Note: Descriptions are shown in the official language in which they were submitted.
~01221
WO 9S/33719 PCT/GB9S
2 ~ ~o9~
SUeSTITUTED PYRROLIDONE, THI~7nLrn~ OR OXAZ0LIDONES AS HERBICIDES
This invention relates to chemical compounds useful as herbicides, to
processes for preparing them, and to herbicidal compositions and processes
utilising them.
Various compounds based upon substituted nitrogen containing
heterocycles are known, for example from DE-A-2212558. AU-A-8656417 also
discloses compounds based upon nitrogen heterocycles and these are said to
be useful as plant growth regulators. There is no mention of herbicidal
activity.
The applicants have found a group of compounds which have a particular
substitution pattern and which are active as herbicides.
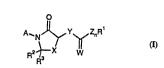
In a first aspect of the present invention there is provided a compound
of general formula I:
wherei n
X is O, S or CR4R5;
Z is 0, 5 or NR4;
n is 0 or 1;
Y is O S NR6 or CR4R5;
each R4 and R5 is, i~)dt~ ..Lly, hydrogen or Cl-C4 alkyl;
R6 is H, OH, CHO, NR16R17 or C1-C1o hydrocarbyl, O-(C1-C10 hydrocarbyl),
either of which may be substituted with one or more substituents chosen
from OR16, COR16, COOR16, OCOR16, CN, halogen, S(O)pR16, NR16R17, NO,
NR16CoR17, NR16CoNR17R18, CoNR16R17 or heterocyclyl; 2
R16, R17 and R18 are each, in~ o,.Lly, hydroyen, Cl-C6 hydrocarbyl or
C1-C6 halohydrocarbyl;
p is 0, 1 or2;
alternatively:
when Y is NR6 or CR4R5, and:
a~ Z is NR4; or
b) n is O;
the substituents of Y and Z or r and R1 may together form a bridge represented
by the formula _Ql_Q2 or _Ql Q2 Q3, where Q1, Q2 and Q3 each in~r ' Lly
represent CR12R13, =CR12, CO, NR14, =N, O or S;
WO 95/33719 2 1 9 0 9 7 9 PCT/GB9S/012~
-- 2 --
each of R12 and R13 independently represents hydrogen, C1-Cq alkyl, OH or
hal ogen;
R14 represents hydrogen or C1-C4 alkyl;
W is O or S;
Rl jS hydrogen or C1-C10 hydrocarbyl or heterocyclyl having 3 to 8 ring
atoms, either of which may optionally be substituted with one or more
substituents chosen from halogen (i.e. chlorine, bromine, fluorine or
iodine) , hydroxy, SO2NRaRb (where Ra and Rb are independently H or C1 6
alkyl), SiRC3 (where each Rc jS independently C1-C4 alkyl or phenyl),
cyano, nitro, amino, mono- and dialkylamino in which the alkyl groups have
from 1 to 6 or more carbon atoms, acylamino, C1 6 alkoxy, C1 6 haloalkoxy,
C1 6 alkylthio, C1 6 alkylsulphinyl, C1 6 alkylsulphonyl, carboxy,
carboxyamide, in which the groups attached to the N atom may be hydrogen or
optionally substituted lower hydrocarbyl; alkoxy carbonyl wherein the
alkoxy group may have from 1 to 6 or more carbon atoms, or aryl such as
phenyl;
R2 and R3 are each independently hydrogen or C1-C4 alkyl;
A is an aromatic or heteroaromatic ring system optionally substituted with one
or more substituents selected from: halogen or C1-C10 hydrocarbyl, -O(C1-C10
hydrocarbyl), -S(C1-C10 hydrocarbyl), -SO(C1-C10 hydrocarbyl) or -SO2(C1-C10
hydrocarbyl), cyano, nitro, SCN, SiRC3 twhere each Rc jS independently Cl-C4
alkyl or phenyl), CoR7, CR7NoR8,NHOH, oNR7R8, SF5, CoOR7, SO2NR7R8, OR9 or
NR1OR11; and in which any ring nitrogen atom may be quaternised or oxidised;
alternatively, two or more substituents of the group A may combine to form a
fused ~- or 6; ~ saturated or partially saturated carbocyclic or
heterocyclic ring in which any carbon or quaternised nitrogen atom may be
substituted with any of the groups mentioned above for A or in which a ring
carbon atom may be part of a carbonyl group or a nitrogen atom may be
oxi di sed;
R7 and R8 are each independently hydrogen or C1-C10 hydrocarbyl;
R9 is hydrogen, C1-C10 hydrocarbyl, 502(C1-C10 hydrocarbyl), CHO,
CO(C1-C10 hydrocarbyl), COO(C1-C10 hydrocarbyl) or CoNR7R8;
R10 and R11 are each independently hydrogen, C1-C10 hydrocarbyl,
O(C1-C10 hydrocarbyl), 5O2(C1-C10 hydrocarbyl), CHO, CO(C1-C10
hydrocarbyl), COO(C1-C10 hydrocarbyl) or CoNR7R8;
WO 9~133719 2 ~ 9 ~ 9 7 9 PCT/GB95/0122.1
-- 3 --
any of the hydrocarbyl groups within the group A may optionally be
substituted with ha10gen ~i.e. chlorine, bromine, fluorine or iodine),
hydroxy, 502NRaRb (where Ra and Rb are indt~.~.,d~.,L1y H or Cl 6 alkyl),
cyano, nitro, amino, mono- and dialkylamino in which the alkyl groups have
from 1 to 6 or more carbon atoms, acylamino, C1 6 alkoxy, C1 6 haloalkoxy,
C1 6 alkylthio, Cl 6 alkylsulphinyl, C1 6 alkylsulphonyl, carboxy,
carboxyamide, in which the groups attached to the N atom may be hydrogen or
lower hydrocarbyl optionally substituted with halogen; alkoxy carbonyl
wherein the alkoxy group may have from 1 to 6 or more carbon atoms, or aryl
such as phenyl;
provided that:
i) when A is a phenyl group or a substituted phenyl group in which no two
adjacent substituents are joined to form a partially or fully saturated
ring and Y is O; then Z is not NR4;
ii) when X is S, R2 and R3 are both H and Y is CH2; then the group (Z)n-R
is other than OH, OC1 4 alkyl, NHN(Cl 2 alkyl)2;
iii) when X is CH2, R~ and R3 are both H, Y is NH or NCH3, A is
unsubstituted phenyl or phenyl substituted with halo, methoxy, CF3 or N02
and n is O; then Rl is other than pyridyl, L~ 1' yl or
di hal ophenyl .
GB 1345159 discloses compounds which are somewhat sim;lar to those of
the present invention. It is suggested in this document that the compounds
may be active as herbicides but there are few examples and no data which
eluidates the degree of activity of the compounds.
WO-A-9413652, published after the priority date of the present
application discloses similar compounds which also have herbicidal
activity. However, there are certain differences in the structure of these
prior art compounds and, in particular, the equiYalent atom to Z of the
present invention is always nitrogen in the compounds of this prior art
document.
The expression "C1-C10 hydrocarbyl" in the foregoing definitions,
whether the expression is used on its own or as part of a larger radical
such as, for example, C1-C10 hydrocarbyloxy, is intended to include
hydrocarbyl radicals of up to ten carbon atoms. Subclasses of such
hydrocarbyl radicals include radicals with up to four or up tn six carbon
atoms. The expression "hydrocarbyl" is intended to include within its
WO 95133719 2 ~ 9 ~ 9 7 9 PCTIGB95/0122.1
scope aliphatic, alicyclic, and aromatic hydrocarbyl groups and
combinations thereof. It thus includes, for example, alkyl, alkenyl, and
~l kynyl radi cal s, CYCl oPropyl, cvcl opropylmethyl, cvcl obutyl, cvcl oPentyl,
and cvclohexyl radicals, the adamantyl radical and the phenyl radical.
The expression "heterocyclyl" in the foregoing definitions is intended
to include both aromatic and non-aromatic radicals. Examples of
heteroaromatic radicals inclquiude pyridyl, pyrimidyl, triazinyl, thienyl,
furyl, oxazolyl, isoxazolyl, and thiazolyl and examples of non-aromatic
radicals include partially and fully saturated variants of the above.
The expression "C1-C6 alkyl" refers to fully saturated straight or
branched hydrocarbon chains having from one to six carbon atoms. Examples
include methyl, ethyl, n-propyl, iso-propyl, n-butyl, t-butyl and n-hexyl.
Expressions such as "alkoxy'l, "cycloalkyl" "alkylthio~' "alkylsulphonyl",
"alkylsulphinyl" and "haloalkyl" should be construed accordingly.
The expression "C2 C6 alkenyl" refers to a straight or branched
hydrocarbon chain having from two to six carbon atoms and at least one
carbon-carbon double bond. Examples include ethenyl, 2-propenyl and
2-hexenyl. Expressions such as cycloalkenyl, alkenyloxy and haloalkenyl
should be construed accordingly.
The expression "C2_C6 alkynyl" refers to a straight or branched
hydrocarbon chain having from two to six carbon atoms and at least one
carbon-carbon triple bond. Examples include ethynyl, 2-propynyl and
2-hexynyl. Expressions such as cycloalkynyl, alkynyloxy and haloalkynyl
should be construed accordingly.
In the context of the present specification the terms "aryl" and
"aromatic ring system" refer to ring systems which may be mono-, bi- or
tricyclic. Eximples of such rings include phenyl, naphthalenyl,
.."~l"dcL..~l or ~. L~ yl. Nitrogen atoms in the ring may be
quaternised or oxidised.
In the context of the present specification, the term "heteroaryl"
refers to an aromatic ring system containing at least one heteroatom and
consisting either of a single ring or of two or more fused rings.
Preferably, single rings will contain up to four and bicyclic systems
up to five heteroatoms which will preferably be chosen from nitrogen,
oxygen and sulphur. Examples of such groups include furyl, thienyl,
pyrrolyl, pyrazolyl, imidazolyl, 1,2,3-triazolyl, 1,2,4-triazolyl,
WO 95133719 2 1 9 0 9 7 9 PCT/GB9S1012~.1
S
tetrazolyl, oxazolyl, isoxazolyl, thiazolyl, isothiazolyl,
1,2,3-oxadiazolyl, 1,2,4-oxadiazolyl, 1,3,4-oxadiazolyl, 1,2,5-oxadiazolyl,
1,2,3-thiadiazolyl, 1,2,4-thiadiazolyl, 1,3,4-thiadiazolyl,
1,2,5-thiadiazolyl, 1,2,3,4-oxatriazolyl, 1,2,3,5-oxatriazolyl,
1,2,3,4-thiatriazolyl, 1,2,3,5-thiatriazolyl, pyridyl, pyrimidinyl,
pyridazinyl, pyrazinyl, 1,2,3-triazinyl, 1,2,4-triazinyl, 1,3,5-triazinyl,
1,2,4,5-tetrazinyl, benzofuryl, benzisofuryl, benzothienyl, benzisothienyl,
indolyl, isoindolyl, indazolyl, benzothiazolyl, benzisothiazolyl,
benzoxazolyl, benzisoxazolyl, benzimidazolyl, quinolinyl, isoquinolinyl,
cinnolinyl, phthalazinyl, quinazolinyl, quinoxalinyl, naphthyridinyl,
benzotriazinyl, purinyl, pteridinyl and indolizinyl.
In the context of the present specification, the term "fused saturated
or partially saturated carbocyclic or heterocyclic ring" refers to a ring
system in which a 5- or 6- membered carbocyclic or heterocyclic ring which
is not of aromatic character is fused to an aromatic or heteroaromatic ring
system. Examples of such systems include benzimidazolinyl, benzoxazolinyl
and benzodioxolyl.
Examples of particular values for substituents of the group A include
methyl, ethyl, n-propyl, iso-propyl, trifluoromethyl, difluoromethyl,
pentafluoroethyl, trichloromethyl, ethoxyvinyl, fluoro, chloro, bromo,
iodo, methoxy, ethoxy, n-propoxy, iso-propoxy, difl~ I,u~y,
trifluoromethoxy, tetrafluu,u~Lhu~y, cyano, nitro, amino, mono- or
dialkylamino in which each alkyl group may have from 1 to 6 or more carbon
atoms, hydroxylamino, acyl (e.g. acetyl or trifluoroacetyl), methylthio,
methylsulphinyl, methylsulphonyl, trifluoromethylthio, 5CN, SF5,
trifluoromethylsulphinyl, trifluoromethylsulphonyl, sulphonamido, carboxy,
alkoxycarbonyl in which the alkoxy group may have from 1 to 6 or more
carbon atoms, carboxyamide in which the groups attached to the N atom may
be hydrogen or optionally substituted lower hydrocarbyl; or acylamino (e.g.
acetamido). When there is more than one substituent, the substituents may
be the same or different.
Preferred substituents of the group A include C1-C4 alkyl, C1-C4
haloalkyl, O(Cl-C4 alkyl), O(Cl-C4 haloalkyl), S(C1-C4 alkyl), S(C1-C4
haloalkyl) and halo. Specific examples of these preferred substituents are
trifluoromethyl, trichloromethyl, trifluù~l Lllu~y, trichlù,. Ll~u~y,
difluur~ Lllù~cy, dichlo,l Ll,..~, fluu,~ Ll,o~y, chl~ll L~ y,
,
21 )097~ --
WO95/33719 PCI'/GB95/01221
-- 6 --
trichloroethoxy, trifluoroethoxy, dichloroethoxy, difluu,u~LlluAy,
fluulu~LllùAy, trifluoromethylthio, ethoxy, methoxy, fluoro, chloro, bromo,
iodo and methyl.
Preferred compounds include those in which Rl is hydrogen or Cl-C6
alkyl, C2 C6 alkenyl, C2 C6 alkynyl, CO(Cl-C6 alkyl), CO(C2 C6 alkenyl),
CO(C2 C6 alkynyl), C3-C8 cycloalkyl, benzyl, phenyl or a 5 or 6 membered
heterocyclic ring. Any of these Rl groups may be substituted with one or
more substituents chosen from halo, SiRC3, CN, COOH, COO(Cl-C4 alkyl), COH
N(Cl-C4 alkyl)~ or CO(Cl-C4 alkyl) and cycloalkyl, benzyl, phenyl or
heterocyclic R groups may, additionally, be substituted with Cl-C6 alkyl,
C2_C6 alkenyl or C2 C6 alkynyl.
Specific examples of preferred groups Rl include optionally
substituted Cl-C6 alkyl, for example methyl, -C(CH3)3, -CH(CH3)2,
-CH2C(CH3)3, -CH2CH3, C(CH3)2CN, -CH2CH(cH3)2l -CH2CHzc(cH3)3l CH2CH2CH3'
CH2C(CH3)2 or C(CH3)2CH2Cl; C2 C6 alkenyl, for example C(CH3)2CH=CH2 and
CH2C(CH3)2CH=CH2; alkynyl, for example CH2C_CH or C(CH3)2C_CH; Cl-C6
alkyl-OH, for example C(CH3)2CH20H; optionally substituted C3-C8
cycloalkyl, for example cyclobutyl, l-methylcyclobutyl,
1-methylcyclopropyl, l-methylcyclûpentyl, 1-methylcyclohexyl,
1-cyanocyclopropyl, l-cyanocyclobutyl, l-cyanocyclopentyl,
1-cyanocyclohexyl, l-acetylenylcyclopropyl, l-acetylenylcyclobutyl,
l-acetylenylcyclopentyl, l-acetylenylcyclohexyl; optionally substituted
phenyl; optionally substituted heterocyclyl, for example pyrrolyl,
methylisoxazolyl or methylpyridyl; COCl-C6 alkyl, for example COC(CH3)3;
Cl-C6 alkylCOO(Cl-C4 alkyl), for example C(CH3)2COOC2H5; or SiR 3, for
example trimethylsilyl.
Other preferred compounds are those in which, ind.,,e,\d..,lly or in any
combination:
X is S, O or CH
Y ls S, O, CH2, CH(CH3) or NR6;
Z is NH or O; or n is O and Z is not present;
R2 and R3 are both hydrogen; or
Ql, Q2 and Q3, when present are CH2 or C=O.
When Y is a group NR6, it is preferred that R6 ;5 hydrogen, -CHO,
Cl-C6 alkyl, Cz C6 alkenyl, C2_C6 alkynyl, C3-C8 cycloalkyl, aryl, for
WO 95133719 9 7 9 PCT/GB95/0122 1
example benzyl which is optionally substituted with CI-C4 haloalkyl, or
C1-C4 haloalkoxy, (C1-C6 alkyl)aryl, (C1-C6 a1kyl)heterocyclyl, -O(CI-C6
alky~), -O(CI-C6 alkyl)aryl, -O(C1-C6 alkyl)heterocyclyl, -CI-C6 alkyl-OH,-(CI-C6 alkyl)-O-(C1-C6 alkyl), -(CI-C6 alkyl)-S-(C1-C6 alkyl)~ -CI-c6
alkyl (OCI-C6 alkyl)z, -CI-C6 alkyl-NH(C1-C6 alkyl), -C1-C6 alkyl-N(C1-C6
alkyl)2, C1-C6 alkyl-COO(CI-C6 alkyl), -CI-C6 alkyl-OCONH(C1-C6 alkyl) and
-C1 -C6 al kyl -OCO (C1 -C6 al kyl ) -
Examples of such preferred R6 groups include hydrogen, CHO, methyl,
ethyl, isopropyl, n-propyl, isobutyl, cyclopropyl, CH2-cyclopropyl, benzyl,
substituted benzyl, for example p-trifl~ ,I,o,.yl.e..~yl, phenyl, methoxy,
2-hydroxyethyl, 2 ~I,o~yeLhyl, 2,2-dimethoxyethyl, 3-propen-1-yl,
3-propyn-1-yl, 2-(N,N-dimethylamino)ethyl, CH2CO2CH3, CH2CO2CH2CH3,
CH2CH20CONHC(CH3)3, CH2CH~COOCH~r~3, 2-acetoxyethyl and 2-thiomethylethyl.
The formula I given above is intended to include tautomeric forms of
the structure drawn, as well as physically distinguishable modifications of
the compounds which may arise, for example, from different ways in which
the molecules are arranged in a crystal lattice, or from the inability of
parts of the molecule to rotate freely in relation to other parts, or from
geometrical isomerism, or from intra-molelcular or inter-molecular hydrogen
bonding, or otherwise.
Some of the compounds of the invention can exist in enantiomeric or
diastereomeric forms. The invention includes all individual forms and
mixtures thereof in all proportions.
Particular examples of compounds of general formula I are listed in
Table I. In all of these compounds, both R2 and R3 are hydrogen. In Table
I, and throughout the specification, Me represents methyl, Et ~ .e..--
ethyl, Pr represents propyl, Ph represents phenyl, Bz l~yl~,E..Ls benzyl and
Ac represents acetyl.
2~ 90q7~ --
WO 95133719 3 PCTIGB9~/017Z~
131,c^l !c_l_o c ~ c= 11 i
' ' ~ ~ ~ N o -- ' ~ _ ~ ' ' C
~, SS S S -- -- S -- -- ~ ,., N = -- ~ ' N S ~ ~ N
~~ S . ~J ,, S ~
NZ--~ -- -- C ^ C _ -- _ _ _ _ ~ _ _ _
_ Z ~ C O _ -- -- O -- -- _ _ _ -- -- _ _ J S _ -- _ -- --
m
N N N N N N N N N N N N N N N N
---- -- -- -- -- -- -- -- -- -- S -- -- -- -- -- -- -- r
E E E E E E E E E E E E E ' E E ' E -- E E E E E E
X X X ~ ,X X ~ X ~ X X X X X~ X X X X X X ~1~ X X
-- N ~I S ~ N r~ S ~ _ _ N N N N N N
SUeSTlTUTE SH ET ~RULE 26)
WO 95133719 2 1 9 0 9 7 9 PCT/CE195/0122J
:~ - C C I C C C C - _ C C o o o o o o _ C ^ C C C ^ o C
~` U -- N ~ ' ~ ' _ r _ ~ r uJ -- ~ r~ . r -
~, N S D.~ J N
~X-- _ '-- U U ~- U U U , _
U
~ Z Z ~. _ _ _ O _ Z
N N __ N N Q~ N ~ 1 0 N N O O N N N N N N ~J
U = -- '~ U U ;J ' : _ U U Z Z U U U U U U Z
_ _ _ _ _ _ _ -- -- -- -- -- O O _ -- -- _ O O O O O O _
X :,, U U U U U U :~J U U U U U U U U U ;)
U U U U U U U _ U U ~ U U ) U O U U J o U U U U O o
E E E E E _ E E E E E E E E E E E E E E E ~ E '
N _ ~ N Q ~ O ~D O C r ~ r N C ^ O Q ~
Ll .'.: X ~ X X X X :~ X X X X X X X X X X X X X X X X X X
O N ~_ r c" O _ N -- r r r~ rr ,~ O -- N r~ D ~ Q r
SUBSTIT~ITE S,~IEET (hULE 26)
` 21 90979
PCT/GB9510 1 22.1
WO 95/33719 I o
~r _ ^ ^ C C ^ O C C ^ ^ ^ C ^ O C C I ^ I C C C ^ ^ C C ^ C C ^
,,
~ r ' r r r ~ ~ ~ ~ N -- -- r w _ N r ~ ~ -- N r~
. _ _ _ U U U U _ U ~ _ U N ' ` -- -- ~ _ U
N -- -- -- -- _ _ C _ ^ ^ , _ _ ~ _ _ _ _ _ , _ _ _ _ _ _ _ _
X -- -- U -- -- -- -- -- -- -- -- -- -- -- -- -- -- -- -- -- -- -- -- -- --
U U U U U ~ ; J ,J ~ _ ` _ UJ U U U U ~ ~ U U U U U U
E E c E E E E E E E E E E E E E E E E ' E ' E E E E E
,n ~ _ -- N N~ N N _ _ _ _ o o ~ _ `D N N N rl -- --
X W W W ~ W ~J ~ W ~ W 'J W W _ W W W W J
N rl ~ J7 ~D -- m r~ o _ N r~ -- In ~ r m r~. o ~ N ~ ~ D r- m a~ o
SUBSTITUTE SHcET ~RULE 26)
WO 9~/33719 2 1 9 Q 9 7 9 PCT/GB9~/0122.1
I I
3 _ c c c ^ _ c _ -- u~ C c o ~ C ~ ~ C -- o o C ^ ~
!
~1: . S _ ~ , :- , ~ J D ~ ~ O _ ~ , _ ., o A~
_ U _ _ U _ _' I ` ~J U U .~ U ~ . U :~J U _
N -- ~ _ z -- _ _ _ _ -- -- -- -- _ _ _ _ _
N N
o _ D ~ ~ D , U ' ~ N _ z z _ ~, `
Z . ~ ` .
1 N ~1 N ~ 1 N ~ 1 ~ N ~ 1 ~ N ~ t~l N ~I t~l N ~1
_ U U U U -- -- U ~i U U U U U U U U _ U U U _ U U U L~ U U U
_ U U U U U U U ~ o C o o U ~ O O O U O ~ O O U U U U U U
r -- r r ~ r ~ , r r ~ r ~ ~ ~ l v r ~ . ~ r r ~c r ~D
~ CJ D D D ~ D D D -- O ~ D D X XD D o X X X C~ ~ D D X X X X X
-- N ~ ~ ~ ~D r ~ o~ o ,~ ~ ~ ~ ~ ~o r c ~ ~ -- ~ I-7 '` ~ ~O r ~ ~
O C ~ =~ ~ O _ o o o _ ~ o o
SUEST'TUTE ~IEET (RULE 26~
2 ~ 9 ~ 9 7 9 PCTIGB951012~.1
WO95133719 - i2-
:~ C o t:2 C ~ C C ^ C o C ^ ^ o C C ~- C I C C _ C o o C
_
U _.J _ 5' , J ~ , _ ~ V _ _ J 7 V 7 ~
N U~,` N ~ ' ~ U -- U U U U _ U _ _ U _ 2 ~ U U U U
-- -- U _ -- :_) U
-- -- -- _ _ _ ~ _ _ _ Z = Z _ ~ Z _ _
-
N
V V V~ N o 11 U O
Z
N N N N N N N N N N N N N N N N N N N N
J U U U U U U U U U U U U U U U U '` U U U ~A
~ _
N N ~ '`1 ~ ~) ~ ~ ~ ~ ~ ~ ~ U ~ U ~ U o o U
-
~ ~ ~ ~ ~ ~ r ~-- ,n o ~ ~ c ol ^ _ N ~-~ O r~
N N ~'V N N N _ N N N N N _~ N N ~
X7 X X X7 X Xv Xv Xv Xv Xv Xv V V Xv Xv V 7 7 V 7 7 7 V V V
~ O _ N r~ D r ~ ~ N N N N N N N N N ~
~ 1 -- -- -- -- -- -- -- -- -- ~ -- -- -- -- '--~ -- -- -- -- -- -- -- -- _ _ _ _
SUBSTITUTE S! IEET (RULE 26~
WO 95133719 2 1 ~ ~ 9 7 9 PCT/GU9S10122 1
- 13 -
3 ~ O ^ C
N
-- -- ~ _ _ _ _ _ ~ ^ _ _ _ A --
~J _ ,.1 ~J ~ _
~J '_~
U _ , _ _ _ _ _ _ _ _ _ _ _ _ _
~A O 5 0 C O O O O O O ~ O O O O
_~ . -- J _ ,_, ' ~ . L
~D ~ iD n N
'' X X , X ,X X X ~ X X ~, X h~ ~X X
O _ _ _ _ _ _ _ _ _ _ _ _ _ _ _
SUBSTIT[JTE S~l'.ET (RULE 26)
WO 95/33719 2 1 9 0 9 7 9 PCT/GB9510122.1
-- 14 --
:~: o O o O o o o o o o o o o o o
_ _ _ _ _ _ _ ~ _ _ _ _ _ _ _ _
_ ~ _ ~ ~ _ ~ ~ _ ~ _ _ _ _
V U U V V V V U U U U U U U U
_ = = = _ = = = = = = = . = = = = Z Z Z Z Z Z Z Z Z Z Z Z Z Z Z
~- o o o o o o o o o o o o o o o o
N N N N N N N N
V U U V V V V V
tq N N N 'r ,
' ' ' ' , ,; , 1
- -- ~ 1 N , L
N al ~ ~D N N ' N ' ~ r D L N
N
O ~ N ~ n U7 o N ~
~I X X X X X X X X X X ` ~D X U~ ~D X X X
r.~ ~ X ~ X
N ~ O _I N
WO g5133719 2 1 9 0 9 7 9 PCT/GB9S10122J
-3 c c ^ - ` c c _ 2 ~
I
---_ v J , ~ . . J
~;S ~- _ _ S _ ~ -- -- _ , -- 7 -- ~
~1U _ _ :J v _ U ~J _` _ _ _ _ _ _
_
~10 0 0 0 0 0 C O O C O C O 0 2 0
IN N N ~ N N 1`1 N N ~ N N N
X;~ U ' ~ _ U U _ U _ _
-- -- ~' -- N ~' --
~ X ~ ~ ~ C
- ,~, . r) N -' E ~ _ -- `
~ ._ N . .-
~_
~,D 3 3 '3 3 3 _ 3 3 ~ D D 3
LlX X X X X X X X X X X X X X .~ X~
U -- -- -- -- -- -- -- -- -- -- -- -- -- _ _
CUBSTITUTE SHEET (RU~E 26)
WO 95/33719 ~ 2 1 9 0 9 7 9 rcT/GB9~/ol22l
~.
:~ ~` O C ~ ^ ^ C G _
N
,~ _.~ _ _ _~, _ ~ ,_, _ ,,~ _ ,,, 'I ~q _ ,~,
U J r ' ,,, ~ .J ~,~ r:
~ ~ Z
J J J
~I N N N N r I r ~ ~ ~ N N ~ N N N N N N N
X ~ J _ _ U ~J
N , ~ - N N N N _ - ' -- r ~ ~
N ~ Q m m
~ ~;, J ~J ~ u N N
N -- ,~ ~ Xl r i - Ni -
~ J
--r- ~ -- -- ~ r~ _, r,~ ~ r- ~ r:
:~x x :~ X x x x x x x x X x
--~ ~D -- -- ~. -- -- r~ _o ~D -- r~ r,r~ o --
C -- r;l r~ . .~ _ _ _ r,~ rr. r. rr rr
SUBSTITUTE SHEET (RU~E 2~3
WO 95133719 2 ~ 9 0 9 7 9 PCT/GB9510~22 1
1 ,
3 C ~ ~ C C I _ C _ ~ I C ^ ~ I C _ _ ^ ~ C O _ _ _ _ --I-- _
N N N N ~ N
N ~ N ~ N ` AJ
-- U U -- =NU " U ~ N _ ' ~ . _
N . '' _ N
_ _ _ _ _ I _
, .
~ ~ 2 _ U U z r u2 _ _ _ ~ _ _
X _ -- _ _ _ _ _ _ _ _ _ _ N _ N N N N N N N N N N N N N N
'_ '~ ) U U U U U U U U U U U U U U C` '~ U U U U l_` U U U U
U J U ,~ u u u u U ~ m ~ c~ U -- a~ ~
O C E ù U U U U U U U E E E E E E c E e E E E E E c E E
-
..
N -A ~ O _ N ~ ~ 1/~ ~D r ~D --, 5 _ N 1~
N ^: N N N N' N N N N N N N N N N N N N N N N N N N N N N
SUaSTITUTE SHEET (RULE 25)
WO 95133719 2 i 9 0 9 7 9 PCr/GB95/01~2J
~ C C o o C I - _ o C C C o C -` ~ C C o C C C ~ o o -- C C C C
~ = = = = = = ^ _l r~
.-1 U3 3 N 1~ N N ~ 3 U ~ _ J N ,~ , ;4
N, U ~ _ _ _ U . _ _ _ _ , ~
U U, ~ ~ _ _ _ -- U U _~ U _ 5' U :J U U - N
_ C, U U _ ~
= Z = = = -- -- -- C O -- -- -- = -- O O -- _ -- -- -- --
_~ .
3 = N N N N N N N N N N N N N N N
_ Z Z ' ' C C U U ~ _ U U U L.) U U U U U U U U U
Z
z
N NN N _ N _ _ _ o o _ _ o o o o o o o o _ _ O O O
U U U U U U ~_` U U U U O U U
m m m m u ~ U O O O O u u ~ , u U ~ _ ' ~ u u
E E E E E E E , u u, u u u ~ '
_ N N -- 1~ -- N r~ r-- r
s~ x x x x x x x x x x x x x x x x x x x x x x x x
3 '' 3 U 3 3 3 3 3 3 3 3 al U 3 C~ 3 3 q~ ~ ~J 3 3 3
O _ N r~ O _ N '`1 ~ =', O _ N ~
N N N N N N N N N N N N N N N N N N N N N N N N N N N N
SUESTITUTE SHEET (RULE 26)
WO 9!;133719 ~91 9 0 9 7 9 PCT/GB9510~22 1
c ~ o O ~ I ~ o O I C C c C c I ^ o o o c o o o r o o c ^ C c o
cl _ r r _ ~J ~ N N ~
. ~ ~ N J ~'~ U N U N N , ~ N ~ U . N N 21 U
U U J U ~. ~. U
~_ _
N ~ = Z Z = = = = ~ z O O z z = l z
~ ~ _
N N N N N N ~ N ~) _ JJ N N N N ~J ) J ~
~ ~ U U Z U ~ . ~ ~ ~ ~ Z U N U U U U U z N N
. U
O O O O O O O O O O O O O O O O O O O O O O O -- -- _ -- -- --
U U U U 'J U
-- _ , _ ~ ~ _ ~ ~ ~ r _ ~ _ _ -- r ~
,N N N N ~N ~"
. u u ~ _ _ u u u u u O ' E = E E C E E E E
-
.
N ~ 11 N N ~ ~ ~ r~ N O ~ --~ O ~
N ~ ~ _ _ _ _ _ r r--
-- x x x x x x x x x x x x x x x x x x x x x ~x, x x x
x x x
~ ~ ~ ~ ~ r ~ cr o _ N ~1 . u~l ~D 1~ =~ ~ O ~ N ~
E ~D n ~ No ~ ~ r r r r ~ -- r r r ~ ~ :D ~ ~ ~ ~D
~ N N N N N N N N N N N N N N N N N N N N N N N N N N N N N
vU~STITUTE SHEET (RULE 26)
WO 95133719 2-1 9 0 9 7 9 PCrlGB9510122.1
2 C ^ C C C ~ ^IC ^ C ^IC C C C O o C C C C ~ C C C C C C C
I
r7 ^7 ~7 ~ r7 ~ L) N r ~ 7 = r'
,~ r .~ J ~ D ~ ~ E U o U ~ U ~ ~ CJ ' r N
N ~ N _ - -- --
N ~_~r; .J ~ .J N ~ N ,~ N ;~ N
N i _ _ _ : _ I _ . , _ _ _, _, : , o _ ~ Z _ , ~ o
- J J -- U G ~ C ~ G
r r J J N r~ ~ ~ N N N 77 Z z S 07 ~ z r7 :~:
Z Z 2
~ ~ ~ u7 a7 o7 07 07 ~77 77 o7 ~7 ol 07 77 77 07 _ N N N =N N N N N N N
s, r r r r ~ ~ ~ ~ r D. r ~:,. r r r r r7 ~7 ^7 N ~7 r7 ~7 r~ ~,N~ r7
"~ r7 ^7 ~ r7 r7 ~ r7 ~,, r7 ~ `7 r7 ~r~ C C C J J C C O J ~ C
E E E É E É E E E E ~ E E E E E E E E E E E E E E ~ E E
~, rC.7 r~7 r7 c7 r~7 r~7 r7 r~7 ~7 ~7 r7 r7 rA7 u7 ~'7 _7 ~7 ~7 o~ N '7 r7 N -7 r~ o7
X X X X X X X X X X X X X X X X X X~ X X X X X ~, X X
~ r~ c7 r,7 I c7 -- N ~7 ~ r7 07 ~- o7 ~ c7 0 0 c7 ~r u7 0 0 rO o -- -- -- ~ --
U N N N ~ N N N N N N r~ ~ N N .'1 ~7 r7 r7 ~ r7 ^7 r7 r7 r7 r7 r7 r7 r7 r7 r7
SUBSTITIJTE SH-~ET ~RULE 26)
WO 95/33719 2 1 9 0 9 7 9 PCT/GB95/0122-1
- :~ c c c c c I ^ c ^ I ^ ^ 5 c c
-
r 7 _ r
,, J ` ~ C _ r , _ -- ~ --
-
N ~ -- ~ -- z C _ , ; z _ _ z
r r
N
N z z N z O O O ^ O N o O
z
N N NN N N N N
X_ _ _ _ _ _ _ _ N N N _ !N~ _1 cA C1 -- _ N N N
N
¢O U ,J ,J ~ , o ~ , N _ __ ~ ._
E E E E ' ~ E E E E E E E -- - N ;,
-- r ~ O
N N ~ N ~ . ..
r N r C 5 ~ N ~ ~7 N r _ ~ 7 ~ ~, r~
X X X X X X X X X X X :C X X '~ ~ X
E u~ -- ~7 r 5 _ N 7
U r ,7 ~7 ~ ~N 7N N7 rN ~ N N N N
SUBSTITUTE SllEET (RULE 26)
WO 9S/33719 2 1 9 0 9 7 9 rcT/GBgSl1~l22~
,. ..
."
_I ' D ~ ~ D D N -- N _ .,
D
,) J
tJ ~ ~
~ == = = = = = = = = = =
_
O O O O O O O O O O O O
N N ~ 1 N N N N N N N N
X UU ~ '` '~ U '` U _ U _ U
~ ,, '. _ -- _ _
N N
_ `-- ~ .~ _ . . _ ~ _ ~ _ _ _
N ~ N _ ~ _~
N
tl
U ~ m ~ _ m m ~7 m m
SU9STITUTE SHEET ~ULE 26~
WO 95/33719 2 1 9 0 ~ 7 9 PCT/GB95/0122.1
- - ~ -- _ ~ _ _ _ _ _ _
~_ -- J
O O _ O O O O O O ^ O O O O
:'1 N N N N N N N N -~ N 1`~ ~'1 N
X--J U ~ ~ -- -- -- -- -- -- -- -- -- --
~ o O I ~ _ _' _ `-
N '. . `~ O I
` ~ ~ _ z N ~ ,.. .
7 ~ ~ O C~ a s,
J ,_ ~ _" _'~ _ -- C N :~ --- N
N
r
SU~STITUTE SHET (RULE 21i)
WO 95/33719 2~1 9 0 9 7 9 PCTIGB95/0122.1
,
~. " ~
Z -- -- ' _ -- -- -- S- -- -- ~ - J
J J _ _ _ _ _ J _ _ _ _ J
8 ~, ~. ~., ~, ,,`
O _ O O O O O O
Z ~ ~ Z _ _
N :~1 N ~1 N r~ ~ N N ~
,J ~ J ~ _ J J
e I ,n
~ e . e
L-- , , , L L C`~ ~ e
~¢ N ~ ~, N
C ~ , ~, C ~ L. ~ ' L _ ~ C ~ _
L~ C
a' a ~ , c ~ ~ - ~ ~ ~ I ~ ~ ~ ~
SUBSTiTUTE SHEET (RULE 26)
WO 95133719 2 i 9 0 9 7 9 PCT/GB9510122.1
Z ~ _ . ~ _ _ ~ _ -- , _
J
~ = = = -- = = = =
_ = = = = = = = = = = = =
O _ O
r r r~ r~ O r~ ~ r~ O rJ
2 -- _ 2 ~J 7 = 2
r~ ~ r r~
~C = ~ = = O O C O O ^ _ ~
o
r~ _ N
_ C~ _ ~ _ _ ~ _ _ _ _ _ - J J
r r~ r N r~
r; r~
Cl
~ , .o
O _ ~, _ _ =' _ _ ~ ~ -- _ , _ _
SUBSTITUTE SI~EET (RULE 26)
WO 95/33719 2 i 9 0 9 7 9 PCTIGB9S/0122.1
- ~6 -
~ _ _ _ _ _ _ _ _ _ _
N
:~. O O :~ -- , O O O ' ~ ~ --
_
N
X O ~ ^ O ^ O _ '`
`~ _ , _ _ _ _ _ ~ ,, _,
N N N N N
C o
~ . . . ...
._ . .r _ ._ _ .~ _ ._ ._ _ ._ ,~ ._ ,-- _ . .. ~ _ _ _
D iO D
,~j
U
~ --. ' D -- = '', = -- N
SUBSTITUTE SI~E~T (RULE 2S)
WO 9~i/33719 i 9 0 9 7 9 PCT/GB9!5/Olt2.1
- 2
C ^ C _ I^ o
r~
v v v v
,, ~J , .
N ~
V ~J JJ J ~ _l V V V r~ r 'J V
o o - --
~ r~ N r~ r~ r r~ r .~ rJ r~ N r~ r
K _ _ ._ _ ~ R o O
~I . ! , _: D V
.r ~ ~ -~ -- -~ _ '` ~ >, " " ., ", _
~ r~ r~ r~ N r~ )
x,_ x ,_ ,_ ,_ ,_ ~ , _ _ , , _ _
J -- -- ~ _ _ - , ._ ._
~ ~ r~ N N N N N N
rl N
D
-- _'l _ _ _ _ C _ N .~ _ u l V r ~ ~
SUaSTlTUTE SH''~T (RULE 2~)
WO 95/33719 2 1 9 0 9 7 9 PCI`IGB95/012~.1
~ C _ ~) _ _ I --
-- 3 ~ 3
, ~ N . ` ~"` U ~
-- -- _ _ _ _ ~ _
~ N ~ '' 3 _ o ~
N N N N N N N N
X -- '.1 _ ~ o N _ -- -- -- ~ O O Vl , ~ _
_
_ ._ _ ,_ ,_ ~ _ ) 3
`. `- ~ '. N N N N N N
' " , C I N N
._ _ _ _ _. _ _ _ _ _ ~ ~ ~ _ _ ~ _
_, ~ .a ~ :~ a ¦ ~ -- N ~ ~ ul ~a
SU~STITUTE SHEET (RllLE 26~
WO 95/33719 2 1 7 o 9 7 9 PCT/GB95/0122.1
O O C C _ ~
~; ---- ' -- '- ' ' ' -- 'J ~ _ _ _ _ _
_ _ _ J ~ _ _ _ _ L) V
= = = = = = = = = = = = = =
=
N N -- ~ oJ N N _ N
., -` -V F - - F - ~-, ~ -v
N ~1 N ~ N N N N
X _ O _ U2 _ _ _ _ ,~ _ _ O O , ~ _ , ~
C _ ~J iU C
, ~ _ _ _
F F :~ :F ~
N N N N N N N
_l u L U _l ~ u N N ~ N N N ~ ~l ~
~, .
~ N -F -~
SUBSTlT~ulTE SHEET ~RULE 26)
WO 95/33~19 2 1 9 0 9 7 9 PCTIGB95/0122.1
- 30 -
,.,
~., ~
r - _ ~ -- -- -! _ - - ~ ~
'1 _ _ O C _ O _ _ O _ C
` ' ` ' ' ' ` ' ` 'J
-J ~7 ~7 n ~7 -7
~ '', '. ~, . , , .
_ J A _ , _, . A . _ , _
N :~! N N ^: Dl ~1 N ~'
~' ' ~ ~ -- -- -- -- -- - ' -- -- ~7 7 ~D D
~ _ _ _ _ _ ~. _ _ ~ _ ~ .~ ~ ~
SUBSTITUTE SHEET (~ULE 2B)
WO 95133~19 ?1 1 9 0 9 7 9 PCT/GB9510122~
.
~, -- -- . .
~.
V
N _ _ _ _ _ _ _ _ _ _ _ _
_ N o N V _ ~ N o = O -- -- N
~ -- Z iJ L~ _
N N
X , _ _ C ; . _ _ _ _ _ _ .
~ ~ ~ ~ ~ :~ N N ~ N
. . __ _ , ~ _ _ _ _ ,_ __ _ ~ _
_
O
._., ~ _
~ . - - _, _ ,~ _ , _
._ ._ .~ ._ ,_ ._ ._ --- ^ -- -- _ -- i _
J ~ N i J
O ,, _ _ ~ _ I _ I . . I _ . _ _ _ _
SUBSTITUTE SHEET (RULE 26)
WO 9S/33719 ~ ~ 9 0 9 7 9 PCTIGB9S10122-1
} _
3 _ _ _ ¦ _ _ _ _ ¦ _ _ _ _ _ _ _
.,~
-- _ ,, , , , . ~ - .
L,
N _ _-- -- _ -- _ _ _ _ _ _
~ _ , ~ N _ _ ~ ~I
~ O _ _ _ _ O ~ O _ ~ _ O
~rJ r~l N r.l ~
X -- ~ U ~ ' ' -- L~ U . ~ ~
,~ , ,
,_ .._ ._~ .~ ._ ' . ' -1 _ ._ ' . ._ ; _ ._1 ',, _ '
c J ~ ~ J ! ~ ~ -- ~ ~
~ .
~. r. _. Lr. ~ _.
LJ - ~ . -- ~ .-- __ ~ ~ _
SUBST,TLITE S!~EET (RIJLE 2~)
2~9
WO 95/33719 9 7 9 PCT/GB95101t2 1
_ o ^ ^ - I ^ o C ~
-- ~ N ~ , N -- . ~ S
L , _ ~ _
= = = = = = = = = = = =
Pl C--AJ ~ ~ C U _ _ U D ~' D ~ ,, -- ~ o
X ~- N N N ~ _ Vl N N N N N N N N N N N N N
_` _ _ _ L` _ U U ' ` L U U _ U U
.~ _ _ _
~, ~ _ _ ~ _ _ ~ Z U :~. -- _ ~ ,,,
'~ -- = = -- = --` = -- _ -- ~ Z N N N N ~.
