Note: Descriptions are shown in the official language in which they were submitted.
CA 02175937 2000-03-07
D-17085
-1-
THERMAL PROCESS FOR REMOVAL OF CONTAMINANTS FROM
PROCESS STREAMS
Background of the Invention
Commercial processes for manufacture of alkylene oxides, e.g.,
ethylene and propylene oxide, are well known. In a typical process, an
alkylene is oxidized over a silver-containing catalyst to the
corresponding oxide, which may be recovered or may be further
reacted, e.g., with water to form the corresponding glycol. In such
oxide manufacturing processes, carbon dioxide and various organic
materials are often formed as unwanted by-products.
In a known process for removing the carbon dioxide, the carbon
dioxide-bearing stream is scrubbed with an aqueous solution of alkali
metal carbonates and/or bicarbonates. Such a process is described in,
e.g., U.S. Pat. No. 3,907,969 and Great Britain Pat. No. 1,415, 036, the
disclosures of which are incorporated herein by reference. The process
of that patent involves the use of vanadium and other catalytic
materials which would be expensive to continually replenish, as well
being undesirable to release to the environment. Accordingly, it is
advantageous to provide an improvement to that process whereby the
depleted scrubbing stream is treated to remove various contaminants,
and is returned to the process.
A method for significantly improving the catalytic oxidation of
the alkylene to the corresponding oxide has recently been disclosed in
U.S. Patent 5, 504,053, the disclosure of which is
incorporated herein by reference. This process utilizes silver
catalysts of the type comprising at least one efficiency-enhancing salt
of a member of a redox-half reaction pair which are employed in
processes in which at least one efficiency-enhancing gaseous member
21'~5~37
D-17085
-2-
of a redox-half reaction pair is present (described hereinbelow). When
the process of this patent application is combined with the carbon
dioxide removal process described above, undesirable nitrate and/or
nitrite, as well as various organic, contaminants may be formed.
It has now been found that organic contaminants, as well as
inorganic contaminants such as those resulting from processes such as
those of the above-cited patent application, can be successfully
removed, or optionally controlled in concentration, without significant
destruction or depletion of the expensive chemicals required for the
carbon dioxide scrubbing procedure, by the method reported below.
Summary of the Invention
The present invention provides a method for controlling in a
process the build-up of contaminants, typically organic contaminants,
in an aqueous process stream bearing such contaminants, comprising:
(a) directing at least some of said aqueous process
stream through a heating means under conditions su~cient
to decompose at least part, and preferably all, of such
contaminants,
(b) directing the effluent from the heating means to at
least one removal means for removing at least part of the
decomposition products of the contaminants from said
effluent,
(c) optionally, dissolving in water at least part of said
effluent after said removal of decomposition products, and
(d) returning said contaminant-reduced effluent to
said aqueous process stream.
Often, the method of this invention will be applied to a process stream
which comprises a stream used for the removal of carbon dioxide from'
the process of which the process stream is a part. When so used, the
n-i7oss 217 5 ~ ~'~
-3-
contaminants likely will be contained in a process stream which
contains alkali metal carbonates and/or bicarbonates.
In a preferred embodiment, the present invention provides a
process for the manufacture of alkylene oxide comprising the catalytic
oxidation of an alkylene, said process further comprising an aqueous
recycle stream bearing dissolved contaminants comprising inorganic
salts and organic materials, the improvement comprising:
(a) directing at least some of said recycle stream
through a heating means under conditions sufficient to
decompose at least part, and preferably all, of the organic
materials and at least part of the inorganic salts,
(b) removing the decomposition products of the
organic materials and inorganic salts,
(c) dissolving the remaining salts in water to form an
aqueous solution, and
(d) returning said aqueous solution to the alkylene
oxide manufacturing process.
In a more specific preferred embodiment, the instant invention
provides a process for the manufacture of alkylene oxide comprising
the oxidation of an alkylene, said process further comprising an
aqueous recycle stream containing dissolved alkali metal carbonate
and/or bicarbonate salts from a C02 absorption step to a C02
desorption step and return, said stream further containing dissolved
contaminants comprising organic impurities and/or nitrogen-
containing salts, the improvement comprising controlling the build-up
of contaminants by:
a) directing at least some of said recycle stream
through a heating means under conditions sufficient to
decompose at least part, and preferably all, of the organic
materials and at least part of the inorganic salts, '
b) removing at least some, preferably all, products of
the decomposition,
D-17085
-4-
c) dissolving remaining salts in water to form an aqueous
solution, and
d) returning said aqueous solution to the aqueous
recycle of alkali metal carbonate/bicarbonate salts.
