Note: Descriptions are shown in the official language in which they were submitted.
WO 95/16995 2 i 7 8 n 8 6 PCTIUS94113824
.
METHOD FOR PRODUCING HEAT
CROSS~ ; TO RELATED APPIIICATIONS
This application is a cnnt;n11~tinn-in-part of application
Serial No. 08/160,941, filed December 3, 1993, which is a
rnnf;n~ tion-in-part of application Serial No. 07/782,558,
filed October 5, 1991, now iqh~nrln
INTRODUCTION
Terhn; cal Field
The field of this invention rnnrPrnR the production of heat
10 as a result of atomic reactions within a metal lattice.
Backqround of ~he Invention
With increasing pop1ll~t;nnR and increasing dependence upon
enerqy utilization to m-;nt~;n societies, the search for
energy sources which are alternatives to the ones used today
15 has been diligently pursued. One of the major efforts has
been ~; rert~d to atomic fusion, wherein atomic plasma is
--;ntA;n~d in a magnetic bottle. The high energies released
by the fusion of elements results in the production of
substantial heat in high energy particles and radiation.
20 While this approach to the proA-lrtinn of energy has many
attractions, as yet it has not been successful in
m3;nt~;n;nr~ the production of heat for an GYt~n~lorl period of
time, it produces radioactive ash, and the success of the
method is still relegated to an uncertain future.
WO 95/16995 2 ~ 7 8 0 8 G PCT/US94113824
Therefore, whik~ ùch prom1se 3till exists for this
approach, for the time being no significant reliance may be
made upon its success.
An alternative approach to fusion, re~erred to as "cold
5 fusion" has also been reported. However, this approach has
received some skepticism in the literature and has not been
shown to be reliable in its repro~7l7rih;lity~ Nevertheless,
a large number of investigators have shown that one can
produce heat by fusion of hydrogen isotopes, with the
10 re8ultant production of tritium, 3He and 4He. There i8 now
3uf f icient evidence in laboratories around the world to
e8tablish that the presence of hydrogen isotopes in a metal
lattice under electrolytic conditions results in the
prn~7.l7~-t i nn of heat beyond that introduced in the
15 electrolytic system, as well as the production of elements
of higher atomic number than the isotopic ~lyur U~ l employed.
The cold fusion systems to date have not 8atisfied the needs
for i ~ve:d reliability, ease of operation, reduced
r7"~r.on~7,~n(-e on materials ior which there i8 inadequate
2Q characterization, and higher ~f~ n~; es of energy
production as compared to energy input.
S7JMM~R~ OF THE lNV~;Nl'lON
Energy i5 produced by directing trAnR;Pnt cavitation bubble
r-nl 1 AraG at a metal surface with A~7anrh~7 hydrogen isotope .
25 The conditions under which the bubble collapses and the
material content of the bubble are selected to provide
excess heat over the energy introduced into the system, as
well as to provide elements of higher and/or lower atomic
number. The system may be 7777;ntAinf~7, in an electromagnetic
30 ~orce field or acoustic field during the reaction. The
resulting heat may be transferred to a heat acceptor or
~ransformed directly into a different form of energy.
Devices are provided for performing the method.
2178086
WO 95/1C995 PCrlUS94/13824
--3 --
3RIEF D~SCRIPTION OF THE DRAWIN~S
Figure l is a schematic of a device for heat production;
Figure 2 is an enlarged view of the reaction vessel shown in
Figure l;
Figure 3 is a projected view of the window and related
~1 ' q as shown in Figure 2 in an P~l r~ od relation;
Figure 4 is an alternative embodiment of the subject device;
Figure 5 is a further alternative: ~ a; t employing a
plurality of cells for heating a flowing exchange fluid;
lO Figurç 6 is a schematic of an alternative embodiment
demonstrating electricity output;
Figure 7 is an alternative embodiment of reduced size for
providing heat;
Figure 8 is a cross section in diay~ tic form of a
l~ reactor; and
Figures 9a, b, c and d are enlarged views of portionG of the
reactor .
DESCRIPTION OF THE SPECIFIC ~1~30DIMENTS
Methods and apparatuses are provided for the production of
20 heat, as well as the production of .ol ntq of higher and/or
lower atomic number than the isotopic hydrogen and other
atomic nuclei which serve as the r~PrtAnts. The method
employs directi~g high energy low atomic number atoms into
a matrix, par~icularly a metal matrix, in which molecules of
2~ at least one 11yd~u~ isotope are Ata~qrlrh~ A significant
number of parameters are involved in ~etermining the
ef f iciency of energy production and the nature and
21781~8~
Wo9~/16995 PCrlUS9~/1382
--4--
ef~iciency of new atomic lsotope production. The parameters
of interest include the manner in which the high energy
bubbles are prDduced, particularly the parameters associated
with the formation and characteristics of transient bubbles
5 and their collapse against a solid surface, which parameters
include the nature of the composition within the bubble, the
size of the bubble, the energy employed in forming the
bubble, the temperature and pressure at which the bubble is
formed and collapses, the pulse cycle, and the direction of
10 the stream of particles ~--n:~t;n~ from the bubble. Other
parameters may include an electromagnetic force field in
which the bubble is formed and collapses, as well as the
solid surface upon which the bubble collapses, the manner of
absorption or adsorption and composition of the element
15 absorbed on the solid sur~ace, the nature of the solid
surface, its fcrmation, and its acoustic properties, as well
as the manner in which the heat is employed. (While
adsorption is frequently considered the manner iIl which a
gas, such as deuterium binds to a metal such as palladium,
20 in the present invention, the gas atoms enter into the metal
lattice and interact in the lattice. In effect, the gas
atoms are absorbed in the metal lattice. ~ithout ;nt-~n~n~
to provide any theoretlcal basis for the events which occur
in the lattice by use of the term absorption, it would
25 appear that absorption better describes the event. )
The first c-nn~ ration will be the composition of the fluid
in which bubbles are formed. For the most part, the ~luid
will include a hydrogen isOtope: hydrogen, deuterium and
tritium and their respective nuclei, which include a proton,
30 deuteron and triton. Also, other low atomic number elements
may be present particularly as ions, such as lithium t6) .
The hydrogen isotope may be present as a diatomic molecule,
as a molecule in which the isotope is bonded to another
atom, such as oxygen, carbon, alkali or other metal,
35 particularly lithium, bismuth, calcium, mercury, uranium,
thorium, and the like, nitrogen, phosphorous, boron, usually
non-metallic ~lements of the first and second rows,
2178086
WO 95/1699S PCrlUS94/13824
--5--
particularly of columns 1 to 5 or metallic elements which
form hydrides. The compositions may include hydrogen
lec~ P, deuterium molecules, water, heavy water
(deuterium oxide), tritium oxide, alkanes of from 1 to 12
5 carbon atoms (methane, butane, etc. ), alkanols of from 1 to
12 carbon atoms ( th~nnl, ethanol, pentanol, etc., silanes,
metal hydrides, and the like. The choice of ~ will
depend upon many factors, such as the temperature and
pressure at which the method is operated, the nature of the
10 surface, 80 a3 to maintain an active surface and avoid
undesirable coatings or corrosion of the surface, and the
like . Individual compnc; t; nnR or combinations of
compositions may be employed, where the isotopes which are
employed may be the same or di~ferent. Under the conditions
15 of the method, the composition will be a mobile li~uid which
is capable of forming bubbles.
