Note: Descriptions are shown in the official language in which they were submitted.
wo gsll3834 ~ 1 7 6 2 ~ 9 PCI7US94/13387
.,
1,
DESCRIPTION
Chi~exic Oliqrn~ eo~ C ""~
Field of the Invention
The present invention relates to antisense oligonu-
cleoside compounds cnnt~;n;n~i modified internucleoside
linkages, and optionally other structural modifications.
The compounds are capable of hybridizing to target nucleic
5 acid sequences and activating RNaseE~-mediated cleavage of
the target.
Related AD~liCati
This int~rn~tinn~l application is a rnnt;nll~tion-in-
part of commonly-assigned U. S . Patent Application Serial
No. 08/238,177, filed May 4, 1994, which is a rnnt;n~
tion-in-part of commonly-assigned U.S. Patent Application
Serial No. 08/233,778, filed April 26, 1994, which is a
cnnt;nl~tion-in-part of commonly-assigned U.S. Patent
Application Serial Nos. 08/154, 013 and 08/154, 014, both
filed NOY ^r 16, 1993. The entire disclosures of all of
these applications are incorporated herein by reference.
B~ckaround o~ the Invention
SnnF~ rable attention hag been directed in recent
years to the design of antisense nucleic acid oligomers
20 or use in studying, treating and ~ nnc;n~ conditions
attributable to ~n~ln~nnus or f oreign nucleic acid se -
guences in living organisms. For example, it is now well
known that a nucleic acid oligomer having suitable anti-
sense complementarity to a target mRNA can hybridize to
25 the target mRNA and, in some cases, disrupt translation of
the mRNA. The antisense approach presents great promise
or the eventual therapeutic treatment of disease condi-
tions caused by foreign te.g., viral) genetic material, or
by misfunctioning or altered eldoyell~us genetic material
30 (e.g., cancer and genêtic disêase conditions).
wo 95/13834 PCr/l~S94113387
21 76259
However, despite the great promise of the antisense
approach, a number of challenge3 still remain. First,
antisense compounds are generally subject to degradation
in the cellular milieu due to endogenous endo- and exonu-
cleases . While a number o~ ~odif ied antisen3e ~tructures
have been described having improved resistance to nuclease
degradation, further i ~ uv~ q are desirable in order
to increase the potency and half-life of the, ~ u-lds.
Second, it is generally required that an antisense com-
pound have a high 3pecificity toward the intended target
nucleic acid so aE to avoid disruption of activity of
unintended native sequences. Although a number of re-
searchers have described approaches designed to increase
the binding af f inity of an antisense compo-.md to a target
sequence, very few results have been reported with respect
to structural r~f; n ts which avoid disruption of the
activity of unintended genetic sequences while still
rf-~ ~; n; ng maximum ef f icacy against the target sequence .
One approach toward disrupting the expression of
undesired target mRNAs involves forming a duplex hybrid
between the target mRNA and an antisense strand, followed
by cleavage of the target mRNA by an ~ldu~:uuus RNaseH.
See Dash, P., et al, Proc. Natl. Acad. Sci. U.S.A.
84 :7896-7990 (1987) . Xowever, because the mode of action
of RNaseH is fairly speciflc, this approach is subject to
a number of constraints. First, }~NaseH enzymes act in
nature to cleave the oligoribonucleic acid strand of an
oligodeoxyribonucleotide-oligoribonucleotide duplex, but
do not cleave DNA-DNA or RNA-RNA duplexes. This ha6 been
attributed, at least in part, to the polar nature of DNA-
RNA hybrids which, in contrast to DNA-DNA and RNA-RNA
hybrids, have 2'-O~I groups on one (but only one) strand.
Crouch, R.J. & Dirksen, M. -L., "Ribonucleases H, ~ in
Nucleases (Linn ~ Roberts, eds . ), Cold Spring Harbor
Laboratory (1982), at 212. As a result, one putative
requirement of the antisense RNaseH cleavage approach is
that at least some of the nucleosides of the antisense
~W0951l3834 ~B2~9 PCT/US94113387
nucleic acid strand must have characteriatics in common
with deoxyribonucleotides (a5 opposed to ribonucleotides~,
particularly, the absence of a polar yroup on the 2 ~ -
position of the antisense nucleoside sugars. Perhaps
5 related to this is the additional requirement that at
leagt some of the sugar groups in the Ant1 ~n~e compound
must be in a 2'-endo (~) conformation as found in deoxy-
ribonucleosides, as opposed to the 3'-endo (~) conforma-
tion found in ribonucleosides. Cook P.D., PCT Publication
No. W0 93/13121 (1993), at 18-19.
It has further been reported that various 2~-position
substituents (e.g., 2~-0-alkyl and 2~-fluoro) will render
the substituted portion of an antisense strand non-acti-
vating to RNaseH, even though binding affinity toward the
target nucleic acid is increased . Inoue , H ., et al ., FEBS
Letters 215 ~2~ :327-330 (1987); Monia, B.P., et al., J.
Biol. Chem. 268 (19) :14514-14522 (1993) . Likewise, the
Monia, et al. report indicates that a minimum of five
consecutive 2 ~ -deoxy residues is required in order to
achieve efficient activation of ~ n (HeLa) RNAaeH,
and that this 2'-deoxy segment (if ~cl~ ;ed by 2'-
substituted residues in the same antisense compound) must
be centered in the oligomer sequence in order to achieve
efficient RNaseH activation in vitro or expression inhibi-
tion in cells.
Another reported requirement of ~the antisense F~NaseH
cleavage approach is that, in order to achieve RNaseH
activation, at least one portion of the ;ntprn~ ]~side
"backbone" of the antisense, ~ olln~ must include charged
(anionic) rhnsph~rus-c-nt~;n;n~ linkage groups. Cook,
P.D., PCT Publication No. W0 93/13121 (1993), at 18. In
studies of chimeric antisense compounds including both
methylphosphonate (uncharged) and rhnsrhr,r~; ester or
phosphorothioate (charged) linkages, Agrawal, et al.
reported that the minimum number of consecutive charged
h~rl~h~n~- linkages required for efficient activation of
r -l;~n RNa5eH in vitro is five. ph~sph~ ;ester linkag-
_ _ _ _ _ _ _ _
Wo 95113834 PCrlUS94/13387
21~2~ `
es positioned in either the t~rm;n~l or center portion of
the oligomers were reportedly more ef f icient than phos-
phorothioate linkages in activating RNase~I, whereas
oligomers rnnt~;n;ng only methylrhnsphnn~te, phn~phnro-N-
morpholidate or phosphoro-N-butylamidate linkages were
inactive . Agrawal , S ., et al ., Proc . Natl . Acad . Sci .
U.S.A. 87:1401-1405 (1990) .
While phosphodiester linkages, being charged, are
suitable to allow activation of RNaseX, they suffer from
the disadvantage of being subject to degradation by
naturally-occurring endo- and/or exonucleases. A variety
of alternative linkage groups, some of which are nuclease-
resistant, have been developed or proposed for use with
antisense compounds. Among these are charged linkage
groups such as phosphorothioate, phosphorodithioate,
phosphoroselenate and phosphorodiselenate linkers. In
general, deoxyribonucleoside antisense oligomers contain-
ing these non-natural linkage groups tend to have lower
binding af f inity toward complementary RNA target strands
than the corresponding phosphodiester-linked antisense
oligomers, although higher af f inity may be achieved where
the antisense strand comprises r; hnnl~rl en~ides or 2 ' -
substituted ribonucleosides (rather than deoxyribonucleo-
sides). See Metelev, V. & Agrawal, S., PCT Publication
No. W0 94/02498 (1994), at 9. Among the uncharged phos-
phorus-containing linkage groups that have been reported
are the alkylrhn~rhnn~te (e.g., methylrhnsphrn~te), aryl
rhn~Fhnn~te, alkyl and aryl rhnsrhnramidate, alkyl and
aryl phosphotriester, 1IydLu~ phosphonate, boranophos-
phate, alkyl and aryl phosphonothioate, phosphoromor-
pholidate, and rhnsrhnropiperazidate linkers. See Cook,
P.D., PCT Publ;r~tjcm No. W0 93/13121 (1993), at 7;
Pederson, T ., et al ., U. S . Patent Nos . 5 ,149, 797 and
5,220,007; Padmapriya, A. & Agrawal S., PCT Publication
No. WO 94/02499 (1994). Non-phosphorus-based linkage
groups have also been reported, ; nrll~li n~ peptide, mor-
pholino, ethy~ene glycol, amide, and other linkers. See
Wo g~/13834 ~ 2 1 7 6 2 ~ g `` PCTtllS94/13387
Reynolds, M.A., et al., PCT Publication No. WO 92/02532
(1992); Cook, P.D., PCT Publication No. WO 93/13121
(1993), at 7. As with the charged phosphorug--~nnt~;n;n~
linkers noted above, many of these other non-natural
5 linkage groups may exhibit lower binding affinity (com-
pared to phosphodiester linkages) toward complementary RNA
target strands, at least in the case of linked 2 ' -unsub-
stituted ~ntiqPnqe nucleotides, and particularly in the
presence of salt ions.
Various workers have attempted to identify combina-
tions of linkage groups and/or structural modifications
for antisense oligome~s that might lead to improved RNaseH
activation, binding affinity, nuclease resistance and/or
target specif icity . Thus, Cohen, et al . have reported
improved half-life for antisense and non-ilnt; q~nqe oligo-
deoYyribonucleotides ~-Qn~;n;n~ at least one phosphoro-
thioate linkage located, for example, at either terminus
of the c ~ In~l, or throughout the compound. Oligomers
cnnt~;n;n~ all phosphorothioate linkages were shown to
have anti-viral (anti-HIV) activity, wherea:
phosphodiester- and methylrhnsrhnn~ tP- linked compounds
were reportedly inactive . Cohen, J . S ., et al ., U . S .
Patent No . 5, 264, 423 . Walder et al . have proposed the use
of a 3'-terminal non-phosphodiester linkage, optionally
combined with a 5 ' -terminal non-phosphodiester linkage or
a 5'-tPrm;n~l "cap" group, to avoid 3'-initiated (and
optionally 5 ' -initiated) exonuclease degradation of
oligodeoxyribonucleotides. RNaseH cleavage activation
reportedly required retention of at least four, and
preferably at least seven, contiguous phflsrhr~; ester
linkages in the antisense oligomer. The preferred com-
pounds contained at least lO, and preferably at least 15,
nucleotides, the majority of which were phosphodiester-
linked. Walder, ~.A., et al., PCT ~Publication No. WO
89/05358 (1989). Padmapraya & Agrawal have reported that
the incorporation of nonionic alkyl or aryl rhn,~phnn~-
thioate liDka~e~, preferably at one or both termini of the
_ _ _ _ . . . , ,, , ... _ ... _ .. _ . ,, . , .. . . _ . ,
WO 95/13834 ' PC rlUS94113387
oligomer, res~lted in; uv~d nuclease resiætance, albeit
with a reduction in Tm of 1-2C/rhnsrhnnnthioate linkage.
PCT Publication No. WO 94/02499 (1994).
Pederson, et al. have reported the use of "mixed
5 phosphate h~rkhnn~" oligomers cnnt~inln,r both a phospho-
diester- or phosphorothioate-linked segment for RNaseH
activation, and one or more non-RNaseH-activating, un-
charged linkage group segments. It was found that a
segment of five or 8iX consecutive phosphodiester linkages
10 was efficient, in a 15-mer compound, to effect RNaseH
cleavage of a target RNA strand, whereas similar compounds
with fewer phosphodiester linkages, or with up to six
consecutive phosphorothioate linkages in place of the
rhnRrhn~liester linkages, had low activity. Pederson, T.,
et al., U.S. Patent Nos. 5,149,797 and 5,220,007.
Giles & Tidd have reported that the target specifici-
ty of an ~nt; ~n~e oligomer can be improved by the use of
a chimeric structure comprising t~rmin~l methylrhnsrhnnn-
diester sections separated by a central RNaseX-activating
20 phosphodiester region having a high A+T to G+C ratio. The
observed reductions in non-specific cleavage were attrib-
uted to the lower Tm caused by the methylphosphonate
segments, the reduced hybridization strength of the small,
A/T-rich phosphodiester region, and the reduced prospects
25 for partially-complementary hybridization at the shortened
RNaseH activation site. Giles, R.V. & Tidd, D.M., Nucl.
Acids Res. 20 (4) :763-770 ~1992) .
Ohtsuka, et al. have described the use of partially
2~-substituted (e.g., 2'-lower alkoxy substituted)
30 oligomers for, site-specific RNaseH cleavage of RNA targets
with or without secondary structure. RNaseH cleavage was
reportedly localized to a site (or sites) on the target
corresponding to the non-substituted (i.e., deoxyribonu-
cleotide) portion of the antisense compound. Single-site
35 cleavage was reportedly optimized by use of a tetradeoxy-
r; hnnllrl Potide segment located centrally in the compound
between two 2'-substituted terminal segments. Inoue, H.,
~76259
Wo 95t13834 Pcr/uS94113387
et al, FEB Letters ~:327-330 (1987); .qh;hAh~ra, S.,
et al., Nucl. Acids Res. ~ :4403-4415 ~1987); Ohtsuka,
E., et al., U.S. Patent No. 5,013,830. The use o~ par-
tially 2'-substituted oligomers additionally rf~ntil;n;ng
5 one or more non-phosphodiester linkages has also been
reported. See .~h;h~hA~a, S., et al., European Patent
Application Publication No. 0 339 842 A2 (1989) (reporting
3'-5' or 2'-5' linked oligomers having phosphorothioate or
other linkages); Cook, P.D., PCT Publication No. WO
93/13121 (1993) (reporting increased binding affinity
attributable to 2'-substitutions, and nuclease resistance
attributable to , e . g ., ph~srhnrothioate and phosphoro-
dithioate linkages); Monia, B . P ., et al ., J . Biol . Chem.
2Ç8(19) :14514-14522 (1993) (reporting effects of 2~-
15 substitutions in phosphorothioate-linked oligomers);
Metelev, V. & Agrawall S., PCT Publication ~o. Wo 94/02498
(1994) (reporting use of 2 ' -substitutions in phosphoro-
thioate- or ~hf~srhnrodithioate-linked oligomers); McGee,
D.P., et al., PCT Publication No. WO 94/02501 (1994)
20 (describing preparation of various 2'-substituted nucleo-
sides and ~h~sph~amidites)~
- of tbe Invention
The present invention relates to improved RNa6eH-
activating Ant;qPnqe oligonucleoside, , ~lq rnntA;n;n~
25 selectively modified ;n~rnllr~eoside linkages, and option-
ally other structural modifications. The compounds
exhibit improved target specificity and potency ~_...~a~ ~d
to other RNaseH-activating antisense ~ . They are
useful both in vivo and in vitro in reducing or ~1 ;m;ni:lt-
30 ing the translation of target mRNA sequences, most prefer-
ably sequences related to disease conditions.
In one aspect, the present, ~ lq incorporate one
or more polynucleoside segments having chirally-pure or
chirally-enriched modified (non-~h~-sph~ ;ester) inter-
35 nucleoside linkages. The chirally-selected linkage
8egments are preferably 8elected to include linkages
,, , , , _ , . . , , _ . .... ,,, . . , _ . _ . .. .
Wo 95113834 2 1 ~ ~ 2 5 9 PCrlUS94113387
;
having R chirality at the asymmetric phoaphorus atom of
one or more of the linkage structures ( "Rp chirality" ) .
Preferably, at least about 40% of the linlcages in a given
chirally-selected segment will be Rp-chiral. Also included
5 are segments selectively including one or more Sp-chiral
linkages. In one preferred embodiment, chirally-selected
segments are situated at the tor~;nAl (3' and 5' ) portions
of the compound, surrounding (fl~nk;ng) a central RNaseH-
activating re~ion . The f lanking chirally- selected seg-
10 ments preferably are subst;~nt;;~lly non-RNaseH-activating.
The RNaseH-activating region, if linked with asymmetric
(chiral) linkage groups, may alternatively or additionally
be chirally selected. In a related embodiment, the
RNaseH-activating region is situated at or near one
15 terminus of the compound, and all or a portion of the
~, ;n~r of the compound is chirally selected and prefer-
ably is non-RNaseH-activating.
The chirally-selected Rp-~nr; rh~l segments of the
invention serve to increase the binding af f inity of the
20 compound as compared to racemic ~ 1uu--ds. In addition,
because the chirally-selected modified linkage structures
are more resistant to degradation by endo- and/or exonu-
cleases than are non-modified phosphodiester linkages, the
chirally-selected segments will tend to protect the
25 compound from degradation in the in vivo environment.
In another aspect, the present ~ __ 'q incorporate
one or more 1 polynucleoside segments comprising mixed
modified (non-phosphodiester) ;nt~rn1l~1eoside linkages.
Two or more ~ifferent ;nt~rnllcl~nR;de linkage structures
30 are ;n~ in the mixed linkage segment, and one or more
of these may be a modified linkage structure. One or more
of the linkage structures in the sequence may be chirally
selected. Preferably, the mixed linkage segment includes
multiple linkage sequence blocks (synthons) each contain-
35 ing two or more different ;ntPrn11clenc;r~ linkage struc-
tures, or a single such synthon that is repeated two or
more times n the mixed linkage segment. Where the
_ _ _
Wo 95/13834 PCT/US94113387
~176259
compound r~ntA;nc more than one mixed linkage segment, the
linkage sequence blocks may be the same or dif f erent in
the respective segments. In one preferred embodiment,
mixed linkage segments are situated at the terminal
(flanking) portions of the compound, surrounding a central
RNaseH-activating region. The RNaseH-activating region
may alternatively or additionally comprise a mixed linkage
segment. The fl~nk;ng mixed linkage segment5 are prefera-
bly non-RNaseH-activating. In a related embodiment, the
RNaseH-activating region is situated at one terminal
portion of the compound, and all or a portion of the
,~ ;n~l~r of the compound contains a mixed linkage segment
and preferably is non-RNaseH-activating.
The mixed linkage segments of the invention may be
racemic or chirally selected; in either case the identity
of the int~rn~ ncide structures and/or the linked
nucleoside substituents can be selected to afford greater
binding affinity to the compound while ~~~;ntc;n;n~ target
specificity and nuclease resistance and increasing poten-
cy. Because the mixed linkage segments of the compound
include one or more modified ;nt~rn~1- leoside linkage
structures that are resistant to degradation by endo-
and/or exonucleases, the ~ ~uul~ds will have higher
potency in the in vivo environment.
In another aspect, the present invention ; n~ Pc
uved RNaseH-activating segments comprising linked
n-1~le~Pides having mixed int~rnllolpr~siclp linkages. In one
preferred : ~ 1; t, the R~aseH-activating segment
;nA1~ c at least five consecutive 2~-unsubstituted li.e.
DNA) n~ residues linked by two or more differe~t
charged (anionic) ;nt~rnll- leoside linkage structures in an
alternating sequence. Preferably, the RNaseH-acti~ating
segment includes at least four such charged ;nt~rnl~rleo-
side linkage structure5. One or more of the internucleo-
side linkage structures in the RNaseH-activating segment
may be chirally selected if an asymmetric phosphorus atom
is present in the linkage 8tructure.
_ _ _ _ _ _ _ _ . .. .. . . ,, _, , .. . _, .... ...
wo 9~/13834 ~ PCTNS94/13387
In another a3pect, the pre3ent invention provide3
chimeric structures for anti3en3e oligonucleoside com-
pounds that maximize activity while r-int~;n;nrJ the
ability to effect 3elective RNaseH-mediated cleavage of
5 the intended target strand These goals are achieved by
structure3 which provide, on the one hand, controlled
binding affinity and, on the other hand, controlled
RNaseH-activation char~rtf~r; Rt; c3 .
Thus, in one ~mhn~;r t, binding a~finity is con-
lO trolled (3electively increa3ed) through the u3e ofchirally-3elected Rp-chiral internucleo3ide linkage3 in one
or more portion3 of the ~ ~CJU~1~. Alternatively or
additionally, one or more Sp linkage3 may be used to
3electively decrea3e binding af f inity In a related
15 embodiment, binding affinity i3 controlled (3electively
increa3ed) through the use of multiple or repeated linkage
seriuence blocks (3ynthon3) in one or more mixed linkage
segments of the compound; the linkage structure3 may be
racemic or chirally-3elected. In another related embodi-
20 ment, binding affinity is controlled (selectively in-
crea3ed) through the u3e of 2'-3ub3tituent3 on one or more
nucleo3ide sugars in the compound, preferably in conjunc-
tion with altPrn~t;n, linkage segments and/or chirally-
selected int~rnllrl ~n~ l ink~ , RNaseH-activating
25 characteristics can simultaneously be controlled (substan-
tially eliminated, or selectively increased) in these
segments of the ~ _ ~1 by the use of 2 ' -substituted or
unsubstituted nucleo3ide sugars and/or by the selection of
uncharged or charged linkage structures for a given
30 segment of the co~n~o11n-1.
Likewise, RNaseH-activation characteristics are
controlled (selectively increased or decreased) by the
selection of mixed or uniform charged ; nt -~nllrl eoside
linkages in the RNaseH-activating region of the compound
35 RNase~I-activating characteristics can be selectively
decreased, particularly in the RNaseH-activating region of
the compound, by the use of linkage structures such as
W0 95/13834 217 6 2 S 9 PcrNS94/13387
~hc~grhnrothioate or especially phosphorodithioate struc^
tures that are poorer substrates f or RNaseH . RNaseH-
activating characteristics are also controlled by the
inclusion of non-R~aseH-activating portions in the com-
r 5 pound such that only a portion of the compound is effec^
tive in activating cleavage of the target genetic se-
quence, f or example by d,U~l u~ ~ iate selection of linkage
structures, 2'-substituents and other features as de-
scribed herein.
Among the highly preferred compounds of the invention
are those having subst~nt;~lly non-RNaseH-activating,
chirally-selected, mixed linkage segments at the two
terminal (flanking) portions of the ~ . uul.d, and an
RNaseH-activating region positioned therebetween. Also
preferred are, ~ A.c: having subst~nt;~lly non-RNaseH-
activating, racemic mixed linkage segments at the two
terminal (flanking) portions of the compound wherein one
or more of the linked nucleosides in the mixed linkage
segments is 2'-substituted, and an RNaseH-activating
region is positioned in the compound between the mixed
linkage segments . Especially pref erred compounds include
those chosen from the following structures:
'-T~r"l~.~l R~ Acti~.r~tlng 3~-T-nnin~l
Portio~ R-~io~ Portio
25 YP ~R) /DE DE IIIP !R) /DE
2 ' OMeMP 1 R ) / 2 ' OMeDE PS 2 2 ' OMeMP ~ R ) / 2 ' OMeDE
YP ~ ~) t 2 ~ OMeMP P S MP ~ R ) / 2 ' OMeMP
llP ~R) eslriched P82/DE MP ~R) enriched
2 ' OMeNP ~R) enriched P8 /DE 2 ' OMeMP ~ R) enriched
3 0 XP ~R) /P8 PS/P82 MP ~R) /PS
2 ' OMeMP (R) /2 ' OMeP8 2 ' OMeMP ~R) /2 ~ OMePS
IIP ~R) /P52 ~P ~R) /P82
2 ' OMeMP ~ R ) / 2 ' OMePS 2 2 ' OMeMP ~ R ) / 2 ~ OMeP8 2
2 ' OMeMP / 2 ' OMeDE 2 ' OMeMP /2 ' OMeDE
3 5 MP/2 ' OMeDI~ MP/2 ' OMeDE
MP (R) /PA~n ISP ~R) /PAIIL
2 ' OMeMP ~R) /2 ' OMePAm 2 ' OMeMP ~R) /2 ' OMePA~
2 ' OMeYP /2 ~ OMePAm 2 ' OMeMP / 2 ' OMePAm
llP/2 ' OMe~Am MP/2 ' OMe~A~
WO 95113834 2 1 7 6 2 5 9 PCTIUS94/13387
,., . ~.
12
MP (R) /TE MP !R) /TE
2 ' OMeMP (R) /2 ' OMeTE 2 ' OMeNP (R) /2 ~ OMeTB
2 ' OMeMP/2 ' OMeTE 2 ~ OMeMP/2 ' OMeTE
MP / 2 ' OMeTI~ MP / 2 ' OMeTE
5 MP (R) /MPS . llP (R) /MP8
2 ' OMeMP (R) /2 ' OMeMPS 2 ' OMeMP (R) /2 ~ OMeMPS
2 ' OMeMP/2 ' OMeMPS 2 ~ OMeMP/2 ' OMeMPS
MP/2 ' OMeMPS MP/2 ' OMeMPS
MP (R) /PF MP (R) /PF
2 ' OMeMP (R) /2 ' OMePF 2 ' OMeMP (R) /2 ' OMePF
2 ' OMeMP/2 ' OMePF 2 ' OMeMP/2 ' OMePF
~P/2 ~ OMePF MP/2 ' OMePF
MP (R) /PB~3 MP (R) /PBB,
2 ' OMeMP (R) /2 ~ OMePBE, 2 ' OMeMP (R) /2 ~ OMePB}I,
2 ' OMeMP/2 ' OMePBM~ 2 ' OMeMP/2 ~ OMePBI}3
MP/2 ' OMePBII, MP/2 ' OMePBX,
MP (R) /Rsi MP (R) /Rsi
2 ' OMeMP (R) /2 ' OMeRSi 2 ' OMeMP (R) /2 ~ OMeRSi
2 ' OMeMP / 2 ' OMeRSi 2 ' OMeMP/2 ' OMeR8 i
2 0 MP/2 ' OMeRSi NP/2 ' OMeRSi
MP (R) /C}I~ MP (R) /C}I,
2 ~ OMeMP (R) /2 ' OMeCHl 2 ~ OMeMP (R) /2 ' OMeOE
2 ~ OMeMP / 2 ' OMe CJIl 2 ' OMeMP / 2 ' OMe CH
MP/2 ' OMeCEIl MP/2 ~ OMeCHl
~y: MP . racemic methylr~ - linkage (between linked
n~lrl~ .e); MP(R) ~ chirally-selected Rp-methylrhnerh~nA~e
lincage; DE - rh~erhn~ Rter linkage; PS = ~ a ~ L~Lh_oate
lin~age; PS2 = PI~JA~I~VL~ h; ~A'-~ li~kage PAm = 1 ~1A to
lincage; TE ~ ~I.oa~1wLLieater linkage; MPS ~ alkyl (particu_arly
metlyl) ~11oa~ ,L~,Lhioate linkage; PF = ~ A~ JLI~1UOr date
lin~age; PB}I~ linkage; Rsi = silyl (espec_ally
alkyl-disuhstituted silyl) linkage, C}~i = f~rr^--e~Al linkage
2'0Me ~ 2'-methoxy-s--hA~ (or cther lower alkoxy, allyloxy
or halo 8~hA~;t~ ) n~rlf.~ o reaidue, linked using the listed
liLkage structurei "enriched`' refers to a segment of li~kages
preferahly ~ n~A;n;ng at least ahout 40~ (and up to 100'6) Rp-
aelected linkages among the linkages in the segment and thus
includes a mixed se~uence of racemic and chirally-selected R
;nr~rn~ rlr~r~A;Ar~ linkage aLLu~.LuL~_; linkage DLLu~ LUL_D grouped
with slashes denote a mixed linkage segment including the listed
linkage DLLuuLULC:s~ optionally in a serie.s of multiple or
repeated mixed linkage seguence blocks.
In another aspect, the present invention includes
improved antisense oligonucleoside compositions useful in
treating or diagnosing diseases or other conditions in
living or~anisms attr;h~l~Ahle to the expression of endoge-
nous or f oreic~n genetic inf ormation . The compounds and
-
Wo 95/13834 PCINS94/13387
~176259
13
compositions are also useful in studying such conditions
in vitro or otherwise. In another aspect, the invention
provides methods for treating, diagnosing or studying such
conditions .
Other aspects and objects of the invention will be
apparent from the following detailed description.
Briel~ De~criDtion of the Draw; n~
FIG~RES 1 and 2 are graphs showing nuclease stability
of various compounds and segments of the present inven-
tion, compared to other mixed linkage compounds, over
time .
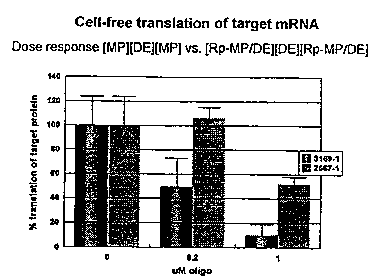
FIG~ S 3 and 4 are bar graphs showing dose-response
activity of a chirally-selected compound of the present
invention, versus a non-chirally-selected ,- ~, in
inhibiting target (Fig. 3) and non-target (Fig. 4) protein
synthesis .
FIG~JRE 5 is a graph showing RNaseX activity of a
chirally-selected compound of the present invention,
versus a non-chirally-selected compound, over time.
FIGllRES 6-lO depict sythong and ;n~l 3;;ltes useful
in constructing ~ ,uullds of the present invention.
FIG~RE ll is a graph showing kinetic data relating to
RNA cleavage by various 2'-sugar-substituted and unsubsti-
tuted ~ ou~ds of the invention.
Detailed De2~cri~tion
A full appreciation of the present invention requires
an understanding of the competing parameters underlying
the present RNaseX cleavage technique. There are a number
of parameters of primary concern, including oligonucleo-
side-target binding affinity, RNaseX cleavage rate,
specificity/mismatch effects, oligonycleoside displacement
by processing ribosomes, and nuclease stability. As will
be seen from the following discussion, a proper balance of
these competing parameters requires that the oligonucleo-
~ide compound have a binding affinity (as quartitated for
.. ... ,,, _, , , _ _ _
WO 95113834 ~ 1 ~ 6 2 ~ ~ PCr/US94/13387
t, ~
14
example by the af f inity constant KA) that is not too large
relative to the RNaseH cleavage rate. The present inven-
tion provides structures that satisfy this requirement as
well as other re~ i ~ outlined below .
The present technique of RNaseH cleavage of a target
genetic sequence requires that the oligonucleoside com-
pound hybridize with the target sequence, and that the
oligonucleoside have a hybridization occupancy time that
i8 sufficiently long to effect cleavage of the target
sequence by the RNaseX enzyme. The initial step of
oligonucleoside-target hybridization is governed, from a
first-order kinetic standpoint, by the forward and reverse
rate constants (kl and k l) that define KA' where KA = k1/k 1-
The rate of cleavage of the target (which is essentially
irreversible) is then governed by the rate constant k2, as
~ollows:
Oligomer + Target Strand
kl I ~ k l
Hybridized Oligomer/Target Strand
k, ~ [RNaseH]
Released Oligomer + Target Cleavage Fragments
Other considerations aside, it would appear that
target cleavage would be optimized by ~-~r;mi7.;n~ both KA
and k2. However, this does not take into account the
problem of non-specific binding (i.e. mismatches) between
the oligonucleoside and lln;nt~on~d nucleic acid sequences
that exist in the cleavage (e . g . cellular) medium which
could result in undesired cleavage of the lln;n~n~
sequences. Nor does this simple approach take into
account the fact that an oligonucleoside with high binding
affinity will typically be displaced from its hybridized
state, and thus will be unable to activate RNaseH-mediated
cleavage, each time the host ribosome processes along the
target ,mRNA sequence.
Wo 95/13834 2 1 7 6 2 5 9 PCr/US94/13387
Consider first the rhilllPnr,e of achieving high target
speci~icity with an antisense cleavage compound. Mammali-
an cells typically contain an RNA population comprising
about 3 x 10' ribonucleotides. By assuming a statistically
5 random distribution of the four naturally-occurring
nucleotides within this pop~ t; nn, the total number of
"match" ser~uences in the population having exact base-by-
base compl~ -t~rity, and the number of "mismatch'~ se-
quences having one or more base mismatches, can be approx-
lO imated for a target sequence of any given length. (O~course, the actual distribution of ri hnnl~r~ eotides in a
given 1 i~n cell population will not be truly random,
but nevertheless such statistical analyses can shed light
on the probabilities of a mismatch sequence occurring. )
15 The following table lists the number of targets that would
exist in such a population as a function of number of
mismatches (zero to five) and target ser~uence length (12,
15 or 18).
M~ ~ trh~ encrth ~rget~
0/12 1 . 8
0/15 2 . 8 x 10-2
1/15 1 . 24
2/15 26
3/15 340
0/18 4 . 4 x 10-~
1/18 2 . 4 x 1o-2
2/18 0 . 62
3/18 9 . 6
4/18 109
30 5/18 930
It will be seen that an appreciable number of pote~tial
mismatch seg,uences may exist even for target sequences as
long as 12 nucleosides, particularly as the number of
single-base mismatches increases. If the K,~ for a given
35 mismatch duplex is sufficiently high as to allow apprecia-
ble hybridization of an ~nt;cPn~e oligomer to a mismatched
Wo 9S/I3834 ~ ~ PcrluS94/13387
2~7~2~
-
16
target, then unintended and undesirable cleavage of the
mismatched target can result.
Take, for example, the case of a one-base mismatch
between a 12-to-18 nllc~ en~ anti3ense oligomer and an
unintended mismatch R~A sequence. The present inventors
have a8certained that the KA for the correct "match"
hybridization typically does not exceed the KA for the
incorrect "mismatch" hybri-l;7at;nn by more than a factor
of one hundred. Furthermore, the forward rate constant of
hybridization (k1) will be approximately the same for both
the match and the mismatch, because the forward hybridiza-
tion i9 typically governed in large part by the physics of
solution-phase intermolecular exposure which tend to
obscure the effect of the single-base mismatch. In this
case, the hybr;~l;7At;nn ~'off rate" (k l) can be no more
than 100 times greater for the mismatch than for the
correct match. It will now be seen that, if the cleavage
rate constant k2 is not subst~nt;~lly smaller than the
reverse rate constant k 1 for the mismatch, then unintended
mismatched nucleic acid sequences will be cleaved (along
with the properly matched target sequence). It will also
be seen that specif icity f or the intended target se~auence
will be optimized if k2 has a value on the order of
k l (match), but much less than k l (mismatch):
k l (match) -- k2 c~ k l (m; F~m-tnll)
In addition, the present invention takes into account
the ribosomal displ ~c t of hybridized oligonucleosides
that typically occurs in the coding region of a target
mRNA during the process of R~A translation. The ribosomal
pron~qsi nn~l rate varies somewhat from RNA to R~A but in
general is calculated to pass any single point on an mR~A
every 10-15 seconds . If the KA (match) for a given oligo-
nucleoside is 101 M~l and the KA(mismatch) is lO~ M~1, then
the half-life hybri~;7~t;nn occupancy times (tl/2) will be
about 28 minutes and 17 seconds, respectively, for the
match and the mismatch. But because the r;hosn--~l proces-
sional rate i~ 80 fast, the correctly-matched oligonu-
Wo 95/l3834 2 17 ~ 2 ~ 9 PCr/US94113387
17
cleoside will be displaced from the target sequenee just
about as frec~uently as the m; I trh~ oligomer, and the
effective oceupancy times will be approximately the same.
The result in this case is that, from a specificity
standpoint, the high affinity constant for the correctly
matched hybridization goes for naught, and nonspecific
cleavage will occur at lea8t as frec~uently as the intended
sequenee-specific cleavage. In fact, nonspecific cleavage
may occur even more frequently if more than one mismatch
sequence exists in the "target" RNA population.
Given considerations such as these, the present
inventors have discoYered that it is beneficial to limit
the binding affinity constant of the subject RNa8eE~-
activating oligonucleoside compounds to values that are
typically no greater than 10l M~l for targets in the coding
region of a target mRNA. Preferred K,~ values for the
present compounds are in the range 107-10l M~l. In such a
ease, beeause the ~off rate" will be relatively high
eompared to compounds with higher binding affinities, it
is possible and desirable to utilize compounds having a
relatively high cleavage rate. Thus, the inventors have
discovered that it is benef icial to control the cleavage
rate constant of the subject compounds to values in the
range of 1 to 10-5 sec~l, preferably 10~1 to 10-~ sec~i, and
most preferably 10-' to 10-3 sec 1 The cleavage rate is
preferably selected to give at least a 3 :1 cleavage rate
of a perfect "match" relative to a 2-mismatch target.
