Note: Descriptions are shown in the official language in which they were submitted.
-
WO 95113069 PCI'IUS94/12816
21 75218
TITLE OF THE INVENTION
PIPERIDINES, PYRROLIDINES AND HEXAHYDRO-lH-
AZEPINES PROMOTE RELEASE OF GROWTH HORMONE
S CROSS REFERENCE TO RELATED APPLICATIONS
This application is a continuation-in-part of copending
application Serial No. 08/323,994, filed October 17, 1994, which is a
continuation-in-part of copending application Serial No. 08/149,441,
filed November 9, 1993; a ~ontinuation-in-part of copending
application Serial No. 08/323,998, filed October 17, 1994, which is a
continuation-in-part of copending application Serial No. 08/165,149,
filed December 10, 1993; and a continuation-in-part of copending
application Serial No. 08/323,988, filed October 17, 1994, which is a
continuation-in-part of copending application Serial No. 08/173,449,
filed December 23, 1993.
BACKGROUND OF THE INVENTION
Growth hormone, which is secreted from the pituitary,
stimulates growth of all tissues of the body that are capable of growing.
In addition, growth horrnone is known to have the following basic effects
on the metabolic processes of the body: (1) Increased rate of protein
sy77thesis in all cells of the body; (2) Decreased rate of carbohydrate
tili~tion in cells of the body; (3) Increased mobilization of free fatty
acids and use of fatty acids for energy. A deficiency in growth hormone
secretion can result in various medical disorders, such as dwarfism.
Various ways are known to release growth hormone. For
example, chemicals such as arginine, L-3,4-dihydroxyphenyl~ nine
(L-DOPA), glucagon, vasopressin, and insulin induced hypoglycemia,
as well as activities such as sleep and exercise, indirectly cause growth
3 hormone to be released from the pituitary by acting in some fashion
" on the hypoth~l~mus perhaps either to decrease somatostatin secretion
or to increase the secretion of the known secretagogue growth
hormone releasing factor (GRP) or an unknown endogenous growth
hormone-releasing hormone or all of these.
W O 9S/13069 2 1 7 5 2 1 8 PC~rrUS94/12816
In cases where increased levels of growth horrnone were
desired, the problem was generally solved by providing exogenous
growth hormone or by ~(lmini~r~tering GRF or a peptidal compound
which stim~ ted growth hormone production and/or release. In either
5 case the peptidyl nature of the compound necessitated that it be
~1mini.~tered by injection. Tniti~lly the source of growth hormone was
the extraction of the pituitary glands of cadavers. This resulted in a
very expensive product and carried with it the risk that a disease
associated with the source of the pituitary gland could be transmitted
0 to the recipient of the growth hormone. Recombinant growth
hormone has become available which, while no longer carrying any
risk of disease tr~n~micsion, is still a very expensive product which
must be given by injection or by a nasal spray.
Other compounds have been developed which stimulate
5 the release of endogenous growth hormone such as analogous peptidyl
compounds related to GRF or the peptides of U.S. Patent 4,411,890.
These peptides, while considerably smaller than growth hormones are
still suscepti~le to various proteases. As with most peptides, their
potential for oral bioavailability is low. Non peptidal growth hormone
20 secretagogues with a benzolactam structure are disclosed in U.S.
Patents S,206,235, 5,283,241, 5,284,841, 5,310,737 and 5,317,017.
The instant compounds are low molecular weight pep~ide analogs for
promoting the release of ~lvwlll hormone which have good stability in
a variety of physiological environments and which may be
25 ~tlmini.stered parenterally, nasally or by the oral route.
~UMMARY OF THE INVENTION
The instant invention is directed to certain piperidine.
pyrrolidine~ and hexahydro-lH-azepine compounds which ha~e the
30 ability to stimulate the release of natural or endogenous growth
hormone. The compounds thus have the ability to be used to treat
conditions which require the stimulation of growth hormone
production or secretion such as in humans with a deficiency of natural
growth hormone or in ~nim~l.s used for food production where the
WO95113069 2 1 752 1 8 ~ s94~l28l6
stimulation of growth hormone will result in a larger, more productive
~nim~l. Thus, it is an object of the instant invention to describe the
piperidine, pyrrolidine, and hexahydro-lH-azepine compounds. It is a
further object of this invention to describe procedures for the
preparation of such compounds. A still further object is to describe
the use of such compounds to increase the secretion of growth
horrnone in hllm~n~ and ~nim~l.c. A still further object of this
invention is to describe compositions cont~inin~ the piperidine,
pyrrolidine, and hexahydro-lH-azepine compounds for the use of
treating hllm~n.c and ~nim~ so as to increase the level of growth
hormone secretions. Further objects will become apparent from a
reading of the following description.
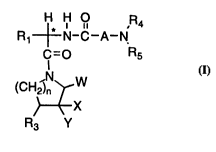
DESCRIPTION OF THE INVENTION
The novel piperidine, pyrrolidine, and hexahydro-lH-
azepine compounds of the instant invention are best described in the
following structural formula I:
H H ,R4
R1~N-C--A--N
C=O R5
(CH2Sn W
~X
R3 Y
2s
Forrnula I
wherein:
Rl is selected from the group consistin_ of:
Cl-Clo aLkyl, aryl, aryl(cl-c6 alkyl), (C3-C7 cycloalkyl)(Cl-C6 alkyl)-
30 (Cl-C~ alkyl)-K-(Cl-C5 alkyl)-, aryl(Co-Cs alkyl)-K-(Cl-Cs aL~yl)-
~and (C3-C7 cycloalkyl)(Co-C5 alkyl)-K-(Cl-Cs alkyl)-, where K is O.
S(O)m, N(R2)c(o)~ C(o)N(R2)~ OC(O), C(O)O, -CR_=CR2-, or-C-C-.
where aryl is selected from: phenyl, naphthyl, indolyl. azaindole,
pyridyl, benzothienyl. benzofuranyl, thiazolyl, and benzimidazolyl. and
R2 and alkvl may be further substituted by l to 9 halogen, S(O)mR2a. l
2 1 7 2 PCI'IUS94/12816
Wogs1l3069 ~ 1 8 ~
to 3 of OR2a or C(O)OR2a, and aryl may be further substituted by 1 to 3
of Cl -C6 alkyl, 1 to 3 of halogen, 1 to 2 of OR2, methylenediox~,
-S(O)mR2, 1 to 2 of-CF3, -OCF3, nitro, -N(R2)c(o)(R2)~ -C(O)OR2,
-C(O)N(R2)(R2), -1H-tetrazol-5-yl, -so2N(R2)(R2)~ -N(R2)so2 phenyl,
5 or-N(R2)S02R2;
R2 is selected from: hydrogen, Cl-C6 aL~cyl, and C3-C7 cycloalkyl, and
where two C l-C6 aLkyl groups are present on one atom, they may be
ophonally joined to forrn a C3-Cg cyclic ring, ophonally including
oxygen, sulfur or NR3a;
R2a is hydrogen, or Cl-C6 aLkyl ophonally substituted by hydroxyl;
R3 is selected from: hydrogen, ~(cH2)rphenyL ~(cH2)rnaphthyl~ -Cl-clo
aLkyl, -C3-C7 cycloalkyl, where ~e phenyl, naphthyl and C3-C7
5 cycloalkyl rings may be substituted by 1 to 3 subs~ituents selected from
the group consisting of: C1-C6 aLkyl, halogen, -OR2, -NHS02CF3,
-(CH2)rOR6, -(cH2)rN(R2)(R6)~ -(cH2)r (R6), -(cH2)rc(o)oR2
-(CH2)rC(O)OR6, -(CH2)rOC(O)R2, -(CH2)rOC(O)R6,
^(CH2)rC(0)R2,-(CH2)rC(0)R6, (cH2)rc(o)N(R2)(R2)~
2 -(CH2)rC(O)N(R2)(R6), -(cH2)rN(R2)c(o)R2 -(cH2)rN(R2)c(o)R6~
-(CH2)rN(R6)C(O)R2, -(cH2)rN(R6)c(o)R6~ -(cH2)rN(R2)c(o)oR2~ -
(CH2)rN(R2)C(O)OR6, -(cH2)rN(R6)c(o)oR2~
-(CH2)rN(R6)C(O)OR6, (CH2)rN(R2)C(O)N(R2)(R6),
-(CH2)rN(R2)C(O)N(R2)(R2). -(cH2)rN(R6)c(o)N(R2)(R6)~
25 (CH2)rN(R2)S02R6,-(CH2)rN(R2)S02R2,-(CH2)rN(R6)S02R2,
CH2)rN(R6)S02R6, (CH2)rOC(O)N(R2)(R6),
-(CH2)rOC(O)N(R2)(R2), -(cH2)rso2N(R2)(R6)~
-(CH2)rS02N(R2)(R2),(CH2)rS02NHC(O)R6, (CH2)rS02NHC(O)R~,
-(CH2)rS02NHC(O)OR6, -(cH2)rso2NHc(o)oR2
3 -(CH2)rC(O)NHC(O)NR2, -(cH2)rc(o)NHc(o)
-(CH2)rC(O)NHC(O)R2 . -(cH2)rcoNHc(o)R6,
-(CH2)rCONHS02R6,~(CH2)rCONHS02R2,
(CH2)rCONHSO2N(R2)R~). -(cH2)rcoNHso~7N(R2)R6)~
') 1 7 r ~ pcrlus94ll28l6
WO 95~13069 ~ 8
.
-(CH2)rN(R2)SO2N(R2)R6), -(cH2)rN(R6)so2N(R2)R6)~
-(CH2)rS(O)mR6. and-(cH2)rs(o)mR2;
R3a is hydrogen, or Cl-C6 alkyl optionally substituted by hydroxyl;
W is selected from the ~roup consisting of: hydrogen,
-CN, -C(O)OR8, -c(o)oR2~ -c(o~o(cH2)laryL -C(O)N(R2)(R2);
-C(O)N(R2)(R8), -c(o)N(R2)(cH2)l aryl, -cH2N(R2)c(o)R8
-CH2N(R2)C(O)(CH2)1aryL -(cH2)roR2~ -CH(OH)R2,
-CH(OH)(CH2)1aryl, -C(O)R2~ -C(O)(CH2)1 aryl, lH-tetrazol-5-yl,
S-arnino-1, 2, 4-oxadiazol-3-yl, and 5-methyl-1, 2, 4-oxadiazol-3-yl,
where R8 is hydrogen, C 1 -C6 alkyl, or C l-C6 aLkyl substituted by
OR2, C(O)OR2, CON(R2)(R2), N(R2)c(o)R2~
N(R2)C(O)N(R2)(R2), and aryl is phenyl, pyndyl, or lH-tetrazol-5-
yl;
X is selected from the group consisting of: hydrogen, -C_N,
-(CH2)qN(R2)C(O)R2, -(CH2)qN(R2)C(O)(CH2)taryl,
-(cH2)qN(R2)so2(cH2)taryl~ -(cH2)qN(~2)so2R2~
2 -(CH2)qN(R2)C(O)N(R2)(CH2)taryl, -(CH2)qN(R2)C(O)N(R2)(R2),
-(CH2)qC(O)N(R2)(R2), -(cH2)qc(o)N(R2)(cH2)tar
-(CH2)qC~O)OR2,-(CH2)qC(O)O(CH2)taF5~ (cH2)qoR2
-(CH2)qOC(O)R2, -(cH2)qoc(o)(c~2)tar
-(cH2)qoc(o)N(R2)(cH2)taryL -(cH2)qoc(o)N(R2)(R2)~
25 -(CH2)qC(O)R2, -(CH2)qC(O)(CH2)taryl,
-(CH2)qN(R2)C(O)OR2. -(cH2)qN(R2)so2N(R2)(R2) .
-(CH2)qS(O)mR2. and -(cH2)qs(o)m(cH2)taryL where an R2~ (CH2)q
and (CH2)t group may be optionally substituted by 1 to 2 Cl -C4 aLIcyl.
hydroxyl, Cl-C4 lower alkoxy, carboxyl, CONH2, S~O)mCH3,
30 carboxylate Cl-C4 alkyl esters, or lH-tetrazol-5-yL and aryl is phenyl,
naphthyl, pyridyl, thiazolyl, or lH-tetrazol-5-yl ~roups which may be
optionally substituted by 1 to 3 halogen, 1 to 3 -OR2, -CON(R2)(R2),
-C(O)OR2, 1 to 3 Cl-C4 aLkyl, -S(O)mR2, or lH-tetrazol-5-yl;
WO95113069 2 ~ 752 ~ 8 ~ 41~2816
Y is selected from the group consisting of:
hydrogen, Cl-Clo aLkyl, -(CH2)taryl,
-(CH2)q(C3-C7 cycloaLkyl), -(CH2)q-K-(Cl-C6 aLkyl),
-(cH2)q-K-(cH2)taryl7-(cH2)q-K-(cH2)t(c3-c7 cycloaLkyl
5 cont~inin~ O, NR2, S), and -(CH2)q-K-(CH2)t(C3-C7 cycloaLkyl), where
K is O, S(O)m, C(O)NR2, CH=CH, C~C, N(R2)C(O), C(O)NR2, C(O)O,
or OC(O), and where the aLkyl, R2, (CH2)q and (CH2)t groups may be
optionally substituted by Cl-C4 aLkyl, hydroxyl, Cl-C4 lower aLkoxy,
carboxyl, -CONH2 or carboxylate Cl -C4 alkyl esters, and aryl is phenyl,
naphthyl, pyridyl, 1-H-tetrazol-5-yl, thiazolyl, irnidazolyl, indolyl,
pyrimidinyl, thi~ 7.olyl, pyrazolyl, oxazolyl, isoxazolyl, thiopheneyl,
quinolinyl, pyITazinyl, or isothiazolyl which is optionally substituted by 1
to 3 halogen, 1 to 3 -OR2, -C(O)OR2, -C(O)N(R2)(R2), nitro, cyano,
benzyl, 1 to 3 Cl-C4 aLkyl, -S(O)mR2, or lH-tetrazol-5-yl;
15 with the proviso that at least one of R3, W, X, and Y are other than
hydrogen;
R4 and R5 are independently hydrogen, Cl-C6 alkyl, substituted Cl-
C6 aLkyl where the substituents may be 1 to 5 halo, 1 to 3 hydroxy, 1
20 to 3 Cl-Clo aLI~anoyloxy, 1 to 3 Cl-C6 aLkoxy, phenyl, phenoxy, 2-
furyl, Cl-C6 aLkoxycarbonyl, S(O)m(Cl-C6 alkyl); or R4 and Rs can
be taken toge~er to folm -(cH2)dLa(cH2)e- where La is C(R2)2, O,
S()m or N(R2)~ d and e are independently 1 to 3 and R2 is as defined
above;
Ais:
F~7 ~7
_(CH2)X--C,--(CH2)y or --z--(CH2)x--C,--(CH2)y
3 o R7a R7a
where x and y are independently 0, 1, 2 or 3;
Z is N-R6a or O, where R6a is hydro~en or C 1 -C6 alkyl;
WO95113069 2 1 752 1 8 ~ u~94~12816
.
R6 is hydrogen, Cl-C6 aLkyl, or (CH2)varyl, wherein the aLkyl and
(CH2)V groups may be optionally substituted by 1-2 O(R2), S(o)mR2
lH-tetrazol-5-yl, C(O)OR2, C(O)N(R2)(R2) or So2N(R2)(R2)~
5 N(R2)C(O)N(R2)(R2),and wherein aryl is phenyl, pyridyl, lH-tetrazol-
5-yl, triazolyl, imidazolyl, thiazolyl, pyrazolyl, thi~ 7.01yl, imidazolone-
l-yl, be~7.imicl~7.01-2-yl, triazolinone-yl optionally substituted with Cl-
C6 alkyl, C3-C6 cycloalkyl, amino, or hydroxyl;
0 R7 and R7a are independently hydrogen, Cl-C6 aLkyl, trifluoromethyl,
phenyl, substituted C l-C6 aLkyl where the substituents are imidazolyl,
phenyl, indolyl, p-hydroxyphenyl, OR2, S(O)mR2, C(O)O(Cl-C6 alkyl),
C3-C7 cycloaLkyl, N(R2)(R2)~ C(O)N(R2)(R2); or R7 and R7a can
independently be joined to one or both of R4 and R5 groups to form
5 aLkylene bridges between the tennin~l nitrogen and the aLlcyl portion of
the R7 or R7a groups, wherein the bridge contains 1 to 5 carbons atoms;
or R7 and R7a can be joined to one another to form a C3-C7 cycloalkyl;
lisO, 1 or2;
20 m is 0, 1, or 2;
nis 1,2,or3;
qisO, 1,2,3,or4;
risO, 1,2,or3;
tisO, 1,2,or3;
visO, 1,or2;
and pharrnaceutically acceptable salts and individual diastereomers
thereof.
When n is 1, a pyrrolidine ring is formed, when n is 2 a
piperidine ring is formed and when n is 3 the ring is desi~n~tefl as a
3 hexahydro- lH-azepine.
In the above s.tructural formula and throughout the instant
specification, the following terms have the indicated me~nin~:
The aLkyl groups specified above are intended to include
those aLkyl groups of the designated length in either a straight or
branched configuration which may optionally contain double or triple
PCr/US94/12816
WO 9S113069
2~752~8
bonds. Exemplary of such aLkyl groups are methyl (Me), ethyl (Et),
propyl (Pr), isopropyl (i-Pr), butyl (Bu), sec-butyl (s-Bu), tertiary
butyl (t-Bu), pentyl, isopentyl, hexyl, isohexyl, allyl, propinyl,
butadienyl, hexenyl and the like.
The aLkoxy groups specified above are intended to
include those aLkoxy groups of the designated length in either a
straight or branched configuration which may optionally contain
double or triple bonds. Exemplary of such aLkoxy groups are
methoxy, ethoxy, propoxy, isopropoxy, butoxy, isobutoxy, tertiary
butoxy, pentoxy, isopentoxy, hexoxy, isohexoxy, allyloxy,
propinyloxy, isobutenyloxy, hexenyloxy and the like.
The terrn "halogen" is intended to include the halogen
atom fluorine, chlorine, bromine and iodine.
The term "aryl" within the present invention, unless
lS otherwise specified, is intended to include aromatic rings, such as
carbocyclic and heterocyclic aromatic rings selected the group consisting
of: phenyl, naphthyl, pyridyl, l-H-tetrazol-5-yl, thiazolyl, irrudazolyl,
indolyl, pyrirnidinyl, thi~ 7olyl, pyrazolyl, oxazolyl, isoxa_olyl,
thiopheneyl, quinolinyl, pyrrazinyl, or isothiazolyl, which may be
optionally substituted by 1 to 3 of C 1 -C6 aLkyl, 1 to 3 of halogen, 1 to 2
of-OR2, methylenedioxy, -S(O)mR2, 1 to 2 of -CF3, -OCF3, nitro,
-N(R2)C(O)(R2), -C(O)OR2, -C(O)N(R2)(R2), - lH-tetrazol-5-yl,
-S02N(R2)(R2), -N(R2)S02 phenyl, or -N(R2)S02R2, wherein R2 is as
defined herein.
2s Certain of the above defined terms may occur more than
once in the above formula or definitions and upon such occurrence,
each term shall be defined independently of the other.
WO95/13069 2 1 752 1 8 ~ 4/12816
I~
g
A first embodiment of the present invention is directed to
the compounds of the structural formula AI:
H H O ,R4
R1~N-C--A--N
l= Rs
(CH -)N~_W
)~X
R Y
Forrnula AI
wherein:
Rl is selected from the group consisting of:
Cl-Clo alkyl, aryl, aryl(Cl-C6 alkyl), (C3-C7 cycloaLkyl)(C1-C6 alkyl)-,
5 (Cl-C5 aLkyl)-K-(cl-c5 alkyl)-, aryl(Co-Cs alkyl)-K-(C1-Cs alkyl)-,
and (C3-C7 cycloalkyl)(Co-C5 alkyl)-K-(C1-Cs alkyl)-, where K is O,
S(O)m, N(R2)C(O), C(o)N(R2)7 OC(O), C(O)O, -CR2=CR2-, or-C-C-,
where aryl is selected from: phenyl, naphthyl, indolyl, ~7~in~1Ole,
pyridyl, benzothienyl, benzofuranyl, thiazolyl, and benzimidazolyl, and
20 R2 and alkyl may be further substituted by 1 to 9 halogen, S(O)mR2a, l
to 3 of OR2a or C(O)OR2a, and aryl may be further substit-lte~ by l to 3
of C 1-C6 alkyl, 1 to 3 of halogen, 1 to 2 of OR2, methylenedioxy,
-S(O)mR2, 1 to 2 of-CF3, -OCF3, nitro, -N(R2)c(o)(R2)~ -C(O)OR2,
-C(O)N(R2)(R2), -1H-tetrazol-5-yl, -S02N(R2)(R2), -N(R2)S02 phenyl,
25 or-N(R2)so2R2;
R~ is selected from: hydrogen, Cl-C6 alkyl, and C3-C7 cycloalkyl, and
where two C l-C6 alkyl ~roups are present on one atom, they may be
optionally joined to form a C3-Cg cyclic ring, optionally includin~
3 oxygen, sulfur or NR3a;
R2a is hydrogen, or Cl-c6 alkyl optionally substituted by hydroxyl;
R3 is selected from: hydro~en, -(CH~)rphenyl, -(CH~)rnaphthyl~ -Cl-Clo
aLkyl, -C3-C7 cycloalkyl, where the phenyl, naphthyl and C3-C7
cycloaLkYl rin~s may be substituted by 1 to 3 substituents selected from
WO95113069 2 1 ~52 1 8 ~ " ~94~28l6
- 10-
the group consisting of: C l-C6 aLkyl, halogen, -OR2, -NHS02CF3,
-(CH2)rOR6, -(cH2)rN(R2)(R6)~ -(cH2)r (R6). -(cH2)rc(o)oR2
-(CH2)rC(O)OR6, -(CH2)rOC(O)R2, -(CH2)rOC(O)R6,
-(CH2)rC(O)R2,-(CH2)rC(O)R6, (cH2)rc(o)N(R2)(R2)~
-(CH2)rC(O)N(R2)(R6), -(cH2)rN(R2)c(o)R2 -(CH2)rN(R2)C(O)R6,
-(CH2)rN(R6)C(O)R2, -(cH2)rN(R6)c(o)R6~ -(CH2)rN(R2)C(O)OR2, -
(CH2)rN(R2)C(O)OR6, -(cH2)rN(R6)c(o)oR2
-(CH2)rN(R6)C(O)OR6, (cH2)rN(R2)c(o)N(R2)(R6)~
-(CH2)rN(R2)C(O)N(R2)(R2). -(cH2)rN(R6)c(o)N(R2)(R6)~
o (CH2)rN(R2)S02R6, -(CH2)rN(R2)S02R2, -(CH2)rN(R6)S02R2,
CH2)rN(R6)S02R6, (CH2)rC(O)N(R2)(R6),
-(CH2)rOC(O)N(R2)(R2) . -(CH2)rs02N(R2)(R6),
-(CH2)rS02N(R2)(R2),(CH2)rS02NHC(O)R6, (cH2)rso2NHc(o)R2
-(CH2)rS02NHC(O)OR6, -(cH2)rso2NHc(o)oR2~
-(CH2)rC(O)NHC(O)NR2, -(CH2)rC(O)NHC(O)NR6,
-(CH2)rC(O)NHC(O)R2 . -(CH2)rCONHC(O)R6,
-(CH2)rCONHS02R6,~(CH2)rCONHs02R2,
(CH2)rCONHS02N(R2)R2) . -(cH2)rcoNHso2N(R2)R6)
-(CH2)rN(R2)S02N(R2)R6).-(cH2)rN(R6)so2N(R2)R6)~
20 -(CH2)rS(O)mR6, and-(cH2~rs(o)mR2;
R3a is hydrogen, or Cl-C6 alkyl op~onally subsatuted by hydroxyl;
W is selected from the group consisting of:
25 -CN, -C(O)OR8, -C(O)OR2, -C(O)O(CH2)1aryl, -C(O)N(R2)(R2);
-C(O)N(R2)(R8), -c(o)N(R2)(cH2)l aryl, -cH2N(R2)c(o)R8
-CH2N(R2)C(O)(CH2)1aryL -(cH2)roR2~ -CH(OH)R2,
-CH(OH)(CH2)1aryL -c(o)R2~ -C(O)(CH2)1 aryl, lH-tetrazol-5-yl,
S-amino-1, 2, 4-oxadiazol-3-yl, and S-methyl-1, 2, 4-oxadiazol-3-yl,
3 where R8 is hydrogen, C 1 -C6 aLkyl, or C 1 -C6 alkyl substituted by
OR2. C(O)OR2, CoN(R2)(R2)~ N(R2)C(O)R2~
N(R2)C(O)N(R2)(R2), and aryl is phenyl, pyridyl, or lH-tetrazol-5-
yl;
WO9S113069 2 ~ 752 1 8 ~ 94/l28l6
.
- 11
X is selected from: hydrogen, -C-N, -(CH2)qN(R2)C(O)R2.
-(cH2)qN(R2)c(o)(cH2)taryl~ -(cH2)qN(R2)so2(cH2)tar
-(CH2)qN(R2)S02R2, -(cH2)qN(R2)c(o)N(R2)(cH2)tar
-(CH2)qN(R2)C(O)N(R2)(R2), -(CH2)qC(O)N(R2)(R2),
5 -(cH2)qc(o)N(R2)(cH2)taryL -(CH2)qC(O)OR2,
-(cH2)qc(o)o(cH2)taryl~ -(cH2)qoR2~ -(cH2)qoc(o)R2
-(cH2)qoc(o)(cH2)taryL -(CH2)qOC(O)N(R2)(CH2)taryl~
-(CH2)qOC(O)N(R2)(R2), -(cH2)qc(o)R2~ -(CH2)qC(O)(CH2)taryl,
-(CH2)qN(R2)C(O)OR2, -(cH2)qN(R2)so2N(R2)(R2)~
0 -(CH2)qS(O)mR2, and -(CH2)qS(O)m(CH2)taryl, where an R2, (CH2)q
and (cH2)t group may be optionally substituted by 1 to 2 C 1 -C4 aLlcyl,
hydroxyl, Cl-c4 lower aL~oxy, carboxyl, CONH2, S(O)mCH3,
carboxylate Cl-C4 aL~yl esters, or lH-tetrazol-S-yl, and aryl is phenyl,
naphthyl, pyridyl, ~iazolyl, or lH-tetrazol-S-yl groups which may be
5 optionally substituted by 1 to 3 halogen, 1 to 3 -OR2, -CON(R2)(R2),
-C(O)OR2, 1 to 3 Cl-C4 aL~yl, -S(O)mR2, or lH-tetrazol-5-yl;
Y is selected from: hydrogen, Cl-Clo aLkyl, -(CH2)taryl,
-(CH2)q(C3-C7 cycloaL~yl), -(cH2)q-K-(cl-c6 aLtcyl),
20 -(cH2)q-K-(cH2)taryL-(cH2)q-K-(cH2)t(c3-c7 cycloaL~yl
cont~nin~ 0, NR2, S), and -(CH2)q-K-(CH2)t(C3-C7 cycloaL~yl), where
K is O, S(O)m, C(o)NR2~ CH=CH, C~C, N(R2)C(O), C(O)NR2, C(O)O,
or OC(O), and where the aLkyl, R2, (CH2)q and (CH2)t groups may be
optionally substituted by C l-C4 alkyl, hydroxyl, C 1 -C4 lower aLkoxy,
carboxyl, -CONH2 or carboxylate C1-C4 alkyl esters, and aryl is phenyh
naphthyl, pyridyl, 1-H-tetrazol-S-yl, thiazolyl, imidazolyl, indolyl,
pyrimidinyl, fhi~ 01yl, pyrazolyl, oxazolyl, isoxazolyl, thiopheneyl,
quinolinyl, pyrrazinyl, or isothiazolyl which is optionally substituted by 1
to 3 halogen, 1 to 3 -OR2, -C(O)OR2, -C(O)N(R2)(R')~ nitro, cyano,
30 benzyl, 1 to 3 Cl-C4 alkyl, -S(O)mR~, or l~-tetrazol-5-yl;
R4 and R5 are independently hydrogen, Cl-c6 aL~yl, substituted Cl-
C6 aLkyl where the substituents may be 1 to 5 halo, 1 to 3 hydroxy, 1
to 3 Cl-Clo aLkanoyloxy, 1 to 3 C1-C6 aL~coxy, phenyl, phenoxy~ ~-
WO 95113069 PCI~/US94/12816
2l~7~?l8 ~
- 12 -
furyl, Cl-C6 alkoxycarbonyl, S(O)m(Cl-C6 alkyl); or R4 and Rs can
be taken together to form -(CH2)dLa(CH2)e- where La is C(R2)2, O,
S()m or N(R2)t d and e are independently 1 to 3 and R2 is as defined
above;
A is:
~7 F~7
_ (C H2)X--C--(C H2)y or _ z--(C H2)X--Ç--(C H2)y
R7a R7a
where x and y are independently 0, 1, 2 or 3;
Z is N-R6a or O, where R6a is hydrogen or Cl-C6 alkyl;
R6 is hydrogen, Cl-C6 aIkyl, or (CH2)varyl, wherein the aL~cyl and
(CH2)v groups may be optionally substituted by 1-2 O(R2), S(O)mR2,
lH-tetrazol-5-yl, C(O)OR2, C(o)N(R2)(R2) or So2N(R2)(R2)7
20 N(R2)C(O)N(R2)(R2),and wherein aryl is phenyl, pyridyl, lH-tetrazol-
5-yl, triazolyl, imi(1s~70lyl, thiazolyl, pyrazolyl, thi~ 7.olyl, imicl~7.010ne-l-yl, ben7:imi~701-2-yl, triazolinone-yl optionally substituted with Cl-
C6 alkyl, C3-C6 cycloalkyl, amino, or hydroxyl;
25 R7 and R7a are independently hydrogen, Cl-C6 aLkyl, trifluoromethyl,
phenyl, substituted Cl-c6 alkyl where the substituents are imicl~7.olyl,
phenyl, indolyl, p-hydroxyphenyl, OR2, S(O)mR2, C(O)O(Cl-C6 aLkyl).
C3-C7 cycloalkyl, N(R2)(R2)~ C(o)N(R2)(R2); or R7 and R7a can
independently be joined to one or both of R4 and R5 ~roups to form
30 alkylene bridges between the terminal nitro~en and the alkyl portion of
the R7 or R7a caroups, wherein the bridc~e contains 1 to 5 carbons atoms:
or R7 and R7a can be joined to one another to form a C3-C7 cycloalkyl:
lisO, l or_;
misO, l,or'';
WO9!i/13069 2 1 752 1 8 ~ 4/12816
nis 1,2,or3;
qisO, 1,2,3,or4;
risO, 1,2,or3;
tisO, 1,2,or3;
visO, 1,or2;
and pharmaceutically acceptable salts and individual diastereomers
thereof.
Preferred compounds within this first embodiment include
those of Forrnula AIa:
H H ,R4
Rl ~N-C--A--N
=O R~;
(CH2~--W
)~X
R3 Y
Formula AIa
wherein:
R1 is selected from the group consisting of:
Cl-Clo aLkyl, aryl (cl-c4 aL~cyl)-, C3-C6 cycloalkyl (Cl-C4 alkyl)-,
(Cl-C4 aLkyl)-K-(cl-c2 aLkyl)-, aryl (Co-C2 aLkyl)-K-(C1-C2 alkyl)-,
and (C3-C7 cycloaLkyl)(Co-c2 aLkyl)-K-(C1-C2 aLkyl)-, where K is O,
S(O)m, OC(O), C(O)O and the alkyl groups may be further substituted
25 by 1 to 7 halogen, S(O)mR2, 1 to 3 OR2 or C(O)OR2 and aryl is phenyl,
naphthyl, indolyl, pyridyl, benzothienyl, or benzofuranyl which may be
further substituted by 1-2 C 1 -C4 aLkyl, 1 to 2 halogen, 1 to 2 OR2,
S(O)mR2 or C(O)OR2;
R2 is hydrogen, Cl-c6 alkyl, or C3-C7 cycloalkyl and where two C 1 -C6
- alkyl groups are present on one atom they may be optionally joined to
form a C4-C7 cyclic ring optionally including oxygen, sulfur or NR3a;
PCr/US94/12816
WO 95/13069
2 ~ 7 ~
- 14-
R3 is hydrogen or phenyl optionally substituted in the ortho position by a
Cl-C6 alkyl group, -NHSO2CF3, -(CH2)r (lH-tetrazol-5-yl),
-(CH2)rC(O)OR2, (CH2)rC(O)N(R2)(R6);
R3a is hydrogen, or Cl-C4 alkyl;
W is -CN, -C(O)OR2, -c(o)N(R2)(R2)~ -C(O)N(R2)(CH2)1 phenyl,
lH-tetrazol-5-yl, or-(CH2)rOR2;
X is hydrogen, -(CH2)qC(O)N(R2)(R6)~ or -(CH2)qC(O)OR2;
Y is hydrogen, C 1 -C8 aL~yl, -(CH2)t phenyl, -(CH2)t pyridyl, or
-(CH2)t~iazolyl;
R4 and R5 are independently hydrogen, C 1 -C6 aLtcyl, or substituted C 1-
C6 aL~yl where the substituents may be 1 to 5 halo, 1 to 3 hydroxyl,
S(O)m (Cl-C6 aL~yl) or phenyl;
R6 is hydrogen, or Cl-C6 aLlcyl;
A is:
R7
(CH2)~C--
2 s R7a
where x is 0, or 1;
R7 and R7a are independently hydrogen Cl-C6 aL~yl, trifluoromethyl,
phenyl, substituted C 1 -C6 aL~yl where the substituents are imidazolyl,
30 phenyl, indolyl, p-hydroxyphenyl, OR2, S(O)mR2~ C(O)O(C1-C6 alkyl)?
C5-C7 cycloaL~yl, N(R2)(R2)~ C(O)N(R2)(R2); or R7 and R7a can
independently be joined to one of R4 or Rs to forrn aL~cylene bridges
between the terminal nitrogen and the aL~yl portion of R7 or R7a groups
to form 5 or 6 membered rings; or R7 and R7a can be joined to one
another to form a C3 cycloaL~yl;
WO 95113069 ~ JS94/12816
~ 2175218
- 15 -
l is O or 1 ;
n is 2;
misO, 1,or2;
r is 0, 1, 2 or 3;
q is O or 1
tisOor 1;
and pharmaceutically acceptable salts and individual diastereomers
thereof.
More preferred compounds within this first embodiment
include those of Formula AIb:
H H ,R4
Rl~--N-C--A--N
1= Rs
N W
~X
~ y
R3
Formula AIb
wherein:
Rl is selected from the group consisting of: Cl-Clo aLkyl,
aryl (Cl-C3 alkyl)-, and aryl (Co-Cl aLkyl)-K-(C1-C2 aLkyl)-, where K is
O or S(O)m and the aryl is phenyl, pyridyl, naphthyl, or indolyl which
are optionally substituted by 1-2 Cl-C4 aLkyl, 1 to 2 halogen, 1 to 2 OR2,
S()m R2 or C(O)OR2;
R2 is hydrogen, C 1-C6 alkyl, or C3-C7 cycloalkyl and where two C 1 -C6
alkyl groups are present on one atom they may be optionally joined to
forrn a C5-C7 cyclic ring optionally including oxygen, sulfur or NR3a;
R3 is hydrogen or phenyl optionally substituted in the ortho position by a
Cl-C3 aLkyl group, (cH2)r(lH-tetrazol-5-yl) or (cH2)rc(o)oR2;
-
WO 9S/13069 PCI~/US94/12816
2175218 ~
- 16-
R3a is hydrogen, or Cl-C4 aLkyl;
W is -CN, -C(O)OR2, or -C(O)N(R2)R2);
X is hydrogen or C(O)OR2;
Y is hydrogen, benzyl, picoyl, or thiazolylmethyl;
R4 and R5 are independently hydrogen, Cl-C3 aLkyl, substituted Cl-C3
aLIcyl where the substituents may be 1 to 2 hydroxyl;
A is
~7
(CH2)~C,--
R7a
where x is 0, or 1;
R7 and R7a are independently hydrogen or Cl-C4 aLkyl;
misO, 1,or2;
risO, 1,or2;
and pharmaceutically acceptable salts and individual diastereomers
thereof.
The most preferred growth hormone releasing
30 compounds within this first embodiment include the following:
PCI~/US94~12816
WO 9S/13069
~ 21752~8
~ 1I NH2 ~N~3~NH
H ~H H ~
~ H2 ~ <~I-N
~ `1I NH2~ C NH2
~ CO2Et ~ CN
u~94ll~8l6
wo 95113069
2175218 ~
1 NH2 ~ r ~;
l o CO2Et H
NH2 ~ `1I NH2
H ~N~f CN ~ ~r
~`N ~ N--N
`1I NH2~¢~N`C~NH
2 5 ~CO2Et ~N~;CO,Et
WO 95/13069 PCI~/US94/12816
2175218
- 19-
~\j~ 1I NH2
H N CN
~CO2Et
and pharmaceutically acceptable salts and individual diastereomers
thereof.
A second embodiment of the present invention is directed
5 to the compounds of the structural formula BI:
H H O ,R4
Rl~N--C-A--N~
C=O Rs
2 o (CH2~
~X
R3 Y
Formula BI
25 wherein:
Rl is selected from the group consisting of:
Cl-Clo aLkyl, aryl, aryl(Cl-C6 alkyl), (C3-C7 cycloaLkyl)(Cl-C6 alkyl)-,
(Cl-Cs alkyl)-K-(Cl-C5 aL~cyl)-, aryl(Co-Cs alkyl)-K-(Cl-Cs alkyl)-,
and (C3-C7 cycloaLkyl)(Co-C5 alkyl)-K-(Cl-Cs alkyl)-, where K is 0,
30 S(O)m, N(R2)C(O), C(O)N(R2), OC(O), C(O)O, -CR2=CR2-, or-C--C-
~where aryl is selected from: phenyl, naphthyl, indolyl, azaindole,
pyridyl, benzothienyl, benzofuranyl, thiazolyl, and benzimidazolyl, and
R2 and alkyl may be further substituted by l to 9 halogen, S(O)mR2a, l
to 3 of OR2a or C(o)oR2a~ and aryl may be further substituted by l to 3
of C l -C6 alkyl, l to 3 of halogen, l to 2 of OR2, methylenedioxy,
WO 95113069 1'~ 9~/12816
2175218 ~
- 20 -
-S(O)rnR2, 1 to 2 of-CF3, -OCF3, nitro, -N(R2)c(o)(R2)~ -C(O)OR2,
-C(O)N(R2)(R2), -1H-tetrazol-5-yl, -S02N(R2)(R2), -N(R2)S02 phenyl,
or -N(R2)S02R2;
R2 is selected from: hydrogen, C l-C6 aL~yl, and C3-C7 cycloalkyl, and
where two C l-C6 aL~yl groups are present on one atom, they may be
optionally joined to form a C3-C8 cyclic ring, optionally includin~
oxygen, sulfur or NR3a, where R3a is hydrogen, or Cl-C6 aL~yl,
optionally subs~ituted by hydroxyl;
R2a is hydrogen, or Cl-C6 aL~yl optionally substituted by hydroxyl;
R3 is selected from: -(CH2)rphenyl, -(CH2)rnaphthyl, -Cl-Clo aL~yl,
-C3-C7 cycloaL~yl, aIld the phenyl, naphthyl and C3-C7 cycloaL~yl rings
may be substituted by 1 to 3 substituents selected from the group
consisting of: Cl-C6 aL~yl, halogen, -OR2, -NHS02CF3, -(CH2)rOR6,
-(CH2)rN(R2)(R6), -(cH2)r (R6), -(cH2)rc(o)oR2~ -(cH2)rc(o)oR6~
-(CH2)rOC(O)R2, -(CH2)rOC(O)R6, -(CH2)rC(O)R2, -(CH2)rC(O)R6,
(CH2)rC(O)N(R2)(R2), -(cH2)rc(o)N(R2)(R6) ~ -(CH2)rN(R2)C(O)R2
-(CH2)rN(R2)C(O)R6, -(CH2)rN(R6)C(O)R2, -(CH2)rN(R6)C(O)R6,
-(CH2)rN(R2)C(O)OR2,-(CH2)rN(R2)C(O)OR6,
-(CH2)rN(R6)C(O)OR2, -(cH2)rN(R6)c(o)oR6~
(CH2)rN(R2)C(O)N(R2)(R6), -(cH2)rN(R2)c(o)N(R2)(R2) .
-(CH2)rN(R6)C(O)N(R2)(R6), (cH2)rN(R2)so2R6
-(CH2)rN(R2)S02R2,-(CH2)rN(R6)S02R2, CH2)rN(R6)so2R6
2 5 (CH2)rOC(O)N(R2)(R6), -(CH2)rOC(O)N(R2)(R2),
-(CH2)rS02N(R2)(R6), -(cH2)rso2N(R2)(R2)~(cH2)rso2NHc(o)R6
(CH2)rS02NHC(O)R,,-(CH2)rS02NHC(O)OR6,
-(CH2)rS02NHC(O)OR2, -(cH2)rc(o)NHc(o)NR2
-(CH2)rC(O)NHC(O)NR6, -(CH2)rC(O)NHC(O)R2,
3 -(CH2)rCONHC(O)R6, -(CH2)rCONHSO2R6,-(CH2)rCONHSO~R-~,
(CH2)rCONHS02N(R2)R2), -(cH2)rcoNHso2N(R2)R6)~
-(CH2)rN(R2)SO2N(R2)R6), -(CH2)rN(R6)SO2N(R2)R6),
-(CH2)rS(O)mR6, and -(CH2)rs(O)mR2;
WO 95113069 2 1 7 5 2 1 8 PCI/US9~112816
R3a is hydrogen, or Cl-C6 aL~yl optionally substituted by hydroxyl;
X is selected from: hydrogen, -C~N, -(CH2)qN(R2)C(O)R2,
-(CH2~qN(R2)C(O)(CH2)taryl, -(cH2)qN(R2)so2(cH2)tar
-(CH2)qN(R2)SO2R2, -(CH2)qN(R2)C(O)N(R2)(CH2)taryl,
-(CH2)qN(R2)C(O)N(R2)(R2), -(cH2)qc(o)N(R2)(R2),
-(cH2)qc(o)N(R2)(cH2)taryL -(CH2)qc(O)oR2
-(cH2)qc(o)o(cH2)taryL -(cH2)qoR2~ -(cH2)qoc(o)R2
-(c H 2)q o c( o)(c H 2)taryl~ -(c H 2)q o c (o) N (R 2)(c H 2)taryl~
0 -(CH2)qOC(O)N(R2)(R2), -(cH2)qc(o)R2~ -(CH2)qC(O)(CH2)taryl,
-(CH2)qN(R2)C(O)OR2. -(cH2)qN(R2)so2N(R2)(R2),
-(CH2)qS(O)mR2, and -(cH2)qs(o)m(cH2)taryL where an R2, (CH2)q
and (CH2)t group may be optionally substituted by 1 to 2 C l-C4 aL~yl,
hydroxyl, Cl-C4 lower alkoxy, carboxyl, CONH2, S(O)mCH3,
carboxylate Cl-C4 aL~yl esters, or lH-tetrazol-5-yl, and aryl is phenyl,
naphthyl, pyridyl, thiazolyl, or lH-tetrazol-5-yl groups which may be
optionally substituted by 1 to 3 halogen, 1 to 3 -OR2, -CON(R2)(R2),
-C(O)OR2, 1 to 3 Cl-C4 aL~yl, -S(O)mR2, or lH-tetrazol-5-yl;
Y is selected from: hydrogen, Cl-Clo aL~yl, -(CH2)taryl,
-(CH2)q(C3-C7 cycloaL~yl), -(CH2)q-K-(Cl-C6 alkyl),
-(cH2)q-K-(cH2)taryl~-(cH2)q-K-(cH2)t(c3-c7 cycloaL~yl
cont~inin~ 0, NR2, S), and -(CH2)q-K-(CH2)t(C3-C7 cycloaL~yl), where
K is O, S(O)m, C(O)NR2, CH=CH, C_C, N(R2)C(O), C(O)NR2, C(O)O,
25 or OC(O), and where the aL~yl, R2, (CH2)q and (CH2)t groups may be
optionally substituted by C 1 -C4 aL~cyl, hydroxyl, C 1 -C4 lower aLlcoxy,
carboxyl, -CONH2 or carboxylate Cl-C4 aL~yl esters, and aryl is phenyl,
naphthyl, pyridyl, l-H-tetrazol-5-yl, thiazolyl, imidazolyl, indolyl,
pyrimidinyl, thiadiazolyl, pyrazolyl, oxazolyl, isoxazolyl, thiopheneyl,
quinolinyl, pyrrazinyl, or isothiazolyl which is optionally substituted by 1
to 3 halogen, 1 to 3 -OR2, 1 to 2 -N(R2)(R2),-C(O)OR2,
-C(O)N(R2)(R2), nitro, -NHC(O)R2,cyano, benzyl, 1 to 3 Cl-C4 aL~yl,
-S(O)mR2, or lH-tetrazol-5-yl;
WO9~/13069 2 1 752 ~ 8 ~ u~94~2816
- 22 -
R4 and R5 are independently hydrogen, Cl-c6 aLkyl, substituted Cl-
C6 aLI~yl where the substituents may be 1 to 5 halo, 1 to 3 hydroxy, 1
to 3 Cl-Clo aLkanoyloxy, 1 to 3 Cl-C6 aIkoxy, phenyl, phenoxy, 2-
furyl, Cl-C6 aLkoxycarbonyl, S(O)m(Cl-C6 alkyl); or R4 and Rs can
5 be taken together to form -(CH2)dLa(CH2)e- where La is C(R2)2, 0,
S()m or N(R2), d and e are independently 1 to 3 and R2 is as defined
above;
Als:
~7 ~7
_ (C H2)X--Ç--(CH2)y or _ z--(C H2)X--Ç--(C H2)y
D r~
~7a r~7a
where x and y are independently 0, 1, 2 or 3;
Z is N-R6a or 0, where R6a is hydrogen or C l-C6 aLIcyl;
20 R6 is hydrogen, Cl-c6 aLkyl, or (CH2)varyl, wherein the aLlcyl and
(CH2)V groups may be optionally substituted by 1-2 O(R2), S(O)mR2,
lH-tetrazol-S-yl, C(o)oR2~ C(o)N(R2)(R2) or S02N(R2)(R2),
N(R2)C(O)N(R2)(R2),and wherein aryl is phenyl, pyridyl, lH-tetrazol-
S-yl, triazolyl, imidazolyl, thiazolyl, pyrazolyl, tl~ 7.olyl, imidazolone-
25 l-yl, oxadiazolyl, benzimidazol-2-yl, triazolinone-yl, optionally
substituted with Cl-C6 aLkyl, C3-C6 cycloaLkyl, amino, or hydroxyl;
R7 and R7a are independently hydrogen, Cl-C6 aLkyl, trifluoromethyl,
phenyl, substituted Cl-C6 aLkyl where the substituents are imidazolyl,
phenyl, indolyl, p-hydroxyphenyl, OR2, S(O)mR2, C(O)OR2, C3-C7
30 cycloaLkyl, N(R2)(R2)~ C(o)N(R2)(R2); or R7 and R7a can
independently be joined to one or both of R4 and R5 croups to form
aLkylene bridges between the terminal nitrogen and the alkyl portion of
the R7 or R7a groups, wherein the bridge contains l to 5 carbons atoms;
or R7 and R7a can be joined to one another to form a C3-C7 cycloalkyl;
WO9S113069 2 1 752 1 8 Pcrrusg4/l28l6
misO, 1,or2;
nis l,2,or3;
qisO, 1,2,30r4;
ris0, 1,2,or3;
5 tisO, 1,2,or3;
visO, 1,or2;
and pharmaceutically acceptable salts and individual diastereomers
thereof.
Preferred compounds within this second embodiment
include those of Formula BIa:
H H R ,R4
R1~N--C -A--N~
C-O R5
(CH2~
) ( X
R3 Y
Formula BIa
wherein:
Rl is selected from the group consisting of:
Cl-Clo aL~yl, aryl (Cl-C4 aLkyl)-, C3-C6 cycloalkyl (Cl-C4 alkyl)-,
(Cl-C4 alkyl)-K-(C1-C2 aIkyl)-, aryl (Co-C2 alkyl)-K-(Cl-C2 aL~cyl)-,
25 and (C3-C7 cycloalkyl)(Co-C2 alkyl)-K-(Cl-C2 aL~cyl)-, where K is 0,
S(O)m, OC(O), or C(O)O, and the alkyl groups may be further
substituted by 1 to 7 halogen, S(O)mR2, 1 to 3 OR2 or C(O)OR2, and
aryl is phenyl, naphthyl, indolyl, pyridyl, benzimidazolyl, azaindoleyl,
benzothienyl or benzofuranyl which may be further substituted by 1-2
30 C1-C4 alkyl, 1 to 2 halogen, 1 to 2 -OR2, -S(O)mR2, or -C(O)OR2;
R2 is hydrogen, C 1-C6 alkyl, C3-C7 cycloalkyl and where two C1-C6
alkyl groups are present on one atom they may be optionally joined to
form a C4-C7 cyclic ring optionally including oxygen, sulfur or NR3a;
WO9S/13069 2 ~ 7 5 2 ~ 8 PCI~/US94/128~6
- 24-
R3 is phenyl which is optionally substituted by 1 to 2 C l-C6 aL~yl
groups, 1 to 2 halogen, or 1 to 2 -OR2, and which may be further
substituted in the ortho position by a substitutent selected from the group
5 consisting of:
-NHSO2CF3, -(cH2)roR6~ -(cH2)rN(R2)(R6)~ -(CH2)r (R6),
-(CH2)rC(0)0~2, -(cH2)rc(o)oR6~ -(CH2)rOC(O)R2,
-(CH2)rOC(O)R6, -(cH2)rc(o)R2~ -(CH2)rC(O)R6,
(CH2)rC(O)N(R2)(R2), -(cH2)rc(o)N(R2)(R6) ~ -(cH2)rN(R2)c(o)R2
0 -(CH2)rN(R2)C(O)R6, -(cH2)rN(R6)c(o)R2~ -(CH2)rN(R6)C(O)R6,
-(CH2)rN(R2)C(O)OR2,~(CH2)rN(R2)C(O)OR6~
-(CH2)rN(R6)C(O)OR2, -(CH2)rN(R6)C(O)OR6,
(CH2)rN(R2)C(O)N(R2)(R6), -(cH2)rN(R2)c(o)N(R2)(R2)~
-(CH2)rN(R6)C(O)N(R2)(R6), (CH2)rN(R2)S02R6,
5 -(CH2)rN(R2)SO2R2, -(cH2)rN(R6)so2R2~ CH2)rN(R6)SO2R6,
(CH2)rOC(O)N(R2)(R6), -(cH2)roc(o)N(R2)(R2)
-(CH2)rS02N(R2)(R6), -(cH2)rso2N(R2)(R2)~(cH2)rso2NHc(o)R6
(CH2)rS02NHC(O)R2, -(CH2)rSO2NHC(O)OR6,
-(CH2)rS02NHC(O)OR2, -(cH2)rc(o)NHc(o)NR2
2 -(CH2)rC(O)NHC(O)NR6, -(CH2)rC(O)NHC(O)R2,
-(CH2)rCONHC(O)R6, -(cH2)rcoNHso2~6~-(cH2)rcoNHso2R2,
(CH2)rCONHS02N(R2)R2), -(cH2)rcoNHso2N(R2)R6)~
-(CH2)rN(R2)SO2N(R2)R6), -(cH2)rN(R6)so2N(R2)R6),
-(CH2)rS(O)mR6, and-(cH2)rs(o)mR2;
R3a is hydrogen, or Cl-C4 aL~yl;
X is selected from: hydro~en, -(CH2)qN(R2)C(O)R2,
-(CH2)qN(R2)C(O)(CH2)taryl, (-cH2)qN(R2)c(o)oR2
3 -(CH2)qN(R2)SO2(cH2)tarYl~ -(CH2)qN(R2)SO2R2.
-(CH2)qN(R2)C(O)N(R2)(CH2)taryl. -(cH2)qN(R2)c(o)N(R2)(R2)~ -
-(CH2)qC(O)N(R2)(R2), -(cH2)qc(o)N(R2)(cH2)tar
-(CH2)qC(O)OR2, -(cH2)qc(o)o(cH2)taryL -(cH2)qoc(o)R2
-(CH2)qOC(O)(CH2)taryl, -(CH2)qS(O)mR2, and
WO 95/13069 2 1 7 ~ 2 1 8 PCr/US94/12816
.
- 25 -
-(cH2)qs(o)m(cH2)taryl7 where an R2 group may be optionally
substituted by hydroxyl, carboxyl, CONH2~ S(O)mCH3, carboxylate Cl-
C4 alkyl esters, or tetrazole and the aryl which is phenyl, naphthyl,
pyridyl or 1-H-tetrazolyl may be optionally substituted by 1 to 2 halogen,
1 to 2 -OR2, -CONH2, -C(O)OR2, 1 to 3 Cl-C4 aL~yl, -S(O)mR2, or lH-
tetrazole-5-yl;
Y is selected from: hydrogen, Cl-c8 alkyl, (cH2)taryL -(cH2)q(c5-c6
cycloaLkyl), -(cH2)q-K-(cl-c6 aLkyl), -(CH2)q-K-(CH2)taryl,
o -(CH2)q-K-(CH2)t(C3-C7 cycloalkyl cont~ining O, NR2, or S), and
-(CH2)q-K-(CH2)t (C5-C6 cycloaL~yl), where K is O or S(O)m and
where the alkyl groups may be optionally substituted by hydroxyl,
carboxyl, CONH2, carboxylate Cl-C4 aLkyl esters or lH-tetrazole-S-yl
and the aryl which is phenyl, naphthyl, pyridyl, 1-H-tetrazolyl, thiazolyl,
imidazolyl, pyrimidinyl, thiadiazolyl, pyrazolyl, oxazolyl, isoxazolyl or
thiopheneyl is optionally substituted by 1 to 3 halogen, 1 to 3 -OR2, 1 to
2 -N(R2)(R2), -C(O)OR2, -C(O)N(R2)(R2), cyano, 1 to 2 C1-C4 alkyl,
benzyl, -S(O)mR2, or lH-tetrazol-5-yl;
R4 and R5 are independently hydrogen, C1-C6 alkyl,or substituted Cl-
C6 aL~yl where the substituents may be 1 to 5 halo, 1 to 3 hydroxyl,
S(O)m (Cl-C6 aLkyl) or phenyl;
R6 is H, Cl-C6 alkyl, or (cH2)varyL wherein the (CH2)V and aLIcyl
groups may be optionally substituted by 1-2 O(R2), S(O)mR2, C(O)OR2,
C(O)N(R2)(R2) or So2N(R2)(R2)~ N(R2)C(O)N(R2)(R2), wherein the
aryl group could be phenyl, pyridyl, 1 H-tetrazol-5-yl, triazolyl,
imidazolyl, thiazolyl, oxadiazolyl, pyrazolyl, thiadiazolyl, benzimidazol-
2-yl, optionally substituted with C1-C6 alkyl, C3-C6 cycloalkyl, amino,
or hydroxyl;
A is :
WO95113069 ~ ~ 7~7 ~ ~ PCI/US9~/12816
- 26 -
F~27
(CH2))~ C--
R7a
5 where x is 0, or 1;
R7 and R7a are independently hydrogen C 1 -C6 aL~cyl, trifluoromethyl,
phenyl, substituted Cl-C6 aLkyl where the substituents are imidazolyl,
phenyl, indolyl, p-hydroxyphenyl, OR2, S(O)mR2, C(O)OR2, C5-q
cycloaL~cyl, N(R2)(R2), C(O)N(R2)(R2); or R7 and R7a can
10 independently be joined to one of R4 or R5 to form aL~cylene bridges
between the terrnin~l nitrogen and the aLkyl portion of R7 or R7a groups
to foIm 5 or 6 membered rings; or R7 and R7a can be joined to one
another to form a C3 cycloaLkyl;
15 nis2;
misO, 1,or2;
risO, 1,2,or3;
qisO, 1,2,or3;
tisO, 1,2,or3;
20 visO, 1,or2,
and pharmaceutically acceptable salts and individual diastereonners
thereof.
wo 9S/13069 2 ~ 7 5 2 1 8 PCI/US94112816
- 27 -
More preferred compounds within this second embodiment
include those of Formula BIb:
H~ H ,R
Rl--C-N--C-A--N~ R4
C=O O
~X
o R3
Formula BIb
wherein:
Rl is selected from the group consisting of: Cl-Clo aL~yl,
aryl (Cl-C3 aL~yl)-, (C3-C7 cycloalkyl)(Cl-C3 aLtcyl)-, and
aryl (Co-Cl alkyl)-K-(Cl-C2 aL~yl)-, where K is O or S(O)m and aryl is
specifically phenyl, pyridyl, naphthyl, indolyl, azaindolyl, or
benzimidazolyl which is optionally substituted by 1-2 C l-C4 aL~yl, 1 to 2
halogen, 1 to 2 OR2, S(O)m R2, or C(O)OR2;
R2 is hydrogen, C l-C6 aL~yl, C3-C7 cycloaL~yl and where two C l-C6
aL~yl groups are present on one atom they may be optionally joined to
form a C5-C7 cyclic ring optionally including oxygen, sulfur or NR3a;
R3 is phenyl optionally substituted by 1 to 2 C1-C6 alkyl groups, 1 to 2
halogen or 1 to 2 OR2, and which may be further substituted in the ortho
position by a substitutent selected from the group consisting of:
-NHS02CF3, -(cH2)roR6~ -(cH2)rN(R2)(R6)~ -(CH2)r (R6),
-(CH2)rC(O)OR6, -(CH2)rOC(O)R2, -(CH2)rOc(O)R6~
3 0 -(CH2)rC(O)R2, -(CH2)rC(O)R6, -(CH2)rC(O)N(R2)(R2),
-(CH2)rC(O)N(R2)(R6), -(cH2)rN(R2)c(o)R2 -(cH2)rN(R2)c(o)R6~
-(CH2)rN(R6)C(O)R2, -(cH2)rN(R6)c(o)R6~ -(cH2)rN(R2)c(o)oR2 . -
(CH2)rN(R2)C(O)OR6, -(cH2)rN(R6)c(o)oR2~
-(CH2)rN(R6)C(O)OR6, (CH2)rN(R2)C(O)N(R2)(R6),
-(CH2)rN(R2)C(O)N(R2)(R2), -(cH2)rN(R6)c(o)N(R2)(R6),
WO 95/13069 2 1 7 5 2 ~ 8 PCr/US94/12816
- 28 -
(CH2)rN(R2)S02R6, -(cH2)rN(R2)so2R27 -(cH2)rN(R6)so2R27
CH2)rN(R6)S02R6, (cH2)roc(o)N(R2)(R6)~
-(CH2)rOC(O)N(R2)(R2), -(cH2)rso2N(R2)(R6)7
-(CH2)rSO2N(R2)(R2),(CH2)rSO2NHC(O)R6, (cH2)rso2NHc(o)R27
-(CH2)rSO2NHC(O)OR6, -(CH2)rS2NHC(O)OR2,
-(CH2)rCONHSO2R6,-(CH2)rCONHSO2R2, -(CH2)rS(O)mR6, and
-(CH2)rS(O)mR2;
R3a is hydrogen, or Cl-C4 alkyl;
X is selected from: hydrogen, -(cH2)qN(R2)c(o)R2~
-(CH2)qN(R2)C(O)(CH2)taryl, -(cH2)q N(R2)so2(cH2)taryL -(CH2)q
N(R2)S02R2, -(CH2)qN(R2)C(O)N(R2)(CH2)taryl,
-(CH2)qN(R2)C(O)N(R2)(R2), -(cH2)qc(o)N(R2)(R2)~
15 -(CH2)qN(R2)C(O)OR2, -(CH2)qC(O)N(R2)(CH2)taryl,
-(CH2)qC(O)OR2, -(cH2)qc(o)o(cH2)taryL -(cH2)qoc(o)R27
-(CH2)qOC(O)(CH2)taryl, -(CH2)qS(O)mR2, and
-(CH2)qS(O)m(CH2)taryl, where an R2 group may be optionally
substituted by hydroxyl, carboxyl, -CONH2, -S(O)mCH3, carboxylate
20 C l-C4 alkyl esters or tetrazole and aryl is phenyl, napthyl or pyridyl
which may be further substituted by 1-2 halogen, 1 to 2 OR2, C(O)OR2,
1 to 3 Cl-C4 aLkyl, S(O)mR2, or lH-tetrazole-S-yl;
Y is selected from: hydrogen, C l-Cg aLkyl, (CH2)taryl, -(CH2)q C5-C7
25 cycloaLkyl, -(CH2)q-K-(Cl -C6 alkyl), -(CH2)q-K-(CH2)taryl, and
-(CH2)q-K-(CH2)t (C5-C6 cycloaLkyl), where K is S(O)m and where the
aLkyl groups may be optionally substituted by hydroxyl, carboxyl,
CONH2, carboxylate Cl-C4 alkyl esters or lH-tetrazole-S-yl and aryl is
speci~lcally phenyl, napthyl, pyridyl, thiazolyl, thiopheneyl, pyrazolyl,
3 oxazolyl, isoxazolyl or imidazolyl which may be optionally substituted
by 1 to 2 halogen, 1 to 2 OR2, 1 to 2 -N(R2)(R2), -CO(OR2), 1 to 2 C1- -
C4 alkyl, S(O)mR2, or lH-tetrazol-S-yl;
WO 95113069 PCI~/US94112816
~ 2175218
- 29 -
R4 and R5 are independently hydrogen, Cl-C4 aLlcyl, substituted Cl-C3
aL~yl where the substituents may be 1 to 2 hydroxyl;
R6 is hydrogen, Cl-C6 alkyl or (CH2)varyl, wherein the Cl-C6 aL~yl and
s the (CH2)varyl groups may be optionally substituted by 1-2 O(R2),
S(O)mR2, C(O)OR2, C(o)N(R2)(R2) or S02N(R2)(R2),
N(R2)C(O)N(R2)(R2), wherein aryl is specifically phenyl, pyridyl, lH-
tetrazol-5-yl, triazolyl, imidazolyl, thiazolyl, oxadiazolyl, pyrazolyl,
thiadiazolyl, benzimidazol-2-yl, optionally substituted with Cl-C6 aL~yl,
C3-C6 cycloaLlcyl, amino, or hydroxyl;
Ais
~7
(CH2)~C,--
R7a
where x is 0, or 1;
20 R7 and R7a are independently hydrogen, Cl-C2 aL~yl, phenyl, substituted
Cl-C6 aL~yl wherein the sub~Lilulel-t is imidazolyl, phenyl, indolyl, p-
hydroxyphenyl, OR2, S(O)mR2; or R7 and R7a can be independently be
joined to one another to form a C3 cycloaL~yl;
25 misO, 1,or2;
risO, 1,2,or3;
qisO, 1,2,or3;
tisO, 1,2,or3;
visO, 1,or2;
3 o and pharmaceutically acceptable salts and individual diastereomers
thereof.
