Note: Descriptions are shown in the official language in which they were submitted.
W O 95113814 _ ~ t ~ ~ ~ ~ ~ PCTlEP94103754
-1-
Microencapsulated 3-Piperidinyl-Substituted 1,2-Benzisoxazoles and
1,2-Benzisothiazoles
Background of the Invention
The present invention relates to microencapsulared 3-piperidinyl-substituted
1,2-benzisoxazoles and 1,2-benzisothiazoles, their preparation and their use
in the
treatment of mental illness.
ll0 U.S. Patent No. 4,804,663 discloses 3-piperidinyl-1,2-benzisothiazoles and
3-piperidinyl-1 ,2~benzisoxazoles that have antipsychotic properties.1n
particular, 3-[2-
[4-(6-fluoro-l,2-benzisoxazol-3-yl)-I -piperidinyl) ethyl]-6,7,8,9-tetrahydro-
2-methyl-
4H-pyrido[1,2-a] pyrimidin-4 one ("risperidone") is disclosed.
U.S. Patent No. 5,158,952 describes 3-piperidinyl-1,2-benzisoxazoles having
long-acting antipsychotic properties. In particular, 3-[2-[4-(6-fluorv-I,2-
benzisoxazol 3
yl)-1-piperidinyl) ethyl]-6,7,8,9-tetrahydro-9-hydroxy-2-methyl-4H-pyrido[1,2-
a]
pyrimidin-4-one ("9-hydroxy-risperidone")is disclosed.
A number of methods are known by wlrich compounds can be encapsulated in the
form
of microparticles. In many of these processes, the material to be encapsulated
is
dispersed in a solvent containing a wall foaming material. At a single stage
of the
process, solvent is removed from the microparticles and thereafter the
microparticle
product is obtained
U. S. Patent No. 3,737,337 discloses the preparation of a wall or shell
forming
polymeric material in a solvent that is only partially miscible in water. A
solid or core
material is dissolved or dispersed in the polymer-containing solution and,
thereafter, the
care material-containing solution is dispersed in an aqueous liquid that is
immiscible in
the organic solvent in order to remove solvent from the microparticles.
Another example of a process in which solvent is removed from microparticles
containing a substance is disclosed in U. S. Patent No. 3,523,906. In this
process a
material to be encapsulated is emulsified in a solution of a polymeric
material in a solvent
that is immiscibIe in water and then the emulsion is emulsified in an aqueous
solution
containing a hydrophilic colloid. Solvent removal from the microparticles is
then
accomplished by evaporation and the product is obtained
In U. S. Patent No. 3,691,090, organic solvent is evaporated from a dispersion
of
microparticles in an aqueous medium, preferably under reduced pressure.
Similarly, U. S. Patent No. 3,891,570 discloses a method in which solvent from
a
dispersion of microparticles in a polyhydric alcohol medium is evaporated from
the
2175370
' WO 95113814 _ PCTlEP94103754
-2-
microparticles by the application of heat or by subjecting the microparticles
to reduced
pressure. Another example of a solvene removal process is shown in U. S.
Patent No.
3,960,757.
U. S. Patents No. 4,389,330 and 4,530,840 describe the preparation of
microparticles
containing an active agent by a method comprising: (a) dissolving or
dispersing an active
agent in a solvent and dissolving a wall forming material in that solvent; (b)
dispersing
the solvent containing the active agent and wall forming material in a
continuous-phase
processing medium; (c) evaporating a portion of the solvent from the
dispersion of step
(b), thereby forming microparticles containing the active agent in the
suspension; and (d)
extracting the remainder of the solvent from the microparticles.
Description of the Invention
The invention relates to a pharntaceutical composition comprising
biodegradable and
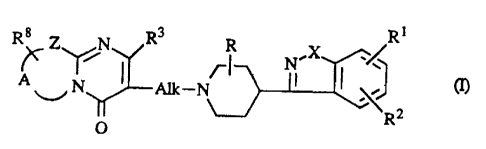
biocompatible microparticles containing a 1,2-benzazole of formula (I)
i
R8 Z N R3 R ,X /R
N I ~~ cn,
~ N Alk-N
R2
O
or a pharmaceutically acceptable acid addition salt thereof, wherein
R is hydrogen or Cl.6alkyl;
RI and RZ independently are hydrogen, halo, hydroxy, Cl~atkyloxy and
Cl_6alkyl;
XisOorS;
Alk is Ct.~alkanediYl; and
R3 is hydrogen or Cl~alkyl;
Z is -S-, -CHZ-, or -CR4=CR5-; where R4 and RS independently are hydrogen or
Cl~alkyl;
A is a bivalent radical -CHZ-CHZ-, -CH2-CHZ-CHZ- or -CR6°CR~-: wherein
R6 and R~
are hydrogen, halo, amino or Cl-6alkyl; and
R8 is hydrogen or hydroxyl.
