Note: Descriptions are shown in the official language in which they were submitted.
21779~7
lo~ryl-5ubst~tuted M~t~l Complexes, ph-rr-~ ~utical Agents
Containing thes~ Complexes, ~rheir Us~ in Di~gnosis, ~8 well as
Process ~or the Production o~ the Complexes And Agents
The invention relates to the objects characterized in the
claims, i.e., new haloaryl-substituted metal complexes,
pharmaceutical agents containing these complexes, their use in
diagnosis as well as process for the production of the complexes
and agents.
Contrast media are indispensable auxiliary agents in modern
diagnosis; thus, many diseases could not be diagnosed without the
use of contrast media. Contrast media are used in all areas of
diagnosis, such as, e.g., diagnostic radiology, radiodiagnosis or
ultrasonic diagnosis or magnetic resonance tomography.
The selection of the respectively preferred methods depends,
i.a., on the diagnostic problem, but is also determined by the
choice of apparatus respectively available to the physician.
Thus, because of the considerable technical and associated high
costs, nuclear spin tomography especially has not yet found the
wide-spread use of other methods , such as , e . g ., methods of
diagnostic radiology.
Also, the selection of the suitable contrast medium varies
as a function of the respective problem. Thus, the suitability
of the contrast medium for a specific object is last but not
least determined by its (concentration) distribution behavior in
the organism.
2177~77
Although great progress has been achieved with respect to
equipment and contrast media, satisfactory solutions are not yet
available for all problems.
Thu6, contrast media that are suitable for all indications
do not yet exist for the various imaging processes. In
particular, to this day, no suitable x-ray contrast medium for
liver diagnosis is available.
In diagnostic radiology, basically contrast media based on
triiodobenzene have been able to gain acceptance, since these
compounds exhibit a high x-ray opacity and a low general and
local toxicity and are very readily water-soluble.
Such compounds are described, e.g., in EP 0 105 725, EP 0
015 867. But the latter do not show any concentration,
6ufficient for imaging, in the liver.
Since the suitability of a ~ d as x-ray contrast
medium, in addition to the concentration behavior in the
respective organ, basically depends on the value of the mass
attenuation coef f icient of the elements that are contained in the
compound, in the diagnostic radiation area, in addition to the
iodine-containing ~ n~l~, metal complexes of an element with a
high atomic number should also be suitable. Such compounds are
widely used in the field of N~ diagnosis. In this case, there
are generally metal complexes, as they are described, e.g., in EP
o 071 564.
Wo 93/16375 descri~es metal complexes, which are linked by
amide bonds to iodine-substituted aromatic compounds. With only
one administration of the contrast medium, these compounds are to
2177977
m~ike it pos~ible to carry out both N~ and x-ray studies. A
combination of the two imaging processes is advantageous in many
cases for a differentiated visualization and a reliable
determination of certain diseases. These compounds are to be
suitable e6pecially for angiography. But as the duplication of
the production examples showed, the compounds show no
concentration, sufficient for x-ray studies, in the area of the
liver .
Liver-specific NI~R contrast media are described in EP 0 405
704. The latter in principle should also be suitable for
diagnostic radiology because of the metal content in the
complexes. A duplication of the experimental examples did not
show sufficient contrasting of the liver in the x-ray picture
even when a high dose was administered tconc.: 1 M/l, dose: 0.5
mmol of Gd/kg intravenously). A sufficient imaging effect in
diagnostic radiology is achieved only with a dose in which the
safety margin is reduced to a no longer justifiable measurement.
It was therefore the object of this invention to make
available very readily compatible and water-soluble contrast
media, which are suitable especially for diagnostic radiology of
the liver.
This object is achieved by this invention.
It has been found that metal complexes that contain at least
one ion of an element of atomic numbers 12, 13, 20-31, 39-42, 44-
50 or 57-83 and a halogen-containing complexing ligand of
21~7~7
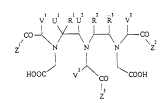
formula I
V U R U R R V
Z~ Ni--
HOOC V J~ ~COOH
CO
Z
in which
R1 stands for a hydrogen atom, a carboxylic acid radical,
a straight-chain or branched C1-C1s alkyl radical, C6-C1s aryl
radical, or a C7-C~s aralkyl radical, which optionally is
substituted by 1-5 hydroxy groups and/or 1-2 carboxy groups
and/or is interrupted by 1-4 oxygen atoms, or in which R' stands
for a radical of general formula II or III,
--CO--NR4RS ( I I )
--CH2--NR6-Co--R7 ( I I I )
in which
R4, Rs, indPrpl ~Pntly of one another, stand for a
hydrogen atom, a straight-chain or branched C1-C15 alkyl
radical, C6-C1s aryl radical, or a C7-C1s aralkyl radical,
which optionally contains 1-5 hydroxy groups, 1-2 carboxy
groups and/or 1-4 oxygen atoms, or in which R4, Rs, together
with incorporation of the nitrogen atom, form a 5- or 6-ring
optionally containing an oxygen atom, another acylated
nitrogen atom or a sulfonyl group, optionally substituted
with 1-3 hydroxy groups,
2177~77
R6 stands for a hydrogen atom, a straight-chain or
branched C~-C~5 alkyl radical, C6-C1s aryl radical or a C7-C1s
aralkyl radical, which optionally contains 1-4 hydroxy
groups, 1-2 carboxy groups and/or 1-2 oxygen atoms, or
in which R6 together with R7, with incorporation of the
nitrogen atom and the carbonyl group, forms a 5- or 6-ring
optionally containing an oxygen atom, another acylated
nitrogen atom or a sulfonyl group, optionally substituted
with 1-3 hydroxy groups, and
R7 stands for a hydrogen atom, a straight-chain or
branched C1-C1s alkyl radical, C6-C1s aryl radical or a C7-C1s
aralkyl radical, which optionally contains l to 2 hydroxy
groups or a carboxy group or
in which R7 together with R6, with incorporation of the
nitrogen atom and the carbonyl group, forms a 5- or 6-ring
optionally containing an oxygen atom, another acylated
nitrogen atom or a sulfonyl group, optLonally substituted
with 1-3 hydroxy groups,
RZ, R3, indep~n~ently of one another, stand for a hydrogen
atom, a C1-C1s alkyl radical, a C6-C1s aryl radical or a C7-C1s
aralkyl radical, which optionally is substituted by 1-5 hydroxy
groups and/or is interrupted by 1-4 oxygen atoms, or together
~orm a trimethylene or tetramethylene group, or have the meaning
indicated for U1 (U2),
Z1, ZZ, Z3, ;n~ r~nr~ntly of one another, stand for a hydroxy
group or a radical -NR17-U1,
in which
2177977
R17 stands for a hydrogen atom, a methyl or methoxyethyl
group, and
U1, U1', U2, V~, VZ and V3, respectively independently of one
another, stand for a hydrogen atom or for a halogenated aromatic
radical of general formula IV,
R
y~R
X
in which
R8, R9, independently of one another, stand for a group
-NR6-Co-R7 and/or have the meaning indicated for R1, with the
exception of a C1-C1s alkyl radical, C6-C1s aryl radical or a
C7-C15 aralkyl radical,
R1C, R11, independently of one another, stand for a
halogen atom or a hydrogen atom,
X stands for a halogen atom or a bridge-type
crosslink of general formula V, and
Y stands for R9 or a bridge-type crosslink of
general formula V
(a)--(CHz) p--(C6H4) n--(L) ~ R (B) (V)
in which
m, n, p, independently of one another, ~;tand for
num~ers O or l,
217~g77
L stands for an oxygen atom, a sulfur atom, a C1-C~,
alkylene radical, a group ~S=O, >SOz or >NR4 with R~ in the
mentioned meaning, and
R1Z stands for a direct bond, a carbonyl, a carboxyl,
a -CO-NR18-, an -NR18-CO-, an -NH-CS- or a CS-NH group, in
which R18 means a hydrogen atom, a straight-chain or
branched C1-C1s alkyl radical, C6-C15 aryl radical, or a C7-C1s
aralkyl radical, which optionally contains 1-4 hydroxy
groups, 1-2 carboxy groups and/or 1-2 oxygen atoms,
or in which R12 stands for a straight-chain or branched
C1-C4 alkylene radical, which optionally contains a carbonyl
group and/or an amino group,
whereby position (~) is linked with the
diethylenetriamine skeleton and position (B) is linked with
the halogenated aromatic compound,
whereby Y stands for a bridge-type crosslink of formula
V, if X is equal to halogen, and Y stands for R9, if X
stands for a bridge-type crosslink of formula V and
at least one of radicals R2, R3, z1, z2, z3, U1, U2, V1, v2 or
V3 stands for the radical of general formula IV or contains the
latter, and optionally free carboxy groups, not required for
complexing the metal ions of the mentioned elements, are present
as salts of an inorganic and/or organic base or amino acid,
with at least one of the following provisions,
that R8 and/or R9 contains an aryl radical, and/or
21779~7
that Z1 and/or ZZ then stand for a radical of general
formula IV only if at least one of substituents R2, R3, U1, U2, V1,
V2 or V3 does not stand for a hydrogen atom, and/or
that if Z3 contains a completely substituted aromatic
_ ~.1 of formula IV, Z1 and/or Z2 do not contain any fully
substituted aromatic ~ ~oun~l~ of formula IV,
and/or
that if all substituents R2, R~, U1, U2, V1, v2 and V3 mean
hydrogen, at least one of radicals R8, R9, R10 or R11 stands for a
hydrogen atom, and/or R8 and/or R9 mean a radical which contains
a carboxylic acid that is not directly bound,
are excellently suited for the production of contrast media for
NMR diagnosis and/or diagnostic radiology, preferably of contrast
media for diagnostic radiology, especially for diagnostic
radiology of the liver.
The complexes according to the invention preferably contain
as a metal ion a r~n~n~e(II) ion, iron(III) ion, iron(II) ion,
praseodymium(III) ion, neodymium(III) ion, samarium(III) ion,
dysprosium(III) ion, an ytterbium(III) ion or a bismuth(III) ion,
especially a gadolinium(III) ion.
As halogen atom(s), the complexes according to the invention
contain (a) chlorine, bromine or iodine atom(s), preferably (a)
bromine or iodine atom(s), but especially (an) iodine atom(s).
Preferred halogenated aromatic compounds of formula IV are
triiodized aromatic compounds, i . e., aromatic compounds in which
X, R10 and R11 stand for iodine, and in which R8 and R9,
independently of one another, stand for a hydrogen atom, for a
2177~77
group -OH, -COOH, -o-CH2-CH(OH) -CH2-OH, -o-CH3, -o-CH2-CH3, -CO-NH-
CH(CH20H)--(CHOH-CH20H), -Co-NR4-CH--(CHzoH)2~ -NR6--CO-CH20H, --Co-NR4-
CH2-CH20H, -CO-NH2, --N (CH3) -CO-CH3, -NH-CO-CH3, -Co-NH-CH3, --N (CH3) -
CO-(CH2)2COOH, -CO-N--(C2Hs)2, -C-N(CH3)-cH2 - cooH~ -CO-NH--(CH2)10-
COOH, -CO-NH-CH2-C6H4-OEt or a group -Co-N(CH3)-CH2-CH(OH)-CH2-OH,
and in which Y 6tands for a bridge-type crosslink of formula V.
Preferred bridge-type crosslinks of formula V are, if one of
radicals
- U stands for a halogenated aromatic radical, the groups
-CH2--, -CH2--C6H4-0-CH2--, -CH2-0-CH2--, -CH2-0-, CH2 CO ,
--CH2--NH-CO-, -CH2-CO-NH-, --CH2-C6H4-O-CH2-CO--NH-, --CH2-0-CO-NH--,
--CH2--NH-CO-NH--, -NH--CO-, --NH-CO-CH2-,
- Z stands for a halogenated aromatic radical, the groups
-NH-CH2-CO-NH-, -NH CH2CH2-CO-NH- or -NH CH2CH2-NH-CO- and that
- V stands for a halogenated aromatic radical, the groups
--CH2--, -CH2--0-, -CH2-0-CH2-, -CH2-0-CO-, -CH2--NH--CO-, --CH2--CO-NH--,
-CH2-o-CO-NH- or -CH2-NH-CO-NH-.
Especially preferred among them are the complexes in which
U1 ~tands for the radical of general formula IV, i.e., c u~Ll,ds
that are substituted with the halogenated aromatic compound in
the ethylene bridge of the polyaminocarboxylic acid and in ~hich
Z stands f or a hydroxy group .
A6 radical R1, there are considered straight-chain or
branched aLlcyl radicals, such as methyl, ethyl, propyl,
isopropyl, butyl and tert-butyl radicals, but preferred are
hydrogen, C1-C4 alkyl and hydroxyalkyl radicals, such as, e.g.,
2177g77
the hydL~ y ~hyl radical as well as alkoxyalkyl radicals, such
as, e.g., the methoxymethyl radical.
As radicals R2, R3, the radicals listed for R1 are
considered, but hydrogen atoms are preferred.
For diagnostic radiology of the liver, especially ionic
complexes are preferred, in which free carboxyl groups that are
present in the molecule ( i . e ., carboxyl groups that are not
required for charge equalization of the metal ions of the
elements of the mentioned atomic numbers) are present as free
acids or as salts of an inorganic and/or organic base or amino
acid .
Suitable cations of inorganic bases are, for example, the
lithium ion, the potassium ion, the calcium ion, the magnesium
ion and especially the sodium ion. Suitable cations of organic
bases are, i.a., those of the primary, secondary or tertiary
amines, such as, e.g., ethanolamine, diethanolamine, morpholine,
glucamine, N,N-dimethyl glucamine and especially N-methyl
glucamine. Suitable cations of amino acids are, for example,
those of lysine, arginine and ornithine.
The production of the complexes according to the invention
can be carried out in varied ways. The various processes as well
as the initial compounds required for them are known in principle
to one skilled in the art. Thus, the compounds generally can be
produced analogously to the already known complexes or complexing
agents, by reaction of a reactive species of the halogenated
aromatic compounds with a reactive species of the complexing
agent in a suitable solvent. The selection of the method of
2177g77
synthesis most suitable in each case depends on the desired
linkage point between the halogenated aromatic compound(s) and
the polyaminopolycarboxylic acid. Accordingly, the complexes can
be divided into three groups. Thus, the halogenated aromatic
cc~: ' (s) can be bound
I) to the ~-carbon atoms of the carboxylic acid (acetic
acid) radical,
II) to the alkylene (ethylene) bridges or
III) to the carboxylic acid group(s)
of the polyaminopolycarboxylic acid.
Complexing agents or complexes of group I can be produced
analogously to the processes that are described in European
Patent Application EP O 230 893. Other universally applicable
possibilities of synthesis for the complexing agents will be
mentioned as examples below.
Thus, by reaction of chloroacetic acid derivatives of
general formula VI
Cl
~--CO~H
R (VI)
in which R13 is the desired halogenated aromatic radical of
general formula IV or an optionally still unhalogenated precursor
21~7~7
of this radical, with polyamines of general formula VII
R3
H~N~N ~ NH.
j~ H 13
R R (VII)
first compounds of general formula VIII can be obtained,
3 RyCO~H
H~N N~/NH
~ H 3
R R ~
which then are reacted in a way known in the art with haloacetic
acid esters, pref erably with bromoacetic acid esters, and then --
if a precursor of the respectively desired aromatic compound is
involved -- are halogenated in a way known in the art and
optionally present ester groups or protective groups are cleaved
in a way known to onc skilled in the art. Disubstitution can
also be achieved with corresponding stoichiometry.
An alternative process for the production of ~-C-substituted
polyaminopolycarboxylic acids starts from an acid-protected
polyaminocarboxylic acid (e.g., from the pentamethyl ester of
diethylenetriaminepentaacetic acid). The latter is reacted with
the lithium salt of a precursor of the desired aromatic compound.
A corresponding lithium salt can be obtained from the benzyl
217797~
halide (e.g., 3-nitrobenzyl chloride, 3,5-dinitrobenzyl chloride,
3-benzyloxybenzyl chloride) by reaction with lithium
diisopropylamine in THF/hexane. Following the coupling, the
aromatic _ ~ull~ is reacted to the desired halogenated aromatic
compound of formula IV, e.g., by the optionally present nitro
groups being reduced to amino groups, which are optionally
reacted with acetyl chloride to the amide; benzyloxy radicals can
be converted to hydroxy radicals, e.g., by catalytic
hydrogenation. The iodization of the aromatic compound is also
carried out in a way known in the art , e . g ., by reaction with
io~ o~ hloride solution in hydrochloric acid medium. Before
the introduction of the iodine atoms, the acid protective groups
of the pentaester are saponified in the basic medium.
An alternative process for the production of iodine-
containing polyaminopolycarboxylic acids starts from an a-amino
acid derivative of general formula XXIII
H`N ~H
V3 ~ CO
Z ~
whose primary amino group optionally can also be present in
protected form (e.g., as monobenzylamine). Thi6 amino group is
dialkylated on a nitrogen atom with an alkylating agent of
21~7~77
formula XXIV
Vl Ul Rl UZ
N Nf
Rl~O
oC (XXIV)
and then -- after cleavage of the optionally present protective
groups -- with an alkylating agent of formula XXV
R2 R3 VZ
~ ~Lco-zZ
Nf N
COOR~
whereby Nf stands for a nucleofuge, such as, e.g., chloride,
bromide, iodide, methanesulfonate or toluenesulfonate, and R14
stands for an acid protective group, such as, e.g., a lower
alkyl, aryl, aralkyl or trialkylsilyl group, and R1, RZ, R3, U1,
UZ, V1, V2, V3, z1, Z2 and Z3 have the previously indicated
meaning .
Synthesis for the production of an ~-C-substituted
polyaminopolycarboxylic acid starts from, e. g., a phenylamino
acid, such as, e.g., 3-aminophenylalanine. The latter is first
halogenated in a way known in the art, the acid group then
protected as ester. The thus obtained int~ liAte product is
reacted with two equivalents of N,N-bis[ (benzyloxycarbonyl) -
methyl]-2-bromomethylamine. Before the cleavage of the acid
15 21779~7
protective groups, the substituents of the aromatic C~l r_ ' are
optionally converted to the de6ired radicals.
The production of the complexing agents of group II can be
carried out analogously to the method5 that are described in EP 0
405 704 as well as DE 43 02 289. Thus, the process starts from,
e.g., known c __I.d6 (DE 37 10 730 and literature cited there)
of general formula IX,
C~
ll J
`f~ Rl R3 R3
0C~\N)~\I~)~N/--
C3~R'"~R1' a~ORI4
in which R1~, R1, R2 and R3 have the indicated meanings, in which
the phenolic OH group is reacted with a reactive form of the
desired halogenated aromatic compound (or its precursor, e.g.,
O 16 2177~7
benzyl halide) o~ formula X
R
~R~
~CI
10 '
R (X),
in which R8', R9', R10 and R1t stand for desired groups R8, R9, R10
and Rl1 or a precursor of the latter. If groups R8, R9, R10 and
R11 stand for precursors of the desired groups, the latter are
generated from those. Acid protective groups R14 are cleaved in
a known way [see, e.g., E. Wiinsch, Methoden der Org. Chemie
[Methods of Org. Chemistry] (Houben Weyl), Vol. XV/l, 4th
Edition, 1974, p. 315 ff~, for example, by hydrolysis,
hydrogenolysis or AlkAl inC~ saponification, generally beforc the
halogenation of the aromatic compound. Both acidic and aqueous-
AlkAl ine reaction conditions can be selected for the cleavage of
the t-butyl esters that are especially advantageous for this
reaction .