Still more preferred compounds within this second
embodiment are realized in Formula BIc:
WO9S~13069 2 1 752 1 8 ~ u~9~2816
- 30 -
H H
Rl--C--N--C -A--N' R4
C=o o R5
~<X
R3
Formula BIc
wherein:
o R1 is selected from the group consisting of:
~3' (1-2)F--~ MeO--~CH2-
H
~ N CH3 ~ N,CH2- ¢~3,CH2-
H
~ CH2- CH2- CH2-
W MeO--~ HO--~l
3~ CH2- 3~o,CH2- 3~o,CH2-
3 0 ~~' (1-2) F--~ (1-2) F--~ ~ ,CH2- J
~~ (1-2) F--~f CH2- ¢~ CH2-
or their regioisomers where not specified;
WO 95/13069 PCr/US94/~2816
2175218
- R2 is hydrogen, Cl-C6 aL~yl, or C3-C7 cycloaL~cyl and where two Cl-C6
aL~yl groups are present on one atom they may be optionally joined to
form a C5-C7 cyclic ring optionally including oxygen, sulfur or NR3a;
R3 is phenyl optionally substituted in the ortho position with a
substitutent selected from the group consisting of:
-NHS02CF3, -(cH2)roR6~ -(cH2)r (R6), -(cH2)rc(o)oR2
-(CH2)rC(O)OR6, -(CH2)rOC(O)R2, -(CH2)rOC(O)R6,
-(CH2)rC(O)R2, -(CH2)rC(O)R6, (cH2)rc(o)N(R2)(R2)~
-(CH2)rC(O)N(R2)(R6), -(cH2)rN(R2)c(o)R2 -(CH2)rN(R2)C(O)R6,
-(CH2)rN(R6)C(O)R2, -(cH2)rN(R6)c(o)R6~ -(CH2)rN(R2)C(O)OR2, -
(CH2)rN(R2)C(O)OR6, -(cH2)rN(R6)c(o)oR2
-(CH2)rN(R6)C(O)OR6, (cH2)rN(R2)c(o)N(R2)(R6)~
15 -(CH2)rN(R2)C(O)N(R2)(R2), -(cH2)rN(R6)c(o)N(R2)(R6) ,
(CH2)rN(R2)S02R6, -(cH2)rN(R2)so2R2~ -(cH2)rN(R6)so2R2
CH2)rN(R6)SO2R6, (CH2)rOC(O)N(R2)(R6),
-(CH2)rOC(O)N(R2)(R2), -(cH2)rso2N(R2)(R6)~
-(CH2)rS02N(R2)(R2),(CH2)rS02NHC(O)R6, (cH2)rso2NHc(o)R2
2 -(CH2)rSO2NHC(O)OR6, -(CH2)rSO2NHC(O)OR2,
-(CH2)rCONHSO2R6,-(CH2)rCONHSO2R2, -(CH2)rS(O)mR6, and
-(CH2)rS(O)mR2;
R3a is hydrogen, or Cl-C4 aL~yl;
X is selected from the group consisting of: hydrogen,
wo 95/13069 PCrlUS9~/12816
2t75218 ~
- 32 -
O O O O CH3
J~ CH3 J~o~ Et J~O ,CH3 J~o CH3
o o o
J~O~`CH3 J~o~ CH3 J~OH
~O~ CH3 J~o~ - H
O O O O CH3
J~ -CH3 J~ N - Et J~ N ~ CH3 ~ N 1CH3
o o o
15 /I~H' CH3 J~H~ CH3 J~NH2
~O ~S -HN N ~OH -HN N,CH3
NH J~'CH3 NH ~O -NH ~-- ~H3
25 -NH `~ 3 NH ,S~CH -NH ~ HN J~ N ~SCH3
-0~ -O CH3 -~l3 -HN ~o,CH3
Y is selected from the group consisting of:
hydrogen, Cl-Cg aLkyl, (CH2)taryl, -(CH2)q C5-C7 cycloalkyl, -(CH2)q-
K-(Cl-C6 alkyl), -(cH2)q-K-(cH2)taryL or -(CH2)q-K-(CH2)t (C5-C6
cycloaLkyl) where K is S(O)m and where the aLkyl groups may be
wo 95rl3069 2 1 7 5 2 1 8 PCI'IUS94/12816
optionally substituted by hydroxyl, carboxyl, CONH2~ carboxylate Cl-
C4 aLkyl esters or lH-tetrazole-5-yl, and where aryl is specifically
phenyl, naphthyl, pyridyl, thiazolyl, thiopheneyl, pyrazolyl, oxazolyl,
isoxazolyl, thi~ 7.olyl, pyrimidinyl, or imidazolyl, which may be
optionally substituted by 1 to 2 halogen, 1 to 2 OR2, CO(OR2), 1 to 2
Cl-C4 aL~cyl, S(O)mR2 or lH-tetrazol-5-yl;
A is selected from the group consisting of:
0 ~<CH3 H3C CH3 l3 ,~CH3 H3CXCH3
H3C~,CH3 ~,NH N~\ ~¢~
R4 and R5 are independently selected from the group consisting of:
--H --CH3 --CH2CH3 /~,CH3 ~CH20H
OH OH
R6 is hydrogen, Cl-C6 aL~cyl or (CH2)varyl wherein the alkyl and
(CH2)v groups may be optionally substituted by halogen, OR2,
25 N(R2)(R2), C3-C6 cycloaLkyl, lH-tetrazol-5-yl, C(O)OR2,
C(O)N(R2)(R2), S02N(R2)(R2) or N(R2)C(O)N(R2)(R2), wherein aryl
is selected from the following aromatic groups and their regioisomers:
wo gS/13069 Pcr/us94/12816
2~ 75218
H
--~' NH ~ Y ~
H
N~ ~N
S 1`1, N~ ~\=N,
O--N N - N O--N
where the aromatic groups are optionally substituted with Cl-C2 aLkyl,
-N(R2)(R2), or hydroxy;
misO, 1,or2;
risO, 1,2,or3;
q is O or 1 ;
tisOor l;
v is O or 1 ;
and pharmaceutically acceptable salts and individual diastereomers
thereof.
3 Representative of the still more preferred compounds
within this second embodiment include the following: -
WO 95/13069 2 1 7 5 2 1 8 PCI~/US94112816
¢~\~N~NH2 ~CO O
~ ~-N
~3~~ ^r b>~NH2 Nb~NH2
C, O O CO O
N N~
~ ~N ,OH [~ SO2NHCH3
WO 95/13069 PClr/US91/12816
2~75218
- 36-
~0 0 ~ CO O
H ~\OEt
10 ~' ~>~NH2 ,~--^r ~NH2
N CO O CO O
~CO2Et ~CO2Et
~N,N ~ H,N
cis dl, cis d2, trans d1, trans d2 cis dl, cis d2, trans d1, trans d2
~NH2 ~ 2
C, O O C, O O
~N~ ~CO2Et
~H ~ NN
cis d1, cis d2, trans d1, trans d2
WO 95/13069 2 1 7 5 2 1 8 PCI/US94/12816
- 37 -
O ¢~, O
H ~ N ~ OH ~CO2Et
~OEt
0 cis d1, cis d2, trans d1, trans d2 cis d1, cis d2, trans d1, trans d2
and pharmaceutically acceptable salts and individual diastereomers
thereof where not otherwise specified.
All of the still more preferred compounds shown above have
5 at least one asymmetric center. Additional asymmetric centers may be
present on the molecule depending upon the nature of the substituents on
the piperidine ring. Each such asymmetric center will produce two
optical isomers and it is intended that all such optical isomers, as
separated, pure or partially puri~led optical isomers, racemic mixtures or
2 diastereomeric mixtures thereof, be included within the ambit of the
present invention.
The most preferred compounds within this second
embodiment include the following:
2 s ~ b~N H2 ~ ~N H2
~ N-N
wo 95113069 ~ 9~ll28l6
217~
- 38 -
~ ~ ~NH2 ~ ~ ~NH2
C,O O C,O O
N ~N~
1~ N ~OH ~--SO2NHCH3
~N~NH ¢~0 o
C~/ ~H ~ OEt
NH2 ~b~NH2
~ ~,CO2Et ~ ~,CO2Et
~ N--N ~/ N--N
[~ 3~N,N ~ N,N
cis dl, cis d2 cis d1, Cis d2
W O 9S113069 2 1 752 1 8 1 ~-1/U~94tl2816
- 39 -
~ Nb~ NH2
CO O Ç
N- N ~CO2Et
[~/</H ' N ~/</H ' N
0
cis d1, CiS d2
--~ ~NH2 ¢ ~ ~NH2
N CO O Ç
H ~f NH~OH ~CO2Et
2o [~ ~OEt
cis d1, Cis d2 cis dl, QiS d2
and their pharmaceutically acceptable salts and individual diasteromers5 thereof where not otherwise specified.
WO 95/13069 PCI~/US94/12816
2~75218 ~
- 40 -
A third embodiment of the present invention is directed to
the compounds of the structural formula CI:
H H o R4
R1~N--C-A--N\
C=O R5
(C~2~
~X
Formula CI
wherein:
Rl is selected from the group consisting of:
5 Cl-Clo aLkyI, aryl, aryl(Cl-C6 aL~yl), (C3-C7 cycloalkyl)(Cl-C6 aL~yl)-,
(Cl-C5 alkyl)-K-(Cl-C5 alkyl)-, aryl(Co-C5 aL~yl)-K-(Cl-Cs alkyl)-,
and (C3-C7 cycloalkyl)(Co-Cs alkyl)-K-(Cl-Cs alkyl)-, where K is O,
S(O)m, N(R2)C(O), C(O)N(R2), OC(O), C(O)O, -CR2=CR2-, or-C=C-,
where aryl is selected from: phenyl, naphthyl, indolyl, azaindole,
20 pyridLyl, benzothienyl, benzofuranyl, thiazolyl, and benzimidazolyl, and
R2 and alkyl may be fur~er substituted by 1 to 9 halogen, S(O)mR2a, 1
to 3 of OR2a or C(O)OR2a, and aryl may be further substituted by l to 3
of C1-C6 alkyl, l to 3 of halogen, l to 2 of OR2, methylenedioxy,
-S(O)mR2, l to 2 of-CF3, -OCF3, nitro, -N(R2)c(o)(R2)7 -C(O)OR2,
2 5 -c(o)N(R2)(R2)~ -1 H-tetrazol-5-yl, -SO2N(R2)(R2), -N(R2)SO2 phenyl,
or -N(R2)S02R2;
R2 is selected from: hydrogen, C1-C6 alkyl, and C3-C7 cycloalkyl, and
where two C 1-C6 alkyl groups are present on one atom, they may be
3 optionally joined to form a C3-Cg cyclic ring, optionally including
oxygen, sulfur or NR3a, where R3a is hydrogen, or Cl-C6 alkyl,
optionally substituted by hydroxyl;
R2a is hydrogen, or C1-C6 alkyl optionally substituted by hydroxyl;
WO 95/13069 PCI~/US9~/12816
~ 21 7521 8
- 41 -
X is selected from: hydrogen, -C--N, -(CH2)qN(R2)C(O)R2,
-(CH2)qN(R2)C(O)(CH2)taryl, -(cH2)qN(R2)so2(cH2)tar
-(CH2)qN(R2)S02R2, -(CH2)qN(R2)C(O)N(R2)(CH2)taryl,
-(CH2)qN(R2)C(O)N(R2)(R2), -(cH2)qc(o)N(R2)(R2)~
5 -(CH2)qC(O)N(R2)(CH2)taryl~ -(CH2)qC(O)OR2,
-(cH2)qc(o)o(cH2)taryL -(cH2)qoR2~ -(cH2)qoc(o)R2
-(c H 2)q o c(o )(c H 2)taryl~ -(c H 2)q o c (o ) N (R 2)(c H 2)taryl~
-(CH2)qOC(O)N(R2)(R2), -(cH2)qc(o)R2~ -(CH2)qC(O)(CH2)taryl,
-(CH2)qN(R2)C(O)OR2, -(cH2)qN(R2)so2N(R2)(R2)~
o -(CH2)qS(O)mR2, and -(CH2)qS(O)m(CH2)taryl~ where an R2, (CH2)q
and (CH2)t group may be optionally substituted by 1 to 2 C l-C4 aL~yl,
hydroxyl, Cl-C4 lower aL~oxy, carboxyl, CONH2, S(O)mCH3,
carboxylate Cl-C4 aL~yl esters, or lH-tetrazol-5-yl, and aryl is phenyl,
naphthyl, pyridyl, thiazolyl, or lH-tetrazol-5-yl groups which may be
5 optionally substituted by 1 to 3 halogen, 1 to 3 -OR2, -CON(R2)(R2),
-C(O)OR2, 1 to 3 Cl-C4 aL~yl, -S(O)mR2, or lH-tetrazol-S-yl;
Y is selected from: hydrogen, Cl-Clo aL~yl, -(CH2)taryl,
-(CH2)q(C3-C7 cycloaL~yl), -(CH2)q-K-(Cl-C6 aL~yl),
20 -(cH2)q-K-(cH2)taryL-(cH2)q-K-(cH2)t(c3-c7 cycloalkyl
cont~inin~ O, NR2, S), and -(CH2)q-K-(CH2)t(C3-C7 cycloaL~yl), where
K is O, S(O)m, C(O)NR2, CH=CH, C-C, N(R2)C(O), C(O)NR2, C(O)O,
or OC(O), and where the aL~yl, R2, (CH2)q and (CH2)t groups may be
optionally substituted by Cl -C4 aL~yl, hydroxyl, C l-C4 lower aLlcoxy,
25 carboxyl, -CONH2 or carboxylate Cl-C4 aL~yl esters, and aryl is phenyl,
naphthyl, pyridyl, l-H-tetrazol-5-yl, thiazolyl, imidazolyl, indolyl,
pyrimidinyl, thiadiazolyl, pyrazolyl, oxazolyl, isoxazolyl, thiopheneyl,
quinolinyl, pyrazinyl, or isothiazolyl which is optionally substituted by l
- to 3 halogen, 1 to 3 -OR2, -C(O)OR2, -C(O)N(R2)(R2), nitro, cyano,
30 benzyl, 1 to 3 Cl-C4 aL~yl, -S(O)mR2, or lH-tetrazol-S-yl, with the
- proviso that if X is hydrogen, Y is other than hydrogen;
R4 and R5 are independently hydrogen, Cl-C6 aL~yl, or substituted Cl-
C6 aL~yl where the substituents may be 1 to 5 halo, 1 to 3 hydroxy, 1 to 3
WO gs113069 2 ~ 7 5 2 1 ~ 94,12816
- 42 -
Cl-Clo aLkanoyloxy, 1 to 3 Cl-C6 aLkoxy, phenyl, phenyloxy, 2-furyl,
Cl-C6 aLkoxycarbonyl, S(O)m(Cl-C6 aLkyl), or R4 and Rs may be taken
together to form -(CH2)d-La(CH2)e- where La is -C(R2)2-, O, S(O)m or
N(R2), d and e are independently 1 to 3 and R2 is as defined above;
Ais:
~7 ~7
(CH2)x--C--(CH2)y or --z--(CH2)X--C--(CH2)y
R7a R7a
where x and y are independently 0, 1, 2 or 3;
Z is N-R6a or 0, where R6a is hydrogen or C 1 -C6 aLkyl;
lS
R7 and R7a are independently hydrogen, Cl-C6 aL~yl, trifluoromethyl,
phenyl, or substituted C l-C6 aLkyl where the substituents are imidazolyl,
phenyl, indolyl, p-hydroxyphenyl, OR2, S(O)mR2, C(O)OR2, C3-C7
cycloaL~yl, N(R2)(R2), C(O)N(R2)(R2), or R7 and R7a may
20 independently be joined to one or both of R4 and Rs groups to form an
aL~cyIene bridge between the termin~l nitrogen and the aLkyl portion of the
R7 or R7a groups, wherein ~e bridge contains 1 to 5 carbons atoms, or
R7 and R7a can be joined to one another to form C3-C7 cycloaL~yl;
misO, 1,or2;
nis 1,2,or3;
qisO, 1,2,3,or4;
tisO, 1,2,or3;
and pharmaceutically acceptable salts and individual diastereomers
thereof.
WO 9S113069 2 1 7 5 2 1 8 ~ 94,~28l6
- 43 -
Preferred compounds within this third embodiment include
those of Formula CIa:
H H
, R4
5Rl-C*-N--C-A--N,
C=O R5
(CH2)n~
~ X
y
Formula CIa
wherein:
Rl is selected from the group consisting of:
Cl-Clo alkyl, aryl (Cl-C4 aL~yl)-, C3-C6 cycloaL~yl (Cl-C4 aL~yl)-,
(Cl-C4 aL~yl)-K-(Cl-C2 aL~yl)-, aryl (CO-C2 aL~yl)-K-(Cl-C2 aL~yl)-,
and (C3-C7 cycloaL~yl)(Co-C2 aL~yl)-K-(Cl-C2 alkyl)-, where K is O,
S(O)m, OC(O), or C(O)O, and the aL~yl groups may be further
substituted by 1 to 7 halogen, S(O)mR2, 1 to 3 OR2 or C(O)OR2, and
20 aryl is phenyl, naphthyl, indolyl, pyridyl, benzimidazolyl, azaindoleyl,
benzothienyl or benzofuranyl which may be further substituted by 1-2
Cl-C4 aL~yl, 1 to 2 halogen, 1 to 2 -OR2, -s(o)mR2~ or -C(O)OR2;
R2 is hydrogen, C l-C6 alkyl, or C3-C7 cycloaL~yl, and where two Cl-C6
25 aL~yl groups are present on one atom they may be optionally joined to
form a C4-C7 cyclic ring optionally including oxygen, sulfur or NR3a;
R3a is hydrogen, or Cl-C4 aL~cyl;
- 30 X is selected from: hydrogen, -(cH2)qN(R2)c(o)R2~
-(CH2)qN(R2)C(O)(CH2)taryl, -(CH2)qN(R2)C(O)OR2,
- -(CH2)qN(R2)S02(CH2)taryl, -(cH2)qN(R2)so2R2
-(CH2)qN(R2)C(O)N(R2)(CH2)taryl, -(cH2)qN(R2)c(o)N(R2)(R2)~
-(CH2)qC(O)N(R2)(R2), -(CH2)qC(O)N(R2)(CH2)taryl,
-(CH2)qC(O)OR2, -(cH2)qc(o)o(cH2)taryL -(cH2)qoc(o)R2
WO 95113069 PCI'IUS94/12816
2175218
-- 44 -
-(CH2)qOC(O)(CH2)taryl, -(CH2)qS(O)mR2, and
-(CH2)qS(O)m(CH2)taryl, where an R2 group may be optionally
substituted by hydroxyl, carboxyl, CONH2, S(O)mCH3, carboxylate C 1-
C4 aL~yl esters, or tetrazole, and aryl is phenyl, naphthyl, pyridyl or 1-H-
5 tetrazolyl which may be optionally substituted by 1 to 2 halogen, 1 to 2
-OR2, -CONH2, -C(O)OR2, 1 to 3 Cl-C4 aL~yl, -S(O)mR2, or lH-
tetrazole-S-yl;
Y is selected from: hydrogen, Cl-c8 alkyl, (cH2)taryL -(cH2)q(cs-c6
cycloaLkyl), -(CH2)q-K-(C l -C6 aLkyl), -(CH2)q-K-(CH2)taryl,
-(CH2)q-K-(CH2)ffC3-C7 cycloaL~yl cont~ining 0, NR2, or S), and
-(CH2)q-K-(CH2)t (C5-C6 cycloaL~yl), where K is O or S(O)m and
where the aL~yl groups may be optionally substituted by hydroxyl,
carboxyl, CONH2, carboxylate Cl-C4 aL~yl esters or lH-tetrazole-S-yl
5 and aryl is phenyl, naphthyl, pyridyl, l-H-tetrazolyl, thiazolyl,
imidazolyl, indolyl, pyrirnidinyl, t~ 7olyl, pyrazolyl, oxazolyl,
isoxazolyl, or thiopheneyl which is optionally substitllterl by 1 to 3
halogen, 1 to 3 -OR2, -C(O)OR2, -C(O)N(R2)(R2), cyano, 1 to 2 Cl-C4
aL~yl, benzyl, -S(O)mR2, or lH-tetrazol-S-yl, with the proviso that if X is
20 hydrogen, Y is other than hydrogen;
R4 and R5 are independently hydrogen, Cl-C6 aL~yl, or substituted Cl-
C6 aL~yl where the substituents may be 1 to 5 halo, 1 to 3 hydroxyl,
S(O)m (C l-C6 aL~cyl) or phenyl;
25 Ais:
~7
(CH2)~Ç--
R7a
where x is 0, or 1 ;
R7 and R7a are independently hydrogen C l-C6 aL~yl, trifluoromethyl,
phenyl, substituted C l-C6 aL~cyl where the substituents are imidazolyl,
phenyl, indolyl, p-hydroxyphenyl, OR2, S(O)mR2, C(O)OR2, C5-C~,~
wo95/13069 2 ~ 752 1 8 ~ us~4tl28l6
- 45 -
cycloaL~yl, -N(R2)(R2), -C(O)N(R2)(R2); or R7 and R7a can
independently be joined to one of R4 or R5 to form aL~ylene bridges
between the terminal nitrogen and the aL~yl portion of R7 or R7a groups
to form S or 6 membered rings; or R7 and R7a can be joined to one
5 another to form a C3 cycloaL~yl;
nis2;
misO, 1,or2;
qisO, 1,2,or3;
t is O, 1, 2, or 3;
and pharmaceutically acceptable salts and individual diastereomers
thereof.
More preferred compounds within this third embodiment
5 include those of Formula CIb:
/ R4
Rl--c~ N--,C, -A--N~
C=O R5
N
~X
y
Formula CIb
wherein:
Rl is selected from the group consisting of: Cl-Clo aL~yl,
aryl (Cl-C3 aL~yl)-, (C3-C7 cycloaL~yl)(Cl-C3 aL~yl)-, and
aryl (Co-Cl aL~yl)-K-(Cl-C2 aL~yl)-, where K is O or S(O)m and the aryl
3 o is phenyl, pyridyl, naphthyl, indolyl, azaindolyl, or benzimidazolyl
which is optionally substituted by 1-2 Cl-C4 aL~yl, 1 to 2 halogen, 1 to 2
OR2, S()m R2, or C(O)OR2;
WO9SI13069 2 1 752 1 8 ~ 4,l28l6
- 46 -
R2 is hydrogen, C l-C6 alkyl, or C3-C7 cycloalkyl, and where two C 1 -C6
aL~yl groups are present on one atom they may be optionally joined to
form a C5-C7 cyclic ring optionally including oxygen, sulfur or NR3a;
5 R3a is hydrogen, or Cl-C4 alkyl;
X is selected from: hydrogen, -(cH2)qN(R2)c(o)R2~
-(CH2)qN(R2)C(O)(CH2)taryl, -(cH2)q N(R2)so2(cH2)taryl~ -(CH2)q
N(R2)S02R2, -(CH2)qN(R2)C(O)N(R2)(CH2)taryl,
o -(CH2)qN(R2)C(O)N(R2)(R2), -(cH2)qc(o)N(R2)(R2)~
-(CH2)qN(R2)C(O)OR2, -(CH2)qC(O)N(R2)(CH2)taryl,
-(CH2)qC(O)OR2, -(cH2)qc(o)o(cH2)taryL -(cH2)qoc(o)R2
-(CH2)qOC(O)(CH2)taryl, -(CH2)qS(O)mR2, and
-(CH2)qS(O)m(CH2)taryl, where an R2 group may be optionally
5 substituted by hydroxyl, carboxyl, -CONH2, -S(O)mCH3, carboxylate
Cl-C4 aLkyl esters or tetrazole and aryl is phenyl, naphthyl or pyridyl
which may be further substituted by 1-2 halogen, 1 to 2 OR2, C(O)OR2,
1 to 3 Cl-C4 aL~yl, S(O)mR2, or lH-tetrazole-5-yl;
20 y is selected from: hydrogen, Cl-C8 alkyl, (CH2)taryl~ -(CH2)q C5-C7
cycloaL~yl, -(CH2)q-K-(Cl-C6 aL~yl), -(CH2)q-K-(CH2)taryl, and
-(CH2)q-K-(CH2)t (C5-C6 cycloalkyl), where K is S(O)m and where the
alkyl groups may be optionally substituted by hydroxyl, carboxyl,
CONH2, carboxylate Cl-C4 alkyl esters or lH-tetrazole-5-yl and aryl is
25 phenyl, napthyl, pyridyl, thiazolyl, thiopheneyl, pyrazolyl, oxazolyl,
isoxazolyl or imidazolyl which may be optionally substituted by 1 to 2
halogen, 1 to 2 OR2, 1 to 2 -N(R2)(R2), CO(OR2), 1 to 2 Cl-C4 aL~yl,
S(O)mR2, or lH-tetrazol-5-yl, with the proviso that if X is hydrogen, Y
is other than hydrogen;
R4 and Rs are independently hydrogen, C l-C4 aL~yl, or substituted C 1-
C3 aL~yl where the substituents may be 1 to 2 hydroxyl;
A iS
WO 95113069 2 1 7 5 2 1 8 PCI~/US94/12816
- 47 -
1j~7
(CH2)~ Cl--
R7a
5 wherexisO,orl;
R7 and R7a are independently hydrogen, Cl-C6 aLkyl, phenyl, substituted
Cl-C6 aLky wherein the substitutent is imidi~olyl, phenyl, indolyl, p-
hydroxyphenyl, OR2, S(O)mR2, or R7 and R7a may be joined to one
another to form a C3 cycloaLkyl;
misO, 1,or2;
qisO, 1,2,or3;
tisO, 1,2,or3;
5 and pharmaceutically acceptable salts and individual diastereomers
thereof.
Still more preferred compounds within this third
embodiment include those of Formula CIc:
H H ,R
2 o R1--,C--N--,C, -A--N~ R4
C,=O O 5
~<X
2 5 Formula CIc
wherein:
Rl is selected from the group consisting of:
WO 95113069 PCr/US94112816
2175218
- 48 -
(1-2)F--~ MeO--~CH2-
H H H
~¢CH2- ~N,CH2- ~,1 N~
H H
~\~ MeO--~\' HO--~ ,CH2-
~f ,CH2- 3~,o,CH2-~ o,CH2-
~ CH2- ~o~CH2~ CH2-
~J (1-2) F--~ (1-2) F--~~'
[3~ (1-2) F--~f CH2-~ ,CH2-
2 5 or their regioisomers where not specified;
X is selected from the group consisting of: hydrogen,
94/l28l6
WO 95113069 2 1 7 5 2 1 8
- 49 -
J~ CH3 ~O, Et /I~o--~CH3 J~ CH3
O O O
J~O'--CH3 J~O ~ CH3 ~OH
J~O--~ CH3 J~O ' J' H ~
O O O CH3
J~ N CH3 J~ N - Et J~ N ,CH3 J~ N 1CH3
O o o
H H 2
~0 ~S -HN N ,OH -HN HN' 3
NH ~CH3 NH ~O -NH ~-- J~H3
i`S' CH3 " ~ "S~ HN~N~sCH3
o$ 3 -O CH3 -J~13 -HN~o'CH3
30 CH3
Y is selected from the group consisting of: hydrogen,
WO95113069 2 ~ 752 1 8 PCrlUS9~ 816
- 50 -
H3C~ CH2 3C~ O~CH2 I~CH ~f CH2-
S 3 EtO2C--~`CH2- 3 1-3halO9en--¢~ CH2-
¢N~J 2 ¢~" CH2- ~ CH3
[3`S- ¢N~J S- ¢~N S- </ ~ S- 3 ~ ~ S
</ ~ CH H C~/ ~ CH </ ~CH2~ </ ~CH2-
CH2- CH2- N~ N CH3
<0_~ CH2- H3C~/ ~ CH2- </ ~CH2~ </ ~CH2-
25 N~ N~ N
< , CH2- H3C~/ CH2- </ ~CH2-
H H H CH3
or their regioisomers whereof where not specified, with the proviso that if
X is hydrogen, Y is other than hydrogen;
A is selected from the group consisting of:
wo 95rl3069 2 1 7 5 2 1 8 PCI/US94/12816
.
- 51 -
H3C CH3 H3C CH3 CH3 ,~CH3 H3CXCH3
H3C ~,CH3 ,~/ ,~/
R4 and R5 are independently selected from the group consisting of:
--H --CH3 --CH2CH3 ,CH3 /~CH20H
OH OH
and pharmaceutically acceptable salts and individual diastereomers
thereof.
The most preferred compounds within this third embodiment
include the following:
¢~ ~NH2 ¢~~r ~<NH2
N C=O O N C=O O
"~OEt ~ C~
wo9S/13069 2 ~ 752 ~ 8 ~ s94/l28l6
F~\r ~NH2 ~ rHN I ~H
N CN;O N N~
~ ""~OEt ~ "~OEt
~0 ~0
10 ~ ~ p ~NH2 ~N~NH
N CN=~O O H CN;O
." ~OEt ~--N ~/
~ ~/ H
N ~ H N
C=O O C=O O
H
1~. "~NH(CH2)2SCH3 ~ "~NHEt
~ 13'
H N~ ~NH2
C=O O N C=O O
~N~ H ~N~
~OEt ~/ ",~OEt
¢~ ~
WO 95/13069 ~ iU~94112816
2175218
- 53 -
~N~NH ¢~ ~r ~NH2
5C=O O H C-O O
""~O(CH2)2SMe ~ ""~OEt
~0 ~/0
H N~NH ~NH2
~ ""~NHEt ~ ",~OEt
~o ~O
~ ~NHMe ~ ~NH2
NH N~ H CN=~O O
~ ""~OEt ~,OEt
¢~ ~ O
~N~NH [~~~r ~NHMe
H CN=~O O H NC' ;
3 ~ ",l~OEt ~/ ""~OEt
- S/~ S/~/
\~N \~N
WO 9S113069 2 1 7 5 2 1 8 PCI'tUS94/12816
- 54 -
O ~
" ~OEt ~ .",I~NHEt
10 ~" ~ NH2 ~--t' o
~ NHEt
~ ""~OEt ~ "'C
s/q~ o ~
\~N ~,N
CH30~N~C~NH ~\~ `ICI NH2
N C, =0 0 C, =0
H ~N~ ~ ~
~ C ~ .,C'Et
~O ~O
~ ~ ~1, C=O O
"~NH(CH2)20H ~OEt
~ WO9~i/13069 2 ~ 752 ~ 8 PCI'IUS94/12816
_ 55 _
3 O~N`C>~NH ¢~ C" NH2
C,=O O C=O O
~N, ~N,
~ ""~OEt ~ NHEt
;~ ~ ~N`C>~NH ~ ~NH2
Cl =O O N C=O O
~N~ H ~N~
~/ ""1~ NHEt j~
S \~N
20 ¢~~~~ C" NH2 ¢~ ~NH2
C=O O N
N~ H ~N~
""~OEt ~ ~--
25</ ~ O [3~ 0
and their pharmaceutically acceptable salts and individual diasteromers
thereof where not otherwise specified.
3 0Throughout the instant application, the following
abbreviations are used with the following meanings:
BOC t-butyloxycarbonyl
BOP Benzotriazol-1-yloxy tris/dimethylamino)-
phosphonium hexafluorophosphate
CBZ Benzyloxycarbonyl
,
WO9S/13069 2 1 752 1 8 ~ s94/l28l6
- 56 -
.
DIBAL-H diisobutylaluminum hydride
DMF N,N-dimethylform~mide
EDC 1-(3-dimethylaminopropyl)-3-ethylcarbodi-
imide hydrochloride
FAB-MS Fastatombombardment-mass spectroscopy
GHRP Growth hormone releasing peptide
HOBT Hydroxybenztriazole
LAH Lithium aluminum hydride
HPLC High pressure liquid chromatography
MHz Megahertz
MPLC Medium pressure liquid chromatography
NMM N-Methylmorpholine
NMR Nuclear Magnetic Resonance
TFA Trifluoroacetic acid
THF Tetrahydrofuran
TLC Thin layer chromatography
TMS Tetramethylsilane
The compounds of the instant invention all have at least
2 one asymmetric center as noted by the asterisk in the structural
Formula I above. Additional asymmetric centers may be present on
the molecule depending upon the nature of the various substituents on
the molecule. Each such asymmetric center will produce two optical
isomers and it is intended that all such optical isomers, as separated,
25 pure or partially purified optical isomers, racemic mixtures or
diastereomeric mixtures thereof, be included within the ambit of the
instant invention. In the case of the asymmetric center represented by
the asterisk in Formula I, it has been found that the absolute
stereochemistry of the more active and thus more preferred isomer is
3 as shown in Formula II. An equivalent representation places Rl and
the N-substituent in the plane of the structure with the C=O group
above. The special configuration of the asymmetric center
corresponds to that in a D-amino acid. In most cases this is also
WO95113069 2 1 752 1 8 PCI-/US94/12816
- 57 -
designated an R-configuration although this will vary according to the
value of Rl used in making B- or S- stereochemical assignments.
H H O ,R4
- R1~N--C-A--N~
C=O R5
(CH2~y
~ ( X
R3 Y
Formula II
The W group may also be present in either R- or S-
configurations. Both afford active growth horrnone secretagogues
although, in general, the R- configuration is more active. In addition,
the W group may be cis- or trans- in respect to substituents X, Y or
R3. In the case of the asymmetric center which bears the X and Y
groups, in most cases, both the R- and S- configurations are consistent
with useful levels of growth hormone secretagogue activity. In
addition, configurations of many of the most preferred compounds of
this invention are indicated. The W, X and Y groups may also be cis-
20 or trans- to the R3 substituent. In some of the most preferred
compounds a cis- or trans- relationship is also specified in respect to
the R3 substitutent. All are within the ambit of this invention and in
some of the most preferred compounds these stereochemical
orientations are indicated. When the carbon atom in Formula I
2 5 bearing an asterisk is of a defined and usually a D-configuration,
diastereomers result according to the absolute configuration at the
carbon atoms bearing the W, X, Y and R3 groups. These
diastereomers are arbitrarily referred to a diastereomers dl, d2, d3, d4,
etc. and if desired, their independent syntheses or chromatographic
3 0 separations may be achieved using standard methods or as described
herein. Their absolute stereochemistry may be determined by the x-
ray crystallography of crystalline products or crystalline intermediates
which are derivatized, if necessary,. with a reagent containing an
asymmetric center of known absolute configuration.
WO 9S/13069 1 ~ /u591/12816
2175218
- 58 -
The instant compounds are generally isolated in the form
of their pharmaceutically acceptable acid addition salts, such as the
salts derived from using inorganic and organic acids. Examples of
such acids are hydrochloric, nitric, sulfuric, phosphoric, formic, acetic,
5 trifluoroacetic, propionic, maleic, succinic, malonic, methane sulfonic
and the like. In addition, certain compounds containing an acidic
function such as a carboxy can be isolated in the form of their
inorganic salt in which the counterion can be selected from sodium,
potassium, lithium, calcium, magnesium and the like, as well as from
organic bases.
The preparation of compounds of Formula I of the present
invention can be carried out in sequential or convergent synthetic routes.
Syntheses detailing the preparation of the compounds of Formula I in a
sequential manner are presented in the following reaction schemes.
The phrase standard peptide coupling reaction conditions is
used repeatedly here, and it means coupling a carboxylic acid with an
amine using an acid activating agent such as EDC, DCC, and BOP in a
inert solvent such as dichloromethane in the presence of a catalyst such as
HOBT. The uses of protective groups for amine and carboxylic acid to
20 facilitate the desired reaction and minimi7.e the undesired reaction are
well documented. Conditions required to remove protecting groups
which may be present and can be found in Greene, T; Wuts, P. G. M.
Protective Groups in Organic Synthesis, John Wiley & Sons, Inc., New
York, NY 1991. CBZ and BOC were used extensively in the synthesis,
25 and their removal conditions are known to those skilled in the art.
Removal of CBZ groups can be achieved by a number of methods known
in the art; for example, catalytic hydrogenation with hydrogen in the
presence of a nobel metal or its oxide such as palladium on activated
carbon in a protic solvent such as ethanol. In cases where catalytic
3 hydrogenation is contraindicated by the presence of other potentially
reactive functionality, removal of CBZ groups can also be achieved by
treatment with a solution of hydrogen bromide in acetic acid, or by
treatment with a mixture of TFA and dimethylsulfide. Removal of BOC
protecting groups is carried out in a solvent such as methylene chloride or
~ WO95/13069 2 1 752 1 8 PCrlUS94/12816
_ 59 _
methanol or ethyl acetate, with a strong acid, such as trifluoroacetic acid
or hydrochloric acid or hydrogen chloride gas.
The protected amino acid derivatives 1 are, in many cases,
commercially available, where the protecting group L is, for example,
5 BOC or CBZ groups. Other protected amino acid derivatives 1 can be
prepared by literature methods (Willi~m~, R. M. Synthesis of Optically
Active a-Amino Acids~ Pergamon Press: Oxford, 1989). Many of the
piperidines, pyrrolidines and hexahydro-lH-azepines of formula 2 are
either commercially available or known in the literature and others can be
0 prepared following literature methods desribed for known compounds,
some of which are described here. The skills required in carrying out the
reaction and purification of the resulting reaction products are known to
those skilled in the art. Purification procedures includes cryst~lli7~tion,
and normal phase or reverse phase chromatography.
wo ss/l306s ~ s~tl28l6
2175218 ~
- 60 -
SCHEME 1
H H
R1~N--L + ( ~X R,~L
2 3
Intermediates of formula 3 may be synthesized as described
in Scheme 1. Coupling of amine of formula 2, whose preparations are
described later if they are not known compounds, to protected amino
acids of formula 1, wherein L is a suitable protecting group, is
conveniently carried out under standard peptide coupling conditions.
SCHEME 2
H H H H
Rl~N--L Rl~N H
C=O C=O
Removal of L
20(CH2~Y (CH )NyW
)~X )~X
3 4
Conversion of 3 to intermediate 4 can be carried out as
illustrated in Scheme 2 by removal of the protecting group L (CBZ, BOC,
etc.) using standard methodology.
~ WO95113069 2 1 752 1 8 PCI~/US94/12816
- 61 -
SCHEME 3
H H ,R4
Rl~N--C-A--N~
,R4 C=O R5
HO-C-A--N
H H ~X
I = 5 R3 Y R4=H
N W H20
0 (CH2)n Y + or /
)~X H H O L
4 Il LR1~ N-C-A--N
HO-C-A--N C=O R5
R5(CH~)Nyw
6 )~X
R3 Y
Interrnediates of formula 5, wherein A is connected to
20 the carbonyl by a carbon atom and thus A is -(CH2)x-C(R7)(R7a)-
(CH2)y- can be prepared as shown in Scheme 3 by coupling
intermediates of formula 4 to amino acids of formula 5 under the standard
peptide coupling reaction conditions. The amino acids 5, as amino acid
1, are either commercially available or can be synthesized. Also if R4 or
2s R5 is a hydrogen then the protected amino acids 6 are employed in the
coupling reaction, wherein L is a protecting group as defined above. The
removal of L in 7 to afford I, where R4 = H, can be carried out under
conditions known in the art.
WO 95/13069 PCI/US94/12816
2175218
- 62 -
SCHEME 4
H H O H H H~ R R4
R1~N--C-A--N R1~N--C-A--N
C=O Rs Reductive alkylation Cl = R5
(CH2)ny orepoxideopening(CH )NyW
) ~ X X
R3 I Y I where R4 is substituted
/unsubstituted alkyl
Compounds of formula I wherein R4 and/or Rs is a
hydrogen can be further elaborated to new compounds I (with most
preferred side chains R4 = CH2-CH(OH)-CH2X, wherein X = H or OH)
15 which are substituted on the amino group as depicted in Scheme 4.
Reductive alkylation of I with an aldehyde is carried out under conditions
known in the art; for example, by catalytic hydrogenation with hydrogen
in the presence of platinum, palladium, or nickel catalysts or with
chemical reducing agents such as sodium cyanoborohydride in a protic
20 solvent such as methanol or ethanol in the present of catalytic amount of
acid. Alternatively, a ~imil~r transformation can be accomplished via an
epoxide opening reaction.
SCHEME S
R1 + N--H X I Z--(CH2)Xj~ (CH2)YN \
Cl=O 8 R7a R5
N W
(CH2)n Y OR ~ Formula I
~ ( X
3 0/ y R7 / R4
4 O= C= N--(CH2)X~ (CH2)yN
R7a \R5
wo 95113069 ~ ~ 7 5 2 1 8 ~ 94/l28l6
- 63 -
Compounds of formula I, wherein A is Z-(CH2)X-
C(R7)(R7a)-(CH2)y and Z is N-R6 or O may be prepared as shown in
Scheme 5 by reacting 4 with reagents 8, wherein X is a good leaving
group such as Cl, Br, I, or imidazole. Alternatively, 4 can be reacted with
5 an isocyanate of formula 9 in an inert solvent such as 1,2-dichloroethane
to provide compounds of formula I where Z is NH.
The compounds of general formula I of the present invention
may also be prepared in a convergent manner as described in Reaction
Schemes 6, 7 and 8.
0 SCHEME 6
H H O R4
Fl4 ~
HOOC -A- N- R5 R 1 ---N--C--A - N- R5
H H 5 COOM
~N--H + OR ~ 11
COOM
L H H O L
HOOC--A - N- R5 R1~ N--C--A- N- R5
6 COOM
11a
The carboxylic acid protected amino acid derivatives l0 are,
in many cases, commercially available where M = methyl, ethyl, or
benzyl esters. Other ester protected amino acids can be prepared by
classical methods f~mili~r to those skilled in the art. Some of these
25 methods include the reaction of the amino acid with an alcohol in the
presence of an acid such as hydrochloric acid or p-toluenesulfonic acid
and azeotropic removal of water. Other methods include~ the reaction of
a protected amino acid with a diazoaLkane, or with an alcohol and an acid
activating agent such as EDC, DCC in the presence of a catalyst such as
3 DMAP and removal of the protecting group L.
Intermediates of formula l l or l la, can be prepared as
shown in Scheme 6 by coupling of amino acid ester l0 to amino acids of
formula 6 or 7. When a urea linkage is present in l l or l la, it can be
introduced as illustrated in Scheme 5.
PCI'IUS94/12816
WO 95113069
2175218 ~
- 64 -
SCHEME 7
R1 ~N-C--A--N- R5 R1 ~ N-C--A--N- R5
COOM COOH
11 .
R1 ~ N- C--A--N- R5 R 1 ~ N- g--A--N--R5
l 0 COOM COOH
1 1a 12a
Conversion of the ester 11 or 1 la to intermediate acids 12 or
12a can be achieved by a number of methods known in the art as
described in Scheme 7; for example, methyl and ethyl esters can be
5 hydrolyzed with lithium hydroxide in a protic solvent like aqueous
methanol. In addition, removal of benzyl groups can be accomplished by
a number of reductive methods including hydrogenation in the presence
of palladium catalyst in a protic solvent such as methanol. An allyl ester
can be cleaved with tetrakis-triphenylphosphine palladium catalyst in the
20 presence of 2-ethylhexanoic acid in a variety of solvents including ethyl
acetate and dichloromethane (see J. Org. Chem.~ 42, 587 (1982)).
SCHEME 8
H H O R4
R1----N--C-A--N- R5
( ,OOH N W
12 (CH2jn Y ~\
)~X ~ Remo~ al of L
H Hl R L R3 Y R4=H
R1~N--C-A--N-R
COOH 2 7
12a
WO 95/13069 2 1 7 5 2 1 8 PCI'IUS94/12816
.
- 65 -
Acid 12 or 12a can then be elaborated to I or compound 7 as
described in Scheme 8. Coupling of piperidines, pyrrolidines or
hexahydro-lH-azepines of formula 2 to acids of formula 12 or 12a,
wherein L is a suitable protecting group, is conveniently carried out under
5 the standard peptide coupling reaction conditions. Transformation of 7 to
I is achieved by removal of the protecting group L. When R4 and/or R5
is H, substituted aL~yl groups may be optionally added to the nitrogen
atom as described in Scheme 4.
The 2-substituted piperidines, pyrrolidines or hexahydro-lH-
azapines are either commercially available or can be prepared by
literature procedures. Illustrated herein are some, but by no means all,
the methods for their preparation.
SCHEME A9
(CH
)~X
R3 A1
1) Na2WO2. H22
2) NaCN, HCI
2 5 ~X TiC13 (CH~ CN
R3 Y 3 A2a
A14 \~ HCI/H20
H
(CH$ ~ Co2H
)~X
R3 Y
A15
WO 95113069 PCrlUS9~tl2816
21 7521 8
- 66 -
According to the protocol developed by S. Murahashi and T.
Shiota (Tetrahedron Lett.. 28, 6469-6472 (1987)) catalytic oxidation of
cyclic amines such as piperidines, pyrrolidines or hexahydro-lH-azapines
with hydrogen peroxide followed by treatment with hydrogen cyanide
5 gives a-hydroxylaminonitriles of formula A14, which upon reduction
(Murahashi, S.-I.; Kodera, Y., Tetrahedron Letters, 26, 4633-4636
(1985)) give o~-aminonitriles of formula A2a. In cases where X and Y
are not both hydrogen and/or n is not 2, regioisomers and
diastereoisomers may arise, and they may be separated by
chromatography methods. Hydrolysis of the amino nitrile under acidic or
basic conditions yields the amino acid. Alternatively, the
hydroxylaminonitrile can be hydrolyzed first, then reduced by palladium
catalyzed hydrogenation to afford the amino acid of formula A15. The
amino acid and their derivatives prepared according to this method are
15 racemic.
Alternatively, the nitrile A2a can be prepared by oxidation
of the compound A13 to the imine as described in the literature (Goti and
Romani in Tetrahedron Letters. ~ 6567-6570 (1994)) followed by
reaction with cyanide. W can also be introduced by direct alkylation of
20 the Boc protected compound A13 by butyl lithium followed by addition
of electrophiles known as the Beak alkylation (Beak and Lee J. Or~.
Chem., 55, 2578-2580 (1990)). Asymmetric introduction of W can also
be achieved by using a chiral catalyst (Kerrick and Beak, J. Am. Chem.
Soc. 113, 9708-9710 (1991)).
SCHEME A10
H H
(CH~ C02H ~X
R3 Y R3 Y
A15 A2
~ wo95/13069 2 1 752 1 8 ~ 4,12816
- 67 -
A15 R2OH, H+ (CH~CO2R2
R3 Y
A2b
L L
(CH2jN~--C02H HOBt, EDC (CH jN~ CONR2R2
; ~ X R2R2NH ,~ ( X
R3 Y R3 Y
A1 6 A2c
(cH2jn ~_CN , tetrazoles
)~X other hetereocycles
R3 Y
A17
The carboxylic acid functionality at the 2-position of
compounds of formula A15 can be converted to ester, amide, nitrile, acyl
sulfonamide, and moieties as defined by W to give compound of general
forrnula 2 according to the conventional methods well documented in the
literature and known to those skilled in the art (The Practice of Peptide
Synthesis, by M. Bodanszky and A. Bodanszky, Springer-Verlag, 1984).
L is an appropriate protecting group such as BOC, CBZ, etc. The
carboxylic acid can also be converted into its next higher homologue, or
to a derivative of the homologous acid, such as amide or ester by an
Arndt-Eistert reaction. Alternatively, the ester can be directly
homologated by the protocol using ynolate anions described by C. J.
Kowalski and R. E. Reddy in J. (~r~. Chem., 57, 7194-7208 (1992). The
resulting acid and/or ester can be converted to the next higher
homologue, and so on and so forth.
WO 9S113069 ~ J~91/12816
2~7521~ ~
- 68 -
SCHEME Al l
(CH~W (CH~W (CH~W
R3 R3 Y R3 Y
A18 A19 A2
Illustrated in Scheme Al 1 is a general method to introduce
y wherein X is an electron withdrawing group such as -CN, -CO2Rg,
where R8 is aLkyl, aryl, and alkyl(Cl-C4)aryl are either known
compounds or may be prepared by methods described above or by
methods analogous to those used for the preparation of known
compounds. Introduction of the Y substitution can be achievedl by first
. ~
reacting compounds of formula Al8 with a strong base such as potassium
bis(trimethylsilyl)amide, lithium diisopropylamide following by addition
of alkylating reagents such as alkyl halides, aryl alkyl halides, acyl
halides, and haloformates in a inert solvent such as THF at temperatures
from -100 to room temperature. Thio derivatives where the sulfur is
2 attached directly to an aL~yl or an aryl group can be prepared similarly by
reacting with a disulfide. The halides used in these reactions are either
commercially available or known compounds in the literature can be
prepared by methods analogous to those used for the preparation of
known compounds. The protecting group L in compounds of formula
B l9 can be removed with conventional chemistry to give compounds of
formula 2.
WO9~;1130692 1 752 1 8 ~ u~4~l28l6
- 69 -
SCHEME A12
~N~ 1) H2021~ ~ 1) HCI/H20 N CO2H
~Xory 2)TMSCN~XorY 2)H2/PtO2 ~XorY
R3 R3 R3
A20 A21 A22
To prepare cis homoproline derivatives, the procedure
described by Shuman et al can be used (Shuman, R. T.; Ornstein, P. L.;
Paschal, J. W.; Gesellchen, P. D., J. Org. Chem., 55, 738-741 (1990))
(Scheme A12). Substituted pyridines of formula A20, many of them
commercially available or prepared by literature procedures, are
converted to their corresponding N-oxide by reacting with hydrogen
l 5 peroxide. Reaction of the pyridine N-oxide with trimethylsilylcyanide
and dimethylcarbamyl chloride gives the 2-nitrile of formula A21. If
regioisomers should arise due to the presence of 3-substitution, they can
be conveniently separated by chromatography. Hydrolysis of the nitrile
to the acid under acidic or basic conditions followed by hydrogenation
20 catalyzed by platinum oxide gives the piperidine carboxylic acid. The
function~li7~tion of the carboxylic acid is described above and in part by
Scheme A10.
The amino acids generated by these synthetic protocols are
racemic. However, procedures for resolving RS-a-amino acids by
25 various methods are known in the literature (Toone, E. J. and Jones, J. B.
Can. J. Chem., 65, 2722 (1987); Okamoto, S.; Hijikato, A. Biochem.
Biophys. Res. Commun., 101, 4~0 (1981); Greenstein, J. P.; Winitz, M.
Chemistry of the Amino Acids; Wiley: New York, 1961, Vol. 1, 715-
760). Therefore, the separated R- and S- isomers can be prepared by this
30 methodology. Alternatively, the racemic piperidine, pyrrolidine and
hexahydro-lH-azepine derivatives can be converted directly to growth
hormone secretagogues or their intermediates, and the resulting
diastereomeric mixtures can be separated at the appropriate stage by
chromatography to yield the enantiomerically pure compounds.
WO 9S/13069 PCr/US9~/12816
21752~8 ~
- 70 -
SCHEME A 13
Ph~ Ph~
CO2Et
~/ + ~ THF-H20 [~ CO2Et ~,CO2Et
A23 ~ R3 A25 R3 A26
Ph ~ Ph~
CO2Et N ~ CO2Et ~ CO2H N ~CO2H
H2/Pd(OH)2/C
R3 A27 A28 R3 A15a 3 A15b
Alternatively, asymmetric synthesis can be carried out to
synthesize optically pure piperidine, pyrrolidine and hexahydro-lH-
azepine derivatives. For example, optically active piperidine-2-
20 carboxylic acid derivatives A15a, AlSb can be prepared by the aza-Diels-
Alder reaction as described by Bailey et al (J. Chem. Soc. Perkin Trans I,
1337-1340 (1991)). Reaction between the chiral irnine A23 and the diene
A24 in the presence of TFA (1 equivalent) and water (catalytic) gives the
adducts A25 and A26 with good diastereoselectivity. The two
25 diastereoisomers can be separated, and each can be hydrogenated to
reduce the double bond and to remove the chiral auxiliary. All four
possible isomers can be achieved by this methodology. Illustrated here
(Scheme A13) is the preparation of the two isomers AlSa and AlSb
which have an S-con~lguration at the chiral center adjacent to the COOH.
30 The two R- isomers at this center can be prepared similarly using
compound A26.
WO 95113069 2 1 7 5 2 1 8 PCI~/US94112816
SCHEME B9
L L
5 ¢~
R3 R3 R3
B13 B14 /~ B15
1 0 \ /
The synthesis of substituted piperidines of formula 2 (n=2)
has been detailed in a number of research articles. For e.g., S. M. N.
Efange et al. (J. Med. Chem., ~6, 1278-1283 (1993() and M. S. Berridge
et al. (J. Med. Chem., 36, 1284-1290 (1993)) have used 4-substituted-
pyridine intermediates B 13 to synthesize 4-substituted
tetrahydropiperidines of formula B 14 (L = methyl) as detailed in Scheme
B9. Removal of L from piperidines of formula B14 can be carried out by
a number of methods familiar to those skilled in the art, including the
20 cyanogen bromide protocol detailed by H. Ong et al. in J. Med. Chem.,
23, 981-986 (1983) and ACE-Cl method as described in R. Olofson et al.
J. Org. Chem., 23, 2795 (1984). For intermediates of formula B 14,
wherein L = Bn, simultaneous removal of the benzyl group and
hydrogenation of the olefin can be accomplished by use of platinum or
25 palladium catalysts in a protic solvent like methanol. Alternatively, B 13
can be directly transformed to piperidines of formula B 15 (L = H) by
carrying out the reduction with platinum oxide in a protic solvent such as
methanol with a catalytic amount of acid.
WO95113069 2 1 7 5 2 1 8 PCI'/US94/12816
- 72 -
SCHEME Bl0
L
h (CH2~n
(CH~_ + R3 - Xa ' )~X
X R3 Y
TfO Y B17 [Xa= B(OH)3 or SnMe3] B18
B16
/
~'
L H
(CH2~> ~ (CH2)n
) ~ X ~ )~X
R3 Y R3 Y
B20 Bl9
Other methods as shown in Scheme B 10 may also be used to
synthesize compounds of formula 2. For example, cross-coupling of enol
20 triflates of formula B 16 (L = protecting group) where X and Y are
defined in formula I with aryl boronic acids of formula B 17 (Xa =
B(OH)3) or aryl or phenyl or naphthyl tin reagents of formula B17 (Xa =
SnMe3) can be accomplished with palladium (II) or palladium (0)
catalysts as detailed in the review article by W. J. Scott and J. E.
25 McMurry Acc. Chem. Res., 21, 47 (1988) to give in their examples
tetrahydropiperidines B 18 (L = protecting group). Various methods exist
for the synthesis of the enol triflate intermediates of formula B 16, phenyl
or naphthyl boronic acids, and phenyl or naphthyl tin reagents of formula
B17 (X = B(OH)3; SnMe3) and are familiar to those skilled in the art.
3 o Removal of the protecting group L furnishes for example, piperidines of
formula B 19 (L = H). Hydrogenation of B 18 followed by removal of the
protection group L also gives saturated derivatives B20. Alternatively,
Bl9 can be transformed to compounds of formula B20 by hydrogenating
the olefin in the presence of platinum or palladium catalysts in a protic
solvent such as methanol.
WO 95/13069 PCI~/US9~/12816
~ 2175218
SCHEME B 11
(CH~ ' R3 ~ ~ X (CH~
B21 B22 Bl 8
~ ~
~X ~ (CH,~X
R3 Y R3 Y
B20 B1 9
Methods for the synthesis of substituted pyrrolidines,
piperidines, and hexahydro-lH-azepines also involve addition of
20 substituted and/or unsubstitllterl alkyl, cycloaLkyl, phenyl or naphthyl
Grignard reagents or lithium reagents to oxo-piperidines, oxo-
pyrrolidines, or oxo-hexahydro-lH-azepines of formula B21 (L = benzyl,
methyl, etc.) to give compounds of formula B22 (L = benzyl, methyl,
etc.). The dehydration of the hydroxyl group of B22 (L = benzyl, methyl,
25 etc.) to yield B18 (L = benzyl, methyl, etc.) can be carried out by treating
it with strong acid or via an elimin~tion reaction of the corresponding
mesylate derived from B22 (L = benzyl, methyl, etc.). Compounds B18
can be transformed to B19 or B20 as described above.
WO9S/13069 2 1 752 1 8 ~ u~4~12816
- 74 -
SCHEME B12
L H
(cH2jn ~ ~ (CH2)n ~ -
) ( X ~ ) ( X
R3 H R3 Y
B21 B1 9
The 3,4-disubstituted piperidines, pyrrolidines and
0 hexahydro-lH-azepines of formula B21 wherein X is an electron
withdrawing group like an ester, ketone, nitrile, etc., can be further
aLkylated, hydroxylated, halogenated by using methods f~ r to those
skilled in the art. Once again, deprotection of the protecting group L can
be carried out by methods f~rnili~r to those skilled in the art.