~ --. -- `' ' -- _ -- W -- ~_, _ -- -- ~ ~ U u~ C ~1 , o ~ y
J _ _ = _ _ _ ,~, _ .
U.
o o o o c N 1'')
SU~STiTUTE SHEET ~RULE 26)
219
3 ~ _ PCrlGB95/0122.1
C C _ _ _ _ ~ i C ¦ C C C _ I _ C C _ I _ _ C _ ¦ O
J . ~ J . J
U ~ N ~ ~ V N N ~ ~ ~ ~ ~ ~ ~ _ N _ ~
C = = = = = = = = = = = = = = = = = = = = = =
r~ = = = = = = = = = = = = = = = = = = = = = = =
z 3 v ~ z Z Z Z l l l ~ ~ ~ ~ S v ~ ~
V
N N N N N N N N N N N N N N N N N N
~ ` v v v v v v r ` v v v v v v v ~
N ~ N N ~" N , ~ N _ ~ ~~ N~ ~~ ~" ~ ~, ~,
--~ ,J v O ~ l o v o ~ , C C ~ ~~
r-- 1 0~ C _ N ~ 7 ~ O _ N r~ 0 ~ O _ N r
_ _ _ N N N N ~ 1 N N N N r.~
SUBSTITUTE SHEET (RULE 26)
WO 95/33719 2 ! 9 9 7 9 PCT/GB9S/0122-1
" _
3 ^ -- --I ^ I ^ I _ I ^ _ ^ _ _ _ _ ^ c -- --I-- c ^ --I-- ^ _ c _ _ --l _ I
_ _ N rl ~I N -- r.7 ~_ _ _ _ _ _ _ _ _ N N N
_ , _ _ _ _ N ~ N N ~.' U l_
t_l U ; U _ N N N N ~ ~ N N ~ ~ S -- ,_ 'r ' .
J O Ll _ U ~J J U ~ ) U N N N N N N N N N N N
Z 2 ~ Z Z -- Z -- _ Z Z 2 _ _ z U U U U U L) U U U ~
X O O O O C O O O O O O O C O O O O ^ O C O O O O O O O O O
_
~ ~, N N N N ~ ~ ~ ~ ' ' ~ N ~ N N
_ r~ ` ` ` ~ O O ~ U ~ J
rl .~ 1'
r. -- ~ N 1'1 ~ .~7 ~V r 5~ I r C _ N r~ ~ ~n ~ r.~ r~ o -- N r7
SUBSTITUTE SHEET (RU!E ~6)
WO 9513371g 2; 1 9 0 9 7 9 PCT/GB9~;101221
~6
~- C ^ C ^ C ~ ~ ^ ^ C C ^ I _ 1 5 c c ^ ~ ~ C C C c O C ^ c c
N -- ~ '7 ~ r~ N N N N
N ~ r ~ ~ J, ~ _ .J _ ~ _ C C N N N
S O , ~, J = N . ~ N N . ~ N ~,~ ` N _ N ~`
;J '-- ~ 'J ~ _ N ~'
N Z _ Z _ _ _ _ _ = = _
,~, N
N N N r~ r~ =N = S ~r ~r ~" c) s al s s -- ~ s s s ~r _
U I J 0 1,) 0 0 CJ Z Z = Z - Z Z = = Z Z = Z Z Z Z Z Z N
N N N N N N N N N N N N N N N N N N N N N N
X C C C ' O C ;~ J U ~
_ r _ r r r -- r ~ r r
N hN, " r "~ ~ ~ ~ ~ s ' ~, ~ r~ ~ r~ ~ r~ r~ r~ r~ r~
J C C C C C -- C C C C ~ r r - r r- r- r~ r~
- ~ r r~ r~ ~I rl r r~ r~
s.
~ ~ r~ -- r~ -- ~ r.~ r.~ r" r,~ r" ~ Q ~ ~D r~ r~ ,N, ~ ~ `D rO~ N r~
SUBSTITUTE SHEET (RU!E 26)
=
~ .2190
WO 95133719 PCr/GB95101~2.1
.
-- 3 o c _ _ _ _ _ _ _ _ ¦ _ I _ _ I _ _ I _ _ c _ o _ o c o _ l _ I
U _ -- U ~I ~ U U _,,
N N ~ J -- D ' N '
= : -- _ _ _ _ _ _ _ _ _ _ _
Z ~ _ _ _
V ` D D D ' D D _ D _ D V V _~ D V D _~ D V _ D --
Z Z Z Z
N N N N N N N - I N N N N N N N N N N N N N N
-- _ D ~ " S _ _ D. D D ~ S _
~ D. ~_ ~ L'~ D 1~ _ _ ^ D~ D. ;~. ~ ~ ~ ~u ~ ~ 1
' ~ U U U U U _ ~ _ D ~D ~D ~,
O O y O O O O _ O O ^~
_I r ) ~ -- r~ r'l ~ -- -- '-- 'J U U U U ~ ~ '1 ~ ~ ' '' ~ ' `
~, .n D r- ~ Ll~ C -- ~ ~ ~ .'1 D -- L~ ~ O ~ N '^ .r U') D ` L'~ Lt,
D D ~D CD ~D D ~ D -- -- _ _ D --D N ND ND ND ~D D ~D D ~D D
S!JBSTITUTE SI~EET ~RULE 2~
WO 9~/33719 2 1 9 0 9 7 9 PCT/GB9~/0122.1
j~
~ _ ~ _ C C _ C C _ C _ ~
~ -- ,
~ _ _ _ _ _ _ _ _
IJ L/ L~ _ ,, IJ ~
Z Z _ _ Z ~ Z ~ C O O O O O o
N N N N N N N N N N N N N N N ~
X~ L~ , ~, ~ O ~, ~, C _
N ' _ , ^ ?
,~ N N N N N ~ , . _ . . , C
_
J -- _ ~ N N
N N ~ --
r
O -- N "t _ .') `D r ~ ~ o -- N ~^ ~ _ . O -- ' ^.
SUBSTITUTE SH'ET (RLILE 26)
WO 95/33719 2 1 9 0 9 7 9 PCTIGB95/0122.1
- 39 -
Compounds of formula I are suitably prepared by a variety of processes
and some of these are discussed below.
For example, compounds of General formula I may be synthesised from
compounds of general formula Il, wherein A, R2, R3 and X are as defined for
General formula I and R15 is OH, SH or NHR6, wherein R6 ;5 as defined for
General formula 1. Compounds of general formula II in which R15 is OH, SH
and NHR6 give rise to compounds of general formula I in which Y is O, S and
RR6 respectively.
Reaction of a ~ompound of general formula II with compounds of general
formula R1COCl, R10C0Cl, R1-N=C=O and R1R4NC0Cl gives rise to compounds of
general formula I in which W is 0 and n is 0, Z is 0, Z is NH and Z is NR4
respectively.
Compounds of general formula I in which W is S and Z is NH may be
prepared by the reaction of a compound of general formula II with a
compound of general formula R1-N=C=S.
Compounds of general formula 11 in which R1S is OH or NR6 may also be
converted to compounds of general formula I in which both Y and W are O by
reaction with a compound of general formula R1O(C=O)O(C=O)OR1.
The conversion of a compound of general formula II to a compound of
general formula I may be carried out in an organic solvent such as
chloroform, dichloromethane or toluene at a, , aLule of from 0 to 50C,
preferably at room ~, ,aLule. The reaction generally proceeds most
favourably in the presence of a base which may be an amine such as
4-N,N dimethylaminopyridine (DMAP) or triethylamine. Alternatively, when
the compound of general formula II is reacted with an isocyanate, the base
may be replaced with a catalytic amount of boron trifluoride etherate.
Compounds of general formula I where X is CR4RS, Z is NR4, W is O, Y
is NR6 and n is 1 may be prepared by treatment of a compound of general
formula II where R15 is NHR6 with a carbamoyl chloride of general formula
R1R4NCOCl in a suitable solvent such as N,N-dimethyl formamide (DMF) in the
presence of a suitable base such as DMAP at an appropriate t, ~aLule
between 0C and 100C, usually at room t~, aLu,e. Compounds of the
formula R1R4NCOCl are known in the art or may be prepared by the treatment
of an amine of formula NHR1R4 with phosgene in a suitable solvent such as
tol uene .
Compounds of general formula II may be synthesised by various routes
.. , .. ,,, . ,, .. , , ,, . ... .. ,, . . .... .. . .. . . .. _ . _ .. .
WO 95133719 ` 2 1 9 0 9 7 9 PCT/GB9510122J
- 40 -
from compounds of gereral formula Ill, wherein A, R2, R3 and X are as
defined for general formula I and R20 is a leaving group such as Cl, Br, 1,
methane sulphonyloxy or toluene sulphonyloxy.
Compounds of general formula II in which Rl5 is NHR6 and R6 jS H may
be prepared from compounds of general formula III by reaction with an
alkali metal azide such as sodium azide to give the equivalent azide
compound followed by reduction of the azide by any known method, for
example using 1,3-propane dithiol in a basic solvent, to give the
appropriate compound of general formula Il. The first step of the reaction
may be carried out at a temperature of from O to 30C, but preferably at
room temperature in a solvent such as dimethylformamide (DMF). The
conversion of the azide to a compound of general formula Il is preferably
carried out under an inert di ~ such as nitrogen at O to 30C, most
suitably at room t~, .dLule~. The solvent may be an amine such as
triethyl ami ne .
Compounds of general formula II in which R1S is NHR6 and R6 jS other
than hydrogen may be prepared from compounds of general formula III by
reaction with a compound of formula NH2R6, wherein R6 j5 as defined in
general formula I. The reaction may be carried out at a temperature of
from O to 80C, preferably from 0C to room tl, ldLuI~ and it is
particularly preferred for the reaction to be initiated at 0C and
subsequently allowed to warm to room t~, .dLUl~ after most of the reactant
has been converted to product. The reaction is generally carried out in an
organic solvent, particularly an ether such as t~L~ ,I,ururan (THF).
Compounds of general formula Il in which R15 is OH may be prepared
from compounds of general formula ~ll, in which R20 is Cl, by reaction with
an aqueous base, typically a weak base such as an alkali metal bicarbonate.
In some cases, however, particularly when the group A is a heterocyclic
group, it is preferable to use mildly acidic conditions which may be
provided by, for example, aqueous potassium hydrogen phosphate. Typically,
a solution of the compound of general formula III in a solvent such as an
ether, for example THF, is stirred with the aqueous reagent at a
temperature of from 0 to 50C, but preferably at room i ,~laLult:.
Compounds of general formula II in which Rl5 is OH may a1so be
prepared from compounds of general formula IV, wherein A, R2, R3 and X are
as defined in general formula 1, by reaction with a strong base such as
WO 95/33719 ~ 1 9 0 9 7 9 PCT/GB9~/0122J
- 41 -
LiN(Si(CH3)3)2 or LiN(CH(CH3)2)2 followed by reaction with a compound
possessiny an actiYe oxygen, such as a compound of formula V in which, for
example, Ar is a p-tolyl group and Ar' is a phenyl group. The reaction is
suitably effected in a solvent such as THF at a, llltUI~ of from about
-100 to 30C, preferably from -80 to 0C.
A method for the preparation of compounds of general formula II in
which R15 is OH and X is CH2 is by the reaction of a derivative of general
formula Vl in which A is as defined for general formula I with a compound
of general formula VII wherein R2 and R3 are as defined for general formula
I, but are preferably hydrogen and R4 and R5 are hydrogen. The reaction
may be conducted in the absence of a solvent and at a temperature of from
about 100 to 300, preferably about 150. This reaction is novel and
forms a further aspect of the invention.
The reaction works particularly well for compounds in which A is
phenyl or substituted phenyl.
Compounds of general formulae Vl and VIl are, in general, readily
available or may be prepared by methods known to those skilled in the art.
50me examples of the synthesis of compounds of general formula Vl are given
in the examples below. Compounds of general formula V may be prepared as
described in J. Ora. Chem., (1988) 53, 2087.
Compounds of general formula III in which R20 is Cl or Br may be
prepared from compounds of general formula IV as defined above by
chlorination or bromination as appropriate. The particular method of
halogenation will depend upon the nature of the groups A and X but an
appropriate route may be determined by the skilled chemist. For example,
when X is S, chlorination may be carried out using an agent such as
sulphuryl chloride, N-chlorosuccinimide or chlorine. The reaction may take
place in a chlorinated solvent such as dichloromethane. The chlorination
reaction will preferably be carried out at a i , 7,~Lu,t: of -15 to 5C,
preferably -5 to 0C. When X is CH2, bromination is typically carried out
by reaction with bromine in the presence of ~hos,l,l,u,us tribromide or with
N-bromosuccinimide in a halogenated solvent. The reaction will often be
conducted at a temperature of from about 70 to 150C and in these
circumstances it will be necessary to use a high boiling solvent such as
chlo,uL~:"~ . An inert atmosphere such as nitrogen will generally be
empl oyed.
. ...... . .
WO 95133719 2 1 9 ~ 9 7 9 PCT/GB9510122.1
- 42 -
Compounds of general formula II in which R15 is OH and X is CH2 may be
converted to compounds of general formula III in which R20 is Cl and X is
CH2 by treatment with a chlorinating agent, particularly thionyl chloride.
The reaction may be initiated at room t~ ,~,aLul~ and maintained at room
tl, ,aLu.e for about 2 to 14 hours before heating, preferably to the
reflux temperature of the solYent.
Compounds of general formula III in which R20 is Br and X is CH2 may
also be prepared from compounds of general formula II in which R15 is OH
and X is CH2. In this case, the compound of general formula II may be
treated with agents such as 1,2-dibromotetrachloroethane and
triphenylphosphine. The solvent employed will preferably be an ether,
particularly diethyl ether and the reaction may be initiated at a
temperature from about -10 to 5C, preferably about 0C and subsequently
allowed to warm to room t ,_~dLU~
Compounds of general formula III wherein R20 is methane sulphonyloxy
or toluene sulphonyloxy may be synthesised from compounds of general
formula II wherein R15 is OH by reaction with methane sulphonyl chloride or
toluene sulphonyl chloride as appropriate. The reaction may be conducted
at a temperature of from 0 to 30C, usually at about 5C in a solvent such
as dichloromethane and in the presence of a base such as triethylamine.
Compounds of general formula III in which R20 is I may be prepared
from compounds of general formula III in which R20 is Cl or Br by reaction
with sodium iodide in a solvent such as acetone.
Compounds of formulae III and IV in which X is CR4R5 may also be
produced by cyclising a compound of formula XXIII in which A, R2, R3, R4
and R5 are as defined in relation to formula I, R26 j5 H, Cl or Br and R27
is halogen such as bromine or iodine in the presence of a base such as an
alkali metal alkoxide or hydride in an appropriate solvent and at
t ,dlu.es of from 0C to 70C, suitably at ambient t~ Lule~. When
the base is an alkali metal alkoxide such as sodium methoxide, then an
alcohol will be a suitable solvent but when an alkali metal hydride, for
example sodium hydride is chosen, it is more appropriate to choose an
aprotic solvent such as THF.
Compounds of formula XXIII may be prepared from compounds of formula
VI, as defined above, by reaction with a compound of formula XXIV in which
R2, R3, R4 and RS are as defined in relation to formula I and R26 and R27
. , . , , _ , . _, , . , _, . .. , _, . ..... ... ......... . .... ....
WO 9S/33719 ' ~ 2 ~ 9 ~ 9 7 9 PCT/GB9S/0122J
- 43 -
are as defined for general formula XXIII, in the presence of a base, such
as triethylamine, in a solvent, such as diethyl ether, at 0C to 100C,
suitably ambient t, I-aLu.e. Compounds of general formula XXIV are readily
available or may be prepared by known methods, for example as described by
Ikuta et al, J. Med. Chem. . 30, 1995 (1987) .
Compounds of general formula IV in which X is S may be prepared from
derivatives of general formula VI, in which A is as defined for general
formula I, by reaction with thioglycolic acid and a compound of general
formula R2R3Co with continuous removal of water from the reaction.
In some cases, however, cyclisation does not occur or is incomplete
and some or all of the product of the reaction is a compound of general
formula XXX, wherein A, R2 and R3 are as defined for general formula I.
Compounds of general formula XXX may be cyclised to give compounds of
general formula IV by treatment with a weak base such as triethylamine in
an organic solvent such as dichloromethane, followed by a halogenating
agent such as thionyl chloride and further treatment with weak base.
Compounds of general formula IV in which X is CH2 may be prepared in
two steps from a compound of general formula VI. Firstly, the compound of
formula VI is reacted with a compound of general formula VIII in which R2
and R3 are as defined for general formula I to give a carboxylic acid of
general formula IX in which R2 and R3 are as defined for general formula I.
The reaction is preferably conducted at a t 'IcLule of from about 15 to
50C in the presence or absence of a solvent. A method for the preparation
of compounds of general formula VIII is described in Or3anic Svnthesis. 60,
66-68 .
The compound of general formula IX may be converted to the compound of
general formula IV by decarboxylation which may be achieved simply by
heating to the melting point and allowing decarboxylation to occur.
Alternatively compounds of formula IV in which X is CR4R5 may be
produced from compounds of formula XXV, where A is as defined in relation
to formula I and R13 is halogen, by reaction with a compound of formula
XXVI where R2, R3, R4 and RS are as defined in relation to formula I, in
the presence of metals or metal oxides, suitably copper or copper I oxide,
at t ,~,dLu,es of 30C to 250C, suitably 130C to lBOC. Alternative
procedures can include treatment of XXV with alkali metal salts of XXVI in
solvents such as dimethyl sulphoxide and at ~ ~cLule~ of 0C to 100C,
. , . . .. , . , , . .... _ . . .. .... .. _ . . . .. .
WO 95/33719 2 1 9 0 9 7 9 PCT/GB9~10122.1
- 44 -
suitably ambient temperatures. This route is particularly useful for
compounds in which A is a heterocyclic group.
Compounds of genera~ formula II in which X is O and R15 is OH may be
prepared from compounds of general formula XIII, wherein A, R2 and R3 are
as defined for general formula I and each Rl9 is, ini~r ' Lly, benzyl or
substituted benzyl; by reduction, suitably hydrogenation over a palladium
or platinum catalyst, in the presence of an acid such as trifluoroacetic
acid .
Compounds of general formula XIII may be prepared from compounds of
general formula XIV, wherein RI9 is as defined for general formula XIII and
A is as defined for general formula I; by reaction with compounds of the
formula R19oCRZR3X where X is halogen, particularly chlorine, and R2, R3
and R19 are as defined above. For optimal results, the reaction is carried
out in a mixed aqueous/organic solvent such as water/dichloromethane and in
the presence of a base, for example sodium hydroxide, and a phase transfer
catalyst, for example tetrabutylammonium iodide.
Compounds of general formula XIV may be prepared from compounds of
general formula Vl by reaction with compounds of general formula XV,
wherein R19 is as defined above for yeneral formula XIII. Usually, the
compound of general formula XV will be converted to the acid chloride using
a chlorinating agent such as oxalyl chloride in the presence of
N,N-dimethylformamide (DMF) before reaction with the compound of general
formula Vl. The reaction may take place in an organic solvent, preferably
a chlorinated solvent such as dichloromethane.
Carboxylic acids of general formula XV may be prepared from esters of
general formula XVI, wherein R19 is as defined for general formula XIII; by
known methods such as treatment with aqueous potassium carbonate in a
solvent such as t~L, ~yd,urula" (THF).
Esters of general formula XVI may be prepared from dichloroacetic acid
by reaction with a mixture of an alcohol of general formula R19OH, where
R19 is as defined above for general formula XIII, and its ..,.~iuu..~ing
alkali metal alkoxide. The reaction will usually be conducted in the
appropriate alcohol. All of the starting materials of this reaction are
readily available.
Compounds of general formula II in which X is O and R15 is OH may,
alternatively be synthesised from compounds of general formula XII, wherein
WO 95/33719 _ 45 0 9 7 9 PCT/GB951012~1
RZ1 jS C1-C6 alkyl and A, R2 and R3 are as defined in general formula 1; by
reaction with an acid such as hydrochloric acid in an organic solvent such
as 1,4-dioxan.
Compounds of general formula XII may be synthesised from compounds of
general formula XVII, wherein A, R2 and R3 are as defined for general
formula 1, R21 is as defined above and R23 jS C1-C6 alkyl; by a two stage
reaction in which the compound is firstly treated with a strong acid such
as trifluoroacetic acid and then heated with a weak base such as sodium
bicarbonate.
In some cases, treatment of a compound of general formula XYII with a
strong acid will result in the production of a compound of general formula
II without it being necessary to isolate the intermediate of general
formul a XI I .
Compounds of general formula XYII may be obtained by the oxidation of
compounds of general formula XVIII, wherein A, R2, R3, R21 and R23 are as
defined above using, for example, an oxidising agent such as sodium
periodate. The reaction is preferably carried out in a polar solvent such
as a mixture of water and an alcohol, for example methanol or ethanol, at a
t, ,~Lu,~ of between 0 and 100C, preferably at room t ILul~.
Compounds of general formula XVIII may be prepared from compounds of
general formula XIX, wherein A and R23 are as defined above-
and compounds of general formula XX, wherein R2 and R3 are as defined for
general formula 1, R21 is as defined for general formula XII and X is a
leaving group, particularly a halogen such as chlorine. The reaction
requires basic conditions which may be provided by, for example, aqueous
sodium hydroxide which may be mixed with an organic solvent such as
dichloromethane. In this case, a phase transfer catalyst may also be
present. Ethers of general formula XX are readily available or can easily
be synthesised by a skilled chemist.
Compounds of general formula XIX may be synthesised by reacting a
compound of general formula Vl with a compound of general formula XXXI,
wherein R23 jS as defined above and R22 is C1-C6 alkyl. Compounds such as
these are readily available. It is greatly preferred that the reaction is
carried out under basic conditions, for example in the presence of sodium
hydride. A polar organic solvent such as DMS0 may be used.
WO 95/33719 ~ 2 1 9 0 9 7 9 PCT/GB9510122.1
- 46 -
Compounds of formula II in which X is S and R15 is OH may prepared by
solvolysis of compounds of formula XXI in which A, R2 and R3 are as defined
in relation to formula I. The reaction is conveniently carried out in the
presence of an aqueous alcohol, such as methanol, in a solvent such as
dichloromethane at t ,.la~ul~S of from O to 50C, preferably ambient
t ,-rdLu~=. The reaction often proceeds more successfully if conducted in
the presence of a weak base such as sodium or potassium bicarbonate.
Compounds of formula XXI may be prepared from compounds of formula
XXII, where A, R2 and R3 are as defined in relation to formula I, by
reaction with trifluoroacetic anhydride in a solvent such as
trifluoroacetic acid at t -la~ules of -10C to 70C, suitably at ambient
temperature .
Compounds of formula XXII may be prepared from compounds of formula IV
in which X is 5, by reaction with, for example, a periodate salt. A
suitable solvent is an aqueous alcohol such as ethanol or methanol and a
suitable temperature is ambient. In cases where A does not contain readily
oxidisable groups, one equivalent of a peracid, suitably m-chlululJ-,L~ oic
acid, in a solvent such as dichloromethane may be used.
Compounds of general formula II in which R1S is SH may be prepared
from compounds of general formula III in which R2U is Br by reaction
firstly with a thioacid of general formula HS(C=O)R1 wherein R1 is as
defined in general formula I to give a compound of general formula I in
which Y is S, W is 0 and n is 0, followed by reaction with ammonia in a
protic solvent such as methanol. The second step is preferably carried out
at a t ,'~dl,ul~ of -10 to +10C, usually about 0C.
An alternative method for the preparation of compounds of general
formula I in which X is CR4R5, Y is 0, W is 0 and Z is NH is by the
cyclisation of a compound of general formula XXVII, wherein A, R1, R2, R3,
R4 and R5 are as defined for general formula I and R25 is halogen such as
chloro, bromo or iodo. The reaction requires basic conditions which may be
provided by, for example, an alkali metal hydride or alkoxide. The solvent
will, to a certain extent, depend on the base which is chosen with solvents
such as THF being preferred for hydride bases and alcoholic solvents being
more appropriate for alkoxide bases. Compounds of general formula XXVII
also show herbicidal activity and form a further aspect of the invention.
Compounds of general formula XXVII may be pre~ared by the reaction of
wo 95133719 2 1 9 0 9 7 9 PCT/GB95/01221
- 47 -
a compound of general formula XXVIII, wherein A, R1, R2, R3, R4 and R5 are
as defined for general formula I dnd R25 ;5 as defined for general formula
XXVII, with a compound of the formula RI-N=C=O.
The reaction is similar to that described above in which a compound of
general formula II is converted to a compound of yeneral formula I by
reaction with a compound of the formula RI-N=C=O and the same or simildr
reaction conditions apply.
In a variation of this process, a compound of general formula I in
which X is CR4R5, Y is O, W is O and Z is NH may be prepared from a
compound of general formula XXVIII using the same steps as described above
but carried out in reverse order. Thus a compound of general formula
XXVIII may be cyclised to give a compound of general formula II in which
RIS is OH using the same reaction conditions as for the cyclisation of the
compound of general formula XXVII. The compound of general formula II may
then be reacted with a compound of the formula R1-N=C=O as described above
to give the required compound of general formula I in which X is CR4R5, Y
is O, W is O and Z is NH.
Compounds of general formula XXVIII may be prepared by the reaction of
an aniline derivative of general formula VI with a compound of general
formula VII, wherein R2, R3, R4 and R5 are as defined in general formula I,
in the presence of a reagent such as boron tribromide, aluminium
trichloride, tin tetrachloride or titanium tetrachloride in a solvent such
as dichloromethane or dichloroethane.
Compounds of general formula XXVII in which R25 jS iodo and A, R2, R3,
R4 and R5 are as defined for general formula I may, alternatively, be
prepared from compounds of formula XXIX, wherein R1, RZ, R3, R4 and R5 are
as defined for general formula I. Firstly, a solution of the compound of
general formula XXIX in a solvent such as dichloromethane is treated
sequentially with trimethylsilyl iodide, trimethylsilyl chloride and
oxalyl chloride in a one pot reaction. A compound of general formula Vl
may then be added to the reaction mixture in a solvent such as
dichloromethane and in the presence of a base such as pyridine and,
optionally, in the presence of dimethylaminopyridine (DMAP) to give a
product of general formula XXVII in which R25 jS I.
Compounds of general formula XXIX may be prepared from compounds of
general formula VII as defined above by treatment with a compound of the
.
wo 95/33719 2 1 9 0 9 7 9 PCI`IGB95/OIZ2.1
- 48 -
formula R1-N=C=0 in dichloromethane and in the presence either of
triethylamine or, preferably, of boron trifluoride etherate. This reaction
is similar to that described for the conYersion of compounds of general
formula II to compounds of general formula I and is carried out under
similar condit;ons.
This route to compounds of general formula XXVII and then to compounds of
general formula I is particularly useful for compounds in which A is a
heterocyclic group and which may be difficult to prepare by other routes.
A route to compounds of general formula I in which Y is 5, W is 0 and
n is 0 has been briefly mentioned above and involves the reaction of a
compound of general formula III in which R20 is Br by reaction with a
thioacid of general formula H5C(=O)Rl. The reaction is preferably carried
out under basic conditions, these being supplied by use of a weak base,
especially an amine base such as triethylamine. The reaction may be
carried out under an inert atmosphere such as nitrogen or argon at a
t~ :latUré of from -20 to 5C, preferably about 0C.
An alternative route to compounds of general formula I is by the
reaction of a compound of qeneral formula IV with a compound of general
formula BrCR4RSC(=O)OR1, wherein R1, R4 and RS are as defined for general
formula I. This reaction produces compounds of general formula I in which
Y is CR4RS, Z is 0 and W is 0. The reaction may be carried out in an
organic solvent, for example an ether such as THF and at a t~ dLule of
from -100 to 30C, most suitably -80 to 0C. It is greatly preferred
that the compound of general formula IV is first reacted with a strong base
and bases such as lithium hexamethyldisilazide have proved to be especially
suitable for the purpose. S ~_E~ Lly the compound of formula
BrCR4R5C(=O)OR1 may be added to the reaction mixture. An inert a r~ .
such as nitrogen or argon may also be necessary for this reaction.
Compounds of general formula I where X is CR4RS, ~ is NR4, W is 0, Y
is NH and n is 1 can be prepared from compounds of general formula XXXII on
treatment with an appropriate amine HNR4R1 in a suitable solvent such as
toluene at an appropriate t- ,_ldLule between 0C and 110C, more typically
70C. Compounds of general formula XXXII can be prepared by a Curtius
rea-,, L reaction from an azide of general formula XXXIII in a
suitable solvent such as toluene at an appropriate t~, ~rdLu,e between 20C
and 120C, more typically between 90C and 100C.
.. ... . . . . . . . . .. . . . . . . ..
WO95133719 ' 2`1 93~79 PCTIGB9510122.J
- 49 -
Compounds of general formula XXXIII can be prepared from compounds of
general formula IX by methods described in the literature (see March,
"Advanced Organic Chemistry: Reactions, Mechanisms & Structure", 4th
Edition, John Wiley ~ Sons, 1992, page 1û92). AlternatiYely compounds of
general formula IX can be converted to compounds of general formula XXXII
in one step on treatment with a reagent such as diphenylphosphoryl azide in
a suitable solvent such as toluene in the presence of a suitable base such
as triethylamine at an appropriate t l raLu,e between 20C and 120C; more
typically between 90C and 100C.
Compounds of general formula I in which Y is NR6 may be prepared from
compounds of formula I in which Y is NH by treatment with a base followed
by alkylation using a compound of formula R6-L, where L is a leaving group.
Compounds of general formula I in which Y is CR4R5 and Z is NR4 may be
prepared from compounds of general formula X in which A, X, RZ, R3, R4 and
RS are as defined in general formula I by reaction with amines of general
formula NHR1R4. It is preferable that the reaction is carried out under
dry conditions in an organic solvent such as dichloromethane and at a
temperature of from -20 to 30C. It is often preferred for the reagents
to be added to one another at about 0C, following which the reaction
mixture may be allowed to warm to room t~ ,_.aLu~e.
Compounds of general formula X may be prepared from the parent acids,
which are compounds of general formula I in which Y is CR4R5, Z is O and
is H, by reaction with an agent such as oxalyl chloride or thionyl
chloride. The reaction will generally take place under dry conditions.
When thionyl chloride is used, it may be added to the compound of genera~
formula I and the mixture heated under reflux. When oxalyl chloride is
used, on the other hand, much colder conditions will generally be used with
the reaction temperature being from about -20 to 20C, generally about
0C. A reaction solvent will also be employed in most cases with a typical
solvent being a halogenated solvent such as chloroform. In many cases the
reaction proceeds more rapidly in the presence of a catalytic amount of
DMF .
The parent acids of general formula I in which Y is CR4RS, Z is O and
Rl is H may be prepared from ~ ,un(ling esters of general formula I in
WO 95/33719 2 i 9 0 9 7 9 PCT/GB9510122.1
- 50 -
which Y is CR4RS, Z is 0 and R1 is an alkyl group. The ester may be
reacted with an organic acid in an organic or aqueous solvent, or with an
inorganic acid in a mixture of an organic solvent, such as an alcohol, and
water. When Rl is a group such as t-butyl, a strong acid such as
trifluoroacetic acid (TFA) is preferred and the reaction may be conducted
in an organic solYent such as dichloromethane or chloroform, or carried out
in the absence of a solvent. The reaction t ,.,~dLUle may be from 0 to
50C with room temperature being preferred.
An alternative method of preparation of parent acids of general
formula I in which Y is CR4R5, Z is 0 and Rl is H is by hydrogenolysis of
the col I ~,uu"~ing esters of general formula I in which Y is CR4RS, Z is 0
and R1 is d benzyl group. The reaction is carried out under an atmosphere
of hydrogen in the presence of a catalyst such as 5% or 10% palladium on
charcoal in an organic solvent such as ethyl acetate, tetrahydrofuran,
dioxane or an alcohol such as methanol or ethanol. The reaction will take
place in the absence of an acid catalyst, but will often proceed more
rapidly in the presence of a catalytic amount of an acid such as
trifluoroacetic acid. Reaction t~ ,_.dLUI~ may range from 0 to 50C with
room t ,...dLule: being preferred.
An alternative synthetic route to the parent acids of general formula
I, in which X is 5, Y is CH2, W is 0, Z is 0 and Rl H, is by reaction of a
derivative of general formula VI with mercaptosuccinic acid and a compound
of general formula R2R3C=o. The reaction may be carried out in a solvent
such as toluene and at the reflux temperature of the solvent.
The esters of general formula I in which Y is CR4RS, W is 0, Z is 0
and R1 is an alkyl group may be converted directly to amides of general
formula I in which Y is CR4RS and Z is NR4 by reaction of an amine in the
presence of a Lewis acid catalyst such as aluminium trichloride. The
reaction should be carried out under dry conditions in an aprotic solvent
such as toluene, dichloromethane or chloroform. In some cases, halogen
exchange may also occur to some extent during the reaction. For example,
when the catalyst is AlCl3 and one of the substituents on group A in the
starting material contains a halogenated moiety such as CF3, the product in
which the substituent is CCl3 may be isolated as well as the CF3 containing
pro~uct .
Esters of general formula I in which W is 0, Z is 0 and Y is CR4RS,
-
WO 95/33719 - 51 - PCT/GB9~/0122,1
with at least one of R4 and RS being other than hydrogen, may be prepared
from the co~,~s~ 'ing esters of general formula I in which Y is CH2 by
reaction with a strong base such as lithium hexamethyldisilazide followed
by reaction with the appropriate compound R4-Hal where Hal is a halo
substituent, typically iodo. The reaction should preferably be carried out
under dry conditions at a reaction tr -laLule of from -100 to 0C,
usually at about -78C. Suitable reaction solvents are aprotic organic
solYents such as THF. The reaction may generate both the required
alkylated product and a product which has been alkylated at a different
site. These products can be separated immediately if necessary but if the
ester is being used as an intermediate to another compound of general
formula I, the remaining reaction steps can be carried out before
separation of the products if this is more convenient. If required,
further alkylation can be carried out with a compound R5-Hal to obtain a
di al kyl ated product .
As already mentioned, esters of general formula I may be prepared from
compounds of general formul a IV .
A further method for the synthesis of compounds of general formula I
in which X is 0, Y is CH2 or CHR4, W is 0, and Z is 0 is from compounds of
general formula XI in which R1 and A are as defined for general formula I.
Compounds of general formula XI may be reacted with compounds of general
formula R2R3C=o in a polar solvent such as DMF and in the presence of a
strong base such as an alkali or alkaline earth metal hydroxide or sodium
hydride. The reaction may be carried out at a temperature of 10 to 50C
but preferably will be conducted at room t~ ,-,a~ul~:. The method is
particularly suitable for the the synthesis of esters of general formula I
in which R1 is an alkyl group. The esters may be converted to other
compounds of general formula I if required by one of the methods given
above .
Fumaric esters of general formula XI where A, Rl and R4 are as
defined in relation to formula I are prepared from the c~ spo ~ing
fumaric monoalkyl esters of general formula XXXV where R1 and R4 are as
defined in relation to formula I. Fumaric monoalkyl esters of general
formula XXXV are reacted with a compound of formula VI where A is defined
in relation to formula I in the presence of a dehydrating agent such as
dicyclohexylcarbodiimide. The reaction is conducted in an aprotic organic
WO 95133719 2 ~ 9 0 9 7 9 PCT/GB9510122.J
- 52 -
solvent such as dichloromethane or chloroform, at temperatures of from 0
to 50C with room temperature being preferred. Alternatively, fumaric
esters of general formula XI where A, R1 and R4 are as defined in relation
to formula I may be prepared by converting fumaric monoalkyl esters of
general formula XXXV where R1 and R4 are as defined in relation to formula
I to the corresponding acid chloride by treatment with thionyl chloride or
oxalyl chloride by methods analogous to those described above for similar
transformations, followed by reaction with the compound of general formula
VI where A is defined in relation to formula I. The reaction is carried
out in an organic solYent such as dichloromethane or chloroform in the
presence of a base such as triethylamine. The reaction may be carried out
at temperatures of from 0 to 70C with room t ,_ldLu~e
bei ng preferred .
Alternatively, fumaric esters of general formula Xl where A and R4 are
as defined in relation to formula I and Rl is benzyl may be prepared from
the corresponding fumaric acids of general formula XI where A and R4 are as
defined in relation to formula I and R1 is hydrogen. Compounds of general
formula Xl in which R1 is hydrogen may be reacted with benzyl alcohol in
the presence of diethyl azodicarboxylate and triphenyl phosphine. The
reaction is preferably conducted in an aprotic organic solvent such as
dichloromethane or chloroform at t~ aLu,-. of from -20 to 50C.
Alternatively, fumaric esters of general formula Xl where A and R4 are
as defined in relation to formula I and R1 is t-butyl may be prepared from
the ~u~ Jullding fumaric acids of general formula Xl where A and R4 are as
defined in relation to formula I and R1 is hydrogen. Compounds of general
formula XI in which R1 is hydrogen may be reacted with dimethylformamide
(bis) t-butyl dimethyl acetal in an organic solvent such as toluene. The
reaction may be carried out at t- rd~u,es of from room t~ .aLu,~ to
20C, preferably from 80 to 100C.
Fumaric acids of general formula XI where A and R4 are as defined in
relation to formula I and R1 is hydrogen may be prepared from fumaric acids
of general formula XI where A and R4 are as defined in relation to formula
I and R1 is alkyl by reaction with an inorganic base such as sodium
hydroxide or potassium hydroxide in an alcohol, preferably methanol or
ethanol. The reaction may be carried out at t-, aLu,e, of from 0 to
100C.
WO 95133719 2 ~ 9 0 9 7 9 PCTIGB9510122.1
Fumaric monoalkyl esters of general formula XXXV where RI and R4 are
as defined in relation to formula I are known compounds, or may be prepared
from known compounds by standard methods.
Compounds of general formula I in which Y is NR6 and Z is NR4 and R4
and R6 form a bridge may be synthesised in a variety of ways.
Compounds in which the bridge is represented by the formula -Q1-C(=O)-
may be synthesised from compounds of general formula 1 in which Z is NH and
Y is N-QI-C(=O)-L in which L is a leaving group such as methoxy, ethoxy,
chloro and Ql is as defined above. The reaction is preferably carried out
in the presence of a strong base such as sodium hydride, suitably in a
solvent such as THF. Usually, the reaction temperature wi11 be in the
range of 0 to 80C, preferably room tl, :l dLur~. They may alternatively
be synthesised from compounds of general formula (Ill) in which RZO is a
leaving group such as I or Br by reaction with an imidazolinedione of
general formula XXXVI where each of RI2 and R13 il)d~.,dcllLly represent
hydrogen or CI-C4 alkyl. The reaction is carried out i~ an organic solvent
such as N,N-dimethylformamide or tetrahydrofuran, in the presence of a
strong base such as sodium hydride.
Compounds in which the bridge is represented by the formula
-C(=O)-C(=O)- or -C(=O)-Q2-C-(=O)- may be synthesised from compounds of
general formula I in which both Y and Z are NH by reaction with a compound
of formula LC(=O)-C(=O)L or LC(=O)-Q2-C(=O)L in which Q2 and L are as
defined above. The reaction may be carried out in an organic solvent such
as toluene at a t~ LI~le of from 30 to 120C. Often, the reaction will
be conducted at a t~ Lule of about 80C.
Compounds in which the bridge is represented by the formula -HC=CH-
may be synthesised from compounds of general formula I in which Z is NH and
Y is NCH2CHL2, wherein L is a leaving group as defined above. The reaction
may be carried out in a solvent such as THF under acidic conditions which
may be provided by the presence of an aqueous inorganic acid such as
hydrochloric acid. The reaction t~, ~raLu~e may be from 5 to 50C but
will, in most cases, be room temperature.
Compounds of general formula I in which the bridge is represented by
the formula -CH=CH- may be converted to compounds of general formula I in
which the bridge is represented by CH2-CH2 by reduction, for example
hydrogenation over a palladium or platinum catalyst. Catalytic
,, _ . . ... . . .. . . ...... . . . . . . . .
21 93979
WO 95133719 ` PCT/GB9510122 1
- 54
hydrogenations may be carried out in a solvent such as ethyl acetate. The
reaction usually proceeds at an acceptable rate at room temperature and at
a pressure of from 1 to S bar.
Compounds in which the bridge is represented by the formula -C(=O)CH2-
may be synthesised from compounds of general formula I in which Y and Z are
both NH by reaction with CH0-CH0. The reaction may be conducted under
acidic conditions which may be provided by the presence of a catalytic
amount of, for example, D-toluene sulphonic acid. An example of a suitable
reaction solvent is toluene and the reaction is preferably carried out
under Dean and Stark conditions at a temperature of from about 80 to
120C, typically at 110C. Similar reaction conditions may also be used
for the synthesis of compounds of general formula I in which the bridge is
represented by the formula -CH2-OCH2-. However, in this case,
paraformaldehyde is used in place of the CH0-CH0. This particular reaction
may be adapted by those skilled in the art for the synthesis of other
bri dged compounds .
Compounds of general formula I may be converted to other compounds of
general formula I, for example by varying the substituents on the group A.
Compounds of general formula I in which X is CR4RS, Y and Z (if
present) are other than S and A is a phenyl group having a substituent
OR28, wherein R28 jS C1-C4 alkyl or haloalkyl, may be converted into
compounds of general formula I in which the phenyl ring is disubstituted
~nd wherein the second substituent is a halo, particularly a chloro, group
by treatment with a halo3enating agent such as N-chlorosuccinimide in a
solvent such as N,N-dimethylformamide (DMF). The reaction may be carried
out at a t, .t,Lu~e of from 15 to 80C, more usually at from 20 to 60C.
When the group OR28 is at the 3-position, the major product of the reaction
is usually the 3,4-substituted compound with the 3,6-substituted compound
being the minor product.
Compounds of general formula I wherein X is CR4R5, Y and Z (if
present) are other than S and the group A is a phenyl group with an S(C1-C4
alkyl) or S(C1-C4 haloalkyl) substituent may be oxidised to give compounds
of general formula I with the cu~ Ju.,~ing sulfoxide substituent on the A
group. The oxidation may be carried out using, for example, one equivalent
of an agent such as metachlo(uy~lbLl~oic acid (MCPBA) in a halogenated
solvent, for example chloroform. The reaction is preferably conducted at a
-
WO 95133719 _ 55 PCIVGB95/0122.1
temperature of from 0C to room t, ldLule.
A similar process may be used to obtain the equivalent sulfone. In
this case, howeYer, two equivalents of MCPBA may be used and the reaction
mixture is preferably heated to a ~, 'IGLur~ of from 30 to 90C, usually
to the reflux t-, ldLure of the solvent employed.
The functional groups C(Z)m Rl may be inter-converted to different
functional groups using techniques of esterification, transesterification,
hydrolysis and amidation some of which are discussed above. Other such
methods are standard procedures well known to the skilled chemist.
Variations of the above ~" -' re~ will be apparent to the skilled
chemist as well as alternative processes for preparing the compounds of the
invention. Other methods for making the compounds of the present invention
are analagous to the methods described in WO-A-9413652.
The compounds of formula I above are active as herbicides, and the
invention therefore provides, in a further aspect, a process for severely
damaging or killing unwanted plants, which process comprises applying to
the plants, or to the growth medium of the plants, a herbicidally effective
amount of a compound of formula I as hereinbefore defined.
The compounds of formula I are active against a broad range of weed
species including monocotyledonous and dicotyledonous species. They show
some selectivity towards certain species; they may be used, for example, as
selective herbicides in soya, rice and maize crops. The compounds of
formula I are applied directly to unwanted plants (post ~ ,y~..ce
application) but they are preferably applied to the soil before the
unwanted plants emerge (prE .y~ ê application).