In another preferred embodiment, the instant invention
provides a process for the manufacture of alkylene oxide comprising
the oxidation of an alkylene, said process further comprising an
aqueous recycle stream containing dissolved alkali metal carbonate
and/or bicarbonate salts from a C02 absorption step to a C02
desorption step and return, said stream further containing dissolved
contaminants comprising organic impurities and/or nitrogen-
containing salts, the improvement comprising controlling the build-up
of contaminants by:
a) directing at least some of said recycle stream
through a heating means under conditions sufficient to take
the salts to substantial dryness,
b) heating the resulting dried salts under conditions
sufficient to decompose at least part, preferably all, of the
organic impurities and at least part, preferably all, of the
nitrogen-containing salts,
c) removing some, preferably all, products of the
decomposition,
d) dissolving remaining salts in water to form an
aqueous solution, and
e) returning said aqueous solution to the aqueous
recycle of alkali metal carbonatelbicarbonate salts.
brief Description of the Fi es
Figure 1 is a flow diagram showing the general relationship of
the present decontamination process to an alkylene oxide
manufacturing process.
D-17085 ~ 1
-5-
Figure 2 is a flow diagram showing the use of a T-Thermal
oxidizer, a preferred device for carrying out the decomposition and
dissolution in the present process.
Description of the Invention
In its broadest embodiment, this invention contemplates the use
of heat to decompose organic and/or inorganic materials which are
present as contaminants, e.g., unwanted by-products, borne by an
aqueous stream which forms part of a manufacturing process.
Decomposition of the contaminants produces decomposition products,
e.g., gases, which can be flashed off or stripped off by means well
known in the art, thereby resulting in a purified or decontaminated
stream which can be returned to the manufacturing process.
While it is expected that most contaminants will be dissolved in
the aqueous stream, the invention is not intended to be limited to
dissolved materials. For instance, organic contaminants could be
borne is suspension or microemulsion form, and inorganic materials
could be in insoluble, particulate form. By removal of at least some,
and preferably all, of the contaminants by the thermal treatment of
this invention, a purified aqueous stream can be returned to the
manufacturing process, thereby controlling the build-up of the
contaminants in the manufacturing process. It will be readily
understood and appreciated that the present method is most
conveniently applied to a manufacturing process through use of a
recycle loop, i.e., the contaminated stream is channeled through the
treatment method of this invention, and the decontaminated effluent
of the present invention is returned to the manufacturing process. It
will likewise be appreciated that the input to, and the output from, the
present treatment method can be situated at any convenient point of ' '_
the manufacturing process.
21753"l
D-17085
-6-
Processes where the method of this invention is particularly
applicable include preparation of alkylene odes, which can be
recovered, or further processed to derivatives, such as glycols,
alkanolamines, polyalkylene oxides and other polymers..
More specifically, one of the preferred embodiments of the
invention relates to known processes for the catalytic conversion of
ethylene to ethylene oxide, with subsequent hydrolysis of the ethylene
oxide to ethylene glycol. Such a process is well known and is described
in general terms in various publications (e.g., Kirk-Othmer
Encyclopedia of Chemical Technology, 4th Ed., vol. 9, pages 915-960
(John Wiley & Sons, New York, 1994)), and in numerous U.S. and non-
U.S. patents. Many variations on such a process, principally
concerned with the catalysis aspects, are also disclosed in the art. See,
for example, U.S. Patent No. 5,187,140 and U.S. Patent Application
Serial No. 08/091,352, filed July 14, 1993, the disclosures of which are
incorporated herein by reference.
