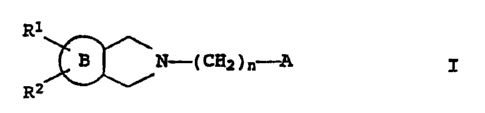Note: Descriptions are shown in the official language in which they were submitted.
CA 02177602 2005-OB-31
1
N-SUBSTITUTED AZABICYCLOALKANE DERIVATIVES AS NEUROLEPTIKA ASF
The invention relates to novel N-substituted azabicycloalkane
derivatives, their preparation and use for preparing pharmaceuti-
cal active compounds.
It has been disclosed that basically substituted butyrophenone
derivatives or benzamide derivatives have neuroleptic or cerebro-
protective activity (US 4 605 655, EP 410 114, DE 12 89 B45,
EP 400 661, DE 29 41 880, EP 190 472, DE 42 19 973).
The observed high affinities to dopamine and~serotonin receptor
s~types appear to play a particular role in this context.
It has now been found that N-substituted 3-azabicycloalkane
derivatives of the formula I
~~N=~ CgZ ) n--~ I ,
R2
where
B is a 3-, 5- or 6-membered ring which is saturated or contains one double
bond and which optionally contains one nitrogen atom and/or one oxygen atom,
R1 is a phenyl group which is unsubstituted or mono- or disub-
stituted .by halogen atoms, cl-caWkYl, trifluoromethyl,
hydroxyl, CI-C4-alkoxy, amino,~monomethylamino, dimethylamino,
cyano or vitro groups,
RZ is a hydrogen atom, a C1-C4-alkyl radical, or a phenyl group
which is unsubstituted or substituted by halogen, methoxy,
hydroxyl or amino,
n is the number 0, 1, 2, 3 or 4,
A is one of the ra~cal.s
CA 02177602 2004-08-26
2~
-CO-RS ar -NR6-CO-R7.
R5. is a phenyl group which is unsubstituted or mono- or disub-
stituted by fluorine, chlorine, bromine , a hydroxyl, nitro,
amino, C1_a-alkanoylamino, Cj_q-alkylamino, Ci-Cq-alkyl or
methoxy group or a naphthyl group which is unsubstituted or
substituted by fluorine or chlorine,
R6 is a hydrogen atom or a methyl group, and
R~ ' is a phenyl group which is mono- or disubstituted by fluo-
rine, chlorine, bromine, Cl-Cq-alkyl, hydroxyl or methoxy or
monosubstituted by nitro, cyano, trifluoromethyl, amine,
C~-Cq-alkylamino or di-C~,-Gq-alkylamino or a thienyl, naph-
thyl, benzothienyl or indenyl group which is unsubstituted or
substituted by fluorine, chlorine or nitro,
and their salts with physiologically tolerable acids, have useful
pharmacological properties.
In the formula T, the substituents Rl to RE and n preferably have
the following meanings:
R1: phenyl, unsubstituted or substituted by fluorine, chlorine,
methoxy, trifluoromethyl, vitro, hydroxyl or amino,
R2: hydrogen, methyl,
n: 2 or 3,
R5: p-fluorophenyl, phenyl, p--chlorophenyl, 1-naphthyl,
WO 95115327 b O z PCT/EP94/03913
3
R6: hydrogen,
R~: phenyl, p-fluorophenyl, o~aminophenyl, o-N-methylaminophenyl,
5-chlorothien-1-yl, 1-naphthyl, 3-indenyl, 3-chloro-1-benzo
thien-2-yl.
The bicyclic ring system in the left moiety of the formula I is
particularly
R1 R1
N ~ N-
RZ R~
R1 R1
'~ .
R~D7\\~N- ,
// Rz~N- ,
Ri
~
N~~N-
~/O
Preferred compounds are in particular those where
R1 is phenyl which is preferably substituted in the p-position
by fluorine or chlorine or in the m-position by fluorine or
chlorine and
R2 is hydrogen or methyl.
The compounds of the formula I according to the invention can be
prepared by reacting a compound of the formula II
Nu-(CHZ)a A II,
where A and n have the~meanings given and Nu is a nucleofugic
leaving group, with a 3-azabicycloalkane derivative of the for-
mula III
Ri
B NB III,
R
WO 95115327 ~ ~ ~ PCf/EP94I03913
Y
4
where B, R1 and R2 have the meaning given above, removing any pro-
tective groups present and if desired converting the compound
thus obtained into the acid addition salt of a physiologically
tolerable acid.
Suitable nucleofugic leaving groups for Nu are preferably halogen
atoms, in particular bromine or chlorine.
The reaction is expediently carried out in the presence of an
inert base, such as triethylamine or potassium carbonate, as acid
acceptor in an inert solvent, such as a cyclic saturated ether,
in particular tetrahydrofuran or dioxane, or a benzene hydrocar-
bon, such as toluene or xylene.
The reaction is in general carried out at from 20 to 150~C, in
particular from 80 to 140~C, and is in general complete within
from 1 to 10 hours.
The compounds of the formula I according to the invention can
either be purified by recrystallization from the customary
organic solvents, preferably from a lower alcohol, such as
ethanol, or by column chromatography.
Racemates can be resolved into the enantiomers in a simple manner
by classical cleavage using optically active carboxylic acids,
eg. tartaric acid derivatives, in an inert solvent, eg. lower
alcohols.
The free 3-azabicycloalkane derivatives of the formula I can be
converted to the acid addition salt of a pharmacologically
tolerable acid in a customary manner, preferably by treating a
solution with an equivalent of the corresponding acid. Pharma-
ceutically tolerable acids are, for example, hydrochloric acid,
phosphoric acid, sulfuric acid, methanesulfonic acid, sulfamic
acid, maleic acid, fumaric acid, oxalic acid, tartaric acid or
citric acid.
The compounds according to the invention have useful pharmaco
logical properties. They can be used as neuroleptics (in partic
ular atypical), antidepressants, sedatives, hypnotics, CNS pro-
tectanta or muscle relaxants. Several of the active qualities can
occur in combination in one compound according to the invention.
Demonstration of the pharmacological action is carried out both
in vivo and in vitro, substance characterization in particular
being possible as a result of the in some cases very high and
selective affinity to receptor subtypes, eg. dopamine Dy, D2, D3
\' ~.. -S'
R'O 95/15327
PCT/EP94ID3913
and especially Dq receptors; serotonin 1A, 1D and 2 receptors,
alpha i and 2 receptors; histamine 1 and muscarine receptors.
The following methods were used for the in vivo characterization:
5
a) Effect on orientation motility
In a new environment, mice show increased exploratory
behavior which is manifested by increased motor activity.
This motor activity is measured in light barrier cages for
0-30 mi.n after the animals (female NMRI mice) have been
placed in the cages.
ED50: dose which reduces the motor activity by 50% in com-
parison with placebo-treated controls.
b) Apomorphine antagonism
Female NMRI mice receive 1.21 mg/kg of apomorphine s.c. At
this dose, apomorphine leads to motor activation which is
manifested by continuous climbing when the animals are kept
in wire mesh cages. The climbing is assessed using a score
(every 2 min for 30 min):
0: animal has four paws on the floor
1: animal has two paws on the wire
Z: animal has four paws on the wire (is climbing).
The climbing behavior can be inhibited by pretreatment with
antipsychotics.
ED50: dose which inhibits the climbing activity of the
animals by 50% in comparison with placebo-treated controls.
c) Methamphetamine antagonism
Female NMRI mice receive 1 mg/kg of methamphetamine p.o. and,
after 30 min, are placed in light barrier cages to measure
the motor activity (2 animals/cage, 4 cages/doae). The test
substances are given orally 30 min before methamphetamine.
The increase in activity due to methamphetamine is calculated
for the period 15 to 60 min after the animals have been
placed in the measurement cages as the difference between
methamphetamine controls and placebo controls and set equal
to 100%. The ED100 is the dose of the test substance which
completely abolishes the increase in activity.