The bubbles are referred to as transient cavitation bubbles,
since they generally survive only for a single cycle. Thus,
the energy density ~nn~ f~nt~ated in the bubble is transferred
20 to the surface without repetitive expansion and contraction
of the bubble. This process increases the energy density in
the collapsing bubble by many orders of magnitude, usually
lo orders or greater, as compared to the original energy of
the bubble. For the most part, the bubbles will be at least
25 about 1 micron and not more than about 250 microns, usually
less than about 100 microns, more usually in the range of
about 10 to loo microns.
In order to provide for nucleation, the li~uid used for the
formation of the bubbles will normally be degassed and then
30 regassed with an appropriate gas. Preferably, inert gases,
particularly noble gases will be used, with the higher
atomic weights providing for great~r: mass and slower heat
tra~sfer. The preferred gases will be hydrogen, deuterium,
nitrogen, helium, argon and xenon, particularly argon and
35 xenon individually or in combination. By initially
degassing with a vacuum, the pressurizing gas may then be
WO 95/16995 2 ~: 7 8 ~ ~ ~; PCT/US94/13824
--6--
introduced at the desired pressure and m:~;ntcin~ at the
~P~ PCtPrl pressure during the run.
Bubbles may be produced by a~wide variety of methods, using
acoustical devices, me~ànical devices, fluia flow devices,
5 and the like. Conveniently, the bubbles are produced by an
induced pressure wave. For acoustical devices, one may use
a snn;~-~tnr~ employing a piezoelectric vibrator tr;lncr~- n Pr,
or other oscillating electronic or mechanical device3.
Alternatively, one may use jets, Venturi tubes, porou8
10 devices providing for flow-through pressure differential,
propellers, rotating or centrifugal devices which produce
turbulence, hydraulic pistons, etc. The particular manner
in which the bubbles are formed is not critical to this
invention, although it has been found that a sonicator is
15 particularly convenient in providing for energy control and
bubble formation. One or more devices may be used 30 that
a plurality of surfaces may be suhjected to cavitation, e.g.
devices on opposite sides of a metal foil serving as a
cavitation surface. The acoustic wave which is produced may
20 be a non-focused wave. Where other than an acoustic device
is used for bubble prnrlllnt;nn, one may augment the energy of
the bubble by using an acoustic device in conjunction with
the other device.
The temperature of the fluid will vary subst~nti~lly from
25 input and output. ,3ince the system generates a substantial
amount of heat, there will be a 5~1hct~ntial rise in
temperature of the l~luid during its resiaency in the
reactor. While one would not require fluid flow, if there
was an efficient way to remove the heat from the liquid, 30
30 as to provide a 3ub3t~nt;~11y isothermal condition in the
reactor, the most convenient way to ~~int~in the temperature
is to control the rate of Elow of the liquid through the
reactor and the temperature of the liquid entering the
reactor. Depending upon the desired e2it temperature and
35 the other parameters associated with the reaction, the
temperature of the Pnt~rin~ fluid may be ambient ~20C) and
WO 95/lC995 2 1 7 8 0 ~ 6 PCrlUS94/13824
up to about 100 C or greater, preferably from about 20C to
80C. The temperature of the entering fluid will depend
upon the temperature of the exiting fluid, the heat t~ ildll!l~
employed, the nature of the composition being used, and the
5 like. The exit temperature which reflects the reactor
temperature will be below about 350C and may be as low as
about 55C, usually not lower than about 75C, again
~P~F-nrli ng upon the general operating conditions of the
reactor and heat exchange. Desirably the exit t, dLu~e
10 will be at least about 75C, preferably at least about
100C, and generally not more than about 250 C, usually not
more than about 200OC. The pressure in the reactor will be
high enough to -~;ntA;n a liquid phase, 80 that the higher
the temperature to which the fluid reaches in the reactor,
15 the higher the pressure required to maintain the composition
in the fluid phase, in relation to the vapor pressure of the
composition in the reactor. For example, deuterium oxide at
about 350C would require about 200 atm. to r-;nt~in a
liquid phase. Therefore, the pressure will generally be at
2 0 least one atm ., usually at least two atms ., and not greater
than about 200 atm., preferably not greater than about 150
atms .
The energy source for the production of bub~les may be
cycled, 50 as to be on a portion of the time and off a
25 portion of the time. It is found that the reaction
~nt;nll~ after the energy source is turned down or off, 80
that one can cycle the energy source while still ret~ i n; n~
energy output. Depending on the degree to which the energy
outp~t can vary during a cycle, the on-off periods can be
30 varied greatly, with one or the other period being of
greater duration . The time ratio of the on period to of f
period will be in the range of about o.OOl - 1000:1, usually
in the range of about 0.01 - 100:1, frequently in the range
of about 0 . 04 - lo: l . By allowing the reaction to proceed
35 in the absence of energy input, a higher ratio of energy
output to energy input may be achieved.
W095/16995 2l78b&6 Pcrll~ss4ll3824
--8--
For prn~ rtinn of bubbles, as already indicated, a sonicator
("tr~3nC~i11rl~r'~ finds particular use. The energy provided by
the sonicator will generally be a leàst about 1 W/cm2,
frequently at least 2 W/cm2, and u~ ùally not greater than
5 about 10 W/cm2, more usually not grèater than about 5 W/cm2,
generally being greater than about 1, usually 3, W/cma, at
the cavitation surface. The frequency will usually be at
least about 5~z, more usually at least about lO~z and could
go to lMEIz or greater, generally not greater than about
10 O lME~z, usually not greater than about lO~Hz. Ranges of
interest include from 5Hz, usually from lO~z to lOR~Iz, and
from 40RHz to about O . lMEIz . The frequency will affect the
size of the bubble, 80 that one can control the energy
density ol~ the bubble and the energy which is rlicc~rate~
15 upon collapse by the energy of the tr~nRr~llr~r, the freruency
of the transducer, and the temperature of the fluid. These
factors are therefore interactive in controlling the energy
at which the bubble collapses.
Other parameters associated with sonication include the
20 cycling schedule, where the sonication ic on ~rom lo to 95~
of the time and of f the other portion of the time where each
cycle will be from about o . 1 to 1200 sec. A more complex
pattern of a non-uniform time for each cycle or cycle
component may be employed. The maximum acoustic power
25 amplitude will generally be in the range of about 1 to 50,
usually about 5 to 20, and preferably about 3 to 6 atms/cm2.
Since the energy at which the bubble collapses can
mechanically erode the metal curface~ this al~io must be
given consideration in the operation of the reactor.
30 Depending upou the nature o~ the surface, it9 ability to
withstand the forces of the collapsing bubble without
erosion, the ease of r.-rl~l , and the cost of the
surface, one may ~ ,, ice the efficiency of the system in
producing heat for ~rt,on~ periods of operation and
35 infrequency of replacement of the solid suri-ace.