In the foregoing definitions, the term "halo" is generic to fluoro, chloro,
bromo, and
iodo; "Ct_balkyl" is meant to include straight and branched chain saturated
hydrocarbon
radicals having from I to 6 carbon atoms, such as, for example, methyl, ethyl,
propyl,
' w0 95113814 , PCT'IEP94103754
-3-
butyl, pentyl, hexyl, and isomers thereof; "Ci.~allcanediyl" is meant to
include bivalent
straight or branched chain alkanediyi radicals having from 1 to 4 carbon
atoms, such as,
for example, methylene, ethylene, propylene, butylene, and isomers thereof.
Preferred compounds within the invention are those wherein R3 is Cl.salkyl and
in
particular is methyl and A is a bivalent radical -CH2-CH2-, -CHZ-CH2-CH2-, or
-CR6=CRS-, wherein R6 and R~ independently are hydrogen or Cl.6allryl.
Particularly preferred compounds are those preferred compounds wherein X is
oxygen,
R is hydrogen, Rt is halo or in particular is hydrogen, and R2 is hydrogen,
halo,
hydroxy, or Cl~alkyloxy.
More particularly preferred compounds are those particulary preferred
compounds
wherein -Z-A- is -CH2-CH2-CH2-CHZ-, -S-CH2-CH2-, -S-(CFi~3-, -S-CR6=CRS-, or
-CH=CH-CR6=CRS-, wherein R6 and R~ independently are hydrogen or methyl and R$
is hydrogen or 9-hydroxy.
The most prefenrd compounds are 3-[2-[4-(6-fluoro-l,2-benzisaxawl-3-yl)-1-
piperidinyl) ethyl]-6,7,8,9-tetrahydro-2-methyl-4H-pyrido[l,2-a] pyrimidin-4-
one
("ltisperidone") and the pharmaceutically acceptable acid addition salts
thereof.
The compounds of formula (1) can generally be prepared by the methods
described in
US-4,804,663 or US-5,158,952.
The compounds of formula (I) have basic properties and, consequently, can be
converted
to their therapeutically active non-toxic acid addition salt forms by
treatment with
appropriate acids, such as, for example, inorganic acids, such as hydrohalic
acid, e.g.,
hydrochloric, hydrobromic, and the Like; sulfuric acid, nitric acid,
phosphoric acid, and
the like; or organic acids, such as, for example, acetic, propanoic,
hydroacetic,
2-hydroxypropanoic, 2-oxopropanoic,ethanedioic,propanedioic, butanedioic,
(Z)-2-butenedioic, (E)-2-butenedioic, 2-hydroxybutanedioic, 2,3-
dihydroxybutanedioic,
2-hydroxy-1,2,3-propanetricarboxylic, methanesulfonic, ethanesulfonic,
benzenesulfonic, toluenesulfonic, cyclohexanesulfamic, 2-hydroxybenwic, 4-
amino-2-
hydroxybenwic, and the like acids.
The compounds of formula (I) are potent antagonists of a series of
neurotransmitters and,
as a result, have useful pharmacological properties. In particular, the
compounds of
formula (I) are combined serotonin and dopamine antagonists. Consequently,
they are
useful as anti-psychotics and in the treatment of a variety of complaints in
which
serotonin release is of predominant importance such as, for example, in the
blocking of
217 ~ 3 7 0 p~~,p94103754
~'O 95113814
-4-
serotonin-induced contractions of bronchial tissues and of blood vessels,
arteries as well
as veins. Therapeutic indications for using the present compounds are mainly
in the CNS
area, i.e. as antipsychotic agents and therefore they can be used in
combatting psychoses,
in particular schizophrenia, aggressive behaviour, anxiety, depression and
migraine.
Additionally the compounds of form (I) are also useful as sedating,
anxiolytic,
anti-aggressive, anti-stress and muscular protectant agents.
The present invention further provides a method of treating warm-blooded
animals
suffering from psychotic disorders, said method comprising the systemic
administration
of a effective amount of a mictnencapsulated compound of formula (1) or a
pharmaceutically acceptable acid addition salt thereof in admixture with a
pharmaceutical
carrier. Or alternatively, there is provided the use for the manufacture of a
medicament of
a microencapsulated compound of formula (1] for treating psychotic disorders.
Or still
alternatively, there is provided the use of microencapsulated compound of
formula ()7 or
a pharmaceutically acceptable acid addition salt thereof in admixture with a
pharmaceutical carrier for treating psychotic disorders. In general, it is
contemplated that
an effective amount of the active ingredient per se would be from 0.01 mg/kg
to 4 mg/kg
body weight, more preferably, from 0.04 mg/kg to 2 mg/kg body weight.
