Note: Descriptions are shown in the official language in which they were submitted.
21 90679
LYMPHOCYTE TACHYKININ
Field of the Invention _ _
This invention relates to the biologically active
peptides known as tachykinins. More particularly, the
invention rela~es to a new tachykinin gene and to a new
tachykinin peptide.
Background of the Invention _ ~
In the description which folIows, references are made
to certain literature citations which are listed at the end
of the specification.
A large number of biologically active peptides have
been identified in higher animals One family of
biologically active peptides is the tachykinin family.
Previously identified mammalian tachykinins are produced in
the nervous system and brain and have therefore been called
neurokinins. Three have been identified, namely, Substance
P (SP), Neurokinin A (NKA; also called Substance K,
neurokinin a, and neuromedin L), and Neurokinin B (NKB;
also called neuroklnin ~, and neuromedin K). Other
tachykinins, founcl in non-r l;~n species,-include
Kassinin, Eledoisin, and Physalaemin (1, 2).
All tachykinin peptides contain the characteristic
carboxy-terminal amino acid motif
Phe-X-Gly-Leu-Met-NH2 or F-X-G-L-M-NH2
Genes which encode the three r 1; ~n neurokinins
have been described (1, 2). One gene, PPT-A, by alternate
splicing encodes three different mRNA transcripts. The
first of fhese, aPPT mRNA, encodes the precursor protein,
preprotachykinln (PPT), from which SP is released by
cleavage.
The other two, ~-PPT and r-PPT, encode a precursor
which yields both SP and an additional amino acid sequence
of 36 or 21 amino acids~ respectively. These peptides are
long forms of NKA which has 10 amiro acids. Both long
forms of NKA are also neurokinins. A second gene, PPT-B,
encodes NKB only.
The previously described neurokinin genes are
expressed=primarily in the nervous system and brain. The
- 21 90679
-- 2 -- =
neuro~inins ara released from nerve en~ings and act on the
immune system. Release of neurokinins is associated with
pain and antagonists or inhibitors cf the neurokinins act
as analgesics (1,2).
A wide variety of additional bioactlvities have been
attributed to the neurokinins, including decrease of blood
pressure, plasma extravasation, smooth muscle contraction,
release of neu~otransmitters, modulation of neuronal
activities, a variety of haematopoietic effects and release
of histamine, prostaglandins and other inflammatory
mediators. These effects are mediate~d by one of three
cellular receptors ~NK-l, NK2, and NK3) which have been
identified on the plasma membrane of a variety of target
cells (l,2).
:
Summary of Drawings _ ~
Certain embodiments of the invention are described,
reference being made to the accompanying drawings, wherein:
Figure 1 shows a Northern blot of poly (A) RNA from
the following cell lines and tissues:
70Z/3:~Pre=B cell line; 70Z/3y RAG=positive variant
of 70Z~3; IIB4 CB17 I.l and 5.1, scid 2.1 and 4.1,
RAG1-14 and -17 and RAG2-5 and -21: fetal liver cell
lines transformed with Abelson murine leukemia virus;
WE~I 231: immature B cell line; J558: myeloma cell
line; RBL 5 and EL4: T lymphoma cells; P338~1 myeloid
lineage cell line; CB 5: erythroid lineage cell line;
NIH 3Y3 and L929: fibroblast cell lines; BMS 2.2:
stromal cell line.
Tissues were normal mouse tissues.
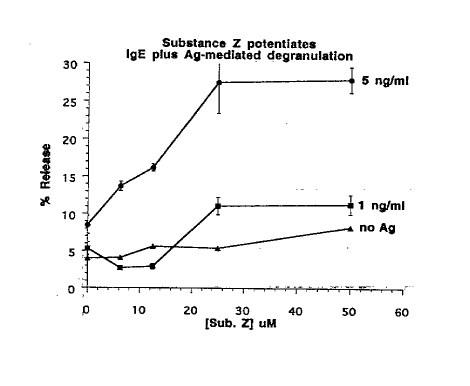
Figure 2 shows the effect of Substance Z on murine
mast cell degranulation. X axis is concentration of
substance Z(,uM) ar,d Y axis is mast cell degranulation
expres~sed as % labelled serotonin release.
Figure 3 shows the activity of Substance Z on human
fibroblasts in a cartilage degradation assay. X axis:
Sample numbers.~ Bars indicate % labelled
glycosaminoglycans released from cartilage disc.
Figure 4 shows the effect of various concentrations of
_, , .. ~
~ 21 90679
- 3 - ~ ~
Substance Z on the proliferation of murine leukemia cells,
proliferation expressed as colonies per 500 plated cells.
Detailed Description of the Invention
The inventors have identified a new mammalian gene,
designated PP~-C, which encodes a pr-acur-sor protein ior a
previously undescribed tachykinin peptide.
The cDNA sequence of the mouse PPT-C gene (Sequence ID
No:l) is shown in Table 1. It compri~es a sequence of 1249
nucleotides including an open reading frame encoding a
sequence of 128 amino acids. The start codon is underlined
in Table 1.
The deduced amino acid sequence (Sequence ID No:2) of :
the new precursor protein, designated substance Z precursor
protein, is shown in Table 2. Its structure shows it to be
a typical tachykinin precursor protein.
The SP precursor protein preprotachykinin, ior
example, includes the amino acid sequence of Substance P,
flanked by cleavage sites which are acted on by proteolytic
enzymes known as convertases=to~release Substance P. The
processing pathway has been described by Harris and Steiner
et al.(3,4). Cleavage takes place at a doublet of basic
amino acids, such as Lys-Arg or Arg-Arg.
Harris has proposed two basic types of recognition
sequence for endoproteolysis: 1) a monobasic amino acid in
close proximity to a cleavage doublet nf basic amino acids;
or 2) a strongly polar amino acid (Glu or Asp) in close
proximity to a cleavage doublet of basic amLno acids. The
neurokinin Substance P is an example of the first type,
with an Arg in front of the cleavage doublet. For this
type of recognition sequence, cleavage may occur either
between the amino acids of the doublet (like Substance P)
or àfter the doublet.
Neurokinin A is an example of the second type, with a
Glu residue in front of the doublet. For the second type,
cleavage usually occurs after the doublet.
Substance Z precursor protein has cleavage sites at
amino acids 55~56 and amino acids 68169 (underlined in
Table 2); these::cleavage sites flank a putative peptide
~ 21 9067q
-- 4 --
having the carboxy terminal motif FXGLM-NH2 characteristic
of all known tachykinins.
The recognition sequence N terminal to the putative
tachykinin could be classified as either type described by
Harris.
By analogy with the other tachykinin precursor
proteins, it is predicted that substance Z precursor
prDtein is cleaved either within the amino acid doublet
55/56 ~R), to give the 11 amino acid=.peptide RSRTRQFYGLM-
0 NH2 (Sequence ID No.:3 and designated herein substance Z) orC terminal to the second basic amino acid of the doublet,
to give the 10 amino acid peptide SRTRQFYGLM-NH2 (designated
substance 2-short form).
The characteristic tachykinin carboxy terminal motL~
of Substance Z is shown above in bold type. The remainder
of its amino acid sequence differs from previously
described tachykinins, as see in Table 3. Substance Z
precursor protein alsD differs considerably from the
precursors of the previously described r ~ n
tachykinIns.
Also included in the scope of the invention are
fragments or analogues of the 11 amino acid peptide
substance Z, including the lO amino a d d peptide, substance
Z-short form, which are agonists retaining the biological
activity of substance Z or act as antagonists of substance
Z.
By analogy with studies on Substance P, it is
predicted that up to about four N terminal amino acids may
be deleted from substance Z while retaining full or
partial agonist activity. The amino acid sequence
comprising the tachykinin carboxy terminal motif is likely
to be useful as an antagonist of substance Z activity.