Wo 95/16995 217 8 0 8 ~ - - PCT/US9~113824
_ g _
There i3 a correlation between the acoustic energy input and
the excess heat produced in microfusion devices that points
to a coupling of the transient cavitation bubbles (TCB) and
- the excess heat. At low and high ~lct rnAl pressures in the
5 reactor there is little if any excess heat g~n~rAt,od. At
low pressures the bubble formation is suppressed with no
effective TCB fnrm-tinn in either case. The TCB formation
and the excess heat formation is dictated by the temperature
and the acoustic enerry in and delivered to the reactor.
10 It is found that the direction of the wave can affect the
direction of the stream of the ~-, P of the bubble.
Therefore, the sound wave front should be directed parallel
to the solid surface, so as to provide for the primary
direction of the bubble stream toward the solid surface.
15 The subj ect method is designed to provide for asymmetric
transient cavitation providing for a violent collapse
directing the bubble rrnt~ntc into the metal.
The reactor may be m-intAin~d in the presence of an
electromagnetic field, where the field is created by an
2 0 electrical or magnetic f ield . The f ield may be produced by
employing two electrodes, where one of the electrodes may be
a metal plate for transmitting sound waves and the opposite
electrode may be grid-like or mesh, which allows for
observation of the solid surface and trAnPm;CPi~n of energy
25 to the solid surface. An electric field in the range of 5-
100 volts may be employed. Alternatively, the field may be
m-;nt~in~d by two poles of a magnet. The field will
generally be of about 100 to 10,000 gauss, more usually of
about 10 0 0 to 6 0 0 0 gaus s .
30 The ~ LI~L~L providing the solid cavitation surface may
take many forms, such as film, foi1, plate, particles, grid,
mesh, e.g. having a "wool-like~ structure, such as steel
wool, etc. The surface may be smooth, crazed, etched,
sputtered, etc., preferably having bubble nuclei forming
3 5 crevices . The hydrogen isotope absorbing material may be
Wo 95tl6995 2 1 7 ~ ~ 8 ~ PCI/~JS94/13824
--10--
used as the s~`material or may be coated onto a different
material, such as a ceramic, thermally 3tahle plastic, metal
alloy, or the like. The surface will comprise a metal that
is capable of A~q~lrh;ng or Ah~30rhin~ a l1Y~1LUY~1 isotope,
5 which includes metals (stable isotope8) of Groups IV and
VIII of the ~?eriodic Chart; specific metals find use, such
as rAl1A~illm (Pd), uranium (IJ), thorium (Th), titanium (Ti),
vanadium (V), chromium (Cr), niobium (Nb), tantalum (Ta),
hafnium (lIf), rlAt;nllm (Pt), rhodium (Rh), iridium (Ir), as
10 well as such metals as Alll~;nl-m (Al), nickel (Ni), bismuth
(Bi), iron (Fe), molybdenum (Mo), tungsten (W) ,etc. The
metal may be a pure metal or an alloy, e.g. steel, stainless
or carbon, or combination of metals, such as the
lAnth;nides, e.g. aluminum lAnth;n;de, misch metals,
15 compri8ing small amounts of cerium and samarium, in
conjunction with larger amounts of such metals as nickel,
V~;~e, etc. Desirably, the metals are capable of high
valency and high ratio of l~y~l~ Uy~l isotope adsorption and
absorption, going from pa~ladium, to t; tAn;l~m~ to zirconium,
20 to vanadium, molybdenum and tungsten.
The volume of the reactor may be varied widely, ~ron~;n~
upon the manner o~ ~formation of the bubbles, the energy
available for formation of the bubbles, the desired size of
the bubbles, the efficiency of heat transfer, the manner of
25 heat transfer, and the like. It would generally be
desirable that the bubbles will expand or travel not more
than about 50011, preferably not more than about 2501~ from
their site of initiation to the surface at which they
collapse. Therefore, the film of liquid between the bubble
30 generator and site of collapse generally will be thin. The
diameter of the collapsing bubble will generally be in the
range of about less than 1 micron to greater than 1 cm, more
usually in the range of about 1 micron to 50 microns. In
addition, fluid may be present surrounaing the lattice to
35 add to the volume and enhance heat transfer. Generally, in
relation to the lattice area, the volume will be ~ . 02 - 10
ml per unit cm2, more usually about 0.2 ml per unit cm2.
WO 95/16995 2 1 7 8 ~) 8 6 PCrlUS94113824
The total circulating volume will usually be at least about
o . 5 ml per unit cm2, more usually at least 2 ml per unit
cm~, and has no upper limit other than one of convenience
and the; nten~l~fl use .
Prior to or during the time of initiation of transient
bubble formation, hydrogen isotope gas will be absorbed to
the lattice surf ace . The presence of the hydrogen isotope
on the surface can be achieved in a variety of ways,
~1~pF.nr~inr upon the nature of the surface. A number of
lO metals, particularly metals such as palladium and titanium
will absorb the hydrogen isotopes on their surface, so that
no additional energy is required. Alternatively,
electrolysis may be carried out, where the lattice surface
may be one electrode and the fluid may comprise a source of
l~ hydrogen isotope atoms. Other ways for providing absorption
include loading under pressure or in the metallurgical
manufacturing process or the like.
The metal surface may provide for compound formation, such
as oxide formation or may be initially formed as the oxide.
20 The oxide will tend to slow the reaction, so that under
conditions where one wishes to have a slower reaction, oxide
formation can be used with advantage.
In carrying out the process, the critical ~1 tc Will be
the (lattice~ collapsing bubble surface, the source of
2~ transient bubble fnrr-t;~n, and the bubble forming fluid.
For illustrative purposes, it will be assumed that the fluid
is deuterium oxide which has been dega3sed and to which a
monatomic inert gas has been added, e.g. argon. The
pressure and temperature of the entering f luid is
30 determined. Besides the reactor volume, that portion of the
f luid that occupies the reactor and is sub; ect to bubble
formation, the total volume of the r~Artir,n fluid may also
be selected, where the total volume may be 2 to lOO, more
usually 3 to lO times the reactor volume. Assuming flow of
3~ the reaction fluid, cirr11lAt;rn of the fluid may begin.
Wo 95/16995 2 1 7 ~ O ~ ~i PCr/USs4/13824 ~
--12--
Where a hydrogen isotope is present in the fluid,
electrolysis may beg~ts provide for ;~ nrhr.rl or ~h~nrh~
l~ydr ~Iy~l~ isotope,'~ the lattice collapsing bubble surface.