:Y: 2: _ ,,'
WO 95115327 217 7 6,~ p,~ , PC'TIEP94103913
6
d) L-5-HTP antagonism
Female Sprague-Dawley rats receive L-5-HTP in a dose of
316 mg/kg i.p. The animals subsequently develop an excitation
syndrome of which the symptoms
- forepaw treading and
- tremor
are assessed with the aid of a score (0 = not present,
1 = moderate, 2 ~ clearly marked) every 10 min in the period
from 2D to 6D min after L-5-HTP administration. on average, a
score of 17 is achieved after L-5-HTP administration. The
test substances are given p.o. 60 min before L-5-HTP. The
ED50 is calculated as the dose which on average decreases the
control score by 508.
The methods mentioned are suitable for characterizing substances
as antipsychotics; in particular, the inhibition of motor stimu-
lation induced by methamphetamine is regarded as predictive of an
antipsychotic effect. A serotonin-antagonistic effect-may be
shown by the inhibition of the L-5-HTP syndrome, a type of effect
which is characteristic of the atypical neuroleptics.
The novel compounds show a good effect in these tests.
The invention accordingly also relates to a therapeutic composi-
tion, which contains a compound of the formula I or its pharmaco-
logically tolerable acid addition salt as active compound in
addition to customary excipients and diluents, and the use of the
novel compounds in the control of diseases.
The compounds according to the invention can be administered in a
customary manner orally or parenterally, intravenously or intra-
muscularly.
The dosage depends on the age, condition and weight of the
patient and on the mode of administration. As a rule, the daily
dose of active compound is from about 1 to I00 mg/kg of body
weight on oral administration and from O.I to 10 mg/kg of body
weight on parenteral administration.
4D
The novel compounds can be used in conventional solid or liquid
pharmaceutical administration forms, eg. as tablets, film
tablets, capsules, powders, granules, coated tablets, supposi-
tories, solutions, ointments; creams or sprays. These are
prepared in a customary manner. The active compounds can in this
case be processed with the customary pharmaceutical auxiliaries
such as tablet binders, fillers, preservatives, tablet
WO 95115327
pCT/EPg4/03913
r. , ~y r~ . ,
disintegrants, flow regulators, plasticizers, wetting agents,
dispersants, emulsifiers, solvents, release-delaying agents,
antioxidants and/or propellant gases (cf. H. Sucker et al.:
Pharmaaeutische Technologie [Pharmaceutical Technology], Thieme-
5 Verlag, Stuttgart, 1978). The administration forms thus obtained
normally contain the active compound in an amount from 1 to 99%
by weight. ,
The substances of the formulae II and III required as starting
10 substances far the synthesis of the novel compounds are known or
can be synthesized from similar starting materials according to
the greparation methods described in the literature.
The following examples serve to illustrate the invention:
Preparation of the precursors
A. exo-2-Phenyl-3-methyl-1,5-cis-3,7-diazabicyclo[3.3.0]octane
a) 6-Phenyl-7-methyl-1,5-cis-3,7-diazabicyclo[3.3.0]-
octane-2,4-dione
17.8 q (200 mmol) of sarcosine, 15.2 ml (150 mmol) of
benzaldehyde and 9.7 g (100 mmol) of maleimide were sus-
pended in 500 ml of toluene and refluxed in a Dean and
Stark apparatus for 3 h. A further 17.8 g (2D0 mmol) of
sarcosihe and 15.2 ml (150 mmol) of benzaldehyde were
then added and the mixture was refluxed for a further
hour. After cooling, 50 g of sodium sulfate were added,
stirred for a few minutes and filtered off. The filtrate
was concentrated and the residual viscous oil (37.2 g)
was purified by column chromatography (silica gel, eluent
dichloromethane/methanol 98:5). In this manner, 4.2 g
(18%) of enriched endo adduct (endo:exo = 80:20) and
10.7 g (47%) of enriched exo adduct (exo:endo = 80:20)
were obtained.
b) exo-2-Phenyl-3-methyl-1,5-cis-3,7-diazabicyclo[3.3.0]-
octane
A solution of 8.5 g (37 mmol) of enriched exo-6rphenyl-
7-methyl-1,5-cis-3,7-diazabicyclo[3.3.0]octane-2,4-dione
in 130 ml of absolute tetrahydrofuran was added dropwise
at room temperature with good stirring in the course of
25 min to a suspension of 7.0 g (185 mmol) of lithium
aluminum hydride in 180 ml of absolute tetrahydrofuran.
After the slightly exothermic reaction had subsided, the
R'O 95115327 ~ ~ '~., PCTIEP94/03913
8
mixture was stirred at room temperature for a further
18 h. While cooling in ice, 70 ml of ten percent sodium
hydroxide solution were then added dropwise with vigorous
stirring and the mixture was allowed to come to room tem-
perature with stirring. The precipitated hydroxides were.
filtered off with suction and washed several times with w
tetrahydrofuran, and the combined filtrates were concen-
trated. 6.3 g (84%) of a pale oil were isolated.
8. endo-2-Phenyl-3-methyl-1,5-cis-3,7-diazabicyclo[3.3.0]octane
In a similar manner to procedure A.b), 3.0 g (81%) of cloudy
oil were obtained from 4.2 g (18 mmol) of enriched
endo-6-phenyl-7-methyl-I,5-cis-3,7-diazabicyclo[3.3.0]-
octane-2,4-dione and 3.5 g (91 mmol) of lithium aluminum
hydride.
C. 3-Phenyl-1,5-cis-3,7-diazabicyclo[3.3.0]octane
a) 7-Benzyl-3-phenyl-1,5-cis-3,7-diazabicyclo[3.3.0]-
octane-2,4-dione
20.1 g (100 mmol) of N-benzylglycine hydrochloride, 7.5 g
(250 mmol) of paraformaldehyde and 8.7 g (50 mmol) of
N-phenylmaleimide were suspended in 500 ml of toluene and
17.4 ml (100 mmol) of N-ethyldiisopropylamine were
finally added. The reaction mixture was refluxed for
min in a Dean and Stark apparatus and then filtered
through sodium sulfate, and the filtrate was concen
30 trated. The residual-viscous oil (18.6 g) was purified by
column chromatography (silica gel, eluent dichlorome-
thane). 8.7 g (56%) of a pale oil were obtained.
b) 7-Benzyl-3-phenyl-1,5-cis-3,7-diazabicyclo[3.3.0]octane
A solution of 8.7 g (28 mmol) of 7-benzyl-3-phenyl-
1,5-cis-3,7-diazabicyclo[3.3.0]octane-2,4-dione in 100 ml
of absolute tetrahydrofuran was slowly added dropwise at
room temperature to a suspension of 2.65 g (7I mmol) of
lithium aluminum hydride in 75 mI of absolute tetrahydro-
furan. The mixture was then stirred under reflux for a
further 2 h. Then, while cooling in ice, 30 ml of ten
percent sodium hydroxide solution were slowly added drop-
wise, and the precipitated hydroxides were filtered off
with suction. Washing with tetrahydrofuran and concentra-
tion of the combined filtrates afforded 7.2 g of a cloudy
oil, which was purified by column chromatography (silica
WO 95/15327
PCTYEP94/03913
9
gel, dichloromethanelmethanol 97:3). Yield: 5.8 g (73%)
of clear oil.
c) 3-Phenyl-1,5-cis-3,7-diazabicyclo[3.3.0]octane
5.8 g (21 mmol) of 7-benzyl-3-phenyl-1,5-cis-3,7-diaza-
bicyclo[3.3.0]octane were dissolved in 170 m1 of metha-
nol, and 0.7 g of palladium on carbon (10%) was added. A
solution of 6.6 g (104 mmol) of ammonium formate in 7 ml
of water was then added dropwise With stirring. The mix-
ture Was then stirred for 3 h at 50~C, a further 0.5 g of
palladium on carbon (10%) was subsequently added, and the
mixture was stirred for a further hour at 50'C. The cata-
lyst was filtered off with suction and washed well with
methanol, and the combined filtrates were concentrated to
dryness. The residue was taken up in water, adjusted to
pH 9-10 and extracted three times with dichloromethane.
Drying and concentration of the organic phase afforded
3.0 g of white solid, which was digested in a little
ether. Filtering off with suction and drying yielded
1.7 g (43%) of colorless, fine crystals.