In the compounds of general formula IX, the aromatic radical
can also be iodized, e.g., with io~ no~hloride, in a way known
in the art. Optionally, the phenolic -OE~ groups can be
etherifled in a way known in the art with alkyl ha;ide/sodium
hydride. The cleavage of the acid protective groups is carried
out in the previously described way.
217~77
An alternative process also star~s from halogen-contalning
chlorinated aromatic compounds of formula X, which are reacted
with a partially protected glycerol, first to the corresponding
dihyclLuxy~rv~yloxy compound of formula XI
R
. 1 10'
! l ~ R OH
t ~0
!w
R OH (XI)
which then are reacted after partial protection of one hydroxy
group and activation of the r~ ining group with sodium azide to
the corresponding azido compound of general formula XII
R
Ir 1 10'
R N
~0
10' 15
R OR ~I)
in which R1s stands for a protective group, such as, e.g., a
benzyl group.
After cleavage cf OH-protective group R15 and activation of
the resulting hydroxy group, e.g., as methanesul~onic acid ester,
the reaction i6 fir6t performed with the . oLLe:,~o~lding
ethylen~;;,~inf~ and then the azide group is reduced in a way
2~77977
known in the art, e.g., with triphenylphosphine, to compounds of
f ormula XI II
R
Ir ¦ lo~
R~ NHl R
R ~ ~NH2
Rw R R2 (Xm)
The latter are reacted in a known way with bromoacetic acid
ester to the corresponding pentaesters . Af ter cleavage of the
acid protective groups, e.g., by reaction with trifluoroacetic
acid and generation of desired groups RB, R9, R10 and Rl~ from
groups R8, R9, R10 and R11, the desired complexing agents are
obta ined .
An alternative process for the production of complexing
agents of group II starts from acid-protected polyaminocarboxylic
acid derivatives of general formula XIV
C~
R R R3
R 00~, ,~ y~,--GOOR
(~2 a~2 ~2
~OR COOR CX)OR
2177977
which are react~d with isocyanato rolnrQ~ln~l~ of general formula XV
R
R~R
R ~\N=C=O
(XV)
to the corresponding urethanes.
As an alternative, the hydroxy group in the compounds of
general forr~ula XIV can also be reacted, e.g., with N-
chlorosuccinimide to the corresponding chloride of formula XVI
'\ R R R3
R 00
~2
COaR ~CR C~OR (XVI~
The latter is then reacted in a way known in the art with a
reactive species of a desired (e.g., hydroxy-group-containing or
carboxy-group-containing) halogenated aromatic compound of
formula XVII
l~R
R ~Y'
R (XVII)
2177977
in which R~, R9, R10 and R11 have the indicated r ni n~s and Y'
stands for an OH or COOH group, to the corresponding ethers or
esters. The cleavage of acid protective groups R14 is carried
out in the above-described way.
As an alternative, the compounds of general formula XVI can
be reacted with an azide (e.g., sodium azide) to the
coLLe-yol,~;n~ azido compound, which then is reduced in a known
way to the amino compound. The latter is then
a) either reacted with an isocyanato compound of formula XV
to the corresponding urea derivative or
b) reacted with a halobenzoyl chloride of formula XVIII
R
Il I lC
R O ~xvm)
to the corr~srnn~l i n~ amide .
An alternative proces~ starts from an aminoethyl alcohol of
formu1a XIX,
R~
~N~
!,6
R (XIX)
in which R16 stands for an amino protective group, preferably a
benzyloxycarbonyl group and R13 means an unhalogenated precursor
2~ 7~77
of the desired aromatic compound, or a "linker, " to which the
desired halogenated aromatic compound is bound in a later
reactiOn step. The aminoethyl alcohol is reacted f irst in a way
known in the art, e.g., with methanesulfonic acid chloride,
toluenesulfonic acid chloride or trifluoroacetic anhydride to the
corresponding mesylate, tosylate or triflate and then it is
reacted with an optionally substituted ethylene~ mi n~ . If, in
the case of R13', an unhalogenated precursor of the desired
aromatic compound of general formula IV is involved, it is
iodized, e.g., with iodomonochloride; if, however, a "linker" is
involved, the latter is brought to reaction with a reactive
species of the desired aromatic compound (or its unhalogenated
precursor) .
Finally, the amino protective groups are cleaved and reacted
with haloacetic acid ester to the desired amino acids (complexing
agents) .
The complexing agents of group III, i . e., complexing agents
in which the halogenated aromatic radical is bound in the form of
an amide bond to the carboxylic acid groups of the
polyaminopolycarboxylic acid, can be produced analogously to the
processes that are described in DE 42 32 925.
Thus, the complexing agents can be produced by partial
conversion of activated carboxyl groups, e . g., of the desired
pentacarboxylic acid, to amide groups. For this process, all
synthesis possibilities known to one skilled in the art are
considered, such as, e.g., the reaction of the acid anhydrides of
2177~77
general formulas XX or XXI wi~h halogenated aromatic compounds of
general formula XXII to the amides according to the invention,
00H
R ~ R
0~ ~1 "N~ ~0
~T~ R2 ~o
O O
R ~ R
HOOC/~N~N~ /\
H~C~ R- ~0
O
R
r I RW'
~`~
R ~2
1~'
R (XX~)
in which R8, R9, R10 and R1~ stand for desired groups R8, R9, R10
and R11 or a precursor of the latter, and Q stands for the
radical of a linker of general formula V. The production of the
217~77
aromatic compounds of general formula XXII is carried out as
described, e.g., in DE 25 23 567.
As radical H2N-Q, there can be mentioned a5 examples an H2N-
CH2--CO-NH, H2N-NH--CO--NH, H2N-CH2CH2--CO-NH, H2N-NH-CO-CH2CH2, H2N-
CH2CH2-NH-co group or an H2N-CH2CH2-N(C0-CH3) group.
This reaction is carried out in liquid phase. Suitable
reaction media are, for example, water, dipolar aprotic solvents,
such as diethyl ether, tetrahydrofuran, dioxane, acetonitrile, N-
methylpyrrolidone, dimethylformamide, dimethylacetamide and the
like cr mixtures thereof. The reaction temperatures are between
about -80C and 160C, and temperatures of 20C to 80C are
preferred. The reaction times are between 0.5 hour and 7 days,
preferably between 1 hour and 36 hours.
The production of acid anhydrides of general formula XX can
be carried out according to known processes , e . g ., according to
the process with acetic anhydride in pyridine that is described
in US 3,660,388 or in DE 16 95 050. But in certain cases, it is
advantageous to undertake gently the dehydration with
carbodiimides in a suitable solvent, such as, e.g.,
dimethylformamide or dimethylacetamide.
The production of the monoanhydrides of general formula XXI
is described in J. Pharm. Sci., 68 (1979) 194.
The halogenated aromatic compounds that are used in the
various processes are known or can be generated easily from the
known compounds.
Thus, e.g., in German laid-open specification DE 29 28 417,
iodized aromatic compound~ are described, which are readily
24
2177~77
reacted with, e.g., thionyl chloride, to the corr~-cpon~in~ acid
chloride group-containing aromatic compounds.
Other aromatic radicals can be produced as described in M.
Sovak; ~adiocontrast Agents, E~andbook of Experimental
Pharmacology Vol. 73 (1984), Springer Verlag, Berlin -
h~rg - New York - Tokyo or in European Patent EP 0 015 867.
Corresponding chlorine or bromine compounds can be produced
as described in patents EP 0 055 689 or DE 10 03 743, EP 0 073
715 or EP 0 118 347 -- or analogously to the compounds that are
described there.
Amino group-containing aromatic compounds, as they are
required, e.g., for the production of acetic acid-substituted
compounds of group III, can be obtained analogously to the
compounds that are described in DE 25 23 567.
The production of the metal complexes according to the
invention from the above-described complexing agents of groups I-
III is carried out in the way as disclosed in patents EP 0 071
564, EP 0 130 934 and DE 34 01 052, by the metal oxide or a metal
salt (for sxample, the nitrate, acetate, carbonate, chloride or
sulfate) of the element of atomic numbers 12, 13, 20-31, 39-42,
44-50 or 57-83 being dissolved or 6uspended in water and/or a
lower alcohol (such as methanol, ethanol, isopropanol and/or N,N-
dimethylformamide) and reacted with the solution or suspension of
the equivalent amount of the complexing agent.
If desired, other acidic hydrogen atoms of acid groups can
then be substituted by cations of inorganic and/or organic bases
or amino acids.
217~7~
As bases, inorganic bases (e.g., hydroxides, carbonates or
bicarbonates) of, e.g., sodium, potassium or lithium and/or
organic bases , such as , i . a ., primary , secondary and tertiary
amines, such as, e.g., ethanolamine, morpholine, glucamine, N-
methyl glucamine and N,N-dimethyl glucamine, as well as basic
amino acids, such as, e.g., lysine, arginine and ornithine, are
suitable .
To provide neutral complex compounds, enough of the desired
bases can be added, for example, to the acid complex salts in
aqueous solution or suspension to ensure that the neutral point
is reached. The solution obtained can then be evaporated to
dryness in a vacuum. Often, it is advantageous to precipitate
the neutral salts formed by adding water-miscible solvents, such
as, e.g., lower alcohols (methanol, ethanol, isopropanol, etc. ),
lower ketones (acetone, etc. ), polar ethers (tetrahydrofuran,
dioxane, 1,2-dimethoxyethane, etc.) and thus to obtain easily
isolated and readily purified crystallizates. It has proven
especially advantageous to add the desired base as early as
during the complexing of the reaction mixture and thus to save a
process step.
If the acid complex compounds contain several free acid
groups, it is often suitable to produce neutral mixed salts,
which contain both inorganic and organic cations as counterions.
This can happen, for example, by the complexing acid being
reacted in aqueous suspension or solution with the oxide or salt
of the element supplying the central ion and half of the amount
of an organic base required for neutralization, the complex salt
26
217~77
formed being isolated, optionally purified and then being mixed
with the required amount of inorganic base for complete
neutralization. The sequence of addition to the base can also be
reversed .
Another object of the invention are agents that contain at
least one of the compounds according to the invention.
The invention further relates to a process for the
production of these agents, which is characterized in that the
paramagnetic complex salt that is dissolved in water is brought
into a form that is suitable for enteral or parenteral
administration with the additives and stabilizers that are
commonly used in galenicals, so that the complex salt is present
at a concentration of 1 to 1500 mmol/l, preferably at a
concentration of 10-1000 mmol/l. The halogen content of the
solutions is usually in the range between 10-400 mg/ml. The
resulting agents are then optionally sterilized. They are
administered generally in a dose of 1-300 ml on the basis of the
halogen content and the diagnostic problem.
Suitable additives are, for example, physiologically
harmless buffers (such as, e.g., tromethamine), small additions
of complexi~g agents (such as, e.g.,
diethylenetriaminepentaacetic acid) or, if necr~cs~y,
electrolytes, guch as, e.g., sodium chloride or, if necessary,
antioxidants, such as, e.g., ascorbic acid.
If su~pensions or solutions of the agents according to the
invention in water or physiological salt solution are desired for
enteral adm inistration or other purposes, they are mixed with one
27
2~77~77
or more adjuvants that are commonly used in galenicals (e.g.,
methyl cellulose, lactose, mannitol) and/or surfactants (e.g.,
lecithins, Tween~R), Myrj~R) and/or flavoring substances for taste
correction (e . g., essential oils) .
In principle, it is also possible to produce the diagnostic
agents according to the invention even without isolating the
complex salts. In each case, special care must be used to
undertake the chelation, so that the salts and salt solutions
according to the invention are virtually free of noncomplexed
metal ions that have a toxic effect.
This can be ensured, for example, using color indicators,
such as xylenol orange, by control titrations during the
production process. The invention therefore relates also to
processes for the production of complex cl olln~ and their
salts. As a final precaution, there remains purification of the
isolated complex salt.
Further objects of the invention are characterized by the
claims .
The substances according to the invention meet the varied
requirementS that are to be set for contrast media in modern
diagnosis. The compounds and agents produced from them are
distinguished by:
-- a high absorption coefficient for x rays,
-- good compatibility, which is necessary to maintain the
noninvasive nature of the studies,
-- high effectiveness, which is necessary to burden the body
with the ss~allest possible amounts of oreign substances,
2~ 77977
-- good water-solubility (this makes it possible to produce
highly concentrated solutions, as they are necessary especially
for use as x-ray contrast media. Thus, the volume burden of the
cycle can be kept within reasonable limits),
-- low viscosity,
-- low osmolality,
-- adYantageous elimination kinetics.
Further, the agents according to the invention exhibit not
only high stability in vitro, but also surprisingly high
stability in vivo, so that a release or an exchange of the ions
that are not covalently bound to the complexes -- toxic in
themselves -- i8 not carried out within the time in which the new
contrast media are again completely excreted.
In addition to the high water 601ubility, which it was
possible to increase, surprisingly enough, in the presence of
paramagnetic metal ions in a range necessary for diagnostic
radiology, the ~ G.~I~ds according to the invention have a
positive efect in diagnostic radiology, in that the complex
compounds according to the invention, surprisingly enough, allow
for studies with more shortwave x-ray radiation than is possible
with conventional contrast media, by which the radiation exposure
of the patient is considerably reduced, since soft radiation, as
is generally known, is a great deal more strongly absorbed by the
tissue than hard radiation (R. Felix, Das Rontgenbild [The X-Ray
Picture ]; rhieme Stuttgart 19 8 0 ) .
Because of the advantageous absorption properties of the
contrast meaia according to the invention in the area of hard x-
2177~77
ray radiation, the media are also especially suited for digitalsubtraction techniques (which operate at higher tube voltages).
Especially to be emphasized is the advantageous in vivo
distribution behavior of the media according to the invention.
This allows for the first time for taking x-ray pictures of high
diagnostic informative value in the area of the liver with a dose
that is commonly used for x-ray contrast media (halogen content:
50-400 mg/ml; dose 0.1 - 1 ml/kg of body weight).
Thus, the complexes according to the invention already
produce an optimum contrast of the liver at a dose of O . 5
mmol/kg. Figure 1, upper picture, shows the liver of a rat
before the contrast medium is administered. The lower picture
shows the liver of the same rat 10 minutes after in~ection of 0. 5
mmol/kg of the c~ : ulld according to the invention produced
according to Example ld).
A picture taken under otherwise identical conditions after
the 6ame dose of the disodium salt of the gadolinium complex of
(4s) 4- (4-ethoxybenzyl) -3, 6, 9-tris (carboxylatomethyl) -3, 6, 9-
triazalln~An~linic acid is administered (EP O 405 704; Example
8c) does not show any diagnostically usable contrasting of the
liver (see Fig. 2, lower picture). The upper figure shows the
liver before the administration of contrast medium.
In comparison, Figure 3 shows the density enhancement (which
can be regarded as a measurement for the effectiveness of a
contrast medium) as a function of time, for a compound according
to the invention (Example ld) and a compound of EP O 405 704
(Example 8c). Accordingly, considerably higher density values
21~77
~re observed in the liver of the rat over the entire period of
study for the compound according to the invention. Thus, the
maximum values for the substance according to the invention are
approximately 60 Houndsfield units (HU~, but only 15 HU for the
comparison substance. The studies were performed on a somatome
plus VD31 (parameter of study: layer thickness = 2 mm, tube
voltage/current = 120 kV/290mA) on female rats (body weight =
200-280 g) after intravenous injection of 0. 5 mmol/kg each of the
re3pective substance.
Also, the complexes that are described in WO 93/16375 do not
show any concentration in the liver that is sufficient for
imaging .
Thus, the isomeric gadolinium complex of 1,13-bis-[5-
(propion-3-ylamido) -2, 4, 6-triiodoisophthalic acid-bis- (2-hydroxy-
l-hydroxymethylethyl) -diamide] -4, 7 ,10-tris- (carboxymethyl ) - (2 ,12-
dioxo)-1,4,7,10,13-pentatriazadecane (Example 17b), in Example 1
of WO 93/16375, was excreted almost completely through the
kidneys. Only 1.3% of the total amount was eliminated in other
ways from the body. Even ~sS~m; n~ that this 1. 3% of the complex
had been completely concentrated in the liver, this amount would
be far below the dose that is required for an imaging effect.
These studies were performed on female rats (90-110 g of
body weight) after intravenous administration of 0. 27 mmol/kg of
c _ul.d 17b). The iodine concentration in the blood, urine,
feces, as well as liver, kidneys, spleen, bones, was measured
with x-ray fluorescence analysis. In addition, the gadolinium
concentration was determined with ICP-atom emission spectroscopy.
2177977
The half-life of o . 32 hour as well aG the distribution volume
show a distribution in the extracellular space with ren21
elimination by glomerular f iltration through the kidneys .
The agents according to the invention, which contain in the
complex a paramagnetic metal ion of an element of atomic numbers
21-29, 42, 44 or 57-70, can be used, in addition to use in
diagnostic radiology, also in N~R diagno6is. This dual nature
opens up further f ields of use . Thus, these agents according to
the invention can always be used advantageously, if a combination
of diagnostic radiology and NMR diagnosis is necessary for
differentiated visualization and reliable determination of
certain diseases. This is true, e.g., in the case of suspicion
of recurrence after tumor operations or radiation therapies. In
these cases, the patient is spared an additional burden resulting
from double administration by using a contrast medium, which is
equally suitable for both techniques.
Taken overall, it has been possible to open up new
possibilities in diagnostic medicine with the above-mentioned
complex compounds.
The following examples are used for a more detailed
explanation of the object of the invention, without intending to
be ll~lted to these ex~mple~.
21~7g~7
Exampl~ 1
Ga~olinium comelex o~ the di80dium ~lt of 3,6,9-triaz7-3,6,9-
trL8-~carbo~y ~hyl~-~-[4-(2,4,6-triiodobenzyloxy)-benzyl]-
nller-r - '; oic ~cid
a) Production Or 2,~,6-tr~edob~n~yl chlorid~
41.6 g (80.1 mmol) of 3-amino-2,4,6-triiodobenzyl chloride
tCollection Czechoslov. Chem. Commun. [Vol. 41] 1976) is
s~p~n~lP~ in 416 ml of glacial acetic acid and mixed in portions
with a suspension of 6 . 08 g (88 .1 mmol) of sodium nitrite in 40
ml of concentrated sulfuric acid while being stirred. The
reaction temperature is held at 25C by cooling. After 30
minutes, the reaction mixture is added to a suspension of 12 g of
copper powder in 416 ml of methanol and stirred until nitrogen
generation is completed at room temperature. Then, the
suspension is cooled to 10C, f iltered, the residue is
absorptively precipitated for 30 minutes with 300 ml of N,N-
dimethylformamide, and the suspension is filtered. The filtrate
is concentrated by evaporation in a vacuum, the residue is
absorptively precipitated with water, filtered off and dried in a
vacuum. The crude product is stirred in hot acetonitrile with
activated carbon, then it is filtered, and the filtrate is cooled
to 0C, whereby a precipitate results. The latter is suctioned
off and dried in a vacuum.