Bubble formation may then begin by providing for transient
5 buhble formation, using a snnir~tnr. The acoustic density,
pulse cycle and power amplitude can then be set to provide
for the deGired energy for the transient bubbles. The
reaction may then be allowed to proceed for ~llfcir;Pnt time,
usually at least about 1 min and generally at least about 5
10 min and maybe 2 weeks or lonyer, depending upon the needs
for the energy, the nature of the system, and the like.
various systems may be employed in conjunction with the heat
formation. By employing thermoelectric devices, which may
be attached to the reactor, particularly on the opposite
15 side from the transient bubble forming device, the heat may
be directly transformed into Pl ertri ri ty. Various devices
which may be employed to produce electricity include thermo-
electric cells, Seebeck devices, bimetallic motor devices,
etc. Alternatively, one may employ a heat exchanger, which
20 may be concentric tubes, where the 1uid ~rom the reactor
flows past the heat receiving fluid. one may also provide
for vanes, where the fluid from the reactors pass through
vanes in a bath, which contains vanes for the heat receiving
fluid, 80 as to r-int~;n the bath at a conatant temperature.
25 Various other heat exchange r ~^h~n; ~ may be employed.
Alternatively, one can uae the 1uid, aa a lir~uid or vapor
to drive various mechanical devices, e.g. tl'rhin~, to
provide mechanical or electrical energy directly, where all
or a portion of the heat g~nPr~tpd in the reactor may be
3 0 dissipated.
There are two different systems which will be considered in
the figurea. The first system is a dual sy3tem, which uses
light water i~a the sonicator or energy tr~n~lr;nrJ portion
and heavy water in the reactiorL system. The heavy water
35 provides the heat production. In the second syatem, a
single system is used where heavy water serves in both the
WO 95/16995 2 1 7 8 ~ 8 ~ PCrlUS94/13824
sonicator and the reaction system, providing heat
i?roduction .
For ~urther understanding o~ the invention, the drawings
will now be considered.
5 FrRrT ~T-TT'RN~TIVE EM;30DIMENT
The apparatus elements shown in Figure 1 can be gathered
into subgroups or systems called " ~ntq which are
convenient for calorimetric mea~u,~ q. These ~ , - q
are:
The reaction volume 14 made up of the elements 18,
20, 22, 26, 36, 38, 39 and 42 monitored by thermocouples 148
and 149.
The sonication volume 16 made up o~ the elements
12, 22, 81, 96, 97, 98, 99 and 100 monitored by
15 thermocouples 152 and 151.
The reaction volume heat P~rh~n~Pr 162 made up of
the Pl R 52, 54, 56 and 44 monitored by thermocouple
154 .
The sonication volume heat exchanger 163 maae up
o~ the elements 70, 77, 79 and 83 monitored by thermocouple
155 .
The reaction volume system pump 165 is element 50
and is monitored by thermocouples 150 and 148.
The sonication volume system pump 164 is element
72 and is monltored by thermocouples 153 and 152.
The experimental apparatus ~or creating the environment ~or
this ~h~l- nn congists o~ two closed cirr~ t; nn systems
that m;:;nt:3;n the proper P~tPrn~l pressure and temperature
WO 95/16995 ~! 1 7 ~ 0 8 ~ PCT/US94/13824
--14--
80 that cavitation bubbles can be produced over el-t--n~
time periods. The larger system is the sonication Gystem in
which water was circulated through a 15 L heat ~ J ~ 83
as shown in~ Fir,ure 1. An ~tPrnAl pressure of air or
5 nitrogen 65 is r-~nt~;n~d to reduce cavitation in the
aonication sy~tem allowing more acoustic energy into the
reaction volume 14 . The latter syatem was rrnr~ntrl c with
the aonication volume 16 and was locat.ed above it. The two
re8ervoir8 o~ the two sy~;tems were.~èparated by a l mm (40
10 mil) stainless steel disk 22. In the 15 ml reaction
re8ervoir, 18, heavy water was circulated at a rate of 300
ml/min by flow meter 51 through a 3.3 L heat .ol~rh~nr~r 162.
The e~tPrn~l preaaure of the gaaes was adjuated to a value
in the reaction volume ayatem to optimize the character of
15 transient cavitation bubblea. The two concentric 7.5 cm
diameter acouatically connected ayatema were run at steady
state temperature conditiona (where input and output power
are r-;nt~in~-l at a ateady atate after an initial atart-up
period) . The reaction reaervoir 18 rr,nt~in,~A the palladium
20 foil 26. Critical temperaturea were monitored at various
points in the two systems, tracking the total energy input
and output with time. The acoustic field was generated by
a 64 mm ~2.5 inch) titanium acoustic horn 12 tuned to 20
Khz. The acoustic ener~y delivered to the Pd foil 26 was
25 about 3 watta/cm2. The rnnt~ or the ~nir~tirn
aystem and horn was 1/2 inch thick ~ n~hl P aluminum split
sliding cylinders g6 and 97, and for the reaction volume,
a 3/4 inch thick stainleas steel ring 20. This describes a
special configuration of the ~n1 r~tr~r volume in Figure 1
30 where one may move the horn 12 with respect to the atainles3
ateel aeparator disk 22 to allow better control of the
acouatic energy delivered to the reaction volume.
The pressure gaugea were digital compound gauges from TIF
Instrument Co., which measured a pres~ure o~ 30 inch lIg(60
35 psig). The gaa mixera 45 and 66 were 25 ml Pyrex bulba.
The gas or riase~3 in the reaction volume were deuterium 62
and/or argon 64. The gaa in the aonication volume was
WO 95/16995 217 ~ PCT/US94113824
-15--
nitrogen 65. The flow meters were from Key In3trument,
(Trevose, Pennsylvania) with an acrylic body and a stainless
steel float.
The circulating pumps 50 and 72 were magnetically driven
5 from Micro Pumps, located in Concord, California, with
Teflon~ gears and a stainless steel body, part 07002-23,
coupled with a variable speed motor, part 07002-45. The
pump's interior material exposure to the cirr~ ;n5 heavy
water was to Teflon~ and stainless steel. The valves were
10 FEP Teflon~ ~rom Galtek Corp., as were the tubing and
fittings. The thermocouples were type K from OMEGA along
with the No. 871 output thermocouple reading devices and the
~H20SW multiprobe switch boxes. The reaction volume
interior material e~ u~ ~ to the circulating heavy water
15 was FEP a~d stainless steel. The input and output ports
were supplied with Teflon~ fittings for the FEP tubing which
was used throughout the apparatus.
The electric isolation of the reaction volume was
accomplished using Teflon~ gaskets 108 shown in Fig. 2 with
20 the sandwiching of reaction volume 14 and separating disk 22
to the sonication volume with six 12.7 mm (1/2 inch) nylon
bolts 38. The s~ni~tinn volume 16 was ~~hin,of~ from an
aluminum block to ~ t~ a 63 . 5 mm (2 . 5 inch) horn 12 .
The heat exchanger 162 for the reaction volume 14, was a
25 polyethylene ~nt~inPr with stirrer 54 for the light water
coolant 56, which received from the reaction volume the hot
heavy water from reaction volume 14, which then passed
through a coiled 1/8 inch 8t~;nl~q heat exchange coil
element 44, then back to t~e reaction volume. The heat
30 ~ lall~r for the sonication volun~e 163 wa6 a polyethylene
~lnt~in~r 83 with stirrer 77 for the light water coolant 70,
which received from the sonication volume the hot light
water 81 which then passed through a coiled 1/4 inch copper
heat exchange coil element 79, then back to the sonication
3 5 volume .
