane-THF complex. After stirring
for one hour, an additional 0.1 ml of Borane-THF complex was added
to the reaction mi~;ture and stirring was continued at room temperature
for I hour more. The reaction mixture was recooled to 0C and
WO 95114025 2 ~ 7 ~ 6 5 0 PCIIUS94/13483
- 103 -
quenched with the dropwise addition of water. After 10 minutes, 0.3
ml of 3N sodium hydroxide solution and 0.3 ml of 30% hydrogen
peroxide solution was added. The resulting mixture was heated to ~0C
for 1 hour and allowed to stand at 23C overnight. The reaction
5 mixture was partitioned between ethyl acetate and brine. The aqueous
phase was extracted with ethyl acetate and the combined organic extracts
were dried (MgSO4) and concentrated. Purification of the crude
reaction product by preparative TLC on silica gel (2:1 hexane-ethyl
acetate) afforded two coll,po,l~llL~. The more polar product (m.p. 232-
o 233C, from methanol) (0.15 mmole) was dissolved in 5 ml ofmethylene chloride and oxidized with pyridinium chlorochromate (2
mmole) at 23C. The reaction mixture was filtered through celite and
the filtrate was applied to 0.25 mm precoated silica gel plates. Elution
with 2:1 hexane-ethyl acetate and recrystallization from methanol
5 afforded the title compound as a white solid: m.p. 210-212C.
NMR: Consistent with structure and verifies presence of solvent
HPLC: >96% pure at 214 nM;
FAB MS: 386 (M++H)
Analysis calculated for C22H27NO3so 3cH2o
C, 67.59; H, 7.12; N, 3.58
Found: C, 67.66; H, 7.22; N, 3.53
EXAMPLE 32
SO2
WO 95/14025 PCI'NS94/13483
~ l 4f~5f~
- 104-
I '-[(g, I 0-dihydro-~, 1 0-ethanoanthracen- 1 ] -yl)sulfonyl] -spiro[ I H-
indane- I .4'-pipericlinel
I'-(Vinylsulfonyl)spiro(lH-indane-1,4'piperidine) (152
mg) was combined with 350 of anthracene m 15 ml of toluene and
5 heated to reflux for 8 days. The solvent and excess reagent was
removed under reduced pressure and the residual oil was
chromatographed on silica gel (4:1 hexane-ethyl acetate elution) to give
the title compound as an amorphous solid.
o NMR: Consistent ~vith structure and verifies presence of solvent
HPLC: >99% pure at 214 nM;
FAB MS: 456 (M~+H)
Analysis calcul~ted for C2gH2gNO2S0.05C H2O 0.25 HC13
C, 72~3; H, 6.08; N, 2.88
Found: C, 72.23; H, 5.92; N, 2.67
EXAMPLE 33
20 l~ ~
SO2~
I '-(spiro[bicyclo[2.2. 1 ]hept-5-ene-7,1 '-cyclopentan]-2-ylsulfonyl)-spiro~ 11 I-indane- I l ~'-piperidinel
l'-(Vinylsulfonyl)spiro(lH-indane-1,4'piperidine) (100
mg) was combined with 500 mg of spirononadiene in 3 ml of toluene
30 and heated to reflux for 17 hours. The solvent and excess reagent were
removed under reduced pressure and the residual oil was chromato-
graphed on silica g~l (4:1 hexane-ethyl acetate elution) to give the title
compound a~ an amorphous solid: m.p. 115-119C.
NMR: Consistent with structure and verifies presence of ~olvent
WO 95/14025 2 1 7 4 ~ 5 0 PCr/US94/13483
- 105-
HPLC: >99% pure at 214 nM;
FAB MS: 398 (M++H)
Analysis ~lC~ d for C24H31N02SO.05CHC13
C, 71.57; H, 7.76; N, 3.47
Found: C, 71.94; H, 7.81; N, 3.42
EXAMPLE 34
~
52/~
I '-(bicyclo[2.2. 1 ]hept-5-en-2-ylsulfonyl)spiro[ 1 H-indane- 1,4'-
piperidinel
I '-(Vinylsulfonyl)spiro( I H-indane- 1 ,4'piperidine) ( l O0
mg) was combined with ten equivalents of cyclohPY~ n-~ in 10 ml of
toluene and heated to reflux for 30 hours. The solvent and excess
reagent were removed under reduced pressure arld the residual oil was
chromagraphed on silica gel (4:1 hexane-ethyl acetate elution) to give
the title compound as an amorphous solid: m.p. 162-166C.
NMR: Consistent with structure and verifies presence of solvent
HPLC: >99% pure at 214 nM;
FAB MS: 358 (M++H)
Analysis calculated for C21H27N02SO.lH20
C, 70.19; H, 7.63; N, 3.90
Found: C, 70.30; H, ~.03; N, 3.61
WO 95114025 PCrlUS94113483
.
2~ 7~55
- 106-
E~AMPLE 35
`o~O~OCH3
1'-[[2'-[[(2-methoxyethoxy)methoxy]methyl]spiro]bicyclo[2.2.1]hept-
5-ene-7.1'-cyclopropanl-2-yllsulfonyll-spirorlH-indane-1 .4'-piperidinel
1 '-(Vinylsulfonyl)spiro( 1 H-indane- 1 ,4'piperidine) (2.45 g)
was combined with 2.2 g of hydroxymethylspirocycloheptadiene and 3
mg of hydroquinone in 8 ml of toluene and heated to reflux for 17
5 hours. The solven~ and excess reagent were removed under reduced
pressure and the residual oil was chromatographed on silica gel (1:2
hexane-ethyl aceta~e elution) to give three components. The least polar
of these products (80 mg) was dissolved in methylene chloride
containing 78 mg of diisopropylethylamine and treated with 49 mg of 2-
20 methoxyethoxy-m~.thyl chloride. The reaction mixture was protected
from moisture and stirred at 23C overnight. The reaction mixture wa~
conl ~ntr~t~d and tile residual material was applied to 0.5 mm precoated
silica gel plates. Elution with 1:1 hexane-ethyl acetate afforded the title
compound.
NMR: Consisten~ with structure and verifies presence of solvent;
PLC: >90% pure at 214 nM;
FAB MS: 4RR (M~ +H)
wo 95/14025 PCr/US94113483
21 7465~
- 107 -
E~XAMPLE 36
~ /
\NJ~N~co2cH3
H CH3
(lS-)-2-[[[exo-2-hydroxy-7,7-dimethyl-1-[(spiro[lH-indene-1 ,4'-
piperidin]-l '-ylsulfonyl)methyl]bicyclo[2.2.1]hept-2-yl]methyl]amino]-
N-(2-methoxy-2-oxoethyl)-N,N-dimethyl-2-oxo-~1l"."~."i"iu"l salt
with trifluoroacetic acid (1:1)
To a stirred solution of (lS)-I'-(((exo-2-hydroxy-endo-2-
aminomethyl-7,7-dimethylbicyclo(2.2. 1 )hept- I -yl)methyl)sulfonyl)-
spiro(lH-indene-1,4'-piperidine) (108 mg, 0.25 mmole) in 5 ml of
20 toluene was added I ml of toluene containing 0.5 mmole of
methyliminodiacetic acid anhydride. Tetrahydrofuran (2 ml) was added
and the white suspension was stirred at 23C overnight. The reaction
mixture was filtered and concentrated. The residue was dissolved in 5
ml of N,N-dimethylformamide and treated in succession ~vith methyl
2s iodide (0.5 ml) and (liisoplu~ylethylamine (0.2 ml). The resulting
solution was protected from moisture and heated for 5 hours at 50C.
An additional 0.5 ml of methyl iodide was added and heating was
continued for 5 hours more. The reaction mixture was concentrated
and the residue was purified via reverse phase preparative HPLC
30 (Vydac Protein & Peptide C-18 column, mobile phase = 0.1% trifluoro-
acetic acid (TFA) in water-acetonitrile). The fractions containing the
title compound were pooled and c~ elltldl~d. The residue was
dissolved in dioxane and this solution was freeze-dried to yield the TFA
salt of the title compound as a white powder:
~Vo 9~114025 PCr/US94113483
.
2~ 74~53
- 108 -
NMR: (~nnsi~t~nt with structure and verifies presence of solvent
HPLC: >99% pure at 214 nM;
FAB MS: 588 (M~-+H)
Analysis calcul~lted for C31H46N3O6S 1.8CF3CO2H 1.0 dioxane
C,52~1;H,6.27;N,4.77
Found: C, 52.~8; H, 6.40; N, 4.76
EXAMPLE 37
[~
S2~
Br>~/ l\
0~
(lR-syn)-1'-[[(1,7-dimethyl-2-oxobicyclo[2.2.1]hept-7-yl)methyl]-
sulfonvll-spiro~ lH-indene-l ~4'-piperidinel
Disso~ved 100 mg (.304 mM; FW=328.23) of (-)-3-
brom~- ~mrhosulfonic acid ~mm~nillm salt in 15 mL of DCM. Added S
25 eq of tllionylchloride (FW=I 18.97; d=1.631; 1.52 mM; 120 mL) and
allowed mixture to react at 0C under a N2 balloon for I hour. After I
hour the mixture ~as concentrated to obtain 63.12 mg of crude acid
chloride (.191 mM).
Dissolved 47.13 mg of indene HCI salt in 20 mL of THF.
30 The acid chloride ~vas added along with 2 eq of DEA, and allowed to
react at room temperature for I hour, then concentrated and washed
with~x~00mLofI NNCI,2x200mLH2Oand2x200mLbrine,
then concentrated and dried over sodium sulfate. Flash chromatography
in 25% EtOAc/petroleum ether.
wo 95114025 PCrlUS94/13483
2~ 74~50
- 109-
EXAMPLE 38
N
O î~
~/~
To a solution of the product of Example 37 (107 mg, 0.300
5 mmol) in glacial acetic acid (10 mL) was added zinc dust (24 mg, 0.367
mmol). The temperature was then increased to reflux. After 30
minutes the mixture was cooled to room ~ yeldtu-~, filtered, then
~u-~ d under reduced pressure. Purification ~y flash
chromatography (35% ethyl acetate in petroleum ether as eluent)
20 afforded 80 mg of the product as a white amorphous foam.
NMR (300 MHz, CDC13): Consistent with structure;
HPLC: (Vydac C18 column, gradient from 95/S to 0/100 H20/CH3CN
with 0.1% TFA, 15 min. gradient, flow rate = 1.5 mL/min.):
25 Purity: 98%, r.t. = 12.67 min.
FAB MS: [M + l]+ at 400.91
Analysis calculated for C23H2gNO3S
C, 69.13; H, 7.32; N, 3.51
Found: C, 69.25; H, 7.30; N, 3.48
~o
WO 95/14025 PC~r/US94113483
21 7~650
- 110 -
EXAMPLE 39
N
SO2
0
I '-[[(7,7-dimethyi-.~-oxobicyclo[2.2. 1 ]hept-l -yl)-methyl]sulfonyl]-3'-
phenvl-spiror I H-indene- I .4'-piperidinel
Ethanolamine (6.66 mL, 0.1103 mmol) was placed in a 3-
neck round bottom flask and heated to reflux. Once at relux, styrene
oxide (6.28 mL, 0.0557 mmol) was added dropwise to the reaction
mixture over 15 m~nutes. The reaction mixture was refluxed an
additional 2 hours and then allowed to cool. The product, N-(2-
hydroxyethyl)-N-(2-hydroxy-2-phenylethyl)amine, was obtained as a
clear oil by vacuunl distillation.
N-(2-hydroxyethyl)-N-(2-hydroxy-2-phenylethyl)amine
(1.30 g, 7.16 mmol) was dissolved in chloroform and then while under
a nitrogen blanket, thionyl chloride (1.05 mL, 14.32 mrnol) was added
dropwise over 10 minutes. The reaction mixture was then refluxed for
I hour and allowed to cool to room temperature. Water was then added
25 to the mixture and allowed to stir I hour. The reaction mixture was
separated and the organic layer was washed three times with 3N HCI.
The aqueous layers were combined and then basified with 40% sodium
hydroxide. The aqueous layer was extracted 3 times with ether, the
organics were combined, and dried over sodium sulfate. The organic
30 layer was filtered and the filtrate concentrated to a yellow oil.