D. exo-6-Phenyl-3-azabicyclo[3.1.0]hexane
a) cis-1,2-bis(Hydroxymethyl)traps-3-phenylcyclopropane
A solution of 20.0 g (85 mmol) of dimethyl trans-
3-phenyl-cis-1,2-cyclopropanedicarboxylate in 250 ml of
absolute tetrahydrofuran was slowly added dropwise to a
suspension of 7.9 g (213 mmol) of lithium aluminum
hydride in 150 ml of absolute tetrahydrofuran while
cooling in ice at O~C. The mixture was slowly allowed to
come to mom temperature and was stirred for 18 h. Then,
70 ml of ten percent sodium hydroxide solution were
slowly added dropwise while cooling in ice, and the
precipitated hydroxides were filtered off with suction
and washed with tetrahydrofuran. Concentration of the
filtrate yielded 14.7 g (97%) of viscous, yellow oil.
b) cis-1,2-bis(Methanesulfonyloxymethyl)traps-3-phenylcyclo-
propane
A solution of 14.7 g (82 mmol) of cis-1,2-bis(hydroxy- '-
methyl)tram-3-phenylcyclopropane in 70 ml of absolute
pyridine was added dropwise at -5~C to a solution of
32.2 g (281 mmol) of methaneaulfonyl chloride in 350 ml
of absolute pyridine such that the internal temperature
21776 r~ ~ ' r-',
R'O 95115327 7~,~ l i ~ ~ PCTYEP94103913
f~"' i' i
did not rise above O~C and the mixture was stirred for a
further 3 h at -S~C. The cold reaction mixture was then
poured onto ice-water to which 60 ml of conc. sulfuric
acid had previously been added. The mixture was stirred
5 for a further 1 h, and the supernatant solution was
decanted from the oily precipitate deposited, the latter
was taken up in a little dimethylformamide and this
solution was poured onto ice-water with stirring. After
stirring for 1 h, the fine crystalline precipitate was
1D filtered off with suction, washed with water and dried.
19.6 g (78%) of pale powder were obtained.
c) 3-(4-Methoxyphenylmethyl)exo-6-phenyl-3-azabicyclo-
[3.1.0]hexane
5.0 g (I6.6 mmol) of cis-I,2-bis(methanesulfonyloxy-
methyl)-trans-3-phenylcyclopropane were introduced into
6.8 g (50 mmol) of Q-methoxybenzylamine and the mixture
was heated at 100~C for 2 h with good stirring. After
cooling, the mixture was dissolved in methylene chloride,
and the organic phase was washed twice with water and
concentrated after drying with sodium sulfate. The crude
product (4.9 g) was purified by column chromatography
(silica gel, eluent methylene chloride/methanol 99:1).
2.2 g (48%) of product were isolated as a yellow oil.
d) exo-6-Phenyl-3-azabicyclo[3.1.0]hexane
2.2 g (7.9 mmol) of 3-(4-methoxyphenylmethyl)exo-
5-phenyl-3-aaabicyclo[3.1.0]hexane were dissolved in
70 ml of methanol and 0.6 g of palladium on carbon (1D%)
was added. A solution of 2.5 g (39 mmol) of ammonium
formats in 3 ml of water was then added dropwise with
stirring. The mixture was then stirred for 1 h at SO~C.
The catalyst was filtered off with suction and washed
well with methanol, and the combined filtrates were con-
centrated to dryness. The residue was taken up in water,
adjusted to pH 9-10 and extracted three times with
dichloromethane. Drying and careful concentration of the
organic phase at a maximum bath temperature of 30~C
afforded 1.1 g (88%) of product as a yellow oil.
E. 6-(4-Fluorophenyl)-1,5-cis-3-azabicyclo[3.3.D]oct-5-one
a) 3-Benzyl-6-(4-fluorophenyl)-6-hydroxy-I,5-cis-3-azabi-
cyclo[3.3.0]octane
.,
R'O 95115327 "~ .. ~ PCT/EP94/03913
11
First I.4 g (56 mmol) of magnesium turnings and then a
solution of 9.4 g (54 mmol) of 4-bromo-1-fluorobenzene in
55 ml of absolute tetrahydrofuran were added dropwise un-
der nitrogen to 20 ml of- absolute tetrahydrofuran. After
the weakly exothermic reaction had subsided, the mixture
was stirred for a further 1 h. A solution of I0.5 g
(49 mmol) of 3-benzyl-6-oxo-1,5-cis-3-azabicyclo[3.3.0]-
octane (R. Miyajima, M. Takemoto and R. Achiwa, Chem.
Pharm. Bull. 39 (I99I), 3175) in 40 ml of absolute tetra-
hydrofuran was then added drapwise and the mixture was
subsequently refluxed for 5 h. 50 ml of a saturated
ammonium chloride solution were then added dropwise while
cooling in ice, and the precipitated hydroxides were
filtered off with suction and washed with tetrahydro-
furan. The combined filtrates were concentrated, and the
residue was taken up in water, adjusted to pH 11 with ten
percent sodium hydroxide solution and extracted twice
with dichloromethane. The organic phase was washed once
with saturated sodium chloride solution, dried over
sodium sulfate and concentrated. The residual oil
(14.8 g) was purified by column chromatography (silica
gel, eluent dichloromethane/methanol 98.5:1.5). 11.5 g
(75%) of yellow oil were obtained.
b) 6-(4-Fluorophenyl)-6-hydroxy-1,5-cis-3-azabicyclo[3.2.0]-
octane
9.0 g (29 mmol) of 3-benzyl-6-(4-fluorophenyl)-6-hydroxy-
1,5-cis-3-aaabicyclo[3.3.0]octane were dissolved in
250 ml of methanol and 2.0 g of palladium on carbon (IO%)
were added. A solution of 9.1 g (145 mmol) of ammonium
formate in 11 mI of water was then added dropwise with
stirring and the mixture was refluxed for a further 3 h
after addition was complete. The catalyst was filtered
off with suction and washed well with methanol, and the
combined filtrates were concentrated to dryness. The
residue was taken up in water, adjusted to pH 9-10 with
ten percent sodium hydroxide solution and extracted twice
with dichloromethane. Drying and concentration of the
organic phase yielded 5.4 g (84%) of yellowish oil.
c) 6-(4-Fluorophenyl)-1,5-cis-3-azabicyclo[3.3.0]oct-6-ene
6.9 g (31 mmol) of 6-(4-fluorophenyl)-6-hydroxy-1,5-cis-
3-aaabicyclo[3.3.0]octane were taken up in 50 ml of half- -
concentrated hydrochloric acid and the mixture was
refluxed for 5 h. It was then diluted with water while
W095/15327 ~ ~ PCT/EP94/03913
~,~ ~~~,~~ 12
cooling in ice, adjusted to pB 11 with concentrated
sodium hydroxide solution and extracted twice with
dichloromethane. Drying and concentration of the organic
phase yielded a dark oil (5.9 g), which was purified by
column chromatography (silica gel, eluent methanol/
aqueous ammonia solution 95:5). 5.3 g (84%) of brown oil
were obtained.
F. 6-(4-Fluorophenyl)-1,5-cis-3-azabicyclo[3.3.0]octane
5.3 g (26.1 mmol) of 6-(4-fluorophenyl)-1,5-cis-3-azabicyclo-
[3.3.0]oct-6-ene were dissolved in 100 ml of methanol and
1.0 g of palladium on carbon (10%) was added. The reaction
mixture was catalytically hydrogenated under normal con-
ditions. The catalyst was filtered off with suction and
washed well with methanol, and the combined filtrates were
concentrated to dryness. 4.4 g (82%) of product was isolated
as an exo/endo diastereomer mixture, which was separated by
column chromatography (silica gel, eluent methanol/ammonium
hydroxide 90:10).