One particularly effective process for the preparation of
ethylene oxide utilizes silver catalysts of the type comprising at least
one efficiency-enhancing salt of a member of a redox-half reaction pair
which are employed in processes in which at least one efficiency-
enhancing gaseous member of a redox-half=reaction pair is present
(described hereinbelow). The term "redox-half reaction" is defined
herein to mean half reactions like those found in equations presented
in tables of standard reduction or oxidation potentials, also known as
standard or single electrode potentials, of the type found in, for
instance, "Handbook of Chemistry', N. A. Large, Editor, McGraw-Hill
Book Company, Inc., pages 1213-1218 (1961) or "CRC Handbook of
Chemistry and Physics" , 65th Edition, CRC Press, Inc., Boca Raton,
Florida, pages D155-162 (1984). The term "redox-half=reaction pair"
refers to the pairs of atoms, molecules or ions or mixtures thereof
which undergo oxidation or reduction in such half reaction equations.
Such terms as redox-half reaction pairs are used herein to include
D-17085 217 5 ~ 3 '~
7-
those members of the class of substances which provide the desired
performance enhancement, rather than a mechanism of the chemistry
occurring. Preferably, such compounds, when associated with the
catalyst as salts of members of a half reaction pair, are salts in which
the anions are oxyanions, preferably an oxyanion of a polyvalent atom;
that is, the atom of the anion to which oxygen is bonded is capable of
existing, when bonded to a dissimilar atom, in different valence states.
Potassium is a preferred ration, although sodium, rubidium and
cesium may also be operable, and among the preferred anions are
nitrate, nitrite and other anions capable of undergoing displacement
or other chemical reaction and forming nitrate anions under
epoxidation conditions. Preferred salts include KN03 and KN02,
with KNOB being most preferred.
The reaction conditions for carrying out the oxidation reaction
are well known and extensively described in the prior art. This
applies to reaction conditions, such as temperature, pressure,
residence time, concentration of reactants, gas-phase diluents (e. g.,
nitrogen, methane and C02), gas-phase inhibitors (e.g., ethylene
chloride and ethylene dichloride), and the like.
The gases fed to the reactor may contain modifiers or inhibitors
or additives such as disclosed in U.S. Patents Nos. 2,279,469 and
2,279,470, such as nitrogen oxides and nitrogen oxide-generating
compounds.
The terms "gaseous member of a redox-half reaction pair,"
"gaseous efficiency-enhancing member of a redox-half reaction pair,"
or like terms referred to herein, have a meaning similar to that for the
"salt of a member of a redox-half reaction pair," or like terms, defined
above. That is, these terms refer to members of half reactions,
represented in standard or single electrode potential tables in
standard reference texts or handbooks which are in a gaseous state
and are substances which, in the reaction equations represented in the
texts, are either oxidized or reduced. The preferred gaseous e~ciency-
D-17085
_g_
enhancing materials are compounds containing an element capable of
existing in more than two valence states, preferably nitrogen and
another element which is, preferably, oxygen. Examples of preferred
gaseous e~ciency-enhancing members of redox-half reaction pairs
include at least one of NO, N02, N204, N203 or any gaseous
substance capable of forming one of the aforementioned gases,
particularly NO and N02, under epoxidation conditions, and mixtures
thereof with one or more of PH3, CO, 503, S02, P205, and P203. NO
is often preferred as the gaseous efficiency-enhancing compound.
The desirability of recycling unreacted feed, or employing a
single-pass system, or using successive reactions to increase
conversion by employing reactors in a series arrangement can be
readily determined by those skilled in the art. The particular mode of
operation selected will usually be dictated by process economics.
Generally, the commercially practiced processes for
manufacturing ethylene oxide are carried out by continuously
introducing a feed stream containing ethylene and oxygen to a
catalyst-containing reactor at a temperature of from about 200°C to
300°C, and a pressure which may vary from about five atmospheres to
about 30 atmospheres depending upon the mass velocity and
productivity desired. Residence times in large-scale reactors are
generally on the order of about 0.1-5 seconds. Oxygen may be supplied
to the reaction in an oxygen-containing stream, such as air or as
commercial oxygen. The resulting ethylene oxide is separated and
recovered from the reaction products using conventional methods.