217~0~S
WO 95/16995 PCTIUS94/13824
--16 -
The apparatus wa3 first cleaned then bolted together and the
reaction system was pressurized with deuterium or argon
testing for tightness and leaks'~, When sati5fied that the
system was tight, the ~lPr~ $l heavy water 121 waa added to
5 the reaction volume lo~ and circulated, removing any
rPm~;n;nr system gas bubbles to the gas mixing bulb 46. The
addition of water to the Elnn1 r~tnr loop and its
pressurization with nitrogen 65 was the next step. The
purpose was to reduce the cavitation in the Snn;rz,tinn
10 volume 81 (the acoustic pressure, which was delivered to the
stainless steel disk, did not produce cavitation damage in
the sonication volume because the formation o~ bubbles was
repressed by the high Pl~tPrn~l pressure) . The apparatus was
brought close to the operating temperature by filling the
15 two heat Pl~r~ngprs~ 162 and 163, with preheated water.
Both the heat exchangers were stirred with stirrers 54 and
77. The two pumps 50 and 72 were turned on circulating the
heavy and light water through their re3pective systems. At
this point, the reaction volume was filled with gas to the
20 appropriate P~rt~rni~l pressure; then, the initial
temperatures were measured, the sonicator 78 was turned on,
the time was noted, and the run was started.
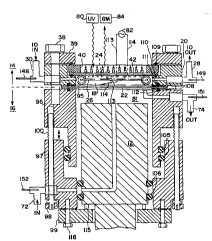
Figure 3 B is the detail of the reactor from Figure 1. A
8t~;nlPc~ gteel reaction volume 20 which consists of a top
25 36 --rh;nP~ of aluminum allowing for viewing through ports
40 and supporting a FEP sealed window 42. The seal is made
by '~O'~ rings llQ. The bottom is a stainless steel disk 22
with two insulating FEP flat gaskets 108 sealed by "O" rings
lll and 112. The window 36 Ls fastened to the reaction
30 volume via stainless steel ring 39 cushioned by `O-t ring
109 .
Figure 3 A shows the reaction volume. Electrodes 32 are
passed through ports 113 and 114. One electrode 32 is
attached to the grid 24 which is insulated from the rest of
35 the system by FEP insulator 25 The other electrode 32 is
Wo 95/1699S ~ 1 7 8 0 ~ 6 PCr/US94113824
attached to the stainless steel disk 22 making it the other
electrode .
The reaction vessel is clamped t~reth~r and to the rest of
the system using nylon bolts 38. The output of the reaction
vessel is 28 and the input is 30.
Referring now to Figure 4, an Al t~rnAtF~ a; ' of the
cold fusion device described above follows.
In this particular embodiment, the heat generated within the
reaction vessel 218 is ~u~, uullded with heat exchange liquid
281.
The system depicted in Figure 4 includes a pi~70Pl rrtric
crystal or ceramic cnn;cAtnr 276, which is positioned
adjacent to the r~Art;nn vessel 218 and which is immersed in
a cooling medium 281. This particular cooling medium 281 is
comparable to the cooling system described in the principal
' ,ra,; Xowever, in this particular emhodiment it
_ let~-l y surrounds the reaction vessel and sonicator,
rather than being just beneath it. In like manner, a
coolant pump 272 will circulate water into and out of this
cooling medium 281. Appropriate heat f~rrhAnr~r and the like
may be affixed to the structure in a manner similar to the
principal ~mhoa;-- , within the rnntA; t vessel 212.
~t~nntA; vessel 212 is used in a descriptive sense to
indicate that the entire structure may be cnnt~;n~ within
one vessel . ) The reaction vessel 218 is positioned in the
center of such cnntA; t ves8el, with the cooling medium
281 surrounding it within the rnntA; t vessel.
Entering into the l~nntA;nm~nt vessel is a control stem 214
which is used to provide conduit means for appropriate
conductors 216 leading to the sonicator 276 . Other control
devices, such as temperature ;n~a~;rAt;nr devices 225 and
pressure indicating devices 224, may be positioned adjacent
to the control stem 214 with the n~c~ccAry electrical leads.
W095/l6995 2l7~n8~i PCTIUS94113824
--18--
In order to provide deuterium oxide and deuterium to the
reaction vessel 218, cnnrllli tc 228 and 230 are provided to
the reaction vessel. These cnnrlll;tq may be used in a manner
similar to the conduits in the principal embodiment to
control to a degree the heat,~ithin the reaction vessel,
should such control be n~n~ ~ry The liquid in reaction
volume 218 is kept circlllAf~n~ by pump 250. The excess heat
generated in reaction volume 218 is ~ with the fluid
281 Qf the ~nnt:: i ' system. On the other hand, it may be
appropriate to keep the structure at a heat ~m~wh~t higher
than in the principal embodiment, thereby rl~p~n~l;n~ upon
coolant medium 281. In that vein, conduit 274 and return
conduit 278 may be provided to the coolant medium 281 so
that water or- heat transfer material may be circulated by
pump 272 to the ~'nnt:~1 t vessel. Finally, an appropriate
power supply 227 is provided to the sonicator 276.
O~eration of the Fir~t Alternate Embodiment
Operation of the alternate embodiment follows generally that
of the principal ~ 9; t, The heat generated in reaction
volume 218 with the interaction of cavitation bubbles on the
surface of Pd wool or wire 226 is removed guickly through
the thin wall 242 and the liquid circulated via pump 250.
Heat exchange liguid is circulated through heat exchange
coil 279 (space heater). The liquid circulation is
accomplished with pump 272 replacing the hot liguid 281 with
cool liquid via conduits 27~ and 278.
gECOND AITERNATE EMBODIMENT
Now referring to Figure 5, the second alternate ' ~ t
of a microfusion device iB shown, consisting of multiple
30 small devices 302 that are closed systems acting in concert.
These devices take the sum of the heat generated in all mini
devices in the flow pipe system 300 and in the fluid 381,
circulate it, and use the heat for some specified purpose.
The heat generated within reaction volume 318 is in the
I WO 9S/16995 217 ~ ~ 8 ~ ~ PCT/US94/13824
--19--
fashion of earlier stated technology. The acoustic energy
i8 supplied by the piezoelectric crystal 376 via electric
conduit 301 which also transfers temperature information to
soni-control 378 for control of the heat transfer for all of
5 the mini devices. The crystal 376 is bonded to the metal
membrane 322 which is in contact with the deuterium oxide in
volume 318 . Also in volume 318 is the pAl l A~ m wire or
wool 326 which provides the surface and lattice for the heat
producing fusion events. The threaded hex sealing nut 310
10 seals the acoustic membrane 322 via "O" ring 307 to the body
342 of the reaction volume. The volume 318 is equipped with
a filling port 330 and pressure release valve 393. The mini
devices are sealed into the tube or heat flow pipe 300 via
threaded element 346 and i,o.. ring 305.