In eontrast, in the non-coding region of a target
mRl~A site (e.g., the 5~-cap region, the 5'-untranslated
region, the initiation codon region, the 3 ' -untranslated
region, splice acceptor or donor sites, intron branch
sites, and polyadenylation site8), inhibition of protein
prorl1lrt; ~n can be achieved prior to the translation
process by suitable hybridization of an antisense oligonu-
eleoside, and r;hc~ pl~ of the hybridized
oligomer generally does not occur. As a result, oligonu-
eleosides having higher binding affinities ~and higher
_ _ _ _ _, _ _ . .. . . ... .. . .. _ . . . _ _ _ _
WO 9S/13834 ~ ~ . PC rlUS94/133~7
2176259
18
half-life occupancy times) can be utilized in the non-
coding region without the 1088 of 6pecif icity described
above with respect to the coding region. In this case, an
upper limit on binding affinity will be imposed by the
lif etime of messages in the mRNA pool relative to the
lifetime of mismatch hybrids. Thus, the lifetime of a
typical mRNA molecular species ~taking into account
repl~n; ~' -nt of the mRNA pool via transcription) is on
the order of five hours. If the hybrid lifetime of
mismatch sequence approaches an hour or more, then the
translation of the mismatched message will be p~LLuLl-t:d by
steric blocking effects apart from any RNaseH cleavage
r -h~n;~T As a result, KA(match) should generally be in
the range 107-1013 M-1. Furthermore, a relatively low
concentration of oligonucleoside is preferably used in
this case so that the total level of mismatch occupancy
over time (in addition to the miamatch hybrid lifetime of
a single m; o~--t.h~d oligonucleoside) is low. (Of course,
the rate of RNaseH-mediated cleavage, k" should still be
much lower than k l(mismatch) for targets in the non-coding
region, just as it is for coding region targets, in order
to avoid non-specific mismatch cleavage. )
Values for KAI k1, k 1 and k, can be ascertained using
methods known in the art. The ~t~orm;n~t;~ of RAI the
equilibrium binding constant, requires the measurement of
the c~n~ n~rations (~h~ol~l~e or relative) of single and
multimeric species, as well as enough time to ensure
complete equilibration. The equilibrium hybridization of
oligomers can be studied by direct methods which physical-
ly separate the single and multi-meric species, such as
gel shift (Lima et al., Biochemistry 31, 1205~-61 (1992) ),
strand cleavage (Young, S., Wagner, R.W., Nucleic Acid
Research 19, 2463-70 (1991) ), filter binding (McGraw, R.A.
et al., BioTechniques 8, 674-678), or equilibrium dialysis
(Bevilacqua, P.C. & Turner, D.E., Biochemistry 30, 10632-
40 (1991) ) . Indirect methods rely on physico-chemical
properties of the multimeric and single-stranded states,
Wo 95~13834 Z 1 ~ ~i 2 5 9 PCrlUS94/13387
-
. .
19
and include method9 6uch a9 optical melting (Albergo, D.D.
et al., Biochemistry 20, 1409-13 (1981)), and differential
scanning calorimetry (Albergo, D.D. et al., op. cit . ) .
These publications are incorporated by reference herein.
t 5 Kinetic meatiuL q of on-rates (kl) and of ~-rates
(k l) uge many of the same detection methods as equilibrium
binding constant determinations, but rely on accurate
correlations of species formation or disappearance with
time. Off-rates can be studied by the direct methods
described above, as well as indirectly by optical methods,
and nuclear magnetic resonance of ~f~ut~rl 1~- exchange of
protons (I,eroy et al, Journal of Molecular Biology 200,
223-38 (1988) ) . On-rates can be determined ~rom Ka and k "
using the e~uation kl = KA x k 1. Mea~uL -nt of oligomer
kl can be measured by specialized kinetic techniques such
as temperature jump k;n~t;--c (Williams, A.P. et al.,
R;c~hf~miEltry 28, 483-4291 (1989), and Turner, D.H. in
Investiqati~nE3 of Rates An~ MF~-~h~n; E ~ o~ Reac~ nE3 6,
141-189) . The foregoing publications are also incorporat-
2 0 ed by ref erence herein .
It will be recognized, in light of the present
disclosure, that the above preferred values ~or binding
and kinetic constants will vary depending on the biologi-
cal system in which the present oligonucleosides are being
used . The values given above represent pref erred values
based on hybridization of the oligonucleoside to a single-
stranded target sequence that does not have substantial
gecondary structure. Where the target sequence is located
in a region of the mR~A molecule that has substantial
8econdary structure, the binding affinity of the oligonu-
cleoside with respect to the secondary-structured target
region may be much lower than that measured with respect
to a non-structured (e.g., synthetic) target sequence
having the same nucleoside sequence. In some cases the ~
for the non-structured strand may be as much as 107-fold
greater than that of the structured strand. If the
resulting ~ with reSpeCt to the intended gE~ ry-
.. .. _ ,, . ,, _ _ _
Wo 95113834 PCrlllS94/13387
217~2S~
structured target is too low relative to, for example, a
non-structured mismatch sequence, problems of specificity
may result.
One preferred approach t~o thi9 situation is to target
5 a region in the target mRNA f or RNaseH-mediated cleavage
that does not have sufficient secondary structure to
adversely af f ect the binding af f inity of the sub; ect
oligonucleoside. The secondary structure of nucleic acids
can be determined directly by the use of nucleases, base
l0 modification chemicals, or sugar-phosphate backbone
modifying reagents, as recently reviewed by Jaeger et al.,
Annual Reviews in R;rl~hPmt~try 62, 255-287 (1993).
Another approach is to utilize two or more antisense
compounds in tandem, at least one of which is a chimeric
15 oligonucleoside of the invention, which antisense com-
pounds have nucleoside base sequences selected to hybrid-
ize to adjacent regions in a secondary-structured mRNA
target region . It is known that adj acently-hybridizing
antisense compounds may be used to disrupt secondary
20 structure of RNA molecules and thus to enhance the effec-
tive KA~ 8 of the respective ~ rt~. By using this
approach, cleavage of target mRNA regions having secondary
structure may be achieved with specificity using oligonu-
cleoside compounds having controlled binding af f inity as
25 taught herein.
As ~ s-~1Rced above in the background section of this
disclosure, a number of workers in the ;Int; C~nCe field
have reporte~l various and disparate efforts to increase
binding af f inity of antisense oligonucleosides, to opti-
30 mize RNaseH activation, to improve nuclease resistance,and to improve target specif icity . It will be seen in
light of the ~ preceding detailed description that many of
these approa~hes involve competing or conflicting consid-
erations. For example, as just discussed, increased
35 binding affinity is not always desirable in view of the
problems it can create for target specificity. Certain
structures that provide increased binding affinity, such
WO95/13834 _ ~t 762$9 PCTNS94/l3387
21
as 2~-methoxy substitutions, or increased nuclease re8is-
tance, such as methylphosphonate ;n~rn~ eoside linkages,
are seemingly incapable of activating RNaseH cleavage.
Conversely, certain 6tructures that provide high RNaseH
5 activation, such as phosphodiester linkages, are nuclease-
unstable while others, such as phosphorothioate linkages
(and also phosphodiester linkage6), may result in cleavage
rates (k2) that approach or exceed the mismatch "Off rate"
(k 1), particularly in longer linkage sequences. The
10 present invention provides improved oligonucleoside
structures that address these competing considerations and
meet other goals as described herein.
The oligonucleoside compounds Of the invention com-
prise linked nucleosides having a base sequence that is
15 complementary to a target region Of the target ribonucleic
acid sequence, and include an RNaseH-activating region and
at least one non-RNaseH-activating region. When used in
conjunction with l;;ln RNaseH (e.g., in l;;ln
cellular systems), the RNaseH-activating region comprises,
2 0 in the pref erred embodiment, a segment of between 5 and
about 9 consecutive 2'-unsubstituted nucleosides linked by
4 to about 8 charged (anionic) internucleoside linkage
structures. When used in conjunction with bacterial
RNaseH (e.g., in bacterial c~ r systems or in antibac-
25 terial therapy in mammals), the R~aseH-activating region
comprises, in the preferred ~ ; , between 3 and
about 7 consecutive 2'-unsubstituted nucleosides linked by
2 to about 6 charged ; nt~rnllrl ~c~ide linkage structures .
The non-RNaseH-activating region comprises, in one
30 preferred ~mho~;- t, a single segment of at least 3
linked nucleosides, and more preferably at least about 5
linked nucleosides, cc~n~;n;n~ one or more chirally-
selected Rp- linkages . In a related second pref erred
embodiment, the non-RNaseH-activating region comprises two
35 separate flanking segments, each segment ront~;n;n~ at
least about 2 linked nucleosides, and more preferably at
lea8t about 4 linked nucleosides (or a total of at least
_ . . . ,,, _
WO 9~113834 PCr/US94/13387
217~259
, ~
- 22
about 8 linked nucleosides in the two separate segments),
wherein one or more of the l;nk~ is a chirally-selected
Rp-linkage. The RNase~-activating region is preferably
f lanked in the compound by two such separate non-RNaseH-
5 activating regions. In a third related preferred embodi-
ment, the non-RNaseH-activating region comprises an
alt~rn~t;n~ se~uence of racemic (non-chirally-selected)
internu~l~n~ linkages comprising ~l) a racemic methyl-
(or lower alkyl- ) phosphonate (NP), methyl- (or lower
lO alkyl-) phosphonothioate (MPS), aminoalkylphosphonate
(AAP) or ~m;nr~lkylphosphonothioate ~AAPS) linkage,
alternating with (2) a negatively-charged phosphate,
phosphorothioate or phosphorodithioate (e.g., DE, PS, or
PS2 ) linkage . In any of the above embodiments, one or
15 more of the nucleosides in the non-RNaseH-activating
region may be 2 ' -substituted, particularly to increase
binding affinity and nuclease resistance while controlling
(selectively decreasing or eliminating) RNaseH-activation
characteristics. It is particularly preferred that one or
20 more, or all, phosphodiester linkages, if present in the
non-RNase~-activity region, be 2 ' -substituted, although
further 2 ' -substitutions may also usefully be employed in
the non-RNaseH-activity region.
As an example, the phosphonate irLternucleosidyl
25 linkages used in oligomers of the present invention may
contain a lower alkyl group replacing one of the two non-
bonding (or non-bridging) oxygens on the phosphorus of a
rh~gph~tl;egter ;nt~rn~ 1eosidyl linkage, wherein the other
non-bonding oxygen remains or is alternatively replaced by
30 sulfur. The replacement of oxygen by lower alkyl creates
a chiral environment around the ph~)srh~rus which can be
designated as either Rp or Sp, depending on which of the
non-bonding oxygens has been replaced with lower alkyl.
The Rp and Sp configurations can be dep~cted as follows:
WO 95/13834 2 1 7 6 2 5 g PCr/uss4~l3387
23
Il 11
o_~3 oi~ ~
wherein X is oxygen or sulfur and R is lower alkyl.
Applicants have discovered that the binding affinity
of the present R~aseH-activating oligonucle~side compounds
can usefully be controlled by selectively incorporating
into the compounds polynucleoside segments crntA;nin~
chirally-selected internucleoside linkage structures.
Such chirally-selected Rp-rich segments afford greater
binding affinity than the corresponding racemic sequences.
Applicants have also discovered that selectively-increased
binding af f inity and improved nuclease resistance can be
acl1ieved in a practical fashion, with or without chiral
enrichment, using multiple or repeated blocks or synthons
comprising both charged (;nrl~ inr phosphodiester) and
uncharged (particularly racemic or chirally-selected
methylphosphonate) internucleoside linkage structures.
Such synthons preferably do not have more than one consec-
utive charged linkage structure in their sequence, partic-
ularly if the charged (anionic) linkage structure is a
phosphodiester bond.
These controllable binding affinity polynucleoside
segments oi the invention provide the benef its of in-
creased nuclease resistance, controllable RNaseH-activa-
tion characteristics and ease of synthesis. Thus, for
example, the linkage structures can be chosen to include
one or more uncharged modified (non-rhn~rl~n~ qter)
linkage structures which will be substAnti~lly non-acti-
vating to RNaseH and al80 nllrl~A~e-resistant~ Use of 2'-
substituents as described herein also leads to increased
nuclea~e re6istan~e of ~ _ t~ including charged linkage
_ _ _ _ _ _ . ,, .. . _ .. .. . . ..
Wo 95113834 ~ ~ 7 ~ 2 5 ~ PCrlUSg4/13387
24
structures, particularly phosphodiester linkages. Fur-
thermore, individual synthon6 can be prelim;n=rily assem-
bled as synthetic blocks which are then readily combined
to provide a controllable binding af f inity segment con-
5 taining two or more dif f erent block structures, or asingle repeated block structure.
While the described technic~ue of chiral selection can
usefully be employed in both the RNaseH-activating and
non-RNaseH-activating regions of the present compounds, it
lO is most advantageously used in the latter region. In
addition, chiral selection is preferably achieved with
multiple or repeated mixed linkage structure blocks as
described h~r~; n= f ter .
A chirally- selected polyn~ segment of the
15 present invention includes a sequence of ;nt~rnllcleoside
linkage structures that is enriched or pure with respect
to Rp chiral linkages. Such a sec~uence is considered
chirally-enriched if at least about 75% of the chiral
(asymmetric) linkage structures in the segment, or alter-=
20 natively at least about 40% of the total linkage struc-
tures in the segment, have Rp chirality A8 shown below,
chiral enrichment of at least about 75% can be achieved
gynth~t;~lly by coupling a series of dimer nucleoside
blocks (synthons) wherein the structure linking the two
25 r)t1~ 1 er-sides of each synthon is a modif ied (non-phospho-
diester) Rp-chiral linking structure, and wherein the
linking strtlcture between the respective synthons is
asymmetric. The coupling reaction between synthons in the
series will, in the simplest case, be carried out racemi-
30 cally, which means that about half of the inter-synthon
linkages will be Rp-chiral and about 75% of all of the
internucleoside linkages in the resulting mixed
chiral/racemic segment will be Rp-chiral. (It should be
noted that the ~racemic~ reaction may be driven more
35 toward one diastereomer in particular cases; for example,
investigations related to the present invention have shown
that coupling of 2~ -O-methyl-substituted methylphosphonate
WO 95/13834 . PCT/US94113387
2-~ 76259
~nn~ ~: leads preferentially to Sp-chiral internucleoside
linkages . )
It will be seen that chiral enrichment in excess of
75~ of the asymmetric linkage5 can be achieved by, for
example, conjugating trimer nucleo5ide synthons wherein
both internucleoside linkages within the block are
Rp-chiral and the respective trimer synthons are conjugated
racemically (or ~rh;r~lly). Synthetic schemes are shown
below f or the preparation of such trimer synthons .
Alternatively, conjugation between individual nucleosides
or between synthon5 can be carried out stereospecif ically
using asymmetric linkage structures, in which case all the
linkages in the segment will be Rp-chiral. While it is not
considered nf~ c~ry to the preferred practice of the
present invention to obtain segments having chiral enrich-
ment in exce8s of about 75~ of the asymmetric linkages (or
about 40% of the total linkages), such highly-enriched
segments will generally exhibit higher binding af f inity
characteristics .
AS seen above, a mixed chirally-selected segment of
the invention may include within it one or more achiral
(non-asymmetric) linkage structures. Thus, in one pre-
ferred structure of~ the invention, a mixed chirally-
selected segment is composed of a1t~rn~t;n~ phosphodiester
(achiral) and Rp-methylphnsrhnnRt~ (or other chiral)
linkage structure5. Such a repeated alternating linkage
sequence segment can be prepared using dimer nucleoside
blocks wherein the structure linking the two nucleosides
of the block is an Rp-chiral methylphosphonate linkage
structure, and where the blocks are conjugated achirally
using a phosphodiester (or other achiral) linkage struc-
ture. It will be seen that a polynucleoside segment
prepared in this manner will be chirally pure ;nz~ h as
all of the chiral linkages in the segment are of the Rp
conformation, whereas subst~nti~l ly 50g~ of the total
linkages will be Rp-chiral.
_ _ , . .. _ _ . . . . . .
Wo 95/13834 PCrlUS94/13387
,21~;25
The inventors have a3certained in investigations
relating to the invention that enrichment of methylphos-
phonate ~p linkages gives an increase in melting tempera-
ture (Tm) of about 0.9 to 1.5 C per internucleosidyl
linkage that is in the Rp conformation as compared to a
random racemic conformation. This translates into an
increase in binding affinity (KA) by a factor of about 1.8
for each additional selected Rp linkage (or a factor of
about 2 . 6 in the case of 2 ' -0-methyl-substituted resi-
dues). It will now be appre~ciated that, by the judicious
use of chirally-selected linkage structure segments in the
present compounds, binding affinity can be controlled in
a manner consistent with the objectives set forth above in
the detailed description. The examples below demonstrate
that increased potency can be achieved with such chirally-
Eielected compounds, as compared to racemic compounds,
while maintaining specif icity against the intended target
sequence .
As P~r~;nP~ above, another objective of the inven-
tion is to provide oligonucleoside structures having
controlled R~aseH activation characteristics. This
objective is obtained in the present invention by provid-
ing in the compound a non-RNaseH-activating polyn~lrlPnc;de
region, or regions, having reduced RNaseH-activation
~r~h; l; ties, along with an RNaseH-activating region
having sufficient RNaseX-activation r~r~hil;ty to effect
RNaseH-mediated cleavage of the target nucleic acid
strand. Preferably, both of these segments of the com-
pound are constructed to be nuclease resistant.
As is also explained above, one putative requirement
of mammalian RNaseH activation is that the antisense
compound must have a sequence of at least f our or f ive
crncerllt~ve charged (anionic) internucleoside linkage
structures (or at least two such linkages in the case of
bacterial RNaseH), wherein the linked nucleosides are 2 ~ -
unsubstituted. Conversely, in the practice of the present
invention, the non-RNaseH-activating segment can usefully
Wo 95/13834 2 1 7 ~ 2 ~ 9 PCr/US94113387
27
include uncharged linkage structures and/or 2 ' -substi-
tuents. By making use in the non-R~aseX-activating region
of modified (non-ph~-srhn~l; PCter) uncharged linkage struc-
tures such as those described herein, the present com-
pounds achieve increased nuclease resistance. Moreover,
the use of 2 ' -substituents as described herein leads to
selectively controllable increa8es in binding af f inity .
Thus, the inventors have ascertained in investigations
relating to the present invention that the use of 2'-0-
methyl nucleosides in methylph~srhnn~te-linked oligomers
results in additional increases in T= o~ about 1C per
substitution of 2'-deoxy with 2'-0-methyl nucleosides.
Furthermore, the inventors have ascertained that the use
of 2~ -substituents on nucleosides linked by phosphodieater
bonds also leads to increased nuclease resistance.
Consistent with these obj ectives, pre~erred 2 ~ -
substituents of the invention include lower (l to about 3
carbons) alkoxy, allyloxy, and halo (preferably fluoro)
substituents. A methoxy group is especially preferred.
In general, 2 ~ -substituents that are electron-withdrawing
are useful in increasing the binding affinity and nuclease
resistance of the present compounds, as such substituents
are believed to create a 3 ' -endo conformation in the
substituted sugar group.
It has further been discovered that a limited propor-
tion of charged linkage structures, ; n~l u~; ng phospho-
diester linkages, may usefully be incorporated into the
non-RNaseH-activating segment, particularly in a linkage
setauence c~r~;n;n~ multiple or repeated blocks of charged
3 0 and uncharged linkage structures . Such segments lead to
controllable increases in binding affinity, nuclease
resi8tance, and controlled RNaseH activation characteris-
tics, and result in compounds having r~nh~nr Pd speci~icity
for the; nl-r~n~r~r~ target nucleic acid se~auence .
Preferred linkage structures and 2~-substituents for
the non-RNaseH-activating se_ c of the invention
include the f ollowing:
. _ _ _ _ _ _, . . , _ . ,,, .. . , _ _
WO95/1383~ PCrlUS94/13387
2 8
MP (R) /DE
2 ' OMeMP (R) /2 ' OMeDE
NP (R) /2 ' OMeNP
NP (R) enriched
2 ' OMeNP ~R) enriched
NP ~R) /PS
2 ' OMeNP ~R) /2 ' OMePS
NP ~R) /PS2
2 ' OMeNP (R) /2 ' OMePS2
2 ' OMeMP/2 ' OMeDE
NP/2 ' OMeDE
MP (R) /PAm
2 ' OMeMP (R) /2 ' OMePAm
2 ' OMeNP / 2 ' OMePAm
NP/2 ' OMePA
MP (R) /TE
2 ' OMeNP (R) /2 ' OMeTE
2 ' OMeNP/2 ' OMeTE
MP/2 ' OMeTE
MP (R) /MPS
2 ' OMeNP (R) /2 ' OMeNPS
2 ' OMeNP/2 ' OMeNPS
NP/2 ' OMeMPS
NP (R) /PF
2 ' OMeNP (R) /2 ' OMePF
2 ' OMeNP/2 ' OMePF
NP/2 ' OMePF
NP ~R) /PB~J
2 ' OMeNP ~R) /2 ' OMePBi}3
3 0 2 ' OMeNP/2 ' OMePBH3
NP/2 ' OMePB~3
NP ~R) /RSi
2 ' OMeMP ~R) /2 ' OMeRSi
2 ' OMeMP/2 ' OMeRSi
MP/2 ' OMeRSi
MP (R) /CII~
2 ' OMeMP (R) /2 ' OMeC~2
wo 9~13834 ~ 1 7 ~ 2 5 9 PCT/US94113387
29
2 ' OMeMP/2 ' OMeCH~
MP/2 ' OMeCH,
3~ey: MP = racemic methylphosphonate linkage ~between
linked nucleoside~); MP(R) = chirally-selected Rp-
methylrhnsrhnn~te linkage; DE = rhnsrhn~;ester
linkage, PS = phosphorothioate linkage; PS2 = phos-
phorodithioate linkage; PAm = phosphoramidate link-
age; T3 = phosphotrieæter linkage; NPS = alkyl
(particularly methyl) phosphorothioate; PF = phos-
phorofluoridate linkage; PBH3 = boranophosphate
linkage; RSi = silyl (especially alkyl-disubstituted
silyl) linkage; CH, = formacetal linkage; 2'0Me - 2'-
methoxy-substituted (or other lower alkoxy, allyloxy
or halo substituted) nucleoside residue, linked using
the listed linkage structure; "enriched" refers to a
segment of linkages preferably rQnt~;n;nr at least
about 4096 (and up to 100~6) Rp-selected 7;nk;~ c among
the linkages in the segment, and thus includes a
mixed -seQIuence of racemic and chirally-selected R
internucleoside linkage structures; linkage struc-
tures grouped with slashes denote a mixed linkage
segment ;nr~ ;nr the listed linkage structures
optionally in a series of multiple or repeated mixed
linkage sequence blocks.
Also preferred are compounds having a segment chosen
from the above listing wherein one or more (or all) of the
methylphosphonate (MP or MP (R) ) linkages are replaced with
lower alkyl-, especially methyl-, phosphonothioate (MPS or
MPS(R) ) linkages, or with ~m;nn~lky~rhn~rhnn~te (AAP or
AaP(R)) or ~m;nn~lkylphosphonothioate (AAPS or AAPS(R))
linkages . Such compounds include 2 ~ -substituted residues
cnnt~;n;nr such linkages, as well as . ~ Jul~ds "enriched"
in these Rp-chiral linkages. Examples of the latter
include compounds having an alternating se~uence of MP
(racemic) and AAP (R) linkages, or an alternating sequence
of MP(R) and AAP (racemic) linkages, or an alt~rn~tins
sequence of ~AP (racemic) and AaP (R) linkages . Also
preferred are compounds chosen from the above listing
wherein one or more (or all) of the Rp-chiral methylphos-
phonate (MP(R)) linkages are replaced with racemic methyl-
phosphonate (MP) linkages, preferably in an alternating
sequence with a second dif f erent linkage structure, and
most preferably in an alternating or other mixed sequence
wo gSrl3834 2 1 ~ ~ 2 5 9 PCTrUS94rl3387
, i.
with phosphodiester, phosphorothioate or phosphorodi-
thioate linkages.
Each of the mixed linkage segments listed above will
contain at least one of each of the linkage structures
5 listed. From a aynthetic standpoint, it may be convenient
to alternate the listed linkage structures or to use a
repeated sequence rnrlt:~;nlng both structuree, although
this is not n-or~q~ry. Two or more of the mixed linkage
segments listed above may be serially combined within a
10 given non-RNaseH-activating region of the compound. In
this case, it may be convenient from a synthetic stand-
point to select discrete synthons from the respective
mixed linkage groups and ~combine them in the single
region .
Thus, it will be seen that the pr~sent invention
provides synthetic oligomers having one or more segments
;nr~ ;nr~ mixed int~rnllrl~osidyl linkages, particularly
oligomers having chirally pure or enriched rhr,sphrn~l~r
; nt~rnllrleosidyl linkages interspersed with single non-
20 rhnqrhrn~te internucleosidyl linkages and methods for
their preparation. Such phosphonate internucleosidyl
linkages include lower alkylphosphonate ; ntPrnllrl eosidyl
linkages of 1 to 3 carbon atoms and lower alkyl rhr~rhr,nn-
thioate (alkylth;r~ph~rhrn~te) internucleosidyl linkages
25 of 1 to 3 carbon atoms. These mixed oligomer segments
preferably have phosphonate ;nt~rnl~rlP~ yl linkages
interspersed between single non-rhnsrhrn~te internucleo-
sidyl linkages in a ratio of from 1 to about 1 to 1 to
about 4 non-phosphonate linkages to rh~l~rhr~n~te linkages.
3 0 According to a pre~erred aspect, such oligomers have
alternating chirally pure phosphonate internucleosidyl
l;nki~r~ which alternate with non-phosphonate ;nt~rnllrleo-
sidyl linkages. Oligomers comprising such segments,
particularly in one or more non-RHaseH-activating regions,
35 may be used to prevent or interfere with expression or
translation of a single-stranded RNA target sequence The
chimeric oligonucleosides have an overall nucleoside base
-
WO 95113834 PCIIUS94/13387
2~762$9
31
seriuence, inrl~ ;nrJ the RHa8eH-activating and non-RHaseH-
activating regions, which is sufficiently complementary to
the RNA target sequence to hybridize therewith.
Preferred chirally pure phosphonate linkages include
5 Rp lower alkylrhr~sphnr)~te linkages, and more preferred are
Rp methylphrsrhnn~te ;nt~rn1~rleo~idyl linkages. Preferred
non-phosphonate linkages include phosphodiester, phos-
phorothioate and phosphorodithioate, while phosphorami-
date, phosphorofluoridate, boranophosphate, formacetal and
10 silyl int.orn11rlPosidyl linkages may also be used. Accord-
ing to an especially preferred aspect, Ry-enriched
oligomers are provided having chirally pure Rp-methyl
phrsrhnn~te linkages which alternate with phosphodiester
linkages in the non-RHaseH-activating regir~n of the
15 compound . These alternating oligomers have been f ound to
exhibit ~nh~nrf~rl binding affinity for an R~A target
ser~uence and also increased nuclease resistance and
specif icity .
The present invention likewise ; nrl ~ chimeric
20 antisense oligomers having ~nhAnrod potency as antisense
inhibitors of gene expression comprising one or more
segments with methylphosphonate ;ntl~rn~1rl~osidyl linkages
F~nh:~nr~d for the E~p configuration which are interspersed
between non-rhr.srhrn~te ;nt~rn11rleositlyl linkages, prefer-
25 ably phosphodiester or alternatively rhn8rhnrothioate orrhr~rhnrodithioate linkages. We have found that chirally
enriched oligomers hybridize more tightly to RNA target
sequences and should show Pnh~nr~tl potency inhibiting
translation of RNA targets as compared with oligomers
30 having racemic MP ;nt~rn11rleosidyl linkages mixed with the
same non-phosphonate internucleosidyl linkages.
As explained above, the RNaseH-activating region of
the present invention can have varying minimum and optimum
lengths ci~r~nrlin3 on the species (~ n or bacterial)
35 of the RNaseH enzyme that is utilized for cleavage. In
either case, the RNaseH-activating region preferably
compri8es a sequence of Consecutive 2 ' -unsubstituted
_ _ _ _ _ _ _ _ _ _ . . . .
WO 95/13834 PCT/US94113387
21~62S~
32 ~
nucleosides linked by charged 1 nt.orn11cleoside linkage
structures. Preferred linkage structures and mixed
linkage structures for the RNaseH-activating region are
selected from among the following:
DE
PS2
PS
PS2 /DE
PS /DE
PS/PS2
One especially preferred linkage structure is the phos-
phorothioate (PS) linkage.
In a related embodiment, two oligonucleosides of the
invention having t~rmin~lly-positioned RNaseH-activating
15 regions may be used in tandem to effect cleavage of a
target mRNA site. The nucleoside base sequences of the
reepective compounds are selected to be complementary to
adj acent regions in the target mRNA strand. The RNaseH-
activating regions may be used in tandem to effect cleav-
20 age of a target mRNA site. The RNaseH-activating regions
are situated at the 5'- t~ n~ and the 3'-terminus of
the respective ~ de such that, upon co-hybridization
to the adj acent regions in the target, the two RNaseH-
activating regions abut one arother and are hybridized to
25 adjacent target subregions in the overall target region of
the mRNA strand. The two R~aseH-activating regions act to
complement one another with respect to RNaseH-mediated
cleavage of the target region. Shorter RNaseH-activating
regions may be used in the two compounds than might
30 otherwise be required, and specificity should be increased
to the extent that dual hybridization is required to
ef f ect cleavage .
Chimeric oligomers of the invention, or segments
thereof, hav~ng a predetermined base sequence of nucleo-
35 sidyl units and having chirally pure rhnsph(~n~t~ inter-
n~1r~ idyl linkageg mixed with non-phosphonate 1; nk~
wherein the phosphonate linkages are interspersed between
Wo 95/13834 2 1 ~ 6 2 5 9 Pc~r/uss4ll33~7
33
single non-phosphonate linkages may be prepared by cou-
pling to one another individual nucleoside dimers, trimers
or tetramers of preselected nucleoside base sequence
having chirally pure or race~ic phosphonate or other
5 int~rn~ idyl linkages.
In this regard, chirally pure or racemic synthons of
the formula:
O Z
X I o ~0 j
-- --n \
O Z
\Cp
lO may be utilized wherein X is oxygen or sulfur, R is lower
alkyl of l to 3 carbon atoms, Bl is a removable blocking
group, Z is hydrogen, alkoxy o~ l to lO carbon atoms,
halogen or alkenyloxy of 3 to 6 carbon atoms; ~3 is an
optionally protected purine or pyrimidine base; n is l, 2
15 or 3 and Cp is a coupling group. The coupling group Cp is
conveniently selected 80 as to give the desired non-
n~te ' nt~ntl~l ensidyl linkage when coupled toanother synthon.
According to one preferred chirally-selective syn-
20 thetic method, nucleoside dimers having a ~hnsrhnn~telinkage connecting the two ~ucleosidyl units of the dimer
are prepared and separated into their Rp and Sj, isomers.
The resulting dimers which have a defined chirality at the
_ _ , .. . . . _ . . . . .. _ _ _ . . . .
2~g
wo 95113834 PcrluS94113387
~ .,
, . .
!-, 34
phosphonate linkage, a~e then derivatized so that they may
be coupled together using an automated DNA synthesizer.
The dimers may have coupling groups which result in any
one of a variety of internucleosidyl linkage6 between
5 dimers. From a stock of 16 dimers, oligomer segments of
any nucleoside base sequence may be synthesized by linking
together the a~Lu~Liate dimers. Dimers are added to the
growing oligomer chain until an oligomer segment having
the desired number of nucleosides is obtained. The
10 resulting oligomer segment has a defined chirality at
every other internucleosidyl linkage (i.e., those linkages
originally derived from the coupled dimeric units). The
L~ ;n;n~ ;ntF~rn~ leo8idyl linkages comprise non-phos-
phonate internl1n1~nR;rlyl linkages, such as phosphodiester,
15 phosphorothioate, phosphorodithioate, morpholino, phos-
phoramidite, phosphorof luoridate, boranophosphate, f orma -
cetal, silyl or other non-rhnRrhnn~te internucleosidyl
linkages .
Alternatively, larger blocks of nucleosides such as
20 trimers and tetramers may be coupled to give a chirally
enriched oligomer. Trimers having two chirally pure
internucleosidyl linkages may be conveniently prepared by
coupling the c-~L-~l~Liate chirally pure dimer synthon to
another nuclec)side and, for example, if Rp chirality is to
25 be selected, then separating the resulting Rp-Rp and Rp-Sp
trimers. The resulting trimer has defined chirality
(i.e., is chirally pure) at both inter-nucleosidyl
linkages. The trimers are then derivatized to give trimer
synthons 80 that they may be coupled together using an
30 automated D~A synthesizer. The trimer synthons have
coupling groups which allow them to be coupled together to
give a chirally enriched rhnsrhnn~t~ oligomer segment.
From a stock of 64 trimers, oligomers of any base se~uence
may be synthesized by linking together the c-~L.,~Liate
35 trimers. Trimers may be seqll~nt;~11y added to the growing
oligomer chain or alternatively coupled with nucleoside
~, dimers and/or tetramers until an oligomer
Wo 95113834 PcrluS94113387
2~62~9
segment having the desired number of nucleosides is
obtained. The reæulting chimeric oligomer has a defined
chirality at those ;ntPrn~ Pnsidyl linkages in the
chirally-seleCted 6egment derived from the internucleo-
5 sidyl linkages of the coupled chirally-selected dimers,
trimers or tetramers. Thus, use of these trimers will
result in an oligomer segment having phosphonate linkages
of def ined chirality at about two out of every three
internucleosidyl linkages. By following analogous tech-
10 ni~ues, tetramers having three chirally pure internucleo-
sidyl linkages may be prepared and coupled to each other
or to other synthons (including monomers) to give other
chirally-selected segments or portions thereof. Alterna-
tively, dimers, trimers and other short oligomers having
15 ;ntGrn1lnl~o~;~lyl linkages of defined chirality (such as
pure Rp) may be coupled together or. to other synthons in
d~L~L~Liate se~uence to give an oligomer segment or
portion thereof of a particular desired se~uence and
length. Such a chirally-selected segment cdn be coupled
20 with additional nucleosides forming a separate segment of
the compound, particularly a segment of consecutive 2'-
unsubstituted nucleosides linked by charged linkage
structures forming an RHaseH-activating region.
According to an alternative synthetic method, cou-
25 pling conditions for nucleoside synthons (or dimers) areused which direct coupling to give an PnhAn~P~ yield of
the desired chiral-configuration. This method may be used
to couple individual n~ nsi~P synthons or alternatively
the chirally pure dimers and, thus, obtained are oligomer
30 segments, particularly non-RHase~I-activating segments,
enriched for the desired chiral configuration at each of
the ~hnsphnn~te ;ntPrn-l~ leosidyl linkages.
The chirally-selected methylE~hn~phnn~te and other
~ - ~, dimers, trimers and the like taught in the
35 examples and Detailed Description herein can be coupled
together by a variety of different methods leading to the
following, non-exclusive, types of ;n~Prnll-~leosidyl
~ ~ = == _ _ _ _ _ _ . . , . . . . _ .. _ .. . _ , .. . .. , _
WO 95/13834 ~ 1 7 6 2 5 9 PCr/US94113387
:. , ~.,,
.. .
36
linkages: phosphodiester, phosphotriester phosphoro-
thioate, phosphorodithioate, phosphoramidate, rh~l~rhf~ro_
fluoridates, boranophosphates, formacetal, and silyl.
Internucl eosidyl phosphodiester linkages can be
obtained by converting the 3'-OH of a chirally-selected or
racemic synthetic unit ~monomer, dimer, trimer, poly-
nucleoside, etc. ) to either a phosphotriester synthon
(Reese, C.B. (1978) Tetrahedron 34, 3142-3179), phosphora-
midite synthon (Beaucage, S.L. and Lyer, R.P. (1992)
Tetrahedron 48, 2223-2311), H-phosphonate synthon
(Froehler, B.C. in Agrawal, S., ed. Protocols for Oligonu-
cleotides and Analogs, Synthesis and Properties, Methods
in Molecular Biology Vol. 20, Humana Press, Totowa, NJ,
1993, pp. 63-80), or phosphoromonochlo~idite reagent
(Hogrefe, R.I. (1987) dissertation, Northwestern Universi-
ty, Evanston, IL).
Tn~rnllrleosidyl phosphorothioate 1 ;nk~rJ-~F can be
obtained by converting the 3 ' -OH of a synthetic unit to
either a phosphotriester synthon (Stec, W.J., et al.
(1991) Nucl. Acids Res. 19, 5883-5888) ), phosphoramidite
synthon (Lyer, R.P., et al. (1990) JACS 112, 1254-1255),
H-phosphonate synthon (Seela, F. and Kretschmer U. (1991)
J. Org. Chem. 56, 3861-3869), or rh~Rrhnromonochloridite
reagent (Hogrefe, R.I. (1987) Dissertation, Northwestern
University, Evanston, I~. ) .