Specifically, ortho-substituted phenyl piperidines of of
formula B22a wherein X,Y = H can be prepared from the phenyl
H
~ N~<X
~ y
~R10
B22a
piperidine intermediate B23 (see S. M. N. Efange et al. J. Med. Chem.,
26, 1278 (1993)).
WO 95113069 PCI'IUS94/12816
~ 21752~8
- 75 -
SCHEME B13
~OCH3 ~OH
B27 B28
~
~-- ~H
B23 B24
H
--~OEt ~OEt
B26 B25
As shown in Scheme B 13, the benzyl alcohol can be
oxidized to aldehyde B24 by a variety of methods familiar to those
skilled in the art. Commonly used methods are manganese dioxide in an
3 inert solvent like chloroform or the Swern protocol. A variety of
functional groups can now be elaborated from B24. For example, an
Emmons reaction with triethylphosphonoacetate in the presence of base
gives the a"B-~-n~tllrated ester B25. Concurrent reduction of the
pyridine unit and the olefin group with a platinum or palladium catalyst
in an alcoholic solvent provides the piperidine of formula B26, wherein
WO95113069 2 1 752 1 8 ~ 9~112816
- 76 -
X, Y = H. The piperidine B26 may be derivatized to ester and acid
bearing compounds of formula I wherein X and Y=H by using chemistry
detailed in Schemes 1-8. Alternatively, B24 can directly be transformed
to a methyl ester B27, wherein X, Y = H, by oxidation of the aldehyde
5 group to an ester with the Corey protocol (NaCN, acetic acid, MnO2, in
methanol) followed by reduction of the pyridine to a piperidine with
platinum or palladium catalysts in a protic solvent like methanol. The
piperidine B27 can be elaborated to compounds of Formula I by using
chemistry detailed in Schemes 1-8. The piperidine unit of B27 can be
protected by a variety of protecting groups L f~rnili~r to those skilled in
the art and the ester unit can be hydrolyzed by well documented methods
to give the acid B28, wherein X, Y = H. The acid intermediate B28 can
be used to prepare compounds bearing a variety of highly function~li7e~1
piperidines that can be transformed to compoundsof formula I.
Highly function~li7e~1 phenyl piperidines of formula B22,
wherein X, Y = H, can be prepared by lltili7ing synthetic methods
detailed below.
WO 95113069 2 1 7 5 2 1 8 PCI/US94/12816
SCHEME B 14
L
--'P`OE~ ~OH
B26 ~/ B29
~NR2R~(~6) ~ N~NR2R2 (R6)
B30 B30a
As depicted in Scheme B 14 the piperidine B26 may also
serve as a key intermediate for the synthesis of a variety of piperidines of
formula B22a, wherein Rlo may be aLkyl and a~yl amides, aLkyl and aryl
20 acylsulfonamides, alkyl and aryl ureas, aLkyl and aryl carbamates, etc.
The piperidine nitrogen of B26 can be protected with a protecting group
L (commonly used groups include BOC, CBZ, FNIOC) by well
documented methods and the ester unit can now be hydrolyzed with
sodium or potassium hydroxide in aqueous or alcoholic media to give
25 B29. Peptide type coupling of B29 with primary and secondary aliphatic
amines, aryl amines, suitably protected amino acids, aLkyl or aryl
sulfonamides provides amides of formula B30, wherein X, Y = H,
followed by removal of the protecting group L. Alternatively, the acid
B29 can be activated with carbonyl diimidazole and subsequently reacted
3 o with primary and secondary aliphatic amines, aryl amines, suitably
protected amino acids, alkyl or aryl sulfonamides in an inert solvent like
tetrahydrofuran or dimethylformamide to give amides of formula B30,
wherein X, Y = H, L is on the nitrogen, and R2 and R6 may be any of the
groups described in the scope of this invention. Ureas of formula B30a,
wherein X, Y = H, L is on the nitrogen and R2 and R6 may be any of the
WO 9S113069 2 1 7 5 2 1 8 PCI'IUS9~112816
- 78 -
groups described in the scope of this invention, can be synthesized from
~329 by carrying out a Curtius rearrangement and trapping the isocyanate
inteImediate with amines of formula HNR2R2 or HNR2R6. The
protecting group L can be removed and elaborated to compounds of
5 Formula I using chemistry presented in Schemes 1-8.
SCHEME B15
o ~ CN
B29 / B31
H
~R11
~ R~ ~ `N
N--N
B32 H
The acid intermediate B29 also serves as a key intermediate
for the synthesis of heterocycle bearing compounds of formula B32,
25 wherein X, Y = H. As shown in Scheme B 15, the acid B29 can be
transformed to a nitrile of formula B3 1, wherein X, Y = H, by a 3-step
sequence involving activation of the acid with ethylchloroformate in the
presence of a base like triethylamine, addition of aqueous ammonia to
yield a primary amide, and dehydration of the amide to a nitrile with
3 0 phosphorous oxychloride in pyridine. The nitrile intermediate B3 1 can
now be transformed to a piperidine of formula B32 wherein X, Y = H and
Rl 1 is a lH-tetrazole, by heating it with trimethyltin azide in an inert
solvent like toluene or xylenes. The protecting group L may be removed
and elaborated to compounds of formula I by using chemistry detailed in
Schemes 1-8.
wo 95113069 ~ 1 7 5 2 1 8 Pcr/uss4ll2sl6
- 79 -
SCHEME B 16
~<X ~<y NH HCI
~" "CN ~OEt
B31 / B33
N / NH~'
~<Y N~
,~!~~R11 H o
lS ~JJ ~N~
B32 N~,OH
Other heterocycle bearing piperidines of formula B32 can
also be prepared from intermediate B31 as shown in Scheme B16.
Treatment of the nitrile B3 1 with anhydrous hydrochloric acid in dry
ethanol gives imino-ether of formula B33. Addition of forrnyl hydrazine
to B33 followed by he~ting of the intermediate in an inert solvent like
toluene provides a piperidine of formula B32, wherein X, Y = H and Rl 1
is a 1,2,4-triazole. Alternatively, carbomethoxyhydrazine can be added
to imino-ether B33 and cyclized to provide B32, wherein X, Y = H and
Rl 1 is a triazolinone. Reaction of B33 with dihydroxyacetone in
methanolic ammonia at high pressure gives B32, wherein X, Y = H and
R 1 1 is a hydroxymethyl imidazole. The protecting group L can be
removed by methods f~mili~r to those skilled in the art and elaborated to
compounds of formula I by using chemistry detailed in Schemes 1-8.
Furthermore, acids, acid chlorides, nitriles, and imino-ethers
serve as key intermediates in the preparation of a number of other alkyl,
phenyl, hydroxy, and amino-substituted heterocycles. Many of the
methods are documented in A.R. E~tri7.ky, Handbook of Heterocyclic
WO 9S/13069 PCrtUS94/12816
2~75218
- 80 -
Chemi~try. Pergamon Press, 1985, New York, New York, and may be
used to synthesize a variety of heterocycle bearing compounds.
SCHEME B 17
L L L
[~ n> Pd/C/H2 ~SN>
Ph O X H ~X ~X
B34 B35 B36
H
deprotection ~N
B37
Other applicable routes for the synthesis of mono- and di-
substituted pyrrolidines, piperidines, and hexahydro-lH-azepines of
formula II (n=l or 2) are known in the literature. For example, J. J. Plati
20 and W. Wenner (J. Org. Chem., 14, 543 (1949)) have demonstrated that
the ketoarnine intermediate B34 may be elaborated to B35 (n=l, 2, 3)
under aldol condensation conditions. Dehydroxylation of B35 can be
achieved by a number of methods including a catalytic hydrogenation
method that utilizes palladium catalysts in a protic solvent like methanol.
25 Removal of L from B36 can be carried out methods, including the ACE-
Cl method as described in R. Olofson et al. (J. Org. Chem., 43, 2795
(1984))-
WO 95/13069 2 ~ 7 5 2 1 8 PCI/US94/12816
- 81 -
SCHEME B 18
L L
N R3MgBr N ~N~
1~ ~ ClC02Et [j' ~ l J~
~CO2Et ~CO2Et ~' CO2Et
B38
~X
R3us
B39
The synthesis of 3,4-disubstituted piperidines of
formula 2 (n=2) can be conveniently prepared by literature procedures.
Illustrated below is one of these general methods. G. T. BorreK has
demonstrated the synthesis of cis 3,4-disubstituted piperidine B39 (US
Patent 4,861,893) from the commercially available ethyl nicotinate and
the Grignard reagent R3MgBr where R3 is defined in formula I. The
ester functionality of B38 may be further modified through conventional
chemistry to provide other functional groups X as defined in the scope of
the invention. Illustrated here are some, but by no means all, the methods
available to prepare functional groups X. For example, the ester of B38
can be hydrolyzed to give the corresponding carboxylic acid B39
(X=CO2H); B39 may then be converted to amides (X=CONR2R2) by a
simple peptide-type coupling reaction, to ureas or carbamates (X=
NC(O)NR2R2, Nc(o)oR2) by the Curtius rearrangement (Smith, Or~.
React.,3,337 (1946)) followed by trapping of the isocyanate
intermediate with amines or alcohols or to an hydroxymethyl unit
(X=CH2OR2) by borane reduction. The acid B39 can also be converted
to a nitrile and then elaborated to heterocyclic compounds (X=tetrazolyl,
triazolyl, triazolinolyl etc.) by the procedures described in Schemes Bl~
and B16. The carboxylic acid B39 (X=C02H) can also be converted into
its higher homologue B39 (X=CH2CO2H) by an Arndt-Eistert reaction
and further derivatized by methods which have been described above.
WO 95113069 ~ ,9~/l28l6
2175218
- 82 -
SCHEME B19
L L
[~CO2Et ~CO2Et [~X
R as R trans R trans
B38 B40 B4 1
The cis 3,4-disubstituted piperidines B38 can be
converted to trans 3,4-disubstituted piperidines B40 as shown in Scheme
l0 B19 by treating B38 with a catalytic amount of base such as sodium
ethoxide in protic solvent. Once again, the ester functional group of B40
can be further modified by methods f~mili~r to those skilled in the art,
including the procedures described in Scheme B 18. The protecting group
L from compounds of formulas B39 and B41 can be removed through
15 conventional chemistry and elaborated to compounds of formula I by
using chemistry described above.
~ Wo9S/13069 2 1 752 1 8 ~ 94/l28l6
- 83 -
SCHEME B20
MgBr 0~~ _ Ll _ L
~ ~ U~ dte ~ N~
ClCO2Et ~CO;~t ~C~t
B42 CuCI ~ J ~B43
~N~ N N
_ ~CO2Et ~ [~X ~ [~X
~CH0 ~;CO2H ~CONR2R2
B44 / / B45~o2Et
L L L
~X ~X ~X
2û ~NHCONR2R2~NHCOOR2 ~--C02R2
As described in Scheme B20, cis 3,4-disubstituted
piperidines of formula B43 can be prepared by the addition of B42 to
ethyl nicotinate by the procedure of G.T. Borrett (U.S. Patent No.
4,861,893). The acetal protecting group can be removed by a number of
methods f~mili~r to those skilled in the art. The resulting aldehyde B44
serves as a key intermediate for the synthesis of highly functionalized
3,4-disubstituted piperidines. The aldehyde B44 can be oxidized to the
corresponding carboxylic acid B45 and then further elaborated to a
variety of functional groups such as amides, ureas, carbamates,
acylsulfonamides and etc. Some examples of these transformations are
discussed in connection with Scheme B 14.
WO 9SI13069 2 1 7 5 2 1 8 ~ ~s9~1281~
- 84-
SCHEME B21
L
~CO2ES ~ ,NCONF(2R2
1 o B44 / \ ~x
L ~/B45 X=CO2Et,~ I ~NCOOR2
~N~ E=CO2Et ~N~ W
l~J~ B31 X=CO2Et, ~x
,R11 ~CONR2R2
Compound B44 can also be converted to an a, ~-
n~ lrated ester or nitrile by an Emmons reaction. The resulting
lln.c~tllrated ester or nitrile can be hydrogenated using a catalytic amount
of palladium or platinum under hydrogen atmosphere. The diester B45
(X=C02Et, E=C02Et) as shown in Scheme B21 can be selectively
hydrolyzed to corresponding acid B45 (X=C02Et, E=C02H) which can
be further elaborated to variety of functional groups by a number of
methods. Compounds of formula B3 1 (X=C02Et, E=CN) can be
transformed to compounds of formula 32 (X=C02Et, Y=H, R1 l=lH-
tetrazole) by heating B31 with trimethyl azide in toluene. Alternately,
the nitrile intermediate B31 (for example, with X=C02Et, E=CN) may
also serves as a synthetic precursor for the synthesis of heterocycle
bearing compounds of formula B32 (X=C02Et). Many of the synthetic
methods as noted above in A.R. Katrizky, Handbook of Heterocyclic
Chemist7y, Pergamon Press, 1985, New York, New York, and are
discussed in connection with Scheme B16.
The 3,4-disubstituted compounds 2 generated by these
synthetic protocols are racemic. Mono and disubstituted pyrrolidines and
~ WO 95tl3069 2 1 7 5 2 1 8 PCI-IUS94/12816
- 85 -
hexahydro-lH-azepines 2 generated by these synthetic protocols are also
racemic. Chiral intermediates of formula 2 are available by numerous
methods including by the classical resolution of racemates. For example
resolution can be achieved by the formation of diastereomeric salts of
5 racemic amines with optically active acids such as D- and L- tartaric acid.
The deterrnination of the absolute stereochemistry can be accomplished
in a number of ways including X-ray crystallography of a suitable
crystalline derivative such as a D- or L- tartaric acid salt. Alternatively,
asymmetric synthesis can be carried out to synthesize optically pure
compounds.
Furthermore, the racemic intermediates of formula 2 can be
derivatized with chiral reagents and these products may be separated by
chromatography and chiral compounds of formula 2 may be regenerated
from them by hydrolysis, or as stated earlier, racemic intermediates of
15 formula 2 can be converted directly to growth hormone secretagogues,
and the resulting diastereomeric mixtures can be separated by
chromatography to yield the enantiomerically pure compounds.
SCHEME C9
¢~ Hydrogenation ~
X(orY) X(or Y)
C13a C13
3-Monosubstituted piperidines of formula C13 can be
prepared by the reduction of pyridine derivatives or their salts by
hydrogenation in a suitable organic solvent such as water, acetic acid,
alcohol, e.g. ethanol, or their mixture, in the presence of a noble metal
catalyst such as platinum or an oxide thereof on a support such as
activated carbon, and conveniently at room temperature and atmospheric
pressure or under elevated temperature and pressure. 3-Monosubstituted
piperidines can also be prepared by modification of the X or Y moiety of
the existing 3-monosubstituted piperidines.
WO95113069 ~1 7521 8 PCI-/US9~/1281~
86
SCHEME C9A
Bn 1) BH3/THF HN
CO2Me 3) HCUROH C1 3b
3-Monosubstituted pyrrolidines are commercially available
or can be conveniently prepared by literature procedures. Shown in
Scheme C9A is an example of the preparation of these compounds via
o pyrrolidine-3-carboxylic acid ester. The commercially available
compound methyl l-benzyl-4-oxo-3-pyrrolidinecarboxylate is reduced by
borane (J. Chem. Soc., 24, 1618-1619). Removal of the benzyl group by
catalytic hydrogenolysis followed by ester exchange in an appropriate
alcohol medium such as ethyl alcohol in the presence of acid gave the
15 compound C13b. The ester functionality may be further modified
through conventional chemistry to other groups as defined by X.
3-Monosubstituted pyrrolidines may also be prepared by catalytic
hydrogenation of 3-substituted pyrroles.
SCHEME C9B
H H
(~ C02H (~ CO2R
C13c
Hexahydro-lH-azepines are commercially available or may
be prepared by the literature procedure. Hexahydro-lH-azepine-3-
carboxylic acid (Krogsgaard-Larsen, P. et al., Acta. Chem. Scand., B32,
327, (1978)) is esterified in an alcohol solvent in the presence of acid.
3 The ester functionality may be further modified through conventional
chemistry to other groups within the definition of X.
wo 95rl3069 PCI/US94/12816
2175218
- 87 -
SCHEME C10
H L
(CH2~ ~ ProteCtion~ (CH2~ ~
\ I X \ I X
C13 C14
Base/
activated Y-
H L
(CH ~)N>removal of L(CH )N~
( X \ ( X
Y Y
C2 C15
Illustrated in Scheme C10 is a general way to prepare di-
substituted piperidines, pyrrolidines, and hexahydro-lH-azepines.
Compounds of Pormula C13 wherein X is an electron withdrawing group
such as -CN, -CO2Rg, where R8 is alkyl, aryl, and (Cl-C4alkyl)aryl are
known compounds or may be prepared by methods analogous to those
2 0 used for the preparation of such known compounds. The secondary
amine of compounds of Formula C13 may be first protected by a
protecting group L such as BOC and CBZ using the conventional
techniques. Introduction of the Y substitution can be achieved by first
reacting compounds of Formula Cl4 with a strong base such as lithium
bis(trimethylsilyl)amide, lithium diisopropylamide following by addition
of alkylating or acylating reagents such as aL~yl halides, aryl alkyl
halides, acyl halides, and haloformates in a inert solvent such as THF at
temperatures from -100 to room temperature. Thio derivatives where
the sulfur is attached directly to an alkyl or an aryl group can be prepared
3 .simil~rly by reacting with a disulfide. The halides used in these
f reactions are either commercially available or known compounds in the
literature or may be prepared by methods analogous to those used for the
preparation of known compounds. The protecting group L in compounds
of formula Cl5 may be removed with conventional chemistry to give
compounds of Formula 2.
WO 9~i113069 PCrlUS94/12816
21752~8
- 88 -
SCHEME C1 1
EtO2C~,CN alkylation EtO2C CN
y . ~ (CH2)n
C16 17 CH2CI
reduction
H
~ N~
C2a
Alternative ways of preparing compounds of Formula 2
include construction of the ring itself (Jacoby, R. L. et al, J. Med. Chem.,
17, 453-455, (1974)). Alkylation of the cyanoacetates of general formula
C16, which are commercially available or may be prepared from
literature procedures, with aLkyl dihalides such as 1-bromo-2-
chloroethane or 1-bromo-3-chloropropane yields the chloride Cl7.
Reduction of the nitriles C17 by borane or by hydrogenation using Raney
20 Ni as a catalyst gives the corresponding primary amines, which upon
refluxing in ethanol to give compounds of Formula 2a.
SCHEME C12
EtO2C~,CN Br(cH2)nco2Et EtO2C~<CN
C16 2 2 C18 CO2Et
reduction
~ of CN
reduction Oq, N
(CH2)n~co Et (CH2)n~co Et
Alternatively, the cyanoacetates of general formula C16 may
be aLIcylated with an ethoxycarbonylaLkyl bromide or reacted with ethyl
~ WO95/13069 2 1 7 52 1 8 r~-l/u~4/l28l6
- 89 -
acrylate to give compounds of Forrnula C18. Reduction of the nitriles
C 18 by borane or by hydrogenation using Raney Ni as a catalyst gives
the corresponding primary amines, which upon refluxing in ethanol gives
lactam C19. Reduction of the lactam C19 by borane gives compounds of
5 Formula C2a.
SCHEME C13
EtO2C CO2Et EtO2C CO2Et
10 ~ ~ (CH2)n
C20 C21 CN
reduction
H H
n(H2C~ reduction n(H2C) ~
y CO2Et y CO2Et
C2a C22
Alternatively, a malonate of general formula C20 may be
aL~ylated with cyanoaL~yl bromide or can be reacted with acrylonitrile to
forrn compounds of forrnula C21. Reduction of the nitriles C21 by
borane or by hydrogenation using Raney Ni as a catalyst gives the
corresponding primary amines, which upon refluxing in ethanol gives
lactam C22. Reduction of the lactam C22 by borane gives compounds of
formula C2a.
J
WO 9S/13069 PCIIUS9~/12816
21 7521 8 ~
- 90 -
SCHEME C14
L L L
(CH2~ ~ ~ (CH2jn ~ (CH ')N ~
\~Lco2Et \ f CO2H \ f CONR2R3
y y y or ester
C15a \ CH2Br2 C23 C15b
\Bases
Arndt-Eistert
L reaction
(CH )N~
~CH2CO2R or
y amide
C15c
The X, Y functionalities in compounds of general structure
C15 may be further elaborated to groups not accessible by direct
15 aL~ylation. For example in Compound C15 when X = C02Et the ester
(provided that this is the only ester group in the molecule) can be
saponified to the carboxylic acid, which can be further derivatized to
amides or other esters. The carboxylic acid can be converted into its next
higher homologue, or to a derivative of the homologous acid, such as
2 o amide or ester by an Arndt-Eistert reaction. Alternatively, the ester can
be directly homologated by the protocol using ynolate anions described
by C. J. Kowalski and R. E. Reddy in J. Org. Chem., 57, 7194-7208
(1992). The resulting acid and/or ester may be converted to the next
higher homologue, and so on and so forth. The protecting group L may
25 be removed through conventional chemistry.
WO 95/13069 PCI-/US94/12816
2175218
- 91 -
SCHEME C15
h reduction
(CH2)n
--~ CO2Et
Y
C15a
L L
(cH2jn > y (CH2)n ~
\~ CH20H \~ CH2O2CR2
Y C18 Y C19
1) MsCI/TEA
, 2)NaN3
L
(CH~ reduction (CH~2~
C20 Y CH2NH2
The ester in C15a may be reduced to an alcohol C18 in a
suitable solvent such as THF or ether with a reducing agent such as
DIBAL-H and conveniently carried out at temperatures from -100C to
0C. The alcohol may be acylated to Compound Cl9 in a suitable
solvent such as dichloromethane using an acyl halide or acid anhydride in
2 5 the presence of a base such as triethyl amine (TEA). The hydroxy group
in C 18 may also be converted to a good leaving group such as mesylate
and displaced by a nucleophile such as cyanide, a thiol or an azide.
Reduction of the azide in compounds of Forrnula C20 to an amine C2 1
can be achieved by hydrogenation in the presence of a noble metal such
3 as palladium or its oxide or Raney nickel in a protic solvent such as
ethanol. The nitrile can be reduced to afford the homologous amine. The
amine of Formula C21 may be further elaborated to amides, ureas
sulfonamides as defined by X through conventional chemistry. The
protecting group L may be removed through conventional chemistry.
WO9S/13069 2 1 752 1 8 ~ usg4/l28l6~
- 92 -
SCHEME C16
Bn Bn
(CH~ ~(C72)n 1
C28 o C29 Y (orX)
acylation
H
( 72)n 1 deprotection (C72)Nl
Y (or X) OCOR
C2b C30Y (or X)
In cases where oxygen is directly attached to the ring, a
convenient method involves the addition reaction by an activated form of
an aL~yl, aryl, alkylaryl group, such as lithium reagent, Grignard reagents,
and the like with a ketone of general formula C28, which is cornmercially
available. Further derivatization of the resulting hydroxy group by
20 acylation, sulfonylation, alkylation, and the like gives compounds as
defined by Y or X through conventional chemistry. Removal of the
benzyl protective group may be carried out under the usual conditions to
give compounds of general formula C2b. Shown in Scheme C 16 is a
general example of acylations.
~ WO 95/13069 2 1 7 5 2 1 8 PCI'JUS94112816
- 93 -
SCHEME C17
L L
(CH2)n~ 1) (COC1)2 (CH2)n~
s \~CO2H 2) NaN3 --~N=C=O
y3) reflux/toluene C31 Y
C23 amines
H20 \or alcohol
or \~
1) BnOH ureas or
0 2) Pd/C ~ carbamates
(CH ')N > acylation (CH ')N ~
\~ NHCOR deprotection \~ NH2
C2c Y C32 Y
In cases where a nitrogen-substituted group is directly attached to
the ring, a convenient method is to use the Curtius rearrangement on the
acid C23 to afford the isocyanate C3 1. Addition of amines or alcohols
give ureas or carbamates respectively which can be deprotected to
remove L to give special cases of compounds of formula C2. Conversion
f the isocyanate to amine by hydrolysis gives compound C32. Further
derivatization of the resulting amine group by acylation, sulfonylation,
alkylation, and the like to give compounds as defined by Y or ~ can be
done through conventional chemistry. Removal of the protective group L
may be carried out under the usual conditions to give compounds of
general formula C2c. Shown in Scheme C17 is a general example of
acylations.
WO9~113069 2 1 752 ~ 8 PCI/US94/12816
- 94 -
.
SCHEME C18
L L L
(CH2)n ~ (CH2)n ~ (CH2)n
I Bu3SnN I HCI
--~X ~ 3 ~X ~ X
~tJétrazole [~ [~CO/ Et
C15c C15b C15d
NaOH
DMSO
22
h
1 5 (CH~
X
-J
~ CONH2
C15e
E~or compounds that are not readily obtainable by direct
aL~ylation as shown in Scheme C10, modifications of easily obtainable
compounds of general formula C15 may be conducted to achieve the
desired substitution through conventional chemistry. For example,
25 compounds with Y being hydroxybenzyl may be prepared by
demethylation of the corresponding compound wherein Y is
methoxybenzyl. Similarly, compounds with Y being aminobenzyl may
be prepared by reduction of the corresponding compound wherein Y is
nitrobenzyl. Shown in Scheme Cl 8 is an example of a procedure that
3 o uses nitrile as a starting point for the preparation of compounds with
different substitutions. Removal of the protective group L gives
compounds of general formula C2 as described in Scheme C10.
Compounds of the general formula C2 prepared in this way
are racemic when X and Y are not identical. Resolution of the two
enatiomers can be conveniently achieved by classical cryst~lli7~tion
WO 95/13069 PCI-/US94/12816
2175218
methods by using a chiral acid such as L- or D-tartaric acid, (+) or (-)-10-
camphorsulfonic acid in a suitable solvent such as acetone, water,
alcohol, ether, acetate or their mixture. Alternatively, the racemic amine
- C2 can be reacted with a chiral auxiliary such as (R) or (S)-O-
5 acetylmandelic acid followed by chromatographic separation of the two
diastereomers, and removal of the chiral auxiliary by hydrolysis.
Alternatively asymmetric alkylation can also be utilized for the synthesis
of optically active intermediate by introducing a removable chiral
auxiliary in X or in place of L with subsequent chromatographic
separation of diastereomers.
In cases where a sulfide is present in the molecule, it may be
oxidized to a sulfoxide or to a sulfone with oxidizing agents such as
sodium periodate, m-chloroperbenzoic acid or Oxone~) in an solvent
such as dichloromethane, alcohol or water or their mixtures.
The compounds of the present invention may also be
prepared from a variety of substituted natural and llnn~tllral amino acids
of formula D46. The preparation of many of these acids is described in
US Patent No. 5,206,237. The preparation of these intermediates in
racemic form is accomplished by classical methods f~mili~r to those
skilled in the art (Willi~ , R. M. "Synthesis of OpticallyActive o~-Amino
Acids" Pergamon Press: Oxford, 1989; Vol. 7). Several methods exist to
resolve (DL)-
H R
R1~N~H
2 5 CO2H
D46
amino acids. One of the common methods is to resolve amino or
carboxyl protected intermediates by cryst~lli7~tion of salts derived from
3 0 optically active acids or amines. Alternatively, the amino group of
carboxyl protected intermediates may be coupled to optically active acids
by using chemistry described earlier. Separation of the individual
diastereomers either by chromatographic techniques or by cryst~lli7~tion
followed by hydrolysis of the chiral amide furnishes resolved amino
acids. Similarly, amino protected intermediates may be converted to a
WO 9S113069 2 1 7 5 2 1 8 PCI~/US94/128~
- 96 -
mixture of chiral diastereomeric esters and amides. Separation of the
mixture using methods described above and hydrolysis of the individual
diastereomers provides (D) and (L) amino acids. Finally, an enzymatic
method to resolve N-acetyl derivatives of (DL)-amino acids has been
reported by Whitesides and coworkers in J. Am. Chem. Soc. 1989, 111,
6354-6364.
When it is desirable to synthesize these intermediates in
optically pure form, established methods include: (1) asymmetric
electrophilic ~min~tion of chiral enolates (J. Am. Chem. Soc. 1986, ~
10 6394-6395, 6395-6397, and 6397-6399), (2) asymmetric nucleophilic
~min~tion of optically active carbonyl derivatives, (J. Am. Chem. Soc.
1992, 114, 1906; Tetrahedron Lett. 1987, 28, 32), (3) diastereoselective
aL~ylation of chiral glycine enolate synthons (J. Am. Chem. Soc. 1991,
113, 9276; J. Org. Chem. 1989, 54, 3916), (4) diastereoselective
15 nucleophilic addition to a chiral electrophilic glycinate synthon (J. Am.
Chem.Soc.1986,108,1103),(5)asymmetrichydrogenationofprochiral
dehydroamino acid derivatives ("Asymmetric Synthesis, Chiral Catalysis;
Morrison, J. D., Ed; Academic Press: Orlando, FL, 1985; Vol S), and (6)
enzymatic syntheses (Angew. Chem. Int. Ed. Engl. 1978, 17, 176).
2s
~ WO 95/13069 PCI-IUS94112816
21 7521 8
- 97 -
SCHEME D14
Ph Ph
NaN(TMS)2, Ph,
' ~o cinnamyl bromide ~ 1
t-Boc t-Boc
D47 Ph
1 ) TFA D48
1 o 2)PdCI2/H2
Ph~ NH2
CO2H
D49
For example, alkylation of the enolate of diphenyloxazinone
D47 (J. Am. Chem. Soc. 1991, 113, 9276) with cinnamyl bromide in the
presence of sodium bis(trimethylsilyl)amide proceeds smoothly to afford
D48 which is converted into the desired (D)-2-amino-5-phenylpentanoic
acid D49 by removing the N-t-butyloxycarbonyl group with
trifluoroacetic acid and hydrogenation over a PdC12 catalyst (Scheme
D14).
SCHEME D15
2 5 Hl NaH/DMF H
HO/~/N~L Ar-CH2-X Ar~O N~L
CO2H CO2H
D50 D51
Intermediates of formula D46 which are O-benzyl-(D)-
serine derivatives D5 1 are conveniently prepared from suitably
substituted benzyl halides and N-protected-(D)-serine D50. The
protecting group L is conveniently a BOC or a CBZ group. Benzylation
of D64 can be achieved by a number of methods well known in the
WO 9S/13069 2 1 7 5 2 ~ 8 PCIIUS9~/1281~
- 98 -
literature including deprotonation with two equivalents of sodium hydride
in an inert solvent such as DMF followed by treatment with one
equivalent of a variety of benzyl halides (Synthesis 1989, 36) as shown in
Scheme D15.
The O-alkyl-(D)-serine derivatives may also be prepared
using an alkylation protocol. Other methods that could be utilized to
prepare (D)-serine derivatives of formula D51 include the acid catalyzed
benzylation of carboxyl protected intermediates derived from D50 with
reagents of formula ArCH20C(=NH)CC13 (O. Yonemitsu et al., Chem.
Pharm. Bull. 1988, 36, 4244). Alternatively, aLkylation of the chiral
gylcine enolates (J. Am. Chem. Soc. 1991, 113, 9276; J. Org. Chem.
1989, 54, 3916) with ArCH2OCH2X where X is a leaving group affords
D51. In addition D,L-O-aryl(aLkyl)serines may be prepared and resolved
by methods described above.
It is noted that in some situations the order of carrying
out the foregoing reaction schemes may be varied to facilitate the
reaction or to avoid unwanted reaction products.
The utility of the compounds of the present invention as
growth hormone secretagogues may be demonstrated by methodology
20 known in the art, such as an assay described by Smith, et al., Science,
260, 1640-1643 (1993) (see text of Figure 2 therein). In particular, all
of the compounds prepared in the following examples had activity as
growth hormone secretagogues in the aforementioned assay. Such a
result is indicative of the intrinsic activity of the present compounds as
25 growth hormone secretagogues.
The growth hormone releasing compounds of Formula I
are useful in vitro as unique tools for understanding how ~rowth
hormone secretion is regulated at the pituitary level. This includes use
in the evaluation of many factors thought or known to influence
30 growth hormone secretion such as age, sex, nutritional factors,
glucose, amino acids, fatty acids, as well as fasting and non-fasting
states. In addition, the compounds of this invention can be used in the
evaluation of how other hormones modify growth hormone releasing
activity. For example, it has already been established that
WO 95113069 PCI'IUS94112816
~ 2175218
99
somatostatin inhibits growth hormone release. Other hormones that
are important and in need of study as to their effect on growth
hormone release include the gonadal hormones, e.g., testosterone,
estradiol, and progesterone; the adrenal hormones, e.g., cortisol and
5 other corticoids, epinephrine and norepinephrine; the pancreatic and
gastrointestinal hormones, e.g., insulin, glucagon, gastrin, secretin; the
vasoactive peptides, e.g., bombesin, the neurokinins; and the thyroid
hormones, e.g., thyroxine and triiodothyronine. The compounds of
Formula I can also be employed to investigate the possible negative or
positive feedback effects of some of the pituitary hormones, e.g.,
growth hormone and endorphin peptides, on the pituitary to modify
growth hormone release. Of particular scientific importance is the use
of these compounds to elucidate the subcellular mechanisms
mediating the release of growth hormone.
The compounds of Formula I may be ~rlmini.~tered to
~nim~l~, including man, to release growth hormone in vivo. For
example, the compounds can be ~(lmini~tered to commercially
important ~nim~l~ such as swine, cattle, sheep and the like to
accelerate and increase their rate and extent of growth, to improve
20 feed efficiency and to increase miLk production in such ~nim~l~. In
addition, these compounds can be ~flmini.~tered to humans in vivo as a
diagnostic tool to directly determine whether the pituitary is capable
of releasing growth hormone. For example, the compounds of
Formula I can be ~lmini.~tered in vivo to children. Serum samples
25 taken before and after such ~-lmini.~tration can be assayed for growth
hormone. Comparison of the amounts of growth hormone in each of
these samples would be a means for directly determining the ability of
the patient's pituitary to release growth hormone.
- Accordingly, the present invention includes within its
3 scope pharmaceutical compositions comprising, as an active
ingredient, at least one of the compounds of Formula I in association
with a pharmaceutical carrier or diluent. Optionally, the active
ingredient of the pharmaceutical compositions can comprise an
anabolic agent in addition to at least one of the compounds of Formula
WO95113069 2 1 752 1 8 PCI'/US9~/12816
- 100-
I or another composition which exhibits a different activity, e.g., an
antibiotic growth permittant or an agent to treat osteoporosis or in
combination with a corticosteroid to minimi7e the catabolic side
effects or with other pharmaceutically active materials wherein the
5 combination enhances efficacy and minimi7es side effects.
Growth promoting and anabolic agents include, but are
not limited to TRH, diethylstilbesterol, estrogens, ~-agonists,
theophylline, anabolic steroids, enkephalins, E series prostaglandins,
compounds disclosed in U.S. Patent No. 3,239,345, e.g., zeranol, and
compounds disclosed in U.S. Patent No. 4,036,979, e.g., sulbenox or
peptides disclosed in U.S. Patent No. 4,411,890.
A still further use of the growth hormone secretagogues
of this invention is in combination with other growth hormone
secretagogues such as the growth hormone releasing peptides GHRP-
5 6, GHRP-l as described in U.S. Patent Nos. 4,411,890 and
publications WO 89/07110, WO 89/07111 and B-HT920 as well as
hexarelin and the newly discovered GHRP-2 as described in WO
93/04081 or growth hormone releasing hormone (GHRH, also
designated GRF) and its analogs or growth hormone and its analogs or
20 somatomedins including IGF-l and IGF-2 or a- adrenergic aginists
such as clonidine or serotonin 5HTID agonists such as sumitriptan or
agents which inhibit somatostatin or its release such as physostigmine
and pyridostigmine.
As is well known to those skilled in the art, the known and
25 potential uses of growth hormone are varied and multitudinous. The
~clmini~tration of the compounds of this invention for purposes of
stimulating the release of endogenous growth hormone can have the same
effects or uses as growth hormone itself. These varied uses of the present
compounds thus may be sllmm~rized as follows: stimulating growth
30 hormone release in elderly humans; treating growth hormone deficient
adults; prevention of catabolic side effects of glucocorticoids; treatment
of osteoporosis; stimulation of the immune system, acceleration of wound
healing; accelerating bone fracture repair; treatment of growth
retardation; treating acute or chronic renal failure or insufficiency;
~ WO 95tl3069 2 1 7 5 2 1 8 PCI~/US94/12816
- 101 -
treatment of physiological short stature, including growth hormone
deficient children; treating short stature associated with chronic illness;
treatment of obesity and growth retardation associated with obesity;
treating growth retardation associated with Prader-Willi syndrome and
5 Turner's syndrome; accelerating the recovery and reducing
hospit~li7~tion of burn patients or ~ollowing major surgery such as
gastrointestinal surgery; treatment of intrauterine growth retardation, and
skeletal dysplasia, treatment of peripheral neuropathies; replacement of
growth hormone in stressed patients; treatment of osteochondrody-
splasias, Noonans syndrome, schizophrenia, depression, Alzheimer's
disease, delayed wound healing, and psychosocial deprivation; treatment
of pulmonary dysfunction and ventilator dependency; attenuation of
protein catabolic response after a major operation; treating malabsorption
syndromes; reducing cachexia and protein loss due to chronic illness such
5 as cancer or AIDS; accelerating weight gain and protein accretion in
patients on TPN (total parenteral nutrition); treatment of
hyperinsulinernia including nesidioblastosis; adjuvant treatment for
ovulation induction and to prevent and treat gastric and duodenal ulcers;
to stimulate thymic development and prevent the age-related decline of
20 thyrnic function; adjunctive therapy for patients on chronic hemodialysis;
treatment of immunosuppressed patients and to enhance antibody
response following vaccination; increasing the total lymphocyte count of
a h~lm~n, in particular, increasing the T4/Tg-cell ratio in a human with a
depressed T4/Tg-cell ratio resulting, for example, from physical trauma,
25 such as closed head injury, or from infection, such as bacterial or viral
infection, especially infection with the human immunodeficiency virus;
improvement in muscle strength, mobility, maintenance of skin thickness,
metabolic homeostasis, renal hemeostasis in the frail elderly; stimulation
- of osteoblasts, bone remodelling, and cartilage growth; stimulation of the
30 immune system in companion ~nim~l~ and treatment of disorders of
aging in companion ~nim~l~; growth promotant in livestock; and
stimulation of wool growth in sheep. Further, the instant compounds are
useful for increasing feed efficiency, promoting growth, increasing milk
production and improving the carcass quality of livestock.
WO 95113069 PCr/US94/12816
2775218
- 102-
In particular, the instant compounds are useful in the
prevention or treatment of a condition selected from the group consisting
of: osteoporosis; catabolic illness; immune deficiency, including that in
individuals with a depressed T4/Tg cell ratio; hip fracture;
musculoskeletal impairment in the elderly; growth hormone deficiency in
adults or in children; obesity; cachexia and protein loss due to chronic
illness such as AIDS or cancer; and treating patients recovering from
major surgery, wounds or burns, in a patient in need thereof.
It will be known to those skilled in the art that there are
numerous compounds now being used in an effort to treat the diseases or
therapeutic indications enumerated above. Combinations of these
therapeutic agents some of which have also been mentioned above with
the growth hormone secretagogues of this invention will bring additional,
complementary, and often synergistic properties to enhance the growth
promotant, anabolic and desirable properties of these various therapeutic
agents. In these combinations, the therapeutic agents and the growth
hormone secretagogues of this invention may be independently present in
dose ranges from one one-hundredth to one times the dose levels which
are effective when these compounds and secretagogues are used singly.
Combined therapy to inhibit bone resorption, prevent
osteoporosis and enhance the he~ling of bone fractures can be illustrated
by combinations of bisphosphonates and the growth hormone
secretagogues of this invention. The use of bisphosphonates for these
utilities has been reviewed, for example, by Hamdy, N.A.T., Role of
Bisphosphonates in Metabolic Bone Diseases, Trends in Endocrinol.
Metab., 4, 19-25 (1993). Bisphosphonates with these utilities include
alendronate, tiludronate, dimethyl-APD, risedronate, etidronate, YM-175,
clodronate, pamidronate, and BM-210995. According to their potency,
oral daily dosage levels of the bisphosphonate of between 0.1 mg and 5 g
3 and daily dosage levels of the growth hormone secretagogues of this
invention of between 0.01 mg/kg to 20 mg/kg of body weight are
~lmini.~tered to patients to obtain effective treatment of osteoporosis.
The compounds of this invention can be ~lmini~tered by
oral, parenteral (e.g., intramuscular, intraperitoneal, intravenous or
WO 95/13069 1 ~ 9-1/12816
2175218
- 103-
subcutaneous injection, or implant), nasal, v~gin~l, rectal, sublingual, or
topical routes of ~lminictration and can be formulated in dosage forms
appropriate for each route of ~minictration.
Solid dosage forms for oral ~dminictration include capsules,
5 tablets, pills, powders and granules. In such solid dosage forms, the
active compound is admixed with at least one inert pharmaceutically
acceptable carrier such as sucrose, lactose, or starch. Such dosage forms
can also comprise, as is normal practice, additional substances other than
inert diluents, e.g., lubricating agents such as magnesium stearate. In the
case of capsules, tablets and pills, the dosage forms may also comprise
buffering agents. Tablets and pills can additionally be prepared with
enteric coatings.
Liquid dosage forms for oral ~flmini.ctration include
pharmaceutically acceptable emulsions, solutions, suspensions,
5 syrups, the elixirs cont~ining inert diluents commonly used in the art,
such as water. Besides such inert diluents, compositions can also
include adjuvants, such as wetting agents, emulsifying and suspending
agents, and sweetening, flavoring, and perfuming agents.
Preparations according to this invention for parenteral
20 ~lminictration include sterile aqueous or non-aqueous solutions,
suspensions, or emulsions. Examples of non-aqueous solvents or
vehicles are propylene glycol, polyethylene glycol, vegetable oils,
such as olive oil and corn oil, gelatin, and injectable organic esters
such as ethyl oleate. Such dosage forms may also contain adjuvants
25 such as preserving, wetting, emulsifying, and dispersing agents. They
may be sterilized by, for example, filtration through a bacteria-
retaining filter, by incorporating sterilizing agents into the
compositions, by irradiating the compositions, or by heating the
compositions. They can also be manufactured in the form of sterile
3 solid compositions which can be dissolved in sterile water, or some
other sterile injectable medium immediately before use.
Compositions for rectal or vaginal ~minictration are
preferably suppositories which may contain, in addition to the active
substance, excipients such as cocoa butter or a suppository wax.
WO 95113069 PCI~/US94/12816
2175218
- 104-
Compositions for nasal or sublingual ~(lmini~tration are
also prepared with standard excipients well known in the art.
The dosage of active ingredient in the compositions of
this invention may be varied; however, it is necessary that the amount
5 of the active ingredient be such that a suitable dosage form is
obtained. The selected dosage depends upon the desired therapeutic
effect, on the route of ~-lmini~tration, and on the duration of the
treatment. Generally, dosage levels of between 0.0001 to 100 mg/kg.
of body weight daily are ~lmini~tered to patients and ~nimal.~, e.g.,
m~mm~l~, to obtain effective release of growth hormone.
The following examples are provided for the purpose of
further illustration only and are not intended to be limitations on the
disclosed invention. As will be apparent, the examples and
intermediates designated "A" correspond to the compounds of the first
5 embodiment, those designated "B" correspond to the compounds of
the second embodiment, and those designated "C" correspond to the
compounds of the third embodiment.
INTERMEDIATE 1
~N~
Step A:
~, NH2
H OBn
To a solution of the commercially available N-t-BOC-D-
tryptophan (25.0 g, 82.2 mmol), benzyl alcohol (10.2 mL, 98.6 mmol),
and DMAP (100 mg) in dichloromethane (200 mL) at 0C, was added
EDC (17.4 g, 90.4 mmol) in several portions over a one hour period. The
WO 95113069 P~ 94/12816
2175218
- 105-
reaction rnixture was stirred at room temperature for six hours and was
poured into water (200 mL), and the organic layer was separated. The
organic solution was washed with a mixture of brine and 3 N
hydrochloric acid, dried over anhydrous m~gnesium sulfate, filtered and
concentrated to give a thick oil, which solidified upon standing.
To a solution of this oil in 30 mL of dichloromethane was
added 20 mL of TFA and stirred for lh. The reaction mixture was
concentrated, neutralized carefully with saturated aqueous sodium
bicarbonate solution, and extracted with dichloromethane (2X100 mL).
The combined organic solution was washed with brine (100 mL), passed
through a short column of silica gel eluting with 5-10% methanol in
dichloromethane to give 23.2 g of the amine as an oil after evaporation.
Step B:
~=0 0
H OBn
To a solution of the above product, HOBT (10.6 g, 78.8
mmol) and N-BOC-a-methyl ~l~nine (19g, 94.5 mmol) in 200 mL of
dichloromethane, was added EDC (19.5 g, 0.102 mol) in several portions
at 0C. After S minutes, the clear reaction mixture became miL~y. After
stirring at room temperature overnight, the reaction mixture was poured
into 200 mL of water and the organic layer was separated. The organic
solution was washed with brine, and with a brine and saturated sodium
bicarbonate solution, dried over anhydrous magnesium sulfate, filtered
and concentrated to give a thicl~ oil, which was purified by flash
30 chromatography eluting with a gradient of 10-40% ethyl acetate in
hexane to give the desired material (28.7 g).
lH NMR (CDCl3, 200 MHz) ~ 8.48 (br.s, lH), 7.54 (br.d, lH), 7.38-7.23
(m, 3H), 7.19 (br.d, 2H), 7.15-7.00 (m, lH), 6.90 (d, lH), 6.86 (d, lH),
5.06 (br.s, 2H), 4.95 (ddd, lH), 3.30 (2dd, 2H), 1.40 (s, lSH)
WO95/13069 2~75218 PCI-/US94/1281~
- 106-
Step C: H
S ~ O
A solution of the material from Step B (28.7g) in 200 mL of
ethanol was stirred at RT under a H2 balloon for 20 minutes in the
presence of 10% palladium on carbon (2 g). The catalyst was filtered off
through a pad of celite and washed with ethyl acetate. The filtrate was
concentrated to give the acid as a slightly pink foam (23.3 g).
lH NMR (CD30D, 400 MHz) o 7.56 (d, J=8 Hz, 1 H), 7.31 (dd, J=1, 8
Hz, 1 H), 7.09 (s, 1 H), 7.07 (dt, J=1, 7 Hz, 1 H), 6.98 (dt, J=1, 7 Hz, 1
H), 4.69 (t, J=6 Hz, 1 H), 3.34-3.23 (m, 2 H), 1.35 (s, 3 H), 1.34 (s, 9 H),
1.29 (s, 3 H).
FAB-MS calc. for C20H27N3os: 389; Found 390 (M+H), 290 (M+H-
100 (BOC)).
INTERMEDIATE 2
3/~0 ~ ~N HBoc
ç=o O
OH
Following the procedures for the preparation of Intermediate
1 using N-t-Boc-O-Benzyl-D-serine in the place of N-t-BOC-D-
tryptophan gave Intermediate 2.
FAB-MS calc. for Cl9H28N2o6: 380; Found 381 (M+H), 325 (M+H-
56 (t-Bu)), 281 (M+H-100 (BOC)).
~WO95113069 2 1 752 ~ 8 PCI~/US94112816
- 107-
INTERMEDIATE 3
¢~ I' ~NHBoc
Ç=o o
OH
Step A: (DL)-N-acetyl-2-amino-5-phenylpentanoic acid
To a solution of sodium (2.3 g, 0.1 mol) in ethanol (60 mL)
under nitrogen at room temperature, was added diethyl
acetamidomalonate. The mixture was stirred at room temperature for one
l0 hour, and then 1-bromo-3-phenylpropane was added dropwisely. After
the addition, the mixture was stirred at room temperature for two hours,
then refluxed overnight. It was cooled to room temperature and
partitioned between water and ethyl acetate. The organic layer was
washed with sodium bicarbonate in water, dried over MgSO4 and
evaporated to give an interrnediate (32.5 g, 97%).
lH NMR (CDC13, 400MHz) 7.26-7.10 (m, 5 H); 6.75 (br. s, 1 H); 4.19
(q,J=7Hz,4H);2.58(t,J=7.9Hz,2H);2.39-2.35(m,2H);2.00(s,3
H); 1.43-1.39 (m, 2 H); 1.20 (t, J=7 Hz, 6 H).
The product above was suspended in 190 mL of 2.5 N
20 NaOH in water and refluxed for two hours. The mixture was cooled to
0C, and it was carefully neutralized with 6 N HCl to pH2. The
precipitate was collected using a sintered glass funnel and washed with a
small amount of cold water and air dried. The solid was then suspended
in 300 mL of water and refluxed for ~our hours. The solution was cooled
2s and acidified to pHl and the solid was collected by filtration (15.3 g,
67%).
lH NMR (CD30D, 400MHz) 7.26-7.12 (m, 5 H); 4.90-4.37 (m, 1 H);
2.65-2.60 (m, 2 H); 1.97 (s, 3 H); 1.87 -1.82 (m, 1 H); 1.73-1.65 (m, 3 H).
30 Step B: (D)-N-acetyl-2-amino-5-phenylpentanoic acid
The racemic intermediate from the previous step (10 g, 42.5
mmol) and CoCl3-6H20 were dissolved in 21 ml of 2 N KOH and 200
mL of water at 40C, and the pH of the solution was adjusted to 8 by the
addition of the several drops of 2 N KOH. Then acylase I (Aspergillus
sp, 0.5 u/mg, from Sigma; 0.9 g) was added with vigorous stirring. The
WO 9S113069 2 1 7 5 2 1 8 Pcrrus94ll28l6~
- 108-
reaction mixture was stirred for one day at 40C and the pH was kept at 8
by the addition of a few drops of KOH. The solid which formed was
filtered off. The filtrate was acidified by 3 N HCl to pH2, and was
extracted with ethyl acetate (200 mLX4). The organic extracts were
5 combined and evaporated to give a white solid (4.64 g, 46%)
1H NMR (CD30D, 400MHz) 7.26-7.12 (m, 5 H); 4.90-4.37 (m, 1 H);
2.65-2.60 (m, 2 H); 1.97 (s, 3 H); 1.87 -1.82 (m, 1 H); 1.73-1.65 (m, 3 H).
Step C: (D)-N-t-Boc-2-amino-5-phenylpentanoic acid
The intermediate from step B (4.2 g, 17.8 mmol) was
suspended in 2 N HCl (100 mL) and refluxed for two hours. The reaction
mixture was evaporated in vacuo to remove water and hydrochloric acid
to yield a white solid. To a solution of this solid in 50 mL of water, was
added 3 N NaOH until the pH 11, then di-t-butyl dicarbonate (4.66 g,
5 21.4 mmol) was added with vigorous stirring. After four hours, the
reaction mixture was acidified to pH2 with 3 N HCl and it was extracted
with ethyl acetate (100 mLX3). The organic extracts were combined and
evaporated to give a white solid (6.56 g, crude) which was used without
purification.
20 1H NMR (CD30D, 400MHz) 7.26-7.12 (m, 5 H); 4.11-4.08 (m, 1 H);
2.65-2.60 (m, 2 H); 1.83-1.62 (m, 4 H); 1.43 (s, 9 H).
Step D: H
~ ~NHBoc
C=O O
OH
Following the procedures for the preparation of Intermediate 1
using (D)-N-t-Boc-2-amino-5-phenylpentanoic acid in the place of N-t-
BOC-D-tryptophan gave Intermediate 3.
1H NMR (CDCl3, 400MHz) 7.24-7.20 (m, 2H), 7.15-7.04 (m, 3H), 4.60-
4.55 (m, lH), 2.62-2.55 (m, 2H), 2.00-1.86 (m, lH), 1.78-1.60 (m, 3H),
1.50 (s, 6H), 1.30 (s, 9H).
WO 95113069 2 ~ 7 5 2 PCr~US94/12816
- 109-
~XAMPLE A 1
~= O O
H ~CO2Et
Step A:
~NHBoc ~ ~NHBoc
C-O O N
H~CO2Et H ~ CO2Et
To a solution of ethyl (dl) pipecolinate (1 g ), HOBT (860
mg) and Intermediate 1 (2.47 g ) in dichloromethane (80 mL) at 0C, was
added EDC (2.3 g). The reaction mixture was stirred at room temperature
overnight. The solution was washed with water, saturated sodium
2 bicarbonate solution, and saturated sodium chloride solution, dried over
anhydrous m~gnesium sulfate; then filtered and concentrated to give a
crude product. The crude product was purified by MPLC eluting with
60% ethyl acetate in hexane to give the product as a mixture of two
diastereomers (2.79 g). Separation of 500 mg of the mixture by MPLC
25 eluting with 50% ethyl acetate in hexane yielded the two individual
diastereomers. The diastereomer which came out of the column first was
designated as d 1 ( 187 mg) and the stereochemistry of the pipecolinic acid
ester was subsequently shown to be R. The one which came out last was
designated as d2 (116 mg) and the stereochemistry of the pipecolinic acid
3 ester in it is S. In addition, there were mixed fractions which were
combined and evaporated to yield 190 mg of a mixture of dl and d2.
dl: FAB-MS calc. for C28H40N4o6: 528; Found: 529 (M+H)
d2: FAB-MS calc. for C28H40N4o6: 528; Found: 529 (M+H)
-
WO 95/13069 ~ IIJS94/12816
217~218 ~
- 110-
Step B:
~,c=o o
H ~CO2Et
A solution of the compound d1 from Step A ( 140 mg) in
ethyl acetate (5 mL) was cooled to 0C. While stirring, hydrogen chloride
gas was bubbled into the mixture until saturation occurred. The reaction
was stirred for 15 minutes. The solution was then concentrated to remove
ethyl acetate. Ihe residue was ~en redissolved in dichloromethane and
hexane followed by evaporation in vacuo to afford the product as a solid
(110 mg).
FAB-MS calc. for C23H32N404: 428; Found: 429 (M+H)
1HNMR (400 MHz, CD30D): compound exists as a mixture of rotamers
(about 2:1). 7.57 (d, 1 H), 7.36 &7.32 ( 2 d, 1 H), 7.14-7.00 (m, 3 H),
20 5.30-5.20 (m), 5.17-5.13 (m), 4.36 (d), 4.21 (q, J=7 Hz), 4.13 (q, J=7 Hz),
4.00 (md), 3.35-3.04 (m), 2.60 (dt), 3.30 ( br. d), 2.70 -2.50 (m), 1.57 (s),
1.55 (s), 1.52 (s), 1.50-1.20 (m), 1.33 (s), 1.27 (t, J=7 Hz), 1.21 (t, J=7
Hz), 1.15-1.10 (m), 0.75-0.65 (m), 0.30-0.20 (rn).
2S EXAMPLE A2
~,C=O O
H ~N~ CO2Et
~
Prepared by the procedure described in Example Al, Step B
from the intermediate d2 from Example A1, step A (40 mg) and HCl gas
at 0C in ethyl acetate (3 mL). Product: 28 mg.
FAB-MS calc. for C23H32N4O4: 428; Found: 429 (M+H)
WO95113069 ~ 1 7 5 2 1 8 PCI-IUS94/12816
- 111 -
lHNMR (400 MHz, CD30D): compound exists as a mixture of rotamers
(about 5: 1). 7.56 (d, J= 8 Hz 5/6 H), 7.50 (d, 1/6 H), 7.34 (d, J=8 Hz, 5/6
H), 7.31 (d, 1/6 H), 7.12-7.00 (m, 3 H), 5.28 (dd, 5/6 H), 5.15-5.11 (m,
1/6 H), 5.11-5.07 (m, 1/6 H), 5.02-4.98 (m, 5/6 H), 4.52-4.45 (m), 4.12
(q, J=7 Hz), 4.25-4.00 (m), 3.65 (m), 3.30-3.05(m), 2.80-2.70 (m), 2.32-
2.25 (m), 2.02-1.97 (m), 1.75-1.65 (m), 1.57 (s), 1.52 (s), 1.51 (s), 1.40-
0.85 (m), 1.22 (t, J= 7 Hz), 0.41-0.30 (m).
EXAMPLE A3
=O O
H ~ CO2Bn
Step A:
~ ~ ~<NHBoc
C=O O
H ~ "~CO2Bn
To a stirred solution of L-proline benzyl ester hydrochloride
25 (155 mg, 0.64 mmole), Intermediate 1 (250 mg, 0.64 mmole), HOBT (1
eq.), and NMM (0.07 mL, 0.64 mmole) in dichloromethane at 0, was
added EDC (246 mg, 1.28 mmole). The reaction mixture was stirred at
0 overnight, and then partitioned between 3 N HCl and ethyl acetate.
The organic layer was washed with brine and saturated sodium
30 bicarbonate and dried and evaporated. MPLC purification eluting with
50% ethyl acetate gave the intermediate tripeptide benzyl ester( 338 mg,
91.5%)-
FAB-MS calc. for C32H40N4o6: 576; Found: 577 (M+H)
WO 9S/13069 PCI/US94/12816
2l75218 ~
- 112-
Step B:
~Ic=o o
H ~ CO2Bn
Prepared by the procedure described in Example Al, Step B
from the intermediate from the previous step (280 mg) and HCl gas in
ethyl acetate (10 mL) at 0C . Reaction time: 25 rninutes. Product: 218
mg.
FAB-MS calc. for C27H32N4O4: 476; Found: 477 (M+H)
lHNMR (400 MHz, CD30D ): 8.20 (d), 7.54 (d, J=7.9 Hz, lH) 7.34-7.00
5 (m, 9H), 5.11 (dd, J=4.2 Hz, 16.5 Hz, 2H), 4.99-4.94 (m, lH), 4.23-4.20
(m, 1 H), 3.58-3.53 (m, lH), 3.31-3.13 (m, 2H), 2.77-2.75 (m, lH), 1.71-
1.60 (m, 3H) 1.55 (s, 3H), 1.51 (s, 3H), 1.37-1.33 (m, lH).
EXAMPLE A4
c o~< 2
H ~rCo2B n
Step A: (dl)-Pipecolinic acid. benzyl ester
A solution of (dl)-pipecolinic acid (25g), p- toluenesulfonic
acid (38g), and benzyl alcohol (84g) in toluene (200 rnL) was refluxed
30 under azeotropic conditions for one day. The solution was cooled to room
temperature and the resulting crystals were collected to give the desired
product (52.4 g). The product was washed with 3 N NaOH to remove
toluenesulfonic acid, and then reacted with HCl gas in ethyl acetate to
convert it to the hydrochloride salt.
WO 95/13069 ~ 2816
2175218
- 113-
Step B:
C, =O O
H ~rCO2B n
Prepared by the procedure described in Example A3, Step A
from (dl)-pipecolinic acid benzyl ester hydrochloride (3.5 g).
Intermediate 1 (5.00 g), HOBt (1.74 g), NMM (1.42 mL) and EDC (3.94
g). Product: 6.32 g
FAB-MS calc. for C33H42N4O6: 590; Found: 591 (M+H)
Step C:
~NH2 HCI
H ~N~,~rC02B n
~
Prepared by the procedure described in Example Al, Step B
from the intermediate from the previous step (250 mg) and HCl gas in
ethyl acetate at 0C to give the title compound (211 mg)
25 FAB-MS calc. for C2gH34N4O4: 490; Found: 491 (M+H)
EXAMPLE A5
~ ~NH2 HCI
H ~N~CO N H Et
~J
-
WO 95/13069 PCI~/US9~/12816
~t75218
- 114-
Step A:
~= o o
H C~rCO2H
A suspension of the product from Fx~mple A4, step B (5.30
g) and 10% palladium on carbon (270 mg) in ethanol (100 mL) was
stirred under a hydrogen balloon for 3 hours. The reaction mixture was
filtered through celite, evaporated to give the acid ( 4.48g).