G. exo-7-Phenyl-1,5-cis-3-azabicyclo[3.3.0]octane
A reaction mixture of 9.9 g (5D mmol) of 3-benzyl-1,5-cis-
3-azabicycla[3.3.0]oct-6-ene (K. Miyajima, M. Takemoto and
R. Achiwa, Chem. Pharm. Bull. 39 (1991) 3175), 14.0 ml
(125 mmol) of iodobenzene, 0.9 g (4.0 mmol) of palladium(II)
acetate, 2.1 g (8.0 mmol) of triphenylphosphine, 4.3 g
(50 mmol) of piperidine and 2.3 g (50 mmol) of formic acid in
100 ml of dimethylformamide was heated at BO~C for 6 h with
good stirring. After concentrating the mixture in an oil pump
vacuum, the residue was partitioned between water and
methylene chloride, the mixture was acidified with ten per-
cent hydrochloric acid, and the organic phase was concen-
trated after drying with sodium sulfate. The crude product
was purified by column chromatography (silica gel, eluent
ethyl acetate/n-hexane 1:1). 3.6 g (26%) of the N-benzyl
derivative were isolated and converted by catalytic hydro-
genation in a similar manner to Example F to the final
product (yellow oil).
The following can be prepared in a similar manner (see
Example O):
exo-7-(p-fluorophenyl)-I,5-cis-3-azabicyclo[3.3.0]octane
wo 9sns3z7
7 6 0 2 PC'f/EP94/03913
13
H. endo-6-p-Fluorophenyl-1,5-cis-3-azabicyclo[4.3.0]nonane
a) endo-3-p-Fluorophenylcyclohex-4~ne-cis-1,2-dicarboxylic
anhydride
53.0 g (358 mmol) of trana-1-p-fluorophenyl-1,3-butadiene
in 100 ml of toluene were slowly heated to 100~C with
34.3 g (350 mmol) of maleic anhydride while stirring well
and the mixture was kept at this temperature for 1.5 h.
After cooling, the mixture was concentrated to one half
and the product was allowed to crystallize while cooling
in an ice bath. The crystals were filtered off with suc-
tion and washed with a little cold toluene. 59 g (69%) of
product of m.p. B8-90~C were isolated.
b) cis-3-p-Fluorophenyl-cis-1,2-bis(hydroxymethyl)-
4-cyclohexene
A solution of 12.0 g (49 mmol) of endo-3-p-fluorophenyl-
cyclohex-4-ene-cis-1,2-dicarboxylic anhydride in 60 ml of
tetrahydrofuran was added dropwise at room temperature
and with good stirring to 3.5 g (92 mmol) of lithium
aluminum hydride in 170 ml of absolute tetrahydrofuran in
the course of 45 min. After stirring for 1.5 h, the
mixture was refluxed for a further 2 h. After cooling,
ten percent sodium hydroxide solution was carefully added
dropwise while cooling in ice and with good stirring, and
the precipitated hydroxides were filtered off with
suction. The filtrate was concentrated to dryness, and
the residue was partitioned between ten percent sodium
hydroxide solution and methyl t-butyl ether. The aqueous
phase was re-extracted twice with methyl t-butyl ether,
and the organic phase was then concentrated after drying
with sodium sulfate. 8.9 g (77%) of product were isolated
as a clear oil.
c) cis-3-p-Fluorophenyl-cis-1,2-bis(methanesulfonyloxy-
methyl)-cyclohex-4-ene
19.7 g (84 mmol) of cis-3-p-fluorophenyl-cis-1,2-bis-
(hydroxymethyl)cyclohex-4~ne in 70 ml of pyridine were
added dropwise at O~C with good stirring to a solution of
28.6 g (250 mmol) of methanesulfonyl chloride in 100 ml
of pyridine and the mixture was stirred for 2 h at O~C.
It was subsequently poured onto ice-water into which
64 ml of concentrated sulfuric acid had been introduced,
and the mixture was extracted twice with methylene
~j7~~o
WO 95/15327 ~ PCT/EP94/039I3
ar~~~'a .
14
chloride. The organic phases were washed twice with ten
percent sulfuric acid and concentrated after drying With
sodium sulfate. 30.3 g (92%) of product were isolated as
a pale oil.
d) 3-Benzylendo-6-p-fluorophenyl-1,5-cis-3-azabi-
cyclo(4.3.0]-non-7-ene
21.8 g (56 mmoI) of cis-3-p-fluorophenyl-cis-1,2-bis-
(methanesulfonyloxymethyl)cyclohex-4~ne were introduced
in portions into 20 ml (183 mmol) of benzylamine with
good stirring (exothermic reaction). The mixture was
subsequently heated at 130~C for a further 2 h. After
cooling, 200 ml of methyl t-butyl ether were added to the
reaction mixture, which was stirred until it crystal-
lized. After filtering off the crystals with suction and
washing with methyl t-butyl ether, the filtrate was
washed twice with aqueous ammonia solution and the
organic phase was concentrated after drying with sodium
sulfate. The crude product (16.5 g) was purified by
column chromatography (silica gel, eluent ethyl acetate/
n-hexane 6:4). 10.5 g (6I%) of product were isolated as a
pale oil.
e) endo-6-p-Fluorophenyl-1,5-cis-3-aaabicyclo(4.3.0]nonane
10.0 g (32 mmol) of 3-benzyl-endo-6-p-fluorophenyl-
1,5-cis-3-azabicyclo[4.3.0]non-7-ene in 200 ml of metha-
nol were catalytically hydrogenated at room temperature
in the presence of 1.3 g of palladium on carbon (10%).
After filtering off the catalyst with suction and washing
with methanol, 7.8 g of crude product were isolated after
concentration as a pale oil which was purified by column
chromatography (silica gel, eluent methanol/agueous
ammonia solution 85:15). 4.6 g (66%) of product of
m.p. 76-78~C were isolated.
The following can be prepared in a similar way:
f) endo-6-phenyl-1,5-cis-3-azabicyclo(4.3.0]nonane
g) endo-6-p-trifluoromethylphenyl-1,5-cis-3-azabicyclo-
[4.3.0]nonane
-,.y . n ~'~
WO 95!15327 C 17 7 6 0 2 PCT/EP94/03913
I. endo-6-p-Fluorophenyl-1,5-cis-3-azabicyclo[4.3.0]non-7-ene
ml of ammonia were injected into a 0.3 1 stirred autoclave
containing 7.6 g (19.4 mmol) of cis-3-p-fluorophenyl-
5 cis-1,2-bis-(methanesulfonyloxymethyl)cyclohex-4-ene in
100 ml of toluene and the mixture was heated at 150~C for 5 h
under autogenous pressure. The reaction mixture was then
poured onto ice-water and the organic phase was washed with
water after filtering off the insoluble components With suc-
10 tion. After drying and concentration, 4.4 g of crude product
were isolated and purified by column chromatography (silica
gel, eluent methanol/aqueous ammonia solution 85:15). 0.9 g
(21%) of product were isolated as a colorless oil.
15 R. 6-p-Fluorophenyl-1,5-cis-3-azabicyclo[4.3.0]non-6-ene
a) endo-3-p-Fluorophenylcyclohex-4-ene-cis-1,2-dicarboximide
50.0 g (338 mmol) of trans-1-p-fluorophenyl-1,3-butadiene
20 in 100 ml of toluene were slowly heated to 100~C with
32.0 g (330 mmol) of maleimide while stirring well and
the mixture was kept at this temperature for 2 h. After
cooling, it was concentrated to one half and the product
was allowed to crystallize while cooling in an ice-bath.
The crystals were filtered off with suction and washed
with a little cold toluene. 69.7 g (86%) of product of
m.p. 184-186~C were isolated.
b) 3-p-Fluorophenylcyclohex-3-ene-cis-1,2-dicarboximide
9.8 g (40 mmol) of endo-3-p-fluorophenylcyclohex-4-ene-
cis-1,2-dicarboximide in 100 ml of dimethylformamide were
treated in portions with 2.4 g (80 mmol) of sodium
hydride (80%) with good stirring (exothermic reaction).
The mixture was stirred for a further 2 h at 45~C and
subsequently poured onto ice-water after cooling. 100 ml
of methyl t-butyl ether were added after acidifying with
ten percent hydrochloric acid and the mixture was stirred
vigorously. The pale solid was filtered off with suction,
washed with a little methyl t-butyl ether and water and
dried under reduced pressure at SO~C. The crude product
was purified by column chromatography (silica gel, eluent
methylene chloride/methanol 97:3). 6.7 g (68%) of product
(main polar zone) of m.p. 197-199~C were isolated.