By the term "administered" as used herein, any method of delivering the 1,2-
benzaaole-
containing microparticles of the invention to a warn blooded animal is
intended, such as,
for example, parenteral (intravenous, intramuscular, or subcutaneous)
administration.
By "microparticles" solid particles that contain an active agent, herein the
1,2-benzawle,
either in solution or in crystalline form are meant. The active agent is
dispersed or
dissolved within the polymer that serves as the matrix of the particle.
In another aspect, the present invention relates to a method of inhibiting
serotonergic or
dopaminergic overstimulation in warm-blooded animals wherein said method
comprises
administration of a biodegradable and biocompatible microparticle composition
comprising a 1,2-benzazole of formula (1) within a polymeric matrix. Or
alternatively>
there is provided the use for the manufacture of a medicament, of a
biodegradable and
biocompatible microparticle composition comprising a 1,2-benzazole of formula
(I)
within a polymeric mattix, for inhibiting serotonergic or dopaminergic
overstimulation in
warm-blooded animals. Or the use of a biodegradable and biocompatible
microparticle
composition comprising a 1,2-benzaaole of formula (1) within a polymeric
matrix far
inhibiting serotonergic or dopaminergic overstimulation in warm-blooded
animals.
2175370
' WO 95/13814 . Pt°lYEP94103754
-5-
1n still another aspect, the invention relates to microparticles made of a
biocompatible and
biodegradable matrix containing a compound of formula (I) or a
pharmaceutically
acceptable acid addition salt thereof.
3 The compositions of this invention are useful for treating mental illness in
warm-blooded
anirttals, preferably mammals, more preferably humans, (hereinafter,
collectively referred
to as "patients") that comprises providing to such patients biodegradable
micropatticles
loaded with a 1,2-benzazole, as described above.
The compositions of the present invention comprise microparticles designed for
the
controlled release from a biocompatible, biodegradable matrix over an extended
period of
time of an effective amount of the 1,2-benzaaole of formula (I). They provide
advantages over compositions lmown in the art, such advantages comprising,
inter alla,
the fact that it is a biodegradable system, an injectable system that prevents
the loss of
IS dose during treatment, the ability to mix microparticles containing
different drugs, and
the ability to program release (multiphasic release patterns) to give faster
or slower rates
of drug release as needed.
In a preferred embodiment, adminisaation of the 1,2-benzazoles to patients is
achieved
by a single administration of the drug loaded microparticles, releasing the
drug in a
constant or pulsed manner into the patient and eliminating the need for
repetitive
injections.
The product of the present invention offers the advantage of having a duration
of action
ranging from 7 to more than 200 days, depending upon the type of microparticle
selected. In a preferred embodiment, the microparticles are designed to afford
treatment
to patienu over a period of 14 to 100 days, in particular 14 to 50 or to 60,
or 30 to 60
days. The duration of action can be controlled by manipulation of the polymer
composition, the polymer:drug ratio, and the micropatticle size.
Another important advantage of the present invention is that practically all
of the active
agent is delivered to the patient because the polymer used is biodegradable,
thereby
permitting all of the entrapped agent to be released into the patient.
The polymeric matrix material of the microparticles of the present invention
is a
biocompatible and biodegradable polymeric material. The term"biocompatible" is
defined
as a polymeric material that is not toxic to the human body, is not
carcinogenic, and does
2175370
' ~JfO 95/13814 PCTIEP94103754
-6-
not significantly induce inflammation in body tissues. The matrix material
should be
biodegradable in the sense that the polymeric material should degrade by
bodily
processes to products readily disposable by the body and should not accumulate
in the
body. The products of the biodegradation should also be biocompatible with the
body in
the same sense that the polymeric matrix is biocompatible with the body.
Suitable examples of polymeric matrix materials include poly(glycoIic acid),
poly-D,L-lactic acid, poly-Irlactic acid, copolymers of the foregoing,
poly(aliphatic
carboxylic acids), copolyoxalates, polycaprolactone, polydioxonone, poly(ortho
carbonates), poly(acetals), poly(lactic acid-caprolactone), polyorthoesters,
poly(glycolic
acid caprolactone), polyanhydrides, and natural polymers including albumin,
casein, and
waxes such as, glycerol mono- and distearate, and the like. The preferred
polymer for
use in the practice of this invention is dl-(polylactide-co-glycolide) i.e. a
copolymer of
poly(glycolic acid) and poly-D,L-lactic acid. It is preferred that the molar
ratio of lactide
to glycolide in such a copolymer be in the range of from about 85:15 to about
35:65,
more in particular from about 75:25 to about 50:50, e.g. 85:15, 75:25, b5:35
or 50:50.