Step B:
C ~< ~= o o
H ~CONHEt H O~ CONHEt
Prepared ~imil~rly by the procedure described in Example
A3, Step A from the acid intermediate from the previous step (200 mg),
ethyl amine hydrochloride (27 mg), HOBt (54 mg), NMM (0.07 mL), and
EDC (154 mg) to give a mixture of two diastereomers, which were
25 separated by MPLC eluting with ethyl acetate. The isomer which came
out of the column first was designated as dl (76 mg), and the isomer
which came out second as d2 (165 mg).
dl FAB-MS calc. for C2gH41N5O5: 527; Found: 528 (M+H)
d2 FAB-MS calc. for C2gH41N5O5: 527; Found: 528 (M+H)
WO 9~113069 2 ~ 7 ~ 2 1 8 PCr/US94/12816
- 115-
- Step C:
~= O O
N ~CONHEt
Prepared ~imil~rly by the procedure described in Example
o A1, Step B from intermediate from the previous step (dl) (60 mg) and
HCl gas in ethyl acetate (5 mL) at 0C to give the title compound (38
mg). Reaction time: 20 minutes.
FAB-MS calc. for C23H33N5o3: 427; Found: 428 (M+H)
HNMR (400 MHz, CD30D ): d 7.63 - 7.00 (m, 5 H), 5.33 (t), 5.40 -
5.25 (m), 5.11 - 5.09 (m), 4.32 (br. d), 4.16 - 4.12 (m), 4.00 (md), 3.35 -
3.03 (m), 2.96 (q, J = 7 Hz), 2.30 (dt), 2.19 (br. d), 1.95 - 1.40 (m), 1.66
(s), 1.64 (s), 1.40 - 1.20 (m), 1.20 -1.00 (m), 1.12 (t, J = 7 Hz), 1.03 (t, J
7 Hz), 0.65 - 0.52 (m), -0.44 - -0.53 (m).
EXAMPLE A6
~NH2 HCI
H ~N~ coN HEt
~J
Prepared simil~rly by the procedure described in Example
A1, Step B from intermediate in Example A5 step B (d2) (100 mg) and
HCl gas in ethyl acetate (5 mL) at 0C to give the title compound (78
30 mg). Reaction time: 20 minutes.
FAB-MS calc. for C23H33NsO3: 427; Found: 428 (M+H)
lHNMR (400 MHz, CD30D ): d 7.54 (d, J = 8 Hz, 1 H), 7.35 (d, J = 8
Hz, 1 H),7.16(s, 1 H),7.13-7.00(m,2H),4.98(dd,J=6Hz, lOHz),
4.93 (d, 4 Hz), 3.53 (br. d, J = 12 Hz, 1 H), 3.35 - 3.22 (m), 3.14 - 3.09
(m, 1 H),2.85(dt,J=3, 13Hz, 1 H),2.02(br.d,J= 12Hz), 1.65(s,3
WO 9~/13069 PCr/lJS94/12816
21752~8
- 116-
H), 1.61 (s, 3 H), 1.10 (t, 7 Hz, 3 H), 1.05- 0.92 (m, 2 H), 0.72- 0.62 (m, 1
H), -0.25 - -0.30 (m, 1 H).
EXAMPLE A7
3~1c=o 0
H ~ C02Et
Step A:
c=o o
H ~ C02Et
Prepared by the procedure described in Example A3, Step A
20 from L-proline ethyl ester hydrochloride ( 115 mg, 0.642 mmole),
Intermediate 1 (250 mg, 0.642 mmole), HOBT (1 eq.), NMM(0.07 mL,
0.642 mmole), and EDC (246 mg, 1.28 rnmole). Product: 330 mg
FAB-MS calc. forC27H40N4o6: 514; Found: 515 (M+H)
25 Step B:
H ~N~ C02Et
Prepared by the procedure described in Example A1, Step B
from the interrnediate from the previous step (280 mg) and HCl gas in
ethyl acetate (10 mL) at 0C. Reaction time: 25 minutes. Product: 220
mg.
WO 95/13069 2 1 7 5 2 1 8 PCI/US94/12816
- 117-
FAB-MS calc. for C22H32N4O4: 414; Found: 415 (M+H)
1HNMR (400 MHz, CD30D ): 7.53 (d, J-7.9 Hz, lH), 7.34 (d, J-8.1 Hz,
lH), 7.14-7.01 (m, 3H), 4.97-4.84 (m, lH), 4.15-4.06 (m, 3H), 3.60-
3.53(m, lH), 3.31-3.13 (m, 2H), 2.77-2.72 (m, lH), 1.72-1.59 (m, 3H),
1.57 (s, 3H), 1.50 (s, 3H), 1.36-1.27 (m, lH), 1.23 (t, J=7.1 Hz, 3H).
EXAMPLE A8
~--~ ~X 2
H ~CN
Step A: 2-Cyano-1-hydroxy-4-phenylpiperidine
To a stirred solution of 4-phenylpiperidine (10 g, 0.062
mole) in methanol (30 mL), was added a solution of sodium tungstate
dihydrate (0.82 g, 2.48 mmole) in water (7 mL). With stirring at 0,
hydrogen peroxide (30%, 13.9 mL, 0.136 mole) was added dropwise.
After complete addition, the reaction mixture was stirred for an additional
3 hours, and then sodium cyanide (4.56 g, 0.093 mole) was added,
followed by 4 N HCl (22 mL, 0.088 mole). The reaction mixture was
stirred overnight during which time it warmed to room temperature. The
solid was collected by filtration through glass sinter funnel, and the
solution was neutralized to pH 7 and was extracted with
dichloromethane. The organic extract was combined with the solid and
- dried over MgSO4 and evaporated. Flash column purification elutingwith 40% ethyl acetate in hexane gave 2-cyano- 1 -hydroxy-4-
phenylpiperidine(8.6 g).
lHNMR (400 MHz, CDCl3 ): 7.35-7.17 (m, 5 H), 6.01 (br. s, lH), 4.34
(br. s, 1 H), 3.31 (td, J=3, 11 Hz, 1 H), 3.09 (dt, J=l l, 3 Hz, 1 H), 2.93-
2.86 (m, lH), 2.20-2.10 (m, 2 H), 1.97-1.80 (m, 2 H).
WO 95/13069 PCI~/US94112816
2~75218 ~
- 118-
Step B: 2-Cyano-4-phenylpiperidine
To a stirred solution of the intermediate from the previous
step (500 mg) in methanol (10 mL) at room temperature, was added
TiCl3 (10% solution in 20-30% hydrochloric acid (3 mL). The mixture
was stirred for 15 minutes and was neutralized by addition of 3 N NaOH.
The residue were extracted with dichloromethane four times and the
organic extracts was combined, dried over MgSO4, and evaporated to
give 450 mg of 2-cyano 4-phenylpiperidine, which was used without
further purification.
Step C:
~= o o
H ~C N
Following the procedure from Example A3, Step A, using
the intermediate from the previous step, afforded two compounds after
25 MPLC purification eluting with 60% ethyl acetate in hexane. The one
which came out of the column first was designated as diastereomer 1, and
the other one as diastereomer 2.
dl: FAB-MS calc. for C32H39NsO4: 557; Found: 558 (M+H)
d2: FAB-MS calc. for C32H39N5O4: 557; Found: 558 (M+H)
-
W095/13069 2 1 752 1 8 PCIIUS94/12816
- 119-
Step D:
~= O O
H ~l,C N
Following the experimental procedure from Example A1,
Step B using products from the previous step and HCl gas in ethyl acetate
at 0C gave the desired products.
dl: FAB-MS calc. for C27H3 lNsO2: 457; Found: 458 (M+H)
d2: FAB-MS calc. for C27H3 lN502: 457; Found: 458 (M+H)
EXAMPLE A9
~ --~NH2 HCI
H [~CO2Et
Step A: 2-Cyano-4-phenylpyridine
To a stirred solution of 4-phenylpyridine N-oxide (25 g,
0.146 mmol) in dichloromethane (200 ml) at room temperature was
30 added trimethylsilyl cyanide (17.4 g), followed by the slow addition of
dimethyl carbamyl chloride (16.2 ml) in dichloromethane (50 ml) over a
30 minute period. The reaction mixture was stirred at room temperature
for one day, and then to it potassium carbonate solution (10%,150 ml)
was added slowly. Stirring continued for an additional 30 minutes, the
WO 9S113069 PCIIUS9~112816
2175218 ~
- 120-
organic layer was separated, and the aqueous layer was extracted with
dichloromethane. The extracts were combined and dried over
magnesium sulfate. Evaporation in vacuo gave a crude reaction product
(35 g) as a white solid. It was used without further purification.
FAB-MS calc. for C 12H8N2: 180; Found: 181 (M+H)
lHNMR (400 MHz, CD30D): 8.71 (dd, 1 H), 8.19 (dd, lH), 7.94 (dd, 1
H),7.81-7.78(m,2H),7.56-7.50(m,3H).
Step B: 4-Phenylpyridine-2-carboxylic acid
A solution of the product from the previous step (25 g) in
100 ml of 6N HCl was refluxed for one day. The solution was cooled to
room temperature, at which time, cryst~lli7~tion started to occur. The
crystals were filtered and collected to give the product (27.5 g, 87%).
Step C: Ethyl 4-phenylpyridine-2-carboxylate hydrochloride
To a solution of the intermediate prepared in the previous
step (5.0 g, 21.2 mmol), ethanol (2 g), DMAP (20 mg) and N-methyl
morpholine (1 eq.) in dichloromethane, was added EDC (1.5 eq.). The
reaction mixture was stirred at 0C overnight. The solution was washed
with saturated sodium bicarbonate, dried over anhydrous magnesium
sulfate; then filtered and concentrated. Purification by MPLC eluting
with 40% ethyl acetate in hexane gave ethyl 4-phenylpyridine-2-
carboxylate (3.71 g, 77%). The compound was converted to its HCl salt
by treatment with HCl gas in ethyl acetate followed by evaporation.
Step D: Ethyl 4-phenylpiperidine-2-carboxylate
A suspension of the product from the previous step (200 mg)
and platinum dioxide (20 mg) in ethanol was stirred under a hydrogen
balloon for three hours. The reaction mixture was then filtered through
celite and evaporated. The resulting material was used without further
puri~lcation.
WO 95tl3069 2 1 7 5 2 1 8 PCI/US94112816
- 121 -
c Step E:
¢~ N~NHBOC
H [~CO2Et
To a solution of the intermediate prepared in the previous
step (200 mg ), and Intermediate 1 (1 eq.), HOBT (1 eq.), and NMM (1
eq.) in dichloromethane was added EDC (1.5 eq.) at 0C. The reaction
mixture was stirred at 0C overnight. The solution was washed with
saturated sodium chloride, dried over anhydrous magnesium sulfate,
~lltered and then concentrated. Purification by MPLC eluting with 50%
ethyl acetate in hexane provided the compound as a diastereomeric
mixture.
20 Step F:
~=0 0
H ~N~CO2Et
~
- To a stirred solution of the intermediate from the previous
step (30 mg) in ethyl acetate (2 mL) at 0C, was bubbled HCl gas until it
was saturated. The reaction mixture was stirred for 15 minutes and was
evaporated to dryness to give the product.
FAB-MS calc. for C29H36N4O4: 504; Found 505 (M+H)
WO 9S113069 PCrlUS9~112816
2~75218 ~
- 122-
The additional Products shown in Table AI were prepared
according to Example A9 Steps E and F, using Intermediate 2 or
Interrnediate 3 and the intermediate from step D.
TABLE AI: ADDITIONAL EXAMPLES
Rl~N~NH2 HCI
C,=O O
1 0 ~,C02Et
Product
entry Rl ~F
FAB-MS (M+l)
Ph(CH2)3- C29H39N304
494
2 PhCH2OCH2- C2gH37N3O5
496
Likewise the compounds shown below are prepared
25 according to Example A9 by introduction of the 2-cyano substitutent to a
variety of readily available substituted 4-phenylpyridines with separation
of isomers where necessary, followed by hydrolysis, reestrification with
anhydrous acidic ethanol and hydrogenation of the pyridine ring to
prepare the following intermediates:
WO 95/13069 PCI'tUS94tl2816
2175218
- 123-
[~XCO2Et ~,CO2Et
~ ~~CO2Et
N CO2Et NXCO2Et
l o ~ N 'N~`N ~ CO2Et
~ H ~ CO2Et
which may be reacted with Intermediate 1 or 3 to give the following
15 compounds respectively.
¢~,= ~C,=O O
H ~NXCO2Et [,N~,CO2Et
`~ CO2Et \~ CO2Et
25 ~ ~NH2 HCI /~J----C,~=O O
H ~,CO2Et ~,CO2Et
3 0 ~, ,CO2Et ~ CO2Et
WO 9Srl3069 2 ~ 7 5 2 ~ 8 PCI'IUS9~112816
- 124-
~NH2 HCI 3'--~ ~NH2 HCI
N C=O O C=O O
H ~,CO2Et N ~' I,N~N
~H ~ ,1H
0 ~NH2 HCI ;~ ~ ~NH2 HCI
H ~N~C02Et ~N~CO2Et
~CO2Et ~CO2Et
~,CO2Et ~~CO2Et
2 o EXAMPLE A 10
~=0 0
H ~N~C02Et
~ cis - d1
Step A: 3-Benzylpyridine N-oxide
A solution of 3-benzylpyridine (25 g, 0.148 mol) in
hydrogen peroxide (30%, 15.1 mL) and acetic acid (100 mL) was
3 0 refluxed for one day. Then more hydrogen peroxide (3 mL) was added
and the resulting mixture was refluxed overnight. The reaction mixture
was then evaporated and partitioned between a mixture of 3 N HCl, brine
and dichloromethane. The organic layer was separated, dried and
evaporated to give the desired compound (27.6 g, 100%).
WO 95/13069 PCI~/US94/12816
2175218
- 125-
Step B: 3-Benzyl-2-cyanopyridine
Prepared according to the procedure in Example A9 step A
from the intermediate from the previous step (27 g). The crude reaction
- product was purified by a SiO2 flash column eluting with 20-40% ethyl
acetate in hexane to give 5-benzyl-2-cyanopyridine (3.0g, 10%) and 3-
benzyl-2-cyanopyridine (24.2 g, 85%).
Step C: 3-Benzylpyridine-2-carboxylic acid hydrochloride
A solution of 3-benzyl-2-cyanopyridine (19.1 g) in
o concentrated hydrochloric acid (S0 mL) and water (50 mL) was refluxed
for two days. The resulting solution was evaporated to give a solid
(30.1g 100%, which contains an equal molar amount of ammonium
chloride).
Step D: Ethyl 3-benzylpyridine-2-carboxylate hydrochloride
Thionyl chloride (15.2 g) was carefully dissolved in ethanol
(300 mL) and the resulting solution was added to the intermediate from
the previous step (20 g). The mixture was refluxed overnight and then
evaporated to give the crude product as hydrochloride salt. The crude
product was dissolved in dichloromethane and washed with saturated
sodium bicarbonate. The organic solution was dried, evaporated and
purified with a short SiO2 column to give the product as free base (18.2
g). To a solution of this intermediate (16.5 g) in ethyl acetate (80 mL),
was bubbled HCl gas until it was saturated. The mixture was then
evaporated to give the HCl salt (18.9 g).
Step E: Ethyl 3-benzylpiperidine-2-carboxylate hydrochloride
A suspension of the product from the previous step ( 1.0 g)
and platinum dioxide (100 mg) in ethanol was stirred under a hydrogen
balloon for five hours. The reaction mixture was then filtered through
celite and evaporated to give the desired compound.
wo 95/13069 PCrrUS94/12816
2175218
- 126-
Step F:
¢~,=0 0
H ~N~CO2Et
cis
\~Bn
To a solution of the interrnediate prepared in the previous
step (180 mg, 0.634 mmol), and Intermediate 1 (1 eq.), HOBT (1 eq.) and
10 NMM (1 eq.) in dichloromethane, was added EDC (1.5 eq.~ at 0C. The
reaction mixture was stirred at 0C overnight. The solution was washed
with saturated sodium chloride, dried over anhydrous m~gnesium sulfate;
then ~lltered and concentrated. Purification by MPLC eluting with 50%
ethyl acetate in hexane provided two enantiomerically pure compounds.
The compound which came out first from the column was designated as
dl (146 mg); and the compound which came out of the column second
was designated as d2 (141 g).
dl FAB-MS calc. for C3sH46N4O6: 618; Found 619 (~I+H)
d2 FAB-MS calc. for C35H46N406: 618; Found 619 (M+H)
Step G:
H ~ H2 HCI
~XB cisd1
To a stirred solution of the intermediate dl from the
30 previous step (130 mg) in ethyl acetate (2 mL) at 0C, was bubbled HCl
gas until it was saturated. The reaction mixture was stirred for 15
minutes and it was evaporated to dryness to give the product ( l 1 1 mg,
95%)
FAB-MS calc. for C30H3gN4O4: 518; Found 519 (M+H)
-
WO 95113069 PCI-/US9-1/12816
2~75218
- 127-
Step H:
¢3~ ~NH2 HCI
H ~NyC02Et
cis d2
Bn
The compound was prepared according to the procedure of
the previous step from the intermediate d2 from Step F (130 mg).
Product: 114 mg, 98%
FAB-MS calc. for C30H38N4o4: 518; Found 519 (M+H)
The additional products shown in Table AII were prepared
5 according to Example A10 Steps F and G, using Intermediate 2 or
Intermediate 3 and the intermediate from step E. No separation of the
diastereoisomers was observed during MPLC purification of the Boc
precursor.
TABLE AII: ADDITIONAL EXAMPLES
H
R1~ N ~NH2 HCI
ç=o o
~NyCO2Et
~ cis
Bn
Product
entry R1 MF
- FAB-MS (M+ 1 )
1 Ph(CH2)3- C30H41N304
- 508
2 PhcH2ocH2- C29H39N305
510
wo 95113069Pcrlus94/12816
21752~8
- 128-
l~XAMPLE A 1 1
~, =O O
H ~N~CO2Et
~CO2Et
Step A:Diethyl piperidine-2~3-(cis)-dicarboxylate
10Hydrogen chloride gas was bubbled into ethanol (400 mL)
until 22 g was absorbed. Pyridine-2,3-dicarboxylic acid (100 g) was
dissolved in this solution and the resulting mixture was refluxed
overnight. The reaction mixture was divided into two portions and each
was shaken with PtO2 (1.4 g) in Parr shakers under 40 psi of hydrogen
5 for 8 hours. The reaction mixture was combined and filtered through
celite and washed with plenty of ethanol. Evaporation gave a gray solid
which was washed with ethyl acetate to give a white solid after filtration
(74.8 g)
Step B: H
~=0 0
H ~N~C02Et
l l
~CO2Et
The compound was prepared according to the procedure of
Example Al Step A from ~he intermediate from the previous step (178
mg) and Intermediate 1. Product: 234 mg FAB-MS calc. for
30 C31H44N4Og: 600; Found 601(M+H)
wo 95tl3069 ~ 4/l28l6
2175218
- 129-
Step C:
~=0 0
H ~N~CO2Et
cis
CO2Et
The compound was prepared according to the procedure of
Example Al Step B from the intermediate from the previous step (230
mg). Product: 215 mg
FAB-MS calc. for C26H36N4O6: 500; Found 501(M+H), 523 (M+Na)
The additional intermediates shown in Table AIII were
prepared from the corresponding pyridine analogs according to the above
5 established procedures from the corresponding pyridine derivatives as
exempli~led in Example Al 1 step A and the fimal products were prepared
according to Steps B and C
TABLE AIII: ADDITIONAL EXAMPLES
H C=O O
2 5 Product
Intermediate (QH) Product
entry MF MF
FAB-MS (M+ 1 ) FAB-MS (M+ 1 )
,H
N~CH3 C24H34N404
443
~CO2Et diastereomeric
mixture
wo 95113069 PCrlUS94112816
2175218 ~
- 130-
2 H
N~CH3 C2sH36N4O4
~ I 457
y~CO2Et diastereomeric
CH3 mixture
3a H
N~CH3 C25H36N404
457
'CO2Et diastereomeric
mixture
lo CH3
a:The intermediate was prepared by epimerization of its all cis isomer
with KHMDS in THF.
EXAMPLE A12
~NH2 HCI
H ~N~co2Et
2 o ~CO2Et
Step A: Diethyl N-Boc-piperidine-(cis)-2.3-dicarboxylate
To a stirred solution of the intermediate from Example Al 1
Step A (10 g, 37.6 mmol) and triethyl~mine (6.4 mL) in dichloromethane
(50 mL), was added di-t-butyl dicarbonate (10.7 g) and the resulting
25 mixture was stirred at room temperature overnight. The reaction mixture
was diluted with dichloromethane and was washed with a mixture of 3 N
HCl and brine. The organic layer was dried, evaporated and purified with
a silica gel column eluting with a gradient of 10-30% ethyl acetate in
hexane to give the desired compound (9.61 g).
Step B:
Boc
N ~,CO2Et
~CO2Et
Bn
WO 95/13069 PCI-/US94/12816
~ 2175218
- 131 -
To a stirred solution of KHMDS (3.79, 19 mmol) in THF
- (150 mL) at -78C under argon was added a solution of diethyl N-Boc-
piperidine-(cis)-2,3-dicarboxylate (5 g, 15.2 mmol) over a 30 minute
period. The solution was allowed to stir an additional 30 minutes at
-78C; then benzyl bromide (2.73 g, 15.9 mmol) was added slowly to the
solution. The reaction mixture was stirred overnight and allowed to
warrn to room temperature. The material was concentrated, then diluted
with water, and e~tracted with ethyl acetate (100 mL). The organic layer
was dried over anhydrous magnesium sulfate, filtered, and concentrated.
Purification by silica gel flash column chromatography, eluting with 20%
ethyl acetate in hexane provided two diastereoisomers. The compound
which came out ~lrst from the column was designated as dl (1.01 g); and
the compound which came out of the column second was designated as
d2 (3.75 g). NMR established the esters are trans in dl and cis in d2.
Step C:
H HCI
~ N ~CO2Et
B CO2Et
The compounds were prepared according to the procedure of
Example A1 Step B from the interrnediates from the previous step.
Intermediate dl (850 mg) yielded the dl title compound (711 mg,98%).
Intermediate d2 (3.2 g) yielded the d2 title compound (2.58 g, 96%)
dl FAB-MS calc. for ClgH25NO4: 319; Found 320(M+H)
d2 FAB-MS calc. for C18H2SNO4: 319, Found 320(M+H)
Step D:
H~
H ~N~CO2Et
--I`CO2Et
WO 95/13069 PCI-IUS94/12816
2175218
- 132-
The compounds were prepared according to the procedure of
Example Al Step A from the intermediates from the previous step.
Intermediate dl (228 mg) yielded a mixture of trans diastereomers (128
mg, 30%)-
Intermediate d2 (228 mg) yielded a mixture of cis diastereomers (164 mg,
30%).
Step E:
0 ~;3~ ~NH2 HCI
H ~N~CO2Et
~CO2Et
The compounds were prepared according to the procedure of
Example Al Step B from the intermediates from the previous step.
Intermediate dl (120 mg) yielded the title compound as mixture of trans
(dl) diastereomers (106 mg, 97%).
Intermediate d2 (155 mg) yielded the title compound as mixture of cis
(d2) diastereomers (135 mg, 96%).
dl FAB-MS calc. for C33H42N4O6: 590; Found S91(M+H)
d2 FAB-MS calc. for C33H42N4O6: 590; Found S91(M+H)
The additional intermediates shown in Table AIV were
prepared according to the above established procedures using N-Boc
intermediates from Table AIII as exemplified in Example A12 Steps A, B
and C and the final products were prepared according to Steps D and E.
WO 9~i113069 ~ 94/12816
~ 2175218
- 133-
TABLE AIV: ADDITIONAL EXAMPLES
~NH2 HCI
Product
Intermediate (QH) Product
entry MF MF
FAB-MS (M+1) FAB-MS (M+l)
H
N CH3 dl:
C31H40N404
CO2Et
H
2 N~,CH3 d2:
C3 l H40N404
E ~ CO2Et 533
H
3 N~CH3 mixture of
diastereomers
B CO2Et C32H42N404
25Likewise the compounds shown below are prepared
according to Example A12 by aLIcylating with 2-picolyl chloride or
4-bromomethylthiazole to give the following intermediates:
- 30 ~CO2Et ~ ~CO2Et
CO2Et CO2Et
1~ s~'
WO9S113069 2 1 752 1 8 PCI~/US94112816
- 134-
which may then be reacted with Intermediates 1 or 2 to give the
following compounds respectively:
¢~,=O O l~,=o O
H ~N~,C02Et H ~N~,C02Et
o --CO2Et CO2Et
15 ~ ~NH2 HCi ~, =o o
2 t ~N~,CO2Et
--CO2Et CO2Et
EXAMPLE A13
H
~ ~NH2 HCI
~N C=O O
H ~CONH(CH2)20H
WO 9S/13069 ~ I 7 5 2 1 8 PCI-IUS94/12816
.
- 135-
- Step A:
~ N ~NHBoc
H ~CONH(CH2)2OH
To a stirred solution of dl-2-pipecolamidoethanol (100 mg,
1.16 mmol), HOBT (78.38 mg, 1.16 mmol) and Intermediate 1 (226.12
mg, 1.16 mmol) in dichloromethane (3ml) at ambient temperature was
added 4-methyl morpholine (63.8 ml, ~1.16 mmol). The mixture was
cooled to 0 C and to which was added EDC (222.3 mg, 2.32 mmol). The
reaction mixture was stirred at room temperature for 16 h. After
5 evaporation, the residue was partitioned in ethyl acetate and lN
hydrochloric acid. The organic layer was washed with saturated sodium
bicarbonate, brine, dried over magnesium sulfate, ~lltered~ and evaporated
to an oily foam which was puri~led by preparative tlc
(acetone/chloroform: 3/7) to give 91 mg of the product (Rf= 0.45).
20 CI-MS: calc. for C2gH41N5O6: 543; Found 544(M+H)
H NMR (400 MHz, CDC13): ~ 8.35 (br.s, lH), 7.57 & 7.55 (2s,
lH),7.35, 7.33, (2s, 2H), 7.17 (t, J= 6.95Hz, lH), 7.15-7.07 (m, 3H), 7.03
(distorted t, J= 4.95 Hz, lH), 5.16 (d, J=4.68 Hz, lH), 4.94 (m, 2H), 3.65
(m, 2H), 3.55-3.10 (m, SH), 2.9-2.62 (m, 4H), 2.3-2.2 (m, lH), 1.43, 1.46
25 and 1.41 (3 s, total 15H), 1.00 (m, lH), 0.83 (m, lH).
Step B:
N~NH HCI
H ~CONH(CH2)20H
WO95113069 2 ~ 752 1 8 ~ 94rl28l6
- 136-
Prepared according to the experimental procédure from
Exarnple Al Step B using product from the previous step and HCl gas in
ethyl acetate at 0C.
C~-MS: calc. for C23H33N504: 443; Found ~1'14 (M+H)
1H NMR (400 MHz, CD30D): ~ ~ 7.54 (d, J=7.7 Hz, lH), 7.36 (d, J=8.1
Hz), 7.12 (distorted t, J=7.5 Hz, lH), 7.03 (distorted t, J=7.5Hz, lH),
4.97-4.92 (m, lH), 3.63 (m, lH), 3.75 (br. d, lH), 2.82 (br. t, J=2.3 Hz,
lH), 2.07 (br. d, J=2.3 Hz, lH), 1.66-1.57 (m, 6H), 1.55-0.88 (m, 4H),
0.70-0.55 (m, lH).
The additional compounds shown in Table AV were
prepared according to Steps A and B using Intermediate 1. The
piperidine intermediates were either comrnercially available or were
prepared according to the above established procedures or from literature
procedures.
TABLE AV: ADDITIONAL EXAMPLES
¢~ ~NH2 HCI
H ~W
Product
W MF FAB(or CI)-MS
(M+l)
-C02CH3 C22H30N404 415
2 -CONH(CH2)20H C23H33N504 444
3 -CONHCH2C(CH3)20H C25H37N504 472
4 -CONHCH2CH(OH)CH3 C24H35N54 458
-C02NH2 C2 l H29N503 399 (EI, M+)
6 -CH2COCH3 C23H32N304 413
7 -CH(OH)Ph-p-Cl C27H33N403C1 497
8 -CH(OH)CH2CH3 C23H34N403 415
wo 95/13069 ~ 941l28l6
2175218
- 137-
9 -CONHBn C2gH35N503 490
-CONH(CH2)2CH3 C24H35N503 442
11 H C25H37N503 456
-CH2' N~o
1 >
12 -CONHPh C27H33N503 476
13 N--N C21 H28N8o2 425
/~ H ,N
14 CH3 C23H30N603 439
//~
,N
The additional compounds shown in Table AVa were
5 prepared according to Steps A and B using Intermediate 3 and some of
intermediates used in the previous table.
TABLE AVa: ADDITIONAL EXAMPLES
~ ~NH2 HCI
ç=o o
~N~,~,W
Product
W MF FAB(or CI)-MS
(M+1)
-C02CH2CH3 C23H35N304 418
3 o 2 -CONHCH2C(CH3)20H C25H40N44 461
3 -CONH(CH3)2 C23H33N503 428
4 -CH(OH)Ph-p-Cl C27H36N303Cl 486
wo 95/13069 ~ 94JI28l6
2175218
- 138-
EXAMPLE B 1
H H ~/
~--~ ~ NH3CI
~ CO O
~1H ~
~/--~ OEt
b,~
Step A:
N
~3 0
~\
To 7.0 g of 2-bromobenzyl alcohol in 7.0 g of dihydropyran
at room temperature was added 2 drops of concentrated hydrochloric acid
and stirred at room temperature for lh. The reaction mixture was diluted
with 150 mL of ether and washed with saturated NaHCO3 (2X100 mL),
brine (150 mL), dried over MgSO4 and concentrated to give a thick oily
material. The residue was purified by flash chromatography with
hexane-EtOAc as eluent to give 10 g of the tetrahydropyranyl ether.
To 260 mL of dry ether at -78C was added 23.6 mL of 1.6
M solution of nBuLi in hexanes. To this solution was added a solution of
7.5 g of the THP compound in 100 mL of ether and stirred at -78C for
30 min. and -40C for an additional 30 min. This solution was added in a
dropwise manner to a mixture of 2.16 g of pyridine and 6.3 mL of t-
butyldimethyl-silyl triflate in 200 mL of ether at -78C. The reaction
mixture was allowed to warm up to room temperature and stirred
overnight. The reaction was quenched with 75 mL of water and oxygen
wo gSrl3069 PCl-tUS9~/12816
~ 2175218
- 139-
gas was bubbled in for 3h. The reaction mixture was diluted with ether
and 3N HCl till the pH = 1 and then the organic layer was separated. The
aqueous layer was basified with 20% NaOH till the pH - 8-9 and then
extracted with chloroform (3X100 mL). The organic layer was washed
with water, brine (200 mL), dried over Na2SO4, filtered, and evaporated.
To 3.42 g of the above compound in 100 mL of CHCl3 was
added 30 g of activated manganese dioxide and stirred overnight. The
solids were filtered off through a pad of celite, and the filtrate was
evaporated.
0 To 2.4 mL of triethylphosphonoacetate in 30 mL of dry THF
at 0C was added 16.3 mL of a solution of sodium hexamethyl~ 7ide
in THF and stirred for 30 min. A solution of the above aldehyde
interInediate in 10 mL of THF was added and stirred for 30 min. The
reaction was quenched with 25 mL of saturated ~IH4Cl solution, and
5 extracted with EtOAc(3X25 mL). The combined organics were washed
with brine, dried over Na2S04, and concentrated. Flash chromatography
of the residue with hexane-EtOAc (4:1) as eluent gave 1.5 g of the
desired product as a pale yellow solid.
1H NMR (CDC13, 400MHz) d 8.63 (d, 2H), 7.68 (dd, lH), 7.60 (d, lH),
20 7.45 7.35 (m, 2H), 7.30 (dd, lH), 7.35 (d, lH), 4.15 (q, 2H), 1.23 (t, 3H).
Step B:
HHCI
O
~3~o
To 1.5 g of the above intermediate in 25 mL of methanol
was added 5 mL of 4M HCl in EtOAc and evaporated to dryness. This
solid was dissolved in 30 mL of methanol and 0.50 g of PtO2 was added
and hydrogenated at 50 psi for Sh. The catalyst was filtered off through a
pad of celite and the filtrate was concentrated to give the title compound.
WO 95113069 PCI~IUS94/12816
2175218
- 140-
lH NMR indicated that this material contained about 5% of the
cyclohexyl-piperidine .
lH NMR (CD30D, 400MHz) d 7.40-7.20 (m, 4H), 4.08 (q, 2H), 3.50 (m,
2H), 3.25-3.10 (m, 3H), 3.00 (t, 2H), 2.60 (t, 2H), 2.03-1.90 (m, 4H),
1.20 (t, 3H)-
Step C:
~/--~-- ~H
g~ o
To a mixture of the above intermediate in 30 mL of CH2cl2
20 was added 0.82 mL of triethylamine, 1.2 mL of NMM, 0.90 g of HOBT,
2.13 g of (2R)-N-tBOC-5-phenylpen~anoic acid (~re~ d as described in
H. K. Chenault et al. J. Am. Chem. Soc., 111, 6354-6364 (1989)), and
finally 1.7 g of EDC and stirred at room temperature for 18h. The
reaction mixture was poured into a saturated NaHCO3 solution and
25 extracted with CH2C12. The combined organics were washed with 0. lN
HCl, brine, dried over Na2SO4, and concentrated.
The above crude material was dissolved in 30 mL of
CH2C12 and 10 mL of TFA was added and stirred at RT for lh. The
solvent was evaporated to dryness and the residue was neutralized with
30 aqueous Na2CO3 solution, and extracted with CH2C12. The combined
organics were washed with brine, dried over K2CO3, and concentrated.
To a mixture of this intermediate in 30 mL of CH2C12 was added 1.04 g
of HOBT, 1.56 g of N-tBOC-a-methyl~l~nine, and finally 1.8 g of EDC
and stirred at room temperature for 4h. The reaction mixture was poured
in saturated NaHCO3 solution and extracted with CH2C12. The
WO 9S/13069 PCI~/US9~/12816
2175218
- 141 -
combined organics were washed with 0. lN HCl, brine, dried over
MgSO4, and concentrated. Flash chromatography of the oily residue
with CH2Cl2-acetone-ether (6: 1: 1) as eluent gave the desired material.
lH NMR (CDCl3, 400MHz) d 7.30-6.98 (m, 9H), 5.00-4.85 (m, 2H),
4.72-4.64 (m, lH), 4.13 (2q, 2H), 4.00-3.82 (m, lH), 3.14-2.85 (m, 4H),
2.7-2.50 (m, 5H), 1.83-1.50 (m, SH), 1.50 (s, 3H), 1.46 (s, l.5H), 1.44 (s,
1.5H), 1.40 (s, 9H), 1.40-1.28 (m, lH), 1.23 (2t, 3H).
Step D:
~~~r ~NH3CI
CO O
N~
~ J
~' O
~3~o--
To 1.70 g of the intermediate in Step C in 30 mL of CH2cl2
was added 10 mL of TFA and stirred at RT for lh. The reaction was
evaporated to dryness, basified with aqueous Na2CO3, and extracted
with CH2C12. The combined organics were washed with brine, dried
over K2CO3, ~lltered, and evaporated to give free base as a thick oil.
This material was dissolved in 5 mL of ether at 0C and 0.50 mL of 4M
HCl in EtOAc was added. The precipitate was ~lltered under an N2
atmosphere and dried to give the title compound.
lH NMR (CD30D, 400MHz) d 7.30-6.98 (m, 9H), 5.00-4.85 (m, 2H),
4.72-4.64 (m, lH), 4.13 (2q, 2H), 4.00-3.82 (m, lH), 3.14-2.85 (m, 4H),
2.7-2.50 (m, 5H), 1.83-1.50 (m, 5H), 1.50 (s, 3H), 1.46 (s, 1.5H), 1.44 (s,
1.5H), 1.40-1.28 (m, lH), 1.23 (2t, 3H).
WO ~S113069 1 ~ 94/12816
2175218
- 142-
F.XAMPLE B2
~~^r ~NH3CI
CO O
N
~OH
Step A:
H H~,
~N~
~OH
To a solution of 0.20 g of the intermediate from Example
B 1, Step C in 5 mL of anhydrous THF was added 46 mg of potassium
trimethylsilanoate. After 2h an additional 46 mg of potassium
trimethylsilanoate and 2 mL of THF were added and stirred at RT
overnight. The reaction was diluted with 10 ml of water and washed with
ether (2X10 mL). The aqueous layer was acidified with 0. lN HCl to
pH=2 and extracted with CH2C12 (2X15 mL). The combined organics
were washed with brine, dried over Na2SO4, filtered and concentrated.
Flash chromatography of the residue with CHC13-MeOH-NH40H
(85:15:1) as the eluent gave 56 mg of the desired material.
WO 9S113069 PCI-IUS94/12816
2t75218
- 143-
lH NMR (CDCl3, 400MHz) d 7.32-7.20 (m, 4H), 7.20-6.98 (m, 5H),
5.10 (bs, lH), 5.00-4.90 (m, lH), 4.65 (bt, lH), 4.90 (dd, lH), 3.10-2,85
(m, 4H), 2.70-2.50 (m, 5H), 1.80-1.50 (m, 5H), 1.50 (s, 4H), 1.46 (s, lH),
1.42 (s, lH), 1.38 (s, 9H), 1.35-1.20 (m, lH).
Step B:
H
~ ~ NH3CI
~ CO O
~3~OH
To the above intermediate at RT was added 2 mL of 4M HCl
in EtOAc m~int~ined at RT for 2h. The reaction was evaporated to
20 dryness and the residue was triturated with ether to give the title
compound as a white solid.
H NMR (CD30D, 400MHz) d 8.15 (t, lH), 7.30-7.00 (m, 9H), 4.90 (m,
lH), 4.60 (bd, lH), 4.05 (d, 1/2H), 3.95 (d, 1/2H), 3.25-3.05 (m, 2H),
3.00 (dt. 2H), 2.80-2.50 (m, SH), 1.85-1.63 (m, 6H), 1.63 (s, 2H), 1.60 (s,
25 4H), 1-60-1.20 (m, 2H).
PCI-/US9~/12816
WO 95tl3069
~75218 ~
- 144-
EXAMPLE B3
H H
~O ~ ~ NH3CI
~ CO O
~H ~
OEt
~
The title compound was prepared as described in Example
B 1 Steps C and D, but commercially available N-t-BOC-O-benzyl-D-
5 serine was substituted for (R)-2-N-t-BOC-S-phenylpentanoic acid.
lH NMR (CD30D, 400MHz) d 8.30 (d, 1/2H), 8.23 (d, 1/2H), 7.40-7.25
(m, SH), 7.20-7.05 (m, 3.5H), 6.88 (d, 1/2H), 5.20 (m, lH), 4.70-4.50 (m,
3H), 4.20-4.05 (m, 3H), 3.84-3.65 (m, 2H), 3.28-2.95 9m, 4H), 2.75 (q,
lH), 2.58 (dt, 2H), 1.85-1.70 (m, 2H), 1.64 (s, 2H), 1.61 (s, 4H), l.SS-
20 1.40 (m, 2H), 1.20 (2t, 3H).
F~AMPLE B4
H
~O~ ~ NH3CI
CO O
~H ~,~
~ OH
~
To 54 mg of the compound prepared in Exarnple B3 was
added 2 mL of 2N aqueous HCl and stirred at RT overnight. The
solvents were removed under reduced pressure and the residue was dried
under vacuum to give the title compound.
WO 95tl3069 ~ U~9~/12816
~ 2~75218
- 145-
lH NMR (CD30D, 400MHz) d 8.30 (d, 1/2H), 8.23 (d, 1/2H), 7.40-7.25
(m, 5H), 7.20-7.05 (m, 3.5H), 6.88 (d, 1/2H), 5.20 (m, lH), 4.70-4.50 (m,
3H), 4.20-4.05 (m, lH), 3.84-3.65 (m, 2H), 3.28-2.95 (m, 4H), 2.75 (q,
lH), 2.58 (dt, 2H), 1.85-1.70 (m, 2H), 1.64 (s, 2H), 1.61 (s, 4H), 1.55-
1.40 (m, 2H)
EXAMPLE B5
~NH2 TFA
CO O
~ ~N~ OH
To a solution of 0.109 g of the interrnediate obtained in Step
20 A Example B2 in 3 mL of CH2C12 was added 0.017 mL of ethanolamine,34 mg of HOBT, and 58 mg of EDC and stirred at RT ovemight. The
reaction mixture was diluted with 10 mL of CH2C12 and washed with 5
mL of 0.10N HCl, 5 mL of saturated aqueous NaHCO3, dried over
MgSO4, and concentrated. Flash chromatograhy of the residue with
25 CH2C12-acetone (3:2) as the eluent gave the coupled product.
As before, the above material was treated with CH2C12-TFA
at RT for 30 min., evaporated to dryness, and triturated with ether to give
the title compound as a pale yellow solid.
lH NMR (CD30D, 400MHz) d 8.15 (t, lH), 7.30-7.00 (m, 9H), 4.95 (m,
30 lH), 4.60 (bd, lH), 4.40 (bs, lH), 4.00 (bdd, lH), 3.60-3.50 (m, 2H),
3.40-3.10 (m, 4H), 3.05-2.90 (m, 2H), 2.85-2.60 (m, SH), 2.52-2.40 (m,
4H), 1.90-1.65 (m, 6H), 1.63 (s, 2H), 1.60 (s, 4H), 1.60-1.20 (m, 2H).
__ ~
WO 9S~13069 ~ llJS9~/12816
21752~8 ~
- 146-
EXAMPLE B6
~f 0~ ~NH2 TFA
CO O
[~H
,~ OH
~
Step A:
HCI
[~3
~~
To 9.0 g of 2-bromophenethyl alcohol in 6.12 rnL of
dihydropyran at room temperature was added 2 drops of concentrated
hydrochloric acid and stirred at room temperature for lh. The reaction
mixture was diluted with 150 mL of ether and washed with saturated
25 NaHCO3 (2X100 mL), brine (150 mL), dried over MgSO4 and
concentrated to give a thick oily material. The residue was purified by
flash chromatography with hexane-EtOAc as eluent to give 10 g of the
ether.
To 200 mL of dry ether at -78C was added 17.7 mL of 1.6
M solution of nBuLi in hexanes. To this solution was added a solution of
8.0 g of the ether intermediate in 100 mL of tetrahydropyranyl ether and
stirred at -78C for 30 min. and -40C for an additional 30 min. This
solution was added in dropwise manner to a mixture of 2.16 g of pyridine
and 6.3 mL of t-butyldimethylsilyl triflate in 200 mL of ether at -78C.
WO 95tl3069 PCI-IUS9:~rl2816
21 ~5218
- 147-
The reaction mixture was allowed to warm up to room temperature and
stirred overnight. The reaction was quenched with 75 mL of water and
oxygen gas was bubbled in for 3h. The reaction mixture was diluted with
- ether and 3N HCl till the pH = 1 and then the organic layer was
5 separated. The aqueous layer was basified with 20% NaOH till the pH =
8-9 and then extracted with chloroform (3X100 mL). The organic layer
was washed with water, brine (200 rnL), dried over Na2S04, filtered, and
evaporated. Flash chromatography of the residue with hexane-
ethylacetate (1:1) as the eluent gave the desired product.
Approximately 0.90 g of the phenyl-pyridine intermediate
prepared as described above was converted to the hydrochloride salt by
treating it with 4M HCl in EtOAc.
lH NMR (CDCl3, 400MHz) d 8.90 (d, 2H), 8.20 (dd, lH), 7.73-7.35 (m,
4H), 3.70 (t, 2H), 2.83 (t, 2H).
Step B:
H
~O~ ~ NH2 TFA
~ CO O
I H
g~ OH
To 0.90 g of the above intermediate in 25 mL of methanol
was added 0.10 g of PtO2 and hydrogenated with pressurized hydrogen at
30 50 psi for Sh. The catalyst was filtered off and the filtrate was
concentrated. The residue was treated with 1.4 g of di-t-butylcarbonate
in 3 mL of dioxane, 1 mL of water, and 1 mL of triethyl~rnine for 18h.
The protected piperidine was separated by flash chromatography with
CH2Cl2-acetone (10:1) as the eluent.
WO 9S113069 PCI-tUS9~112816
2~752~8 ~
- 148-
To 0.25 g of protected piperidine intermediate synthesized
above was added 2 mL of CH2C12 and 0.50 mL of TFA and s~irred at RT
for 30 min. The reaction was evaporated to dryness and azeotroped with
toluene.
To a solution of the above residue in 2 mL of CH2Cl2 was
added 0.079 g of HOBT, 0.14 g of Intermediate 2, 0.070 mL of NMM,
and 0.090 g of EDC and stirred at RT overnight. The reaction mixture
was poured into saturated NaHCO3 and extracted with CH2Cl2. The
combined organics were washed with 0.5N HCl, brine, dried over
MgSO4, and concentrated. Flash chromatography of the residue with
CH2C12-acetone (9: 1) as the eluent gave the coupled product.
Deprotechon of the N-t-butoxycarbonyl group was carried
out by treating the above intermediate with 1 mL of TFA in 2 mL of
CH2C12 for 2h. Concentration of the reaction mixture, trituration with
ether and drying under vacuum gave the title compound as a colorless
solid.
1H NMR (CD30D, 400MHz) d 7.40-6.88 (m, 9H), 5.17 (bs, lH), 4.77-
4.50 (m, 3H), 4.18 (bd, lH), 3.80-3.65 (m, 4H), 3.30-3.05 (m, 4H), 2.95-
2.70 (m, 2H), 1.85-1.60 (m, 2H), 1.60 (s, 2H), 1.58 (s, 4H), 1.70-1.45 (m,
20 2H).
EXAMPLE B7
~ ~NH2 HCI
N C,O O
~H
~ ~
Step A:
WO 9SI~3069 PCI'IUS9~12816
2175218
- 149-
BOC
~3~O
The PtO2 reduction of the phenyl-piperidine intermediate
prepared in Step A, Example B 1 was attempted in different solvents like
ethanol and methanol in the presence and absence of conc. HCl.
Transesterification as well as unselective reduction of the pyridine was
observed. Several of these reactions were combined and treated with
excess di-t-butylcarbonate in CH2Cl2 and triethylamine. Approximately
5-0 g of the crude material thereby obtained after acid work-up was
treated with 1.6 g of NaOH in 100 mL of methanol and 10 mL of water
for 2h. The reaction mixture was diluted with water and washed with
ether. The aqueous layer was acidi~led with 0.50N HCl till acidic and
extracted with CHC13. The combined organics were washed with brine,
20 dried over Na2SO4, ~lltered, and concentrated. To about 4.0 g of this
piperidine acid in 150 mL of CH2C12 at RT was added 1.86 rnL of
benzyl alcohol, 1.90 g of HOBT, 3.45 g of EDC and a catalytic amount of
DMAP, and stirred at RT overnight. The reaction mixture was washed
with saturated NaHCO3, 0.50N HCl, brine, dried over Na2SO4, filtered
25 and concentrated. The desired material was obtained after purification
via flash chromatography.
lH NMR (CDC13, 400MHz) d 7.40-7.28 (m, SH), 7.22-7.10 (m, 4H),
5.12 (s, 2H), 4.25 (bs, 2H), 3.04 (t, 2H), 2.94-2.70 (m, 3H), 2.67 9t, 7H),
1.75-1.60 (m, 3H), 1.53 (s, 9H), 1.33-1.20 (m, lH).
wo 95/13069 PCr/US9~/12816
~175218 ~
- 150-
Step B:
s ~H
~H
~--0
To a solution of 0.70 g of the above intermediate in 2.5 mL
of CH2C12 was added 1 mL of TFA and the reaction mixture was stirred
at RT for lh. The reaction mixture was evaporated to dryness, dissolved
5 in saturated aqueous NaHCO3, and extracted with CH2Cl2. The
combined organics were washed with brine, dried over K2CO3, and
concentrated. The residue was reacted with Intermediate 1 as described
in Step B, Example B6. Flash chromatography of the residue with
hexane-acetone-ether (6:1:1) as the eluent gave 0.47 g of the desired
20 material.
Step C:
~ ~NH2 HCI
H N
3~ o
To a solution of 0.20 g of the above intermediate in EtOAc
at 0C was bubbled in HCl gas for about 10 seconds. The reaction
mixture was capped and stirred for 30 min. Ether was added and the
WO 9~/13069 PCI/US94112816
2175218
- 151 -
precipitate was ~lltered under an N2 atmosphere. This gave 0.195 g of
the title compound as a white solid.
The NMR indicated a 2: 1 mixture of rotamers. lH NMR
- (CD30D, 400MHz) d 8.30 and 8.20 (2d, lH), 7.53 and 7.45 (2d, lH),
7.40 and 7.35 (2d, lH), 7.30-7.00 (m, 11 andl/3), 6.54 (d, 2/3H), 5.30-
5.18 (m, lH), 5.09 and 5.05 (2s, 2H), 4.60 and 4.55 (2d, lH), 3.90 (2d,
lH), 3.35 (dd, lH), 3.20 (dd, lH), 3.00-2.85 (m, 3H), 2.75-2.40 (4H),
1.64 (s, 6H), 1.40 (d, 2/3H), 1.06 (d, 2/3H), 0.73 (dt, 1/3H), -0.03 (dt,
1/3H).
PXAMPLE B8
~ ~NH2 HCI
[~2H
~ OH
To a solution of 0.19 g of ~e intermediate from Step C,
Example B7 in 3 mL of dioxane was added 50 mg of 10% Pd/C and
2s hydrogenated under H2 balloon for 3h. The reaction was slow so about
50 mg of 20% Pd(OH)2/C was added and hydrogenated overnight. The
catalyst was filtered off through a pad of celite and washed with dioxane.
Evaporation of the filtrate gave the title compound as a pink solid.
The NMR indicated a 2: 1 mixture of rotamers. lH NMR
30 (CD30D, 400MHz) d 8.30 and 8.20 (2d, lH), 7.53 and 7.45 (2d, lH),
7.40 and 7.35 (2d, lH), 7.20-7.00 (m, 6 and 1/3), 6.54 (d, 2/3H), 5.30-
5.18 (m, lH), 4.60 and 4.55 (2d, lH), 3.90 (2d, lH), 3.35 (dd, lH), 3.20
(dd, lH), 3.00-2.85 (m, 3H), 2.75-2.40 (4H), 1.64 (s, 6H), 1.40 (d, 213H),
1.06 (d, 2/3H), 0.73 (dt, 1/3H), -0.03 (dt, 1/3H).
WO 95~13069 PCI/US9~/12816
2175218
- 152-
EXAMPLE B9
~ ~ ~XNH2 HCI
CO O
H
--OH
To 0.20 g of the benzyl alcohol-pyridine intermediate
synthesized in Step A of Example B 1 was added 2 mL of dry acetone and
0.10 mL of benzyl bromide and stirred at room temperature for 1 h. The
volatiles were removed on the rotary evaporator and the residue was
azeotroped with toluene. The residue was dissolved in methanol and
treated with 0.10 g of sodium borohydride for lh. The reaction rnixture
was diluted with water and extracted with CH2C12. The combined
organics were washed with brine, dried over magnesium sulfate, filtered~
and evaporated. This gave a mixture of N-benzyl-tetrahydropyridines.
which was hydrogenated in ethanol for Sh with 10% Pd/C as the catalyst.
The catalyst was filtered off and the filtrate was concentrated.
Purification of the residue with CH2Cl2-methanol (90:10) as the eluent
~ave 70 mg of a mixture of tetrahydro- and hexahydropyridines. To a
solution of 7~) mg of the above rnixture in 5 mL of CH2C12 was added
0.10 g of Intermediate 3, 0.040 g of HOBT and 0.070 g of EDC and
stirred at RT overnight. The reaction mixture was poured into saturated
aqueous NaHCO3 and extracted with CH2C12. The combined organics
were washed with brine, dried over Na2SO4, and concentrated.
Purification of the residue by flash chromatography with hexane-EtOAc
(4:1) as the eluent gave 0.090 g of the coupled product as a mixture of
diastereomers.
The above coupled product was hydrogenated in ethanol
with 10% Pd/C as the catalyst for 18h. The catalyst was filtered off
WO 95/13069 PCI-/US9~/12816
2175218
- 153-
through a pad of celite and the filtrate was concentrated. Flash
chromatography of the residue with CH2Cl2-ether (6: 1) as the eluent
gave 90 mg of the desired product.
A final deprotection of the above interrnediate was carried
5 out in methanol (2 mL) in the presence of 1 mL of concentrated HCl for
Sh. The reaction mixture was evaporated to dryness and the residue was
triturated with ether to give a solid. Purification of this material by
MPLC on an LH20 column with methanol as the eluent gave 34 mg of
the title compound as a white solid.
0 lH NMR (CD30D, 400MHz) d 7.35-7.04 (m, 9H), 4.95 (m, lH), 4.69 (d,
2H), 4.60 (d, lH), 3.97 (dd, lH), 3.30-3.10 9m, 3H), 2.82-2.60 9m, 4H),
1.90-1.70 (m, SH), 1.63 9s, 2H), 1.60 (s, 4H), 1.55-1.40 (m, lH).
EXAMPLE B10 (cis. dl+d2)
~ H H Me H
~ NH2 HCI
N CO o
H ~N~
~ 1
~ - . CO2Et
~3s
The intermediate prepared from Example B12, Step B (930
25 mg, mixture of two diastereomers) was dissolved in methanol and
hydrogenated over Pd(OH)2 at one atmosphere for 12 hours. The
mixture was filtered through Celite and the filtrate concentrated under
vacuum to give 700 mg of deprotected product. To the residue (S.S mg)
, in 0.5 ml of methylene chloride was added N-BOC-(D)-~l~nine (4.9 mg),
- 30 EDC (5.0 mg) and HOBt (3.5 mg). After stirring overnight, the mixture
was poured into water, exacted with methylene chloride and washed with
brine. The organic layer was dried over sodium sulfate, filtered and
concentrated. The residure was purified by PLC (hexaneslethyl
acetate=1/1) to give coupling product. A final deprotection of the
WO 9S/13069 ~ 11JS9~112816
21 7521 8 . ~
- 154-
coupled intermediate was carried out by following the procedure
described in Example B 19, Step B to give 7.8 mg of desired compound.
lH NMR (400 MHz, CD30D, mixture of diastereomers and rotamers):
7.59 (m, 1 H), 7.39-7.01 (m, 9 H), 5.37 (m, 1/2 H), 5.18 (m, 1/2 H), 4.61
(m, 1 H), 4.30 (m, 1/2 H), 4.02-3.61 (m, 3 H), 3.35-2.35 (m, 7 1/2 H),
1.60 (m, 1 H), 1.56 (d, 7 Hz 3/2H), 1.50 (m, 3/2H), 0.95 (m, 3/2 H), 0.88
(m, 3/2 H). FAB-MS: 491.0 (M+1).
E~XAMPLE B 11
~NH2 HCI
CO O
j~H~ `N
To a solution of 0.10g of the compound prepared in Step A
20 of Example B15 in 2mL of chloroforrn at 0Cwas added 0.018g of 5-
aminomethyltetrazole, 0.027g of HOBT, 0.65mL of triethyl~rnine and
0.048g of EDC and stirred for 10 min. at 0C. lmL of DMF was added
to the suspension and stirred overni~ht The reaction mixture was
concentrated and the residue was separated by prep TLC (lmm plate)
2s with CHCl3-MeOH-NH40H (90:10:1) as the eluent to give the desired
material. FAB MS m/e calcd. for C34H46NgO5 646.36; found 647.2
(m+1).
To a cooled solution of 0.025g of the above product in lmL
of ethyl acetate was bubbled in HCl(gas) till it was saturated and allowed
30 to stand at rt for 30 min. The reaction was concentrated to give the title
compound.
lH NMR (200MHz; CD30D) indicated a mixture of rotamers; 7.91 (d,
J=8 Hz); 7.35-7.06 (m); 5.14 (bs); 4.65-4.48 (m); 3.92 (bt, J=13); 3.72-
3.04 (m); 2.76-2.58 (m); 1.95-1.68 (m); 1.61 (s); 1.28 (s). FAB MS Calc.
for C34H46NgO5: MVV=546.31; found m/e = (m+1) 547.1.
WO 95tl3069 PCI-/US94/12816
2t75218
- 155-
EXAMPLE B 12 (cis~ dl )
r ~N~NH2 HCI
N CO o
H ,N~
~CO2Et
lo ~cis, d1
Step A-l:
IBOC
~CO2Et
~3
To a solution of 3-ethoxycarbonyl-4-piperidone hydro-
chloride (11.4 g, 54.9 mmole) in 82 ml of lN aqueous sodium hydroxide
25 was added di-t-butyl-dicarbonate (12.2 g, 56.0 mmole) in 82 ml of
dioxane at room temperature. After 12 hours, the mixture was diluted
with ethyl acetate and washed with 0.5 N hydrochloric acid and brine.
The organic layer was dried over magnesium sulfate, filtered and
concentrated. To the crude residue in 200 ml of methylene chloride there
3 o was added diisopropylethyl~mine (14.3 ml, 82.3 mmole) and triflic
anhydride (10.1 ml, 60.4 mmole) at -78C. After 1/2 hour, the mixture
was poured into saturated sodium bicarbonate solution and extracted with
methylene chloride. The organic layer was washed with lN
hydrochloride, brine and dried over magnesium sulfate. The organic
layer was concentrated to give the vinyl trifl~te (21.0 g, 95%). To a
WO 95113069 PCI'IUS94112816
2175218 e
- 156-
solution of the vinyl triflate (4.67 g, 11.6 mmole) in 100 rnl of methylene
chloride and 100 rnl of 1-methyl-2-pyrrolidinone was added
phenyltrimethyltin (2.1 ml, 11.6 mmole), and palladium acetate (0.13 g,
0.58 mmole) at room temperature. After a couple of hours, the reaction
5 was poured into water and extracted with ether (3X). The organic layers
were washed with water (3X), brine and dried over magnesium sulfate.
After concentration and purification (MPLC, hexanes/ethyl
acetate=10/1), the desired compound was isolated in 83% yield (3.2 g).
l0 Step A:
BOC
[~\.CO2Et
~cls
Prepared from the intermediate obtained from Step A-1 (3.2
g, 9.66 mrnole) which was dissolved in 100 ml of methanol,
hydrogenated over PtO2 at one atmosphere for a couple of hours (very
slow reaction) and then a portion of Pd/C was added under hydrogen.
The mixture was stirred for 72 hours and then filtered through Celite.
The filtrate was concentrated under vacuum. The residue was purified by
MPLC (hexanes/ethyl acetate=10/1) to give the cis compound (1.9 g).
WO 95113069 PCI'lUS9~tl2816
2175218
- 157-
Step B:
~CO
~ 02Et
To intermediate prepared from Step A (200 mg, 0.6 mmole)
there was added 2 ml of T~A. After 10 minutes, the mixture was
concentrated and azeotroped with toluene (3X). The residue was
dissolved in ethyl acetate and washed with sodium bicarbonate. The
organic layer was concentrated. To the residue in 10 ml of methylene
chloride there was added N-CBZ-D-tryptophan (223 mg, 0.66 mmole),
EDC (138 mg, 0.72 mmole), and HOBt (89 mg, 0.66 mmole). After a
20 couple of hours, the reaction was poured into water and extracted with
methylene chloride, dried over sodium sulfate, filtered and concentrated.
The residue was purified by MPLC (hexanes/ethyl acetate=2/1) to give
two diastereomers in total 66% yield (the less polar diastereomer dl, 82
mg; and the more polar diastereomer d2, 138 mg).