Internucleosidyl rh~rh~rodithioate linkages can be
prepared as by the disclosures herein and by U . S . Patent
No. 5, 213, 088 to Gorenstein et al . Internucleosidyl
phosphotriester linkages can be obtained by converting the
3 ' -OH of a synthetic unit to either a phosphotriester
synthon (ReeGe, C B. (1978) Tetrahedron 34, 3143-3179),
phosphoram.idite synthon (Beaucage, S.~. and Lyer, R.P.
(1992) Tetrahedron 48, 2223-2311), H-phosphonate synthon
(Froehler, B.C. in Agrawal, S., ed. Protocols for
Oligonucleotides and Analogs, Synthesis and Properties,
Methods in MrlPrlll~r Biology Vol. 20, Humana Press,
Totowa, NJ, 1993 , pp . 63-80), rh~ h~romonochloridite
Wo 95/13834 2 1 7 6 2 5 g PCTIUS94/13387
37
reagent (Hogrefe, R. I . (1987) Di8sertation, Northwestern
University, Evan5ton, IL.), or post synth~t;r~lly (see
U.S. Patent No. 5,023,243 to Tullis.
Int~rn~lrlensidyl phosphoramidate, phosphorofluor-
5 idate, boranophosphate, formacetal, and silyl linkages canbe obtained by converting the 3'-OH of a 8ynthetic unit to
the ~L-~Liate synthons. (See Agrawal, S., ed. Protocols
f or Oligonucleotides and Analogs, Synthesis and Proper-
ties, Methods in Molecular Bioloqy Vol. 20, Humana Press,
10 Totowa, NJ, 1993, for synthetic protocols to obtain
synthons for each of the above. )
Chemical structures for synthons and reactive inter-
mediates useful in the present invention are depicted in
FIGS. 6-10, and are discussed in further detail in U.S.
Patent Application Serial Nos. 08/154,013 and 08/154,014.
The following example8 demonstrate various signifi-
cant aspects of the present invention, but are examples
only, and should not be considered as limiting the scope
of the present invention.
r 1,,~
r le 1
Pre~aration of MP(R~) /DE and ME ~l?s) /MP Dimer Svnthr~nc
A. Preparation o~ a ~CT) Dimer Havinq a ~h;~all~ pl~re
Methvl~hosl~honate Internudeosidvl T-; nk~qe Usinq
Solution Pha8e Chemistrv
Into a 2 L roto-evaporator flask was placed 10 . 0 g
(28 mM) of 3 ' -tert-butyldimethylsilyl thymidine and 26 .1
g ( 3 5 mM ) of 5 ~ - dime thoxytri tyl - NÇ - i 8 obutyryl - 3 ~ - me thyl -
N, N- di i sopropyl ~m; noFhr~sphoramidite - 2 ~ - deoxycytidi~e . The
solids were dissolved in 500 ml of acetonitrile and
evaporated to dryness under vacuum. This process was
repeated with another 500 ml of acetonitrile and then the
flask was released under argon and stoppered with a rubber
~epta .
... . . . . . . .
W095/13834 21 762S9 PCr/Uss4ll3387
.
38
This dry solid foam was then dissolved in 500 ml of
acetonitrile ( "ACN" ), and with manual stirring, treated
all at once with 404 ml tetrazole (180 mM, 0.45 M
tetrazole in THF) . Manual stirring is ~ nt;n~ l for 30
seconds and then the flask is allowed to stand for another
2.5 minutes, after which time the reaction mix is treated
all at once with 275 ml of an oxidizer solution
(I2/H20/lutidine/THF; 25 g/2.5 ml/100 ml/900 ml). The
solution was stirred manually and allowed to stand at room
temperature for 15 minutes. The resulting dark amber
solution was then treated with bisulfite (2 g/25 ml H20),
which upon addition, turned the solution light amber as it
reacted with the excess iodide. The reaction mix was then
concentrated to a thick oil and taken up in ethyl acetate
~"EtOAc") (500 ml) and washed with saturated sodium
bicarbonate (2 X 250 ml) and H20 (2 x 250 ml). The organic
phase was dried over MgSOi, filtered and ~ nrf~ntrated to a
light colored solid foam, which upon further drying
yielded 35 grams of crude dimer.
The crude dimer was run on HPLC (reverse phase,
Waters C18 b~n~r~k) with a program (ACNMETH) starting
with 5096 acetonitrile and O.1 M triethylammonium acetate
(TEA~, pH ~ 7.0) which increased to 1009~i acetonitrile over
20 minutes with a linear gradient. Two major peaks were
resolved, one at 4.5 minutes, which is residual lutidine
and the other at 14 . 5 minutes which is the mixture of Rp
and Sp diastereomers. The ratio of Rp and Sp was determined
tauantitatively by taking a 5 mg ali~[uot of the crude
product and dissolving it in 1. 5 ml of acetonitrile along
with 0.5 ml of tetrabutyli 11-m fluoride (TBAF, 1 M
solution in THF) . After standing at room temperature for
10 minutes the sample was run on HPLC Two new peaks were
observed at 6.5 and 7.1 minutes and the later eluting peak
was gone. The first new peak, which is believed to be the
Sp diastereomer, represented 66~6 (2/1) of the normalized
value for the two peaks. The crude product was also
analyzed by the (normal phase silica plate) in 75/25
-
~ 2~76~
WO 95113834 - . PCrlUS94113387
39
EtOAc/CH2Cl2 ("75/25") with 5~ methanol added. The TLC
6howed two spots with Rf's of 0.45 and 0.64, respectively;
the faster running product (believed to be the Rp form~ was
less intenge than the slower moving one.
The Rp diastereomer was separated on normal phase
silica using a methanol step gradient in 75/25
EtOAc/CH2Cl2 . A 7 . 5 cm by 60 cm column, was loaded with
700 g of silica (first slurried in 2.5 L of neat 75/25
EtOAc/CH2Cl2) . The crude dimer was then dissolved in 75 ml
of 75/25 EtOAc/CH,Cl2 and loaded onto the column. The
column was started with 1~ methanol and increased to 29~
and finally 3~ where the Rp dimer began to elute. The Rp
dimer eluted cleanly over several bed volumes while
m~;nt~;n;n~ 3~ methanol in the eluent. The Sp dimer was
eluted later with 30~ methanol. The Rp dimer yield was
11. O grams, while the Sp yield was 17 . 8 grams . HPLC
analysis (ACNMETH) was performed on the Rp dimer and one
peak was observed at 14.5 minutes. The TLC (75/25
EtOAc/CH2Cl2, 5~ methanol) o~ this product, revealed a
single spot product with an Rf of 0.55 which, upon treat-
ment with 10~ sulfuric acid in ethanol and heat, was both
trityl and 6ugar positive.
The newly resolved Rp dimer, 11. 0 g (O . 011 M) was
dissolved in 110 ml of ACN and treated all at once at room
temperature with 22 ml of TBAF (O . 022 M, 1 M in THF) . The
reaction mixture was allowed to stand overnight at ambient
temperature. The next morning the reaction was determined
to be complete by TLC (75/25, EtOAc/CH2Cl2 with 10~
methanol); no starting material was detected but a small
amount of 5'-DMT-dT was observed, which runs considerably
faster on normal phase silica than the 3'-OH of the dimer.
The reaction mixture was .-nncl~ntrated on a rotary evapora-
tor to a thick oi~ which was then dissolved in CH2Cl2 (200
ml) and washed with saturated sodium bicarbonate (2 x 100
ml) and H,O (2 x 100 ml) . The organic phase was dried over
MgSO~, filtered, and c~lnr~ontrated to a light yellow solid
foam, which was puri~ied on 100 grams of silica ~75/25,
_, . . ... . . .... _ _ _ .. _ . .
Wo 9~13834 , ~' PCT/US94/13387
X1~ 9
EtOAc/CH2Cl2 with 596 methanol). The 5'-DMT-dT was removed
but an impurity at 13 . 5 minutes (HPLC, ACNMETH) was
detected which was first believed to be unreacted starting
material (t-BDMS on) but after ~;t;nni:ll treatment with
TBAF this was found not to be the case. A second column,
using 100 g of silica and the same eluent was run and
smaller fractions were taken; the column was able to
successfully separate the two spots. The pure CT-Rp dimer
fractions were pooled and cnn~ntrated to yield 5.5 grams
of a nearly white solid foam.
B. Pre~aration of a ChirallY Pure ~CT) MP(R~) /DE Dimer
SYnthon
Into a 100 ml round bottom flask was placed 0.5 g
(0.55 mMol) CT-3'-OH dimer (product of Example lA) which
was rendered anhydrous by 3 x 20 ml co-evaporations with
pyridine. The flask was released from the vacuum system
under argon gas and stoppered with a rubber septa. The
compound was redissolved in 10 ml acetonitrile and 200 /Ll
( 1. 4 mMol, 2 . 5 eq) TEA were added . To the resulting
mixture at room temperature and with manual stirring, was
added in one portion 200 ~l (0.90 mmol, 1.6 eq.) 2'-
cyanoethyl-N,N-diisopropylchlorophosphoramidite. The
reaction mixture was allowed to sit at room temperature
bef ore being analyzed by reverse phase HP~C . The HP~C
(Beckman System Gold, C18 bnn~r~lk, ACN method; Solution
A was 50/50 ACN/0.1 M TEAA in water, pH 7 and Solution B
was ACN; a gradient of 0 to 1009~ Solution B was run at a
rate of 1 ml/minute over 25 minutes) showed complete
conversion of starting material and a crude purity of
greater than 90 percent. The diastereomers of the phos-
phoramidite were not resolved. The reaction mixture was
n~Pntrated under vacuum to a light yell solid foam. The
foam was purified immediately by L:I1LI tngraphy on 20 g of
normal flash grade silica equilibrated with 5/1/5 ethyl
acetate/ acetonitrile/methylene chloride with 2~6 TEA to
give 0.5 g (82~ yield) of the above-identified product as
~76259
WO 95/13834 PCr/US94/13387
41
an off-which solid foam having a purity of 99.39~ as
determined by HPLC.
C. Preparation of a ChirallY Pure (CT~ MP (RF) ~MP Dimer
SYnthon
The CT-3 ' -OH dimer, 5 . 5 g (6 mM), prepared as de-
scribed in part A above, was rendered anhydrous with two
co-evaporations with pyridine. The resulting solid foam
was released f rom the rotary evaporator with argon and
stoppered with a rubber septa . The solid f oam was dis -
solved in 100 ml of 9/1, ACN/CH2Cl" then treated with 1.7
ml triethylamine (TEA, 12 mM). lqith magnetic stirring,
the reaction mix was treated dropwise at room temperature
with 1.5 ml chloromethyl-N,N-diisopropylamino phosphine
(Cl-MAP, 8 mM) . The reaction was monitored on HPLC
(ACNMETH) and a~ter 1. 5 hours was complete, showing two
main products, one at 3.5 minutes which was pyridine and
a second at 14 . 3 minutes which was the desired amidite .
The reaction mixture was cnn~n~rated on a rotarY
evaporator using a partial vacuum; the flask which con-
tained the resulting light amber sludge was released under
argon and capped. The crude product was immediately
passed through a flash column ~nn~1nin~ 60 grams of
silica (first equilibrated in 1/1/1 ACN/EtOAc/CH2Cl2 with
3~ TEA). The product was eluted quickly with this eluent
and all U.V. positive fraction5 were pooled and concen-
trated. The resulting 501id foam wa5 co-evaporated with
ACN to remove any residual TEA, then dried overnight under
full vacuum. The fi~al product, an off white solid foam,
weight 5. 0 grams.
3 0 E~cam~le 2
Preparation of (CU) 2'-0-MethYl MP(R~)/2'-0-MethY1 DE and
2' -Q-MethYl MP (R~ /2~ -O-MethYl MP Dimer SYnthons
A. Pre~aration of 2 ' -0-MethYl C Monomer
A 5.0 g (8 mmol) portion of 2'-0 methyl cytidine was
re~ldered anhydrous with pyridine co-evaporations (3 X 25
ml~ and then dissolved in 50 ml acetonitrile. The solu-
WO 95113834 . PCTNS94/13387
~1762~9
42
tion was treated with 1. 65 ml triethylamine ( "TEA" ) (12
mmol, 1. 5 eq. ) and cooled in an ice bath. The solution
was then treated with dropwise addition of 1.65 ml chloro-
methyl-N,N-diisopropylamino phosphine ( "Cl-M~p" ) over two
5 minutes. The ice bath was removed and the reaction
mixture stirred for two hours. The reaction mixture
(reaction was determined to be complete by HPLC) was
concentrated to dryness. The residue was dissolved in 20
ml ethyl acetate/heptane (1:1) with 4% TEA, then loaded
10 onto 40 g silica gel equilibrated with the same solvent
system. All W absorbing eluent from the column was
collected and pooled, then concentrated to give 5 . 5 g of
the above-identified product (yield about 90%).
B . Pre~aration of Silyl-Protected 2 ' -0-Methvl Uridine
Into a 250 ml round bottom flask was placed 5 . 0 g
(9.0 mmol) 5'-DMT, 2'0-methyl uridine which was rendered
anhydrous with dimethylformamide (DMF) co-evaporations (3
X 25 ml). The resulting dry foam was taken up in 50 ml
DMF, then treated all at once with 2.4 g (35 mmol, 3.9
eq. ) imidazole, followed by dropwise addition of 3 . 0 ml
(12 mmol, 1.3 eq. ) t-butyldiphenylsilyl chloride. The
reaction mixture was stirred at room temperature over-
night .
The progress of the reaction was checked by HPLC (ACN
method (Solution A was 50/50 ACN/0.1 M TEAA in water, pH
7 and Solution B was ACN; a gradient of 0 to 100% Solution
B was run at a rate of 1 ml/minute over 25 minutes) and
thin layer chromatography ("T~C") using 5% methanol in
methylene chloride, and determined to be complete (no
starting material was evident). The reaction mixture was
then poured into ice water. and taken up in methylene
chloride, then washed several times with aqueous sodium
bi~rhrn~te and water. The organic phase was dried over
magnesium sulfate, filtered and then ~ n~ ntrated to give
7 . 2 g of a solid foam which gave a single spot on TLC.
The solid foam was then dissolved in 70 ml methylene
WO 95113834 - PCr/US94/13387
~76259
43
chloride and treated (with rapid magnetic stirring) all at
once with 70 ml benzene sulfonic acid, 2~ by weight in 2:1
methylene chloride/methanol. After stirring for 15
minutes at room temperature, the reaction mixture was
quenched with 10 ml TEA. The resulting detritlylated
compound was stripped down to a thick amber oil which was
then loaded onto 150 g. 8ilica gel equilibrated in heat
methylene chloride . The product was eluted f rom the
column using 296 methanol (in methylene chloride). After
drying, 3.51 g of the above i~Ph~;~;.od product were
obtained (yield about 80~).
C . Pre~aration of (CU~ 2 ~ -O-MethYl 25P (P~ ) Dimer
The silyl-protected 2 ' -0-methyl uridine monomer
(product of Example 2B) (3 . 0 g, 6 mmol) was taken up in 30
ml anhydrous ACN . The 2 ' -0 methyl cytidine amidite
monomer (product of Example 2A) (5.5g, 7 mmol, 1.2 eq.)
separately, was taken up in 55 ml ACN. Both solutions
were allowed to stand over 3 A molecular sieves overnight
at room temperature.
The two solutions were carefully decanted into a
single flask and treated with 94 ml tetrazole (0.45 M in
ACN, 42 mmol, 7 e~). The resulting mixture was stirred
for 4 minutes and then oxidized by addition of 1.5 ml (1.2
eq. ) cumene hydroperoxide. The reaction mixture was
r~ln~~~ntrated to dryness, then taken up in methylene
chloride and washed with aqueous sodium bicarbonate and
water. The organic phase was dried over magnesium sul-
fate, filtered and concentrated to give 7.5 g. of a solid
f oam . The diastereomeric ratio as determined by HP~C by
comparison of areas under peaks was 57/43 Sp to Rp.
The Rp diastereomer was isolated by column chromatog-
raphy using two silica columns (100:1, silica to crude
product, equilibrated in 3:1 ethylacetate/methyl chloride
with an increasing methanol gradient irom 1 to 5~). A
total of 1. 07 g oi pure Rp dimer was isolated.
, _ _ _ _, _ . , . . . . . . . -
Wo95113834 2i7625la PCr/US94/1338
44
D . DeDrotection of (CH~ 2 ' -0-Methvl Dimer
A 1.07 g (0.90 mmol) portion of the 2'-0 methyl CU
dimer (product of Example 2C) was dis501ved in 10 ml THF
and treated all at once with 1.5 ml (1 m in THF, 1.5 eq.)
tetrabutylammonium fluoride ("TBAF"). The reaction
mixture wae stirred at room temperature of r 3 0 minutes
after which time HP3-C revealed complete deprotection of
the silyl group had been achieved. The reaction mixture
was concentrated and the ,-"n,Pntrate purified on 10 g
silica gel, eluting with 3 :1 ethyl acetate/methylene
chloride with 596 th;ln~ll. The clean fractions were
rnnr-Pntrated to give 550 mg of the above-identified pure
5 ' -OH aimer .
E. Pre~aration of a ChirallY Pure (CU) 2'-0-Methvl
(NP/DE) Dimer Svnthon
A 230 mg portion of 2'-0-methyl CU 3'-0~ dimer (prod-
uct of ExampIe 2D) was rendered anhydrous by 2 X ~ ml co-
evaporations in ACN. The resulting dry solid foam was
dissolved in 2.5 ml ACN and then 73 ~1 (2.5 eq.) triethyl-
amine ("TEA") and 94 ~Ll (2.0 eq.) 2'-cyanoethyl-N,N-
diisopropyl chlorophosphoramidite (,~CNE) were added. The
reaction mixture was stirred at room temperature for 2
hours at which time EIP~C analysis determined the reaction
to be complete. The reaction mixture was fl; RR~l VP~ in
eluent (3/1/1 ethylacetate/acetonitrile/methylene chloride
with 4~6 TEA) and loaded onto 2 g silica gel equilibrated
with 3/1/1 ethylacetate/acetonitrile/methylene chloride
with 496 TEA. The column was run using 3/1/1
ethylacetate/acetonitrile/methylene chloride with 196 TEA.
The clean fractions, 3 to 25, were concentrated, redis-
solved in acetonitrile and concentrated again to a solid
foam. The foam was dried overnight under full vacuum to
give 200 mg of the above-irlpnt;r;ed product.
W0 95/l3834 2 ~ ~ ~ 2 S 9 PCINS94/13387
-
F. Pre~arPtion of Ch;rally Pure (C~J) 2'-0-~ethyl
ID?~R;) /NP Dimer Svnthon
Into a 100 ml round bottom flask was placed 400 mg
(0.372 mmole) of 2'-0 methyl CU dimer (product of Example
2D); it was rendered anhydrous by 1 X 5 ml co-evaporation
with acetonitrile. The dry foam was then released from
the vacuum system under argon gas, dissolved in 4 ml ACN
and stoppered with a rubber septa. The solution was
treated with 2 equivalents TE~A (103 ~Ll, 0.744 mmol),
followed by 1.75 equivalents chloro-methyl-N,N-diisopropyl
rh~Srh;n~ ("Cl-MAP") (118 ILl, 0.651 mmol). The reaction
mixture was stirred for 1 hour at room temperature, after
which time HPLC showed about 50/50 starting
material/product. An additional 50 ~l TEA and 70 ~l Cl-
M~P were then added and the mixture stirred for an hour.
When HPLC showed only 80~ conversion, an additional 30 ~l
TEA and 3 0 1ll Cl-MAP were added and the re8ulting mixture
stirred another hour. At this time HPLC revealed 6~
starting material. The reaction mixture was concentrated
to dryness. The residue was dissolved in 500 ml 3/1/3
e~hylacetate/acetonitrile/methylene chloride with 496 TEA
and loaded onto 5 g silica equilibrated in the same
solvent system. Fractions were collected. The early
fractions were c~- nt~m;n~tPd with a yellow impurity and,
thus, were pooled and concentrated separately. The
product from those fractions was then repurified by
chromatography using the same conditions and pooled with
the clean product isolated from the first column. The
~ .' in~d products were co-evaporated with ACN (3 X 5 ml)
and dried overnight under full vacuum to give 350 mg (77~
yield) of the above i ~l.ont; f i ed product which HPLC Rhowed
to be 95.59~ pure.
Wo95113834 ~ ~. PCr/Uss4/l3387
æ~6~5~
- 46
rnnle 3 -
Pre~aration of 2 ' -O-MethYl MpS (1~ ) / 2 ' -O-MethYl-DE and 2 ' -
Q-Methvl ~SPS (R~) / 2' -O-Methvl-MP Dimer SYnthons
These dimer sYnthons are prepared ky f ollowing the
procedures described in Example 2, except that in Para-
graph C, an equivalent amount of 3H-1,2-benzodithiole-3-
one, l, 1-dioxide (Beaucage reagent) is substituted for
cumene l~y~L~ ide~ The L~ .,ceduL ::8 of Paragraphs 2E and
2F, respectively, lead to the phosphodiester and methyl-
phosphothioate linkage combinations.
r le ~ ~
Pre~aration of MPS (R~) /DE Dimer SYnthons
These dlmer synthons are prepared by following the
procedures of Example 1, except in Paragraph A, an e~uiva-
lent amount 3-E-l~2-benzodithiole-3-one~ 1,1-dioxide
(Beaucage reagent) is substituted for the oxidizer solu-
tion (I~/H2O/lutidine/THF) .
.nr)le 5
Pre~aration of MP (~ ) /PS2 Dimer Svnthons
The MP(Rp)/PS2 dimer synthons are prepared as follows.
Isometrically pure Rp dinucleosides having a free 3'-OH are
prepared according to the methods described in Example lA.
The dinucleoside is converted to the corrPspr~n~l;n~
th1 nph~sph~.ramidite using procedures such as those of
Plotto et al. (Plotto et al, Tetrahedron 47:2449-61
(1991)) or Gorenstein et al., U.S. Patent No. 5,218,088.
The dinucleoside is co-evaporated three times with anhy-
drous pyridine, followed by three co-evaporations with
toluene. A portion of dinucleoside (10 mmoles) is dis-
solved in 200 ml anhydrous dichloromethane, then three
equivalents of anhydrous diisopropylethylamine followed ~y
1.5 equivalents of ~ chloro-N,N-diisopropylamino-
th; ~ th~yphosphine are added at 0C with stirring. The
reaction is monitored by TLC until ~pt~orm;npd to be
complete.
WO 95/13834 2 1 7 ~ ~ ~ 9 PCI/US94/13387
47
The product i8 worked up and purif ied using the
procedures of Example lB for isolation of the MP (Rp) /DE
phosphoramidite .
Examl~le ~i
PreParation of ~SPS (R~) /PS2 Dimer SYnthons
The MPS (Rp) /PS2 dimer synthon6 are prepared as
follows. The isometrically pure Rp dinucleoside with a
free 3'-OH is prepared according to the methods of Example
4. Using the dinucleoside, the dimer synthon is prepared
by the methods of Example 5.
rnle 7 - =
Pre~aration of ~tPS(R~)/2'-0 Me~hY1 DE D;r-r ~Ynthon~
The MPS (Rp) /2 ~ -O-methyl DE dimer sYnthons are prepared
using procedure6 analogous to those of Examples 1 and 3
but using the c.~L~,pLiate protected 2'-deoxynucleoside and
protected 2 ' -O-methyl nucleosidea .
Exam~le 8
Prel:1aration of a PQlv-CT Oliqomer Xav;n~r Alternatinq
MP (R~) /DE InternucleosidYl J,; nkAqes
An oligomer having the sequence 5~ - (C T) - (C'T) - (C-T) -
(C-T) - (C-T) - (C-T) - (C T) -A-3' was prepared using a C-T
MP (P~) /DE dimer synthon prepared according to Example 1.
The grouped dinucleosides indicate where the stereochemis-
try is fixed as the fast eluting isomer on 8ilica gel
(putative Rp) and the asterisks indicate the chirally pure
linkages .
Manual couplings were used to synthesize the oligomer
to conserve reagent, although the process can be done on
an automated DNA synthesizer. The sequence was synthe-
sized from the 3'-term;nl~q starting with methacrylate
support bound deoxyar~-~nn~;n,~.
The protected dinucleoside methylphosFhnn~m;~;te (22
mg each per required co~rl;n~) freshly co-evaporated with
pyridine and toluene to ensure dryness were placed into
_ _ _ _ _ ~ ., . . , . . _ .. . . _ .. ... . .. . .
~ 217~2Sg
WO 95/13834 ~ ~' PCT/US94/13387
48
dried l ml glass autosampler vials and dissolved in
anhydrous acetonitrile to a cnn~ ntration of O.l M (200 ~l
per coupling). The vessels were purged with argon and
tightly sealed with screw caps with tef lon septa .
A l ~mole scale DNA synthesis column (Milligen) was
f illed with 1 ILmole of methacrylate support bound deo~;y-
Arlf~nns;nf-. The column was attached to a ring stand in a
vertical orientation . A male-male luer ~f itting was
attached to the bottom along with an 18 gauge needle to
control the effluent. The column was washed with lO ml
acetonitrile using a syringe. The support bound nucleo-
side was detritylated by passing 3 ml of 29~ dichloroacetic
acid in dichloromethane through the column over l. 5
minutes. The orange, dimethoxytrityl cation bearing
solution was reserved. The column was washed twice with
ml each of anhydrous acetonitrile.
The $irst collr1;n~ was accomplished as follows: lO
ml more anhydrous acetonitrile was passed through the
column. Then, 200 ~Ll of the CT methylrh~srhnnAm;~ite was
drawn into a 1 ml syringe. Next, 200 Ill of 0.45 M tetra-
zole in anhydrous acetonitrile was likewise drawn into the
syringe c~nt~;nlng the methylphosrh~nAm;~;te. The re-
agents were rapidly mixed in the syringe, then slowly
passed through the column dropwise over three minutes,
being sure to lightly draw the plunger up and down to
ensure adequate mixing with the support. A$ter 3 minutes,
1 ml of the rn~;~l;7;n~ reagent (O.l M I, in 73~ tetrahydro-
furan, 259~ 2, 6-lutidine and 29~ water) was passed through
the column over one minute. The column was washed with 20
ml acetonitrile and then treated with 600 ILl of a solution
~ntA;n;n~ 205~ (v/v) acetic anhydride, 30~6 (v/v) acetoni-
trile, 509~ (v/v) pyridine and 0.312~ (w/v) dimethylamino-
pyridine. The column was then washed with 20 ml acetoni-
trile .
The above-described synthetic cycle was repeated
until the synthesis was completed. The overall coupling
Wo 9S/13834 ~ 1 7 ~ 2 S 9 PCr/lJS94/13387
49
efficiency based on dimethoxytrityl absorbance was 95.7~,
for an average of 99.3~ per coupling.
The oligomer was then cleaved from the support and
'deprotected. The support bound oligomer was removed from
the synthesis cartridge and placed in a glass 1 dram vial
with a screw top. The support was treated for 30 minutes
at room temperature with 1 ml of a solution of acetoni-
trile/ethanol/NH~OH ~9/9/1). Then, 1 ml of ethyl-~nf~1Ar-;n~
was added to the reaction vessel and the reaction allowed
to sit for 6 hours at ambient temperature in order to go
to completion. The sUp~rnAt~nt cnn~A;n;n~ the oligomer
was then removed f rom the support and the support was
rinsed twice with 2 ml of 1/1 acetonitrile/water; the
washings were combined with the supernatant. The combined
solution was diluted to 30 ml total volume with water and
neutralized with apprn~;r-~t~ly 4 ml of 6 N HCL. The
neutralized solution was desalted using a W~ters C-18 Sep-
Pak cartridge which was pre-equilibrated with 10 ml
acetonitrile, 10 ml of SO~ acetonitrile/100 mM triethyl-
ammonium birArh~nAte~ and 10 ml of 25 mM triethylammonium
birArhnnAte, seql~n~;A11y. After the reaction solution
was passed through the column, it was washed with 30 ml of
water. The product was then eluted with 5 ml of 1/1
acetonitrile/water .
The oligomer was purified on HPLC using a Beckman
Ultrasphere-reverse pha5e 4 . 5 X 250 mm column with an
increasing gradient of acetonitrile in O . 5 M triethyl -
ammonium acetate (0~ to 403~ over 40 minutes). The isolat-
ed yield was 41 OD,60 units (35~). The compound was
characteri~ed by electron spray mass ~e- LL, ~ (calc .
4391/found 4391).
Alternatively, the above-identi~ied oligomer can be
synthesized on an automated DNA synthesizer. In this case
the c.~Lu~liate dimer synthons (as used above in the
manual synthesis) are dissolved in acetonitrile to a
rnnrC~n~ration of O.1 M as described above. The amidite
so~ utions are placed in conical vessels on a Millipore
Wo 9~/13834 PCrlUS94/13387
7~2~ ~o
Expedite DNA Synthesizer. All other reagentæ (oxidizer,
deblock, capping reagents and activator) are prepared as
described above for the manual synthesis, and applied to
the d~ ,L,Liate positions on the instrument as instructed
in the manual. P'U~L 'nq parameters for one synthesis
cycle are as given in Table I in U. S . Patent Application
Serial No. 08/158, 014. The deprotection and purification
of the oligomer is carried out as described above f or the
manually synthesized oligomer.
Exam~le 9
PrenAration of a Polv-CU Qliqomer Havinq Alternatinq 2'-O-
MethYl NP(~) /2' -O-Methvl DE and 2~ -O-Methvl NP(R,~ /2' -O-
Methvl NP Internucleosidvl T,; nk;~qe5
An oligomer having the sequence= 5' (C~U) - (C`U) - (C-U) -
( C'U) - ( C'U ) - ( C'U ) - ( C U) - A- 3 ' was prepared us ing 2 ' - O - me thyl
NP(Rp)/2'-O-methyl DE dimer synthons prepared according to
Example 2 hereinabove.
The d~ Liate dimer synthons were dissolved in
acetonitrile to a ron~-~ontration of 0.1 M. All other
reagents used were as described in Example 8.
A 1 llmole scale DNA synthesis column (Millipore) was
f illed with 1 ~Lmole of methacrylate support bound deoxy-
adenosine. The dimer synthons were coupled geq~ nt;Ally
from the 3'-terminu~ as described in Example 8 except that
the co-lrl; n~ time was ~t~n~ to two minutes . The
overall ~ o~rl;n~ efficier,cy based on dimethoxytrityl
~hp~rhAn~e was 50%, for an average of ~919~ per coupling.
The dimethoxytrityl group was removed from the oligomer at
the end of the synthesis.
3 o The deprotection was carried out as described in
Example 8. The crude yield was 103 OD26~ units. The
oligomer was purif ied on XPI C with a Beckman Ultrasphere-Rp
using an increasing gradient of acetonitrile in 0 . S M
triethylammonium acetate (10~ to 30~6 over 30 minutes).
35 The isolated yield was 39 OD26~ units (3896). The compound
WO 95l~3834 PCTIUS94/13387
2~7l~2~ig
51
was characterized by electron spray mass spectrometry
(calc. 4713/found 4712)
This oligomer can also be synthesized on an automated
DNA synthesizer as follows. The d~L~Liate dimer
8ynthons (as used above in the manual synthesis are
dissolved in acetonitrile as described in Example 8. The
amidite solutions are placed in conical vessels on the
Millipore Expedite DNA synthP~i7Pr. All other reagents
(oxidizer, deblock, capping reagents and activator) are
prepared as described in Example 8, and are applied to the
a~lJL~L iate positions on the in8trument as instructed by
the manual. The same coupling program a8 described in
Example 8 is used except that the coupling time is extend-
ed to 2 minutes.
The deprotection is carried out as described in
Example 8. The oligomer can be purified on HPLC using as
described above for the manual synthesis.
Using similar procedures as described in detail in
Example 8 of U. S . Patent Application Serial No .
08/154, 013, the oligomer 5' - (C*U) - (C*U) - (C*U) - (C*U) - (C~U) -
(C*U)-(C*U)-A-3' having 2~-0-methyl MP(R~)/2'-0-methyl MP
(racemic) mixed linkages was prepared. The product was
also characterized by electron spray mass spectroscopy
(calc . 4699 . 5/found 4701) . Automated synthesis may also
be employed as explained above.
le lO
Pre~aration of 5' - (T A) - (G C~ - (T T) - (C C) - (T T) - (A ::) - (C'T) -
(C'C) - (T'~) -C-3' }Iavinq Re~eated MP(R~) ~MP ~,1nkAqe Struc-
tures
The grouped dinucleosides indicate coupled dimers and
the asterisk indicates where the ster~nrhPm; ~try is fixed
(chirally de~ined or chirally pure) as the fast eluting
isomer on silica gel (identified as Rp).
An oligomer having this se~uence was synthesized
using the d~L~Liate protected dinucleotide methylphos-
ph-~n~m; tii te~ prepared using methods such as those de-
wo 95/13834 ; ! ~ I Pcr/Uss4ll3387
~ I
~1~62~9 52
scribed in Examples lA and lC above. Manual couplings
were used to synthesize the oligomer to conserve reagent,
although the process can be done on an automated DNA
synthesizer from the 3 ' terminus starting with support-
5 bound cytidine.
Each of the desired protected 7; n~ ntide methyl-
rhnsrhnn;7m; dites ~22 mg each per required coupling), T'A,
G'C, T T (2x), C C (2x), A G, C T, and T G, freshly co-
evaporated with pyridine and toluene to ensure dryness,
10 was placed into a dried 1 ml glass ;7llt-~ ler vial and
dissolved with anhydrous acetonitrile to give a concentra-
tion of 0.1 M (200 ~Ll were used per coupling). The vials
were purged with argon and tightly sealed with screw caps
with tef lon septa .
A 1 ~mole scale Milligen DNA synthesis column was
filled with 1 ~mole of support bound cytidine. The column
was attached to a ring stand irL a vertical orientation.
A male-male leur fitting was attached to the bottom along
with an 18 gauge needle to control the f~l 17~nt . The
column was washed with 10 ml of ACN using a syringe. The
support bound nucleoside was then detritylated by passing
3 ml of 29~ dichloroacetic acid in dichloromethane through
the column over 1. 5 minutes . The orange, dimethoxytrityl
cation bearing solution was reserved. The column was
washed twice with 10 ml each of ACN ~anhydrous).
The first coupling was accomplished by passing 10 ml
more ACN (anhydrous) through the column. Then, 200 1~l of
the TG methylphosphonamidite was drawn into a 1 ml sy-
ringe. Next, 200 ~ of 0.45 M tetrazole in anhydrous ACN
was likewise drawn into the syringe cnnt;7;n;n~ the methyl-
rhn9phnn;7m; rl 7 te . The reagents were rapidly mixed in the
syringe, then slowly passed through the column dropwise
over 3 minutes, being sure to lightly draw the plunger up
and down to ensure adequate mixing with the support.
After 3 minutes, 1 ml of the n~r;r7.;7;ng reagent (0.1 M I2 in
74.259~ THF, 259~ 2,6-lutidine, and 0.2596 water) as passed
through the column over 1 minute. The column was then
WO95/13834 ~17B2~9 PCrlUS94/13387
53
washed with 20 ml of ACN. The column was then treated for
1 minute with 600 ~il of a solution rnntA;n;n~ 20~ (v/v)
acetic anhydride, 3096 (v/v) ACN, 50~ (v/v) pyridine, and
O . 312~ (w/v) dimethyaminopyridine. The column was washed
with 2 0 ml of ACN .
The synthetic cycle was then repeated with each
dinucleotide methylpho9~hnn~m;~l;te until t~,e synthesis was
completed . The order of addition of dimers af ter the
initial T G cmlrl;nr, was C C, C T, A G, T T, C C, T T, G C,
and T'A.
The dimethoxytrityl group was removed from the oligo-
mer at the end of the synthesis.
The oligomer was the~ cleaved from the support and
deprotected The support bound oligomer was removed f rom
the synthesis cartridge and placed in a glass 1 dram vial
with a screw top. The support was treated for 30 minutes
at room temperature with 1 ml of a solution of acetoni-
trile/ethanol/NH~OH (9/9/1) . Then, 1 ml of ethylPnP-l;Am;nP
was added to the reaction vessel and the reaction mixture
allowed to sit for 6 hours at ambient temperature in order
to go to completion. The ~llr~rn~t~nt cnntA;n;~r~ the
oligomer was then removed from the support and the support
was rinsed twice with 1 ml of 1/1 acetonitrile/water; the
w-~h;n~c were ' inP~l with the supPrnAtAnt The combined
solution was diluted to 50 ml total volume with water and
neutralized with approximately 1 7 ml of glacial acetic
acid. The neutralized solution was desalted using a
Waters C-18 Sep-Pak cartridge which was pre-e~uilibrated
with 5 ml acetonitrile, 5 ml of 50~ acetonitrile/water,
and 5 ml of water, sequPnt;Ally After the reaction
solution was passed through the column, it was washed with
50 ml of water. The product was then eluted with 2 ml of
1/1 acetonitrile/water.