Yield: 29.8 g (73.8~) of light beiqe solid
33 21~977
Analy~is (relative to solventless subYtance):
Cld: C 16. 67 H 0 . 80 Cl 7 . 03 I 7S . 50
Fnd: C 16.82 H 0.95 Cl 7.14 I 75.41
b) 3,6,9-Triaz~-3,6,9-tris-(t~rt-butoxyc~rbonylm~thyl)-4-[4-
(2,~,6-tri~ocl~h~n~yloYy)-benzyl]-l 'ec~ns~ioic ~cid-~i-t~rt-
butyl aster
15.6 g (20.0 mmol) of 3,6,9-triaza-3,6,9-tris-(tert-
butoxycarbonylmethyl)-4-(4-hydroxy-benzyl)-undecanedioic acid-di-
tert-butyl ester (Example 9f of DE 3710730) is mixed in
tetrahydrofuran at 0C with 660 mg (22 . 0 mmol) of 80% sodium
hydride suspension in mineral oil. 12 . 4 g (22 . o mmol) of the
2 , 4 , 6-triiodobenzyl chloride, produced according to Example la),
i8 added to the above and stirred for 3 hours. Then, the
solution is mixed with water, tetrahydrofuran is distilled off,
and the aqueous emulsion is extracted with diethyl ether. The
organic phase is washed with water, dried on NazSO4 and
concentrated by evaporation.
The residue is chromatographed on silica gel 60 (Merck) with
hexane/methyl acetate/triethylamine, the product fractions are
concentrated by evaporation and dried in a vacuum.
Yield: 22.8 g (91.296 of theory) of yellowish oil
Analysis (relative to solventless substance):
Cld: C 46.20 H 5.82 I 30.51 N 3.37 0 14.10
Fnd: C 46.37 H 5.93 I 30.44 N 3.35
34
2177977
c) 3,6,9-~riasa-3,6,9-tris-~carboxylssthyl)-4-t4-~2,~,6-
triiodobon8yloxy)-benzyl]-~ndecAne~ioic ~cid
22.8 q (18.3 mmol) of the tert-butyl ester described in
Example lb) i6 dissolved in 250 ml of trifluoroacetic acid and
stirred for 1 hour at room temperature. Then, the solution is
mixed with tert-butyl methyl ether, the precipitate is suctioned
off, washed with tert-butyl methyl ether and dried at 40C in a
vacuum on phosphorus pentoxide. The crude product is
absorptively precipitated in water, filtered off and dried in a
vacuum .
Yield: 15.4 g (86.8% of theory) of light beige solid
Analysis (relative to anhydrous substance):
Cld: C 34.77 H 3.33 I 39.36 N 4.34 o 18.19
Fnd: C 34.63 H 3.56 I 39.28 N 4.38
d) G~dolinium complex of tho disodium ~alt of 3,6,9-triaza-
3,6,9-tris-~c~lrboxymothyl)-4-t~ 2,~,~-triiodobenzyloxy)-
b~nzyl]-~n~ec~n~;oic acid
A suspension of 11.8 g (12.2 mmol) of the penta acid,
produced according to Example lc), in 118 ml of water is mixed
with 2 . 21 g (6.1 mmol) of gadolinium oxide and stirred at 80C
for 2 hour6 . Then, 24 . 4 ml of lN sodium hydroxide solution is
added with a microburette and stirred for 1 more hour. Then,
after 0 . 5 g of activated carbon is added, the solution is stirred
for 2 hours at 80C and filtered. After freeze-drying, the
filtrate yields a colorless solid.
2177977
Yield: 13.1 g (91.89~ of theory)
Analysis (relative to anhydrous substance):
Cld: C 28 . 86 H 2 . 34 I 32 . 67 N 3 . 61 O 15 .10 Gd 13 . 49 Na 3 . 95
Fnd: C 28.66 H 2.43 I 32.70 N 3.49 Gd 13.28 Na 4.16
ExamplQ 2
Gadol~nium complex of the -;^o~ lt of 3,6,9-triaza-3,6,9-
tris-tc~LLv~ethy~ [4-(N-~cctyl-3-methylamino-2~4~6
triiodobenzyloxy)-benzyl]-unaecanedioic ~cid
a) N-Ac~tyl-3-methyl~mino-2, ~, 6-triiollobenzyl chloride
42.5 g (79.7 mmol) of 3-methylamino-2,4,6-triiodobenzyl
chloride (Collection Czechoslov. Chem. Commun. [Vol. 41] 1976) is
dissolved in 180 ml of N,N-dimethylacetamide and mixed drop by
drop with 13.7 ml (191.3 mmol) of acelyl chloride while being
cooled with ice. After 30 minutes of stirring at about 0C, it
is stirred for 12 hours at room temperature and the dark brown
solution is introduced in water while being stirred. A
precipitate is precipitated, which is suctioned off and dried in
a vacuum.
Yield: 44 . 6 g (99 . 6~ of theory) of light beige solid
Analysis (relative to anhydrous substance):
Cld: C 20 . 88 H 1. 58 Cl 6 .16 I 66 .17 N 2 . 43 O 2 . 78
Fnd: C 20.98 H 1.69 C1 6.04 I 66.18 N 2.52
36
217 977
b~ 3,6,9-q!riaz~-3,6,9-tris-(tert-butoxyc~rbonylmethyl)-~-[~-tN-
acetyl-3-methylamino-2, 4, 6-triiodobenzyloxy) -benzyl] -
~nl~c~ngl~;oiC acid-di-tert-butyl diester
15.6 g (20.0 mmol) of 3,6,9-triaza-3,6,9-tris-(tert-
butoxycarbonylmethyl) -4- (4-hydroxybenzyl) -undecanedioic acid-di-
tert-butyl ester (Example 9f of DE 3710730) is mixed in
tetrahydrofuran at 0C with 660 mg (22.0 mmol) of 80% sodium
hydride suspension in mineral oil. 12.66 g (22.0 mmol) of the
compound, produced according to Example 2a), is added to the
above and stirred for 3 hours. Then, the solution is mixed with
water, tetrahydrofuran is distilled off, and the aqueous emulsion
is extracted with diethyl ether. The organic phase is washed
with water, dried on Na2SO4 and concentrated by evaporation. The
residue is chromatographed on silica gel 60 (Merck) with
hexane/methyl acetate/triethylamine, the product fractions are
concentrated by evaporation and dried in a vacuum.
Yield: 23 . 5 g (89 . 2% of theory) of yellowish oil
Analysis (relative to solventless substance):
Cld: C 46 . 45 H 5 . 89 I 28 . 87 N 4 . 25 0 18 . 49
Fnd: C 46 . 63 H 5 . 96 I 28 . 72 N 4 .18
c) 3,6,9-Triaza-3,6,9-tris-(carboXymethyl)-4-[4-(N-aCetyl-3-
methyla~ino-2, ~, 6-triiodobenzyloxy) -benzyl] -unde~anedioic
~c id
21.9 g (16.6 mmol) of the tert-butyl ester described in
Example 2b) is dissolved in 250 ml of trifluoroacetic acid and
~ 2177977
stirred for 1 hour at room temperature. Then, the solution is
mixed with diethyl ether; the precipitate is suctioned off,
washed with diethyl ether and dried at 40C in a vacuum on
phosphoru8 pentoxide. The crude product i5 absorptively
precipitated in water, filtered off and dried in a vacuum.
Yield: 16.2 g (94.1% of theory) of light beige solid
Analysis (relative to anhydrous substance):
Cld: C 35.86 H 3.59 I 36.67 N 5.40 0 18.49
Fnd: C 35.73 H 3.75 I 36.81 N 5.41
d) G~olinium complex of the disodium sAlt of 3,6,9-triaza-
3,6,9-tris-(c~rboxymethyl) -~.-t4-(N-acetyl-3-methylAmino-
2,4,6-tri;odob~n~yloxy~-benzyl]-undecanedioic ACid
A suspension of 14.8 g (14.3 mmol) of the penta acid,
produced according to Example 2c), in 150 ml of water is mixed
with 2 . 58 g (7 .13 mmol) of gadolinium oxide and stirred at 80C
for two hours. Then, 28 . 5 ml of lN sodium hydroxide solution is
added with a microburette and stirred f or one more hour . Then,
after 0 . 8 g of activated carbon is added, the solution is stirred
for two hours at 80C and filtered. After concentration by
evaporation, the ~iltrate yielded a colorless ~olid.
Yield: 16.4 g (93.3~c of theory)
Analysis (relative to anhydrous substance):
Cld: C 30.11 H 2.61 I 30.79 N 4.53 0 15.53 Gd 12.72 Na 3.7a
Fnd: C 30.00 H 2.82 I 30.58 N 4.67 Gd 12.79 Na 3.82
38
21779~7
Ex~mpl~ 3
Gadolinium compl~3x of th~ trisodium s~lt of 3,6,9-triaza-3,6,9-
tris- (carboxymethyl) -{~-[N- (3-car~oxypropionyl) -3-methylamino-
2, ~, 6-triiodol~nzyloxy] -benzyl} -ll nrler~n e~ ~ oic acid
a) N- (5-Oxa-1, ~-dioxoheptyl) -3-methyl~mino-2, 4, 6-triiodo~enzyl
chloride
24.7 g (150 mmol) of succinic acid chloride monoethyl ester
is added at room temperature to a suspension of 53 . 3 g (100 mmol)
of 3-methylamino-2, 4, 6-triiodobenzyl chloride (Collection
Czechoslov. Chem. Commun. tVol. 41] 1976) in 200 ml of anhydrous
dioxane, stirred with exclusion of moisture. The batch is
refluxed for several hours until no more feedstock can be
detected according to thin-layer chromatography; then, it is
concentrated by evaporation, the residue is taken up in
dichloromethane and shaken out with saturated aqueous sodium
bicarbonate solution. Af~er drying on anhydrous magnesium
sulfate, th~ organic phase is concentrated }~y evaporation, and
the residue is recrystallized from ethyl acetate/tert-butyl
methyl ether.
Yield: 58.4 g (88.396 of theory) of colorless solid
Analys~ s (relative to solventless substance):
Cld: C 25.42 H 2.29 Cl 5.36 I 57.56 N 2.12 0 7.26
Fnd: C 25.31 H 2.49 C1 5.43 I 57.50 N 2.17
1- 2177977
b) 3~6~9-Triaz~-3~6~9-tris-~t~rt-butoxycarbonylmethy~ -{~-tN
~5-ox~ -dioxo-heptyl) -3-methyl~mino-2,~, 6-
tr~odob~n~yloxy~-benzyl}-~n~ ec-ne~ ioic aci~
15 . 6 q (20 . 0 mmol) of 3, 6, 9-triaza-3, 6, 9-tris- (tert-
butoxycarbonylmethyl)-4-(4-hydroxybenzyl)-undecanedioic acid-di-
tert-butyl ester (Example 9f of DE 3710730~ is mixed in
tetrahydrofuran at 0C with 660 mg (22 . 0 mmol) of 80% sodium
hydride suspension in mineral oil. 14.55 g (22.0 mmol) of the
compound, produced according to Example 3a), is added to the
above and stirred for 3 hours. ~hen, the solution is mixed with
water, tetrahydrofuran is distilled off, and the aqueous emulsion
is extracted with diethyl ether. The organic phase is washed
with water, dried on sodium sulfate and concentrated by
evaporation. The residue is chromatographed on silica gel 60
(Merck) wit~l hexane/ethyl acetate/triethylamine, the product
fractions are concentrated by evaporation and dried in a vacuum.
Yield: 22.9 g (81.6~c of theory) of yellowish oil
Analy ~is (relative to solventless substance):
Cld: Y 47.02 E~ 5.95 I 27.10 N 3.99 0 15.94
Fnd: ~ 46.86 H 6.13 I 26.98 N 3.84
c) 3,6,9--~rri~z~-3,6,9-tris-~carboxymethyl)-{~-[N-~3-
c~rbo~propionyl) -3-methylamino-2,4,6-triiodobenzyloxy]-
benzyl~---nC~ec~n~ ioic ~cid
20.4 ~ (14.5 mmol) of the hexaester described in Example 3b)
is dissolv~ in 100 ml of methanol and mixed with 87 ml of 2N
~1 77977
sodium hydroxide solution. It i8 refluxed for about 2 hours, the
methanol is drawn off in a vacuum and, after 100 ml of water is
added, it is stirred for another 2 hours at 60CC. By adjusting
to pH 1-2 with semiconcentrated hydrochloric acid, a colorless
precipitate results, which is suctioned off and dried in a
vacuum .
Yield: 15 . 3 g (96 . 0% of theory~ of colorless solid
Analysis (relative to anhydrous substance):
Cld: C 36.15 H 3.59 I 34.72 N 5.11 0 20.43
Fnd: C 36.23 H 3.65 I 34.58 N 5.05
d) G~aolinium complex of the trisodium salt of 3,6,9-triaz~-
3,C,9-tris-~c~rboxy~ethyl)-{4-[N-(3-c~rb~YyroL~ionyl1 ~3~
methylzlmino-2,~, 6-triioaobenzyloxy]-benzyl}-~n~ec~ne~ioic
acia
A suspension of 14.1 g (12.9 mmol) of the hexa acid,
produced according to Example 3c), in 150 ml of water is mixed
with 2.33 g (6.43 mmol) of gadolinium oxide and stirred at 80C
for 2 hours . Then, 38 . 6 ml of lN sodium hydroxide solution is
added with a microburette and stirred for 1 more hour. Then,
after 0. 8 g of activated carbon is added, the solution is stirred
at 80C for 2 hours and filtered. After freeze-drying, the
filtrate yielded a colorless solid.
Yield: 16.3 g (96.4~6 of theory)
41
1-- 2~ 77977
Analysis (relative to anhydrous substance):
Cld: C 30.11 H 2.53 I 28.92 N 4.26 0 17.01 Gd 11.94 Na 5.24
Fnd: C 30 . 01 H 2 . 64 I 28 . 88 N 4 . 34 Gd 11. 86 Na 5 . 02
ExAmple ~
G~dolinium compleY o~ 3,6,9-tri~za-3,6,9-tris-~caLl,~,A~ ~hyl)-~-
t3,5-diiod-~-ethoxybenzyl)--ln~ec-n~oic acid, di30dium s~lt
a) 3,6,9-Triaza-3,6,9-tris-tcarbOXymethyl)-4-t4-hydroxybenzyl)-
ioiC ~cid
7.8 g (10 mmol) of 3,6,9-triaza-3,6,9-tris-(tert-
butoxycarbonylmethyl)-4-(4-hydroxybenzyl)-undecanedioic acld-di-
tert-butyl ester (Example 9f of DE 3710730) is dissolved in 100
ml of trif luoroacetic acid and stirred f or 1. 5 hours at room
temperature. Then, it is diluted with diethyl ether, and the
precipitate is suctioned off. It is washed with diethyl ether
and dried at 50C in a vacuum. The crude product is dissolved in
water and treated with activated carbon. The filtered solution
is freeze-dried several times to remove residual trifluoroacetic
acid .
Yield: 4 . 0 g (80 .1~ of theory) of colorless lyophilizate.
Elemen~ary analysis (taking into consideration the solvent
content):
Cld: C 50.50 H 5.85 N 8.41 0 35.24
Fnd: C 50 . 68 H 5. 99 N 8 . 25
42
~ 21~7977
b) 3,6,9-~riaza-3,6,9-tri8-(carboYym~thyl)-4-~3,5-aiioao-~-
L~ ~L ;)nybCnZy~ -n-lecA n e~l~ QiC acid
3.2 g (6.4 mmol) of the phenol of Example 4a) is suspended
in 50 ml of water and mixed with solid sodium hydroxide up to the
neutral point . The batch is stirred at 50C, and 5 . 7 ml ( 14 .1
mmol) of a 40% hydrochloric acid-iodomonochloride solution is
added in drops. After 20 hours at 50C, the excess iodine is
reduced with sodium disulfite, the precipitate is suctioned off
and washed with water.
Yield: 4.15 g (8696 of theory) of pale yellow solid
Elementary analysis (taking into consideration the solvent
content):
Cld: C 33.57 H 3.62 I 33.78 N 5.59 o 23.43
Fnd: C 33.71 H 3.86 I 33.41 N 5.65
c) 3,6,9-Triaza-3,6,9-tris-~carboYymethyl)-5-(3,5-diio~o-4-
ethoYybenzyl)-undecaneaioic acia
3 . 0 g (4 mmol) of the diiodophenol of Example 4b) is mixed
in 25 ml of anhydrous tetrahydrofuran with 0.792 ~ (26.4 mmol) of
80~6 sodium hydride suspension in mineral oil. 4.1 g (26.4 mmol)
of ethyl iodide is added to this suspension, and the reaction
mixture is stirred for 6 hours at room temperature. Then, it is
mixed with 30 ml of 2N sodium hydroxide solution, evaporated to
dryness and the residue is taken up in water. The aqueous
solution is acidified with concentrated hydrochloric acid, the
2177977
precipitate is suctioned of f and washed with water . For
purification, the crude product is recrystallized from ethanol.
Yield: 2 . 35 g (7S . 4% of theory) of colorless crystals .
Elementary analysis (taking into consideration the solvent
content ~:
Cld: C 35.45 H 4.01 I 32.57 N 5.39 0 22.58
Fnd: C 35.59 H 3.94 I 32.39 N 5.23
d) G~lolinium complex of 3,6,9-triaz~-3,6,9-tri~-
(C~LLO~y -thyl)-4-~3,5-diiodo-4-ethoxy~enzyl)-~1n~ler~ ioic
~c$d
1. 75 g (2 . 2 mmol) of the pentacarboxylic acid of Example 4c)
is suspended in 55 ml of water and mixed at 60C with 407 mg (1.1
mmol) of gadolinium oxide. After 4 hours, the clear solution is
treated with activated carbon. Then, it is polished with a
cellulose-mem3~rane filter (0.2 mm, Sartorius) and freeze-dried.
Yield: 1.95 g (94.996 of theory) o~ colorless lyophilizate.
Ele~nentary analysis (taking into consideration the solvent
conte nt):
Cld: C 29.59 H 3.02 Gd 16.84 I 27.19 0 18.85
~nd: C 29.64 H 3.25 Gd 16.66 I 26.93
44
~ 2177'J77
e) Gadol$nl~m comploY of 3,6,9-trisz~-3,6,9-tris-
~c~rboxymethyl)-4-~3,5-diiodo-4-ethoxybenzyl~ n~lec-ne~ioic
~cid, disodium salt
1.5 g (1.6 mmol) of the complex described in the above
example i5 dissolved in 120 ml of water and mixed using a
microburette with 3 . 2 ml of a lN sodium hydroxide 601ution .
After freeze-drying, the disodium salt is obtained as colorless
lyophilizate .
Yield: 1.55 g (99% of theory) of colorless lyophilizate.
Elementary analysis (taking into consideration the solvent
content ):
Cld: C 28.26 H 2.68 Gd 16.09 I 25.96 N 4.30 Na 4.70 0 18.00
Fnd: C 28.03 H 2.91 Gd 15.86 I 25.72 N 4.09 Na 4.4s
Ex~mple 5
Gadolinium complex of the disodium salt of 4- tN-acetyl-3-
methylamino-2,~,6-triiodobenzyloxymethyl)-3,6,9-triaza-3,6,9-
tris- ~ c~rbo~ymethyl ) -~ ~ n eA i o ic acid
a) N-Ac~tyl-N-methyl-3-[ ~2,2-dimethyl-1,3-dioxolan-4-yl)-
metho~methyl ~ -2, ~, 6-triiodoani1 ine
20.0 g (36.3 mmol) of the compound produced under Example
2a), 5.8 g (43.6 mmol) of 2,3-0-isopropylidene glycerol, 0.41 g
(1.8 mmol) of N-benzyl-N,N,N-triethylammoniUm chloride and 4.1 g
(72.7 mmol) of ground potassium hydroxide are refluxed in 35 ml
of toluene _or 6 hours. Then, the organic phase is separated,
shaken out -~.ith saturated aqueous common salt solution and dried
2177977
on magnQsium sulfate. After the filtrate i6 filtered and
concentrated by evaporation, an oily residue is obtained, which
is chromatographed on silica gel with toluene/ethyl acetate.