Substance Z or fragments or.analogues thereof may be
prepared by any suitable peptide synthetic method.
Chemical synthesis may be employed, for example
standard solid-phase peptide synthetic techniques may be
used. In standard solid phase peptide synthesis, peptides
of varying length can be prepared using commercially
available equipment. This equipment can be obtained from
~ 21 90679
Applied Biosystems (Foster City, CA.). The reaction
conditions in peptide synthesi3 are optimized to. prevent
isomerization of stereochemical centres, to prevent side
reactions and to obtain high yields. The peptides are
synthesized using standard autPmated protocols, using t-
butoxycarbonyl-alpha-amino acids, and ~ollowing the
manufacturer's instructions for blocking interfering
groups, protecting the amino acid to be reacted, coupling,
deprotecting and capping of unreacted residues. The solid
support is generally based on a polystyrene resin, the
resln acting both as a support for the growing peptide
chain, and as a protective group for the carboxy terminus.
Cleavage from the resin yields the free carboxylic acid~
Peptides are purified by HPLC techniques, for example on a
preparative Cl~ reverse phase column, using acetonitrile
gradients in 0.1% trifIuoroacetic acid, followed by vacuum
drying.
The peptides of the invention may also be produced by
recombinant synthesis. A DNA sequence encoding the desired
peptide is prepared, for example by cloning the required
fragment from the DNA sequence encoding the complete
precursor protein, and subcloning into an expression
plasmid DNA. Suitable mammalian express-ion plasmids
include pRC/CMV from Invitrogen Inc. The gene construct is
expressad in a suitable cell line, such as a Cos or CHO
cell line and the expressed peptide is extracted and
purified by conventional methods. Suitable methods for I
recombinant synthesis of peptides are readily available
(5).
Analogues of substance Z may be prepared by similar
synthetic methods. The term "analogue" extends to any
functional and/or chemical equivalent of substance Z and
includes peptides having one or more conservative amino
acid substitutions, peptides incorporating unnatural amino
acids and peptides having modified side chains.
Examples of side chain modifications contemplated by
the present inventlon include modification of amino groups
such as by reductive alkylation by reaction with an
aldehyde followed by reduction with NaBH4; amidation with
~ 21 qO67q
-- 6 --
methylacetimidate; acetylation with acetic anhydride;
carbamylation of amino groups with cyanate;
trinitrobenzylation of amino groups with 2, 4, 6,
trinitrobenzene sulfonic acid (TNBS); alkylation of amino
groups with succinic anhydride and tetrahydrophthalic
anhydride; and pyridoxylation of lysine with pyridoxal-5'-
phosphate followed by reduction with NaBH4.
The guanidino group of arginine residues may be
modified by the formation of heterocyclic condensation
products with reagents such as 2, 3-butanedione,
phenylglyoxal and glyoxal.
The carboxyl group may be modified by carbodiimide
activation via -acylisourea formati'on fol'lowed by
subsequent derivatisation, for example, to a corresponding
amide.
Tyrosine residues may be altered by nitration with
tetranitromethane to form a 3-nitrotyrosine derivative.
Examples of incorporating unnatural amino acids and
derivatives during peptide synthesis include, but are not
limited to, use of norleucine, 4-amino butyric acid, 4-
amino-3-hydroxy-5-phenylpentanoic acid, 6-aminohexanoic
acid-, t-butylglycine, norvaline, phenylglycine, ornithine,
sarc-osin-e, 4-amino-3-hydroxy-6-methylheptanoic acid, 2-
thienyl alanine and/or D-isomers or amino acids.
Examples of conservative amino acid substitutions are
substitutions within the folIowing five groups of amino
acids:
Group 1: F Y W
Group 2: V L I
30 Group 3:' H K R
Group= 4: ' M S T P A G
Group 5: D E
For creation of antagonist compounds, C terminal
residues are fre'quently susceptible targets. For example,
in substance Z, Phe and/or Gly may be substituted with D-
Trp and/or Met may be substituted with Leu.
Additionally, metabolically stable, non-peptide small
molecules may be useful substance Z antagonists, by analogy
with the previous described non-peptide antagonists of
~ 21 90679
-- 7 --
Substance P (1).
Fragments or analogues of substance Z may be
conveniently screened for their effectiveness as agonists
or antagonists. For example, agonlst activity may be
assessed in the cartilage degradation assay described
herein. Identification of antagonists is discussed further
below.
The gene PPT-~ appears to be expressed in
haematopoietic cells, for example in pre-B cells, but not
in tissues such as brain, lung, heart, liver, spleen, tymus
and kidney, as can be seen in Figure 1. PPT-C expression
hag also been fsund in isolated fetal liver cells, where
expression was increased by administration of IL-7.
The following embodiments of the present invention are
pro~ided.
1. IsolAted Nucleic Acids
In accordance with one series of embodiments, the
pres:ent invention provides isolated nucleic acids
corresponding to, or related to, the PTT-C nucleic acid
sequence disclosed herein.
In accordance with a first embodiment, an isolated
nucleic acid sequence is provided which encodes substance Z
precursor pro:tein. The invention includes degeneracy
equivalents of the disclosed nucleic acid sequences ~and
sequences which hybridize to the disclosed sequences under
stringent conditions~.
In addition to the disclosed nucleic acid sequences,
one of ordinary~skilI in the art is now enabled to identify
and isolate nucleic acids representinq PPT-C genes or cDNAs
allelic to~the disclosed sequ-ence-s or which are_homologues
of the disclosed se;~uences. One of ordinary skill in the
art may now screen preparations of genomic or cDNA from any
selected organism, including humans, other mammals,
bacteria, viruses or yeasts or from genomic or -cDNA
librarles, using-probes or PCR primers to identify allelic
or homologous sequences. In particular, the pre5ent
invention enables the identification of the human homologue
of the murine gene identified herein. ~It is also
contemplated that additional PPT-C nucleic acid sequences
_ _ _ _ _ _ _ _ . _ _ . , . . . _ ,
~ 21 9067q
-- 8 --
will be isolated from human subjects suffering from a
variety of disorders, enabling the identification of gene
mutations which may contribute to these disorders.
Additionally, homologues of the mammalian PPT-C gene
identified in lower organisms such as yeast, invertebrates
or insects, may~provide suitable means for drug screening.
As will be understood by those in the art, allelic or
homologous nucleic acid sequ-ences ~ay be identified and
isolated using standard hybridization screening or=PCR
techniques, using short portions of~the nucleic acid
sequences disclosed herein.
The present invention further provides portions of the
disclosed nucleic acids sequences which are useful as
probes and PCR primers, for example for identification of
homologous ganes.
2. Substantially Pure Proteins
The present invention further provides for
substantially pure preparations of substance Z precursor
20' protein or frag'ments thereof. ~-
Substance Z precursor protein may be produced by
recombinant methods, as will be understood by those skilled
in the art.
3. Antibodies
The present invention also provides antibodies, and
methods of making antibodies, which selectively bind to
substance Z.
The antibodies of the invention may be polyclonal or
monoclonal, or may be antibody fragments, including Eab
fragmen-ts'an'd sIngle chain antibody fragments. In
addition, recombinant antibodies may be generated, as well
as humanized antibodies based upon non=human antibodies to
substance Z. Antibody preparation techniques are generally
described in references such as Antibody Engineering: A
Practical Guide, (6), or Antibody Engineering, (7).
In order to prepare polyclonal antibodies to substance
Z, substance Z peptide may be conjugated to a carrier
protein or m~y-be expressed recombinantly as a fusion
protein which contains the peptide sequence of substance Z.
~ 2~ qO679
Preferably, the carrier protei~ or fused protein is
conjugated to~ the carboxy terminal end of substance Z
peptide.