The oligomer was purified by HPLC on a reverse phase
column (Poros II R/H 4 . 6 x 100 mm) using a gradient of
acetonitrile in water.
wo 95113834 ~17 ~ 9 PCrNS94/l3387
.
54
Coupling efficlencies are set forth in the table
below .
Coupling Efficiencie~ of Dinucleotide
Methylrh~ph~nnm~ ~ teEI
Dinucleotide Coupling Efficiency
T G 99.7
C'C 90.2
C T 91. 8~6
A G 85.5
T T 97 . 8
C'C 83 . 696
T'T 1009~
G'C 86 . 296
T'A 92 . 496
,
5~.Y~mnle 11
Pre~aration of 5' - (G T) - (C T) - (T C) - (C A) - (T G) - ~C A) - (T'G) -
(T'T)-(G'T)-C-3' Havinq RePeated MP(R~)/MP T.;nk~e Struc-
tures
The grouped dinucleotides indicate coupled dimers and
20 the asterisk indicates where the stereochemistry is fixed.
This sec~uence was synthesized using the appropriate
protected Rp dinucleotide methylrh~cnh~n~m;~l;tes prepared
and isolated using procedures such as those described in
Examples lA and lC above. Manual couplings were used to
25 synthesize the oligomer in order to conserve reagent.
However, if desired, the process can be done on an auto-
mated DNA synthesizer from the 3 ~ terminus starting with
methacrylate jsupport bound 2~-deoxycytidine.
Each of the desired protected dinucleotiae methyl-
30 rh~c~h-~n~m;dites (100 mg), G T, T T, T'G, C'A, T'G, C'A, T'C,
C-T, and G~T was placed into a dried 3 ml glass conical
vial and dissolved with anhydrous acetonitrile to a
c~nrPntration of 0 ~ M. Molecular sieves (3 A) (0 . 5 ml
Wo 9sll3834 2 1 ~ 6 2 ~ 9 PCrlU594113387
volume) were added to each vessel, the vessels purged with
argon, and tightly sealed with screw caps with teflon
6epta. The reagent9 were allowed to stand overnight prior
to use.
A 1 ~lmole scale 1~; l l ;3-~n DNA synthesis column was
f illed with 1 ~Lmole of methacrylate support bound 2 ' -
deoxycytidine. The column was attached to a ring 8tand in
a vertical orientation. A male-male luer fitting was
attached to the bottom along with an 18 gauge needle to
control the effluent. The column was washed with lO ml of
ACN using a syringe. The support bound nucleoside was
then detritylated by passing 3 ml of 2 . 5~ dichloroacetic
acid in dichloromethane through the column over 3 . 0
minutes. The orange, 1; hnl~ytrityl cation bearing
solution was reserved. The column was washed twice with
lO ml each of ACN ~anhydrous).
The first coupling was accomplished by passing lO ml
more ACN (anhydrous) through the column. Then 200 ~l of
the G~T methylphosphoramidite was drawn into a l ml
syringe. Next, 200 1ll of 0.4~ M tetrazole in anhydrous
ACN was likewise drawn into the syringe rnntA;n;nrJ the
methyl~hnsphnn~m;dite. The reagents were rapidly mixed in
the syringe, then slowly passed through the column drop-
wise over l minute, being sure to lightly draw the plunger
up and down to ensure ade~uate mixing with the support.
Af ter 3 minutes, 1 ml of the n~ ; ng reagent ( 0 .1 M I2 in
74.25~ THF, 25~ 2,6-lutidine, and 0.25~ water) was passed
through the column over 1 minute. The column was then
washed with 20 ml of ACN. The column was then treated for
1 minute with 600 ~l of a solution rnn~A;n;n~ 209~ (v/v)
acetic anhydride, 30~ (v/v) ACN, 50% (v/v) pyridine, and
0.31296 (w/v) dimethyaminopyridine. The column was washed
with 2 0 ml of ACN .
The synthetic cycle was then repeated with each
tl;nllr1f~otide methylrhn~phon~m;dite until the synthesis was
completed . The order of addition of dimers af ter the
WO 95/13834 ?. i~ 5 9 PCT/US94/13387
56
initial G T coupling was T T, T G, C A, T G, C A, T C, C T and
G T.
The dimethoxytrityl group was removed from the oligo-
mer at the end of the synthesis.
The oligcmer was then cleaved from the support and
deprotected. The support bound oligomer was removed from
the synthesis cartridge and placed in a glass l dram vial
with a screw top . The support was treated f or 3 0 minutes
at room temperature with l ml of a solution of acetoni-
trile/ethanol/NH~OH (9/9/l) . Then, l m~ of ethyl PnPr~ mi nP
was added to the reaction vessel and the reaction allowed
6 hours to go to completion. The supernatant ~ nnt~;n;ng
the oligomer was then removed from the support and the
support was rinsed twice with l ml of l/l
acetonitrile/water; the w~ch;n~C were combined with the
supernatant. The ,- ;nPd solution was diluted to 30 ml
total volume with water and neutralized with approximately
l . 7 ml of glacial acetic acid. The neutralized solution
was desalted using a Waters C-18 Sep-Pak cartridge which
was pre-equilibrated with 5 ml acetonitrile, 5 ml of 50~6
acetonitrile/water, and 5 ml of water, sPql1Pnt;~lly.
Af ter the reaction solution was passed through the column
it was washed with 5 ml of water. The product was then
eluted with 2 ml of l/l acetonitrile/water.
The oligomer was purif ied by HPLC on a reverse phase
column (Poros II R/H 4 . 6 x l00 mm) using a gradient of
acetonitrile in water.
r ~le 12
Pre~a~atiQrl of 5' - (G A) - (G G) - (A G) - (G A) - (G G) - (A'G) - (G'A) -
(A~G) -G-3 ' Havinq RePeated MP (R~) /MP Linka3e Structures
The grouped dinucleosides indicate the coupled dimers
and the asterisks indicates where the stereochemistry is
fixed (chirally defined or chirally pure) as the fast
eluting dimer isomer on silica gel (;~lpnt;~ied as E~p).
9~/13834 ,! 1 ~ ~i 2 5 9 PCr/US94/13387
Thia oligomer was prepared using automated synthesis
coupling G A, G G and A G MP (Rp) /MP dimer synthons prepared
according to the procedures of Examples lA and lC.
An amount of G A, G G and A G dimer 6ynthon6 was
dissolved in acetonitrile to give a concentration of 0.1
M and stored over 3 A molecular sieves (Millipore,
Milford, MA) overnight.
The di6solved dimers, with molecular sieves, were
placed in conical vessels on a Millipore ~xpedite DNA
Syn~h~R; 7Pr which as equipped with end-line filters to
remove particulates. All other reagents ~oxidizer,
deblock, capping reagents and activator) were prepared and
applied to the c.~L.,~Liate positions on the instrument as
instructed in the manual. The coupling program was
modified to place the oxidizing step immediately subse-
quent to the ~ r~; ng step in order to reduce h~ khnn~
cleavage prior to oxidation. (See Hogrefe, R. I , et al .
"An Improved Method for the 5ynthesis and Deprotection of
Methyl rh~Rrh~n~te Oligonucleotides " in ~ethods in Molecu-
lar Biolo~v, vol. 20: Protscol5 for Oliqonucleotides and
Analoqs (ed. Agrawal, S.) pages 143-164, Humana Press,
Totowa N.Y. ~1983). The ~L~yL n~ parameter6 for one
synthesis cycle ~"Syn4all-1 /lmol~) are set forth in Table
II of U.S. Patent Application Serial No. 08/154, 013 .
A 1 ~mole scale DNA 6ynthesi6 column ~Millipore) was
filled with 1 ~Lmol o~ methacrylate support-bound deoxy-
gll~n~Rinf~ and was placed on the DNA synthe6izer. The
dimers were coupled 6eqll~n~i~1ly from the 3' t~rm;nl1R.
The dimethoxytrityl protecting group was removed from the
oligomer at the end of the synthe6is.
The oligomer was then cleaved from the support and
deprotected. The support bound oligomer wa~ removed from
the synthesis cartridge and placed in a gla6s 1 dram vial
with a screw top . The support was treated ~or 3 0 minutes
at room temperature with 1 ml of a solution of acetoni-
trile/ethanol/NH40H ~9/9/1) . Then, 1 ml of ethyl~n~A;~m;n-~
wa6 added to the reaction vessel and the reaction allowed
Wo95/13834 2~ 2S;9 PCrlUS94/13387
58
6 hours to go to completion. The S~l~Prn~tAnt ~-nnti~in;n~
the oligomer was then removed from the support and the
support rinsed twice with 1 ml of 1/1 acetonitrile/water,
when ~'nml~; nPtl with the supPrn~t~n~ . The ~ '; nPd solution
was diluted to 50 ml total volume with water and neutral-
ized with approximately 1. 7 ml of glacial acetic acid.
The neutralized solution was desalted using a Waters C-18
Sep-Pak cartridge which was pre-es~uilibrated with 5 ml
acetonitrile, 5 ml of 50~ acetonitrile/water, and 5 ml of
water, se~uentially. After the reaction solution was
passed through the column, it was washed with 5 ml of
water. The product was then eluted with 1. 8 ml of 1/1
acetonitrile/water .
The crude yield was 87 OD260 units. The Oligomers was
purified on ~IPLC using a ~-cyclobond standard phase 4.5 X
250 mm column (Azetec, Inc. Whippany, NJ) ~.vith a decreas-
ing gradient (80% to 4096) of acetonitrile in 0 . 05 M
triethylammonium acetate (pH 7 ) . The isolated yield was
22 OD26~ units (25~). The product was characteri2ed by
electron spray mass spectrometry (calc. 5407/found 5401).
F le 13
Pre~aration of an Oliqomer Havinq Alternatinq MP ~R~) /PS
InternucleosidYl Linkaqes
An oligomer having altPrn~t; n~ MP (R~) /PS; nt~rnllcl eo-
25 sidyl linkages is prepared using dimer synthons. All theparameters of the synthesis, deprotection and purification
are as described in Example 8, except that the oxidizing
reagent is replaced by a 0 .1 M solution of 3H-1, 2-benzo-
dithiole-3-one, 1,1-dioxide or a 0.1 M solution of sulfur
30 in 1/1 carbon disulfide/diisopropylethylamine.
F le 14
Preparation of an Oliqomer Havinq AltPrnPtln~ MPS(R~)/DE
TnternucleosidYl Linkaqes
An oligomer having alternating MPS (R~) /DE internucleo-
35 sidyl linkages is prepared using the dimer synthons of
wo 95/13834 2 i 7 6 2 S g PCT/US94/13387
59
Example 4. All other parameters of synthesis,
deprotection and purification are as described in Example
8.
Exam~le 15
5 Pre~aration of an Oliqomer Havi n~ ~l t~rn~t; nq MPS (RF) /PS
Tnterrll-rlensidvl Linkaqes
An oligomer having alt~orn~t; n~ NPS (Rp) /PS; nt~rnll~ l eo-
sidyl linkages is prepared using the dimer synthons of
E~cample 4. All of the parameters of synthesis, depro-
10 tection and purification are as described in Example 8,except that the nl~1~1i7;n~ reagent is replaced by a 0.1 M
solution of 3_-1,2-benzodithiole-3-one, 1,1-dioxide or a
0.1 M solution of 9ul~ur in 1/1 carbon
disulf ide/diisopropylethylamine .
~YAr'-le 16
Pre~aration Qf an Oliqomer Hav;n~ Alternatinq NP(R~)/PS2
Internucleosidvl Linkaqes
An oligomer having alt~orn~t; n~ NP (Rp) /PS2; nt~rnllcl eo-
sidyl linkages i8 prepared using the dimer synthons of
2 o Example 5 . All of the parameters of synthesis, depro-
tection and purification are as described in Example 15.
F le 17
Pre~aration o~ an Oliqomer Havinq Alternatinq NPS (~) /PS2
Internucleosidvl Linkaqes
An oligomer having alternating NPS (R~) /PS2 inter-
nucleosidyl l; nk~PR is prepared using the dimer synthons
of Example 6. All of the parameters of synthesis, depro-
tection and purification are as described in Example 16.
ExamDle 17A
Prel~aration of an oliqomer Havinq Altorn~t;n~ MP (R~) /2 ~ -0-
Meth~l ~E Internucleosidvl Linkaqes
An oligomer having alt~rn~t; n~ NP (R9) /2 ' -0-Methyl DE
int~rnucleo_idyl linkages is prepared using dimer synthons
2sg
Wo 95/13834 . ~ , PCr/US94/13387
.
similar to those of Example 7. All other parameters of
synthesis, deprotection and purification are as described
in Example 9.
T;~ le 18
Pre~aration o~ an Oliqomer Havinq Alternatinq MP (Rq) /MPS
Internucleosidvl :I.inkaqe8
The preparation of an oligomer having alternating
MP (R7) /MPS internucleosidyl linkages is aL~ h~d using
dimer synthons prepared according to Examples lA and lC
and dissolved and stored over molecular 6ieves. The
oxidizing reagent is a 0 .1 M solution of 3H-1, 2-benzo-
dithiole-3-one, l,l-dioxide ("Beaucage Reagent", see Iyer,
R.P. et al., JACS 112:1254-1255 (1990)) or a 0.1 M solu-
tion of sulfur in 1/1 carbon disulfide/ diisopropylethyl-
amine, with synthesis proceeding generally as described in
Example 12 .
~Y~ e 19
P~eT~aratiorl of ;~n Oliqomer Havinq 2 ' -O-Methvl Nucleosidvl
Unite and Alternatinq MP(R~)/MPS Internucleosidvl rink~es
This oligomer is prepared using the dimer synthons as
described in Examples 2A-2D and 2F and ~ollowing the
general synthetic procedures of Bxample 8 of U. S . Patent
Application Serial No. 08/154,013, except that the oxidiz-
ing reagent described therein is a 0 . lM aolution of 3~-
1l2-benzodithiole-3-onel 1,1-dioxide or a 0.1 M solution
on 1/1 carbon diaulf ide/diisopropylamine .
r - ~le 20
Pre~aration of an Oliqomer Havinq 2'-O-Methvl Nucleosidvl
TTn;ta and Alternatinq MPS(F~)/MP Internucleoaidvl ~inkaqe~
This oligomer is ~~ epa, e~ using dimer synthons a~
described in Example 3 above and by f ollowing the parame -
ters of synthesis, deprotection and purification of
Example 19.
-
WO 9~/13834 ~ I ~ 6 2 ~ 9 PCr/US94113387
61
F le 21
Pre~aration of an Oliqomer Havinq Alternatinq MPS (~ ) /MP
InternucleosidYl J,i nkA~re$
This oligomer i5 prepared using dimer synthons pre-
pared according to Examples lA and lC, substituting
Beaucage reagent for the oxidizer in Example lA, and by
following the parameter8 of synthesis, deprotection and
purif ication as described above in Example 12 .
F le 22
Pre~aration of an Oliqomer Xavinq Alternatinq ~PS ~R~,l /lLPS
InternurleosidYl T,inkAqeg
This oligomer is prepared using dimer synthons as
referred to in Example 21 and by following the parameters
of synthesis, deprotection and purification as described
above in Example 12, except that the oxidizing reagent
used therein is replaced by a 0.1 M Rn11lt;nn of 3_-1,2-
benzodithiole, l,l-dioxide or a 0.1 M solution of sulfur
in 1/1 carbon disulfide/ diisopropylethylamine.
~A mn 1 e 23
Prel~aration o~ 2'-F Dimer Syntl~nnR
Dimer 8ynthons useful in the preparation of the
oligomers of the present invention may be prepared using
2~-fluoronucleosides. Methods for preparation of 2~-
fluoronucleosides have been reported and are known to
those skilled i~ the art. lSee, e.g.: Codington, JOC
Vol. 29 (1964) ~2'-F U); Mangel, Angew. Chem. 96:557-558
(1978) and Doen, JOC 32:1462-1471 (1967) (2'-F C);
Ikehara, Chem. Pharm. Bull. 29:1034-1038 (1981) (2'-F G);
Ikehara, J. Carbohydrates, Nucleo8ides, Nucleotides 1:131-
140 (1980) (2'-F A), and also Krug, A, Nucleosides &
Nucleotides 8:1473-1483 (1989).)
The preparation of dimer synthons using 2'-fluoro-
nllrler,R; ~1~R may be accomplishing using the procedures
analogous to those de8cribed for the 2 ' -O-methyl dimer
35 sYnthons (See, e.g., ~xamples 2, 3, and 7~. The resulting
7 &2S9
W095/13834 2i PCrlUS94113387
62
dimer synthons may be used to prepare oligomers using
methods analogous to the methods used f or the 2 ~ -O-methyl
dimer synthons such as in Example 9.
~ mn 1 e 2 4
5 Pre~aration of MP(~)/MP(R,)/DE and MP(R~)/MP(R~)/MP Trimer
5yr~thons
The above- identif ied trimer synthons are prepared
using the MP (Rp) /MP dime~ synthons of Example lC. The
dimer synthon is coupled to a 5'-hydroxy, 3'-silylated
10 nucleoside according to the methods of Example lA for the
coupling of the 3' -nucleoside: to the monomer phosphorami-
di te
The selected 5'-hydroxy, 3'-silylated nucleoside (1
equivalent ) and isomerically pure Rp dimer methylphos -
15 phonamide (1. 25 equivalents) are weighed into a roundbottom flask and dried by co-evaporation with acetoni-
trile. The resulting foam is dissolved in acetonitrile
and treated with a solution of O . 45 M tetrazole in aceto-
nitrile (4.5 equivalents) . After 3 minutes, the reaction
20 mixture is oxidized and the reaction product i8 worked up
as described in Example lA. The diastereoisomers of the
3'-silylated trimer are resolved on a silica gel column as
described in Example lA f or resolution of the dimer
isomers . The conf iguration of the separated diastereo-
25 isomers iB determined using 2-D nmr (ROSE~) . The trimer
having the desired chiral conf iguration (Rp/R;,~ of the two
internucleosidyl linkages i8 converted to a trimer synthon
by reaction with chloro-j~-cyanoethoxy-N,N-diisopropyl-
amin~.L,h~ hn~ c~midite using methods as described in Example
30 lB. The trimer synthon is worked up and purified using
methods as described in Example lB to achieve the
MP t}~) /MP (Rp) /DE trimer .
Using similar procedures, an MP(Rp)/MP(Rp)/MP phos-
phoramidite synthon may be obtained by using chloromethyl-
35 N,N-diisopropyl~m;nnrhn~phine in the fi:nal reaction as
described in Example lC for the corresponding dimer
Wo 9S/13834 ~ ~ 7 6 2 ~ 9 PCr/uSg4113387
. .
63
synthon. Workup and purification are as described in
Example lC .
F le 25 - ~
PrsPaxation Of 2 ' -O-Allyl p;r and Trimer SvnthnnR And
Their Use in Oliqomer ,3vnthes; R
The dimer and trimer 9ynthons described, for example,
in Examples 1 and 24 can be prepared using 2 ' -0-allyl
nucleosides . The preparation of 2 ' -O-allyl nucleoside6
and their use in the preparation of oligomers has been
reported (see e.g. Iribarren, et al. (19gO) Proc. Natl.
Acad. Sci. (USA~ 87:7747-51; and I,esnik et al. ~1983),
Biorh~m; Rtrv 32: 7832-8), and such substituted nucleosides
are commercially available. The nucleosides are used to
prepare dimer and trimer fiynthons using procedures de-
scribed hereinabove. The synthons are used to prepare
oligomers using methods such as those described in Exam-
ples 10, 11, 12, 13 and others above.
le 25
Pre~aration of an OlicrnmPr Hav; n~ MP ll~, ) /MP~DE Internllr] eo-
sidvl Tl;nk~re8
The above-; ~l~on~ Ol; ~ r is prepared using the
trimer synthons of Example 24, or by tho8e in Example 20
of U.S. Patent Application Serial No. 08/l54, 014, and by
following the methods described in Example 8, substituting
the trimer synthons for dimer synthons. All other parame-
ters o~ synthesis, deprotection and pur;firA'r;nn are as
described in Example 8.
Wo 95/l3834 2 1 7 6 2 5 ~ PCT/US94/13387
64
le 27
Pre~aration of an Oliaomer Havinq MP(~,)/Mp(R~)/MP Inter-
nucleosidYl Linkaaes
The above-;~lrnt;f;ed oligomer is prepared using the
5 procedures described in Example 14 of IJ.S. Patent Applica-
tion Serial No. 08/154, 013 .
F le 28
Pre~aration of Olicro~ibonucleosides
O1igor;h~n1~r~ tides used in the present examples may
10 be synth~ ; 7~rl using general procedures such as described
below .
The appropriate 5'-D-dimethoxytrityl-2'-0-tert-
butyldimethylsilyl - 3 ' -0 -N, N- diisopropyl - ,B -cyanoethylphos -
phoramidite nucleosides (Millipore, E~ilford, MA) were used
15 or synthesis . Syntheses were . done on a 1 l~mole scale
with a Milligen 8750 automated DNA synthesizer using
standard Milligen phosphoramidite procedures with the
exception that the coupling times were ~lct~n~ to 12
minutes to allow ader~uate time for the more sterically
20 hindered 2'-0-tert-butyldimethylsilyl RNA monomers to
react. The syntheses were begun on control-pore glass
bound 2'-0-ter~-butyldimethylsilyl r;hon-~c~eosides pur-
chased from Millipore. All other oligonucleotide synthe-
sis reagents were as described in Millipore' 8 standard
25 protocols.
After synthesis, the olis~n~rlr~tides were handled
under sterile, RNase-free conditions. Water was steril-
ized by overnight treatment with 0 . 5~ diethYlpyrocarbonate
followed by autoclaving. All glassware was baked for at
30 least 4 hours at 300C.
The oligonucleotides were deprotected and cleaved
f rom the support by f irst treating the support bound
oligomer with 3/1 ammonium hydroxide/ethanol for 15 hours
at 55C. The supernatant, which rfnt;l;nl~rl the oligonucle-
35 otide, was then ~ nt~d and evaporated to dryness. Theresultant residue was then treated with 0 . 6 mL of 1 M
Wo 95113834 2 i 7 6 2 ~ 9 PCr/US94113387
tetrabutylammonium fluoride in tetrahydrofuran (which
t-"ntsiin~c~ 5~ or less water) for 24 hours at room tempera-
ture. The reaction was c~uenched by the addition of 0.6 mL
of aqueous 2 M triethylammonium acetate, pH 7. De6alting
5 of the reaction mixture was accomplished by passing the
solution through a Bio-Rad l~DG column uæing sterile
water. The desalted oligonucleotide was then dried.
Purif ication of the oligoribonucleotides waæ carried
out by polyacrylamide gel electrophoresis ~PAGE) contain-
ing 1596 19/l polyacrylamide/bis-acrylamide and 7 M urea
using ~tandard procedures (See Maniatis, T. et al.,
Molecul~r l~ nin~: A Labor~torY MAnllAl, pages 184-185
(Cold Spring Harbor 1982) ) . The gels were 20 cm wide by
40 cm long and 6 mm in width. The oligoribonucleotides
~60 OD Units) were dissolved in 200 /LL of water C~ntAining
1.25~ bro--~rh~nnl blue and loaded onto the gel. The gels
were 1-un overnight at 300 V. The product bands were
visualized by W bA~-k~hArl~wing and excised, and the
product eluted with 0 . 5 M sodium acetate overnight . The
product was desalted with a Waters C18 Sep-Pak cartridge
using the manu~acturer supplied protocol. The product was
then 3~P l~h~llecl by kinA~;n~ and analyzed by PAGE.
r le 29
pre~ara~ion of Rac~m; c Methvl~hoqnh~n~te Olic~onucleotides
Various racemic oligomers were synthesized using 5 ~ -
(dimethoxytrityl) deoxynucleoside-3' - [ (N,N-diisopro-
pylamino) methyl] -phosrh~nm~m; dite ~ ~ . Solid-phase
synthesis was p~LLo, 1 on methacrylate polymer supports
with a Biosearch Model 8750 DNA syrJth~s; ~or according to
the manufacturer's r~ ~At;r)n~ except for the fol-
lowing modifications: the monomers were dissolved in
acetonitrile at a ~onr~ontrations of 100 mM, except dG,
which was dissolved in 1/l acetonitrile/dichloromethane at
100 mM. DEBLOCK reagent = 2 . 5~ dichloroacetic acid in
dichl~ thAnf~ OXIDIZER reagent = 25 g/L iodine in
0.259~ water, 259~ 2,6-lutidine, 72.596 tetrahydrofuran. CAP
Wo 95/l3834 2 1 7 6 2 ~ 9 . . PcrrUsg4rl3387
66
A = 10% acetic anhydride in acetonitrile . CAP B = O . 625
N,N-dimethylaminopyridine in pyridine.
The dimethoxytrityl group was removed f rom the
oligonucleotide at the end of the synthesis.
The oligonucleotide was then cleaved from the support
and deprotected. The support bound oligonucleotide was
removed from the synthe5i5 cartridge and placed in a glass
1 dram vial with a screw top. The support was treated for
3 0 minutes at room temperature with l ml of a solution of
acetonitrile/ethanol/NH,OH (9/9/1). Then, 1 ml of ethyl-
l;Am;nf~ wag added to the reaction vessel and the
reaction allowed 6 hours to go to completion. The super-
natant cnnti~;n;nq the oligonucleotide was then removed
f rom the support and the support rinsed twice with 2 ml of
1/l acetonitrile/water, when combined with the superna-
tant. The combined solution was diluted to 30 ml total
volume with water and neutralized with approximately 4 ml
of 6 N HCl. The neutralized solution was desalted using
a Waters C-18 Sep-Pak cartridge which was pre-equilibrated
with lO ml acetonitrile, 10 ml of 509~i acetonitrile/lOo mM
triethylammonium ~icarbonate, and lO ml of 25 mM triethyl-
ammonium bicarbonate, se~l~nt;;-lly. After the reaction
solution was passed through the column it was washed with
3 0 ml of water . The product was then eluted with 5 ml of
1/l acetonitrile/water.
The oligonucleotide was purified by HPLC on a reverse
phase column ~Whatman RAC II) using a gradient of acetoni-
trile in 50 mM triethylammonium acetate.
F le 30
~'h; -riC oliqonucleo~ide Assemblv From MP ~R~) /MP and
~5P (R~) /DE ~imer Synthnn~ and PhosPhoramidite and MethYl-
~hosl~honamidite Monomer SYnthons
MP (R~) /MP dimer synthons contained a methylphosphor-
amidite coupling group at the 3 ' end. When coupled
together to make an oligomer, these synthons give racemic
methylphosrhnr-~t-~ linkages at every other position.
Wo95113834 '~ ;9. PCrlUS94/13387
67
NP (Rp) /DE dimer synthons contained a ,~-cyanoethyl phos-
phoramidite coupling group at the 3 ' -end. Both types of
dimer synthons were synthe9ized as described in ~xample 1.
Methylphosphonamidite monomer synthons were synthesized at
5 ~BL Scientific ~San Luis Obispo, CA). Betacyanoethyl
phosphoramidite monomer synthons were purchased from
Milligen/Biosearch .
All synthons were coupled using a Milligen Expedite'R
automated DNA synthesizer The coupling ~LU~L~ for each
10 8ynthon were as tabulated below. To generate a phosphoro-
thioate bond during a coupling step, the program "Thioate-
511M" was used with either a dimer or monomer synthon
containing a ,B-cyanoethyl phosphoramidite coupling group.
WO 95/13834 2 1 ~ 6 2 ~ g - PCI/US94/13387
68
DIESTER -- 5 IIM
_ _ _ _ _ _ _
/i Function Morde Amount Time(sec) nl~ 1r~1nn
/ i /Argl /Arg2
5 /i
~ri
Sn..hl nr-r ~ n
0; /i ;r ~ault i/ d~IT l S ~a;t"
l4 ~i ~hotometer S ~ .TAET data rn~ inn
/i blk i/ ~-TLSE 1 1 0 'lblk to column"
6 /i ~blk ~ SE 20n 49 '~eblock"
/i qsh A to Cl i/ ~ ISE 8n ~ ~lush sy~temwith Wsh A"
15 l.. _ /i 'hotometer S i/ I 1 ~MTOP data rnll~r~inn~
/i r,as A to Cl i/ ? I,SE 1 0 ~r-as A to Cl waste~
-.- /t Adv~nce Frar i/ X ~ ~ O ' vent Out r~FF
/i qsh A i/ ?J~SE 20 o n sh A"
$Couplilg
_ /i qsh i/ ~U.S l0 n Flush system with Wsh"
/i ~ct i/ ~r . 1 n ~Flu~h syr;tem with Act"
l /i ~ + Act i/ 'U, O IlMonomer + Act to column"
+ Act i/ YCr. l~l 61' ~Couple monomer"
/i ~ct i/ ~; . ll~ nCouple monomer"
/~ qsh i/ ~J, I 56 - "Couple monomer~
/i qsh i/ ~, so ~Flush ~yst~m with W3h~
~Capping
2 /i Wsh A i/ PUI,SE 25 O Caphs to columr"
5r;xidizing i/ PUI,SE lS0 0 'End oi rycle wash"
5 /i Ox i/ PU~SE 50 30 "Ox"
z /i Wsh A i/ PULSE 50 0 ~Flush system with Wsh A"
5 rlpping
353 /t Caps i/ pu~SE 25 0 "Caps to column~
2 /i Wsh A i/ PUI,SE 50 0 'Wsh A"
~ /~ w-h A ~/ I'lll.SE 5~ ~ EIId o~ cycle ~ ~h~
.
wo 95113834 ~ ~ 7 ~ 2 ~ 9 ~1US94/13387
69
T~IOATE -- 5 ,~dY.'
/~ Function Mode Amount Time(sec) npnr~irtirn
/~ /Argl /Arg2
5 /"
/~ ~
_ _ _ _
Snrh~
14~ .dvanc~ PrAc ~ n~vent out ON~
l0 ~ e~ault ~/ ~IT l. " Irlit~'
1'. /~ ~hotometer S ~/ A _ " TART dzta rrllPrr~;rn
blk ~ ~SB l n 1blk to column"
.r /~ blk ~/ ~I~SE 20r~ 41 '-eblock~
~ /~ 'sh A to Cl i/ '~SE 8 ) " 'lush sy6temwith Wsh A~
15 l~ hotometer 5 ~/ I. r _ n~:TOP data rnll~rt~rn~
,. /t l.as A to Cl ~/ ? I,SE 1 ~ ~r,aS A to Cl war~te"
dvanc~ Frac ~ v~nt Out OFF"
sh A ~/ ? ~SE 20 ~ ~ sh A"
$roupling
20 . /t Wsh f/ '~r 5_ 10 0 n`lush system with Wsh"
/~ Lct ~/ ~U,~ lr n n ~lush 6ystem with Act"
2 /i ~ + Act ~/ ' .. r; n n onomer + Act to column"
Z /~ + Act ~ 6~ ouple monomer"
/` .. =t t/ ~ 1 nrouple monomer~
25/~ sh ~ ,r,, 55, ~ n cuple monomer~
. /i sh ~/ ~.'J 50 I n lush system with Wsh"
$rapping
_3 /~ Caps ~/ PULSE 25 0 "Caps to column~
.2 /~ Wsh A i/ PUI.SE s0 o "Wsh A"
30.2 /~ Wsh A ~/ PU~SE l50 o "End of cycle wash~
1;1 mr~ r1i 7~ nrJ
.7 /~ Aux ~/ PULSE 5 0 "SOx"
.7 /i Aux ~/ PU~SE 45 60 "SOx~
:.2 /~ Wsh A ~/ PULSE 50 o "Flush system with Wr;h A"
35S~ apping
_3 /t Caps ~/ PUIISE 25 0 "Caps to columnn
,2 /~ Wsh A ~/ PU3 SE s0 0 "Wsh A"
.2 /i Wsh A ~/ PUISE l50 0 "End of =ycle wash"
2176259
WO 9S113834 PCllllS94/13387
M~T . v ~ Us ~lU_._!TE - - 5 UM
_ _ _ _ _ _ _ _ _ _ _ _ _ _ _
/t Functlon Mode Amount Time~sec) nrRr~;r~
/t /Argl /Arg2
5 /t
/t
__ !
~n~.hlor~;ng
14. /~ ~.dvanr/e Fra~ t/ ~ I vent out ON~
10 ~ /t )e~ault t/ q,~IT ~ 'aitr
~' /t ~hotometer !9 t/ ~,'. ' ' ~ TART data ~ollection"
; /t ~blk t/ ~ LSE 1 ~ " )blk to rlolumn~
,f' /t ~blk t/ ~ ~LSE 20n 4 I ~Neblock'
I /t Ish A to Cl t/ ~ LSE 8n n ~ lush systemwith Wsh A"
15 1.. : /t ~hotomet~r S t/ '' r ~ TOP data collection"
/t ;as A to Cl t/ ~ LSE 1 ' aa A to Cl waste~
.~. /t ~dvance Frac t/ ~ (I r v~nt Out OFF"
.~ /t qsh A t/ ~ LSE 2 0 r. I' sh A'
5Coupli-g
20 /t qsh t/ ~U.- 1n D 'FluRh ~ystem with Wsh"
/t ~ct t/ ~ 1 n DFlush system with Act"
1 /t ~ ~ Act t/ ~ n ~Monomer + Act to column"
19 /t A ~ Act t/ ~U. 1' 6 "Couple monomer~
, /t ~ct t/ ~ Couple monomer~
25 /~ qsh t/ ~ ' 56 ~Couple monomer"
/t ~qSh t/ ~ 50 'Flush system with Wsh
,5 /t OX t/ PULSE 50 30 'Ox"
.2 /t Wsh A t/ PUI.SE 50 0 'Flush system with Wsh A~
3 0 $ apping
- 3 /t Cap6 t/ PUI,SE 25 0 'Cap6 to column"
2 /t Nsh A t/ PULSE 50 0 ~Wsh A~
.2 /t Wsh A t/ P~LSE 150 0 ~End oi cycle wash'
~76259
WO 95/13834 ~ PCT/US94113387
71
MP ~R,) /~P -- 5 UM
/'Function Mode Amount Time(rlec) D~rr1rttrn
/~ /Argl /Arg2
/~
/~ ~
_ _ _ _ _
$n~.hl rr~ nrJ
14 /~ ~dvance Frac ~/ ~ : '''vent out ON"
10 /~ 1efault i/ ~IT l.~ ~'~ait-
hotom~ter S ~/ X~ : TART data rnll~r~;r
)blk ~ lIgB 1 " Iblk to column-
r~ blk ~/ lnLSE 20n 4 ~ "neblock~
~ /~ 1sh A to Cl ~/ ~1~5E 8~ r ~lush systemwith Wsh A"
15 1 /~ ~hotometer S ~ n 1~ ~TOP d~ta rrl~rr~
/~ ~as A to Cl ~ ? LSE l~ .as A to Cl waste"
1-4 /~ dvance Frac ~/ ~ n r vent Out OFF"
'` /~ sh A ~/ ~ LSE 20 n 11 sh A"
$r~ouplilg
20/~ ~sh ~/ 'U_~ l0 n ~1 ~lush gystem with Wr~h
/~ ~ct ~/ 'U. l n 'lush system with Act"
Act ~/ ~J- n ~1 onomer 1 Act to column"
Act ~ l. 6r R( ouple monomer"
ct~ lr) 1~1 ouple monomer"
25/~ ~sh ~/ ' .. ' 56 - "~ ou l m om 1'
/f ~sh ~/ 1 .. 50 p e on er
$r~71~1~7in
S /~ Ox ~/ PUIISE 50 30 1'Ox1'
2 /~ Wsh A ~/ PULSE 50 0 "Flush system with Wsh A"
3 0S ~pping
3 /~ Caps i/ PULSE 2s 0 Cap& to column"
2 /~ Wsh A ~/ PULSE 50 0 ~Wsh A~
2 /~ Wsh A ~/ PULSE l50 o End of cycle wash~
WO 95113834 2 ~ ~ ~ 2 5 9 PCINS94/13387
72
NP ~R,,) /DE -- 5 U16
/i~ Function Mode Amount Time 1sec) Description
/~ /Argl /Arg2
/~
/
n - hl nr r; T`
l4. /~ .~.dvzmce Fr~c i/ ~ 1 "Event out ON"
101 /i efault ~/ ~IT 0 l. "iait~
'hotometer S ~ TA~T data rr~ r ~ i nn n
)blk i/ ~SE l 1 " )blk to column~
6 /~ blk ~/ ~ ~SE 20ri 4 ~ n ;eblock"
/~ sh A to Cl ~/ '~SE 8 ~ ) n ~lUGh system with Wsh A"
15 l~ hotometr~r 5 ~ TOP dat~ collection~
.as A to Cl ~/ ~SE ln "~-as A to Cl waste
l~c /~ . dvance Frac ~ n ~ vent Out OFF"
_ /~ sh A i/ nsE 20 n ~ sh A"
5Coupli Ig
20. /~ ~sh ~/ ~Tr.5 ln O RFlush system with W3h"
/~ ~ct ~/ ~tr, 1 I nFlULh system with Act"
Act ~/ ' . "Monomer ~ Act to columr"
Act ~/ ', l, 6r1 nCouple monomer~
~ct ~/ '. . ln "Couple monomer"
2~sh ~/ '',_ ' 56. nCouple monomer"
Bh ~/ '' ._ sn "Flush GyGtem with Wsh"
Sn7,~ ~ r ' n~
7 /~ UX t/ PUDSE 50 30 i'Aux"
.2 /~ sh A ~/ PUnSE 50 0 "Flush system with Wsh A"
3 0$ apping
.3 /~ CapG ~/ PlnSE 25 0 "Caps to column"
.2 /~ Wsh A '/ PlnSE 50 0 Wsh A"
.2 /~ Wsh A ~/ PUnSE lS0 0 "End of cycle wash"
~17625g
Wo 95/13834 PCrtUS94tl3387
73
Applying one or more of these co-lrl 1 n~ routines with the
u~r iate dimer or monomer synthons, one akilled in the
art can recognize that each of the chimeric oligomers
described in sub8e~lue~t examples can be synthesized.