2~~7f0~.
WO 95115327 PCT/EP94/03913
Z6
c) 6-p-Fluorophenyl-1,5-cis-3-azabicyclo[4.3.0]non-6-ene
A solution of 4.0 g (16.4 romol) of 3-p-fluorophenyl-
cyclohex-3-ene-cis-1,2-dicarboximide in 50 ml of tetra-
s hydrofuran was added dropwise at raom temperature and
with good stirring to 1.96 g (51 mmol) of lithium
aluminum hydride in 60 ml of absolute tetrahydrofuran in
the course of 45 min. After stirring for 1.5 h the mix-
ture was refluxed for a further 3 h. After cooling, ten
percent sodium hydroxide solution was carefully added
dropwise with ice-cooling stirring and the precipitated
hydroxides were filtered off with suction. The filtrate
was concentrated to dryness, and the residue was parti-
tioned between water and methyl t-butyl ether at pH a 10.
The organic phase was extracted with five percent hydro-
chloric acid, rendered alkaline with concentrated sodium
hydroxide solution and extracted twice with methyl
t-butyl ether. After drying and concentration, 1.6 g
(45%) of product were obtained as a pale oil.
ao
The following can be prepared in a similar manner:
d) 6-phenyl-1,5-cis-3-azabicyclo[4.3.0]non-6-ene
L. exo-6-p-Fluorophenyl-1,5-cis-3-aaabicyclo[4.3.0]nonane
a) exo-3-p-Fluorophenylcyclohex-4-ene-cis-1,2-dicarboximide
15.0 g (6I mmol) of endo-3-p-fluorophenylcyclohex-4-ene-
cis-1,2-dicarboximide in 100 ml of dimethylformamide were
treated in portions with good stirring with 12.9 g
(94 mmol) of finely pulverized potassium carbonate and
the mixture was kept for 2 h at 100~C and then poured
onto iceJwater after cooling. After acidifying with con-
centrated hydrochloric acid, it was extracted with methyl
t-butyl ether and the organic phase was washed with ten
percent hydrochloric acid. After drying and concentra-
tion, 18.1 g of crude product were isolated, which was
washed with 50 ml of ether by stirring. 12.1 g (81%) of
colorless crystals of m.p. 119-12I~C were isolated. The
configuration of the product was confirmed by the crystal
structure analysis.
R'O 95/15327 ~ ~ ~ ~ PCTBP94103913
17
b) exo-6-p-Fluorophenyl-1,5-cis-3-azabicyclo[4.3.0]non-7-ene
A solution of 6.5 g (26 mmol) of 3-p-fluorophenyl-
cyclohex-4-ene-cis-1,2-dicarboximide in 60 ml of tetra-
s hydrofuran was added dropwise at room temperature and
with good stirring to 3.2 g (84 mmol) of lithium aluminum
hydride in 120 ml of absolute tetrahydrofuran within the
course of 45 min. After stirring for 1.5 h, the mixture
was refluxed for a further 3 h. After cooling, ten
percent sodium hydroxide solution was carefully added
dropwise with good stirring and with ice cooling and the
precipitated hydroxides were filtered off with suction.
The filtrate was concentrated to dryness, and the residue
was partitioned between ten percent hydrochloric acid and
methyl t-butyl ether. The aqueous phase was washed with
methyl t-butyl ether, rendered alkaline with concentrated
sodium hydroxide solution and extracted twice with methyl
t-butyl ether. After drying and concentration, 3.6 g
(64%) of product were isolated as a pale oil.
c) exo-6-p-Fluorophenyl-1,5-cis-3-azabicyclo[4.3.0]nonane
Obtained by catalytic hydrogenation of exo-6-p-fluoro
phenyl-1,5-cis-3-azabicyclo[4.3.0]non-7-ene according to
the procedure of Example F: yield 89%.
The following can be prepared in a similar manner:
d) exo-6-phenyl-1,5-cis-3-azabicyclo[4.3.0]nonane
M. exo-7-Phenyl-1,5-cis-3-azabicyclo[4.3.0]non-8-ene
a) exo-4-Phenylcyclohex-5-ene-cis-1,2-dicarboximide
A mixture of 15.1 g (100 mmol) of cyclohex-4-ene-
cis-1,2-dicarboximide, 28.0 ml (250 mmol) of iodobenzene,
I.8 g (8.0 mmol) of palladium(II) acetate, 2.1 g
(8.0 mmol) of triphenylphosphine, 8.5 g (100 mmol) of
piperidine and 4.6 g (100 mmol) of formic acid in 200 ml
of dimethylformamide was heated at 80~C for 6 h with good
stirring. After concentration of the mixture in an oil
pump vacuum, the residue was partitioned between water
and methylene chloride, the mixture was acidified with
ten percent hydrochloric acid and the organic phase was
concentrated after drying with sodium sulfate. The crude
product (31 g) was purified by column chromatography
(silica gel, eluent ethyl acetate/n-hexane 1:1). 2 main
R'0 95115327 ~ ~ ~ ~ ~ ~~~ -~: ~ ' ) ,'y PCTIEP94I03913
18
fractions were isolated: the polar zone yielded 3.9 g
(I7%) of product as a yellowish oil.
b) exo-7-Phenyl-1,5-cis-3-aaabicyclo[4.3.0]non-8-ene
1.0 g (26 mmol) of lithium aluminum hydride was added in
portions to 3.0 g (17.2 mmol) of exo-4-phenylcyclo-
hex-5-ene-cis-1,2-dicarboximide in 150 ml of tetrahydro-
furan at room temperature and with good stirring. After
stirring for 0.5 h, the mixture was refluxed for a
further 3 h. After cooling, ten percent sodium hydroxide
solution was carefully added dropwise with good stirring
and with ice cooling and the precipitated hydroxides were
filtered off with auction and washed with tetrahydro-
furan. The filtrate was concentrated to dryness, and the
residue was partitioned between water and methyl t-butyl
ether at pH ~ 10. After drying and concentration, 3.2 g
of crude product were isolated as a dark oil. The crude
product was purified by column chromatography (silica
gel, eluent methylene chloride/methanol 1:1). 1.1 g (32%)
of product were isolated as a pale oil.
N. 7-Phenyl-3-azabicyclo[4.3.0]non-1-ene
a) 4-Phenylcyclohex-1-ene-1,2-dicarboximide
The non-polar main fraction from column chromatography of
Example M.a) yielded 3.2 g (15%) of product as a color-
less oil/crystal mixture.
b) 7-Phenyl-3-azabicyclo[4.3.0)non-1-ene
In a similar manner to Example M.b), reduction with
lithium aluminum hydride yielded 0.7 g (25%) of product
as a pale oil.
O. exo-7-p-Fluorophenyl-1-,5-cis-3-azabicyclo[4.3.0]nonane
a) 4-p-Fluorophenylcyclohex-1-ene-1,2-dicarboximide
A reaction mixture of 45.3 g (300 mmol) of cyclo-
hex-4-ene-cis-1,2-dicarboximide, 82.5 ml (750 mmol) of
p-bromofluorobenaene, 5.4 g (24 mmol) of palladium(II)
acetate, 6.3 g (24 mmol) of triphenylphosphine, 29.7 ml
(300 mmol) of piperidine and 1I.4 ml (300 mmol) of formic
acid in 600 ml of dimethylformam;de was heated at 95 to
IOO~C,for 6 h with good stirring. After concentrating the
W095I15327 ~ ~ ~ ~ 6 Q2'~,, ~ ~ 'pCT/EP94103913
19
mixture in an oil pump vacuum, the residue was parti-
tinned between water and methylene chloride, the mixture
was acidified with ten percent hydrochloric acid, the or-
ganic phase was washed with ten percent hydrochloric acid
and the organic phase was concentrated after drying with
sodium sulfate. The crude product (71 g) was stirred in
350 ml of ethyl acetate and the.brown solid was filtered
off with suction and washed with ethyl acetate. Concen-
tration of the filtrate afforded 56 g of crude product,
which was purified by column chromatography (silica gel,
eluent ethyl acetate/n-hexane 40:60). 2 main fractions
were isolated: the non-polar zone yielded 13.3 g of
product which was washed with a 1:1 mixture of ethyl
acetate and n-hexane by stirring. 8.9 g (12%) of product
Z5 of m.p. 136-137~C were isolated.