As has been indicated, a typical process for the production of
alkylene oxides produces significant amounts of carbon dioxide as a
by-product. It is desirable to remove this material because
concentrations of carbon dioxide much in excess of about 15 mole
percent adversely affect the activity of the ethylene oxide catalyst. A~ '
preferred procedure for this removal is that described in U.S. Patent
No. 3,907,969, referred to above. This process for scrubbing carbon
D-17085 ~ ~ 7 J
-9-
dioxide-containing aqueous streams, and the special chemical streams
used for such scrubbing, are well known in the industry and are called
the "Benfield process" and the "Benfield solution," respectively, owned
and licensed by UOP, Inc., of Des Plaines, Illinois... Accordingly, the
present invention will be described, for convenience, principally with
reference to the Benfield solution.
In a typical commercial design, the effluent stream from the
carbon dioxide scrubbing column using the Benfield solution as the
scrubbing agent goes largely (or completely depending where the
removal unit takeoff stream is) to the C02 desorber. Economical and
environmentally sound utilization of the Benfield process requires
recycle of the effluent stream from the carbon dioxide desorbing
column back to the carbon dioxide absorbing column. When
contaminants build up in the Benfield solution to the point that the
system no longer operates efficiently, the solution is taken in whole or
in part to the decontamination unit, i.e., the unit which implements
the method of this invention, where impurities as described previously
are removed. From there, the solution is brought back to the Benfield
scrubber and returned to the scrubber recycle stream. The organics
present are primarily the acids formic and oxalic. It is important to
remove such organics since they form acid salts with.potassium and so
tie up potassium that otherwise would be present as carbonate and
available for removing carbon dioxide.
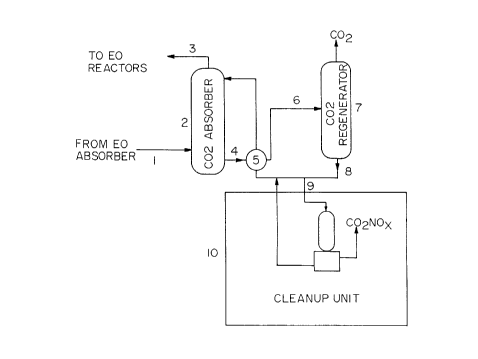
More specifically, referring to Fig. 1, C02-bearing gaseous
stream 1 is fed to C02 absorber column 2 where it is contacted .
countercurrently with Benfield solution, i.e., an aqueous potassium
carbonate/bicarbonate solution promoted with vanadium oxide and
boric acid. Overhead stream 3, from which the C02 has been
removed, is returned to the alkylene oxide manufacturing process.
Bottoms stream 4 passes through heat exchanger 5 from which it is ''
sent via stream 6 to a C02 regenerating column 7, in which C02 is
separated and released to the atmosphere. Bottoms stream 8 from
CA 02175937 2000-03-07
D-17085
-10-
column 7 comprises an aqueous potassium carbonate/bicarbonate
solution, promoted with vanadium oxide and boric acid, and
contaminated with potassium organic acid salts, small amounts of
alkylene glycol and, in the practice of certain embodiments of the
method of U.S. Patent 5,504,053, mentioned above, also potassium
nitrate/nitrite. Bottoms stream 8 is returned to the system until such
time as the build-up of contaminants is found to be interfering with
the efficient absorption of COZ. At that time, slip stream 9 is diverted
to contaminant removal (or "clean-up" unit) 10 for the practice of the
present method. In general terms, the clean-up unit comprises
heating means, separating means, and dissolution means, to perform
the steps previously mentioned.
A preferred embodiment of clean-up unit 10 is depicted is Fig. 2,
which relates to a T-Thermal SUB-X oxidizer (sold by T-Thermal, Inc.,
Conshohocken, Pennsylvania) and associated equipment. To describe
this device in general terms, the solution to be treated is sprayed into
a high-temperature, flame-heated zone, water is evaporated, and the
resulting particles of salt are pyrolyzed, all within a few seconds. A
quench tank below the oxidizer scrubs the effluent gas to remove
particulates, reconstituting the solution, now regenerated. Any
molten salts hitting the wall run down with gravity into the quench.
After the quench, the gas flows through a Venturi scrubber for final
particulates removal and out through a stack. An advantage of the
system is that solution is fed and solution is pumped out. No handling
of solids nor operation of large pieces of rotating equipment is
required. The system offers the added potential advantage that an situ
NOx reduction may be practiced simply by adding ammonia to the
feed; thus, regeneration and NOx reduction are combined in one
operation.