Step C:
6~ ~H
H N
'f CO2Et
,~!~ cis, d1
W
WO 9S113069 2 1 7 5 2 1 8 ~ l/lJ~g~ 28l6
- 158-
The less polar diastereomer from Step B (82 mg) was
dissolved in S ml of methanol and hydrogenated over Pd/C at one
atmosphere for a couple of hours (monitored by TLC). The mixture was
~lltered through Celite and the filtrate concentrated under vacuum. To the
5 residue in 5 ml of chloroform was added N-CBZ-a-methyl~l~nine (38
mg), EDC (31 mg) and HOBt (21 mg). After 3 hours stirring at room
temperature, the mixture was poured into water, extracted with methylene
chloride, and washed with brine. The organic layer was dried over
sodium sulfate, filtered and concentrated. The residue was puIified by
chromatatron (hexanes/ethyl acetate=l/l) to give ~e desired compound
in 69% yield (60 mg).
Step D:
~N ~NH2 HCI
N Ç
N~
2 0 y`CO2Et
[~clis, d1
The intermediate obtained from Step C was dissolved in 3
ml of methanol and hydrogenated over Pd(OH)2/C at one atmosphere for
an hour (monitored by TLC). The mixture was filtered through Celite
and the filtrate concentrated under vacuum. The residue was acidified
with HCl in ether to give a white precipitate (dl, 40 mg).
lH NMR (400 MHz, CD30D mixture of rotamers): 7.64 (d, 8 Hz, 1/2 H),
7.57(d,8Hz, 1/2H),7.37-7.01 (m,9H),5.28(dd,8,5Hz, 1/2H),5.18
(t, 7 Hz, 1/2 H), 4.76 (m, 1 H), 4.30 (m, 1/2 H), 4.15 (m, 1/2 H), 3.81 (m,
21/2H),3.35(m,1/2H),3.16(m,21/2H),3.02(m,1 1/2H),2.98(m,
1/2 H), 2.45 (m, 1 H), 2.25 (m, 1/2 H), 1.74 (m, 1/2 H), 1.63 (m, 1/2 H),
WO 9~i113069 PCI-IUS94/128~6
2 1 752 1 8
- 159-
1.57 (s, 3/2H), 1.52 (s, 3/2H), 1.49 (s, 3/2H), 1.34 (s, 3/2H), 0.98 (t, 7 Hz,
3l2 H), 0.90 (t, 7 Hz, 3/2 H). FAB-MS: 505.6 (M+1).
EXAMPLE B 13 (cis. d2)
~`~,~NH~
H N
0 ~ CO2Et
~3cis, d2
Prepared from the intermediate obtained from the more polar
diastereomer of Example B 12, Step B (93 mg) by the procedure
described in Example B 12 Steps C and D to give the desired compound
(d2, 56 mg).
o lH NMR (400 MHz, CDCl3, mixture of rotamers): 7.57(m, 1 H), 7.35-
6.94(m, 9 H), 5.37(t, 7 Hz, 2/3 H), 5.17 ~m, 1/3 H), 4.61 (m, 1 H), 4.28
(m, 1/3 H), 4.06 (m, 2/3 H), 3.84-3.53 (m, 2 H), 3.28-2.80 (M, 5 H), 2.53
(M, 1 H), 1.61 (S, 2 H), 1.51 (S, 1 H), 1.47 (S, 2 H), 1.29 (S, 1 H), 0.95
(t, 7 Hz, 2 H), 0.80 (t, 7 Hz, 1 H). FAB-MS: 505.7.
FXAMPLE B14 ~trans. dl+d2)
~NH2 HCI
N CO O
H N
~trans 2
~1
_
WO 95/13069 PCr/US94112816
21 7521 8 ~
- 160-
Step A:
BOC
~N~
l J,
~' CO2Et
g3trans
A small piece of sodium was added to 2.5 ml of anhydrous
ethanol. When the sodium was dissolved, the intermediate from
Example B12, Step A (40 mg) was added to the reaction mixture and
placed in an 80C oil bath for 2 hours. This rnixture was poured into
0. lN HCl and extracted with ether. The organic layer was dried over
sodium sulfate, ~lltered and concentrated. The residue was purified by
PLC (hexanes/ethyl acetate_5/1) to give the trans isomer (26 mg).
Step B:
~ ;XNH2 HCI
2 5 ~`CO2Et
~rans
Prepared from the intermediate obtained from Step A (24
mg) arld Intermediate 1 according to the procedures described in Example
B7, Steps B and C to give 5.4 mg of product as the hydrochloride salt.
lH NMR (400 MHz, CD30D, mixture of diastereomers and rotamers):
7.63-7.35 (m, 2 H), 7.24-6.75 (m, 8 H), S.01 (m, 1 H), 4.60 (m, 1 H),
4.08-3.68 (m, 3 1/3 H), 3.39-2.41 (m, 5 2/3 H), 1.78-0.96 (1 1/3 H), 1.62
WO 95/13~69 PCrlUS94/1Z816
-
~ 21 7521 8
- 161 -
(s,3H), 1.61 (s,3H),0.86(m,3H),0.66(m, 1/3H),-0.10(m, 1/3H).
FAB-MS: 505.6 (M+l).
- EXAMPLE B 15
H H \ /
NH3CI
CO O
~N~
~ o
~OH
5 Step A:
NHtBOC
CO O
~N~
\~ o
~OH
Approximately 0.250g of the piperidine intermediate
prepared in Step A of Example B44 was reacted with 0.39g of
Intermediate 3, 0.152g of HOBT, 0.17mL of N-methylmorpholine, and
0.225g of EDC in l5mL of chloroform for 18h. The reaction mixture
was washed with 0.50N HCl (lOmL), saturated aqueous NaHCO3
30 (lOmL), dried over MgSO4 and concentrated. The crude was purified by
flash chromatography with hexane-EtOAc (4: 1) as the eluent.
To 0.136g of this material in 10mL a 1:1 rnixture of
methanol-water was added 25mg of lithium hydroxide and stirred
overnight. The reaction mixture was diluted with 10mL of water and
wo 9SI13069 ~ 59~112816
2~ 75218
- 162-
washed with water, the aqueous layer was acidified to pH=2 with 0.50 N
HCl and extracted with ether (3XlOmL). The combined organics were
washed with brine, dried over MgSO4 and concentrated to give the
desired matelial as a white solid.
Step B:
H \/
NH3CI
~ CO O
~N~l
[~OH
The title compound was prepared from the compound made
in Step A by treating it with a saturated solution of HCl(gas) in ethyl
acetate for 30min. at RT. Ether was added and the precipitate was
filtered and dried.
20 lH NMR (400 MHz, CD30D mixtllre of rotamers): 8.10 (t, lH), 7.78
(dd, lH), 7.50-7.00 (m, 8H), 4.90 (m, lH), 4.55 (d, lH), 3.94 and 3.90 (2
doublets, lH), 3.80-3.60 (m, lH), 3.05 (dt, lH), 2.70-2.50 (m, 4H), 1.90-
1.50 (m, 6H), 1.55 (s, 3H), 1.50 (s, 3H), 1.40 (m, lH).
2 5 EXAMPLE B 16
¢~NH2 HCI
H ~N~
~C02Et
~3
WO 95113069 PCI-/US9-~/12816
2175218
.
- 163-
Step A:
I BOC
,N~
5 ~J~Me
~ CO2Et
To a solution of the intermediate obtained from Example
B 12, Step A (89 mg, 0.267 mmole) in 2 ml of THF there was added
potassium bis(trimethylsilyl)amide (0.5 M, 800 ml, 0.4 mmole) at -78C.
After 1/2 hour, methyl iodide (22 ml, 0.34 mmole) was added to reaction
mixture. This reaction was slowly warmed up to room temperature and
stirred for additional 12 hours. The mixture was poured into water and
then extracted with ether. The organic layer was dried over sodium
sulfate, filtered and concentrated. The residue was purified by a
chromatatron (hexanes/ethyl acetate=l/l) to give the desired compound
20 (91 mg, 98%).
Step B:
~ ~NH2 HCI
N CO o
H N
~1~ Me
~ CO2Et
~
Prepared from the intermediate obtained from Step A (91
mg) by the procedure described in Example B 12, Steps B, C, and D to
give the desired compound.
WO 9S/13069 2 ~ 7 5 2 1 8 PCI-IUS94/12816
- 164-
lH NMR (400 MHz, CD30D, mixture of diastereomers and rotamers):
7.58 (m, 1 H), 7.37-7.00 (m, 9 H), 5.40-5.23 (m, 1 H),4.60 (m, 1 H),
4.20-3.73 (m, 3 H), 3.40 (m, 1/2 H), 3.15 (m, 2 H), 2.82 (m, 1 H), 2.61-
2.30 (m, 2 1/2 H), 1.72 (m, 1/2 H), 1.63-1.29 (m, 6 H), 1.13-0.84 (m, 6
5 H). EI-MS: 518.2 (M)-
EXAMPLE B17(cis~ dl+d2)
~ N~NH TFA
f
~CO2Bn
[~cis
Step A:
IBOC
ycj\sCO2Bn
To a stirred solution of the intermediate prepared from
Example B 12, Step A-l (1.0 g, 3.02 mmole) in 4 ml of ethanol there was
30 added 4N sodium hydroxide (4 ml). The reaction was stirred at room
temperature for 16 hours and evaporated in vacuo. The residue was
diluted with water and acidified with 0.5N hydrochloric acid and then
exacted with ether. The organic layer was dried over sodium sulfate,
filtered and concentrated. The crude residue was dissolved in methanol
and hydrogenated over Pd(oH)2 at one atmosphere for 16 hours. ~he
WO 9S/13069 2 1 7 5 2 1 8 PCI'IUS94/12816
- 165-
mixture was filtered through Celite and the filtrate concentrated under
vacuum. To crude acid in 10 ml of chloroform there was added benzyl
alcohol (341 ml), EDC (750 mg) and a catalytic amount of DMAP. After
16 hours, the mixture was diluted with methylene chloride and then
5 washed with water and brine. The organic layer was dried over
magnesium sulfate, filtered and concentrated. The residue was purified
by MPLC (hexanes/ethyl acetate=5/1) to give the desired compound (459
mg, 38%).
0 Step B:
~H
H N
~CO2Bn
(~
To intermediate prepared from Step A (459 mg, 1.16
mmole) there was added 2 ml of TFA at room temperature. After 10
minutes, the reaction mixture was concentrated and azeotroped with
25 toluene (3X). To the residue in 10 ml of chloroform there was added
Intermediate 1 (433 mg), EDC (265 mg), HOBt (172 mg), and
triethylamine (194 ml). The reaction was stirred at room temperature for
3 hours and poured into water. The mixture was extracted with
methylene chloride, and dried over sodium sulfate. Concentration and
30 purification (MPLC, hexanes/ethyl acetate=1.5/1) gave the coupling
product (574 mg) in 76% yield.
Step C:
WO gS/13069 ~ ll28l6
2175218
- 166-
~NH2 TFA
N C0 0
H ~N~
~CO2Bn
~3cis
To intermediate (10 mg) obtained from Step B there was
added TFA at room temperature. After 10 minutes, the mixture was
concentrated to give the desired compound (3 mg).
lH NMR (400 MHz, CD30D, mixture of diastereomers and rotamers):
15 7.62 (m, 1 H), 7.37-6.81 (m, 14 H), 5.42-5.15 (m 1 H), 4.79 (m, 2 H),
4.65 (m, 1 H), 4.32 (m, 1/2 H), 4.12 (m, 1/2 H), 3.27-2.85 (m, 5 1/2 H),
2.55-2.27 (m, 1 1/2 H), 1.74 (m, 1 H), 1.60-1.29 (m, 6 H). FAB-MS:
567.0 (M+l).
F.XAMPLE B 18 (cis~d 1 +d2)
~NH2 TFA
H N
C02H
cis
~ 3
WO95113069 2 1 7 52 1 8 PCI`/US94/12816
- 167-
Step A:
~ ~H
CO2H
1 cis
~3
Prepared from the intermediate obtained from Example B17,
Step B (20 mg) by the procedure described in Example B8 to give the
5 desired compound.
Step B:
~ XNH2 TFA
~CO2H
0
Prepared from the intermediate obtained from Step A by the
procedure described in Example B 17, Step C to give the desired
30 compound (10 mg).
lH NMR (400 MHz, CD30D, mixture of diastereomers and rotamers):
7.62 (m, 1 H), 7.37-6.98 (m, 9 H), 5.36-5.21 (m 1 H), 4.69 (m, 1/2 H),
4.58 (m, 1/2 H), 4.27-3.91 (m, 2 H), 3.27-2.75 (m, 5 H), 2.51-2.34 (m, 2
H), 1.72 (m, 1 H), 1.58-1.21 (m, 6 H). FAB-MS: 576.9 (M+1).
wo 95113069 ~ /u~94tl28l6
. 21 75218
- 168-
EXAMPLE Bl9 (cis. dl+d2) ,
~3~ ~NH2 HCI
N C,O O
~--`S~
~isO
Step A:
l S ~ ~H
~~--~
Prepared from the interrnediate obtained from Example B 18,
25 Step A (142 mg) in 3 ml of methylene chloride to which there was added
2-(methylthio)ethanol (22 ml), EDC (57 mg) and a catalytic amount of
DMAP. After 3 hours, the rnixture was diluted with methylene chloride
and then washed with water and brine. The organic layer was dried over
magnesium sulfate, filtered and concentrated. The residue was purified
30 by PLC (hexanes/ethyl acetate=l/l) to give the desired product (69 mg,
43%).
WO 95/13069 PCI-/US94/12816
2175218
- 169-
Step B:
~NH2 HCI
N CO o
H ~N~
~--`S~
~lso
W
Prepared from the intermediate obtained from Step A (50
mg) in 2 ml of ether into which there was bubbled HCl gas at 0C. After
30 seconds, the mixture was concentrated to give the white solid (41 mg).
lH NMR (400 MHz, CD30D, mixture of diastereomers and rotamers):
7.61(m, 1 H), 7.37-6.97 (m, 9 H), 5.38-5.18 (m 1 H), 4.83-4.54 (m, 1 H),
4.37-3.77 (m, 3 H), 3.57-2.83 (m, 6 H), 2.55-2.21 (m, 3 H), 2.14-1.84 (m,
3 H), 1.72 (m, 1 H), 1.61-1.29 (m, 6 H). FAB-MS: 551.0 (M+1).
EXAMPLE B20 (cis~ dl+d2)
~NH2 TFA
N C0 o
2s H N
~CONHEt
cis
~3
Prepared from the intermediate obtained from Example B 18,
Step A (52 mg) in 3 ml of methylene chloride to which there was added
ethylamine hydrochloride (9 mg), EDC (21 mg), triethyl~mine (15 ml)
and a catalytic amount of DMAP. After 3 hours, the mixture was diluted
WO 9S113069 2 1 7 5 2 1 8 PCI-/US9-1/12816
- 170-
with methylene chloride and then washed with water and brine. The
organic layer was dried over magnesium sulfate, filtered and
concentrated. The residue was purified by PLC (methylene
chloride/methanol=20/1) to give the coupling product (25 mg). This
5 intermediate by the procedure described in Example B 17, Step C gave the
desired compound t25 mg).
lH NMR (400 MHz, CD30D, mixture of diastereomers and rotamers):
7.68-6.93 (m, 10 H), 5.34-5.12 (m 1 H), 4.75-4.30 (m, 2 H), 3.50-2.60
(m, 8 H), 1.72-1.17 (m, 8 H), 0.83-0.68 (m, 3 H). FAB-MS: 504.0
o (M+1).
EXAMPLE B21 (cis. d 1 +d2)
~ ~NH2 TFA
H ~CNO
~OH
Step A:
IBOC
- ~,OH
-
To a solution of the intermediate obtained from Example
B12, Step A-1 (950 mg, 2.87 mmole) in 10 ml of THF there was added
diisobutylaluminum hydride (1.0 N in methylene chloride, 8 ml, 8.0
WO 95/13069 PCI-/US9-1/12816
21 7521 8
- 171 -
mmole) at -78C. The mixture was stirred at 0C for 1 hour and then
slowly warmed to room temperature. The mixture was quenched with 1 N
sodium hydroxide, and extracted with ether (3X). The organic layer was
dried over sodium sulfate, filtered and concentrated. The residue was
5 purified by MPLC (hexanes/ethyl acetate=2/1) to give 617 mg of
reduction product.
StepB:
IBOC
~,OH
1 cis
0
Prepared from the intermediate obtained from Step A (57
mg) by hydrogenation under the conditions described in Example B 12,
Step A to give the desired compound (13 mg).
Step C:
XNH2 TFA
N C,O O
H ~N~
~OH
cis
~ 3
Prepared from the intermediate obtained from Step B (13
mg) by the procedure described in Example B17, Steps B and C to give
the desired compound (12 mg).
- ~
WO 9~113069 PCI~/US94112816
2175218 ~
- 172-
lH NMR (400 MHz, CD30D, mixture of diastereomers and rotamers):
7.74-6.80 (m, 10 H), 5.55 (m 1/2 H), 5.20 (m, 1/2 H), 4.66(m, 1 H), 4.11
(m, 1/2 H), 3.93 (m, 1/2 H), 3.20 (m, 3 H), 3.00-2.82 (m, 2 1/2 H), 2.69-
2.45 (m, 2 1/2 H), 2.05-1.84 (m, 1 H), 1.68 (s, 3/2 H), 1.61 (s, 3/2 H),
1.60 (s, 3/2 H), 1.47 (s, 3/2 H), 0.90 (m, 1/2 H), 0.17 (m, 1/2 H). FAB-
MS: 463.0 (M+l).
EXAMPLE B22 (cis, dl+d2)
~NH2 HCI
H N
1 5 ~1~OAC
~is
20 Step A:
tBOC
~1,OAC
~is
Prepared from the intermediate obtained from Example B21,
30 Step A (330 mg, 1.14 mmole) in 10 ml of methylene chloride to which
there was added acetic anhydride (130 ml), triethylamine (240 ml), and a
catalytic amount of DMAP at 0C. After 1 hour, water was added to the
mixture and it was stirred an additional 1 hour at room temperature. The
mixture was extracted with methylene chloride and then washed
sequentially with lN sodium hydroxide and brine. The organic layer was
WO 9S/13069 1"-llUS9~/12816
-
~ 2175218
- 173-
dried over magnesium sulfate, filtered and concentrated. The residue was
" hydrogenated under the conditions described in Example B 12, Step A to
give the desired compound.
Step B:
~Cy O
H N
~OAc
I cis
[~
Prepared from the intermediate obtained from Step A (24
mg) by the procedure described in Example B 17, Step B and Example
Bl9, Step B to give the desired compound (23 mg).
0 lH NMR (400 MHz, CD30D, mixture of diastereomers and rotamers):
7.74-6.87 (m, 10 H), 5.55-5.16 (m 1 H), 4.65 (m, 1 H), 3.96 (m, 1 H),
3.81 (m, 1/2 H), 3.20 (m, 3 H), 2.86 (m, 1 H), 2.61 (m, 1 H), 2.46 (m, 1/2
H), 2.27 (m, 1/2 H), 2.13 (m, 1 H), 1.98 (s, 1/2 H), 1.93 (s, 1 H), 1.90 (s,
1 H), 1.85 (s, 1/2 H), 1.73-1.30 (m, 7 1/2 H), 0.85 (m, 1/2 H), 0.12(m, 1/2
25 H). FAB-MS: 505.3 (M+l).
WO 9~/13069 PCr/US94tl2816
~752~8 ~I
- 174-
EXAMPLE B23 (cis dl)
~--~ ~NH2 HCI
CO O
~j CO2Et
W
Step A:
H H
~N`tBOC
CO
~\CO2Et
~lcis
To intermediate prepared from Example B 12, Step A (87
mg) there was added 1 ml of TFA. After 10 minutes, the mixture was
2 5 concentrated and azeotroped with toluene (3X). The residue was
dissolved in ethyl acetate and washed with sodium bicarbonate. The
organic layer was concentrated. To the residue in 3 ml of methylene
chloride there was added N-BOC-(2R)-amino-S-phenylpentanoic acid (70
mg), EDC (55 mg), and HOBt (35 mg). After a couple of hours, the
3 reaction was poured into water and extracted with methylene chloride,
dried over sodium sulfate, filtered and concentrated.
WO 9~/13069 PCTIUS94/12816
2175218
- 175-
Step B:
~ ,N~N,tBOC
\~CO2Et
0
To intermediate prepared from Step A there was added 1 ml
of TFA. After 10 rninutes, the mixture was concentrated and azeotroped
with toluene (3X). To the residue in 3 ml methylene chloride there was
5 added BOC-a-methyl~l~nine, EDC, HOBt, and triethyl~mine. After a
couple of hours, the reaction was poured into water and extracted with
methylene chloride, dried over sodium sulfate, filtered and concentrated.
I'he residue was purified by MPLC (hexanes/ethyl acetate=2/1 ) to give
two diastereomers in 75% yield (the less polar diastereomer dl, 54 mg;
20 the more polar diastereomer d2, 53 mg).
Step C:
~ NH2 HCI
,CO O
\~CO2Et
30 ~s, d1
To the less polar diastereomer prepared from Step B (54 mg)
was added 1 ml of T~A. After 10 minllte.s, the mixture was concentrated
WO 95~13069 PCrlUS9~112816
2175218
- 176-
and azeotroped with toluene (3X). The residue was dissolved in ethyl
acetate and washed with sodium bicarbonate. The organic layer was
dried over sodium sulfate, filtered and concentrated. The residue was
dissolved in ether to which was added HCl in ether to give a white solid
5 (dl, 40 mg).
1H NMR (400 MHz, CD30D mixture of rotamers): 7.23 (m, 10 H), 5.08
(m, 1 H), 4.76 (m, 1 H), 4.21 (m, 1 H), 3.80 (m, 2 1/2 H), 3.47 (m, 1/2
H), 3.26-2.99 (m, 4 H),2.86 (m, 1/2 H), 2.63 (m, 2 H), 2.40 (m, 1/2 H),
1.75 (m, 4 H), 1.63 (s, 2 H), 1.60(s, 2 H), 1.57 (s, 2 H), 0.95 (t, 7 Hz, 2
o H), 0.87 (t, 7 Hz, 1 H). FAB-MS: 494.1 (M+1).
EXAMPLE B24 (cis~ d2)
¢~---- ~NHz HCI
CO O
. ' CO2Et
~s, d2
The desired d2 compound (40 mg) was prepared from the
more polar diastereomer obtained in Example B23, Step B (53 mg) by the
25 procedure described in Example B23, Step C.
1H NMR (400 MHz, CD30D mixture of rotamers): 7.23 (m, 10 H), 4.91
(m, 1 H), 4.75 (m, 1 H), 4.03 (m, 1 H), 3.81 (m, 2 H), 3.45 (m, 1/2 H),
3.26-2.96 (m, 4 H), 2.71 (m, 2 1/2 H), 2.40 (m, 1 H), 1.90-1.64 (m, 4 H),
1.63 (s, 2 H), 1.61 (s, 3 H), 1.59 (s, 3 H), 0.93 (t, 7 Hz, 3 H). FAB-MS:
30 494.3 (M+1).
WO 95/13069 PCr/US9.1/12816
2175218
- 177-
EX~MPLE B25 (cis~ dl+d2)
~ ~NHz HCI
CO O
[~?\CONHEt
CIS
lo ~3
Step A:
IBOC
lS N
T coNHEt
[~
To a stirred solution of the intermediate prepared from
Example B 12, Step A-l in 4 ml of ethanol there was added 4N sodium
hydroxide (4 ml). The reaction was stirred at room temperature for 16
25 hours and evaporated in vacuo. The residue was diluted with water and
acidified with 0.5N hydrochloric acid and then extracted with ether. The
organic layer was dried over sodium sulfate, filtered and concentrated.
The residue ( 100 mg) in 3 ml of methylene chloride therewas added
ethylamine hydrochloride (74 mg), EDC (115 mg), HOBt (49 mg) and
3 0 triethylamine (83 ml). After a couple of hours, the reaction was poured
into water and extracted with methylene chloride, dried over sodium
sulfate, filtered and concentrated. The residue was puri~led by MPLC
(hexanes/ethyl acetate=l/l) to give desired compound (74 mg).
wo 95113069 PCrlUS9~/12816
2175218 ~
- 178-
Step B:
IBOC
f
\~\CONHEt
g~S
Prepared from the intermediate obtained from Step A (74
mg) by the procedure described in Example B 12, Step A to give desired
compound (60 mg).
Step C:
~~^r ~NH2 HCI
CO O
N~
~l
CONHEt
~S
Prepared from the intermediate obtained from Step B (60
mg) by the procedure described in Example B23, Steps A, B, and C to
give the desired compound (15 mg).
1H NMR (400 MHz, CD30D mixture of diastereomers and rotamers):
0 7.27 (m, 10 H), 4.91 (m, 1 H), 4.67 (m, 1 H), 3.96 (m, 1 H), 3.42 (m, 1/2
H), 3.26-2.59 (m, 9 1/2 H),1.90-1.64 (m, 4 H), 1.64-1.57 (m, 6 H), 0.79
(t, 7 Hz, 3/2 H), 0.77 (t, 7 Hz, 3/2 H). FAB-MS: 493.3 (M+1).
WO 9S113069 ~ ~594/l2816
2175218
- 179-
INTERMEDIATE 4
~NHtBOC
N CO O
1H
~OH
To a solution of 0.80g of the compound prepared in Step B
of Example B7 in 20mL of ethanol was added 0.080g of 20% pallium
hydroxide/C and hydrogenated at atmospheric pressure for 3h. The
catalyst was filtered through a pad of celite and the filtrate was
concentrated to give the title compound.
~XAMPLES B26. B27~ B28. B29
The following compounds shown in Table B 1 were
20 prepared in two steps from Intermediate 4. The acid intermediate in a
methylene chloride solution was coupled with alcohols or amines in the
presence of EDC and DMAP at ambient temperature and these
intermediates were purified and treated with hydrochloric acid(gas) in
ethyl acetate to provide the compounds shown in Table B 1.
-
, pcrlUs9~ll28l6
.. Vo9s,l3069 21752~8
- 180-
TABLE B 1
~'`~N~NH2 HCI
H 1~1
~ O
10Exa~, R molecularformula FAB h/lS (m~1~
B26 ~ocH(cH3)2 C31H42N404 ~ 569 l 570.2
15B27 ~ O(GH2~3cH3 C33H44N404 l 560 ~ 5
B28 ~ N\J0 C33H43NsO4 ~ 573.33 ~ 574.1
20B29 ~ NHcH2cH3 C31H41NaO3 ~ 531 ¦ 532.3
-
WO 95113069 2 1 7 5 2 1 8 PCI-/US94112816
.
- 181 -
EXAMPLE B30
~NH2 HCI
CO O
~~
Step A:
~ N O J<
1 5 ~N~
~/ O
~~
To a solution of 1. lg of the piperidine intermediate prepared
in Exarnple B7, Step A in SmL of ethyl acetate at room temperature was
bubbled in HCl (gas) for 10 seconds and stirred for 30 min. The solvent
was removed and the oily residue was basified with aqueous sodium
bicarbonate solution and extracted with CH2Cl2. The combined organics
2s were washed with brine, dried over K2CO3, filtered, and concentrated to
give 0.90g of the amine as a thick oil. To a solution of the above
intermediate in 20mL of CH2Cl2 was added 0.97g of (2R)-N-t-BOC-5-
phenylpentanoic acid, 0.45g of HOBT, and 0.80g of EDC and stirred at
, RT overnight. The reaction rnixture was poured into saturated aqueous
- 3 sodium bicarbonate solution and extracted with CH2cl2. The combined
organics were washed with 0.50N hydrochloric acid solution, brine, dried
over MgSO4, filtered and concentrated. The residue was purified by
flash chromatography with hexane-acetone (5:1) as the eluent to yield
about 2.0g of the coupled product.
wo 95113069 PcrnJsg~1l28l6
2175218
- 182-
The above interrnediate was treated with 2mL of trifluoroacetic
acid in 20mL of CH2C12 at room temperature for lh. The volatiles were
removed on the rotary evaporater and the residue was basified with
aqueous NaHC03 and extracted with CH2C12. The combined organics
5 were dried over K2C03, filtered and concentrated. The residue was
dissolved in CH2Cl2 and coupled with 0.60g of N-t-BOC-a-
methyl~l~nine in the presence of 0.40g of HOBT and 0.70g of EDC. The
reaction was stirred overnight and worked up as described above. The
residue was purified by flash chromatography using hexane-acetone (S: 1)
as the eluent to give the title compound as a colorless foam.
lH NMR (400 MHz, CDC13 mixture of rotamers): 7.40-6.85 (m, 14 H),
5.10 (s, 2H), 5.05-4.88 (m, 2H), 4.70-4.60 (m, lH), 3.93 (d, 1/2H), 3.85
(d, 1/2H), 3.10-2.85 (m, 4H), 2.70-2.50 (m, SH), 1.85-1.60 (m, 7H), 1.50
(s, 3H), 1.48 and 1.47 (2s, 3H), 1.42 (s, 9H), 1.40-1.20 (m, lH).
Step B:
H H \ /
--~ ~ NH3CI
~ CO O
N~
~~
Approximately 0.050g of the intermediate from Step A was
dissolved in lmL of ethyl acetate and lmL of saturated HCl(gas) in ethyl
acetate was added and stirred for at room temperature for 30rnin. The
reaction mixture was cooled to 0C and etner was added and the solvents
30 were evaporated to leave the derired product as a foam.
lH NMR (400 MHz, CD30D mixture of rotamers): 7.40-7.00 (m, 14H),
5.10 (s, 2H), 4.90 (m, lH), 4.58 (d, lH), 3.95 and 3.90 (2 doublets, lH),
3.20-2.95 (m, 4H), 2.80-2.60 (m, 5H), 1.85-1.60 (m, 9H), 1.62 (s, 3H),
1.60 (s, lH), 1.40 (m, lH).
WO 9~/13069 PCI-/US9~/12816
21 7521 ~
- 183-
EXAMPLE B3 1
H NHb~
C10 O
~N~
NH2
Step A:
H H \ /
NHtBOC
~ c,o o
~ O
~\J`OH
To O.90g of the intermediate prepared in Step A Example
B30 in SmL of methanol was added O.lOg of 20% palladium hydroxide
and hydrogenated at atmospheric pressure overnight. The catalyst was
filtered off through a pad of celite and washed with methanol. The
filtrate was concentrated and the residue was dried under vacuum to
provide the acid as a colorless foam that was used without purification.
Step B:
~ ~NH3C
C,O O
W~NH2
WO 9!i/13069 PCI~/US9-1/12816
2175218 ~
- 184-
To a solution of 0.30g of the acid intermediate prepared in
Step A in 10mL of dry THF was added 0.14mL of triethyl~mine and
0.07mL of ethylchloroformate and stirred for lh. The reaction was
quenched with 2rnL of aqueous ammonium hydroxide solution and
5 extracted with CH2Cl2. The combined organics were washed with 0.50N
hydrochloric acid, dried over MgSO4 and concentrated. The residue was
purified by flash chromatography with chloroform-methanol (95:5) as the
eluent to provide a solid that was deprotected with HCl in ethyl acetate as
described above to give the title compound as a white solid.
H NMR (400 MHz, CD30D mixture of rotamers): 8.15 (t, lH), 7.30-
7.00 (m, 9H), 4.90 (m, lH), 4.70 (d, lH), 4.05 and 3.95 (2 doublets, lH),
3.30-2.95 (m, 4H), 2.90-2.60 (m, 3H), 2.50 (bs, 2H), 1.90-1.65 (m, 7H),
1.60 (2 singlets, 6H), 1.48 (m, lH).
EXAMPLES 32-35 and 49
The compounds described in Table B2 were prepared from
intermediate synthesized in Step A of Example B31 by taking advantage
of chemistry used to prepare the title compound in Example B5. Other
20 amines as depicted below were used in place of ethanolamine and the
final deprotection was carried in ethyl acetate and dry hydrochloric acid.
Ether was generally used to precipitate the hydrochloride salt.
PCT~JS9~1~2816
WO 95113069 2 1 7 5 2 1 8
.
- 185-
TAE3~E B2
~NH2
CO O
~) O
,J~R
Example Rmolecular formula F~B MS mle found
No. mle calc.(m+1 )
B32 N(CH3)2C31H44N4O3 520 521~2
B33 NHtBu C33H4sN4O3 548 ~49.2
B34 N~'3 C33H46N4O3s 578 579.2
B49 NHCH2CH3C31H44N4O3 520 521.2
~XAMPLE B35
H H \/
~,N~ NH3CI
~ ~0 0
;~H H
To a so~ution of 0.5ûg of the acid intermediate prepared in
Step A of Example B31 in ~ml of 1,2-dichloroethane was added 0.16g of
carbonyldiimidazole and stirred at 60C ~or 30min. The reaction was
Wo9Srl3069 2 1 7 52 ~ 8 ~ u~g~lz8~6
- 186-
cooled to RT, half of it was then treated with 0.12g of 2-aminopyrazole
and heated at 60C for lh, cooled to RT and stirred for 2 days. The
reaction mixture was poured into 0.50N aqueous hydrochloric acid and
extracted with CH2Cl2. The combined organics were washed with brine,
5 dried over MgSO4, concentrated and the residue was purified by flash
chromatography with hexane-acetone (1:1) as the eluent. The purified
material was deprotected with the HCl/EtOAc protocol as described
above to give the title compound as a white solid.
FAB MS m/e cacl. (for C32H42N603) 558; found 559.2 (m+1)
EXAMPLE B36
NH3C
CO O
N
~ ~0
The title compound was prepared as described in Example
B5 but morpholine was used in place of ethanol~mine.
1H NMR (400 MHz, CD30D mixture of rotamers): 7.30-6.95 (m, 9H),
4.95 (m, lH), 4.68 (d, lH), 4.00 and 3.95 (2 doublets, lH), 3.59 (m, 4H),
25 3.35 (m, 4H), 3.25-2.90 (m, 4H), 2.80-2.50 (m, SH), 1.90-1.65 (m, 7H),
1.63 (s, 3H), 1.60 (s, 3H), 1.47 (m, lH).
wo 9S/13069 Pcr/us94112816
~ 2~75218
- 187-
EXAMPLE B37
~~r ~NH3CI
CO O
t 5 ~N~
~NHS02Ph
Step A:
CBZ
~ OH
To a stirred solution of 5.0g of the piperidine intermediate
20 prepared in Example Bl Step B was added SmL of triethylamine at 0C
and 2.8mL of CBZ-Cl. The reaction was allowed to warm up to Rt and
stir overnight. The reaction rnixture was poured into aqueous arnmonium
chloride solution and extracted with CH2Cl2. The organic layer was
washed with 0.50N HCl solution, dried over MgSO4 and concentrated.
2 This crude residue was dissolved in 25 mL of methanol-water and 3eq. of
sodium hydroxide was added and stirred for 2h. The reaction mixture
was acidi~led to pH=2 with 2N HCl and extracted with EtOAc. The
combined organics were washed with brine, dried over Na2SO4 anà
concentrated to give the acid as a foam.
WO 93/13069 2 1 7 5 2 1 8 PCrlUS94112816
- 188-
Step B:
~~ ~r ~NH3CI
CO O
~ ~NHSO2Ph
To a solution of 0.225g of the above acid intermediate in
10mL of CH2Cl2 was added 0.12g of benzenesulfonamide, 0.093g of
DMAP and 0.164g of EDC and stirred overnight. The reaction mixture
was washed with 0.50N HCl (2XlOmL), dried over Na2SO4 and
concentrated. The crude residue was dissolved in 10mL of methanol and
0.10g of 10% Pd/C and hydrogenated at 40psi overnight. The catalyst
was filtered off through a pad of celite and the filtrate was concentrated
to provide the piperidine that was used without purification.
The piperidine intermediate was now coupled to
20 Intermediate 3 and deprotected with HCl/EtOAC as described above to
give the title compound as a white solid.
FAB MS m/e cacl. (for C3sH44N4OsS) 632; found 633.1 (m+l)
EXAMPLE B38
~ ~NH3CI
C,O O
~,~ J~--N
WO 95/13069 2 7 7 5 2 1 8 PCIIUS9.1/12816
.
- 189-
Step A:
BOC
,,N~
~, "COOH
This interrnediate was prepared as described in Step A of
Example B37 but di-t-butylcarbonate was used in place of CBZ-Cl.
Step B:
BOC
1 5 ~"~,CN
To a stirred solution of 2.90g of the acid prepared in Step A
in 30mL of dry THF was added 2.5mL of triethylamine and 1.25mL of
20 ethylchloroformate and stirred for 30 min. 10mL of the reaction mixture
was removed. The rem~ining mixture was quenched with 20mL of
aqueous ammonium hydroxide solution, stirred for 30 min., and extracted
with EtOAc. The combined organics were washed with 0.50N HCl,
brine, dried over Na2SO4, filtered and evaporated to give an oily residue.
2 5 This material was dissolved in 20mL of CH2C12 and 20mL of pyridine at
0C and 1. lmL of POCl3 was added and stirred for 30min. The reaction
mixture was poured into brine and washed with 0.50N HCl solution,
saturated NaHCO3 solution, brine, dried over Na2SO4 and concentrated.
Flash chromatography of the residue with hexane-ethyl acetate (5:1) as
3 the eluent gave the desired product.
WO 95/13069 PCrlUS9 ~/12816
2175218 ~
- 190-
Step C: -
¢~ ~NHtBOC
CO O
,N~
~N
To a solution of 1.0g of the nitrile intermediate prepared in
Step B in 20mL of toluene was added 1.96g of trimethyltin azide and
heated at reflux for 1 8h. The excess azide that precipitated upon cooling
to room temperature was filtered off. The filtrate was concentrated and
15 spilt in half. To this half was added 10mL of EtOAc and a trace of
methanol and HCl(gas) was bubbled in for 5 minutes and stirred for lh.
Ether was added and concentrated to give a gummy material that was
washed with ether and dried under vacuum to give a brownish solid.
400MHz NMR (CD30D) revealed that this was the desired tetrazole
20 intermediate.
To 0.30g of the piperidine hydrochloride synthesized above
in 10mL of chloroforrn was added 0.47g of Intermediate 3, 0.16g of
HOBT, 0.45mL of N-methylmorpholine, and 0.29g of EDC and stirred
overnight. The reaction mixture was poured into 0.50N HCl solution and
25 extracted with CHC13. The combined oraganics were washed with brine,
dried over Na2SO4, and concentrated to give a gummy residue that was
puri~led by flash chromatography with CHC13-MeOH-NH4OH (85:15:1 )
as the eluent. This provided 0.15g of the desired product.
wo 9S/13069 ~ /osg4/l28l6
~ 2175218
- 191 -
Step D:
~ ~NH3CI
,, CO O
N--N
~H
This material was prepared from the intermediate prepared
in Step C by the EtOAc/HCl protocol described above.
lH NMR (400 MHz, CD30D mixture of rotamers): 8.15 (t, lH), 7.60-
7.05 (m, 9H), 4.90 (m, lH), 4.60 (d, lH), 4.05 and 3.95 (2 doublets, lH),
3.30-3.10 (m, 4H), 3.10-2.60 (m, SH), 1.90-1.65 (m, 9H), 1.60 (s, 6H),
1.50 (m, lH).
EXAMPLE B39
~ b~NH3CI
~N
~N,N CH3
~
To a solution of 0.030g of the intermediate prepared in Step
C of Example B38 in 2mL of dry acetone was added 13mg of powdered
potassium carbonate and 0.006mL of methyl iodide and stirred at RT
3 o overnight. The reaction mixture was poured into brine and extracted with
CHCl3. The combined organics were washed with brine, dried over
Na2S04, filtered and evaporated to give the alkylated product that was
deprotected by the EtOAclHCl protocol without further purification.
This gave 0.006g of the title compound as a mixture of isomers.
FAB MS m/e cacl. (for C3OH4lN7O2) 531; found 532.3 (m+l)
WO 9~ 3069 PCI'IUS9~/12816
2175218 0
- 192-
EXAMPLE B40
CO O ,,
~N
~ N ~J
Step A:
~Z
~,CN
This intermediate was prepared in an analogous manner to
the BOC material prepared in Step B of Example B38.
Step B:
NH3CI
CO O
~ N--N
~Hi
3 0 To a stirred solution of 1 .0g of the nitrile from Step A in
10mL of dry ethanol at 0C was bubbled in HCl(gas) for lh. The
reaction was capped and stored in the freezer overr~ight. The excess
HCl(gas) was removed by bubbling N2 gas for lh and ether was added to
induce precipitation of the imino-ether interrnediate, but only an oily
material formed. Hence, the solvents were removed on the rotary
WO 9S113069 PCI'JUS94/12816
~ 21~5218
- 193-
evaporator and the gurnmy residue was dissolved in CH2C12 and
evaporated twice. Ether was now added and this provided the imino-
ether hydrochloride as a foam.
To 0.20g of the above intermediate in 5mL of
dichloroethane was addedO.073mL of diisopropylethylamine and 0.030g
of fonnylhydrazine and stirred at room temperature overnight. The
reaction rnixture was poured into water and extracted with CH2Cl2. The
combined organics were washed with brine, dried over Na2SO4 and
concentrated. The residue ~hereby obtained was dissolved in 5mL of
xylenes and heated at reflux for several hours. The reaction mixture was
cooled to room temperature and the xylenes were evaporated. The
residue was hydrogenated for 2h in 2mL of methanol and 40mg of 20Yo
palladium hydroxide catalyst. The piperidine thereby obtained was
coupled with Intermediate 3 under the standard EDC/HOBT conditions
described earlier. The crude product was purified by flash
chromatography with CH2C12-MeOH-NH40H (95:5:1) as the eluent.
Removal of the BOC protecting group under the EtOAc/HCl conditions
gave the title compound as a white solid.
lH NMR (400 MHz, CD30D mixture of rotamers): 9.15 (s, lH), 8.16
(bS, lH), 7.30-7.00 (m, 9H), 4.90 (m, lH), 4.60 (bs, lH), 4.10 and 3.95 (2
doublets, lH), 3.30-3.00 (m, 4H), 3.00-2.60 (m, 5H), 1.90-1.60 (m, 9H)~
1.62 (s, 3H), 1.60 (s, 3H), 1.40 (m, lH).
EXAMPLE B41
2 5 ~ NH3C
CO O
~N~
I~J ~N
~N~O
H
WO 95/13069 PCI'tUS94112816
2175218 ~
- 194-
The title compound was prepared in an analogous manner to
Exarnple B40 but N-carbomethoxyhydrazine was used in place of N-
formylhydrazine .
lH NMR (400 MHz, CD30D mixture of rotamers): 7.30-7.02 (m, 9H),
4.90 (m, lH), 4.60 (d, lH), 4.05 and 3.95 (2 doublets, lH), 3.30-2.95 (m,
SH), 2.80-2.60 (m, 4H), 1.90-1.70 (m, 9H), 1.60 (s, 3H), l.S9 (s, 3H),
1.39 (m, lH).
EXAMPLE B42
1 0 b~NH3CI
CO O
~ H H
~N~N~
o
Step A:
CBZ
~
H H
[~ 3~,N~N~
o
To a solution of 3.0g of the acid intermediate prepared in
Step A of Example B37 in SOmL of benzene was added 0.70mL of oxalyl
chloride and 3 drops of DMF and stirred at RT for 2h. The benzene was
evaporated off and the residue was dissolved in acetone at 0C. A
30 solution of l.S9g of sodium azide in SmL of water was added at stirred at
0C for lh. The reaction was diluted wi~ ether and water and the
organic layer was separated. The organics were washed with brine, dried
over Na2S04 and concentrated to give an oily residue. This material was
dissolved in dry toluene and heated at reflux for 4h. The reaction mixture
WO 95tl3069 PCr/US9~112816
~ 2~752~8
- 195-
was concentrated and the isocyanate thereby obtained was storred in the
- refrigerator.
To 0.40g of the isocyanate in toluene was added 0.80mL of
triethyl~mine and 0.20g of methylamine hydrochloride and stirred for
5 overnight. The reaction mixture was poured into aqueous NaHC03
solution and extracted with EtOAc. The combined organics were washed
with brine, dried over MgSO4 and concentrated to give the methylurea
that was used without purification.
0 Step B:
NH3C
CO O
~N~
~,J
H H
~,N~,N~
o
The piperidine intermediate prepared in Step A was
hydrogenated with Pd(OH)2 in methanol to remove the CBZ protecting
group, coupled with Intermediate 3, purified and deprotected with the
EtOAc/HCl protocol as described above to give the title compound.
1H NMR (400 MHz, CD30D mixture of rotamers): 8.10 (m, lH), 7.40-
7-00 (m, 9H), 4.95 (m, lH), 4.63 (d, lH), 4.10 and 4.00 (2 doublets, lH),
3.40-3.10 (m, 4H), 2.85-2.90 (m, 2H), 2.70 (s, 3H), 2.80-2.60 (m, 3H),
1.90-1.62 (m, 7H), 1.63 (s, 3H), 1.60 (s, 3H), 1.40(m, lH).
wo 95113069 ~ 94/12816
2~ 752~8
- 196-
EXAMPLE B43
¢~~r b>~NH3
C10 0
~---- SO2CH3
The isocyanate intermediate prepared (0.20g) in Step A of
Example B42 was refluxed in 5mL of 6N aqueous HCl overnight. The
reaction mixture was washed with ether and the ether layer was
discarded. The aqueous layer was basified to pH=10 with aqueous
potassium carbonate solution and extracted with CH2C12. The combined
organics were washed with brine, dried over K2C03 and concentrated.
This crude amine was converted to the methanesulfonamide by treating it
with methanesulfonyl chloride and triethylamine in dichloromethane.
After standard work-up the CBZ group was removed by hydrogenation
20 and elaborated to the title compound as discussed previously.
lH NMR (400 MHz, CD30D mixture of rotamers): 7.30-7.00 (m, 9H),
4.85 (m, lH), 4.55 (d, lH), 4.00 and 3.90 (2 doublets, lH), 3.30-3.10 (m,
4H), 2.95-2.83 (m, 2H), 2.80 (2 s, 3H), 2.80-2.60 (m, 3H), 1.90-1.65 (m,
9H), 1.60 (s, 3H), 1.56 (s, 3H), 1.55 (m, lH).
EXAMPLE B44
H \/
NH3CI
~ C,O O
~N~
O
~OCH3
~tep A:
WO 95/13069 PCI~/US94/12816
2~ 75218
- 197-
HHCI
.' ~0
~ OCH3
To a solution of S.Og of the pyridine aldehyde intermediate
prepared in Step A of Example B 1 in lOOmL of methanol was added 4.0g
of sodium cyanide, SmL of glacial acetic acid and 20g of manganese
dioxide and stirred for 2h. The solids were filtered off through a pad of
celite and the filtrate was concentrated. The residue was taken up in
lOOrnL of saturated sodium bicarbonate solution and extracted with
3XlOOmL of ethyl acetate. The combined organics were washed with
brine, dried over Na2S04 and concentrated to provide the pyridine
methyl ester. This m~ter~l was dissolved in methanol and 5mL of
saturated HCl in ethyl acetate was added and concentrated to give the
hydrochloride salt.
To 2g of the above pyridine hydrochloride salt in 15mL of
methanol was added 0.225g of platinum oxide and hydrogenated at SOpsi
on the Parr shaker for 2h. The catalyst was filtered off through a pad of
celite and washed with methanol. The filtrate was concentrated to give
2.17 of the piperidine hydrochloride as a foam.
Step B: ~NH3CI
~OCH3
-
The title compound was prepared from the compound made
in Step A and Intermediate 3 as described previously.
WO 95/13069 PCI'IUS9-1/12816
2~752~8 ~
- 198-
lH NMR (400 MHz, CD30D mixture of rotamers): 8.10 (t, lH), 7.78
(dd, lH), 7.50-7.00 (m, 8H), 4.90 (m, lH), 4.55 (d, lH), 3.94 and 3.90 (2
doublets, lH), 3.85 (s, 3H), 3.80-3.60 (m, lH), 3.05 (dt, lH), 2.70-2.50
(m, 4H), 1.90-1.50 (m, 6H), 1.55 (s, 3H), 1.50 9s, 3H), 1.40 (m, lH).
EXAMPLE B45
>~NH3
CO O
~N~
[~ H ,N
The title compound was prepared from the ester intermediate
prepared in Step A of Example B44 in an analogous manner to the
tetrazole compound prepared in Example B38.
lH NMR (400 MHz, CD30D mixture of rotamers): 7.60-7.45 (m, 2H),
20 7.45-7.38 (m, 2H), 7.30-7.10 (m, 5H), 4.90 (rn, lH), 3.95 and 3.90 (2
doublets, lH), 3.30-3.00 (m, 2H), 2.80-2.55 (m, 4H), 1.90-1.63 (m, 7H),
1.65-1.50 (4 singlets, 6H), 1.40 (m, lH).
EXAMPLE B46
~ ~N~NH3CI
Co o
1 ~ N--N
~3f H~N_I I
wo 95113069 PCrlUS9~112816
~ 2175218
- 199-
Step A:
NOC
~OH
This compound was prepared in an analogous manner to the
o protected piperidine acid compound synthesized in Step A of Example
B37.
Step B:
HHCI
~N~
O
Ll N--N
,~H~ ll
This intermediate was prepared from the compound synthesized in
Step A by using the carbonyl~liimi-l~7.ole method descibed in Example
B35, but amino-tetrazole was used in place of aminopyrazole.
Step C:
2 5 b~NH3CI
C10 0
~N~
3 0 ~ 11~ N--N
,~ H~/ 1 1
wo 95~13069 f~ /US94/12816
2175218 ~
- 200 -
This compound was synthesized from the piperidine
intermediate made in Step B and Intermediate 3 by using chemistry
presented above.
FAB MS m/e cacl. (for C2gH36NgO3) 532; found 533.1 (m+l)
EXAMPLE B47
1~ , b~NH3CI
N
~OH
Step A:
2 o ~3l~,OH
To a solution of 0.30g of the imino-ether intermediate
25 prepared in Step B of Example B40 in lOmL of ethanol was added
0.124g of dihydroxyacetone and heated at 60C under an ammonia
atmosphere in a bomb for 16h. The reaction was cooled to room
temperature and the solvent was evaporated. The residue was purified by
flash chromatography to give 0.129g of the desired product that was still
30 cont~min~ted with other impurities.
WO ~S/13069 PCI'JU59~/12816
2~75~18
- 201 -
Step B:
~N~NH Cl
CO o
C~ N
,~,OH
The intermediate prepared in Step A was elaborated to the
title compound after removal of the CBZ protecting group, coupling with
Intermediate 3, purification, and a final deprotection with the EtOAc/HCl
protocol described earlier.
H NMR (400 MHz, CD30D mixture of rotamers): 8.18 (2 triplets, lH),
7.30 (s, lH), 7.30-7.00 (m, 9H), 4.90 (m, lH), 4.56-4.55 (singlet
overlapping a doublet, 3H), 4.05-3.95 (2 doublets, lH), 3.30-2.95 (m,
4H), 2.95-2.60 (m, SH), 1.90-1.65 (m, 7H), 1.63 (s, 3H), 1.60 (s, 3H),
1.45 (m, lH).
EXAMPLE B48
H N
CO O
~3"' N'
3 (~
WO 95/13069 ~ /USg~/12816
2~75218 ~
- 202 -
~tep A:
~C
~3
To a solution of 1.0g of the ester prepared in Step B of
Example B 1 in 50mL of dry THF at 0C was added 0.20g of lithium
0 aluminum hydride and stirred at room temperature overnight. The
reaction was quenched at 0C with 10mL of water and 10rnL of 30%
aqueous sodium hydroxide solution. The precipitate was filtered and
washed with EtOAc. The ethyl acetate extracts were washed with brine,
dried over Na2SO4 and concentrated. The crude alcohol was dissolved in
5 30mL of CH2C12 and 1.3mL of triethylamine and 1.4g of di-t-
butylcarbonate was added at 0C and then stirred at RT for 2h. The
reaction was poured into saturated NaHCO3 solution and extracted with
CH2Cl2. The combined organics were washed with 0.50N HCl, brine,
dried over Na2S04 and concentrated. This material was purified by flash
20 chromatography with hexane-acetone (5:1) as the eluent.
The alcohol obtained above was dissolved in 10mL of
CH2C12 at 0C and 0.45mL of triethyl~mine and 0.14mL of
methanesulfonyl chloride were added and stirred for lh. The reaction
was diluted with water and extracted with CH2C12. The combined
25 organics were washed with 0.50N HCl, brine, dried over Na2SO4 and
concentrated. The crude mesylate was heated at 60C with 0.20g of the
sodium salt of 1,2,4-triazole in 10mL of dry DMF for 3h. The reaction
was cooled to RT and quenched with aqueous ammonium chloride
solution. The reaction mixture was extracted with ether (3X15mL). The
30 combined organics were washed with brine, dried over Na2SO4, filtered
and concentrated. This gave the triazole product that was used without
puri~lcation.
WO 95/13069 PCr/US9~112816
2~75218
- 203 -
Step B:
H H \ /
J ~ N ~ NH3CI
~ CO O
~
N,
The BOC protecting from the piperidine synthesized in Step
A was removed with the TFA procedure as described previously and
elaborated to the title compound by coupling with Intermediate 3,
purification and a fimal deprotection with the EtOAc/HCl protocol.
FAB MS m/e cacl. (for C3lH42N602) 530; found 531.4 (m+1)
EXAMPLE B50
~NH3CI
N CO O
H N
N N
N
l~,b H
Prepared as described in Fx~mple B40 Step B but
Intermediate 1 was used in place of Intermediate 3.
1H NMR (400 MHz, CD30D mixture of rotamers): 8.32 and 8.20 (2
doublets, lH), 7.65 and 7.58 (2 doublets, lH), 7.40 and 7.35 (2 doublets,
30 lH), 7.25-7.00 (m, 6H), 6.50 (d, lH), 5.30-5.20 (m, lH), 4.58 and 4.55 (2
doublets, lH), 4.10 and 3.95 (2 doublets, 1/2H), 3.90 (d, 1/2H), 3.40-3.00
(m, 7H), 2.70-2.45 (m, 3H), 2.80-2.50 (m, 2H~, 1.60 (s, 6H), 1.34 (d, lH),
0.95 (d, 1/2H), 0.70 (dt, 1/2H).
_
WO 9S/13069 ' PCI/US9~112816
2175218 ~
- 204 -
EXAMPLE B50 A
rN b~NH2 HCI
CO O
~N~
~ O
1~ N~OH
~ H
To a solution of 0.330g of the acid intermediate prepared in
Step A of Example B15 in 3.3mL of dry THF was added 0.196g of
carbonyldiiimidazole and heated to 60C for 2h. A small aliquot of the
reaction m~xture was removed and to the rem~ining solution was added
15 - lOmL of 4-aminobutanol and heated for 2h. The reaction mixture was
concentrated, taken up in chloroform, washed twice with water, once
with lM K2HPO4, brine, dried over MgSO4, filtered and concentrated to
provide a residue that was separated by prep TLC (lrnm plate ) with
CHC13-MeOH-NH40H (90:10:1) as the eluent to give the desired
20 intermediate.
To a solution of 0.20g of the above material in 2mL of
anisole was added 3-4mL of TFA and allowed to stand at rt for 30 min.
The volatiles were removed under reduced pressure and the residue was
partitioned between chloroform and lM K2HPO4 and basified to pH>9
25 with NaOH. The organic phase was separated and the aqueous phase was
extracted with chloroform, The combined organics were washed with
brine, dried over MgSO4, filtered and concentrated to provide a gum that
was separated by prep TLC (lmm plate ) with CHC13-MeOH-NH40H
(90: 10: 1) as the eluent to give the desired product.
30 lH NMR (200MHz; CDC13 mixture of rotamers): 8.24 (d, J=8); 7.42-
7.07 (m); 6.16 ("dd", J=12, 4); 4.97-4.8 (m); 4.69 (bd, J=13); 3.93 ("bt",
J~10); 3.75-3.64 (m); 3.54-3.4 (m); 3.35-3.16 (m); 3.07 (quart., J=13);
2.77-2.5 (m); 1.97-1.42 (m); 1.34 (s). .FAB MS Calc. for C31H44N404
: MW=536.34; found m/e = (m+l) 537.1.
WO 9S113069 PCr/US94/12816
2175218
- 205 -
A solution of O.l50g of the above free base was lyophillized
from 0.50mL of acetic acid and 0.030mL of conc. HCl to give title
compound.
EXAMPLE B50B
H H \/
~NH2 HCI
C10 0
~N~
~ o
,OH
H
Prepared in an analogous manner to the compound prepared
in Example B50A but ethanolamine was used in place of 4-aminobutanol.
lH NMR (200MHz; CDCl3 mixture of rotamers): 8.22 (d, J=8), 7.45-
7.05 (m); 6.58 (dt, J=16, 5); 4.88 (bs); 4.64 (bd, J=12); 3.90 (t, J=11);
3.79 (bs); 3.65-3.50 (m); 3.25-3.15 (m); 3.05 (quart., J=12); 2.8-2.5 (m);
2~ 2.32 (vbs); 2.0-1.77 (m); 1.77-1.45 (m); 1.35 (s). FAB MS Calc. for
C2gH40N4o4: MW=508.30; found rn/e = (m+l) 509.2.
A solution of 0.029g of the above free base was lyophillized
from 0.50mL of acetic acid and O.OlOmL of conc. HCl to give title
compound.
FXAMPEE B50C
H H \/
~NH2 HCI
~ CO O
~ O
~--OJ~H'--
WO 9S/13069 PCI~/US94112816
2175218 ~
- 206 -
Step A:
H
r
[~ OH
To a solution of 0.379g of the free base (prepared by
basification to pH>9 with NaOH and extraction with CHC13) of the
0 intermediate prepared in Step A of Example B44 in 20mL of dry THF
was added 5.5mL of lM solution of lithium aluminum hydride in THF
and stirred overnight. The reaction was quenched with 10mL of 30%
aqueous NaOH, the organic phase was decanted, and the paste was
extracted with ethyl acetate. The combined organics were dried over
5 MgSO4 and concentrated. Purification of the residue by prep TLC (lmm
plate) gave the desired amino alcohol.
lH NMR (200MHz; CDC13 rnixture of rotamers): 7.38-7.13 (m); 4.75
(s); 3.22 (bd, J=12 Hz); 3.1-2.92 (m); 2.81 (td, J=10, 4 Hz); 2.13 (bs);
1.85-1.6 (m).
Step B:
~ H
~N~Il,OtBU
~ C,O O
~N~
~3~ H
The intermediate prepared in Step A was coupled with (2R)-
N-t-BOC-5-phenyl pentanoic acid under the standard EDC/HOBT
protocol as described above and purified by prep TLC (lmm plate).
To a solution of 0.145g of the above coupled product in 2mL
of CDC13 was added 0.50mL of 2-chloroethylisocyanate and was heated
WO 95~13069 PCI~/US9~/12816
1~ 2175218
- 207 -
at 60C for 6h and allowed to stand at RT overnight. Prep TLC of this
mixture with hexane-EtOAc (1:1) as the eluent gave 0.1 lg of the desired
carbamate.
1H NMR (200MHz; CDC13 mixture of rotamers): 7.45-7.05 (m); 5.50
(bd, J=6); 5.19 (s); 5.14 (bs); 4.86-4.45 (bdd?); 4.11 (bd, J=7); 3.93 (bt,
J=12); 3.72-3.42 (bm); 3.2-2.88 (m); 2.8-2.5 (bm); 1.95-1.55 (m); 1.44
(s).
Step C:
H H \ /
--~ ~ NHtBOC
~ CO O
~ O
g3~' H
The intermediate prepared in Step B was deprotected with
20 EtOAc/HCl and the hydrochloride salt thereby obtained was coupled ith
N-t-BOC-a-methyl~l~nine under standard EDC/HOBT conditions. This
material was purified by prep TLC (lmm plate) with hexane-EtOAc (1: 1)
as the eluent. lH NMR (200MHz; CDCl3 mixture of rotamers): 7.40-
7.04 (m); 5.19 (s); 5.17 (bs); 4.98 (s); 4.92 (bs); 4.72 (bd, ~=13); 4.54 (bd,
25 J=13); 4.18-4.04 (m); 3.95 (bt, J=13); 3.68-3.45 (m); 3.2-2.85 (m); 2.78-
2.47 (m); 2.0-1.6 (m); 1.6-1.4 (m); 1.44 (s).