The amount of active agent incorporated in the microparticles usually ranges
from about 1
wt °Jo to about 90 wt. %, preferably 30 to 50 wt. 9b, more preferably
35 to 40 wt. % . By
weight % is meant parts of agent per total weight of microparticle. For
example, 10 wt.
96 agent would mean 10 parts agent and 90 parts polymer by weight.
The molecular weight of the polymeric matrix material is of some importance.
The
molecular weight should be high enough to permit the formation of satisfactory
polymer
coatings, i.e., the polymer should be a good film former. Usually, a
satisfactory
molecular weight is in the range of 5,000 to 500,000 daltons, preferably from
50,000 to
400,000, more preferably from 100,000 to 300,000, in particular from 100,000
to
200,000 and especially about 150,000 daltons. However since the properties of
the film
are also partially dependent on the particular polymeric material being used,
it is very
difficult to specify an appropriate molecular weight range for all polymers.
The molecular
weight of a polymer is also important from the point of view of its influence
upon the
biodegtadation rate of the polymer. For a diffusional mechanism of drug
release, the
polymer should remain intact until all of the drug is released form the
microparticles and
then degrade. The drug can also be released from the micropatticles as the
polymeric
excipient bioerodes. By an appropriaae selection of polymeric materials, a
microparticle
formulation can be made in which the resulting microparticles exhibit both
diffusional
release and biodegradation release properties. This is useful in affording
multiphasic
w0 J5113814 PCT1EP94/03754
release patnerns.
The microparticle produce of the present invention can be prepared by any
method
capable of producing micaoparticles in a size range acceptable for use in an
injectable
composition such as the methods described in U.S: 4,389,330 and U.S:
4,530,840.
One preferred method of preparation is that described in the former patent and
comprises
dissolving or dispersing the active agent in an appropriate solvent. To the
agent
containing medium is added the polymeric matrix material in an amount relative
to the
active ingredient that provides a product having the desired loading of active
agent.
Optionally, all of the ingredients of the micropatticle product can be blended
together in
the solvent medium.
Solvents for the agent and the polymeric matrix material that can be employed
in the
practice of the present invention include organic solvents, such as acetone;
halogenated
hydrocarbons, such as chloroform, methylene chloride, and the Iike; aromatic
hydrocarbon compounds; halogenated aromatic hydrocarbon compounds; cyclic
ethers;
alcohols, such as, benzyl alcohol; ethyl acetate; and the like. A preferred
solvent is a
mixture of benzyl alcohol and ethyl acetate.
The mixture of ingredienes in the solvent is emulsifed in a continuous-phase
processing
medium; the continuous-phase medium being such that a dispersion of
microdroplets
containing the indicated ingredients is formed in the continuous-phase medium.
Naturally, the continuous-phase processing medium and the ~ganic phase must be
largely immiscible. The continuous-phase processing medium most commonly
employed
is water, although nonaqueous media, such as, xylene, toluene, and synthetic
and natural
oils can be used.
Usually, a surfactant is added to the continuous phase processing medium to
prevent the
microparticles from agglomerating and to control the size of the solvent
microdroplets in
the emulsion. A preferred surfactant-dispersing medium combination is a 0.1 to
10 wt.
96, more preferably 0.5 to 2 wt. % solution of polyvinyl alcohol) in water.
The
dispersion is formed by mechanical agitation of the mixed materials. An
emulsion can
also be formed by adding small drops of the active agent-wall forming material
solution
to the continuous phase processing medium.
The temperature during the formation of the emulsion is not especially
critical, but can
influence the size and quality of the microparticles and the solubility of the
agent in the
' WO 95/13814 ~_ ~ ~ 5 3 7 ~ PCT/EP94103754
_g_
continuous phase. Of course, it is desirable to have as little of the agent in
the continuous
phase as possible. Moreover, depending on the solvent and continuous-phase
processing
medium employed, the temperature must not be too low or the solvent and
processing
medium will solidify or become too viscous for practical purposes. On the
other hand, it
must not be so high that the processing medium will evaporate or that the
liquid
processing medium will not be maintained. Moreover, the temperature of the
medium
cannot be so high that the stability of the particular active agent being
incorporated in the
microparticles is adversely affected. Accordingly,the dispersion process can
be
conducted at any temperature that maintains stable operating conditions,
preferably about
20°C to about 60°C, depending upon the agent and excipient
selected.