E. coli expression systems such as lacZ fusions using
the pU~ series of vectors and trpE fusions using the pATH
vectors may, for example, be used to produce substance Z
fusion proteins. The expressed protein can then be
purified, coupled to a carrier protein ~f desired, and
mixed with Freund's adjuvant and injected into rabbits or
other appropriate laboratory animals. Following booster
injections at weekly intervals, the rabbits or other
laboratory animals are then bled and the sera isolated.
The sera can be used directly or purified by conventional
methods, such as affinity chromato~graphy. The sera can be
used as probes to ldentify substance Z on gels of protein
extracts from cells and tissues.
Monoclonal antibodles (MAbs) against substance Z may
be raised in a number of animals, such as mice. Methods
for making MAbs are well known and are described in
publications such as that by Harlow and Lane(8). As an
example, one such method is described.
Peptide Z is first coupled with the carrier protein,
keyhole limpet haemocyanin, using carbodimide. The peptide-
protein conjugate is then separated from free peptide by
dialysis. The conjugate is injected into mice (typically
2-6 mice) at a dose~of 50~g per mouse with complete
Freund's adjuvant. At two-week intervals, the mice receive
a second and third booster. Test bleeds are performed 10
days after each booster to assess the development of the
antibody response to substance Z. Antibody capture enzyme
immunoassay may be used to determine the anti-Substance Z
antibody titer. In one embodiment of this assay,
polyvinylchloride wells are coated with 50~1 of synthetic
substance Z at a concentration of 2mg/ml. The wells are
~then blocked with 8% BSA/PBS. Serum obtained from the
immunized mice:is serially diluted and 50~1 of samples of
each dilution are added to the wells. Unbound antibodies
are removed by washing and the presence of mouse anti-
Substance Z antiboclies is then detected using horseradish
~ 21 9067q
-- 10 --
peroxidase-labelled rabbit anti-mouse lmmunoglobulin
antibody. The antibody titer is determined by the highest
dilution of the serum which shows the presence of anti-
Substance Z antibo1ies.
A mouse with a high anti-Substance Z titer is selected
for hybridoma production. After a final booster, spleen
cells are obtained from the mouse and fused with myeloma
cells such as sp2/0. Following fusion, the resultant
hybridoma cells are diluted and plated in multi-well
culture dishes. Supernatants of the cultures are screened
for the presence of anti-Substance Z antibodies. Cells
from positive wells are single-cell cloned. MAbs produced
by these cloned hybridoma lines may be harvested from
tissue culture supernatants or ascitic fluid.
Production of monoclonal antibodies as starting
materials for obtaining humanised antibodies is well known
(9). There are se~eral examples of humanisation of MAbs in
the literature (10). Typically, a humanised antibody
contains a binding portion obtained from non-human cells ~.
20 . (e.g., mouse cells) and one or more human portions,
particularly framework portions of antibody obtained from
human sources Particular examples are offered in the
patent literature; United States Patent No. 5,558,864,
issued September 24, 1996 to Bendig et al. described
humanised and chimeric anti-epidermal growth factor
receptor monoclonal antibodies; and United States Patent
~o. 5,482,85~, issued January 9, 1996 to Fell, Jr. et al.
described production of chimeric antibodies by homologous
recombination. The specifications of both of these patent
references and references mentioned therein are
incorporated herein by reference.
Once a humanised antibody is obtaired, it should be
tested in one or more anlmal models (ll-lb). Testing for
toxic effects should also be conducted. For example, a
single dosage (between 0.1 and 1 mg per kilogram of body
weight) of antibody is administered intraperitoneally to
mice and~or guinea pigs. The animals are observed for a
week or so for adverse effects such as weight change and
other obvious signs of toxicity. Immunohistological
~ 21 qO679
studies involving human tissues can be carri~ed out. For
example, the reactivity of an antibody is evaluated using
immunoperoxida5e staining on a varièty of normal human
tissues. High dosage pharmacology/toxicology studies in
adult chimpanze~es can be carri~d out. Analysis of blood
chemistry, hematology, and urinalysis:is conducted.
Further, an assay for immunocompetence is conducted.
Animals are challenged with different strengths of
dinitrochlorobenzene in acetone and the extent of response
to DNCB is evaLuated.
A dosage r-egimen used for treating a patient will be
determined by the attending physician considering various
factors which affect drug action, e.g., the condition, body
weight, sex and diet of the patient, the severity of the
disease, time of administration and other clinical factors.
For example, for treatment of rheumatoid arthritis, a
r~ n~d dosage is likely to be in the range of. 10 to
100 mg, over a period of a week or 50 (17). Such
recommendatian must be based on an objective study, and
particularly a stu~y which measures the level of agent in
patient serum over time. Development of recommended
dosages would likely be preceded by analysis of plasma
levels of MAb in chimpanzees.
The antibadies of the invention may be labelled or
conjugated for diagnostic and/or therapeutic uses. ~For
example, they may be coupled to radionuclides, fluorescent
compounds or enzymes for imaging or therapy or may be
incorporated into liposomes for targeting to a specific
tissue site.
The antibodies of the invention have utility as
laboratory reagents, for example, for Western blotting to
identify cells:or tissues~cxpressing~the PPT-C gene or
immunocytochemistry or immunofluoresence techniques to
identify the subcellular location of the precursor protein.
~ Additionally, the antibodies of the invention may be
used as therapeutic agents to selectively bind and inhibit
the activity of suastance Z for treatment of disorders
associated with excess or inappropriate production of
substance Z. For example, cancer cells such as leukemia
.
21 ~067~
- 12 -
cells, whose growth is stimulated by substance Z, may be
inhibited or suppres3ed by administration of antibodies
against substan:ce Z.
~imilarly, any cell type which is dependent for growth
or activation on stimulation by substance Z may be
controlled by administration of anti-substance Z
antibodies.
4. Trnnsgenic Animnl Models
The present invention also provides for the production
of transgenic~non-human animal models for the study of the
effects of over expression of the PPT-C gene and over-
production of substance Z, for the screening of candidate
compounds as potential antagonists of substance Z and for
the evaluation of potential therapeutic interventions.
The transgenic ani~als of the invention also provide
models of disease conditions associated with abnormalities
of substance Z production. For example, the transgenic
animals of the invention may provide an animal model of at
least some aspects of rheumatoid arthritis.
Animal species suitable for use in the animal models
of the inventlon include mice, ra-ts, rabbits, dogs, cats,
goats, sheep, pigs and non-human primates.
Animal models may be produced by inserting a selected
nucleic acid sequence into a germ line cell~or a stem cell
using previously described techniques such as oocyte
microinjection o-r transfection or microinjection lnto
embryonic stem cells. Alternatively, an endogenous PPT-C
gene may be inactivated or replaced by homologous
recombination within embryonic stem cells to produce
"knock-out" or "knock-in" animal models. Techniques for
obtaining transgenic an-imals are widely available in the
literature. For example, laboratory techniques for=the
production of transgenic mice is described in Hogan et al.
(18).
In accordance with one embodiment of the invention,
transgenic- animals generated by the introduction of a PPT-C
transgene into a fertili~ed animal oocyte, with subsequent
growth of the embryo to birth as a live animal. The PPT-C
transgene is a transcription~unit which directs the
~ 21 qo67q
expression of PPT-C gene in eukaryotic cells. To create the
transgene, PPT-C gene is ligated with an eukaryotic
expression module. The basic eukaryotic expression module
contains a promoter element to mediate transcription of
PPT-C sequences and signals required for efficlent for
termination and polyadenylation of the transcript.
Additional elements of the module may include enhancers
which stimulate transcription of PPT-C sequences. The most
frequently utilized termination and polyadenylation signals
are those ~erived from SV40 (5). The choice of promoter and
enhancer elements to be incorporated into the PPT-C
transgene is determined by the cell types in which PPT-C
gene is to be expressed. To achieve expression in a broad
range of cells, promoter and enhancer elements derived from
viruses may be utilized, such as the herpes simplex virus
thymidine promoter and polyoma enhancer (19). To achieve
exclusive expression in a particular cell type, such as B
cells, specific~promoter and enhancer:elements could be
used, such as the promoter of the mb-1 gene and the
intronic enhanc~er of the immunoglobuiin heavy chain gene
(20).