Referring to Fig. 2, bottoms stream 9 from Fig. 1 is sprayed into
radiant zone 10 of T-Thermal device 11, from which it passes into
pyrolysis (oxidation) zone 12 for combustion with an appropriate fuel,
D-17085
11-
such as natural gas. Ammonium hydroxide or ammonia may
optionally be introduced into zone 10 to provide ammonia to assist in
reduction of any NOx present. Introduction of ammonia or ammonium
hydroxide is particularly advantageous to increase the decomposition
of nitrites. In zone 12, the organic acid salts will be converted to
potassium carbonate and carbon dioxide, and glycol will be converted
to carbon dioxide. Any potassium nitrite/nitrate will be regenerated to
potassium carbonate and a gaseous nitrogen species. If ammonia (or
ammonium hydroxide) has been introduced, the nitrogen species will
be largely molecular nitrogen. If ammonium hydroxide (or ammonia)
has not been introduced, the nitrogen species will be largely nitrogen
oxides. The molten salts pyrolysis products run down the surface of
oxidation zone 12 and pass into aqueous quench tank 13 where the
regenerated salts are dissolved to form a solution which is routed to
separation section 14, comprising aqueous scrubbers 15, Venturi
scrubber 16, stack 17 to the atmosphere, and optionally filter 18 to
remove any residual partieulates. Separated gases, i.e., C02 and NOx ,
(if produced) will be released through stack 17. Regenerated
potassium carbonate solution 19 will be returned to the C02
absorption train.
Oxidation zone 12 is lined with refractory brick. It has been
found that molten Benfield salts will react with and degrade some
types of refractory brick, and will penetrate others. The best choice for
refractory brick to resist degradation is believed to be a magnesium
oxide brick known as "OXIBAK H," available from Harbison-Walker
Refractories, Pittsburgh, PA. However, since there is some
penetration of OXIBAK H, it is considered useful to back it with a less
penetrable brick such as "Greenal-90" from A. P. Green Industries,
Inc. of Mexico, MO) Temperature in the oxidation zone should be kept
reasonably constant, e.g., within about ~10° C, to minimize thermal '
'.
cycling degradation of the refractory bricks.
CA 02175937 2000-03-07
D-17085
-12-
While the present invention is described in relation to the
Benfield process, it will be readily understood, however, that the
procedures disclosed and claimed herein can also be effectively applied
to processes other than the Benfield process, if appropriate.
In a preferred embodiment of the present invention, the method
and materials of U.S. Patent 5,504,053, mentioned above, are
combined with those of the Benfield process. Under these conditions,
it has been observed that a build-up of alkali nitrates and nitrites
occurs in the Benfield solution, particularly when a nitrogen
compound, e.g., an oxide of nitrogen, is used as a promoter for the
ethylene oxide catalyst by conversion within the catalyst bed to
nitrogen oxides. Such contamination should be removed because the
nitrates, nitrites, and their inorganic acids reduce the effectiveness of
the Benfield solution for carbon dioxide removal.
The percentage of the inorganic salts present which is
decomposed is not critical; however, in order for the process to be
operated under commercially efficient conditions of recycle, it is
recommended that at least about 50 weight percent of the nitrates and
at least about 50 weight percent of the nitrites be decomposed before
return of the decontaminated recvcle stream. It will be understood
that the overall objective of the removal of the contaminants is to
prevent their uncontrolled build-up in the process streams to
concentrations which significantly interfere with the efficiency of the
carbon dioxide absorption in the scrubbing column. It is recommended
that the concentration of contaminants in continuous circulation be
limited to no more than about 10%, based on the weight of the
solution, preferably no more than about 5% nitrates, no more than
about 10%, preferably no more than about 5% nitrites, and no more
than about 10%, preferably no more than about 5% organics.
Accordingly, the size of the stream sent to heat treatment should be
determined so as to permit these steady-state concentration limits to
be met after return of the decontaminated stream to the process.