The compounds of formula I may be used on their own to kill or
severely damage plants, but are preferably used in the form of a
composition comprising a compound of formula I in admixture with a carrier
comprising a solid or liquid diluent.
Compositions containing compounds of formula I include both dilute
compositions, which are ready for immediate use, and ~Ollc~Llc~Led
compositions, which require to be diluted before use, usually with water.
Preferably the compositions contain from 0.01% to 90% by weight of the
active ingredient. Dilute compositions ready for use preferably contain
from 0.01 to 2% of active ingredient, whi1e co"ce"L,dLed compositions may
WO 95133719 2 1 9 0 9 7 9 PCTIGB9510122.1
- 56 -
contain from 20 to 90% of active ingredient, although from 20 to 70% is
usually preferred.
The solid compositions may be in the form of granules, or dusting
powders wherein the active ingredient is mixed with a finely divided solid
diluent, e.g. kaolin, bentonite, kieselguhr, dolomite, calcium carbonate,
talc, powdered magnesia, Fuller's earth and qypsum. They may also be in
the form of dispersible powders or grains, comprising a wetting agent to
facilitate the dispersion of the powder or grains in liquid. Solid
compositions in the form of a powder may be applied as foliar dusts.
Liquid compositions may comprise a solution or dispersion of an active
ingredient in water optionally containing a surface-active agent, or may
comprise a solution or dispersion of an active ingredient in a
water-immiscible organic solvent which is dispersed as droplets in water.
Surface-active agents may be of the cationic, anionic, or non-ionic
type or mixtures thereof. The cationic agents are, for example, quaternary
ammonium compounds (e.g. cetyltrimethylammonium bromide). Suitable anionic
agents are soaps; salts of aliphatic mono ester of sulphuric acid, for
example sodium lauryl sulpha~e; and salts of sulphonated aromatic
compounds, for example sodium dodecylbenzenesulphonate, sodium, calcium,
and ammonium lignosulphonate, butylnaphthalene sulphonate, and a mixture of
the sodium salts of diisopropyl and triisopropylnaphthalenesulphonic acid.
Suitable non-ionic agents are the condensation products of ethylene oxide
with fatty alcohols such as oleyl alcohol and cetyl alcohol, or with
alkylphenols such as octyl- or nonyl- phenol (e.g. Agral goTM) or
octyl-cresol. Other non-ionic agents are the partial esters derived from
long chain fatty acids and hexitol anhydrides, for example sorbitan
monolaurate; the condensation products of the partial ester with ethylene
oxide; the lecithins; and silicone surface active agents (water soluble
surface active agents having a skeleton which comprises a siloxane chain
e.g. Silwet L77TM~. A suitable mixture in mineral oil is Atplus 411FTM.
The aqueous solutions or dispersions may be prepared by dissolving the
active ingredient in water or an organic solvent optionally containing
wetting or dispersing agent(s) and then, when organic solvents are used,
adding the mixture so obtained to water optionally containing wetting or
dispersing agent(s~. Suitable organic solvents include, for example,
ethylene di-chloride, isopropyl alcohol, propylene glycol, diacetone
. . . _ ~
WO 95/33719 2 1 9 0 9 7 9 PCT/GB9S/0122.1
- 57 -
alcohol, toluene, kerosene, methylnaphthalene, the xylenes and
trichloroethylene.
The compositions for use in the form of aqueous solutions or
dispersions are generally supplied in the form of a cu,.,~"L,cLe containing
a high proportion of the active ingredient, and the concentrate is then
diluted with water before use. The concentrates are usually required to
withstand storage for prolonyed periods and after such storage, to be
capable of dilution with water to form aqueous preparations which remain
for a sufficient time to enable them to be applied by
conventional spray equipment. Concentrates conveniently contain ZO-90%,
preferably 20-70%, by weight of the active ingredient(s). Dilute
preparations ready for use may contain varying amounts of the active
ingredient(s) depending upon the intended purpose; amounts of 0.01% to
10.0% and preferably 0.1% to 2%, by weight of active ingredient(s) are
normal ly used .
A preferred form of concentrated composition comprises the active
ingredient which has been finely divided and which has been dispersed in
water in the presence of a surface-active agent and a suspending agent.
Suitable suspending agents are hydrophilic colloids and include, for
example, polyvinylpyrrolidone and sodium Cal~JA~. Ll,ylcellulose, and the
vegetable gums, for example gum acacia and gum tragacanth. Preferred
suspending agents are those which impart thixotropic properties to, and
increase the viscosity of the ~ L,.l~e. Examples of preferred
suspending agents include hydrated colloidal mineral silicates, such as
montmorillonite, beidellite, nontronite, hectorite, saponite, and
saucorite. Bentonite is especially preferred. Other suspending agents
include cellulose derivatives and polyvinyl alcohol.
The rate of application of the compounds of the invention will depend
on a number of factors including, for example, the compound chosen for use,
the identity of the plants whose growth is to be inhibited, the
formulations selected for use and whether the compound is to be applied for
foliage or root uptake. As a general guide, however, an application rate
of from 0.001 to 20 kilograms per hectare is suitable while from 0.025 to
10 kilograms per hectare may be preferred.
The compositions of the invention may comprise, in addition to one or
more compounds of the invention, one or more compounds not of the invention
.
WO 95/33719 2 1 9 0 9 7 9 PCT/GB9510122J
- 58 -
but which possess biological activity. Accordingly in yet a still further
embodiment the invention provides a herbicidal composition comprising a
mixture of at least one herbicidal compound of formula I as hereinbefore
defined with at least one other herbicide.
The other herbicide may be any herbicide not having the formula I. It
will generally be a herbicide having a complementary action in the
particular application.
Examples of useful complementary herbicides include:
A. benzo-2,1,3-thiadiazin-4-one-2,2-dioxides such as bentazone;
B. hormone herbicides, particularly the phenoxy alkanoic acids such
as MCPA, MCPA-thioethyl, dichlorprop, 2,4,5-T, MCPB, 2,4-D,
2,4-DB, mecoprop, trichlopyr, clopyralid, and their derivatives
(eg. salts, esters and amides);
C. 1,3 dimethylpyrazole derivatives such as pyrazoxyfen, pyrazolate
and benzofenap;
D. Dinitrophenols and their derivatives (eg. acetates) such as
dinoterb, dinoseb and its ester, dinoseb acetate;
E. dinitroaniline herbicides such as dinitramine, trifluralin,
ethalflurolin, pendimethalin, oryzalin;
F. arylurea herbicides such as diuron, flumeturon, metoxuron,
neburon, isoproturon, chlorotoluron, chloroxuron, linuron,
monolinuron, chlu,ui,,, ,u", daimuron, methabenzthiazuron;
G. phenylcarbamoyloxyphenylcarbamates such as phenmedipham and
desmedipham;
H. 2-phenylpyridazin-3-ones such as chloridazon and norflurazon;
1. uracil herbicides such as lenacil, bromacil and terbacil;
J. triazine herbicides such as atrazine, simazine, aziprotryne,
cyanazine, prometryn, dimethametryn, simetryne, and terbutryn;
K. p~,~,,,ul~uLl,ioate herbicides such as piperophos, bensulide, and
butamifos;
L. thiolcarbamate herbicides such as cycloate, vernolate, molinate,
thiobencarb, butylate, EPTC, tri-allate, di-allate, esprocarb,
tiocarbazil, pyridate, and dimepiperate;
M. 1,2,4-triazin-~-one herbicides such as metamitron and
metri buz i n;
WO 95/33719 2 ~ 9 0 9 7 9 PCT/GB95/0122.1
- 59 -
N. benzoic acid herbicides such as 2,3,6-TBA, dicamba and
chl oramben;
0. ani1ide herbicides such as pretildchlor, butachlor, alachlor,
propachlor, propanil, metazachlor, metolachlor, acetochlor, and
dimethachlor;
P. dihalobenzonitrile herbicides such as dichlobenil, bromoxynil
and i oxyn i l;
Q. haloalkanoic herbicides such as dalapon, TCA and salts thereof;
R. diphenylether herbicides such as lactofen, fluroglycofen or
salts or ester thereof, nitrofen, bifenox, aciflurofen and salts
and esters thereof, oxyfluorfen, fomesafen, chlornitrofen and
chlomethoxyfen;
s. r -r yp~ U.~yplUuionate herbicides such as diclofop and esters
thereof such as the methyl ester, fluazifop and esters thereof,
haloxyfop and esters thereof, quizalofop and esters thereof and
fenoxaprop and esters thereof such as the ethyl ester;
T. cyclohexanedione herbicides such as alloxydim and salts thereof,
sethoxydim, cycloxydim, tralkoxydim, and clethodim;
U. sulfonyl urea herbicides such as chlorosulfuron, sulfometuron,
metsulfuron and esters thereof; benzsulfuron and esters thereof
such as DPX-M6313, chlorimuron and esters such as the ethyl
ester thereof pirimisulfuron and esters such as the methyl ester
thereof, 2-[3-(4-methoxy-6-methyl-1,3,5-
triazir--zyl)-~-me~hyt~reidosulphonyl) benzoic acid estérs such
as the methyl ester thereof (DPX-LS300) and pyrazosulfuron;
V. imidazolidinone herbicides such as imazaquin, imazamethabenz,
imazapyr and isopropylammonium salts thereof, imazethapyr;
W. arylanilide herbicides such as flamprop and esters thereof,
benzoylprop-ethyl ~ diflufenican;
X. amino acid herbicides such as glyphosate and glufosinate and
their salts and esters, sulphosate and bialaphos;
Y. organoarsenical herbicides such as monosodium methanearsonate
(MSMA);
Z. herbicidal amide derivative such as napropamide, propyzamide,
carbetamide, tebutam, bromobutide, isoxaben, naproanilide and
naptal am;
,. .. . ....
WO 95133719 2 1 9 0 9 7 9 PCTIGB9510122.1
- 60 ~
AA. miscellaneous herbicides including ~LI~ur. -ate, cinmethylin,
difenzoquat and salts thereof such as the methyl sulphate salt,
clomazone, oxadiazon, b" r~"Lxim, barban, tridiphane,
flurochloridone, quinchlorac, mefanacet, and triketone
herbicides such as sulcotrione;
BB. Examples of useful contact herbicides include:
bipyridylium herbicides such as those in which the active entity
is paraquat and those in which the active entity is diquat,
* These compounds are preferably employed in combination with
a safener such as dichlormid.
The invention is illustrated by the following Examples. The
abbreviations used in the Examples have the following meanings:
-THF = tetrahydrofuran
-DMF = N,N-dimethylformamide
-HPLC= High Performance Liquid Chromatography
-NMR = Nuclear Magnetic Resonance (performed at 270MHz and in CDC13 as
solvent unless otherwise stated). The following abbreviations are used to
indicate the multiplicity of the peaks in the NMR spectrum: s (singlet); d
(doublet); t (triplet); q (quartet); quin (quintet); m (multiplet); br
(broad) .
-ppm = parts per million
-m.p. = melting point
-Chromatography on columns of silica gel, unless specified
-Solutions dried over magnesium sulfate, unless specified
-Solutions were cu,,L,:,,L~dLed under reduced pressure
-IR spectrum: infra-red absorption spectrum.
-MS: mass spectrum
-GC: gas ~ll" Loyl dp~
-TLC: thin layer chromatography
-b . p: boi 1 i ng poi nt
WO 95/33719 2 ~ 9 0 9 7 9 PCTIGB95/0122.1
- 61 -
EXAMPI F 1 Preparation of Compound 1: 5-t-~utylc~rb~moylamino-3-(3-tr;-fluoromethyl)phenyl-4-thia~olidinone.
SteD 1 Preparation of 3-(3-trifluoromethyl)phenyl-4-thiazolidinone
A stirred solution of 3-trifluoromethylaniline (43.509) in toluene (275ml)
was treated with thioglycolic acid (24.909). After 10 minutes, the
solution was treated dropwise with 37% aqueous formaldehyde (20.8ml),
fo110wed by p-toluenesulphonic acid (30mg). The mixture was then heated
under reflux, and water was collected in a Dean and Stark apparatus. After
23.5ml of water had been collected, the mixture was cooled, extracted with
ether (2xlOOml) and the combined extracts washed with saturated aqueous
sodium bicarbonate solution (lOOml) and dried (Mgso4). Evaporation under
reduced pressure left a yellow oil, which afforded the title compound as a
white solid on trituration with hexane, yield 44.509, mp 59-60C.
H nmr (CDC13): ~ 3.76 (2H, s), 4.85 (2H, s), 7.47-7.58 (2H, m), 7.6B-7.76
(2H, m).
Step 2 Preparation of 5-chloro-3-(3-trifluoromethyl)phenyl-4-
thiazolidinone.
A stirred solution of 3-(3-trifluoromethyl)phenyl-4-thiazolidinone
(prepared as in Step 1 above) (10.009) in dichloromethane (150ml) was
cooled in an ice bath. A stream of nitrogen was bubbled through the
solution, and a solution of sulphuryl chloride (5.479) in dichloromethane
(5ml) was added dropwise. After the addition the solution was allowed to
warm to room t, '~d~UI~, and was stirred for a further 1 hour whilst
maintaining the nitrogen flow. The solution was evaporated under reduced
pressure to leave the product as a solid residue. This product was used
directly in subsequent rea~tions.
1H nmr (CDC13): ~4.72(1H,d`; 5.24(1H,d); 5.77(1H,s); 7.50-7.61(2H,m);
7 .70-7 .82 (2H,m) .
WO 95133719 2 1 9 0 9 7 9 PCTIGB9510122.1
- 62 -
Step 3 Preparation of 5-azido-3-(3-trifluoromethyl)phenyl-4-
thiazol idinone.
A stirred solution of 5-chloro-3-(3-trifluoromethylphenyl)-4-thiazolidinone
from Step 2 (3.809) in dimethylformamide (40ml) was treated with sodium
azide (0.889). After stirring for 10 minutes brine was added, and the
resulting mixture was extracted with diethylether (2x50ml). The combined
ether extracts were washed with brine, dried (MgS04) and evaporated ln
y~ç~ to leave a brown oil. This was separated by silica gel
chromatography, eluting with ethyl acetate/hexane mixtures, to afford the
title compound, yield 3.609.
1H NMR (CDC13): ~ 4.73(1H,d); 5.07(1H,d); 5.40(1H,s); 7.51-7.60(2H,m);
7.69-7.77(2H,m). MS: m/e 288(M ).
Preparation of 5-amino-3-(3-trifluoromethyl)phenyl-4-thiazolidinone
A solution of 5-azido-N-trifluoromethyl)phenyl-4-thiazolidinone (prepared
as in Step 3) (1.009) in triethylamine (1.4ml) was stirred under a nitrogen
atmosphere, and treated with 1,3-propanedithiol (l.OOml). The mixture was
stirred for a further 1 hour, and was then dissolved in diethyl ether.
This solution was washed with brine, dried (MgS04) and evaporated in vacuo
to leave an oil. This was separated by silica-gel chromatography eluting
with ethyl acetate/hexane mixtures, to afford the title compound as a
colourless oil which slowly crystallised on standing, yield 0.779. lH NMR
(CDC13): ~ 2.12(2H,broad s), 4.72(1H,d); 4.83(1H,d); 5.08(1H,s);
7.49-7.60(2H,m); 7.70-7.79(2H,m).
SteD ~ Preparation of 5-t-butylcarbamoylamino-3-(3-trifluoromethyl)
phenyl-4-thiazolidinone
A stirred solution of 5-amino-3-(3-trifluoromethyl)phenyl-4-thiazolidinone
from Step 4 (0.6559) in triethylamine (0.38ml) was treated with t-butyl
isocyanate (0.2729). After stirring for a further 30 minutes, the solid
was broken up under hexane, and filtered off to leave an off-white solid,
which was recrystallised from toluene. The resultant solid was added to
...... .. . . _ . .. . . _ . . .. .. . .. . _ . _ . _ . _ .... . .. ... _ .. . _ .. .
WO 95/33719 2 1 9 0 ~ 7 9 PCT/GB9S/0122.1
- 63 -
chloroform (40ml), and the mixture was heated to reflux when the insoluble
material (A) was filtered off. On cooling the filtrate, a precipitate
formed, which was filtered off (B). Solid samples (A) and (B), the tit1e
compound, were combined, yield 0.2499, m.p. 203-204C. 1H NMR (CDCl3):
1.30(9H,s); 4.78(1H,d); 4.89(1H,d); 5.53(1H,s); 5.73(1H,d); 6.42(1H,d);
7.45-7.58(2H,m); 7.69(1H,d); 7.82(1H,d).
The p,ucedu(., described in Steps 3 to 5 were also used to prepare
Compounds 28 (m.p. 175-176C), 53 (m.p. 175-176.5C) and 55 (m.p.
155-157C) of Table I.
EXAMPLE 2 Preparation of Compound 2: 5-IsopropyluA~ ' ,1amino-3-(3-
-trif~uoromethyl)phenyl -4-thiazolidinone
A solutiûn of 5-amino-3-(3-trifluoromethyl)phenyl-4-thiazolidinone
(prepared as in steps 1 to 4 of Example 1) (0.6559) in dry toluene (2.5ml)
was stirred under a nitrogen di ~ , and treated with triethylamine
(0.3Bml). The resultant solution was cooled to 0C, and a solution of
isûpropyl chloroformate in toluene (1.OM; Z.5ml) was added. A precipitate
began to form. The mixture was allowed to warm to room tl ,~ldLul~, and
the precipitate was filtered off and dried to afford the title compound as
a white solid, yield 0.3509, m.p. 187-188C. 1H NMR (CDC13): ~ 1.23(3H,d);
1.25(3H,d); 4.78(1H,d); 4.87-S.ûO(2H,m); 5.61 (ZH,s); 7.49-7.59(2H,m);
7.67-7.80(2H,m). MS: m/e 348 (M ).
EXAMPLE 3 Prep~ration of Compound 3: 5-t-~utanoylamino-3-(3-tri-
fluoromethyl)phenyl-4-thiazolidinone
A stirred solution of 5-amino-3-(3-trifluoromethyl)phenyl-4-thiazolidinone
(prepared as in steps 1 to 4 of Example 1) (0.8009) in toluene (Sml) was
treated sequentially with triethylamine (0.42ml) then pivaloyl chloride
(0.37ml). The mixture was stirred for 4 hours, then the precipitate was
filtered off. This was recrystallised from chloroform/petrol to remove
triethylammonium hydrochloride, and the material from the mother liquors
was recrystallised from chloroform/petrol to afford the title compound as a
white solid, yield 0.2859, m.p. 136-137C. 1H NMR (CDCl3): i~ 1.24(9H,s);
WO 95/33719 2 1 9 0 9 7 9 PCT/G~95/0122.1
- 64 -
4.78(1H,d); 4.97(1H,d); 5.62(1H,d); 6.58(1H,d); 7.47-7.58(2H,m);
7.71(1H,m); 7.78(1H,s).
EXAMPLF 4 Prepar~tion of Compound 4:
5-t-Butanoyloxy-3-(3-trifluoromethyl)phenyl-4-thiazolidinone.
SteD I Preparati on of 5-hydroxy-3- (3-tri f 1 uoromethyl ) phenyl -
-4-thi azol i dinone
A stirred solution of 5-chloro-3-(3-trifluoromethyl)phenyl-4-thiazolidinone
(from Step 2 of Example 1) in L~L~cd~ uru~a~ (lOOml) was treated with
aqueous sodium bicarbonate solution (lOOml), and the mixture was stirred
vigorously for 3 hours. The organic layer was separated, diluted with
ethyl acetate (SOml), washed with brine (50ml), then dried (MgS04).
EYaporation of the solvent under reduced pressure left a gum. Trituration
with hexane afforded a buff solid, which was recrystallised from ethyl
acetate/hexane to give the title compound as a white crystalline solid,
yield 7.089, mp 87-88C. lH NMR (CDCl3): ~ 4.70(1H,d); 5.00(1H,d);
5.74(1H,s); 7.48-7.59(2H,m); 7.64-7.76(2H,m).
$teo 2 Preparation of 5-t-Butanoyloxy-3-(3-trjfluoromethyl)-
phenyl-4-thiazol idinone.
A stirred solution of 5-hydroxy-3-(3-trifluoromethyl)phenyl-4-thiazolidione
from Step 1 (0.1109) in chloroform (lOml) was cooled to 0C and treated
with triethylamine (0.056ml) followed by pivaloyl chloride (0.052ml). The
mixture was stirred for 3 hours, then washed with 2M hydrochloric acid and
saturated sodium bicarbonate, then dried (MgS04). Evaporation of the
solvent irL vacuo left the title compound as a clear gum, yield 0.1219. 1H
NMR (CDC13): ~ 1.25(9H,s); 4.69(1H,d); 5.12(1H,dd); 6.18(1H,d);
7.49-7.61(2H,m); 7.73-7.80(2H,m).
WO 95/33719 2 1 9 ~ 9 7 9 PCI`/GB95/0122J
- 65 -
EXAMPLE S Prep~r~tion of Compound S:
5-(3,3-Dimethylbut~noyloxyJ-3-(3-trifluoromethyl)phenyl-4-thiazo1idinone
By a procedure similar to that described in Example 4 but using
5-hydroxy-3-(3-trifluoromethyl)phenyl-4-thiazolidinone (prepared as in Step
1 of Example 4) (0.2009), 3,3-dimethylbutanoyl chloride (0.106ml),
triethylamine (0.106ml) and dichloromethane (lOml) as solvent, the title
compound was obtained as a clear gum, yield 0.2579. 1H NMR (CDCl3):
1.07(9H,s); 2.31(2H,s); 4.70(1H,d); 5.09(1H,dd); 6.12(1H,d);
7 . 49-7 . 59 (2H, m); 7 . 72-7 . 79 (2H, m) .
The procedure of this example was also used for the synthesis of Compounds
29 (m.p. 138-139C), 51 (m.p. 66.5-68.5C), 54 (m.p. 135-136C), 63 (gum),
72 (gum), 89 (m.p. 72-73C), 96 (m.p. 115-116C), 97 (m.p. 87-88C), 102
(m.p. 69-71C), 103 (m.p. 91-93C), 104 (gum), 110 (gum), 113 (sol id gum),
123, 125, 126 305, 306, 307, 310, 313, 322, 324 and 325 of Table 1.
EXAMPLE 6 Prepar~tion of Co~pound 6: N-t-Butyl-~3-(3-trifluoromethyl)
phenyl-4-thiazolidil 5 yl]dcetamide
SteD 1 Preparation of [3-(3-trifluoromethyl)phenyl-4-thiazolidinone-S-
yl]acetyl chloride.
2-[3-(3-trifluoromethylphenyl)-4-thiazolidinone-S-yl]acetic acid (1.459)
(prepared as in Example 14 below) was placed in a flask equipped with a
stirrer bar, a reflux condenser and a silica gel drying tube. Thionyl
chloride (2.25ml) was added and the reaction mixture taken slowly to
reflux. After 1.5 hours at reflux, the dark coloured reaction mixture was
allowed to cool to room t ,_.~Lul~ and the excess thionyl chloride was
removed under reduced pressure to give the crude acid chloride as a dark
oil. 1H NMR (CDCl3): ~ 3.55(1H,dd); 3.74(1H,dd); 4.29(1H,m); 4.82(2H,m);
7.50-7.75(4H,m). IR (film): 1790cm~1, 1690cm 1
Step 2 Preparation of N-t-3utyl-[3-(3-trifluoromethyl)phenyl-4-
thiazol idinone-S-yl]acetamide.
The crude acid chloride from Step 1 was dissolved in dry dichloromethane
(30ml) and was stirred at 0C. t-Butylamine (1.05ml) was then added. Much
_ . . . . . . . . .
21 90979
WO 95/337t9 . PCTIGB95/0122.1
- 66 -
fuming occurred and the reaction mixture was allowed to stir at room
t ~.ldLul~ overnight. The following day, a further portion of
t-butylamine (0.5ml) was added and after 1 hour, the reaction mixture was
diluted with dichloromethane and washed successively with 2M aqueous HCl,
sat; NaHC03 (aq) and water. The organic layer was dried (MgS04) and
evaporated in Yacuo to give the crude product as a brown solid.
Purification by flash .I". Loy,~ on silica, eluting with ethyl acetate
in hexane (a gradient of 35% to 40% ethyl acetate), gave a yellow solid
which was recrystallised from ethyl acetate/hexane to give the pure title
compound as a light yellow so1id (0.569) m.p. 144.5-145.5C. lH NMR
(CDC13): ô 1.35(9H,s); 2.69(1H,dd), 2.98(1H,dd); 4.29(1H,m); 4.80(2H,m);
5.52(1H,brs); 7.53(2H,m); 7.74(2H,m). M.S. m/e.
EXAMPLE 7 Preparation of Compound 7: t-Butyl [1-(3-trifluoromethyl)
phenyl-2-pyrrolidinone-3-yl]acetate
Step 1 Preparation of 1-(3-trifluoromethyl)phenyl-2-pyrrolidinone-3-
carboxyl i c aci d
A suspension of 6,6-dimethyl-5,7-dioxaspiro[2.5]octane-4,8-dione (prepared
as described in Or~anic S~Yntheses. Volume 60, p66-68) (8.009) in 3-
trifluoromethylaniline (8.05g) was stirred at room,, ~.dLu,e for 24
hours. The mixture was filtered, and the insoluble solid was washed with
chloroform. The combined filtrates were washed with 2M hydrochloric acid,
brine and then dried (MgS04). Evaporation of the solvent under reduced
pressure left a brown solid, which was recrystallised from
chloroform/hexane to give the product as a white, crystalline solid, yield
4.10g, mp 135-136C (dec).
H nmr (CDC13): ~ 2.47-2.67 (2H, m), 3.70 (lH, t), 3.92-4.01 (2H, m), 7.00
(broad), 7.45-7.60 (2H, m), 7.81-7.90 (2H, m)
5~ Preparation of 1-(3-trifluoromethyl)phenyl-2-pyrrolidinone
1-(3-trifluoromethyl)phenyl-2-pyrrolidinone-3-carboxylic acid from Step 1
(3.609) was heated to its melting point, and heating was continued until
effervescence ceased (ca 50 minutes). The melt was cooled, dissolved in
=. . .. .... .. , = = _ . ...
WO 95/33719 ' PCI/GB95/0122.1
diethyl ether, and treated with decolourising charcoal. The charcoal was
filtered off, and the solvent was removed under reduced pressure to leave a
solid residue. This was recrystallised from hexane to give the product as
colourless needles, yield 2.209, mp 67-68C.
H nmr (CDCl3): ~2.19 (2H, quin), 2.62 (2H, t), 3.89 (2H, t), 7.35-7.53
(2H, m), 7.81-7.93 (2H, m)
MS: m/e 229 (M )
Stel~ 3 Preparation of t-Butyl-[1-(3-trifluoromethyl)phenyl-2-
pyrrol i di none-3-yl ] acetate
A solution of lithium hexamethyldisilazide (7.86ml, l.OM) in THF was addled
via syringe to 1-(3-Trifluoromethyl)phenyl-2-pyrrolidinone from Step 2
(1.59) in THF (15ml) at -78C under a nitrogen di rl ~. The yellow
solution was stirred at -78C for 30 minutes, followed by rapid addition of
t-butylbromoacetate (1.58ml) yia syringe. After 20 minutes, the reaction
was allowed to warm slowly to room t, ,~,Lu,~ before pouring into water
(lOOml) and extracting with ethyl acetate (2xlOOml). The combined extracts
were washed with brine (lOOml), dried (Na2S04) and evaporated in vacuo.
Purification of the residue by flash chromatography on silica, eluting with
40af ethyl acetate in hexane, gave the title compound as a yellow oil
(0.919). lH NMR (CDCl3): ~ 1.46(9H,s); 1.94(1H,m); 2.48(1H,dd) overlapping
Z.49(1H,m); 2.89(1H,dd); 3.13(1H,m); 3.84(2H,m); 7.39(1H,d); 7.49(1H,dd);
7.89(1H,s) overlapping 7.90(1H,d). M.S. m/e 344 (MH ).
EXAMPLE 8 PrepAr~tion of Compound 8: N-t-Butyl-[1-(3-tr~fluoromethyl)
phenyl -2-pyrrolidinone-3-yl]~cet~mide
Oxalyl chloride (0.127ml) was added yla syringe to a suspension of
2-[1-(3-trifluoromethyl)phenyl-2-pyrrolidinone-3-yl]acetic acid (0.3809)
(prepared as in Example 13 below) in chloroform (15ml) at room temperature.
Dimethylformamide (1 drop) was added and stirring continued for 90 minutes
during which time the effervescence stopped and the solid dissolved.
t-Butyl amine (0.41ml) was added dropwise at 0C causing an instant
precipitate. The reaction mixture was diluted with chloroform (lOOml) and
WO 95/33719 2 1 9 0 9 7 9 PCTIGB95/012~J
- 68 -
washed with water (lOOml), dried over Na2S04 and evaporated in y~yQ.
Purification by flash chromatography on silica, eluting with ethyl acetate
gave the amide contaminated with a little of the starting acid. The solid
was dissolved in ether (lOOml) washed with sat. NaHC03 (aq) (SOml), dried
(Na2504) and evaporated ln vacuo to give the pure title compound (0.2709)
as a colourless crystalline solid, m.p;. 127-lZ9C. lH NMR (CDC13): ~
1.33(9H,s); 1.99(1H,m); 2.37(1H,dd); 2.51(1H,m); 2.71(1H,dd); 3.05(1H,m);
3.83(2H,m); 5.98(1H,brs); 7.41(1H,d); 7.49(1H,t); 7.87(1H,d) overlapping
7.89(1H,s). M.S.: m/e 342 (M ).
The procedure of this example was also used jn the synthesis of Compound 16
(m.p. 128-129C), Compound 22 (m.p. 137-138C), Compound 23 (m.p. 110C)
and Compound 31 (m.p. 87-90C) of Table l.
EXAMPI F 9 Prep~r~tion of Compound 9: 3-(2-Pyrrolylc~rbonyloxy)-1-
(3-tri f 1 uoromethyl ) phenyl -2-pyrrol i di none
Step 1 Preparation of 3-hydroxy-1-(3-trifluoromethyl)phenyl-2-
pyrrol idinone
A stirred solution of 1-(3-trifluoromethyl)phenyl-2-pyrrolidinone (prepared
as in Steps 1 and 2 of Example 7 above) (1.109) in dry tetr~h~ ru-
(5ml) was cooled to -70C under a nitrogen t-i r~ l and a solution of
lithium hexamethyldisilazide in hexane (l.OM, 4.9ml) was added dropwise.
The resultant pale yellow suspension was then treated with a solution of N-
toluenesulphonyl-3-phenyloxaziridine (prepared as described in Journal of
Or~anic rhpmistrv~ (1988), 53, 2087) (2.009) in dry t~LI.lh.rlluru, (5ml).
The resultant pale yellow solution was allowed to warm to room t, 'idLu~,
and was then quenched with water and acidified to pHS using 2M hydrochloric
acid. The mixture was extracted with diethyl ether (x2), and the combined
extracts were washed with water, dried (MgS04) and evaporated under reduced
pressure to leave an oil. Purification by silica gel chromatography,
eluting with ethyl acetate/hexane mixtures, afforded the title compound as
a clear gum, yield 0.269.
WO gS/33719 2 1 9 0 9 7 q PCTIGB95/0122.~
- 69 -
H nmr (CDCl3): ~1.62 (lH,broad s); 2.12(1H, m); 2.63(1H, m); 3.72-3.90
(2H, m); 4.51(1H, m); 7.39-7.5B(2H, m); 7.77-8.02(2H, m).
MS: m/e 245 (M )
SteD la Alternative Preparation of 3-hydroxy-1-(3-trifluoromethyl)phenyl-2-
-pyrrol idinone
Alpha-hydroxy-delta-butyrolactone (2.049) and 3-trifluoromethylaniline
(2.74ml) were heated without solvent to 100C with stirring. After 4
hours, the t, .dLult: was raised to 150C (oil bath t ,- ~Lu,~:) and
stirring was continued for a further 20 hours. After cooling, the dark red
liquid was taken up in dichloromethane (Sml) and applied to a silica flash
column. Elution with ethyl acetate in hexane (a gradient of 40-60% ethyl
acetate) gave the title compound as a pale orange crystalline solid
(2 . 429) .
Physical data identical to that observed for the material prepared in Step
1.
SteD lb Further alternative route for the preparation of
3-hydroxy-1(3-trifluoromethylphenyl)pyrrolidin-2-one
i) Preparation of 4-chloro-2-hydroxy-N(3-trifluoromethylphenyl)butanamide
Titanium tetrachloride (11.Oml, 1.0M solution of d;chloromethane) was
added dropwise to a stirred solution of 3-hydroxyteL~ ' ~u,uru, 2-one
(1.09) and 3-trifluoromethylanine (1.SBg) in dry 1,2-dichloroethane (20ml).
After the initial exotherm had subsided, the mixture was heated under
reflux for five hours, cooled and stirred vigorously for thirty minutes
with an aqueous solution of ethylenediaminetetraacetic acid. It was then
extracted several times with dichloromethane. The extracts were washed
with hydrochloric acid (2M) and brine, dried over magnesium sulphate and
evaporated under reduced pressure. The residue was chromatographed on
silica, using dichloromethane-ethanol (49:1) to give the title compound
(0.639, m.p. 9B-100C). NMR (CDCl3): ~ 2.2(1H,m); 2.5(1H,m); 3.35(1H,bd);
3.B(2H,m); 4.5(1H,m); ?.4(2H,m); 7.75(1H,d); 7.9(1H,s); B.7(1H,bs).
WO 95/33719 2 ;1 9 0 9 7 9 PCT/GB95101Z2.1
- 70 -
The L(",.i,uu,.ding diol (0.089) was d150 obtained as a colourless gum.
It too can be conceived of as an intermediate. The use of other Lewis
acids gave similar results: aluminium chloride gave chloride-diol (1:4),
stannic chloride and titanium tetraisopropoxide gave diol, zinc chloride
gave chloride-diol (1:2) and magnesium bromide have bromide-diol (9:1).
ii) Preparation of 4-bromo-2-hydroxy-N(3-trifluoromethylphenyl)butanamide.
Boron tribromide (11.Oml, 1.0M solution in dichloromethane was added
dropwise to a stirred solution of 3-hydroxytetrdl,y.ll~ru,d,~-2-one (1.09) and
3-trifluoromethylphenylaniline (1.589) in 1,2-dichloroethane (20ml). The
mixture was stirred overnight at room t ,..d~Ule:, poured on to water and
extracted with dichloromethane. The extracts were washed with hydrochloric
acid (2M) and brine, dried over magnesium sulphate, and evaporated under
reduced pressure. The residue was chromatographed on silica, using
dichloromethane ethanol (49:1) as eluant, to give the title compound
(0.749, m.p. 67-69C). NMR (CDC13): ~ 2.3(1H,m); 2.6(1H,m); 3.5(1H,bs);
3.6(2H,dd); 4.5(1H,dd); 7.4(2H,m); 7.7 (lH,d); 7.9(1H,s); 8.7(1H,bs). MS:
M 325, 327.
iii) Preparation of 3-hydroxy-1(3-trifluoromethylphenyl)pyrrolidin-2-one.
Sodium hydride (0.0169, 60% suspensiûn in mineral oil) was added to a
stirred solution of the substrate (0.109, prepared as described in Step (i)
above) in dry t~LI`uhydluru~ (lOml), whilst maintaining the, _~dLul~
below 5C. The mixture was stirred for fifteen minutes, diluted with water
and extracted with dichloromethane. The extracts were washed with brine,
dried over magnesium sulphate, and evaporated under reduced pressure to
give the title compound (0.089). NMR (CDC13): ~ 2.1(1H,m); 2.6(1H,m);
3.4(1H,bs); 3.75(2H,m); 4.5(1H,t); 7.4~2H,m); 7.8(2H,m). This material was
identical to that prepared by an alternative method in WOg4/13652,
Preparative Example 42, Step 3.
The bromoalcohol, prepared as described in Step (ii) above, can be
used in similar fashion.
WO 95/33719 9 7 9 PCT/GB9510122.1
SteD 2 Preparation of 2-Pyrrole carboxylic acid chloride.
Oxalyl chloride (0.48ml) was added to a suspension of 2-pyrrole carboxylic
acid (0.459) in chloroform (lOml) at room t-, dlul~. After 2 hours,
effervescence had ceased and the solvent was evaporated in vacuo to give a
solid. Trituration with hexane left the crude crystalline acid chloride
which was used directly.
5teD 3 Preparation of 3-(2-Pyrrolylcarbonyloxy)-1-(3-trifluoromethyl)-
phenyl -2-pyrrol idinone.
2-Pyrrole carboxylic acid chloride from Step 2 (0.259) was dissolved in
dichloromethane (lOml) along with 3-hydroxy-1-(3-trifluoro-
methyl)phenyl-2-pyrrolidinone (0.389) from Step 1. Triethylamine (0.26ml)
was added. The solution turned reddish-orange and was left stirring
overnight at room t~ ,_,cLul~. After diluting with dichloromethane
(lOOml), the solution was washed with sat. NaHC03 (aq) (2xSOml), brine
(SOml), dried (Na2504) and evaporated. Purification of the residue by
flash chromatography on silica, eluting with 3096 ethyl acetate in hexane,
gave a pale yellow oil which crystallised. Re-crystallisation from ethyl
acetate/hexane gave the pure title compound (0.219) as colourless crystals,
m.p. 127.5-128C. lH NMR (CDC13): ~ 2.30(1H,m); 2.81(1H,m); 3.94(2H,m);
5.68(1H,t); 6.30(1H,m); 7.00(1H,m); 7.04(1H,m); 7.45(1H,d); 7.53(1H,t);
7.94(1H,d) overlapping 7.95(1H,s); 9.20(1H,brs).
EXAMPLE 10 Prep~ration of Compound 10: 3-(t-Butylc~rb~moyl-H-
methyl)~mino-1-(3-trifluoromethyl)phenyl-2-pyrrolidinone.
SteD 1 Preparation of 3-bromo-1-(3-trifluoromethyl)phenyl-2-pyrrolidinone
A 200ml 3 necked flask was equipped with stoppers, dropping funel,
thermometer, nitrogen bubbler and magnetic stirrer. The flask was charged
with lS.Og of 1-(3-trifluoromethyl)phenyl-2-pyrrolidinone (prepared as
described in Steps 1 and 2 of Example 7), ~ ' ,u, tribromide (1.Oml) and
chlulu~ell~L~- (65ml). This was heated to 105C. The dropping flJnnel was
charged with bromine (10.69) and this was added over a 70 minute period.
WO 95/33719 . 1 2 1 9 0 9 7 9 PCTIGB95/0122.1
- 72
After 165 minutes, the reaction was cooled and left at room tl, ldLule for
16 hours. Tlc indicated incomplete reaction, so the mixture was heated to
105C and a further quantity of bromine (0.739) added. After 100 minutes,
the reaction was cooled and washed with 75% Na2S2O3 solution (3x25ml),
dried over MgS04 and evaporated to give 17.79 of material which
crystallised. The solid was recrystallised twice from 25ml of methanol
giving the pure title compound as a crystalline solid (9.09), mp 82-87C.
.SteP la Alternative Preparation of 3-bromo-1-l3-trifluoromethyl)-
phenyl -2-pyrrol idinone
By a procedure similar to that of Example 68 below, but using
3-trifluoromethylaniline, were prepared successively 2,4-dibromo-
N(3-trifluoromethylphenyl)butanamide and 3-bromo-1(3-trifluoromethyl-
phenylpyrrolidin-2-one). The latter compound was identified by an NMR
spectrum which was identical to that of the product of Step l above.
.Ste~ Z Preparation of 3-(t-Butylcarbamoyl-N-methyl)amino-1-(3-
tri f l uoromethyl ) phenyl -2-pyrrol i di none .
Methylamine gas was bubbled through a solution of 3-bromo-1-(3-
trifluoromethyl)phenyl-2-pyrrolidinone (0.209) from Step 1 in THF (25ml) at
0C. After 15 minutes the reaction was allowed to warm to room
t ,,.dLule. After 1 hour, with methylamine still bubbling through the
so1ution, tlc showed no remaining bromide. The solvent was evaporated in
yacuo and the residue re-dissolved in dichloromethane (lOml).
t-Butylisocyanate (0.22ml) and triethylamine ~0.27ml) were added and the
yellow solution was stirred overnight at room t~, ~Lulc. The solvent was
then evaporated in vacuo and the residue purified by flash chromatography
on silica, eluting with 50% ethyl acetate in hexane. The title compound
was obtained as a colourless solid (0.1859). 1H NMR (CDCl3): ~ 1.37(9H,s),
2.14(1H,m); 2.49(1H,m); 2.85(3H,s); 3.B2(2H,m); 4.44(1H,brs); 5.23(1H,dd);
7.40(1H,d); 7.49(1H,t); 7.,1(lH,d) overlapping 7.92(1H,s). MS m/e 357(M ).
The procedure of this example was also used in the synthesis of Compounds
20 (m.p. 146-147C), 36 (m.p. 132-133C), 37 (using phenylisocyanate in
WO 95133719 2 t 9 0 9 7 9 PCT/GB9~/0122 1
- 73 -
place of t-butylisocyanate), (m.p. 167-169C), 43 (m.p. 127-128C), 44
(m.p. 136-137C), 52 (m.p. 135.5-136.5C), 60 (m.p. 134-135C), 70 (m.p.
133-135C), 71 (m.p. 122-123C), 124 and 282 of Tab1e 1.
EXAMPLF 11 Preparation of Compound 11: N-t3-Methyl;soxazol-5-yl)-
[1- (3-trifluoromethyl)phenyl -2-pyrrol idinone-3-yl] acetamide.
Oxalyl chloride (0.127ml) was added to a suspension of
[1-(3-trifluoromethyl)phenyl-2-pyrrolidinone-3-yl]acetic acid (prepared as
in Example I3 below) (0.389) in chloroform (6ml) at room t ,~ldLule.
Dimethylformamide (1 drop) was added, causing effervescence. After 2
hours, the reaction was cooled to 0C and 5-amino-3-methylisoxazole (0.149)
was added, followed by triethylamine (0.36ml). The reaction darkened and a
brown solid appeared. After 6 hours, the mixture was poured into ethyl
acetate (lOOml) and washed with sat. NaHCO3 (aq) (lOOml), brine (lOOml) and
lN HCl (aq) (50ml) brine (lOOml) and then dried (Na2S04). Evaporation of
the solvent in vacuo and purification of the residue by flash
chromatography on silica, eluting with ethyl acetate/hexane (a gradient of
40-50% ethyl acetate) gave the crude product. Recrystallisation from ethyl
acetate/hexane gave the title compound (0.149) as a colourless solid, m.p.
182-183C .
lH nmr (CDCl3): 1.91-2.06(1H,m); 2.26(3H,s); 2.49-2.63(1H,m);
2.67(1H,dd); 3.01(1H,dd); 3.10-3.24(1H,m); 3.83-4.00(2H,s); 7.46(1H,br d);
7.54(1H,br t); 7.84(1H,br s), 7.92(1H,br d).
EXAMPLE 12 Prepardtion of Compound lZ: 2-([1-(3-trifluoromethyl)
phenyl-2-pyrrolidinone-3-yl]acetylamino)-2,2-dimethylethanol
Oxalyl chloride (0.19Oml) was added to a stirred suspension of
[1-(3-trifluoromethyl)phenyl-2-pyrrolidinone-3-yl]acetic acid (prepared as
in Example 13 below) (0.5709) in chloroform (lOml) at room t~ ule.
Dimethylformamide (1 drop) was added and stirring continued for 2 hours.
The solvent was evaporated in vacuo and the residue re-dissolved in
dichloromethane (20ml) and cooled to 0C. Triethylamine (0.55ml) was
added, followed by 2-amino-2,2-dimethylethanol (0.55ml) and the mixture was
,, , , . . .. .. .. . . _ . . .. .. .. . . . . . _ .... . ..