The PPT-C transgene is inserted into a plasmid vector,
such as pBR322 for amplification. The entire PPT-C
transgene is then released from the plasmid by enzyme
digestion, purlfied and injected into an oocyte. The
oocyte is subsequently implanted into a psèudopregnant
female animal. Southern blot analysis or other approaches
are used to determined the genotype of the ~ounder animals
and animals generated in the subsequent backcross and
intercross. ~
The PPT-C nucleic acid sequences of the invention may
also be utilized in the creation of transgenic mice
deficient in the production of Substance Z by homologous
recombination.
Methods of disrupting the genes of an animal are well
established and are described in publications such as
Kitamura et al. (21). As the first step, the genomic
sequence which gives rise to the PPT-C mRNA is pulled out
by screening a mouse genomic library wi~h the PPT-C cDNA
21 ~0679
- 14 -
molecule as a probe. A fragment containing the coding
sequence for Substance Z peptide and some flanking
sequences is cloned into a plasmid vector such as pBR322. A
lkb pMClneo fragment contalning the neomycin resistant gene
is then insert~ed into the sequences encoding Substance Z
peptide. This resultant construct is linearized, and
introduced into D3 embryonic stem (ES) cells by
electroporation. Neomycin-resistant colonies are selected
and expanded. Homologous recombination events are
identified by PCR and Southern blotting. ES cell clones
carrying the disrupted PPT-C genes are in~ected into
blastocysts of C57BL/6 mice, and the resulting male
chimeras are m~ted to C57BLj6 females. Agouti offspring are
analyzed by Southern blotting for the presence of the
mutant PPT-C gene. Heterozygous mice are intercrossed, and
homozygous PPT-C-mutant mice are identified by Southern
blotting.
These mice will provide a model for study of the
effects of substance Z deficiency and the inter-
relationship between substance Z and other factors inmaintenance Q~ health, including the maintenance of a
normal immune response. These animals will also provide
tools for scree-ning candidate compounds for their
interaction with substance Z or the signalling pathway
activated by substance Z.
5 . .5~- e~n; nq for Druqs Which Affect Expression o~ the
PPT-C Gene
The present invention also enables the analysis of
factors affectIng the expression of the PPT-C gene in
humans or in animal models. The invention further provides
a system for screening caffdidate compounds for their
ability to turn on or turn off expression of the PPT-C
gene.
~ For example, pre-B cells may be isolated from a mammal
and grown in culture in the presence of IL-7 (43-45). Such
a cell culture system can be used to-Ldentify compounds
which activate productiDn of substance Z or, once substance
z production has been activated in the cells, they can be
.. . _ _ ~ .. . . .
- 15 - 2 1 9 0 67q
used to rdentify compounds which lead to suppression or
switching off o~ substance~Z production.
Compounds thus identified are useful as therapeutics
in conditions where substance Z production is deficient or
is excessive. 5~
6. Identirication o~ Antagoni3ts
The present invention enables also a screening method
ior compounds of therapeutlc utility as antagonists of the
biological activity of substance Z. Such antagonist
compounds are useful, for example, to reduce or prevent
tissue damage resulting from activatlon of synovial ~.
fibroblasts by substance Z, for example in conditions such
as rheumatoid arthritis, and to reduce or prevent symptoms
or tissue damage resulting from mast cell activation by
substance Z, for example in conditions such as acute
allergy.
Those skiLled in the art will be able to devise a
number of possible scIeening methods for screening
candidate compounds for substance Z antagonism.
~ For example, candidate compounds may be screened for
biological activity and for antagonist activity in the
cartilage degradation assay described herein.
A screening method may also be based on binding to the
substance 2 receptor. Such competitive binding assays are
well known to those skilled in the art. Once binding has
been established for a particular compound, a biological
activity assay is employed to determine agonist or
antagonist potential.
7. Diagnostic Methods
. The present invention enabies the identification of
disorders associated with overproduction or underproduction
of substance Z by assay of substance Z in appropriate
tissue samples.
Substance Z may be assayed by a variety of methods,
immunoassay being ~referred. Many types of immunoassay are
described in the literature. For example, radioimmunoassay
may be employed (22).
In accordance with one embodiment, substance Z is
labelled with 12sI by chloramine T, as described in Example
~ 21 ~067q
-~16 -
1, for use in radLoimmunoassay.
A 100~1 al'iquot of serially diluted substance Z
standard or sample is mixed with 100~1 of l2sI-labelled
substance Z and 100~1 of a dilution of anti-substance Z MAb
that gives approximately 50% binding in the absence of
unlabelled peptide. One ml. o~ a mixture of 6mg/ml Norit A
~Amend Drug and Chemical, Irvington, NJ) and 0.75mg/ml
Dextran 70 in 0.25~ BSA-Dulbecco's ~BS buffer is added_
The tubes are vortexed and centrifuged. A lml aliquot is
counted in a gamma counter. Standard concentration is
plotted against (cpm bound in the pres-ence of
standard)/(cpm bound in the absence of standard).
~oncent'ra~ion of substance Z in tissue samples is
determined by reference to the standard curve. A normal
range of subs~tance Z levels is obtained by assay of a
numoer of normal subject tissue samples, as is understood
by those skilled in the art. ~ -
8. ~h~r~r~l-tic Methods
The present invention further enables therapeutic
intervention in disorders ass-ociated with an inappropriate
level or location of substance Z.
Such interYentiOnS include .=
~ a) in conditions associated with undesired
biological activity of substance Z, inhibition of its
activity by administration of antagonist compounds or anti-
substance Z antibodies.
For example, tumour cells whose growth is responsive
to substance Z may be inhibited or suppressed by
administration of substance Z antagonists.
In condltions such as rheumatoid arthritis or acute
allergy, where cells are activated to produce substance Z
leading to tissue damage, therapeutic intervention may be
achieved by administration of substance Z antagonists to
reduce substance Z activity.
Alternatively, substance Z-producing cells may be
targeted and destroyed to reduce undesired substance Z
production.
~b) conditions associated with a deficiency of
substance Z may be treated by administration of
- 17 - 2~qO67q
pharmaceutical compositions lncluding substance Z.
Substance:2 or therapeutically effective analogues or
fragments thereof may be be administered therapeutically by
injection or by oral, nasal, buccal, rectal, vaginal,
transdermal or ocular routes in a variety of formulations,
as is known to those in the art.
For oral administration of peptides, various
techniques can be used to improve stability, based for
example on chemical modification, formulation and use of
protease inhibitors. Stability can be improved if
synthetic amino acids are used, such as peptoids or
betidamino acids, or if metabolically stable analogues are
prepared.
Formulation may be, for example, in water/oil emulsion
or in liposomes for improved stability. Oral
administration of peptides may be accompanied by protease
inhibitors such as aprotinin, soybean trypsin inhibitor or
FK-448, to provide protection for the peptide. Suitable
methods for preparation of oral formulations of peptide
drugs have been described, for example, by Saffran et al.,
(23~ (use~of trasyloI protease inhibitor); Lundin et al.
(24) and Vilhardt et al., (25).
Due to the high surface area and extensive vascular
network, the nasal cavity provides a good site for
absorption of both lipophilic and hydrophilic drugs,
especially when coadministered with absorption enhancers.
The nasal absorption of peptide-based drugs can be improved
by using aminoboronic acid derivatiYes, amastatin, and
other enzyme inhibitors as absorption enhancers and by
using surfactan~s such as sodium glycolate, as described in
Amidon et al., (26).