Concentration by evaporation of the product fractions yields a
colorless oil, which is dried in a vacuum.
Yield: 21. 3 g (87 . 2% of theory)
Analysis (relative to solventless substance):
Cld: C 28.64 H 3.00 I 56.73 N 2.09 0 9.54
Fnd: C 28 . 60 H 3 . 09 I 56 . 72 N 2 .11
b) N-Acetyl-N-methyl-3- t (2, 3-dihydroxypropyloxy) -methyl] -2, 4, 6-
triio~o~n i ~ i n~
20.2 g (30.1 mmol) of the compound produced according to
Example 5a) is introduced in a mixture of 60 ml of ethanol and 10
ml of concentrated sulfuric acid. After 12 hours of stirring at
30C, the batch is taken up in dichloromethane, and the organic
phase is shaken out once with concentrated sodium chloride
solution and twice with concentrated sodium bicarbonate solution.
The organic phase is dried on anhydrous magnesium sulfate,
f lltered and concentrated by evaporation . The residue is
chromatographed on silica gel 60 (MercX) with
dichloromethane/methanol. After the product fractions are
concentrated by evaporation, a colorless oil is obtained, which
is dried in a vacuum.
Yield: 16.9 g (89.29~ of theory)
46
~ 2177977
Analysis (relative to solventless substance):
Cld: C 24.75 H 2.56 I 60.34 N 2.22 O 10.14
Fnd: C 24.86 H 2.69 I 60.12 N 2.34
c) N-Ac~tyl-N-methyl-3- [ ( 3 -b~nzoyloxy-2 -hydroxypropyloxy) -
methyl~ -2, ~., 6-triio-io~ n~ 1 ~ n~
15.2 g (24.1 mmol) of the compound produced in Example Sb)
is stirred in 150 ml of dichloromethane under argon and mixed
first with 4.0 ml (28.9 mmol) of triethylamine, then at 0C drop
by drop with 3 . 47 g (26 . 5 mmol) of benzoyl cyanide. After 12
hours of stirring at 0C, the batch is diluted with
dichloromethane and shaken out against saturated sodium
bicarbonate solution. The organic phase is dried on magnesium
sulfate, filtered and concentrated by evaporation, and the
residue is chromatoqraphed on silica gel 60 (Merclc) with
dichloromethane/methanol. After concentration by evaporation,
the product fractions yield a colorless oil.
Yield: 13 . 9 g (78 . 4% of theory)
Analy~;is (relative to solventless substance):
Cld: C 32.68 H 2.74 I 51.79 N 1.91 O 10.88
Fnd: C 32.54 H 2.88 I 51.83 N 1.74
d) N-Ac~t~l-N-mothyl-3-[ ~3-benzoyloxy-2-
meth~nesul~onyl~ryLoy~loxy) methyl]-2,4,6-triiorlo~ n~1~rt
13.4 g (18.2 mmol) of the compound produced under Example
5c) is stirTed in 80 ml of dichloromethane under argon and mixed
47
2177977
first with 3 . o ml (21. 9 mmol) of triethylamine, then at oC drop
by drop with 1. 56 ml t20 .1 mmol) of methanesulfonic acid
chloride. The reaction temperature is allowed to increase within
3 hours to room temperature and then shaken out against saturated
sodium bicarbonate solution. The organic phase is dried on
magnesium sulfate, filtered and concentrated by evaporation. The
oily residue is chromatographed on silica gel 60 (~erck) with
dichloromethane, the product fractions are concentrated by
evaporatiOn, and the residue is dried in a vacuum.
Yield: 12.8 g (86.2% of theory) of yellowish foam
Analysis (relative to solventless substance):
Cld: C 31.02 H 2.73 I 46.82 N 1.72 o 13.77 s 3.94
Fnd: C 31.20 H 2.89 I 46.67 N 1.83 S 4.02
e ) N-Acetyl-N-methyl-3 - [ ~ 3 -benzoyloxy-2 -az idopropyloxy ) -
methyl]-2~ 6-triiodoaniline
11.8 g (14.5 mmol) of the compound produced in Example 5d)
is stirred in 50 ml of N,N-dimethylformamide together with 2.83 g
(43 . 5 mmol) of sodium azide for one hour at 85CC under argon.
Then, it is concentrated by evaporation in a vacuum, and the
residue is shaken out with dichloromethane/saturated sodium
bicarbonate solution. The organic phase is dried on magnesium
sulfate, filtered and concentrated by evaporation in a vacuum.
Yield: 10. 2 g (92 .19~ of theory) of yellowish foam
48
2177977
Analysis (relative to 601ventless free substance):
Cld: C 31.60 H 2.52 I so.os N 7.37 o 8.42
Fnd: C 31.59 H 2.63 I 49.87 N 7.49
f) N-Acetyl-N-methyl-3-t ~2-~zido-3-hydroxypropyloxy) methyl]-
2,~,6--triiodc~-ni 1 in~
9 . 58 g (12 . 6 mmol) of the compound produced according to
Example 5e) is di6solved in 60 ml of methanol. After 40 ml of 2N
sodium hydroxide solution is added, it is stirred for one hour at
50C bath temperature and after the cooling, it is neutralized
with 2N hydrochloric acid . The methanol is drawn of f in a
vacuum, and the residue is dispersed between dichloromethane and
saturated sodium bicarbonate solution. The organic phase is
dried on magnesium sulfate, filtered, concentrated by
evaporation, the residue is chromatographed on silica gel 60
(l~erck), and the product fractions are concentrated by
evaporation in a vacuum.
Yield: 7.47 g (90.396 of theory)
Analysis ~relative to solventless substance):
Cld: C 23.80 H 2.31 I 58.04 N 8.54 0 7.32
Fnd: C 23.92 H 2.50 I 57.85 N 8.6
g) N-Acetyl-N-methyl-3-t (2-~zido-3-
moth~e~ulfonyloxypropyloxy)-methyl]-2~4~6-triiollo~n;
-
49
217~977
7.22 g (11.0 mmol) of the hydroxy compound produced
according to Example Sf ) is reacted to the corresponding mesylate
under the conditions described in Example 5d).
Yield: 7.46 g (92 4% of theory)
Analysis (relative to solventless substance):
Cld: C 22.91 H 2.33 I 51.86 N 7.63 0 10.90 S 4.37
~nd: C 23.01 H 2.58 I 51.63 N 7.75 S 4.49
h) N-Acetyl-N-methyl-3-(6,9-diaz~-4-~zido-2-oxanonyl)-2,4,6-
triiodo~ni ~ inQ, dibydrochloride
7 . 21 g (9 . 82 mmol) of the mesylate described in Example 5g)
is dissolved in 50 ml of methanol, and after 150 ml of 1,2-
diaminoethane is added, it is stirred f or 15 hours at room
temperature. Then, the batch is concentrated by evaporation and
dispersed between dichloromethane and saturated sodium
bicarbonate solution. The aqueous phase is extracted several
times with dichloromethane, the combined organic phases are dried
on sodium sulfate, filtered and concentrated by evaporation. The
residue is taken up in tert-butyl methyl ether/methanol and
adjusted to pH 2 with concentrated hydrochloric acid, whereby a
colorless precipitate precipitates. The latter is separated and
dried in a vacuum.
Yield: 7.33 g (96.8% of theory)
so
2177977
Analysis (relative to solventles6 6ubstance):
Cld: C 23 . 37 H 3 . 01 I 49 . 38 N 10. 90 0 4 . lS Cl 9 . 20
Fnd: C 23.28 H 3.22 I 49.39 N 11.02 Cl 9.37
i) N-Acetyl-N-methyl-3-(4-amino-6,9-diaz~-2-oxanonyl)-2,4,6-
tri ~ oc'l~- n ~ l i ne, trihydrochlorid~
7 . 04 g (9 .13 mmol) of the dihydrochloride produced under
Example 5h) is taken up in 70 ml of a 4: l mixture of
dioxane/water and mixed with 12.0 g (4S.7 mmol) of
triphenylphosphine. The batch is allowed to stir for 3 days at
room temperature under argon, the organic solvent is evaporated,
and precipitate is filtered out. The precipitate is washed with
2N hydrochloric acid; the combined filtrates are concentrated by
evaporation, and the residue is recrystallized from
methanol/tert-butyl methyl ether.
Yield: 6.12 g (8S.8% of theory)
Analysis (relative to solventless substance):
Cld: C 23.05 H 3.35 I 48.72 N 7.17 0 4.10 Cl 13.61
Fnd: C 23 . 28 H 3 . 60 I 48 . 49 N 7 . 43 Cl 13 . 88
; ) ~- (N-Acetyl-3-methyl~mino-2, ~, 6-triiodobenzylo~ymethyl) -
3,6,9-tris-~tert-butyloxycarbonylmethyl~ -3,6,9-
triaz~un~e~nedi oic ~cid-di-tert-butyl ester
5 . 98 g (7 . 6S mmol) of the trihydrochloride produced under
Example 5i) is stirred in 60 ml of N,N-dimethylformamide under
argon at room temperature and mixed with 10 . 6 g (76 . 5 mmol) Of
51
217797~
pOtassium earbonate and 7.46 g (38.3 mmol) of bromoacetie aeid-
tert-butyl ester. After 12 hours of stirring, it is filtered,
coneentrated by evaporation in a vacuum, and the residue is
dispersed between ethyl acetate and saturated sodium biearbonate
601ution. The organie phase is dried on sodium sulfate,
filtered, eoncentrated by evaporation, and the residue is
chromatographed on siliea gel 60 (Merek) with hexane/ethyl
acetate. After the product fractions are concentrated by
evaporatiOn, a yellowish oil is obtained.
Yield: 8.94 g (98.5~c of theory)
Analysis (relative to solventless substance):
Cld: C 41.50 H 5.52 I 32.08 N 4.72 O 16.18
Fnd: C 41.52 H 5.73 I 31.96 N 4.68
k) ~~ (N-Acetyl-3-methylamino-2, 4, 6-triiodobenzyloxymethyl) -
3,C,9-~riAzA-3,6,9-tris-(c~rboxymethyl~-undec~nedioic ACid
8 . 50 g (7 .16 mmol) of the pentaester produced under Example
5; ) is eonverted to the eorreseonding penta acid under the
conditions deseribed in Example lc).
Yield: 5.80 g (84.19~ of theory) of light beige solid
Analys-- (relative to anhydrous substance):
Cld: e 31.20 H 3.46 I 39.56 N 5.82 O 19.95
Fnd: C 31.25 H 3.66 I 39.42 N 5.~3
52
2t 77977
l) Gadolinium compleY of the qisodium s~lt o~ 4- (N-~c~tyl-3-
methylamino-2, ~, 6-triioao~enzyloxymethyl ] -3, 6, 9-tri~z~-
3, 6, 9-tris-tc~rboxymethyl) -~nde~nadioic ~cid
5.69 g (6.19 mmol) of the penta acid produced in Example 5k)
ls complexed with gadolinium oxide under the conditions described
in Example ld) and converted to the corresponding disodium salt.
Yield: 6.81 g (94.896 of theory) of colorless lyophilizate
Analysis (relative to anhydrous substance):
Cld: C 25.88 H 2.43 I 32.81 N 4.83 0 16.55 Gd 13.55 Na 3.96
Fnd: C 25.94 H 2.61 I 32.78 N 4.85 Gd 13.44 Na 4.00
~xample 6
1,19-Bis-~3-carboxy-2,4,6-triiodophenyl~ -7,10,13-tri~-
(carboYymethyl) -2,5,15,18-tetraoxo-1,4,7,10,13,16,19-
hepta~z~non~decane, g~dolinium complex, disodium s~lt
a) 1,19-Bi~-~3-c~rboYy-2,4,6-triiodophenyl)-7,10,13-tris-
(carboYym~thyl)-2,5,15,18-tetraoxo-1,4,7,10,13,16,19-
hept~ no~ --lqc -ne
11.42 g (20 mmol) of 3-glycylamino-2,4,6-triiodobenzoic acid
(DE 2523567~ is dissolved in 60 ml of N,N-dimethylformamide while
being heate~. It is mixed at room temperature with 6 . 9 ml of
triethylamine and 3.6 g (10 mmol) of N,N-bis-[2,6-
dioxomorpholino) ethyl] -glycine, and the reaction mixture is
stirred for 15 hours at room temperature. Then, it is evaporated
to dryness, the residue is taken up in water and acidified with
concentrate~ hydrochloric acid. The settled precipitate is
2177g77
suctioned of f and washed with water . The crude product is
purified by an RP 18 chromatography on silica gel.
Yield: 9.~ g (63% of theory) o~ colorless solid.
Elementary analysis (taking into consideration the solvent
content):
Cld: C 25.60 H 2.22 I 50.73 N 6.53 0 13.18
Fnd: C 25.53 H 2.35 I 50.52 N 6.29
b) 1,ls-Bis-~3-cJ~rboxy-2,~,6-triio~ophenyl)-7,10,13-tris-
(c~rboxymethyl)-2,5,15,18-tetraoxo-1,4,7,10,13~16,19-
hepta~ r~0n~d~c~ , gadolinium complex, disodium s~lt
7 . 2 g (48 mmol) of the ligand of Example 6a) is suspended in
50 ml of wa,er and mixed in portions with 1.74 g (4.8 mmol) of
gadolinium oxide at 50-60C. After the complexing is completed,
the pH is aljusted to seven with lN sodium hydroxide solution,
filtered, ~d the aqueous solution is freeze-dried.
Yield: 7. 6 g (93% of theory) of colorless lyophilizate.
Elemer--ary analysis (taking into consideration the solvent
content~:
Cl~: - 22.62 ~{ 1.66 Gd 9.25 I 44.81 N 5.77 Na 2.71 0 13.18
Fn~: _ 22.43 H 1.85 Gd 9.07 I 44.71 N 5.63 Na 2.49
~ 54
217797~
Bxampl~ 7
l~lg-Bis-{3-~(lo-carboxydecyl)-~A~rh-r yl~-2,4,6-triiodophenyl}-
7,10,13-tris-(carboYymethyl~ -2,5,15,18-tetraoxo-
1, 4, 7, 1 0, 1 3, 1 6, 1 9 h e p ~ n ~ c ~ , g a d o 1 i n i um c o mp l e x,
disodium salt
a) ~ 3 _7~n i nA AcetylrAmido ) -N- ( 10 -carboYydecyl ) -2, 4, 6 -
tri ;Od~AhAn70iC acid amide
lO.9 g (15 mmol) of 3-phthalimidoacetylamino-2,4,6-
triiodobenzoic acid chloride (DE 2523567) is dissolved in 60 ml
of N,N-dimethylacetamide and reacted at 80C with 1.98 g (16
mmol) of ll-Am;nolln~ An~ic acid. The reaction mixture is
stirred for 32 hours at this temperature, and then hydrochloride
is filtered out. The filtrate is evaporated to dryness, the
residue is suspended in 40 ml of water and reacted with 4 . 5 g (90
mmol) of hydrazine hydrate. After three hours of stirring at
65C, the reaction mixture is allowed to cool, and the settled
precipitate is suctioned off. The product is rewashed with ample
water, and the solid is dried at 50C in a vacuum.
Yield: 9.8 g (8796 of theory) of pale yellow crystals.
Elementary analysis (taking into consideration the solvent
content):
Cld: C 34.73 H 4.05 I 40.77 N 6.75 0 13.71
Fnd: C 34 . 90 H 3 . 92 I 40 . 68 N 6 . 51
2177977
b) l,l9-Bis-{3-t(10-c_rboxydecyl)-~ArbA--yl~-2,4,6-
trilodophenyl}-7~lo~l3-tris-(carboxymethyl~ -2,5,15,18-
t~tr~oYo- l , ~., 7, 1 0 ,13 ,16 ,19 -heptaa zanonadecane
8.75 g (11.6 mmol) of the amine of Example 7a) is reacted
analogously to Example 6a) with 2.14 g (6 mmol) of N,N-bis-[2-
(2,6-di~ ~holino)ethyl]glycine and purified in a similar way
by a eolumn ehromatography on RP 18.
Yield: 17 . 4 g (809~ of theory) of pale yellow solid.
Elementary analysis (taking into consideration the solvent
content):
Cld: C 34.73 H 4.05 I 40.77 N 6.75 0 13.71
Fnd: C 34.90 H 3.92 I 40.68 N 6.51
c) l,l9-Bis-{3-t(10-carboYydecyl)-carbamoyl]-2,4,6-
triiodophenyl}-7~10~13-tris-(carboxymethyl)-2~5~15~18-
tetr_oYo-1,~,7,10,13,16,19-heptaazanonAde~ne, gadolinium
co~ples, di~odium ~alt
15 g (8 mmol) of the ligand of Example 7b) is complexed
aeeording to Example 6b) with 2.9 g (8 mmol) of gadolinium oxide
and eonverted with lN Godium hydroxide solution to the disodium
salt .
Yield: 15.6 g (95% of theory) of colorless lyophilizate.
56
2177~77
Elementary analysis (talcing into consideration the solvent
content):
Cld: C 31.40 H 3.42 Gd 7.61 I 36.86 N 6.10 Na 2.23 0 12.39
Fnd: C 31.28 H 3.63 Gd 7.56 I 36.61 N 5.89 Na 1.97
Exampl~ 8
~;~dolinium complex of th~ Cisodium sAlt of 4-(3-~cetylamino-
2, 4, 6-tr~; o-lol~ Qyl-aminomethyl) -3, 6, 9-triaza-3, 6, 9-tris-
(CaLLOAy ~hyl)-4-mcthyluntleCanedioic ~cid
a) 2, ~-Dimethyl-4-methanesulfonyloxymethyl-2-oxazo1ine
40.8 g (316 mmol) of 2,4-dimethyl-4-hydroxymethyl-2-
oxazoline (J. Nys and J. Libeer, Bull. Soc. Chim. Belg., 65, 377
(1956) ) is stirred in 400 ml of dichloromethane and 52.5 ml (379
mmol) of triethylamine at oC under nitrogen and mixed drop by
drop with 39.8 q (347 mmol) of methanesulfonic acid chloride.
The reaction temperature is allowed to increase to room
temperature within 3 hours, and the batch is shaken out with
saturated sodium bicarbonate solution. The organic phase is
dried on sodium sulfate, filtered and concentrated by
evaporation .
Yield: 58.5 g (89.4% of theory) of yellowish oil
Analysis (relative to solventless substance):
Cld: C 40.57 H 6.32 N 6.76 0 30.88 S 15.47
Fnd: C 40.49 H 6.48 N 6.83 S 15.30
217797~
b) ~- (2, 5-Diazapentyl) -z, ~-~imethyl-2-oxazoline,
~ihy~rochlori~l~
A solution of 36.7 g (177 mmol) of the compound, produced
according to Example 8a), in 100 ml of methanol is added drop by
drop to 291 ml (4427 mmol) of 1,2-diaminoethane. The reaction
mixture i5 6tirred for 3 hours at 50C and for another 12 hours
at room temperature. Then, the batch is completely concentrated
by evaporation in a vacuum. A solution of the residue in
methanol is adjusted to pH 1. 5 at 0C with concentrated
hydrochloric acid. Ethyl~n~ mi n~-dihydrochloride precipitating
in this connection is separated by filtration. By adding tert-
butyl methyl ether in drops to the filtrate, a colorless
precipitate results, which is suctioned off and dried in a
vacuum .