The dispersion formed is stable and from this dispersion the organic phase
fluid can be
partially removed in the first step of the solvent removal process. The
solvent can easily
be removed by common techniques, such as heating, the application of a reduced
pressure, or a combination of both. The temperature employed to evaporate
solvent from
the microdroplets is not critical, but should not be so high as to degrade the
agent
employed in the preparation of a given microparticle or to evaporate solvent
at a rate rapid
enough to cause defects in the wall forming material. Generally, from 10 to 90
~Yo,
preferably 40 to 60 R6 of the solvent is removed in the first solvent removal
step.
After the first stage, the dispersed micrtaparticles in the solvent immiscible
fluid medium
are isolated from the fluid medium by any convenient means of separation.
Thus, for
example, the fluid can be decanted from the microparticles or the
micropaaticle
suspension can be faltered. Various other combinations of separation
techniques can be
used, if desired.
Following the isolation of the microparticles ianm the continuous phase
processing
medium, the remainder of the solvent in the microparticles is removed by
extraction. In
this step, the microparticles can be suspended in the same continuous-phase
processing
medium used in step one, with or without surfactant, or in another liquid. The
extraction
medium removes the solvent from the microparticles, but does not dissolve
them. During
the exaaction, the extraction medium containing dissolved solvent must be
removed and
replaced with fresh extraction medium. This is best done on a continual or
continuous
basis where tha rate of exriaction medium replenishment is critical. If the
rate is too slow,
agent crystals may protrude from the microparticles or grow in the extraction
medium.
Obviously, the rate of extraction medium replenishment for a given process is
a variable
that can easily be determined at the time the process is performed and,
therefoae, no
precise limits for the rate may be predetermined After the remainder of the
solvent has
217537
t ' WO 95113814 _ PG17EP94103754
-9-
been removed, the microparticles are dried by exposure to air or by other
conventional
drying techniques, such as, vacuum drying, drying over a desiccant, or the
like. This
process is very efficient in encapsulating the agent since core loadings of up
to 80 wt. 3'0,
preferably up to 50 wt. % can be obtained.
A more preferred method of encapsulating the active agent to form the
controlled release
microparticles of the present invention involves the use of static mixers.
Static or
motionless mixers consist of a conduit or tube in which is received a number
of static
mixing elements. Static mixers provide homogeneous mixing in a relatively
short length
of conduit, and in a relatively short period of time. With static mixers, the
fluid moves
through the mixer, rather than some part of the mixer, such as a blade, moving
through
the fluid. A static mixer is more fully described in U.S. Patent No.
4,511,258.
When using a static mixer to form an emulsion, a variety of factors determine
emulsion
particle size. These factors include the density and viscosity of the various
solutions or
phases to be mixed, volume ratio of the phases, interfacial tension between
the phases,
static mixer parameters (conduit diameter, length of mixing. element; number
of mixing
elements), and linear veloaty through the static mixer. Tcmperotttre is a
variable because
it affects density, viscosity, and interfacial tension. The controlling
variables are linear
velocity, shear rate, and pressure drop per unit length of static mixer.
Particularly,
droplet size decreases as linear velocity increases and droplet size increases
as pressure
drop decreases. Droplets will reach an equilibrium size after a fixed number
of elements
for a given flow rate. The higher the flow rate, the fewer elements needed
Because of
these relationships, scaling from laboratory batch sizes to commercial batch
sizes is
reliable and accrsate, and the same equipment can be used for laboratory and
commercial
batch sizes.
In order to create microparticles containing an active agent, an organic phase
and an
aqueous phase are combined The organic and aqueous phases are largely or
substantially
immiscible, with the aqueous phase constituting the continuous phase of the
emulsion.
The organic phase includes an active agent as well as a wall forming polymer
or
polymeric matrix material. The organic phase can be prepared by dissolving an
active
agent in an organic or other suitable soivent, or by forming a dispersion or
an emulsion
containing the active agent Preferably, the organic phase and the aqueous
phase are
pumped so that the two phases are simultaneously flowing through a static
mixer,
thereby fomting an emulsion, which comprises micropatticIes containing the
active agent
encapsulated in the polymeric matrix material. The organic and aqueous phases
are
2175370
' w0 95/13814 , PCTlEP94103754
-10-
pumped through the static mixer into a large volume of quench liquid The
quench liquid
may be plain water, a water solution, or other suitable liquid Organic solvent
may be
removed from the microparticles while they are being washed or being stirred
in the
quench liquid. After the microparticles are washed in a quench to extract or
remove the
organic solvent, they are isolated, as through a sieve, and dried
A laboratory set up for carrying out a static mixer process is illustrated in
Figure 1. An
organic ar oil phase 30 is prepared by dissolving and, optionally, heating an
active agent
and a polymeric matrix material or polymer in a stored pot 32 on a hot plate.
However,
the process of the present invention is not limited to preparing organic phase
30 by
dissolving an active agent. Alternatively, organic phase 30 may be prepared by
dispersing an active agent in a solution containing a polymeric matrix
material. In such a
dispersion, the active agent is only slightly soluble in organic phase 3U.