D-17085
-13-
Surprisingly, the decomposition temperatures of alkali metal
nitrites and nitrates is not well known. For example, decomposition
temperatures ranging from about 400 to about 1,000° C have been
reported for sodium and potassium nitrate (see C.M. Kramer,
"Intrinsic Decomposition of Sodium Nitrate and Potassium Nitrate,"
Thesis, University of California, Davis, December 1980). While the
operating temperature of the heating means is not narrowly critical, it
has been found that for the streams being treated in the present
invention, the heating means should be operated so as to subject the
dry solids and organics to a temperature of at least about 300° C, and
preferably at least about 350° C. Below about 300° C the removal
of
contaminants begins to take an unacceptably long time. Upper
temperature is determined primarily by equipment limitations and
expense rather than reaction rates or products. An upper temperature
due to equipment limitations might be in the range of about 1700°C.
Preferred temperature is in the range of about 350 to about 1400°
C.
In addition to the other inorganic salts, the stream will also
contain a substantial concentration of carbonates, e.g., alkali
carbonates, resulting from the reaction of the carbon dioxide with the
Benfield solution. It is not critical to the method of this invention
whether or not such carbonates are decomposed. If they are
decomposed, they go to hydroxides or oxides, which are effective in
C02 removal. If they are not decomposed, they stay as carbonates
which also are effective in C02 removal.
While the preferred thermal treatment device is the T-Thermal
oxidizer described above, the heating means can be any suitable device
for applying the necessary heat while maintaining the materials in
handleable condition. For example, an oven or series of ovens could' be
employed, if appropriately designed to avoid melting of the salts. By
way of guidance but not limitation, it has been found that simple
pyrolysis in air at about 600° C will remove essentially all nitrites
and
organics, but will provide little or no significant removal of nitrates.
D-17085
-14-
There may also be conversion of some nitrites to nitrates. The nitrates
can bye removed by treatment at about 750° C or more; however, there
is the likelihood of producing molten products which could be difficult
to handle in an industrial facility. ..
A useful alternative to simple pyrolysis is the use of spray
drying. Any of the numerous available spray drying devices should be
satisfactory to take the contaminated Benfield solution to
substantially dry powder. The powder can then be recovered and
subjected to pyrolysis, as described above. Other alternative means of
heat treatment include rotary calcination, band calcination and
microwave treatment. Such methods, however, have the disadvantage
of requiring either handling of solids, operation of large pieces of
rotating equipment, or both. Care should also be taken to make sure
the resulting solids are substantially dry, to avoid sticking or
damming on the hot surfaces of the equipment. By "substantially dry"
is meant the essential absence of a liquid phase.
It is considered to be desirable to conduct the heating in an
inert, oxygen-lean atmosphere. Use of an inert atmosphere appears to
reduce somewhat the temperatures needed to achieve decomposition.
While any inert gas should be useful, the preferred gases are nitrogen
and carbon dioxide. Complete absence of oxygen may, however, result
in charring of organics; accordingly, an oxygen-lean environment is
preferred to an oxygen-free one. The oxygen concentration should be
maintained at a level of at least about 1% by volume to facilitate the
decomposition of organics, and preferably in the range of about 3% to
about 5%. Under these conditions, temperatures in the range of about
500-600° C should be satisfactory to accomplish the decomposition of
the nitrogen-bearing salts and the organics.
D-17085 217 5 9 3 '~
-15-
Examples
Example 1
Into a four-foot diameter by eight-foot high refractory-lined
chamber maintained at a temperature of 1880° F, an oxygen level of
5.0 vol. %, and a pressure of 5 psig with an internal gas-fired burner,
was atomized at a rate of 90 pounds per hour an aqueous solution
comprising a solution of potassium carbonate and potassium
bicarbonate in water with proprietary promoters used for removal of
carbon dioxide from an ethylene oxide process, 90 wt. %, ethylene
glycol, 5 wt. %, potassium nitrate, 4.5 wt. %, and potassium nitrite, 0.5
wt. %. In use, the potassium carbonate/potassium bicarbonate
solution had built up organic acid salts to an extent such that the feed
solution contained about 10,500 parts per million by weight of formate
ion and about 5,000 parts per million by weight of oxalate ion. From
the chamber downstream of the injection point was pulled a sample of
vapor and atomized salts, the salts having been in the heated chamber
for a period of about 3.5 seconds. The salts were recovered by
scrubbing in water, then the water was analyzed for nitrate, nitrite,
formate, oxalate and potassium carbonate. Table 1 shows the results
of analyses of the feed and of the water used to scrub the sample of
vapor and salts. Adjusting the scrubber solution concentrations to the
same carbonate level as the feed shows that formate and oxalate were
completely decomposed, nitrate was 98+% decomposed, and nitrite
was 42% decomposed. Nitrogen oxides were detected in the gaseous
effluent from the unit in amount corresponding to 0.64 mole of
nitrogen oxides per 1.00 mole of nitrite plus nitrate fed.