The yellow oil was dissolved in ether and then di-t-butyl
dicarbonate (1.50 g, 6.87 mmol) was added along with triethylamine
(500 ml, 3.59 mmol).The mixture was allowed to stir overnight and
then it was washed with H2O (2x), 0.1 N HCI (2x) and sodium
bicarbonate (2x). The organic layer was dried over sodium sulf~te,
WO 95/14025 PCT/US94/13483
21 7455i3
filtered, and the filtrate concentrated to a yellow oil. The yellow oil
was dissolved in CH2C12 and poured onto a silica gel column. The
column was eluted with 1:1 CH2C12/hexane. The product fractions
- were combined and concentrated to dryness to yield the product, N-(2-5 hydroxyethyl)-N-(2-hydroxy-2-phenylethyl)-t-butyl-carbamate.
To a solution of indene (268 ml, 2.30 mmol) in dry THF (2
mL) cooled in an ice bath and m~int~in~d under a nitrogen blanket was
added lithium bis(trimethylsilyl)amide (IM solution in THF, 4.6 mL,
4.60 mmol) over 10 minutes. The mixture was stirred in the cold bath
o for 30 minutes, then added over 10 minutes to a solution of N-(2-
hydroxyethyl)-N-(2-hydroxy-2-phenylethyl)-t-butylcarbamate (694 mg,
2.30 mmol) stirred in an ice bath. The mixture was stirred for 2 hours
in the cold and for 30 minutes at 25C under nitrogen, then
concentrated to an orange oil. 10% ethyl acetate in hexane was added
15 and the resulting mixture poured onto a silica gel column packed with
10% ethyl acetate in hexane. Elution was with the sarne solvent and the
product fraction were cnncçntr~t~d to provide l'-(t-butyloxycarbonyl)-
spiro-(indene-3 '-phenyl - I ,4'-piperidine .
1 '-(t-Butyloxycarbonyl)spiro(indene-3-'phenyl- 1,4'-
20 piperidine) (290.9 mg, 0.842 mrnol) in ethyl acetate was stirred in anice bath and saturated with HCI (g) for 30 minutes. The mixture was
concentrated to dryness and reconcentrated from ether three times to
yield spiro ( I H-indene-3 -phenyl- I ,4'-piperidine)-hydrochloride.
Spiro( I H-indene-3'-phenyl- 1 ,4'-piperidine)hydrochloride
25 (205.2 mg, 0.690 mmol) and (+)-10-camphorsulfonyl chloride (196 mg,
0.782 mmol) were combined in THF and the pH was adjusted to 9 with
triethylarnine (196 mL, 1.41 mmol). The reaction mixture was stirred
ovemight at 25C, then concentrated to dryness, and redissolved in
CH2C12, then poured onto a silica gel column and eluted with CH2C12
30 The product fractions were combined and evaporated to dryness to
provide the title compound which was crystallized from ether and dried
in vacuo overnight.
m.p.: 1~3 - 215C
WO 95~14025 PC~r/US94/13483
2~ i74~
- 112 -
NMR: Consistent with structure
HPLC: >95% pure
MS: M + H 476.3 (FAB)
Analysis calcul~lted for C29H33N03S SOH20
C, 71.~6; H, 7.07; N, 2.89
Found: C, 71.~R; H, 7.03; N, 2.87
EXAMPLE 40
~
SO2 y
N,LO
H
(I S(la,2a,4a))-2-h~droxy-7,7-dimethyl-1 -((spiro-(lH-indene,l ,4'-
yiperidin)- I '-yl-sull-onvl)methyl)-bicyclo-(2.2. 1 )heptane-2-acetic acid
(100 mg, 0.217 mmol) diphenylphosphoryl azide (51.~ mL.
0.239 mmol), and triethylamine (66.2 mL, 0.471 mmol) were combined
in DMF. The mixtllre was allowed to stir overnight and then it was
concentrated to dryness. The resulting residue was dissolved in
CH2C12, poured onto a silica gel column, and eluted with 5% methanol
in CH2C12. The product fractions were combined and concentrated to
dryness. The title compound was obtained as a white solid from ether
and dried in vacuo overnight.
NMR: Consistent with structure
HPLC: >93% pure
MS: M + H 4~7.3 (FAB)
Analysis caiculated for C2sH32N2O4S
C, 6~.76; H, 7.06; N, 6.14
Found: C, 65.76; H, 7.42; N, 5.80
.
wo g5,l4025 2 1 7 4 6 5 0 PCT/IJS94/13483
- 113 -
EXAMPLE 41
~2
- ~ /
H ~
(lS-exo)-[[2--hydroxy-7,7-dimethyl-1 -[(spiro[lH-indene-1,2'-
piperidin)-1'-ylsulfonyl)methyl]bicyclo2.2.1]hept-2-yl]carbarnic acid
l.l-dimethylethyl ester
( 1 S)- I '-(((7,7-dimethyl-2-oxobicyclo-(2.2. 1 )-hept- I -yl)-
methyl)sulfonyl)spiro(lH-indene-1,4'-piperidine) (1.92 g, 4.81 mmol)
was combined with zinc iodide (47 mg, 0.15 mmol) in 2 mL of toluene.
While under a nitrogen atmosphere, the mixture was treated with
trimethylsilyl cyanide (960 mL, 7.21 mmol) dropwise. The reaction
mixture was then heated to 100C for 3.5 hours. The mixture was
allowed to cool and diluted with 10 mL of dry THF. Then lithium
~ min~lm hydride (IM in THF; 8.6 mL, 8.6 mmol) was added dropwise
over 5 minutes and the mixture was allowed to stir at 25C for 2 hours.
The reaction mixture was then diluted with ether (N50 mL) and 10%
sodium hydroxide solution was added dropwise until a gray precipitate
stopped forming. The mixture was now filtered, the filtrate was washed
with sodium bicarbonate and brine, the organic layer was separated and
dried over sodium sulfate. The or~anic layer was filtered, the filtrate
concentrated and the residue dissolved in CH2cl2. This was poured
onto a silica gel column, eluted with 97/3/0.3 of CH2C12/methanol/
ammonium hydroxide and product fractions collected. The product
fractions were combined and concentrated to dryness. The desired
product, ( I S~- I '-(((~,7-dimethyl-(2-endo-aminomethyl-2-exo-hydroxy)-
WO gS~14025 PCrlUS94113483
.
21 ~465G
- 114
bicyclo-(2.2.1)-hept-1-yl)-methyl)-sulfonyl)spiro(lH-indene-1 ,4'-
piperidine), was obtained as a white foam from ether.
(I S)- I '-(((7,7-dimethyl-(2-endo-aminomethyl-2-exo-
hydroxy)-bicycylo (2.2.1)-hept-1-yl)-methyl)-sulfonyl)spiro(lH-indene-
1,4'-piperidine) (44.9 mg, 0.104 mmol) and di-t-butyl dicarbonate (25
mg, 0.115 mmo~) were combined in S mL of CH2cl2. The reaction
mixture was treate~3 with triethylarnine (18.1 mL, 0.13 mmol) and then
stirred for 30 minutes at 25C. The reaction mixture was poured onto
a silica gel column and eluted with 15% ethyl acetate in hexane. The
0 product fractions were combined and evaporated to dryness. The title
compound was obtained as a white solid from CH2C12/hexane and was
dried in ~asl overnight.
m.p.: 71C-1 15C
NMR: Consistent with structure
HPLC: >95% pure
Analysis calculated for C2gH42N2OsS 0.45
C, 6~I4; H, 8.73; N, 5.03
Found: C, 66.r'1; H, 8.52; N, 4.75
EXAMPLE 42
N r
\SO2 H o OH
N~
H /
~`OH
WO 9~/14025 PCr/Usg4113483
21 74~
- 115 -
(lS-exo)-3-hydroxy-N-[[2-hydroxy-7,7-dimethyl-1-[(spiro[lH-indene-
1 ,4'-piperidin]- 1 '-ylsulfonyl)methyl]bicyclo[2.2. 1 ]hept-2-yl]methyl] -2-
(hvdroxymethyl)-2-methyl-propanamide
(lS)-1 '-(((7,7-dimethyl-(2-endo-aminomethyl-2-exo-
5 hydroxy)-bicicylo-(2.2. 1 )-hept- I -yl)-methyl)-sulfonyl)spiro( 1 H-indene-
1,4'-piperdine) (51 mg, 0.118 mmol), 2,2-bis(hydroxymethyl)-
propionic acid (19 mg, 0.142 mmol), 1-hydroxybenzotriazole hydrate
(HBT) (19 mg, 0.142 mmol), and 1-ethyl-3-(dimethylaminopropyl)
carbodiimideâHCl(EDC) (27 mg, 0.142 mmol) were all combined in 5
o mL of DMF. Then, triethylamine (46 mL, 0.33 mmol) was added to
adjust the pH to 9, and the mixture was allowed to stir overnight at
25C. The reaction mixture was concentrated to dryness and treated
with 5% citric acid solution. The water layer was then basified with
saturated sodium bicarbonate and extracted 3 times with ethyl acetate.
5 The organics were combined and dried over sodium sulfate. The
organic layer was filtered, the filtrate concentrated, and the resulting
residue dissolved in CH2C12. This was then poured onto a silica gel
column and eluted with 3% methanol in CH2cl2. The product fractions
were combined and evaporated to dryness. The title compound was
20 obtained as a white solid from CH2cl2thexane
and was dried in vacuo overnight.
m.p.: 167-170C
NMR: Consistent with structure
Z5 HPLC: >99% pure
MS: M + H 547.3 (FAB)
Analysis calculated for C2gH42N2O6S
C, 63.71; H, 7.74; N, 5.12
Found: C, 63.65; H, 7.59; N, 4.88
wo 95/14025 PCr/usg4ll3483
2 1 7~50
- 116-
EXAMPLE 43
- OH
,~
(lS-exo)-4-benzoyl-N-[[2-hydroxy-7,7-dimethyl-1-[(spiro[lH-indene-
1,4'-piperidin]-1'-ylsulfonyl)methyl]bicyclo[2.2.1]hept-2-yl]methyl]-
benzamide
The procedure of Example 42 was carried out using 49.6
mg, 0.115 mmol of (lS)-(((7,7-dimethyl-(2-endo-aTninomethyl-2-exo-
hydroxy)-bicicylo-(2.2.1)-hept-1-yl)-methyl)sulfonyl)spiro(lH-indene-
1,4'-piperidine),26.~ mg, 0.138 mmol of EDC, 18.7 mg, 0.138 mmol of
HBT, 48 mL, 0.345 rnmol of triethylamine, and substituting 4-
benzoylbenzoic acid (31.3 mg, 0.138 mmol) for 2,2-bis-(hydroxy-
methyl)-propionic acid. Chromatographic elution was with 2%
methanol in CH2C12. The title compound was obtained and dried in
vacuo, ovemight.
m.p.: 110-135C
NMR: Consistent vvith structure
HPLC: >94% pure
MS: M + H 639 (FAB)
Analysis calculated for C3gH42N2O~S0.40C6H14
C, 72.C6; H, 7.13; N, 4.16
Found: C, 72.CI; H, 7.14; N, 4.18
WO 95/14025 PCT/US94/13483
21 74650
.
- 117-
EXAMPLE 44
D02 ~
- OH,p )H
`N~N~1~0~<
1 '-(bicyclo[2.2. 1 ]hept-5-en-2-ylsulfonyl)spiro[ I H-indane- 1,4'-
piperidinel
The procedure of Example 42 was carried out using 94.5
mg, 0.22 mmol of (IS)-(((7,7-dimethyl-(2-endo-amino-methyl-2-exo-
hydroxy)-bicicylo-(2.2. 1 )-hept- I -yl)-methyl)-sulfonyl)spiro( I H-indene-
1,4'-piperidine), 49.8 mg, 0.26 mmol of EDC, 35.2 mg, 0.26 mmol of
HBT, 90 mL, 0.66 mmol of triethylamine, and substituting t-boc-L-
20 serine(bzl) (76.8 mg, 0.26 mmol) for 2,2-bis-(hydroxymethyl)-
propionic acid. Chromatographic elution was with 98/2 of
CH2C12/methanol/ammonium hydroxide. The title compound was
obtained as a white solid from ether and dried in vacuo overnight.