Alternatively,
organic phase 30 may be prepared by preparing an emulsion crontairung an
active agent
and a polymeric matrix material (double emulsion process). In the double
emulsion
process, a primary emulsion is prepared which contains an active agent and a
polymeric
matrix material (organic phase 30). The primary emulsion may be a water-in-oil
emulsion, an oil-in-water emulsion, ar any suitable emulsion. The primary
emulsion
(organic phase 30) and an aqueous phase are then pumped through a static mixer
to form
a second emulsion which comprises microparticles containing the active agent
encapsulated in the polymeric matrix material.
Organic phase 30 is pumped out of stirred pot 32 by a magnetically driven gear
pump 34.
The discharge of pump 34 feeds a "Y" connection 36. One branch 361 of "Y"
connection 36 returns to pot 32 for recirculation flow. The other branch 362
feeds into an
in-Iine static mixer 10. Aqueous or water phase 40 is prepared in like manner,
with a
stirred pot 42, a magnetically driven gear pump 44, and a "Y" connection 46.
One branch
461 of "Y" connection 46 returns to pot 42 for recirculation flow. The other
branch 462
feeds into in-line static mixer 10. Organic phase 3~ and aqueous phase 40 are
substantially immiscible.
Branches 362 and 462 from each solution which feed in-line static mixer 10 are
joined by
another "Y" connection 50 and feed through mixer inlet line 51 into static
mixer 10. Static
mixer 10 discharges through mixer outlet line 52 into wash tank 60. Silicone
tubing and
polypropylene fittings are used in the system illustrated in Figure I.
Silicone tubing
having 9.53 mm ll~ is used for all Iines except mixer outlet line 52. Smaller
diameter
tubing (4.76 mm 1D) is used for mixer outlet line 52 to prevent collapse of
the emulsion
2175370
R'O 95113814 , PCTYEP94I03754
-I I-
both in mixer outlet line 52 and upon entering wash tank 60.
In one embodiment of the process, pumps 34 and 44 are started in recirculation
mode and
desired flow rates are set for organic phase 30 and water phase 40. The flow
rate of
water phase 40 is preferably greater than the flow rate of organic phase 30.
However, the
two flow rates may be substantially the same. The ratio of the flow rate of
water phase 40
to the flow rate of organic phase 30 is preferably in the range of 1:1 to
i0:1. "Y"
connection 46 is then switched so that water phase 40 flows through branch 462
to static
mixer 10. Once water phase 40 fills mixer inlet line 51, static mixer 10, and
mixer outlet
line 52, "Y" connection 36 is switched so that organic phase 30 flows through
branch
362 to static mixer 10. Organic phase 30 and aqueous phase 40 are now flowing
simultaneously through static mixer 10. When the desired volume of organic
phase has
been pumped to static mixer 10, "Y" connection 36 is switched to recirculation
through
branch 361. Water phase 40 continues to flow for a short time to clean out any
organic
phase remaining in mixer inlet line SI, static mixer I0, and mixer outlet line
52. "Y"
connection 46 is then switched to recirculation through branch 461.
Organic phase 30 and aqueous phase 40 are mixed in static mixer 10 to form an
emulsion. The emulsion formed comprises microparticles containing the active
agent
encapsulated in the polymeric matrix material.
The microparticles praluced by the method of the present invention are usually
of a
spherical shape, although they may be irregularly shaped. The microparticles
produced
by the method of the present invention can vary in size, ranging from
submicron to
millimeter diameters.1n a preferted embodiment of the present invention,
static mixing
elements 14 of static mixer 10 are selected so that the resulting
microparticles range in
size from i to 500 microns (pro), preferably 25 to 180 microns in particularly
60 to 120
microns, e.g. 90 microns, whereby administration of the microparticles can be
carried
out with a standard gauge needle. The microparticles may be stirred in wash
tank 60
which contains a quench liquid. The microparricles may be isolated from the
quench
liquid, such as by using a sieve column. The microparticles may be dried using
conventional drying techniques, and further size isolation may be done.
The active agent bearing microparticles are obtained and stored as a dry
material. Prior to
administration to a patient, the dry microparticles can be suspended in an
acceptable
pharmaceutical liquid vehicle, preferably a 2.5 wt. °.6 solution of
carboxymethyl
cellulose, whereupon the suspension is injected into the desired portion of
the body.
The microparticles can be mixed by size or by type so as to provide for the
delivery of
*rB
CA 02175370 2005-O1-10
WO 95/13814 , . PCT/EP94/03754
-12-
active agent to the patient in a multiphasic manner and/or in a manner that
provides
different agents to the patient at different times, or a mixture of agents at
the same time.