~ he transdermal route provides good control of
delivery and maintenance of the therapeutic level of drug
over a prolonged period of time. A means of increasing
skin permeability is desirable, to provi-de for systemic
access of peptides. For example, iontophoresis can be used
as an active driving force for. charged peptides or chemical
enhancers such as the nonionic surfactant n-decylmethyl
sulfoxide (NDMS) can be used. --
~ 2l ~067~
- 18 -
Transdermal delivery of peptides is described in
Amidon et al. (26) and Choi et aI. (2i).
Peptides may also be conjugated with water soluble
polymers such as polyethylene glycol, dextran or albumin or
incorporated into drug delivery systems such as polymeric
matrices to increase plasma half-liie. ~
More generally, formulations suitable fer particular .
modes of administration of peptides are descrlbed, for
example, in Remington's Pharmaceutical Sciences (28).
As an alte~native therapy in substance Z deficiency,
gene theraFy may be carried out, comprising administration
of a PPT-C gene to a substance Z deficient subject.
Appropriate techniques may be employed to target the
introduced gene to a desired target tissue. Gene therapy
has the potential to avoid life lo~g administration of
exogenous peptides and may provide for a more
physiologically-appropriate level of substance Z than
exogenous ~administration.
EXAMPLES
The examples are described for the purposes of
illustration and are not intended to~limit the scope of the
invention.
Methods of molecular genetics, protein and peptide
biochemistry and immunology referred~to but not explicitly
described in this disclosure and exampies are reported in
the scientific literature and are well known to those
skilled in the art.
Example 1: Isolation Qf PPT~-C cDNA ,_
RNA prep~ration and Northern analysis
Total RNA was isolated from CDl mouse tissues and
cultured cell lines as described by Bergman et al. (29) and
Sambrook et al.(5). Poly (A) RNA was selected by passage
over oligo(dT)-cellulose (Pharmacia). For Northern
anaIysis, 5~g o~ poly(A)+RNA was separated on 1% agarose
gels containing 20mM NaHPO4, and lM formaldehyde,
transferred onto Hybond-N nyion membranes (A~ersham), UV-
immobilized, and hybridized with 32P-labelled probes
prepared by a random hexamer-primed method (30).
2 1 90(~7~
- 19 -
Hybridization was at 42 C in 5X SSPE (750mM NaCl, 5mM EDTA,
50mM NaH2PO4, pH 7.4), 2% SDS, 5X Denhart's solution, 100~g
of sheared/boiled salmon sperm DNA, 100~g of poly A, and
50% formamide. washIng was in 0.1X SSC~(i5mM NaCl, 1.5mM
sodium citrate, pH 7.0), 0.1% SDS at 65 .
~;ff~ntial displ~y PCR
Differential display PCR was performed following the
method described by Liang and Pardee t31) with GenHunter
Kit ~Brookline, MA). Poly (A)+RNA (0.2~g) from IIB4 and
70Z/3 cells (both transformed murine pre-B cell lines) was
used for first strand cDNA synthesis with each of the four
modified oligo(dT) primers (T12MN). The synthesized first
strand cDNA was used as a template in the subsequent PCR
- reaction. In a 0.2ml PCR tube the following were added:
2~1 of 10X PCR buffer (500mM KCl, i5mM MgCi2, 100mM Tris-HCl
at pH 8.3), 5'-arbitrary 10mer ¦2~M), T12 MN (10,uM, same as
used in cDNA synthesis), cDNA synthesisl, cDNA template,
1.6~1 of dNTP mix (25~M), 12.5~Ci 2sS-dA~P (100 Ci/mmole), 1
U of Taq DNA polymerase (Perkin Elmer), and 9-2~1 of dH2O.
PCR was performed as follows: 94 C, 30s; 40 C, 2min; 72 C,
30s for 40 ~ycles. Four microliters of the PCR products
from the two starting cells were run side by side on a 6%
urea:acrylamide sequencing gel. ~The dried gel was exposed
to an X-ray film and the autoradiogram was analyzed for
differentialiy displayed bands. These bands were cut out
from the gel, and the DNA was eluted by soaking the gel
slices in 100~1 of TE buffer for 10mIn and then boiling for
10min. The eluted DNA was precipitated using glycogen and
ethanol, air-dried, and redissolved in 10~1 of dH2O. This
DNA was reamplified with the same comoination of primers
used in the first PCR. The reamplified DNA was gel-
purifLed and used as a probe in Northern analysis. Once
the differential expression was confirmed, the DNA was
cloned using the TA Cloning Kit (Invitrogen, CA).
I cDNA library construction ~nd screening
A 70~/3 cDNA library was constructed using standard
procedures es~entially as described by Sambrook et al.,
(5). Reverse transcription was carried out on 5~g of
_ _ . _ _ _ _ _ _ . .. .. . . .. .
~ 21 qO67q
- 20 -
poly(A) RNA to qenerate first strand cDNA using an
oligo~dT)12-1~ primer. The mRNA-cDNA hybrid was treated
- with Rnase H. Remnants of mRNA served as primers for the
synthesis of second strand cDNA. The double strand cDNA
was cleaved with Klenow to create blunt-ends, and then
ligated to an EcoR I/Not I adapter. This adapter-tailed
cDNA was purifled to remove the unligated adapters, and
then inserted into lambda ZAPII vectors ~Stratagene, CA).
The constructs were packaged into infectious phage
partLcles, amplified in E. coli strain XL1-Blue. The ratio
of recombinants in the library was over 85~. The total
yield of the recombinants was -4x106. The size of cDNA
inserts from 12 randomly picked up clones ranged from 0.8-
4.5kb with an average of 1.4kb. Using the 440bp
differentIal display PCR fragment as a probe, 2X106 plaques
were screened. Up to 20 positive clones were isolated by
three rounds of screening. The in vivo excision procedure
was followed to release pBluescript plasmid from the lambda
ZAPII vector. The insert size of the ten clones varied
from 0.5-1 1 kb. Nucleotide sequence of each clone from
both strands was determined by the dideoxynucleotide chain
termination method ~6).
5' RACE of n~A
The largest cDNA clone which was isolated from the
above mentioned ~library still lacked the 5' end of the
gene. Therefore the 5' end of PPT-C cDNA was amplified
using the method of 5' RACE ~rapid amplification of
complementary DNA ends) using standard protocols included
with a reagent kit from Clontech iPalo Alto, CA). The PPT-
C gene specific primer sequence used was5'GGACAGAGAG~CA~ lC-3'. The amplified PCR product was
cloned using the TA Cloning kit ~Invitrogen, CA).
Nucleotide sequences were determined by dideoxynucleotide
chain reaction termination method.
~ The nucleotide sequence of the cDNA is shown in Table
1 and the amino acld sequencé deduced ~rom the open reading
frame is shown in Table 2.
Exnmple 2: Expression of PPT-C gene
_ _ _ _ , _ _ _ _ _ _ _ _
~1 2' 90679
- 21 -
RNA was extracted from cells and tissues, and poly
(A)~ RNA was isolated by passage over cligo (dT) -
cellulose. Northern blot analysis was performed as
described in Example l. 7G9 was a probe derived from PPT-C
cDNA. L32, a ribosomal protein-coding gene, was used as a
loading control. II4B, CB17 l.1 and 5.1, scid 2.1 and 4.1,
RAGl 14 and 17, RAG2 5 and 21 are fetal liver cell lines
transformed with Abelson Murine Leukemia virus. 70Z/3 is a
pre-B cell line. 70Z/3y is a RAG-positive variant of 70Z/3.
WEHI 231 is an immature B cell line. J558 is a myeloma cell
line. RBL5 and EL4 are T lymphoma cells. P33BD1 and CB5
are cell lines of myeloid and erythroid lineage
respectively. NIH3T3 and L929 are fibrobLasts. BMS2.2 is a 5
stromal cell line. Mouse tissues included in the scheme
were brain, lung, heart, liver, spleen, thymus and kidney.