Deprotection and purif ication of each chimeric
oligomer was done essentially as described in Examples 8
through 12.
The identitie5 of certain chimeric oligomers made ac-
cording to this Example, as well as other compounds, were
confirmed by electrospray mass spectrometry as shown in
the following table:
WO 95/13834 ~ i 7 6 2 5 9 PCT/IJS94113387
74
Seq. 1~ Sequence Backbone MW MW
Predictod 3:ound
2624-1 3--CTGTTG TACGT ACCTTCTG-5' Racemic MP 5725 5726
2371-1 3--CTGTTG TACGT ACCTTCTG.5' 75%MP(R~) 5725 5725
3130-3 3'-CCTGTTG TACGT ACCTTCTG-5' MP(R~)IDE 6028 6029
2366-1 3'-CCTGTTG TACGT ACCTTCTG-5' PS 6354 6357.9
2567-1 3'-CCTGTTG(TACGT)ACCTTCTG-5' [MP][DE]IMpl 6022 6018
2687-1 3'-CCTGTT(GTACG)TACCTTCTG-5' [7~ ' JLt~k~ oRpMP] 6022 6022
3169-1 3'-CCTGTTG(TACGT)ACCTTCTG-5' [MP(P~,,)lnF]lnF]r ''(Pp)/DE] 6033 6034
3214-1 3'-CCTGTTG(TACGT)ACCTTCTG-5' [MP(P~p)IDE][PSIDE][MP(P~p)IDE]6082 6081
10 3257-1 3'-CCTGTTG(TACGTAC)CTTCTG-5' [MP(R~,)IDE][PSIDE][MP(Rp)IDE]6100 6100
3256-1 3'-CCTGTTG(TACGT)ACCTTCTG-5' [MP(R,)IDE][PS][MP(R~)IDE] 6113 6114
3258-1 3'-CGTCCTCGATT(CCTTC)GATGGTAC-5' [MP(R;)IDE][PS DE][MP(Rp)IDE]7300 7299
3260-1 3'-CGTCCTCGATT(CCTTC)GATGGTAC-5' [MP(R~)IDE][PS][MP(P~")IDE] 7331 7331
3261-13'-~ ~lA(GTGAC)CTATATGG-5' [MP(P~")IDE][PSIDE][MP(R,)IDE]7313 7310
15 3262-13'-(~ lA(GTGAC)CTATATGG-5' [MP(R;)IDE][PS][MP(Pp)IDE] 7345 7346
3269-1 3'-ACGTCTGATCA(GTAAC)TAACTCAC-3' [MP(Rp)/Dy[PS/DE][MP(Rp/DE])7309 7308
3270-1 3'-ACGTCTGATCA(GTAAC)TAACTCAC-5' [MP(Rp)lDE][PS][MP(Rp)lDq 7341 7340
I . (T', ' ` - 1~1 the portion that activates RNAseH; thc linkage on the 5 '-side of the indic~ted nucleoside
is charged.
.
Wo 95113834 2 1 7 6 2 5 9 PCr/US94/13387
le 31
~ucl~qe Stabilitv Studies of Various R~kh-~n~ Mo~; fied
(Non-Chimeric) Oliqomers
In each of the experiments described in this example,
5 various bSrkh~ modified oligomers were evaluated having
the f ollowing sequence: 5 ' - ~l Cl cl C'l'~ 'l'A- 3 ' ( f or 2 ' -
deoxy sugars); or 5'-W~U~:U~:uW~u~uA-3' tfor 2'-O-methyl
sugars). The all-diester (DE) h~l~kh~ oligomer was
purchased from Oligos Etc. The other ba/ kh(~n~ oligomers
10 were synthesized as described in the preceding examples.
(a) Stability studie~ in the ~ of puri~ied
snake venom rh~ hn~l~ e~tera~e . Snake venom phosphodies-
terase I (PDE-I) from crotalus adamanteus was purchased
from US Biochemicals, Inc Aliquots of each oligomer
(0 . 075 A260 units) were pipetted into polypropylene
microcentrifuge tubes and dried in a Speed-Vac'M vacuum
centrifuge (Savant, Inc.). Next, the tubes were placed on
ice and aliquots of PDE-I were added to each tube (0.1
unit/mL in 95 ~L of 10 mM Tris-HCl, pH 8-8, 2 mM MgCl2,
0.4~6 glycerol). The zero time point samples were diluted
immediately with acetonitrile (3511L), frozen in a dry
ice/isopropanol bath, and stored at -20C for analysis at
a later time . The l~ i n; n~ samples were then placed in
a water bath at 37C. Samples for each specified time
point were then removed from the water bath, diluted with
acetonitrile and frozen as described for the zero time
point samples.
At the conclusion of the nuclease degradation exper-
iment, the samples were individually thawed and analyzed
; ~ t~ly by L~vel~e~ phase HPLC using a Beckman System
Gold apparatus with a Model 126 binary gradient pump
module and a Model 168 Diode Array Detector. The samples
were injected onto the column u6ing a manual injector with
a 2000 I~L sample loop. A Vydac C4 Protein column was used
for these experiments (Vydac cat. no. 901019, 4.6 mm i.d.
X 250 mm long). Elution was done with a dual solvent
8y8tem: Buffer A = 196 acetonitrile in 50 mM triêthyl
_ _ _ _ = = = _ . . .. . . .. .. . . ......
wo g5,l3834 2 ~ 6 2 ~ 9 PCr/US94/13387
' ' ' ~ ' '' 1 ' '
76
ammonium acetate ~TEAA, pH 7.0); Buffer B = 50% acetoni-
trile in 50 mM TEAA (pE~ 7.0). Solvent flow rates were
increased rom 0 . 05 to 1. 0 mL/min . over the irst minute
of the run and then held at 1. 0 mL/min. for the rf~m~; n~
5 of the run. G~adient condition5 for each backbone were as
follows: All-DE b~Ckhnn~o- 5-2596 Buffer B (2.5 - 9 min.),
25-45~ Buffer B (9.0 - 22.5 min.) 45-100~ Buffer B (22.5 -
28.0 min.); 2'-deoxy MP(Rp)/DI!: h~kh~n~- 5-3596 Buffer B
(2.5 - 12.5 min.), 35-50% Buffer B (12.5 - 22.5 min.), 50-
10096 Buffer B ~22.5 - 27.5 min.); 2'-0-methyl MP(Rp)/DE~
bac~hone- 5-50~ Buffer B (2.5 - 17.5 min.), 50-659~ Buffer
B (17.5 - 27.5 min), 65-10096 Buffer B (27.5 - 31.0 min.).
Average retention times for each h~--khnn~ oligomer
(undegraded) were as follows:
A11-DE: 15 . 7 min .
2 ' -deoxy MP ~Rp) /DE: 18 . 5 min .
2'-O-methyl MP(R~)/2'-O-methyl DE: 18.6 min.
Degradation was determined by the appearance of earlier
eluting peaks and a decrease in area (or complete loss) of
the peak corr~cpnn~;ns to the full-length oligomer.
(b) Stability studies in lleLa cell lysates. HeLa
cell CytQrl~elm;c lysate was purchased from ~ndotronics,
Inc. (Minneapolis, MN) . This preparation is a hypotonic
dounce lysis in 5 X the packed cell volume. It was
buffered to pH 6 . 0 by adding 0 .4 mL of 2- (N-morpholino)
eth~n.o~l~lfonate (MES, 0.5 M solution, pH 6.0) to 3.6 mL of
cell lysate on ice and mixing with mild agitation.
Alisluots of oligomer were dried and then diluted with HeLa
cell lysate (95 ~LL) as described in the preceding example.
Samples were then ;ncllh~t-~ at 37C and analyzed by
reversed-phase HPLC exactly as described in the preceding
example .
(c) Stability studies in cell lysate ~rom African
Green Monkey ~idney COS-7 cells. COS-7 cell lysate for
these experiments was p~a-~d as follows. COS-7 cells
were grown to 90% confluency and then harvested in the
presence of 0.2596 trypsin. The cell pellets were washed
-
Wo 95/13834 2 1 7 ~ 2 5 9 PCrlUSs4ll3387
twice with phosphate buffered saline and then frozen
overnight at -20C. Next, the pellets were resll~pon~1~d in
approximately an equal volume of lysis buffer (2.5 mM
HEPES, pH 7.2, Z.0 mM MgCl" 0.19c NP-40), drawn up and down
ten times through a sterile 1 mL polypropylene pipette,
and then centrifuged at 10,000 x G for 5 minutes. Approx-
imately 4096 of the resulting sUpernatant was then used to
lyse the cell pellet in a dounce homogenizer (Type A
pestle) with twenty strokes. This suspension was then
centrifuged as above and the sllr~rn~t~nt was, '~in~d with
the rest of the supernatant from the first resuspension.
The resulting solution represents pr~ n~nt ly cytosolic
lysate without any nuclear debris and is approximately 1-
1. 5 times the volume of the original packed cell pellet .
Aliquots from the resulting cell lysate were buffered with
either 25 mM Tris-acetate (final pH 7.4) or 25 mM MES
(final p~ 6.0) prior to ;n, llh~tinn with oligomer.
Ali~auots of each oligomer (0 075 A260 unit) were dried in
sterile polypropylene microcentrifuge tubes and then
resuspended in 10 ~L of COS-7 cell iysate on ice. Water
(90 ~LL) and acetonitrile (35 ~L) were added; -';~tely to
the zero time samples and they were frozen in a dry
ice/ethanol bath and stored at -20C for later analysis.
The 1, ;n;n~ samples were then incubated in a water bath
at 37C. At specified time points, samples were removed
from the water bath, dilutea with water and acetonitrile,
and frozen exactly as described for the zero time point
controls. Following the incubations with cell lysate, the
samples were individually thawed, diluted with water (535
IlL) and analyzed immediately by reversed phase HPLC as
described above.
(d) Stability ~tudie~ cell ly~ate from Esche-
richi~ coli E. coli cell lysate was prepared as follows.
Approximately 2 x 101l cells were pelleted by centrifuga-
tion, r~ p~nri~d in 10 mL of Tris-HC1 (50 mM, pH 7.5) and
;nrllhatF.d at room temperature for five minutes. Next,
dithiothreitol and lysozyme were added to final cnnn~on~ra-
WO 95/13834 , - PCr/US94113387
2~7~2~
tions of 2 mM and l mg/mL, respectively, and the resulting
suspension was incubated at 37C for 30 min. The mixture
was then sonicated briefly ten times on ice and centri-
fuged at 7, 000 rpm for 20 min. Based on visual inspec-
tion, it was estimated that this procedure had not suffi-
ciently lysed the cells, so the S~ rn~t~nt (vol. = 5 mL)
was collected and stored at 4 C and the cell pellet was
resuspended in in l mL of Tris-HCl (50 mM, pH 7 . 5) . The
res-1~p~nt8~ cell pellet was exposed to five rounds of
freeze/thaw, sonicated briefly to break up the chromosomal
DNA, and then centrifuged at 8, 000 rpm for 5 min. The
resulting sup~rn;~t~nt (approx. 700 ,LL) was then . ` in~
with the supernatant from the previous step (approx. 5 mL)
and centrifuged at 6, 000 x G for 5 min. to pellet any
residual debris . The f inal supernatant was estimated to
contain approximately 50~ lysed cells in approximately 57
times the original cell pellet volume (lO0 ILL). Alir~uots
of the oligomers (0.050 A26~ units) were dried in sterile
polypropylene microcentrifuge tubes and resuspended in 95
~LL of cell lysate on ice. Incubations at 37C, HPLC
analysis, and ~r~uantitation of oligomer degradation were
done exactly as described aboYe.
(et) Stability studies in cell ly~ate from St~p~ylo-
cocc~l aureuf;. S. aureu~ cell lysate was prepared as
described above for E. coli except with the following
modifications: (i) the lysis was conducted with a cell
pellet containing approximately 4 x lO10 cells; (ii)
lysostaphin was used instead of lysozyme (500 units,
Sigma, Inc. ); and (iii) a total of lO freeze/thaw cycles
were used instead of five. Incubation with oligomers at
37C, HPI-C analysis and det~rD~;n~t;rn of oligomer degra-
dation from the chromatograms were rnn~ ct~tl exactly as
described for the experiment with ~. coli in the example
above .
Results. Percent degradation was determined by
comparing the peak heights and peak areas f or each time
point in each experiment to the 2ero time point controls.
wo 95/13834 ~ 1 7 ~ 2 ~ 9 PCT/US94113387
The half-lives for each oligomer in the presence of PDE-I
were then determined by plotting log(~ full-length) versus
time and finding the value corresponding to log (50~) =
l . 699 . The following table ~ummarizes the results from
5 these experiments:
Meta7Dolic Degradation Rate8 of pA~kh~ln~ Analog~ in Bio-
logical Sy~tems.
Alternating
~tl~-llf~ of Normal 2~-0-Met_yl MP(R~,)/D3i: 2~-0-Mcthyl
Annlos Phospho- ~NA Alterrating 7fP ~
diester 2 ' -0-methyl
10109~ Fetal Calf
Serum, p~l 8 12 min. 40 min. 5 Ers. ~ 300 ~rs.
Green Monkey
Kidney Cell c 10 mi~. ~ 5 }7rs. - 25 Elrs. Stable*
Lysate, pEI 6. O
15Green Monkey
Kidney Cell ~ S min. ~ S E~rs. - 20 }Irs. sta
Lysate, p}~ 7 . 4
E. co7~ Cell 1-3 min. _.2 7Irs. ~ 65 ~rs. Stahle~
Lysate
S. Aureus Cell 13 min. ~ 20 Elrs. ~ 75 71rs. Stable*
Lysate
Snake Ve~om 15 min . 2 . 5 min . 167 min . Stahle~
Phospho -
die~7tt~r:la9~Aae
* No ~t~ctAh~ r~ t~nn after 24 hour ~nrl~h5lt~r~n,
r ~le 32
HYbri~;~Ation of 6~h;rAlly 7~nriGhed Antl Non-6~h;ra
Oliqomers to RNA Tarqets
Chirally enriched all-pyrimidine (C'T) 7A and all-
30 puri~e ~A'G) ~T MP-oligomers were prepared using either Rp-
or Sp-dimeric units. Control o1;~ s were also prepared
using the individual I - ic units. The asterisks
indicate the positions of def ined chirality.
Each oligomer was annealed to a complementary syn-
35 thetic RNA target and then monitored by ~hqorhAn~-~ at 260
nm as a function of temperature. Sigmoidal transitions
were observed corr~ro~;ng to the7-mal denaturation o~ the
hybridization complexes. The Tm values were r~ta~rrn;nf~ at
. , . . , . , _, _ _
WO 95/13834 - PCr/US94/13387
2~9 - i
the midpoint of each sigmoidàl transition. Previously, we
have shown that ~CT) B r~l; S forms a double-stranded
complex with RNA at neutral pX, whereas (AG) 8 Oligomer
forms a triple-stranded complex. Thus, we anticipated
5 that the data for each chirally enriched seriea would be
applicable to double-stranded and triple-stranded MP/RNA
helices, respectively. The Tm data is summarized below:
Al ~ernatinq ~ CT),
(A)
10 Oliaomer No. Seauence Confiquration'
2286-1 5'-c t-c t-c t-c t-c t-c t-c t-a-3' (Rp)
2288-1 5 ' ctctctctctctct-a-3 ' (R, S)
2287-1 5'-c't-c~t-c't-c't-c't-c't-c't-a-3' (Sp)
(B)
Oliqo Tm ~l:l.RNA) 4Tm~RNA)
2286-1 45.5C +10.4C
2288-1 35. 1C ----------
2287-1 25.4C -9.7C
Alternatinq (AG)7
2 0 (A)
Oligomer
No . Seauence Conf iquration'
2323-1 5 ' -a'g-a g-a g-a g-a g-a g-a g-t-3 ' (Rp)
2 2 5 3 -1 5 ' - agagagagagagag - t - 3 ' ( R, S )
2252-1 5 ' -a'g-a'g-a'g-a'g-a'g-a'g-a'g-t-3 ' (Sp)
(B)
Qli~o Tm ~l:l.RNA) ~Tm(RNA)
2323-1 55.2C +7.2C
2253-1 ~ 48.0C -------
2252-1 40 . 0C -8 . 0C
As shown in the tables above, the Rp-enriched preparations
have higher Tms with RNA targets. On the other hand, Sp-
enriched preparations have lower Tms with RNA targets.
In separate experiments, we conf irmed that the
35 chirally-enriched (C T) ~A and (A G) ,T MP-oligomers form
-
WO 9~113834 PCTNS94113387
~17~25~
81
double- and triple-stranded complexes with RNA at neutral
pH, respectively.
These experiments demonstrate that chiral enrichment
can dramatically effect the binding affinities of MP-
5 oligomers in both a duplex and triplex motif.
F le 33
Tm Com~arisons for Methvlphos~honate Oliqomer9 ContAin;na
Eithe~ R~-~n~iched or Racemic Backbones
Racemic methylphosphonate oligomers and complementary
10 RNA targets were synthesized according to the methods
described in Examples 28 and 29 . The MP (R~) ~MP oligomers
were synthesized according to the methods described herein
by coupling MP (E~,) /MP dimers . Each coupled MP (Rp) /MP dimer
is indicated by parentheses in the table below, wherein
15 asterisks indicate chirally pure linkages.
z~nn~ ;nr reaction mixtures rr,nt~;n~ equimolar
amounts of methylphosphonate oligomer and RNA target
oligomer (2 . 4 llM total strand rrnr~ntration), 20 mM
potassium phosphate (pH 7.2), 100 mM sodium chloride, 0.1
20 mM EDTA and O . 039~ potassium sarkosylate . The reaction
mixtures were heated to 80C and then slowly cooled to 4C
over apprrY~-t~y 4 to 6 hours. The annealed samples
were then transferred to 1 cm quartz cuvettes and absor-
bance at 260 nm as a ~unction of temperature was monitored
25 using a Varian Cary Model 3E Spectrophotometer rclnt~;n;n~
a 6 x 6 temperature controlled sample holder and which
interfaced with an IBM compatible PC t~r. The
temperature was varied from 5C to 80C at a ramp rate of
1C/minute. The Tm for each melt profile is defined at
30 the point corresponding to the first deri~ative (of the
A260-temperature function) . The following table summarizes
data obtained for a number of pairs of racemic versus Rp-
enriched methylphosphonate oligomers. Based on the
observed increases in Tm, Rp-enrichment u9ing the MP(Rp) /MP
35 dimer coupling method described herein leads to signifi-
~6~5~ ~
Wo 95113834 - , PCrlUS94/13387
82
cant r~nh~3nc t in the binding energy between a methyl-
phosphonate oligomer and its RNA target.
Comparison of Tm's for MP(R,j)/MP Enriched and Racemic
MethYlPhos~honate Oliqomers
Sequence Sequence Tnn ~Tm
number
2288-1 5'-CT-CT-CT-CT-CT-CT-CT-A-3' 34.4C
2286-1 5'-(C-T)(C-T)(C'T)(C'T)(C-T)(C T)(C~)-A-3' 44.0C 9.6C
2253-1 5'-AGA-GAG-AGA-GAG-AG-T-3' 48.9C
2323-1 5'-(A G)(A G)(A G)(A G)(A G)(A G)(A G)-T-3' 56.3C 7.4C
2517-1 5'-GTG-TGT-GTG-TGT-GTG-TA-3'-3' 41.0C
2516-1 5'-(G-T)(G-T)(G~T)(G-T)(G'T)(G-T)(GT)(GT)-A-3' 48.8C 7.8C
1634-1 5'-TAG-CTT-CCT-TAG-CTC-CTG-3' 38.2C
2570-1 5'-(T-A)(G C)(T-T)(C C)(l-r)(A G)(C-T)(C C)(T G)-C-3' 46.9C 8.7C
2688-1 5'-ATG-GTGTCT-GTT-TGA-GGT-T-3' 40.0C
2662-2 5'-(A'T~(G-G)~T-G)(T-C)(T-G)(T~(T G)(A G)(G-T~-T-3' 47.5C 7.5C
2624-1 5'-GTC-TTC-CAT-GCA-TGT-TGT-C-3' 38.6C
2571-1 5'~G T)(C T)(T C)(C A)(T G)(C A)~G)(T'T)(G-T)-C-3' 46.3C 8.2C
2625-1 5'-GCT-TCC-ATC-TTC-CTC-GTC-C-3' 42.9C
2 0 2574-1 5'-(G C)(T-T)(C C)(A'T)(C-T)(T C)(C T)(C G)(T C)-C-3' 51.8C 8.9C
F le 34
R;nrlino Stabilitv of Various Backbone Modified Oliqomers
Havinq a (CT) .A Model Seouence to Com~lementarv Svnthetic
RNA ~arqets
Racemic methylphosphonate oligomers and complementary
RNA target oligomers were synthesized as described in
previous applications. A series of oligomers having the
same seo,uence but with different b~rkhnnr~C was prepared as
described ~1 rewh~re in this application. Rp- (CT) dimers
were used to make the 755~ Rp-enriched all-methylphosphonate
and the 2'-deoxy MP(Rp)/2'-deoxy DE oligomers. Rp-(CU)
dimers were used to make the 2~-0-methyl MP(Rp)/2'-0-methyl
Wo 95/13834 ~ `~ PCrlUS~4113387
~176259
83
DE oligomer. Oligomers cnntA;n;n~ ph~Erhnr-othioate
linkages mixed with other 1; nkA~ were synthesized
according to the general procedures described in Example
30 and other examples above. Control oligomers cnnt~;n;ng
5 either a normal phosphodiester (2 ' -deoxy all-DE) backbone
or a 2'-O-methyl phosphodiester h~rkh~nnc (2'-O-methyl DE),
and all-phosphorothioate oligomers, were purchased from
Oligos Etc. Where 2'-deoxy or 2'-O-methyl substitutions
are indicated below, these structures occur on all of the
10 residues in the alt~orn~;ng or repeated sequence.
~ nn~ l ;n~ reactions cnnt~;n~l e~uimolar amounts of
b~rkhnn~-modified oligomer and RNA target oligomer (2.4 ~lM
total strand concentration), 20 mM potassium phosphate (pH
7.2), lO0 mM sodium chloride, O.1 mM EDTA and O.039~
15 potassium sarkosylate. These reactions were heated to
80C and then slowly cooled to 4C over a time period of
approximately 4 - 6 hours . Next, the annealed samples were
transferred to l cm quartz cuvettes and monitored by
absorbance at 260 nm as a function of temperature in a
20 Varian Cary Model 3E Spectrophotometer cnn~;n;nS a
temperature controlled 6 x 6 sample holder and interfaced
to an IBM compatible PC computer. The temperature was
varied from 5C to 80C at a ramp rate of 1C/min. The Tm
is defined as the point corr~cronr~; n~ to the maximum o~
25 the first derivative of the thermal dissociation profile.
The binding constants at 37C (K"(37C) ) were lot~rm;n~od by
a non-linear least s~uares fit of the thermal dissociation
data assuming a two-state model for the melting process.
The f ollowing table summarizes the -esults:
.
WO 95/13834 2 1 ~ ~ 2 ~ ~ PCrlUS94/13387
84
Sequence = 5'-~L~lClCl~ lA-3'
~egu-nc~,
nu~ r BAcl~bone typ- Tm(C) 1~(37-
C)
2288-1 ~acemic all-MP 34.0 8.3 x
105
5 2781-1 2'-0-Methyl racemic all-MP 37.1 2.1 x
10'
2782-1 ~lt~7nAt;n~ racer~ic MP/DE 40.6 6.3 x
106
2286-1 75% '.~,-enriched all-MP 44.0 2.6 x
10'
3253-1 Alternating 2'-deoxy YP(Rp)/PS 47.3 1.8 x
109
2768-1 2~-0-Methyl 75% ~,-enriched all-15P 47.4 3.9 x
10'
10 2793-1 All-PS 50.4 4.3 x
10'
2760-1 Alternating 2~-deoxy MP(Rp)/DE 53.8 7.9 x
109
2784-1 .7~1t~.rnAtin~ 2~-0-Methyl racemic- 59.
MP/2~-0-methyl DE 10~3 x
2795-1 2'-Deoxy all-DE 60.8 7.1 x
101l
2765-1 Z8t~rnAt;n~ 2~-0-Methyl XP(R~)/2'-0- 67.9 5.2 x
methyl DE 1ol'
15 2792-1 2'-0-Methyl all-DE 75.0 5.3 x
1 ol~
According to this data, a dramatic; _ .,v- t in
binding stability for an RNA target is achieved with the
various backbone modif ications to the original racemic
all-MP oligomer.
20 Exam~le 35 ~ ~
B.;nr~;n~r Aff;n;ties of Various Chimeric Backbone Oliqomers
~lementarv RNA Tarqets
The following oli~n~ P~ tides were tested for their
ability to hybridize to a complementary synthetic RNA
25 target .
WO 95/13834 2 ~ ~ 6 2 ~ 9 PCT/US94/13387
I.D. # Sequence !:~escription
2~67-1 5'-GTCTTCCA~TGCAT)GTTGTCC-3' rMP] [DE] rMP]
2681-1 5'-GTCTTCCA(TGCAT)GTTGTCC-3' [MP] [PS/P~] [MP]
2687-1 5'-GTCTTCCAT(GC~TG)TTGTCC-3' [75'~MP(R") 3 [Dl!] r75~MP(Rp)]
- 5 3169-1 5~-GTCTTCCA(TGCAT)GTTGTCC-3' [MP(7,,)~DE] [D~] [MP(R~)/DB]
3214-1 5'-GTCTTCCA(TGCAT)GTTGTCC-3' [llP(Rp)/DI!] [PS/DE] [MP(Rp)/~E]
3257-1 5'-GTCTTC(CATGCAT)GTTGTCC-3' [MP(Ep)/DE3 [PS/DE] [MP(P7)/DE]
3 2 5 6 -1 5 ' - GTCTTCCA (TGCAT ) GTTGTCC- 3 ' [MP (RD ) /DE] [ PS ] [MP ( Rl,) /DE]
The bases shown in par.onth~q~o~ contain the ba~kh~
10 modi~ication indicated in the middle set of brackets for
each description, and likewise the terminal portions of
the oligomers contain linkage structures as shown in the
terminal sets of brackets. The PS/DE notation indicates
an alternating array of bases beginning with a phos-
15 phorothioate linkage . For example, if there are f ivebases in a sequence denoted as P6/DE, they include three
phosphorothioate (PS) bonds and two phosphodiester (DE)
bond8 .
Each oligomer was mixed with its complementary
2 0 synthetic 3~NA target in a 1:1 molar ratio in a buf f er
system con6isting of 20 mM sodium ~ht~s~h~te buffer (pH
7.2), 100 mM NaCl, 0. 03~ potassium sarkosylate and 0.1 mM
EDTA; total strand concentration = 2 . 4 micromolar . The
resulting solutions were heated to 70C and slowly cooled
25 to 4C over a time period of approximately 4-6 hours.
Next, the annealed oligomers were monitored at 260 nm over
an increasing temperature gradient of 1C/minute using a
Varian Cary Model 3E W/Visible SpeuL,u~ otometer equipped
with a thermostat multicell holder, temperature controller
30 and temperature probe accessories. Data was recorded and
processed using a PC computer interface. The Tm values
were determined from the first deriYative of the sigmoidal
melt transition. The binding constants at 37C (KA(37C) )
were determined by applying a non-linear least squares fit
35 to the data and assuming a two-state model for the dena-
_ _ _ _ _ _
WO 95113834 2 1 7 6 2 ~ ~ PCTNSg4113387
86
turation process. These values are shown in the tablebelow:
I.D. # Tint C) ~A(370C)
2567-1 45 . 6 2 . 9 x 10~
5 2681-1 44.1 2.1 x 107
2687-1 52.8 2.6 x 109
3169-1 62 . 6 6 . 0 x 10l4
3214-1 61.0 2.3 x lO
3257-1 60.9 2.1 x lOli
3256-1 60 . 1 5 . 5 x 10l3
Studies with other chimeric b~khnnP oligomers
further demonstrated that compounds cnnt;l;n;n~ Rp-chiral
methylphosphonate bonds have higher net binding stabili-
ties with RNA targets compared to oligomers having the
same compositions but with racemic methylphosphonate
bonds. Determination of Tm values was done generally as
described above . Data f or a variety of sequences, some
having varying sizes in their RNaseH-activating regions as
well as selected 2~-sugar substitutions, were obtained as
follows. ~Linkage structures separated by slashes indi-
cate an alternating sequence of the listed l; nkil~c; thus,
in the case of the 5-base PS/DE core of compound 2681-1,
a linkage sequence -PS-DE-PS-DE-PS- appears. Uridine
residues were substituted f or thymidine residues in the
bracketed portions of the compounds below having 2 ~ -o-
methyl substitutions. 2'-O-methyl sugar substituents were
incorporated on each of the methylphosphonate- and phos-
phodiester-linked nucleoside sugars of the terminal non-
RNaseH-activating regions of these compounds ~numbers
3341, 3336, 3339, 3337, 3382 and 3386), except for the 3'-
terminal residues that were separately bound to the solid
support prior to dimer synthon addition ~cf. Example 44
below) . )
2i7~5~
WO 95/13834 - . : . PCT/US94/13387
87
Sequence TYPe I
5-base core: 5' [GTCTTCCA](TGCAT)[GTTGTCC] 3'
7-base corc: 5' [GTCTTC](CATGCAT)[GTTGTCC] 3 '
Tm
5ComPound r '-L ! ' Structure ~,~
2681-1 [MP(racemic)]-(PS~DE)~-[MP(racemic)] 44.1
2567-1 [MP(racemic)]-(DE)5-[MP(racemic)] 45.6
2687-1 [75% MP(R~)]-(DE)5-[75% MP(ll~)] 52.8
3256-1 [MP(I~)/DE]-(PS)5-[MP(R")/DE] 60.1
10 3214-1 [MP(II?)/DE]-(PS/DE)5-[MP(Rp)/DE] 61.0
3169-1 [MP(Rp)/DE]-(DE)s-[MP(RF)/DE] 62.6
3257-1 [MP(RpyDE]-(PS/DE)7-[MP(R")/DE] 60.9
3341-1 [2'0Me{MP(Rp)/DE}]-(PS),-[2'0Me{MP(Elp)/DE}] 65.8
3336-1 [2'0Me{MP(Rp)~DE}]-(PS/DE)7-[2'0Me{MP(Rp)/DE}] 66.8
'.5Seauence TYDe 2
5-base core: 5' [GCTTGGCTA](TTGCT)[TCCATCTTCC] 3'
7-base core: 5' [GCTTGGCTA](TTGCTTC)[CATCTTCC] 3'
Tm
Compound r ~ ~ ! ' StrucPJre (C. RNA)
2 03234-2 [MPIRpyDE]-(PS/DE)5-[MP(Rp)/l)E] 62.0
3233-1 [MP(Rp)/DE]-(DE)5-[MP(R,yDE] 63.6
3330-1 [MP[Rp)/DE]-(PS/DE)7-[MP(RpyDE] 61.3
3339-1 [2'0Me{MP(R,~/DE}]-(PS/DE)7-[2'0Me{MP(RpyDE}] 68.8
3337-1 [2'0Me{MP(I~)/DE}]-(PS)7-[2'0Me{MP~pyDE}] 70.3
25Sequence TYPe 3
5-bas~ core: 5' [GGTATATC](CAGTG)[A~ U~:U l~;lUl 3'
Tm
ComPound E ' Lin,a~e Structure (C RNAl
3383-1 [r ~ .. iL)/2'0MeDE](PS)5[~ .. ;. )/2'0MeDE] 59.6
30 3382-1 [2'0Me{MP(rac.)/DE}](PS)5[2'0Me{MP(rac)/DE}] 64.4
3386-1 [2'0Me{MP(Rp)/DE}](PS}~[2'0Me{MP(Rp)/DE}] 64.4
The data 8h3wed that a 8ignificant ~nhAn~ ' in
binding affi~ity results when racemic methy1rh~,~phnnA~e
WO95/13834 ~17G25~. . PCr/US94113387 ~
88
linkages are replaced with R~-chiral methylphosphonate6.
This observation applie9 to nucleo9ides rnntA;n;nr 2'-
deoxy ribofuranose 6ugars as well as to bases cnntA;n;ng
2'-O-methyl ribofuranose sugars. Oligomers cnntA;n;n~
5 regions of alternating MP (R~) /DE linkages have higher
binding affinities than oligomers having alternating
MP (R~,) /MP (racemic) linkages . A further binding ~onhAnr
results when 2 ' -O-methyl ribofuranose sugars are substi-
tuted f or 2 ' -deoxy sugar9 . Ba9ed on the data presented
lO above, it is estimated that the Tm increases by about 0.5-
0 . 6 C per substitution .
r le 36
Demonstration of the Abilitv of Various Chimeric Oliqomers
to Activate RNaseH from He~a Cell Nuclear E~tract
The following oligomers were tested for their ability
to activate endogenous eukaryotic RNaseH derived from He~a
cell nuclear extracts.
r.D~ ~ S~nc~ ~ D~scrw~rn
2498-1 5'-GTCTTCCATGCATGTTGTCC-3' AII-DE
2 o 2566-1 5'-GTCTTCCATGCATGTTGTCC-3' AII-PS
3130-1 5'-GTCTTCCATGCATGTTGTCC-3' rlPal,)/l)E Al~errlting a~on-Chimeric)
3169-1 5'-GTCTTCCA(TGCAT)GTTGTCC-3' IMP/Rr!~ R']
3214-1 5'-GTCTTCCA(TGCAT)GTTGTCC-3' IMI'(I~,)/DEI[PS/DE]~MP(Rp)/DE]
3256-1 5'-GTCTTCCA(TGCAT)GTTGTCC-3' IMP(R~,)I.,_". ,~ ' ~p)lDE]
25 Each o~ these oligomers (lO IlM) was annealed to its
complementary synthetic RNA target (l ~M) in a buf fer
system rnn~A;n;ns 50 mM Tris-HCl (pH 8.0), 20 mM KCl, 9 mM
MgCl2, 1 mM ,B-mercaptoethanol, 250 l~g/m~ bovine serum
albumin, and 25-lO0 units/mL of RNasin (Promega, Corp.,
30 Madison, WI) . RA~;nlAhPl.of~ RNA having 3~P at the 5'-
terminus was prepared using [ y-32P] -ATP (New England
Nuclear/DuPont, Boston, MA) and T4-polynucleotide kinase
(Stratagene, Inc., San Diego, CA) according to standard
procedures. Approximately 200, 000 dpms of 3'P-labeled RNA
. , , _ _ _ _ _ _ _ _ _ _ , , . . . , , , , . . . , , , . , . . , , . : , . . , . : . . . . . . . .