25 m.p. 75-95C
NMR: Consistent with structure
HPLC: >9~% pure
MS: M + H 639 (FAB)
Analysis ~ t~d for C3gHs3N3O7S
3~ C, 66.16; H, 7.55; N, 5.94
Found: C, 66.04; H, 7.68; N, 5.84
WO 95/14025 PCr/US94/13483
2i7~65`a
- 118-
EXAMPLE 4
OHII
0 N OH
H
[lS-[l.alpha., 2 Beta., 2(R*), 4. beta.]]-2-amino-N[[1-[[(2,3-dihydro
spiro[ 1 H-indene- I ,4'-piperidin] -yl)sulfonyl]methyl] -2-hydroxy-7,7 -
dimethylbicyclor2.2.1 lhept-2-lylmethvll-3-hydroxy-propanamide
The product of Example 44 (120 mg, 0.17 mmol) was
dissolved in 4% formic acid in methanol. Palladium hydroxide catalyst
(25 mg) was added to the solution and it was placed on a PARR
apparatus under sa~ p.s.i. of hydrogen, overnight. The reaction mixlure
20 was filtered over solka floc and the filtrate was concentrated to a clear
oil. This clear oil ~as dissolved in 2 mL of ethyl acetate and chilled to
0C. At this point, ~ mL of prechilled sat'd HCI/ethyl acetate solution
was added dropwise. The reaction mixture was allowed to stir for I
hour and then concentrated to dryness. The resulting residue was
25 partitioned between ethyl acetate and saturated sodium bicarbonate, and
after extracting 3 times with ethyl acetate, the organics were combined
and dried over sodium sulfate. The organic layer was filtered, the
filtrate was concen~:rated, and the resulting residue was dissolved in
CH2C12. This was then poured onto a silica gel column and eluted witl
30 95/5/0 5 of CH2C12/methanol/ammonium hydroxide. The product
fractions were combined and evaporated to dryness. The title
compound was obtained as a white .solid from ether and dried in vacuo
overnight.
WO 9~11402~ PCr~US94/13483
211465~
- 119-
m.p.: 70-1 lO~C
NMR: Consistent with structure
HPLC: >93% pure
- MS: M +H 520.3 (FAB)
Analysis calculated for C27H41N3OsS0.25 C4Hlo
C, 61.31; H, 8.20; N, 7.67
Found: C, 61.35; H, ~S.11; N, 7.58
EXAMPLE 46
SOz
- ~
H
HO2C
3-[[[[2-hydroxy-7,7-dimethyl-1-[(spiro[lH-indene-1,4'-piperidin]-1'-
ylsulfonyl)methyl]bicyclo[2.2.1]-hept-2-yl]methyl]amino]carbonyl]-
bicyclor2.2. 1 lhept-5-ene-2-carboxylic acid
( 1 S)-(((7 ,7-dimethyl-(2-endo-aminomethyl-2-exo-
hydroxy)-bicicylo-(2.2. 1 )~hept- I -yl)-methyl)sulfonyl)spiro( I H-indene-
1,4'-piperidine) (98.5 mg, 0.23 mmol) was combined with bicyclo
(2.2.1)-5-heptene-2,3-dicarboxylic anhydride (43 mg, 0.26 mmol) in 5
mL of THF. Triethylamine (50 mL, 0.36 mmol) was added to the
reaction dropwise to adjust the pH to 9. The reaction mixture was
stirred overnight and then concentr~ted to dryness. The resulting
re.sidue was dissolved in CH2cl2~ poured onto a silica gel column, and
eluted with 971310.3 of chloroform/methanol/acetic acid. The product
fractions were combined and concentrated to dryness. The title
WO 95114025 PCT/US94/13483
0
- 120-
compound was obtained as a white solid from ether and dried in vacuo
overnight.
m.p.: 128-16~C
5 NMR: Consistent ~vith structure
HPLC: >95% pure
MS: M + H 595 (FAB)
Analysis r~lc~ tt~d for C33H42N206SO.10 CH2C12
C, 64.74- H, 7.12- N, 4.56
Found: C, 64.76, H, 7.07, N, 4.66
EXAMPLE 47
1 5
SO2 OH
NH~
o
( 1 S)- I '-(((2-endo-cyanomethyl-7,7-dimethyl-exo-2-hydroxybicyclo-
25 (2.2.1)-hept-1-yl)methyl)sulfonvl)-spiro(lH-indene-1.4'-piperidine)
Lithium bis(trimethylsilyl)amide (21.3 ml of a 1.0M THF
solution, 21.3 mmol) was cooled to -7~C under N2, treated with
acetonitrile (1.07 ml, 20.4 mmol) and stirred 15 mmutes. A THF
solution (30 mL) oi` ( I S)- I '-(((7,7-dimethyl-2-oxobicyclo( 2.1 )-hept- I -
30 yl)methyl)sulfonyl)spiro(lH-indene-l .4'-piperidine) (40 gm. 10 mmol)
was added dropwise and stirred 10 minutes after addition was complete.
6N HCI (2.R ml) w~ls added to the cold reaction all at once and the
mixture was allowed to warm to 25C.
The mixture was diluted with H2O (25 ml) and extracted
with EtOAc (3 x 100 ml). The or~anic layers were combined~ washed
WO 9~/14025 Pcr/uss4ll34s3
21 7~5~
- 121 -
with H20 (25 ml) and brine (25 ml), dried over Na2SO4, filtered and
concentrated to dryness in vacuo. Trituration with ether gave the title
compound (4.17 gm, 93% yield) as a crystalline solid (m.p. 199-202C).
TLC: Rf = 0.69, Silica GF (7% Et2O in CH2cl2)
PMR: Consistent with structure
( I S)- 1 '-(((2-endo-aminoethyl-7,7 -dimethyl-2-exo-hydroxybicyclo-
(2.2.1 )-hept- I -yl)methyl)~ulfonyl)spiro( I H-indene- I ~4'-piperidine)
IJnder a blanket of nitrogen, Lithium Alllminllm Hydride
(8.0 ml, 8.0 mmol, of a 1.0 M THF solution) was cooled to 0C, treated
dropwise with a THF solutioP (37 ml) of (IS)-1'-(((2-endo-cyano-
methyl-7 ,7-dimethyl-2-hydroxybicyclo-(2.2. 1 )-hept- I -yl)methyl)-
sulfonyl)spiro(lH-indene-1,4'-piperidine) (4.4 gm, 10.0 mmol) and the
reaction stirred 10 minutes. With rapid stirring, the reaction was
treated with H20 (0.37 ml), 20% NaOH (aq) (0.40 ml) and H2O (1.46
ml) followed by extraction with EtOAc (3 x 150 ml). The organic
layers were combined, washed with H2O (25 ml) and brine (25 ml),
dried over Na2SO4, filtered and cu~ ld~d to dryness in vacuo.
Flash chromatography on silica gel (90/10/1/1 of
CH2C12/MeOH/H20/HOAc) provided product fractions which were
pooled and concentrated to dryness. The residue was treated with sat'd
Na2CO3 (aq) and extracted with EtOAc (3 x 100 ml). The orgarlic
layers were combined, washed with H2O (25 ml) and brine (25 ml),
25 dried over Na2SO4, filtered and coricentrated to dryness to give the title
compound (2.55 gm, 57% yield) as a white foam/solid.
TLC: Rf = 0.27 Silica GF (90/1011/1 of CH2cl2lMeoHlH2olHoAc) PMR: Consistent with structure
(lS)-I '-(((2-endo-aminoethyl-7,7-dimethyl-2-exo-
hydroxybicyclo-(1.2. 1 )-hept- I -yl)methyl)sulfonyl)-spiro( I H-indene-
1,4'-piperidine) (6û mg. 0.135 mmol) was dissolved in CH2C12 (I ml),
treated with 4-iodobenzoyl chloride (3~.0 mg, 0.135 mmol) and Et3N
_ . . . ..
WO 95114025 PCTNS94113483
2l7~sa
- 122-
to adjust the pH to 9.5. The reaction was stirred 30 minutes at 25C and
flash chromatograplhed on silica gel (8% Et2O in CH2C12) to give the
title compound (45 mg, 50% yield) as a white foam from Et2O (m.p.
98-138C, shrinks).
TLC: Rf = 0.35 Silica CF (10% Et20 in CH2cl2)
PMR: Consistent ~lith structure
HPLC: 95.8% pure
M.S.: (FAB) M + H + Thioglycerol = 783
o Analysis calculated for C32H3gIN2O4s0 30c4Hloo;o 35 H2O
C, 56 70; H, 6.12; N, 3.98
Found: C, 56 73; H, 6.11; N, 4.01
EXAMPLE 48
~`0~
= OH A
NH~
2S O
(lS-exo-N-[2-[2-hy~roxy-7,7-dimethyl-1 -[(spiro[lH-indene-l ,4'-
piperidin] I '-ylsulfonyl)methyl]bicyclo[2.2. 1 ]hept-2-yl]ethyl]-cyclo-
propanecarboxamide
The title compound was prepared according to the
30 procedure of Exarnple 47 except that cyclopropylcarbonyl chloride
(12.4 ml, 0.135 mmol) was s~-h~tih-t~-d for 4-iodobenzoyl chloride.
Flash chromatography of the reaction mixhure on silica gel
(15% Et2O in CH2C12) gave the title compound (36.6 mg, 53% yield)
as a white foam from Ether (m.p. 93-105C shrinl; + foam).
t
WO 95114025 PCrlUS94113483
21 74~50
- 123 -
TLC: Rf= 0.22 Silica GF (15% Et2O in CH2cl2)
PMR: Consistent with Structure
HPLC: 96.8%
- M.S.: (FAB) M + H + Thioglycerol = 621
Analysis calculated for Calc'd for C2gH40N2O4S0.20C7H1oO
C, 67.84; H, 8.03; N, 5.31
Found: C, 67.69; H, 8.06; N, 5.06
EXAMPLE 49
52~ OH
N~
(lS-exo)-N-[2-[2-hydroxy-7,7-dimethyl-1-[(spiro[lH-indene-1 ,4'-
piperidin[- I '-ylsulfonyl)Methyl]bicyclo[2.2. 1 ]hept-2-yl]ethyl] -2,2-
dimethyl -propanamide
The title compound was prepared according to the
25 procedure of Example 47 except that trimethyl acetyl chloride (16.6 ml7
0.135 mmol) was s~lb~titllt~d for 4-iodobenzoyl chloride.
Flash chromatography of the reaction on silica gel (13%
Et2O in CH2C12) gave the title compound (32.8 mg, 46% yield) as a
white foam from Et2O (m.p. 78-104C, shrink + foam).
TLC: Rf = 0.2~ Silica GF (15% Et2O in CH2cl2)
PMR: Consistent with structure
HPLC: 97.6%
M.S.: (FAB) M + H + Thioglycerol = 637
Analysi~ calculated for C30H44N2O4S0.20C4HIoO0.30 H2O =
WO 95114025 PCTIIJS94/13483
2~ 74~
- 124 -
C, 67.38; H, 8.56; N, 5.10
Found: C, 67.37; H, 8.52; N, 5.02
EXAMPLE 50
SO2
OH ~ ~NOz
HN~
(IS-exo)-N-[2-[2-hydroxy-7,7-dimethyl-1-[(spiro[lH-indene-1,4'-
piperidin] - I '-ylsulfonyl)methyl]bicyclo[2.2. 1 ]hept-2-yl]ethyl] 4-
methoxy-benzamide ~._
The title compound was prepared according to the
20 procedure of Exam]?le 47 except that 4-nitrobenzoyl chloride (25.1 mg,
0.135 mmol) wa~ sllhstitl-t~d for4-iodobenzoyl chloride.
Flash chromatography of the reaction on silica gel (10%
Et2O in CH2C12) ga~e the title compound (41.6 mg, 52% yield) as a
white foam from Ether (m.p. 11 8-33C, shrink + foam).
TLC: Rf= 0.26 Silica GF (10% Et2O in CH2C12).
PMR: Consistent with structure
HPLC: 97.1%
M.S.: (FAB) M + ~1 + Thioglycerol = 702.
30 Analysis ~ t~d for C32H3gN3O6S0.30C4H10O0.20 H20
C, 64.35; H, 6.90; N, 6.7
Found: C, 64.32; H, 6.9~; N, 6.63
wo 9~/1402~ Pcrlus94ll3483
2~ 74~5~
- 125 -
EXAMPLE 51
OCH3
HN~
(IS-exo)-N-[2-[2-hydroxy-7,7-dimethyl-1-[(spiro[lH-indene-1 ,4'-
piperidin]-l'-ylsulfonyl)methyl]ethyl]bicyclo[2.2.1]hept-2-yl]ethyl[-4-
5 methoxy-benzamide
The title compound was prepared according
to the procedure of Example 47 except that p-anisoyl chloride (18.3 ml,
0.135 mmol) was s-lhstitllt.od for 4-iodobenzoyl chloride.