Approximately 85 mg (0.13 mmoles) of the above
productwas taken up in 1.0 mL of DMSO-d6 to which was added 38 mg
(0.37 mmoles) of LiOAc.2H2O, and 30 mg (0.2 moles) of NaI; the
30 solution was heated in an 80 C oil bath over night. The reaction mixture
was then taken to a gum under a nitrogen stream. It was then partitioned
in a mixture of CHCl3 and water, the organic phase separated, dried with
anhydrous MgSO4, filtered, and after concentration to a gum under
reduced pressure, purified by preparative tlc on one 8"x 8" x 1,000m
plate in 1: 1 EtOAc: hexane to give 85 mg of the title compound.
wo 95113069 PCrrusg4ll28l6
21752~8
- 208 -
lH NMR (200MHz; CDC13 mixture of rotamers) :7.40-7.04 (m); 5.19
(s); 5.17 (bs); 4.98 (s); 4.92 (bs); 4.72 (bd, J=13); 4.54 (bd, J=13); 4.18-
4.04 (m); 3.95 (bt, J=13); 3.68-3.45 (m); 3.2-2.85 (m); 2.78-2.47 (m);
2.0-1.6 (m); 1.6-1.4 (m); 1.44 (s).: 7.4-7.0 (m); 7.88-7.69 (bm); 5.4 (s);
5.14 (s); 4.95-4.74 (m); 4.67 (bd, J=12); 4.38 (bd, J=13); 4.15-4.02 (m);
3.93 (bt, J=14); 3.50-3.30 (m); 3.18-2.8 (m); 2.75-2.35 (bm); 2.01 (s);
1.9-1.7 (bm); 1.5-1.3 (m); 1.40 (s).
Step D:
--~r ~NH2 HCI
CO O
N
~ O
~f OJ~ N ~~
To 49 mg of the intermediate from Step D in 0.5 m~ of
20 methanol was added 1-2mL of conc. H2SO4. After standing over night a
lM solution of K2HP04 was added and the reaction mixcure was taken to
dryness under a stream of nitrogen and the residue was partitioned
between C~Cl3 and lM K2HP04, adjusted to pH >9 with NaOH. The
organic phase was removed and the aqueous phase extracted several more
25 times with CHCl3 The combined organic phases were dried with
anhydrous MgSO4, filtered, and concentrated under reduced pressure.
The resultant gum was subjected to preparative tlc on one 8" x 8" x
1,000m silica gel GF plate using 1:10:90 (conc. NH40H:MeOH:CHCl3);
two major bands were observed. Isolation of the faster band afforded the
30 title compound. lH NMR (200MHz; CDCl3 mixture of rotamers): 7.4-
7.05 (m); 5.44-5.12 (m); 5.18 (s); 5.12-4.8 (m); 5.05 (s); 4.69 (bd, J=lL2);
4.52 (bd, J=12); 4.12 (bs); 3.93 (bt, J=12); 3.78-3.63 (m);3.44-3.24 (bm);
3.24-2.83 (m); 2.83-2.5 (m); 2.01-1.6 (m); 1.6-1.35 (m); 1.45 (s). FAB
MS Calc. for C35H50N4O7: MW- 638.37; found m/e = (m+l) 639.3.
WO 95113069 PCI-IUS9.1112816
2~75218
- 209 -
A solution of 23 mg ( 0.042 mmoles) of the above free base
in 0.5 mL of acetic acid in a vial was treated with 0.005 mL (0.06
mmoles) of conc. HCl, shell frozen, and lyophyllized overnight to give
the title compound.
EXAMPLE B5 1 (cis~ d 1 )
~lC O
H N
~i ~
T ~--N O
~3 cis, dl
Step A:
BOC
tN~
~ S C02H
~3
To a solution of 4. lg of the intermediate prepared in
Example B12 Step A-l in 25 ml of ethanol was added 25 ml of 6 N
NaOH and stirred 12 hours. The mixture was diluted with water and
extracted with ether. The organic layer was discarded. The aqueous
layer was cooled to 0C and acidified with conc. HCl and then extracted
with ether. The organic layer was dried over sodium sulfate, filtered and
concentrated to give 2.57g of the crude acid. The crude acid (438 mg)
3 was dissolved in methanol and hydrogenated over Pd(QH)2 at one
atmosphere for 16 hours. The mixture was filtered though Celite and the
filtrate was concentrated under vacuum to give the desired compound
(370 mg)-
Wo 95/13069 }~ 4/l28l6
2175218 ~1
- 210 -
Step B:
I OC
~ NAO
~3
To the intermediate prepared in Step A (100 mg) in
chloroform was added morpholine (0.35 ml), EDC (95 mg), and HOBt
(49 mg). The reaction was stirred for 12 hours at room temperature and
was diluted with methylene chloride and then washed with water and
brine. The organic layer was dried over sodium sulfate, filtered and
concentrated. The residue was purified by prep TLC (hexanes/ethyl
acetate=l/l) to give the desired product (71 mg).
Step C:
~ ~NH-BOC
2 0 N C O O
~-N~O
,~
2 5 ~ cis,dl +d2
To the intermediate prepared in Step B (71 mg) in ethyl
acetate was bubbled in HCl(g) at 0C for 15 seconds. The mixture was
allowed to stand at room temperature for 30min., concentrated to give a
3 0 solid (64 mg). To this crude material (32 mg) in 2 ml of chloroform was
added Intermediate 1(43 mg), EDC (29 mg), HOBt (15 mg) and
triethylamine (21 mL). The reaction was stirred at room temperature for
3 hours and poured into water and extracted with methylene chloride,
dried over sodium sulfate and concentrated. Purification (prep TLC,
WO 95113069 PCrrUS94112816
~ 2~75218
- 211 -
methylene chloride/methanol=20/1) gave two diastereomers (dl, the less
polar diastereomer, 14 mg; d2, the more polar diastereomer, 16 mg).
Step D:
~NH2HCI
~C--N/--\O
~ 3 cis, d1
The less polar diastereomer (dl, 14 mg) prepared in Step C
15 was dissolved in ethyl acetate and treated wi~ HCl(g) at 0C for 15
seconds. After 30 minutes at room temperature the mixture was
concentrated to give the desired product (10 mg).
FAB-MS: 546.3 (M+1)
2 0 EXAMPLE B52 (cis~ d?)
~ ~NH2HCI
~Nl
T &--N
cis, d2
The title compound (12 mg) was prepared from the more
polar diastereomer (d2, 16 mg) obtained in Example B51, Step C by the
procedure described in Example B51, Step D.
FAB-MS: 546.3 (M+1)
_ _
W095113069 2 1 752 1 ~ PCI'IUS94112816
- 212 -
The compounds 1-7 shown in Table B3 were prepared
according to the procedures reported above (using different amines in the
coupling step). Details are available in Example B51 Steps B, C and D.
TABLE B3
~C~ O
~ ~
~C--R
cis O
~3
R FAB-MS (M+1)
1 d1+d2 thiomorpholine 562.2
2 d1+d2 pyrrolidine 530.2
3 d 1+d2 N-methylpiperazine 559.3
4 dl+d2 piperidine 544.3
dl+d2 ethanolamine 520.2
6 dl+d2 dimethyl~rnine 504.3
7 dl+d2 glycine ethyl ester 562.3
EXAMPLE B53 (cis~ dl)
~ N ~X
. ~N~
3 0 ~--N~ ~0
~3 cis, dl
WO 9S~13069 ~ /u~g4/l28l6
~ 2175218
- 213 -
Step A:
~ CO
~N~
--N O
~3 cis
The intermediate prepared from Example B5 1, Step B in
ethyl acetate was treated with HCl(g) at 0C for 15 seconds and allowed
to stand at room temperature for 30 min. The mixture was concentrated
to dryness to give the crude material. To this crude material (99 mg) in 3
ml of chloroform was added N-t-BOC-O-benzyl-D-serine (107 mg), EDC
(92 mg), HOBt (47 mg) and triethylamine (67 ml) and stirred at room
temperature for 3 hours and poured into water. The mixture was
extracted with methylene chloride and dried over sodium sulfate, and
concentrated. ~lrific~tion of the residue by RPLC (chromatatron,
20 methylene chloride/methanol=20/1) gave the desired product (97 mg).
Step B:
CO o
~N~
~C--N/--\O
fS~ cis, dl and d2
~
The intermediate prepared from Step B (97 mg) in ethyl
acetate was treated with HCl(g) at 0C for 15 seconds and allowed to
stand at room temperaturte for 30 minutes. The reaction mixture was
concentrated to give a residue that was dissolved in 2 ml of chloroform
WO 9S113069 PCrrUS94/12816
2175218
- 214 -
and reacted with N-t-BOC-a-methyl~l~nine (52 mg) in the presence of
EDC (62 mg), HOBt (36 mg) and triethyl~mine (40 ml). After 64 hours
at room temperature the reac~ion mixture was poured into water and
extracted with methylene chloride, The combined extracts were dried
5 over sodium sulfate, filtered and concentrated to give a residue that was
purified by RPLC (chromatatron, methylene chloride/methanol=20/1) to
give two diastereomers (dl, the less polar diastereomer, 65 mg; d2, the
more polar diastereomer, 23 mg).
Step C:
H ~X
-r C--N O
~3 cis, dl
The less polar diastereomer (dl, 65 mg) prepared from Step
B in ethyl acetate was treated with HCl(g) at 0C for 15 seconds. After
st~nclin~ at room temperature for 30 minllte~, the mixture was
concentrated to give the desired product (58 mg).
FAB-MS: 537.4 (M+l)
EXAMPLE B54 (cis. d2)
H ~ /
~ C O
3 0 ~N~
--N~O
~3 cis, d2
WO95/13069 2 1 7 52 1 8 ~ u~9~2816
- 215 -
The title compound (20 mg) was prepared from the more
polar diastereomer (d2, 23 mg) obtained in Example B53, Step C by the
procedure described in Example B5 1, Step D.
FAB-MS: 537.3 (M+l).
The compounds 1-6 shown in Table B4 were prepared as
described above (wi~ different amines). The details of the syntheses are
available in Example B51, Step B and Example B53, Steps A, B and C.
TABLE B4
~f O~ ~XNH2HCI
~N~
~O--R
G~ ds
R FAB-MS (M+l)
d l+d2 thiomorpholine 553.3
2 dl+d2 pyrrolidine 521.3
3 dl N-methylpiperazine 550.4
4 d2 N-methylpiperazine 550.4
dl+d2 piperidine 535.4
6 dl+d2 dimethyl~n~ine 495.2
wo 95/13069 PCrlUS94/12816
2175218
- 216 -
EXAMPLE B55 (cis~ d
~ ~XNH2HCI
~N~
~C-NAO
--/
~3 ds
The intermediate prepared from Example B5 1, Step B in
ethyl acetate was treated with HCl(g) at 0C for 15 seconds. The reaction
mixture was allowed to stand at room temperature for 30 minutes and
concentrated to give the crude product. To this material (209 mg) in 10
ml of chloroform was added Tntennediate 3 (295 mg), EDC (202 mg),
HOBt (105 mg) and triethyl~mine (147 ml) and stirred at room
temperature for 16 hours and poured into water. The mixture was
extracted with methylene chloride and dried over sodium sulfate,
concentrated and the residue was purified by RPLC (chromatatron,
20 methylene chloride/methanol=20/1) to give the desired product (387 mg).
This mixture of diastereomers in ethyl acetate was treated with HCl(g) at
0C for 15 seconds and allowed to stand at room te~ ,elature for 30
minutes. The reaction mixture was concentrated to give the desired
product (330 mg).
25 FAB-MS: 535.3
The compounds shown in Table B5 were prepared according
to established procedures (with ethanol~mine instead of morpholine) as
exemplified in Example B5 l, Step B and Example B53, Steps A, B and C
3 o using Intermediate 3.
wo 93/13069 2 1 7 5 2 1 8 Pcr~usg4/l2816
.
- 217 -
TABLE B5
~N~
~ as
R FAB-MS (M+1)
d 1ethanolamine 509.1
2 d2ethanolamine 509.2
EXAMPLE B56 (cis. d_ ~
~0
~ ~ O
2 0 ~ NH NH ~~
25 Step A:
N o C
NCO
3 0
- To the interme~ te prepared in Example B5 1, Step A ( 1.15
g) in benzene (80 ml) was added oxalyl chloride (365 ml) and DMF (2
drops) at 0C and stirred at 0C for 10 minlltes and room temperature for
2 hours and concentrated to give the acyl chloride. To a solution of acyl
-
WO 9S/13069 PCT~US94/12816
2~752~8 ~
- 218 -
chloride at 0C in acetone (10 ml) was added sodium azide (741 mg) in
water (3 ml) and stirred at room temperature for 45 minutes. The mixture
was extracted with ether, washed with water, brine, dried over MgSO4,
filtered and evaporated to give the acyl azide which was dissolved in
toluene (35 ml) and was refluxed 12 hours to give the isocyanate (1.02 g).
Step B:
IH HCI
~ H H ~~
~3 ds
A solution of ~e intermediate prepared in Step A (55 mg)
and 2-(methylthio)ethylamine (147 mg) in toluene (5 ml) was refluxed
for one hour. The reaction was quenched with lN HCI and extracted
with ether and then dried over sodium sulfate. Concentration and
p ~fication (chromatatron, methylene chloride/methanol=20/1) gave the
20 deslred urea. Deprotection of the BOC protecting group under conditions
described above gave the desired product (40 mg).
Step C:
2s ~NH2Hc
O
~ H NH ~~
To the intermediate prepared in Step B (20 mg) in 2 ml of
chloroform was added Intermediate 1 (28 mg), EDC (19 mg), HOBt (10
mg) and ~iethyl~mine (14 ml). The reaction was stirred at room
WO 95113069 ~ /U~94112816
~ 21 7521 8
- 219 -
temperature for 16 hours and poured into water. The mixture was
~- extracted with methylene chloride and dried over sodium sulfate. Concentration and purification (chromatatron, methylene
chloride/methanol=20/1) gave desired product The mixture was treated
5 with HCl in EtOAc to give the final product (6 mg).
FAB-MS: 565.3 (M+l)
The compounds shown in Table B6 were prepared according
to established procedures (with different arnines or alcohol).
TABLE B6
~XNH2HCI
N ~CO Q
H N
~ H R
R FAB-MS (M+ 1 )
dl+d2 ethanol 52~.3
2 dl+d2 morpholine 561.4
3 dl+d2 ethanol~mine 535 3
4 dl+d2 ethylamine 519.2
EXAMPLE B57 (cis. dl+d2)
~C
~NJ~N~S~
WO 9S113069 PCI'IUS9.~112816
2~75218 ~
- 220 -
To a solution of the intermediate prepared from Example
B56, Step B (20 mg) in 1 ml of chloroform was added Intermediate 3 (28
mg), EDC (19 mg), HOBt (10 mg) and triethyl~mine (14 ml). The
reaction was stirred at room tempel~t~lre for 16 hours and poured into
5 water. The mixture was extracted with methylene chloride and dried over
sodium sulfate. Concentration and purification (chromatatron, methylene
chloride/methanol=20/1) gave the desired product Deprotection of this
diastereomeric mixture with HCl/EtOAc gave the ~mal product (8 mg).
FAB-MS: 554.4 (M+l)
The compounds shown in Table B7 were prepared according
the above-described procedures (with ethanol and different amines).
TABLE B7
~ H
~ ~ NH2HCI
~HN R
R FAB-MS (M+ 1 )
2s 1 dl+d2 ethanol 509.3
2 dl+d2 morpholine 550.4
3 dl+d2 ethanolamine 524.3
4 dl+d2 thiomorpholine 566.2
,.
- -
WO ~tS/13069 PCI'~US94112816
~ 21752~8
- 221 -
EXAMPLE BS8 (trans~ dl+d2)
~ ~NH2HCI
N C O o
,N ~OH
~ trans
Step A:
70c
~I--CO2H
1 5 [~trans
To a solution of the intermediate prepared from Example
B 14, Step A (2.52 g) in ethanol was added 6N NaOH. The rnixture was
2 refluxed for 3 hours and then concentrated. The residue was diluted with
water and acidified with 0.5 N hydrochloric acid and extracted with ether.
The organic layer was dried over sodium sulfate, filtered and
concentrated to give the desired product (2.12 g).
25 SteP B:
BOC
, N ~O H
3 ~3 trans
- To a solution of the intermediate prepared from Step A (15
mg) in 1 ml of chloroform was added 4-amino-1-butanol (9 ml), EDC (19
mg), and HOBt (7.~ mg). The reaction was stirred at room temperature
WO 9~/13069 2 t 7 5 2 1 8 PCI-/US9~/12816
- 222 -
for 2 hours and poured into water. The mixture was extracted with
methylene chloride and dried over sodium sulfate. Concentration and
purification (chromatatron, methylene chloride/methanol=20/1) gave the
desired product.
Step C:
~ ~NH2HCI
H N
H
~,N~ 0 H
~3 trans
The interme~ te prepared from Step B was deprotected with
the HCI/EtOAc protocol. To this crude material in 1 ml of chloroform
was added Intermediate 1 (18 mg), EDC (19 mg), HOBt (7.5 mg) and
triethylamine (20 ml). The reaction was stirred at room temperature for 4
20 hours and poured into water. The mixture was extracted with methylene
chloride and dried over sodium sulfate. Concentration and purification
(PLC, methylene chloride/methanol=10/1) gave the desired product (20
mg, unseparable diastereomer mixture) ~at was treated with HCl (gas) in
EtOAc to give the desired product (18 mg).
25 1H NMR (400 MHz, CD30D, mixture of diastereomers and rotamers):
7.73 (d, 8 ~Iz, 1/2 H), 7.65 (d,8 Hz, 1/2 H), 7.54-6.98 (m, 7 1/2 H), 6.87
(t, 7 Hz, 1 1/2 Hz), 5.25 (m, 1 H), 4.53 (m, 1 H), 3.89 (m, 1 H), 3.39-2.47
(m, 10 H), 1.71-0.93 (m, 5 H), 1.61 (s, 3/2H), 1.60 (s, 3 H), 1.58 (s, 3/2
H), 0.41 (m, 1/2 H), 0.11 (m, 1/2 H).
30 FAB-MS: 548.2 (M+1).
The compounds shown in Table B8 were prepared according
to the above procedures (with different amines).
WO 95/13069 1~ /U~i9-1112816
~ 2175218
-223-
TABLE B8
XNH2HCI
~0--R
~3 trans
R FAB-MS(M +l)
1 dl+d2 e~yl~mine 504.3
2 dl+d2 morpholine 546.3
3 dl+d2 ethanol~mine 520.2
4 dl H2N ~ ~ 556.1
NH
d2 H2N~ 556.1
EXAMPLE BS9 (trans. d2)
2 5 H ~
~, N ~C02Et
~ trans, d2
~ ~
WO 95113069 2 1 7 5 2 1 8 PCIIUS94112816
- 224 -
Step A:
BOC
~CH3
~C,O ~N,CH3
~3 trans Ph CH3
To a solution of the intermediate prepared from Example
B58, Step A (91S mg) in chloroform was added (lR, 2R)-N-methyl
pseudoephedrine (590 ml), EDC (1.14 g), and a catalytic amount of
DMAP. The reaction was stirred at room ternperature for 12 hours and
poured into water. The mixture was extracte~ with methylene chloride
and dried over sodium sulfate. Concentration and purification (MPLC,
hexanes/ethyl acetate=3/1) gave two diastereomers (dl, the less polar
diastereomer, 316 mg; d2, the more polar diastereomer, 138 mg).
Step B:
BOC
~co2H
~rans, d2
A solution of the more polar intermediate prepared in Step A
(138 mg) in methanol was hydrogenated with Pd(OH)2/C at one
atmosphere for a couple of hours. The mixture was filtered through
Celite and the filtrate was concentrated. The residue was redissolved in
ether and washed with lN hydrochloric acid. The aqueous layer was
3 discarded. The organic layer was dried over sodium sulfate, filtrated and
concentrated to give the desired product (84 mg).
WO 95113069 PCI'IUS9~/12816
~ 2175218
- 225 -
Step C:
BOC
, N ~C02Et
~3 trans, d2
To the intermediate prepared from Step B (16 mg) in
0 chloroform was added glycine ethyl ester hydrochloride salt (21 mg),
EDC (19 mg), HOBt (13 mg) and triethylamine (35 ml). After 3 hours at
room temperature the mixture was diluted with methylene chloride and
then washed with water and brine. The organic layer was dried over
sodium sulfate, filtered and concentrated. The residue was purified by
prep TLC (hexanes/ethyl acetate=l/l) to give the desired product (16
mg).
Step D:
~ ~XNH2HCI
NH ICO O
~C' N ~C02Et
~3 trans, d2
The intermediate prepared in Step C (8 mg) was treated with
HCl(gas) in EtOAc to give a crude hydrochloride. To this crude material
3 0 in 1 ml of chloroform was added intermediate 1 (8 mg), EDC (8 mg),
HOBt (5 mg) and triethyl~mine (8 ml). The reaction was stirred at room
temperature for 12 hours and poured into water. The mixture was
extracted with methylene chloride and dried over sodium sulfate.
Concentration and purification (PLC, hexanes/ethyl acetate=1/2) gave the
WO 95113069 PCI/US9~112816
2175218 ~
- 226 -
desired product which was deblocked with the HCl/EtOAc protocol to
give the desired product (11 mg).
lH NMR (400 MHz, CD30D, mixture of rotamers): 7.73 (d, 8 Hz, 1/2
H), 7.54 (d, 8 Hz, 1/2 H), 7.38-6.99 (m, 8 H), 6.84 (d, 7 Hz, 1 H), 5.28-
5.05 (m, 2 H), 4.80-4.52 (m, 1 H), 4.09 (m, 3 H), 3.59 (m, 1 1/2 H), 3.34
(m, 1 1/2 H), 3.24 (m, 1 H), 2.98 (m, 1 H), 2.70-2.48 (m, 2 1/2 H), 1.70-
1.55 (m, 1 1/2 H), 1.61 (s, 3 H), 1.60 (s, 3 H), 1.22 (t, 7 Hz, 3 H), 1.00
(m, 1/2 H), 0.57 (m, 1/2 H).
FAB-MS: 562.3 (M+1)
The compounds shown in Table B9 were prepared according
to the above procedure shown in Example B59.
TABLE B9
~ ~NH2HCI
H 7
~O~
trans, d2
R FAB-MS (M+1)
1 d2 ~-alanine ethyl ester 576.3
2 d2 L-alaninemethyl ester 562.3
Wo 95113069 PCrtUS9~/12816
~ 2175218
- 227 -
EXAMPLE B60 (trans~ d2)
N
lCO O
[~ H CO Et
~3 trans, d2
The intermediate prepared in Example B59, Step C (8 mg) in
ethyl acetate was treated with HCl(g) at 0C for 15 seconds and
m~int~ined at room temperature for 30 minlltes, concentrated to dryness
to give the crude material. To this crude material in 1 ml of chloroform
was added intermediate 3 (8 mg), EDC (8 mg), HOBt (5 mg) and
triethy~mine (8 ml). The reaction was stirred at room temperature for 12
hours and poured into water. The mixture was extracted with methylene
chloride and dried over sodium sulfate. Concentration and purification
(PLC, hexanes/ethyl acetate=1/2) gave the desired product which was
20 treated with HCl(gas) in EtOAc to provide the title compound (11 mg).
FAB-MS: 551.4 (M+1)
The compounds shown in Table B 10 were prepared
according to the above procedure (coupled with different arnino acids).
TABLE B 10
~ -- ~ ~y 2
CO O
~N~
~o
3 trans, d2
W0 9S113069 PCr/US94/12816
2175218 ~
- 228 -
R FAB-MS (M+ 1 )
d2 ~-alanine ethyl ester 565.4
2 d2 L~ nine methyl ester 551.4
FXAMPLE B61 (trans~ dl +d2~
~ ~XNH2HCI
~y cl o o
~N~
~CO2Et
~3 trans
15 Step A:
qoc
~\CO2Me
2 0 ~ trans
To a solution of the interm~diate prepared in Example B 12,
Step A (200 mg) in me~anol was added a catalytic amount of sodium
methoxide in methanol and refluxed for a couple of hours. The mixture
25 was poured into 0.1 N hydrochloric acid and extracted with ether. The
organic layer was dried over sodium sulfate, filtered and concentrated to
give the desired product (190 mg).
WO 95/13069 PCr/US9~112816
~ 2175218
- 229 -
Step B:
BOC
T CHO
~ trans
To a solution of the intermediate from Step A (120 mg) in 2
0 ml of toluene was added diisobutylaluminum hydride ( lN in hexanes,
0.49 ml) at -78C. After the reaction was stirred at -78C for 1 hour it
was quenched with methanol and then poured into 0.5 N hydrochloric
acid solution. The mixture was extracted with ether. The organic layer
was dried over sodium sulfate, filtered and concentrated. The residue
was purified by PLC (hexanes/ethyl acetate=3/1) to give the desired
product (60 mg).
Step C:
BOC
2 o ~N~
~CO2Et
trans
To a solution of triethyl phosphonoacetate in THF (5 ml)
was added potassium bis(trimethylsilyl)amide (0.5 N in toluene, 1.45 ml)
at 0C. After 1 hour at room temperature the intermediate from Step B
(42 mg) in THF (1 ml) was added to the phosphorane solution and
refluxed for an hour. This mixture was concentrated and the residue was
purified by PLC (hexanes/ethyl acetate=4/1) to give the desired product
(50 mg)-
,
-
WO~S113069 2 1 752 ~ 8 PCI-/US9~112816
- 230 -
Step D:
~XNH~HCI
~CO2Et
trans
lo ~3
To the intermediate prepared in Step C (50 mg) was added
0.5 ml of TFA at room temperature. After lQ minutes, the mixture was
concentrated and azeotroped with toluene (3X). To a solution of the
residue in 1 ml of chloroforrn was added Intermediate 1 (62 mg), EDC
(53 mg), HOBt(23 mg) and triethylamine (58 ml). The mixture was
stirred at room temperature for 3 hours and poured into water. The
mixture was extracted with methylene chloride, and dried over sodium
sulfate. Purification (PLC, hexanes/ethyl acetate=1/1) of the residue gave
2 o the coupled product (65 mg).
Step E:
~ --~NH2HCI
H CO O
~--CO2Et
trans
~
A solution of the intermediate prepared from Step D (50 mg)
in methanol was hydrogenated with Pd(OH)2/C at one atmosphere for a
couple of hours. The mixture was filtered through Celite and the filtrate
WO9S/13069 2 t 752 1 8 ~ u~9~128~6
.
- 231 -
was concentrated. The residue was treated with HCl(gas) in EtOAc to
give the desired product (36 mg).
FAB-MS: 533.3 (M+l)
EXAMPLE B62 (cis. dl)
~NH2HCI
cl o o
1 0 ~N~
~C02Et
~_,CO2Et
15 Step A:
CO2Et
EtO2CJ~ 0--
~
Ethyl chloroformate (12.9 ml) was added to a stirred
suspension of cuprous chloride (1.35 g) in THF (200 ml). At 0C, a
solution of ethyl nicotinate was added slowly followed by the addition of
2S Grignard reagent (prepared from 2-bromobenzaldehyde (25 g), 1,3-
propandiol (20 ml), and magnesium (4.9 g) by the procedure described in
J. Org. Chem., 51,3490 (1986)). The reaction was stirred for an hour and
poured into a saturated ammonium chloride/ammonia solution (ltl) and
extracted with ethyl acetate. The organic layer was washed with 1 N
3 hydrochloric acid and brine and dried over sodium sulfate. Evaporation
of the solvent gave the desired product. Cryst~lli7~tion of this material
from ethyl acetate gave 25g of the desired material.
WO 93/13069 PCI-IUS9~/12816
2175218
- 232 -
Step B:
H
EtOzCJ~ O--
~0
The intermediate prepared in Step A (25 g) was dissolved in
hot ethyl acetate (500 ml) and then cooled down to room temperature.
This organic solution was hydrogenated with PtO2 at one atmosphere for
a couple of hours (monitored by TLC). The mixture was ~lltered through
Celite and the filtrate concentrated under vacuum. The residue was
dissolved in hot ethanol (150 ml) was treated with 6N NaOH (75 ml) at
reflux for 10 rninutes. The mixture was concentrated under vacuum and
to the residue was added water and stirred at room temperature for 10
minutes. The pale white solid was collected by filtration. The filtrate
was extracted with methylene chloride and washed with brine and dried
over sodium sulfate. The solvent was concentrated and combined with
20 pale white solid to give 13.6 g of desired product.
Step C:
CBZ
EtO2C~CiS o~~
~o
To a solution of the intermediate prepared in Step B ( 10.1 g)
30 in THF (300 ml) at 0C was added a catalytic amount of indicator
(bromocresol green) and NaCNBH3 (64 mmole). To this reaction
mixture was added 1 N hydrochloric acid till a yellow color persisted
(pH=4.0). After an hour, the mixture was poured into 1 N NaOH and
extracted with chloroform. The organic layer was washed with brine
wo 9SI13069 ~ S94/12816
~ 2175218
- 233 -
dried over sodium sulfate and concentrated. The residue was purified by
filtration through silica gel with methylene chloride/methanol=10/1 to
remove very polar material. The material obtained after concentration of
the filtrate was dissolved in chloroform and to this mixture was added
triethylarnine (6 ml) and CBZ-Cl (4.6 ml) at 0C. After stirring for 15
minutes, the reaction was poured into water and extracted with methylene
chloride. The organic layer was washed with brine, dried over sodium
sulfate. Concentration and purification (MPLC, hexanes/ethyl
acetate=5/1) gave the desired product (6.4 g).
Step D:
~CBZ
EtO2C~
~C H O
To a solution of the intermediate prepared from Step C (2.17
20 g) in methanol (30 ml) was added 1 N hydrochloric acid (5 ml) and
stirred for an hour. The mixture was poured into lN NaOH solution and
extracted with ether. The organic layer was washed with brine and dried
over magnesium sulfate. Purification of the residue (chromatatron,
hexanes/ethyl acetate=S/1) gave the desired product (1.56 g).
Step E:
CBZ
3 o EtO2C~
~CO2Et
To a solution of triethyl phosphonoacetate in THF (25 ml)
was added potassium bis(trimethylsilyl)amide (0.5 N in toluene, 4.56 ml)
WO 9~/13069 PCI'IUS9~112816
2175218 ~
- 234 -
at 0C. After stirring an hour at room temperature the intermediate from
Step D (860 mg) in THF (10 rnl) was added to the phosphorane solution
at room temperature. The mixture was stirred at room temperature for an
hour and then quenched with 1 N hydrochloric acid. This mixture was
5 extracted with ether, washed with brine, and dried over magnesium
sulfate. Purification of the residue (chromatatron, hexanes/ethyl
acetate=5/1) gave the desired product (873 mg).
Step F:
1- H \/
~ ~NH-BOC
~ CO O
~--CO2Et
~CO2Et
The intermediate prepared in Step E (870 mg) was dissolved
in methanol and hydrogenated with Pd(OH)2/C at one atmosphere for
one and one-half hours. The mixture was filtered through Celite and the
filtrate was concentrated under vacuum. To the residue in chloroform
was added intermediate 3 (749 mg), EDC (714 mg) and HOBt (276 mg)
and stirred for 2h. The mixtllre was concentrated and purified
(chromatatron, hexanes/ethyl acetate=2/1) to give two diastereomers (~45
mg, the less polar diastereomer, dl; 500 mg the more polar diastereomer,
d2).
WO 9S/13069 ~ 94/12816
2175218
.
- 235 -
Step G:
O
~CO2Et
1 0 ~C02Et
To a solution of the less polar diastereomer prepared in Step
F (200 mg) in ethyl acetate was bubbled in HCl(g) at 0C for 15 seconds.
After standing for 30 minutes at room temperature the mixture was
concentrated and purified (LH-20, 100% methanol) to give the cis, dl
product as a white solid (100 mg).
lH NMR (400 M[Hz, CD30D, mixture rotamers): 7.28-7.06 (m, 9 H),
5.09 (m, 1/2 H), 4.85-4.55 (m, 1 1/2 H), 4.17 (m, 1 H), 4.10 (q, 7 Hz, 2
H),3.77(m,2H),3.46(m, 1 1/2H),3.25(m, 1/2H),3.15-2.39(m,9H),
1.89-1.60 (m, 5 H), 1.65 (s, 2 H), 1.62 (s, 2 H), 1.57 (s, 2 H), 1.21 (t, 7
20 Hz,3H),0.91 (t,7Hz,3/2H),0.85(t,7Hz,3/2H).
FAB-MS: 594.3 (M+l)
EXAMPLE B63 (cis~ d2)
NC~ O O
~CO2Et
~",CO2Et
WO 95/13069 PCr~US94tl2816
2175218 ~
- 236 -
The desired cis, d2 product (3.3 mg) was obtained from the
more polar diastereomer obtained in Example B62, Step F by the
procedure described in Example B62, Step G.
lH NMR (400 MHz, CD30D, mixture rotamers): 7.90-7.03 (m, 9 H),
4.92-4.61 (m, 2 H), 4.10 (q, 7 Hz, 2 H), 4.07 (m, 1 H), 3.79 (m, 2 H),
3.45 (m, 1 1/2 H), 3.25 (m, 1/2 H), 3.07-2.38 (m, 9H), 1.94-1.69 (m, 4
H), 1.63 (s, 3/2 H), 1.61 (s, 3/2 H), 1.60 (s, 3/2 H), 1.59 (s, 3/2 H), 1.20
(t, 7 Hz, 3 H), 0.91 (t, 7 Hz, 3 H).
FAB-MS: 594.3 (M+1).
EXAMPLE B64 (cis. dl)
~ bX 2
Cl O O
1~ ~N~
~CO2Et
cis,d1 O
~ 3~C~ ~0 H
Step A:
H ~/
~/~ ~NH-BOC
~ C10 0
2 5 ~N~
~COzEt
cis,d1
~3~C02H
To the less polar diastereomer prepared in Example B62,
Step F (30 mg) in ethanol (1 ml) was added 6 N NaOH (30 ml) at room
temperature. After stirring for an hour the mixture was concentrated. To
the residue was added 1 N hydrochloric acid and extracted with ethyl
WO 93113069 PCI-/US9~/12816
~ 2175218
- 237 -
acetate. The organic layer was washed with brine, dried over sodium
- sulfate and concentrated to give the desired product (20 mg).
Step B:
r 5 C
1 0 ~CO2Et
cis,dl Q
~3~C~ ~0 H
To a solution of the intermediate prepared in Step A (6 mg)
in 0.5 ml of chloroform was added ethanolamine (0.8 ml), EDC (3.5 mg),
and HOBt (1.8 mg). The reaction was stirred at room temperature for a
couple of hours and poured into water. The mixture was extracted with
methylene chloride and dried over sodium sulfate. Concentration and
puri~lcation (PLC, methylene chloride/methanol=20/1) provided the
coupled product. This material was deprotected with HCl in EtOAc to
give the desired cis, dl product (1.6 mg).
1H NMR (400 MHz, CD30D, mixture rotamers): 7.28-7.07 (m, 9 H),
5.09 (m, 1/2 H), 4.85-4.62 (m, 1 1/2 H), 4.19 (m, 1 H), 3.75 (m, 2 H),
3-55 (t, 6 Hz, 2 H), 3.45 (m, 1 H), 3.34-2.84 (m, 6 1/2 H), 2.73-2.45 (m, 5
1/2 H), 1.85-1.57 (m, 5 H), 1.65 (s, 3/2 H), 1.62 (s, 3/2 H), 1.57 (s, 3/2
H), 1.56 (s, 3/2 H), 0.92 (t, 7 Hz, 3/2 H), 0.85 (t, 7 Hz, 3/2 H).
FAB-MS: 609.2 (M+ 1)
WO 9~113069 PCI'/US94112816
~ 1 752 1 8 ~
- 238 -
EXAMPLE B65 (cis. dl)
~N~
CO2Et
,CONHEt
To a solution of the intermediate prepared in Example B64
Step A (6 mg) in 0.5 ml of chloroform was added ethyl~mine
hydrochloride salt (1 mg), EDC (3.5 mg), triethylamine (4 ml) and HOBt
(1.8 mg). The reaction was stirred at room temperature for a couple of
hours and poured into water, and e~;tr~cted with methylene chloride and
dried over sodium sulfate. Purification of the residue (PLC, methylene
chloride/methanol=20/1) gave the coupled product. This material was
treated with HCl in EtOAc to yield the desired cis, dl product (1.5 mg).
lH NMR (400 MHz, CD30D, mixture rotamers): 7.28-7.07 (m, 9 H),
20 5.09 (m, 1/2 H), 4.83-4.62 (m, 1 1/2 H), 4.17 (m, 1 H), 3.75 (m, 2 H),
3.50 (m, 1 1/2 H), 3.25-2.84 (m, 6 1/2 H), 2.72-2.39 (m, 5 H), 1.89-1.58
(m, 5 H), 1.65 (s, 3/2 H), 1.61 (s, 3/2 H), 1.57 (s, 3/2 H), 1.56 (s, 3/2 H),
1.06(t,7Hz,3H),0.91 (t,7Hz,3/2H),0.85(t,7Hz,3/2H).
FAB-MS: 593.3 (M+l)
EXAMPLE B66 (cis. dl)
~ ~NH2HCI
~N~
~CO2Et
WO 9S/13069 PCI'/US94/12816
2~75218
- 239 -
To a solution of the intermediate prepared from Example
B64, Step A (8 mg) in methylene chloride (1 ml) was added ethyl
chloroformate (2.3 ml) and triethyl~rnine (5 ml) at 0C. The reaction
mixture was stirred at 0C for 10 minutes and room temperature for an
5 hour. The mixture was poured into saturated sodium bicarbonate and
extracted with methylene chloride. The organic layer was washed with
brine and dried over sodium sulfate, and concentrated. To the residue in
acetone (0.5 ml) was added sodium azide (2.3 mg) in water (0.2 ml) at
0C. After stirring at room temperature for an hour the mixture was
extracted with ether, washed with water and brine, and dried over
MgSO4. Filtration and evaporation gave acyl azide which was dissolved
in toluene (1 ml) and refluxed for 3 hours to give the isocyanate. The
toluene solution was cooled down to room temperature and methylamine
(40% in water, 9 ml) was added. After stirring for 12 hours in room
15 temperature, the reaction was quenched with 1 N HCl and extracted with
methylene chloride and then dried over sodium sulfate and concentrated.
Purification of the residue (PLC, methylene chloride/methanol=20/1 )
gave desired urea which was deprotected with HCl in EtOAc to yield the
desired product (3.5 mg).
20 FAB-MS: 594.3 (M+l).
EXAMPLE B67 (cis. dl+d2)
~ XNH2HCI
~C02Et
~CO2Et
~
The intermediate prepared in Example B62, Step E (17 mg)
was dissolved in methanol and hydrogenated with Pd(OH)2/C at one
atmosphere for one and half hours. The mixture was filtered through
Celite and the filtrate was concentrated under vacuum to give the free
WO gS/13069 PCI~/US94112816
21 7521 8 ~
- 240 -
amine (11 mg). To this free amine (5.5 mg) in chloroform was added
Intermediate 1 (7 mg), EDC (6 r eg) and HOBt (4 mg). After 12 hours,
the mixture was concentrated an~ purified (chromatatron, hexanes/ethyl
acetate=2/1 ) to give an inseparable mixture of di- stereomers. This
5 diastereomeric mixture in ethyl acetate was treated with HCl(g) at 0C
for 15 seconds. After standing for 30 minutes at room temperature, the
rnixture was concentrated to give a white solid (8 mg).
,, FAB-MS: 605.3 (M+l)
EXAMPLE B68 (cis~ d 1 )
~ ~XNH2HCI
~--N~
Step A:
EtO2C~
2 5 ~,~C N
To a solution of diethyl cyanomethyl phosphonate in THF
(25 ml) was added potassium bis(trimethylsilyl)amide (0.5 N in toluene,
3.44 ml) at 0C. After stirring an hour at room temperature the
3 intermediate from Example B62, Step D (65i g) in THF (10 ml) was
added to the phosphorane solution at room temperature. The mixture was
stirred at room temperature for an hour and then quenched with 1 N
hydrochloric acid. This mixture was extracted with ether and washed
with brine, and dried over magnesium sulfate and concentrated.
-
WO g5113069 PCI'IUS9~/12816
~ 2175278
- 241 -
Purification (chromatatron, hexanes/ethyl acetate=S/l) gave the oc"B-
un~ rated nitrile (trans, 466 mg; cis, 124 mg).
Step B:
~N~;XNH soc
7o o
1 0 ~CO2Et
cis,d1
~3~C N
The intermediate prepared in Step A (590 mg) was dissolved
in methanol and hydrogenated with Pd(OH)2/C at one atmosphere for
one and half hours. The mixture was filtered through Celite and the
filtrate was concentrated under vacuum. To the residue in chloroform
was added intermediate 3 (560 mg), EDC (560 mg) and HOBt (208 mg).
After a couple of hours, the mixture was concentrated and purified
(chromatatron, hexanes/ethyl acetate=l/l) to give two diastereomers (220
mg, the less polar diastereomer, dl; 260 mg the more polar diastereomer,
d2).
Step C:
2 5 ~ ~N H - B O C
7o o
~CO2Et N--N~
~H
To the less polar diastereomer prepared in Step B (220 mg)
in toluene (S ml) was added trimethyltin azide (206 mg) and refluxed for
WO 95113069 PCrrUS9-1112816
21 752 1 8 ~
- 242 -
6 1/2 hours. The solvent was removed under vacuum. The residue was
redissolved in methylene chloride/methanol/acetic acid=20/1/0.1 (20 ml)
and allowed to stand at room temperature for 12 hours and the solvent
was removed under vacuum. The residue was purified by PLC >
(methylene chloride/methanol/acetic acid=20/1/0.1) to give the desired
product (120 mg).
Step D:
- N ~
~ ~ NH2HCI
~ CO O
1 5 ~,N
The intermediate in Step C (120 mg) was treated with HCl
in EtOAc to give the desired cis, dl product as a white solid (98 mg).
20 lH NMR (400 MHz, CD30D, mixture of rotamers): 7.28-7.08 (m, 9 H),
5.08 (m, 1/2 H), 4.84-4.53 (m, 1 1/2 H), 4.18 (m, 1 H), 3.78 (m, 3 H),
3.27-3.03 (m, 6 H), 2.85-2.30 (m, 4 H), 1.90-1.38 (m, 5 H), 1.65 (s, 3/2
H), 1.61 (s, 3/2 H), 1.57 (s, 3/2 H), 1.56 (s, 3/2 H), 0.90 (t, 7 Hz, 3/2 H),
0.85 (t, 7 Hz, 3/2 H).
25 FAB-MS: 590.2 (M+l).
WO 93tl3069 PCIJUS94112816
2 1 7 5 2 1 8
- 243 -
EXAMPLE B69 (cis~ d2)
cl o o
--N
The desired product (2 mg) was prepared from the more
polar diastereomer (6.8 mg) obtained in Example B68, Step B by the
procedure described in Example B68, Step C and D.
FAB-MS: 590.4 (M~ l ).
EXAMPLE B70 (cis~ dl)
~ ~NH2HCI
2 0 ~N~
~H
Step A:
~,NH
N Cl O
3 0 H N
~CO2Et
~C N
WO 9~113069 PCI~/US9~112816
2175218 ~
- 244 -
The intermediate prepared in ~xample B68, Step A (782
mg) was dissolved in methanol and hydrogenated with Pd(OH)2/C at one
atmosphere for one and one-half hours. The rnixture was ~lltered through
Celite and the filtrate was concentrated under vacuum. To the residue in
chloroforrn was added Boc-D-Tryptophan (468 mg), EDC (534 mg) and
HO~3t (207 mg). After a couple o~ hours, the mixture was concentrated
and purified (MPLC, hexanes/ethyl acetate=1/1) to give two
diastereomers (316 mg, the less polar diastereomer, dl; 300 mg the more
polar diastereomer, d2).
Step B:
~NH-BOC
~CO2Et
C N
The less polar diastereomer from Step A (316 mg) in ethyl acetate
was treated with HCl(g) at 0C for 15 seconds. After standing 30
minutes at room temperature, the mixture was concentrated to dried to
give crude material. To the residue in 5 ml of chloroform was added N-
25 Boc-a-methyl~l~nine (1~8 rng), EDC (149 mg), triethyl~mine (217 ml)
and HOBt (77 mg) and stirred for 12 hours at room temperature. The
mixture was poured into water and extracted with methylene chloride and
washed with brine. The organic layer was dried over sodium sulfate,
filtered and concentrated. The residue was purified by chromatatron
30 (hexanes/ethyl acetate=1/2) to give the desired compound (287 mg).
wo 95113069 ~ ,94l~28l6
~ 2 1 752 1 8
- 245 -
Step C:
N Cl O O
~CO2Et N--N~
1 cis,dl 1l N
~--~ H
The desired cis, dl product (135 mg) was prepared from
above intermediate (287 mg) prepared in Example B70, Step B by the
procedure described in Example B68, Steps C and D.
1H NMR (400 MHz, CD30D, mixture rotamers): 8.09 (d, 8 Hz, 1/2 H),
15 7.80(d,8Hz, 1/2H),7.64(d,8Hz, 1/2H),7.57(d,8Hz, 1/2H),7.35(d,
7 Hz, 1 H), 7.22-7.00 (m, 7 H), 5.31-5.20 (m, 1 H), 4.71 (d, 12 Hz, 1/2
H), 4.41 (d, 12 Hz, 1/2 H), 4.15 (m, 1/2 H), 3.92-3.67 (m, 2 1/2 H), 3.43-
3.03 (m, 8 1/2 H), 2.80 (m, 1 H), 2.52-2.25 (m, 1 1/2 H), 1.59 (s, 3/2 H),
1.54 (s, 3/2 H), 1.50 (s, 3/2 H), 1.35 (3/2 H), 1.43 (m, 1 H), 0.93 (t, 7 Hz,
20 3/2H),0.84(t,7Hz,3/2H).
FAB-MS: 601.1 (M+l).
EXAMPLE B71 (cis~ d2)
~ ~YNH2HCI
H IC O O
~CO2Et N--N~
¦ cis,d2 D N
~~~ H
,.
The title product (125 mg) was prepared from the more polar
diastereomer (300 mg) prepared in Example B70, Step A by the
WO gStl3069 PCr/US9~112816
2175218
- 246 -
procedure described in Example B70, Step B and Example B68, Steps C
andD.
lH NMR (400 MHz, CD30D, mixture rotamers): 8.24 (d, 8 Hz, 1/2 H),
8.09 (d, 8 Hz, 1/2 H), 7.59 (d, 8 Hz, 1/2 H), 7.54 (d, 8 Hz, 1/2 H), 7.34-
6.92(m,8H),5.40(m, 1/2H),5.15 (m, 1/2H),4.64(d, 13Hz, 1/2H),
4.55 (d, 13 Hz, 1/2 H), 4.22 (m, 1/2 H), 4.09 (m, 1/2 H), 3.81-3.58 (m, 2
1/2 H), 3.40-2.84 (m, 9 1/2 H), 2.71-2.32 (m, 1 1/2 H), 1.63 (s, 3/2 H),
1.52 (s, 3/2 H), 1.48 (s, 3/2 H), 1.29 (3/2 H), 1.53 (m, 1/2 H), 1.32 (m, 1/2
H),O.90(t,7Hz,3/2H),0.79(t,7Hz,3/2H).
FAB-MS: 601.2 (M+ 1).
EXAMPLE B72 (cis. dl +d2)
~ ~NH2HCI
~CO2Et
~as Me
r Ir
Step A:
I BZ
2 5 ,~N~
EtO2C ~r ~
~f O H
To a solution of the intermediate prepared in Example B62,
Step C (235 mg) in methanol (3 ml) was added 1 N hydrochloric acid
(0.5 ml) and stirred for an hour. To the resulting mixture was added
NaCNBH3 (1.0 N in THF, 0.7 ml) and after 5 minlltes the reaction
mixture was poured into 1 N hydrochloric acid and extracted with ether.
The organic layer was washed with brine, dried over sodium sulfate and
wo 9~/13069 ~ 94/l2816
_.
~ 2175218
- 247 -
concentrated. The residue was purified (chromatatron, hexanes/ethyl
acetate=1/1) to give the desired product (142 mg).
Step B:
- 5 H HCI
EtO2C~ds
~M e
L~
To a solution of the interrnediate prepared in Step A (142
mg) in methanol was added HCl in ether and Pd(OH)2 and stirred under
an hydrogen atmosphere for 12 hours. The mixture was filtered through
15 Celite and the filtrate was concentrated to give the desired product ( 105
mg).
Step C:
~NH2HCI
~CO2Et
~,Me
To the intermediate (105 mg) prepared in Step B in 2 ml of
chloroform was added Interrnediate 1 (81 mg), EDC (54 mg), HOBt (28
30 mg) and triethylamine (53 ml) and the reaction was stirred at room
temperature for 12 hours and poured into water. The mixture was
extracted with methylene chloride, dried over sodium sulfate and
concentrated. Puri~lcation of the residue (chromatatron, hexanes/ethyl
acetate=1/1) gave the desired product which was treated with HCl in
EtOAc to give the desired product (56 mg).
WO 95J13069 PCI~/US91/12816
2175218
- 248 -
FAB-MS: 519.2 (M+1)
FXAMPLE B73 (cis~ dl+d2)
H ~N~
~ CO2Et
~,CO2Me
Step A:
~ BZ
EtO2CJ~.
as
3~CO2H
To the intermediate prepared in Example B72, Step A (100
mg) at 0C in acetone was added Jones reagent (4 N, 0.2 ml). After
stirring for 16 hours in room temperature the rnixture was quenched with
25 isopropanol, filtered through celite. The filtrate was extracted with ethyl
acetate. The organics were washed with brine and dried over sodium
sulfate and concentrated to give ~e desired product (100 mg).
Step B:
C~ BZ
EtO2C~,
cs
~ 3~co2M e
WO 9~/13069 PCI-NS9~/12816
2~7~2~
- 249 -
To the intermediate prepared in Step A (50 mg) in ether at
0C was added diazomethane (Blatt, Org. Syn. Collective Vol. 4, p225) .
The mixture was slowly warmed up to room temperature and stirred for
12 hours. Concentration and purification of the residue (PLC,
5 hexanes/ethyl acetate=3/1) gave the desired product (50 mg).
Step C:
~ XNH2HCi
~CO2Et
~,CO2M e
~
The intermediate prepared in Step B (50 mg) was dissolved
in methanol and hydrogenated over Pd(OH)2/C at one atmosphere for
one and half hours. The mixture was filtered through Celite and the
20 filtrate was concentrated under vacuum. To the residue in chloroform
was added interrnediate 1 (48 mg), EDC (45 mg) and HOBt (24 mg).
After a couple of hours, the mixture was concentrated and purified
(chromatatron, hexanes/ethyl acetate=l/l) to give the coupled product.
Deprotection of this material by the HCl/EtOAc protocol gave the desired
25 product (47 mg).
FAB-MS: 563 .1 (M+ 1 )
PcrtrJss~/l28l6
Wo 9S/13069 2 1 7 5 2 1 8
- 250 -
EXAMPLE B74 (cis~ dl+d2)
~YNH2HCI
N Co o
H N
C02Et
ds Q
~ N CO2Et
Step A:
CBZ
~"C~ ~CO E
To the intermediate prepared in Example B73, Step A (50
mg) in chloroform was added glycine ethyl ester HCl salt (51 mg), EDC
20 (46 mg) triethylamine (84 ml), and HOBt (32 mg). The reaction was
stirred at room temperature for 12 hours and poured into water. The
mixture was extracted with methylene chloride and dried over sodium
sulfate, concentrated and purified (PLC, hexanes/ethyl acetate=l/l) to
give the coupled product (45 mg).
Step B:
~ ~NH2HCI
30 H N
~C02Et
ds Q
3~ H CO2Et
Wo 9S113069 PCr/US94112816
~ 21752~8
- 251 -
The title product (43 mg) was prepared from the
intermediate (45 mg) obtained in Step A by the procedure desired in
Example B73, Step C.
FAB-MS: 634.2 (M+1).
EXAMPLE B75 (cis~ dl+d2)
NH2HCI
co o
~C02Et
cis O
~C~ ~0 H
Step A:
Cl BZ
EtO2CJ~.
as Q
~ 3~ H
To intermediate prepared in Example B73, Step A (53 mg)
in chloroform was added ethanolamine (12 ml), EDC (37 mg) and HOBt
25 (19 mg) and stirred at room temperature for 12 hours and poured into
water. The mixture was extracted with methylene chloride and dried over
sodium sulfate and concentrated. The residue was puri~led
~chromatatron, methylene chloride/methanol=20/1) to give the coupled
product (29 mg).
Step B:
2 1 7 5 2 1 8 ~ uS94~28~6
~O 95113069
- 252 -
H ~/
N
N
~CO2Et
H
Th d siredproduct(16.8 mg) wasprepared
intermediate (29 mg) by the procedure described in Example B62, Steps
FandG.
FAB-~S: 581.2 (~1).
~X~PLE B76
Q~_ ~
NH CNO
[~C02Et
~J
To a solution of the intermediate prepared in Example B 1'',
Step ~-1 (100 mg) in acetic acid was added PtO2 and hydrogenated at
tmospherefori~4hourfl(tratewascor~centrated~ d ~ ed
azeotroped with toluene. The residue was dissolved m TFA an s lrr
for 20 minutes at room temperature. The reaction mixture was
ted and the residUe w diate 1(15 rng) ~DC (
mg) and triethylamine (11 ml). The mixture was stirred at room
temperature ~or 3 hours and poured into water. The mixture was
extracted with methylene chloride, dried over sodium sulfate and
WO 95/13069 PCr/US9-~/12816
~ 2175218
- 253 -
concentrated. Purification of the residue (PLC, hexanes/ethyl
acetate=1/1) gave the coupled product which was treated with HCl in
EtOAc to yield the desired product (8 mg).
FAB-MS: 511.1 (M+1)
EXAMPLE B77 (cis~ dl+d2)
NH2
y`CO2Et
[~
The intermediate prepared in Example B 12, Step B was
dissolved in methanol and hydrogenated over Pd(OH)2 at one
atmosphere for a couple of hours. The mixture was filtered through celite
and the filtrate was concentrated under vacuum. To the residue (88 mg)
20 in chloroform (1 rnl) was added N-Boc-,B,~-dimethyl-~-alanine (48 mg,
W.R. Schoen etc., J. Med. Chem., 37, 897 (1994)), EDC (48 mg), and
HOBt (30 mg), stirred for 12 hours and the mixture was poured into
water. The mixture was extracted with methylene chloride, dried over
sodium sulfate and concentrated. Purification of the residue
25 (chromatatron, hexanes/ethyl acetate=1/1) gave the coupled product that
was deblocked with HCl in EtOAc to give the desired product (58 mg).
FAB-MS: 519.2 (M+1)
WO ~/13069 2 1 7 5 2 1 8 PCI~/US9~/12816
- 254-
EXAMPLE B78 (cis. dl+d2)
N HCI
~CO2Et
CiS
~
Step A:
O-THP
H
To a solution of methyl (R)-lactate (1 ml) in dihydropyran (5
ml) was added one drop of concentrated hydrochloric acid at room
temperature. The reaction was stirr~ for an hour, concentrated and
purified by chromatatron (hexanes/ethyl acetate=3/1) to give the desired
O product (1.49 g). To the THP protected lactate (500 mg) in toluene (10
ml) was added diisobutylal-lmin-lm hydride (lN in cyclohexane, 3.45 ml)
at -78C and after one and half hours, the reaction was quenched with
methanol at low temperature. The mixture was poured into 5% aqueous
citric acid and extracted with ether. The organic layer was washed with
brine, dried over sodium sulfate and concentrated to give the protected
aldehyde.
WO 95113069 PCIrUS9~112816
217~8
- 255 -
Step B:
~N~N H
~CO2Et
cis
~ ~
To a solution of the product (25 mg) prepared in Example
B77 in methanol (0.5 ml) was added the intermediate (36 mg) prepared in
Step A and sodium acetate (18 mg) and stirred at room temperature for an
hour. To the mixture was added NaCNBH3 (lN in THF, 90 ml) slowly
5 and stirred for 16 hours and concentrated. The residue was purified by
chromatatron (methylene chloride/methanol/ammonium
hydroxide=10/1/0.1) to give the desired product which was dissolved in
methanol (0.5 ml) and was treated with 9 N hydrochloric acid (0.2 ml).
After stirring for 2 hours, the mixture was concentrated and dried to give
20 the desired product (10.5 mg).
FAB-MS: 577.4 (M+1).
WO 95/13069 2 1 7 5 2 1 8 PCI'IUS9~/12816
- 256 -
EXAMPLE C1
~NH2 HCI
H ~N~
~,OEt
~
Step A:
Boc
~OEt
To a stirred solution of ethyl nipecotate (1~ g, 95.4 mmol)
20 and DMAP (0.05 eq.) in dichloromethane at 0C was added dropwise by
an addition funnel di-tert-butyl dicarbonate (21.8 g, 100 mrnol) in
dichloromethane (200 m~ ). The mixture was stirred for 2-3 hours. The
solution was washed with 3 N HCl and saturated sodium chloride, dried
over anhydrous magnesium sulfate; then filtered and concentrated to give
25 the desired product (18.7 g, 88%).
Step B:
Noc
3 ~OEt
¢~
WO 95~13069 PCr/US94/12816
2i75218
- 257 -
To a stirred solution of ethyl N-t-Boc nipecotate (7 g, 26.90
mmol) in THF (100 mL) at -78C under argon was added LHMDS (28
mL, 28 mmol) over a 10 minute period. The solution was allowed to stir
an additional 30 minutes at -78C; then benzyl bromide (4.8 g, 28 mmol)
- 5 was added slowly to ~e solution. The reaction mixture was stirred
overnight and allowed to warm to room temperature. The material was
concentrated, then diluted with water, and extracted usin~ ethyl acetate (2
x 200 mL). The organic layer was dried over anhydrous magnesium
sulfate, filtered, and concentrated. Purification by silica gel flash column
o chromatography, eluting with 20% ethyl acetate in hexane, provided the
title compound. (8.32 g, 88%).
FAB-MS calc. for C20H29NO4: 347; Found 348 (M+H)
Step C:
H HCi
~N~
~OEt
~ O
~
A solution of the intermediate from Step B (8 g, 23.02
mmol) in ethyl acetate (80 mL) was cooled to 0C. While stirring,
hydrogen chloride gas was bubbled into the mixture until saturation
25 occurred. The reaction was stirred for 40 minutes, until TLC analysis
indicated that the reaction was complete. The solution was then
concentrated to remove the ethyl acetate to afford the product (6.53 g,
99%).
lH NMR (CDCl3, 400MHz) ~ 7.25-7.19 (m, 3 H), 7.04-7.01 (m, 2 H),
30 5.35(v.br.s,2H),4.22-4.10(m,2H),3.44(d,J=13Hz,lH),3.21(br.
d, J = 12.7 Hz, 1 H), 2.95 (d, J = 13.5 Hz, 1 H), 2.76-2.68 (m, 3 H), 2.22
(br. d, J = 13 Hz, 1 H), 1.73-1.71 (m, 1 H), 1.61-1.48 (m, 2 H), 1.18 (t, J
= 7 Hz, 3 H).
FAB-MS calc. for Cl5H2lNo2: 247; Found 248 (M+H)
WO 9S/13069 ~ /U:~9~/12816
.
2175218
- 258 -
Step D: -
5 ¢~ ~NHBoc ~ ~NHBoc
H ~N~ H ~N~
~,OEt l~ ",~OEt
3/ and [[~
To a solution of the intermediate prepared in the previous
step (1.2 g, 4.23 rnmol), and Intermedate 1 (l eq.), HOBT (1 eq.), and N-
5 methyl morpholine (1 eq.) in dichloromethane cooled to 0C was added
EDC (1.5 eq.). The reaction mixture was stirred at 0C overnight. The
solution was washed with saturated sodium chloride, dried over
anhydrous magnesium sulfate; then filtered and concentrated.