WO 95/13834 PCTNS94113387
2~7~259
89
was included in each reaction as a radiotracer. These
samples were ~nnP;~ by heating to 65C and slowly
cooling to 4C over a period of apprn~ t.oly 4-6 hours.
Stock solutions c^nt~;n;n~ EIeLa cell nuclear extract
were prepared as follows. HeLa cell nuclear extract
(Promega Corp., Madison, WI, Catalog # E3521, 5 mg/mL
protein) wa5 diluted 250-fold in a buffer consisting of 20
mM HEPES (pH 8.0), 20% glycerol, 0.1 M KCl, 0.2 mM para-
methylphenylsulfonyl fluoride (PM.~3F) and 0 . 5 mM dithio-
threitol.
RNaseH cleavage reactions were initiated by adding
diluted HeLa cell nuclear extract (5 ,I~L) to each of the
annealed oligomer samples (lO~L) and then the samples were
;n~llh~t~d at 37C for either fifteen minutes or two hours.
At the end of the specified incubation time, each cleavage
reaction was terminated by addition of 1.5 IlL of EDTA (125
mM, pH 8) and then quickly frozen on dry ice and stored at
-20C. When all of the cleavage reactions had been termi-
nated they were removed from the freezer for analysis by
polyacrylamide gel electrophoresis. Aliquots (5 /lL) were
withdrawn from each reaction and diluted with gel loading
buffer (5 ~L, 90% formamide/lxTBE buffer/0.19~ bromphenol
blue/0.1% xylene cyanole blue). The resulting samples
were loaded onto a 15% polyacrylamide/7 M urea gel (20 cm
X 30 cm g 0.5 mm thick) prepared in lX TBE buffer (pH
8.2). The gel was electrophoresed at 1200 volts for 1.5
hours. Bands on the gel corresponding to full length and
cleaved RNA products were rlPte~-ted by phosphorimager
analysis using a Bio-Rad Model GS-250 Molecular Imager
3 0 (Bio-Rad Laboratories, Hercules, CA) . The amount of
cleavage that occurred in each reaction was determined by
comparing the phosphorimager counts for the full length
band to the total counts per lane. The results are
summarized below:
Wo 95/13834 PCrlUS9411338~
~762~
Oligomer I.D. # ~Na~e}~ Cleav~ge after 2 ~Ir~.
At 37C
2498-1 24.4%
2566-1 10 . 59~
3130-1 None detected
3169-1 52 . 0~6
3214-l 38 . 096
3256-1 18 . 7~
The length of each cleavage fragment was estimated from
the electrophoretic mobility of its associated radioactive
10 band. From this analysis, it was determined that cleavage
occurs selectively in the middle of heteroduplexes derived
f rom the chimeric oligomers . More numerous cleavage
products were observed with the all-phosphodiester (DE)
and all-phosphorothioate ~PS) oligomers, as expected.
15 This data shows that the replacement of PS for DE linkages
results in a reduction in the rate of RNaseH-mediated
cleavage. There was no cleavage observed in the sample
rrn~z~;n;nr an alternating MP(Rp)/DE bArkhr,ne.
r ~le 37
20 S~AhilitV of Various Chimeric Oliqomers to Nucl~oAqe
Dicrestion in the Presence of Sl-~n~nllrlease
The following oligomers were tested for nuclease
stability in the presence of S1-Pn-~nnl~rl ease.
LD. # Sequencc Dcscription
2567-l 5'-GTCTTCCA(TGCAT)GTTGTCC-3' ~P][DE][~]
2681-l 5'-GTCTTCCA(TGCAT)GTTGTCC-3' lMP][PSll)E [lMP]
3169-l 5'-GTCTTCCA(TGCAT)GTTGTCC-3' ~Pff~p)/DE ~E][~(Rp)/DE]
3214- l 5 '-GTCTTCCAtTGCAT)GTTGTCC-3 ' ~lP~)/DE PSIDE] [1\1P(Rp)/DE]
3256-l 5'-GTCTTCCAtTGCAT)GTTGTCC-3' ~P(Rp)/DE PS][MP(Rp)/l)E]
30 Sl-~n~r~nllr~ ] ease was purchased from Promega Corp. (Catalog
# E576B, Madison, WI). Aliquots of each chimeric oligomer
(0.05 - 0.075 OD260 units) were individually added to
polypropylene microcentrifuge tubes cr~nt~;n;nr~ S1-endonu-
clease tO.5 units/mL) in 30 mM sodium acetate (pH 5.0), 50
~WO95113834 2 1 ~ 6 ~ 5 9 PCTIUS94113387
.,
91
mM NaCl, 1. 0 mM zinc acetate and 5~ glycerol; total
reaction volume = 10 ~LL. These tubes were incubated at
37C for specified time periods, quickly frozen in dry ice
and then stored in a freezer at -20C. The samples were
5 then analyzed by reversed-phase ~PLC using a Beckman
System Gold chromatography system equipped with a Model
126 Solvent Module and a Model 168 Diode Array Detector.
Column = Vydac Protein C4 (catalog #214TP54, 4.9 mm i.d.
x 250 mm long) . Buffer A = 50 mM triethylammonium acetate
(pH 7) /196 acetonitrile; Buffer B = 50 mM trietbyl ;llm
acetate (pH 7) /50~; acetonitrile. The elution profile was
5-35% Buffer B (2.5 - 12.5 min.); 35-509G Buffer B (12.5 -
22.5 min.); 50-100~ Buffer B (22.5 - 27.5 min.); flow rate
= 1.5 mL/min. The samples were diluted with water (50 ,uL)
15 and injected onto the column using a 100 ~L sample loop.
Peaks corresponding to full length oligomer and its
degradation products were detected by monitoring at 260
nm. The amount of degradation occurring in each reaction
was determined by measuring the r~ t;r~n in peak area for
20 the full-length oligomer (;d~nt;f;~1 by comparison to an
~rt~rn~l control and/or by coinjecting undigested oligomer
as an internal control). The data is shown in tabular
format below, and in graphic format in FIG. ~.
OligomerLD. # ~alf-LifeforD~h -
2567-1 1.7 Hrs.
2681-1 12.2 Hrs.
3 1 69-1 0.9 Hrs.
3214-l 5.0 Hrs.
3256-1 12.5 E~s.
30 * Detorm;ned as the point where 50% full length oligomer
has been digested based on a least-squares fit of the
data .
This data shows that the rep~ of
~h~"3phr~rothioate (PS) bonds for phosphodiester (DE) bonds
_ _ , .. _ .. . , . _ . . . _ _ _ . , _
Wo 95113834 PCrlUS94113387
~1762~9
92
imparts a resistance to nuclease degradation catalyzed by
Sl--~n~ nll~1 ease.
E le 38
St~hilitv of Various Chimeric Oliqomers to Nuclease
5 DiqestiQn in the Presence of lO96 Fetal Calf Serum
Multiple ~ samples of each chimeric oligomer were
prepared in l . 5 mL polypropylene microcentrifuge tubes on
ice. Each sample ~ ntz~;n~rl oligomer (O.l OD260 unit), lO9
fetal calf 8erum (FCS, Gemini Bioproduct8, ~ lAh;lc;~, CA~,
20 mM HEPES (pH 8.0), 0.2g~ paramethylsulfonyl fluoride
(PMSF), 175 mM KCl, O.l mM dithiothreitol, O.l mM EDTA, 2
mM MgCl2 and 49~ glycerol -- total volume = lO0 IlL. The
samples were incubated at 37C for specified time periods
and then diluted with 0.49~ NP-40/acetonitrile (35 ~
15 quickly frozen on dry ice and stored at -20C. Samples
were then individually thawed, diluted with water (635 IlJJ)
and analyzed immediately by reversed-phase HP~C according
to the method given in the preceding example (except that
a 2 mL sample 1QP was used to load the samples Qnto the
20 column). Results are shown in tabular format below, and
in graphical format in FI~. 2.
Oligomcr l.D. # Half-~ if e for D.~j. ' *
2567-1 5.8 Hrs.
2681-1 8.1 Hrs.
25 3169-1 3.4 Hrs.
3214-1 4.3 Hrs.
3256-1 16.2 Hrs.
* Determined as the point where 50~ full length oligomer
has been digested based on a least-squares fit of the
30 f irst three time points in each data set .
This example indicates a similar enhancement in
stability to nuclease degradation when PS linkages are
used in place of DE linkages.
~WO 95113834 93 PCTIUS94/13387
~Y~m~le 3 9
ACtivitY of rMP] rDEl rMP] oliqomer 2567-1 and rMP(R~)/DEl-
rDEl - rMP(RF) ~DEl oliaomer 3169-1 o~ cell-free tr~n~lation
of tarqet mRNA
A target mRNA having complementarity to these
oligomers at the initiation codon region was prepared by
standard cloning techniques with reverse-transcription
catalyzed by T7 polymerase (Promega MEGAscript kit for
uncapped RNA), according to the manufacturer' 8 protocol .
Control CAT mRNA was obtained from GIBCO as a control for
specif icity .
Target mRNA and control CAT mRNA were translated in
a cell-free translation assay in rabbit reticulocyte
lysates (Promega), in the presence of 35 [S] -Cys
(NEN/DuPont) following the m-nllf~n~llrer's directions.
Oligos 2567-1 and 3169-1 were added to individual trans-
lation reactions at 0, 0.2, or 1.0 M, final concentra-
tions. RNAse-H (Promega Corp. ) was added to all the
translation reactions at 0 . 04 units/ul . Each condition
was run in triplicate. Translation reactions were incu-
bated at 37 C for 1 hour. At the end of the translation
reactions, proteins were denatured with ~aemmli Sample
Puffer (Novex) and the amounts of target proteins synthe-
sized in each case were evaluated after immunoprecipi-
tation with an hyperimmune antibody serum followed by gel
fractionation of the protein products (10-20% gradient
SDS-PAGE~ gels, Novex) and phosphoimage analysis. The
amount of control CAT protein synthesized in each case was
evaluated after gel frant;nn~inn of one aliquot of the
denatured translation reaction (10-20 ~ gradient SDS-PAGE
gels, Novex) and phospho-image analysis.
As shown in FIGS. 3 and 4, oligomer 3169-1 produced
approximately 50% and 90% inhibition of target mRNA
translation when pre8ent at 0 . 2 or 1 ~LM, respectively.
Oligomer 2567-1 produced approximately 0% and 5096 inhi-
bition of target mRNA translation when present at 0 . 2 or
1 IlM, respe~tively. Both oligo8 produced little inhibi-
WO 95/13834 21 ~ 6 2 5 9 ~ PCT/US94/13387
94
tion of control CAT mRNA translation, indicating good
specif icity .
This result indicates that replacement of racemic MP
ends by chirally-selected MP (R~) /DE linkage segments
5 significantly increases the ability of an oligomer to
block cell-free translation of the target mRNA.
~m~le 4
Cleavaqe of tarqet mRNA. in the ~resence of RN~qeH, o~
r~lPl rDEl rMPl oliqomer 2567-1 and rMP(R9) /DEl - rDEl -
rMP ~R~) /DEl oliqomer 3169-1
A target mRNA having complementarity to these
oligomer6 at the initiation codon region was prepared by
standard cloning techni~ues with transcription using a T7
polymerase cell-free assay tPromega MEGAscript kit for
uncapped RNA), according to the manufacturer~ s protocol .
The resulting mRNA transcript is approximately 340 nt in
length .
The ability to cleave this target mRNA, in the
presence of RNAseH and either of oligomers 2567-1 ~ [MP] -
2C [DE] - [MP] } and 3169-1 { [MP (Rp) /DE] - [DE] - [MP (R~) /DE] } was
t,orm;n--~ as follows.
Cell-free transcribed mRNA (100 nM) was incubated at
37 C, in a cell-free translation buffer (rnnt~;n;n~ 3 5
mM MgCl~, 25 mM KCl, 70 mM NaCl and 20 mM potassium
acetate), in the presence of 0 . 04 units/~Ll of RNAseH
(Promega) and either of oligomers 2S67-1 or 3169-1 at 0,
0.01, 0.1, 1, or 10 IlM. After 30 minutes, the RNA was
extracted, denatured and run in a denaturing gel. After
the run, the RNA was stained with ethidium bromide and its
integrity was determined by visual observation of the RNA
bands present in the gel.
As shown in the table below, a good dose-response
effect was obtained for both oligomers at the concentra-
tions tested. Oligomer 3169-1 was more active than
oligomers 2567-1 ~3169-1, at 1 ~M, cut ~98 96 of the target
mRNA present in the reaction, while oligomer 2567-1, at
~wo 95/l3834 2 ~ ~ ~ 2 5 ~ PCrlUS94/13387
the 8ame concentration, cut -50~ of the target mRNA
present in the reaction). Both oligomers showed good
specificity, cleaving the target mRNA in one position.
Cle~ivage of t~rget mRNA, in the presence of RNAseH,
of IMP]IDE~[MPI oligomer 2567-1 and
[MP(Rp)tDE]-[DE]-[MP(Rp)/DEI oligomer 3169-1
Oligomer 2~67-1 3169-1
Backbone [MP]-[DE]-[MP] [MP(I~)/DE][DEIIMP(}~,)/DE
Ol~gomerconcen- 0.01 0.1 1 iO 0.01 0.1 1 10
tr~tion (yM)
/. of tsrf~et mRN~ ~ 15 50 80 5 40 9 100
clenvn~e~
) Estimated values obtained by visual inspection of the ~el
Exam~le 4 1
15 ;rnh;hitiQn of Protein Svnthesis in a Cell Culture With
t~hir-ric .~nt-i qense Oliaomers T~rqeted to a Non-~llk~rvotic
Re~orter Gene, Chll h~ni col Tr~nqfer~qe
The following example shows the ability of chimeric
antisense oligomers to selectively inhihit protei~ syn-
20 thesis in a eukaryotic cell culture 8ystem. COS-7 cells
were transiently transfected with plasmids encoding either
a target reporter gene or a control non-target reporter
gene. These cells were then treated with various chimeric
antisense or control oligomers and then assayed f or the
25 expression of the reporter genes.
Plasmi~q
The following plasmids were used in this example.
pG1035: Splicer CAT, in8erted i~to a pRc/CMV vector
pG1036: Wild-type ~AT, in8erted into a pRc/CMV vector
. , . _ . . .. . . . . . . .. _ _ _ _ _ .
21 ~2~9 0
WO 95/13834 : ~ PCrNS94113387
, 96:
pGl040: UCAT, inserted into a pRc/CMV vector
pGL2: Lucifera9e expressing plasmid (Promega)
pSV~ galactosidase expressing plasmid (Clonetech)
A description of plasmids pGl035, pGl036 and pGl040
5 follows.
l. pGl035 (SplicerCAT) and pGl036 (wild-type CAT)
and the sequences of the synthetic splice sites:
A. Sequence of the wild type CAT gene used to create
plasmid pGl 0 3 6:
+409 +410
GCC UAU WC CCU AW IJCC CUA AAG GGU WA WG AGA A~A ~ ~
B. Full sequence of the intron inserted within the
CAT coding sequence to create SplicerCAT and plasmid
l~ pGl035:
+409 l
... UAU WC CCU AW UCC CUA i~AGI quq aqu qac uaa cua ac,u
39
cqa cuq caq acu aqu cau ua(~ ) uuq aqu qua aca aga ccg gau
~7 +410
auc uuc qaa ccu cuc ucu cuc ucu c~a GGU WA WG AGA ...
The region of the CAT gene into which the intron was
inserted is shown in sequence A above. Wild type CAT DNA
(Pharmacia) was inserted into pRc/CMV (Invitrogen) to
create plasmid pGl036. The sequence is shown as the mRN~.
Bases 409 and 410 are labeled for comparison to pGl035.
A synthetic intron, shown as sec~uence B above, was insert-
ed into the CAT DNA to create plasmid pGl035. Mature mRNA
sequences are shown uppercase, intronic sequences are
lower case. The canonical guanosine of the splice donor
is labeled +409, which corresponds to base 409 of the CAT
open reading ~rame . The f irst base of the intron is
labeled l. The canonical branchpoint ;~ n~;nF~ is base 39
~Wo 95113834 ~ 1 7 6 2 ~ 9 PCrlU594/13387
97
and the canonical intronic splice acceptor guanosine i8
base 87 of the intron. Base 410 marks the resumption of
the CAT open reading frame The sequences against which
the oligomers are targeted are underlined The consensus
splice site bases are given in bold face italics (Smith et
al. 1989; Green 1986) .
The clDne pG1035 was created using synthetic DNA PCR
primers to create a Hind III-Spe I 5'fragment cfmtA;n;n~
the first 2/3 of the open reading frame and half of the
synthetic intron and an Spe I-Not I fragment containing
the second half of the intron and the last 1/3 of the open
reading frame. These were ~ ' ;n~d with Hind III-Not I
cut pRc/CMV in a 3-way ligation to yield the final plas-
mid. The artificial CAT gene ~nntA;n;n~ the intron is
named SplicerCAT. References applicable to the foregoing
include Smith CWJ/ Patton JG/ and Nadal-Ginard B/ (1989) /
"Alternative splicing in the control of gene expression, "
Annual Reviews in Genetics 23: 527-77; Green, MR (1986),
"Pre-mRNA splicing, " Annual Reviews in Genetics 20: 671-
708.
Wo 95/l3834 2 1~ 6 25 9 Pc~rluS94/13387
98
2. pG1040 (UCAT) 5' untranslated regions and amino
terminus:
Wild-t~e CAT:
5' ~1
ll-t Glu Ly~ Ly~ B-r aly
uuu uc~ gga gcu aag gaa gcu aaa aug gag aaa aaa ayc acu gga
3'
Tyr Thr Thr
uau acc acc
l8G104 0 . UCAT:
5' +1
15~t Glu LyEI Ly~ S~r Gly
agu qca qqa qcu aaq qaa qCu acc auq qaq aaq aaq auc acu qqa
3258-1 3 AUG 31te
3260-1
3'
Tyr Thr Thr
uaU aCc acc
The se auences of wild type and pG1040 UCAT around the
AUG start co~on are shown . The target sites f or the
oligomers are named and underlined, and the numbers of the
chimeric oligomers against each target site are shown
beneath .
UCAT was made from wild-type CAT DNA (Pharmacia)
using synthetic DNA PCR primers . The resulting f ragment
was cloned as a Hind III (5' end), Not I (3' end) fragment
into the vector pRc/CMV (Invitrogen) . The first ;~ n~sin~
oi the open reading frame is designated +1. The amino
acid changes between wild-type and pG1040 are conserva-
tive .
Chimeric Oliqonucleotides were as follows.
5' AUG oliqomers (~osition -21 to +3):
3258-1, 24~er, ~MP(R~)/DE) (PS/DE) (MP~Rp)/DE):
5' cat ggt ag(c ttc c) tt agc tcc tgc 3'
~Wo 95113834 2 1 7 6 2 ~ 9 PCrlVS94113387
99
3260-1, 24mer, (MP(Rp)/DE) (PS) (MP(R5)/DE):
5 ' cat ggt ag (c ttc c) tt agc tcc tgc 3 '
3 ' AYG oliqome~s (position +4 to +27):
3261-1, 24mer, (MP(Rp)/DE) (PS/DE) (MP(R~)/DE):
5 ' ggt ata tc (c agt g) at ctt ctt ctc 3 '
3262-1, 24mer, (MP(R7)/DE) (PS) (~P(Rp)/DE):
5 ' ggt ata tc (c agt g) at ctt ctt ctc 3 '
3636-1, 24mer, (MP(R~)/DE) (PS) (MP(Rp)/DE):
5' ggt a (ta tcc) agt gat ctt ctt ctc 3 '
3638-1, 24mer, (MP(R~)/DE) (PS) (MP(Rp)/DE):
5 ' ggt ata tcc agt (gat ct) t ctt ctc 3 '
3637-1, 24mer, (MP (R~) /DE) (PS) (MP (Rp) /DE):
5 ' ggt ata tcc agt gat c (tt ctt) ctc 3 '
3640-1, 24m~r, (MP(Rp)/DE) (PS) (MP(Rp)/DE):
5 ' ggt ata tc (a agt g) at ctt ctt ctc 3 '
3639-1, 24mer, (MP(R")/DE) (PS) (MP(Rp)/DE):
5 ' ggt ata tc (g agt g) at ctt ctt ctc 3 '
S~lice ~o~or oli~omers:
3264-1, 24mer, (MP (Rp) /DE) (PS) (MP (Rp) /DE):
5 ' cac tca cct t (ta ggg) aaa tag gcc 3 '
3263-1, 24mer, (MP(Rp)/DE) (PS/DE) (MP(R~)/DE):
5' cac tca cct t(ta ggg) aaa tag gcc 3'
XV-5, 24mer, all rhn~rhn. othioate:
5 ' cac tca cct tta ggg aaa tag gcc 3 '
Wo 95/13834 2 1 7 6 2 5 g PCrlUS94/13387
100
S~lice branch Point olicomers:
3269-1, 24mer, (MP(Rp)/DE) (PS/DE) (MP(Rp)/DE~:
5' cac tca at (c aat g) ac tag tct gca 3
3270-1, 24mer, (MP(R~)/DE) (PS) (MP(R~)/DE):
5 ' cac tca at (c aat g) ac tag tct gca 3 '
XV-6, 24mer, all rh~ h-- ~,thioate:
5 ' cac tca atc aat gac tag tct gca 3 '
S~lice acce~tor site oliqomers:
3265-1, 24mer, (MP(}I~)/DE) (PS/DE) (MP(Rp)/DE):
5 ' ccc tga ga (g aga g) ag aga ggt tcg 3
3266-1, 24mer, (MP(Rp)/DE) (PS) (MP(Rp)/D13):
5 ' ccc tga ga (g aga g) ag aga ggt tcg 3 ~
3387-1, 24mer, t2~oNe(Mp(Rp)/DE)] (PS) t2'0Me(MP(Rp)/DE)]:
5 ' ccc tga ga (g aga gag) aga ggt tcg 3 '
XV-7, 24mer, all rh~ -) oLhioate:
5' ccc tga gag aga gag aga ggt tcg 3
Cell Pre~aration and Treatment
COS 7 cells were plated at 1. 5 x 105 cells/well in a
12 well plate format on the day before trans-fections
20 ~egan. All cultures were r-;nt~;n~d at 37C. On the next
day, the transfection mixes were prepared. For each well
of a 12 well plate, 1.0 IlM oligomer was c ' in~od with 1 ~Lg
pGL2 or pSV,B + 1 ~g of the target CAT plasmid in 0 . 5 ml of
Optimem (Gibco/BRL) and 18.75 ~Lg Tran~fectam (for chimeric
25 oligomers, Promega) or ~ipo~ectamine (for all PS
oligomers, Promega) also in 0 . 5 ml of Optimem. These
quantities gave a 6.9 or 4.5 or 2.0 to 1 cationic lipid to
oligomer plus DNA ratio, respectively, in one milliliter
total. pGL2 and pSV,B servea as transfection and oligomer
30 specificity controls.
~Wo 95/13834 ~ ~ 7 6 2 5 9 t PCrrUS94/13387
101
The culture medium was aspirated of f and the cells
were rinsed twice in one ml Optimem (Gibco/BRL) per well,
and then one ml of tranfection mix was added to each well.
The cells were cultured in the transfection mix for 16
5 hours. The mix was removed and replaced with one ml of
complete culture medium (DMEM plus 109~ fetal bovine serum
and 1/100 dilution of penicillin/streptomyciri stock, all
from Gibco/BRL) and the cells were incubated another 5
hours .
Cell lysates were prepared by rinsing twice in PBS
and then treated with 0.5 ml of lX Reporter Lysis Buffer
(Promega). The released and lysed cells were pipetted
into 1.5 ml tubes and frozen in CO2/EtOH once and thawed.
The crude lysate was then centrifuged 10 minutes to pellet
cell debris, and the supernatant was recovered and assayed
direct ly or f roz en at - 2 0 C .
The cell lysates were then assayed for CAT, and
luciferase or ,~ ~t~se activity, and the total protein
rf~nrPntration was detprm; nP~l as described below.
t'hl ~ Pn; col Acetvltr~ns~ferase (CAT) AssaY ProtocQl
This assay was performed generally as follows.
First, the following reaction mixture was prepared for
each 8ample:
65ml 0.25M Tris, pH8/0.57~ BSA,
4~ 4C-Chloramphenicol, 50 nCi/~ll (Dupont), and
5~L1 5 mg/ml n-Butyryl Coenzyme A (Pharmacia)
A CAT standard curYe was ~re~red by serially fl;lllt;n~ CAT
stock (Promage) 1:1000, 1:10,000 and 1:90,000 in 0.25M
Tris, pH8/0.596 BSA. The original stock CAT was at 7000
Units/ml. CAT lysate was then added in a labeled tube
with Tris/BSA buffer for final volume of 50 ml.
74 ml of reaction mixture was then added to each
tube, which was then incubated for, typically, approxi-
mately 1 hour in a 37C oven. The reaction was terminated
by adding 500 ~Ll Pristane/Mixed Xylenes (2:1) (Sigma) to
each tube. The tubes were then vortexed for 2 minutes and
WO95/1383~ ` 21 76~g ~ '` 1 `; PCT/US94/1338~
102
spun for 5 minutes. 400 ml of the upper phase was trans-
ferred to a srintillAtion vial with 5 ml Scintiverse
(Fisher). The sample was then counted in a Packard
srl nt; 11 Ation counter.
Luciferase Assav Protocol
This assay was performed generally as follows~ ac-
cording to standard procedures. 20 /11 of lysate was
combined with lO0 /11 of luciferase assay reagent ~Promega)
and counted in a srint;llAt;on counter (Packard) within 20
seconds (as r~r ~ 1 by Promega) .
~-Galactosidase Assav Protocol
This assay was performed generally as follows. A ~-
gal standard curve was prepared by serially diluting
1:1,000 and 1 9,000 in 0.25M Tris-HC1, plI8.0/0.59~ BSA.
Stock ~-gal was 1, 000 Units/ml (Promega) . Thus, for the
1:1,000 dilution, 1 ~l stock ~-gal enzyme was diluted in
1000 ~l Tris/BSA buffer, and for the l:9,000 dilution, 100
,ul of the 1:1,000 dilution was further diluted in lO00 ,ul
Tris/BSA buf f er .
75 ~Ll of lysate per well (untreated microtiter plate,
Corning) was then added. 75 1ll 2X ,B-gal RF'~rt;nJl Buf~er
(Promega) was added to each tube. TnrllhAt;nn proceeded
for, typically, apprn~ t~ly 1-1.5 hours in a 37C oven.
Plates were read at A~os (405 nm) on a microplate reader
(Molecular Devices).
prot~; n Aggav Protocol
Samples were prepared in an untreated microtiter
plate (Corning). A series of protein standards were
prepared in duplicate as follows.
l. 6 ILl lX Reporter Lysis Buffer (Promega)
2. 6 ~l 75mg/ml BSA (Promega)
3. 6 ~11 lOOmg/ml BSA
4. 6 1ll 250mg/ml BSA
5. 6 ~11 400mg/ml BSA
6. 6 1ll 500mg/ml BSA
7. 6 111 looomg/ml BSA
~Wo 95/138~4 2 ~ ~ 6 2 ~ 9 PCT/US94/13387
103
8. 6 ~l 1500mg/ml BSA
Six ~1l of lysate per well was added, followed by 300
~l Coomassie Protein Assay Reagent (Pierce) per well. The
individual sample plates were then read at As70 on a
5 microplate reader (Molecular Devices ) . CAT activity
values were normalized to the protein content of the
lysate and other parameters as given.
The results of these experiments were as follows.
Anti-s~lice site oliqomers versus ~C71035 ~n~ ~Gl036
lO (splici~g inhibition by antisense oligomers):
pG1035=spiicing pG10~6 .. ,.. 5, '' _
Oligomer
chemistry Donor Branch Acceptor Donor Branch Acceptor
PS/DE 3263-1 3269-1 3265-1 3263-1 3269-1 3265-1center 65ill% ?2+1% 90+5% o~O oo~O o%
15PS3264-1 3270-1 3266-1 3264-1 3270-1 3266-1
center 59+2% 56+7% 53+2% o% o% 0%
A11 PS XV-5 XV-6 XV-7 XV-5 XV-6 XV-7
32+1% 23+15% 17_6% 35+1% 30+4% 20+4%
PS center, N.D. N.D. 3387-1 N.D. N.D. 3387-1
2'0Me ends 98i2% o%
20 Oligomers were transfected into COS-7 cells and
lysates were made and assayed as described previ-
ously, All oligomers were at l . 0 ~M f inal in the
culture medium. The results are given as percent
inhibition + std error N.D. = not ~i~tf~ ;n~d. All
samples were perormed in triplicate. In the case of
the chimeric oligomers (PS/DE center and PS center)
the expression of the non-splicing pGl036 CAT was
slightly higher in oligomer treated versus untreated
cells, so the expression of pGl035 was normalized to
pGl036 expression. All results were r~ormalized ~o
total protein and luciferase counts.
The results show specif ic inhibition of CAT expres-
sion when the splice site sequences are targeted using the
WO 95/13834 ~ f ' PCTIUS94/13387
25~
104
chimeric oligomers. In the case of all phosphorothioate
oligomers, pG1036 expression was inhibited approximately
as well as pG1035, revealing large non-specific effects on
gene expression. In addition, the incorporation of 2 ' -O-
5 methyl groups in the fl~nk;n~ t~rminz~l portions of thesplice site acceptor oligomer 3387-1 and lengthening the
PS center from five to seven r~nt;n~ phosphorothioate
h;lrkhnm~ linkages increases the antisense activity against
the splice acceptor site target significantly but does not
10 increase non-specific activity against the control target.
(~hi - ic ;oli~omerg tarqeted a~ainst the AUG of CAT
inhibit ex~ression:
5'AUG Target 3'AUG Target Control No
No oligomer
Target
Oligomer 3258-l 3260-l 3261-l 3262-l 3269-l None
Chemistry PS/DE PS PS/DE PS PS/DE No
center center center center center treatment
15 % Inhibition 43+l9% 72+28% 96+7% 97+4% 4+14% 0+15%
oligomers were transfected into COS-7 cells and
lysates made and assayed as described previously.
A11 oligomers were at 1. O ~LM f inal in the culture
medium. Oligomer 3269-1 was a control that does not
have a target site in pGl040, because the CAT gene
does not contain a splice site. Results are ex-
pressed as ~ inhibition + error. Each oligomer was
tested in triplicate.
Chimeric oligomers targeted against the 5 ' AUG site
(3258-1, 3260-1) were effective at blocking expression of
the CAT mRNA (43-729~ inhibition, respectively). Chimeric
oligomers targeted against the 3' AUG site (3261-1, 3262-
1) were even more effective, giving 96 and 97~ inhibition,
respectively. The control oligomer (3269-1) gave no
WO 9S/13834 2 1 7 6 2 5 9 PCT/US94/13387
105
inhibition, demonstrating that the inhibition observed for
the chimeras that match the pG1040 mRNA was specific.
In conclusion, these results indicate the ability to
down-regulate CAT activity using chimeric oligomers
5 introduced into cultured COS - 7 cells via cationic lipids .
The targets have been AUG sites ~present in both the
pre-mRNA and mature mRNA) and intronic sites (present only
in pre-mR~A in the nucleu8 of any cell). The chimeric
oligomers with both PS/DE and PS centers have proven to be
10 more specific than all-PS oligomers and control chimeraE.
Both target-specific and r~ , -specific controls were
included, demonstrating that the results are based on
se~uence- specif ic antisense ef ~ects .
r le 42
15 S~ecificitv Determln~tion
Singly and multiply mismatched, complementary gene
targets and oligomers allow cross-over experiments to
estimate oligomer discrimination of perfect match targets
from imperfect non-specific targets. The present example
20 shows the preparation of C~T mRNA targets having 0- or 4-
ba~3e mismatches with respect to the oligomers used in
~xample 41, as well as the effect of various mismatches on
the specificity and activity of oligomers of the inven-
tion .
WO 95113834 . - PCrlU594/13387
2~ i2~9 106
~ Gl040 (UCAT) and ~Gl042 (UCAT 4mm) 5' untran81ated
reqions and Z~m;nn term;n; ;In~1 oliqomers:
Wild-t~e CAT:
5~ +1 3
H t Glu Ly~ Ly- Il~ S-r Gly Tyr Thr Thr
uuu uca gga gcu aag gaa gcu a~a aug gag aaa aaa auc acu gga uau acc acc
~G1040, UCAT:
5' +1 3'
M~t alu Ly~ Ly~ :~1-- 8~r aly Tyr Thr Thr
10agu qCa qqa qcu a~q qa~ qcu acc auq qaq aaq aaq auc acu qqa uau acc acc
3~ (cgt cct cga ttc ctt cga tgg tac) (ctc ttc ttc tag tg~ cct ata tgg) 5'
XV-l XV-2
~G10 4 2 . UC~T 4 mi ll:mat ch:
5' ~1 3'
1 5 t ~ t
M~t A p Arg Ly~ Thr Gly Tyr Thr T~r
' (cgt tCt caa cgC ctt cga tgg taUacq) ~cqtgc atqCcq attaq tauu aCq qqa uau acc acc
XV-3 XV-4
Mismatches between pGl040 (UCAT) and pGlOg2 (UCAT) 4mm are
marked with asterisks ( * ) . All other bases in the mRNAs
produced by these plasmids are identical. The sequence of
the wild-type CAT gene is shown for comparison. The first
adenosine of the open reading frame is designated +l. The
oligomer target sites are underlined.
Plasmids pGl040 and pGl042 were created using syn-
thetic DNA PCR primers to amplify precisely mutated DNA
~1,, tC, The fragments were then cloned as Hind III (5'
end), Not I (3 ' end) ~1 ~ R into the vector pRc/CMV
(Invitrogen) and positive clones were ;~l~nt;fied. -
It will be noted that, for a given oligomer against
either of these target genes, a control target is provided
having a precisely def ined degree of mismatch . This
allows testing of one oligomer against a perfect match and
precisely-defined mismatch targets, as, ,1;fied by the
f ollowing:
WO 95113834 ~ 2 5 9 PCTIUS94/13387
107
4 0, ~JCAT: --
s~ +
Agu gca gga gcu aag gaa gCu aCc aug ga; ig ja jag al lag jaTT IATT lgg3 TaT ~aTc ~cc
~ctc ttc ttc tag tga cct ata tgg~
xv-2
042, IJCAT 4 mi~match:
Agu gca Aga guu gcg gaa gÇu aCc aug igiA~c jasjs alalg ITU lalcg gigl TlaT laTT ACC
~ctc ttc ttc tag tga cct ata tgg)
xv-2
In this case, the oligomer XV-2 i8 a perfect match to
pG1040, but has four mismatches to pG1042. The relative
ef f ects of this one oligomer against two target mRNAs that
are identical except in the four known mismatch bases can
thus be determined.
In addition, mismatches in the target gene can be
precisely controlled by the sec~uence of the PCR primers
used in the amplification procedure, and a defined se-
quence of precise mismatches can be created such as a
series in the region just 5 ' of the AUG codon . This is
shown in the following example:
s~ +
X-t alu Ly~ Ly~ 8-r Gly Tyr Thr Thr
AgU gca gga gcu aag gaA gcu acc aug gag aag aag auc acu gga uau acc acc
3 ~ cct cga ttc ctt cga tgg tac s ~
1 mil 'ch:
Agu gca gga gcu aag gaa gcu ccc aug gag aag aag auc acu gga uau ACC acc
3~ cct cga ttc ctt cga Tgg tac
3 0 2 m; rh~c
~gu gca gga gcu ~ag gaa ACU CCC aug gag Aag aag auc acu gga uau acc ACC
3~ cct cg2 ttc ctt cga Tgg tac
3 m~ -rh ~:
Agu gca gga gcu aag ~a~ ACU CCC ~ug gag Aag aag AUC ACU gga uau acc acc
3~ cct cga ttc ctt cga Tgg tac
4 mi ' rh~5:
agU gcA gga gcu Gag IJaa ACU CCC ~ug gAg aag aag auc acu gga uaU acc ACC
3 ' cct c:ga Ttc ctt cga Tgg tac S '
S m~ rh ~
~0 agu gca gga ccu Gag ~laa ACU CCC aug gag aag aag auc ACU gga uau acc ACC
3~ cct cgA Ttc ctt Cga Tgg tac s~
Wo 95/13834 : PCTNS94113387
76259
108
Here, the target sequenCe within the mRNA to be studied
extends from -18 to +3. Mismatches in mutant mRNAs
relative to the top sequence are shown in bold upper case.
The oligomer sequence in this example, a 21mer, is shown
5 beneath each mRNA and is invariant. Mismatches in the
oligomer to each subsequent IrRNA are shown in upper case.