Flash chromatography of the reaction on silica gel (15 %
20 Et2O in CH2C12) gave the title compound (36 mg, 46% yield) as a
white solid from Et20 (m.p. 98-121~C, shrink).
TLC: Rf= 0.26 Silica GF (15% Et2O in CH2C12)
PMR: Consistent with structure
HPLC: 955%
M.S.: M + H + Thioglycerol = 687
Analysis calculated for C33H42N20sS0.25 C4HloO
C, 68.36; H, 7.51; N, 4.69
Found: C, 68.14; H, 7.67; N, 4.55
WO 95/14025 PCIIUS94/13483
21 746~0
- 126-
EXAMPLE 52
HN~ NH~,~
O
(lS-exo)-1'-[[[2-[2-[[[(1 ,1 -dimethylethyl)amino]-carbonyl]amino]-
ethyl] -2-hydroxy-7,7-dimethylbicyclo-[2.2. 1 ]hept- I -yl]methyl] -
sulfonyll-spirorlH-indene-1~4'-piperidinel
(IS)-I '-[[[endo-aminoethyl-7,7-dimethyl-exo-hydroxy-
bicyclo(2.2.1)-hept l-yl]methyl]sulfonyl]spiro(lH-indene-1,4'-
piperidine) (60 mg, 0.135 mmol) was dissolved in THF (2 ml), treated
with t-butyl isocyanate (15.4 ml, 0.135 mmol) and stirred 30 minutes at
25C
After removal of the solvent in vacuQ, the residue was flash
chromatographed on silica gel (25% Et2O in CH2C12 to give the title
compound (27.9 m~, 38% yield) as a white solid from Et2O (m.p. 97-
119C, shlink and foam).
TLC: Rf= 0.21 Silica GF (20% Et2O in CH2C12)PMR: Consistent with structure
HPLC: 95 1%
M.S.: (FAB) M + 11 + Thioglycerol = 652
Analysis calculated for C30H4sN3O4S0.25C4H10O0.30 H2O
C, 65.58; H, 8.54; N, 7.40
Found: C, 65.70; H, 8.74; N, 7.05
WO 95/14025 PCrrUS94rl3483
21 74650
- 127-
EXAMPLE 53
5 ~
~; ~
~`S~O
O ~No~~
To a stirred solution of 10 ml of dry N,N-dirnethyl-
formamide Gonts~inin~ endo(lS)-1'-(((2-amino-7,7-dimethyl-
bicyclo(2.2.1) hept-1-yl)methyl)sulfonyl)-spiro(lH-indane-1,4'-
piperidine) (670 mg, 1.66 mmole) and Na-tert-butyloxycarbonyl-5,5-
dimethyl-L-thiazolidine-4-carboxylic acid (500 mg, 1.91 mmole) was
added 932 mg (2.11 mmole) of be~ ol-l-yloxytris (dimethyl-
amino)phosphonil~m hexafluorophosphate. The pH of the reaction
mixture was adjusted to 8.~ with diisopropylethylamine and the reaction
2s mixture was stirred at 23C overnight. The reaction mixture was
filtered and concentrated under reduced pressure. The residue was
dissolved in ethyl acetate and this solution was washed in succession with
saturated sodium bicarbonate solution (3 X 25 ml) and brine. The
organic phase was dried (sodium sulfate) and concentrated to yield a
3 brown solid which was purified by chromatography on silica gel
(chloroform-methanol-concentrated ammonium hydroxide elution,
98:2:0.2 v/v) to yield a white solid. This material was dissolved in 10
ml of chloroform. The solution was cooled in an icebath and treated
dropwise with a solution of 5 ml of chloroforrn containing 2.2
wo 9~/14025 PCr~S94/13483
211~6~0
- 128 -
equivalents of m-chloroperoxy-benzoic acid. After addition was
complete the ice bath was removed and stirring was continued for 24
hours. The reactioll mixture was diluted to 100 ml with choroform and
was washed with lhl sodium hydroxide solution (2 X 30 ml) and brine.
5 The organic phase was dried (sodium sulfate) and concentrated under
reduced pressure. The crude product was purified by chromatography
on silica gel (chloroform-methanol-concentrated ammonium hydroxide
elution, 98:2:0.2 v/~) to yield the title compound:
o NMR: Consistent with structure and verifies presence of solvent;
HPLC: >94% pure at 214 nM;
FAB MS: 678 (M+ + H);
Elem Analysis calculated for C34HslN3O7S2-0.8CHC13
~, 54.04; H, 6.75; N, 5.43
Found: ~, 54.11; H, 6.64; N, 5.48
EXAMPLE 54
~2
2s 1 ~ ~
O~NH2
HN ~ ~N
COOCH
WO 95/14025 PCr/US94113483
21 74650
- 129-
Endo(lS)-1 '-(((2-amino-7,7-dimethylbicyclo(2.2.1)hept-1-
yl)methyl)sulfonyl)spiro(l H-indane- I ,4'-piperidine)(660 mg, 2.05
mmole) and (S)-3-[(tert-butyloxycarbonyl)amino]-2-oxo-1-pyrrolidine-
(R)-2-methylcarboxysuccinic acid (826.5 mg, 2.05 mmole) were
combined with 392 mg (2.05 mmole) of 1-ethyl-3-(3-dimethylamino-
propyl) carbodiimide hydrochloride and 275 mg (2.05 mmole) of 1-
hydroxybenzotriazole hydrate in 20 ml of dry methylene chloride at
room ~ eldLul~ under nitrogen. The pH of the reaction mixture was
adjusted to 8.5 with triethylamine and the resulting solution was stirred
o for 12 hours. The reaction mixture was diluted with methylene
chloride (150 ml) and the resulting solution was washed with saturated
sodium bicarbonate solution (2 X 40 ml), 10% citric acid solution (2 X
40 ml), and brine, then dried (m~gn~ m sulfate) and concentrated to
give the crude product. The analytically pure material was obtained via
preparative HPLC chromatography employing a Vydac C-18 column
(4.5 X 150 mm, water-acetonitrile-1% trifluoroacetic acid 45 minute
gradient). The homogeneous fractions c--nt~inin~ product were pooled
and cullc~ d. The residue was dissolved in methylene chloride
containing trifluoroacetic acid (50%)at room temperature. After 2
hours, the volatiles were removed under reduced pressure to yield the
title compound as a trifluoroacetate salt:
NMR: Consistent with structure and confirms presence of solvent;
HPLC: > 97% pure at 214 nm;
FABMs:6l5(M++H);
Elem Analysis calculated for C34H47F3N4OgS-0.3 H20-0.8TFA:
C, 51.59; H, 5.88; N, 6.74.
Found: C, 51.59; H, 5.89; N, 6.81.
WO 95/~4025 PCTIIIS94/13483
217~0
- 130 -
EXAMPLE 55
o~,7
HN--~--I`NH~.~O~
o
Endo-( I S)-l '-(((2-amino-7,7-dimethylbicyclo(2.2. 1 ) hept-
20 1-yl)methyl)sulfon~ll)spiro(lH-indane-1,4'-piperidine)(216 mg, 0.53
mmole) and Na-terl-butyloxycarbonyl-D~L-(3-thienyl)alanine (160 mg,
0.59 mmole) were combined with 113 mg (0.59 mmole) of 1-ethyl-3-
(3-dimethylaminop~opyl) carbodiimide hydrochloride and 79 mg (0.59
mmole) of l-h~dlu~y~ lz~ vle in 6 ml of dry methylene chloride at
25 room te,l.l,~.a~u~ mder nitrogen. The pH of the reaction mixture was
adjusted to 9 with triethylamine and the resulting solution was stirred
for 12 hours. An additional 20 mg of tert-butyloxycarbonyl-D,L-(3-
thienyl)alanine, 11 mg of 1-ethyl-3-(3-dimethylaminopropyl)-
carbodiimide hydrochloride were added and stirring was continued for
30 6 hours more keeping the pH of the medium between 8 and 9. The
reaction mixture wa.~ diluted with methylene chloride (150 ml) and the
resulting solution was washed with saturated sodium bicarbonate
solution (2 X 40 ml), 10% citric acid solution (2 X 40 ml), and brine,
then dried (magnesium sulfate) and concentrated to give the crude
product. The analytically pure material was obtained via flash column
WO 95/14025 PCT/US94/13483
21 7~50
- 131 -
chromatography on silica gel (chloroform-methanol-concentrated
ammonium hydroxide elution, 95:5:0.5 v/v):
NMR: Consistent with structure and confirms presence of solvent;
5 HPLC: > 99% pure at 214 nm;
FAB MS: 656 (M+ + H);
Elem Analysis calc'd for C3sH49N3sS2 -5 H2O:
C, 63.22; H, 7.58; N, 6.32.
Found: C, 63.26; H, 7.54; N, 6.10.
EXAMPLE 56
15 ~J
S
CO2CH2CH~
Endo( I S)- I '-(((2-amino-7 ,7-dimethylbicyclo(2.2. 1 ) hept- I -
30 yl)methyl)sulfonyl)spiro(lH-indane-1,4'-piperidine)(67 mg, 0.156
mmole) and ~I-ethyl-carboxymethyl-5,5-dimethyl-L-thiazolidine-4-
carboxylic acid (57 mg, 0.23 mmole) were combined with 44 mg (0.23
mmole) of l-ethyl-3-(3-dimethylaminopropyl) carbodiimide
hydrochloride and 31 mg (0.23 mmole) of l-hydroxybenzotriazole
wo 95/1402s PCrlUS94113483
.
- 132 -
hydrate in 5 ml of dry methylene chloride at room temperature under
nitrogen. The pH of the reaction mixture was adjusted to 8.5 with
triethylamine and the resulting solution was stirred for 3 hours. The
reaction mi~ture was diluted with methylene chloride (50 ml) and the
5 resulting solution was washed with saturated sodium bicarbonate
solution (2 X 40 ml), 10% citric acid solution (2 X 40 ml), and brine,
then dried (m~gJn~ m sulfate) and concentrated to give the crude
product. The analytically pure material was obtained via preparative
thick layer chromatography (2 X 20 X 20 mm, pre-coated SiO2 plates,
o hexane-ethyl acetate elution, 2:1 v/v). The analytical product was
isolated in homogeneous form as a solid:
NMR: Consistent with structure and confirms presence of solvent;
FAB MS: 660 (M+ + H);
Elem. Analysis calc'd for C34H47N3O6S2-0.85 H2O-O.lEtOAc:
C, 60.40; H, 7.59; N, 6.14.
Found: C, 60.37; H, 7.36; N, 6.13.
lEXAMPLE 57
`T'
H~
.
-
WO9~/14025 21 i~b~ PcTruss4rl34s3
- 133 -
To a stirred solution of 50 ml of dry N,N-dimethyl-
formamide containing endo(lS)-1'-(((2-amino-7,7-dimethyl-
- bicyclo(2.2. 1 ) hept- I -yl)methyl) sulfonyl)spiro(1 H-indane- 1,4'-
5 piperidine) (4.0g, 9.9 mmole) and Na-tert-butyloxycarbonyl-L-
methioninesulfone (2.84 g, 11.9 mmole) was added 5.65 g (12.87
mmole) of benzotriazol-l-yloxytris(dimethylamino) phosphonium
hexafluorophosphate. The pH of the reaction mixture was adjusted to
8.5 with diisopropylethylamine and the reaction mixture was stirred at
o 23C for two hours. The reaction mixture was filtered and
concentrated under reduced pressure to give a solid. This material was
dissolved in ethyl acetate and this solution was washed in succession with
saturated sodium bicarbonate solution (3 X 25 ml), 10% citric acid
solution (3 X 25 ml) and brine. The organic phase was dried (sodium
5 sulfate) and concentrated to yield 2-tert-butyloxycarbonylamino-N-[I-
[[(2,3-dihydrospiro[lH-indene-1,4'-piperidin]-1'-yl)sulfonyl]methyl]-
7,7-dimethylbicyclo[2.2. 1 ]hept-2-yl]-4-(methylsulfonyl)-k~ n~mi~
which was purified by chromatography on silica gel (chloroforrn-
methanol-concentrated ammonium hydroxide elution, 95:5:0.5 v/v) to
20 yield a solid. This material was dissolved irl 50 ml of ethyl acetate and
the resulting solution was cooled to 0C and treated with a continuous
stream of hydrogen chloride gas for 15 minutes. The ice bath was
removed, the reaction vessel was capped, and stirring was continued for
45 minutes more at room ~ lp~ldtul~. The solvent and excess
25 hydrogen chloride were removed under reduced pressure to give 2-
amino-N-[ I -[[(2,3-dihydrospiro[ I H-indene- I ,4'-piperidin]- I '-yl)-
sulfonyl]methyl]-7,7-dimethyl-bicyclo[2.2. 1 ]hept-2-yl]-4-(methyl-
sulfonyl)butanamide hydrochloride as an off-white solid (6.5 g).