219
WO 95133719 . 9 7 9 PCTIGB9!i10122.1
- 74 -
stirred at 0C for 1 hour before dllowing to warm to room t~ dLule. The
reaction was diluted with ethyl acetate (lOOml) and washed with water
(2x50ml), brine (SOml), dried (Na2504) and evaporated. The residue was
purified by flash chromatoyraphy on silica, eluting first with ethyl
acetate and then with 5% methanol in ethyl acetate to give the title
compound as a yellow gum. 1H NMR (CDCl3): ~ 1.30(6H,s); 1.90-2.05(1H,m);
2.48(1H,dd); overlapping 2.49(1H,m); 3.04-3.15(1H,m); 3.53(1H, v broad d);
3.67(brd), 3.78-3.94(2H,m); 4.72(1H, brm); 6.44(1H,brs); 7.42(1H,d);
7.50(1H,t); 7.85-7.90(2H,m).
The procedure of this example was also used in the synthesis of Compounds
26 (m.p. 128-131C), 27 (oil), 30 (oil) and 33 (oil) of Table 1.
EXAMPLE 13 Preparation of Compound 13: [1-(3-trifluoromethyl)phenyl-
2-pyrrolidinone-3-yl]~cetic acid.
Trifluoroacetic acid (lml) was added to a solution of t-Butyl-[l-
(3-trifluoromethyl)phenyl-2-pyrrolidinone-3-yl~acetate (prepared as
described in Example 7) (0.709) in dichloromethane (lOml) at room
t , ~,aLu,e. The yellow solution was stirred for 24 hours and the solvent
then evaporated in vacuo. Trituration of the residue with ether/hexane
caused a palê yellow crystalline solid to separatê out. This was filterêd
off to give the pure title compound (0.48g),m.p. 126-128C.
lH NMR (d6DMSO): ~ 1.80(1H,m); 2.28(1H,m); 2.41(1H,dd); 2.74(1H,dd);
2.91(1H,m); 3.78(2H,m); 7.42(1H,d); 7.56(1H,t); 7.75(1H,d); 8.16(1H,brs).
MS m/e 287(M )
EXAMPLF ~4 Preparation of Com,oound 14: ~3-~3-trifluoromethyl)phenyl
-4-thi~zolidinone-5-yl]Acetic ~cid.
Mercaptosuccinic acid (7.59) was weighed into a 3-necked flask equippêdwith a Dean and Stark trap and a condensor. 3-Trifluoromethyl anilinê
(8.059), toluene (lOOml) and 37% aqueous formaldehyde solution (4.25ml)
were introduced together with a stirrer bar. The stirrêd reaction mixture
was slowly brought to reflux during which time all the remaining solid
dissolved. Water was collected in the Dean and Stark trap once reflux was
WO 95/33719 2 ~ 9 0 9 7 9 PCTIGB95/0122.1
attained, and after 15 minutes a solid began to precipitate. After 3h at
reflux the hot solution was filtered through a sinter and the filtrate
allowed to cool to room t~, ,dLul~. The solid that then precipitated was
collected at the pump (7.349) and was recrystallised from hot toluene to
give the title compound (3.789). IH NMR (CDCl3/d6DMS0): 2.95(1H,dd);
3.19(1H,dd); 4.27(1H,m); 4.82(2H,m); 7.53(2H,m); 7.71(1H,m); 7.79(1H,s).
IR (Nujol mull): 3500-2500(broad), 1700cm~1 (broad). MS: m/e 305(M ).
EXAMPLE 15 Preparation of Compound 15: Ethyl ~3-(3-trifluoromethyl)-
phenyl -4-oxazol idinone-5-yl] acetate
A solution of the ethyl ester of N-(3-trifluoromethylphenyl) fumaric acid
amide (10.09) in DMF (50ml) was added to a stirred suspension of sodium
hydride (0.149 of a 609s oil dispersion) in DMF (25ml). The reaction
mixture turned bright orange. Paraformaldehyde (5.809) was then added in
one portion. The reaction turned brown. After 15 minutes, the reaction
was poured into water (250ml) with approximately 10ml of 2N HCl (aq). The
aqueous mixture was extracted with ether (2x200ml). The combined organic
extracts were washed with brine and then dried over MgS04, filtered and the
solvent removed under reduced pressure to give the oxazolidinone as a brown
oil (10.8589, 98%) which solidified on standing. The procedure of this
example was also used in the preparation of Compounds 45, 62 242 and 243 of
Table 1.
EXAMPLE 16 Preparation of Compound 17 and Compound 18: N-(1,1-dimethyl-propyl)-~1-(3-trifluoromethyl)phenyl-Z-pyrrolidinone-3-yl~acetamide and
N-(l,l-dimethylprop-2-enyl)-~1-(3-trifluoromethyl)phenyl-2-pyrrolidinone-3-
yl] dcetamide .
The propargyl amide (Compound 16, prepared by a method similar to thatdescribed in Example 8) (0.4079) was dissolved in ethyl acetate (20ml). 5%
Palladium on calcium carbonate, poisoned with lead (0.049) in ethyl acetate
(5ml) was added. With stirring, the flask was eYacuated and flushed with
hydrogen via a balloon. This procedure was repeated twice. After 2 hours
hydrogenation at one dL ,'e.~, the reaction was filtered through a pad of
HyfloTM to remove catalyst. Evaporation of the filtrate gave a solid which
.. . , , , . . . . . . . . . .... . . . .. .. . _ . . .. _ . _ ... . . . .. .
- 21 90979
WO 95/33719 PCT/GB95/0122 1
- 76 -
was purified by HPLC, eluting with 25% acetone in hexane to give firstly
Compound 17 as a colourless solid (0.0959), m.p 117C; and then Compound 18
as a colourless solid (0.1599), m.p. 111C. This procedure was also used
in the preparation of Compound 34 (m.p. 104-105C), Compound 35 (m.p.
120-121C), Compound 46, Compound 47, Compound 48, Compound 49, Compound 56
(m.p. 98-99C), Compound 57 (m.p. 171-172C), Compound 74 (m.p. 77-81C),
Compound 79, Compound 80 (m.p. 83-84.5C), Compound 98 (m.p.
111.5-112.5C), Compound 118 (m.p. 114.5-116C), Compound 240, Compound 255
and Compounds 273-277 of Table I.
EXAMPLE 17 Prepar~tion of Compounds 40 and 39: H-(1,1-dimethylpropynyl)-
tl-(3-trifluoromethyl)phenyl-3-oxazolidinone-5-yl]acetamide and
N-(1,1-dimethylpropynyl)-[1-(3-trichloromethyl)phenyl-3-oxazolidinone-5-yl~
acet~mi de
The ester (Compound 15, prepared as in Example 15) (0.89) was dissolved in
dry dichloromethane (20ml) and aluminium trichloride (0.849) added. To
this suspension was added dropwise 1,1-dimethylpropargylamine (1.169).
When the effervescence had ceased and exotherm of 15C died down, the
mixture was stirred at room t~ LI~le for 1 hour. The mixture was then
carefully quenched with 2M HCl(aq), the layers separated and the aqueous
layer extracted with dichloromethane. The combined organics were washed
with water, dried over MgS04 and evaporated. The solid was recrystallised
and the crystallisation residue purified by HPLC and then preparative TLC
to give Compound 39 as a white solid (0.1689) and Compound 40 as a white
solid (0.089). This procedure was also used in the preparation of
Compounds 21, 50, 66, 67 and 68 of Table I.
XAMPI F 18 Prepar~tion of Compound 24: N-cyclobutyl-~1-(3-tr~flu~ro-
methyl)phenyl-2-pyrrolidinone-3-yl]~cetamide.
The acid chloride (prepared as described in Example 8 from the
,eipo"ding acid, Compound 13, 0.59) was dissolved in dichloromethane
(18ml). Sodium carbonate (0.4639) was dissolved in water (18ml) and added
to cyclobutylamine hydrochloride (0.2079). After the effervescence had
subsided, this solution was added to the acid chloride. After vigorous
WO 95133719 2 1 9 0 9 7 9 PCTIGB95/0122.1
stirring, the reaction was left to stand overnight. The reaction was
diluted with dichloromethane (20ml) and washed with NaHC03 (aq). The
aqueous ldyer was re-extracted with dichloromethane and then with brine
before drying over MgS04. Evaporation gave a yellow oil which was purified
by column chromatography, eluting with 90% ethyl acetate in hexane tû give
Compound Z4 as a white solid (0.4759), m.p. 146-147C. This procedure was
also used for the preparation of Compound 25 of Table I (m.p. 96.5-99C).
EXAMPI F 19 Prep~ration of Compound 32:N-t-Butyl-2-[1-(3-trifluoromethyl
phenyl ) -2-pyrro1 i di none-3-yl ~ propami de
Ste~ I Preparation of t-butyl 2-[1-(3-trifluoromethylphenyl)-2-
pyrrol idinone-3-yl] propanoate
The ester (Compound 7, 2.1129) was dissolved in THF (30ml) at -78C.
Lithium hexamethyldisilazide (6.79ml of a 1.0m THF solution) was added via
syringe. After 30 minutes at -78C, iodomethane (1.7529) was added. This
was left to stir at -78C for 20 minutes and then allowed to warm to room
t~, r.lLu~e. After 10 minutes at room temperature, the reaction was poured
into water (30ml). The aqueous layer was re-extracted with ether (x2) and
the combined organic layers were washed with sodium thiosulphate solution
and dried over MgS04. The solvent was e.~.r~"~,L~d and the residue purified
by flash chromatography, eluting with 25% ethyl acetate in hexane to give a
(1:1) mixture of the required product and the 3-methylpyrrolidone product
(1 .09) .
SteD 2
The mixture of two esters obtained in Step 1 was hydrolysed according
to the method in Example 13, to give a mixture of the ~ull~-r 'ing acids
(0 . 7079) -
Stec 3
The mixture of acids obtained in Step 2 was reacted with oxalylchloride and then t-butylamine as in the method for Example lO. The
mixture of amide products was separated by HPLC to give the title compound
as a white solid (0.2089), m.p. 107.5-111C.
WO 95/33719 ~ l q 0 q 7 9 PCT/GB95/0122
- 78 ~-
EXAMPLE 20 Prepar~tion of Compound 41: 3-(t-Butylcarbamoylthio)-1-(3-tri-
fluoromethyl)phenyl -2-pyrrolidinone .
To a solution of thioacetic acid (0.04939) in ether (5mls) at 0C
under nitrogen d' ,' .~ was added triethylamine (0.06569) dropwise. The
bromo-pyrrolidinone (prepared as in Step 1 of Example 10), (0.29) in ether
(3ml) was added. The mixture was allowed to warm to room t- ,-raLu.e and
then heated to reflux for 4 hours. The mixture was diluted with ether,
washed with 2M HCl(aq) solution (x3), water (x2), brine and dried over
MgS04. The solvent was evaporated and the residue purified by
Luy~ y, eluting with ethyl acetate in hexane (27:73) to give the
thioacetate (0.2119).
~2
The thioacetate, prepared as in Step 1 (0.039) was dissolved in
methanol and cooled to 0C. Ammonia gas was bubbled through the mixture
for 1~ minutes. The mixture was L~dLed to dryness. Chromatography,
eluting with ethyl acetate/hexane (2~:75) gave the thiol compound.
SteD 3
The thiol compound obtained in Step 2 was reacted with
t-Butylisocyanate according to Example 10 to give the thiocarbamate
compound 41 as a colourless solid, m.p. 104-106C. A similar procedure was
used to prepare Compounds 42 (m.p. 119-120C), 75 (m.p. 111-112.5C), 76
(m.p. 83-86C), 127, 309, 314, 319, 320, 323 and 327 of Table 1.
EXAMPLF 21 Preparation of Compound 61: 3-(t-Butyl~,A~I, ,l-N-methyl)-
amino-1-~3-trifluoromethyl)phenyl-2-pyrrolidinone.
TD a solution of Compound 10 (0.409) prepared as in Example 10 in
dioxane/water (1:1,6ml) was added triethylamine (0.32ml) and then Boc-on.
The reaction was left at room t ,_.dLu~e overnight and then partitioned
between ethyl acetate/water. The aqueous layer was re-extracted with ethyl
acetate and the organics washed with water (x2), O.~N NdOH (aq), wdter and
brine and then dried (Na2S04). Evaporation and ci~(~ tOy~a,u~ eluting
.
WO 9'.5133719 2 1 9 0 9 7 9 PCT/GB95/012~.1
- 79
with ethyl acetate/hexane (30:70) to give the carbamate, Compound 61, as a
colour1ess solid (0.515g), m.p. 135-136C.
A similar procedure was used to prepare Compounds 58 (m.p. 106-107C),
59 (m.p. 136-137C), 69 (m.p. 147-148C) and 73 (m.p. 139-140C) of Table
1.
EXAMPLE 2Z Prepar~tion of Compound 77: 3-(t-Butylu~ca~' ~loxy)-1-(3-tri-
f l . ~ ' Yy) phenyl -2-pyrrol i di none
t-Butyloxycarbonyl anhydride (Boc-anhydride) (0.48ml) was added to a
solution of the hydroxy pyrrolidinone (prepared as in step 1 or la of
Example 9 (0.50g) in dichloromethane (lOml), followed by DMAP (0.0249).
After lO minutes, effervescence started. The solvent was evaporated to
half volume after 1~ hours and the residue purified by chromatography,
eluting with 20% ethyl acetate/hexane to give the carbamate 77 as a
colourless solid (0.62g), m.p. 80-92C. This procedure was also used for
the preparation of Compound 81 of Table I (m.p. 105-108C).
EXAMPLE 23 Preparation of Compound g9: 3-(t-autylcarb~moyl-N-ethoxy-
carbonylmethyl~mino)-1-(3-trifluoromethyl)phenyl-2-pyrrolidinone.
Ste~ l Preparation of 3-(N-ethoxycarbonylmethyl)amino-1-(3-trifluoro-
methyl)phenyl-2-pyrrolidinone.
Ethylbromoacetate (0.155ml) was added yia syringe to a solution of
3-amino-1-(3-trifluoromethyl)phenyl-2-pyrrolidinone (prepared by a similar
method to that described in Example 1, Step 4) (0.319) and triethylamine
(0.176ml) in THF (5ml) at room tl, ~.~LI~le. After 16 hours, the mixture
was poured into saturated NaHC03(aq) and extracted with ethyl acetate (x2).
Combined extracts were dried (Na2S04), ~.,, .aL~d and the residue purified
by flash chromatography (eluting with ethyl acetate) to give
3-(N-ethoxycarbonylmethyl)amino-1-(3-trifluoromethyl)phenyl-Z-pyrrolidinone
as a colourless solid (0.3159).
Steo 2 Preparation of 3-(t-Butylcarbamoyl-N-ethoxycarbonyl-methyl)-
amino-1-(3-trifluoromethyl)phenyl-2-pyrrolidinone.
3-(N-Ethoxycarbonylmethyl)amino-1-(3-trifluoromethyl)phenyl-2-pyrrolid
,, . , .... . , .. .. ., . . , .. _ ... . . ........ . . . . . . ..... ........ .... ... .. ....
WO 95/33719 2 1 9 0 9 7 9 PCTIGB95/0122J
- 80 -
inone (0.2159) (prepared as described in Step 1) was dissolved in
dichloromethane (3ml). Triethylamine (0.5ml) and t-Butylisocyanate
(0.25ml) were added and the reaction left at room temperature for 16 hours.
After this time, the solvent was evaporated, the residue purified by flash
chromatography (eluting with ethyl acetate) to give the crude product.
This was recrystallised from ethyl acetate/hexane to give the pure title
compound (0.139) as colourless crystals, m.p. 144-146C.
A similar procedure was used to prepare Compound 119 (m.p. 130.5-132.5C).
EXAMPLE 24 Preparation of Compound 86: 3-(t-Butylcarbamoyl
amino-1-(3-trifluoromethyl)phenyl-2-pyrrolidinone.
SteD l Preparation of 3-(N-methoxy)amino-1-(3-trifluoromethyl)phenyl-2--pyrrol idi none .
Methoxylamine hydrochloride (3.399) and sodium carbonate (4.299) were
stirred together in methanol (20ml) for S minutes. This mixture was then
added to a solution of 3-bromo-1-(3-trifluoromethyl)phenyl-2-pyrrolidinone
(2.59) (prepared as described in Example 10, Step 1) in methanol (30 ml) at
room t~, dLulc. The mixture was heated to reflux overnight. Further
methoxylamine hydrochloride (3.399) was added and heating continued for a
further 24 hours. After cooling, the reaction was poured into water and
extracted with dichloromethane (x3). The comDined extracts were washed
with brine and dried (Na2504). Chromatography (eluting with 40% ethyl
acetate/hexane) gave the product as a pale yellow oil which crystallised.
This proved to be a 4:1 mixture of the title compound and the c~,,c, 'ing
3-hydroxyl-(3-trifluoromethyl)phenyl-2-pyrrolidinone (0.0689) and was used
directly in the next step.
Step 2 Preparation of 3-(t-Butylcarbamoyl ~ "u,~y)amino-l-
-(3-trifluoromethyl)phenyl-2-pyrrolidinone.
A solution of 3-(1~-methoxy)amino-1-(3-trifluoromethyl)phenyl-2-
-pyrrolidinone (0.49) (prepared as described in Step 1), triethylamine
(O.Sml) and t-butylisocyanate (O.Sml) in dichloromethane (lml) was stirred
at room temperature for 20 hours. After this time, further quantities of
wo 9sl3371g 2 ~ 9 0 9 7 9 PCT/GBg~/0l221
- 81 -
triethylamine (0.25ml) and t-butylisocyanate (0.25ml) were added and
stirring continued for 3 hours. The solvent was evaporated and the
product purified by flash chromatography, eluting with 40-50% ethyl acetate
in hexane. The product was obtained as a brown oil which crystallised to
give the title compound (0.275g), m.p. 127.5-128.5C.
A similar procedure was used to prepared Compound 88 (m.p. 102-104C).
EXAMPLE 25 Preparation of Compound 123: 3-(3,3-dimethylbutanoyl-N-formyl)-
amino-1-(3-trifl .~ i' y)phenyl-2-pyrrolidinone.
SteD 1 Preparation of 3-methylsulfonyloxy-1-(3-trifluoromethoxy)phenyl-2-
pyrrol idinone.
3-Hydroxy-1-(3-trifluo,, y)phenyl-2-pyrrolidinone (5.229) (prepared by
a simi~ar procedure to that described in Step 1 or Step la of Example 9)
was dissolved in CH2Cl2 (75ml) and cooled to 5C. Triethylamine (3.48ml)
was added followed by dropwise addition of methane sulfonyl chloride
(1.64ml) over 15 minutes. The reaction was left to stir for 20 minutes at
5C and then allowed to warm to room t ~,~Lu~ and stirred for 2 hours.
After diluting with CH2Cl2, the mixture was washed with water (x2) and
dried over MgS04. Concentration gave 5.63g of the mesylate as a pale brown
sol id.
SteD 2 Preparation of 3-(N-formyl)amino-1-(3-trifluo" .I,ù~)phenyl-2-
pyrrol idinone.
Sodium hydride (0.339 of an 80% oil dispersion, washed with 40-60 petrol)
was suspended in dry DMF (2ml). Formamide (lOml) was added dropwise over
15 min, with ice bath cooling. The mesylate (prepared as in Step 1)
(3.399) was added in DMF (15ml) over 10 min at 20C and the resulting dark
yellow solution heated to 50c for 3 hours. After cooling, the reaction
was added to water and extracted with ethyl acetate (x3). The combined
extracts were washed with water (x3), dried over MgS04, cu,,,e,,~.~L~d and
triturated with ether/petrol to give the N-formyl cûmpound as a pink solid
(O . 979) -
WO 95/33719 2 ~ 9 0 9 7 9 PCT/GB9~/0122.1
- 82 -
SteD 3 Preparation of 3-(3,3-dimethylbutanoyl-N-formyl)amino-1-1(3-tri-fluo,. Ll,u,~y)phenyl-2-pyrrolidinone.
The N-formyl compound (prepared as in Step 2) (0.979) was dissolved in
CH2C12 (20ml). triethylamine (0.51ml) was added, followed by
3,3-dimethylbutanoyl chloride (0.51ml) in CH2C12 (5ml) which was added
dropwise over 5 min. The reaction was stirred at room t~ ~,aLu,~ for 17
hours, whereupon further triethylamine (0.25ml) and 3,3-dimethylbutanoyl
chloride (0.25ml) were added. After a further 3 hours, the reaction was
diluted with CH2C12 and washed with water. After drying over MgS04, the
solvent was evaporated to give the crude product as an orange oil.
Chromatography, eluting with ether gave a pale yellow product which was
triturated with ether/petrol to give the title compound as a white solid
(0.579) -
EXAMPLF 26 Preparation of Compound 94: 3-(t-ButylthiocArb~moyl-N-methyl)-
llmino-1-(3-difl .~ ,' y)phenyl-2-pyrrolidinone.
3-(~-methyl)amino-1-(3-difluu,, Ll,uxy)phenyl-2-pyrrolidinone (0.209)
(prepared in a similar procedure to that described in Step 2 of Example 9)
was dissolved in CH2C12 (Sml). t-Butyl isothiocyanate (0.13ml) and
triethylamine (0.16ml) were added and the reaction stirred at room
temperature overnight. The solvent was evaporated and the residue purified
by column chromatography, eluting with 60~ ethyl acetate/hexane to give the
thiourea title compound as a colourless solid (0.2539), m.p. 141-143C. A
similar procedure was used to prepared Compounds 95 (m.p. 141-144C), 100
(m.p. 140-14ZC), 107 (m.p. 100-107C), 109 (m.p. 148.5-150C), 112 (m.p.
130-132C), 114 (m.p. 108-109C) and 115 (m.p. 14Z-143C).
21 90979
WO 95/33719 PCT/GB9~10122
- B3 -
EXAMPL Z7 Preparation of Compound 93: 3-(t-~utylcarbamoyl-N-allyl)-
amino-1-(3-trifl , . :' y)phenyl-2-pyrrolidinone.
SteD 1 Preparation of 3-(N-allyl)amino-1-(3-trifl , ~,v,~y)-
phenyl -2-pyrrol idinone.
3-Bromo-1-(3-trifluoromethoxy)phenyl-2-pyrrol jdinone (0.809) (prepared by a
method similar to that described in Example 10, Step 1) was dissolved in
THF. To this solution was added allylamine (0.709) and the mixture stirred
at room tl, ,~Lu~e for 4 hours and then left to stand overnight. The
reaction was diluted with ethyl acetate, washed with water (x3), brine and
dried over MgS04. Filtration and evaporation of the solvent gave a crude
product that was purified by column chromatography (eluting with 90-100%
ethyl acetate in hexane) to give the pure N-allyl compound (0.689).
SteD 2 Preparation of 3-(t-Butylcarbamoyl-N-allyl)amjno-1~(3-trifluoro-methoxy)phenyl -2-pyrrol idinone.
The N-allyl derivative (0.239) (prepared as descr;bed in Step 1) was
dissolved in CH2Cl2 (3ml) and treated with triethylamine (0.16ml) followed
by t-butylisocyanate (O.Z2ml). After 48 hours at room t~ u,~, the
reaction was diluted with CH2Cl2, washed with 2N HCl (aq) (x2), brine and
dried over MgS04. The mixture was filtered and - Ll~ed and the
residue purified by flash chromatography, eluting with 40g6 ethyl acetate in
hexane. The urea title compound was obtained as colourless solid (0.2939),
m.p. 107-108C.
A similar procedure was used to prepare Compounds 19 (m.p. 112-114C), 38
(m.p. 157-158C), 64 (108.5-110C), 65 (m.p. 146-148C), 78 (m.p. 110-111.5
C), 8Z (m.p. 136.5-138.5C), 83 (gum), 84 (m.p. 111-112C), 85
(m.p.137-138C), 87, 90 (m.p. 95-96.5C), 91 (m.p. 136.5-137.5C), 92
(solid gum), 93 (m.p. 107-108C) 101 (m.p. 111.5-112.5C), 105 (m.p.
137-138.5C), 106 (m.p. 84-87C), 108 (m.p. 107-109C), 111, 116 (m.p.
172-173.5C), 120 (m.p. 105-107.5C), 121 (m.p. 117-118.5C), 122 (m.p.
106-107.5C), 241, 242, 254, 312, 316, 317 and 318 of Table I.
WO 95/33719 2 1 9 0 9 7 9 PCT/GB95/0122.1
- 84 -
EXAMPLE 28 Prep~ration of Compound 117: 3-~t-~utylcarbamoyl-N-~cetoxy-
ethyl)amino-1-(3-trifluoromethyl)phenyl -2-pyrrolidinone
Compound 82 (0.259) (prepared by a similar method to that described in
Example 27) was dissolved in CH2Cl2 (Sml). To this was added triethylamine
(0.09lml) followed by acetyl chloride (0.046ml). The reaction was stirred
at room temperature for 45 minutes, diluted with CH2Cl2 and washed with 2N
HCl (aq) (x2), brine and dried over MgS04. Filtration and evaporation gave
the crude product which was purified by column chromatography, eluting with
ethyl acetate in hexane (1:1). The acetoxy compound was obtained as a
colourless solid, m.p. 129-131C.
EXAMPLE 2q Prepar~tion of Compound 128:
Compound 99 (prepared as described in Example 23) (0.1109) was dissolved in
THF (Sml) and sodium hydride (30mg of a 60% oil dispersion) added. The
reaction effervesced and turned yellow, then pinkish orange. After 4
hours, the mixture was poured into water and extracted with ethyl acetate
(x3). The combined extracts were dried (MgS04) and eYaporated.
Purification by flash chromatography (eluting with ethyl acetate) gave the
cyclic compound 128 as a pale yellow gum (0.0769).
EXAMPLF 30 Preparation of Compound 129
To an ice-cooled solution of Compound 55 (prepared by a similar procedure
to that described in Example 1) (0.1009) in toluene (Sml) was added oxalyl
chloride (0.027ml). The mixture was removed from the ice-bath and stirred
at room tr, :a~ure for ~ hour and then 80C for 1 hour. The solvent was
evaporated to give the cyclic compound 129.
A similar procedure was also used to prepare Compound 133.
EXAMPLE 31 Preparation of Compound 130
Compound 101 (prepared by a similar procedure so that described in Example
27) (0.1009) was dissolved in THF (2ml) and 2N HCl (2ml) added. The
reaction was stirred at room t-, ~r~LUle oYernight. The mixture was poured
WO 95133719 2 ~ 9 0 9 7 9 PCTIGB95/0122.1
- 85 -
into ethyl acetate. The organic phase was separated, dried (Mg504) and
evaporated to give Compound 130 as a colourless solid.
EXAMPI F 32 Prep~ration of Compound 131
Compound 28 (prepared by a similar method to that described in Example 1)
(0.1009) was dissolved in toluene (lOcm3). Glyoxal. 3H O (0.0339) and PTSA
(catalytic amount) were added and the mixture heated under Dean and Stark
conditions for 4 hours. Further glyoxal. 3H20 (0.0339) was added and the
mixture heated under reflux for a further 4 hours. After evaporation of
the solvent, the residue was purified by chromatography, eluting with ethyl
acetate in hexane (1:2) to give Compound 131 as a colourless solid
(O .0439) -
EXAMP~F 33 Preparation of Compound 132
Compound 28 ~prepared by a similar method so that described in Example 1)(0.1159) was dissolved in toluene (lOml). Paraformaldehyde (0.0239) and
PTSA (catalytic amount) were added and the mixture heated under Dean and
Stark conditions for 8 hours. Additional paraformaldehyde (0.0239) was
added at 2 hour intervals over this time. The mixture was allowed to cool
and the solvent evaporated. The residue was purified by .~". Luy~ ,'y
(eluting with ethyl acetate/hexane 1:2) to give Compound 132 (0.0189).
A similar procedure was also used to prepare Compound 134.
EXAMPI.F 34 Prep~r~tion of Compound 135: 5-t-ButylcArb~moyloxy-3(3-
trifluoromethylphenyl)oxazolidin-4-one
Step 1 Preparation of benzyl dibenzyloxyacetate
A solution of dichloroacetic acid (12.8gg) in benzyl alcohol (50ml)
was added to a solution of sodium benzyloxide, from sodium hydride (13.539,
55% dispersion in mineral oil) in benzyl alcohol (lSOml). The resultant
mixture was heated at 190C for four hours, then the solvent distilled off
.... . . . _ .. .. . . . .. . .
WO 95/33719 ? 1 9 o q 7 9 PCT/GB9S/0122J
- 86 -
under reduced pressure. The residue was triturated with ether, the solid
removed by filtration and distributed between hydrochloric acid (2N) and
ether. The extracts were dried over magnesium sulphate and evaporated
under reduced pressure. The residue was chrYmatographed on silica, using
dichloromethane as eluant, to give benzyl dibenzyloxyacetate (12.509) as a
colourless oil. None of the expected cu",, 'ing acid was eluted with
more polar solvents.
NMR (CDC~3): ~ 4.7(4H,dd), 5.1(1H,s), 5.2(2H,s), 7.3(15H,m). MS: M~ 362.
NB When the residue was triturated with ether, it appears that sûme
of the ester product may have been lost; the work-up procedure should be
modified in view of ester, rather than acid, being produced.
SteD Z Preparation of dibenzyloxyacetic acid
Water (2ûml) and potassium carbonate (10.64g) were added to a solution
of benzyl dibenzyloxyacetate (11.15g), from Step 1 above, in
teLr~llydluful (80ml) and the mixture heated under reflux for twenty-four
hours. It was allowed to cool, poured into water, extracted with ether,
acidified with cu,.~ellLl~Led hydrochloric acid and again extracted with
ether. The extract from acidic solution was washed with brine, dried over
magnesium sulphate and evaporated under reduced pressure to give the title
compound (8.129), used crude in Step 3.
NMR (CDCl3): ~ 4.7(4H,m), 5.1(1H,bs), 7.3(10H,m), 9.2(1H,bs).
SteD ~ Preparation of 2,2-dibenzyloxy-N(3-trifluoromethylphenyl)acetamide
A stirred solution of dibenzyloxyacetic acid (4.09), from Step 2, in
dichloromethane (40ml) was cooled to 0C and treated dropwise with,
successively, N,N-dimethylformamide (lOOmg) and oxalyl chloride (2.0g).
After thirty minutes, pyridine (3.52g), 3-trifluoromethylaniline (2.649)
and 4-dimethylaminopyridine (lOOmg) were added. The mixture was stirred at
0C for a further thirty minutes, then allowed to warm to room i, .,,Lu,~.
After three hours, it was poured into water, extracted with ethyl acetate
and the extracts washed successively with dilute hydrochloric acid, water,
aqueous sodium bicarbonate sYlution, and brine. After drying over
magnesium sulphate, the extracts were evaporated under reduced pressure to
WO 95/33719 2 1 9 0 9 7 9 PCT/GBgS/0l22~
- 87 -
give the title compound (5.629) as an orange gum, sufficiently pure to be
used in Step 4.
NMR (CDC13): ~ 4.7(4H,dd), 5.1(1H,s), 7.3(12H,m), 7.8(1H,dd), 7.85(1H,s),
8.5(1H,bs) .
SteD 4 Preparation of 2,2-dibenzyloxy-N-benzyloxymethyl-N(3-tri-
fluoromethylphenyl)acetamide
2,2-Dibenzyloxy-N(3-trifluoromethylphenyl)acetamide, (4.759), from
Step 3, benzyl chloromethylether (1.799) and tetrabutylammonium iodide
(lOOmg) were added successively to a vigorously stirred mixture of aqueous
sodium hydroxide solution (lOOml, 50%) and dichloromethane (lOOml). Af$er
stirring for eighteen hours, the mixture was extracted several times with
dichloromethane and the extracts washed with brine. After drying over
magnesium sulphate, the extracts were evaporated under reduced pressure.
The residue was chromatographed on silica, using hexane-ethyl acetate (4:1)
as eluant, to give the title compound (2.879).
NMR (CDCl3): ~ 4.6(6H,m), 4.9(1H,bs), 5.15(2H,bs), 7.3(18H,m), 7.55(1H,dd).
SteD S Preparation of S-hydroxy-3(3-trifluoromethylphenyl)ox2zolidin-4-one
A mixture of 2,2-dibenzyloxy-N-benzyloxymethyl-N(3-trifluoromethyl-
phenyl)acetamide (0.279), prepared as described in Step 4, 10% palladium on
carbon (50mg), trifluoroacetic acid (lml) and dichloromethane (50ml) was
stirred under an d' r~~ ~ of hydrogen for five hours. It was filtered
through Hyflo Supercel TM, evaporated under reduced pressure and
chromatographed on silica, using dichloromethane-ethanol (49:1) as eluant,
to give the title compound (0.079) as a waxy solid, m.p. 75-76C.
NMR (CDC13): ~ 5.35(1H,bs), 5.45(1H,d), 5.7(2H,m), 7.5(2H,m), 7.65(1H,d),
7.7(1Hj,s) .
SteD 6 Preparation of 5-t-Butylcarbamoyloxy-3(3-trifluoro-
methylphenyl )oxazol idin-4-one
A stirred solution of 5-hydroxy-3(3-trifluoromethylphenyl)-
oxazolidin-4-one, prepared as described in Step 5, can be converted into
the Title compound, by treatment with t-butyl isocyanate in the presence of
triethylamine as described in Step 5 of Example 1.
WO 95/33719 2 t 9 ~ 9 7 9 PCT/GB951012~ 1 --
- 88 -
EXAMPLE 35 Prep~r~tion of Compound 136: 5-t-Butylc~rb~moyloxy-3(5-
trifluoromethyl-1,3,4-thi~di~zol-2-y1)thiazolidin-4-one
S~L L Preparation of 3(5-trifluoromethyl-1,3,4-thiadiazol-2-yl)-
th i azol i d i n -4-one
A stirred so1ution of 2-amino-5-trifluoromethyl-1,3,4-thiadiazole (S.Og) in
toluene (50ml) was treated successively with thioglycolic acid (2.779), 37%
aqueous formaldehyde solution (2.45ml) and p-toluenesulphonic acid
(0.0259). The reaction mixture was heated under reflux, water being
collected in a Dean and Stark apparatus. After four hours it was cooled,
washed with saturated aqueous sodium bicarbonate solution and brine, dried
over magnesium sulphate and evaporated under reduced pressure to give the
title compound (4.359, m.p. 99-100C). NMR (CDCl3): ~ 3.9(2H,s);
5.25(2H,s). MS: M 255.
Ste~ 2 Preparation of 5-chloro-3(5-trifluoromethyl-1,3,4-thiadiazol-
-2-yl ) th i azol i d i n-4-one
Sulphuryl chloride (0.27ml) was added dropwise to a stirred solution of3(5-trifluoromethyl-1,3,4-thiadiazol-2-yl)thiazolidin-4-one (0.59),
prepared as described in Step 1 above, in dichloromethane (ZOml), cooled in
an ice-salt bath. The mixture was stirred for a further two hours, then
allowed to warm to room t~ Lurc over one hour. It was then evaporated
under reduced pressure and the residue chromatographed on silica, using
hexane-ethyl acetate (3:1) as eluant, to give the title compound (0.349) as
a white solid. NMR (CDCl3): ~ 5.4(2H,s); S.9(1H,s).
Ste~ 3 Preparation of 5-hydroxy-3(5-trifluoromethyl-1,3,4-thiadiazol-
-2-yl ) thi azol i din-4-one
A solution of 5-chloro-3(5-trifluoromethyl-1,3,4-thiadiazol-Z-yl)-
thiazolidin-4-one (1.725), prepared as described in Step 2 above, in
tetl-al,y-l,uru~ (ZOml) was added dropwise to a vigorously stirred aqueous
WO 95/33719 ; 2 ~ 9 0 9 7 9 PCT/GB9510122~
- 89 -
solution of potassium dihydrogen phosphate buffer (20ml, to retain pH 4.5).
The reaction mixture was stirred for twenty hours at room t ,.,~ u~e,
allowed to stand overnight and diluted with water. The white precipitate
was filtered off and dried in vacuo to give the title compound (1.319, m.p.
176-177C). NMR (DMS0-d6): ~ 5.15 (2H,dd); 5.9(1H,d); 7.4(1H,d). M5: M
271 .
SteD 4 Preparation of 5-t-butylcarbamoyloxy-3(5-trifluoromethyl-1,3,4-
thiadiazol-2-yl)thiazolidin-4-one
A stirred solution of 5-hydroxy-3(5-trifluoromethyl-1,3,4-thiadiazol-
-2-yl)thiazolidin-4-one (0.919), from Step 3, in dichloromethane (25ml) was
cooled in ice and treated successively with t-butyl isocyanate (0.379),
dropwise, and triethylamine (0.379). The mixture was allowed to warm,
stirred for eighteen hours at room t~ Lule, then evaporated under
reduced pressure. The residue was dissolved in ethyl acetate, washed with
water and brine, dried over magnesium sulphate and evaporated under reduced
pressure. Chromatography on silica, using hexane-diethyl ether (3:1) as
eluant, gave the title compound (0.949, m.p. 151-152C). NMR (CDCl3):
1.3(gH,s); 4.85(1H,bs); 5.3(2H,m); 6.3(1H,s). MS: MH 371.
EXAMPLE 36 Prepar~tion of Compound 137: S-t-Butylcarbamoyloxy-3(5-methyl-
1,3,4-thiddi~zol-2-yl)thi~zolidin-4-one
SteD 1 Preparation of 3(5-methyl-1,3,4-thiadiazol-2-yl)thiazolidin-4-one
By a procedure similar to that described in Step I of Example 35, but using
2-amino-5-methyl-1,3,4-thiadiazole (5.09), thioglycolic acid (4.09), 37~f
aqueous formaldehyde solution (3.52ml), p-toluenesulphonic acid (0.0259)
and toluene (60ml). The toluene layer was decanted and evaporated under
reduced pressure to give the title compound (6.049, m.p. 139-140C). NMR
(CDCl3): ~ 2.7(3H,s); 3.8(2H,s); 5.15(2H,s). MS: M 201.
SteD 2 Preparation of 5-hydroxy-3(5-methyl-1,3,4-thiadiazol-2-yl)-
thiazolidin-4-one
N-chlorosuccinimide (2.849) was added portionwise to a stirred solution of
WO 95/33719 2 1 9 0 9 7 9 PCT/GB9~/0122J
- 90 -
3(5-methyl-1,3,4-thiadiazo1-Z-yl)thiazo1idin-4-one (4.279), prepdred as
described in Step 1 above, in dichloromethane (30ml). After twenty hours,
the solvent was removed under reduced pressure. The residue was converted
into the title compound by treatment with aqueous potassium dihydrogen
phosphate solution in tetrahydrofuran by a procedure similar to that
described in Example 35, Step 3. The reaction mixture was diluted with
water and extracted with ethyl acetate. The extracts were washed, dried
over magnesium sulphate, evaporated under reduced pressure and
chromatographed on silica, using dichloromethane-ethanol (49:1) as eluant,
to give the title compound (1.479, m.p. 154-156C). NMR (DMS0-d6):
2.6(3H,s); 5.0(ZH,m); 5.75(1H,bd); 7.25(1H,bd). MS: M 217.
Step 3 Preparation of 5-t-butylcarbamoyloxy-3(5-methyl-1,3,4-
thiadiazol-2-yl)thiazolidin-4-one
The title compound was prepared by a procedure similar to that described in
Example 35, but using 5-hydroxy-3(5-methyl-1,3,4-thiadiazol-2-yl)
thiazolidin-4-one (0.59), from Step Z, t-butyl isocyanate (0.259),
triethylamine (0.269) and dichloromethane (lSml). Chl~ ioyld~JI.y on
silica, using dichloromethane-ethanol (49:1) as eluant, gave the title
compound (0.329, m.p. 160-161C). NMR (CDCl3): ~ 1.3(9H,s); 2.75(3H,s);
4.85(1H,bs); 5.2(2H,m); 6.3(1H,s). MS: MH 317.
EXAMPLE 37 Prepar~tion of Compound 138: 5-t-Butylc~rb~moyloxy-3(6-
trifluoromethylpyridin-Z-y1)thi~zolidin-4-one
Ste~ I Preparation of 3(6-trifluoromethylpyridin-2-yl)thiazolidin-4-one
By a procedure similar to that described in Step 1 of Example 35, but using
Z-amino-6-trifluoromethylpyridine (5.09), thioglycolic acid (3.19), 37%
aqueous formaldehyde solution (Z.7ml), p-toluenesulphonic acid (O.OZ5g) and
toluene (50ml). The crude product (5.769, m.p. 87-88C) was sufficiently
pure for use in Step Z. NMR (CDC13): ~ 3.8(2H,s); 5.15(2H,s); 7.5 (lH,d);
7.9(1H,t); 8.55(1H,d). MS: M 24~3.
-
WO 95/33719 "2 1 9 0 9 7 9 PCT/GB95/0122.1
- 91 -
SteD 2 Preparation of 5-chloro-3(6-trifluoromethylpyridin-2-yl)-
thi azol idin-4-one
A stirred solution of 3(6-trifluoromethylpyridin-2-yl)thiazolidin-4-one(4.759), prepared as described in Step 1 above, in dichloromethane (lOOml)
was cooled to 0C and treated dropwise with sulphuryl chloride (1.299).
After one hour, the mixture was allowed to warm to room t, ,~Lul~ and
stirred for a further three hours. It was then again cooled to 0C and
treated dropwise with more sulphuryl chloride (1.299). After thirty
minutes, the mixture was evaporated under reduced pressure and the residue
chromatographed on silica, using hexane-ethyl acetate (3:1) as eluant, to
give the title compound (3.039) used directly in the following step. NMR
(CDCl3): ~ 5.3(2H,m); 5.8(1H,s); 7.5(1H,d); 7.95(1H,t); 8.6(1H,d).
Step 3 Preparation of 5-hydroxy-3(6-trifluoromethylpyridin-2-yl)-
thiazolidin-4-one
5-Chloro-3(6-trifluoromethylpyridin-2-yl)thiazolidin-4-one (3.039),
prepared as described in Step 2 above, was hydrolysed using aqueous
potassium dihydrogen phosphate solution in t~:LI~ yllOrur~... by a procedure
similar to that described in Example 36, Step 2. The crude product (2.589,
m.p. 120C) was sufficiently pure for use in Step 4 and Example 38. NMR
(CDCl3): ~ 3.5(1H, very broad), 5.2(2H,m); 5.75(1H,bs); 7.5(1H,d);
7.95(1H,t); 8.6(1H,d). MS: MH 265.
SteD 4 Preparation of 5-t-butylcarbamoyloxy-3(6-trifluoromethylpyridin-2-yl) thiazolidin-4-one
The title compound was prepared by a procedure similar to that described in
Example 35, but using 5-hydroxy-3(6-trifluoromethylpyridin-2-yl)
thiazolidin-4-one (1.09), from Step 3, t-butyl isocyanate (0.419),
triethylamine (0.429) and dichloromethane (30ml). ChY, ~Uyl~pil~ on
silica, using hexane-ethyl acetate (3:1) as eluant, gave the title compound
(1.029, m.p. 118C). NMR (CDCl3): ~ 1.3(9H,s); 4.8(1H,bs); 5.2(2H,s);
6.3(1H,s); 7.5(1H,d); 7.95(1H,t); 8.65(1H,d). MS: MH 364.
WO 95/33719 ' P 9 7 9 PCT/GB95/0122.1
- 92
EXAMPLF 38 Prepar~tion of Compound 139: 5-N(1,1-dimethylprop-2-ynyl)-
carbamoyloxy-3(6-trifluoromethylpyridin-2-yl)thi~zolidin-4-one
Step I Preparation of 1,1-dimethylprop-2-ynyl isocyanate
A stirred solution of 1-amino-1,1-dimethylpropyne (14.30g) in toluene
(25ml) was cooled to ODC and solutions of phosgene in toluene (1.93M,
88.3ml) and sodium hydroxide (13.709) in water (SOml) were added
simultaneously over twenty minutes. The resultant suspension was stirred
for a further five minutes, then filtered through phase-separating paper.