.r
21753'7
D-17085
- 16 -
ai
~o 0 0
O ~ ~ °
0 0~
s
0
w ~ ° o
cdl o
c~
z~
-I ~ cso
z~
of
. ,
U 31
G
as
o
O O
~
+~
d
v~ ~ ~ 3
o
c~
'~ . v
~ ~
~
~
o
U U U
b~
~
O
s
w ra r~ ca
~
r?
D-17085 2 i ~ ~ g 3 "~
-17-
Example 2
This example shows the effect of ammonium hydroxide.
Into the chamber of Example 1 maintained at a temperature
of 1880° F, an excess oxygen amount of 2.0 vol. %, and a pressure of 5
psig, was atomized the solution fed in Example 1, 132 pounds per
hour, mixed with 25 wt. % aqueous ammonium hydroxide, 18.3 pounds
per hour. The amount of ammonium hydroxide corresponded to 2.8
moles of ammonium hydroxide per 1.0 mole of nitrite plus nitrate.
From the chamber downstream of the injection point was taken a
sample of vapor and atomized salts, the salts having been in the
heated chamber for a period of about 2.4 seconds. The salts were
isolated by scrubbing in water, then the water was analyzed for
nitrate, nitrite, formats, oxalate and potassium carbonate. Table 2
shows the results of analyses of the feed prior to mixing with
ammonium hydroxide and of the water used to scrub the sample of
vapor and salts. Adjusting the scrubber solution concentrations to the
same carbonate level as the feed prior to mixing with ammonium
hydroxide shows that formats and oxalate were completely
decomposed, nitrate was 99+% decomposed, and nitrite was 95+%
decomposed. Nitrogen oxides were detected in the gaseous effluent
from the unit in amount corresponding to 0.16 mole of nitrogen oxides
per 1.00 mole of nitrite plus nitrate fed. Under the same conditions
without ammonium hydroxide, nitrogen oxides were detected in the
effluent in amount of 0.66 mole of nitrogen oxides per 1.00 mole of
nitrite plus nitrate fed. Thus nitrogen oxides were reduced by 76%
when ammonium hydroxide was fed. Also, the conversion of nitrite
was increased from 42% (Example 1) to 95+% when ammonium
hydroxide was fed.
2175~3'~
-- D-17085
--18 -
ai
~ ct~
~0 0 o s
O ~ ~ °
0 0~
° o
~
o
z
z~ ~
o
0
o~ o
0
v
~~
0 0~
"bo
~ v '
b~
-
~ ~ ,~~o~
w
D-17085 217 5 ~ 3 '~
-19-
Example 3
This example shows the effect of a lower amount of ammonium
hydroxide and higher temperature.
Into the chamber of Example 1 maintained at a temperature of
2000° F, an excess oxygen level of 1.2 vol. %, and a pressure of 5
psig,
was sprayed the solution fed in Example 1, 90 pounds per hour, mixed
with 25 wt. % aqueous ammonium hydroxide, 8.5 pounds per hour.