wo 9S/14025 PCrlUs94ll34S3
2~
- 134-
~o~ i \ 0~,0
HN _~/
N
o
2-arnillo-N-[1-[~(2,3-dihydrospiro[lH-indene-1,4'-
piperidin]-1'-yl)sulfonyl]methyl]-7,7-dimethylbicyclo[2.2.1]hept-2-yl]-
4-(methylsulfonyl)-butanamide hydrochloride was chromatographed on
20 silica gel (chloroform-methanol-concentrated ammonium hydroxide
elution, 90:10:1 v/~/) to afford a white solid which was dissolved in 15
ml of methanol cf-n~:linin~ 1% acetic acid. To this reaction mixture wa~
added 3 ml of aqueous formaldehyde solution and 1.56 g (24.8 mmole)
of sodium cyanoborohydride. The resulting reaction mixture was
25 stirred at room ~ ueldLu~ overnight. The reaction mixture was
concentrated in vacuo and the residue was partitioned between ethyl
acetate and saturated sodium bicarbonate solution. The phases were
separated and the organic phase was washed with brine, dried (sodium
sulfate), and conc~ntrated under reduced pressure. The crude product
3~ was purified by ~ ul~latography on silica gel (chloroform-methanol-
concentrated ammonium hydroxide elution, 95:5:0.5 v/v) to yield 2-
dimethylamino-N-[1-[1(2,3-dihydrospiro[lH-indene-1,4'-piperidin]-1'-
yl)sulfonyl]methyl]-7,7-dimethylbicyclo[2.2. 1 ]hept-2-yl] -4-(methyl-
sulfonyl)bllt~n~mi~
WO95/14025 PCI/US94/13483
21 74650
- 135 -
NMR: Consistent with structure and verifies presence of solvent;
HPLC: >98% pure at 214 nM;
FAB MS: 594(M+ + H);
Elem. Analysis calc'd for C30H47N3OsS2-0.1CHC13:
C, 59.67; H, 7.84; N, 6.94.
Found: C, 59.90; H, 7.24; N, 6.93.
~XAMPLE 58
~X~
N
0,~
OH
HN~ ~N b,O~¦
To a stirred solution of (IS)-1'-(((2-exo-hydroxy-2-endo-
aminomethyl-7 ,7-dimethylbicyclo(2.2. 1 )hept- l -yl)methyl)sulfonyl)-
spiro(lH-indene-1,4'-piperidine) (300 mg, 0.694 mmole) in 10 ml of
dry, degassed N,N-dimethylformamide was added Na-tert-butyloxy-
carbonyl-S-methyl-L-cysteine (212 mg, 0.832 mmole) and 398 mg
(0.902 mmole) of benzotriazol-l-yloxy tris(dimethylamino)-
phosphonium hexafluorophosphate (BOP) at room I~ UI~. The
resulting reaction mixture was protected from moisture and the pH wa~
adjusted to 8-9 with diisopropylethylamine. After 24 hours all volatile
wo 95/14025 Pcr/usg4/l3483
2 ~ a
- 136-
components were removed under reduced pressure and the residue was
partitioned between ethyl acetate and sodium bicarbonate solution. The
phases were separated and the organic phase was washed with saturated
sodium bicarbonate solution (3 X 40 ml) and brine, then dried
5 (m:l~nP~illm sulfate) and concentrated. The crude product was
chromatographed on silica gel (hexane-ethyl acetate elution, 3:1 v/v) to
give the title compound:
NMR: Consistent with structure and verifies presence of solvent;
o HPLC: >98% pure at 214 nM;
FAB MS: 650 (M+ ~ H);
Elem. Analysis calc'd for C33H50N3o6s~:
C, 61.07; H, 7.71; N, 6.47.
Found: C, 61.24; H, 7.98; N, 6.13.
EXAMPLE 59
~
2s lsJz,~
O
HN ~NH
~<
O HN~,
o
To a stirred solution of (lS)-1'-(((2-exo-hydro~y-2-endo-
aminomethyl-7,7-dimethylbicyclo(2.~.1 )hept- I -yl)methyl)sulfonyl)-
spiro(lH-indene-1,1'-piperidine) (125 mg~ 0.311 mmole) in 7 ml of
WO 9511402~ PCr/lJS94/13483
21 74~5~
- 137 -
dry, degassed N,N-dimethylformamide was added S-hydantoinacetic
acid (59 mg, 0.372 mmole) and 178 mg (0.404 mmole) of benzotriazol-
1-yloxy tris(dimethylamino)pho~ ulliu ll hexafluorophosphate (BOP)
at room ~ e~dlu~. The resulting reaction mixture was protected
5 from moisture and the pH was adjusted to 8-9 with diisopropyl-
ethylamine. After 24 hours all volatile components were removed
under reduced pressure and the residue was partitioned between ethyl
acetate (100 ml) and sodium bicarbonate solution. The phases were
separated and the organic phase was washed with saturated sodium
bicarbonate solution (3 X 40 m]) and brine, then dried (magnesium
sulfate) and concentrated. The crude product was initially flash
chromatographed on silica gel (chloroform-methanol elution, 96:4, v/v)
to give semi-pure material from which the title compound was obtained
as a white solid after preparative thick layer chromatography on silica
15 gel (chloroforrn-methanol-concentrated :~mmonillm hydroxide elution,
96:4:0.4, v/v) and trituration with ether-petroleum ether:
NMR: Consistent with structure and verifies presence of solvent;
FAB MS: 543 (M+ + H);
Elem. Analysis calculated for C28H48N4O5S
C, 61.96; H, 7.00; N, 10.32
Found: C, 61.70; H, 7.36; N, 10.09
wo 95114025 Pcrlusg4ll3483
2 1 74~0
- 138 -
EX~MPLE 60
10 \ ~
¦ H
HN
15 o~ N~/~
N
0~0~
(1S)-1~-(((7,7-dimethyl-2-endo-(4-nitrophenyloxycarbonylamino)-
bicyclo-(2.2.1)-hept-1 -yl)-methyl)-sulfonyl)spiro(lH-indan-l ,4'-
~iperidine
(lS)-1'-(((7,7-dimethyl-(2-~n~ mino)-bicyclo(2.2.1)-hept-
I-yl)-methyl)sulfonyl)spiro(lH-indene-1,4-piperdine)[3.47mmol] and 4-
Nitrophenyl chloro~ormate [3.64mrnol] were combined in THF. The
reaction mixture W~IS treated with triethylamine [4.54 mmol] and
allowed to stir for 3 hours. The reaction mixture was concentrated to
dryness and the reslulting residue was purified by a silica gel column,
while eluting with l % ethylacetate in methylene chloride. The product
fractions were combined and concentrated to dryness in vacuo. (IS)-I'-
(((7,7-dimethyl-(4-~itrophenyloxycarbonyl-2-endo~minr )-bicyclo-
(2.21 )-hept- I -yl)-miethy~)-sulfonyl)spiro( I H-indene- 1 ,4'-piperidine was
obtained as a white solid from ether.
(lS)-I '-(((7,7-dimethyl-2-endo-(4-nitro-phenyloxy-
carbonylamino)-bicicylo-(2.2.1)-hept-1-yl)-methyl)sulfonyl)spiro(ll~-
wo 95/14025 Pcr/usg4/l3483
21 74~
- 139-
indene-1,4'-piperdine)-[0.278mmol] and (benzyloxycarbonyl) piperazic
acid [0.334mmol] were combined in DMF. The reaction mixture was
treated with triethylamine [0.401mmol] and allowed to stir for 2 hours.
The reaction mixture was concentrated to dryness and the resulting
5 residue was dissolved in CH2cl2. This solution was placed on a silica
gel column and eluted with 5% methanol in CH2cl2 and then with
96/4/0.4 of CH2C12/methanol/acetic acid. The product fractions were
combined and evaporated to dryness. The title compoumd was obtained
as a white solid from ether and was dried in vacuo overnight.
m p.: 9o-l2ol~c
NMR: Consistent with structure
HPLC: >98% Pure
MS: M-->H+=693.6
Analysis calculated for C37H4gN4O7S-0.20mol
C4HloO-0 25mol H20
C, 63.74; H, 7.15; N, 7.87
Found: C, 63.78; H, 7.08; N, 7.81
EXAMPLE 61
HN
~ N ~O
~VO 95/14025 PCr/US94113483
2174650
- 140-
The procedure of Example 60. second paragraph was
carried out using (lS)-1'-(((7,7-dimethyl-2-endo-(4-nitrophenyloxy-
carbonylarnino)-bicyclo-(2.2. 1 )-hept- 1 -yl)-methyl)sulfonyl)spiro( 1 H-
indan- 1 ,4'-piperidine)[0. 1 93rnmol], triethylamine [0.29mmol~, and
5 ~ "l;"g 3-(t-butoxycarbonylarnino-methyl)piperidine [0.212mrnol]
for (benzyloxycarbonyl) piperazic acid. Chromatographic elution was
with 5% ether in CH2C12, 10% ether in CH2cl27 and then 1% methanol
in CH2C12. The title compound was obtained from ether and dried in
vacuo, overnight.
o m p 95-105C
NMR: Consistent with structure
HPLC: >97% Pure
MS: M+H+=643.4
Analysis calculated for C3sHs4N4OsS-0 20 H2O
C, 65~2; H, 8.48; N, 8.67
Found: C, 64.~8; H, 8.43; N, 8.86
WO 95J140~5 PCr/US94/13483
2174~50
- 141 -
EXAMPLE 62
Endo-( I S)- I '-(((2-(L-4(tert,butoxycarbonylamino)glutaramyl)amino-
7,7-dimethylbicyclo(2.2. 1 )hept- I -yl)-methyl)-sulfonyl)spiro-( I H)-
indan- I .4'.piperidine
N r7`
~
H '~NH2
In an oven dried flask (50ml) under nitrogen was dissolved
endo-(lS)-1'-(((2-amino-7,7-dimethyl-bicyclo(2.2.1)hept-1 -yl)-methyl)-
sulfonyl)spiro-(lH)-indan-1,4'-piperidine (50mg, 0.125mmol), L-
4(tert-butoxycarbonyl)glutaramic acid (34mg, 0.14mmol), 1-ethyl-3-(3-
dimethyla..li-.op.vl yl)carbodiimide hydrochloride (30mg, 0.15mmol),
and l-hydroxybenz-triazole hydrate (20mg 0.15mmol) in dimethyl
formamide (Iml). Triethylamine (50ml) was added and a white solid
separated. After stirring for I hour, the solvent was removed under
reduced pressure. The residue was dissolved in ether:methylene
chloride (3:1). The cloudy solution was washed with sodium
bicarbonate (sat., aqueous), water, potassium hydrogen sulfate (10%,
aqueous) and brine. After drying over sodium sulfate, the organic
material was filtered and concentrated. Silica "flash" chromatography
using methylene chloride methanol (9:1) gave the product which was
isolated as an oil upon evaporation of the solvents. Hexane:ether
WO 9~i/140~i PCIIUS94/13483
2 l ~ 4 63
- 142-
addition and remov~ll under reduced pressure produced the title
compound as a white foam.
HPLC: >99%
MS: M+H+=63 1.3
NMR consictpnt with structure
Analysis calcula~ed for C33HsoN4O6S-0.5 DMF-0.5 H20
C, 61.26; H, 8.12; N, 9.32
Found: C, 6127; H, 8.05; N, 9.31
EXAMPLE 63
Endo-( l S)- I '-(((2-(4 (imidazole-2-ethylacetyl)amino-7,7 -dimethyl-
bicyclo(2.2. 1 )hept- l -yl)-methyl)spiro-( 1 H)-indan-( I ,4'),piperdine
1~ hydrochloride
N
2s H ,CH3
CH -
HN J 2
0
Step 1: Endo-( I S)- I '-(((2-4(3-methylbenzyloxy)imida~ole)-2-
[ethyl acetyl)amino-7,7-dimethylbicyclo- (2.2.1 )hept- I -
vl)-methyl)-sulfonyl)spiro( I H)-indan-( I .4')-pi~eridine
Into an over dried flask (50ml) under nitrogen was placed
dimethylformamide (4ml). Endo( I S)- I '-((2-amino-7 ,7-dimethyl-
WO 95/140~5 PCT/US94/13483
21 ~46~
- 143 -
bicyclo(2.2.1)hept-1-yl)-methyl)sulfonyl)spiro(lH)-indan-(1,4')-
piperidine (300mg,0.74mmol), 4(3-methylbenzyloxy)imidazole-2-
ethylacetic acid (219mg, 0.8mmol), 1-ethyl-3-methyl-(3-dimethyl-
aminopropyl)carbodiimidehydrochloride-(153mg, 0.8mmol) and 1-
hydroxybenztriazolehydrate-(108mg, 0.83mmol) were added and the
mixture was stirred until solution was achieved. Triethylamine (400ml)
was added to ma~e the solution basic (pH 9) and a solid separated.