The results are shown in Figure 1. Expresslon of PPT-C
mRNA was restricted to cells o~ B lineage at its early
developmental stage.
To test PPT-C expresslon in primary cells
representing pre-B cells, RT-PCR was used to amplify PPT-C
mRNA (5) from fresh mouse.fetal liver celis and from mouse
fetal liver cells expanded with IL-7 for~enrichment of B
lineage cells. The primers used were 5'-
TAACCACCAGCAACGAGA-3' and 5'-ATGGCTGAGGAAGCTACCT-3'. PCR
products were blotted on a nylon membrane, and probed with
7G9.
PPT-C was expressed in fetal liver cells and culturing
of the cells in the presence of IL-7 further increased PPT-
C expression ~data not shown).
Ex~mple 3: prepAration of Subst~nce Z
Peptides~RSRTRQFYGLM (substance Z) and SRTRQFYGLM
(substance Z - short form) were synthesised using
conventional solid phase peptide synthesis (33).
Analytical and preparative high-performance liquid
chromatography were utilized to characterize and isolate
the final compounds. Fast bombardment mass spectrometry
was used to confirm the molecular weight.
Synthetic substance Z was used for the studies of
~ 21 90679
biological activity described in the following examples.
Ex~le 4: Efl~ect of Substance Z on cartilage degradation
by human fibroblasts
Generation of Synovial Fibroblast Lines
The ability of 3 human fibroblast cell lines, synovial
fibroblast lines RA1 and RA2 and skin fibroblast cell line
CCD-967, to degrade cartilage discs~ was tested.
RA1 and RA2 were synovial fibroblast lines derived
from synovium obtained from Rheumatoid Arthritis patients
~according to ACR criteria ~34)) undergoing knee
replacement. They were established by placing finely
minced synovial tissue into a 25cm tissue culture flask
with medium, consisting of OPTI-MEM ~Gibco, Grand Island,
NY., USA) supplemented with 10% FCS ~Gibco, Grand Island,
NY, USA), antibiotic-antimycotic mix ~penicillin G sodium
[1000 units]/streptomycin sulfate [1000 unitsl/amphotericin
B [2.5 mg~ml]) ¦Gibco, Grand Island, NY, USA), 5.5 x 10 sM
b-mercaptoethanol ~Sigma, St. Louse, MO, USA) and 2.4 g/L
.sodaum carbonate ~Mallinckrodt Inc., Point-Claire, Quebec,
Canada) . Minced synovial tissue was left in the culture
flask for a period of 1 week to allow the fibroblasts to
grow out of the tissue and onto the s rface of the culture
flask, at which point the tissue was rerrLoved. Although
25: there was variation from line to line, in general, cells
were passaged every 2 weeks and media was replenished every
3 days. The distinct morphology of fibroblasts along with
their unique ability to survive multiple passages in the
absence of added growth factors in vitro was used to assign
30 a lineage to these cells.
Skin fibroblast cell line CCD-967 ~Skin 1) was
obtained from ATCC and cultured exactly as described for
the synovial fibroblast c~ll lines.
The macrophage cell line U937 was used to generate a
35 monditioned medium. To prepare U93i-conditioned medium,
U937 cells were ~grown to a concentration of approximately
lxl05/ml of OPTI-MEM. This medium was subsequently
centrifuged ~30a0rpm for 15rn n at 4 C) and filtered ~0.2mm
millex-G~I filter, I~ pore, Bedford, MA, USA) prior to use
~ 21 9n67q
in the assay. If the conditioned medium was not used
immediately, it was stored immediately at -70 C.
Cartilagc degr~dAtion ~ss~y
The cartilage degradation assay originally described
by Steinberg et al. (35), and modified by Janusz and Hare
(36) was~used with human cell lines and human normal
cartilage. In brief, measurement of degradation of
cartilage was performed by culturing of fibroblasts in the
presence of radiolabelled human cartilage discs. Cartilage
discs (qmm x lmm~ were prepared from normal human femoral
cartilage using a 4mm cork bore. Femoral cartilage was
obtained from normal appearing cartilage in patients with
osteoarthritis-undergoing ~oint arthroplasty. Discs were
incubated overnight with OPTI-MEM containing S33 Na~S04
(~OmCi/ml~ (Amersham, Oakville, ON, Canada~. Label was
incorporated into the glycosaminoglycan side chains of the
proteoglycan within the cartilage. The discs were then
washed (x5) with sterile PBS to remove unincorporated
r~diosotDpe. Discs were then freeze-thawed 5 times and
heated at 65 C for 15 min. to inactivate endogenous enzymes
and cytokine activity. The discs were store~ at -20 C prior
to use. Inco~poration of radionuclide was normally found
to be between 50,000 to 100,000 dpms/disc.
Adherent fibroblasts to be cocultured were trypsinized
from culture flasks with 0.05% trypsiniO.53 mM EDTA 4Na
(Gibco, Grand Island, NY, USA). 1 x 104 fibroblasts were
cultured together with a radioactive cartilage disc, either
in U937-conditionea medium or with l nM substance Z for=7
days in 96 well Nunclon~plates (Nunc, Roskilde, Denmark).
On day 3, t~e original medium was removed and replaced with
200mI fresh medium, supplemented as before. In some
experiments, cells were:cultured with U937-conditioned
medium or with substance Z in transwell tissue culture
inserts for 96 well tissue culture piates with a 0.2mm
anapore membrane (Nunc, Roskilde, Denmark) between disc and
fibroblasts.
On day 7, 200 ml of medium was removed and added to
3ml scintillation fluid (Beckman Instruments Inc.,
Fullerton, CA, USA) and counted in a scintillation counter
- 24 _ 2 1 q 0 6 7 9
(Beckman Instruments InC., Fullerton, CA, USA 151071). The
remaining isotope in the cartilage di5c was measured by
completely digesting the disc in 0.5ml of tissue
solubilizer (Beckman Instruments Inc., Fullerton, CA, USA).
Solubili~ed discs were counted in scintiilation counter
using 3ml scintillation fIuid (Beckman Instruments Inc.,
Fullerton,~CA, USA). Data were expressed~as % of S35
released into the supernatant using the equation: %= (dpm
in supernatant)/(dpm in supernatant + disc) X 100.
In all experiments, culture of the radio-labeled disc
alone was performed as a control. All experiments reported
were ~arried out in quadruplicate and the % release of S35
calculated for each individuai replicate.
The results are shown in Figure 3.
The results show that synovial fibroblast lines
degLaded cartilage if both contact with cartilage discs
(Contact) and U937-conditioned medium (U937) were present.
In the absence of either i-actor, degradation did not
occur. Substance Z, however, stimulated the degradation of
cartilage by synovial fibroblast3 when neither contact nor
conditioned medium was present.
Skin fibroblasts did not degrade cartilage when
provided with contact and U937-conditioned medium or when
challenged with Substance Z.
: ~ ~
Ex~m~le 5: Effect of S~L~ L~n~e Z on growth of 1 enk~mi A
C~
Growth of 70Z/~3 T.~~1k~mi J- Cell~
70Z/3 murine leukemia cells were maintained in liquid
culture using the supplemented OPTI-~EM medlum described in
Example 4, except that 5% FCS was used instead of 10%. To
examine the effects of Substance Z on the growth of 70Z~3,
cells were cloned in medium containing 0.3% melted agar
(Bacto Agar, Gibco) following standard procedures described
by Sauter and Paige (37). Briefly this consisted of
pouring lml of medium contalning 0.3% melted agar into a
35mm tissue culture:plate. This layer was allowed to gel
for 2~min at room temperature after which a second lmi
layer, containing 70Z/3 cells (300 - I0~D/plate) in medium
.... . . . .. . _ . . . . _ _ . .... . . .