Using this method of increasing the number of pre-
cisely known mismatches in otherwise identical targets,
one can accurately determine the specif icity of various
10 oligomer chemistries (e.g. rh~3p~ rothioates versus
chimeras) and modes of action (e.g. steric blockers versus
RNaseH cleavers ) .
Tests were undertaken to study the effects on ac-
tivity and specificity caused by variations in the loca-
15 tion of the charged-h~-kh~-n~ RNasH-activating region
within a chimeric oligonucleoside, and by various mis-
matches incorporated into the base sequence of an oligo-
nucleoside and/or in the target mRNA. The chimeric
compounds listed below (see also Example 41) were assayed
20 for antisense activity against both the pG1040 (UCAT)
target and the pG1042 (UCAT) 4-base mismatch control. The
ol1 s r sequences were aa follows .
pG1040 (UCAT) tarqet mRNA and antisense oliqomers:
+1 +4 +27
l l l
Met Glu LYR Ly~ 3--r Gly Tyr T}lr
mRNA aug gag aag aag auc acu gga uau acc
3637-1 3' ete tte tte t~g tg~ eet at~ tgg S'
363b-1 3' ete tte tte ta~l tg- eet ~t~ tgg S'
3262-5 3' ete tte tte ta tClA cet t~ tgg S'
3636-1 3 ' ete tte tte t~g tg~ eet ~t-- tgg S '
3639-1 3' ete tte tte tag t~ cet t~ tgg S'
3640-1 3' etc ttc tte t~sl t~ ~et ~ta tgg S'
XV-2 3 ete ttc tte t~q tq/ eet ~tA tc~ S'
wo 95113834 PCrlUS94113387
21762$9
109
The phosphorothioate linkages in these chimeric oligomer8
are immediately 5' of the underlined bases. It will be
seen that the position of the phosphorothioate core is
seq-l.on~ ly shifted in position with respect to the
5 target mRNA.
Antisense activity was assayed against both pGl041
(UCAT) and pGl042 (UCAT) using procedures as generally
described irl Example 41, except that 0 . 5 ~M oligomer was
used. It was demonstrated that mismatches in the phos-
lO phorothioate core and the position of the core in chimericoligomers greatly affected antisense activity. The
following table sets forth the percentage of gene ex-
pression (t error) measured for each of the tested
oligomers .
01igomer number
15 Target 3637-1 3638-1 3262-5 3636-1 3639-1 3640-1
79+5% 37i3% 35i:7% 70i3% 98iS% 103iS%
pG1 040
pG1042 89i3% 102i2% 88~4% 120i8% 93i2% 115i3%
The results show the effect of moving the RNAseH-
activating rh~ h~rothioate core within the oligomer. The
2 0 position of the phosphorothioate core and/or the base
composition of the phosphorothioate core has a large
effect on antisense activity, as seen by comparing 3637-l,
3 6 3 8 - l, 3 2 6 2 - 5 and 3 6 3 6 - l . A more cent ral pos i t ion wi t~in
the chimera is most active, but some activity is detected
25 even when the core is near the ends of the chimera.
A single base mismatch (denoted by an "x" above the
sequences shown above) within the RNaseH phosphorothioate
core se~uence of the chimeric oligomers e' iminates anti-
sen8e activity in this eukaryotic cell culture assay, as
30 8een by comparing 3639-l and 3640-l with 3262-5. In a
WO95113834 ; f: i ~' i " .. PCrf~S94/13387
2l~6259
110
separate experiment using the Game assay system, the all-
phosphorothioate 24mer XV-2 gave 909~ inhibition of pG1040
(UCAT) expression and approximately 50g~ inhibition against
pG1042 (UCAT) even though there were four mismatches in
5 the case of the latter target. This indicates that all-
phosphorothioate oligomers are far less specific than
chimeric oligomers rnnt~;n;n f short regions of phosphoro-
thioate 1 ;nk~c, ;n~l nh as even a single mismatch
between the chimeric oligomers 3639 and 3640 and the
10 pG1040 target abolished activity, whereas four mismatches
in the case of XV-2 and pG1042 reduced activity by less
than 5 0 9~ .
Exam~le 4 3
IncrP~ed RNaseH Cleavaqe Rate with Chimeras rnnt;l;n;nq
15 (~h; r~ 1 1Y Enriched Oliqonucleoside Methvl l~hns~hnn~te End-
The present example demonstrates that chimericoligomers with ~nh;~nn~ binding affinity promote RNaseH
cleavage of RNA target strands at a f aster rate than lower
20 affinity oligomers having the same base sequence. Chime-
ric oligonucleoside~ cnnt~;n;ng either racemic or chirally
pure (Rp) methylE~hr~sr~hr~n~tes were P~m;n~d for their
ability to activate RNaseH.
The following chimeric oligomers were used in this
25 example:
Sequence = 3 ' - [CCTGTTG] [TACGT] [ACCTTCTG] -5 '
2681-1 [MP] [PS/DE] [MP]
3214-1 [MP (Rp) /DE] [PS/DE] [MP (R~,) /DE]
Each of these chimeric oligomers was synthesized according
30 to the method described in Example 30. A complementary
synthetic RNA target was prepared according to the method
given in Example 28. This oligomer has the following
secfuence:
WO 9~/13834 2 1 7 6 2 ~ 9 PCrtUS94/~3387
111
5 ~ -G~.~ c~TTGCA~GGAAGAC-3 '
- A 32P-label was coupled to the 5'-end of this oligomer
using [~-32P]-ATP and T4 polynucleotide kinase according to
a procedure commonly known in the art.
RNaseH from bacterial ~. coli was purchased from
Promega Corp. (Madison, WI) . Buffer A, used for the RNaseH
reactions ~nnt~;n~rl 20 mM KCl, 9 mM MgC1~, 1 mM 2-mercapto-
ethanol, 250 f~g/ml of BSA ~Promega Corp. ) and 100 u/ml of
RNasin (Promega Corp. ) .
A mixture of 5' -32P-labelled RNA target (approximately
80,000 dpms, 5 x 10-1 M) was mixed w~ith 1 molar equivalent
of either chimeric oligomer in reaction Buffer A (total
volume = 98 microliters). This mixture was incubated at
37C for 1 hour. Next, RNaseH (1.1 microliters, 30
units/mL, final concentration = 2 x 10-9 M) was added and
the resulting mixture was incubated at 37C. Aliquots (15
microliters) were removed at specified time intervals,
diluted with EDTA (0.5 M, 3 microliters) frozen on dry ice
and then stored at -20C. The products of RNA cleavage
were analyzed by gel electrophoresi6 uæing a 1590 poly-
acrylamide/7 M urea gel (20 cm x 30 cm x 0.5 mm i.d.)
equilibrated in 1 X TBE buffer (p~ 8.2). The gel was
electrophoresed at 1200 volts for approximately three
hourE. Bands on the wet gel were visualized by phosphor-
imager analysis using a Bio-Rad Model GS-250 Molecular
Imager (C~1 ~h~ , CA) .
Site-specific RNAse~I-r~';~t~od cleavage was observed
with both chimeric oligomers. The lengths of the frag-
ments were estimated according to their electrophoretic
3 0 mobility . According to thi3 analysis, it was determined
that cleavage was limited to the center of the RNA target
sequence. That is, cleavage was limited to the position
of the RNA strand complementary to the negatively charged
segment of each chimeric oligomer. A difference in the
rate of RNase~l mediated cleavage was detected for the two
dif f erent chimeric oligomers as 8hown in FIG . 5 .
WO95/13834 ~ 59 ,-, PCrrUS94/13387
112
It is seen that the rate of RNA hydrolysis in the
presence of chimeric oligomer 3124 -1 (Cnnt~; n; n~ alter-
nating MP(R~)/DE backbone segments at the 3'- and 5'-ends)
i8 about 10 tlmes faster than that for the other chimeric
5 oligomer 2681-1 (rr~tA;n;n~ racemic MP harkhr,nP seqments) .
~,rAml21e 44
Effect of 2'-Suqar Substitution Location on Chimeric
Oliqomer Cleavaqe Activitv
The effect of the location of 2'-sugar substituents
10 relative to the RNaseH-activating region of the present
oligomers was studied by measuring the cleavage activity
of differently-substituted chimeric oligomers against a
target RNA sequence. A synthetic 20mer RNA molecule,
designated 3593, containing an AUG sequence near the
15 targeted cleavage site was prepared having the following
se~uence:
3593 (target RNA): 5' AG AGA GAG AUG CAG AGA GAG 3'
Chimeric 20mer RNaseH-activating ol;rnn~rleosides 3463,
3465 and 3466 were synthesized using appropriate dimer
20 synthon methods as generally described above. These
c ,~ ds included a central RNaseH-activating region
comprising five consecutive phosphorothioate-linked
deoxyribonucleosides (shown in parentheses below) flanked
by non-RNaseH-activating regions linked by alternating
25 ME' (Rp) /DE linkages . Selected nucleoside sugars in the
~ nk;nrj regions of chimeras 3463 and 3465 rnntA;nP~l 2'-O-
methyl substitutions, indicated by the underlined capital-
ized nucleoside abbreviation letters below (the target
3593 sequence is also depicted to show target complement-
3 0 arity):
.
Wo 95113834 ~ 1 ~ 6 2 ~;9 PCrrUS94/13387
113
3593: 5~ AG AGA GA G AUG C AG AGA GAG 3~ (target RNA)
3463: 3' uc UcY cU(c tac g)Uc UcU clrc 5~
3465: 3' uc UC~ u(c tac g)uc lJC~J C~C 5'
3466: 3' uc ucu cu(c tac g)uc ucu cuc 5~
5 As with other chimeric oligomer compounds disclosed
herein, the charged (here, phosphorothioate) linkages
associated with the R~aseH-activating region are situated
5' to each of the nucleosides shown in parPnthPRPq~ Thus,
compound8 3463, 3465 and 3466 above each i~clude a stretch
of five consecutive, central phosphorothioate ( {P~} )
linkages, flanked on either side by a chirally-selected Rp-
methylrh~qph~n~te ( {MP(R~) } ) linkage, as follows (shown 3
to 5~ ) :
. . . C~DII~}U{~SP (}1~) 3 (c{P5~t{P8}a{PS}c{PS~g) ~PS}u{~ ~ C{DE~U . . .
The underlined phosphorothioate linkage shown above [in
the 6egmert . . .u{MP(Rp) } (c{PS}t. . .] can be incorporated
into the compounds using dimer synthon methods as de-
scribed, for example, in ~xample 13 above. The rr--;ninr,
non-RNaseH-activating portions of the chimeric compounds
include alt-~rn~t;nr, ~P(Pp)tDE linkage q~_ ~ incorporat-
ed, for example, by successive addition of appropriate
dimers following the support-bound 'lucr dinucleotide
ser~uence at the 3'-tPrm;nllq of the compounds (see, e.g.,
Examples 8, 9 and 17A above). Thus, 2~-sugar substitu-
tions shown above for compounds 3463 and 3465 can be
achieved by successively incorporating suitable
2'0Me~{~P(R~) }c~DE} or 2'0MeU~MP(Rp) }2'0Me{DE} dimers into
the respective oligomers.
To assess the RNaseH cleavage activity O$ the fore-
going chimeric oligomers, 320 /~l of a mixture of 5~ 32p_
labelled RNA target c~ ~,IUUlld 3593 (160 dpm) and the
selected test oligomer (1:1 molar ratio; rr~nr~ntrations
0.5 nM) was incubated in Buf$er A at 37C for one hour to
achieve cOmp~ementary complex formation a~d equilibration.
,,,,, ,, . .. . . ... , ~
wo 9S/l3834 2 1 7 6 2 5 9 ~ PCrlUS94/13387
114
(suffer A: 20 mM KCl, 9 mM MgCl2, 1 mM 2-mercaptoethanol,
250 llg/ml BSA [Promega], 100 u/ml RNasin [Promega] . ) A 20
111 aliquot was removed as a time zero sample and 3 . 3 ~Ll of
a 2 nM solution of bacterial (E. coli) RNa5eH (Promega) in
5 Buffer A was added (final concentration of enzyme in
solution was 0 . 022 nM) . The reaction mixture was kept at
37C. Twenty microliter aliquots were removed from the
mixture at appropriate time intervals and the reaction was
stopped by adding 2 ILl of 0 . 5 M sodium EDTA solution and
10 then freezing on dry ice. The products of RNA cleavage
were analyzed in 1596 PAGE (20 cm x 30 cm x O . 5 mm) con-
taining 7 M urea and lx TBE buffer ~pH 8.1). Gels were
run at 1200 V for 2 hours. Quantitative kinetic data were
obtained by integration of the volumes of the bands by
15 means of Phosphor-image analysiE.
The kinetic curves for this example are shown in FIG.
11. A significant decrease (about 10-fold) in the overall
rate of RNA cleavage was found when 2'-O-methyl nucleoside
units were positioned next to the central phosphorothioate
20 RNaseH-activating region (compound 3463, triangle data
points) as compared to the chimeric compound rnnt:~in;nrj
all 2'-H nucleosides (~ , -JUlld 3466, circles) . The
initial number of cleavage products was reduced for
compound 3463 as compared to compound 3466 (2 instead of
25 3). When a 2'-H nucleoside instead of a 2'-O-methyl
nucleoside was incorporated on the border of the alternat-
ing methylphosphonate/phosphodiester 5 ' -end-block and
pl~r,5rhnrothioate regions (compound 3465, r~ ), no
sirjn; f; r~nt decrease in cleavage rate was found, and the
3 0 number of cleavage products also did not change as com-
pared to that obtained with ~ , olln~ 3463 .
This example demonstrates that the presence of a non-
hydroxy 2~-sugar substituent adjacent to the RNaseH
cleavage site has a significant ~l;m;nllt;ve effect on
35 RNaseH cleavage activity and that even a single 2 ~ -O-
methyl substituent may be responsible for the reduction in
cleavage activity. In contrast, the use of a 2'-substitu-
W0 95/13834 ~ :1 rt 6 2 5 9 PCT/US94/13387
115
tion that i8 removed from the RNaseH-activating region by
one or two nucleosides has a negligible ef f ect on RNaseH
binding and/or cleavage activation.
r ~le 45
Activitv of Chimeric Oliaonucleoside Com~o~1n~lc A~A;nct HPV
Taraets
This example describes experiments using various
chimeric oligonucleosides of the invention targeted
against human papilloma virus (HPV) gene seauences.
A. Preparation of Plasmid E~pressing a Polyciatronic
E 6 /E7 mRNA
An expression vector having an insert coding f or
HPVll E6/E7 was prepared usin~ the expres6ion vector
pRc/CMV ( Invitrogen) . The plaGmid pRC/CMV was linearized
with Eind III. The recessed 3' ends were filled with the
5 ' -3 ' polymerase activity of T, DNA polymerase . A full
length clone of HPV-11 cloned at the BamEI Site in pBR322
was digested with the restriction enzymes B6t II and Hinf
I. The 873 base pair LL _ nt ~)rtAinlnS the E6 and E7
open reading frames was purified on agarose gel. The
restriction ends of this fragment were modified by filling
in the recessed 3 ' -ends with T, DNA polymerase .
The vector and insert were ligated with T4 DNA ligase
and transformed into DH5~Y E. Coli. Recombinants were
screened for c-~L-,~Liate insert and or;,~ntAti-~n as well as
E6/E7 transcription and translation activity.
This plasmid (pRc/CMVII-E6/E7) was used in the cell
free trAncl Atir~n system described below.
B. Preparation of Pla~mid HAving an E2 Insert
An expression vector having an HPV-11 E2 insert was
prepared using pRc/CMV (Invitrogen). The plasmid was
linearized with Eind III, followed by treatment with calf
thymus ~1 kA 1, nl~ phosphatase . To isolate the E2 open
reading frame, a full length clone of HPV-11, cloned at
Wo 95/13834 2 i 7 6 ~ ~ 9 PCTN594113387
116
the Bam HI site in pBR322, was digested with the restric-
tion enzymes ~nmI and SspI. The recessed 3 ' ends were
filled in with the 5'-3' polymerase activity of the Klenow
fragment of DNA polymerase I. Hlnd III linkers were then
5 added. The 1309 base pair fragment containing the com-
plete E2 ORF was agarose gel purif ied. The modif ied
vector and B2 insert were ligated with Ts DNA ligase and
transformed into DH5~ E. Coli. Recombinants were screened
for appropriate insert, transcription and translation.
This plasmid (pRc/CMVII-E2) was used in the cell-free
translation system described below.
C. Preparation of Plasmid Eaving Nonoci6tronic E7 Insert
An expression vector having an HPV- 11 E7 insert was
prepared using pcDNA-1 ( Invitrogen) . The plasmid pcDNA
was digested with Bam HI and with Xba I. A Cla~ t
nt~;n;n~ the complete open reading frame of HPV-11 (from
-30 through the termination codon) flanked by Bam HI and
Xoa I restriction sites was prepared by PCR using standard
protocols. The digested vector and fragment were ligated
with T~ DNA ligase and transformed into MC 1061/P3 cells.
R~-_ 'I;nAntF: were screened for appropriate insert, tran-
scription and translation.
This plasmid (pcDNA E7) was used in the cell-free
translation system and in the transient expression assay
described below.
D. Demon~tr~tion of Activity o~ ~nt; ~er~e Chimeric
ol; ~ " Targeted to EPV-11 E7 in Cell Free Tr~ms-
l~tion Extr~ct~3
Mono-cistronic (100 nM) HPV-11 E7 or polycistronic
(50 nM) HPV-l1 E6/E7 RNA was co-translated with chloram-
phenicol acetyl transferase (CAT) RNA (2 to 10 nM) in
cell-free rabbit reticulocyte extracts (Promega). The
c~nt~nts of e~ch assay system was as follows.
i
Wo 9~13834 2 1 7 ~ 2 5 ~ PCr/USs4/l3387
117
COMPOI~ T FINAL l ~N~ ~:Nl~TlON
In vitro transcribed un- (As noted above)
capped RNA
3ss-cysteine o . 8 mCi/mI,
5 Amino acids mixture, cys- 2011 each
teine def icient
Rabbit reticulocyte lysate 72 by volume
RNAsin (Promega) 0.5 units/~LL
Oligomer . 1 to 10~LM
Cell free translation was performed at 37C for 60
minutes and was stopped by addition of SDS gel loading
buffer and ;nllhation at 95 for 3 minuteG. Translation
of E7 was evaluated after; ~recipitation with aE7
goat antiserum and protein A sepharose, followed by SDS-
15 PAGE and phosphoimage analysis. This protocol was also
used in the cell-free translations referred to below.
E . D L~ .-tion of Activity of ~nt; r -e O~; L 8 in
Cell-Free RNAseH Cleavag~ A~say
In vitro transcribed, llnc~rp~d mono-cistronic RNA was
20 prepared by transcribing plasmid pcDNAllE7 with RNA
polymerase (Ambion MegaScript ) . The E7 RNA was incubated
at a c~ n~nt~ation of 100 nM in the presence of 0 . 04 units
l~uL E. Coli. RNAseH (Promega), 3.5 mM MgCl" 25 mM KCl, 70
mM NaCl and 20 mM potassium acetate at 37C for 30 min-
25 utes. Reactions were stopped by addition of formamide gel
loading buffer followed by heating to 100C for 5 minutes.
Samples were analyzed by 4~6 Urea-PAGE analysis,
followed by 8taining with e~h;~ m bromide. Percentages
of cleavage of E7 MRNA, in the presence of RNAseH, of
30 methylphosphonate chimeric oligomers 2657-1, 316g-1,
WO 95/13834 2t ~ ~ 2 5 g PCr/~'S94/13387
118
3214-1, 3257-1, 3241-1 and 3236-1 are shown in the table
below. Good dose response effects were obtained for all
the oligomers at the concentrations tested. The order of
potency was 3169-1 - 3257-1 ~ 3214-1 - 2657-1 ~ 3236-l ~
5 3241-1. All oligomers showed good specificity, cleaving
E7 mRNA in one position.
Oligomer
(ILM)
Oligomer Backbone 0.0 l 0. l 1 10
3169-l [MP(R~)/DE]-[DE]s-[MP(Rp)/DE] 7 45 85 loO
3214-l [MP(Rp)/DE]-[PS/DE]s-[MP(l~?)lDE] 2 20 50 ~0
103257-l [MP(Rp)/DE]-[PS/DE]7-[MP(R")/DE] 4 40 75 lO0
334'.-l 2'0Me[MP(R")/DE]-[PS],-2'0Me[MP~/DE] 5 40 60 60
3336-l 2'0Me[MP(Rp)/DE]-[PS/DE],-2'0Me[MP(Rp)/DE] 5 50 60 65
Results are percentage of cleavage of E7 m'~NA.
Estimated values were obtained by visual inspection of the
gel.
F. Demon~tr~tion of Activity o$ Ant;n~nne 01;3 -~'D in
TrAnDiently Tr~nDfected COS-7 CellD
COS-7 cells were seeded at 1 X 105 cells/well in 24
well plates and then cultured overnight in cell culture
media (90% DMEM, 10% fetal bovine serum and 50 I.U./ml
penicillin, 50 mg/ml streptomycin and 0.25 llg/ml ampho-
tericin B). After 24 hours the cells were approximately
80 to 90% confluent. A transfection cocktail of 2.5 ~g/mL
pcDNA 1 E7, 50 ~g/mL Transfectam (Promega) and varying
rnnr~ntrations of oligomer was prepared and incubated for
15 minutes at room temperature after a 2 second vortex
mix .
Wo 95/l3834 ~ 2 ~ PCTIUS94/13387
119
Cells were washed on the plates two times, 1 ml/well
with Optimem (Gibco-BRL) . Then 0.5 mL tran6fection
cocktail per well was applied to duplicate wells. The
plates were incubated for 4 hours in 5~ CO2 at 37C. After
5 incubation cells were washed two times, 1 mL/well with
cell culture media and cultured overnight. Then cells
were washed twice, 1 mL/well with cysteine rl~f;~ nt DMEM
and then ;nt'llhê~t~'~ for 309 minutes in cysteine deficient
DMEM under cell culture conditions. Cells were labelled
by incubation with 250 IlCi of 35S-cysteine/well in 500 ~L
cysteine def icient DMEM without serum f or 5 hours . The
cells were then washed twice, 1 mL/well with 1 X rhnsrh~te
buffered saline and then lysed with 100 ~ SDS sample
buffer (50 mM Tris-C1 [pH 6.8], 100 nM dithiothreitol, 296
sodium dodecyl sulfate, 0.1~ bL- Lh~nnl blue, 10g~ glycer-
ol). Wells were washed with 100 ~L~ RIPA buffer (10 mM
Tris-Cl [pH 7.4], 150 mM NaCl, 19~ Triton X-100, 0.1~
sodium dodecyl sulfate, 0.59~ sodium deoxycholate) and
combined with sample buf f er lysate .
2 o B7 synthesis was evaluated by immunoprecipitation of
E7 protein with goat anti-HPV-11 E7 serum and protein A
sepharose beads (Sigma). T nrrecipitated E7 protein
was quantitated by SDS-PAGE and rhn~rhr~ir-~e analysis.
Total protein synthesis was evaluated by SDS-PAGE and
phosphoimage analysis of a fraction of the transfected
cell lysate bef ore immunoprecipitation .
Representative experiments were performed as follows.
E7 expression plasmia pcDNAllE7 (5~g/ml) and different
amounts of antisense ol i ~r nl-rle - tide were transf ected into
COS-7 cells in the presence of Transfectam' (Promega).
Cells were incubated with transfection mixture for 4
hours, allowed to recover in media plus serum overnight,
and labeled with 35S-cysteine for 5 hours before harvest-
ing. Cells were lysed and E7 protein synthesis was
evaluated by i nprecipitation with ~E7 serum followed
by SDS-PAGE gel fractionation of protein products and
phosphoimage analysis. Total protein synthesis was
W095ll3834 21 7~25g (` i ` ~ PCr/US94113387
120
analyzed by SDS-PAGE separation of an aliquot of the cell
extract, autoradiography and phosphoimage quantitation of
all the proteins present in each lane. The following
table summarizes the IC50 and IC90 values obtained with
chimeric oligomers 3169-1, 3214-l, 3256-l, 3257-l and
3336-1 .
POTENÇY OF OLIGQ~Rq TARÇETED TO E~PV-11 E7 IN A CELL
BASED ASS~Y
Cell-based assay
Oligomer Backbone IC50 IC90
103169-2 [MP(R")/DE]-[DE]-~MP(I~)/DE] >2 ~LM >>10 I.M
3214-1 [MP(Rp)/DE:]-[DE/PS]-[MP(E~7)/DE] 0.2 IIM I ILM
3256-1 [MP(E~p)/DE]-[PS]-[MP(Rp)/DE] 0.12 1 ~M
yM
3257-1 [MP(Rp3/DE]-[DE/PS]-[MP(R")/DE] 0.06 <0,3 I~M
/LM
3336-1 2'0Me[MP(Rp)/DE]-[DE/PS]-2'0Me[MP(ilp)/DE] 0.4 ILM ~2 ~LM
It is clear from this example that chimeric oligo-
nucleotides 3214-1, 3257-1 and 3256-1, which contain all
rhngrhnrothioate ( [PS] ) or alternating phosphorothioate/
phosphodiester ( [PS/DE] ) linkage in the middle and chiral
methylphorothioate/methylrhnsrhnn~t~ dimers linked by
phosphodiester linkages ([MP(P~)/DE]) as end-blocks, are
potent inhibitors of transient expre3~ion of HPV E7
protein in COS-7 cells.
Chimeric oligonucleotides with rhn~rhnrii ester link-
ages in the middle, such as 3169-1, were not potent in the
cell-based assay, although they proved to be very potent
in the cell-free assay. This difference may be due to the
Wo 95113834 2 1 7 6 2 5 9 PCrlUS94/13387
_ . .
121
intracPl 1 1ll Ar instability of the phosphodiester linkage .
Finally, oligonucleotides c~ntA;n;n~ 2'0Me modification in
the sugar of the nucleosides present at the ends (see
3336-1) were les6 potent than the corrPAp~-n~; n~ chimeras
5 with [MP (Rp) /DE] ends .
G. Demonstration of Oligomer Activity by Microinjection
in VERO CellR
(i) lIicro in~ection
oligomers were microinjected together with E2
(pRc/CMV 11-E2~ or E7 (pcDNAE7) expression plasmids at 50
g/l~l into the cytoplasm of VERO cells according to the
following procedure. On the day preceding injection, VERO
cells (approximately 2 X 105 cells/ml) were plated on
coverslipæ. Plasmid DNA was diluted in PBS to a r~n~pnt~a-
tion of 20 ng/~ul (E7) or 50 ng/~Ll (E2) in an Eppendorf
tube. The tube~3 c~ntA;n;n~ plasmid DNA were centrifuged
for 15 minutes at 1,400 rpm. The tubes were set on ice
prior to microinjection. A 2 ~LL aliquot of plasmid DNA
solution was loaded onto a fem to top. The tip was set
with the coverslip at 45. The pre6sure on the micro-
injector was set at 80 and the injection was performed.
The coverslips were incubated at 37C overnight after
insertion. At 16 hours post-injection, cells were fixed
and immunostained with goat anti-E7 polyclonal antibody,
as explained below.
(ii) Indirect Fluore~3cence T -- vv_y
Prior to use in this assay, goat anti-HPV-11 E7 or
HPV-11 E2 serum was preabsorbed with VERO cells as fol-
lows. ~mfluPnt VERO cells from two T-150 fla~ks were
scraped and then washed twice with P8S. 200 1ll serum was
then added to the cell pellet and mixed at ~0C overnight.
The mixture was centrifuged and the 8upernatant was
removed to a new tube. The preabsorbed 8erum was stored
in 5 0 ~ glycerol at - 2 0 C .
WO 95/13834 ~ PCTIUS94/13387
122
Expression level of E2 or E7 was assessed using a
fluorescent antibody assay. Coverslips were fixed in 1096
formaldehyde in PBS for 20 minutes at room temperature and
then washed twice with PBS, followed by incubation with
goat anti-HPV-11 E7 or l;PV-11 E2 protein serum preabsorbed
as set forth above at a 1:1000 dilution in PBS for two
hours at room temperature. The coverslips were then
washed with PBS three times, five minutes per wash, and
incubated with FITC-conjugated Donkey Anti-Goat IgGAb
tJackson, T ~R~qearch, Cat #705-095-147) at 1:200
dilution in PBS. The coverslips were then washed with PBS
three times, air-dried, and mounted with 509~ glycerol on
slide glass. Examination was done under W lights.
Results are presented in the following tables.
POTENCY OF OLIGOMERS TARGETED TO HPV-I I E7
Cell-free assay Vero cells
Oligomer Backbone Tm IC50 IC90 CAT
Inhibition
2687-1[75/~MP(l~j][DE]175%MPal~,)] 52.8 ~0.04 1 IIM 20%, 5 N-3+ (0.5 IIM)
,uM nM
3169-1IMP(Rp)/I'F][l~F]~[~- (R")/DE] 62.6 ~0.04 0.8 /IM 20%, 5 C-3+ (2 ~I~M)
~M ~LM
3214-1[MP(RV)/T ,, ' S]~IMP~B,,)/DE] 61.0 ~0.2 ~M 4 IIM No inh., C-3+ (I iLM)
10 IIM:
25%, 5
~M
3257-1 [MP(Rp)/Dy[DElPS]7lMP(R7)/DE] 60.9 ~0.06 0.6 yM 50%, 2 C-3+ (0.5 ILM)
uM rM
3256-1[MP(RV)' ,~ .,J~[MP(R,)/DE] 60.1 ~0.25 5 uM No inh., C-3+ (0.5 ~.M)
uM 5 ~IM
3336-12'0MelMP(Rp)/DE]lPS]7- 66.8 N/D
2'0MetMP(R,)/DE]
3341-12~oMe[Mp(R7)lDE][ps]7- 65.8 2 ~M >~10 N/D
2'0MclMP~R,)/DE] IIM
WO 95113834 2 1 7 6 2 ~ 9 PCI'IUS94/13387
123
o
+
~ ' _
r ~ o o
~_ +
~ o
~ ~r~ ~ ~r~
o
O,
~2 " Z +
Z r~
O
,
r --`
'" _ i _ ~ ~
,
" ~ ~; 2 2
O
r ~ ~.
r~ r o . , _
o ,,~ r~ , r
~;1
U~ o
.1
2~7~59
Wo 95113834 PCrrUS94/13387
124
E. Demonstr~tion of Activity of ~nt; ~n~e O
T~rgeted to E2 in Cell-Free Translation Extract~
E2 RNA was prepared by transcribing plasmid pRc/CMV-
llE2 with T7 RNA polymerase using an Ambion MegaScript
5 kit, following the nn-n~lfA~t-lrer's directions.
In vitro transcribed E2 mRNA was cell-free translated
in rabbit reticulocyte lysates (Promega). The final
~nn,!~ntrations of each, ~ n~nt of the assay system was
as f ollows:
In vitro ~r~nerr~h-~ uncapped RNA: 50 nM
'sS-M~h i rn; n~ 1. 3 I~Ci/
Potassiu= Acetate: 20 mM
Amino ncid mixtures, m~thionine deiicient: 50 ~IM
Rabhit Reticulocyt~ Lys~te: ~ 339s vol~vol
RNAsin: None or 0.5 units/lll
Cell-free translation was performed at 37C for one
hour and was stopped by addition of SDS gel loading buffer
and incubation at 95C for 3 minutes. Translation of E2
was evaluated after separation of the translation mix by
SDS-PAGE analysis, followed by r~n~rl~n;r-~e analysis. To
determine the effect of oligomers targeted to the transla-
tion initiation codon of E2, in vl tro transcribed E2 mRNA
was translated in the presence of 0.02 or 0.04 units/~l of
RNAseH, and using oligonucleotide concentrations ranging
from 0.01 to 10 I~M. CAT mRNA was co-translated, or trans-
lated in ; n~or~nrl~nt trAn~lAt; nn reactions as control .
As shown in the following table, mea~uL~ ~ of E2
cell-free translation inhibition and of specificity with
respect to CAT control mRNA were obtained with the
oligomers 3170, 3233 and 3234. Parallel studies showed
that these end-blocked chimeric ol; 3 s were more
specific than all-phosphodiester oligomers.
~7~2ag
WO 95/13834 PCTIUS94113387
125
POTENCY OF OLIGOMERS TARGETED TO
HPV- 11 E2 IN CELL FREE ASSAY
Cell-free assay
Oligomer Target Backbone IC50 IC90 CAT-IC50
3170-1 AUG-12 [MP(Rp)/DE][DE]5[MP(R")/DE] ~0.06 ~M ~1 I-M 20% (5
I~M)
3233-1 AUG-4 [MP(R")/DE][DE]5[MP(Rp)/DE] ~0.1 ~IM ~1 /IM 20% (5
I.M)
3234-1 AUG-4 [MP(Rp)/DE][DE/PS]5[MP(I~)/DE] ~0.1 !-M ~1 ~LM 15% (10
I~M)
I. Demonstration of Activity Of Anti~enEle Oligo~er~
T~rgeted to E6 in Cell-Free Tranalation Extr~ct~
Polycistronic E6/E7 mRNA was prepared by transcribing
the plasmid pRc/CMV11-E6/E7 with T7 RNA polymerase using
an A~nbion MegaScript kit, following the manufacturer~ s
directions. Tn vitro transcribed E6/E7 mRNA ~50nM) was
cell-free translated in rabbit reticulocyte lysates
(Promega) as described in part D above. Cell-free trans-
lation was performed at 37C for one hour and was stopped
by addition of SDS gel loading buf f er and incubation at
95C for 3 minutes. Translation of E6 was evaluated after
separation of the translation mix by SDS-PAGE analysis,
followed by rhnpphn;r-~e analysis.
To determine the effect of oligomers targeted to the
translation initiation codon of E6, in vi tro transcribed
E6/E7 mRNA was translated in the presence or absence of
the oligonucleotides shown below. Translations were
performed in the presence of O . 02 or O . 04 units/lLl of
RNA8e H, and using oligomer cnnr~nt~ations ranging from
O . 01 to 10 ~M. CAT mRNa was co-translated as control . As
8hown in the table below, the be8t re8ults were obtained
WO 95113834 PCT/13S94/l3387
~17~
126
with oligomer 3215-1, a 20mer chimeric methylphosphonate
oligomer targeted to AUG-10.
Cell-~ee assay
Oligomer Target Backbone ICSO IC90 CAT
inhibi-
tion
3255-l AUG-IO [MP(Rp)/DE][DEI[MP(Rp)~DE] I 5 IIM No inh
~M (lo ~IM)
5 3215-l AUG-Io [MP~)/DE][PS/DE][MP(RD)/DE] 0.3 2 ,~ M no inh.
.M (lO ~LM)
~ , uullds 3255 and 3215 are as follows:
3 2 5 5 - 1: 3 ' - CTGCTCC ( GTAAT ) ACCTTTCA- 5 '
3 2 15 - 1: 3 ' - CTGCTCC ( GTAAT) ACCTTTCA- 5 '
Following are a set of examples relating to certain
10 chemistry useful in the synthesis of chirally pure 2'-0-Me
dimers. The preparation of two dimers are discussed in
Examples 46 and 47 to further illustrate the utility of
the P(III) coupling chemistry through either a 5' or 3'
phosphoramidite monomer. These two examples also demon-
15 strate the ability to oxidize (with retention) inter-
nucleoside methyl rhnRrhnramidite l; nk~P~ using either
cumene hydroperoxide or camphorsulfonyl oxaziridine to
yield the desired methylrhn~rhnn~tP linkage. Although
either or both reagents may be used, our preference is to
20 use camphorsulfonyl oxaziridine because it does not have
the hazards associated with cumene l~ydLu~eLu~ide. Example
48 describes the synthesis of a 2'-O-Me-gl:~nn1:;nP 5'-OE~,
and is a general scheme applicable tû the preparation of
other 5'-OH nucleosides. Example 49 describes the phos-
.
Wo 9S/13834 ~ ~ 7 ~ 2 5 9: PC'r/US94113387
127
phitylation of a 2 -O-Me UC dimer with B-cyanoet_yl ("CE")
~hnsphnramidite .
le 46
Pre~aration oi a 2 -O-Me GG (5 O-DMT, 3 o-BcE. N2I8U)
5 NP(Rq) D~m~r Via 5'methvl~hos~hnn~m;tl;te MQnomer.
Into a 500 ml RBF was placed 30 .5 g (O . 05 M) of 2 ' -
OMe, G(3'O-tBDPS,5'-OH, N2IBU) which was rendered anhydrous
with 1 x 100 ml pyridine and 2 x 100 ml acetontrile
(ACN) . The resulting dry foam was released from the roto-
evaporator with argon and treated with 300 ml anhydrous
ACN, 10.5 ml triethylamine (0.075 M, 1.5 eq.). The flask
was stoppered with a rubber septa and treated (dropwise)
with 10.9 ml chloro, methyl-N,N-diisopropyl;~m;nnrhnsphine
(0.06 M, 1.2 eq.) . The reaction was allowed to stir
overnight at room temperature.