After stirring ovemight room l~,l,pe,d~ul~, t'ne solvent was removed
under reduced pressure. The resulting ~um was dissolved in
o methyenechloride:ether (1:3) and the cloudy solution was extracted with
sodium bicarboante (sat., aqueous) and brine. After drying the organic
layers over sodium sulfate, the solution was filtered and was
concentrated. The product was purified by silica gel "flash"
chromatography using methylene chloride, methanol, ammonium
hydroxide (9:1:1) as solvents. The product fractions were concentrated
to give the title compound as white foam.
HPLC: >95% (42% + 53%)
MS: M+H+=659.3
NMR (CDC13) cnn~ist.-nt with structure
Arlalysis Ç~ tPd for C3gHsoN404S-0.65H20
C,68.05; H,7.71; N,8.36
Found: C,68.03; H,7.87; N,8.60
2S Nitrogen was bubbled into a solution of Endo(lS)-1'-(((2-
4-(3-methylbenzyloxyimidazole)-2-ethylacetylamino-7 ,7-dimethyl -
bicyclo(2.2.1)hept-I-yl)-methyl)-sulfonyl)spiro-(1H)-indan-
(lm4')piperidine (50mg, 0.076mmol) in absolute ethanol (20ml)-acetic
acid (Sml). After 5 minutes, 10%Pd/C(SOmg) was added and the
mixture was hydrogentated at 55psi ovemight. The catalyst was
- removed by filtration and the solvents were concentrated to dryness
under reduced pressure. The residue was purified by silica gel "flash"
chromatography using methylenechloride, methanol,ammonium
hydroxide (9:1:1) as solvent. The product fractions were collected and
concentrated. The gum was redissolved in methylene chloride and wa.s
WO 95/14025 PCTIUS94/13483
21 74~
- 144 -
filtered through a sintered glass funnel. After the solution was
concentrated, the re.~idue was dissolved in ethyl acetate (5ml). The
solution was cooled to 0 and it was saturated with hydrogen chloride
gas (bubbling 5 minutes). The solvent was removed under reduced
5 pressure and ether was added and was removed to give the title
compound as a white solid.
HPLC: >90% (42% + 45%)
MS: M+H+=539.2 (freebase)
o NMR (CDC13) consistent with structure
Analysis calcula~ed for C30H42N4O3S-HC1 0.25 ether- 1 30H2O
C, 60.33; H, 7.86; N-9.08
Found: C, 60.33; H, 7.48; N-9.04
EXAMPLE 64
H H
HN~N~
o
3 SO2CH~;
To a stirred solution of the product of Example A (500 mg.
1.24 mrnol) in DMF (10 mL) was added the HCI salt of 2-(1-benzyloxy-
methylimidazoly-5-yl)-4-methylsulfonylbutanoic acid (506 mg; 1.30
WO 95/14025 PCrllJS94113483
21 74650
- 145-
mmol), DIEA (0.70 mL; 4.0 mmol), and BOP (600 mg; 1.35 mmol).
After being stirred at ambient temperature for 14 h, the solvent was
removed under reduced pressure. The residue was dissolved in EtOAc
(75 mL) and washed with aqueous NaHC03 (3 x 50 mL). The or~anic
5 phase was dried (MgS04), filtered, and the solvent was removed under
reduced pressure. The benzyloxymethyl protecting group was removed
by dissolving the residue in a 4:1 solution of EtOH-HOAc (10 mL) and
hydrogenating over palladium black (75 mg) under I atm of hydrogen
for 24 h. The catalyst was removed by filtration through Celite and the
o filtrate solvents were removed under reduced pressure. The residue
was purified by pressurized silica gel column chromatography using a
50:50:7 by volume mixture of CHC13, EtOAc, and 4% NH40H-MeOH
as eluant. Two diastereomers were obtained and each was Iyophilized
from water containing 1% TFA. The title compound is the lower Rf
15 isomer.
Higher Rf Isomer:
Analysis calculated for (C31H44N40sS2)-1.0 TFA-l.l H20
C, 52.45; H, 6.24; N, 7.37
20 Found: C, 52.69; H, 6.12; N, 7.32
TLC: Rf 0.33 (50:50:7 CHC13:EtOAc:4%NH40H-MeOH)
HPLC (method A): retention time 8.96 min
FABMS:m/z617(M++H)
25 IH NMR (300 MHz, CDC13): free base: d 7.70 (s, lH), 7.55 (br d, lH),
7.15-7.25 (m, 4H), 7.00 (s, IH), 4.35 (m, lH), 2.90 (s, 3H), 1.00 (s,
3H), 0.98 (s, 3H)
Lower Rf Isomer:0 Analy~is calculated for (C31H44N405S2) 1 0 TFA-1-3 H20
C7 52.55; H 6.36; N 7.43
Found: C. 52.89; H 6.13; N 7.43
TLC Rf 0.28 (50:50:7 CHC13:EtOAc:4%NH40H-MeOH)
HPLC (method A): retention time 9.15 min
wo 9511402~ Pc~!r/Usg41l3483
~ 1-/ 4 ~
- 146 -
FAB MS: m/z 617 (M+ + H)
IH NM~ (300 MHz, CDC13): free base: d 7.72 (s, IH), 7.34 (br d, lH),
7.15-7.25 (m, 4H), ~.98 (s, IH), 4.35 (m, IH), 2.90 (s, 3H), 1.00 (s,
3H), 0.96 (s, 3H)
LXAMPLE 65
S~
H
HN
~,NH2
O ~
SO2CH3
To a 0C stirred solution of the product of E~ample A
(1.10 g; 2.74 mmol) in CHC13 (100 mL) was added DIEA (0.75 mL;
4.3 mmol) and benzyl chloroformate (0.51 g; 3.0 mmol). The solution
was stirred at 0C for 1 h and then at ambient ~ eldlul~ for 14 h.
The reaction mixtule was concentrated under reduced pressure and the
residue was dissolved in 25 mL of a solution of 10;1 MeOH-NH40H.
The mixture was concentrated under reduced pressure, the residue was
dissolved in CHCI~ (100 mL) and washed with 5% aqueous
HCI (2 x 50 mL) a~d aqueous NaHC03 (100 mL). The organic phase
was dried (MgS04), filtered, and the solvent was removed urlder
reduced pressure. The residue was purified by pressurized silica gel
WO 9511402~ PCI~/US94/13483
21 74~5~
- 147-
column chromatography using 1:4 EtOAc-hexanes as eluant. The
benzyl urethane was obtained as a white foam.
TLC: Rf 0.40 (1:3 EtO~r h~Y~n~s)
5 HPLC (method A): retention time 12.25 min
FAB MS: m/z 537 (M+ + H)
To a 0C stirred solution of the benzyl urethane (1.25 g;
2.33 mmol) in DMF (20 mL) was added iodomethane (0.435 mL; 7.00
mmol) and sodium hydride (0.140 mg of a 60% dispersion in mineral
oil; 3.50 mmol). The solution was stirrred at 0C for 1 h and then at
ambient temperature for 18 h. The reaction mixture was treated with
HOAc (1 mL) and the solvents were removed under reduced pressure.
The residue was dissolved in EtOAc (100 mL) and washed with aqueous
NaHCO3 (2 x 50 mL). The organic phase was dried (MgSO4), filtered,
15 and the solvent was removed under reduced pressure. The residue was
purified by plcs~u~ d silica gel column chromatography using 1:5
EtOAc-hexanes as eluant. The N-methyl benzyl urethane was obtained
as a white foam.
TLC: Rf 0.34 (1:4 EtOAc:hexanes)
20 HPLC (method A): retention time 12.71 min
FAB MS: m/z 551 (M+ + H)
To a stirred, argon purged solution of the N-methyl benzyl
urethane (1.00 g; 1.82 mmol) in 96:4 MeoH-Hco2H (25 mL) was
added p~ lm black (0.37 g). The reaction mixture was stirrred for
25 16 h at ambient te.ll~e.~lul~. The catalyst was removed by filtration
through Celite, and the filtrate solvents were removed under reduced
pressure. The residue was purified by pressurized silica gel column
chromatography using 95:5:0.5 CHC13:MeOH:NH40H as eluant. The
N-methyl endo-amine product was obtained as a white foam.
3 TLC: Rf 0.35 (92:~:0.8 CHC13:MeOH:NH40H)
HPLC (method A): retention time 7.~8 mir
FAB MS: m/z 417 (M+ + H)
To a stirred solution of the N-methyl endo-amine (0.650 g;
1.56 mmol) in CHC13 (~0 mL) was added the acid fluoride of Na-Boc-
WO 95/14025 PCT/US94/13483
b'3~
- 148 -
L-methionine sulfolIe (510 mg; 1.80 mmol) and DIEA (0.35 mL; 2.0
mmol). The mixtul-e was stirred at ambient temperature for 48 h, and
then extracted with 10% aqueous citric acid solution (2 x 30 mL), water
(30 mL), and aqueous NaHCO3 (2 x 30 mL~. The organic phase was
5 dried (MgSO4), filtered, and the solvent was removed under reduced
pressure. The residue was dissolved in CH2C12 (10 mL), and to the
solution was added TFA (6 mL). The mixture was stirred at ambient
U~ UI~ for 1.5 h. The solvents were removed under reduced
pressure and the residue was purified by preparative reverse phase
HPLC using a water-acetonitrile gradient containing 0.1% TFA. The
TFA salt of the title compound was obtained as a Iyophili~ed powder.
Analysis calculated for (C2gH4sN3OsS~ 0.4 TFA-0.5 EtOAc
C 57.0~ H 7.55 N 6.28
Found: C 57.0~ H 7.44 N 6.28
TLC: Rf 0.18 (95:5:0.5 CHC13:MeOH:NH40H)
HPLC (method A): retention time 9.80 min
FAB MS: m/~ 580 (M+ + H)
IH NMR (300 MH;~, CDC13): d 7.15-7.25 (m, 4H), 5.20 (m, 2H), 3.17
20 (s, 3H), 2.92 (s, 3H), 1.06 (s, 3H), 0.96 (s, 3H)
LX~MPLE 66
To a stirred solution of endo-(lS)-1'(((2-amino-7,7-
25 dimethylbicyclo(2 2.1 )-hept- I -yl)-methyl)-sulfonyl)spiro( I H-indan-
1,4'-piperidine (94 Ing; 0.17 mmol) in ethanol (20 mL) was added N-
(3-bromopropyl)-phth~limi~1P (69 mg; 0.255 mmol) followed by
diisopropylethyl amine (0.044 mL; 0.255 mm~ol). The temperature wa~
then increased to 50C. After i~ u~ lately 18 hr the ~olution was
30 cooled then concentrated under reduced pressure. The residue was
dissolved in methyl~ne chloride (150 mL), washed with IM HCI (2 x
150 mL) then dried over sodium sulfate and concentrated. Purification
by flash chromatography (5% methanol in methylene chloride) afforded
the title compound (45 mg; 40%) as a white solid.
WO 95/14025 PCT/US94/13483
2~ ~46~û
- 149-
Analysis ~ t~d for (C39H50N4O6S)-0-10 H20 0 15 CH2C12
C, 65.54; H, 7.10; N, 7.81
Found: C, 65.52; H, 7.09; N, 7.71
HPLC: (Vydac C18 Column; gradient from 95/5 to /100 H20/CH3CN
with 0.1% TFA. 15 min. gradient, flow rate = 1.5 ml/min.)