In vitro dissolution studies measuring the release of risperidone from
microparticles of
the invention showed an almost constant release of risperidone during a
sustained period
of time. Similarly, in vivo studies in dogs being dosed intramuscularly with
microparticle formulations of the invention, in particular with the
formulations described
thereinafter in the examples; showed almost constant and long-lasting plasma
concentrations of active agent.
The following examples further describe the materials and methods used in
carrying out
the invention. The examples are not intended to limit the invention in any
manner.
Example 1: Preparation of 35% Theoretically Loaded Risperidone Microparticles
(Batch
ProdeX 2)
First, the aqueous phase (solution A) is prepared by weighing and mixing 906.1
g 1%
polyvinyl alcohol), (Vinyl 205"'', Air Products and Chemical Inc.), 29.7 g
benzyl
alcohol end 65.3 g ethyl acetate. Then the organic phase (solution B) is
prepared by
dissolving 29.3 g of high viscosity 75 :25 dl (polylactide-co-glycolide), in
108.7 g ethyl
acetate and 108:4 g benzyl alcohol. Once the polymer is completely dissolved,
15.7 g
risperidone base is added and dissolved in the polymer solution. The exposure
time of
the dissolved risperidone with the polymer is kept to a minimum ( < 10
minutes).
Solutions A and B are then pumped through a 6.35 mm diameter static mixer
(Cole
Parmer L04667-14) via a gear drive pump and head (Cole Parmer L07149-04,
L07002-16) at flow rates of 198 and 24 ml/minute, respectively, into a quench
composed
of 55 liters of water for injection containing 1276.0 g of ethyl acetate, 92.3
g (0.02
Molar) of anhydrous sodium bicarbonate, and 116.2 g (0.02 Molar) of anhydrous
sodium carbonate at 11 °C. The rnicroparticles are allowed to stir in
the first wash for
1.75 hours, then isolated by sieving with a 25 micron sieve. The product
retained by the
sieve is transferred to a 20-liter wash at 13°C. After stirring in the
sieved wash for 2.25
hours, the microparticles are isolated and size fractionated by sieving
through a stainless
steel sieve colurnn composed of 25- and 180-micron mesh sizes. The
microparticles are
dried overnight, then collected and weighed.
Example 2: Preparation of 40% Theoretically Loaded Risperidone Microparticles
(Batch
Prodex 3)
First, the aqueous phase {solution A) is prepared by weighing and mixing 904.4
g 1 %
* trademark
2775370
' W095/13814 . PC1YEP94/03754
-13-
poly (vinyl alcohol), (Vinyl 205'2', Air Products and Chemical Inc.), 30.1 g
benzyl
alcohol and 65.8 g ethyl acetate. Then the organic phase (solution B) is
prepared by
dissolving 27.1 g of high viscosity 75 :25 dl (polylactide-co-glycolide), in
99.3 g ethyl,
acetate and 99.1 g benzyl alcohol. Once the polymer is completely dissolved,
18.1 g
tisperidone base is added and dissolved in the polymer solution. The exposure
time of
the dissolved risperidone with the polymer is kept to a minimum (c 10
minutes).
Solutions A and B are then pumped through a 6.35 mm diameter static mixer
(Cole
Parmer L04667-14) via a gear drive pump and head (Cole Parmer L07149-04,
L07002-16) at flow rates of 198 and 24 ml/minute, respectively, and into a
quench
composed of 55 liters of water for injection containing 1375.6 g of ethyl
acetate, 92.4 g
(0.02 Molar) of anhydrous sodium bicarbonate, and 116.6 g (0.02 Molar) of
anhydrous
sodium carbonate at 12'C. The microparticIes are allowed to stir in the first
wash for 2
hours, then isolated by sieving with a 25-micron sieve. The product retained
by the sieve
is transferred to a 20 liter wash at 12'C. After stirring in the sieved wash
for 3 hours, the
microparticles are isolated and size fractionated by sieving through a
stainless-steel sieve
column composed of 25- and 180 micron mesh sizes. The microparticles are dried
overnight, then collected and weighed.