b) 7-p-Fiuorophenyl-3-azabicyclo[4.3.0]non-I-ene
4.5 g (118 mmol) of lithium aluminum hydride were added
in portions to 9.4 g (38 mmol) of 4-p-fluorophenylcyclo-
hex-1-ene-1,2-dicarboximide in 3DD ml of tetrahydrofuran
during the course of 45 min at room temperature and with
good stirring. After stirring for 0.5 h, the mixture
was
refluxed for a further 6 h. After cooling, ten percent
sodium hydroxide solution was carefully added dropwise
with good stirring and with ice cooling and the precip-
itated hydroxides were filtered off with suction and
washed with tetrahydrofuran. The filtrate was concen-
trated to dryness and the residue was partitioned between
water and methyl t-butyl ether at pH = 10. The organic
phase was subsequently extracted twice with ten percent
hydrochloric acid and the acidic aqueous phase was ren-
dered alkaline again after this with concentrated sodium
hydroxide solution. It was then extracted twice with
methyl t-butyl ether. After drying and concentration,
1.8 g (22%) of product were isolated as a pale oil.
c) exo-4-p-Fluorophenylcyclohex-5-ene-cis-1,2-dicarboximide
The polar main fraction from column chromatography of
Example O.a) yielded 9.2 g of product, which was digested
in a little ether: 4.3 g (6%) of product of m.p. 139 to
142~C were isolated.
WO 95115327 2 ,' T 7 S ~~'~~ -' PCTIEP94103913
d) exo-7-p-Fluorophenyl-1,5-cis-3-azabicyclo[4.3.0]-
non-8-ene
In a similar manner to Example O.b), reduction with
5 lithium aluminum hydride yielded 2.4 g (63%) of product
as a pale oil.
e) exo-7-p-Fluorophenyl-1,5-cis-3-azabicyclo[4.3.0]nonane
10 Catalytic hydrogenation in a similar manner to Example F
yielded 2.2 g (92%) of product as a yellowish oil.
P. 6-Phenyl-1,5-cis-8-oxa-3,7-diazabicyclo[3.3.0]oct-6-ene
15 5.0 g (41 mmol) of benzaldehyde oxime in 50 ml of methylene
chloride were treated with 6.8 g (41 mmoI) of 1-trifluoro-
acetyl-3-pyrroline. 36.9 g (74 mmol) of a five percent sodium
hypochlorite solution were then added dropwise while stirring
well (exothermic reaction). After stirring for 2 h, the
20 mixture was poured onto ice-water, rendered alkaline with
concentrated ammonia solution and extracted twice with
methylene chloride. The combined organic phases were washed
with water and concentrated after drying with sodium sulfate.
The crude product (13.1 g) was purified by column chroma-
tograghy (silica gel, eluent methylene chloride). 3.5 g (30%)
of the trifluoroacetyl derivative were isolated and were
hydrolyzed during a further 30 min at room temperature in 2 N
methanolic sodium hydroxide solution to give the final prod-
uct of m.p. 78-BO~C.
Reduction with sodium cyanoborohydride afforded exo/endo-
6-phenyl-1,5-cis-8-oxa-3,7-diazabicyclo[3.3.0]octane as a
pale oil.
The following can be prepared in a similar manner:
6-p-fluorophenyl-1,5-cis-8-oxa-3,7-diazabicyclo(3.3.0]oct-6-ene
exo/endo-6-p-fluorophenyl-1,5-cis-B-oxa-3,7-diazabicyclo[3.3.0]-
octane
W095115327 ~ ;,~'~~ PCT'/EP9q~03913
21
Example 1
N-(2-[exo-6-Phenyl-7-methyl-1,5-cis-3,7-diazabicyclo[3.3.0]octan-
3-yl]ethyl)-4-fluorobenzamide
2.5 g (12.4 mmol) of exo-6-phenyl-7-methyl-1,5-cis-3,7-diaza-
bicyclo-[3.3.D]octan in 50 ml xylene were treated with 2.5 g
(12.4 mmol) of N-(2-chloroethyl)-4-fluorobenzamide and with 1.7 g
(12.4 mmol) of finely pulverized potassium carbonate and 0.5 g of
potassium iodide and refluxed with good stirring for 2 h.
After cooling, the mixture was concentrated on a rotary evapora-
for and the residue was partitioned between methylene chloride
and water.
The aqueous phase was re-extracted twice with methylene chloride
and the organic phase was then concentrated after drying with
sodium sulfate. The crude product (4.9 g) was purified by column
chromatography (silica gel, eluent methylene chloride/methanol
95:5). 2.5 g (55%) of product of m.p. 108-110~C (hydrochloride)
were obtained.
The .following can be prepared in a similar manner:
2. N-(2-[endo-6-phenyl-7-methyl-1,5-cis-3,7-diazabicyclo(3.3.0]-
octan-3-yl]ethyl)-4-fluorobenzamide,
3. N-(2-[exo-6-p-fluorophenyl-7-methyl-1,5-cis-3,7-diazabi-
cyclo[3.3.0]octan-3-y1]ethyl)-4-fluorobenzamide,
4. N-(2-[exo-6-p-trifluoromethylphenyl-7-methyl-1,5-cis
3,7-diazabicyclo[3.3.D]octan-3-yl]ethyl)benzamide,
5. N-(2-[exo-6-m-chlorophenyl-7-methyl-I,5-cis-3,7-diazabi-
cyclo(3.3.0]octan-3-yl]ethyl)benzamide,
6. N-(2-[exo-6-p-methoxyphenyl-7-methyl-1,5-cis-3,7-diazabi-
cyclo[3.3.0]octan-3-yl]ethyl)-4-chlorobenzamide,
7. N-(2-[exo-6-phenyl-7-methyl-1,5-cis-3,7-diaaabicyclo-
[3.3.0]octan-3-yl]ethyl)naphthalene-1-carboxamide,
B. 1-(4-fluorophenyl)-4-[exo-6-p-fluorophenyl-7-methyl-
1,5-cis-3,7-diazabicyclo[3.3.0]octan-3-yl]butan-1-one,
W095115327 ~ ~~'~~y~ f 's PCTIEP94103913
U~(J~~. 22
9. 1-(4-fluorophenyl)-4-[exo-6-p-fluorophenyl-7-methyl-
1,5-cis-3,7-diazabicyclo[3.3.0]octan-3-yl]butan-1-ol.
Example 10
N-(2-[exo-6-p-Fluorophenyl-1,5-cis-3-azabicyclo[4.3.0]non-3-yl]-
ethyl)benzamide
1.6 g (7.3 mmol) of exo-6-p-fluorophenyl-1,5-cis-3-aaabicyclo-
iD [4.3.0]nonane in 50 ml of xylene were treated with 2.7 g
(14.6 mmol) of N-(2-chloroethyl)benzamide and With 1.4 g
(10.0 mmol) of finely pulverized potassium carbonate and 0.5 g of
potassium iodide and refluxed with good stirring for 8 h.
After cooling, the mixture was concentrated on a rotary evapora-
tor and the residue was partitioned between methylene chloride
and water (pH = 10).
The aqueous phase was re-extracted twice with methylene chloride
and the organic phase was then concentrated after drying with
sodium sulfate. The crude product (4.0 g) was purified by column
chromatography (silica gel, eluent methylene chloride/methanol
93:7). 1.3 g (49%) of product of m.p. 160-162~C (maleate) were
obtained.