Rt= 13.81 min. Purity= 100%
IHNMR: Consistent with structure
FABMS: m/~ = 703 (M+ + H)
EXAMPLE 67
5 ~ >
S2 1~OH O O~,NH2
NH J~"N ~,~
Valine methyl ester (2.5g, 14.9 mmol), t-BOC-methionine
(3.73 g,l4.9 mmol), HBT (2.01 g, 14.9 mmol) and EDC (2.33g, 14.91
mmol) were suspended in methylene chloride (60ml). The pH of the
reaction was adjusted using E. Merck strips to about 9 with Et3N. After
stirring ovemight at ambient ~ a~UI~, the reaction was evaporated
and the residue was dissolved in ethyl acetate (60m~). The ethyl acetate
solution was extracted three times with 5M citric acid (20 ml), three
times with I M sodium bicarbonate (20 ml), one time with water and one
time with saturated sodium chloride (20ml). The ethyl acetate solution
was then dried over anhydrous sodium sulfate, filtered and evaporated
to dryness.
The residue was dissolved in methyl iodide (25ml) and
stirred overnight, and the resulting mixture was evaporated to dryness.
The resultin~ methyl sulfonium salt (4.72mmol) was dissolved in a 1:1
.... . . .. . . ... . . . . .. , ... ... , . .. , . . . .. . _
WO 9~i114025 PCrlUS94/13483
2~ 5~
- 150-
mixture of DMF and methylene chloride (150ml). After cooling to zero
degrees in a saltwater/ice bath, sodium hydride (60% in mineral oil:
0.57g, 9.44mmol) ~ivas added. After stirring for 3hr, the reaction was
treated with glacial acetic acid (I ml) followed by water (I ml). The
5 mixture was stirred for 15min. at ambient temperature, then
evaporated. The crllde product was dissolved in water (150 ml) and
neutralized with aqlleous sodium bicarbonate. The mineral oil and
neutral by-products were removed by extracting three times with
methylene chloride (50 ml). The aqueous layer was then acidified with
citric acid and extracted three times with methylene chloride (50ml).
The combined metJtlylene chloride layers were dried over sodium
sulfate, filtered and evaporated. Purification of the crude product was
carried out by prep~lrative HPLC using a Waters C-18 column in a
SepTech ST/Lab 800C ill~llUIll~lll with a water/acetonitrile gradient
15 increasing from lO'Yo to ca. 60% acetonitrile.
The pr~duct so obtained (60 mg, 0.20 mmol) was combined
with (lS)~ (((7,7-dimethyl-(2-endo-aminomethyl-2-exo-hydroxy)-
bicyclo-(2.2. 1 )-hept- I -yl)-methyl)sulfonyl)-spiro-( I H-indene- 1,4'-
piperidine) (86 mg, 0.20 rnmol), HBT (27 mg, 0.20 mmol) and EDC
20 (38 mg, 0.20 mmol) in methylene chloride (10 ml). The pH of the
mixture was adjust~d to ca. 9 with triethylamine. After stirring
overnight, the reaction was evaporated and the residue was dissolved
in ethyl acetate (25ml). The ethyl acetate solution was extracted three
times with 5M citric acid (2ml), three times with IM sodium
25 bicarbonate (2ml), ~ne time with water and one time with saturated
sodium chloride (2ml). The ethyl acetate solution was then dried over
anhydrous sodium sulfate, filtered and evaporated to dryness. The
product was dissol~ed in a 40% solution of trifluoroacetic acid in
methylene chloride (3 ml) and stirred for Ihr at ambient temperature.
30 The solvent was then evaporated, and the residue was dissolved in
methanol and puri~led by preparative HPLC using the conditions
described.
NMR: Consistent with structure
WO 95114025 PCTNS94/13483
21 74650
- 151 -
FAB MS: 613 (M+ + H)
HPLC: >99% pure at 214 nM
Analysis calc'd for C33H47N405 2.0 C2HF3o2 l.4H2o:
Calc'd: C, 51.31; H, 6.15; N, 6.47
Fourld: C, 51.31; H, 6.00; N, 6.66.
EXAMPLE 68
10 ~ "
SO2 ~~,~
NH2
To a solution of (IS)-1'-(((7,7-dimethyl-(2-endo-amino-
methyl-2-exo-hydroxy)-bicyclo-(2.2. 1 )-hept- I -yl)-methyl)sulfonyl)-
spiro(lH-indene-1,4"-piperdine (100 mg; 0.23 mmol) in DMF was
added t-boc-L-valine (60 mg; 0.28 mmol), I-hydroxybenzotriazole (37
mg; 0.28 mmol), and 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide
25 (53 mg; 0.28 mmol). The pH was adjusted to 9 with triethylamine (96
uL; 0.69 mmol). After stirring for 1.5 hrs., the solution was
con~erltr~l~d to dryness and treated with 5% citric acid solution. This
water layer was then basified with saturated sodium bicarbonate and
extracted 3 times with ethyl acetate. The organics were combined and
3C dried over sodium sulfate, filtered, and concentrated to yield a white
oily foam. This white foam wa~ dissolved in ethyl acetate, cooled to
0C and treated with saturated sollution of HCI in ethyl acetate. The
reaction mixture was stirred for 45 min., purged with N2 for 30 min.,
and then concentrated to dryness. The resulting solid was partitioned
between ethyl acetate and ~aturated ~odium bicarbonate. The a~lueous
WO 95114025 PCT/US94/13483
2 1 l4~0
- 152-
layer was extracted 3 times with ethyl acetate, and the combined organic
fractions were dried over sodium sulfate, filtered, and concentrated in
vacuo. The residue was puri~led by flash chromatography (eluted witll
98/2/0.2 of methylene chloride/methanol/ammonium hydroxide) to yield
5 the title compound as a white foam.
m.p.: not available
NMR: consistent ~vith structure
HPLC > 97% pure
0 MS MH-530.3 ~AB)]
CHN Analysis calculated for C29H43N3O4S
C, 65.75; H, 8.18; N, 7.93
Found: C, 65.75; H, 8.18; N, 7.93
EXAMPLE 69
2 ¦~OH o
~NH~
NH2
To a solution of (IS)-1'-(((7,7-dimethyl-(2-endo-amino-
methyl-2-exo-hydr~;)xy)-bicyclo-(2.2.1)-hept-1-yl)-methyl)sulfonyl)-
spiro(lH-indene-l,~"-piperdine) 51 mg; 0.12 mmol) in DMF was added
30 t-boc-L-leucine (36 mg; 0.14 mmol), I-hydroxybenzotraizole (19 mg;
0.14 mmol), and 1 -(3-dimethylaminopropyl)-3-ethylcarbodiimide (28
mg; 0.14 mmol). The pH was adjusted to 9 with triethylamine (50 ~LL,
0.36 mmol). After stirling for 18 hrs., the solution was concentrated to
dryness and treated with 5% citric acid solution. The water layer was
then basified with .saturated sodium bicarbonate and extracted 3 times
WO 9!5/14025 PCr/US94/13483
21 74651~
- 153 -
with ethyl acetate. The organics were combined amd dried over sodium
sulfate, filtered, and concentrated to yield a yellow oil. This yellow oil
was dissolved in ethyl acetate, cooled to 0C and treated with a saturated
solution of HCl in ethyl acetate. The reaction mixture was stirred for
5 45 min., purged with N2 for 30 min., and then concentrated to dryness.
The resulting solid was partitioned between ethyl acetate and saturated
sodium l,:cd~ atc the aqueous layer was extracted 3 times with ethyl
acetate and the combined organic fractions were dried over sodium
sulfate, filtered, and concentrated in vacuo. The residue purified by
flash chromatography (eluted with 98/2/0.2 of methylene
chioride/methanol/qmmnnillm hydroxide) to yield the title compound as
a white foam.
m.p.: 88 - 110 C
15 NMR: consistent with structure
HPLC: > 98% pure
MS: MH-544.3 (FAB)
CHN Analysis cq1~ 1l1qt~d for C30H4sN304SØ35H20:
C, 6550; H, 8.37; N, 7.64
Found: C, 65.48; H, 8.39; N, 7.60
EXAMPLE 70
SO2 ~OH O O~ NH2
3 0 ~ NJ~
To a ~olution of (lS)-1'-(((7,7-dimethyl-(2-endo-amino-
methyl-2-exo-hydroxy)-bicyclo-(2.2. 1 )-hept- I -yl)-methyl)sulfonyl)-
spiro(lH-indene-1,4"-piperdine) (72 mg; 0.17 mrnol) in DMF was
WO 95/14025 PCTIIIS94/13483
21 74~50
- 154 -
added 2-(3-amino-2-oxopyrrolidin-1-yl)acetic acid (52 mg; 0.20 mmol),
1-hydroxybenzotria~ole (27 mg; 0.20 mmol), and 1-(3-dimethyl-
aminopropyl)-3-ethylcarbodiimide (38 mg; 0.20 mmol). The pH was
adjusted to 9 with triethylamine (70 IlL; 0.50 mmol). After stirrin~ for
5 18 hrs., the solution was concentrated to dryness and treated with 5%
citric acid solution. The water layer was then basified with saturated
sodium bicarbonate and extracted 3 times with ethyl acetate. The
organics were combined and dried over sodium sulfate, filtered, and
concentrated to yield a yellow oil which was purified by flash
o chromatography (eluted with 98/2/0.2 of methylene chloride/met~lanol/
ammonium hydroxide). The product was dissolved in ethyl acetate,
cooled to 0C, and treated with a saturated solution of HCI in ethyl
acetate. The reaction mixture was concentrated to dryness. The
resulting solid was partitioned between ethyl acetate and saturated
5 sodium bicarbonate. The aqueous layer was extracted 3 times with ethyl
acetate and the organic fractions were combined, and dried over sodium
sulfate, filtered and concentrated in vacuo. The residue was purified by
flash chromato~raphy (eluted with 9713/0.3 of methylene
chloride/methanol/ammonium hydroxide) to yield the title compound a~
20 a white foam.
m.p.: 115- 155C
NMR: consistent with structure
HPLC: > 98% pur~
25 MS: M+H-571 (FAB)
CHN Analysis c~lr~ d for C3oH42N4o5s.o.8sH2o-o~25
C4HIoO:
C, 61.58; H, 7.70; N, 9.27
Found: C, 61.56, H, 7.44; N, 9.23
E~A~PLE 7 1
-
WO9~/14025 2 1 74 ~5~ PCINS94113483
~\ N ~
0 H N'~<
,~0+
o
Endo ( 1 S)- I '(((2-~minomf~thyl-7 ,7-dimethylbicyclo-
(2.2.1 )hept- I -yl)methyl)sulfonyl)spiro(indene- 1 ,4'-piperidine) (337 mg,
5 0.78 mmole) and N-isobutyl-oxycarbonyl-5,5-dimethyl-L-thiazolidine-
4-carboxylic acid (230 mg, 0.90 mmole) were combined with 438 mg
(0.99 mmole) of benzotriazol-l-yloxytris(dimethylamino)phosphonium
hexafluorosphosphate in 5 ml of dry dimethyl formamide at room
,e-~lul~ with protection from moisture (calcium sulfate drying
20 tube). The pH of mixture was adjusted to 8.5 with diisopropyl-
ethylamine. The resulting solution was allowed to stir at room
e-dlul~ for two hours. The dimethyl~llllalllide wa3 then removed
under reduced pressure, and the residue taken up in 150 ml of ethyl
acetate and washed with saturated sodium bicarbonate (2 x 25 ml) and
25 brine (I x 25 ml). The ethyl acetate was dried over sodium sulfate,
filtered, and ~O~ la~ed to give 941 mg of an oil. The analyticâlly
pure material was obtained via chromatography on precoated silica gel
plates (1.5 mm x 20 x 20 cm) developed with ethyl acetate in hexane
(35:65). The product was obtained as an oil.
NMR: consistent with structure
FAB MS: M++H=574 (Compound minus t-BOC)
Analysis calculated for C35H51N3O6S2-1.3 CHC13
C, 52.58; H, 5.35; N, 5.07
Found: C, 52.62; H, 6 ,40; N, 4.99
WO 9~/14025 PCI/IIS94/13483
21 74~G
- 156 -
EX~MPLE 72
SO~-->H
N J~S~
H NH
The product of the preceding Example 71 was dissolved in
ethyl acetate, and the solution was cooled to 0C. HCI (g) was bubbled
into the solution for three minutes, and then let stand at 0C for 30
mirlutes. The ethyl acetate was removed under vacuum and the residue
20 evaporated twice from ethyl acetate. The residue was chromatographed
on precoated silica gel plates 6.5 mm x 20 cm x 20 cm) using
chloroform-methanol-ammonium hydroxide (97-3-0.3/v-v-v) to
develop the plates, The product was obtained as an amophous solid.