The filtrate, a toluene solution containing the title compound (2240cm~1),
was used directly in subsequent reactions.
Step 2 Preparation of 5-N(1,1-dimethylprop-2-ynyl)carbamoyloxy-
3(6-trifluoromethylpyridin-2-yl)thiazolidin-4-one
The title compound was prepared by a procedure similar to that described in
Example 35, but using 5-hydroxy-3~6-trifluoromethylpyridin-2-yl)
thiazolidin-4-one (1.389), prepared as described in Example 37, Steps 1-3,
1,1-dimethylprop-2-ynyl isocyanate (14.38ml, solution in toluene), from
Step 1, triethylamine (0.589) and dichloromethane (30ml). Chromatography
on silica gel, using hexane-ethyl acetate (3:1) as eluant, gave the title
compound (1.399, m.p. 85-86C). NMR (CDC13): ~ 1.6(6H,s); 2.35(1H,s);
5.15(1H,bs); 5.2(2H,s); 6.3(1H,s); 7.5(1H,d); 7.95(1H,t); 8.6(1H,d). MS:
MH 374.
EXAMPLE 39 Prep~r~tion of Compound 140: 5-N~1,1-dimethylprop-2-eayl)
~rbamoyloxy-3(6-trifluoromethylpyridin-2-yl)thi~zolidin-4-one
A solution of S-N(1,1-dimethylprop-2-ynyl)carbamoyloxy-3(6-trifluoro-
methylpyridin-2-yl)thiazolidin-4-one (O.Sg), prepared as described in
Example 38, in dichloromethane (lOml) was hydrogenated over 5% palladium on
carbon catalyst (0.059). Af~.er nine hours, the mixture was filtered and
the filtrate evaporated under reduced pressure. The residue was
WO 95133719 2 1 9 0 9 7 9 PCTIGB9510122.1
- 93
chromatographed on silica, using hexane-ethyl acetate (3:1) as eluant, to
give the title compound (0.429) as a colourless gum. NMR (CDC13): ~
1.4(6H,s); 4.95(1H,bs); 5.1(2H,m); 5.2(2H,s); S.95(1H,dd); 6.3(1H,s);
7.5(1H,d); 7.95(1H,t); 8.6(1H,d). MS: MH 376.
EXAMPLE 40 Preparation of Compound 141: 3(4,6-Bis-trifluoromethylpyridin-
2-yl) -5-t-bublcarbamoyloxy-thiazolidin-4-one
Step I Preparation of 3(4,6-Bis-trifluoromethylpyridine-2-yl)
th i azo 1 i d i n -4-one
The title compound was prepared by a procedure similar to that described in
Step 1 of Example 35, but using 2-amino-4,6-bis-trifluoromethylpyridine
(S.Og), thioglycolic acid (2.09), 37% aqueous formaldehyde solution
(1.8ml), p-toluenesulphonic acid (0.0259) and toluene (SOml). The crude
product (4.329), isolated as a viscous orange oil which solidified on
standing, was sufficiently pure for use in Step 2. NMR (CDC13):
3.8(2H,s); 5.15(2H,s); 7.65(1H,s); 8.9(1H,s). MS: M 316.
SteD 2 Preparation of 3(4,6-bis-trifluoromethylpyridin-2-yl)
-5-hydroxythiazol idin-4-one
The title compound was prepared by a procedure similar to that described in
Example 37, Steps 2 and 3, but using 3(4,6-bis-trifluoromethylpyridin-2-yl)
thiazolidin-4-one (3.539), prepared as described in Step 1 aboYe, sulphuryl
chloride (0.46ml and 0.4ml) and dichloromethane (30ml). The reaction
mixture was evaporated under reduced pressure to give a mixture (3.93g) of
the desired S-chloro derivative, the S,S-dichloro analogue and hydrolysis
products. NMR (CDC13): ~ for the 5-chloro compound only: ~ 5.35(2H,m);
5.8(1H,s); 7.7(1H,s); 8.9(1H,s). A solution of the mixture (3.939) in
tetrahydrofuran was hydrolysed in a manner similar to that described in
Example 36, Step 2. The crude product was chromatographed on silica, using
hexane-ethyl acetate (3:1) as eluant, to give the title compound (0.39g,
m.p. 99-101C). NMR (CDC13): ~ 3.5(1H,d); 5.2(2H,m); 5.75(1H,d);
7.7(1H,s); 9.0(1H,s).
. _
WO 95/33719 .2 1 9 0 9 7 9 PCTIGB95/0122~
- 94 -
Step 3 Preparation of 3(4,b-Bis-trifluoromethylpyridjn-Z-yl)-5-t-butyl-carbamoyloxy-thiazolidin-4-one
The title compound was prepared by a procedure similar to that described in
Example 35, but using 3(4,6-bis-trifluoromethylpyridin-2-yl)-5-
hydroxythiazolidin-4-one (0.369), from Step 2, t-butyl isocyanate (0.129),
triethylamine (0.12g) and dichloromethane (20ml). Chromatography on silica
gel, using hexane-ethyl acetate (4:1) as eluant, gave the title compound
(0.309) as a pale yellow gum. NMR (CDC13): ~ 1.3~ (9H,s); 4.8(1H,bs);
5.2(2H,s); 6.3(1H,s); 7.7(1H,s); 9.0(1H,s). MS: MH 432.
EXAMPLE 41 Preparation of Compound 142: 5-t-Butylc~rbamoyloxy-3(5-
trifluoromethylpyridin-3-yl)thi~olidin-4-one
SteD 1 Preparation of 3(5-trifluoromethylpyridin-3-yl)thiazolidin-4-one.
The title compound was prepared by a procedure similar to that described in
Step 1 of Example 35, but using 3-amino-5-trifluoromethylpyridine (5.09),
thioglycolic acid (3.19), 37% aqueous formaldehyde solution (2.7ml),
p-toluenesulphonic acid (0.0259) and toluene (50ml). The toluene layer was
decanted, washed with sodium bicarbonate solution then brine, dried over
magnesium sulphate and evaporated under reduced pressure to give the title
compound (2.769, m.p. 82-84C). NMR (CDCl3): ~ 3.75(2H,s); 4.9(2H,s);
8.3(1H,t); 8.75(1H,bs); 8.9(1H,d). MS: M 248.
SteD 2 Preparation of 5-hydroxy-3(5-trifluoromethylpyridin-3-yl)
thiazol idin-4-one.
The title compound was prepared by a procedure similar to that described in
Example 35, but using 3(5-trifluoromethylpyridin-3-yl)thiazolidin-4-one
(2.259), prepared as described in Step 1 above, in dichloromethane (30ml)
and adding sulphuryl chloride (0.73ml) dropwise at 0C. lmmediate
precipitation occurred. The mixture was stirred at 5C for thirty minutes
and evaporated under reduced pressure. The residue was hydrolysed ~irectly
~ WO 95/33719 2 1 9 0 9 7 9 pCT/GB95/0~221
- 95 -
using aqueous potassium dihydrogen phosphate solution in tetrahydrofuran by
a procedure similar to that described in Example 36, Step 2. The crude
product was chromatographed on silica, using dichloromethane-ethanol (19:1)
as eluant, to give the title compound (1.169) as a pale yellow gum. NMR
(CDCl3): ~ 4.75(1H,d); 5.1(1H,d); 5.75(1H,d); 8.3(1H,d); 8.75(1H,s);
8.95(1H,d). MS: MH 265.
SteD 3 Preparation of 5-t-butylcarbamoyloxy-3(5-trifluoromethylpyridin
-3-yl)-thiazolidin-4-one
The title compound was prepared by a procedure similar to that described in
Example 35, Step 1 but using 5-hydroxy-3(5-trifluoromethylpyridin-3-yl)
thiazolidin-4-one (0.509) from Step 2, t-butyl isocyanate (0.219),
triethylamine (0.219) and dichloromethane (lOml). Chromatography on
silica, using dichloromethane-ethanol (24:1) as eluant, gave the title
compound (0.549, m.p. 161-163C). NMR (CDC13): ~ 1.35(9H,s); 4.7(1H,d);
4.9(1H,bs); 5.1(1H,dd); 6.2(1H,s); 8.3(1H,m); 8.8(1H,m); 8.95(1H,d). MS:
MH 364.
EXAMPLE 42 Preparation of Compound 143: 5-t-Butylcarbamoyloxy-3(2-
trifluoromethylpyridin-4-yl)thiazolidin-4-one
SteD 1 Preparation of ((Z-trifluoromethylpyridin-4-yl)aminomethylthio)-acet i c ac i d .
The title compound was prepared by a procedure similar to that described in
Example 35, but using 4-amino-2-trifluoromethylpyridine (2.359),
thioglycolic acid (1.339), 37% aqueous formaldehyde solution (1.189),
p-toluenesulphonic acid (0.0259) and toluene (9Oml). After heating for
ninety minutes, the mixture was cooled and the precipitate filtered off.
This was washed with toluene, then hexane, and dried under reduced pressure
to give the title compound as a white solid (3.229), sufficiently pure for
use in Step 2 below. NMR (DMSO-d6/CDCl3): ~ 3.27(2H,s); 4.57(2H,d);
6.82(1H,dd); 7.06(1H,d); 7.68(1H,m); 8.23(1H,d).
WO 95/33719 2 1 9 3 9 7 9 PCT/GB95/0122-1 ~
- 96 -
The filtrate was evdporated under reduced pressure and the residue
chll ~Uylap~.i on silica, using hexane-ethyl acetate (2:1) as eluant, to
give the title compound (0.18g, m.p. 63-65C) of Step 2 below. NMR as
bel ow.
SteD 2 Preparation of 3(2-trifluoromethylpyridin-4 yl)thiazolidin-4-one
The title compound was prepared by a procedure similar to that described in
Example 47, Step 2 below, but using ((2-trifluoromethylpyridin-4-yl)amino-
methylthio)acetic acid (2.919), prepared as described in Step 1 above,
thionyl chloride (1.309) triethylamine (2x 1.119) and dichloromethane
(50ml). The reaction mixture was evaporated under reduced pressure,
treated with water and extracted with ethyl acetate. The extracts were
washed with aqueous sodium bicarbonate solution to remove starting
material, then brine, dried over magnesium sulphate and evaporated under
reduced pressure to give the title compound (1.679, m.p. 62-64C). NMR
(CDC13): ô 3.79 (2H,s); 4.89 (2H,s); 7.75(1H,dd); 7.97(1H,d); 8.71(1H,d).
M5: M 248.
5te~ 3 Preparation of 5-hydroxy-3(2-trifluoromethylpyridin-4-yl)-
thiazolidin-4-one.
The title compound was prepared by a procedure similar to that described in
Example 40, 5tep 2, but using 3(2-trifluoromethylpyridine-4-yl)thiazolidin-
4-one (1.679), sulphuryl chloride (0.83gJ and dichloromethane (30ml). The
reaction was followed by hydrolysis with an aqueous solution of potassium
dihydrogen phosphate in t~l h~,uru~ . The crude product was
Luylu~Jh~l on silica, using ethyl acetate-hexane (1:1) as eluant, to
give the title compound (0.6359) as a yellow oil. NMR (CDC13): ~
3.58(1H,bs); 4.79(1H,d); 5.09(1H,d); 5.71(1H,s); 7.81(1H,dd); 8.02(1H,d);
8.74(1H,d) .
WO gS/33719 2 1 9 0 9 7 9 PCTIGB95/0122.1
- 97 -
SteD 4 Preparation of 5-t-butylcarbamoyloxy-3(2-trjfluoromethyl-
pyridin-4-yl)thiazolidin-4-one
The title compound was prepared by a procedure similar to that
described in Example 35, but using
5-hydroxy-3(2-trifluoromethylpyridin-4-yl) thiazolidin-4-one (0.639), from
Step 3, t-butyl isocyanate (0.479), triethylamine (0.489) and
dichloromethane (30ml). The reaction mixture was allowed to stand for
twenty hours, evaporated under reduced pressure and chromatographed on
silica, using ethyl acetate-hexane (1:3) as eluant, to give the title
compound (0.359, m.p. 124-126C). NMR (CDCl3): ~ 1.34(9H,s); 4.75(1H,d);
4.86(1H,bs); 5.11(1H,d); 6.20(1H,s); 7.81(1H,dd); 8.00(1H,d); 8.75(1H,d).
MS: MH 364.
EXAMPLE 43 Preparation of Compound 144: S-t-Butylcarbamoyloxy-3(4-
trifluoromethylpyridin-2-yl)thiazolidin-4-one
SteD 1 Preparation of 3(4-trifluoromethylpyridin-2-yl)thiazolidin-4-one
The title compound was prepared by a procedure similar to that described in
Example 35, but using 2-amino-4-trifluoromethylpyridine (10.09),
thioglycolic acid (5.709), 37% aqueous formaldehyde solution (4.80ml) and
toluene (lOOml). No p-toluenesulphonic acid catalyst was used. The crude
product was chromatographed on silica, using ethyl acetate-hexane mixtures
as eluant, to give the title compound as a pale yellow oil (2.809). NMR
(CDCl3): ~ 3.81(2H,s); 5.13(2H,s); 7.31(1H,d); 8.52(1H,d); 8.65(1H,s).
SteD 2 Preparation of 5-hydroxy-3(4-trifluoromethylpyridin-2-yl)
thi azol idin-4-one
A stirred solution of 3(4-trifluoromethylpyridin-2-yl)thiazolidin-4-one(2.309), prepared as described in Step l above, in dichloromethane (25ml)
was cooled in an ice-bath and treated with sulphuryl chloride (0.75ml) over
a period of two minutes. The solution was allowed to warm to room
t~ tUIc:, then treated with saturated sodium bicarbonate solution
(50ml). The mixture was stirred for one hour, the organic layer separated,
dried over magnesium sulphate and evaporated under reduced pressure to give
wo 9S/33719 2 ~ 9 0 9 7 9 PCTlCBgS/0l22~ ~
- 9B -
the intermediate 5-chloro derivdtive. This was dissolved in
teL,.,.,~rJ,uru~ (25ml) and treated with saturated sodium bicarbonate
solution (50ml). The mixture was stirred vigorously for four hours,
diluted with water and extracted with ether. The extracts were washed with
brine, dried over magnesium sulphate and evaporated under reduced pressure.
The residue was recrystallised from chloroform-hexane to give the title
compound (2.009, m.p. 109-110C, dec). NMR (CDC13): ~ 4.19(1H,bs); S.O9
(lH,d); 5.22(1H,d); 5.78(1H,s); 7.35(1H,d); 8.52(1H,d); 8.67(1H,s).
Ste~ 3 Preparation of
5-t-butylcarbamoyloxy-3(4-trifluoromethylpyridin-2-yl)- thiazolidin-4-one
The title compound was prepared by a procedure similar to that described in
Example 35, but using 5-hydroxy-3(4-trifluoromethylpyridin-
2-yl)thiazolidin-4-one (0.809), from Step 2, t-butyl isocyanate (0.309),
triethylamine (0.42ml), and dichloromethane (5ml). The crude product was
triturated with ether-hexane to give the title compound (0.909). NMR
(CDC13): ~ 1.33(9H,s); 4.87(1H,bs); 5.12-5.20(2H,m); 6.29(1H,s);
7.36(1H,d); 8.53(1H,d); 8.71(1H,s). MS: MH 363.
EXAMPLF 44 Prep~ration of Compound 145: 5-N(1,1-timethylprop-2-ynyl)-
carb~moyloxy-3t4-trifluoromethylpyridin-2-yl)thi~olidin-4-one
The title compound was prepared by a procedure similar to that described in
Example 35, but using
5-hydroxy-3(4-trifluoromethylpyridin-2-yl)thiazolidin- 4-one (1.109),
prepared as described in Example 43, Steps 1 and 2, triethylamine (0.58ml)
and a solution (7.0ml) of 1,1-dimethylprop-2-ynyl isocyanate in toluene,
prepared as described in Example 38, Step 1. After three days reaction was
incomplete, so a further aliquot (lOml) of the isocyanate solution was
added. The reaction was worked up in the usual way. The crude product was
chromatographed on silica, using ethyl acetate-hexane mixtures as eluant.
Further recrystallisation from carbon tetrachloride-hexane gave the title
compound (0.649, m.p. 119-120C). NMR (CDC13): ~ 1.549 (6H,s); 2.34(1H,s);
WO 95/33719 1 9 9 7 9 PCTIGB95/0122 1
5.10(1H,bs); 5.1Z-5.21(2H,m); 6.32(1H,s); 7.37(1H,dd); 8.55(1H,d);
8.71 (lH,d) .
EXAMPLE 45 Prepar~tion of Compounds 146 ~nd 147: S-N(l,l-dimethylpropyl)
carb~moyloxy-3(4-trifluoromethylpyridin-2-yl) thi~zolidin-4-one (Compound
146) and 5-N(l,l-dimethylprop-2-enylcarb~moyloxy-3(4-trifluoromethyl-
pyridin-2-yl)thiazolidin-4-one (Compound 147)
The title compounds were prepared by a procedure similar to that described
in Example 39, but initially using
5-N(1,1-dimethylprop-2-ynyl)carbamoyloxy- 3(4-trifluoromethylpyridin-2-yl)
thiazolidin-4-one (0.3199), prepared as described in Example 44, and
Lindlar's catalyst (0.0329) in ethyl acetate (5ml). After three hours no
reaction had occurred, so 10~ palladium on carbon catalyst (0.039) was
added. The resultant mixture was hydrogenated for thirty minutes and
worked up in the usual manner. The residue was .1", ~ "~ on silica,
using ethyl acetate-hexane mixtures as eluant, to give firstly title
compound 146 (0.0499). NMR (CDC13): ~ 0.87 (3H,t); 1.25(3H,s); 1.27(3H,s);
1.67(2H,m); 4.78(1H,bs); 5.13-5.20(2H,m); 6.29(1H,s); 7.35(1H,dd);
8.56(1H,d); 8.71(1H,d). The second component, tit1e compound 147 (0.21gg,
m.p. 90-91C) had NMR (CDC13): ~ 1.41(6H,s); 4.98(1H,bs); 5.03-5.19(4H,m);
5.gS(lH,dd); 6.30(1H,s); 7.36(1H,dd); 8.53(1H,d); 8.70(1H,d).
EXAMPLE 46 Preparation of Compound 148: 3-t-Butylc~rbAmoyloxy-1(4-
i ~, . i di n-3-yl ) pyrrol idi n-2-one
Preparation of 5(4-chlorobutanoylamino)-2 L~lVA~Pjl idine
A solution of 5-amino-2 Ll,u~pyl idine (2.09) in ether (20ml) was treated,
dropwise at 0C, successively with 4-chlorobutanoyl chloride (2.779) and
triethylamine (1.7gg). The mixture was allowed to warm to room
temperature, stirred for two hours, poured into water and extracted with
ethyl acetate. The extracts were dried over magnesium sulphate and
evaporated under reduced pressure to give the title compound (3.709, m.p.
gl-g5C) .
WO 95133719 2 1 9 0 9 7 9 PCTIGB95/0122.1
- 100 -
SteD 2 Preparation of 1('1: L~lu~cypyridin-3-yl)pyrrolidin-2-one
5(4-chlor~obutanoylamino-2-methoxypyridine (3.509), prepared as described in
Step 1 above, was added portionwise to a solution of sodium methoxide (from
0.35g sodium) in methanol (30ml), stirred under nitrogen. The reaction
mixture was stirred for two hours, diluted with ether and filtered. The
filtrate was evaporated under reduced pressure and the residue triturated
with ether to give the title compound (2.839, m.p. 64-65C). NMR (CDCl3):
~ 2.20(2H,p); 2.60(2H,t); 3.84(2H,t); 3.94(3H,s); 6.78(1H,d); 8.10(1H,dd);
8.19(1H,d) .
SteP 3 Preparation of 3-hydroxy-1(4-methoxypyridin-3-yl)pyrrolidin-2-one
Lithium bis(trimethylsilyl)amide (5.72ml, lM solution in te~r~hy.lluru,~,,)
was added dropwise to a stirred suspension of 1('1 Ll,o,~y,uyl idin-3-yl)-
pyrrolidin-2-one (0.59), prepared as described in Step 2 above, in
tetrahydrofuran (20ml), under nitrogen at -78C. The reaction mixture was
stirred for thirty minutes at -78C, allowed to warm to 0C, then treated
with a stream of oxygen. After one hour, the mixture was poured into
saturated aqueous sodium sulphite solution and shaken vigorously for five
minutes. It was then extracted with ether and the extracts washed with
aqueous sodium sulphite solution then brine, dried over sodium sulphate and
evaporated under reduced pressure. The residue was chromatographed on
silica, using ethyl acetate as eluant, to give crude product, further
recrystallised from ethyl acetate to give the title compound (0.1559, m.p.
121-123C). NMR (CDCl3): ~ 2.14(1H,m); 2.64(1H,d); 3.12(1H,bs);
3.76(2H,m~; 3.93(3H,s); 4.49(1H,dd); 6.79(1H,d); 8.11(1H,dd); 8.22(1H,d).
SteD 4 Preparatiûn of 3-t-butylcarbamoyloxy-1(~ - Il.ù,~y~yl idin-3-yl)pyrrol idin-2-one
The title compound was prepared by a procedure similar to that described in
Example 35, Step 1, but using
3-hydroxy-1(4-methoxypyridin-3-yl)pyrrolidin-2-one (0.1459), from Step 3,
triethylamine (0.39ml), t-butyl isocyanate (0.32ml) and dichlûromethane
(5ml). After eighteen hûurs reactiûn was incomplete, so further aliquots
WO 95/33719 2 1 9 0 9 7 9 PCT/GB95/0122 t
- 101 -
of triethylamine and isocyanate were added. After a further twenty-four
hours the reaction was worked up in the usual way and the crude product
chromatographed on s i l i ca, us i ng hexane-ethy l acetate ( 10: 3 ) as e l uant, to
give the title compound (0.107g, m.p. 110-111C). NMR (CDCl3): ~
1.34(9H,s); 2.15(1H,m); 2.75(1H,m); 3.80(2H,2d); 3.94(3H,s); 4.90(1H,bs);
5.33(1H,dd); 6.78(1H,d); 8.14(1H,dd); B.24(1H,d). MS: M 307.
EXAMPLE 47 Preparation of Compound 149: 5-t-Butylc~rbamoyloxy-3(4,6-
dimethylpyrimidin-2-yl)thi~zolidin-4-one
Step 1 Preparation of ((4,6-dimethylpyrimidin-2-yl)aminomethylthio)acetic
acid
The title compound was prepared by a procedure similar to that described in
Example 35, but using 2-amino-4,6-dimethylpyrimidine (5.09), thioglycolic
acid (3.749), 37% aqueous formaldehyde solution (3.29ml),
p-toluenesulphonic acid (0.025g) and toluene (70ml). After heating for
three hours, the mixture was allowed to cool. The precipitate was filtered
off and dried under reduced pressure. Soluble in aqueous sodium
bicarbonate solution, it was shown to be the uncyclized title compound
(4.0g, m.p. 164-165C). NMR (DMS0-d6/CDCl3): ~ 2.3(6H,s); 3.35(2H,s);
4.7(2H,d); 6.4(1H,s); 6.85(1H,t).
SteD Z Preparation of 3(4,6-dimethylpyrimidin-2-yl)thiazolidin-4-one
A stirred suspension of ((4,6-dimethylpyrimidin-2-yl)aminomethylthio)
acetic acid (4.049), from Step 1 above, in dichloromethane (30ml) was
cooled to 5~C, treated with triethylamine (1.909) them dropwise with
thionyl chloride (2.239). The mixture was stirred at 5C for two hours,
treated with more triethylamine (1.909), then allowed to warm and stand
overnight at room temperature. It was diluted with dichloromethane, washed
with water and brine, dried over magnesium sulphate and evaporated under
reduced pressure. The residue was chromatographed on silica using
dichloromethane-ethanol (10:1) as eluant, to give the title compound
(2.27g, mp 85-86C). NMR (CDCl3): ~ 2.5(6H,s); 3.8(2H,s); 5.05(2H,s);
6.85(1H,s). MS: MH 210.
WO 9~/33719 ` 2 1 9 0 9 7 9 PCT/GB95/0122~ --
- 102 -
Ste~ 3 Preparation of 3(4,6-dimethylpyrimidin-2-yl)-5-hydroxy
thiazol idin-4-one
The title compound was prepared by a procedure similar to that described in
Example 37 Step 2, but using 3(4,6-dimethylpyrimidin-2-yl)thiazolidin-4-one
(0.5g),from Step 2 above, sulphuryl chloride (O.lOml and O.O9ml) and
dichloromethane (lOml). The crude chloro derivative was dissolved in
tetrahydrofuran and treated with an aqueous solution of potassium
dihydrogen phosphate in a procedure similar to that described in Example
3b, Step 2. The crude product was chr, Lu~a~h.~ on silica, using
dichloromethane-ethanol (19:1) as eluant, to give the title compound
(0.109). NMR (CDC13): ~ 2.5(6H,s); 5.1(2H,m); 5.7(1H,s); 6.9(1H,s). MS:
MH 226.
SteD 4 Preparation of 5-t-butylcarbamoyloxy 3(4,6-dimethylpyrimidin-2-yl)
thiazolidin-4-one
The title compound was prepared by a procedure similar to that described in
Example 35, but using 3(4,6-dimethylpyrimidin-2-yl)-5-hydroxythiazolidin-
4-one (0.109), from Step 3, t-butyl isocyanate (0.0489), triethylamine
(0.059) and dichloromethane (lOml). Chromatography on silica, using
dichloromethane-ethanol (49:1) as eluant, gave the title compound (0.109,
m.p. 153-154C). NMR (CDC13): ~ 1.3(9H,s); 2.5~6H,s); 4.9(1H,bs);
5.1(2H,m); 6.2(1H,s); 6.9(1H,s). MS: MH 325.
EXAMPLF 48 Prepar~tion of Compound 150: 3(5 ~.. Ll,i~zol-Z-yl)-5-t-
butylc~rbamoylaxy-thi~zolidin-4-one
Step I Preparation of 3(5-bromothiazol-2-yl)thiazolidin-4-one
The title compound was prepared by a procedure similar to that described in
Example 43 Step 1, but using Z-amino-5-bromothiazole (5.09), thioglycolic
acid (2.609), 37% aqueous formaldehyde solution (Z.24ml) and toluene
(lOOml). The title compound was obtained as a crystalline solid (0.629).
NMR (CDC13): ~ 3.81(2H,s); 5.08(2H,s); 7.42(1H,s).
-
WO 95133719 2 1 9 0 9 7 9 PCT/GB9510122.~
103 -
.
Step 2 Preparation of 3(5-bromothiazol-2-yl)-5-hydroxythiazolidin-4-one
A stirred solution of 3(5-bromothiazol-2-yl)thiazolidin-4-one (0.629,
prepared as described in Step 1 above) in dichloromethane (lOml) was
treated with sulphuryl chloride (0.19ml). The resultant suspension was
stirred for one hour, giYing a green solution, then evaporated under
reduced pressure. The residue was dissolved in tetrahydrofuran (25ml),
treated with saturated sodium bicarbonate solution (35ml) and the mixture
stirred vigorously for five hours. It was then diluted with water and
extracted with chloroform (Zx50ml). The extracts were washed with water,
dried over magnesium sulphate, and evaporated under reduced pressure. The
residue was chromatographed on silica, using ethyl acetate-hexane mixtures
as eluant, to give the title compound as a pale yellow solid (0.3509).
SteP 3 Preparation of 3(5-Bromothiazol-2-yl)-5-t-butylcarbamoyloxy-
thiazolidin-4-one
The title compound was prepared by a procedure similar to that described in
Example 35, but us;ng 3(5-bromothiazol-2-yl)-5 ~ ."~AyLI,iazolidin-4-one
(0.359), from Step 2, t-butyl isocyanate (0.1239), triethylamine (0.17ml)
and d i ch l oromethane ( 10ml ) . The crude product was recrystal l i sed f romethyl acetate-hexane to give the title compound as white needles (0.2209).
NMR (CDCl3): ~ 1.32(9H,s); 4.83(1H,bs); 5.05-5.13(2H,m); 6.31(1H,s);
7.44(1H,s). MS: M (Br=79) 379.
EXAMPLE 49 Preparation of Compound 151: 5-t-Butylc~rbamoyloxy-3-
(4-chl".,~ Ll~iazol-2-yl)thi~olidin-4-one
Step 1 Preparation of ((4-chl~,u~ uL~,iazol-2-yl)aminomethylthio)aceticacid
The title compound was prepared by a procedure similar to that described in
Example 47, Step 1, but using 2-amino-4-chlorobenzothiazole (9.209),
thioglycolic acid (4.609), 37% aqueous formaldehyde solution (3.9ml) and
toluene (75ml). No p-toluenesulphonic acid catalyst was used. The
.. , . .. .. . . .. .. . _ _ . . . . . . . . .
WO 95133719 2 1 ~ 0 9 7 ~ PCT/GB95~01221
- 104 -
precipitate formed on cooling was filtered off, washed with ethyl acetate
and dried to give the title compound as a white solid (8.009). NMR
(DM50-d6): ~ 3.63 (2H,s); 4.80(2H,s); 7.19(1H,t); 7.46 (lH,d); 7.82(1H,d);
9.10(1H,broad), 12.78(1H,broad).
SteP 2 Preparation of 3(4-chlorobenzothiazol-2-yl)thiazolidin_4-one
A stirred suspension of ((4-chlù,u~c..~uL~liazol-2-yl)aminomethylthio)aCetiC
acid (7.60g),from Step 1, in dichloromethane (70ml) WdS treated with
thionyl chloride (2.1ml). After stirring for thirty minutes, triethylamine
(7.6ml) was added and stirring continued for a further one hour. The
mixture was diluted with water and extracted with dichloromethane. The
extracts were dried over magnesium sulphate, evaporated under reduced
pressure and the residue recrystallised from ethyl acetate to give the
title compound as a crystalline solid (2.60g, m.p. 229C). NMR (CDC13):
3.85(2H,s); 5.28(2H,s); 7.25(1H,t); 7.45(1H,d); 7.71(1H,d).
~tep 3 Preparation of 3(4-chlo,JL~ uL~liazol-2-yl)-s ~ ,u~ythiazolidin-
-4-one
The title compound was prepared by a procedure similar to that described in
Example 48 Step 2, but using 3(4-chlo,l' Ll,iazol-2-yl)thiazolidin-4-one
(3.50g) from Step 2 above, sulphuryl chloride (1.05ml) and dichloromethane
(50ml), followed by t~:Lld~,yd,uru,~,, (50ml) and saturated aqueous sodium
bicarbonate solution (50ml). The crude product was recrystallised from
toluene to give the title compound (0.459, m.p. 220C). NMR (CDCl3):
5.27(1H,d); 5.38(1H,d); 5.77(1H,d); 7.11(1H,d); 7.28(1H,t); 7.49(1H,d);
7.76(1H,d) .
Steo 4 Preparation of 5-t-butylcarbamoyloxy-3(4-chlù~ui~e~uL~liazol-2-yl)
th i azol i din-4-one
The title compound was prepared by a procedure similar to that described in
Example 35 but using
3(4-chlu~u~ uL~iazol-2-yl)-5-hydroxythiazolidin-4-one (û.3ûûg), from 5tep
3, t-butyl isocyanate (û.104g), triethylamine (û~14ml) and dichloromethane
WO 95/33719 2 1 9 0 9 7 9 PCl'/GB9510122.1
- 105 -
(lOml). The crude product was recrysta31ised from carbon
tetrachloride-hexane to give the title compound as a white crystalline
solid (0.19g). NMR (CDCl3): ô 1.34(9H,s); 4.83(1H,bs); 5.31(1H,d);
5.39(1H,d); 6.35(1H,d); 7.28(1H,t); 7.49(1H,d); 7.73(1H,d).
EXAMPI F SO Prepar~tion of Compound 152: 3-t-Butylcarbamoyloxy-1(2-
chlorothien-4-yl)pyrrol itin-Z-one
SteD 1 Preparation of 1(2-chlorothien-4-yl)pyrrolidin-2-one
A stirred mixture of 4-bromo-2-chlorothiophene (15.809), pyrrolidin-2-one
(6.80g) and cuprous oxide (11.40g) was heated to 130C under a nitrogen
d' "~,~,e for eight hours. The mixture was cooled and filtered, washing
through thoroughly with chloroform. The filtrate was evaporated under
reduced pressure and the residue chromatographed on silica, using ethyl
acetate-hexane mixtures as eluant, to give the title compound as a white
solid. 90% pure, it was used in the next step without further
purification. NMR (CDC13): inter alia, ~ 2.10-2.21(2H,m); 2.54(2H,t);
3.76(2H,t); 6.90(1H,d); 7.46(1H,d).
Stel~ 2 Preparation of 1(2-chlorothien-4-yl)-3-l,yJ,u~y~,y,,ulidin-2-one
A stirred solution of 1(2-chlorothien-4-yl)pyrrolidin-2-one (1.85g), from
Step 1 above, in dry teLI.h~dluruldll (ZOml) was cooled to -74C under a
nitrogen di ~ and treated dropwise with a solution of lithium
bis(trimethylsilyl)amide (ll.Oml, lM solution in toluene). The mixture was
stirred for ten minutes, then solid 3-phenyl-N-toluenesulphonyloxaziridine
(2.78g), prepared as described in J. Ora. Chem., (1988) 53, 2087, was added
in one portion. The mixture was stirred at -74C for ten minutes, then
allowed to warm to room t .dLrrle. The reaction was quenched with a
mixture of 2M hydrochloric acid (llml) and brine (SOml), and extracted with
ethyl acetate (2x20ml). The extracts were washed with brine, dried over
magnesium sulphate and eYaporated under reduced pressure. The residue was
chromatographed on silica, using ethyl acetate-hexane mixtures, then
tetrd~,yd,urural~-hexane mixtures, as eluants. The crude product was
triturated with ether to give the title compound as a white crystalline
. .. . . . . . . . ... . . . . . . .. _ .. . . .. . .... _ . . . . . . . _
21 90979
WO 95133719 PC'r/GB9510122 1
_ 106 -
solid (O.Z60g). NMR (CDC13): S 2.09(1H,m); Z.54(1H,m); 3.63(1H,m),
3.74(1H,m); 4.82(1H,d); 6.g5(1H,d); 7.49(1H,m).
Preparation of 3-t-butylcarbamoyloxy-1(2-chlorothien-4-yl)
pyrrol idin-2-one
A stirred suspension of 1(2-chlorothien-4-yl)-3-~1ydru~p~,,ulidin-2-one(0.269), from Step 2, in dichloromethane (lOml) was treated with t-butyl
isocyanate (0.1189) and triethylamine (0.17ml). After three hours, no
reaction had occurred. Tt:Lrdl~ydr~rula" (Sml) was added and stirring
continued for sixteen hours. Further quantities of isocyanate (0.14ml,
0.4ml and 0.2ml) were added immediately, then after further periods of
twenty hours and sixty-eight hours. After a final five hours, the mixture
was evaporated under reduced pressure and the residue .~ lo~" h ' on
silica, using ethyl acetate-hexane mixtures as eluant, to give after
recrystallisation from ethyl acetate-hexane the title compound (0.129) as a
white crystalline solid. NMR (CDC13): S 1.32(9H,s); 2.11(1H,m);
2.71(1H,m); 3.63-3.84(2H,m); 4.78(1H,bs); 5.41(1H,t); 6.99(1H,d);
7.43(1H,d) .
EXAMPLE Sl Preparation of Compound No.153: 5-t-Butylcarbamoyloxy-3
(pyridin-3-yl)thiazolidin-4-one
SterJ 1 Preparation of 3(pyridin-3-yl)thiazolidin-4-one
The compound was prepared by a procedure similar to that described in
Example 36, Steps 1 and 2, but using 3-aminopyridine (5.09), thioglycolic
acid (4.99), 37% aqueous formaldehyde solution (4.35ml), p-toluenesulphonic
acid (0.0259) and toluene (70ml). The toluene layer was decanted and
evaporated under reduced pressure to give the title compound (2.799) as a
pale red solid sufficiently pure for use in 5tep 2. NMR (CDC13): S
3.75(2H,s); 4.85(2H,s); 7.4(1H,m); 8.0(1H,dd); 8.5(1H,dd); 8.7(1H,d).
~ 2 1 909 79
WO 9S/33719 PCT/GB9S/0122 1
-- 107 -
SteD 2 Preparation of 3(pyridin-3-yl)thiazolidjn 4_one S-oxide
A solution of 3(pyridin-3-yl)thiazolidin-4-one (2.799, prepared as
described in Step 1 above) in ethanol (30ml) was added to a stirred
solution of sodium periodate (3.359) in water (30ml). The mixture was
stirred for four hours, allowed to stand overnight at room temperature and
evaporated under reduced pressure. The dry residue was stirred overnight
with ethanol and filtered. The extracts were evaporated under reduced
pressure to give the title compound (2.759) as a pale yellow solid
sufficiently pure to be used in Step 3. NMR (DMSO-d5): ~ 3.6(1H,dd);
4.15(1H,d); 4.85(1H,dd); 5.1(1H,d); 7.45(1H,dd); 7.95(1H,m); 8.4(1H,dd);
8.7(1H,d) .
SteD 3 Preparation of 5-hydroxy-3(pyridin-3-yl)thiazolidin-4-one
A stirred solution of the sulphoxide (2.759, prepared as described in
Step 2 above) in trifluoroacetic acid (30ml) was treated dropwise with
trifluoroacetic anhydride (3.219), whilst maintaining the t, (~Lu.e below
5C. The mixture was stirred for a further thirty minutes at 0-5~C, for
four hours at 20C, allowed to stand overnight, then evaporated under
reduced pressure. The residue was dissolved in dichloromethane, treated
with solid sodium carbonate (portionwise), water (dropwise) and methanol
(lOml) until effervescence ceased. The mixture was dried with magnesium
sulphate, filtered through 'Hyflo-Supercel' TM and evaporated under reduced
pressure. The residue was ch" tGy~ on silica, using
dichloromethane-ethanol (19:1) as eluant, to give the title compound
(1.019, m.p. 150-152C). NMR (CDCl3): ~ 4.7(1H,d); 5.1(1H,dd); 5.6(1H,d);
7.0(1H,d); 7.4(1H,dd); 8.0(1H,m); 8.5(1H,dd); 8.8(1H,d).
Sten 4 5-t-Butylcarbamoyloxy-3(pyridin-3-yl)thiazolidin-4-one
By a procedure similar to that described in Example 35, the alcohol
(0.509, prepared as described in Step 3 above) was treated with t-butyl
isocyanate, triethylamine and dichloromethane to give, after
cllr, Loy., ' ~, Compound 153 (0.529, m.p. 59-61C) . NMR (CDCl3):
1.3(9H,s); 4.7(1H,d); 5.1(1H,dd); 6.2(1H,d); 7.4(1H,dd); 8.0(1H,m);
8.55(1H,dd); 8.7(1H,d). MS: MH 296.
21 qO979
WO 95133719 PCl'IGB95/012~1
- 108 -
EXAMPLF 57 Preparation of Compound 154: S-t-~utylc~rbamoyloxy-
3(pyrazin-2-yl)thiazolidin-4-one
Step I Preparation of 3(pyrazin-2-yl)thiazolidin-4-one
By a procedure similar to that described in Example 51 was obtained,
from 2-aminopyrazine (5.09), the title compound (1.859, m.p. 115-116C).
NMR (CDC13): ~ 3.8(2H,s); 5.1(2H,s); 8.35(2H,m); 9.7(1H,d). MS: M 181.
5t~p 2 Preparation of 3(pyrazin-2-yl)thiazolidin-4-one S-oxide
The thiazolidinone (0.59, prepared as described in Step 1 above) was
oxidised by a procedure similar to that described in Example 51 to give the
co" c,uu~.iing sulphoxide (0.409, m.p. 170-171C) . NMR (CDC13): ~
3.9(2H,m); 5.0(1H,d); 5.3(1H,dd); 8.35(1H,m); 8.45(1H,d); 8.7(1H,d). MS:
M 197.
$tep 3 Preparation of 5-hydroxy-3(pyrazin-2-yl)thiazolidin-4-one
The sulphoxide (1.119, prepared as described in Step 2 above) was
treated with trifluoroacetic anhydride in trifluoroacetic dcid, and the
resulting trifluoroacetate hydrolysed, both following procedures similar to
those described in Example 51. The product (0.209, m.p. 125-lZ7C) was
isolated by chromatography on silica, using dichloromethane-ethanol (19:1)
as eluant. NMR (CDCl ): ~ 5.1(2H,m); 5.75(1H,s); 8.4(ZH,m); 8.7(1H,d).
MS: MH+ 198 3
SteD 4 Preparation of 5-t-butylcarbamoyloxy-3(pyrazin-2-yl)-
th i azol i d i n -4-one
By a procedure similar to that described in Example 51, the alcohol
(0.189, prepared as described in Step 3 above) was converted into Compound
154 (0.159, m.p. 121-12ZC). NMR (CDCl3): ~ 1.4(9H,s); 4.85(1H,bs);
5.1(ZH,m); 6.3(1H,s); 8.35(~H,t); 8.4(1H,d); 9.7(1H,d). MS: MH 297.
21 90979
WO 95133719 PCTIGB9S/0122.J
- 109 -
EXAMPLE 53 Prepdrdtion of Compound 155: 5-t-~utylcarbamoyloxy-
3(~-trifluoromethylpyridin-Z-yl)thiazolidin-4-one
,SteP 1 Prepdration of 3(5-trifluoromethylpyrjdjn-2-yl)thiazolidin-4-one
Treatment of 2-amino-5-trifluoromethylpyridine (S.Og) with
thioglycolic acid (3.19), 37~ aqueous formaldehyde solution (2.7ml),
p-toluenesulphonic acid (0.0259) and toluene (70ml), in a manner similar to
that described in Example 51, gave a mixture (7.139) of the title compound
and ((S-trifluoromethylpyridin-2-yl)aminomethylthio)acetic acid in a ratio
of 1:2. The mixture was dissolved in dichloromethane (SOml), treated with
triethylamine (1.449), cooled to below 5C, and the stirred solution
treated with thionyl chloride (1.749) then, after two hours, with
triethylamine (1.449). The mixture was allowed to stand overnight at room
tl, .dLul~, diluted with dichloromethane and washed with water, then
brine. The organic phase was dried over magnesium sulphate, t~dpu,dL.d
under reduced pressure and .I,r, t~yl ~ ' on silica, using
dichloromethane-ethanol (49:1) as eluant, to give the title compound
(3.339, m.p. 77-78C). NMR (CDC13): ~ 3.8(2H,s); 5.1(2H,s); 7.95(1H,dd);
8.5(1H,d); 8.6(1H,bd). MS: M 248.
SteD 2 Preparation of 3(5-trifluoromethylpyridin-2-yl)thiazolidin-4-oneS-oxide
The thiazolidinone (3.25g, prepared as described in Step 1 above) was
oxidised with sodium periodate by a procedure similar to thdt described in
Example 51 to give the cu,,,.,uù.,~inq sulphoxide (3.389, m.p. 131-133C).
NMR (CDCl3): ~ 3.9(2H,m); 5.0(1H,d); 5.4(1H,dd); 8.0(1H,dd); 8.55(1H,d);
8.65(1H,d). MS: MH 265.