O~eration of the Second ~l ternate Embodiment
Operation of the second alternate: ' ~.li follows
generally that of the first alternative ~mhotli~Ant. The heat
generated in reaction volume 318 from the interaction of
cavitation bubbles on the surface of Pd wool or wire 326 is
removed quickly through the wall of 342. The liquid in 302
relies on convection from the wire 326 to the liquid
~ ntAin,~ in reaction volume 318, then through the wall of
body 342. Here the heat is transferred to the circ~ t;ng
liquid 381 and carried to the point of use. In summary, the
heat generated by fusion events is transported by pipe 300
circ1-lAt;n~ liquid 381 at a rate controlled by valves 371
to a device similar to that found in the first alternate
embodiment. The mini microfusion cells embedded in the pipe
300 serve to provide constant and even heat to the
cirr1llAt;n~ liquid which can be extracted at some point
downstream for the users' benefit, then returned as cool
liquid for reheating and reuse.
WO95/16ggs 2~-8(3~i PCTIUS94/13824 -
--20 -
THI~D ~T-'rT'RI`T~TE EM30DIMENT
Now referring to Figure 6, the third alternate: ~;m~nt of
a fusion device is shown. In this .omhorl; - , the heat
generated within the reaction vessel 418 is converted to
electricity- or electrlcal current by means of a
thermoelectric device 402 (TED~ using the heat differential
developed between the pAl 1 Arli lattice 426 and the heat
exchange tluid 470 . The TED 4t~2 ca~ take the configuration
shown schematically in Figure 6, which is a serieA
aLL,.~J . ~t. In Figure 6, there is a tem~erature gradient
between the two elements 426 and 442. The system depicted
in Figure 6 includes a so~icator 476 mounted on metal
membrane 422 forming a wall of reaction volume 418. The
reaction volume 418 is immersed in a heat exchange liquid
470 r~ntA;n,~l in insulated box 412.
The entire syfitem is cnntA;n~l in the box 412 80 as to
capture mo~t of the heat g~onf~rAt~d by all factor~3
(cavitation, electronics, and lattice events). The
Sf~n; rAtr~ 476 i9 protected, as is the power supply and
control for the sonicator 424 and the temperature sensing
and control 425, from the liquid 470 by shield 497, keeping
the electrical ~ therein dry. The TED 402 i8 a
sealed volume which consists of the rAl l A~l; llm 426 and the
outer wall 447, and can be filled with deuterium ga~ 462.
2~ The reaction volume is situated in the center of confinement
vessel 412 with the heat exchange 1 iquid ~LL, L~ .ling it
within the cnntA; vessel_ E~tering into the
rr~ntA; ~ volume is stem 414 which is used to provide
conduit means ~or appropriate conductors 416 leading to the
electric i~put for the Gonicator 476. Another conduit 466
performs the functiQn of (1) allowing deuterium pressure to
both the reaction volume 418 and the TED volume 447 for the
purpose of keeping the deuterium at equilibrium pressure in
the pAl l Ar~ m lattice, and (2~ acting as a conduit for the
35 electric energy transported to the outside of r~nt;l; t
412 and 447 by leads 406. The energy generated by TED i~3
Wo gS/l6995 2 1 7 8 ~ PCr/US94113824
--21--
carried by 406 to the collection and dlstribution device
444. Other control devices for measuring pressure and
temperature can be placed near either conduit 466 or 414 as
a matter of practicality.
5 In order to provide deuterium oxide to the reaction volume
418, the conduit 466 can be used. The circ~ t;nn of
deuterium oxide through the reaction volume 418 is via
convection with the hot liquid in 418 rising into volume
446, then settling down after cooling through conduit 430
10 and traveling back into the reaction volume 418 through the
bottom. The control of the temperature of the system is
m-int~in~ at a steady state to r~int~in the best
environment for cavitation. It may be appropriate to keep
the fluid 470 cool by circulation to the outside environment
15 for heat exchange via conduits 478 and 474 by pump 472.
OT~eration of the Third Al ternate T'm~r,diment
Operation of this alternate ' ~(1i follows generally
that of the first alternate embodiment; however, in this
instance, a sonicator is operated adjacent the reaction
20 vessel 418, thereby causing microfusion to occur in the
m-faced thermoelectric device TED 402. Microfusion
events raise the temperature at the hot junction 426 of the
TED creating an ell~rtrir~l current within the system. Such
current is tapped off through lead 406. A positive
25 deuterium pressure is m-int~in.o~ in the TED rnnt~inm~nt 447.
FOURTH AT TE~NATE EMBODIM~NT
Ref erring now to Figure 7, ln the f ourth alternate
t pump 602 circulates D2O into reactor 600 through
conduits 606 and 608. Where conduits 606 and 608 empty into
3(~ reaction volume 604 are heaters 614 and 616 located outside
of the acoustic field generated by sonicators 610 and 612.
Above and below reaction volume 6~4 are sonicators 610 and
612. ~ocated in the reaction volume 604 is metal lattice
Wo 95JI6995 217 8 ~3 8 6 PCTt7i~Sg 7tl3824
--22 -
618. D2O flows out of reactor 600 through conduits 620 and
622 through flow meter 624 and into bubbler 626. Pressure
devices 628 control the pressure in the bubbler 626 and
reactor 600. 7~2O flows back to the pump 602 whereby it i8
5 recirculated into the reactor 600.
. . .
In Figure 8 is depicted a cross section in di~ayL tic form
of a reactor 600. The reactor 600 has ah upper aluminum
ring 632 and a lower ;71l7m;nl1m ring 634. ~h upper sonicator
612 and lower snn; ~ 7tnr 610 are emplo~ed to provide for
lo transient bubble formation above and below metal foil 618.
The metal foil 618 is placed in the reactor area 604. The
reactor area 604 has input ports 604 and 606 and output
ports 620 and 622. Heaters 636 and 638 are provided for
heating the ; n~; n~ liquid to the desired temperature .
15 Upper and lower ring insulators 636 and 638 respectively are
provided for insulating lower and upper metal (e.g.
8t-7inlP~7 steel [S.S.] )electrodes 640 and 642, which
electrodes also serve to transmit the e7 ergy from the
sonicator to the fluid in reactor volume 604. The
electrodes 6~0 and 642 are connec:ted to circuit 644 to
provide for a rnntinllnus electrical field during the course
of the reaction.
The reactor is shown in greater detail in Figures 9a, b, and
c.
The following examples are offered by way of illustration
and not by way of limitation.
7~pr7? Tr~ENTA:l~
The apparatus employed was sub6tantially as described for
Figure 1 The apparatus comprised two closed ~-irClll;7tinn
systems, in which the proper P~tPrn;71 pressure and
temperature were Ir-int,7;nPd to produce cavitation bubbles
over PxtPnr7Pd time periods at steady state conditions.
Bubbles were produced on the surface of a palladium foil by
WO 95116995 ~ ~ 7 ~ D 8 5- PCTIUS94/13824
an acoustic generator operating at 20Khz providing a non-
focused acoustic field with an average intensity on the foil
of about 3W/cm~ and an amplitude of about 3 atms.