2 1 qO67q
- 25 - - ~
supplemented with 0.3% agar was poured. After 7 days of
culture, the colonies tFoci containlng at least 50 cells)
were enumerated visually with the aid of a
stereomicroscope. ~ ~
The results are 5hown in Figure .4 and show that
leukemia -cell growth was augmented by Substance Z.
XxAmple 6: ~nhAn~ Ma~t Cell D~ An~lAtion by Sub~t~nce Z
Primary mast cells were obtained by culturing bone
~marrow cells in IL-3-containing WEHI-3 conditioned medium
as described by Berger (38~. By 6 weeks, >99% of the cells
in the culture were mast cells. Experiments were carried
out generally as described by Berger (38), except that
degranulation wàs measured by checking the release of 3H-
serotonin lnstead of that of b-hrxos~m~n;dase.
Mast cells were cultured overnight in Opti-MEM (Gibco)
+ 5~FCS + b-mercaptoethanol + WEHI-3 conditioned medium
plus 3.5mCi/ml of 3H-serotonin. 3H-serotonin would be
preferentially incorporated into granules of mast cells
during the cuiture. The cells were then washed to remove
excess serotonin, and cultured in fresh medium for 15 min.
Subsequently, cells were washed and resuspended in Tyrode's
buffer (lOmM Hepes" pH7.4, 130mM NaCl, 5mM KCl, 1.4mM CaCl2,
lmM MgCl2, 5.6mM glucose, 0.1% BSA). Aliquots of 2~105 cells
were incubated with 5mg/ml SPE-7 anti-DNP monoclonal IgE
antibodies ( Sigma Chemicals Co.) for 30-~0 min, washed and
treated with DNP-HSA antigen (Sigma Chemicals Co.) at a
concentration of 0, 1 and 5ng/ml for=30-6Q min at 37 C.
Substance Z was included in the finai incubation at a
:concentratior Eanging from Q-50mM. Cells were pelleted at
3000rpm for 5 min. An aliquot of supernatant was removed
and placed in scintillation vials with appropriate
scintillation fluid. ~emaining supernatant was discarded,
cell pellet was lysed ln an equlvalent volume of Tyrode's +
0.5% Triton X-100 and 1/5th of the iysate was transferred
to a fresh scintillation vial with scintillation fluid
Samples were counted in scintillation counter.
~ Degranulation = counts in super~atant x 100
,_ . . = i, . , _ ~ .
~ 21 9067q
- 26 -
counts in (Supernatant + pellet)
The results are shown in Figure 2.
Mast cells express high affinity receptors for IgE
antibodies ~rass-linking of surface-baund IgE by antigen,
typicaLly at high concentrations, leads to the activation
of these cells and the release of granules containing
inflammatory mediators. This process of degranulation is
also regulated by other signals~. Substance P, for example,
has been shown to increase the sensitivity of mast cells to
degranulation stimulated by exposure to antigen (39).
As demonstrated here, substance Z also enhanced IgE-
mediated degranulation of mast cells
Example 7: Isolation of ~_C~yLOI ~or S~L~...ce Z
For identification and isolation of the receptor for
substance Z, substance Z is labelled with an easily
detectable marker or label. For example, substance Z is
labelled with a radio-label such as I by a conventional
method such as those described by Harlow and Lane (8). One
such method employs~chloramine T. 10mg of synthetic
Substance Z in 25ml of 0.5M sodium phosphate (pH 7.5) ls
mixed with 500mci of Na12sI and 25ml of 2mg/ml chloramine T.
After a incubation of Z0sec, 50ml of stop solution
(2.4mg/ml sodium metabisulfite, 10mg/ml tyrosine, 10
glyceroL, 0.1% xylene cylanol in PBS) ~s added. The
iodinated Substance Z is subsequently separated from the
iodotyrosine on gel filtration column.
Labeled Substance Z is utilized to determine the
distribution of its receptor in various cells and tissues,
and to elucida~te the binding affinity and kinetics of
Substance Z with its receptor (1).
Various approaches may be employed for the isolation
of the receptor gere for Substance Z. Expression cloning is
one of the most frequently applied strategies in receptor
cloning (41, 42). For example, RNA is extracted from the
cells or tissues which express Substance Z receptor as
determined by using l2sI -labeled Substance Z. Poly(A)+ RNA
is isolated using cligo-dT cellulose, cDNA is synthesized
and ligated into the mammalian expression vector pMET7
~ ~1 9067q
- 27 ~
(42). Ligated DNA is EtOH precipitated and resuspended in
dH~O at 25ng/ml. DNA (lml) is used to transform each of 40ml
of competent DHlOB E. coli cells by electroporation. Based
on the titers of the cDNA transformations, 96-well plates .
are inoculated~with 150cfu per well in lml of LB-Amp.
Cultures are~grawn for 15-16hr at 37 C. 100ml of each
culture is:removed and added to 100ml of 50% Glycerol,
mixed, and stored at -80 C. Plasmid DNA is prepared from the
rest of the culture.:The library is thus produced as pools
of 150 clones.
To screen the library, plasmid DNA is transiently
transfected into a 10cm dish of COS cells with
lipofectamine. DNA from eight pools is used for each
transfection. After 48hr, the cells, just at or before
conf~uence, are transferred onto culture slides, and then
incubated with 0.SnM l2sI -labeled Substance Z. After two
washes , the slides are exposed to film. Positive pools
are subsequently broken down to subpools of 15Q clones
each. The positive subpools are further divided until a
single clone encoding a Substance Z-binding activity is
identified.
The nucleotide sequences of the isolated clones are
determined, and deduced amino acld sequences are obtained.
In order to confirm that the isolated clones encode
receptor specific for Substance= Z, COS cells are
transfected with each of the individual clones. Substance Z
binding characteListics conferred by these clones should be
comparable with that observed in receptor-bearing cells.
In addition, the value of the dissoc~ation constant (KD) as
determined by Scatchard analysis should be similar, and the
binding of l~sId-labeled Substance Z to the transfected cell
should be ,able to be completely blocked by cold Substance
Z, but not by other tachykinins.
The present invention is not limited to the features
of the embodiments described herein, but includes all
variations and modifications within the scope of the
claims.
~ 2l 90679
- 28 -
Re~erences
All of the listed references ~e inco~porated herein
by reference.
1. Regoli, D. Bodon, A. And Fauchere J-L. (1994)
Pharmacological Reviews v. 46, pp. 551-599.
2. Nakanishi, S., (1987), Physiological Reviews v. 117,
p. 1142.
3. HarLis Arch, (1989), Biochem. Biophys. v. 275, pp.
315-333.~
4. Steiner et al. (1992), J. BioI. Chem. v. 267, pp.
23435-23438.
5. Sambronk, J., Fritsch, E.F., and Maniatis, T. (198)
Molecular Cloning, A Laboratory Manual, Cold Spring
Harbor Laboratory, Cold Spring Harbor, NY.
6. Antibody Enqineering: A Practical ~Gyide, (1992~, _
Borrebaek, Ed., W.H. Freeman & Company, N.Y.
7. Antibody Engineering, (1995), 2nd Ed., Borrebaek, Ed.,
Oxfo~d University Press, Oxford.
8. Harlow and Lane, Antibodies, a Laboratory Manual, (1988)
Col~ Spring Harbor Laboratory, New York.
9. Tami J.A., Parr, M.D., Brown, J.S., (1986), Am. J. Hosp.
Pharm., v. 43, p. 2816-25.
10. Gorman, S.D., Clark, M.R. (1990), Semin Immunol., v. 2,
pp. 457-466.
11. Halloran, M.M., Szekanecz, Z., Barquin, N., Haines,
G.K., Koch, A. (1996), Arthritis Rheum., v. 39, pp. 81-
19 .