The next morning, the reaction was founa to contain
no starting material, as determined by HPLC (Beckman Gold,
RP, Waters C18 bnn~Ar~k; A254 nm, 20 min. program 50/50
ACN/O .1 M TEAA to 1009~ ACN. ) . The reaction mix was
rnnr.ont~ated then purified on 225 g silica in 3 :1 ethyl
acetate/heptane rnnt~1n;n~ 256 TEA. Product was pooled and
rnnr~ntri~ted to obtain 25 g (67~) of solid foam that was
86~ pure by HPLC. This product was taken up in ACN to
give a 1096 solution of the desired amidite, which was
stored over molecular sieves.
Into a 500 ml flamed dried RBF with argon balloon
overhead, was transferred via an addition funnel with
glass wool, 100 ml (10 g, 0.013 M, 1.25 eq.) of stock
solution of 2'0-Me G(5'-amidite, 3'-tBDPS, N2IBU) along
with 71.1 ml (7.1 g, O . 011 M, 1. 0 eq. ) of stock solution
of 2 ' O-Me, G ( 5 ' DMT, 3 ' OH, N2IBU) . The reaction mixture was
then treated all at once with 30 . 9 ml (259t by weight sol .
in ACN, 5 . O eg. ) of ethylthio-tetrazole (ETT) and stirred
at room temperature for 5 minutes, after which time cumene
lly~lLuyeI~-~ide (2.1 ml, tech., 80~6) was added all at once.
~he reaction was guenched 5 minutes later with 2 0 ml
Wo 9s/l3834 2 1 7 ~ 2 5 9 PCrn3S94113387
128
saturated sodium bisulfite. The reaction mixture was
analyzed by HPLC and determined to be 86~ dimer with a
ratio of 1. 2/1. 0 (Sp/Rp) . The reaction mixture was then
placed on a roto-evaporator and the ACN was removed. The
resulting concentrate was then taken up in 150 ml dichlor-
omethane (DCM), and washed using 2 x 75 ml sat. NaHCO3 and
1 x 75 ml water. The aqueous wa5hes were combined and
then extracted with 1 x 75 ml DCM and combined with the
original organic phase and dried over NaSO4, f iltered and
concentrated to a light amber solid foam.
The solid foam, 12.6 g (0.0094 M, 1.0 eq.) of 2'-0-
Me, GG15'DMT, 3 tBDPS, N2-iBU) MP(Rp/S;,) product was taken
up in 120 ml of THF and treated all at once with 14 . 2 ml
TBAF (1 M in THF, 0.014 M, 1.5 eq.) and allowed to stand
at room temperature overnight. The next morning desily-
lation was determined to be complete by HPLC with a purity
of 84% (4496 Sp and 4096 Rp). A small amount of silica gel
was added to the reaction mixture and after stirring for
10 min. the reaction mix was passed through a glass
sintered funnel rrnt~;n;nrJ a small bed o~ silica gel. The
product was eluted off the bed with 500 ml 1096 methanol in
DCM. The filtrate was rr,nr~ntrated, taken up in DCM and
washed using 2 x 75 ml sat. NaHCO3 and 1 x 75 ml brine.
The organic layer was dried over MgSO" f iltered and
rrnr~ntrated to a thick oil, which weighed 14 g but had a
strong cumene hydroperoxide odor.
The oil was taken up in ACN to give a 23~ by weight
solution and purified on a 2 inch preparative HPLC column
(Beckman Gold, RP, Kromasil C18, 10u, A295nm, 60 ml/min.,
isocratic 459~ ACN and 559~ H20). Three separate runs were
made and the pure Rp fractions were pooled and concentrated
to yield 3.3 g of 1009~ pure GG(3 -OH) MP(Rp) dimer.
~lO 95/l383~ ~ ~ 7 6 2 ~ 9 PCr~94/13387
129
Exam~le 4 7
Pre~aration of 2 -O-Me. CU(5 -DMT. 3 -OH, N4IBU) MP(R~)
Dimer Via a 3 ' -methYl~horh~ n~m;~1; te Monsmer.
50 g (0 . 082 M, 1. 0 eq. ) of the 2 -O-Me, DMT protected
- 5 cytidine was rendered anhydrous with 3 x 100 ml pyridine
and 1 x 100 ml ACN co-evaporations. The flask was re-
leased with argon and to it was added a stir bar, 500 ml
ACN, 22 . 7 ml TEA (0 .163 M, 2 eq. ) and a septa with an
argon ballon overhead. The solution was treated dropwise
with 19.2 ml (0.11 M, 1.3 eq.) of Cl-MAP via a 20 ml
plastic syringe and stirred overnight at room temperature.
The reaction was checked the next morning on EIPLC and
starting material was gone. The reaction mixture was
concentrated and purified on 300 g silica gel with 50/50,
EtOAc/Heptane, with 29t TEA. Four liters of the eluent
was passed through the column and all U.V. positive
material was pooled and ~-nc~ntrated to a solid foam ~52
g, 9596 purity (HPLC), 84~ recovery). The product was
taken up in ACN to give a 10~ solution by weight of the
desired 3'-methylphos~hr~n~m;tl;te and to this solution was
added molecular sieves.
After sitting over molecular sieves for one night,
100 ml (10 g, 0.013 M, 1.25 eq.) of this stock solution
was added to a flame dried 500 ml RBF along with 51 ml of
a stock solution of U, 5'0H (5.1 g, 0.01 M, 1.0 eq.). The
ETT (67 ml, 10~ solution in ACN over molecular sieves, 6.7
g, 0.052 M, 5.0 eq.) was added all at once via an addition
funnel and the reaction was stirred for 5 minutes at room
temperature. The phosphite int, --l;Ate was then oxidized
with 36 ml camphorsulfonyl oxaziridine (CSO) solution (10~6
in ACN over molecular sieves) for 5 minutes. The reaction
mixture was checked by HPLC and found to contain 79~ dimer
with a ratio of 1. 2/1. 0 ~Sp/Rp) . The reaction mix was
c~- n~-~ntl^ated to a solid foam, taken up in 150 ml DCM and
worked up as described above in Example 46. The resulting
solid foam was 89~ dimer by HPLC and was desilylated (see
below) without f urther purif ication .
=
WO 95113834 PCTIUS94/13387
2~2~9
130
The solid foam, 2 -O-Me, CUl5'-ODMT, 3'-OtBDPS,
N~IBU), MP (Sp/Rp) dimer, was taken up in 100 ml THF then
treated all at once with 12 . 3 ml tetrabutyl ammonium
fluoride ~1 M in THF, 0.012 M, 1.5 eq.). The reaction was
checked 1 hr. later by ~IPLC and det~rrrl; n~d to be complete
by the disappearance of starting material. The reaction
mix was concentrated and purified on silica gel (10 :1)
with 3:1 EtOAc:DCM with 10~ methanol. The purified dimer
(8 g, 1.5/1.0, Sp/Rp) was then purified by preparative
HPLC, which following two separate runs produced 3.3 g of
pure Rp dimer, 3'-OH.
Exam~le 4 8
Pre~aration of 2 ' -OMe, G (5 -OH, 3 -OtBDPS, N2IBU) Via the
DMT Protected 3'-OH.
25 g of DMT protected 2'-0-Me guanosine was rendered
anhydrous with 3 x 100 ml DMF co-evaporations. The solid
foam was released from the roto-evaporator via argon and
dissolved in 250 ml anhydrous DMF. The solution was then
treated with 15.3 g t-butyldiphenylsilyl chloride (0.056
M, 1.5 eq.) and 10.1 g imidazole (0.15 M, 4.0 eq.), then
stirred manual ly until the solution was ~ ~, ^ous and
allowed to let stand overnight at room temperature. The
reaction was checked by HPLC the next morning and found to
contain no starting material. The reaction mix was then
poured into 300 ml ice water while manually stirring. The
solids were rol 1 e~-t~ in a Buchner funnel and rinsed with
cold water and then dissolved in 250 ml DCM and washed
using 3 x 200 ml sat. NaHCO3, 1 x 100 ml water. The
combined aqueous phases were extracted with 2 x 100 ml
3 0 DCM . The organic phases were combined and dried over
NaSO~, filtered and cnncPn~rated to a solid foam, obtaining
35 g of newly silylated product (slightly more than the
theoretical yield).
The solid foam, 2 -O-Me, G(5'-ODMT, 3'-OtBDPS, N'IBU)
was dissolved in 150 ml DCM and with magnetic stirring was
treated all at once with 260 ml benzene sulfonic acid (0.1
Wo 95/13834 2 ~ ~ ~ 2 5 9 PCrrUS94/13387
131
M solution in 75~25 DCM/MeOH, 0 . 026 M, 0 . 67 eq. ) . Reac-
tion proceeded for 10 minutes after which time a TLC in 5~
MeOH in DCM revealed that complete desilylation had
occurred. The reaction was; ~ tPly quenched with 20
ml TEA at which time the solution changed f rom a deep
clear amber color to a light clear yellow color. The
solution was concentrated to a thick oil and then loaded
onto 250 g silica gel equilibrated in 0 . 59~ MeOH in DCM.
The free trityl was removed with the same eluent and the
product was then removed with 6~6 MeOX in DCM. The frac-
tions crnt;3ln;n~ product were pooled and concentrated to
obtain 21.8 g (98.5% pure by XPLC, 9196 yield overall) of
the titled compound.
Exam~le 4 9
Pre~ara~ion of 2 -O-Me UC(5 -ODMT, N4IB~-3'CE Phos~hor-
;:lm; dite Via UC, 3 ' -OH
980 mg 2'-O-Me 17C (5'DMT, 3'0X, N4IBU) MP(R~) dimer
was rendered anhydrous with 3 x 10 ml ACN co-evaporations.
The resulting dry foam was then taken up in 10 ml anhy-
drous ACN and to it was added 325 ~l TEA (2.32 mmol, 2.25
eq. ), followed by dropwise addition (via a 1 ml glass
syr inge ) o f 4 6 0 ~ l 2 ' - cyanoethyl - N, N - di i s opropyl chloro -
phosphoramidite (2 . 06 mmol, 2 . 0 eq. ) . The reaction was
allowed to stir overnight, after which time a TLC and HPLC
showed the reaction to be complete. The reaction mix was
concentrated and loaded onto a 1.5 x 20 cm column contain-
ing 30 g of silica equilibrated in 3:1:1, EtOAc: DCM:ACN,
with 1% TEA. The product was eluted in the same and the
fractions with pure product were pooled and rrmr-pnt~ated
to yield 600 mg of pure amidite.
Pharmaceutical compositions utilizing the compounds
of the present invention, and methods of fo, ll~t;ng the
same, are known in the art, and appropriate composition
and formulation techniques are further described in U.S.
Wo 95113834 : PCTIUS94113387
2~ 7~2~9
132
Patent Application Serial Nos. 08/1~4, 013 and 08/154, 014.
Likewise, applicable methods of using the present com-
pounds and compositionæ, for-example in l;;~n disease
treatment, are disclosed in those applications, which are
5 incorporated herein by reference.
While the foregoing examples and description fiet
forth the preferred embodiments and various ways of
accomplishing the present invention, they are not ;n~n~
to be limiting as to the scope of the invention, which i5
10 as set forth in the following claims. Moreover, it will
be recognized in view of the foregoing disclosure that the
invention embraces alternative ~ 1; ts and structures
that are the lawful equivalents of those described herein.
WO 9~113834 ~ 1 7 6 2 ~ 9 PCTIUS94/13387
~ , . . .
,
133
SEQUENOE LISTING
~1 ) GENERAL INPORMATION:
(i) APPLICA~T: Arnold Jr., Lyle
Reynolds, Mark A
Giachetti, Christina
~ii) TITLE OF INVBNTION: Chimeric ~l;J~n~ 1PAC;~ Compounds
(iii) NUMBER OF SEQUENCES: 2
(iv) U~ UNL/~Y~ ADDRESS:
10 A ~nnl?~q~q~R Lyon h Lyon
B STREET 611 We~t sixth St
C CIT-v: Los Angeles
D STATE: CA
E COUNTRY: U.S A
15 Fl ZIP: 90017
(v) CC'M'UTER READABLE FORM
A MEDIUM TYPE: Floppy disk
B COMPllTER: IBM PC, t i hl rl
C OPER~TING SYST13M: PC-DOS/MS-DOS
D~ SOFTW'ARE: PatemtIn Release #l o, Version #1 2s
(vi ) CURRE~IT APPLICATION DATA
(A) APPLICATION NUMBER: US 08/239,177
(B) FILING DATE: 04-MAY-1994
(C) CLASSIFICATION: 03B1/0712
(viii) ATTORN~EY/AGENT lN~U.. _.IlUN:
(A) NAME: Meier, P~ul EI
(B) REGISTRATION NUMSER: 32,274
(C) ~sr:;~L~u~;/DOCRET NUMBER. Z07/174
(ix) TEL ù IN I r .~7~TO~ lNrU.~ lUN:
(A) TELEPhONE 213/489-1600
(B) TELEFAX: il3/955-0440
(C) TELEX: 67-3510
(2) INFORMATION FOR SEQ ID NO:l:
(i) SEQ-~ENCE ~r7~v~ T~ lr~
A LENGTE~: 15 base pairs
B TYPE: ~ucleic acid
C ~ q: single
Dl TOPOLOGY: linear
(ii) MOLECULE TYPE: other nucleic acid
(iii~ nY~ L:
(iv) ANTI-SENSE: yes
( ix ) FEATURE:
(A) NAME/};EY: CT oligomers 2286-1, 2288-1, 2287-1,
2781-1, 2782-1, 3253-1, 2768-1, 2793-1
2760-1 2784-1, 2795-1, 2765-1, 2792-1
(C) l~llrl~llUN MÉTI~OD: synthesig P~Pr~; C
(D) OTE~ER lNr~ :1, lUN: ~ 1 l y to synthetic RNA
target
WO 95/13834 = ` ,~ i PCI/IJS94113387
~ 7~2~
-
134
~Xi) SEQUENCE DESC~IPTION: SEQ ID NO:1:
~1.1~l~1 CTCTA 1
(3) INFORMATION FOR SEQ ID NO:2:
(i) SEQ~ENCE ~TDDr~
~ A. LENGTH: 15 ba8e Pair8
~ B TYPE: nUC1eiC aCid
I C STRr~T~RI~ ~C: 8ing1e
~'D, TOPOLOGY: 1inear
(ii) MOLECV~E TYPE: Other nUC1eiC aCid
(iii) ~YJ'U~r~11W~L: nO
(iV) ANTI-SENSE: YeS
( iX ) FEATVRE:
(~) NAME/KEY: CU O1igOmer
(C) 1~ 11n1~ TION METHOD: SYntheSiS Prr r;
(Xi) SEQUENCE ~ llUN: SEQ ID NO:2:
- ll CUCUA . 15
(4) INFORMATION FOR SEQ ID NO:3:
(i) SEQVENCE ~7r~r-~'T~I~TCTICS:
IA) LENGTH: 19 baSe Pair8
IB) r'PE: nUC1eiC aCid
C ) S~ r A- ing1 e
D) TOPOLOGY: 1inear
(ii) MOLEC~LE TYPE: Other nUC1eiC aCid
(iii) ~Y~U,~1~L: nO
(iV) ANTI-SENSE: YeS
( iX) FEATURE:
(A) NAME/KEY: O1igOmerS 1634-1, 2570-1
(C) 1L~ L1~1~ Jn METHOD: SYntheSiS PYrPr; a
(D) OTHER INFORMATION: ~ _ 1 Pn1Pn~:~rY tO 5YnthetiC RNA
3 0 target
(Xi) SEQVENCE DESCRIPTION: SEQ ID NO:3:
TAGCTTCCTT AGCTCCTGC 19
(5) INFORMATION FOR SEQ ID NO:4:
(i) SEQUENOE ~r~ 11U~;
IA,~ LENGTH: 19 baSe Pair8
B I TYPE: nUC1eiC aCid
I C I CTAr : Sing1e
D TOPOLOGY: 1inear
(ii) MOLECIILE TYPE: Other nUO1eiC aCid
(iii) ~Y~u~ll~L: nO
(iV) ANTI-SENSE: Ye8
WO 95/13834 2 1 ~ ~ 2 S 9 PCT/US94/133~7
135
( ix ) PEATURE:
~A) NAME/KEY: oligomers 2624-1, Z571-l
(C) LL)~ TION M~THOD: synthesis experiments
(D) OTEIER lNl~ ~J~N:, ,1. Ary to 3ynthetic RNA
target
(xi) SEOUENCE l~L:b~ llUl~: SEQ ID NO:4:
~1.ll.~1~ CATGTTGTC 19
(6) lNr~ T FOR SEQ ID NO:5:
( i ) SE QUENCE ~T~ T cTIcs:
A) LENGI~I: 17 base pairs
i3) TYPE: nucleic acid
C) slrR~NnFn~cc Gingle
D) TOPOLOGY: linear
(ii) MOLECULE TYPE: other nucleic acid
(iii) liY~Ul~llC:AL: no
(iv) ANTI-SENSE: ye6
) FEATURE:
lX (A) NAME/REY: GAG oligomer
(C) lL~wllr'L~TION MET~OD: 3ynthesiG oYr ~;
(xi) SEQUENCE L)~;~GKI~lluN: SEQ ID NO:5:
r~ AGGAAGG 17
(7) 1N~I _ilUN FOR SEQ ID NO:6:
( i ) SE QtlENCE ~T~ T~ G ~ . l L ~
Al LENGTE~: 20 base pairs
~B TYPE: nucleic acid
'C STRG~T~Rn~ - ..c: single
ID TOPOLOGY: linear
(ii) MOLECULE TYPE: other nucleic acid
(iii) hY~ul~lL~L: no
(iv) ANTI-SENSE: yes
( ix ) FEATURE:
(A) NAME/REY: oligomers 3130-~, 2566-1, 2567-1, 2687-
3169-1, 3214-1, 3257-1, 3256-1, 2681-1,
2498-1, 3130-3
(C) LlJ~NlL~LW~TION M~TEIOD: syntheGi5 oYr~or; ' -
(D) OTHER LN~ T~l~: cleave target mRNA and inhibit
mRNA T ~An c l A ~-; nn
(xi) SEQUENOE L)~3CKL~lLUN: SEQ ID NO:6:
GTCTTCCATG CAi~ lCC 20
4 0 ( 8 ) INFORMATION FOR SEQ ID NO: 7:
(i) SEQ~ENCE ~r~T~ T~ilLu~:
A LENGTE~: 24 base pairG
B TYPE: nucleic acid
~ C. sTl~D~Tr~T:nNFcc: single
1:) TOPOLOGY: linear
-
WO 95/13834 ~ PCT/US94/13387
~7~2~
-
136
(ii~ MOLECULE TYPE: other nucleic acid
~iii) ~Y~U~ UAL: no
(iv) ANTI-SENSE: yeq
( ix ) FEATURE:
(A) NAME/KEY: oligomers 32~8-1, 3260-1, XV-l
(C) lL~ TION MET~OD: aynthesis experiments
(D) OT~ER INFORMATION: inhibit target mRNA trAnqlAt;nn
(xi) SEQUENCE L~ :l~Kl~llUN: 8EQ ID NO:7:
CATGGTAGCT TCCTT~GCTC CTGC 24
( 9 ) INFORMATION FOR SEQ ID NO: 8:
(i) SEQ;IENOE r~T7~D~rT~DTCTICS:
A I LENGTII: 24 base pairs
B ~ TYPE: nucleic acid
C ST~ ~: single
D TOPOLOGY: linear
(ii) MOLECULE TYPE: other nucleic acid
(iii) ~Y~Ul~;ll~L: no
(iv) A`wTI-SENSE: yes
( ix ) FEATURE:
(A) NAME/KEY: oligomers 3261-1, 3262-1, XV-2
(C) lY~c.wll~ TION METE~OD: aynthesis experiments
(D) OTE}ER INFORMATION: ~nhibit target mRNA trAnqlAt~nn
(xi) SEQUENCE U~ Kl~llUI~: SEQ ID NO:8:
Wl~l~iU~ GTGATCTTCT TCTC ==~= 24
(10) lN~ --Tr-~r POR SEQ ID NO:9:
(i) SEQ'~ENCE rTT~D~rT~DTqTT~.C
A LENGT~I: 24 base pairs
B TYPE: nucleic acid
c sTDr~n~cc: single
3 0 D TOPOLOGY: linear
(ii) MOLECULE TYPE: other nucleic acid
(iii) IIY~ul~ll AL: no
(iv) A~TI-SENSE: yes
(ix) FEATURE:
(A) NAME/KEY: oligomers 3269-1, 3270-1, XV-6
(C) lU ~ ATION METEIOD: synthe8i8 f.rr~r~ ~ q
(D) OTXER INFORMATION: inhibit target mD~NA trAnq1At;nn
(xi) SEQUENCE l~Kll:'llUN: SEQ ID NO:9:
CACTCAATCA ATGACTAGTC TGCA 24
(11) lNr~ ~Tt-l~ FOR SEQ ID NO:10:
(i) SEQ~ENCE r~D~ -llu~
(A) LENGTE~ base pairs
(B) TYPE: nucleic acid
WO 95tl3834 2 1 7 6 2 S 9 PCTIUS94/13387
137
(c~ sT~ n~qc: single
(D) TOPOLOGY linear
(ii) MOLEC~LE TYPE other nucleic acid
(iii) ~Y~o~ L: no
5 (iv) ANTI-SENSE ye6
( iX ) FEATURE
(A) NAME/KEY 01igOmerq 2323-1, 22~3-1, 2252-1
(C) 11~ 1~IION MET~OD synthesis experiment_
(D) OTEIE}~ mT~)N: . ,1- ~ y to synthetic RNA
target
(Xi) SEQIJENCE l,/~ ~KJ ~llOh SE~ ID NO 10
P~ AGAGT 15
(12) INFORMATION FOR SEQ ID NO:11:
( i ) SEQUENCE r~
,A. LENGTE~ 17 base pairs
B ~ TYPE nucleic acid
C sT~ nNRcc: single
ID TOPOLOGY: linear
(ii) MOLECULE TYPE other llucleic acid
(iii) ~Y~ l~L: no
(iV) ANTI-SENSE: ye~
(iX) FEATURE
(A) NAME/~EY GT oligomers 2517-1, 2516-1
(C) IDENTIFICATION METHOD ~ynthesis '-~T''r;
(D) OTEER l~r~ 1~: 1~ Ary to synthetic RNA
target
(Xi) SEQUENCE L)~ ~Kll'llO~J SEQ ID NO 11
1 GTGTGTA 17
(13) INFORMATION FOR SEQ ID NO:12:
3 0 ( i ) SEQ-JENCE ~
A LENGT~ 19 ba3e pairs
'B TYPE nucleic acid
C, ST~ : single
D. TOPOLOGY linear
(ii) MOLECULE TYPE other nucleic acid
( i i i ) ~ y ~ L: no
- (iV) ANTI-SENSE: yes
( iX) FEATURE
(A) NAME/}~EY oligomers 2688-1, 2662-2
4 0 (C) 1~ ION METIIOD 3ynthegis nT~--1 q
(D) OT~ER lNI''~ mTfl~ ,1 ` y to synthetic RNA
target
(Xi~ SEQUENCE Jl;~Kl~lloDl SEQ ID NO 12
; TTTGAGGTT 19
WO 95/13834 PCT/US94/13387
(14) INFORMATION FOR SEQ ID NO:13:
~i) SEQT~ENCE f~~ rTT;~l~TcTIcs:
A LENGT~I: l9 base pairs
B TYPE: nucleic acid
C l sT~ nN~cc: single
ID: TOPOLOGY: linear
(ii) MOLECULE TYPE: other nucleic acid
(iii) ~YPOTEIETIC~L: no
(iv) AaTI-SENSE yes
(ix) FEATT~RE:
(A) NAME/REY: oligomers 2625-1 2574-1
(C) lJ~rl~:ATION METlIOD: synthesis ~r~n~n~
(D) OTE~ER INFORMATION: complementary to synthetic RNA
target
(xi) SEQUENCE DESCRIPTION: SEQ ID NO:13:
GCTTCCATCT TCCTCGTCC . - 19
( 15 ) INFORMATION FOR SEQ ID NO :14:
(i) SE:QJENCE CT~D~ I 'iLl~'i
A~ LENGTEI: 39 base pa~rs
B TYPE: nucleic acid
C ~ . c: single
D I TOPOLOGY: linear
(ii) MOLECULE TYPE: mR~A
(iii) ~YPOTE~ETICAL: no
(iv) A~TI-SENSE: no
( ix ) FEATURE:
(A) NAME/XEY: wild-type CAT gene portion (as mRNA)
(D) OTHER INFORMATION: pG1036 insert (as TnKNA)
(xi) SEQllENCE 1~ Kl~llUN: SEQ ID NO:14:
3 0 GCCUAUUITCC ~ 7` r GIlrlTT~ I~UGAGAAUA 3 9
( 16 ) INFORMATION FOR SEQ ID NO :15:
(i) SEQUENCE ~T~
A~ LENGTE~: 120 base pairs
B TYPE: nucleic acid
C sT~ n--~C: single
D TOPOLOGY: linear
(ii) MOLECT~LE TYPE: ToKNA
(iii) hY~ ~L: no
(iv) A~TI-SENSE: no
(ix) FEATT~RE:
(A) NAME/XEY: CAT gene portion with intron (as mKNA)
(D) OT~;ER INFORMATION: pG1035 insert (as mRNA)
(xi) SEQ~ENCE DESCRIPTION: SEQ ID NO:15-
WO 95/13834 ~! ~ 7 6 2 ~ 9 : PCTIUS94113387
139
TT~TTrTUrCrTTD U;.~UCCWADA r.~;rTr.DrlTr.Dr TT7~~TT~ rrJc r~rP~~~ TT7~TrDrTUr.~ 60
UTT~'`~Tr~U'`" rD~ rrr~ TT~TTrTTTTrr~r~ ~WWCAGGG riTjTT~TTUr~ 70
(17) lN~( - rrM FOR SEQ ID NO:16:
(i) SEQ',JENCE r~TrnD~
1 A LENGT~: 54 base pairs
B I TYPE: nucleic acid
C~ STD~'-)T~nMrqq: 3inyle
I D'~ TOPOLOGY: linear
(ii) MOLEWLE TYPE: TnD~NA
10 (iii) liY~ol~ll~L: no
(iv) AMTI-SENSE: no
(ix) FEATURE:
(A) NAME/KEY: ~qild-type CAT gene portion ~as T~D~NA)
(xi) SEQUENOE IJ~O~ll:'~lUN: SEQ ID NO:16:
T~UrD~~-`-- rTTT D~~'`7`~~ UA~A ArLTG GAG A~A A~A AUC AW GGA UAU ACC 51
Met Glu LYB LyG Ile Ser Gly Tyr Thr
ACC 54
Thr
ao 1O
(18) INFORMATION FOD~ SEQ ID NO:17:
(i) S~QJENCE rr~nDDr~T.DTqTICS
A LENGTE~: 54 base pairs
B: TYPE : nucleic acid
C sTDDMnr~nMr.~.qq siLyle
D I TOPOLOGY: linear
(ii) MOLEWLE TYPE: T~D.NA
(iii) ~Y~ols~ll~ ~L: no
(iv) AMTI-SEN8E: no
(ix) FEATURE:
(A) NAME/~tEY: pG1040 insert (as ToD~NA)
(Xi) SEQrlJENcE J~ N: SEQ ID NO:17:
rGrr~ rTTDD~~7~rr~ UACC ArTG GAG AAG AAG AlTC ACU GGA UAU ACC 51
Met Glu Lys Lys Ile Ser Gly Tyr Thr
ACC 54
Thr
(19) lNr~l --TOM FOR SEQ ID NO:18:
(i) SEQ~7CE rS~DD~ . ~ TI~
(A) LENGTE}: 24 base pairs
(B) TYPE: nucleic acid
(C) STD~ : single
WO 95/13834 ~ 2 5 9 PCT/US94/13387
140
(D) TOPOLOGY: linear
(ii) MO~ECULE TYPE: other nucleic acid
(iii) nY~uln~ AL: no
(iv) ANTI-SENSE: yes
( ix) FEATURE:
(A) NAME/KEY: oligomers 3264-1, XV-5
(C) l~ lUATION METXOD: synthetic oTn.~rlm~ntc
(D) OThER INFORMATION: inhibit target mRNA trAnqlAt;rn
(xi) SEQUENOE DESCRIPTION: SEQ ID NO:18:
10 CACTCACCTT T~r~rr7~ T~ GGCC 24
(20) INFOKMATION FOR SEQ ID NO:1g:
(i) SEQ-~ENCE rT7DrJ~rTr.'r~TqTICS:
A LENGTX: 24 ~ase pairG
B I TYPE: nucleic acid
IC sTr~r~nNr~lcc: single
IDI TOPOLOGY: linear
(ii) MOLECULE TYPE: other nucleic acid
(iii) XYPOTHETICAL: no
(iv) ANTI-SENSE: yes
20 (ix) FE~TURE:
(A) NAME/KEY: oligomers 3265-1, 3266-1, J~V-7
(C) llr~ ATION METXOD: synthetic experiments
(D) OTHER lNr~ mTr,N: inhibit target mRNA trAnqlAt;rn
(xi) SEQUENOE Ll~sb~KI~IluN: SEQ ID NO:19:
25 CCCTGAGAGA r7 r~ `r~rr, TTCG 24
(21) lN~'Unl_.llUN FOR SEQ ID NO:20:
(i) SEQ-~ENCE r~rT~r~rTr~T~TcTIcs:
A LENGTX: 54 base pairs
B TYPE: nucleic acid
~C ~,~ r~ N~:.`. 5: single
~D TOPOLOGY: linear
(ii) MOLECULE TYPE: m.RNA
(iii) nYl~Ul~~ AL: no
(iv) ANTI-SENSE: no
35 (ix) FEATUKE:
(A) NAME/KEY: pG1042 mismatch insert (as m.~NA)
(D) OTHER lNlC -lUN: controlled mismatch oligomer
screening
(xi) SEQUENCE YJK~UKll~llUN: SEQ ID NO:2~:
~rrJr~r7~r~ UrJl3r-cr~7lr-r UACC ~7G GAC AGG A~G A~U ACG GGA UAU ACC 51
Met Asp Arg ~yc Ile Thr Gly Tyr Thr
WO gS/13834 ~ PCTIUS9~/1338
1~1
ACC
Thr 54
(22) INFO~MATION FOR SEQ ID NO:21:
(i) SEQ~ENCE rT-Tr~n;~rTT.~TiT~TIcs~
A LENGT~I: 54 base pair3
f' B TYPE: nucleic acid
C sTT~r~TnT~nNlpcc: single
ID TOPOLOGY: linear
(ii) MOLECULE TYPE: TCRNA
( iii) t- yJ~ ~L: yes
(iv) ANTI-SENSE: no
(ix) FEATURE:
(A) NAME/}~EY: mismatch insert (as m~NA)
(D) OT}~ER INFORMATION: controlled mismatch oligomer
screening
(xi) SEQT~ENCE Lm;~s~l~ll~N: SEQ ID NO:21:
p~-T-TfTrDr-r~D~- C'TTD anrT T ' '` urrrDTTG~ '' P D~ TTrD rTT~-rT ~TT T~r CACC 54
(23) INFORMPTION FOR SEQ ID NO:22:
(i) SEQTJENCE r~TDT rrTT~T~TC TICS:
A LENGTEI: 54 base pair~
B: TYPE: nucleic acid
C sTT~r : single
D TOPOLOGY: linear
(ii) MOLECULE TYPE: mRNA
(iii) ~iY~Jl~lL~AL: yes
(iv) ANTI-SENSE: no
(ix) FEATURE:
(A) NAT~E/KEY: mismatch insert (as mRNA)
3 0 (D) OT}~ER lNr'~ _.I1UN: controlled mismatch oligomer
screenLng
~xi) SEQUENCE DESC~IPTION: SEQ ID NO:22:
DrTJ13rD~::Dn ~TTT.7~"--T7~r ur~rrDTT~rpr D7~"T7~"~TTrD rTT~ TT~TTDr CACC 54
~24) lNr~ ~TrN FOR SEQ ID NO:23:
3 5 ~ i ) SEQ~ENCE riTT~ n D ' . . .~ I '. 11~:
I A LENGT~: 54 base pair6
B TYPE: nucleic acid
' STT~r ~: single
D TOPOLOGY: linear
~ii) MOLECULE TYPE: mRNA
Y~Clrlr;l l~L: yes
( iv) ANTI - SENSE: no
( ix ) FEATURE:
(A) NAMi~/XEY: mismatch insert (as mRNA)
WO 95/13834 2 ~ ~ 2 ~ 9 PCTIUS94/13387
142
(D) OTdER INFORMDTION: controlled mismatch oligomer
screening
(xi) SEQUENCE Lll:;~Kl~llUN: SEQ ID NO:23:
rrrTqrDr~r. rTTDD--rTDDrr urrrT~TTrr~-- Pr--~r`--7'TTrD rTTr~ATT7~T~r CACC 54
~25) INFORMATION~ FOR SEQ ID NO:24:
(i) SEQ-~ENCE r~TD~Dl . r.~ I .'ill~:
A LENGTd: 54 base pairs
B TYPE: nucleic acid
' C b ~ C: single
~D TOPOLOGY: linear
(ii) MOLECULE TYPE: mRNA
(iii) nY~ULd~~ L: yes
( iv) ANTI - SENSE: no
( ix) FEATURE:
(A) NAME/EEY: mismatch insert (as mRNA)
(D) OTdER INFORMATION: controlled mismatch oligomer
screening
(xi) SEQUENCE DESCRIPTION: SEQ ID NO:24:
Dr.lTr,rDr-7~- rTT~ Trrrr urrrDTT--~- r7~ rTrD rTT--"TT~rT~- CACC 54
(26) INFORMATION FOR SEQ ID NO:25:
(i) SEQ'~ENCE rlT~D- ~ .Ll~
A LENGTd: 54 baoe pairs
B I TYPE: nucleic acid
lc sT~r c: single
2!~ D: TOPOI,OGY: linear
(ii) MOLECULE TYPE: mRNA
(iii) dYfUld~ L: yes
(iv~ ANTI-SENSE: no
( ix) FEATURE:
(A) NAME/KEY: mismatch insert (as mRNA)
(D) OT}~ER INFORMATION: controlled mismatch oligomer
screening
(xi) SEgTJENCE J~ lrllUN: SEQ ID NO:25:
Dr~UGrr~ -- rTT--~--TT~ r TTrrrDTTrr.Dr. D~ rr~rTrD rTT----~rT~rT~-- CACC 54
(27) IN-FOR~TION FOR SEQ ID NO:26:
(i) SEQUBNCE r~TDT~DrTFTlTcTIcs
(A) LENGTd: 21 base pairs
(B) TYPE: nucleic acid
(C) ::, : single
(D) TOPOLOGY: linear
(ii) MOLEC~lLE TYPE: other nucleic acid
( iii ) d~ ~u ~ d~ l~ AL: yes
WO 95/13834 217 6 2 5 9 PCT/US94/13387
-
143
~iv~ ANTI-SENSE: yes
(ix) FEATUE:
(A) NAME/KEY: mismatch oligomer
~D) OTE~ER INFORMATION: mismatch oligomer to target
m.~NA SEQ ID NOS: 21-25
(xi) SEQUENCE DESCRIPTION: SEQ ID NO:26:
CATGGTAGCT TCCTTAGCTC C 21
(28) INFORMATION FOR SEQ ID NO:27:
(i) SEQUENCE ~Tr~rTT~.T~TICS:
A) LENGTE~: 20 base pairs
B) TYPE: nucleic acid
C) ~ .c: single
D) TOPOLOGY: liLear
(ii) MO~ECULE rYPE: other nucleic acid
l~i (iii) XYPOrHETICAL: no
(iv) ANTI-SENSE: no
(ix) F13ATU.~E:
(A) NAME/~EY: PNA target oligomer
(C) ll~ ~TION METHOD: synthetic experiment
(D) OTHER INFORMATION: target ~or oligomers 26B1-1
3214-1
(xi) SEQ~ENCE J~ UN: SEQ ID NO:27:
r~ .TT~ ~TT7~r~T~r~ ~ 20