SteQ 3 Preparation of 5-hydroxy-3(5-trifluoromethylpyridin-2-yl)-
thi azol i di n-4-one
The sulphoxide (2.09, prepared as described in Step 2 above) was
treated with trifluoroaceti~ anhydride in trifluoroacetic acid and the
resultant 5-trifluoroacetate hydrolysed, both according to pluc~lules
........ . . . . . .. ...... . . .. .. . .. _ . . . . .. . .. . . . .. _ .. _
21 9~79
WO 95t33'~1g PCT/GB95/012~ 1
- 110 -
similar to those described in Example 51. The product (1.199, m.p.
130-131C~ was iso1ated by chromatography on silica, using
dichloromethane-ethanol (49:1) as eluant. NMR (CDC13): S 5.2(2H,m);
5.75(1H,s); 8.0(1H,dd); 8.55(1H,d); 8.65(1H,bd). MS: MH 265.
Ste~ 4 Preparation of S-t-butylcarbamoyloxy-3(5 trifluoromethylpyridin--2-yl)thiazol idin-4-one
By a procedure similar to that described in Example 51, the alcohol
(O.Sg, prepared as described in Step 3 above) was converted into Compound
155 ~0.359, m.p. 189-190C). NMR (CDCl3): ~ 1.3(gH,s~; 4.8(1H,bs);
5.2(2H,s); 6.3(1H,s); 8.0(1H,dd); 8.6(1H,d~; 8.65(1H,d). MS: MH 364.
EXAMPI F 54 Preparation of Compound 156: 5-t-Buty1c~rbamoyloxy-
3(4-trifluoromethylpyrimidin-2-yl)thi~zolidin-4-one
~Ll Preparation of 3(4-trifluoromethylpyrimidin-2-yl)thiazolidin-4-one
2-Amino-4-trifluoromethylpyrimidine (5.09) was treated with
thioglycolic acid, aqueous formaldehyde solution, p-toluenesulphonic acid
and toluene in a manner similar to that described in Example 51. The crude
product (8.189) was, howeYer, largely uncyclized ((4-trifluoromethyl-
pyrimidin-2-yl)aminomethylthio)acetic acid. NMR (CDC13): ~ 3.4(2H,s);
4.8(2H,s); 8.4(1H,d); 8.6(1H,d). MS: MH+ 268. This material was cyclized
in a manner similar to that described in Example 51 to give the title
compound (0.759, m.p. 117C), following chromatoyraphy on silica using
dichloromethane-ethanol (49:1) as eluant. NMR (CDC13): ~ 3.8(2H,s);
5.1(2H,s); 7.45(1H,d); 9.05(1H,d). MS: MH 249.
Step Z Preparation of S-hydroxy-3(4-trifluoromethylpyrimidin-2-yl)-
thiazol idin-4-one
The thiazolidinone (0.38g, prepared as described in Step 1 above) was
chlorinated with sulphuryl chloride, then the total product hydrolysed with
potassium dil~yd,., ,' ,'-~te buffer, both using a procedure similar to
that described in Example 35, Steps 1-3. The second reaction mixture was
-
: 2190979
WO 9~/33719 PCT/GB9~/0122J
- 111 -
extracted with ethyl acetate, the extracts dried and evaporated under
reduced pressure and the residue chromatographed on silica, using
dichloromethane-ethanol (49:1) as eluant, to giYe the title compound
(0.219, m.p. 150-151C). NMR (CDC13): ~ 5.2(2H,m); 5.7(1H,s); 7.5(1H,d);
9.0(1H,d). MS: MH 266.
SteD 3 Preparation of
5-t-butylcarbamoyloxy-3(4-trifluoromethylpyrimidin-2-yl)thiazolidin-4-one
By a procedure similar to that described in Example 51, the alcohol
(0.209, prepared as described in Step 2 above) was converted into Compound
156 (0.199, m.p. 63C) after chromatography on silica, using
dichloromethane-ethanol (97:3) as eluant. NMR (CDC13): ~ 1.3(9H,s);
4.8(1H,bs); 5.1(2H,m); 6.2(1H,s); 7.5(1H,d); 9.0(1H,d). MS: MH 365.
EXAMPLE SS Preparation of Compound 157: 5-t-Butylcarb~moyloxy-
3(6-trifluoromethylpyrimidin-4-yl)thiazolidin-4-one
SteD 1 Preparation of 3(6-trifluoromethylpyrimidin-4-yl)thiazolidin-4-one
Following the p,~ S described in Example 54,
4-amino-6-trifluoromethylpyrimidine (2.169) was converted into crude
((6-trifluoromethylpyrimidin-4-yl)aminomethylthio)acetic acid (3.899) and
thence into the title compound (1.559, m.p. 90-91C) following
chromatography on silica, using dichloromethane-ethanol (49:1) as eluant.
NMR (CDC13): ~ 3.8(2H,s); 5.1(2H,s); 8.75(1H,s); 9.1(1H,s). MS: M 249.
SteD Z Preparation of 5-hydroxy-3(6-trifluoromethylpyrimidin-4-yl)-
thiazolidin-4-one
The thiazolidinone (1.449, prepared as described in Step 1 above) was
converted into the 5-chloro analogue and thence the 5-hydroxy analogue
using p,.~ ' ,es similar to those described in Example 54. The title
compound (1.219, m.p. 89-91~C) was isolated by ch" ~yl~ph~ on silica,
using dichloromethane-ethanol (49:1) as eluant. NMR (CDC13): ~ 5.1(2H,m);
5.75(1H,s); 8.8(1H,d); 9.15(1H,s). MS: MH 266.
.. . . .. . . . . . . . .. . . .
79
WO 95/33719 PCTIGB9S/0122.1
- 112 -
Step 3 Preparation of 5-t-butylcarbamoyloxy-3(6-trifluoromethyl-
pyridin-4-yl)thiazol idin-4-one
3y a procedure similar to that described in Example 51, the alcohol
(0.509, prepared as described in Step 2 above) was converted into Compound
157 (0.199, m.p. 140-142C), after chromatography on silica using
dichloromethane as eluant. NMR (CDCl3): ~ 1.3(9H,s); 4.9(1H,bs);
5.1(2H,m); 6.25(1H,s); 8.8(1H,d); 9.1(1H,s). MS: MH 365.
EXAMPI F 56 Preparation of Compound 158: 5-t-Butylcarb~moyloxy-
3(2,6-bistrifluoromethylpyrimid1n-4-yl)thiazolidin-4-one
SteP I Preparation of 3(2,6-bistrifluoromethylpyridin-4-yl)-
thiazol idin-4-one
3y a procedure similar to that described in Example 35, but using
4-amino-2,6-bistrifluoromethylpyridine (0.839) and appropriate amounts of
the other reagents, was obtained the title compound (0.329), after
Loy,~ ,y on silica, using hexane-ethyl acetate (6:1) as eluant. NMR
(CDC13): ~ 3.81(2H,s); 4.93(2H,s); 8.15(2H,s). MS: M 316.
Preparation of S-hydroxy-3(2,6-bistrifluoromethylpyridin-4-yl)-
thiazol idin-4-one
By ,ul~_ ' e~ similar to those described in Example 35, Steps 2 and 3
but using the thiazolidinone (0.299, prepared as described in Step 1
above), was obtained the title compound (0.159), following .1." L~
on silica using hexane-ethyl acetate (3:1) as eluant. NMR (CDC13): ~
4.16(1H,bs); 4.82(2H,d); 5.14(1H,d); 5.73(1H,s); 8.19(2H,s). MS: M 332.
Step 3 Preparation of 5-t-butylcarbamoyloxy-3(2,6-bistrifluoromethyl_
pyrid;n-4-yl)thiazolidin-4-one
By a procedure similar to that described in Example 51, the alcohol
(0.119, prepared as described in Step 2 above) was converted into Compound
2 1 90979
WO 95/33719 PCI`IGB95/01~21
- 113 -
158 (0.068, m.p. 122-124C), after Llll~ Loy-a~lly on silica using
hexane-ethyl acetate (6:1) as e~uant. NMR (CDCl3): ~ 1.34(9H,s);
4.79(1H,d); 4.86(1H,bs); 5.16(1H,dd); 6.18 (lH,s); 8.19(2H,s). MS: M 431.
EXAMPLE ~7 Prepar~tion of Compound 15g: 5-t-Butylc~rb~moyloxy-
3(2,2-difluoro-1,3-benzodioxol-5-yl)thi~zolidin-4-one
Step 1 Preparation of 3(2,2-difluoro-1,3-benzodioxol-5-yl)-
thiazol idin-4-one
By a procedure similar to that described in Example 35, Step 1, but
using 5-amino-2,Z-difluoro-l~3-benzodioxole (1.509), thioglycolic acid
(0.809), 37% aqueous formaldehyde solution (0.709), p-toluenesulphonic acid
(0.0259) and toluene (9Oml). After heating for ninety minutes, the mixture
was evaporated under reduced pressure, diluted with ether and washed
successively with hydrochloric acid (2M), water, aqueous sodium bicarbonate
solution, and brine. The organic layer was dried over magnesium sulphate,
evaporated under reduced pressure and the residue chromatographed on
silica, using hexane-ethyl acetate (4:1) as eluant, to give the title
compound (0.529, m.p. 105-107C). NMR (CDC13): ~ 3.75(2H,d); 4.79(2H,d);
7.06(2H,m); 7.33(1H,d). MS: M 259.
SteD 2 Preparation of 5-hydroxy-3(2,2-difluoro-1,3-benzodioxol-5-yl)-
th i azol i di n-4-one
The title compound was prepared by a procedure similar to that
described in Example 35, Steps 2 and 3, but using
3(2,2-difluoro-1,3-benzodioxol-5-yl)thiazolidin-4-one (0.499, prepared as
described in Step 1 above), sulphuryl chloride (0.259) and dichloromethane
(20ml). The reaction mixture was evaporated under reduced pressure and the
residue hydrolysed with an aqueous solution of potassium dihydrogen
phosphate in tetr~,y.i~ru~d,l. The crude product was triturated with hexane
to give the title compound (0.449, m.p. 98-100C). NMR (CDC13): ~
4.69(1H,d); 4.95(1H,dd); 5.71(1H,d); 7.10(2H,m); 7.37(1H,d). MS: M 275.
21 9097q
WO 95/33719 PCT/GB95/0122
- 114 -
Ste~ 3 Preparation of S-t-butylcarbamoyloxy-3(z,2-difluoro-1,3-benzo-
dioxol-5-yl)thiazolidin-4-one
The title compound was prepared by a procedure similar to that
described in Examp1e 35, Step 4, but using the alcohol (0.109), prepared as
described in Step 2 aboYe, t-butylisocyanate (0.0369), triethylamine
(0.037g) and dichloromethane (Sml). Cl," LOY~ on silica, using
hexane-ethyl acetate (4:1) as eluant, gave Compound 159 (0.0649, m.p.
159-161C). NMR (CDC13): ~ 1.34(9H,s); 4.62(1H,d); 4.88(1H,bs);
4.96(1H,dd); 6.17(1H,d); 7.10(2H,m); 7.35(1H,d). MS: M+ 374.
EXAMPLF 58 Prep~ration of Compound 160: 5-N(1,1-dimethylprop-2-ynyl)
carbamoyloxy-3~2,2-difluoro-1,3-benzodioxol-5-yl)thiazolidin-4-one
By a procedure similar to that described in Example 57, Step 3, but
using 1,1-dimethylprop-2-ynyl isocyanate in place of t-butyl isocyanate,
the substrate alcohol (0.309, prepared as described in Example 57, Step 2)
gave Compound 160 (0.219, m.p. 111-115C). NMR (CDC13): ~ 1.72(6H,s);
2.37(1H,s); 4.62(1H,d); 4.98(1H,dd); 5.15(1H,bs); 6.Z1(1H,d); 7.10(2H,m);
7.35(1H,d). MS: M 384.
EXAMPLE S9 Prepar~tion of Compound 161: 3-t-Butylc~rb~moyloxy-
1(2,6-dichloropyridin-4-yl)pyrrolidin-2-one
Ste,o 1 Preparation of 3-hydroxy-1(2,6-dichloropyridin-4-yl)pyrrolidin-2-one
A suspension of 4-amino-2,6-dichloropgridine (4.29) in 3-hydroxy-
tet,~ lr~r,-,.,.. 2-one (9.79) was heated at 180C for twenty hours and
allowed to cool. The residue was dissolved in dichloromethane, washed with
water, hydrochloric acid (lM) and brine, dried over magnesium sulphate and
evaporated under reduced pressure. This residue was triturated with a
little dichloromethane to give the title compound (1.09), sufficiently pure
for use in the next stage. NMR (DMS0-d6): ~ 1.8(1H,m); 2.4(1H,m);
3.6(1H,m); 3.8(1H,m); 4.3(1H,m); 5.9(1H,d); 7.8(2H,s).
WO 95/33719 2 1 9 0 9 ~ 9 PCT/GB95/0122~
- 115 -
5teo 2 Preparation of 3-t-butylcarbamoyloxy-1(2,6-dichloropyridin-4-yl)-
pyrrol idin-2-one
A solution of the alcohol (0.809, prepared as described in Step 1
above), triethylamine (0.369) and t-butylisocyanate (0.369) in
dichloromethane (150ml) was allowed to stand overnight at room t dLu~C.
Additional aliquots of triethylamine and t-butylisocyanate were added at
one day intervals. After four days in total, the mixture was evaporated
under reduced pressure. The residue was chromatoy,~ d on silica, using
dichloromethane-ethanol (49:1) as eluant, to give Compound 161 (0.389, m.p.
164-166C). NMR (CDC13): ~ 1.35(9H,s); 2.Z(lH,m); 2.75(1H,m); 4.3(2H,m);
4.9(1H,bs); 5.35(1H,t); 7.75(2H,s). MS: MH 346, 348, 350.
EXAMPLE 60 Prep~r~tion of Compound 162: 3-t-Butylc~rb~moyloxy-
1(4-trifluoromethylpyridin-2-yl)pyrrolidin-2-one
By a procedure similar to that described in Example S9, 2-amino-4-
trifluoromethylpyridine (3.249) was treated with excess
3 ~ r~l~YtcLl ~dluru~... 2-one to give, after chromatography on silica
using hexane-ethyl acetate (3:1) as eluant, 3-hydroxy-1(4-trifluoro-
methylpyridin-2-yl)pyrrolidin-Z-one (0.169). This material was converted
into the title compound by a procedure similar to that described in Example
59. Preparative layer chromatography on silica, using chlùluro\,.. ~ ane-
methanol (91:5:4) as eluant, and recrystallisation from chloroform-hexane
gave Compound 162 (0.0249, m.p. 112-114C). NMR essentially identical to
that given in Example 62, describing an alternative synthesis.
EXAMPLE 61 Prep~r~tion of Compounds 163-165
By p.~cc~,c, similar to those described in Example 59, the
appropriate heterocyclic amines were converted into the
l~;llu~ ulidinones and thence into their t-butylcarbamates.
2 1 90979
WO 95/33719 PCI`IGB9~/0122.1
- ~116 -
Compound 163
2-Amino-4-trifluoromethylthiazole (1.09) gave
3-hydroxy-1(4-trifluoromethylthiazol-2-yl)pyrrolidin-2-one (0.359, m.p.
140-141C). NMR (CDCl3): ~ 2.2(1H,m); 2.7(1H,m); 3.0(1H,bs); 4.0(1H,m);
4.3(1H,m); 4.65(1H,t); 7.5(1H,s). MS: M+ 252. The t-butylcarbamate,
Compound 163, had m.p. 157C. NMR (CDC13): ~ 1.3(9H,s); 2.2(1H,m);
2.8(1H,m); 4.0(1H,m); 4.3(1H,m); 4.85(1H,bs); 5.5(1H,t); 7.5(1H,s). MS:
MH 352.
Compound 164
4-Amino-6-trifluoromethylpyrimidine (5.09) gave
3-hydroxy-1(6-trifluoromethylpyrimidin-4-yl)pyrrolidin-2-one (0.369, m.p.
178-180C). NMR (CDCl3): ~ 2.15(1H,m); 2.7(1H,m); 3.05(1H,bs); 3.8(1H,m);
4.3(1H,m); 4.6(1H,m); 8.75(1H,s); 9.1(1H,s). MS: M+ Z47. The
t-butylcarbamate, Compound 164 had m.p. 136-137C. NMR (CDCl3): ~
1.35(9H,s); 2.12(1H,m); 2.7(1H,m); 3.85(1H,m); 4.3(1H,m); 4.9(1H,bs);
5.5(1H,t); 8.8(1H,s); 9.1(1H,s). MS: MH 347.
Compound 165
2-Amino-5-trifluoromethyl-1,3,4-thiadiazole (2.09) gave
3-hydroxy-1(5-trifluoromethyl-1,3,4-thiadiazol-2-yl)pyrrolidin-2-one
(0.429). NMR (CDCl3): ~ 2.3(1H,m); 2.8(1H,m); 4.1(1H,m); 4.4(1H,m);
4.7(1H,t). MS: MH 254. The t-butylcarbamate, Compound 165, had m.p.
184-186C. NMR (CDCl3): ~ 1.35(9H,s); 2.3(1H,m); 2.85(1H,m); 4.15(1H,m);
4.45(1H,m); 4.9(1H,bs); 5.5(1H,t). MS: MH 353.
WO 95/33719 2 1 9 0 9 7 9 PCT/GB9510122.1
- 117 -
EXAMPLE 62 A general route to 3-hydrocarbyl-carbamoyloxypyrrolidinones
exemplified with Compound 162: 3-t-Butylcarbamoyloxy-1(4-trifluoromethyl
- pyridin-Z-yl)pyrrolidin-Z-one
SteD 1 Preparation of 3-t-butylcarbamoyloxy-tetrahydrofuran-2-one
Boron trifluoride diethyl etherate (1.389) was added dropwise, over d
period of fifteen minutes, to a stirred solution of
3-hydroxytetrdl,yd,uru,dl~-2-one (lû.Og) and t-butylisocyanate (9.79) in dry
dichloromethane (300ml), whilst maintaining the t, ldLul~ below 10C.
The mixture was stirred at room t - dLul~ for a further four hours,
treated with brine and sufficient aqueous sodium bicarbonate solution to
render the aqueous phase basic, then extracted several times with
dichloromethane. The extracts were washed with brine, dried over magnesium
sulphate and evaporated under reduced pressure to give the title compound
(18.59, m.p. 104-106C). NMR (CDCl3): ~ 1.34(9H,s); 2.28(1H,m);
2.73(1H,m); 4.28(1H,dt); 4.46(1H,dt); 4.90(1H,bs); 5.31(1H,t). MS: M 201.
The addition can alsû be catalysed using triethylamine or gaseous
hydrogen chloride in place of boron trifluoride. However a real, ,
product can be formed in variable amounts which can necess;tate
purification of the desired material, for example by ~I", Luyl~Jllj on
silica using hexane-ethyl acetate (3:1) as eluant.
Step 2 Preparation of 2-t-butylcarbamoyloxy-4-iodo-N(4-trifluoromethyl-
pyridin-2-yl)butanamide.
A stirred solution of 3-t-butylcarbamoyloxy-t~L~ Jdluru, 2-one
(1.Og, prepared as described in Step 1 above) in dry dichloromethane (25ml)
was placed under nitrogen and kept dark with an aluminium foil shroud. It
was treated dropwise with i~JuLI Ll,~lsilane (1.Og), allowed to stand
overnight at room t-, ,dLu,~, treated with chlu,uLl .I,~lsilane (1.09g)
and stirred for a further three hours. lt was then cooled to 0C and
treated dropwise with oxalyl chloride (0.639) and N,N-dimethylformamide
(0.059). After stirring for thirty minutes at 0C and a further two hours
at 20C, the mixture was evaporated under reduced pressure. The residue
was dissolved in dichloromet~ane (25ml) and treated successively, with
stirring, with pyridine (2.36g), 4-dimethylaminopyridine (0.06g) and
.. ..
WO 95133719 2 1 9 0 9 7 9 PCT/GB951012~J
- 118 -
2-amino-4-trifluoromethylpyridine (0.899). The mixture was allowed to
stand overnight at room temperature, diluted with dichloromethane, washed
with hydrochloric acid (2M) and brine, dried over magnesium sulphate and
evaporated under reduced pressure. The residue was .llr, Log~l,.d on
silica, using dichloromethane-ethanol (99:1) as eluant, to give the title
compound (1.119, m.p. 83-85C). NMR (CDC13): ~ 1.35(9H,s); 2.5(2H,m);
3.25(2H,t); 5.0(1H,bs); 5.3(1H,dd); 7.3(1H,dd); 7.45(1H,d); 8.55(1H,bs);
8.8 (lH,bs). MS: M 473.
Step 3 Preparation of 3-t-butylcarbamoyloxy-1(4-trifluoromethyl-
pyridin-2-yl)pyrrolidin-2-one
Sodium hydride (0.0909, 55% suspension in mineral oil) was added
portionwise to a stirred solution of 2-t-butylcarbamoyloxy-4-iodo-N-
(4-trifluoromethylpyridin-2-yl)butanamide (0.979, prepared as described in
Step 2 above) in dry te~l~hjJlurul (lOml). After stirring for a further
fifteen minutes, the mixture was poured on to water and extracted with
ethyl acetate. The extracts were washed with brine, dried over magnesium
sulphate and evaporated under reduced pressure. The residue was
chromatographed on silica, using dichloromethane-ethanol (49:1) as eluant,
to give Compound 162 (0.459, m.p. 115.5-116.5C). NMR (CDC13): ~
1.35(9H,s); 2.15(1H,m); 2.7(1H,m); 3.9(1H,m); 4.25(1H,m); 4.9(1H,bs);
5.45(1H,t); 7.3(1H,dd); 8.55(1H,d); 8.75(1H,s). MS: MH 346.
EXAMPLE 63 Prep~ration of Compounds 166-171
By y., ' ~, similar to those described in Example 62, the
appropriate heterocyclic amines were converted into the pyrrûlidinone
carbamates via the open-chain iodo-amides.
Compound 166
4-Amino-2-trifluoromethylpyridine (1.209), scaled to 3-t-butyl-
carbamoyloxy-tet,cl,yJ,uru.~.. 2-one (1.509) and Culle.~. ding quantities of
other reagents/solvents, gave 2-t-butylcarbamoyloxy-4-iodo-N(2-trifluoro-
methylpyridin-4-yl)butanamide (1.159, contaminated with starting lactone).
WO 95133719 ` 2 1 9 0 9 7 9 PCTIGB9510122 1
- 119 -
NMR (CDC13) for product only: ~ 1.39(9H,s); 2.41(2H,m); 3.26(2H,m);
5.03(1H,bs); S.Zl(lH,m); 7.67(1H,dd); 7.83(1H,d); 8.58(1H,d); 8.93(1H,bs).
Cyclis2tion of this crude material with sodium hydride in tet~ah~ld~ururd"
gave Compound 166 (0.20g, m.p. 101-104C). NMR (CDC13): ô 1.35(9H,s);
2.20(1H,m); 2.77(1H,m); 3.82(1H,m); 3.93(1H,dt); 4.90(1H,s); 5.40(1H,t);
7.86(1H,dd); 8.03(1H,d); 8.69(1H,d). MS: M+ 345.
Compound 167
4-Amino-2-chloropyridine (0.329), scaled to
3-t-butylcarbamoyloxy-tetrahydrofuran-2-one (0.509) etc., gave
2-t-butylcarbamoyloxy-4-iodo-N(2-chloropyrjdin-4-yl)butanamide (û.65g, m.p.
65-67C). NMR (CDC13): ~ 1.35(9H,s); 2.4(2H,m); 3.25(2H,m); 5.05(1H,bs);
5.2(1H,t); 7.3(1H,dd); 7.55(1H,d); 8.2(1H,d); 8.9(1H,bs). MS: MH 440,
442. Base catalysed cyclisation of this material (0.58g) gave Compound 167
(0.189, m.p. 152-154C). NMR (CDC13): ~ 1.35(9H,s); 2.15(1H,m);
2.75(1H,m); 3.75(2H,m); 4.9(1H,bs); 5.4(1H,t); 7.65(2H,m); 8.35(1H,m). MS:
MH 312,314.
Compound 168
2-Amino-4-chloropyridine (0.409), scaled to lactonecarbamate (0.639~
etc., gave 2-t-butylcarbamoyloxy-4-iodo-N(4-chloropyridin-2-yl)butanamide
(0.215g, m.p. 39-42C). NMR (CDC13): ~ 1.35(9H,s); 2.47(2H,m); 3.22(2H,t);
4.98(1H,bs); 5.24(1H,dd); 7.10(1H,dd); 8.18(1H,d); 8.32(1H,d); 8.62(1H,bs).
MS: M+ 439, 441. 8ase-catalysed cyclisation of this material (0.179) gave
Compound 168 (0.055g, m.p. 133-135C). NMR (CDC13): ~ 1.37 (9H,s);
2.09(1H,m); 2.68(1H,m); 3.85(1H,m); 4.23(1H,dt); 4.90(1H,bs) 5.42(1H,t);
7.09(1H,dd); 8.26(1H,d); 8.52(1H,d). MS: M 311, 313.
Compound 169
4-Amino-2-iodopyridine (0.909), scaled to lactonecarbamate (1.09)
etc., gave the corresponding iodo-amide (0.269, m.p. 76-77~C). NMR
(CDCl3): ~ 1.3(9H,s); 2.35(2H,m); 3.25(2H,m); 5.15(1H,t); 5.2(1H,bs);
7.4(1H,dd); 7.8(1H,d); 8.15(1H,d); 9.15(1H,bs). MS: MH 532. Base-
. , .. , . ... ... , . ,, . , . . ... . , . . . , . _ . .. . ... ..
WO 95133719 2 1 9 0 9 7 q PCT/GB9510122.1
- 120 -
catalysed cyclisation of this material (0.22g) gave Compound 169 (0.149,
m.p. 69-70C). NMR (CDC13): ~ 1.35(9H,s); 2.15(1H,m); 2.7(1H,m);
3.8(2H,m); 5.0(1H,bs); 5.35(1H,t); 7.7(1H,dd); 8.0(1H,d); 8.3(1H,d). MS:
MH 404.
Compound 170
2-Amino-4,6-bistrifluoromethylpyridine (1.729), scaled to
lactonecarbamate (1.509) etc, gave the C~ ,u"~ing iodo-amide (0.949,
m.p. 127-131C). NMR (CDCl3): ~ 1.39(9H,s); 2.46(2H,m); 3.24(2H,t);
5.03(1H,bs); 5.26(1H,dd); 7.64(1H,d); 8.74(1H,d); 8.86(1H,bs). MS: M 541.
8ase-catalysed cyc~isation of this material (0.159) gave Compound 170
(0.0989, m.p. 123-126C). NMR (CDC13): ~ 1.37(9H,s); 2.15(1H,m);
2.72(1H,m); 3.92(1H,m); 4.33(1H,dt); 4.90(1H,bs); 5.48(1H,t); 7.63(1H,s);
8.99(1H,s). MS: MH 414.
Compound 171
2-Amino-6-chloro-4-trifluoromethylpyridjne (1.089), scaled to
lactonecarbamate (1.09) etc.,gave the co,,..~,u,lding iodo-amide (1.149, m.p.
115-116~C). NMR (CDC13): ~ 1.35(9H,s); 2.5(2H,m); 3.2(2H,t); 5.0(1H,bs);
5.25(1H,dd); 7.35(1H,s); 8.45(1H,s); 8.7(1H,bs). MS: MH+ 508, S10. Base-
catalysed cyclisation of this material (O.Sog) gave Compound 171 (0.219,
m.p. 149-151C). NMR (CDC13): S 1.35(9H,s); 2.1(1H,m); 2.7(1H,m);
3.85(1H,m); 4.25(1H,m); 4.9(1H,bs); 5.45(1H,t); 7.3(1H,s); 8.7(1H,s). MS:
MH 379, 381.
EXAMPLE 64 Preparation of Compound 172: 3-t-Butylc~rb~moyloxy-
1(pyridin-3-yl)pyrrolidin-2-one
By a procedure similar to that described in Example 62,
3-aminopyridine (0.479), scaled to 3-t-butylcarbamoyloxytetrahydro-
furan-2-one (1.09) etc., gave a crude product (3.19) containing
approximately 20 mole % of 2-t-butylcarbamoyloxy-4-iodo-N(pyridin-3-yl)-
butanamide. The desired product was apparently unstable in the mixture and
to chromatography on silica. NMR (CDC13) for product only: ~ 1.38(9H,s);
,
WO 95/33719 2 1 9 0 9 7 9 PCT/GB95101~
- 121 -
2.46(2H,m); 3.25(2H,t); 5.12(1H,bs); 5.25(1H,dd); 7.29(1H,m): 8.17(1H,dd);
8.36(1H,dd); 8.58(1H,d). 8ase-catalysed cyclisation of this crude material
gave Compound 172 (0.18g, m.p. 129-131C) after several chromatographic
separations on silica using dichloromethane-ethanol (19:1) as eluant. NMR
(CDCl3): ~ 1.35(9H,s); 2.12(1H,m); 2.77(1H,m); 3.86(2H,m); 4.90(1H,bs);
5.36(1H,t); 7.33(1H,dd); 8.28(1H,m); 8.44(1H,dd); 8.76(1H,d). M5: M 277.
EXAMPlE 65 Preparation of Compound 173: 3-t-Butylcarbamoyloxy-
1(pyridin-3-yl)pyrrolidin-2-one N-oxide
A stirred solution of the pyridine (0.09Og, prepared as described in
Example 64) in dichloromethane (lOml) was treated with m-chl~,u~.L~"~uic
acid (0.129, 55%). After being allowed to stand overnight at room
temperature, the mixture was diluted with dichloromethane, washed with
aqueous sodium bicarbonate solution and brine, dried over magnesium
sulphate and evaporated under reduced pressure. The residue was
chromatographed on silica, using dichloromethane-ethanol (19:1) as eluant,
to give Compound 173 (0.0709, m.p. 224-2z5~C). NMR (CDC13): ~ 1.35(9H,s);
2.17(1H,m); 2.75(1H,m); 3.72(2H,m); 4.92(1H,bs); 5.34(1H,t); 7.27(1H,dd);
7.85(1H,d); 8.05(1H,dd); 8.63(1H,t). MS: M 293.
EXAMPLF 66 Preparation of Compounds 174-182 and
Compound 164 (alternative method)
By ~,.u,~dulc, similar to those described in Example 62, the
appropriate heterocyclic amines were converted into the pyrrolidinone
carbamates via the open-chain iodo-amides.
Compound 174 2-Amino-4-trifluoromethylpyrimidine (0.419), scaled to
lactonecarbamate (0.509) etc., gave the cu,,~.~rl 'ing iodo-amide, (0.169,
contaminated with starting lactonecarbamate). NMR (CDCl3) for product
only: ~ 1.3(9H,s); 2.3(2H,m); 3.3(2H,t); 4.9(1H,bs); 5.3(1H,t); 7.4(1H,d);
8.95(1H,d); 8.8(1H,bs). Base-catalysed cyclisation of this material gave
Compound 174 (0.0189, m.p. 100-101C). NMR (CDCl3): ~ 1.35(9H,s);
2.1(1H,m); 2.7(1H,m); 3.9(1H,m); 4.3(1H,m); 4.9(1H,bs); 5.4(1H,t);
7.4(1H,d); 9.0(1H,d). M5: MH 347.
... _ . . , .. _ ... .. .. .. .
WO 95/33719 2 1 9 0 9 7 9 PCT/GB9510122 1
- 122 -
Compound 175
5-Aminopyrimidine (0.529), scaled to lactonecarbamate (1.09) etc.,
gave the urle~pun~ing iodo-amide (0.389, m.p. 77-79C). NMR (CDCl3): ~ -1.35(9H,s); 2.4(2H,m); 3.25(2H,m); ~.05(1H,bs); 5.25(1H,dd); 8.65(1H,bs);
9.0(3H,s). MS: MH 407. Base catalysed cyclisation of this material
(0.349) gave Compound 175 (0.179, m.p. 171-173C) . NMR (CDCl3) : ~
1.35(9H,s); 2.2(1H,m); 2.8(1H,m); 3.85(ZH,m)- 5.0(1H,bs)- 5.4(1H,t);
9.05(1H,s); 9.15(2H,s). MS: MH 279.
Compound 176
2-Aminûpyrazine (0.719), scaled to lactonecarbamate (1.509) etc., gave
the cu.l, r 'ing iodo-amide (0.329, m.p. 113-115C). NMR (CDCl3): ~
1.38(9H,s); 2.47(2H,m); 3.33(ZH,t); 4.97(iH,bs); 5.28(1H,dd); 8.27(1H,dd);
8.39(1H,d); 8.56(1H,bs); 9.56(1H,d). MS: M+ 406. Base-catalysed
cyclisation of this material (0.269) gave Compound 176 (0.119, m.p.
146-149C). NMR (CDC13): ~ 1.36(9H,s); 2.14(1H,m); 2.74(1H,m); 3.B4(1H,m);
4.18(1H,dt); 4.92(1H,s); 5.45(1H,t); 8.35(2H,m); 9.76(1H,d). MS: M 278.
Compound 177
4-Amino-6-chloropyrimidine (0.719), scaled to lactonecarbamate (1.09)
etc., gave the c~"~,,uu"ding iodo-amide (0.659, m.p. 175-176C). NMR
(COCl3): ~ 1.35(9H,s); 2.45(2H,m); 3.2(2H,t); 5.0(1H,bs); 5.25(1H,dd);
8.25(1H,s); 8.65(1H,s); 8.8(1H,bs). MS: MH 441, 443. Base-catalysed
cyclisation ûf this material (0.549) gave Compound 177 (0.169, m.p. 117C).
NMR (CDC13): ~ 1.35(1.35(9H,s); 2.15(1H,m); 2.7(1H,m); 3.8(1H,m);
4.25(1H,m); 4.9(1H,s); 5.4(1H,t); 8.5(1H,d); 8.75(1H,s). MS: M 312, 314.
Compound 178
4-Amino-6-chloro-2-methylthiopyrimidine (1.029), scaled to
lactonecarbamate (1.09) etc., gave the cu.,~,~u,,ding iodo-amide (1.089,
m.p. 131-132C). NMR (CDCl3): ~ 1.35(9H,s); 2.3(2H,m); 2.55(3H,s);
,
2190
WO 95133719 PCT/CB95/0122.1
- 123 -
3.2(2H,t); 5.0(1H,bs); 5.2(1H,dd); 7.9(1H,s); 8.7(1H,bs). MS: MH 486,
488. Base-catalysed cyclisation of this material (0.889) gave Compound 178
(0.0659, m.p. 165-167C). NMR (CDCl3): ~ 1.35(9H,s); 2.1(1H,m);
2.55(3H,s); 2.65(1H,m); 3.8(1H,m); 4.25(1H,m); 4.9(1H,bs); 5.4(1H,t);
8.1(1H,s). MS: MH 359, 361.
Compound 164 (Alternative method)
4-Amino-6-trifluoromethylpyrimidine (1.069), scaled to
lactonecarbamate (1.09) etc., gave the co"~iuu"ding iodo-amide (0.769,
m.p. 169-171C). NMR (DMS0-d6): ~ 1.3(9H,s); 2.4(2H,m); 3.3(2H,t);
5.15(1H,dd); 6.1(1H,bs); 8.55(1H,s); 9.0(1H,s); 10.9(1H,bs). MS: MH 475.
Base-catalysed cyclisation of this material (0.609) gave Compound 164
(0.219, m.p. 137C). NMR and MS were identical to values given for this
compound, prepared as described in Example 61.
Compound 179
4-Amino-6(2,2-diflu~,,uc~ho~y)pyrimidjne (0.869, m.p. 127C) was made
by treating 4-amino-6-chloropyrimidine (2.509) with sodium
2,2-difluoroethoxide in tetra,,~d,uru,d,,. Reaction of it (0.919), scaled to
lactonecarbamate (1.09) etc., gave the cul,c-r 'ing iodo-amide (1.289,
m.p. 42-44C). NMR (CDC13): ~ 1.35(9H,s); Z.5(2H,m); 3.2(2H,t);
4.6(2H,dt); S.O(lH,bs); 5.2(1H,dd); 6.1(1H,tt); 7.65(1H,s); 8.5(1H,s);
8.7(1H,bs). MS: MH 487. Base-catalysed cyclisation of this material
(1.099) gave Compound 179 (0.439, m.p. 49-51C). NMR (CDC13): ~ 1.3(9H,s);
2.1(1H,m); 2.7(1H,m); 3.8(1H,m); 4.25(1H,m); 4.6(2H,dt); 4.9(1H,bs);
5.4(1H,t); 6.1(1H,tt); 7.9(1H,s); 8.6(1H,s). MS: MH 359.
Compound 18û
4-Amino-6(2,2,2-trifl ,u~LI,u~y)pyrimidine (0.619, m.p. 113C) was
made by treating 4-amino-6-chloropyrimidine (1.09) with sodium
2,2,2-trifluoroethoxide in N,N-dimethylformamide. Reaction of it (0.599),
scaled to lactonecarbamate (0.589) etc., gave the corresponding iodo-amide
(0.559, m.p. 46-47C). NMR (CDC13): ~ 1.4(9H,s); 2.5(2H,m); 3.2(2H,t);
.. , . . . , .. .. _ . , _ . . , . ,,,, . .. .. . , . , ... _ , . , _ . _ ... , , , _ ,
WO 95133719 2 1 9 0 9 7 9 PCTIGB9510122.1
- 124 -
4.8(2H,m); 4.95(1H,bs); 5.25(1H,dd); 7.7(1H,s); 8.5(1H,s); 8.65(1H,bs).
MS: MH 504. Base-catalysed cyclisation of this material (0.44g) gave
Compound 180 (O.Zl, m.p. 100-101C). NMR (CDC13): ~ 1.3(9H,s); 2.1(1H,m);
2.7(1H,m); 3.8(1H,m); 4.25(1H,m); 4.8(2H,q); 4.9(1H,bs); 5.4(1H,t);
7.9(1H,s); 8.6(1H,s). MS: MH 377.
Compound 181
4-Amino-6-difluo,. Ll,v~pyl imidine (0.179, m.p. 152-154C) was made
by passing chlorodifluoromethane into a solution of
4-amino-6-hydroxypyrimidine (O.Sg) in aqueous dioxan at 70C, in the
presence of sodium hydroxide. Reaction of it (0.949), scaled to
lactonecarbamate (1.06g) etc., gave the CGllc r ding iodo-amide (1.019,
pale yellow gum). NMR (CDC13): ~ 1.4(9H,s); 2.5(2H,m); 3.2(2H,t);
5.0(1H,bs); 5.25(1H,dd); 7.48(1H,t); 7.75(1H,s); 8.5(1H,s); 8.75(1H,bs).
MS: MH 473. Base-catalysed cyclisation of this material (0.809) gave
Compound 181 (0.23g, m.p. 140-141C). NMR (CDC13): ~ 1.3(9H,s); 2.1(1H,m);
2.7(1H,m); 3.8(1H,m); 4.3(1H,m); 4.9(1H,bs); 5.4(1H,t); 7.5(1H,t);
8.0(1H,s); 9.6(1H,s). MS: MH+ 345.
Compound 18Z
4-Amino-6-difluo,. Lhv~y-2; ~ ~py~ imidine (1.739, m.p. 112-113C)
was ~ade by passing chlorodifluoromethane into a solution of
4-amino-6-hydroxy-2 Li,v~y, illlidine (4.09) in aqueous dioxan at 70C, in
the presence of sodium hydroxide. Reaction of it (0.849), scaled to
lactonecarbamate (0.809) etc., gave the cull. r 'ing iodo-amide (0.33g,
m.p. 54-55C). NMR (CDC13): ~ 1.35(9H,s); 2.4(2H,m); 3.2(ZH,t);
3.95(3H,s); 4.95(1H,bs); 5.2(1H,dd); 7.4(1H,s); 7.45(1H,t); 8.6(1H,s). MS:
M+ 502. Base-catalysed cyclisation of this material (0.29g) gave Compound
182 (0.12g, m.p. 107-108C). NMR (CDC13): ~ 1.35(9H,s); 2.1(1H,m);
2.7(1H,m); 3.8(1H,m); 4.0(3H,s); 4.25(1H,m); 4.95(1H,bs); 5.4(1H,t);
7.45(1H,t); 7.6(1H,s). MS: MH 375.
wo 95/33719 1 9 0 9 7 q PCT/GB9~/0122.1
125 -
EXAMPLE 67 Prep~r~tion of Compounds 183-189
By l~,u-edu~es simildr to those described in Example 6Z, the
appropriate heterocyclic amines were converted into the pyrrolidinone
carbamates via the open-chain iodo-amides.
Compound 183
2-Amino-5-bromothiazole (0.459), scaled to lactonecarbamate (0.5ûg)
etc., gave the col,~,u"~ing iodo-amide (0.459, m.p. 59-61C). NMR
(CDCl3): ~ 1.3(9H,s); 2.5(2H,m); 3.2(2H,t); 4.9(1H,bs); 5.3(1H,dd);
7.4(1H,s); 10.0(1H,vbs). MS: MH 490, 492. Base-catalysed cyclisation of
this material (0.409) gave Compound 183 (0.149, m.p. 193-194C). NMR
(CDCl3): ~ 1.35(9H,s); 2.2(1H,m); 2.8(1H,m); 3.9(1H,m); 4.2(1H,m);
4.9(1H,bs); 5.5(1H,t); 7.4(1H,s). MS: M 361, 363.
Compound 184
2-Amino-5-trifluoromethylthiazole (5.579 of hydrochloride salt after
appropriate work-up) was made by treating 2-aminothiazole 5-carboxylic acid
(8.209) with sulphur tetrafluoride and hydrogen fluoride at 120C. The
anhydrous free base (0.429), liberated from the hydrochloride salt with
aqueous sodium bicarbonate solution, scaled to lactonecarbamate (0.509)
etc, gave the .~"..~,~",ding iodo-amide (0.529, m.p. 50-52C). NMR (CDCl3):
1.3(9H,s); 2.5(2H,m); 3.25(2H,t); 4.95(1H,bs); 5.3(1H,dd); 7.85(1H,s);
10.6(1H,bs). MS: MH 48û. Base-catalysed cyclisation of this material
(0.459) gave Compound 184 (D.13g, m.p. 189-19DC). NMR (CDCl3): ~
1.35(9H,s); 2.25(1H,m); 2.8(1H,m); 4.D(lH,m); 4.3(1H,m); 4.9(1H,bs);
5.5(1H,t); 7.8(1H,m). MS: MH 352.
Compound 185
2-Amino-5-iodothiazole (1.309, as hydrochloride salt), scaled to
lactonecarbamate (1.Dg) etc., gave the cur,e.~.u,,ding iodo-amide (0.299,
m.p. 50-60C, decomp). NMR (CDCl3): ~ 1.32(9H,s); 2.45(2H,m); 3.22(2H,t);
4.85(1H,bs); 5.3D(lH,dd); 7.56(1H,s). MS: MH 538. Base-catalysed
.. . . . . . . . _ .. . . . _ . . . .. .. . . .
WO 95133719 2 1 9 0 9 7 9 PCl`IGB9~10122.1
- 126 -
cyclisation of this material (0.229) gave Compound 185 (0.149, m.p.
199-201C). NMR (CDCl3): ~ 1.34(9H,s); 2.20(1H,m); 2.77(1H,m); 3.93(1H,m);
4.24(1H,dt); 4.87(1H,bs); 5.48(1H,t); 7.53(1H,s). MS: M 409.
Compound 186
2-Amino-S-chlorothiazole (0.859, as hydrochloride salt), scaled to
ldctonecarbamate (l.Og) etc., gave the Lolle~lJullding iodo-amide (O.SOg,
m.p. 119-122C). NMR (CDCl3): ~ 1.34(9H,s); 2.46(2H,m); 3.22(2H,t);
4.90(1H,bs); 5.32(1H,dd); 7.34(1H,s). Base-catalysed cyclisation of this
material (0.399) gave Compound 186 (0.199, m.p. 191-192C). NMR (CDC13):
1.35(9H,s); 2.20(1H,m); 2.77(1H,m); 3.92(1H,m); 4.24(1H,dt); 4.87(1H,bs);
5.48(1H,t); 7.32(1H,s). MS: M 317, 319.