Yield: 38.2 g (88.49~ of theory)
Analysis (relative to solventless substance):
Cld: C 39.35 ~ 7.84 N 17.21 0 6.55 C1 29.04
Fnd: C 39.40 H 7.78 N 17.09 Cl 29.11
c) 2-Amino-~.~ 7-aiAz~-2-methylheptan-1-ol, trihy~rochlori~c
30.8 g (126 mmol) of the dihydrochloride produced under
Example 8b) is taken up in 150 ml of ethanol. After 31 ml of
concentrated hydrochloric acid is added, it is refluxed for 4
hours. After the cooling, the batch is concentrated by
evaporation on a vacuum and stirred in 300 ml of isopropanol.
~ 58 2177~77
The precipitate is suctioned of f, washed with isopropanol and
diethyl ether and dried in a vacuum.
Yield: 29.6 g (91.4 of theory)
Analysis (relative to solventless substance):
Cld: C 28.08 H 7.86 N 16.38 0 6.24 Cl 41.45
Fnd: C 28.23 ~ 7.95 N 16.46 Cl 41.19
d) 3,6,9-q~ri~z~-3,6,9-tris-ttcrt-butyloxyc~rbonylmethyl)-4
hydro~lymethyl-~-methyl-~ln~ec-n~ i oic ~cid-di-tert-butyl
~ster
18.9 g (73.7 mmol) of the trihydrochloride produced under
Example 8c) is added to a solution of 51.2 g ~369 mmol) of
potassium carbonate in 60 ml of water. While being stirred
vigorously, 60. 0 ml (369 mmol) of bromoacetic acid-tert-butyl
ester, dissolved in 60 ml of tetrahydrofuran, is now added and
stirred for 6 hours at 60C. After the cooling, ethyl acetate
and water are added and shaken out; the aqueous phase is
eYtracted several times with ethyl acetate. The combined organic
phases are dried on sodium sulfate, filtered, and the filtrate is
concentrated by evaporation in a vacuum.
Yield: 52 . 2 g 198 . 896 of theory)
Analysis (relative to solventless substance):
Cld: C 60.23 H 9.41 N 5.85 0 24.51
Fnd: C 60.11 H 9.62 N 5.67
2177g77
e) ~-Chlorom~thyl-4-methyl-3, 6, 9-tris- (tert-
butyloYyc~rbonylmethyl) -3, 6, 9-tri~zaundecanedioic acid-ai-
tert-butyl ester
A solution of 22.3 g (31.0 mmol) of the alcohol, produced
under Example 8d), in 100 ml of dichloromethane is mixed with
8 . 91 g (34 . 0 mmol) of triphenylphosphine and, after cooling to
0C, mixed with 4.54 g (34.0 mmol) of N-chlorosuccinimide. After
2 hours of stirring at 0C, it i6 6tirred up with 200 ml of
diethyl ether, the solid is separated and discarded. The ether
phase is concentrated by evaporation, and the re6idue is
chromatographed on silica gel 60 (Merck) with hexane/ethyl
acetate ( 2 :1 ) .
After concentration by evaporation in a vacuum, the product
fractions yield a yellowish oil.
Yield: 18.7 g (81.7% of theory)
Analysis (relative to solventless substance):
Cld: C 58.72 H 9.03 Cl 4.81 N 5.71 0 21.73
Fnd: C 58 . 68 H 9 . 23 Cl 4 . 98 N 5 . 64
f ) 3, 6, 9--~rriA za-~-~zi~omethyl-3, 6, 9 -tris- ( tert-
butylcsycarbonylmethyl)-4-methylundecanedioic acid-di-tert-
butyl ester
A soll=tion of 18. 6 g (25 . 3 mmol) of the chloride, produced
under Exa3i~le 8e), in 70 ml of N,N-dimethylformamide is mixed
with 4.92 S (75.8 mmol) of sodium azide and stirred for 6 hours
at 50~C. ~en, it is concentrated by evaporation in a vacuum,
-
2177977
and the residue is dispersed between ethyl acetate and saturated
sodium bicarbonate solution. After the organic phase is dried on
magnesium sulfate, filtration and concentration by evaporation, a
yellowish oil is obtained.
Yield: 18 . 2 g (97 . 0% of theory~
Analysis (relative to solventles6 substance):
Cld: C 58.20 H 8.95 N 11.31 0 21.73
Fnd: C 58.15 H 8.72 N 11.18
g) ~-Aminomethyl-3, 6, 9-triaz~-3, 6, 9-tris- ~ tert-
butyloxycArbonylmethyl~-4-methyllln~ec~ne~ioic Acid-di-t~rt-
butyl ester
A solution of 18.0 g (24.2 mmol) of the azide, produced in
Example 8f), in 180 ml of ethanol is vigorously shaken after
0.90 g of palladium on activated carbon (10% by weight of
palladium, manufacturer Degussa) is added under hydrogen
at~ ^re, until no more hydrogen absorption can be observed.
Then, catalyst is filtered out, and the filtrate is concentrated
by evaporation in a vacuum.
Yield: 17.4 g (99.996 of theory) of yellowish oil
Analysis (relative to solventless substance):
Cld: C 60.31 H 9.56 N 7.82 0 22.32
Fnd: C 60.2Z H 9.78 N 8.03
61
217797~
h) ~-~3-Acetylamino-2~,6-triio~obenzoyl~m~r -thyl)-3,6,9-
triilz~-3,C,9-tri~-ttert-butyloxyc~rbonylmothyl)-4-
methyllln~e~ne~ioic ~cid-di-tert-butyl ester
A solution of 17.2 g (24.0 mmol) of the amine, produced
under Example 8g), in 70 ml of N, N-dimethylacetamide is mixed
with 3.99 ml (28.8 mmol) of triethylamine and 15.2 g (26.4 mmol)
of 3-acetylamino-2, 4, 6-triiodobenzoyl chloride (H. Priewe et al .,
Chem. Ber. 87, 651 (1954) ), and it is stirred for 6 hours at room
temperature. ~hen, it is concentrated by evaporation in a
Yacuum, the residue is dispersed between ethyl acetate and
6aturated sodium bicarbonate solution, and the organic phase is
dried on sodium sulfate. After filtration, the filtrate is
concentrated by evaporation, and the residue is chromatographed
on silica gel 60 (Merck) with hexane/ethyl acetate (3 :1) . After
the product fractions are concentrated by evaporation, a
yellowish oil is obtained.
Yield: 27 . 9 g (92 . 6~ of theory)
Analysis (relative to solventless substance):
Cld: C 43.04 H 5.78 I 30.32 N 5.58 O 15.29
Fnd: C 43.21 H 5.86 I 30.19 N 5.63
i) 4-~3-Acetylamino-2,~,6-triiodobenzoyl~mino~ethyl)-3,6,9-
tri~zl-3,6,9-tri3-~c~ Lo..y~t;hyl)-4-methyl~ln~e~An~ioic _ci~
26.6 g (21.2 mmol~ of the pentaester described under Example
8h) is converted to the corresponding penta acid under the
conditions described in Example lc).
62 2177977
Yield: 19.5 g (94.4%) of light beige solid
Analysi6 lrelative to anhydrous substance):
Cld: C 30.79 H 2.32 I 39.04 N 7.18 Na 3.92 0 19.69
Fnd: C 30.98 H 2.40 I 38.84 N 7.24 Na 4.04
j ) Gadolinium compleY of the disodium s_lt o~ 4- (3-_cetylamino-
2,~,6-triiodobenzoyl-~min~ ~thyl~-3,6,9-triAza-3,6,9-tris-
(carboxymethyl)-4-methyl-1-n~l~cAn~;oic ~cid
18.8 g (19.3 mmol) of the penta acid described in Example
8i) is converted to the title compound under the conditions
described in Example lc).
Yield: 20.7 g (91.5% of theory) of colorless solid
Analysis (relative to anhydrous substance):
Cld: C 25.59 H 2.32 Gd 13.50 I 32.44 N 5.97 Na 3.92 0 16.36
Fnd: C 25.64 H 2.40 Gd 13.29 I 32.27 N 6.08 Na 4.04
BYampl~ 9
Gadolinium complex of the disodium salt of 4- ~3-acetyl_mino-
2,~,6-triio.lob~n7oyl-oYymathyl)-3,6,9-triazA-3,6,9-tris-
(c_rboyymethyl~-4-methylundec_nedioic ~cid
a ) ~ - ~ 3 -~cety lam ino - 2, ~, 6 -tr i iodoben zoy loxymethy l ) - ~ -methy l -
3,6,9--tris-~tert-bUtyloxy-c_rboYymethyl)-3~6~9-
tri~z~undecanedioic acid-di-tert-butyl ester
A solution of 14.7 g (20.0 mmol) of the chlorine c , u--d,
described -~n Example 8e), i~ 30 ml of N,N-dimethylacetamide is
63 2i77g77
added at room temperature to a solution of 16.9 g (29.9 mmol) o~
the sodium salt of 3-acetylamino-2, 4, 6-triiodobenzoic acid
(Wallingford et al., J. Am. Chem. soc. 74, 4365 (1952) ) in 50 ml
of N, N-dimethylacetamide . The reaction mixture is stirred f or 6
hours at 80C, then concentrated by cvaporation in a vacuum and
shaken out with ethyl acetate and saturated sodium bicarbonate
solution. The organic phase is dried on sodium sulfate, filtered
and concentrated by evaporation, the residue is chromatographed
on silica gel 60 ~lerck) with hexane/ethyl acetate (3:1). After
the product fractions are concentrated by evaporation, a light
beige oil remains.
Yield: 18.0 g t71.9% of theory~
Analysis (relative to solventless substance):
Cld: C 43.01 H 5.59 I 30.29 N 4.46 0 16.55
Fnd: C 43.11 H 5.84 I 30.42 N 4.48
b) ~-~3-Acctylamino-2,~,6-triiodooenZoyloXymethyl)-4-methyl-
3,6,9-triazl~-3,6,9-tris-~tert-~utyloxy-c~lL~ I.hyl)-
nl~eCIInt~(~;oiC aci~
17.7 g (14.1 mmol) o~ the pentaester described under Example
ga) i8 converted to the correspondin~ penta acid under the
conditions described in Example ld).
Yield: 12.7 g (92.7% of theory) of light beige solid
217~977
Analysis (relative to anhydrous substance):
Cld: C 30.76 H 3.20 I 39.00 N 5.74 0 21.30
Fnd: C 30 . 81 H 3 . 48 I 38 . 90 N 5 . 77
c) GadolLnium complex of th~ di~odium salt of 4- (3-acetylamino-
2,~,6-triiodoh,~n~oyl-oxymethyl)-3,6,9-triaza-3,6,9-tris-
~carboxymethyl)-~-methyl~ndec~n~d~oic acid
9 . 26 g (9 . 48 mmol) of the penta acid described under Example
9b) is converted to the title compound analogously to the
conditions described in Exa~ple ld).
Yield: 9.88 (88.7% of theory) of colorless 601id
Analysis (relative to anhydrous substance):
Cld: C 2S.14 H 2.29 I 33.21 N 4.89 0 16.75 Gd 13.72 Na 4.01
Fnd: C 25.13 H 2.38 I 33.11 N 4.93 Gd 13.67 Na 4.11
EYampl~ 10
Gadolinium complex of the ~isod$um 8:~ lt of 3, 6, 9-triaza-3, 6, 9-
tris- (carbo~yml~thyl) -4-methyl-~- (3-methylc~ h-- yl-2, 4, 6-
triiodophenyloxymethyl)-l~n~ec~ne~;oic acid
a) 3, 6, 9-~rriaza-3, 6, g-tris- (tert-butyloxycarbonylmethyl) -4-
methyl-4-(3-m~thyl-~ yl-2,4,6-triiodophenyloxymethyl)-
undQcanedioic acid-di-tcrt-butyl ester
A sol~tion of 18.9 g (25.7 mmol) of the chlorine compound,
described a~cording to Example 8e), in 35 ml of N,N-
dimethylace~amide is added at room temperature to a solution of
16.3 g (30.3 mmol) of 3-hydroxy-2,4,6-triiodobenzoic acid-
~ 2177~77
methylamide (P.L. Conturior, Ann. Chim II 10 (1938) 559) and 1.72g (30.8 mmol) of potassium hydroxide in 30 ml of N,N-
dimethylformamide. The reaction mixture is stirred for 8 hours
at 60C, then concentrated by evaporation in a vacuum, and the
residue is shaken out with ethyl acetate and saturated sodium
biearbonate golution. The organic phase is dried on sodium
sulfate, filtered and concentrated by evaporation, the residue is
chromatoqraphed on siliea gel 60 (Merck) with hexane/ethyl
acetate (3 :1) . After the product fractions are concentrated by
evaporatiOn, a light beige oil remains.
Yield: 21.9 g (69.496 of theory)
Anal~-sis (relative to solventless substance):
Cld: C 43.01 H 5.82 I 30.98 N 4.56 0 15.63
Fnd: C 43.20 H 5.97 I 30.~2 N 4.60
b) 3~ 6, 9--TriAza-3~6~s-tri~-(cAr~o~yL.~l~hyl) -4-methyl-4-~3-
~ethyl~rh~- yl-2,~,6-triiodophenyloxymethyl)-unaecanedloic
~ci~l
20 . 6 (17 . 0 mmol) of the pentaester described in Example
lOa) is cc~verted to the corresponding penta acid under the
conditions ~escribed in ~xample lc).
Yielc~: 13.9 g (86.3% of theory) of light beige solid
66
217~77
Analysis (relative to anhydrous substance):
Cld: C 30.40 H 3.30 I 40.15 N 5.91 o 20.25
Fnd: C 30.64 H 3.52 I 39.94 N 6.04
c) Gadolinium complex of the disodium s~lt of 3,6,9-tri~zA-
3, 6, 9-tris- (carboxymothyl) -~-methyl-4- (3-methylcarb~moyl-
2,~,6-triiodophenyloxymethyl)-~n-'lec~ne~ioic acid
13.2 g (13.9 mmol) of the penta acid described under Example
lOb) is converted to the title compound analogously to the
conditions described in Example ld).
Yield: 15 . O g (94 .196 of theory) of colorless lyophilizate
Analysis (relative to anhydrous substance):
Cld: C 25.14 H 2.29 I 33.21 N 4.89 0 16.75 Gd 13.72 Na 4.01
Fnd: C 25.13 H 2.38 I 33.11 N 4.93 Gd 13.67 Na 4.11
EYampl~ 11
Gadolinium comple~c of the disodium salt of N,N-bis-[2-[N',N'-bis-
~c~L~ Lhyl)-amino~-ethyl]-3-acetylamino-2,~,6-
triiodophenyl ,.1 ~n ~ ne
a) 3-Amino-2,~,6-triiodophenylAl~n~n~, hydrochloride
A solution of 32 . 5 g (150 mmol) of 3-aminophenylalanine
hydrochloride (Jenninqs, J. Chem. Soc., 1957, 1512) in 300 ml of
water is added in drops at 50C to a mixture of 300 ml of
concentrated hydrochloric acid and 240 ml of 2N potassium iodine
dichloride solution in 6. 0 1 of water while being stirred slowly.
After another 3.5 hours, the hot clouded solution is filtered and
67
217~977
concentrated by evaporation in a vacuum until the crystallizatio"
starts. Then, it is also well-cooled in ice, suctioned off,
absorptively precipitated with water and dried on phosphorus
pentoxide .
Yield: 50.7 g (67.0% of theory) of light beige solid
Analysis (relative to anhydrous substance):
Cld: C 18.19 H 1.70 I 64.06 N 4.71 O 5.38 Cl 5.97
Fnd: C 18.38 H 1.94 I 63.82 N 4.83 Cl 6.11
b) 3-Amino-2,~,6-triiodopbenyl~lAn1nPthyle9ter hydrochloride
30 . 8 g (51. 8 mmol) of the amino acid produced according to
Example lla) is refluxed in a mixture of 150 ml of ethanol and
4.1 ml (57 mmol) of thionyl chloride for 10 minutes, then stirred
for 12 hours at room temperature. The batch i5 then concentrated
by evaporation, and the residue is dried in a vacuum.
Yield: 32. 3 g (100% of theory) of light beige solid
Analysis (relative to anhydrous substance):
Cld: C 21.23 H 2.27 I 61.17 N 4.50 O 5.14 Cl 5.70
Fnd: C 21.44 H 2.38 I 60.93 N 4.62 Cl 5.89
c) ~,N-Bis-[2-[N',N'-bis-[tbenzyloxycarbonyl)-methyl]- mino]-
~thyl]-3-Amino-2,4,6-triiodophenylAl~ninethyle~ter
20.4 g (32.7 mmol) of the amine produced according to
Example llb) and 31.0 g (73.7 mmol) of N,N-bis-
[ (benzyloxycarbonyl) -methyl]-2-bromomethylamine (M. Williams and
68 2177977
H. ~apoport, J. Org. Chem. 58, 1151 (1993) ~ are introduced in 50
ml of acetonitrile and mixed with 20 ml of 2N phosphate buffer
solution (pH 8. 0) . The batch is vigorously stirred at room
temperature for 24 hours, whereby the aqueous phosphate buffer
phase is exchanged after 2 and 8 hours for fresh buffer solution.
Then, the organic phase is concentrated by evaporation in a
vacuum, and the re6idue is chromatographed on silica gel with
hexane/ethyl acetate/triethylamine (3:1:0.01). The product-
containing fractions are concentrated by evaporation in a vacuum.
Yield: 25.8 g (62.396 of theory) of yellowish oil.
Analysis (relative to solventless substance):
Cld: C 48.43 H 4.38 I 30.10 N 4.43 0 12.65
Fnd: C 48.50 H 4.45 I 30.01 N 4.44
d) N~N-Bi!i-t2-[N~N~-bis-[t~enzyloxycnrbonyl)-methyl]-amino]-
ethyl]-3-Acotyl mino-2,4,6-triiodopheny-~lAn;n~thyle:3ter
13.7 g (10.8 mmol) of the compound described in Example llc)
is dissolved in 30 ml of N,N-dimethylacetamide, and after 1.80 ml
(13.0 mmol) of triethylamine and 0.85 ml (11.9 mmol) of acetyl
chloride are added, it is stirred for 12 hours at room
temperature with exclusion of moisture. Then, it is concentrated
by evaporation in a vacuum, and the residue is dispersed between
ethyl aceta~e and sodium bicarbonate solution. The organic phase
is dried on sodium sulfate, filtered and concentrated by
evaporation .