25 m p 126 -129C
NMR: consistent with structure
FAB MS: M++H = 574.3
Analysis calculated for C30H43N304S2-0.15 CHC13
C,61.19;H,7.35;N,7.10
30 Found: C, 61.]0; H, 7.33; N, 7.12
EXAMPLE 73
WO 95/14025 PC~rlUS94113483
21 74~5a
- 157 -
[~ ~OH
S2 H J~ N~
H
To a solution of (lS)-1'-(((7,7-dimethyl-(2-endo-amino-
methyl-2-exo-hydroxy)-bicyclo-(2.2. 1 )-hept- I -yl)-methyl)sulfonyl)-
spiro(lH-indene-1,4-piperdine) (108 mg; 0.25 mmol) in degassed DMF
was added 4-imidazoleacrylic acid (41.44 mg; 0.30 mmol) and
benzotriazol-l-yloxy-tris(dimethylamino)phosphonium
hexafluorophosphate (132.69 mg; 0.30 mmol). The pH of the reaction
mixture was adjusted to 9 with diisopropylethylamine. After stirring
for 14 hr, the reaction mixture was concentrated to dryness. The
resulting solid was dissolved in methanol and then purified by reverse
phase preparative HPLC utilizing a Water C-18 column (45 minute
gradient, 5% to 70% acetonitrile/water containing 0.1% trifluoroacetic
acid, 13.5 ml/min flow rate). The product containing fractions were
combined and Iyophilized to give an amorphous white solid.
NMR: consistent with structure;
HPLC: > 97% pure (254 nM)
FAB MS: M+H+ thioglycerol matrix = 659
CHN Analysis calculated for C30H38N4o4s-l~6cF3co2H
Calc'd: C, 54.39; H, 5.44; N, 7.64.
Found: C, 54.50; H, 5.20; N, 7.53.
In addition to those compounds specifically exemplified
above, additional compounds of the present invention are set forth in
tabular forrn belo~. These compounds are synthesized by use of the
synthetic routes and methods described in the above Schemes and
WO 95/14025 PCIIUS94/13483
21 7~50
- 158 -
Exarnples and variations thereof well known to those of ordinary skill
in the art, and not requiring undue e~ ion. All variables listed
in the Tables belo~v are with reference to the following generic
structure:
T~BLES
10 ~
R= (CH2)
I R~R6
~CH2)p =~ 2)q
L\
Variable
0 95/1402~; PCT/US94/13483
~74G~
- 159 -
TABLE I
Variable=
NH,SO2~ R H~J~F
0
NH 'S2\
R` SO2 H 1~'
NH R~N~
o
NH ~
R~NH~SO2\¢~ R N~3
WO 95/14025 PCT/US94/13483
21 7~650
- 160-
TA.BLE I (~ONT'D)
5 R, ,so2~ ,J~I H
R NH~~ R~
10 o OH
--NH J~ NH~OCH3
--NH~3 R N~
NH~--~ R~
N~ ,~Br NH~
O C1
R ~ N ~f ~CI N HJ~N
Cl'l~S94/13483
WO 95/1402!i P
21 7~65~
- 161 -
TABLE 2
O O ~
\--NHJ~ NHJ~ J~J
\--NHJ~ H~
`N~ R~N~CI
R`NJ~OCH3 NJ~ J~OCH3
H
--NH~ R ~OCH3
WO 95/14025 PCT/US94/13483
21 74~5~ ~
- 162-
TABLE 2 CONT'D
R NH2 R ,~
0 R~--CI N~1113
--N~--\/ R~CO2H
H~f~No2 CH3
R ~ N ~3 R N
WO gS/14025 PCI~/US94/1348
21 7~6~
- 163 -
TABI,E 3
R ~ N ~S2~/~ R --~
H ~`OCH3 H
OH --H~
10 ~ O
N~J~OH ~N
O
o R`f NH2
NH~COOH N~
r
WO 95/14025 PCI/US941134~3
2~7~
- 164 -
TABLE 3 CONTrD
R~,N~--COOH ~ N~NH
H CH3 ~ ~ R~N~
NH
R~,COOH ~'H~
R~ O`/ `J'` NH2
H~
WO 95/14025 PCT/US94/13483
2l 7~55
- 165-
TABLE 4
5 NH~ --HJ~
¢~0 0 O~,NH2
R J~ N~ ~H
~ ~ NH
R N ~ C H3
-
WO 95/14025 PCT/US94/13483
2~ 746~0
- 166-
RLF 4 CONT'~
~,~ R N~ ,NH2
O R ~ N ~,CO2H
R N)~ o
HCO2~ R N~
O ~ O SO3H
RJ~Hi N
N ~ HN J~----N
WO 9~/14025 2 1 7 4 6 3 0 PCI'IUS94/13483
- 167-
TABLE S
R~N J~o~0 R~,N~
R~N ~,~ S J~s><
15 H oHcHis ~ o
R~N~
O COOH
R~ N ~?N R~ N~ +~
O COOH CO2CH3
3 o
H~ 3 R~N~N~CO2H
O NH2 CH3
WO 9~/14025 PCT/US94/13483
21 /4~0
- 168 -
TABLE 6
o
N ~ ~ \~ X NH
COOCH3
OH
NH ~ R~ ~=
O COOC,~, R~ Q~=¦
O o ~
~ NH J~N~ NH2 R~ N J~
OH
H J~ J~l
OH `~S
WC 9S114025 PCTNS94/13483
21 7465G
- 169 -
TABLE 7
R~ J~N~
sl ~o
J~ N J~S
OH NH2
15 R~NH~--Nf'l~S~o R`N'~ NH2
o=( N H2
0~
O O
O COOCH3 NH NHJ~O'¦\
NH\I~o~<
R~ ~X~H3O ` NH/J~ N H
NH2
R~N ~NH2
WO 95114025 PCTIUS94/13483
2 ~ 3 ~
- 170-
TABLE 8
5 ~ J3 R`N~NH2
NH~J o~S~
10 o O R`
R~ J~NHJ~O~ NHJ~ X
S~ o,
CO2CH3 NH/~HJ`~
R~N /~\N~ NH N
WO 95114025 PCTIUS94/13483
21 7~5G
- 171 -
TABLE 9
R~ NH~ R~ '~N~?
NH2 o o
R~ J~\'^`OH R~ ~N~
NH I R~NH~N
R "S"O R~ NH--~O~
-S~ ~`S~;
R~NH~ J~ ~NH2
H
~f ~
NH
WO 95/14025 PCI'NS94/13483
21 74~50
- 172 -
TABI~E~ 10
R J~, N ~X
R "S"O R~N~
R~ ~N N~/~N~
o,S ~o
o NH~--NH ~
R~H~X ~~
SO~2
R~ ~NH2
R\~N~ NH ~--~ NH ~S
O O
WO 9~/1402~ PCr/US94/13483
21 74~50
- 173 -
EXAMPLE 74
RADIOLICAND BINDING ASSAYS
The high affinity binding of [3H]oxytocin (OT) to uterine
5 tissue and [3H]arginine vasopressin (AVP) to liver (AVP-Vla site) and
kidney (AVP-V2 site) tissue was determined using crude membrane
preparations as described previously [Pettibone, D.J., et al., J.
Pharmacol. and Exper. The7-., 256(1): 304-308 (1991)]. Uterine tissue
was taken from nonpregnant adult Sprague-Dawley rats (Taconic
Farms, Germantown, NY) pretreated (18-24 h) with diethylstilbestrol
propionate (DES; 300 llg/kg, i.p.). Uterine tissue (full thickness) was
also taken with informed consent from nonlabor pregnant women
undergoing cesarean section at 38 to 39 weeks gestation (Oregon Health
Sciences Center, Portland, OR). Liver and kidney medulla samples
5 were taken from male rats and from human surgical and early
postmortem donors (National Disease Research Interchange,
Philadelphia PA; Analytical Biological Services, Wilmin~tr)n, DE).
Competition studies were conducted at equilibrium using
1 nM [3H]oT or 0.5 nM [3H]AVP in the following buffer: 50 mM Tris,
20 5 mM MgC12, 0.1% bovine serum albumin. Nonspecific binding was
~ t~rminf d using I ~lM unlabeled OT or AVP in their respective assay~.
The binding reactions were initiated by the addition of tissue
preparation and terminated by filtration using a Skatron cell harvester
(model 7019, Skatron, Inc., Sterling, VA). Ki values were calculated
25 for each compound using three to six separate IC50 d~ dlions
(K;=lCso/[l- c/Kd]); [Cheng, Y-C; Prusoff, W.H.; Biochem Pharmacol
22:3099 (1973)] with mean Kd values obtained from replicate (n = 3)
equilibrium saturation binding assays (10 point, 100 fold concentration
range): [3H]oT rat utenus, 0.69 nM; human myometrium, I.l nM;
30 [3H]AVP: rat liver, 0.21 nM; rat kidney, 0.27 nM; human liver, 0.27
nM; human kidney, 1.4 nM. Computer analysis of the saturation assays
by EBDA/LIGAND [McPher.son, G.A.; Kinetic, Ebda, Ligand, Lowry:
A Collection of Radioligand Binding Analysis Programs, Elsevier
Science Publishers, Amsterdam ( 1985)] indicated that both radioligands
apparently bound to single sites in all tissues examined. The final
WO 95/14025 PCr/US94113483
2 i 7 ~ b ~ G
- 174-
protein concentration for the various tissues in each assay ranged from
150 to 300 ,ug/ml [Lowry, P.H.; Rosebrough, N.J.; Farr, A.L.; Randall,
R.J.; J. Biol. Chem., 193 265-275 (1951)].
IC50 ~alues were determined for the [3H]oT and [3H]AVP
5 binding assays by linear regression of the relation log concentration of
compound vs. percent inhibition of specific binding. Data is either
reported as a given percentage of inhibition at a specified concentration,
or if an IC50 was c~ r~-l, as a nanomolar concentration.
Rc;~ e IC50 values of the compounds of the instant invention
for rat L3H]oT are given below.
l~xample Result For l_HlOT
70% inhib. at 1000nM
2 1100nM
3 50% inhib. at 1000nM
4 620nM
5 190nM
6 60% inhib. at 1000nM
20 7 54% inhib. at 1000nM
8 140nM
9 68% inhib. at 1000nM
10 140nM
I 1 69% inhib. at 1000nM
2512 37% inhib. at 100nM
13 1 80nM
14 76% inhib. at 1000nM
15 160nM
16 61nM
3017 45nM
1~ 52% inhib. at 100nM
19 150nM
20 50% inhib. at 100nM
21 61nM
WO 95/14025 PCIIUS94/13483
-- 21 74~5~
- 175 -
Example Result For l-HlOT
22 58.8nM
26% inhib. at 10000nM
26 34% inhib. at 1000nM
27 47nM
28 82nM
29 31nM
45nM
32 36% iohib. at 10000nM
33 30% inhib. at 1000nM
34 48% inhib. at 1000nM
43% inhib. at 1000nM
36 67nM
38 33% inhib. at 1000nM
39 8500nM
71% inhib. at 10000nM
41 120nM
42 96.5nM
43 4300nM
44 39% inhib. at 100nM
25% inhib. at 100nM
46 36% inhib. at 100nM
47 4600nM
48 90nM
49 54nM
so 71% irlhib. at 1000nM
51 84% inhib. at 1000nM
52 290nM
67 7.6 nM
73 45 nM
WO 95/14025 PCTIUS94113483
2l 74~5~ --
- 176-
Binding (Ki) of compoumds to Hulnan OT, Human AVP-VIa and
Human AVP-V2:
~xample OT AVP-V I a AVP-V~
s 67 ~~ 3.7 nM 1.4 nM 1600 nM
73 3.8nM 2.7 nM --
While the invention has been described and illustrated wit~
reference to certail1 preferred embodiments thereof, those skilled in th~
art will appreciate that various changes, modifications and substitutions
can be made therein without departing from the spirit and scope of the
invention. For example, effective dosages other than the preferred
dosages as set forth hereinabove may be applicable as a consequence of
variations in the responsiveness of the mammal being treated for
prevention of preterm labor, or for other indications for the compounds
of the invention indicated above. Likewise, the specific
pharmacological r~sponses observed may vary according to and
depending upon the particular active compound selected or whether
there are present pharmaceutical carriers, as well as the type of
20 formulation and mode of administration employed, and such expected
variations or differences in the results are contemplated in accordance
with the objects and practices of the present invention. It is intended,
therefore, that the invention be limited only by the scope of the claim~
which follow and that such claims be interpreted as broadly as is
25 reaSO!lable-