B~am~l : Lyophilisation and Gamma Irradiation of Microparticles from Batches
Prodex
2 and Prodex 3 (Samples Prodex 4A, Prodex 4B, and Prodex 4C)
Microparticles from batches Prodex 2 and Prodex 3 were lyophilized. The
microparticles
were weighed into 5 cc serum vials. Then an aqueous vehicle composed of
0.759!o CMC,
596 Mannitol, and 0.196 Tween 80~"'' was added to the vials. The
microparticles were
suspended in the vehicle by agitation, then quickly frozen in a dry
ice/acetone bath. The
vials were then lyophilized in a pilot-scale lyophilizer employing a camped
30'C
maximum temperature cycle for 50 hours. Samples Prodex 4A and Prodex 4C were
lyophilized samples from Prodex 2 and Prodex 3, respectively. Sample Prodex 4B
was
lyophilized from Prodex 2 that had bean subsequently sterilized by 2.2 MRad
gamma
irradiation from a 60Co source.
x le 4 : In vivo study
The duration of action of the micropatticle-based risperidone formulations in
the
apomorphine-induced emesis test in dogs were studied. Neuroleptics are known
to
antagonize apomorphine-induced emesis by blocking dopamine D2 receptors in the
area
postrema of the fourth ventricle. The test is generally used to predict the
onset and
2175370
WO 95113814 ~ PGTIEP94103754
-14-
duration of antipsychotic action of neuroIeptics in man (Janssen et al.,
Arzneim. Forsch.IDrug Res. I5: 1 I96-1206 (1965); Niemegeers et al., Life Sci.
24:2201-2216 (1979)).
9-OH-risperidone has a pharmacological profile that is virtually identical to
that of
risperidone. Both constitute together the "active moiety" that determines the
biological
activity of risperidone.
Apomorphine was administered subcutaneously at 0.31 mg/kg to the dogs twice a
week,
during the whole course of the experianent. The dogs were observed for
vomiting durang
a I-hour period after the administration of apomorphine. Complete absence of
emesis for
1 hour after apomorphine challenge was considered to reflect significant anti-
emetic
activity. The duration of the anti-emetic action was defined as the time
interval during
which 2 out of 3 dogs were protected from emesis.
The formulations were injected in a volume of OS ml into the biceps femoralis
of one of
the hind limbs at the level of the thigh. At several time intervals after the
intramuscular
injection, blood samples were taken and, immediately thereafter, the dogs were
challenged with a dose of apomorphine. Complete absence of emesis within 1 h
after
apomorphine challenge (which is never observed in control animals; n> 1000)
was
considered to reflect significant antiemetic activity.
Table 1 indicates whether the dogs were protected (+) or not protected (-)
from
apomorphine-induced emesis at the various time intervals after intramuscular
injection of
the depot formulations. All formulations showed an immediate onset of anti-
emetic
action.
2175370
WO 95113814 . PCTIEP94103754
-15-
N
~ + + + +
O N -~-+ + + + + + .+ +
W .d.
d W ' ~ o ~ E + + I- +
i
~ e - + + + + + + + +
~ ._
d ~ N
.,~. ~ in v E + + + + + + + +
A' C N
+ + + +
~ ~
u
'n "' ~ ~ + + + + + + +
N
, + + + + , +
O +
~
O
b ~ ~ ~ h h ~
+
~ LC ~ O N ~ + ' ' + + + + + + +
Cr
o n Pn ~n
v. ; E ' + + ' '
o ~, + + + + + +
,d .o
U ~O N
'a o N ~ + + + ' ' + + + + + + +
.
L~ rQ aV
C~~ ~ O N ~ + + + + + + + + + + + +
Ti
fr
~ C
~
pp O
G4 N ~
, ~ O N + + + + + + + + + + + +
'~" O . n
U ~
O ~
~ O tV
+ + + + + + + + + + + + +
v O
e3 'C7 cn
O N ~ + + + ' + + f + + + + + +
a
p ~ o N F + + + + ' + +
~,
~ + + + + + +
00
os o N ~ + + + + + + +
CS q N
h h Vt
w ~ ~ ~ ~ ~ N + + + ~ + + + +
_ i
O N
+' + +
, N + + +
V OD
~
,.O ~ ~ V 7 ~ 7 .C t 'p 'D 'p9 'C'O 'O 'O~ 'O
y ' ~
O
V p ' q ~ ~ V' 1~~ V 00
" ~ E C N N N rt
E
SU$SlItUTE SHEEP (AUL~ 2~
2175370
' R'0 95113814 PC1'1EP94103754
-16-
+ , , , ,
d
.r
+ + + + + , o
a,
,a + + + + ,
~
a
W
U d
~, ~ n
H
W
O
H
O
b ~ n n , $
.
.~ ~W H
',
, , n
U r
r
d
a'n , . , ,
,
ab
p
~
W
d + n n a
N
~y OyN"+ + ,
W W
~
.,.,
,~
p ..d.
w t +
~ d
i7 v~
.:.0
0
a b
w
m , . g
a ~
~
+ + +
p W
~
O
C~'
U
+a~ + , ,
V
~ + n O
'~
~~
.Uu
4
CC
d 9 TJ'O 'O 'p'O
W m V'Q' ef N Vhf
H
SJBSTiTUTE SHEET (RULE 26~
*rB