The cr~nt~;nPr comprising the sonicator provided for water
5 circ-llAt jt~n at a rate of 600ml/min through a 15 L heat
exchanger. An ~YtPrnAl pressure of about 6 atm of nitrogen
was ~;nt~;nPd in the sonicator flow system to reduce
cavitation in this system. The fluid in the reaction vessel
and the fluid in the c~n;~t;on vessel are separated by a
10 lmm stainless steel disc. The reaction volume in the
reactor is 15ml and the reaction medium is circulated at a
rate of 300ml/min through a 3.3~ heat exchanger. The
PYtPrn ~l pressures of gases were varied in the reaction
volume system. The acoustic field was generated by a 64mm
15 titanium acoustic horn tuned to 20 KHz. The sonication
vessel was a 13mm thick aluminum cylinder, while the reactor
vessel was a l9mm thick stainless steel cylinder. The
;ntPrnAl walls of the reaction vessel were either FEP or
stainless steel. Electric isolation of the reaction volume
20 was accomplished using Teflong gaskets which sandwiched the
reaction volume and disc to the ~ n;~t;~n unit. the
p~ foil was 50x50xO . lmm ~Johnson Matthey Chemicals
Iltd. ) weighing 3g, and 99 . 9975~ pure . The foil was
suspended by being held at its corners by an FEP supporter
25 in a plane parallel to that of the stainless steel disc.
The heavy water employed was 99 . 9~ pure, was (1P~ Prl and
pressurized with deuterium and/or argon, prior to use. The
deuterium was shown to have 14 ppm 4He.
In carrying out the process, the apparatus was ~irst cleaned
30 and then bolted to~PthPr. The reaction system was then
pressurized with deuterium and/or argon and tested for
tightness and leaks. The reaction medium was then added to
the reactor and circulation system, carrying ~ln~--ntP~l gas
bubbles to the gas phase of the gas mixing bulb where the
35 bubbles were removed. water was then added to the sonicator
system and pressurized with nitrogen. The apparatus was
217~Q8~.
WO 95/16995 ,~ , PCTIUS9~/13824
--24--
then brought close to the operating temperature by f illing
the two heat ~Yt~h~n~rS with preheated water, with stirring
of the water in the heat exchangers. The pumps were then
turned on to circulate the f luids in the two systems . The
5 reaction volume was then pressurized with deuterium and/or
argon, where different ~Ytc.rn~l gas prQssures were employed
for different runs. After pressurizing the system, the
initial temperatures were measured, the sonicator turned on,
and the run begun. ThP ~uu~les were employed to determine
the temperature of each ~ in each run . For the
first 2 to 3 hours of each run, the temperatures of all
components increased and then leveled off as the system
approached steady state. Excess heat was determined by a
steady-state measurement technique by measuring the heat
15 output from each , -~t of a system in a single run. The
following table indicates the results of a number of
8tudies .
WO 95/1699~ 2 1 7 8 ~ ~ 6 pCllUS94/13824
--25--
.C
~ N~ i ~ N ~i ~r) N ~Y) (n ~i
N N N ('7 ~'1 ~ 1`7 N ~) N ~1
O
. .
H ~ h ,i o N N N r ~ c o 3
N N N N ~`; N N N N ~1 ~i 3 3
~ o a) r c~ D ~ m
r ~ g E~ Ln v
-
oU o
U~ O N N ~ C~ r~ ~i N ~ i ~ E~ '
O
:~ ~ a
~i ~1 u~ a) N
'i+~ +~ ~ +~ +~ +~ +
oo z ,~ O ~ m
m ri
H _ f~) (`7 (~ ~ O ~ 0 o ~ ~r ~ U r
~ CQ 'i 'I u~ z V S E~ m
N ~ o ~ i ~i ~i ~1 0 LD LD O ~a 3 ~: C
X 3
,¢ LU i O
~i Z H i~ I z p~ ~ I~ . ~ ~N i U
- Z Y l¢ ~ ~,)
~ Li V '~ ID L~i
Wo 95/16995 2 1 7 8 ~ ~ 6 PCT/US94/13824
--26--
On many or~A~;nn~ following heavy water-deuterium runs, the
palladium foil was found to be discolored and ~ f~ ~
where on some ocrAAinn~ the foil partially melted,
displaying both high discoloration and ~" 'nPnt holefi about
5 5mm in diameter. The monitoring of steady state heat energy
output (about 400 watts over 24 ~to 72 hours) ; nri; nAt~l that
heavy water-deuterium runs were~characterized by an output
energy a3 high as 10 0 w3~s more than that f ound in
comparable light water-lLy~ L runs. A comparison of heavy
10 water runs I, J, L, M, R and 5 to light water run K, as well
as a comparison of heavy water runs A and B to the light
water run C ;n~ At~-q a correlation between excess heat and
4He production.
In order to measure the 4He produced, a 35ml gas ~ 1 in~
5 bulb, equipped with 2 isolation valves and a gas syringe
port, was inserted between ;~nlAt;r~n valves in the reaction
volume system at the end of each heavy water run. A portion
of the gas was transferred to the ,l;n~ bulb. The bulb,
~nntA;n;n~ wet gageg, was then removed from the system and
20 1 to 3ml of the gas transferred through the syringe port to
a valved gas syringe . The gas f rom the gas valved syringe
was then injected into the port of a mass qre~ t~ for
analysis. 1~L1 of pure 4He was iniected directly into the
mass 3pectrometer and served as a rough quantitative
25 standard. The re301ution of the maas ~e.:L,- ter was about
o . 01 mass units . This resolving power easily resolved the
mass peaks, 4He and D2. The potential for nnntAm;nAtion of
the gas with 4He from the argon and/or deuterium is
unlikely, because in those runs where heat was not produced,
30 4He could not be detected.
The pAl 1 Atiium Foil Analvsis
The 4He found by MS analysis may not account ~or all the
excess heat . If DD fusion events occurred in the pAl l A~ m
lattice, then perhaps there were other ~usion events that
35 followed, energized by the DD events, causing small changes
WO 95/16995 2 1 7=8 o ~ ~ PCrlUS94/13824
--27--
in the pAllA~;llm lattice isotope distribution and perhaps
some trAnp~-~lt~tinn. Other possibilities for the generation
of excess heat Q(x), such as trAn~ ~t;ons in the
cavitation exposed palladium lattice, could be found by
5 analyzing the exposed palladium foil, using an Inductively
Coupled Plasma Mass Spectrometer (ICP-MS). In an
; ntl~r.on~ nt laboratory, a comparative analysi3 was made of
before and after cavitation-exposed palladium sample using
a Perkin Elmer Sciex Elan 500 ICP-MS, with a resolution of
10 one mass unit. This analysis looked for changes in the
before and after ~ U~ULC of the metal rAl l A~ m lattice
samples, and was done six months after the completion of the
above study.
The p~llA~ m foil, which had a purity of 99.9975~, used in
15 runs I, J, K, ~, M, R, and S had a certified ~ Al
analysis by the vendor for 72 .~l~ A. The lattice
impurities of interest for this particular analysis were
rhodium at less than 3 ppm, silver at less than 1 ppm, and
cadmium at less than 1 ppm (below the level of detection of
20 the certified rl ~ Al analygis) . The ICP-MS analysis
measured small differences in isotope rnnrrntrpt;nnR of
metals in the palladium foil lattice with masses similar to
those of the palladium isotopes . The two r~l l A~ m samples
were dissolved in nitric acid, O . 024 gm before and O . 022 gm
25 after exposure, and analyzed.