Purification by MPLC eluting with 40% ethyl acetate in hexane provided
20 two enantiomerically pure compounds. The compound which came out
first from the column was de~i~n~ted as dl (1.14g), which has an R-
absolute stereochemistry at the 3-position of the nipecotate; and the
compound which came out of the column second was designated as d2
(1.08 g), which has an S-absolute stereochemistry (see Example C2 for
25 assignment) at the 3-position of the nipecotate.
dl FAB-MS calc. for C35H46N406: 618; Found 619 (M+H)
d2 FAB-MS calc. for C35H46N406: 618; Found 619 (M+H)
Step E:
WO 9S113069 ~ l/u~9~tl2816
2175218
- 259 -
- ¢~ ~NH2 HCI
N C=O O
H ~N~
l,OEt
~- o
Prepared by the procedure described in Step C from the
intermediates dl from the previous step (1 g) in ethyl acetate (20 mL) and
HCl gas at 0C for 1.5 hours. Product: 860 mg, 91%.
FAB-MS calc. for C30H38N4o4: 518; Found 519 (M+H)
F~xAMpLE ClA
H C=o 0
~ ",l~OEt
~ O
~
Prepared by the procedure described in Step C of Example
C 1 from 1 g of the d2 intermediates from the Step D of Example C 1 in
ethyl acetate (20 mL) by bubbling HCl at 0C until saturated and then
evaporated after 30 minutes to give the title compound (878 mg, 93%).
- 30 FAB-MS calc~ for C30H3gN404: 518; Found 519 (M+H)
lH NMR (CD30D, 400MHz) compound exists in two rotamers in
approximately a 5/3 ratio that slowly interconvert relative to the NMR
time scale ~ 7.60 (d, J = 7.9 Hz, 5/8 H), 7.55 (d, J = 7.9 Hz, 3/8 H), 7.34-
6.93 (m, 9H), 5.36 (dd, J = 5.2Hz, 9.7 Hz, 3/8 H), 5.31 (dd, J = 6.7 Hz,
WO 95/13069 PCI'/US9 1112816
2~75218
- 260 -
8.8 Hz, 5/8 H), 4.23 (br. d, J = 13.7 Hz, 3/8 H), 4.10-4.00 (m, 6/8 H),
4.04-3.98 (m, 3/8 H), 3.96-3.82 (m, 10/8 H), 3.80 (br. d, J = 13.5 Hz, 5/8
H), 3.36 (br. d, J = 13.3 Hz, 5/8 H), 3.29-3.22, 3.17-3.10, (2m, 2H), 3.20
(br. d, J = 14.5 Hz, 3/8 H), 3.10-2.96 (m, 5/8 H), 2.90 (s, 6/8 H), 2.60 (d,
J = 13.4 Hz, 5/8), 2.41 (d, J = 13.4 Hz, 5/8 H), 2.19-2.12, 1.82-1.70, 1.68-
1.60, 1.50-1.40, 1.34-1.25, 1.05-0.95 (6m, 4 H),1.55 (s, 9/8 H), 1.50 (s,
15/8 H), 1.09 (t, J = 7.1 Hz, 3 H).
The additional intermediates shown in Table CI were
prepared according to the above established procedures as exemplified in
Example C1 steps A through C. The final compounds were prepared
according to Example C1 Steps D and E, and Example CIa using
Intermediate 1.
TABLE CI: ADDITIONAL EXAMPLES
H
~ ~NH2 HCI
H ~ N Ç=
~<CO2Et ~<CO2Et
Intermediate Product
Intermediate Product
entry Y MF MF isomera
FAB-MS (M+1) FAB-MS (M+ 1)
Me CgHl7No2 C24H34N4O4 dl
171 (M+, EI-MS) d2
2 Et C1oH19NO2 C2sH36N4O4 dl
185 (M+, EI-MS) d2
3 n-Pr Cl 1H21NO2 C26H3sN4O4 dl
199 (M+,EI-MS) d2
WO 9S113069 PCI'IUS94112816
2~752~8
- 261 -
4 allyl Cl lHlgNo2 C26H36N404 dl
198 d2
n-Bu C12H23N02 C27H40N404 dl
- 213 (M+,EI-MS) d2
- 5 6 -CH20Et Cl lH2lNo3 C26H38N405 RS
216
7 cyclohexane- C15H27N02 C30H44N404 dl
methyl 254 d2
o 8 Ph(CH2)2- C16H23N02 C31H40N404 dl
261 (M+,EI-MS) d2
g Ph(CH2)3- C17H25N02 C32H42N404 dl
275 (M+,EI-MS) d2
o-MeOBn- C16H23N03 C31H40N405 dl
278 d2
11 m-MeOBn- C16H23N03 C31H40N40s dl
278 d2
12 p-MeOBn- Cl 6H23N3 C3 1H40N405 dl
278 d2
o-Me-Bn- C16H23N02 C31H40N404 dl
262 d2
14 m-Me-Bn- C16H23N02 C31H40N404 dl
262 d2
p-Me-Bn- C16H23N02 C3lH40N404 dl
2 5 262 d2
16 o-Cl-Bn- ~l5H2oNo2cl C30H37N4o4cl dl
282,284 (3:1) 554,556 (3:1) d2
17 m-Cl-Bn- C15H20No2cl C30H37N4o4cl dl
282,284 (3:1) 554,556 (3:1) d2
p-Cl-Bn Cl5H20No2cl C30H37N4o4cl dl
282,284 (3:1) 554,556 (3:1) d2
16 2,6-di-Cl-Bn- C15HlgN02Cl2 C3oH36N4o4cl2
3 16,318,320 587,589,591
17 p-Br-Bn- Cl5H20No2Br C30H37N404Br dl
326,328 (1:1) 597,599 (1:1) d2
- - -
WO 95/13069 PCI~/US9~rl;!816
2175218
- 262 -
18 m-Br-Bn- Cl5H20No2Br C30H37N4o4Br dl
326,328 (1:1) 597,599 (1:1) d2
19 o-nitro-Bn- C15H20N204 C30H37N506 dl
293 564 d2
m-nitro-Bn- C15H20N204 C30H37N506 dl
293 564 d2
21 p-nitro-Bn- Cl5H20N204 C30H37N506 dl
293 564 d2
22 l-naphthylmethyl Cl9H23No2 C34H40N404 dl
298 569 d2
23 ~ Cl3HlgNo2scl C28H3sN4o4scl dl
Cl S CH2- 288,290 (3: 1) 559,561(3: 1) d2
24 Bn02C- C16H21N04 C31H38N406 RS
292 563
Et02C- Cl lH19N04 C26H36N406 RS
230 501
26 p-Ph-Bn- C21H25N04 C36H42N404 dl
324 595 d2
27 O~CH2- C 16H20N04Cl C31 H37N406Cl d 1
<o~l~l~ 326, 328(3:1) 597, 599(3:1) d2
28 ~0 C17H20N204 C32H37N506 dl
~NCH2- 317 588 d2
0
a: In tE~s and in subsequent tables for isomer designation: R or S means
the stereochemistry at the carbon atom to which X and Y are attached, RS
means it is a mixture of the two isomers at this center; dl or d2 means
the two diastereomers were separated and are as defined in Fx~mple Cl
step D.
The additional examples shown in Table CIa were prepared
according to Example Cl Steps D and E, using Intermediate 1 and
commercially available interme~ tes.
WO 95113069 PCrJUS9~/12816
~ 2175218
- 263 -
TABLE CIa: ADDITIONAL EXAMPLES
H¢~ ~NH2 HCI
~X H ~`X
Intermediate Product
Product
entry X MF isomer
FAB-MS (M+l)
-OH C20H28N403 RS
373
2 -CH20H C2lH30N43 RS
387
3 -C02Et C23H32N404 RS
429
4 C02Bn C28H34N44 RS
491
2 o 5 CONMe2 C23H33N53 RS
428
The additional Products shown in Table CIb were prepared
according to F.x~mple Cl Steps D and E, using Intermediate 3 and some
25 of the intermediates shown in Table C1.
wo ~5113069 PCrlUS9~112816
2175218
- 264 -
TABLE CIb: ADDITIONAL EXAMPLES
Ph~ ~NH2 HCI
Ç=O O
~<CO2Et
Product
entry Y MF isomer
FAB-MS (M+ 1 )
Bn C30H41N304 R
508
2 Bn C30H41N304 S
508
3 Ph(CH2)2 C31H43N304 dl
d2
4 Ph(CH2)3 C32H45N34 dl
d2
l-naphthylmethyl 554H43N304 RS
6 _~ C28H38N304SCl RS
Cl S~CH2- 548,550 (3:1)
7 p-Ph-Bn- C36H45N34 RS
584
8 BnO2C- C31H41N306 RS
552
g o ~CH2- C31H40N306Cl dl
<o~l~ 586,588 (3:1) d2
The additional products shown in Table CIc were prepared
according to Example C 1 Steps D and E, using Intermediate 2 and some
of the intermediates shown in Table CI.
WO 9S/13069 ~ 9~/12816
21 752~
- 265 -
TABLE CIc: ADDITIONAL EXAMPLES
Ph~O N~NH2 HCI
C=O O
N
~CO2Et
y
entry Y MF isomer
FAB-MS (M+l)
Bn C29H39N305 R
510
2 Bn C29H39N305 S
510
3 Et C24H37N305 RS
Ph(CH2)2 C30H41N305 dl
524 d2
Ph(CH2)3 C3 lH43N305 dl
538 d2
6 H C22H33N305 RS
420
FXAMPLE C2
~ ~NH2 HCI
2 5 N C=O O
H ~N~
1~ ",~OEt
Step A:
WO 9S~13069 ~ 4/~2816
2175218
- 266 -
H L-tartaric acid
~N~
~ "~OEt
~ O
The intermediate from Example C1, Step C (50.8 g) was
dissolved in dichloromethane and it was washed with 3N NaOH. The
aqueous layer was extracted with dichloromethane and the combined
solution was dried (MgSO4) and evaporated to give the free amine as an
oil. The ethyI 3-benzyl nipecotate and D-tartaric acid (31 g) were
dissolved in 880 mT . of water/acetone (1:4) solution with heating. The
solution was left in the refrigerator overnight and the crystals which were
formed were filtered off. Recryst~11i7~tion in 470 mL of water/acetone
(1:4) at room temperature gave the ethyl 3-(R)-benzyl nipecotate D-
tartaric acid salt (21 g).
The structure of this compound was determined by X-Ray
20 crystallographic analysis. With the configuration of D-tartaric acid
known to be S,S, the configuration of the chiral site in this ethyl 3-
benzylmipecotate salt was determined to be R.
The combined mother liquor was evaporated and to it was
added 3N NaOH and dichloromethane, the mixture was stirred for 30
25 minutes and the organic layer was separated. The aqueous was extracted
twice with dichloromethane and the combined organic extracts were
dried over MgSO4 and evaporated to give 24.4 g of the S-isomer
enriched compound. It was cryst~lli7~-1 with L-tartaric acid (14.8 g) in
400 mL of water/acetone (1:4) at room temperature to give ethyl 3 (S)-
O benzyl nipecotate L-tartaric acid salt (27.3 g).
lH NMR (CD30D, 400MHz) ~ 7.31-7.22 (m, 3 H), 7.12-7.09 (m, 2 H),
4.40 (s, 2 H, from tartaric acid), 4.30-4.10 (m, 2 H), 3.49 (br. d, J = 13
Hz, 1 H), 3.06 (d, J = 13.6 Hz, 1 H), 2.98 (d, J = 13 Hz, 1 H), 2.92 (dt, J =
3.3 Hz, 13 Hz, 1 H), 2.82 ( d, J = 13.6 Hz, 1 H), 2.30 (d, J = 12.4 Hz, 1
WO 95/13069 PCI`IUS9~/12816
~ 2175218
- 267 -
H), 1.88 (td, J = 3 Hz, 14.5 Hz, 1 H), 1.69 (dt, J = 3 Hz, 13 Hz, 1 H),
1.63-1.51 (m, 1 H), 1.25b (q, J = 7.1 Hz, 3 H).
- Step B:
NHBoc
H ~N~
." fOEt
~/ 'O
Ethyl 3 (S)-benzyl nipecotate L-tartaric acid salt (39.74 g)
was suspended in 70 mL of 3N NaOH and 70 mL of water, followed by
extraction with dichloromethane. The extracts were combined, dried, and
evaporated to give a thick oil. To a stirred solution of the oil, N-t-Boc D-
TrpOH (30.43 g) and HOBt (13.5 g) in dichloromethane (200 mL) at
20 0C, was added EDC (23 g) in several portions. The rnixture was stirred
overnight and it was poured into water and 3 N HCl and was extracted
with dichloromethane. The organic layer was washed with brine,
saturated sodium bicarbonate, dried over MgSO4 and evaporated to give
a crude product (67.7 g), which was used without further purification.
FAB-MS calc. for C31H39N3O5: 533; Found 534 (M+H)
Step C:
~;~3~ NH2
H ~N~
WO 9S113069 PCIIUS94112816
2175218
- 268 -
To a solution of the intermediate from the previous step
(67.7 g crude) in ethyl acetate (100 mL) at 0C, was bubbled HCl gas
until it was saturated. The reaction mixture was stirred at 0C for 30
minutes and was evaporated to remove excess HCl and ethyl acetate.
5 The residue was suspended in dichloromethane and was washed with a
mixture of 3 N NaOH (70 mL) and water (100 mL). The organic layer
was dried (MgSO4), evaporated io a small volume and used in next step
without further puri~lcation.
FAB-MS calc. for C26H3 1N3O3: 433; Found 434 (M+H)
Step D:
~~r ~NHBoc
H ~N~
~/.",~OEt
~/ o
~
A solution cont~inin~ the intermediate obtained in the last
step, N-Boc-oc-Me-AlaOH (20.3 g), and DMAP (200 mg) in
2 5 dichloromethane ( 100 mL) was stirred at room temperature and to it was
added EDC (24 g) in several portions. The reaction mixture was stirred
for 3 hours and was worked up by diluting it with dichloromethane and
washing with 3 N HCl, brine, and saturated sodium bicarbonate solution.
The organic layer was dried over MgSO4, and evaporated to give a thick
3 o oil. This oil was passed through a pad of silica gel, eluting with 60%
ethyl acetate in hexane to remove some very polar impurities, to give the
desired compound (54.2 g)
FAB-MS calc. for C35H46N406: 618; Found 619 (M+H)
Step F:
WO 95/13069 PCI~/IJS91/12816
~ 2175218
- 269 -
~=0 0
- H ~N~
""~OEt
[3~ o
To a solution of the intermediate from the previous step
(54.2 g) in ethyl acetate (100 mL) at 0C, was bubbled HCl gas until it
was saturated. The reaction mixture was stirred at 0C for 15 minutes
and was evaporated to remove excess HCl and ethyl acetate. The residue
was first dissolved in dichloromethane (100 mL) and then ethyl acetate
(300 mL) was added. The mixture was stirred overnight and the solid
was collected by filtration to give the title compound (34 g). Further
evaporation of the mother liquor to a small volume gave the second crop
20 of product (10.1 g).
MS and NMR identical with Example ClA.
The additional products shown in Table CII were prepared
according to Example C2, Steps B through F, using the readily available
25 Boc protected amino acids instead of N-t-Boc-D-TrpOH.
TABLE CII
R~=O O
~CO2Et
Bn
Product
WO95113069 2 1 752 1 8 PCl-tUS9~/12816
- 270 -
entry R MF
FAB-MS (M~1)
4-F C30H37N404F
537
2 S-F C30H37N404F
537
3 6-F C30H37N404F
537
4 l-Me C31H40N404
0 533
5-MeO C31H40N405
549
6 5-HO C30H38N405
535
7 6-MeO C31H40N405
549
EXAMPLE C3
2 0 H ~NH2 HCI
CO2Et
3/
Step A:
Boc
~N~,
~
CO2Et
To a stirred solution of ethyl 3-pyrrolidinecarboxylate
hydrochloride (J. Chem. Soc., 24, 1618-1619; 10 g, 69.8 mmol),
triethyl~mine (7~75 rnL) and DMAP (857 mg) in dichloromethane (40
mL), was slowly added di-t-butyl dicarboxylate (18.3 g, 83.7 mmol) and
WO 9S/13069 PCI-/US94/12816
~ 2~75218
- 271 -
the resulting mixture was stirred at room temperature for three days. The
mixture was then concentrated, washed with 3 N HCl and dried and
evaporated to give the intermediate.
5 SteP B:
Boc
CO2Et
,~
W
Prepared by the procedure described in Example C 1, Step B
from the intermediate obtained from previous step (500 mg, 2.05 mmol),
KHMDS (512 mg, 2.57 mmol) and benzyl bromide (371 mg, 2.16 mmol).
Purification by silica gel flash column eluting with 5-20% ethyl acetate in
hexane provided the title compound (385 mg, 56%).
Step C:
H HCi
~
CO2Et
¢~
Prepared by the procedure described in Example C1, Step C
from the intermediate from the previous step (385 mg, 1.16 mmol) in
ethyl acetate (5 mL) and HCl gas at 0C for 15 minll~es (306 mg, 98%).
30
WO 95113069 PCI'tUS9~112816
21752~8
- 272 -
Step D: H
s H ~NHE~oc
CO2Et
¢~
Prepared by the procedure described in Example Cl, Step D
from the intermediate prepared in the previous step (138 mg, 0.514
mmol), intermediate 1 (200 mg, 0.514 mmol), HOBT (1 eq.), N-methyl
morpholine (1 eq.), and EDC (2 eq.). Purification by ~hPLC, eluting with
60% ethyl acetate in hexane gave the product (250 mg, 80%)
FAB-MS calc. for C34H44N406: 604; Found 605 (M+H).
Step E:
~~,r='O O
H C~
CO2Et
¢~]/
Prepared by the procedure described in Example Cl, Step C
from the intermediate from the previous step (250 mg, 0.036 mmol) in
ethyl acetate (3 mL) and HCl gas at 0C for 10 minutes.
FAB-MS calc. for C29H36N404: 504; Found 505 (M+H).
WO 9S113069 PCI~/US9~/12816
2~75218
- 273 -
EXAMPLE C4
~NH2 HCI
H ~N~
~OEt
,S o
Step A:
Boc
~N~
OEt
,S o
To a stirred solution of ethyl N-t-Boc nipecotate (4 g, 15.7
mmol)) in THF (100 mL) at -78C under argon was added LHMDS (1 M,
32 mL, 32 mmol) over a 10 minllte period. The solution was allowed to
20 stir an additional 30 minutes at -78C; then methyl disulfide (1.92 g,
20.37 mmol) was added slowly to the solution. The reaction mixture was
stirred overnight and allowed to warm to room temperature. The material
was concentrated, then diluted with water, and extracted using ethyl
acetate (2 x 200 mL). The organic layer was dried over anhydrous
2S magnesium sulfate, filtered, and concentrated. Purification by silica gel
flash column chromatography eluting with 20% ethyl acetate in hexane
provided the title compound.
FAB-MS calc. for C14H25N04S: 271; Found 272 (M+H)
30 Step B:
H HCI
~N~
~OEt
_ o
WO 95113069 PCI'IIJS9~/12816
2175218
- 274 -
Prepared by the procedure described in Example Cl, Step C
from the intermediate from the previous step (1 g, 3.3 mmol) in ethyl
acetate (25 mL) and HCl gas at 0C for 35 minutes to yield the product
(783 mg, 99%).
FAB-MS calc. for CgH17NO2S: 171; Found 271 (M+H)
Step C:
~ ~NHBoc
H ~N~
~OEt
~ O
Prepared by the procedure described in Example Cl, Step D
from the intermediate prepared in the previous step (123 mg, 0.514
mmol), Intermediate 1 (1 eq.), HOBT (1 eq.), NMM (1 eq.), and EDC
20 (197 mg, 1.028 mmol). Purification by MPLC provided diastereomers.
The compound which came out first from the column was designated as
dl (109 mg, 37%); and the compound which came out of the column
second was desi~n~te-l as d2 (88 mg, 30%),
dl FAB-MS calc. for C29H42N4O6S: 574; Found 575 (M+H)
25 d2 FAB-MS calc. for C29H42N4O6S: 574; Found 575 (M+H)
Step D:
~=O~
H ~N~
~OEt
~'
WO 95113069 PCI`/US9~/12816
~ 2~752~8
- 275 -
Prepared by the procedure described in Example Cl, Step C
- ~rom the intermediates dl(80 mg) and d2 (80 mg) separately from the
previous step in ethyl acetate (5 mL each) and HCl gas at 0C for 20
minutes.
dl: (71 mg, 99%)
d2: (70 mg, 98%)
dl lH NMR (CD30D, 400MHz): The compound exists in two rotamers
in approximately a 1:1 ratio. o 7.71 (d, J = 7.2 Hz, 1/2 H), 7.56 (d, J =
7.2Hz, 1/2H),7.38(d,~=8.0Hz, 1/2H),7.33(d,J=7.5Hz, 1/2H),
7.14-7.01 (m,3H),5.44(dd,J=6Hz,8Hz, 1/2H),4.30-4.10(m,5/2
H),3.92(d,J= 13.3Hz, 1/2H),3.81 (d,J= 13.3Hz, 1/2H),3.67(d,J=
13.3 Hz, 1/2 H), 3.48-3.40 (m, 1/2 H), 3.28-3.09 (m, 7/2 H), 2.55 (dt, 1/2
H), 2.26-2.20 (br. d, 1/2 H), 2.05 (s, 3 H), 1.80-1.70 (m, 1/2 H), 1.67,
1.59, 1.55, 1.43 (4s, 6 H), 1.27 (t, J = 7.0 Hz, 3/2 H), 1.19 (t, J = 7.0 Hz,
3/2 H), 0.90-0.85 (m, 1/2 H).
d2 lH NMR (CD30D, 400MHz): The compound exists in two rotamers
in approximately a 1: 1 ratio. ~ 7.77 (d, J = 7.5 Hz, 1/2 H), 7.56 (d, J =
7.9 Hz, 1/2 H), 7.35-7.30 (m, 1 H), 7.13-6.98 (m, 3 H), 5.53 (dd, J = 5.5
Hz,8Hz, 1/2H),5.24(app.t,J=7Hz, 1/2H),4.30(br.d,J=14Hz, 1/2
H), 4.20-4.10 (m, 2 H), 3.90-3.85 (m, 1/2 H), 3.86 (d, J = 13.2 Hz, 1/2
H), 3.70 (d, J = 13.7 Hz, 1/2 H), 3.35-3.10 (m, 4 H), 2.30-2.20 (m, 1/2
H), 2.12, 2.04 (2s, 3 H), 2.04-2.00 (m, 1/2 H), 1.80-1.70 (m, 3/2 H~, 1.54,
1.50, 1.43, 1.26 (4s, 6 H), 1.23 (t, J = 6.7 Hz, 3 H), 0.90-0.84 (m, 1/2 H).
dl FAB-MS calc. for C24H34N4O4S: 474; Found 475 (M+H)
d2 FAB-MS calc. for C24H34N4O4S: 474; Found 475 (M+H)
The additional intermediates shown in Table CIII were
prepared according to the above established procedure as exemplified in
Example C4 steps A and B. The final compounds were prepared
according to Example C4 Steps C and D, using Intermediate 1.
.,
WO 9Srl3069 PCrlUS9'1tl2816
2~752~8
- 276 -
TABLE CIII
N H ~NH2 HCI
~<CO2Et ~<CO2Et
Interrnediate Product
Intermediate Product
entry Y MF MF isomer
FAB-MS (M+l) FAB-MS (M+l)
PhS- C14Hl9NO2S C29H36N4O4S dl
266 537 d2
2 BnS- Cl5H21NO2S C30H3gN4O4S dl
280 551 d2
3 2-pyridyl~io- C13Hl8N202S C28H3sNsO4S RS
267 538
4 N CH3 C27H35N5O4S2 RS
-S~/ 3~ 558
S
EXAMPLE C5
~ ~NH2 HCI
H ~N~
~OEt
3 o
O O
wo 95113069 ~ 594/l28l6
21 75218
- 277 -
Step A:
-
Boc
~N~
~OEt
, ,- o
o
To a stirred solution of NaIO4 (316.5 mg, 1.48 mmol) in
water (5 mL) and ethanol (5 mL) was added the intermediate from
0 Example C4, Step A (300 mg, 0.99 mmol). The mixture was stirred for S
hours at room temperature, then concentrated to remove ethanol. The
material was then extracted with ethyl ~cet~te. (2 x 10 mL). The organic
layer was dried over magnesium sulfate and concentrated to give the title
compound (286 mg, 90.5%).
5 FAB-MS calc. for C14H25NO5S: 319; Found 320 (M+H)
Step B:
H HCI
~N~
~OEt
~i O
o
Prepared by the procedure described in Example Cl, Step C
from the intermediate from the previous step (230 g, 0.72 mmol) in ethyl
acetate (10 mL) and HCl gas at 0C for 25 minutes (197 mg, 100%).
FAB-MS calc. for CgH17NO3S: 219; Found 220 (M+H)
WO 9S~13069 2 1 7 5 2 1 8 ~ /u~94112816
- 278 -
Step C:
3~ N~NHBoc
H ~N~
~OEt
O
Prepared by the procedure described in Example C l, Step D
from the intermediate prepared in the previous step (140 mg, 0.547
mmol), Intermediate 1 (l eq.), HOBT (l eq.), N-me~yl morpholine (1
eq.), and EDC (210 mg, 1.094 mmol). Purification by MPLC provided a
15 diastereomeric mixture of compounds (177 mg, 55%).
FAB-MS calc. for C29H42N407S: 590; Found 59l (M+H)
~tep D:
~'N~NH2 HCI
H ~N~
~OEt
2~ ~S
o o
Prepared by the procedure described in Example Cl, Step C
30 from the intermediate from the previous step (lS0 mg, 0.254 mmol) in
ethyl acetate (l0 mL) and HCl gas at 0C for 20 min11tes (l 18 mg, 90%).
FAB-MS calc. for C24H34N405S: 490; Found 491 (M+H~
wo 95113069 ~ 94/128l6
~ 2175218
- 279 -
EXAMPLE C6
H
~ ~NH2 HCI
~N C=O O
H ~N~
,OEt
,o,S~OO
Step A:
Boc
~N~
,OEt
,,S~
o u
To a stirred solution of Oxone (910 mg, 1.48 mmol) in water
(5 mL) and methanol (5 mL) was added the intermediate from Example
20 C4, Step A (300 mg, 0.99 mmol). The mixture was stirred for 4 hours at
room temperature, then concentrated to remove methanol. The residue
was then extracted with ethyl acetate (2 x 10 mL). The organic layer was
dried over m~gnesium sulfate and concentrated to give the title
compound (321 mg, 97%).
25 FAB-MS calc. for C14H25NO6S: 335; Found 336 (M+H) [Found 236
(M- t-Boc)]
StepB:
H HCI
~N~
,OEt
,oS~8
wo9S/13069 2 1 7 52 1 8 ~ 94,12816
- 280-
Prepared by the procedure described in Example C1, Step C
from the intermediate ifrom ~e previous step (221 mg, 0.66 mmol) in
ethyl acetate (10 mL) and HCl gas at 0C for 25 minutes (192 mg, 99%).
FAB-MS calc. for CgH17N04S: 235; Found 236 (M+H)
Step C:
¢~ ~NHBoc
H N~
~,OEt
,oS~8
Prepared by the procedure described in Example C1, Step D
from ~e intermediate prepared in the previous step (140 mg, 0.515
mmol), Tntetmediate 1 (l eq.), HOBT (1 eq.), N-methyl morpholine (1
eq.), and EDC (197 mg, 1.03 mmol.). Purification by MPLC provided a
20 diastereomeric mixture of compounds (251 mg, 80%).
FAB-MS calc. for C29H42N4OgS: 606; Found 607 (M+H)
Step D:
2 5 ~' ~NH2 HCI
H ~N~
,OEt
~''S``8
O
Prepared by the procedure described in Example Cl, Step C
from the intermediate from the previous step (210 mg, 0.317 mmol) in
wo 95113069 ~ 94/12816
i~ 2~75218
- 281 -
ethyl acetate (10 mL) and HCl gas at 0C for 30 minutes (193 mg,
98.5%) FAB-MS calc. for C24H34N4OgS: 506; Found 507 (M+H)
- FxAMpLE C7
~;~ N~NH2 HCI
H ~N~
~OEt
~ O
~N HCI
Step A:
Boc
fN~
~OEt
~ O
~N
2s To a stirred solution of ethyl N-t-Boc nipecotate (50 g, 0.196mol) in THF (600 mL) at -78C under argon was added KHMDS (0.5 M
in toluene, 298 mL, 0.298 mol) over a 30 minute period. The solution
was allowed to stir an additional 30 minutes at -78C. Meanwhile, a
suspension of 2-picolyl chloride hydrochloride (25 g) in dichloromethane
3 o was washed wi~ a mixture of 3 N NaOH and brine to remove the
hydrochloride. The organic layer was dried over MgSO4 and evaporated
to yield a brown oil and it was added slowly to the solution at -78C.
The reaction mixture was stirred overnight and allowed to warm to room
temperature. The material was concentrated, then diluted with water, and
extracted using ethyl acetate. The organic layer was dried over
wog5rl3069 2 1 152 1 8 ~ /US9~,l28l6
- 282 -
anhydrous magnesium sulfate, filtered, and concentrated. Purification by
silica gel flash column chromatography eluting with a solvent gradient of
20-80% ethyl acetate in hexane provided the title compound. (54.8 g,
80%).1H NMR (CD30D, 400MHz) ~ 8.45 (dd, J = 1.5 Hz, 5 Hz, l H),
7.52 (app dt, J = 2 Hz, 8 Hz, 1 H), 7.07 (dd, J = S Hz, 6.6 Hz, 1 H), 7.05
(d,J=8Hz, lH),4.09-4.04(br.m,2H),3.92(br.d, lH),3.46(br.m, 1
H), 3.30-3.10 (br. m, l H), 3.06 (d, J = 13.7 Hz, 1 ~I), 2.95 (d, J = 13. 7
Hz, 1 H), 2.01-1.91 (br. m, 1 H), 1.63-1.50 (br. m, 3 H), 1.36 (v. br. s, 9
H), 1.13 (t, 7.1 Hz, 3 H). FAB-MS calc. for C lgH2gN2O4: 348; Found
o 349 (M+H~
Step B:
H HCI
~N~
~OEt
~ O
~ NHCI
Prepared by the procedure described in Example C 1, Step C
from the intermediate from the previous step (6.36g, 18.2 mmol) in ethyl
~cet~te (100 mL) and HCl at 0C for 4~ minllte.s (6.10g, 100%.)
FAB-MS calc. for C14H20N2O2: 248; Found 249 (M~H)
25 Step C:
~ ~NHBoc
3~ H ~N~
~OEt
¢~
N
WO 95/13069 PCI~/US94/12816
2175218
- 283 -
Prepared by the procedure described in Example C 1, Step D
from the compound prepared in the previous step (500 mg, 1.556 mmol),
Intermediate 1(1 eq.), HOBT (1 eq.), N-methyl morpholine (2 eq.), and
EDC (597 mg, 3.11 mmol). Purification by MPLC eluting with ethyl
acetate provided the title compound (883 mg, 91.5%).
FAB-MS calc. for C34H45N5O6: 619; Found 620 (M+H)
Step D:
~NH2 HCI
H ~N~
~OEt
~/ o
~N HCI
Prepared by the procedure described in Example C1, Step C
20 from the intermediate from the previous step (250 mg, 0.404 mmol) in
ethyl acetate (25 mL) and HCl gas at 0C for 25 minutes (204 mg, 85%)
FAB-MS calc. for C29H37N5O4: 519; Found 520 (M+H)
The additional intermediates shown in Table CIV were
prepared according to the above established procedure as exemplified in
25 Example C7 step A and B. The final compounds were prepared
according to Example C7 Steps C and D, using Intermediate 1.
TABLE CIV
¢~`,r'=O O
~CO2Et ~CO2Et
Intermediate Product
wo 9S113069 2 1 7 5 2 1 8 ~ 9~1.28~
- 284 -
.
Intermediate Product
entry Y MF MF isomer
FAB-MS (M+l) FAB-MS (M+l)
1 3-picolyl C14H20N202 C29H37N504 RS
2 4-picolyl C 14H20N202 C29H37N504 RS
3 2-quinoline- ClgH22N202 C33H39N504 RS
me~yl 298 569
4 N C16H21N302 C3 lH38N604 dl
,~ CH2- 288 d2
NJ3~CH ClgH25N302 C34H42N604 RS
6 [~CH3 C15H22N202 5330H39N504 RS
N CH2-
The additional compounds shown in Table CIVa were
20 prepared according to Example C7 Steps C and D, using some of ~e
interrnediates shown in Table CIV and Interrnediate 3 instead of
Interrnediate 1.
TABLE CIVa: ADDITIONAL EXAMPLES
Ph~ ~NH2 HCI
C=O O
3 o ~<CO2Et
Y
entry Y MF isomer
FAB-MS (M+l)
PCI/US94/12816
WO95/13069 2 1 7 5 2 1 8
- 285 -
0 1 2-picolyl C29H40N404 ddl
2 3-picolyl C29H40N404 d2
3 4-picolyl C29H40N404 d2
4 ~ C33H42N404 RS
H C3 lH41N504 dl
o 5 ~ /~ CH2- d2
6 ~CH3 C30H42N404 dl
~ ~ 523,545(M+Na) d2
N CH2-
EXAMPLE C8
H
2 0 ~ ~NH2 HCI
H ~N~
~ "~OEt
~f O
I~N HCI
WO 95/13069 PCI'IUS9~112816
2~75218 ~
- 286 -
Step A:
OAc OAc
~ ~
¢~ ~ ~OEt
Prepared by the procedure described in Fx~n~r)le Cl, Step D
from the intermediate (6g, 18.67 mmol) prepared in Example C7, Step B,
and using (R)-(-)-(O)-acetyl m~n~lelic acid (l eq.), HOBT (1 eq.), N-
methyl morpholine (2 eq.), and EDC (7.16 g, 37.34 mmol). Purification
5 by MPLC eluting with 80% ethyl acetate in hexane provided two
enantiomerically pure compounds. The isomer which came out of the
column ~lrst was designated as dl (3.92 g, 99%) and the isomer which
came out of the colurnn second as d2 (3.69 g, 93%)
FA13-MS calc. forC24H2gN2Os Found425. The structureof
20 intermediate dl was determined by x-ray crystallography. Given the
absolute stereochemistry of (R) -O- acetylmandelic acid, the
stereochemistry at the piperidine 3-position was ~si~ned (S)- in dl.
Step B:
H HCI
~N~
~
The intermediate dl from the previous step (2.91g, 6.86
mmol) in ethanol (30 mL) and concentrated HCl (25 m~ ) was refluxed
for five hours. The reaction mixture was evaporated in vacuo and the
wo95rl3069 2 1 752 1 8 ~ 94/12816
- 287 -
residue was purified by silica gel flash column chromatography eluting
with a solvent gradient of 1:10:90 to 2:20:80 ammonium
hydroxide:methanol:chloroform to provide the compound (dl, 1.52g,
70%). lH NMR (CD30D, 400MHz) o 8.84 (app. d, J = 6 Hz, 1 H), 8.60
(app.dt,J=1.5Hz,8Hz, lH),8.04(t,J=6Hz, lH),7.94(d,J=8Hz,
1 H), 4.34-4.27 (m, 1 H), 4.23-4.17 (m, 1 H), 3.75 (d, J = 13 Hz, 1 H),
3.46 (d, J = 13.3 Hz, 1 H), 3.40 (d, J = 13.3 Hz, 1 H), 3.31-3.29 (m, 2 H),
3.20 (d, J = 13 Hz, 1 H), 3.03 (app dt, J = 3.1 Hz, 12.8 Hz, 1 H), 2.24 (br.
d, 1 H), 2.00-1.93 (m, 1 H), 1.88 (dd, J = 3.7 Hz, 13.5 Hz, 1 H), 1.63-1.60
0 (m, 1 H), 1.23 (t, 7.1 Hz, 3 H). FAB-MS calc. for C14H20N2o2: 248;
Found 249 (M+H)
Step C:
~,NHBoc
H ~N~
~ ",~OEt
[~ O
N
Prepared by the procedure described in Example C 1, Step D
from the intermediate prepared in Step B of this example (dl, 1.50g, 4.67
25 mmol), N-t-Boc-D-Trp (1 eq.), HOBT (1 eq.), and EDC (1.53g, 8.00
mmol). Purification by MPLC eluting with ethyl acetate provided the
title compound (1.764 g, 71%). FAB-MS calc. for C30H3gN4O5: 534;
Found 535 (M+H)
WO9S113069 2 ~ 752 ~ 8 ~ u~g4~l28~6
- 288 -
Step D:
~, NH2 HCI
H ~N~
~ "~OEt
~0
~ N HCI
Prepared by the procedure described in Example C3, Step C
from the intermediate from the previous step (1.658 g, 3.11 mmol) in
e~yl acetate (50 mL) and HCl gas at 0C for 35 minutes (1.56 g, 99%).
FAB-MS calc. for C25H30N403:434; Found 435 (M+H)
Step E:
~ ~NHBoc
2 o N q
H ~N~
~ "~OEt
~ O
~N
Prepared by the procedure described in Example C1, Step D
from the intermediate prepared in Step D of this example (1.5 g, 2.96
mmol), N-t-Boc-a-methyl~l~nine (l.l eq.) DMAP (0.15 eq.), N-methyl
30 morpholine (1 eq.), and EDC (1.135 g, 5.92 mmol). Purification by
MPLC provided the title compound. (1 .488g, 81 %)
FAB-MS calc. for C34H4sNsO6: 619; Found 620 (M+H)
Wo 95/13069 2 1 7 5 2 1 8 PCI~/US9~112816
- 289 -
, StepF:
~7~C=O O
H ~N~
~/~""~OEt
~0
~N
Prepared by the procedure described in Example Cl, Step C
from the intermediate from Step E (1.40 g,2.26 mmol) in ethyl acetate
(100 mL) and HCl gas at 0C for 1 hour (1.388 g, 100%).
lH NMR (CD30D,400 MHz):8.79-8.78 (M, lH), 8.56-8.48 (M, 24),
8.0-7.96 (M, lH),7.72 (d, J=8.21 Hz, lH) 7.53 (d, J=7.98, Hz, lH) 7.25-
7.22 (M, 2H) 6.89-6.86 (M, lH) 5.48-5.43 (M, lH) 3.89 (1, J=7.1 Hz,
2H) 2.30 (d, J=14.3 Hz, lH) 1.85 (d, J=14.4 Hz, lH) 1.01 (t, J=7.1 Hz,
3H) FAB-MS calc. for C29H37N5O4: 519; Found 520 (M+H)
EXAMPLE C9
H
~ ~NH2 HCI
H ~N~
,OEt
~ O
~N
The title compound was similarly prepared from the
intermediate d2 from Example C8, Step A. FAB-MS calc. for
C29H37N5O4: 519; Found 520 (M+H)
WO 95J~3069 ~ 94112816
2~75218 ~
- 290 -
EXAMPLE C10 J
s ~N I ~NH2HCI
H ~N~
~OBn
~' O
Step A:
Boc
~OH
To a stirred solution of nipecotic acid (5 g, 38.7 rnrnol) in
NaOH (2 eq.) in water was added di-tert-butyl dicarbonate (10 g, 46.44
mrnol). The mixture was stirred at room temperature for 2 days. The
mixture was ~en slowly acidified to pH=3 and s~irred for two hours. The
solution was extracted with ethyl acetate, dried, and concentrated to give
white solid (6.25 g, 70%).
Step B:
~N~
~fOBn
O
To a solution of the intermediate from the previous step
(6.25 g, 27.3 rnmol), benzyl alcohol (3.4 mL, 32.7 mmol) and DMAP (33
mg, 0.273 mmol) in dichloromethane at 0C, was added EDC (6.9 g, 35.4
mmol). The reaction mixture was stirred at room temperature for 7
WO 95/13069 ~ g4/12816
2175218
- 291 -
hours. It was washed with a mixture of brine and 3 N HCl, dried over
anhydrous magnesium sulfate, filtered and concentrated. Purification by
silica gel flash column eluting with a gradient of 10-30% ethyl acetate in
hexane provided the benzyl ester (7.41 g,85%).
Step C:
Boc
~N~
o ~OBn
~/0
P-~ared by the procedure described in Fx~mple C1, Step B
5 from benzyl N-t-Boc-nipecotate (7.12 g, 22.2 mmol), LHMDS in THF
(33.3 mL, 33.3 mmol) and benzyl bromide (4.0 g, 33.3 mmol).
Purification by silica gel flash column chromatography eluting with 5-
20% ethyl acetate in hexane provided the title compound. (9.10 g, 100%)
lH NMR (CDCl3, 400MHz) ~ 7.33-7.28 (m, 3 H), 7.23-7.17 (m, 5 H),
20 7.01-6.98 (m, 2 H), 5.00 (br. ABq, JAB = 12 Hz, 2 H), 4.00 (br. s, 1 H),
3.55-3.50 (m, 1 H), 3.18 (d, J = 13 Hz) 3.14 (v. br. s, 1 H), 2.92 (d, J =
13.5 Hz), 2.74 (d, J = 13.4 Hz), 2.03-1.99 (m, 1 H), 1.63-1.50 (m, 3H),
1.39 (s, 9 H).
25 Step D:
H HCI
~N~
OBn
~/ O
r
Prepared by the procedure described in Example C1, Step C
from the intermediate from the previous step (3.08 g, 7.52 mmol) in ethyl
~ et~te (40 mL) and HCl gas at 0C for 15 minutes (2.65 g, 100%).
WO 9S/13069 ~ 12816
2~ 752~8
- 292 -
FAB-MS calc. for C20H23No2: 309; Found 310 (M+H)
Step 3~:
¢~ ~NHBoc
H ~N~
o ~OBn
3/o
Prepared by ~e procedure described in F.x~n~rle Cl, Step D
15 from the intermP~ te prepared in the previous step (768 mg, 2.22 mmol),
Interrnediate 1 (720 mg, 1.85 mmol), HOBT (1 eq.), N-methyl
morpholine (1 eq.), and EDC (2 eq.). Purification by MPLC, eluting with
50% ethyl acetate in hexane, provided two diastereomers. The isomer
which carne out first was desi~n~te(l as dl (504 mg, 40%) and ~e one
20 which eluted second was de~i~n~te~l as d2 (474 mg, 38%)
dl FAB-MS calc. for C40H4gN4O6: 680; Found 681 (M+H)
d2 FAB-MS calc. for C40H4gN4O6: 680; Found 681 (M+H)
Step F:
~NH2 HCI
H ~N~ t
3 ~,OBn
~' O
WO 9S/13069 1 ~ )S94112816
2175218
- 293 -
Prepared by the procedure described in Example Cl, Step C
- from the intermediate dl from Step E (25 mg, 0.036 mmol) in ethyl
acetate (3 mL) and HCl gas at 0C for 10 minutes (20.2 mg, 91%).
FAB-MS calc. for C35H40N4O4: 580; Found 581 (M+H)
EXAMPLE Cl 1
~ ~NH2 HCI
H ~N~
""~OBn
¢~ O
Prepared by the procedure described in Example C 1, Step C
from the intermediate d2 (20.1 mg, 0.03 mmol) of F.x~mple C10, Step E
in ethyl acetate (3 mL) and HCl gas at 0C for 10 min~ltes (12.8 mg,
20 70%).
FAB-MS calc. for C35H40N4O4: 580; Found 581 (M~H)
EXAMPLE C12
H
¢~ ~NH2 HCI
H ~N~
3 I,OH
r ~/ O
.~ ~
Step A:
WO 95/13069 1~ 94/12816
2175218 ~--
- 294 -
~ 3~ N ~NHBoc
H ~N~
~OH
A suspension of 10% palladium on carbon (60 mg) and ~e
int~diate (dl) from Fx~mple C10, Step E (442.6 mg, 0.65 mmol) in
ethanol (20 rnL) was vigorously stirred under a hydrogen atrnosphere for
30 minutes. The reaction mixture was then filtered through celite and
evaporated to give the product (376.0 mg, 98%).
5 dl FAB-MS calc. for C33H42N406: 590; Found 591 (M+H)
Step B:
~~r ~NH2 HCI
H ~N~
~,OH
¢~ O
Prepared by the procedure described in Example Cl, Step C
from ~e intermediate from the previous step (211 mg, 0.357 mmol) and
30 HCl gas in ethyl acetate (15 mL) at 0C for 10 minutes (175.6 mg, 93%).
lH NMR (CD30D, 400MHz): The compound exists in two rotamers in
approximately a 1: 1 ratio. ~ 7.~7-7.54 (m, 1 H), 7.38 (d, J = 8.2 Hz, 1/2
H), 7.33 (d, J = 8.2 Hz, 1/2 H), 7.25-7.00 (m, 8 H), 6.81-6.79 (m, 1 H),
5.36 (dd, J = 6 Hz, 8.5 Hz, 1/2 H), 5.18 (app t, J = 7.5 Hz, 1/2 H), 4.32
(br.d,J= 13Hz, 1/2H),4.00(br.d,J= 13Hz, 1/2H),3.78(br.d,J- 13
WO 9!!itl3069 ~ 94/12816
2175218
- 295 -
Hz, 1/2 H), 3.26-3.02 (m, 11/2 H), 2.86 (d, J = 13.4 Hz, 1/2 H), 2.80 (d, J
= 13.4 Hz, 1/2 H), 2.53 (d, J = 13.4 Hz, 1/2 H), 2.46 (d, J = 13.4 Hz, 1/2
H), 2.29 (dt, 1/2 H), 2.09 (d, J = 12.7 Hz, 1/2 H), 1.92-1.88 (m, 1/2 H),
l.S5, 1.50, 1.44 (3s, 6 H), 1.40-1.25 (m, 1 H), 1.20-1.12 (m,l/2 H).
dl FAB-MS calc. for C2gH34N4O4: 490; Found 491 (M+H)
EXAMPLE C13
~ ~NH2 HCI
H ~N~
."~OH
~ O
Step A:
~1'=
H ~N~
~.",~OH
¢~ O
Prepared simil~rly from the intermediate d2 (224.2 mg, 0.33
mmol) from Example C10, Step E (169.3 mg, 87%).
d2 FAB-MS calc. for C33H42N4O6: 590; Found 591 (M+H)
wo ssrl3069 ~ 4ll28l6
2t752~8
- - 296 -
Step B:
~NH2 HCI
H ~N~
~ "~OH
~ O
~
Prepared by the procedure described in Example Cl, Step C
from the intermediate from the previous step (139 mg, 0.235 mmol)) and
HCl gas in e~yl ~et~te (15 mL) at 0C for 10 minutes (122.7 mg, 99%).
H NMR (CD30D, 400MHz): The compound exists as two rotamers in
approximately a 1:1 ratio. ~ 8.21 (d, J = 7.4 Hz, 1/2 H), 7.91 (d, J - 7.4
Hz, 1/2H),7.62(d,J=7.9Hz, 1/2H),7.50(d,J=7.9Hz, 1/2H),7.34-
6.90 (m, 9 H), 5.40-5.34 (m, 1 H), 4.40 (d, J = 13.7 Hz, 1/2 H), 4.13 (d, J
= 126Hz, 1/2H),3.63 (d,J= 13.3Hz, 1/2H),3.50(d,J= 13.3Hz, 1/2
20 H), 3-30-3.10 (m, 7/2 H), 2.93 (ABq, lH), 2.88 (v. br. d, 1/2 H), 2.60 (d, J
= 13 Hz, 1/2 H), 2.40 (d, J = 13 Hz, 1/2 H), 2.19-2.16 (m, 1/2 H), 1.78-
1.75 (m, 1 H), 1.60-1.40 (m, 3/2 H), 1.20-1.10 (m, 1/2 H), 1.58, 1.50,
1.47, 1.15 (4s, 6 H), 1.00-0.90 ~m, 1/2 H).
d2 FAB-MS calc. for C2gH34N4O4: 490; Found 491 (M+H)
The additional examples shown in Table CV were prepared
according to Examples C10 through C12 using Inte~nediate 3 and the
intermediate obtained in Example C10 Step D.
pCI'IUS94112816
wo951l3069 2 1 7 5 2 1 8
- 297 -
BLE C~ ADDITlO~
Ph ~N~NH2 HC~
C=O
~E~n
=~
entry X FAB-~S (~1~
C35H43N304 l~S
co2Bn 570
C28H37N304 E~S
2 Co2H 480
FX~.MPL~ C14
W~
H ,l`J~ o
~OEt
BNoc
~OEt
,~
W
added to a s~ed~ ~
wo 95/13069 ~ 4112816
2 1 752 1 8
- 298 -
separate flask, a stirred mixture of ethyl N-t-Boc-3-benzyl-nipecotate
(Example Cl, Step B, 1.73 g, 5 mmol) and CH2Br2 (0.78 mL, 2.15 g,
12.4 mmol) in THF (20 mL) was cooled to -78C, and the T ithillm salt
solution of TMP solution just prepared was then added over a 15 minllte
5 period at a tempelatule below -65C. After 10 minllte, a solution of
LHMDS (11.2 mL, 11.2 mmol) was added over a 10 minlltes period at
-78C. Following the addition, the cooling bath was removed and the
mixture was allowed to warm gradually to 0C. The mixture was cooled
with an ice bath, and a solution of n-BuLi in hexane (13.5 mL, 33.7
rnmol) was added at a temperature below 5C over a 15 min~ltes period.
The mixture was warmed to room tempeldl~le and stirred for 45 mimltes.
The mixture was cooled to -78C and quenched over a 50 minllte period
by ~ 1ing it into a stirred solution of acidic ethanol (30 mL) at 0C. The
mixture was evaporated to dryness and suspended in dichlorometh~ne
5 (100 mL), to which was added triethyl~mine (0.7 mL, 5.0 mmol) and di-
tert-butyl dicarbonate (1.09 g, 5.0 mmol) while stirrin.~. After 1 hour of
stirring at room temperature, ~e material was washed with brine, dried,
and concentrated. Purification by silica gel flash column
chromatography, eluting with 10-30% ethyl acetate in hexane, provided
20 ~e compound (1.44 g, 80%).
Step B:
H HCI
~
OEt
3 Prepared by the procedure described in Example Cl, Step C,
from the inte.rmediate from the previous step (1.30 g, 3.56 mmol) and
HCl gas in ethyl acetate (50 mL) at 0C for 4S minutes (975 mg, 91%).
FAB-MS calc. for C16H23N02: 261; Found 262 (M+H)
PCI~/US94/12816
WO 95/13069
~ 2~752~8
- 299 -
" Step C:
~NHBoc
H ~N~ Q
--Jl`OEt
~
Prepared by the procedure described in Example C 1, Step D
from the intermediate prepared in the previous step (55 mg, 0.21 mmol),
Intermediate 1 (1 eq.), HOBT (1 eq.), N-methyl morpholine (1 eq.), and
EDC (80 mg, 0.42 mmol). Purification by MPLC eluting with 60% ethyl
acetate in hexane provided the compound (77 mg, 61.5%).
Step E:
~,~O O
H ~N~ O
~OEt
3/
Prepared by the procedure described in Example Cl, Step C
from the intermediate from the previous step (77 mg, 0.13 mmol) and
HCl gas in ethyl acetate (8 mL) at 0C for 15 minutes (59 mg, 85%).
FAB-MS calc. for C3 lH40N4O4: 532; Found 533 (M+H)
PCr~lJS9~12816
~vo95113069 2~75218
300 -
EXA~PL~ C~5
~NH2 HCI
H ~ ~
~ NHEt
BNoc
~OH
¢~
2s 3 15 3 05 Cbr m~ 2 H)i H); 1 70-1-45 (m~ 3 H)~ 1 42 (s~ 9
f ~ C18H25No4: 319; ~ound 319 (
3 0 BNoc
~ ~ NHEt
¢~
WO 95/13069 1~ JSg4/12816
~ 2175218
- 301 -
To a solution of the intermediate from the previous step
(320.4 mg, 1.0 mmol) in dichloromethane cont~inin~ ethyl amine
hydrochloride (163 mg, 2.0 mmol), DMAP (1.0 eq.), and N-methyl
morpholine (2 eq.), was added EDC (2 eq.). The reaction mixture was
5 stirred at room temperature overnight. The solution was washed with 3 N
HCl and brine, dried over anhydrous magnesium sulfate, then filtered and
concentrated. Puri~lcation by silica gel flash column eluting with a
gradient of 60-100% ethyl acetate in hexane provided the title compound
(262 mg, 76%). lH NMR (CDCl3, 400 MHz) ~ 7.21-7.13 (m, 3 H), 7.03
(d, 2 H), 6.68 (br. s, 1 H), 4.18 (br. d, 1 H), 3.96 (br. d, 1 H), 3.12-3.00
(m, 4 H), 2.70-2.40 (br. m, 5 H), 1.60-1.50 (m, 1 H), 1.37 (s, 9 H), 1.20-
1.30 (m, lH), 0.90 (q, J = 7.3 Hz, 3 H). EI-MS calc. for C20H30N23:
346; Found 346 (M+)
5 Step C:
H HCI
~N~
NHEt
~/ ll
Prepared by the procedure described in Example C1, Step C
from the intermediate from the previous step (262 mg, 0.76 mmol) and
25 HCl gas in ethyl acetate (5 mL) at 0C for 1 hour (194 mg, 90%).
lH NMR (CD30D, 400MHz) ~ 8.28 (br. s, 1 H), 7.30-7.24 (m, 3 H),
7.14-7.12 (m, 2 H), 3.43 (d, J = 12 Hz, 1 H), 3.34-3.28 (m, 2 H), 3.26-
3.20 (br. d, 1 H), 3.11 (d, J = 14 Hz, 1 H), 2.88 (dt, J = 3.2 Hz, 13 Hz,
lH),2.81(d,J=12.5Hz,lH),2.77(d,J=14Hz,lH),2.24(d,J=13
30 Hz, 1 H), 1.87 (td, J = 2.8 Hz, 14 Hz, 1 H), 1.75 (dt, J = 3.3 Hz, 13.5 Hz,
1 H), 1.64-1.55 (m, 1 H), 1.17 (t, J = 7 Hz, 3 H).
u~9~ll28l6
WO 95/13069 2 1 7 5 2 1 8 ~1
- 302 -
Step D:
¢$~ ~NHBoc
H ~N~
~,NI IEt
¢~/
Prepared by the procedure described in Example Cl, Step D
from interm~ te prepared in the previous step (62.2 mg, 0.22 mmol),
Intermediate 1 (1 eq.), HOBT (1 eq.), N-methyl molpholine (1 eq.), and
EDC (2 eq.). Purification by MPLC eluting with e~yl acetate provided
two diastereomers, the one which was eluted out of the column first was
desi~n~tetl as dl (35.8 mg, 26%) and the one came out second was
desi~n~t~d as d2 (43.8 mg, 31%).
d2. lH NMR (CD30D, 400 MHz): The compound exists in two
20 rotamers in approxim~tely a 1:1 ratio. ~ 8.16 (br. s, 1/2 H), 7.53 (d, J =
8.7 Hz, l H), 7.32 (d, J = 8.1 Hz, l H), 7.25-6.96 (m, 8 H), 6.69 (br. s, 1/2
H), 5.28-5.12 (m, 1/2 H), 4.94 (v. br. m, 1/2 H), 4.31 (br. d, J = 14.6 Hz,
1/2H),3.49(v.br.d,J= 13Hz, 1/2H),3.22(dd,J=4.7Hz, 14.3Hz, 1/2
H), 3.03-2.97 (m, 2 H), 2.90 (d, J = 13.4 Hz, 1/2 H), 2.40 (br. d, 1/2 H),
25 2.36 (d, J = 13.3 Hz, 1/2 H), 2.10 (d, J = 13.5 Hz, 1/2 H), 1.92-1.82 (br.
m, 3/2 H), 1.47 (s, 3 H), 1.41 (s, 9 H), 1.38 (s, 3 H), 1.32-1.20 (m), 1.10-
1.00 (dt, 1/2 H), 0.85 (t, J = 7.2 Hz, 3 H).
dl FAB-MS calc. for C35H47N505: 617; Found 618 (M+H)
d2 FAB-MS calc. for C35H47N505: 617; Found 618 (M+H)
pC~russ4ll28l6
wo 9sll3069 2 1 7 5 2 1 ~
- 303 -
~';
~NHEt
,.5 100'Y). ~C
E~.XA~lPLE C16
/~CHz HCI
~/ ."~ Nl lEt
2s ¢~ O
ocedure descnbed in ~xatnple 0 066
oC~65
tamers in ap~ 7 ~ J = 7.8 HZ- 115 HH' i/5 Hj, 7.26-6 98 (In~
WO 95113069 PCI~/US94/12816
2175218
- 304 -
8 H), 5.46-5.40 (m, 1/5 H), 5.25-5.20 (m, 4/5 H), 4.00 (br. d, 4/5 H), 3.85
(br. d, 1/5 H), 3.65 (br. d, J = 13.2 Hz, 4/5 H), 3.60-3.54 (m, 1/5 H), 3.36
(br. d, 1/5 H), 3.30-3.03 (m), 2.99-2.90 (m), 2.82-2.62 (m), 2.46 (d, J =
13.3 Hz, 8/5 H), 2.08 (br. d, 4/5 H), 1.90-1.84 (m, 1/5 H), 1.76-1.65 (m),-
1.51, 1.49 (2s, 6 H), 1.40-1.20 (m), 1.00 (t, J = 7.2 Hz, 3/5 H), 0.88 (t, J =
7.2 Hz, 12/5 H). FAB-MS calc. for C30H39N5O3: 517; Found 518
(M+H)
E~XAMPLE C 17
~NH2 HCI
C=O O
~N~
¢~ ."~ NHEt
Step A
Boc
~N~
"~OH
~ O
~
To a suspension of the S- isomer intermediate of Step A of
Example C2 (27.3 g, 68.8 mmol) in 3 N sodium hydroxide (25 mL),
dichloromethane (200 mL) and water (100 mL), was slowly added di-t-
30 butyl dicarbonate (18 g, 1.2 equiv.). The mixture was stirred for anadditional 5 hours after the addition, it was acidified to pH 3 carefully
and then extracted with ethyl acetate three times. The organic extracts
were combined, dried, and concentrated to give a white solid (23.7 g).
WO 95113069 PCrlUS9~/12816
~ 21752~8
- 305 -
A solution of this intermediate (11.5 g, 33.l mmol) and 3 N NaOH (30
mL) in ethanol (200 mL) and water (10 mL) was refluxed for one day.
The solution was evaporated to remove ethanol, and then acidi~led with 3
N HCl to pH=3 and extracted wi~ ethyl acetate. The extract was dried,
evaporated and purified by a short silica gel column, initially eluting with
20% ethyl acetate in hexane, then with ethyl acetate to give the product
(8.76 g, 83%). NMR and MS were identical to Example C15 stepA.
Step B
Boc
~N~
"~NHEt
O
To a mixture of the interrnediate from the previous step (660
mg, 2.07 mmol), ethylamine hydrochloride (251 mg, 1.5 equiv.), NMM
(0.23 mL, 1 equiv.) and HOBT (1 eq) in dichloromethane and DMF (l:l,
l0 mL) was added EDC. The mixture was stirred at room temperature
for two days, heated at reflux for 2 hours, and was poured into a dilute
HCl and brine mixture. It was extracted with ethyl acetate, and the
organic layer was washed with dilute NaOH, dried and evaporated.
Purification by flash column eluting with 20-80% ethyl acetate in hexane
gave the product (540 mg, 75%). NMR and MS were identical to
Example C15 Step B.