Compound 187
S-Amino-3-trifluoromethylisoxazole (0.769), scaled to lactonecarbamate
(1.09) etc., gave the c~,,c,~ 'ing iodo-amide (0.919, m.p. 100-102C).
NMR (CDC13): ~ 1.35 (9H,s); 2.4(2H,m); 3.25(2H,m); S.OS(lH,bs); 5.3(1H,dd);
6.65(1H,s). MS: MH 464. Base-cataiysed cyclisation of this material
(0.799) gave Compound 187 (0.199, m.p. 181-182C). NMR (CDC13): ~
1.3(9H,s); 2.2(1H,m); 2.8(1H,m); 3.9(1H,m); 4.15(1H,m); 4.9(1H,bs);
5.4(1H,t); 6.8(1H,s). MS: M+ 335.
Compound 188
2-Amino-4-trifluoromethyloxazole (0.809), scaled to lactonecarbamate
(2.09) etc., gave the co..~,~u.,ding iodo-amide (0.309, brown oil).
NMR(CDC13): ~ 1.32(9H,s); 2.40~2H,m); 3.22(2H,m); S.O(lH,s); 5.27(1H,t);
7.79(1H,s~; 9.30(1H,s). MS: M 463. Base-catalysed cyclisation of this
material (0.209) gave Compound 188 (O .095g, m.p. 150-151C). NMR (CDC13):
ô 1.32 (9H,s); 2.21(1H,m); 2.72(1H,m); 3.90(1H,m); 4.13(1H,m); 4.88(1H,bs);
5.35(1H,t); 7.83(1H,s).
-
90979
WO 95/33719 2 1 PCT/GB95/0122.1
- 127 -
Compound 189
-5-Amino-2,2-difluoro-1,3-benzodioxole (0.7sg), scaled to
lactonecarbamate (1.Og) etc., gave the col,es~.u..ding iodo-amide (0.539,
m.p. 135-135C). NMR (CDC13): ~ 1.37(9H,s); 2.43(2H,m); 3.25(2H,t);
4.94(1H,bs); 5.20(1H,dd); 6.99(2H,m); 7.58(1H,dd); 8.32(1H,bs). MS: M
484. Base-catalysed cyclisation of this material (0.479) gave Compound 189
(0.209 m.p. 147-148C). NMR (CDC13): ~ 1.34(9H,s); 2.08(1H,m); 2.74(1H,m);
3.80(2H,m); 4.90(1H,bs); 5.35(1H,t); 7.05(1H,d); 7.14(1H,dd); 7.71(1H,d),
MS: M 356.
EXAMPlE 68 A general route to 3 (N (h.~ . L ~ , l ) al kyl ami no) - and
3(N(alkanoyl)alkylamino)-pyrrolidinones exempl;fied by Compounds l9û and
191
CompDund 190: 3(N(t-butylcarbamoyl)methylamino-1(4-trifluoromethyl-
pyridin-2-yl)pyrrolidin-2-one
SteD I Preparation of 2,4-dibromo-N(4-trifluoromethylpyridin-2-yl)-
butanami de .
A solution of 2-amino-4-trifluoromethylpyridine (5.009) and
triethylamine (3.439) in dry teLr_~y~,uru~a,, (50ml) was added dropwise,
over ten minutes, to a stirred solution of 2,4-dibromobutanoyl chloride
(9.519) in dry tetr~ d,uru, (50ml), whilst maintaining the t~ ~laLul~
below 5C. The mixture was allowed to stir overnight at room t ,;,aLul~,
diluted with hydrochloric acid (lM) and extracted with ethyl acetate. The
extracts were washed with brine, dried over magnesium sulphate and
evaporated under reduced pressure. The residue was chromatographed on
silica, using hexane-ethyl acetate (5:1) as eluant, to give the title
compound (10.099, yellow gum) sufficiently pure for use in Step 2 below.
Rechromatographed material had NMR (CDC13): ~ 2.57(1H,m); 2.74(1H,m);
3.62(2H,m); 4.71(1H,dd); 7.33(1H,dd); 8.49(3H,d+s); 8.84(1H,bs). MS: M+
388.
WO 9S133719 2 1 9 0 9 7 9 PC~/GB9S10122-1
- 128 -
SteP 2 Preparation of 3-bromo-1(4-trifluoromethylpyridin-2-yl)pyrrolidin-
-2-one
-
Sodium hydride (0.829, 55-65% dispersion in mineral oil) was added
portlonwise to a stirred solution of the substrate (7.289, prepared as
described in Step 1 above) in dry tetrahydrofuran (lSOml). The mixture was
stirred for one hour, diluted carefully with water and extracted with ethyl
acetate. The extracts were washed with brine, dried over magnesium
sulphate, and evaporated under reduced pressure. The residue was
chromatographed on silica, using hexane-ethyl acetate (7:1) as eluant, to
give the title compound (3.909, m.p. 43-47C). NMR (CDC13): ~ 2.48(1H,m);
2.74(1H,m); 4.21(2H,m); 4.66(1H,dd); 7.32(1H,dd); 8.56(1H,d); 8.74(1H,s).
MS: M 30~3,310.
Ste~ 3 Preparation of 3-methylamino-1(4-trifluoromethyl-
pyridin-2-yl)pyrrolidinone
Gaseous methylamine was bubbled through a stirred solution of the
substrate (2.159, prepared as described in Step 2 above) in dry
tet~l,yd,ururall (lOOml) for one hour. The mixture was diluted with water,
and extracted with ethyl acetate. The extracts were washed with brine,
dried over magnesium sulphate, and evaporated under reduced pressure. The
residue was chromatographed on silica, using dichloromethane-ethanol (19:1)
as eluant, to give the title compound (1.30g, m.p. 79-81C). IIMR (CDC13):
~ 1.96(1H,m); 2.50(1H,m); 2.56(3H,s); 3.61(1H,dd); 3.86(1H,m); 4.24(1H,m);
7.26(1H,dd); 8.51(1H,d); 8.74(1H,d). MS: M 259.
SteP 4 Preparation of 3(N(t-butylcarbamoyl)methylamino)-1(4-trifluoro-
methylpyridin-2-yl)pyrrolidin-2-one
A stirred solution of substrate (0.30g, prepared as described in Step
3 above) in dichloromethane (20ml) was treated successively with
triethylamine (0.129) and t-butylisocyanate (0.1159). The residue was
allowed to stir for one hour, diluted with dichloromethane and washed with
water and brine. The extracts were dried over magnesium sulphate and
evaporated under reduced pressure. The residue was chromatographed on
,, , _ ,, _,,
2 1 9 0 9 7 9 PCT/GB95/0122~
WO 95/33719
- 129 -
silica, using hexane-ethyl acetate (1:1) as eluant, to give Compound 1gO
(0.289, m.p. 152-155C). NMR (CDC13): ~ 1.38(9H,s); Z.13(1H,m),
2.45(1H,m); 2.85(3H,s); 3.81(lH,m); 4.30(1H,m); 4.44(1H,bs); 5.29(1H,dd);
7.26(1H,dd); 8.53(1H,d); 8.78(1H,s). MS: M 358
CompDund 191:
3((N(3,3-dimethylbutanDyl))~ethylamino) -1(4-trifluoro~ethylpyridin-2-yl) -
pyrrolidin-2-one
A stirred solution of 3-methylamino-1(4-trifluoromethylpyridin-2-yl)-
pyrrolidin-2-one (0.30g, prepared as described in Example 68, Step 3,
above) in dichloromethane (20ml) was treated successively with
triethylamine (0.139) and 3,3-dimethylbutanoyl chloride (0.169). After one
hour, the mixture was diluted with dichloromethane, washed with water and
brine, dried over magnesium sulphate and evaporated under reduced pressure.
The residue was chromatographed on silica, using hexane-ethyl acetate (1:1)
as eluant, to give Compound 191 (0.31g, m.p. 47-53C). NMR (CDC13): ~
1.31(9H,s); 2.31(4H,m+s); 3.09(3H,s); 3,88(1H,m); 4.32(1H,m); 5.22(1H,t);
7.26(1H,dd); 8.52(1H,d); 8.78(1H,s). MS: M+ 357.
EXAMPLE 69 Prepar~tion of Compounds 192 and 193
The title compounds were prepared by IJlu~edul~ similar to those
described in Example 68 but using 2-amino-5-trifluoromethylthiazole
(prepared as described in Example 67) in Step 1. This amine (2.179) gave
2,4-dibromo-N(5-trifluoromethylthiazol-2-yl)butanamide (4.509, m.p.
113-115C). NMR (CDC13): ~ 2.55(2H,m); 3.65(2H,t); 4.8(1H,dd); 8.0(1H,d).
MS: MH 395, 397, 399. Base-catalysed cyclisation of this material (4.27g)
gave 3-bromo-1(5-trifluoromethylthiazol-2-yl)pyrrolidin-2-one (2.689, m.p.
105-106C). NMR (CDC13): ~ 2.6(1H,m); 2.85(1H,m); 4.25(2H,m); 4.7(1H,dd);
7.8(1H,s). MS: M 314, 316. This material (1.09) was treated with
methylamine in tet, ' ~drorul to give 3-methylamino-1(5-trifluoro-
methylthiazol-2-yl)pyrrolidin-2-one (0.249, m.p. 108-109C). NMR (CDC13):
~ 2.1(1H,m); 2.6(4H,m); 3.7(1H,t); 4.0(1H,m); 4.3(1H,m); 7.8(1H,s). MS: M
265.
2~ 90979
WO 95133719 PCTIGB9510122.1
- 130 -
Sdmples of this amine (0.129) were treated with t-butyl isocyanate to
give Compound 192 (0.07g, m.p. 186-187C) and with 3,3-dimethylbutanoyl
chloride to give Compound 193 (0.159, m.p. 123C). Compound l9Z had NMR
(CDCl3): ~ 1.35(9H,s); 2.3(1H,m); 2.6(1H,m); 2.9(3H,s); 3.9(1H,m);
4.35(1H,m); 4.45(1H,bs); S.l(lH,dd); 7.8(1H,d). MS: M+ 364. Compound 193
had NMR (CDC13): ~ 1.1(9H,s); 2.35(1H,m); 2.6(1H,m); 3.15(3H,s); 4.0(1H,m);
4.35(1H,m); 4.85(1H,bs); 7.B(lH,d). M5: MH 364.
EXAMPLE 70 Prepar~tion of Compounds 194 and 195
The title compounds were prepared by ~l,uc~:du.,s similar to these
described in Example 68 but using S-amino-2,2-difluoro-1,3-benzodioxole in
Step 1. In this case the intermediate b., ,,y,,ulidine (and chloro
contaminant) was converted into the c~.,,, 'ing iodide, by treatment with
sodium iodide in acetone, before introduction of the alkylamine
functionality. In some cases, higher yields can be obtained.
The aminobenzodioxole (2.009) gave the dibromobutanamide (2.469). NMR
(CDCl3): ~ 2.56(1H,m); 2.76(1H,m); 3.63(2H,m); 4.69(1H,dd); 7.04(2H,s);
7.60(1H,t); 8.0(1H,bs). (This material can be contaminated by varying
amounts of the 2-chloro analogue). Base-catalysed cyclisation of this
material (2.469) gave the 3-lJI. ,~.,ulidinone (1.669). NMR (CDCl3): ~
2.51(1H,m); 2.76(1H,m); 3.81(1H,dt); 4.04(1H,m); 4.59(1H,dd); 7.06(1H,d);
7.17(1H,dd); 7.68(1H,d). MS: M 319, 321). (This material can be
contaminated by varying amounts of the 3-chloro analogue). Treatment of
the bromide (1.669) with sodium iodide in acetone gave the
iodopyrrolidinone (1.82g, m.p. 71-74C). NMR (CDCl3): ~ 2.39(1H,m);
2.62(1H,m); 3.71(1H,dt); 3.92(1H,m); 4.72(1H,dd); 7.05(1H,d); 7.16(1H,dd);
7.67(1H,d). MS: M 367. Further treatment of this material (l.Og) with
gaseous methylamine in tetrahydrofuran gave the 3-methylaminopyrrolidinone
(0.749, m.p. 65-69C). NMR (CDCl3): ~ 1.98(1H,m); 2.49(1H,m); 2.53(3H,s);
2.81(1H,d); 3.53(1H,dd); 3.77(2H,m); 7.D3(1H,d); 7.12(1H,dd); 7.68(1H,d).
MS: M 270.
Samples of this amine (0.209) were treated with t-butyl isocyanate to
give Compound 194 (0.229, m.p. 155-157C) and with 3,3-dimethylbutanoyl
chloride to give Compound l9S (0.169, m.p. 111-112C). Compound 194 had
NMR (CDCl3): ~ 1.37(9H,s); 2.13(1H,m); 2.47(1H,m); Z.84(3H,s); 3.75(2H,m);
- - -
2 1 9 0 9 7 9 9510122~1
WO 95~33719 PCT/GB
- 131 -
4.44(1H,bs); 5.17(1H,dd); 7.04(1H,d); 7.12(1H,dd); 7.73(1H,d). MS: M 369.
Compound 195 had NMR (CDCl3): ~ 1.09(9H,s); 2.20(1H,d); 2.33(2H,d);
2.44(1H,m); 2.88(0.3H,s); 3.07(2.7H,s); 3.80(2H,m); 5.14(1H,t); 7.03(1H,d);
7.12(1H,dd); 7.71(1H,d). This spectrum is complicated by effects arising
from restricted rotation. MS: M+ 368.
EXAMPI F 71 Prepar~tion of Compound 196: 3-t-Butylcarb~moyloxy-
1(2-chlorothien-5-yl)pyrrolidin-2-one
The title compound was prepared by plûceuu,~s similar to those
described in Example 50.
S-Bromo-2-chlorothiophene (15.89) was treated with pyrrolidin-2-one and
cuprous oxide to give 1(2-chlorothien-S-yl)pyrrolidin-2-one (3.259, m.p.
152-154C). NMR (CDCl3): ~ 2.25(2H,m); 2.63(2H,m); 3.83(2H,t); 6.21(1H,d);
6.70(1H,d). Oxidation of this material (3.259) gave the c~",..pu,.ding
3-l~ydro~cyuy,,ulidin-2-one (0.349, m.p. 159-161C). NMR (CDC13): ~
2.20(1H,m); 2.65(1H,m); 3.70(1H,m); 3.87(1H,m); 3.0(1H,bs); 4.55(1H,t);
6.30(1H,d); 6.72(1H,d). MS: M Z17, Zl9. Treatment of this material
(0.Z0g) with t-butylisocyanate gave Compound 196 (0.049, m.p. 177-179C).
NMR (CDCl3): ~ 1.37(9H,s); 2.Z0(lH,m); Z.78(1H,m); 3.75(1H,m); 3.87(1H,m);
4.87(1H,bs); 5.40(1H,t); 6.30(1H,d); 6.7Z(lH,d). MS: M 316, 318.
EXAMPLE 72 Prep~ration of Compound 197: 3-(N,N-diisopropylcarb~moyl)-
~mino-1-(3-trifluoromethyl)phenyl-2-pyrrolidinone.
3-Amino-1-(3-trifluoromethyl)phenyl-2-pyrrolidinone (0.3059), prepared in a
similar manner to that described in Step 4 of Example 1, diisopropyl-
carbamoylchloride (0.4099), and 4-N,N-dimethylaminopyridine (0.1529) were
dissolved in DMF (2ml) and stirred at room temperature for 3 days. The
reaction mixture was then poured into water and extracted into ethyl
acetate (3 times). The combined ethyl acetate extracts were washed with
water (twice) before drying (MgS04) and cu..c~ Ling to an oil. Column
chromatography eluting with ethyl acetate / hexanes (Z:1) gave the product
as a gum (0.099, 199~).
WO 95/33719 2 1 9 0 9 7 9 PCI/GB9510122 1
- - 132 -
EXAMPLF 73 Prep~r~tion of Compound 198: 3-(N-methyl,N-t-butylcarbamoyl)-
dmi no-1- (3 -tri f 1 . . i ' y) phenyl -2-pyrrol i di none .
1-(3-Trifluoromethoxy)phenyl-2-pyrrolidinone-3-carboxylic acid (O.Sg),
prepared as described in Example 7, Step 1, and triethylamine (0.24ml) were
dissolved in toluene (lOml). Diphenylphosphoryl azide (0.384ml) was then
added and the mixture heated to 90-100C for l.Z5 hours. Nitrogen gas was
evolved during the first 15 mins. The reaction mixture was then allowed to
cool to room tl ,,ldLule, t-Butylmethylamine (0.2ml) was added and the
reaction stirred at room ~ , -, dLule for 1.5 hours. During this time a
white precipitate formed which was filtered off. t-Butylmethylamine
(0.4ml) was added to the toluene solution and the reaction mixture heated
to 7DC for 7 hours before allowing to cool overnight. The mixture was
then poured into water and extracted into ethyl acetate 3 times. The
combined organic layers were washed with brine, dried over MgS04, and
concentrated to give the crude product. Co1umn chromatography eluting with
ethyl acetate / hexanes 1:1 gave the product as a white powder (150mg,
23%). M.p. 136 - 138C
EXAMPI F 74 Preparati on of Compound 234: 3 - ( (N -ethyl ) dimethyl ami noacetyl )
amino-1- (3-tri f 1 uoromethyl ) phenyl -2 -pyrrol i dinone
A solution of 3-(~-ethyl)amino-1-(3-trifluoromethyl)phenyl-2-pyrrolidinone
(0.105 9) in dichloromethane (10 ml) was treated with chloroacetyl chloride
(0.031ml) and left to stand for 1 hour. The solution was then washed with
dilute hydrochloric acid followed by saturated sodium bicarbonate, then
dried (MgS04) and evaporated under reduced pressure. The residue, which
contained crude 3-((N-ethyl)chloroacetyl)amino-1-(3-
trifluoromethyl)phenyl-2-pyrrolidinone, was dissolved in te~ hJ~lu~u
(10 ml) and treated with a 40% aqueous solution of dimethylamine (2 ml)
together with sodium iodide (ca. 5mg). The resultant mixture was stirred
at room t' ,~dLule for 2 hours, then water was added, and the mixture was
extracted with ethyl acetate. The ethyl acetate extract was dried (MgS04)
and evaporated under reduced pressure, to leave the title compound
(0.065 9).
2 1 9 0 9 7 9 GB9s/olzz~
WO 95/33719 PCT/
- 133 -
EXAMPI F 7~ Prep~r~tion of Compounts 235, 236 237 ~nd 238
A stirred solution of Compound 53 (0.200 g) (prepared by a method sim;lar
to that described in Example 1) in toluene (10 ml) was treated with
glyoxy1ic acid Dhy~ldte (0.042 9) and the mixture was heated under
reflux for 4 hours, with the water produced being removed using a Dean and
Stark trap. The resultant solution was evaporated under reduced pressure
to dryness, and the residue was separated by chromatography on silica gel,
eluting with ethyl acetate/hexane mixtures, to afford each of the title
compounds in a pure state. Compounds 23q and 235 are diastereomers, as are
Compounds Z36 and 277.
EXAMPLE 76 Prep~r~tion of Compound 239: N-(l,l-dimethylprop~rgyl) [3-(3-
trifl,,~,. ;' ~y)phenyl-4-oxazolidinDne-5-yl~cetamite
Step 1 Preparation of Benzyl [3-(3-trifluu,, LhoAy)phenyl-4-
oxazolidinone-5-yl]acetate
A solution of methyl [3-(3-trifl ~l~ Ll,oAy)phenyl-4-oxazolidinone-5-yl]
acetate (prepared by a method similar to that described in Example 15)
(0.719 9) in benzyl alcohol (10 ml) was treated with 1 drop of concentrated
sulfuric acid, and stirred for 48 hours, after which time gas
chromatrography indicated that the reaction had gone to 2% completion. A
further drop of concentrated sulfuric acid was added, and the mixture was
stirred for a further 13 days, when gas chromatography indicated that the
reaction had gone to 86% completion. The mixture was diluted with diethyl
ether, and the solution was washed with water, dried (MgS04) and evalJu~tll.d
under reduced pressure to leave a colourless liquid, which contained benzyl
alcohol. Kugelrohr distillation at 100C at 0.01 mm Hg removed the benzyl
alcohol, leaving the crude title compound as a clear liquid, which
contained ca. 12% of the methyl ester starting material. This was used in
the next step without further purification.
21 90979
WO 9~133719 PCT/GB9~10122-1
- 134 -
Ste~ 2 Preparation of [3-(3-trifluoromethoxy)phenyl-4-oxazolidinone-
5-yl]acetic acid
A solution of benzyl Z-[3-(3-trifluoromethoxy)phenyl-4-oxazolidinone-
5-yl]acetate from Step 1 (0.7509) in ethanol (8 ml) containing
trifluoroacetic acid (5 drops) was hydrogenated over a 5% palladium on
charcoal catalyst for 29 hours, and the mixture was then filtered through
Hyflo (washing through with ethanol). The filtrate was evaporated under
reduced pressure to give a brown oil, which was purified by chromatography
over silica-gel, eluting with a 60:40 mixture of hexane/ethyl acetate, to
afford the tit~e compound as a white solid (0.3009).
SteD 3 Preparatjon of [3-(3-trifluoromethoxy)phenyl-4-oxazolidinone-
5-y l ] acety l ch l ori de
A stirred suspension of [3-(3-triflu~,. L~,u,~y)phenyl-4-oXaZolidinone-5-yl]acetic acid from Step 2 above (0.290 9) in carbon tetrachloride (3 ml)
was treated with oxalyl chloride (0.130 9), and the mixture was heated
under gentle reflux for 2 hours. The resultant colourless solution was
cooled and evaporated under reduced pressure to leave the title cûmpound
(0.295 9).
Preparation of N-(1,1-dimethylpropargyl) [3-(3-trifluo,~ u~y)
phenyl-4-oxazol idinone-5-yl]acetamide
A stirred solution of l,l-dimethylpropargylamine (131 mg) in diethyl ether
(1 ml) was treated with a solution of [3-(3-trifluu" .' y)phenyl-4-
oxazolidinone-5-yl]acetyl chloride from Step 3 above (0.220 9), and the
resultant suspension was stirred for a further l hour. The mixture was
filtered, and the filtrate WdS evaporated under reduced pressure to leave
the crude title compound as a white solid. This was purified by
preparative thin layer chromatography, eluting with ethyl acetatelhexane
(1:1), to afford the pure title compound as a white solid (0.214 9).
21 90979
WO 95/33719 PCT/GB95/0122 1
- 135 -
EXAMPLF 77 Prep~ration of Compound 244: N-(t-butyl)~3-(3-trifluoromethyl-
4-fluoro)phenyl-4-oxazoli.' ' yl]acetamide
Stev 1 preparation of the ethyl ester of N-(3-trifluoromethyl-4-
fluoro)phenyl fumaric acid amide
A stirred solution of 3-trifluoromethyl-4-fluoroaniline (21.10 9) and
monoethyl fumarate (17.9û 9) in t~LIuhyJ.,arll,d,, (100 ml) was treated slowly
with a solution of dicyclohexyl carbodiimide (24.30 9) in tet.al,yi,uru,~"
(50 ml). The resultant thick white suspension was stirred for a further 1
hour, and was then filtered. The filtrate was eYaporated under reduced
pressure, the residue was triturated with hexane, and the white precipitate
was filtered off, washed with hexane, and dried, affording the title
compound as a white solid (18.70 9). A further 6.30 9 of this compound was
obtained by concentration of the mother liquors from the trituration under
reduced pressure. I
~_Z Preparation of N-(3-trifluoromethyl-4-fluoro)phenyl fumaric acid
amide
A stirred slurry of the ethyl ester from Step 1 above (23.50 9) in ethanol
(80 ml) was treated with a solution of sodium hydroxide (3.10 9) in water
(20 ml). The mixture was stirred for 30 minutes, acidified to pH 1 using
2M hydrochloric acid. The resultant white precipitate was filtered off,
washed with water and dried, affording the title compound as a white solid
(20 .409) .
Ster~ 3 Preparation of the t-butyl ester of N-(3-trifluoromethyl-4-
fluoro)phenyl fumaric acid amide
A suspension of the acid from step 2 (10.00 9) in toluene (150 ml) was
stirred at 70C, and treated dropwise over 30 minutes with dimethyl
formamide (bis)-t-butyl acetal (34.5ml). The mixture was allowed to cool
and was filtered. The filtrate was washed with water, saturated sodium
bicarbonate and brine, dried (MgS04) and evaporated under
reduced pressure to leave a brown solid. This was purified by silica-gel
WO 9~i133719 2 1 9 0 9 7 9 PCTIGB9S10112.1
- 136 -
chromatography, eluting with ethyl acetate/hexane mixtures (1:9), and then
recrystallised from carbon tetrachloride to afford the title compound as a
white solid (5.00 9).
SteP 4 Preparation of t-butyl [3-(3-trifluoromethyl-4-fluoro)phenyl-
4-oxazol idinone-5-yl]acetate
A stirred solution of the ester from Step 3 aboYe (4.50 9) in dimethyl
formamide (2~ ml) was treated with a 80% dispersion of sodium hydride in
oil (0.0429), followed by paraformaldehyde (2.10 9). The white suspension
was stirred for 1 hour, and the reaction was then quenched by the addition
of 2M hydrochloric acid. The resultant mixture was extracted with diethyl
ether (2x) and the combined ether extracts were washed with brine, dried
(MgS04) and eYaporated under reduced pressure to leaYe an off-white solid
residue. This was recrystallised from diethyl ether/hexane to afford the
title compound (3.70 9) as colourless crystals.
Preparation of [3-(3-trifluoromethyl-4-fluoro)phenyl-4-
oxazolidinone-5-yl]acetic acid (Compound 243)
The t-butyl ester from Step 4 aboYe (3.60 9) was dissolYed in
dichloromethane (25 ml) and the solution was treated with trifluoroacetic
acid (7.60 ml). The clear solution was stirred for 1 hour, then washed
with brine, dried (MgS04) and eYaporated under reduced pressure to leaYe an
o;l which crystallised on standing. Purification by silica-gel
chromatography, eluting with ethyl acetate/hexane (1:1) afforded the title
compound as a white solid (3.05 9).
Ste~ 6 Preparation of [3-(3-trifluoromethyl-4-fluoro)phenyl-4-
oxazolidinone-5-yl]acetyl chloride
A solution of the acid from Step 5 aboYe (2.60 9) in carbon tetrachloride
(10 ml) was treated with oxalyl chloride (2.2 ml). The mixture was then
heated under gentle reflux for 1 hour, then cooled and eYaporated under
reduced pressure to leaYe the crude title compound as a pale brown oil
(2.75 9). This was used in the next stage without further purification.
WO 95133719 2 ~ 9 0 9 7 9 Pr~lGB9~/0l22~
- 137 -
~e~ Preparation of N-(t-butyl)[3-(3-trifluoromethyl-4-fluoro)phenyl-
4-oxazolidonone-5- yl]acetamide
A solution of the acid chloride from Step 6 above (0.390 g) in diethyl
ether (1 ml) was added to a stirred solution of t-butylamine (û.204 9) in
diethyl ether (3 ml). The thick suspension was stirred for 1 hour, then
water was added and the mixture was extracted with ethyl acetate. The
ethyl acetate extract was dried (MgS04) and evaporated under reduced
pressure to give an off-white solid, which was recrystallised from diethyl
ether to afford the title compound as a white solid (0.080 9). The mother
liquors from the recrystallisation were evaporated under reduced pressure,
and the residue was recrystallised from diethyl ether/hexane to give a
further crop of the title compound (0.200 9).
EXAMPLE 78 Preparation of Compound 253: 3-((N-Z-(t-butylcarbamoyloxy)
ethyl ) - t-butyl carbamoyl ) ami no-1- (3 -tri fl I ' y) phenyl -2-
pyrrol i di none
A stirred solution of 3-(N-2-1,yd~o~cLl,jl)amino-1-(3-trifluu,, Lho~y)
phenyl-Z-pyrrolidinone (prepared by a method analogous to that described in
Example 27, Step 1, but using 2-11j~ru,~J,Ll,anol in place of allylamine)
(0.069 9) in dichloromethane (S ml) was treated with t-butyl isocyanate
(0.022 ml) and triethylamine (0.028 ml), and the resultant solution was
stirred for 16 hour. The mixture was then evaporated to dryness, and the
residual mixture was separated by silica-gel ~11.. LUY~ Y, eluting with
ethyl acetate/hexane (1:2), to afford the title compound (0.064 9).
wo 95133719 2 1 9 0 9 7 9 PCTIGB95/012~.1
- 138 -
EXAMPLE 79 Preparation of Compound 280: 2-[1-(3-[trifluoromethyl]
phenyl)oxazolidin-2-on-3-yl]propanamide, and also of Compounds 278
and 279, the two dia~.. , of benzyl Z-[1-(3-[trifluoromethyl]phenyl)
OXAZOl i di n-2 -on-3 -yl] ~..1, ~e .
5te~ 1
A solution of 3-aminobenzotrifluoride (16.2g) in diethyl ether (50ml) was
added dropwise to a stirred solution of citraconic anhydride (11.29) in dry
diethyl ether (75ml) under nitrogen over a period of 30 minutes. During
the addition, the temperature was maintained at 15-20 C by cooling in an
ice-bath. A white precipitate was formed. Following the addition, the
reaction mixture was stirred or allowed to stand at room t~ ,-ld~UI~ for a
total of about 48 hours. The precipitate was then filtered off and
air-dried to give (Z)-N-[3-(trifluoromethyl)phenyl]-3- carboxybut-2-enamiae
(25.69) as a white powder, m.p. 135-7 C, lH NMR: ~ 1.95 (3H, s), 6.05 (lH,
s), 7.36 (lH, d), 7.50 (lH, t), 7.68 (lH, d), 8.08 (lH, s), 10.40 (lH, s),
12.80 (lH, br s) ppm.
Ste~ 2
A mixture of the amide from step 1 (5.46g) and diethyl
azodicarboxylate (3.489) in dry THF (80ml) was stirred at room tr ,.~c-Lu,~
under nitrogen, producing a yellow solution. A solution of
triphenylphosphine (5.259) and methanol (0.649) in dry THF (40ml) was then
added dropwise over a period of 45 minutes (mildly exothermic). Following
the addition, the reaction mixture was stirred at room ~ Lule for 4
hours. It was then stripped down under reduced pressure to leave a
colourless paste, which was purified by ~i", Luyl ,'J using a 1: 1
mixture of ethyl acetate and hexane as eluent to give
(Z)-N-[3-(trifluoromethyl)phenyl]-3- (methoxycarbonyl)but-2-enamide (3.299)
as a white, waxy solid, lH NMR: ~ 2.06 13H, d), 3.86 (3H, s), 6.08 (lH, d),
7.34 (2H, m), 7.75 (lH, d), 7.84 (lH, s), 8.84 (lH, br s) ppm.
A mixture of the amide from step 2 (1.609) and N-~" inimide
(99mg) in carbon tetrachloride (30ml) was heated under reflux for 38 hours.
Further portions of N-bl~ inimide (50mg) were added during heating,
.
WO 95133719 2 1 9 0 9 7 9 PCT/GB9~/0122.1
- 139 -
after 7 and 14 hours. The volatiles were then removed under reduced
pressure, and the residue was chromatographed using a 1: 1 mixture of
ethyl acetate and hexane as eluent to give a pale yellow solid (1.3Zg),
which was combined with material from a parallel reaction on the same
scale. The resulting material, a pale yellow solid (2.569), was shown to
contain about 609~ (E)-N-[3-(trifluoromethyl)phenyl]-3-
(methoxycarbonyl)but-2-enamide. This impure material was used directly in
step 4.
SteD 4
Aqueous sodium hydroxide (50ml, 2M solution) was added to a stirred
solution of the crude amide from step 3 (2.56g) in t~L~ dlurul~n (75ml),
producing a deep purple solution. After about 2 hours, the reaction
mixture was poured into water. The resulting mixture was washed with ethyl
acetate, acidified with concentrated hydrochloric acid, and then extracted
with dichloromethane. The dichloromethane extracts were washed with water
and dried. Removal of the solvent under reduced
pressure then gave a sticky solid which, on trituration with diethyl ether,
yielded a pale cream-coloured powder (1.309). Analysis by proton NMR
showed that this powder was a ca. 2: 1 mixture of the (E)- and (.)-isomers
of N-[3-(trifluoromethyl)phenyl]-3-carboxybut-2-enamide.
5teD 5
The amide from step 4 (1.289) and diethyl azodicarboxylate (0.8179)
were stirred as a mixture in dry THF (40ml) under nitrogen. A solution of
triphenylphosphine (1.239) and benzyl alcohol (0.5079) in dry THF (20ml)
was added dropwise over 10 minutes (mild exotherm). An hour later, the
solvent was stripped off under reduced pressure and the residue was
triturated with diethyl ether. The insoluble material, which was
1,2-dicdr~c~ J,~ine (0.529), was discarded. The soluble material was
chromatographed using a 1: 2 mixture of ethyl acetate and hexane as eluent
to give
(E)-N-[3-(trifluoromethyl)phenyl]-3-(benzyloxycarbonyl)but-2-enamide
(0.6979) as a white powder, m.p. 116-8 C, lH NMR: ~ 2.40 (3H, d), 5.27
(2H, s), 6.94 (lH, d), 7.40-(7H, m), 7.70 (2H, m), 7.89 (lH, s) ppm.
WO 9S/33719 2 1 9 0 9 7 9 PCT/GB9510122.1
- 140 -
A solution of the amide from step 5 (0.6369) in dry DMF (lOml) was
added dropwise over 10 minutes to a stirred suspension of sodium hydride
(7mg of a 60% oil dispersion) in dry DMF (lOml) under nitrogen. The
reaction mixture turned dark red. After 5 minutes, paraformaldehyde
(0.2639) was added as a solid in one portion, and the resulting mixture was
stirred at room t ,~ldLule for 2 hours, during which time the colour
fdded. The reaction mixture was poured into water (50ml) containing 2M
hydrochloric acid (2ml), and this aqueous mixture was then extracted with
diethyl ether. The extracts were washed with brine, dried, - LlaLed
and chromatographed using a 1: 3 mixture of ethyl acetate and hexane as
eluent to give a roughly 1: 1 mixture of dia,L~ of benzyl
2-[1-(3-[trifluoromethyl]phenyl)oxazolidjn-2-on-3-yl]propanoate (0.3569) as
a viscous colourless oil. HPLC on Sorbsil C30TM silica gel using a 1: 5
mixture of ethyl acetate and hexane as eluent then separated these
diastereomeric products (the configurations of these products were not
assigned). Dia,Le,c f A, eluted first, was a viscous, colourless gum
(0.1079), lH NMR as in Table III; diastereomer B, eluted second, was a
white powder (0.1189), lH NMR as in Table lII.
Ste~ 7
Trifluoroacetic acid (1 drop) and 10% palladium on carbon (lOmg) were
added successively to a stirred solution of diastereomer A of the
propanoate from step 6 (0.1059) in ethyl acetate (15ml). The resulting
mixture was treated with hydrogen at ~i , ', ic pressure for 6 hours at
room t ,~YdLule. The ai ~' ~e of hydrogen was then replaced with
nitrogen, and the reaction mixture was allowed to stand overnight and was
then filtered through 'Hyflo'TM. The filtrate was cu,,~e,,L~dLed to leave a
single diastereomer of 2-[1-(3-[trifluoromethyl]phenyl)oxazolidin-2-on-
3-yl]propanoic acid as a white solid (73mg), lH NMR: ~ 1.33 (3H, d), 3.17
(lH, m), 4.92 (lH, m), 5.54 (2H, s), 7.52 (2H, m), 7.80 (2H, m) ppm.
WO 95/33~19 2 1 9 0 9 7 9 PCTIGB9~/0122~
- 141 -
St~ 8
A mixture of the propanoic acid from step 7 (72mg) and oxalyl chloride
(5ml) was stirred at room t, ,a~ule for 1 hour, then all the volatiles
were stripped off under reduced pressure. The residue was taken up in dry
dichloromethane (8ml) and stirred under nitrogen, and a solution of
t-butylamine (17mg) and triethylamine (26mg) in dry dichloromethane (2ml)
was added dropwise over 2 minutes (white fumes and mild exotherm). The
resulting mixture was stirred at room tl, e, aLu.c for 3.5 hours, then
washed with water, dried, cu.,cE..LluLed and chromatographed using a 1: 1
mixture of ethyl acetate and hexane as eluent to give a single diastereomer
of N-t-butyl-[1-(3-[trifluoromethyl]
phenyl)oxazolidin-2-on-3-yl]propanamide, containing small amounts of the
starting acid and its corresponding anhydride, as a colourless gum (46mg).
After this sample had been allowed to stand for a week, analysis showed
that the anhydride had gone, presumably having been hydrolysed to the acid.
The sample then comprised the single diastereomer of N-t-butyl
2-[1-(3-[trifluoromethyl]phenyl)oxazolidin-2-on-3-yl]propanamide containing
about 25% of the starting acid. This material was a solid, with lH NMR for
the amide as shown in Table III.
EXAMPLE ~30 Prep~ration of Compound 285: N-[pyrid-2-ylmethyl]-N-~1-(3-
[trifluoromethyl]phenyl)pyrrolidin-2-on-3-yl]-3,3-dimethylbut~n~mide.
SteD 1
Bromine (4.3ml) was added over a period of 30 minutes, below the
surface of the reaction mixture, to a stirred mixture of
gamma-butyrolactone (8.609) and ~ ,u, tribromide (0.2ml), heating the
reaction mixture throughout to a t, .aLule between 100 and 110 C.
Following the addition, the mixture was heated with stirring at 100C for 2
hours, then allowed to cool to 50 C. Dry DMF (O.Olml) was added, the
mixture was heated to 90 C, thionyl chloride (8.6ml) was added dropwise
over 20 minutes (effervescence), and the resulting mixture was heated at
100C for a further 3 hours. After cooling, the reaction mixture was
subjected to short-path distillation (Kugelrohr), and all material which
distilled at less than 80C at 1-2 mbar was collected. This distillate was
... , . . . . . . ... ... .. . .... _ ... _ . . _ . .... .. . . . . . ..
WO 95/33719 2 1 q O q 7 q PCT/GB95/01221
- 142 -
a dark yellow oil (17.59), containing roughly 50% (by lH NMR)
2,4-dibromobutanoyl chloride. This crude material was Lsed directly for
step 2.
SteD 2
A solution of 3-amilloL~ uL, if luoride (5.329) and triethylamine
(3.679) in dry THF (50ml) was added dropwise over 3û minutes to a solution
of the crude acid chloride from step 1 (17.59) in dry THF (75ml), cooled in
an ice-water bath to keep the t, ~Lure below 10C (a precipitate was
formed). The resulting mixture was allowed to warm to room temperature,
and it was then stirred for 5 hours and allowed to stand overnight. The
mixture was poured into lM hydrochloric acid (lOOml), and then extracted
with ethyl acetate. The extracts were washed with brine, dried,
concentrated, then .~"1 Loy,ap~ using a 1: 3 mixture of ethyl acetate
and hexane as eluent to give almost pure
N-[3-(trifluoromethyl)phenyl]-2,4-dibromobutanamide as a very pale brown
solid (11.959). lH NMR: ~ 2.57 (lH, m), 2.73 (lH, m), 3.59 (2H, m), 4.70
(lH, dd), 7.46 (2H, m), 7.73 (lH, m), 7.85 (lH, s), 8.23 (lH, br s) ppm.
~L~
Sodium hydride (1.29 of a 60% dispersion in oil) was added in portions
over 10 minutes to a stirred solution of the amide from step 2 (11.679) in
dry THF (150ml) under nitrogen (effervescence and mild exotherm). The
resulting mixture was stirred at room t, .r~Lule for 4 hours, then further
sodium hydride (0.259 of a 60% dispersion in oil) was added, and the
mixture was stirred for a further 2 hours. Water was carefully added to
the reaction mixture, and it was extracted with ethyl acetate. The
extracts were washed with brine, dried, filtered and ,1", Lrryl~rh_.. using
a 1: 3 mixture of ethyl acetate and hexane as eluent to give
3-bromo-1-[3-(trifluoromethyl)phenyl]pyrrolidin-2-one (5.709) as a white
powder. lH NMR: ~ 2.48 (lH, m), 2.78 (lH, m), 3.87 (lH, m), 4.08 (lH, m),
4.60 (lH, m), 7.48 (2H, m), 7.90 (2H, m) ppm.
Step 4
A solution of 2-(aminomethyl)pyridine (1.959) and triethylamine
(0.919) in dry THF (25ml) was added dropwise over 1 hour to a stirred
wo 9s/337l9 i ` 2 1 9 0 9 7 9 PCrlGB95/0122~
- 143 -
so1ution of the bl~ ,y~lolidinone from step 3 (0.924g) in dry refluxing
THF (25ml) under nitrogen. The reaction mixture was then heated under
reflux for 24 hours and allowed to cool. The solYent was stripped off
under reduced pressure, and the residue was taken up in ethyl acetate,
~ashed with water, dried, concentrdted under reduced pressure, and
chromatographed using a 9: 1 mixture of chloroform and ethanol as eluent
to give 3-(pyrid-2-ylmethylamino)-1-[3-(trifluoromèthyl)phenyl]
pyrrolidin-2-one as a mobile yellow oil (1.089) which set to a waxy solid,
m.p. 82-3 C, on standing. lH NMR: i~ 2.05 (lH, m), 2.50 (2H, m), 3.70 (lH,
dd), 3.84 (2H, m), 4.10 (2H, 'AB quartet'), 7.19 (lH, m), 7.40 (2H, m),
7.49 (lH, m), 7.69 (lH, dt), 7.90 (2H, m), 8.58 (lH, m) ppm.
Steo 5
A solution of the aminopyrrolidinone from step 4 (0.335g) and
triethylamine (0.1119) in dry dichloromethane (6ml) was added dropwise over
10 minutes to a stirred solution of 3,3-dimethylbutanoyl chloride (0.14~9)
in dry dichloromethane (2ml) at room t" .~ e under nitrogen (white
fumes and exotherm). The resulting mixture was stirred for 7 hours and
allowed to stand overnight. It was then washed with water, dried,
concentrated and chromatographed using a 9: 1 mixture of ethyl acetate and
ethanol as eluent to give N-[pyrid-2-ylmethyl]-N-[1-(3-
[trifluoromethyl]phenyl)pyrrolidin-2-on-3-yl]-3,3-dimethylbutanamide as a
pale yellow gum (0.3559), with lH NMR as in Table III.
The following compounds were prepared from 3-bromo-1-[3-
(trifluoromethyl)phenyl]pyrrolidin-2-one (the product of step 3) using
analogous routes: Compounds 287, 289 and 283.
EXAMPLE 81 Prepar~tion of Compound 286: N-[pyrid-2-ylmethyl]-N-[1-(3-
[trifluoromethyl]phenyl)pyrrolidin-2-on-3-yl]-N'-~t-butyl]urea
Step 1
t-Butyl isocyanate (0.2979) was added to a stirred solution of
3-(pyrid-2-ylmethylamino) -1-[3-(trifluoromethyl)phenyl]pyrrol idin-2-one
(0.3359, prepared as described in Example 80) and triethylamine (0.3049) in
dry dichloromethane (8ml) under nitrogen. The resulting mixture was then
... ... .... . .... _ . . . . .. _ .... . , . . _ ,, _ _ . .
wo 95133719 2 1 9 0 9 7 9 PCT/GB9~10122.1
- 144 -
stirred for 7 hours, and allowed to stand overnight. The volatiles were
stripped off under reduced pressure, and the residue was purified by
chromatography, using a 9: 1 mixture of ethyl acetate and ethanol as
eluent, to give N-[pyrid-2-ylmethyl]-N-[1-(3-[trifluoromethyl]phenyl)
pyrrolidin-2-on-3-yl]-N'-[t-butyl]urea as a white powder (0.2909), m.p.