12. Joosten, L.A.B., Nelsen, M.M.A., van de Loo, F.A.S.,
van den Berg, W.B., (1996), Arthrltis Rheum., v. 39, pp.
3Q 797-8~:g;
13. Jasin, H.E., Lightfoor, E., David. L.S., Rothlien, R.,
Foanes, R.B., Lipsky, P., (1992),~~Arthritis Rheum, v. 35,
pp. 541-549.
14. Geiler, T., Kriegsmann, J., Keyszer, G.M., Gay, R.,
Gay, S., (1994), v. 37, pp. 1664-1671.
15. Yoshina, S., Quattrochhi, E., Weiner, H.L., (1995),
Arthritis Rheum., v. 38, pp. 1091-1~96.
16. Caccesse, R.G., Zimmerman, J.L., Carlson, R.P., ~I992),
Mediators ~lamm., v. 1, pp. 273-279.
~ 21 9067q
- 29 -
17. Dëla~uente, ~.C., Resman-Targo~, B.H., ~1999), Annals
of Pharmacotherapy, v. 28, pp. 650-654.
18. Hogan et al. (1986) Manipulating the Mouse Embryoc
Coldspring Harbour Laboratory Press, Coldspring Harbour,
~ N.Y.
l9. Thomas and Capecchi, Cell, (1987), v. 51, pp. 503-512.
20. Sun Cell, ~19g4), v. 79, pp. 853-900.''
21. Kitamura et al. Cell (1992), v. 69,'pp. 823-831.
22. ~einstock et al., J. Immunol. ~1588J, v. 141, pp. 961-
23. Saffran et'''al., (1979) (use of'trasylol proteaseinhibitor~;
24. 1undin et al. (1986);
25. Vilhardt et al., (17) (1986), Gen. Pharmacol., v. 17,
~ pp. 481-483.: -
26. Amidon et al., tl994), Ann. Rev. Pharmacol.~Toxicol.,
v. 34, pp. 321-341.
27. Choi et al.', (l990),.Pharm. ~es., v. 7, pp. 1099-1106.
28. Remington's Pharmaceutical Sciences.
29. Bergman; Y., Stewart, S.J., Levy, S., and Levy, R.
(1983) J. Immunol. v. 131, pp. 1876-1881.
30. Feinberg, A.P. and Vogelstein, B. (1983), Anal,
Biochem. v. 192, pp. 6-13.
31. 1iang, P. and Pardee, A.B. 1992) Science v. 257, pp.
967:-970.~
32. Sanger, F., Nicklen, S., and Coulson, A.R. (1977) Proc. =
Natl. Acad. Sci USA v. 74, pp. 5463-5467.
33. Barany and Merrifield, In the Peptides~ Analysis,
Synthesis,=Biolo.7y, ed. by Gross and Meienhofer (1979),
v. 2, pp. 1-~34, Acadëmic Press, New York.
34. Arnett F.C., Edworth, S.M., Bloch, D.A., (1988),
Arthritis ~heum. v. 31, pp. 315-324_
35~ Steinberg, J., Tsukamoto, S., Sledge, C.B. (I979) Arth.
Rheum.
35 36. Janusz, M.J., Hare, M. (1993) J. Immunoi., v. 150(5),
pp. 1922-1931. _ __
37. Sauter, H. and Paige, C.J. (i98~c), Proc. Natl. Acad.
Sci USA, v. 84, pp. 4989-4993.
~ 21 90679
- 30 -
38. Berger et al. J. Exp. Med. (1994), v. 180, pp. 471-476.
39. Johnson A.R., Erdcs~ E.G., (1973), Proc. Soc. Exp.
Biol. Med., v. 142, pp. 1252-1256.
40. Kluxen et al. Proc. Natl. Acad. Sci USA, (1992), v.
89, pp. 4618-4622.
41. Rose et aL_ (1995), ~. Biol. Chem. v. 270, pp. 22661-
22664. ~~ ~ :
42. Takebe et al. Mol. Cell Biol. (1988~, v. 8, pp. 466-
472.
43. Kee, B., Cumano, A., Iscove, N., and Paige, C.J.,
-(1994), Internat. Immunog, v. 6, pp. 4~f-407.
44. Ray, R.J., Eurlonger, C., Williams, D.E., Paige, C.J.,
(1996), Eur; J. Immunol. , v, 26, pp. 1~-16.
45. Marshall, A.J., Wu, G.E., Paige, C.J., (1996), J.
Immunol., v. 156, pp. 2077-284.
. .
~ 21 90679
~ 31 -
TABLE 1 ~
1 ggagaggctg tcccatgaag cagtgcaaag gtggggtgag gcataggaag gaagcagaga
61 gtctaggaca catctcagcc ac4tacaagt ggccccgtgg ggaccggagc ctgctggagg
121 aggtggcatc aggctgagtg gggctccctg gacagagaag cacctgctca ccatqttgcc
181 tctccttgcc ctgcttct-c tgatcgggcc atcagtgtgc,actacagcag gagacaqaga
241 ggaactggct tt~ggtgcag aggcagagtc ctgggtgacc gtg~acctga agggaatccc
301 cgtccccagc attgaactta agcttcagga gttgaagaga agcaggactc gccagttcta
361 tggtctgatg gggaagcggg tgggagggta tcagctggga cgt.atagtgc aggatctcct
421 tggcacgaga ggtttgtcca tagaagggac ctgcagacag gcggcgagtc aacagagggc
481 acgacccgga gcagtgacca gagaaagcct ccagagtcga gaggaagatg aggctcccct
541 aaccaccagc aacgtgtagc actctgccac ccatctctcc ccagaacatc acagctgagg
601 agcctcagcc aa~aqtcctg tcttttgtcc tcaacttggg tatttcctgc caggccctcg
661 tcacactctg cat~tttctcc caqgactcac tcttggcatc tggtagcaac gacacacaaa
721 gcacggctgc ctcctiacaa gctggactga cccagctctc ccttgtccct acagcaaagc
781 ttcacactct gtatcccagg cctagcacac agtagggctc aatcaacgac gagttagca~
841 taataggtag cttcctcagc catgcagggt agagggtggg atgtcagaaa agc:agagacc
901 aacattacac agcccaggtc taatgtctaa tccttgggtg ggaagagagc ttggggcctg
961 gctaaaacgg caatagaaac aagaaaaaga ccctggtgga~gaagacgctg catgtattgt
1021 atttgggggc ggggtcagga gccttcttcc ctttagattc ctgatgttgc taccaacaga
1081 catctccctc tgtgctgagg ctatggctca gtgggctaag gcgattgtgg ccaagcctga
1141 caacttgagt tcaatcccca ggacccacaa agtggaagaa aaaaattgtc cttttacctc
1201 tgcatgtgtc atggcatat.g ttacacaaaa taaaaagaca gtttggaaa
~ 21 ~067~
-- 32 --
TABLL 2
ULPLLALLLL :IGPS~CTTAG DREELAFGAE AESWVTVNLK GIPVPSIELK
10 20 30 40 50
I,QF.T,~K~ ~h~G _ V GGYQLGRIVQ DLLGTRGLSI: EGTCRQAASQ
60 :~0 80 90 100
QRARPGAVTR F..qT.Q.5RFF.nF APLTTSNV
11~ 120 128
Substance Z peptide is shown in ~old.
~ 21 9067~
~ABLL 3
j An tachykininS
NAME
Substance PR P K P Q Q F F G L M-NH~
Neurokinin AH K T D S F V G L M-NH2
Neurokinin 8D M H D F F V G L M-NH2
Substance ZR S R T R Q F Y G L M-NHz
Non~1iAn tachykinins
NAME
Physalaemin E A D P N K F Y G L M-NH2
Eledoisin E P s K A F I G L M-NH2
Kassinin D V P K s D G F V G L M-NH2