Yield: 13.8 g (97.6% of theory) of yellowish oil
69
2177977
Analysis (relative to solventless substance~:
Cld: C 48.71 H 4.40 I 29.13 N 4.29 0 13.47
Fnd: C 48.83 H 4.67 I 29.02 N 4.38
e) N-N-Big-t2-tN',N'-bis-~eArboxymethyl)-amino]-ethyl]-3-
ac~tylamino-2~6-triiodopheny~ An;nn
12 . 8 g (9 . 80 mmol) of the pentaester deseribed in Example
lld) i8 dissolved in 75 ml of methanol and mixed with 49 ml of 2N
sodium hydroxide solution. It is refluxed for about 2 hours, and
the methanol is drawn off in a vacuum. ~3y ad~usting to pH 1-2
with semieoneentrated hydrochlorie aeid, a eolorless precipitate
results, which is suctioned off and dried in a vacuum.
Yield: 7.80 g (86.796 of theory)
Analysis (relative to anhydrous substance):
Cld: C 30.09 H 3.18 I 41.46 N 6.10 0 19.17
Fnd: C 30.22 H 3.31 I 41.39 N 6.17
f) GAdol~nium complex of the disoditlm salt o~ N,N-bis-t2-
tN' ,~' -bi8- (carboxymethyl) -amino] -ethyl]-3-acetylamino-
2,~, 6--triiodophenylalA~;ne
7.42 5 (8.08 mmol) of the penta acid described in Example
lle) is ee~verted to the title eompound analogously to the
eondition~ deseribed in Example ld).
Yiel~= 8.72 g (96.796 of theory)
217~977
Analysis (relative eO 2nhydrous substance):
Cld: C 24.75 H 2.17 Gd 14.07 I 34.10 N 5.02 Na 4.12 O 15.76
Fnd: C 24 . 64 ~ 2 . 38 Gd 13 . 83 I 33 . 94 N 5 . 08 Na 3 . 89
Exampl~ ~ 2
2-~3-Ac~tamido-2, 1,6-triiodobenzyl)-3,6,~-triaza-3,6,9-tris-
(ca.Lo,~y ~hy~ nr~e~ np~ ic ACid, ga~olinium complex, ~isoaium
8~1t
a) 3,6,9-Triaza-3,6,9-tris-(methoxyc~rbonylmethyl)-
nl~Pc~nel-ioic aci~ methyl e~ter ~JOC 55, 2868, 1990)
20.6 g (52.4 mmol) of diethylenetriaminepentaacetic acld in
618 ml of methanol is introduCed at 0C and mixed drop by drop
with 38.2 ml (0.524 mol) of thionyl chloride within 30 minutes.
Then, the reaction mixture is stirred for 16 hours at room
temperature. After the reaction is completed, it i5 concentrated
by evaporation on a rotary evaporator, and the whitish solid is
suspended in 300 ml of diethyl ether. The suspension is mixed at
0C with 200 ml of saturated sodium bicarbonate solution, the
organic phase is separated, and the aqueou3 phase is extracted
three times with 100 ml each of diethyl ether. The extract is
dried on potassium carbonate and evaporated to dryness after
f iltration . The product is dried overnight in a vacuum on
phosphorus pentoxide.
Yield: 19.7 g (81% of theory) of colorless oil.
71
217~377
Elementary analysis (taking into consideration the solvent
content):
Cld: C 49.24 H 7.18 N 9.07 0 34.52
Fnd: C 49 37 H 7.26 N 8.85
b) 3,6,9-Tri~za-3,6,9-tris-(methoxycarbonylmethyl)-2-(3-
nitrobenzyl)---n~lec~n~ oic ~cid ~imethyl ester
6. 64 ml (47. 3 mmol) of diisopropylamine in 200 ml of
anhydrous tetrahydrofuran is introduced in a light argon stream
at 0C and mixed drop by drop with 22.2 ml (52 mmol) of
butyllithium (159~ in hexane) within 15 minutes. Then, it Ls
cooled to -78C, and 18 . 4 g (40 mmol) of pentaester (Example 12a)
dissolved in 300 ml of anhydrous tetrahydrofuran is added in
portions to it. After 30 minutes of stirring at this
temperature, a solution of 8 .11 g (47 . 3 mmol) of 3-nitrobenzyl
chloride and 1.38 ml (11.44 mmol) of 1,3-dimethyl-3,4,5,6-
tetrahydro-2 (lH) -pyrimidinone in 180 ml of anhydrous
tetrahydrofuran is added within 30 minutes. The reaction mixture
is allowed to reach room temperature overnight, and the solution
is concentrated by evaporation on a rotary evaporator. The oily
residue is taken up in 150 ml of ethyl acetate and mixed with 50
ml of ice ~-ater. The organic phase is separated, and the aqueous
phase is eYtracted three times with 75 ml of ethyl acetate each.
The combine~ organic phases are dried on potassium carbonate,
filtered and evaporated to dryness. To purify the crude product,
the substa}Ece is chromatographed on silica gel 60 (Merck).
Yield: 13.2 g (55% of theory) of pale yellow oil.
21779~7
Elementary analysis (taking into consideration the solvent
content):
Cld: C S2.17 H 6.40 N 9.36 0 32.07
Fnd: C S2.01 H 6.23 N 9.48
c) 2-13-~n~inob~n~yl)-3,6,9-triaza-3,6,9-tris-
~ methoxycarbonylmethyl)-undecanedioic acid dimethyl e3ter
A methanolic solution of 12.7 g (21.2 mmol) of the nitro
compound of Example 12b) is hydrogenated at room temperature with
the addition of 1.35 g of palladium on carbon (10%) at 4 bars.
After 5 hours, the hydrogenation is completed, and catalyst is
filtered out. The filtrate is evaporated to dryness and used in
the next stage without further purification.
Yield: 11. 35 g (94% of theory) of colorless oil .
d) 2-~3-~"-inob--nsyl)-3,6,9-triaza-3,6,9-Sri~-(carboxymethyl)-
undecanedioic ~cid
10.8 g (19 mmol) of the pentaester of Example 12c) is
saponified ~ith 60 ml of 2N sodium hydroxide solution at 40C.
After the reaction is completed, the solution is mixed with
concentrate~ hydrochloric acid until the acid is completely
precipitate~ . The precipitate is suctioned of f and washed
neutral wit~ water. The product is dried overnight at 50C in a
vacuum .
Yield: 9.75 g (96% o~ theory) of colorless solid.
73
2177g77
Elementary analysis (taking into consideration the solvent
content ):
Cld: C 47.15 H 5.84 Cl 6.63 N 10.47 O 29.91
Fnd: C 47.04 H 6.12 Cl 6.35 N 10.59
e) 2-t3-Amiuo-2,4,6-triiodobenZyl)-3,6,9-triaZ~-3,6,9-tris-
~ c~lLo,.~ -thyl)-l~n~e~ne~t~ic ~cid
9 . 55 g (18 mmol) of pentacarboxylic acid (Example 12d) is
suspended in 50 ml of water and mixed drop by drop with 22 . 8 ml
(56 . 4 mmol) of a hydrochloric acid 4096 iodomonochloride solution.
The reaction mixture is allowed to stir for 16 hours at 65C, and
the iodine excess is reduced with diluted sodium disulfite
solution. The settled precipitate is 6uctioned off and rewashed
with water The dried solid is taken up in concentrated ammonia
solution, ~iltered and precipitated with concentrated
hydrochloric acid. The precipitate is washed until neutrality of
the wash w;~ter i8 reached. The product is dried for 18 hours at
50C in a ~cuum until a constant weight is reached,
Yield: 13.1 g (8396 of theory) of light yellow solid.
Eleme~tary analysis (taking into consideration the solvent
content ):
Cld: C 28.79 H 3.11 I 43.45 N 6.39 O 18.26
Fnd: C 28.93 H 3.37 I 43.32 N 6.48
74
217~9~7
f) 2-~3-Ac~tyl~mino-2~6-triiodo3~nnq~yl)-3~6~9-tri
~carboxymethyl)-3,6,9-triaz~undecanedioic acid
12.7 g (14.5 mmol) of triio~o~nil;ne of Example 12e) is
dissolYed in 30 ml of N,N-dimethylaCetamide and mixed with 2.465
ml (34.8 mmol) of acetyl chloride while being cooled with ice.
It is allowed to reach room temperature overnight, and the
reactiOn mixture is stirred in ice water. The precipitate is
SUCtioned off, washed with water and dried at 50C in a vacuum.
Yield: 11.55 g (879~ of theory) of colorless solid.
Elementary analysis (taking into consideration the 301vent
content):
Cld: C 30.09 H 3.18 I 41.46 N 6.10 O 19.17
Fnd: C 29.88 H 3.26 I 41.29 N 6.02
g) 2-(3-Acetamido-2,4,6-triiodobenZyl)-3,6,9-triaZa-3,6,9-tris-
~carbo~cymethyl)-undecanedioic acid, g~dolinium complex,
~isodium ~JIlt
11.2 g (12.2 mmol) of complexing agent of Example 12f) is
reacted acc~rding to the method of Example 4d) with gadolinium
oxide a~ 5C~c. After the complexing is completed, the
intermedia--~ product is converted to disodium salt with lN sodium
hydroxi~e ~lution . The resulting solution is purif ied with 1. 2
g of act~ ed carbon, filtered with a 0.2 ,um membrane-cellulose
f ilter ~ hen freeze-dried.
Yi~'d: 12.6 g (92.5~ of theory) of colorless lyophilizate.
2177977
Elementary analysis (taking into consideration the solvent
content ):
Cld: C 24.75 H 2.11 Gd 14.09 I 34.10 N 5.02 Na 4.12 0 15.76
Fnd: C 24.89 H 2.23 Gd 13.88 I 34.02 N 4.87 Na 4.03
Examp le~ 13
Gadolinium complex of the disodium salt of 3,6,9-triaza-3,6,9-
tri~-(carboxymQthyl~ -~-[3-[N,N'-dimethyl-N,N'-bis-(2,3-
dihydr.",y~r~.pyl) -3, 5-dicarbamoyl-2, 4, 6-
triiodophenylc~ ~lmethoxy~-benzyl]-unaecanedioic acid
a) N,0-Bis-(benzyloxycarbonyl~-3-hydroXyphenylAlAn;ne-N-[2-
(benzyloxycarbonylamino~ -ethyl~ -amide
168.54 g (375 mmol) of N,0-bis-(benzyloxycarbonyl)-3-
hydroxyphenylalanine (de Castiglione, Bosisio, Gazz. Chim. Ital.,
97, 1858 (1967) ) is dissolved in 3.0 l of tetrahydrofuran and
cooled to 0C. After 72.8 ml (525 mmol) of triethylamine is
added, 36.7 ml (383 mmol) of chloroformic acid ethyl ester is
added in drops. After 20 minutes, 75. 8 g (390 mmol) of
benzyloxycarbonyl-(2-aminoethyl)-amide (G. Atwell, W. Denny,
Synthesis, 1032-33 (1984) ) in 500 ml of tetrahydrofuran is added.
After stirring overnight, the resulting precipitate is suctioned
of f, the f iltrate is concentrated by evaporation and dried in a
vacuum .
Yield: 183 . 7 g (78 . 3% of theory) of colorless solid.
2177~77
Analysis (relative to solventless substance):
Cld: C 67.19 H 5.64 N 6.72 0 20.46
Fnd: C 67.07 H 5.78 N 6.84
b) 3 -Hydroxyphenyl A 1 A n; n ~- ~ 2 -aminoethyl ) - mide
62.57 g (100 mmol) of the compound described in Example 13a~
is &llcppn~d in 1. 5 l of methanol and after 6 . 3 g of palladium on
carbon (10% by weight of palladium, Degussa) is added, it is
hydrogenated with hydrogen at normal pressure. Then, it is
filtered, the filtrate is concentrated by evaporation, and the
residue is absorptively precipitated in diisopropyl ether. After
suctioning off and drying in a vacuum, a colorless solid is
obta ined .
Yield: 21. 4 g (95 . 8% of theory)
Analysis (relative to solventless substance~:
Cld: C 59 .17 H 7 . 67 N 18 . 82 0 14 . 33
Fnd: C 59.28 H 7.73 N 18.74
c) 1, 5-Diamino-3-aza-2- ~3-ethoxybenzyl) -pentane,
trihy~rochloride
20.1 g (90 mmol) of the compound described in Example 13b)
is taken up in 135 ml of tetrahydrofuran and mixed drop by drop
at 0C under argon with 180 ml o~ 1 m boron hydride solution in
tetrahydrofuran. After 30 minutes of stirring at 0C, stirring
is continue for 120 hours at 60C. After the cooling, 100 ml of
methanol is added in drops, the reaction mixture is saturated
~ 77
~ 2177977
with hydrogen chloride, and the resulting acid suspension is
stirred for 6 hours. Then, the precipitate is suctioned off and
dried at 50C in a vacuum.
Yield: 25. 0 g (87 . 2~) of colorless solid
Analysis (relatiYe to solventless substance):
Cld: C 41.46 H 6.96 Cl 33.38 N 13.19 0 5.02
Fnd: C 41.40 H 7.04 Cl 33.47 N 13.08
d) 3,6,9-Tri~z~ 3-hydroxybcn~yl)-3,6,9-tris-~tert-
buto~rycArbonylmethyl)-~n~ec~ne~;oic ~cid-di-tert-butyl ester
15.1 g (47.4 mmol) of the compound described in Example 13c)
is s~l~pon~r-tl in 500 ml of tetrahydrofuran and mixed with 25 ml of
water and 34 . 5 g (249 mmol) of potassium carbonate. After 52 . 3
ml (356 mmol) of bromoacetic acid-tert-butyl ester is added in
drops, it is stirred for 3 days at 60C. After the cooling, it
is filtered, concentrated by evaporation in a vacuum, and the
residue is chromatographed on silica gel with diethyl
ether/hexane/triethylamine (70:20:5). The product fractions are
concentrated by evaporation in a vacuum.
Yield: 27 . 2 g (73 . 3% of theory) of yellowish oil
Analysis (relative to solventless substance):
Cld: C 63 .13 H 8 . 92 N 5. 39 0 22 . 56
Fnd: C 63.2g H 8.88 N 5.43
78
2177977
e ) S -Chloroacetylamino-2, 4, 6 -tr i iodoi sophthal ic a¢ id- [N, N ' -
dimethyl-N,N'-bi~-~2,2-dimethyl-1,3-dioxolan-4-ylmethyl) ]-
diamide
81.0 g (100 mmol) of 5-chloroacetylamino-2,4,6-
triiodoisophthalic acid-N,N'-dimethyl-N,N'-bis-(2,3-
dihydroxypropyl)-diamide (D~ 2928417) is introduced in 500 ml of
tetrahydrofuran and mixed with 0. 95 g (5. 0 mmol) of p-
tolu-~n~Qulfonic acid monohydrate and 22.9 g (220 mmol) of 2,2-
dimethoxypropane. Then, it is refluxed for 12 hours,
Concentrated by evaporation in a vacuum, and the residue is
dispersed between ethyl acetate and sodium bicarbonate solution.
The organic phase is dried on magnesium sulfate and filtered,
concentrated by evaporation, and the residue is absorptively
precipitated with tert-butyl methyl ether. After filtering, the
residue is dried in a vacuum.
Yield: 79.7 g (89.69~ of theory) of colorless solid
Analysis (relative to solventless substance):
Cld: C 32.40 H 3.51 Cl 3.99 I 42.79 N 4.72 0 12.5
Fnd: C 32.38 H 3.62 Cl 4.04 I 42.70 N 4.63
f) 3,6,9-Triaza-3,6,9-tris-ttert-butoxycarbonylmethyl~-4-[3-
[N,N'-dimethyl-N,N'-bis-~2,2-dimethyl-1,3-dioxolan-4-
ylmethyl) -3, 5-dicarbamoyl-Z, ~, 6-triiodophenyl-carbamoyl-
metho~]-benzyl]-~nde ~ne~oic acid-di-tert-butyl ester
12.8 g (16.4 mmol) of the hydroxy compound described in
Example 13d) is dissolved in 50 ml of N,N-dimethylformamide under
217797~
argon and mixed with 0.59 g (19.7 mmol) of 8096 sodium hydride
suspension in mlneral oil. After 30 minutes of stirring at room
temperature, 19 . 0 g (21. 3 mmol) of the compound described in
Example 13e) is added, and the batch is allowed to stir for 12
hours at 50C. Then, it is concentrated by evaporation in a
vacuum, and the residue is chromatographed on silica gel 60
(Merck) with diethyl ether/hexane/triethylamine (70:20:5). The
product fractions are concentrated by evaporation in a vacuum.
Yield: 16. 8 g (62 . 6% of theory) of viscous oil
Analysis (relative to solventless substance):
Cld: C 47.80 H 6.11 I 23.31 N 5.15 0 17.63
Fnd: C 47.63 H 6.05 I 23.24 N 5.24
g) 3~6~9-Triaza-3~6~9-tris-(carboxymethyl)-4-[3-[N~N~-dimeth
N,N'-bis-(2,3-dihydroxypropyl)-3,5-dicarbamoyl-2,4,6-
triioaophenylcarbamoylmethoXy]-benzyl]-UndeCanedioiC ~cid
16 .1 g (9 . 86 mmol) of the compound described in Example 13f )
is freed of all protective groups under the conditions described
in Example lc) and converted to the title compound.
Yield: 12.4 g (98.7% of theory) of light beige solid
Analysis (relative to anhydrous substance):
Cld: C 36.81 H 4.04 I 29.92 N 6.60 0 22.63
Fnd: C 36.94 H 4.05 I 29.86 N 6.53
2~7~'17
h) G~tolinium complox of tho disod$um s~lt of 3,6,9-triaz~-
3,6,9-tris-(carboxymethyl) -4-t3-[N,N'-di~ethyl-N,N'-bis-
~2~3-dih~ yyroyyl)-3,5-dicarbamoyl-Z,4,6-triiodophenyl-
carbamoylmethoxy]-benzyl~ n~lec~ne-~10ic ~cid
11.9 g (9.35 mmol) of the penta acid described under Example
13g i6 converted to the title compound analogously to the
conditions described in Example ld).
Yield: 13 .1 g (95. 5% of theory) of colorless lyophilizate
Analysis (relative to anhydrous substance):
Cld: C 31.85 H 3.15 I 25.88 N 5.71 0 19.58 Gd 10.69 Na 3.13
Fnd: C 31.92 H 3.20 T 25.76 N 5.73 Gd 10.58 Na 3.20
Examp l e. 1~
Gadolinium complex of the disodium salt of 3,6,9-tri~za-3,6,9-
tris-tcarb~"~y ~hyl)-4-~3-methoxy-2,-1,6-triiodobenzyl)-
er-net~;oic ~cid
a) 3~ 6, 9-Tri~z~-3, 6, 9-tris- (c~rboxymethyl) -4- (3-hydroxybenzyl) -
undecanedioic acid
13.2 g (16.9 mmol) of the compound described in Example 13d)
i8 converted to the corresponding penta acid under the conditions
described in Example lc).
Yield: 8.20 g (97.0% of theory)
Analysis (relative to anhydrous substance):
Cld: C 50.50 H 5.85 N 8.41 0 35.24
Fnd: C 50.41 H 5.93 N 8.49
81
J~ 2177~77
b) 3,6,9-Tri~z~-3,6,9-tris-tcaLLo,~y -thyl)-4-~3-hydroxy-~,4,6-
tr i1odoben 7yl)_11n-~ e~ n~ iOic aci~
8 .11 g (16. 2 mmol) of the compound described in Example 14a)
is dissolved in 80 ml of 5N aqueous ammonia. 26 . 8 ml of 2N
potassium iodine dichloride solution is now slowly added in drops
while being stirred, and stirring is continued for 12 hours. It
is adjusted to pH 1. 5 with concentrated hydrochloric acid and
sodium disulfite is added until a light suspension is present;
this is stirred for 6 hours and filtered. The residue is washed
with 2N hydrochloric acid and dried in a vacuum.