The amount of ammonium hydroxide corresponded to 1.6 moles of
ammonium hydroxide per 1.0 mole of nitrite plus nitrate. From the
chamber downstream of the injection point was taken a sample of
vapor and atomized salts, the salts having been in the heated chamber
for a period of about 2.4 seconds. The salts were isolated by scrubbing
in water, then the water was analyzed for nitrate, nitrite, formate,
oxalate and potassium carbonate. Table 3 shows the results of
analysis of the feed prior to mixing with ammonium hydroxide and of
the water used to scrub the sample of vapor and salts. Adjusting the
scrubber solution concentrations to the same carbonate level as the
feed prior to mixing with ammonium hydroxide shows that formate
and oxalate were completely decomposed, nitrate was 99+%
decomposed, and nitrite was 90% decomposed. Nitrogen oxides were
detected in the gaseous e$luent from the unit in amount
corresponding to 0.55 mole of nitrogen oxides per 1.00 mole of nitrite
plus nitrate fed. Under the same conditions without ammonium
hydroxide added to the feed, nitrogen oxides were detected in the
effluent in amount of 0.78 mole of nitrogen oxides per 1.00 mole of
nitrite plus nitrate fed. Thus, nitrogen oxides were reduced by 29%
when ammonium hydroxide was fed at the level indicated.
2175937
D-17485
- 24 -
to 0 0
O ~ ~ °
0 00
w ~ ° o
m
z~
'"
z
~ 0
o ~
o~ o
0
U ~
~
A
~ ~
~
o
O O
~
~
v~ '
+~ ~ ,
r
d .
w
~
m
~
~ v U
~
, ~ l c~
~
'.
_ D-17085 21 '~ 5 ~ 3 7
-21-
ExamRle 44
This example shows the effect of ammonia and higher pressure.
Into the chamber of Example 1 maintained at a temperature of
1930° F, an excess oxygen amount of 1.2 vol. %, and a pressure of 11
psig, was sprayed the solution fed in Example 1, 90 pounds per hour,
mixed with 100 wt. % ammonia, 1.16 pounds per hour. The amount of
ammonia corresponded to 1.7 moles of ammonium hydroxide per 1.0
mole of nitrite plus nitrate. From the chamber downstream of the
injection point was taken a sample of vapor and atomized salts, the
salts having been in the heated chamber for a period of about 3.5
seconds. The salts were isolated by scrubbing in water, then the water
was analyzed for nitrate, nitrite, formate, oxalate and potassium
carbonate. Table 4 shows the results of analysis of the feed prior to
mixing with ammonia and of the water used to scrub the sample of
vapor and salts. Adjusting the scrubber solution concentrations to the
same carbonate level as the feed prior to mixing with ammonium
hydroxide shows that formate and oxalate were completely
decomposed, nitrate was 99+% decomposed, and nitrite was 90%
decomposed. Nitrogen oxides were detected in the gaseous effluent
from the unit in amount corresponding to 0.24 mole of nitrogen oxides
per 1.00 mole of nitrite plus nitrate fed. Under the same conditions
without ammonia added to the feed, nitrogen oxides were detected in
the effluent in amount of 0.86 mole of nitrogen oxides per 1.00 mole of
nitrite plus nitrate fed. Thus, nitrogen oxides were reduced by 72%
when ammonia was fed.
n-moss 217 ~ ~ 3'~
- 22 -
,~ cal ~o 0 0
O W "~ o
ai
0 os
0
o ~ o
w
ai
z~
H '
-
z~ ..
0
~U
a~
0 0
o
~
,~ crJ
o
U
A
0
O O
~
~ ob ~
;S
r~ 1i ir N
~
W
~
V C~
b~
'
O
~i
D-17085 217 5 ~ 3 ~
-23-
Bxam lie 5_
Into a 6.5-inch diameter by 11.5-foot long rotary calciner heated
at 620°C were fed over 52 minutes approximately 13 pounds of dried
salts from Benfield solution. The salts contained 3.10 wt. % nitrate
ion, 3.35 wt. % nitrite ion, 1.94 wt. % oxalate ion, and 5.1~ wt. %
carbonate ion. Salt residence time within the calciner was
approximately one-half hour. Nitrogen at 3 cu. ft. per min. was fed
countercurrently relative to salts through the calciner. The oxygen
level in the gas exiting the calciner was 9 volume percent. Calcined
product, 8.1 pounds, was obtained, which contained from grab sample
analysis 0.12 wt. % nitrite ion, 1.57 wt. % nitrate ion, 0.00% oxalate
ion, and 0.00 wt. % formate ion. Nitrite decomposition was 96%,
nitrate decomposition was 53%, oxalate and formats decompositions
were 100%.