Step C
H HCI
N~
f
",~ NHEt
~ O
WO 95tl3069 PCI'/US94112816
2~ 752~8
- 306-
Prepared by the procedure described in Example Cl, Step C
from the intermediate from the previous step ( 0.33 g, 0.95 mmol) in
ethyl acetate (5 mL) and HCl gas at 0C for 15 minutes (0.279 mg,
100%). FAB-MS calc. for C15H22N2O: 246; Found 247 (M+H)
Step D
H \/
~\'--~ ~ NHBoc
Ç=O O
o ~N~
~ "~NHEt
¢~/ O
15 Prepared by the procedure described in Example Cl, Step D
from the intermediate prepared in the previous step (100 mg, 0.354
mmol), Intermediate 3 (134 mg, 0.354 mmol), HOBT (48 mg, 1 eq.), N-
methyl morpholine (0.039 mL, 1 eq.), and EDC (102 mg, 1.5 eq.).
Purification by MPLC, eluting with ethyl acetate, provided the
20 intermediate (140 mg, 65%). FAB-MS calc. for C35H50N4O5: 606;
Found 607 (M+H)
~tep E H
~ ~NH2 HCI
C,=O O
~N~
",~NHEt
~0
Prepared by the procedure described in Example Cl, Step C
from ~e intermediate from ~e previous step (132 mg, 0.217 mmol) and
HCl gas in ethyl acetate (5 mL) at 0C for 10 minlltes (113.3 mg, 96%).
WO 95/13069 PCI'IUS94/12816
~ 2~75218
- 307 -
dl FAB-MS calc. ~or C30H42N403: 506, Found: 507 (M+H)
The additional irltermediates shown in Table CVIa were
prepared according to the above established procedure as exempli~led in
5 Example Cl5, and Example C17 steps A through C,. The final
compounds were ~lc~ d according to Example C17 Steps D and E,
using Tnterm~diate 1.
TABLE CVIa: ADDITIONAL EXAMPLES
H W~l=
~)<B~ [~<X
Intermediate Product
Intermediate Product
entry X MF MF isomer
FAB-MS (M+l) FAB-MS (M+l)
-CO(morpholino) C17H24N202 C32H41N504 S
288 (M+, EI) 560
2 -CONHCH3 C14H20N20 C29H37N503 S
233 504
3 -CONH- C17H24N203 C32H41N50s S
CH2C02Et 304 (M+, EI) 576
4 -C02CH2C02Et C17H23N4 C32H40N406 R
306 577 S
-C02(CH2)2SMe Cl6H23No2s C31H40N404S R
294 565 S
6 -CON(CH3)2 C15H22N20 C30H39N503 dl
247 518 d2
7 -CONH- C15H22N202 C30H39N504 S
(cH2)2oH 263 534
WO95113069 2 1 752 1 8 ~ u~94~12816
- 308 -
Likewise using 3-aminopropanol or 2-(ethylthio)ethylamine
it is possible to prepare the compounds shown in Table CVIb using
Intermediate 1.
TABLE CVIb
;;3~N~NH
H ~N~
~/""X
entry X
-CONH(CH2)30H
2 -coNHcH2cH2scH3
The additional compounds shown in Table CVIc were
prepared according to Examplel7 Steps C and D, using some of the
intermediates shown in Table CVIa and Intermediate 3.
TABLE CVIc: ADDITIONAL EXAMPLES
Ph~ ~NH2 HCI
C,=O O
~N~
'--X
entry Y MF isomer
FAB-MS (M+l)
wo 95113069 ~ 94112816
~ ' 2175~18
- 309 -
-CO(morpholino) C32H44N404 S
549
2 -CONHCH3 C29H40N4O3 S
493
3 -CONH- C32H44N405 S
CH2CO2Et 565
4 -CONH- C30H42N404 S
(CH2)2OH 523
0Likewise using 3-aminopropanol and 2-(methylthio)-
ethylamine it is possible to prepare the compounds shown in Table CVId.
TABLE CVId
H ~
15 ~~~ ~ NH2
C=O O
~N~
~/""X
¢~
entry X
-CONH(CH2)30H
2 52 -CONHCH2CH2SCH3
The additional compounds shown in Table CVIe were
prepared according to Examplel7 Steps C and D, using some of the
intermediates shown in Table CVI and Intermediate 2.
WO9Stl3069 2 ~ 752 ~ 8 1~ 9~/12816
- 310-
TABLE CVIe: ADDITIONAL EXAMPLES
Ph~O~N~NH2 HCI
C,=O O
X
~
entry Y MF isomer FAB-MS (M+l)
-CO(morpholino) C31H42N40s S
2 -CONHCH3 C28H38N404 S
495
3 -CONH- C3 1 H42N406 S
CH2C02Et 567
EXAMPLE C18
¢~1 =
H ,N~
~,OH
~ .,
W095/13069 2 1 7 5 PCrr[JS94112816
- 311 -
Step A:
Boc
~N~
OH
To a stirred solution of ethyl N-t-Boc-3-phenylmethyl
nipecotate (10.4 g, 29.93 mmol) in dichloromethane (100 mL) at -78C
was added DIBAL (lM, 45 mL). The reaction was stirred at -78C for 4
hours and quenched by the addition of methanol (5 mL). The reaction
mixture was washed carefully with tartaric acid water solution and brine,
dried over MgSO4 and evaporated. Silica gel flash chromatography
15 elu~ing with a gradient of 40-80% ethyl acetate in hexane yielded the
product (6.81 g, 75%).
EI-MS calc. for ClgH27NO3: 305; Found 305 (M+)
Step B: 3-Phenylmethyl-3-piperidinemethanol hydrochloride
H HCI
~N~
~,OH
~
A solution of the intermediate from the previous step (770
mg, 2.52 mmol) in ethanol (20 mL) and concentrated HCl (1 mL) was
refluxed for 3 hours. The reaction mixture was cooled to room
30 temperature and evaporated to give the title compound as a white solid.
(609.0 mg, 100%)
lH NMR (CD30D, 400 MHz) ~ 7.31-7.19 (m, 5 H), 3.45 (ABq, J = 11
Hz, 2 H), 3.18 (d, J = 13 Hz, 1 H), 3.19-3.13 (m, 1 H), 3.03-2.99 (m, 1
H), 2.96 (d, J = 13 Hz, 1 H), 2.72 (s, 1 H), 1.92-1.84 (m, 2 H), 1.60-1.50
(m, 2 H).
wo gS/13069 2 ~ ~ 5 2 PCr~US9411281~
- 312 -
EI-MS calc. forC12H17NO: 191; Found 191 (M+,)
StepC:
¢~~r ~NHBoc
H ~N~
1o OH
[3/
Prepared by the procedure described in Example Cl, Step D
15 from the intermediate prepared in the previous step (142 mg, 0.587
mmol), Intermediate 1 (0.8 eq.), HOBT (1 eq.), N-methyl morpholine (1
eq.), and EDC (2 eq.). Purification by MPLC eluting with ethyl acetate
gave two compounds; the compound which came out of the column first
is designated as dl (98.5 mg, 58%) and the compound which came out of
20 the column next as d2 (34.5 mg, 12%)
dl F~B-MS calc. for C32H42N405: 562; Found 563 (M+H)
d2 FAB-MS calc. for C32H42N405: 562; Found 563 (M+H)
~tep D:
H
~NH2 HCI
H ~N~
3 o l ,OH
-
WO 95/13069 2 1 7 5 2 1 8 PCI~/US94/12816
- 313 -
The intermediate (dl) from the previous step (60 mg, 0.104
mmol) was treated with HCl gas at 0C in ethyl acetate (3 mL) for five
minlltes. Evaporation gave the diastereomer 1 of the title compound.
dl FAB-MS calc. for C2gH36N4O3: 476; Found 477 (M+H)
The intermediate (d2) from Step C (20 mg) was treated with
HCl gas at 0C in ethyl acetate (3 mL) for five minutes. Evaporation
gave the diastereomer 2 of the title compound.
d2 FAB-MS calc. for C2gH36N4O3: 476; Found 477 (M+H)
o EXAMPLE Cl9
~ ~ ~NH2 HCi
H~N~
H
, N `S~
~ 0""0
Step A:
iNoc
~ `
N3
.. ~
3 0 To a stirred solution of intermediate from Example C 18,
Step A (5.12 g, 16.8 mmol), and triethylamine (4.7 mL) in
dichloromethane at 0C was added mesyl chloride (1.95 mL). The
reaction mixture was stirred for 2 hours. The solution was poured into a
mixture of brine and 3 N HCl and extracted with ethyl acetate. The
organic layer was washed with saturated sodium bicarbonate, dried over
WO 9Stl3069 2 1 7 5 2 1 8 PCI'IUS9~1/1281~
- 314 -
magnesium sulfate and evaporated to yield the mesylate. The mesylate
was heated with sodium azide (2.2 g, 33.6 mmol) in DMSO (20 mL) at
80C for two weeks. The mixture was poured into ice water and
extracted with ethyl acetate. The organic layer was washed with
S saturated sodium bicarbonate and brine; it was dried, and evaporated.
Purification by silica gel flash colurnn chromatography provided the
azide (4.14 g, 75%).
lH NMR (CDC13, 200 MHz) o 7.29-7.13 (m, 5 H), 3.61-3.57 (br. m, 1
H), 3.47 (d, J - 12 Hz, 1 H), 3.20-3.10 (v. br. s, 2 H), 3.10-2.96 (v. br. d,
o 1 H), 2.60-2.45 (br. m, 2 H), 1.65-i.48 (m, 4 H), 1.44 (s, 9 H), 1.41-1.35
(m, 1 H).
FAB-MS calc. for ClgH26N4O2: 330; Found 331 (M+H)
Step C:
Boc
~N~
~, NH2
~
The azide from the previous step (1.60 g, 4.84 mmol) was
hydroge~te~l over 10% p~ 1ium on carbon (160 mg) in ethanol (25
rnL) under a 1 atrn hydrogen balloon for 2 hours. The reaction mixture
25 was filtered through celite and evaporated to give the amine (1.42 g,
96%). FAB-MS calc. for ClgH2gN2O2: 304; Found 305 (M+H)
Step D:
Boc
~, NHCbz
¢~
wo 9~113069 ~ Js941l28l6
2 1 752 1 8
- 315 -
To a stiIred solution of the amine from the previous step
(1.30 g, 4.27 mmol) in dichloromethane (20 mL) which also contained
DMAP (20 mg) and triethylamine (1 mL) at 0C, was added CbzCl (0.73
mL, 5.12 mmol). The reaction mixhlre was stirred for 2 hours. The
5 solution was poured into a mixture of brine and 3 N HCl and extracted
with e~yl acetate. The organic layer was washed with saturated sodium
bicarbonate, dried over m~nesium sulfate and evaporated to give a
residue which was puri~led by flash chromatography, eluting with 20%
e~yl acetate in hexane, to yield the product (1.52 g).
FAB-MS calc. for C26H34N204: 438, Found 439 (M+H)
Step E:
H HCI
~N~
NHCbz
¢~
To a stirred solution of the intermediate from the previous
step (1.50 g, 3.42 mmol) in ethyl acetate (50 mL) at 0C was bubbled
HCl until it was saturated. The reaction mixture was stirred for one hour
and evaporated to yield the salt (1.32 g, 100%).
lH NMR (CD30D, 400MHz) ~ 7.40-7.18 (m, 10 H), 5.14 (s, 2 H), 3.43,
2s 3.42 (2 d, J = 14. 8 Hz, 1 H), 3.23 (td, J = 4 Hz, 12.1 Hz, 1 H), 3.00 (d, J
= 13 Hz, 1 H), 2.94 (d, J = 14.7 Hz, 1 H), 2.83 (dt, J = 3.4 Hz, 12 Hz, 1
H),2.74(d,J=13Hz,lH),2.68(d,J=13.6Hz,lH),2.62(d,J=13.6
Hz, 1 H), 2.00-1.90 (m, 1 H), 1.92-1.88 (m, 1 H), 1.59-1.52 (m, 1 H),
1.47-1.44 (m, lH).
FAB-MS calc. for C21H26N2O2: 338; Found 339 (M+H)
WO 9S113069 2 1 7 5 2 1 8 ~ u~9~128l6
- 316 -
Step F:
~NHBoc
H ~N~
NHCbz
~
Prepared by the procedure described in Example C1, Step D
from the intermediate prepared in the previous step (1.00 g, 2.67 mmol),
Intermediate 1 (1.04 g, 1 eq.), HOBT (1 eq.), N-methyl morpholine (2
eq.), and EDC (820 mg, 4.27 mmol). Purification by MPLC, eluting with
60% ethyl acetate in hexane, provided the compound. (1.54 g, 81%)
Step G:
~N~NHBoc
H ~N~
NH2
0~
The intermediate from the previous step (1.30 g, 1.83 mmol)
30 was hydrogenated over 10% palladium on carbon (100 mg) in ethanol (15
mL) under a hydrogen balloon. The reaction mixture was filtered
through celite and evaporated to yield the amine (1.20 g, 100%).
FAB-MS calc. for C33H45N504: 575; Found 576 (M+H)
WQ 95/13069 1 ~1/U594/12816
2175218
- 317 -
Step H:
~NHBoc
H ~N~
NHMs
~
To a stirred solution of the intermediate prepared the
previous step (286 mg, 0.497 mmol), DMAP (10 mg) and N-methyl
morpholine (0.109 mL) in dichloromethane (10 mL) at 0C was added
15 mesyl chloride (0.042 mL). The reaction mixture was stirred for 2 hours.
The solution was poured into a mixture of brine and 3 N HCl and
extracted with ethyl acetate. The organic layer was washed with
saturated sodium bicarbonate dried over m~gnçsium sulfate and
evaporated to give a residue which was purified by flash
20 chromatography, eluting with 90% ethyl acetate in hexane, to give the
product (285.9 mg, 88%). FAB-MS calc. for C34H47N506S: 653;
Found 654 (M+H)
Step I:
H
¢~ ~NH2 HCI
H ~N~
NHMs
WO 9S113069 2 1 7 5 2 ~ 8 PCI/US9~12816
- 318 -
Prepared by the procedure described in Fx~mple Cl, Step C
from the intermediate from the previous step (265 mg, 0.405 mmol) and
HCl gas in ethyl acetate (8 mL) at 0C for 30 minlltes (189 mg, 79%)
FAB-MS calc. for C29H39N504S: 553; ~ound 554 (M+H)
FXAMPLE C20
~ ~NH2 HCI
H ~N~
~, NHCbz
13'
Prepared by the procedure described in Example C 1, Step C
from the intermediate from Example Cl9, Step F (109 mg, 0.154 mmol)
20 and HCl gas in ethyl acetate (4 mL) at 0C for 30 minutes (90 mg, 90%).
FAB-MS calc. for C36H43N504: 609; Found 610 (M+H)
EXAMPLE C2 1
~' ~NH2 HCI
H ~N~
~ NHAc
wo 95113069 ~ 94/12816
2175218
- 319 -
Step A:
~NHBoc
H ~N~
, NHAc
~
The mixture of the intermediates from Example C 19, Step G
(208 mg, 0.362 mmol) and pyridine (2 mL) and acetic anhydride (2 mL)
was heated at 60C for 30 minutes. The mixture was then evaporated
15 under vacuum. MPLC purification eluting with 80% ethyl acetate in
hexane yielded the product (202 mg, 90%).
FAB-MS calc. for C3sH47NsOs: 617; Found 618 (M+H)
Step B:
~NH2 HCI
H ~N~
~, NHAc
Prepared by the procedure described in Example C 1, Step C
from the intermediate from ~e previous step (192 mg, 0.311 mmol) and
HCl gas in ethyl acetate (4 mL) at 0C for 30 minntes (168.1 mg, 98.5%).
FAB-MS calc. for C30H3gNsO3: 517; Found 518 (M+H)
WO ~?S/13069 ~ 94/12816
2~752~
- 320-
EXAMPLE C22
~--1' ~NH2 HCI
H ~N~ O
W~OEt
Step A:
H HCI
[~OEt
A suspension of platinum (IV) oxide (200 mg), ethyl 3-
pyridylacetate (5.0 g, 30.3 rnmol) and concetrated hydrochloric acid (10
mL) in ethanol (50 mL) was stir~ed under a hydrogen balloon overnight.
The mixture was filtered through celite and evaporated to yield a residue,
which was refluxed with anhydrous acidic ethanol for 30 minlltes.
Evaporation yielded the product (6.28 g, 100%).
1H NMR (CD30D, 400MHz) ~ 4.13 (q, J = 7.2 Hz, 2 H), 3.40 (dd, J =
3.5 Hz, 12Hz, 1 H), 3.35 (br. d, 1 H), 2.90 (br. t, 1 H), 2.73 (t,J= 12Hz,
1 H), 2.35 (d, J = 7.5 Hz, 2 H), 2.26-2.17 (m 1 H), 1.96-1.80 (br. m, 2 H),
1.80-1.70 (m, 1 H), 1.37-1.26 (m, 1 H), 1.24 (t, J = 7.1 Hz, 3H).
Step B:
~NHBoc
O
~OEt
WO 95113069 PCI~/US94112816
2 1 752 1 8
- 321 -
Prepared by the procedure described in Example Cl, Step D
from the intermediate prepared in the previous step (128 mg, 0.617
mmol), Intermediate 1 (200 mg, 0.514 mmol), HOBT (1 eq.), N-methyl
morpholine (1 eq.), and EDC (200 mg). Purification by MPLC eluting
5 with 80% ethyl acetate in hexane provided the compound. (247 mg,
89%)
Step C:
0 ~ ~NH2 HCI
H ~N~ o
~OEt
Prepared by the procedure described in Example Cl, Step C
from the intermediate from the previous step (225 mg, 0.415 mmol) and
HCl gas in ethyl acetate (5 mL) at 0C for 15 minutes (184 mg, 100%).
20 FAB-MS calc. for C24H34N404: 442; Found 443 (M+H)
F~AMPLE C23
~--~ ~NH2 HCI
~/ ""~OH
~/ o
N HCI
wo95113069 2 ~ 752 1 8 ~ /U~94/12816
- 322 -
Step A:
Boc
~0
~N
The less polar (dl) intermediate from Example C8 step A (
7.25g, 17.08 mmol) was refluxed for 8 hours in ethanol (20 ml) and 10N
NaOH (8.5 mL). The mixture was then cooled to room temperature and
slowly treated with 3 N HCl to pH=11. To this stirred solution was
added di-tert-butyl dicarbonate in dioxane (20 mL) and stirred for two
hours. The solution was acidified to pH 4 and then neutralized to pH 7
and extracted with ethyl acetate three times. The organic extracts were
combined, dried, and concentrated to give white solid (6.80g).
FAB-MS calc. for C17H24N204: 320, Found: 321 (M+H)
Step B:
Boc
~N~
lf
[~
N
To a solution of the intermediate from the last step (6.5 g),
benzyl alcohol (2 equiv.), and DMAP (20 mg) in dichloromethane (100
rnL), was added EDC (1.2 equiv.). The mixture was stirred at room
3 o temperature for three days, and was poured into dilute NaHCO3 solution.
It was extracted with ethyl acetate three times, and dried over MgSO4.
Evaporation and purification by a flash column eluting with 40% ethyl
acetate in hexane gave the desired product.(6.53 g, 78%).
FAB-MS calc. for C24H30N2o4: 410; Found 411 (M+H); 311 (M+-
Boc(100)) .
WO 95/13069 2 1 7 5 2 1 8 PCI-/US94/12816
- 323 -
~, Step C:
H HCI
~ `
~ f OBn
~N HCI
Prepared by the procedure described in Fx~rnrle C1, Step C
0 from the intermediate from the previous step ( 1.0 g, 2.44 mmol) in ethyl
acetate (40 mL) and HCl gas at 0C for 15 minutes (935 mg, 99%).
FAB-MS calc. for C19H22N2O2: 310; Found 311 (M~H)
Step D:
, ~J ;~ , ~NHBoc
~ ", fOBn
~/ o
~N
Prepared by the procedure described in Example Cl, Step D
from the intermediate prepared in the previous step (800 mg,2.09 mmol),
intermediate 1 (812 mg, 2.09 mmol), HOBT (1 eq.), N-methyl
morpholine (1 eq.), and EDC (2 eq.). Purification by MPLC, eluting with
80% ethyl acetate in hexane, provided the Intermediate (1.10g, 77%)
30 dl FAB-MS calc. for C3gH47N5o6: 681; Found 682 (M~H)
WO 95tl3069 2 ~ 7 5 2 ~ 8 PCT/US9411281~
- 324-
Step E:
~ ~NHBoc
f OH
~ O
~N
A suspension of 10% palladium on carbon (150 mg) and the
intermediate from previous step ( 1.05 g, 1.54 mmol) in ethanol (20 mL)
was vigorously stirred under a hydrogen atmosphere for 30 minutes. The
reaction mixture was then filtered through celite and evaporated to give
the product (828 mg, 91%). dl FAB-MS calc. for C32H41N5O6: 591;
Found 592 (M+H)
Step F:
H
H C, =O O
~ ",~OH
~ O
~N
P~ ed by the procedure described in Example Cl, Step C
30 from the intermediate from the previous step (211 mg, 0.357 mmol) and
HCl gas in ethyl acetate (15 mL) at 0C for 10 minutes (175.6 mg, 93%).
dl FAB-MS calc. for C27H33N504: 491 Found 492 (M+H)
wo95113069 2 1 752 1 8 ~ 94,12816
- 325 -
EXAMPLE C24
~NHz HCI
H ~N~
~ o(cH2)2scH3
~0
N HCI
o Step A:
Boc
~N~
~ o(cH2)2scH3
~ O
~N
To a stirred solution of the product from Example C23, step
A (5.79 g, 18.1 mmol), 2-(methylthio)ethanol (2.49 g, 27.1 mrnol),
DMAP (220 mg) in dichloromethane (100 mL) was added EDC and the
mixture was stirred for one day. The reaction mixture was washed with
brine, dried, evaporated, and purified on silica gel column eluting with
60% ethyl acetate in hexane to give the desired compound (6.64 g, 94%)
FAB-MS calc. for C20H30N204S: 394, Found: 395 (M+H)
25 Step B:
H HCI
~N~
o(cH2)2scH3
, ~ O
3 ~ N HCI
Prepared by the procedure described in Example Cl, Step C
from the intermediate from the previous step(6.12 g, 15.5 mmol) in ethyl
acetate (30 mL) and HCl gas at 0C for 30 minutes (5.38 g, 95%).
FAB-MS calc. for C15H22N2O2S: 294; Found 295 (M~H)
WO 9!i113069 2 1 7 5 2 ~ 8 PCI'IUS94tl281~
- 326-
Step C:
s ~ O
o(cH2)2scH3
~0
~N
Prepared by the procedure described in Example Cl, Step D
from the interm~ te prepared in the previous step (2.0 g, 5.44 mmol),
Tntçrmediate 1 (2.12, 5.44 mmol), HOBT (1 eq.), N-me~yl morpholine
(1 eq.), and EDC (1.5 eq.). Purification by MPLC, eluting with 80-100%
ethyl acetate in hexane, provided the intermediate (3.44g, 95%)
FAB-MS calc. for C35H47N5O6S: 665; Found 666(M+H)
Step D:
¢~ ~ 2
~ " ~O(cH2)2scH3
~ N HCI
Prepared by the procedure described in Example C 1, Step C
from the intermediate from ~e previous step(2.94 g, 4.42 mmol) in ethyl
acetate (10 mL) and HCl gas at 0C for 20 minutes (2.80 g, 99%).
FAB-MS calc. for C30H39N5O4S: 565; Found 566(M+H)
The additional int~rmediates shown in Table CVII were
prepared according to the above established procedure as exemplified in
Example C24, steps A and B,. The final compounds were prepared
according to Example C17 Steps D and E, using Intermediate 1.
wo 95/13069 PCrrUS9~112816
~ 2175218
- 327 -
TABLE CVII
¢~~r ~NH2 HCI
N ~ N N;
~X ~X
~ ~
Intermediate Final Product
entry X Intermediate Final Product isomer
MF MF
FAB-MS(M+l) FAB-MS(M+l)
CO2(CH2)2SMe ClsH22N202s C30H3gNsO4S R
2 C02Bn ClgH22N202 C34H39N504 R
3 C02Bn ClgH22N202 C34H39N504 S
4 C02(CH2)3CH3 C16H24N202 C31H41N504 RS
C02(CH2)2CH3 C15H22N202 C30H39N504 RS
6 Co2cH(cH3)2 C15H22N202 C30H39N504 RS
7 CONH(CH2)3CH3 2C7l66H25N3 C3lH42N603 RS
8 CONHCH(CH3)2 2C6125H23N3 C30H40N603 RS
9 2 2 2 306 C31H39N506 RS
CONHEt C14H2lN3 C29H38N63 RS
3 o 11 CONHCH2CO2Et 3Co76H23N3o3 C3 lH40N605 RS
Note: RS compounds were prepared by using racemic intermediates
instead of chiral ones.
WO 95/13069 2 1 7 5 2 ~ 8 ~ 94,l28l~
- 328 -
EXAMPLE C24A
H ~NH2
~ ",~O(CH2)2S(O)CH3
~0
o To a stirred solution of the ~mal product from Example C24
(120 mg,0.188 mmol) in ethanol/water (3/2 mL), was added sodium
periodate (100 mg, 0.467 mmol) and the resulting mixture was stirred at
room temperature for six hours. The reaction mixtllre was then poured
into saturated sodium bicarbonate solution (10 mL) and extracted with
dichloromethane (10 nnL,3 times). The organic extracts were combined
and evaporated to give the desired compound (89 mg,81 %).
FAB-MS calc. for C30H39N5O5S: 581; Found 582(M+H)
H
¢~, =O O
~ o(cH2)3sMe
~ O
~ N HCI
WO 9Stl3069 2 1 7 ~ 2 1 8 ~ 4,l28,6
- 329 -
Step A:
H ~NHBoc
~ ",~,O(CH2)3SMe
~0
N
o To a stirred solution of the intermediate from Example C23,
step E (100 mg, 0.17 mmol), 3-(methylthio) propanol (18 mg, 0.17
mmol) and DMAP (3 mg) in dichloromethane (15 mL) was added EDC
(1.5 equiv.), and the mixture was stirred at room temperature for one day.
The reaction mixture was washed wi~ water and brine, dried, evaporated
and purified by MPLC eluting with 80% ethyl acetate in hexane to give
the desired compound (88 mg). FAB-MS calc. for C36H49N5O6S: 679;
Found 680(M+H)
Step B:
Ll
~
2~ ~/""~O(CH2)3SMe
~ O
~ N HCI
Prepared by the procedure described in Example C 1, Step C
30 from the intermediate from the previous step(85 mg, 0.125 mmol) in
ethyl acetate (3 mL) and HCl gas at 0C for 20 minutes ( 74 mg, 95%).
FAB-MS calc. for C3 1H41N5O4S: 579; Found 580(M+H)
wo 9SI13069 ~ .Sg4112816
21 75218 ~
- 330 -
The compounds shown in Table CVIII were prepared
according to the above established procedure as exemplified in Example
C25 using a~r~riate amines and alcohols.
TA}3LE CVIII
~NH2 HCI
0 H ~N~
~ "'X
~N
entry X MF
FAB-MS(M+l)
CO(morpholine) ~3lH40N604
560 (M+, EI MS)
2 C02(CH2)4SMe C32H43N54S
594
3 CoNH(cH2)2sMe C3oH4oN6o3s
565
4 CONHEt C2gH38N603
519
S CONH(CH2)20H C29H38N604
535
Likewise using the intermediate from Example C23, Step C
and following the procedures described in Step D and E using
~n~errnediate 3 instead of Intermediate 1, ~e compounds shown in Table
CVIIIa were prepared according to ~e established procedures as
30 exemplified in Example C25 using ~plopliate amines.
W095ll3069 2 1 752 1 8 ~ "2816
- 331 -
, TABLE CVIIIa
H
~ ~NH2 HCI
~ C=O O
~N~
~""X
~N
entry X MF
FAB-MS(M+l)
CONHEt C29H41N503
lS 508
2 CONH(CH2)20H C29H41NS04
524
Likewise using ~e intermediate from Example C23, Step E
and following the procedure described above; or the intermediate from
Example C23, step A and following the procedure described in Example
20 C24 steps A through D using Interme~ tP 1 or Interrnediate 3 the
compounds shown in Table CVIIIb may be prepared.
TABLE CVIIIb
H
Rl~N~NH2
C=O O
"X
~'
~N
entry Rl X
/u:~94/12816
WO 9S113069
21 752~8
- 332 -
~ CH2- -CONHCH3
2 ~ (CH2)3-
~ -CONHCH3
3 ¢~ ,CH2- -CONH(CH2)30H
H
4 3, (CH2)3- -CONH(CH2)30H
S ~ (CH2)3- -CONHCH2CH2SCH3
~
EXAMPLE C26
2 0 ¢~ ~NH2 HCI
H ~N~ o
~N~/
Step A: 3-Carbobenzyloxyaminopyridine
To a solution of 3-aminopyridine (10 g, 0.106 mol) and
triethyl amine (16.3 ml, 0.117 mol) in dichloromethane (100 mL) at 0C,
was added benzyl chloroformate (15.2 mL, 0.106 mol) slowly. The
reaction mixture was stirred overnight and was washed with water,
saturated NaHCO3, dried over MgSO4, and evaporated. The residue was
purified on a silica gel column to give the product (9.51 g)
FAB-MS calc. for C13H12N2O2: 228; Found 229(M+H)
Step B: 3-Carbobenzyloxyaminopiperidine
~ WO 9S/13069 2 t 7 ~ 2 1 8 ~ s94~l28l6
- 333 -
A solution of the intermediate from the previous step (9.51
g, 41.7 mmol) and hydrochloric acid (3.5 mL, 41.7 mmol) in ethanol (300
mL) was hydrogenated over PtO2 (0.9 g) and hydrogen (1 atm)
overnight. Filtration and evaporation gave the product as a brown solid.
FAB-MS calc. for C13HlgN2O2: 234; Found 235(M+H)
Step C: H
m~ ~NHBoc
o N C=O
\~ NHCBZ
To a solution of the intermediate from the previous step
lS (4.65 g, 17.2 mmol), Tntermediate 1 (6.68 g, equiv.), HOBT (2.32 g, 1
equiv.) and NMM (2.1 mL, 1 equiv.) in dichloromethane (100 mL), was
added EDC (3.94 g, 1.2 equiv.). The reaction mixture was stirred
overnight and worked up by w~hin~ with water, saturated NaHCO3,
dried over MgSO4 and evaporated. Purification on a SiO2 column gave
20 2.5 g of the desired product.
FAB-MS calc. for C33H43N5O6: 605; Found 606(M+H)
Step D:
2s ~ ~ HBoc
NH2
A suspension of the intermediate from the previous step (2.5
g) and Pd(OH)2/C (250 mg, 10 ~o) in methanol (60 mL) was stirred under
H2 ( 1 atm) for three days. The reaction mixture was filtered through
celite and evaporated to give the desired material.
FAB-MS calc. for C25H37N5O4: 471; Found 472(M+H)
WO95113069 2 ~ 752 1 8 1~-l",~9~,l28l~
- 334-
Step E:
~=0 o
~1 N J~/
To a solution of the intermediate from the previous step (236
0 mg, 0.5 mmol) and N,N-diisopropylethyl~min~ (0.11 mL, 0.6 mmol) in
dichloromethane (10 mL), was added isobutyryl chloride (0.053 mL, 0.5
mmol) at 0. The reaction mixture was stirred for 2 hours and was
washed with water, bline, dried over MgS04 and evaporated. SiO2 flash
column chromatography eluting wi~ 90-lOO~o ethyl acetate in hexane
15 yielded the product.
FAB-MS calc. for C29H43N505: 541; Found 542(M+H)
Step F:
¢~ ~NH2 HCI
2 5 ~ H ~/
To a solution of the intermediate from the previous step in
ethyl acetate (5 mL) at 0C was bubbled HCl until it was saturated. The
mixture was stirred for 30 minutes and evaporated to dryness to give the
product. FAB-MS calc. for C24H35N503: 441; Found 442(M+H)
Simil~rly the following compounds were prepared according
to the same procedure as described above, but using different acylating
reagents.
- -
752 ~ 8 pcr~sg4ll28l6
~o 95113069
- 335 -
TABLE CIX
-- ~ r ~HZHcl
~1Y
entry Acylatingagent Y FAB ~S(~l)
o Ac20 AcNH C22H31N503
2 ChxCOCl ChXcoNH C27H39N503
CH C0Cl ChxcH2coNH C268 4
BZCI BzNH C27H33N503
PhSo2cl Phso2NH C26H33N504S
6 isO-PrNC0 p ~IcoNH C24H36N603
te Chx cyclohexYI~ Bz: benZY
~XA~pLE (~27
~%NH2 HCI
/ "'CO2Et
/~
~ N ~lCI
Step A:
WO 9S/13069 1~ ~594/12816
2175218
- 336-
Boc
~--CO2Et
S/~/
~N
To a stirred solution of KHMDS (27.4 g, 0.138 mol) in THF
(500 mL) at -78C under argon was added ethyl N-t-Boc nipecotate (28.3
g, 0.11 mol) in THF (100 mL) over a 20 minute period. The solution was
o allowed to stir an additional 30 minutes at -78C. Then, a solution of 4-
bromomethylthiazole or 4-chloromethylthiazole in THF (100 mL) was
added slowly to the reaction mixture. 4-Bromomethylthiazole was
prepared by refluxing 4-methylthiazole (10 mL, 0.11 mmol), N-
bromosuccinimide (19.6 g, 0.11 mol) and AIBN (0.2 g) in CC14 (300
mL) for 2 hours, cooled to room temperature, filtered and evaporated; 4-
chloromethylthiazole can be prepared as described by Hsiao, C-H et al,
Synthetic Communications, 20 (22), 3507-3417 (1990) and Caldwell, W
and Fox, S.M. J. Am. Chem. Soc. 73, 2935 (1955). The resulting black
mixture was stirred overnight and allowed to warm to room temperature.
20 The material was concentrated, then diluted with water, and extracted
using ethyl ~eet~te. The organic layer was dried over anhydrous
m~gnesium sulfate, filtered, and concentrated. Purification by silica gel
flash column chromatography eluting with a solvent gradient of 30-65%
ethyl acetate in hexane provided the title compound. (7.58 g, 20%).
25 FAB-MS calc. for C17H26N2O4S 354; Found 355 (M+H)
Step B:
H HCI
--`CO2Et
,~
S~ N HCI
WO95113069 2 1 7 5 2 1 8 PCI'IUS94112816
- 337 -
To a solution of the intermediate from the previous step (7.0
g, 19.8 rnmol) in ethyl acetate (100 mL) at 0C, was bubbled hydrogen
chloride gas until saturation occurred. The reaction was stirred for 30
minutes, and then concentrated to remove the ethyl acetate to afford the
product (5.3g, 93%). lH NMR (CDC13, 400MHz) o 9.67 (s, 1 H), 7.75
(s, 1 H), 4.34-4.15 (2 m, 2 H), 3.67 (d, J=12.8 Hz, 1 H), 3.34 (d, J=15 Hz,
1 H), 3.28 (d, J=12.5 Hz, 1 H), 3.21 (d, J=15 Hz, 1 H), 3.01 (dt, J=3.0,
12.5 Hz, 1 H), 2.26 (br. d, J=13.7 Hz, 1 H), 1.97-1.92 (m, 1 H), 1.80 (dt,
J=3.5, 13 Hz, 1 H), 1.78-1.58 (m, 1 H), 1.26 (t, J=7.2 Hz, 3 H). FAB-MS
0 calc. for C12HlgN2O2S: 254; Found 255 (M+H)
Step C:
OAc OAc
~N~ ~N~
'CO2Et l CO2Et
S~ N S~ N
To a stirred solution of the intermediate (6g, 18.67 mmol)
prepared in Step B, (R)-(-)-(O)-acetyl mandelic acid (1 eq.), HOBT (1
eq.) and N~M (2 eq.) at 0C was added EDC (7.16g, 37.34 mmol). The
reaction mixture was stirred overnight during which time it was allowed
25 to warm to room temperature. The solution was poured into brine and
extracted with CH2C12. The organic layer was dried over MgSO4,
evaporated and purified with a SiO2 flash column eluting with 40-80%
ethyl acetate in hexane to provided two enantiomerically pure
compounds. The isomer which came out of the column first was
30 designated as dl (2.17 g, 30%) and the isomer which came out of the
column second as d2 (0.87 g, 12%) and mixed fractions (700 mg). The
initial stereochemistry assignment was made by NMR comparison of
these compounds with the intermediates obtained in Example C8 Step A.
The absolute stereochemistry of those intermediates was established by
X-ray analysis. The assignment was later con~mned by an X-ray analysis
-
wo95rl3069 2 1 752 1 8 PCItUS91/1281~
- 338 -
of Intermediate 1. FAB-MS calc. for C22H26N2O5S: 430; Found
431(M+H) dl: lH NMR (CDC13, 400MHz) indicated the compound
exists as a mixture of two conformers o 8.77, 8.65 (2 s, 1 H), 7.46-7.34
(m, 5 H), 7.07, 7.02 (2 s, 1 H), 6.64, 6.23 (2s, 1 H), 4.29 (br. d, J=13.9
Hz, 1/2 H), 4.10-4.02 (m, 3/2 H), 3.92-3.87 (m, 3/2 H), 3.61 (d, J=13.5
Hz, 1/2 H), 3.46 (d, J=14 Hz, 1/2 H), 3.40-3.32 (m, 1/2 H), 3.25-3.21 (m,
1/2 H), 3.18 (d, J=14 Hz, 1/2 H), 3.06 (d, J=14 Hz, 1/2 H), 2.96 (d, J=14
Hz, 1/2 H), 2.84 (d, J=14 Hz, 1/2 H), 2.85-2.7~ (br. m, 1/2 H), 2.14,2.11
(2s, 3 H), 1.90-1.82 (m, 1 1/2 H), 1.80-1.75 (m, 1 H), 1.61-1.55 (m, l H),
0 1.50-1.40 (br. m, 1/2 H), 1.14 (t, J= 7 Hz, 3/2 H), 1.03 (t, J= 7 Hz, 3/2 H).
d2: lH NMR (CDCl3, 400MHz) indicated the compound exists as a
mixture of two conformers. ~ 8.71, 8.68 (2d, J=1.8 Hz, 1 H), 7.41-7.34
(m, S H, 7.06, 6.83 (2 d, J=1.8 Hz, 1 H), 6.41 6.20 (2s, 1 H), 4.46 (br. d,
J=13.4 Hz, 1/2 H), 4.24-3.93 (m, 3 H), 3.41 (d, J=13.5 Hz, 1/2 H), 3.31-
15 3.28 m, 1 H), 3.13 (d, J=14.2 Hz, 1/2 H), 3.04 (d, J=14.2 Hz, 1/2 H), 3.04(d, J=14.2 Hz, 1/2 H), 2.92 (d, J=14 Hz, 1/2 H), 2.73 (d, J=14 Hz, 1/2 H),
2.54 ( d, J=13.8 Hz, 1 H), 2.30 ( br. d, J=13 Hz), 2.15, 2.09 (2 s, 3 H),
2.00-1.95 (m, 1/2 H), 1.65-1.49 (m, 2 H), 1.37 (dt, J=4, 12.8 Hz, 1/2 H),
1.17-1.10 (m, 3 H).
Step D: H
~N~
1~/ 'C02Et
S/q/
~N
A solution of the intermediate dl from the previous step (2.0
g, 4.65 mmol) concentrated hydrochloric acid (25 mL) and ethanol (25
30 mL) was refluxed for 3 hours and was evaporated to dryness. The
residue was neutralized by ammonium hydroxide and extracted by
dichloromethane, and then was puri~led by SiO2 flash column eluting
with 1:10:90 NH40H:MeOH:CHC13 to yield the product (0.72 g, 61%).
lH N~R (CD30D, 400MHz) o 8.88 (d, J=2 Hz, 1 H), 7.21 (d, J=2 Hz, 1
H), 4.20-4.07 (m, 2 H), 3.28 (br. d, 1 H), 3.06 (d, JAB=14 Hz, 1 H), 2.97
wo 95113069 2 ~ 7 5 2 1 8 ~ 94,l28l6
- 339 -
(d, JBA=14 Hz, 1 H), 2.92-2.80 (md, 1 H), 2.61-2.57 (m, 2 H), 2.21-2.16
(br. d, 1 H), 1.66-1.40 (m, 3 H), 1.20 (t, J=7.3 Hz, 3 H).
FAB-MS calc. for C12HlgN2O2S: 254; Found 255 (M+H)
5 Step E:
¢~ ~NHBoc
H ~N~
~ 'CO2Et
,~
S~N
To a stirred solution of the intermediate from the previous
step (163 mg, 0.642 mmol), Intermediate 1 (250 mg, 0.642 mmol) and
HOBT (87 mg, 0.642 mmol) in dichloromethane (20 mL) was added
EDC (247 mg, 1.28 mmol) at 0C. The reaction mixture was stirred
overnight and allowed to warm to room temperature. The solution was
washed with saturated sodium chloride, dried over anhydrous magnesium
sulfate; then filtered and concentrated. Purification by MPLC eluting
with 60% ethyl acetate in hexane provided the desired compound (285
mg, 71%). FAB-MS calc. for C32H43N5O6S: 625; Found 626 (M+H);
526 (M+-Boc(100)).
Step F:
~ ~NH2 HCI
H
'CO2Et
S/q~
\~ N HCI
WO95/13069 2 1 752 1 8 ~ s54/l28l~
- 340 -
Hydrogen chloride gas was bubbled into a solution of the
intermediate from the previous step (270 mg, 0.43 mmol) in ethyl acetate
(10 mL) at 0C until it was saturated. The reaction was stirred for 30
minutes, and evaporated to remove the ethyl acetate to afford the product
(226 mg, 93%). lH NMR (CD30D, 400MHz):
9.90 (d, J=2.2Hz, 4/SH), 9.S (d, J=2.2 Hz, l/SH), 8.48 (d, J=7.15, 4/SH),
8.15 (d, 7.15, l/SH), 7.70 (d, J=2.2 Hz, 4/SH), 7.68 (d, J=2.2, l/SH), 7.55
(d, J=7.89 Hz, lH), 7.27 (d, J=8.0 Hz, lH), 7.06-6.95 (M, lH), 3.94 (q,
J=7.1 Hz, 2H), 3.94 (q, J=7.1 Hz, 2H), 2.37 (d, J=14.9, lH), 1.90 (d,
J=14.9, lH), 1.60(s, 6H), 1.07 (t, J=7.1 Hz, 3H),
FAB-MS calc. for C27H35N5O4S: 525; Found 526 (M+H)
EXAM[PLE C27A
H
~NH2 HCI
H ~N~
2 o l CO2Et
~q/
\~ N HCI
Following the same procedures as in Example C27 and
using the product d2 from step C, the title compound was prepared.
FAB-MS calc. for C27H35N5O4S: 525; Found 526 (l\I+H)
The additional intermediates shown in Table CX were
prepared with the corresponding aLlcylating agents according to the above
established procedure as exemplified in Example C27 steps A and B.
The final compounds were prepared according to Example Cl Steps D
and E, using Intermediate 1.
PCI'IUS94~2816
wo9sl-30~;9 2 1 7 5 2 1 8
.
- 341 -
TABLE CX
H C ~ H2 HCI
~<CO2Et H ~<CO2Et
Intennediate Product
Intermediate P~oduct
entry Y MF MF isomer
FAB-MS (~1) F~B-MS (M~l)
H3C CH2- C14H22N203 C2gH39N505 RS
~ 267 538
Ct~3
2 2-~ia~olylmethyl Cl2Hl8N2o2s C27H35~504~ RS
~55 526
3 4-~iazolylmethyl Cl2Hl8N2o2s C27H3sNs04S RS
2~5 526
4 5-~iazolylmethyl Cl2~l8N2o2s C27H3sN5o4s RS
255 526
(4-rne~yl-2- C13H2~N202S C2gH37NsO4S RS
thia~olyl~methyl 269 540
6 ~2-methyl-4- C13H20N202S C2sH37NsO4S l~S
~ia~olyl~methyl 269 540
7 (4-methyl-5- C13H20N2o2s C28H37Nso4s RS
~ia~olyl)methyl 269 540
8 (5-methyl-4- C13H20N202S C2gE~37NsO4S RS
~ia~o~yl~methyl 269 540
3~)
WO 95/13069 2 1 7 5 2 1 8 PCI~/US9~/1281~
- 342 -
EXAMPLE C28
~-
H ~N~
~/ 'CO2H
S~
\~N
o A solution of the ~mal product of Example C27 (50 mg),
NaOH (3 N, 5 equiv.) in an mixture of ethanol/water (3:1, 5 mL) was
stirred at 60C for two days. The reaction mixture as then evaporated in
vacuo to remove ethanol. The residue was acidified by hydrochloric acid
to pH=l and then evaporated to dryness. The white residue was purified
by silica gel column eluting with 3/30/70 NH40H/MeOH/CHC13 to give
the desired product (25 mg). FAB-MS calc. for C25H3 lN504S: 497;
Found 498(M+H)
EXAMPLE C29
~NH2 HCI
H ~N~
~ 'CO2Bn
~q/
S~ N HCI
Step A:
3 o Boc
~ ""I~OH
\~N
WO 95J13069 1 ~, 1IUS~4/12816
21 752~8
- 343 -
The less polar (dl) intermediate from Example C27 step C
(1.5 g, 3.48 mmol) was refluxed for 2 hours in ethanol (10 ml) and 5 N
NaOH (3.5 mL). The mixture was then cooled to room temperature and
slowly treated with 3 N HCl to pH--11. To this stirred solution was
5 added di-tert-butyl dicarbonate (1.52 g, 7 mmol) and stirred for two
hours. The solution was acidified to pH 4 and then neutralized to pH 7
and extracted with ethyl acetate three times. The organic extracts were
combined, dried, and concentrated to give white solid (810 mg).
Step B:
Boc
OBn
\~N
To a solution of the intermediate from the last step (800 mg),
benzyl alcohol (1.27 mL), and DMAP (30 mg) in dichloromethane (40
mL), was added EDC (935 mg, 4.9 mmol). The rnixture was stirred at
2 o room temperature for three days, and was poured into dilute NaHCO3
solution. It was extracted with ethyl acetate three times, and dried over
MgSO4. Evaporation and purification by a flash column eluting with 20-
40% ethyl acetate in hexane gave the desired product.(145 mg).
Step C H
~N~
~ ""~OBn
s/q~ o
\~N
Prepared by the procedure described in Example C 1, Step C
from the intermediate from the previous step (140 mg) in ethyl acetate
(20 mL) and HCl gas at 0C for 15 rninutes. After evaporation, the
wo 95/13069 1'~-1/U~9~tl2816
2~752~8
- 344-
residue was dissolved in dichloromethane and the solution was washed
with NH40H. The organic layer was dried evaporated to give the
product.
5 Step D:
~, =O O
o H ~N~
~ ",~OBn
S/q/
\~N
Prepared by the procedure described in Example Cl, Step D
from the intermediate prepared in the previous step (140 mg, 0.443
mmol), Intermediate 1 (172 mg, 0.443 mmol), HOBT (60 mg.) and EDC
(170 mg). Purification by MPLC, eluting with 80% ethyl acetate in
20 hexane, provided the intermediate (210 mg).
Step E:
¢~ 0 0
lf
S~
\~ N HCI
3 Prepared by the procedure described in Example Cl, Step C
from the intermediate from the previous step (12 mg, 0.018 mmol) and
HCl gas in ethyl acetate (3 mL) at 0C for 10 minutes.
-
WO95/13069 2 1752 18 ~1_r/U:~94tl2816
- 345 -
Likewise it is possible to prepare the compounds shown in
Table CXa according to this example by reacting the intermediate from
Example C29, Step A, with methylamine, ethyl~rnine, ethanolamine, 3-
aminopropanol or 2-(methylthio)ethyl~mine instead of benzyl alcohol in
5 Step B, and using Intermediate 1 or Intermediate 3 in Step D.
TABLE CXa
H
R1,rN~NH2
C,=O O
~N~
~/""X
1 5 S/q~
entry Rl X
/~CH2-
2 o a~3 -CONHCH3
2 ~ (CH2)3-
W -CONHCH3
3 ~ ,CH2-
2 5 ll~ -CONHCH2CH3
4 ~, (CH2)3-
W -CONHCH2CH3
3 S ~,CH2-
~ N 3 -coNHcH2cH2oH
_
WO95113069 2 1 752 1 8 ~ g~tl281~
- 346 -
6 ~ (CH2)3-
W -CONHCH2CH20H
7 ~,CH2-
W N~ -CONH(CH2)30H
8 ~(CH2)3- -CONH(CH2)3OH
9 ~ ;~3,CH2- -CONH(CH2)2SCH3
~ (CH2)3- -CONH(CH2)2SCH3
~
EXAMPLE C30
--~ ~NH2 HCI
7=o o
C "CO2Et
S/
Step A:
~~~ ~NHBOC
Cl=O O
~N~
\/ 'CO2Et
S~
-
PCIIUS94/12816
WO 95113069
2175218
- 347 -
Prepared by the procedure described in Example C1, Step D
from the intermediate prepared in Example C27 Step D (134 mg, 0.528
mmol), Intermediate 3 (200 mg, 0.528 mmol), HOBT (71 mg, 1 eq.), and
EDC (200 mg, 2 eq.). Purification by MPLC, eluting with 60% ethyl
5 acetate in hexane provided the intermediate (160 mg, 49%)
FAB-MS calc. for C32H46N4O6S: 606; Found 607 (M+H)
StepB: H
~NH2 HCI
C=O O
N~
~/ 'CO2Et
\~ N HCI
Prepared by the procedure described in Example C 1, Step C
from the intermediate from the previous step (155 mg, 0.252 mmol) and
20 HCl gas in ethyl acetate (5 mL) at 0C for 10 minlltes (142 mg, 96%).
FAB-MS calc. for C27H3gN4O4S: 506; Found 507 (M+H)
EXAMPLE C3 1
¢~~1.'O~ 2
H ~N~
~ NHCO2Me
~J/
WO 95113069 1 ~ 4/12816
2~ 7521 8
- 348 -
Step A:
Boc
~N~
~NCO
3~
To a stirred solution of the product from example 15 step A
(2.00 g, 6..26 mmol) and DMF (3 drops) in benzene (20 mL) at 0C, was
10 added oxalyl chloride (0.89 g, 6.89 mmol) slowly. The reaction was
stirred at 0C for 10 minlltes and another 20 minutes at room
tempel~lule. The reaction mixture was evaporated in vacuo to give the
acyl chloride and it was used for the next reaction without further
purification. To a stilTed solution of the residue in acetone (20 mL) at 5
C, was added sodium azide (1.22 g, 18.8 mmol) in water (3 mL) and the
resulting mixture was stirred at room temperature for 30 minutes. The
reaction mixture was evaporated to remove acetone, and was diluted with
water and extracted with ether. The ether extracts were combined and
dried over MgSO4. Filtration and evaporation gave the crude azide and it
20 was used without further purification. The resulting material was
dissolved in toluene (70 mL) and was refluxed overnight to give the
isocyanate toluene solution. FAB-MS calc. for C1gH24N2O3: 316;
Found 217 (M+H-BOC(100)).
25 Step B:
Boc
~N~
~` NHCO2Me
¢~
A solution of methanol (5 mL) and the solution obtained
from the last step (15 mL out of 70 mL total, 1.3 mmol) was refluxed
wo ssrl306s ~ Jss4/12816
2175218
- 349 -
overnight. The reaction mixture was evaporated to give a white solid
(331 mg). FAB-MS calc. for C19H2gN2O4: 348; Found 349 (M+H).
Step C:
H HCI
~N~
~ NHCO2Me
~
To a solution of the intermediate from the previous step (271
mg) in ethyl acetate (15 mL) at 0C, was bubbled hydrogen chloride gas
until saturation occurred. The reaction was stirred for 30 minutes, until
5 TLC analysis indicated that the reaction was complete. The solution was
then concentrated to remove the ethyl acetate to afford the product (284
mg). FAB-MS calc. for C14H20N2o2: 248; Found 249 (M+H)
Step D: H
~ N ~NHBOC
H ~N~
~ NHCO2Me
Prepared by the procedure described in Example C 1, Step D
from the intermediate prepared in the previous step (0.2g4 g, 1 mmol),
30 Intermediate 1 (0.388 g, 1 mmol), HOBT (1 eq.), N-methyl morpholine
(1 eq.), and EDC (1.~ eq.). Purification by MPLC, eluting with 60%
ethyl acetate in hexane, provided the intermediate (0.35 g).
FAB-MS calc. for C34H45N506: 619; Found 620 (M+H)
-
WO 9S/13069 PCrrUS94/12816
2175218
- 350 -
Step E: H
H2 HCI
H ~N~
~ NHCO2Me
To a solution of the interrnediate from the previous step (200
mg, mmol) in ethyl acetate (10 mL) at 0C, was bubbled hydrogen
chloride gas until saturation occurred. The reaction was stirred for 30
minutes, and then concentrated to remove the ethyl acetate to afford the
product (158mg). FAB-MS calc. for C29H37N504: 519; Found 520
(M+H)
EXAMPLE C32
¢~<
~ "~OEt
~ O
Step A: H
~=~ O NHBOC
~ ",~OEt
~ O
WO 9S113069 2 1 7 5 2 1 8 ~ 94/l2816
- 351 -
To a stirred suspension of the intermediate obtained in
Example C2, step C (HCl salt, 2.51 g, 5.34 mmol), N-Boc-,~-amino-,B-
Me-butyricacid(1.16g, 1 equiv.),NMM(0.6mL, 1 equiv.)andDMAP
(33 mg,0.05 equiv.) in dichloromethane (30 m~ ,), was added EDC (1.55
5 g, 1.5 equiv.) in several portions. The reaction mixture quickly became
clear and it was stirred for 3 hours and was worked up by diluting it with
dichloromethane and washing with 3 N HCl, brine, and saturated sodium
bicarbonate solution. The organic layer was dried over MgSO4,
evaporated and purified by silica gel column chromatography, eluting
o with 60% ethyl acetate in hexane to give the desired compound (3.40 g,
100%). FAB-MS calc. for C36H4gN4O6: 632; Found 633 (M+H)
Step B: H
H C,=O O NH2 HCI
~ "~OEt
13' o
To a stirred solution of the intermediate from the previous
step (3.28 g, 5.18 mmol) in ethyl ~cet~t~ (30 mL) at 0C, was bubbled
HCl gas until it was saturated. The reaction was stirred for 10 minutes,
25 and was evaporated to dryness. The residue was dissolved in
dichloromethane, and to which ether was ~ le-l- The solid which formed
was collected by filtration, and it was air dried and left under high
vacuum overnight to give the product (2.44g, 83%).
FAB-MS calc. for C31H40N4O4: 532; Found 533 (M+H)
Similarly the following compounds were prepared according
to the same procedure as described above, but using different Boc
protected amino acids which were subsequently deprotected as described
above.
WO9S~13069 2 t 752 1 8 ~ s~l28l~
- 352 -
TABLE CXI
N :~ R
~ ",l~OEt
~0
~
entry Rll MF
FAB-MS (M+ 1 )
D-Ala- C29H36N404
505
2 L-Ala- C29H36N404
505
3 ~-Ala- C29H36N404
505
4 DL-oc-Me-Ser- C30H38N405
535
V C30H36N404
s~slf NH2 517
6 f ~I C33H42N404
~J 559
5SS~ NH2
7 D-Pro- C31H38N404
531
8 N-Me-Aib- C31H40N404
533
WO 95/13069 2 ~ 7 5 2 ~ 8 PCI'IUS94/12816
.
- 353 -
EXAMPLE C33
H H -~
~ N~N~~O
~ "~OEt
~0
~
To a stirred solution of the product from Example C32 (808
mg, 1.42 mmol), (R)-glyceraldehyde acetonide (923 mg, 5 equiv.) and
sodium acetate (582 mg, 5 equiv.) in methanol (15 mL) at 0C, was
slowly added sodium cyanoborohydride (134 mg, 1.5 equiv.) and the
resulting mixture was stirred at room temperature overnight. The mixture
was evaporated to remove methanol and partitioned between sodium
bicarbonate solution and dichloromethane. The organic layer was
separated and the aqueous layer was extracted two more times with
20 dichloromethane. The combined organic extracts were dried over
mz~pnesium sulfate and purified by a silica gel column, eluting with 5-
10% methanol in dichloromethane to give the product (835 mg, 91%)
FAB-MS calc. for C37H50N4o6: 646; Found 647 (M+H)
EXAMPLE C34
H H OH
~=Ol~N ~,OH
3 ~/ "~OEt
¢~ 0
To a solution of the product from Example C33 (367 mg,
0.566 mmol) in methanol (10 mL) was added hydrochloric acid (3 N, 1
WO 95/13069 2 ~ 7 5 2 1 8 ~ 4/128~
- 354 -
mL) and the resulting mixture was stirred at room temperature for one
day. The reaction mixture was evaporated in vacuo, and toluene was
added and evaporated in vacuo again to remove the residual water to give
the product (350 mg, 99~o). FAB-MS calc. for C34H46N406: 606;
Found 607 (M+H)
FXAMpLE C35
o ~ ~NH2 HCI
--CO2Et
--~ Rl2
Additional benzyl substituted intermediates and products as
shown in Table CXII were prepared according to procedures described in
Example Cl Steps A and B using appropriately substituted benzyl halides
in the aLkylation step. Functional groups changes as needed were made at
the intermediate Step B stage to convert as needed cyano groups to
carbox~mi(les, esters and tetrazoles, nitro groups to amines and
acetyl~min~s and esters to acids (at step D) according to standard
literature procedures.
WO 95tl3069 2 1 7 ~ 2 ~ 8 PCI/US94/12816
- 355 -
TABLE C~II: ADDITIONAL EXAMPLES
s H ~N` ~NH2 HCI
~N~
--CO2Et l--CO2Et
-2 ~12
Intermediate Product
Tntçnnediate Product
entry Rl2 MF MF isomer
FAB-MS (M+l) FAB-MS (M+l)
o-cyano- C16H20N202 C31H37N504 dl
273 544 d2
2 m-cyano- C16H20N202 C31H37N504 dl
273 544 d2
3 p-cyano- C16H20N202 C31H37N504 dl
273 544 d2
4 p-NH20C- C16H22N203 C31H37N504 RS
291 562
p-EtO2C- C18H25N04 C33H42N406 dl
320 591 d2
6 p-H02C- C3 lH38N406 dl
563 d2
7 p-(lH-tetrazole-5- Cl6H2lNso2 C3 lH38N8o4 RS
yl) 316 587
8 m-NH20C- C16H22N203 C31H37N504 RS
291 562
g m-EtO2C~- ClgH25N04 C33H42N406 dl
320 591 d2
m-H02C- C31H38N406 dl
563 d2
-
WO 95113069 ~ ~ 7 5 ~ ~ 8 ~ 94,1281~
- 356 -
11 m-(lH-tetrazole-5- C16H21N52 C31H38N804 RS
yl) 316 587
12 o-NH20C- C16H22N203 C31H37N504 RS
291 562
13 o-EtO2C- C18H25N04 C33H42N406 dl
320 591 d2
14 o-H02C- C31H38N406 dl
563 d2
o-(lH-tetrazole-5- C16H21N52 C31H38N804 RS
yl 316 587
16 p-AcNH- C17H24N203 C32H41NSO5 RS
305 576
17 m-AcNH- C17H24N203 C32H41N50s RS
305 576
While the invention has been described and illustrated with
reference to certain particular embodiments thereof, those skilled in the
art will appreciate ~at various adaptations, changes, modifications,
substitutions, deletions, or additions of procedures and protocols may be
made without departing from the spirit and scope of the invention. For
exarnple, effective dosages other than the particular dosages as set forth
herein above may be applicable as a consequence of variations in the
responsiveness of the m~mm~l being treated for any of the indications
with the compounds of the invention indicated above. Likewise, the
specific pharrnacological responses observed may vary according to and
depending upon the particular active compounds selected or whether
there are present ph~ ceutical carriers, as well as the type of
formulation and mode of ~(lmini~tration employed, and such expected
variations or differences in the results are contemplated in accordance
with the objects and practices of the present invention. It is intended,
therefore, that the invention be defined by the scope of the claims which
follow and that such claims be intel~reted as broadly as is reasonable.