Yield: 12.1 g (84.89~ of theory)
Analysis (relative to anhydrous substance):
Cld: C 28.75 H 2.99 I 43.40 N 4.79 0 20.06
Fnd: C 28.81 H 2.83 I 43.43 N 4.62
c) 3,6,9-~riaza-3,6,9-tri~-(carboxymethyl)-4-~3-methoxy-2,4,6-
triiodobenzyl) _lln~lec~n~A i oic acid
11. 6 g (13 . 2 mmol) of the compound described in Example 14b)
is mixed in 60 ml of tetrahydrofuran at 0C with 2 . 77 g (92 . 6
mmol) of 80% sodium hydride suspension in mineral oil. 13.1 g
(92.6 mmol) of iodomethane is added to it and stirred for 30
minutes. Then, the solution is mixed with 60 ml of 2N sodium
hydroxide solution and refluxed for 30 minutes. After the
cooling, the organic solvent is drawn off in a vacuum, and the
rF-~qin;nq a~ueous solution is adjusted to pH 1.5 with
2177~77
COncentrated hydrochloric acid. A precipitate precipitates,
which i8 suctioned of f and dried in a vacuum .
Yield: 10.7 g (91.4% of theory)
Analysis (relative to anhydrous substance):
Cld: C 29.65 H 3.17 I 42.72 N 4.72 O 19.75
Fnd: C 29.74 H 3.23 I 42.65 N 4.63
d) Gadoliniu_ complex of the di~odium salt o~ 3,6,9-tri~za-
3, 6, 9 -tri~- ~ CA rboxymethyl ) -4 - t 3 -methoxy-2, 4, 6 -
triio~ob~n7yl)--lln ~A~ec-n~A i oic acid
10.2 g (11.4 mmol) of the penta acid described in Example
14c) is converted to the title compound as described in Example
ld) .
Yield: 11.7 g (94.096 of theory) of colorless lyophilizate
Analysis (relative to anhydrous substance):
Cld: C 24.26 H 2.13 I 34.95 N 3.86 O 16.15 Gd 14.43 Na 4.22
Fnd: C 24.30 H 2.10 I 34.91 N 3.90 Gd 14.50 Na 4.28
~x~npl~ 15
GAdoliniwn complex o~ the di-20Aillm salt o~ 3,6,9-triaza-4-~3-
diethylca.rbamoyl-2,~,6-triiodophenylcarbamoyloxymethyl) -3,6,9-
tris- (cArboxymethyl) -llnAec~necli oic acid
a) 3-IsocyAmlto-2,~,6-triiodobenzoic acid diethylamide
57.00 g (100.0 mmol) of 3-amino-2,4,6-triiodobenzoic acid
diethylamide (CA 54, P 20987i (1960)) is mixed under nitrogen
83 2i77977
atmosphere at room temperature with 250 ml of 2N toluenic
phosgene 601ution. After 0.5 ml of N,N-dimethylformamide is
added, the batch is allowed to stir for 5 hours at 60C and then
evaporated to dryness.
Yield: 59.60 g (100.0~6 of theory) of yellowish solid
Analysis (relative to solventless substance):
Cld: C 24.18 H 1.86 I 63.88 N 4.70 0 5.37
Fnd: C 24 . 07 H 1. 92 I 63 . 80 N 4 . 66
b) ~- (3-Diethylc-- ~' y1-2, 4, 6-
triiodophenyl~rh~- yloxymethyl) -3, 6, 9-tris- (tert-
butyloYycarbonylmethyl)-3,6,9-tr;~r-~n~er~ne~ioic acid-~i-
tert-butyl ester
A solution of 14.68 g (20.85 mmol) of 3,6,9-tria~a-3,6,9-
tris- (tert-butyloxy-carbonylmethyl) -4-hydroxymethyl-undecanedioic
acid-di-tert-butyl ester (DE 3806795) in 100 ml of anhydrous
pyridine is mixed with exclusion of moisture with 12.42 g (20.85
mmol) of the isocyanate described in Example 15a), and it is
stirred overnight at room temperature. ~hen, it is completely
concentrated by evaporation, and the residue is chromatographed
on silica gel (eluant: hexane/ethyl acetate). After the
fractions containing the product are concentrated by evaporation,
a yellowish oil is obtained as residue.
Yield: 24.29 g (89.6% of theory)
84
217~
Analysis (relative to solventless substance):
Cld: C 43.43 H 5.98 I 29.29 N 5.39 0 16.00
Fnd: C 43.42 H 6.11 I 29.25 N 5.44
c) 3,6,9-Triaza-4-(3-diethylcArbamoyl-2,~,6-
triiodophenylcarbamoyloxymethyl)-3,6,9-triS-(carboxymethyl)-
un~l-?o~n~ oic ~cid
21. 62 g (16. 63 mmol) of the pentaester described in Example
15b) is converted to the corrPCp~nrl i n~ penta acid under the
conditions described in Example lc).
Yield: 16.17 g t95 . 4% of theory~ of colorless solid
Analysis (relative to solventless substance):
Cld: C 31.81 H 3.56 I 37.35 N 6.87 0 20.40
Fnd: C 31.73 H 3.64 I 37.25 N 6.72
d) G~olinium complex of the disodium SAlt of 3~6~9-tri~ZA-~.-
(3-~i~thyl~ ~ yl-2, 4, 6-triiodophenyloarbamoyloxymethyl) -
3, 6, 9-tri~- (calLv.5y ~ thyl) -llnd~c ~n~i oic acid
15.52 g (15.22 mmol) of the penta acid described in Example
15c) is converted to the title compound as described in Example
ld) .
Yield: 17.44 g ~94.196 of theory) of colorless lyophilizate
Analysis (relative to anhydrous substance):
Cld: C 24,. 26 H 2 .13 I 34 . 95 N 3 . 86 0 16 .15 Gd 14 . 43 Na 4 . 22
Fnd: C 24.30 H 2.10 I 34.91 N 3.90 Gd 14.50 Na 4.28
21~7~77
Exampl~ 16
G~olinium complex of the disodium salt of 3,6,9-triaza-3,6,9-
tris-~carboxymethyl)-4-~N-c~rboxymethyl-3-methylamino-2,~,6-
triiodophenylureyl~nemethyl)-~-methyl~ ec~n~ioic acid
A ) 3-I~ocyan~to-2, ~, 6-triiodobenzoic acid-N-
( ethoxycarbonylmethyl ) -methylami~e
61.40 g tlO0.0 mmol) of 3-amino-2,4,6-triiodobenzoic acid-N-
(ethoxycarbonylmethyl) -methylamide (CA 54 P 20987i (1960) ) is
mixed under nitrogen atmosphere at room temperature with 250 ml
of 2N toluenic phosgene solution. After 0.5 ml of N,N-
dimethylformamide is added, the batch is allowed to stir for 5
hours at 60C and then evaporated to dryness.
Yield: 64 . 00 g (100. 0% of theory) of yellowish solid
Analysis (relative to solventless substance):
Cld: C 24.40 H 1.73 I 59.49 N 4.38 0 10.00
Fnd: C 24.37 H 1.82 I 59.53 N 4.26
b) 3,6,9-~l!riaza-3,6,9-tris-ttert-butyloxycarbonylmethyl)-4-(N-
cthoxycarbonylmethyl-3-methylamino-2, 4, 6-
tri~o~oph~ylureylenemethyl)-4-methylundecanedioic acid-di-
tert-butyl ester
A solution of 16 . 41 g (22 . 89 mmol) of the pentaester,
described in Example 8g), in 100 ml of anhydrous pyridine is
mixed with exclusion of moisture with 14 . 65 g (22 . 89 mmol) of the
isocyanate described in Example 16a) and stirred overnight at
room temperature. ~hen, it is completely concentrated by
217~977
evaporation, and the residue i8 chromatographed on silica gel
(eluant: hexane/ethyl acetate). After the fractions containing
the product are concentrated by evaporation, a yellowish oil is
obtained as residue.
Yield: 27 . 39 g (88 . 2% of theory)
Analysis (relative to solventless substance):
Cld: C 43.37 H 5.87 I 28.06 N 6.19 0 16.51
Fnd: C 43.49 H 6.01 I 28.22 N 6.14
c) 3, 6, 9 -TriA z~-3, 6, 9 -tris- ( c~r~oxymethyl ) -4 - ( N-carboxymethyl -
3-methylilmino-2, ~, 6-triiodophenylureylenemethyl ) -~ -
methylundec~ne~lioic ~cid
26.68 g (19.66 mmol) of the hexaester described in Example
16b) is converted to the corresponding hexa acid under the
conditions described in Example 3c).
Yield: 19.19 g (93.19~ of theory) of colorles6 solid
Analysis (relative to solventless substance):
Cld: C 30.92 H 3.37 I 36.32 N 8.02 0 21.37
Fnd: C 31.03 H 3.48 I 36.21 N 8.14
~ 87 2177977
d) Gadolinlum complex of the di~odium ~alt of 3,6,9-triaza-
3,6,9-tris-tcJ~lLv~y l.hyl)-4-~N-carboxymethyl-3-methylamino-
2,~,6-triiodophenyl-ureylenemethyl) -4-methylundecanedioic
acid
24.32 g (23.19 mmol) of the penta acid described in Example
16c) i6 converted to the title compound as described in Example
ld) .
Yield: 27.84 (94.6~ of theory) of colorless lyophilizate
Analysis (relative to anhydrous substance):
Cld: C 25.57 H 2.30 I 30.01 N 6.63 0 17.66 Gd 12.40 Na 5.44
Fnd: C 25 . 62 H 2 . 34 I 29 . 94 N 6 . 58 Gd 12 . 35 Na 5 . 38
Example 17
Comparison tegt: igomer to Examp~e 1 of W0 93/16375
1,13-Bis[5-~propion-3-ylamido]-2,4,6-triiodoisophthalic acid-
bis ~ 2 -hydroxy-l-L~ .,y,..cthylethyl) -diami~e l -4, 7 ,10 -
tris(carbox-ymethyl)-2,12-dioxo)-1,~,7,10,13-pentaazatridec~ne,
gadolinium complex
a) 1,13-Bis[5-(propion-3-ylAmido]-2,4,6-triiodoisophthalic
~cid-bis (2-hy~roxy-1-hydroxymethylethyl) -diamide] -4, 7,10-
tris~carboxymethyl)-2,12-dioxo)-1,~,7,10,13-
pentaazatridecane
16.5 g (21.2 mmol) of 5-(3-aminopropionamido~-2,4,6-
triiodoisophthalic acid-bis(2-hydroxy-1-hydroxymethylethyl)-
diamide is dissolved at 120C bath temperature in 82 . 5 ml of DMF.
It is mixec~ at room temperature with 7 . 38 ml (53 . 25 mmol) of
2177977
triethylamine and then with 3.8 g (10.64 mmol) of N,N'-bis[2-
(2,6-dioxomorpholino)ethyl]glycerol. The reaction mixture is
stirred overnight at room temperature. The solvent is evaporated
in a vacuum, and the residue is foamed up in the oil pump. The
solid is etirred with 200 ml of ethanol for two hours at room
temperature, suctioned off and dried at 50C in a vacuum. Then,
the residue is taken up in a little water and chromatographed on
silica gel RP 18 (eluant: water/methanol). After the product
fractions are concentrated by evaporation, the title compound is
obtained as a colorless solid.
Yield: 17 . 24 g (4296 of theory)
Analysis (relative to anhydrous substance):
Cld: C 30.19 H 3.43 I 39.8~3 N 8.07 0 18.43
Fnd: C 29.88 H 3.30 I 40.21 N 7.95
b) 1~l3-Bl~[s-(proplon-3-ylamldo]-2~6-trilo~oi~ophth~lic
acid-bis ( 2-hydroxy-1-hydroxymethylethyl ) -diamide] -~, 7, 10-
tris ~ c~rboxy3lethyl ) -2 ,12 -d ioxo ) -1 , 4, 7 ,1 0 ,13 -
penta~ tridec~ne, gadolinium complex
400 mg of the compound described in Example 17a) is mixed in
portions with 17.8 ml of a 0.01 molar gadolinium acetate
solution. I'he pH is moved with triethylamine into the neutral
range, and the aqueous solution is stirred for one hour at room
temperature with activated carbon. Af~er filtration and freeze-
drying, the gadolinium complex is obtained as a colorless solid.
89
2177~77
Yield: 145 mg (30. 5% of theory)
Analysis (relative to anhydrous 6ubstance):
Cld: C 27.91 H 3.03 I 36.90 N 7.47 O 17.06 Gd 7.62
Fnd: C 27.77 H 2.99 I 36.72 N 7.15 Gd 7.38
Example 18
Ga~Solinium complex of 3,6,9-triaza-3,6,9-tris-(carboxymethyl)-4-
~3-io~o-~-ethoxybenzyl)-1lndec~ne~;0ic Acid, disodium ~alt
a) N-Benzyloxycarbonyl-3- t4-ethoxyPhenyl) -2-aminopropanol
31. 8 g (848 . 4 mmol) of sodium borohydride is added to a
solution of 221.41 g (605.9 mmol) of N-benzyloxycarbonyl-O-
ethyltyrosine methyl ester in 1. 5 l of tetrahydrofuran at room
temperature. 279 ml of methanol is added to it in drops within
two hours while being stirred. Then, the tetrahydrofuran is
distilled o~f in a vacuum, the residue is taken up in 1 l of
water and extracted three times with 700 ml of ethyl acetate.
~he combined organic phase is washed with water, dried with
sodium sulfate and concentrated. It is recrystallized from ethyl
acetate/hexane .
Yield: 187.0 g (93.7~6 of theory)
Analys~ s (relative to solventless substance):
Cld: C 69.28 H 7.04 N 4.25 O 19.43
Fnd: C 69.11 H 7.20 N 4.13
9o
2177~7~
b) 1-Ac~toxy-N-benzyloYyc~rbonyl-3 - ( 3 - iodo-~-ethoxyphenyl ) -2 -
aminopropAno
29.4 g (89.3 mmol) of N-benzyloxycarbonyl-3-(4-
ethoxyphenyl)-2-aminopropanol is dissolved in 88 ml of glacial
acetic acid and mixed drop by drop with 22.2 g (134 mmol) of
iodomonochloride in 35. 5 ml of glacial acetic acid at room
temperature. The reaction mixture is allowed to stir overnight
at room temperature and poured in 1.1 1 of ice water for working-
up. The aqueous phase is extracted several times with ethyl
acetate, the organic phase is washed with water, sodium
bicarbonate solution and sodium bisulfite solution and then dried
on sodium sulfate. After the solvent is evaporated, a yellowish
oil is obtained, which is slowly thoroughly cry6tallized.
Yield: 34 . 5 g (77 . 7~c of theory)
Analys is (relative to solventless substance):
Cld: C 50.72 H 4.86 I 25.52 N 2.82 0 16.08
Fnd: C 50.53 H 4.98 I 25.38 N 2.74
c) N-Be~yloyycarbonyl-3-(3-iodo-4-ethoYyphenyl)-2-
A ~ op~ no l
2g.8 -- (60 mmol) of 1-acetoxy-N-benzyloxycarbonyl-3-(3-iodo-
4-etho~.~h~yl)-2-aminopropane is suspended in 150 ml of methanol
and miY~ 5t room temperature with 4.94 g (60 mmol) of anhydrous
sodium a ze--~te. The batch is stirred for 6 hours at 60C,
evapora-~ to dryness and the residue is taken up in ethyl
acetate. ~'le precipitated salt is suctioned off, washed with
2177977
ethyl acetate, and the filtrate i3 concentrated by evaporation.
The viscous oil obtained is used in the next stage without
further purification.
Yield: 27.3 g (100% of theory)
d) 1-MothAno~ulfonyloXy-N-bonzyloxycArbonyl-3- (3-iodo-4-
othoxyphenyl) -2 -aminopropane
26.5 g (58 mmol) of N-benzyloxycarbonyl-3-(3-iodo-4-
ethoxyphenyl)-2-aminopropanol is dissolved in 130 ml of
dichloromethane, mixed with 24.1 ml of triethylamine and brought
to reaction at 0C with 6.78 ml (87 mmol) of methanesulfonic acid
chloride . Af ter 3 0 minutes at room temperature, no more initial
material can be detected according to TLC. The batch is diluted
with dichloromethane and washed with sodium bicarbonate and with
water. After the organic phase is dried, the solvent is
evaporated, and the product is obtained as pale yellow oil.
Yield: 30 . 4 g (98% of theory)
Analysis (relative to solventless substance):
Cld: C 45 . 04 H 4 . 54 I 23 . 79 N 2 . 63 0 18 . 00 S 6 . 01
Fnd: C 45.13 H 4.72 I 23.58 N 2.74 S 5.88
e) N-Benzyloxycar~onyl-1- (3-iodo-4-ethoxybenzyl) -N'- (2-
inoothyl) ~thy~ m; ng
28 . 2 g (53 mmol) of 1-methanesulfonyloxy-N-
benzyloxycarbonyl-3-(3-iodo-4-ethoxyphenyl)-2-aminopropane is
dissolved at room temperature in 140 ml of tetrahydrofuran and
92
~- 217797~
mixed with 143 ml (2.12 mol) of ethylenediamine. After 22 hours
of stirring, the batch i6 concentrated by evaporation to an oil,
and the residue is dissolved in ethyl acetate. It is washed
neutral with water and the organic phase is dried on sodium
sulfate. After concentration by evaporation, a yellow oil is
obtained .
Yield: 25. 9 g t98~ of theory)
Analysis (relative to solventless substance):
Cld: C 50 . 71 H 5 . 67 I 25 . 51 N 8 . 45 0 9 . 65
Fnd: C 50.87 H 5.78 I 25.40 N 8.74
f ) 1- (3-Iodo-4-~thoxybenzyl) -N' - (2-aminoethyl) ethyl~ne~ i~m; ne,
trihydrobromide
25 g (50 mmol) of N-benzyloxycarbonyl-1-(3-iodo-4-
ethoxybenzyl)-N'-(2-aminoethyl)-ethylenediamine is suspended at
room temperature in 20 ml of glacial acetic acid and mixed with
61.1 ml (250 mmol) of 33% hydrobromic acid in glacial acetic
acid. After 40 hours at room temperature, the feedstock is
completely consumed. ~he reaction mixture is concentrated by
evaporation in an oil pump vacuum, and the residue is
subsequently distilled three times with toluene. The solid is
mixed with water and evaporated to dryness. To remove residual
traces of water, the residue is concentrated by evaporation three
times with dichloromethane. The crude product is recrystallized
from ethanol.
Yield: 24.6 (81.2% of theory) of colorless crystals.
93
2177~77
Analy3ig ~relative to solventless substance):
Cld: C 25.77 H 4.16 I 20.94 N 6.93 0 2.64 Br 39.56
Fnd: C 25.89 H 4.41 I 20.73 N 6.74 Br 39.29
g) ~- ~ 3 -Iodo-~-ethoxybenzyl ) -3, 6, 9 -trLs ~ tert-
butoxyc~rbonylmethyl)-3,6,9-tri~ ln~lec~nR-l,ll-dioic aci~l,
di- (tert-butyl~ -~ster
23.2 g (38.3 mmol) of 1-(3-iodo-4-ethoxybenzyl)-N'-(2-
aminoethyl)ethyl~nF~fl;Am;ne~ trihydrobromide is suspended in 450
ml of tetrahydrofuran and mixed drop by drop with 43 . 6 g (316
mmol) of potassium carbonate and 18 ml of water, as well as with
42.13 ml (287.2 mmol) of bromoacetic acid-tert-butyl ester. It
is stirred for 10 hours at 60C and then overnight at room
temperature. The salt is suctLoned off, washed with
tetrahydrofuran, and the filtrate is concentrated by evaporation.