Some of the suspect stable trAnl ~1 isotopes in the mass
range of interest were blocked from analysis by the high
concentrations of the stable isotopes of pAllallillm foil.
The rnP~;hlf~ stable trAn~ Ptinnq orig;n~t;nr, from
30 pAl l A~ lm changes in the lattice and impurities that might
be pre~ent are blocked to any ICP-MS analysis of low
cnnrl~ntrAt;nn transmutations or impurities, because of the
- high rnnrF~ntration o~ Pd isotopes. The low rnnr-~ntration
isotopes can be analyzed below Pd mass number 100 and above
35 mass numher 112. The isotope Cdll4 is the only isotope
deiinitely found in excess when compared to the unexposed
WO 95/16995 PCT/US94/13824
21~118~
--28--
r~ 7m The strip i~l the lower right defines the scope
of the 'cransmutation analysis.
Small differences in ion counts from the two dissolved
ps:llArl;~ 3amples, before and after, were measurable only
5 for those isotopes that were not blocke~d by the r~
isotopes. It would be of intere8t to measùre any changes i~
the palladium i30tope distribution, b~t~ these changes would
be 80 small in the r~ m ric,~,i system and therefore
-~C;hle to detect. This blocking limited detection of
10 possible tr atinnq to the two cadmium isotopes Cdll2
and Cdll4, with the i~otopes Cdll3 and Cdll6 not likely
tr~n~ ~t;on candidates. Only for the isotope Cdll4 was
the difference in ion counts, equivalent to 30 :: 10 ion
counts, statistically significant as an analytical re8ult.
WO 95/16995 2 1 ~ 8 ~ 8 ~ PCT~S94113824
--29--
Ul ~` ~D In
r ~` N
N ~ N C)
~I N ~I N
~D
~ U
cq
N ~~. E-l H H +l +l
'¢ ~ N ~ ~1
U~ ~
~: Z E~
O
~ ~ Z ~ I` ~ ~ ~
O ~ ~ ,~ o +l -H +l H
t` t~ N N
N ~ ~ ~1
Nz +i -H +I t~
X
O O ~1
O ~ ~ ~ O
Z D. .¢ ~1, Z
W095/16995 2l7~n8~i PCr/US94/13824 -
--30--
In Table 2, the IC~P-,D~S: anaiysis of the llnhl ork~d cadmium
isotopes found in the exposed and unexposed p~ m foil
is shown. Column 1 is a list of fusion reactions of a hot
alpha or a deuteron with a p~ m lattice isotope forming
5 a cadmium isotope. Column 2 is the ion count of the acid
used to dissolve the sample. Column 3 is the ion count of
the p~l l A~ m foil before the e~o~u~ ~: to the cavitation
process. Column 4 is the ion count Qf the palladium foil
after the ~O~U1C: to the cavitation process.
10 Column 5 is the change in ion counts between columns 2 and
3 taken together when compared to column 4. (The change in
the ion count is close to ppm values for cadmium in the
palladium ~oil. ) The relative sensitivities of Cd and Pd,
which were not measured during the analysis, are related to
15 their relative average irni7:~tion potentials which are close
in value.
Column 6 is the list of cadmium isotopes. Column 7 is the
natural ~hlln~l~nro of some of the stable cadmium isotopes.
The ion counts for the metal isotopes are close to the ppm
20 Cl-nr~ntration that existed in the 3 r~m palladium foil,
before and after the runs I-S in Table 1. No Cdll6 was
found in the exposed sample and little, if any, Cdll2 and
Cdll3. The analysis did find Cdll4 at a level 30~10 counts,
which relates to about 30ilO ppm in the exposed sample when
25 1 _ ~d to the unexposed p~ m sample.
The above result could be attributed to a redistribution of
a cadmium cnnt~mi n~nt in the system in the course of heavy
water-deuterium runs. If such c~nt~m;n~t;on was present, it
would have led to an increase in all cadmium isotopes. In
30 particular, if a level of the isotope Cdll6 had been found,
rrnt;tmi n:qt j rn could explain the analytical result . On the
other hand, there being no Pdll2 isotope, no Cdll6 could
have been formed through a tr~n~ at i r,n process of
r~ m in the after sample. The latter was the case;
35 there were nP ~ hl ,~ rli fff.~f-nr~ in the ICP-MS ion
2~78086
WO 95/16995 PCrNS94113824
--31--
counts for the isotope Cdll6. The presence of Cdll4,
without the presence of Cdll6, points to a trqnl ~tinn
-h;~n;r~ rather than to a cadmium cnnt~min~tinn source.
In the next study, palladium and titanium foils were
5 employed, where the titanium foil had a size of 50mm x 50mm
x 0.2mm (-2 grams). The ~LuceduL=8 employed have been
already described a~ove, f~ r;f;5~ conditions being provided
in the following table.
WO 95/16995 2 1 ~ 8 0 8 6 PCT/US94113U4
--32--
' X
V ~ "
, .. ...
~ O ~ ~ o ~ _
~1 e
O
rl ~
C O .~ "
-dX 3 ~ ~
. ~ e
~il ~ I_
~1
WO 95/16995 2 1 7 ~ ~) 8 5 PCrlUS94113824
--33--
The above results demonstrate a number o:E f actors . First,
there is substantially greater heat being produced as
compared to the amount of heat which is introduced into the
- system as various forms of energy. Thus, the total energy
5 which is obtained from the sy3tem is greater than the total
energy which is introduced into the system in the various
forms of heat, -h~nicAl energy and electrical energy.
Secondly, helium is produced in amounts substAntiAl 1y
greater than can be ~rlA;n~d by ~nnt~m;n~tinn of the
10 system. The exce3s helium is produced substwntiA7~y
reproducibly and correlate3 with the amount of heat produced
with the system. Furthermore, trAnl At; nn appears to
occur in the fnrm-t;nn of cadmium from rAll~ m Finally,
using titanium foil, a substAntiAlly ~nh~n~-~d ratio of 31Ie
15 to 4~Ie i8 obtained, which requires that there be production
of 3~e from the interaction between deuterium and the
titAnil~m foil. The subject invention therefore provides a
number of important r~pAh; 1 i ties, in that heat can be
produced, novel isotopes can be produced, and most
20 importantly, energy which is employed can be amplified in a
safe way with simple devices u~ing ;nPyr~nqive' clean
material~ .
All publications and patent applications cited in this
specification are herein incorporated by reference as if
2~ each individual publication or patent application were
specifically and individually indicated to be incorporated
by ref erence .
Although the foregoing invention has been described in some
detail by way of illustration and example for purpose3 o~
30 clarity of understanding, it will be readily apparent to
those of ordinary skill in the art in light o~ the tF-a~h;n~
of this invention that certain changes and modifications may
be made thereto without departing from the spirit or scope
of the <l~e.lded claims.