Example 11
1-(2-[exo-6-p-Fluorophenyl-1,5-cis-3-azabicyclo[4.3.0]non-3-yl]-
ethyl)naphthalene
3D
1.6 g (7.3 mmol) of exo-6-p-fluorophenyl-1,5-cis-3-aaabicyclo-
[4.3.D]nonane in 50 m1 of xylene were treated with 1.7 g
(7.3 mmol) of 1-(2-bromo)ethylnaphthalene and with 1.4 g
(10.0 mmol) of finely pulverised potassium carbonate and 0.5 g of
potassium iodide and refluxed with good stirring for 9 h.
After cooling, the mixture was concentrated on a rotary evapora-
tor and the residue was partitioned between methylene chloride
and water (pH = 10).
The aqueous phase was re-extracted twice with methylene chloride
and the organic phase was then concentrated after drying with
sodium sulfate. The crude product (3.5 g) was purified by column
chromatography (silica gel, eluent methylene chloride/methanol
96:4). 1.8 g (66%) of product of m.p. 99-100~C (decomposition,
fumarate) were obtained.
WO 95115327 ~ O ~~~ ' i' p~/gp9q~p3913
23
The following can be prepared in a simmilar manner:
12. 1-{4-fluorophenyl}-4-[exo-6-p-fluorophenyl-1,5-cis-
3-azabicyclo[4.3.0]non-3-yl]butan-1-one,
13. 1-(4-fluorophenyl}-4-[exo-6-p-fluorophenyl-1,5-cis-
3-azabicyclo[4.3.0]non-3-yl]butan-1-ol,
14. N-(2-[exo-6-p-fluorophenyl-1,5-cis-3-azabicyclo[4.3.0]non-
3-yl]ethyl)-2-(N-methyl)sminobenzamide,
15. N-(2-[exo-6-p-fluorophenyl-1,5-cis-3-azabicyclo[4.3.0]non-
3-yl]ethyl)-5-chlorothien-2-ylcarboxamide,
i5 16. N-(2-[exo-6-p-fluorophenyl-1,5-cis-3-azabicyclo[4.3.0]non-
3-yl]ethyl)inden-3-carboxamide,
17. N-(2-[exo-6-phenyl-1,5-cis-3-azabicyclo[4.3.0]non-3-yl]-
ethyl)-4-fluorobenzamide,
I8. N-(2-[exo-6-phenyl-1,5-cis-3-azabicyclo[4.3.0]non-3-yl]-
ethyl)naphthalene-1-carboxamide,
19. 1-(2-[exo-6-p-fluorophenyl-1,5-cis-3-azabicyclo[4.3.0]non-
7-en-3-yl]ethyl)naphthalene,
20. 1-(4-fluorophenyl)-4-[exo-6-p-fluorophenyl-1,5-cis-3-aza-
bicyclo[4.3.0]non-7-en-3-yl]butan-1-one,
21. N-(2-[exo-6-p-fluorophenyl-1,5-cis-3-azabicyclo[4.3.0]non-
7-en-3-yl]ethyl)benzamide,
22. N-(2-[exo-6-p-fluorophenyl-1,5-cis-3-azabicyclo[4.3.0]non-
7-en-3-yl]ethyl)inden-3-carboxamide,
23. N-(2-jexo-6-p-fluorophenyl-1,5-cis-3-azabicyclo[4.3.0]non-
7-en-3-yl]ethyl)naphthalene-1-carboxamide,
24. N-(2-[exo-6-phenyl-1,5-cis-3-azabicyclo[4.3.0]non-7-en-3-yl]-
ethyl)-4-fluorobenzamide,
25. N-(2-[exo-6-phenyl-1,5-cis-3-azabicyclo[4.3.0]non-7-en-3-yl]-
ethyl)-3-chloro-1-benzothien-2-ylcarboxamide,
26. 1-(2-[6-p-fluorophenyl-I,5-cis-3-azabicyclo[4.3.0]non-6-en-
3-yl]ethyl)naphthalene,
2.i 7 7 6 0 2 ' pC'T/Ep94/03913
WO 95115327
24
27. I-(4-fluorophenyl}-4-[6-p-fluorophenyl-1,5-cis-3-azabicyclo-
[4.3.0]non-6-en-3-yl]butan-1-one,
2B. 1-(4-fluorophenyl)-4-[6-p-fluorophenyl-1,5-cis-3-azabicyclo-
[4.3.0]non-6-en-3-yl]butan-I-ol,
29. N-(2-[6-p-fluorophenyl-1,5-cis-3-azabicyclo[4.3.0]non-6-en-
3-yI]ethyl)-2-(N-methyl)aminobenzamide,
30. N-(2-[6-p-fluorophenyl-I,5-cis-3-azabicyclo[4.3.0]non-6-en-
3-yl]ethyl)-5-chlorothien-2-yl-carboxamide,
31. N-(2-[6-p-fluorophenyl-1,5-cis-3-azabicycloj4.3.0]non-6-en-
3-y1]ethyl)benzamide,
32. N-(2-[6-phenyl-1,5-cis-3-azabicyclo[4.3.0]non-6-en-3-yl]-
ethyl)-3-chloro-1-benzothien-2-ylcarboxamide,
33. N-(2-[6-p-trifluoromethylphenyl-1,5-cis-3-azabicyclo[4.3.0]-
non-6-en-3-yl]ethyl)naphthalene-1-carboxamide,
34. N-(2-[6-p-fluorophenyl-1,5-cis-3-azabicyclo[4.3.0]non-6-en-
3-yl]ethyl)inden-3-carboximide,
35. N-(2-j6-p-fluorophenyl-1,5-cis-3-azabicyclo[4.3.0]non-6-en-
3-yl]ethyl)napthalene-1-carboxamide,
36. 1-(2-[endo-6-p-fluorophenyl-1,5-cis-3-azabicyclo[4.3.0]non-
3-yl]ethyl)naphthalene,
37. 1-(4-fluorophenyl)-4-[endo-6-p-fluorophenyl-1,5-cis-3-aza-
bicyclo[4.3.0]non-3-y1]butan-1-one,
38. 1-(4-fluorophenyl}-4-[endo-6-p-fluorophenyl-1,5-cis-3-aza-
bicyclo[4.3.0]non-3-yl]butan-1-ol,
39. N-(2-[endo-6-p-fluorophenyl-1,5-cis-3-aaabicyclo[4.3.0]non-
3-yl]-2-(N-methyl)aminobenzamide,
40. N-(2-[endo-6-p-fluorophenyl-1,5-cis-3-azabicyclo[4.3.0]non-
3-yl]ethyl)benzamide,
41. N-(2-[endo-6-p-fluorophenyl-1,5-cis-3-azabicyclo[4.3.0]non-
3-yl]ethyl)-5-chlorothien-2-ylcarboxamide,
WO 95!15327 2 ~ 7 7 b 0 ~ ' 'f '' PCT/EP94/03913
30
42. N-(2-[endo-6-phenyl-1,5-cis-3-azabicyclo[4.3.0]non-3-yl]-
ethyl)-4-fluorobenzamide,
43. N-(2-[endo-6-p-triouoromethy!phenyl-1,5-cis-3-azabicyclo-
5 [4.3.0]non-3-yl]ethyl)inden-3-carboxamide,
44. 2-(2-[endo-6-p-fluorophenyl-1,5-cis-3-azabicyclo(4.3.0]non-
7-en-3-yl]ethyl)naphthalene,
10 45. N-(2-[endo-6-p-fluorophenyl-1,5-cis-3-aaabicyclo[4.3.0]non-
7-en-3-yl]ethyl)benzamide,
46. N-(2-[endo-6-p-fluorophenyl-1,5-cis-3-azabicyclo[4.3.0]non-
7-en-3-yl]ethyl)-2-(N-methyl)aminobenzamide,
47. N-(2-[endo-6-p-fluorophenyl-1,5-cis-3-azabicyclo[4.3.0]non-