The crude product i5 purified using a silica gel column (mobile
solvent: dichloromethane/methanol).
Yield: 34 . 6 g (96 . 7g~ of theory)
Analys is (relative to solventless substance):
Cld: C 55.30 H 7.77 I 13.59 N 4.50 0 18.84
Fnd: C 55.37 H 7.95 I 13.44 N 4.43
217797~
h) ~- ~ 3-Iodo-~-~thv~y~ yl ) -3, 6, 9 -tris ( c~rboxyl~tomethyl ~ -
3,6,9-triA~al~n~e~n~-l,ll-~ioic acid, ga~lolinium complex,
Ciso~ium salt
17.6 g (18.8 mmol) of 4-(3-iodo-4-ethoxybenzyl)-3,6,9-
tris (tert-butoxycarbonylmethyl) -3, 6, g-triazal~nd~r In~-l ,11-dioic
acid, di-(tert-butyl)-ester is dissolved in 130 ml of methanol at
room temperature and mixed with 6. 03 g (150 . 4 mmol) of sodium
hydroxide in 10 ml of water. The batch is stirred for five days
at 60C, evaporated to dryness and subsequently distilled twice
with water. The residue is taken up in 180 ml of water and
ad~usted to pH 2.9 with acid ion exchanger. 3.41 g (9.4 mmol) of
gadolinium oxide is added, and the reaction mixture is heated to
80C. After the complexing is completed, it is adjusted to pH
7 . 2 with cation exchanger (Na~ form), and the clear solution is
freeze-dried .
Yield: 14.75 g (92% of theory)
Analysis (relative to anhydrous substance):
Cld: C 32.44 H 3.20 I 14.90 N 4.93 0 20.67 Gd 18.46 Na 5.40
Fnd: C 32.23 H 3.47 I 14.76 N 4.88 Gd 18.36 Na 5.17
2177~77
~x~pl- lg
Gadolinium complex of 3, 6, 9 -tri~z~ -3, 6, 9 -tris- ~ c~rboxymethyl ) -4 -
~3-bromo-,1-othoxybenzyl)-1lnde~ne~oic ~c:id, di30dium salt
a) l-Acstoxy-N-benzyloxyc~rbonyl-3- (3-bromo-4-ethoxyphenyl) -2-
~minoprop~ ne
20.0 g (61 mmol) of the title compound of Example 18a isdissolved in 200 ml of glacial acetic acid and mixed with 0 . 2 g
of iron powder. After the reaction solution is cooled to 10C,
it is mixed at this temperature drop by drop with 12.48 g (78
mmol) of bromine. The reaction mixture is allowed to stir
overnight at room temperature, and it is poured into 750 ml of
ice water for working-up. The aqueous phase is extracted several
times with ethyl acetate, the organic phase is washed with water,
sodium bicarbonate solution and sodium bisulfite solution and
then dried on sodium sulfate. After the solvent is evaporated,
yellowish-colored crystals are obtained.
Yield: 19.50 g (71.0% of theory)
Analysis (relative to solventless substance):
Cld: C 56.01 H 5.37 Br 17.74 N 3.11
Fnd: C 56.13 H 5.42 Br 17.84 N 3.16
b) N-BsnzyloxycJ~rbonyl-3- ~3-bromo-4-ethoxyphenyl) -2-
minoprop~nol
18.0 g (39.8 mmol) of 1-acetoxy-N-benzyloxycarbonyl-3-(3-
bromo-4-ethoyyphenyl)-2-aminopropane is suspended in 100 ml of
methanol and mixed at room temperature wi~h 3.30 g (40 mmol) of
,~
~ 217~g7~
anhydrous sodium acetate. The batch is ~tirred for 6 hours at
60C, evaporated to dryness, and the residue is taken up in ethyl
acetate . The precipitated salt is suctioned of f, washed with
ethyl acetate, and the filtrate is concentrated by evaporation.
The viscous oil obtained is used in the next stage without
further purification.
Yield: 16 . 0 g (98 . 2% of theory)
c) l-~feth~ne~ul~onyloxy-~-benzyloXyc~ rbonyl-3 - ( 3 -bromo-4 -
~thoxyphenyl ) -2 -Yminopropane
15. 5 g (38 mmol) of N-benzyloxycarbonyl-3- (3-bromo-4-
ethoxyphenyl)-2-aminopropanol is dissolved in 100 ml of
dichloromethane, mixed with 6. 3 ml (45 mmol) of triethylamine and
brought to reaction at oDC with 5. 04 ml (52 mmol) of
methanesulfonic acid chloride. After 30 minutes at room
temperature, no more initial material can be detected according
to TLC. The batch is diluted with dichloromethane, washed with
sodium bicarbonate and with water. After the organic phase is
dried, the solvent is evaporated, and the product is obtained as
pale yello~ oil.
Yield: 18.1 g (98% of theory)
Analysis (relative to solventless substance):
Cld: C 49.39 H 4.97 Br 16.43 N 2.88 S 6.59
Fnd: C 49 . 21 H 4 . 92 Br 16 . 40 N 2 . 79 S 6 . 54
97
~ 21~7977
d) N-B~nzyloxycarbonyl-l-t3-bromo-4-etboxybenzyl)-N'-(2-
am~ no~tbyl) ethylen~ j Am; nf~
17.0 g (34.9 mmol) of 1-methanesulfonyloxy-N-
benzyloxycarbonyl-3-(3-bromo-4-ethoxyphenyl)-2-aminopropane is
dissolved at room temperature in 100 ml of tetrahydrofuran and
mixed with 100 ml (1.48 mol) of ethyl~nP~;~min~. After 36 hours
of stirring, the batch is concentrated by evaporation to an oil,
and the residue is dissolved in ethyl acetate. It is washed
neutral with water, and the organic phase is dried on sodium
sulfate. After concentration by evaporation, a yellow oil is
obtained .
Yield: 14 . 9 g (95% of theory)
Analysis (relative to solventless substance):
Cld: C 56.00 H 6.27 Br 17.74 N 9.33
Fnd: C 56.21 H 6.32 Br 17.81 N 9.38
e) 1-(3-Bromo-~-ethoxybenzyl)-N'-~2-~minoethyl)ethylenetl;t~mine,
trihyC~robromide
14 g (31 mmol) of N-benzyloxycarbonyl-1-(3-bromo-4-
ethoxybenzyl)-N~-(2-aminoethyl)ethyl~nP~ m~n~ is suspended at
room temperature in 39.1 ml (160 mmol) of glacial acetic acid,
and it is mixed with 39.1 ml (160 mmol) of 33% hydrobromic acid
in glacial acetic acid. After 24 hours at room temperature, the
feedstock is completely consumed. The reaction mixture is
concentrated by evaporation in an oil pump vacuum, and the
residue i5 subsequently distilled three times with toluene. The
98
2177977
Bolid i8 mixed with water and evaporated to dryness. To remove
residual traces of water, the residue is concentrated by
evaporation three times with dichloromethane. The crude product
is recrystallized from ethanol.
Yield: 16 . O g (92 . 496 of theory) of colorless crystals
Analysis (relative to solventless substance):
Cld: C 27.93 H 4.51 Br 39.56 N 7.52
Fnd: C 27 . 90 H 4 . 48 Br 39 . 29 N 7 . 49
f ) 4- ~3-Bromo-4-ethoxybenzyl) -3, 6, 9-tr i8 ( tert-
butoxycarbonylmethyl) -3, 6, 9-triazaundecane-1, 11-dioic ~cid,
~i- (tert-butyl) -e~ter
15.0 g (26.8 mmol) of 1-(3-bromo-4-ethoxyben~yl)-N'-(2-
aminoethyl)ethylener~i~mi~R~ trihydrobromide is suspended in 400
ml of tetrahydrofuran, and it is mixed drop by drop with 29. 6 g
(215 mmol) of potassium carbonate and 15 ml of water, as well as
with 36 . 67 ml (250 mmol) of bromoacetic acid-tert-butyl ester.
It is stirred for 12 hours at 60C and then overnight at room
temperature . The salt is suctioned of f, washed with
tetrahydrofuran, and the filtrate is concentrated by evaporation.
l'he crude product is purified using a silica gel column (mobile
solvent: dichloromethane/methanol).
Yield: 22.4 g (94.2% of theory)
99
217~977
Analysis (rel~tive to solventles3 substance):
Cld: C 58.23 H 8.18 Br 9.01 N 4.74
Fnd: C 58.19 E 8.12 Br 8.96 N 4.70
g) ~-t3-Elromo-~-ethoxybenzyl)-3,6,9-tri~(carboxylatomethyl)-
3, 6, 9-tr~ r~ o-llnAqc-n~ ioic ~cid
20. 0 g (22 . 5 mmol) of 4- (3-bromo-4-ethoxybenzyl) -3, 6, 9-
tris (tert-butoxycarbonylmethyl) -3, 6, 9-triazaundecane-1, ll-dioic
acid, di-(tert-butyl)-ester is dissolved in a mixture of 400 ml
of tetrahydrofuran and 40 ml of water, and it is mixed drop by
drop at room temperature with a solution of 6. 4 g (160. 8 mmol) of
sodium hydroxide in 20 ml of water. After a reaction time of 48
hours at room temperature, the reaction mixture is concentrated
by evaporation in a vacuum, and the residue is subsequently
distilled twice with water. The r~ i n; n~ residue is taken up in
300 ml of water and adjusted to pH 2 . 0 wi~h cation exchanger (E~
form) . After freeze-drying, the f iltrate obtained after the
suctioning-off from the ion exchanger yields the pentacarboxylic
acid as colorless powder.
Yield: 11.3 g (83~6 of theory)
Analysis (relative to anhydrous substance):
Cld: C 45.55 H 5.31 Br 13.17 N 6.93
Fnd: C 45.62 E 5.38 Br 13.41 N 7.02
- 100
217797~
h) ~-~3-Bromo-~-~thoYybenzyl~-3,6,9-tri~carboxylatomethyl)-
3,6,9-tr~Az~l~n~ec~ne-l,ll-dioic acid, gadolinium compleY,
disodium salt
10.0 g (16.5 mmol) of 4-(3-bromo-4-ethoxybenzyl)-3,6,9-
tris (carboxylatomethyl) -3, 6, 9-triazaundecane-1, ll-dioic acid is
dissolved in 200 ml of water and mixed at 80C with 3 . 0 g (8 . 25
mmol) of gadolinium oxide. After a reaction time of 1 hour at
80C, the now almost clear reaction solution is brought to room
temperature and adjusted to pH 7 . 2 by mixing drop by drop with
0. 2 molar sodium hydroxide solution. The reaction mixture i8
freeze-dried after f iltration.
Yield: 12.7 g (9696 of theory~
Analysis (relative to anhydrous substance):
Cld: C 34.33 H 3.38 Br 9.93 N 5.22 Gd 19.54 Na 5.71
Fnd: C 34.26 H 3.34 Br 9.90 N 5.21 Gd 19.50 Na 5.68
Example 20
Gadolinium compleY of the disodium salt of N,N-bis-t2-[N',N'-bis-
~carboyymethyl) -amino] -ethyl]-p-iodophenylAlAnine
a ) p-IodophenylA l ~ n ~ n ~ ~opropylester, hydrochloride
50 ml of isopropanol is stirred at 0C under argon and mixed
drop by drc~p with 3.12 ml (41.6 mmol) of thionyl chloride. 30
minutes later, 10.0 g (34.4 mmol) of p-iodophenylalanine is added
in portionc, stirred for one hour at room temperature, and the
batch is t~en allowed to reflux for two hours. After the coolinq
to room tem~perature, the batch is allowed to stand overnight at
101
~ 2177977
0C, and then the precipitated, colorless precipitate is
suctioned of f .
Yield: 12.4 g (97.896 of theory)
Analy6is:
Cld: C 38.99 H 4.64 I 34.33 N 3.79 O 8.66 Cl 9.S9
Fnd: C 38.85 H 4.70 I 34.29 N 3.78 Cl 9.66
b) N,N-Bis-[2-tN'tN'-bis-[ (tert-butyloxyc~rbonyl)-methyl]-
amino] -ethyl] -p-iodophenyl al ~Ini n; sopropyle~ter
12.1 g (32.7 mmol) of the amine produced according to
Example 20a) and 25.4 g (72.0 mmol) of N,N-bis-[ (tert-
butyloxycarbonyl)-methyl]-2-bromoethylamine (M. Williams and H.
Rapoport, J. Org. Chem. 58, llS1 (1993) ) are introduced in 50 ml
of acetonitrile and mixed with 20 ml of 2N phosphate buffer
solution (pH 8.0). The batch is vigorously stirred at room
temperature for 24 hours, whereby the aqueous phosphate buffer
phase is exchanged after 2 and 8 hours for fresh buffer solution.
Then, the organic phase is concentrated by evaporation in a
vacuum, and the residue is chromatographed on silica gel with
hexane/ethyl acetate/triethylamine (3: 1: 0 . O1) . The product-
containing fractions are concentrated by evaporation in a vacuum.
Yield: 17.9 g (62.3~6 of theory) of yellowish oil.
Analysis (relative to solventless substance):
Cld: C s4.85 H 7.60 I 14.49 N 4.80 O 18.28
Fnd: C 54.80 H 7.65 I 14.41 N 4.74
102
~ 2177~77
c) N,N~ 2-[N',N'-bis--(carboxymQthyl)-amino]-ethyl~-p-
iodophenyl ~1 ~n ~ n~
17.1 g (19.5 mmol) of the tert-butyl ester described in
Example 20b) is dissolved in 250 ml of trifluoroacetic acid, and
it i6 stirred for 1 hour at room temperature. Then, the solution
is concentrated by evaporation, the residue is absorptively
precipitated in water, filtered off and dried in a vacuum.
Yield: 11.5 g (96.8% of theory) of light beige solid
Analysis (relative to anhydrous substance):
Cld: C 41.39 H 4.63 I 20.82 N 6.90 0 26.26
Fnd: C 41.33 11 4.56 I 2~.78 N 6.93
d) G~dolinium complex of the disodium salt of N,N-bis-[2-
tN' ,N' -bi5- ~c~rboxymethyl) -amino] -ethyl] -p-iodophenylA 1 Ani nR
A suspension of 7.43 g (12.2 mmol) of the penta acid,
produced according to Example 20c), in 118 ml of water is mixed
with 2.21 g (6.1 mmol) of gadolinium oxide and stirred at 80C
for 2 hours. Then, 24.4 ml of lN sodium hydroxide solution is
added with a microburette and stirred for 1 more hour. Then,
after O.S g of activated carbon is added, the solution is stirred
at 80C for 2 hours and filtered. After freeze-drying, the
filtrate yields a colorless solid.
Yield: 9.12 g (92.6% of theory)
103
2177977
Analysis (rel~tive to anhydrous 6ubstance):
Cld: C 31.23 H 2.87 I 15.72 N 5.20 O 19.81 Gd 19.47 Na 5.69
Fnd: C 31.26 H 2.95 I 15.70 N 5.13 Gd 19.36 Na 5.74
Example 2 1
~dolinil~m compl~x of the monosodium salt of N,N-bi3-[2-[N',N'-
bi~-tca.~ y~ hyl)- mino]-ethyl]-glycine-N''-[3,5-bis-(N'''-~2-
hy~ yethyl)-c~ ~yl)-2~4~6-triiodophenyl-c~rbamoylmethyl]
a~nide
a) N,N-Bi3-[2-[N',N'-bis-[ ~tert-butyloxyc~rbonyl) -methyl]-
amino]-ethyl]-glycine-N''-[3,5-bi3-(N'''-(2-hy~roxyethyl)-
c~rb moyl) -Z, ~, 6-triiodophenylc~rh~-~ylmethyl] -amide
22.1 g (31.5 mmol) of 5-aminoacetylamino-2,4,6-
triiodoisophthalic acid-N,N'-bis-(2-hydroxyethyl)-diamide and
24.4 g (69.3 mmol) of N,N-bis-[ (tert-butyloxycarbonyl)-methyl]-2-
bromoethylamine (M. Williams and H. Rapoport, J. Org. Chem. 58,
1151 (1993) ) are introduced in 50 ml of acetonitrile and mixed
with 20 ml of 2N phosphate buffer solution (pH 8 . 0) . The batch
i5 vigorously stirred at room temperature for 24 hours, and the
aqueous phosphate buffer phase is exchanged after 2 and 8 hours
for fresh buffer solution. ~hen, the organic phase is
concentrated by evaporation in a vacuum, and the residue is
chromatographed on silica gel with dichloromethane/methanol
(95:S). The product-containing fractions are concentrated by
evaporation in a vacuum.
Yield: 21.9 g (55.8% of theory) of yellowish oil.
217~977
Analy6is (relative to solventless substance):
Cld: C 40.53 H 5.43 I 30.59 N 6.75 O 16.71
Fnd: C 40.50 H 5.44 I 30.52 N 6.79
b~ N,N-Bis-[2-[N',N'-bis-tc~rboxymethyl)-amino]-ethyl]-glycine-
N~-[3~5-bis-~N~-(2-hy~L~ y~:thyl)-carb~moyl)-2~4~6-
triio~ophenylc~rbamoylnethyl] -amide
20.8 g (16.7 mmol) of the tert-butyl ester described in
Example 21a) i8 dissolved in 250 ml of trifluoroacetic acid and
stirred for 1 hour at room temperature. Then, the solution is
mixed with tert-butyl methyl ether, the precipitate is separated,
washed with tert-butyl methyl ether and dried in a vacuum.
Yield: 16.9 g (98.9% of theory)
Analysis:
Cld: C 30.61 H 3.46 I 37.31 N 8.24 O 20.38
Fnd: C 30.77 H 3.58 I 37.25 N 8.28
c) G~dolinium complex of the nono~odit~ salt of N,N-bis-[2-
[N',N'-bis-~c~rboxymethyl) -~nino]-ethyl]-glycine-N' '-[3,5-
bis- (N~ ~ ~ - (2-hy~roxyethyl~ -c~rbamoyl) -2, 4, 6-
triiodophenylc-- -ylmethyl] -~mide
A suspension of 16.9 g (16.6 mmol) of the penta acid,
produced according to Example 21b), in 130 ml of water is mixed
with 3.00 g (8.28 mmol) of gadolinium oxide and stirred at 80C
for 2- hours. Then, 16.6 ml of lN sodium hydroxide solution is
added with a microburette and stirred for 1 more hour. Then,
105
2177977
after 0 . 5 g of activated carbon is added, the solution is stirred
at 8~C for 2 hours and filtered. After freeze-dryin~, the
filtrate yields a colorless solid.
Yield: 18.4 g (93.0% of theory)
Analysis (relative to anhydrous substance):
Cld: C 26.10 H 2.61 I 31.82 N 7.02 0 17.38 Gd 13.14 Na 1.92
Fnd: C 26.11 ~ 2.74 I 31.84 N 7.06 Gd 13.10 Na 1.93