7-en-3-yl]ethyl)naphthalene-1-carboxamide,
48. 1-(2-[exo-7-p-fluorophenyl-1,5-cis-3-azabicyclo[4.3.0]non-
20 8-en-3-yl]ethyl)naphthalene,
49. 1-(4-fluorophenyl)-4-[exo-7-p-fluorophenyl-1,5-cis-3-aza-
bicyclo[4.3.0]non-8-en-3-yl]butan-1-one,
25 50. N-(2-[exo-7-p-fluorophenyl-1,5-cis-3-azabicyclo[4.3.0]non-
8-en-3-yl]ethyl)benzamide,
51. N-(2-[exo-7-p-fluorophenyl-1,5-cis-3-azabicyclo[4.3.0]non-
B-en-3-yl]ethyl)-5-chlorothien-2-ylcarboxamide,
52. N-(2-[exo-7-p-fluorophenyl-I,5-cis-3-azabicyclo[4.3.0]non
8-en-3-yl]ethyl)-3-chloro-1-benzothien-2-ylcarboxamide,
53. N-(2-[exo-7-phenyl-1,5-cis-3-azabicyclo[4.3.0]non-B-en-
3-yl]ethyl)inden-3-carboxamide,
54. N-(2-[exo-7-phenyl-1,5-cis-3-azabicyclo[4.3.0]non-8-en-
3-yl]ethyl)naphthalene-1-carboxamide,
55. 1-(2-[exo-7-p-fluorophenyl-1,5-cis-3-azabicyclo[4.3.0]non-
3-yl]ethyl)naphthalene,
56. 1-(4-fluorophenyl)-4-[exo-7-p-fluornphenyl-1,5-cis-3-aza-
bicycto[4.3.0]non-3-yl]butan-1-one,
21176' p2 ' 1
WO 95/15327 PCT/EP94103913
26
57. 1-(4-fluorophenyl)-4-[exo-7-p-fluorophenyl-1,5-cis-3-aza-
bicyclo[4.3.0]nan-3-yl]butan-1-ol,
58. N-(2-[exo-7-p-fluorophenyl-1,5-cis-3-azabicyclo[4.3.0]non-
3-yl]ethyl)-2-(N-methyl)aminobenzamide,
59. N-(2-[exo-7-p-fluorophenyl-1,5-cis-3-azabicyclo[4.3.0]non-
3-yl]ethyl)benzamide,
60. N-(2-[exo-7-p-fluorophenyl-1,5-cis-3-azabicyclo[4.3.0]non-
3-yl]ethyl)-5-chlorothien-2-ylcarboxamide,
61. N-(2-[exo-7-p-fluorophenyl-1,5-cis-3-azabicyclo[4.3.0]non-
3-yl]ethyl)naphthalene-1-carboxamide,
IS
62. N-(2-[exo-7-p-fluorophenyl-1,5-cis-3-azabicyclo[4.3.0]non-
3-y1]ethyl)inden-3-carboxamide,
63. I-(2-[6-p-fluorophenyl-1,5-cis-3-azabicyclo[3.3.0]oct-6-en-
3-yl]ethyl)naphthalene,
64. 1-(4-fluorophenyl)-4-[6-p-fluorophenyl-1,5-cis-3-azabicyclo-
[3.3.0]oct-6-en-3-yl]butan-1-one,
65. N-(2-[6-p-fluorophenyl-1,5-cis-3-azabicyclo[3.3.0]oct-6-en-
3-yl]ethyl)benzamide,
66. N-(2-[6-p-fluorophenyl-1,5-cis-3-azabicyclo[3.3.0]oct-6-en-
3-yl]ethyl)naphthalene-1-carboxamide,
67. 1-(2-[exo-6-p-fluorophenyl-1,5-cis-3-azabicyclo[3.3.0]octan-
3-yl]ethyl)naphthalene,
68. 1-(4-fluorophenyl)-4-[exo-6-p-fluorophenyl-I,5-cis-3-aza-
bicyclo[3.3.0]octan-3-yl]butan-1-ol,
69. N-(2-[exo-6-p-fluorophenyl-1,5-cis-3-aaabicyclo[3.3.0]octan-
3-yl]ethyl)benzamide,
70. N-(2-[exo-6-phenyl-1,5-cis-3-azabicyclo[3.3.0]octan-3-y1]-
ethyl)naphthalene-I-carboxamide,
71. N-(2-[exo-6-phenyl-1,5-cis-3-azabicyclo[3.3.0]octan-3-yl]-
ethyl)inden-3-carboxamide,
21776,02 .
WO 95115327 PCT/EP94/03913
27
72. 1-(2-[endo-6-p-fluorophenyl-1,5-cis-3-azabicyclo[3.3.0]octan-
3-yl]ethyl)naphthalene,
73. 1-(4-fluorophenyl)-4-[endo-6-p-fluorophenyl-1,5-cis-3-aza-
bicyclo[3.3.0]octan-3-yl]butan-1-one,
74. N-(2-[endo-6-p-fluorophenyl-1,5-cis-3-azabicyclo[3.3.0]octan-
3-yljethyl)benzamide,
75. N-(2-[endo-6-phenyl-1,5-cis-3-azabicyclo[3.3.0]octan-3-yl]-
ethyl)naphthalene-1-carboxamide,
76. 1-(2-[exo-7-p-fluorophenyl-1,5-cis-3-azabicyclo[3.3.Ojoctan-
3-yl]ethyl)naphthalene,
77. 1-(4-fluorophenyl)-4-[exo-7-p-fluorophenyl-1,5-cis-3-aza-
bicyclo[3.3.D]octan-3-yljbutan-1-one,
78. N-(2-[exo-7-p-fluorophenyl-1,5-cis-3-azabicyclo[3.3.0]octan-
3-yl]ethyl)benzamide,
79. N-(2-[exo-7-phenyl-1,5-cis-3-azabicyclo[3.3.0]octan-3-ylj-
ethyl)naphthalene-1-carboxamide,
80. N-(2-[exo-7-phenyl-1,5-cis-3-azabicyclo[3.3.0]octan-3-yl]-
ethyl)inden-3-carboxamide,
81. N-(2-[exo-6-p-fluorophenyl-3-azabicyclo[3.1.0]hexan-3-yl]-
ethyl)-4-fluorabenzamide,
82. N-(2-[exo-6-phenyl-3-azabicyclo[3.1.0]hexan-3-yljethyl)-
5-chlorothien-2-ylcarboxamide.
83. N-(2-[6,6-diphenyl-3-azabicyclo[3.1.0]hexan-3-yl]-ethyl)-
naphth-1-ylcarboxamide,
84. N-(2-[exo-6-p-fluorophenyl-3-azabicyclo[3.1.0jhexan-3-yl]-
ethyl)-3-chloro-1-benzothien-2-ylcarboxamide,
85. N-(2-[exo-6-m-chlorophenyl-3-azabicyclo[3.1.0]hexan-3-yl]-
ethyl)-4-chlorobenzamide,
86. 1-(2-[6-p-fluorophenyl-1-5-cis-8-oxa-3,7-diazabicyclo[3.3.0]-
oct-6-en-3-yl]ethyl)naphthalene,
WO 95!15327 217 7 6 p 2 PCT~P94103913
28
g7, N-(2-[6-p-fluorophenyl-I-5-cis-8-oxa-3,7-diazabicyclo(3.3.0]-
oct-6-en-3-yl]ethyljbenzamide,
88. N-(2-[6-p-fluorophenyl-I-5-cis-8-oxa-3,7-diazabicyclo[3.3.0]-
oct-6-en-3-yl]ethyl)naphthalene-1-carboxamide,
89. N-(2-[6-p-fluorophenyl-1-5-cis-8-oxa-3,7-diazabicyclo[3.3.0]-
oct-6-en-3-yl]ethyl)inden-3-carboxamide,
90. 1-(2-[exo/endo-6-p-fluorophenyl-1-5-cis-B-oxa-3,7-diaza-
bicyclo[3.3.D]octan-3-yl]ethyl)naphthalene,
91. N-(2-[exo/endo-6-p-fluorophenyl-1-5-cis-8-oxa-3,7-diaza-
bicyclo[3.3.0]octan-3-yl]ethyl)benzamide,
92. 1-(2-[6-phenyl-I-5-cis-B-oxa-3,7-diazabicyclo(3.3.0]oct-
6-en-3-yl]ethyl)naphthalene,
93. N-(2-[6-phenyl-1-5-cis-8-oxa-3,7-diazabicyclo[3.3.0]oct-
6-en-3-yl]ethyl)benzamide.
30
40