147-9C, with lH NMR data as in Table III. The following compounds were
prepared by analogous methods: Compounds 288, 290 and 284.
EXAMPLE B2 Preparation of Compound 281: N-[methyl]-N-[1-(3-
~trifl..~, ~h~]phenyl)pyrrolidin-2-on-3-yl]-N'-[2-(trifluoromethyl)prop-
2-yl] urea
A solution of phosgene in toluene (4.0ml of a 1.93M solution) was
added to 2-(trifluoromethyl)prop-2-ylamine hydrochloride (0.4209, prepared
from 2-aminoisobutyric acid according to the procedure described in J.
Or~anic Chem., (1961), ~, 1406) in toluene (4.0ml), and the resulting
mixture was heated with stirring at 60C for 2 hours. Most of the solid
amine hydrochloride dissolved during this time. The mixture was allowed to
cool to room tr, ~dLule and the excess phosgene was removed under reduced
pressure. 3-(Methylamino)-1-[3-(trifl ~., 'I y)phenyl]pyrrolidin-2-one
(0.509, prepared ir, analogy to the procedure for the preparation of
3-(Methylamino)-1-[3-(trifluoromethyl)phenyl] pyrrolidin-2-one describêd in
Example 10) and triethylamine (0.1859) were then added successively with
stirring, and the mixture was stirred for 4 hours at room t~ dLule and
allowed to stand overnight. The reaction mixture was applied to the top of
a column of silica gel, and eluted with a 7: 3 mixture of ethyl acetate
and hexane to give N-[methyl]-N-[1-(3-[trifluv, L~.v~y~phenyl)pyrrolidin-
2-on-3-yl]-N'-[2-(trifluoromethyl) prop-2-yl]urea as a white solid
(0.2209), m.p. 115-6 C, with lH NMR data as in Table III.
WO 95133719 2 ~ 9 0 9 7 9 PCTIGB95/0122~
- 145 -
EXAMPL F 83 Prep~r~ti on of Compound 301: N- (propargyl ) -N- [3- (3-
trifluoromethyl)phenylthiazolidin 4 5 yl]-li'-(neopentyl)ure~
5teD 1
Propargylamine (4.0619) was ddded to a stirred solution of
5-chloro-3-[3-(trifluoromethyl)phenyl]thiazolidin-4-one (5.1969, prepared
as described in Example 1); the reaction mixture darkened, and a thick
precipitate formed. The mixture was stirred for 9 hours and the volatiles
were stripped off under reduced pressure. The residue was taken up in
t-butyl methyl ether (orange solution plus solid) and washed successively
with water (x3) and brine, then dried, co~ ..L~àLed and chromatographed to
give 5-propargylamino-3-[3-(trifluoromethyl)phenyl] thiazolidin-4-one as a
viscous brown oil (3.7209), lH NMR: ~ 2.28 (2H, m), 3.66 (2H, dd), 4.77
(lH, d), 4.88 (lH, d), 5.19 (lH, s), 7.50 (2H, m), 7.75 (2H, m) ppm.
~Z
A mixture of 3,3-dimethylbutanoic acid (0.5779), diphenylphosphoryl
azide (1.3699) and triethylamine (0.5039) in dry toluene (15ml) was heated
with stirring at about 80 C under nitrogen for 2 hours. Effervescence
began to occur after about 20 minutes, and had ceased after 2 hours. The
mixture was allowed to cool to room tl, atul~, and the thiazolidinone
from step 1 (0.5009) and triethylamine (0.1679) were added with stirring.
The following morning, the solvent was removed under reduced pressure, and
the residue was chromatographed using a 3: 2 mixture of hexane and ethyl
acetate as eluent to give N-(propargyl)-N-
[3-(3-trifluoromethyl)phenylthiazolidin-4-on-5-yl]-N'-(neopentyl)urea as a
solid (0.4849), almost pure by NMR spectroscopy. Recrystallisation from a
mixture of ethyl acetate and hexane gave the product as a yellow
crystalline solid (0.2679), m.p. 145-145.5C, with lH NMR data as in Table
lII .
The urea Compound 302 was prepared from 5-propargylamino-3-[3-
(trifluoromethyl)phenyl]thiazolidin-4-one, the product of step 1 above, and
3-chloro-2,2-dimethylpropanoic acid in the same way. The urea, Compound
WO 95/33719 2 1 9 0 9 7 9 PCT/GB95/0122.1
- 146 -
298 was made from 5-propargylamino-3-[3-(trifluoromethyl)
phenyl]thiazolidin-4-one and the available t-butyl isocyanate using the
method described for a related pyrrolidinone in Example 27. The amide
Compound 297 was prepared from 5-propargylamino-3-[3-(trifluoromethyl)
phenyl]thiazolidin-4-one and 3,3-dimethylbutanoyl chloride by a
conventional method similar to that described for related compounds in
Examples 3 and 25.
The following ureas and amides were prepared from 5-chloro-3-[3-
(trifluoromethyl)phenyl]thiazolidin-4-one using appropriate amines and then
isocyanates (either directly or produced ln situ by the Curtius
red"~, L of appropriate acids using the method described above) or
acid chlorides according to the steps described above: Compounds 124, 125,
291, 292, 293, 294, 295, 296 and 300.
EXAMPLE 84 Prepar~tion of Compound 299: N-(methoxy)-N-~3-(3-
trifluoromethyl)phenylthiazolidin 4-~ 5 yl]-N'-(t-butyl)ure~
.Step 1
A viscous mixture of methoxylamine hydrochloride (1.4829) and
triethylamine (1.7939) in DMF (lOml) was stirred at room t ,_,cLul~: for 5
minutes. 5-Chloro-3-[3-(trifluoromethyl)phenyl]thiazolidin-4-one (1.0009,
prepared as described in Example 1) was added, and the mixture turned pink.
It was stirred for 3 hours, then diluted with water and extracted with
diethyl ether. The extracts were washed with water and then brine, dried,
cu,,.~llLldLed and chromatographed using a 1: 1 mixture of ethyl acetate and
hexane as eluent to give a dark oil (0.5799) containing
5-methoxyamino-3-[3-(trifluoromethyl)phenyl]thiazolidin-4-one. This oil
was then combined with similar material from a second ~ il L which had
been performed on 4 x the scale described aboYe. Further ,IIl. Luyl~uily
using the same eluent then gave a roughly 1.1: I mixture of
5-methoxyamino-3-[3-(trifluoromethyl)phenyl]thiazolidin-4-one and
5-hydroxy-3-[3-(trifluoromethyl)phenyl]thiazolidin-4-one as a yellow oil
(3.5429); this was treated without further purification with t-butyl
isocyanate and triethylamine in dichloromethane in the way described in
wo 95/33719 ~ 2 1 q 0 9 7 9 PCT/GB9~/0122~
- 147 -
Example 81 above to give a 35% yield of N-[methoxy]-N-[3-(3-
[trifluoromethyl]phenyl)thiazolidin-4-on-5-yl]-N'-[t-butyl]urea as a white
solid, m.p. 128.5-lZ9.5C, and IH NMR data as in Table III.
The amide, Compound 126, was prepared from 5-methoxyamino-3-[3-
(trifluoromethyl)phenyl]thiazolidin-4-one and 3,3-dimethylbutanoic acid and
triethylamine using a conventional method similar to that described for
related compounds in Examples 3 and 25.
EXAMPLE 85 Preparation of Compound 303: 3-t-butyl-1-{[3-
(trifluoromethyl)phenyl~thiazolidin-4-on-S-yl}-i~idazolidin-2,4-dione
Step 1
Triethylamine (1.9749) was added to a stirred solution of
5-chloro-3-[3-(trifluoromethyl)phenyl]thiazolidin-4-one (2.5009, prepared
as described in Example 1) and the hydrochloride salt of the methyl ester
of glycine (1.3369) in THF (30ml). The reaction mixture, which became
bright yellow, was stirred or allowed to stand for a total of about a week.
The solvent was then removed under reduced pressure and the residue was
partitioned between ethyl ace~ate and water. The ethyl acetate layer was
washed with water and then brine, dried, c~ ed and chromatographed
using a 2: 3 mixture of hexane and ethyl acetate as eluent to give
5-[(methoxycarbonyl)methylamino]-3-[3-(trifluoromethyl)
phenyl]thiazolidin-4-one as a yellow solid (1.0149), lH NMR: ~ 2.60 (lH,
s), 3.56 (lH, d), 3.69 (lH, d), 3.75 (3H, s), 4.75 (lH, d), 4.82 (lH, dd),
5.19 (lH, s), 7.55 (2H, m), 7.72 (2H, m) ppm.
SteD 2
Triethylamine (0.2939) and t-butyl isocyanate (0.2879) were added to a
stirred solution of the thiazolidinone from step 1 (0.9709) in
dichloromethane. The resulting mixture was either stirred or allowed to
stand at room t~ u,~: for a total of about 6 days. The solvent was
then removed and toluene was added to the residue. The mixture was then
heated at 80C under nitrogen for a total of 8 hours, with intervening
periods at room t ,_,~u~t. Extensive chromatography and HPLC, using
_ _ _ _ _ _ _ _ _ . . . .
WO 95133719 2 1 9 0 9 7 q PCTIGB9510122.1
- 148 -
mixtures of ethyl acetate and hexane or t-butyl methyl ether and hexane as
eluents, followed by crystallisation and recrystallisation from mixtures of
ethyl acetate and hexane, then gave 3-t-butyl-1-{[3-
(trifluoromethyl)phenyl]thiazolidin-4-on-5-yl}-imidazolidin-2,4-dione as a
white crystalline solid (56mg), m.p. 199.5-Z01C, with lH NMR data as in
Table Ill.
EXAMPLE 86 Preparation of Compound 304: S-(t-butylaminocarbonylthio)-
3-[3-(trifluoromethyl)phenyl]thiazolidin-4-one
Step 1
A mixture of 5-chloro-3-[3-(trifluoromethyl)phenyl]thiazolidin-4-one
(5.0009, prepared as described in Example 1) and potassium thioacetate
(2.2279) were stirred together in DMF (50ml) (mild exotherm on mixing, and
mixture became dark brown). After about an hour, the mixture was diluted
with water and extracted with diethyl ether. The extracts were washed
successively with water and brine, then dried, ~ LI~Led and
chromatographed using a 2: 1 mixture of hexane and ethyl acetate as eluent
to give 5-acetylthio-3-[3-(trifluoromethyl)phenyl]thiazolidin- 4-one as a
dark oil (5.4239), lH NMR: ~ 2.41 (3H, s), 4.79 (lH, d), 5.04 (lH, dd),
5.49 (lH, d), 7.51- 7.77 (4H, m) ppm.
~L~
Gaseous ammonia was bubbled for 15 minutes through a stirred solution
of 5-acetylthio-3-[3-(trifluoromethyl)phenyl]thiazolidin-4-one (1.3009) in
methanol (15ml), cooled in an ice bath. The reaction mixture was then
allowed to warm to room t~ iule and stir for a further 3 hours.
Volatiles were removed under reduced pressure and the residue was
chromatographed using a 1: 1 mixture of ethyl acetate and hexane as eluent
to give a roughly 7: 3 mixture of 5-mercapto-3-[3-(trifluoromethyl)
phenyl]thiazolidin-4-one and the cu,IL,r Jing dimeric disulfide,
respectively, as a viscous yellow oil (0.9769). This was used without
further purification.
2190
WO 95/33719 PCT/GB95/0122.1
- 149 -
SteD 3
- t-Butyl isocyanate (0.2919) and triethylamine (0.2479) were ddded
successively to a stirred solution of the crude thiol from step 2 (0.6839)
in dichloromethane (7ml), and the mixture darkened to an orange colour.
After 40 minutes, the reaction mixture was applied to a column of silica
gel and eluted with a 1: 1 mixture of t-butyl methyl ether and hexane to
give S-(t-butylaminocarbonylthio)-3-[3-(trifluoromethyl)
phenyl]thiazolidin-q-one as a yellow solid (0.5319) containing as an
impurity roughly 209~ N,N'-di-t-butylurea. Recrystallisation from a mixture
of ethyl acetate and hexane gave 5-(t-butylaminocarbonylthio)-
3-[3-(trifluoromethyl)phenyl]thiazolidin-4-one as a yellow crystalline
solid (0.23gg), m.p.149.6-150.6C, with lH NMR data as in Table III.
EXAMPLE 87 Prep~r~tion of Compound 328: 3-(4,4,4-Trifluorobutanoyl-N-
methy~)amino-1-(3-trifl .~ )phenyl-2-pyrrolidinone
4,4,4-trifluorobutyric acid (0.319) was dissolved in CHzCl2 (lOmls),
cooled in an ice/water bath and stirred under a nitrogen ~L ;,' .e. To
this was added triethylamine (0.31mls) then pivaloylchloride (O.Z7ml)
dropwise. Stirring was continued for 60 minutes. A fine white needle
precipitate formed over this time. A solution of the amino pyrrolidinone
(prepared in a similar manner to that described in Step 2 of Example 10)
(0.49), DMAP (SOmg) and triethylamine (0.2ml) in CH2Cl2 (lOmls) was added
to the cooled reaction mixture. After 30 minutes at this I , ~a~u,~, the
reaction was allowed to warm to room t ,,~.~LIlle and stirred for 60
minutes. The mixture was diluted with CH2Cl2, washed with 2N HCl (aq)
(xZ), brine (xZ), dried (MgS04), filtered and evaporated. CI", LuyI~Jhy
on silica gave the amide as a gum (0.5439).
EXAMPIF 88 Preparation of Co~pound 326: 3-(Pentafl_ ..,.~ l-N-
methyl)~mino-1-(3-trifl_ .. ' .r)phenyl-2-pyrrolidinone
The amino-pyrrolidone (prepared in a similar manner to that described
in Step 2 of Example 10) (0.49) was dissolved in CHzClz (ZOml). To this
was added DMAP (50mg) and triethylamine (O.Zlml) with the reaction cooled
.. .. .. .... . . . _ _ . _ .. . . .. . . .
WO 9~;/33'719 2 1 9 0 9 7 9 PCIIGB95J0122`1
- 150 -
in an ice-water bath. Pentaf1uoropropionjc anhydride (0.3ml) was added
dropwise and the mixture stirred for 40 minutes with cooling. The mixture
was diluted with CH2Cl2, washed with 2N HCl (aq), brine (x1), dried
(MgSO4), filtered dnd .u.,cL,,L,~ILed. Purification of the residue by
chromatography on silica gave the amide as d $olid (0.59). m.p.
65 . S-67 . 5C .
EXAMPLE 89 Prep~ration of Compound 311: 3-(3-t-Butylimidazoline-
2,4-dione-1-yl)-1-(3-trifl .~ ' y)phenyl-2-pyrrolidinone
A solution of Compound 130 (prepared as described in Example 31)
(0.5239) in ethylacetate (30ml) containing 10% Pd/C (lOOmg) was stirred
under an ai r~.e of hydrûgen at room t~ ,.,dLUl~. After 21 hours, the
catalyst was filtered and sûlvent evaporated. Analysis of the residue
showed that the reaction was only 40% complete. The residue was
re-dissolved in ethylacetate and SOmg of 10% Pd/C added. Hydrogenation was
achieved at 3 bar pressure using a hydrogenator for six hours. The
catalyst was filtered off and the filtratê evapûrated. This process was
repeated, except using a pressure of 4 bar hydrogen fûr 4 hours. After
filtering the catalyst and evaporation of solvent, the residue was purified
using chromatography on silica, eluting with 60% ethyl acetate in hexane,
to give the reduced product as a colourless solid (0.3859) m.p 153-154C.
EXAMPLE 90 Prep~ration of Compound 308: 3-(2,2,2-TrichlL ~ L Jl-
N-methyl)~mino-1(3-trifluoromethyl)phenyl-2-pyrrolidinone
To a solution of the amino-pyrrolidinone (prepared as described in
Step 2 of Example 10) (0.1389) in CH2Cl2 (5ml) at 0C (ice/water b`ath
cooling) was added triethylamine (0.096ml) followed by
2,2,2-trichloroethylchloroformate (0.088ml). The clear solution was
stirred at 0C for 1 hour. The mixture was diluted with CH2Cl2, washed
with water and dried (MgS04). Evaporation of the solvent and purification
of the residue by chromatography on silica gave the carbamate as a
colourless gum (0.209).
WO 95/33719 2 1 9 0 9 7 9 PCT/GB9510122-1
- 151 -
EXAMPLF 91 Preparation of Compound 267: 5-(3-ally1-1-t-butyl-3-ureido)-3-
(3-trifluoromethylphenyl)oxazolidin-4-one.
Stev 1 Preparation of 2-(methylthio)-N-(3-trifluoromethylphenyl)acetamide
3-Trifluoromethylaniline (16.1g) was added dropwise to a rapidly
stirred suspension of ~ ched sodium hydride (4.09, 60% in mineral
oil) in dimethylsulphoxide (50ml) under a nitrogen atmosphere, with water
bath cooling to 20C. After 30 minutes ethyl (methylthio)acetate (14.79)
was added dropwise with cooling tQ 20C. After stirring for 3 hours
half-saturated aqueous potassium dihydrogen phosphate (300ml) was added
cautiously with cooling to 20C. The mixture was extracted with diethyl
ether (5xlOOml), the extract washed with water (2x50ml), dried over sodium
sulphate, filtered and ether evaporated under reduced pressure to leave the
crude product as a yellow solid (23.89). A sample was recrystallised from
hexane solution for analysis. m.p. 75-77C. lH NMR (CDCl3):
2.21(3H,s); 3.38(2H,s); 7.45-7.85(4H,m); 8.85(1H,bs).
SteD 2 Preparation of N-(Ethoxymethyl)-N-(3-trifluoromethyl-
phenyl)-2-(methylthio)acetamide
Chloromethyl ethylether (18.19) was added dropwise, during 20 minutes,
to a vigorously stirred mixture of crude product of Step 1 (21.89)
dissolved in dichloromethane (SOml~, 52% aqueous sodium hydroxide (349) and
benzyl triethylammonium chloride (0.29), with water-bath cooling to 20C.
After 30 minutes the mixture was treated with saturated aqueous potassium
dihydrogen phosphate until pH 8, at 20C, extracted with dichloromethane
(5xlOOml), the extract dried over sodium sulphate, filtered and
concentrated under reduced pressure to give the crude product as a yellow
oil (27.59). 1H NMR (CDCl3): ~ 1.23(3H,t); 2.21(3H,s); 3.0(2H,s);
3.68(2H,bq); 5.1(2H,s); 7.58(4H,m).
wo ss/337ls 2 1 9 0 9 7 9 PCIIGB9510122~ --
- lSZ -
SteD 3 Preparation of N-(Ethoxymethyl)-N-(3-trifluoromethyl-
phenyl)-2-~methylsulphinyl)acetamide
A solution of sodium periodate (20.59) in water (19Oml) WdS added
dropwise to a stirred solution of crude product of Step 2 in ethanol
(850ml) at 5C. The mixture was allowed to reach 20C gradually and
stirred for 24 hours, than cu.,,c,,~,~Led under reduced pressure. The
concentrate was extracted with dichloromethane (500ml), the extract dried
oYer sodium sulphate, filtered and cull,c,,~,aLed under reduced pressure to
give the crude product as a brown oil (27.59). lH NMR (CDC13):
1.24(3H,t); 2.76(3H,s); 3.56(2H,s); 3.67(2H,q); 5.12(2H,s);
7 .51-7 .69(4H,m) .
SteD 4 Preparation of 5-Hydroxy-3-(3-trifluoromethyl-
phenyl)-oxazolidin-4-one
Trifluoroacetic anhydride (17.69) was added dropwise to a stirred
solution of crude product of Step 3 in tetl- ' ~.I,uru,~... (22ûml) with water
bath cooling to 20C. After 2 hours the mixture of left to stand for 20
hours. A solution of sodium hydrogen carbonate (14.19) in water (220ml)
was added during 5 minutes with stirring and cooling to 20C. After 30
minutes the mixture was refluxed for 43 hours, cooled to 25C, extracted
with dichloromethane (3x300ml), the extract dried over magnesium sulphate,
filtered and cu..,~..LT~L~ under reduced pressure to give a brown oil
(21.49). The brown oil (20.099) was dissolved in 1,4-dioxane (SOOml) and
hydrogen chloride yas bubbled in for 3~ hours, with stirring, at 23C. The
mixture was left in a stoppared flask for 20 hours, LraLed under
reduced pressure, dissolved in dichloromethane (400ml), neutralised with a
minimum of saturated aqueous sodium hydrogen carbonate, dried over
magnesium sulphate, filtered and cu,,.~.,LIa~ed under reduced pressure to a
brown oil (21.99). The oil was subjected to column chromatography on
silica gel, gradient eluting with dichloromethane/t-butyl methylether
mixtures to give the crude product as a yellow gum (4.79). The gum was
recrystallised from hexane solution to give a yellow solid. 1H NMR
(CDC13): ~ 4.43(1H,bs); 5.49(1H,s); 5.68(2H,m); 7.49-7.8(4H,m).
WO 95/33719 2 1 9 0 9 7 9 PCT/GB95/0122.1
- 153 -
SteD 5 Preparation of 5-chloro-3-(3-trifluoromethylphenyl~oxazolidin-4-one
- Methanesulphonyl chloride (16mg) was added to a stirred solution of
5-hydroxy-3-(3-trifluoromethylphenyl)oxazolidin 4 one (27mg, from Step 4)
in diethyl ether (lml). After 5 minutes triethylamine (18mg) was added and
the mixture stirred for 20 hours. Water (lml) was added, the mixture
extracted with ether (3x5ml), the extract dried over magnesium sulphate,
filtered and concentrated under reduced pressure to give the crude product
as a yellow oil (22mg). 1H NMR (CDCl3): ~ 5.56(1H,d); 5.72(1H,d);
6.3(1H,s); 7.5-7.B(4H,m).
SteDs 5 and 6 Preparation of 5-(allylamino)-3-(3-trifluoromethyl-
phenyl)oxazolidin-4-one
Methanesulphonyl chloride (0.99) dissolved in diethylether (2ml) was
added to a stirred solution of 5-hydroxy-3-(3-trifluoromethylphenyl)-
oxazolidin-4-one (1.09, from Step 4) in dichloromethane (6ml).
Trie~hylamine (0.89) dissolved in ether (2ml) was added and the mixture
allowed to exotherm to 35C. After 2~ hours the mixture was cooled in an
ice-water bath and a solution of allylamine (0.929) in ether (2ml) added
dropwise. After 1 hour the mixture was treated with aqueous sodium
chloride (20ml), extracted with ether (3x80ml), the extract dried over
magnesium sulphate, filtered and concentrated under reduced pressure to
give the crude product as a yellow gum (1.39). lH NMR (CDCl3): ~
3.49(2H,d); 5.13(1H,dd); 5.21(1H,s); 5.3(1H,m); 5.44(1H,d); 5.48(1H,dd);
5.9(1H,m); 7.5-7.8(4H,m).
Ster 7 Preparation of 5-(3-allyl-1-t-butyl-3-ureido)-3-(3-trifluoro-
methylphenyl)oxazolidin-4-one
A solution of the product of Step 6 (D.43g) in t-butylisocyanate (2ml)
was stirred for 2 hours then left for 20 hours. The mixture was
concentrated under reduced pressure to give a yellow gum which was
subjected to column chromatography on silica gel, eluting with
dichloromethane: t-butylmethylether 98:2. This gave a yellow solid which
was recrystallised from hexane solution to give the product as a white
, . .. . . . .. . , _ . , . ...... _ . .. . .. ... . . . . . . .. .
WO 95/33719 2 1 9 0 9 7 9 PCT/GB95/0l2Z~
- 154 -
solid (0.21g). m.p.l49-150C. 1H NMR (CDC13): ~ 1.31(9H,s); 3.86(2H,m);
4.73(1H,s); 5.32(1H,d); 5.45(1H,d); 5.46(1H,dd); 5.56(1H,t); 5.88(1H,s);
5.95(1H,m); 7.5-7.8(4H,m).
EXAMPLE 92 Preparcation of Compound 266: 5-[N-(N-~llyl-2-t-butylacetamido]-
3-(3-trifluoromethylphenyl)oxazolidin-4-one
Pyridine (0.249) was added dropwise to a stirred solution of
t-butylacetylchloride (0.49) in dichloromethane (2ml) and the resulting
solution added dropwise to a stirred solution of 5-(allylamino)-3-(3-tri-
fluoromethylphenyl)oxazolidin-4-one (0.439, Example 91 Step 6) in
dichloromethane (8ml) at 7C. After stirring at 7C for 2 hours aqueous
sodium chloride (lOml) was added, the mixture extracted with diethylether
(3x50ml), the extract dried over magnesium sulphate, filtered and
concentrated under reduced pressure to give a yellow gum. The gum was
subjected to column chromatography on silica gel eluting with
dichloromethane:t-butyl methylether 99:1 t3 give a yellow solid which
yielded the product as a white solid (.269) on tritriation with cold
hexane. m.p. 92-93C. 1H NMR (CDCl3): ~ 1.07(9H,s); 2.26t2H,q);
4.13(2H,d); 5.32(1H,d); 5.33(1H,s); 5.47(1H,d); 5.48(1H,d); 5.62(1H,s);
5.89(1H,m); 7.4-7.8(4H,m).
A similar method was also used to prepare the compounds listed below.
Compound 269: 5-[N-(2-t-butyl-N-methylacetamido)]-3-(3-trifluu,, L~IV~
phenyl)oxazolidin-4-one;
NMR(CDCl3) : ~ 1.09(9H,s); 2.32(ZH,s); 3.12(3H,s); 5.47(1H,s); 5.58(1H,s);
5.95(1H,bs); 7.03-7.17(1H,m); 7.41-7.48(2H,m); 7.61(1H,s).
MPt: 99.5-102CC.
Compound 272: 5[N-(2-t-butyl-N-ethylacetamido)]-3-(3- trifl ,. LIIUA~
phenyl ) oxazol i di n-4-one;
2 1 9 ~ 9 7 9 s,0l22l
WO 95133719 PCT/GB9
- 155 -
NMR(CDCl3) : ~ 1.08(9H,s); 1.32(3H,t); 2.29(2H,d); 3.43-3.64(2H,m);
5.30(1H,bs); 5.45tlHrs); 5.58(11H,bs); 7.0-7.19(1H,m); 7.39-7.q5(2H,m);
7 .60(1H,bs) .
MPt: 95-97~C.
Compound 271: 5-[N-(2-t-butylacetamido)]-3-(3-trifl~ I,u,~
phenyl)oxazolidin-4-one;
NMR(CDCl3) : ~ 1.07(9H,s); 2/14(2H,s); 5.45(1H,s); 5.55(2H,m); 6.39(1H,bd);
7.05-7.13(1H,m); 7.38-7.44(2H,m); 7.60(1H,s).
MPt: 145-148C (dec).
EXAMPI F 93 Prepar~tion of Compound 265: 5-(3-methyl-1-t-butyl-3-
uveido)-3-(3-trifluoromethylphenyl)oxazolidin-4-one
This compound was prepared by a method analogous to Example 91.
m.p. 114-116C. 1H NMR (CDCl3): ~ 1.37(9H,s); 2.91(3H,s); 4.53(1H,bs);
5.47(1H,t); 5.56(1H,t); 6.08(1H,s); 7.5-7.9(4H,m).
The precursor compound was 5-(methylaminû)-3-(3-trifluoromethyl-
phenyl)oxazolidin-4-one. lH NMR (CDCl3): ~ 2.57(3H,m); 5.2(1H,d);
5.46(1H,d); 5.5(1H,dd); 7.5-7.9(4H,m).
EXAMPLE 94 Prep~ration of Compound 262: 5-[N-(2-t-butyl-N-
methyl~cet~mido]-3-(3-trifluoromethylphenyl)ox~zolidin-4-one
This compound was prepared by a method analogous to Example 92. m.p.
116-117C. 1H NMR (CDCl3): Major rotomer: ~ 1.09(9H,s); 2.32~2H,s);
3.13(3H,s); 5.5(1H,d); 5.6(1H,t); 5.9(1H,bs); 7.5-7.8(4H,m). The precursor
compound is detailed in Example 92.
WO 95/33719 2 1 9 0 9 7 9 PCI'IGB9510122J
- 156 -
EXAMPLE 95 Prepar~tion of Compound Z70: 5-[N-(2-t-butylacetamido]-3-
(3-trifluoromethylphenyl)oxazolidin-4-one
This compound was prepared by a method analogous to Example 92. m.p.
159-161C. 1H NMR (CDCl3): ~ 1/07(9H,s); 2.14(2H,s); 5.49(1H,d);
5.53(1H,dd); 5.59(1H,t); 6.41(1H,bd); 7.5-7.8(4H,m).
The precursor compound was 5-amino-(3-trifluoromethylphenyl)-
oxazolidin-4-one. IH NMR (CDCl3): ~ 2.35(2H,bd); 5.23(1H,t); 5.42(1H,d);
5.49(1H,dd); 7.5-7.8(4H,m).
EXAMPLF 96 Prepar~tion of Compound 260: S-[N-(1-methylcyclobutyl)-1-
Acetamide]3-(3-trifluoromethylphenyl)oxazolidin-4-one
Preparation of (3-trifluoromethylphenylamide)ethyl fumarate.
3-Trifluoromethylaniline (32.29) and ethyl fumarate (30.329) were
mixed and dissolved in tetrahyd,uru,d,, (65ml). A solution of
dicyclohexylcarbodimide (041.29) in tt:L. 'y~,uru, (lOOml) was then added
dropwise. The resultant mixture was left to stand overnight, it was then
filtered and the filtrate evaporated under reduced pressure to leave a wet
yellow solid which was re-crystallised from ether to give the title
compound (33.95g) as a white solid. NMR (CDC13): ~ 1.36(3H,t); 4.20(2H,q);
7.12(1H,d); 7.20(1H,d); 7.42(1H,d); 7.50(1H,t); 7.86(1H,s); 7.95(1H,d);
8.35(1H,bs) .
SteD Z Preparation of (3-trifluoromethylphenylamido)fumaric acid
Sodium hydroxide (2.78g) in water (120ml) was added to a solution of
(3-trifluoromethylphenylamido)ethyl fumarate (109 as prepared in 5tep 1) in
iso-propanol (180ml). The resultant mixture was left to stand over night
then evaporated under reduced pressure. The residue was acidified with
hydrochloric acid (2N) and extracted with ether. The extracts were dried
over magnesium sulphate and evaporated under reduced pressure to leave the
title compound (6.949) as an off white solid. NMR (CDCl3): ~ 6.89(1H,d);
7.20(1H,d); 7.33(1H,d); 7.43(1H,t); 7.92(1H,d); 8.06(1H,s).
WO95/33719 2 1 90979 PCIIGB95/0122.1
- 157 -
SteD 3 Preparation of (3-trifluoromethy1phenylamido)tert-butoxy carbonyl
- N,N-Dimethylformamide di-tert-butyl acetal was added to a suspension
of (3,trifluoromethyl phenyl amido)fumaric acid (8.249 as prepared in Step
2) in toluene 50ml at 75C. The resultant mixture was heated under reflux
under N2 for thirty minutes. It was allowed to cool then was washed with
water, saturated sodium bicarbonate and brine. The organic layer was dried
over magnesium sulphate and evaporated under reduced pressure. The residue
was chromatographed on silica using h~Yi Ll,,, (2:1) as eluant to give the
title compound (5.459) as a white solid. m.p. 104-5-106.5C. NMR (CDCl3):
~ 1.52(9H,s); 6.90(1H,d); 7.0(1H,d); 7.40(1H,d); 7.46(1H,t); 7.83(1H,d);
7.89(2H,s) .
Step 4 Preparation of 5-[methylene tert-butoxy carbonyl]3-trifluoromethyl-
phenyl)oxazolidin-4-one
A solution of (3-trifluoromethylphenyl amido)tert-butoxy carbonyl
(5.159 as prepared in Step 3) in dimethylformamide (25ml) was added
dropwise to a stirred suspension of sodium hydride (0.0659, 60% dispersion
in mineral oil) in dimethyl formamide (lOml). Paraformaldehyde (2.79) was
then added in one portion. After thirty minutes the resultant mixture was
poured into water and extracted with ether. The extracts were washed with
water and brine, dried over magnesium sulphate and evaporated under reduced
pressure to give the title compound (5.499) as a yellow solid. m.p.
78-81C. NMR (CDCl3): ~ 1.44(9H,s); 2.86(2H,m); 4.75(1H,m); 5.51(1H,s);
7.48(1H,d); 7.55(1H,t); 7.79(1H,d); 7.83(1H,s).
SteD S Preparation of 5-[Methylene carboxy]-3-(3-trifluoromethylphenyl)-
oxazol idin-4-one
Trifluoroacetic acid (lOml) was added to a solution of 5-[methylene
tert-butoxy carbonyl]-3-(3-trifluoromethylphenyl)oxazolidin-4-one (4.499,
as prepared in Step 4) in dichloromethane (75ml). The resultant mixture
was left to stand oYer night then poured into water and extracted with
ether. The extracts were washed with water, dried oYer magnesium sulphate
WO 95f33719 2 1 9 0 9 7 9 PCI'/GB95/0122.1
- 158 -
and evaporated under reduced pressure to give the title compound (5.329) as
a brown oil, sufficiently pure to be used in 5tep 8.
Ste~ 6 Preparation of l-methylcyclobutyl-1-acetamjde
Methylene cyclobutane (lOg) was added to a solution of acetonitrile
(6.629), glacial acetic acid (73.5ml) and cu,,~,,L.dled sulphuric acid
(14.7ml). After one hour the resultant mixture was cooled, diluted with
water and made basic by the addition of potassium carbonate, then extracted
with ether. The extracts were dried over magnesium sulphate and evaporated
under reduced pressure to give the title compound (11.459) as a mixture of
a solid and an oil, sufficiently pure to be used in Step 7. NMR (CDCl3):
1.39(3H,s); 1.~9-1.82(3H,m); 1.82(3H,s); 2.12-2.30(3H,m); 5.73(1H,bs).
Step 7 Preparation of 1-methylcyclobutyl-1-amino chloride
A solution of 1-methylcyclobutyl-1-acetamide (11.459 as prepared in
Step 6) in cull.ellLldLed sulphuric acid was heated under reflux for fifty
five hours. The resultant muxture was allowed to cool and was washed with
ether. Theaqueous phase was made strongly basic with 50% sodium hydroxide
and extracted with ether. The extracts were dried over potassium
hydroxide. Hydrogen chloride (g) was bubbled through the extracts to give
the title compound (5.639) as a white solid. m.p. 247-250C (dec). NMR
(CDCl3): ~ 1.34(3H,s); 1.69-1.88(4H,m); 2.12-2.29(2H,m); 8.34(3H,bs).
Preparation of 5-[N-(1-methylcyclobutyl)-1-acetamido]-3-(3-tri-
fluoromethylphenyl)oxazolidin-4-one
Oxalyl chloride (3.5ml) was added to 5-[methylenecarboxy]-3-(3-tri-
fluoromethylphenyl)oxazolidin-4-one (0.8749 as prepared in Step 5). After
two hours the resultant mixture was evaporated under reduced pressure. The
residue was suspended ;n ether (9ml) and cooled. A suspension of 1-methyl
cyclobutyl-1-amino chloride (0.3209 as prepared in Step 7) and
triethylamine (0.5319) in ether (6ml) was added. After twenty-four hours
the resultant mixture was filtered and washed with ether. The filtrate was
dried over magnesium sulphate and evaporated under reduced pressure. The
WO 95/33719 2 1 9 0 9 7 9 P~/GB95/0l22~
- 159 -
residue was Lllll Loyl~Jh~ on silica, ~sing hexane-ethylacetate to give
the title compound (0.1669) as a yellow solid. [MPt 12g.5-135.5C].
NMR (CDCl3); ~ 1.47(3h,s); 1.74-1.93(Zh,m); 1.96-2.08(2H,m);
2.22-2.36(2H,m); 2.73(2H,m); 2.83(1H,m); 5.50(2H,s); 5.86(1H,bs);
7.45(1H,d); 7.53(1H,t); 7.76(1H,d); 7.84(1h,s).
The compounds listed below were prepared by analdgous methods.
Compound 259: 5-[N-(l-methylcyclopentyl)-1-acetamido]-3-(3-
trifluoromethylphenyl)oxazolidin-4-one;
NMR (CDCl3); ~1.41(3H,s); 1.65(6H,m); 1.94(2H,m); 2.73(2H,m), 4.80(1H,m);
5.49(2H,s); 5.70(1H,bs); 7.47(1H,d); 7.54(1H,t), 7.76(1H,d), 7.83(1H,s).
Compound 261: 5-[N-(1-ethyl-1-cyclopropyl-1-acetamide]-3-(3-
trifluoromethylphenyl)oxazolidin-4-one;
NMR(CDCl3): ~0-0.36(4h,m), 0.53-0.67(2H,m) 0.98(3H,t); 2.58(2H,m);
3.20(1H,q); 4.61(1H,m); 5.30(2H,s); 5.59(1H,bd); 7.25(1H,d); 7.32(1H,t);
7.56(1H,d); 7.63(1H,s).
Compound 263: 5-[N-(1-methylcyclohexyl)-1-acetamido]-3-(3-
trifluoromethylphenyl)oxazolidin-4-one;
NMR (CDCl3): ~ 11.330-11.55(8H,m); 1.35(3H,s); 1.90-204(2H,m); 2.75(2H,m);
4.80(1H,m); 5.50(2H,s); 5.50(1H,bs); 7.46(1H,d); 7.53(1H,t); 7.76(1H,d);
7.84(1H,s) .
Compound 264: 5- [N- (neopentyl ) -l -acetami de] -3- (3-tri fl uoromethyl
phenyl)oxazolidin-4-one;
NMR (CDCl3): ~ O.90(9H,s); 2.83(2H,m); 4.81(1H,m); 5.51(2H,s); 5.59(1H,bs);
7.46(1H,d); 7.53(1H,t); 7.77(1H,d); 7.82(1H,s).
Mpt: 117.5-lZ2C.
Compound 268: 5-[N-(1-methylcyclopropyl)-1-acetamido]-3-(3-
trifluoromethylphenyl)oxazol idin-4-one;
NMR (CDCl3): ~ 0.54(2H,m); 0.64(2H,m); 1.38(3H,s); 2.72(2H,m); 4.78(1H,m);
2190
WO 95133719 PCT/GB95/OIZ2.1
- 160 -
5.50(2H,s); 6.08(1H,bs); 7.45(1H,d); 7.53(1h,T); 7.77(1h,D); 7.85(1h,5).
mpt: 182.5-184.5c (dec).
EXAMPLE 97 An alternative route to 3(3-hydrocarbyl-2,4-dioxoimidazoli-
din-1-yl)pyrrolidin-2-one (Compare Example 29) exemplified by Co~pound 334:
3(3-t-Butyl-2,4-dioxoimidazolidin-1-yl)-1(2,2-d jfluoro-1,3-benzodi-
oxol -5-yl ) pyrrol i di n-2 -one
Sodium hydride (0.0249, 55% dispersion in mineral oil) was added to a
stirred solution of 3-t-butyl-imidazolidine-2,4-dione (0.0859) in
N,N-dimethylformamide (lOml) and the mixture allowed to stir for thirty
minutes at room tl, .~Lul~:. The stirred mixture was cooled to 0C,
treated with a solution of 1(2,2-difluoro-1,3-benzodioxol-5-yl)-3-iodo-
pyrrolidinone(O.20g) in N,N-dimethylformamide (lOml). allowed to warm to
room temperature, then to stir for a further two hours. It was then
diluted with water and extracted with ethyl acetate. The extracts were
washed with water and brine, dried over magnesium sulphate and evaporated
under redu~ed pressure. The residue was chromatographed on silica, using
dichloromethane-ethanol (4g:1), then ethyl acetate-hexane (1:1), as
eluants, to give the title compound (0.0449, m.p. 120-124C). NMR (CDCl3):
~ 1.63(9H,s); 2.18(1H,m); 2.56(1H,m); 3.84(2H,q); 3.84(2H,m); 5.00(1H,dd);
7.06(1H,d); 7.13(1H,dd); 7.69(1H,d). MS: M+ 395. Confirmation that the
imidazolidine ring was not attached in an alternative manner was provided
by 13C NMR (pyrrolidinone methine carbon, 54.1ppm).
Intermediates and Analapous Methods of PreDaration
EXAMPLF 98 PrepAration of 3-(2,2,2-Trifluoroethylamino)-1-(3-
trifl ~ ,' ,1-2-pyrrolidinone
SteD I Preparation of 3-iodo-1-(3-trifluo,, ' y)phenyl-2-pyrrolidinone
3-bromo-1-(3-trifluoromethoxy)phenyl-2-pyrrolidinone (prepared in a
similar manner to that described in Step 1 of Example 10) (1.09) was
dissolved in acetone (20ml). To this solution was added sodium iodide
~0.469~ and the rea~tion was stirred at room temperature for 4 hours under
WO 95/33719 2 1 9 0 9; 9 PCT/GB95/0122J
- 161 -
a nitrogen di ~ . A further 0.0469 of sodium iodide was added and
reaction left to stand for 12 hours. The precipitate was filtered off
through a pad of Hyflo, washing with acetone. After evaporating the
solvent the residue was dissolved in ethyl acetate, washed with brine (x2),
dried (MgS04), filtered and concentrated to give the crude
3-iodopyrrolidinone (1.069) which was used directly in Step 2.
Preparation of 3-(2,2,2-Trifluoroethylamino)-1-(3-trifluoro-
methoxy)phenyl-2-pyrrol idine
3-lodo-1-(3-trifluu,, i' y)phenyl-2-pyrrolidinone (prepared in Step
1) (1.069) was mixed with 2,2,2-trifluoroethylamine (S.Og) at room
temperature and then cooled to 0C (ice/water bath cooling) and stirred for
17 hours. After standing at room temperature for 96 hours, the mixture was
diluted with ethyl acetate, washed with water (x2), brine (xl), dried
(MgS04), filtered and cullc~ Lldtc~ to give the crude amino pyrrolidinone
title compound as an oil (0.9249).
EXAMPLE 99 Prepar~tion of 3(N-methyl~nino)-1-(3-trifluoromethylthio)-
phenyl -2-pyrrol i di none
Step 1 Preparation of 3-chloro-1-(3-trifluoromethylthio)-
phenyl -2-pyrrol i di none
A solution of 3-hydroxy-1-(3-trifluoromethylthio)-
phenyl-2-pyrrolidinone (prepared by a similar method to that described in
Example 9) (1.09) in thionyl chloride (5ml) was stirred at room t, ~ILUlt:
for two hours before heating to reflux for 16 hours. After cooling, the
excess thionyl chloride was removed in vacuo and the residue purified by
flash chromatography, eluting with 30% ethyl acetate/hexane to give the
3-chloropyrrolidinone as a pale yellow oil (0.819).
SteD 2
A solution of 3-chloro-1-(3-trifluoromethylthio)phenyl-2-
pyrrolidinone (prepared as in Step 1) (0.809) in THF (30ml) was treated
WO 95/33719 2 1 9 0 9 7 9 PCTIGB9510122.) ~
- 162 -
with a continuous stream of methylamine gas at room temperature for 1 hour.
The reaction was then heated to reflux, maintaining the flow of
methylamine. After 4 hours, the reaction was left to stand for 72 hours
and then poured into saturated NaHC03 (aq) and extracted with ethyl acetate
(x2). After drying (MgS04), the solvent was evaporated and the residue
purified by chromatography, eluting with ethyl acetate then 30%
methanol/ethyl acetate to give the amine title compound as a pale yellow
oil which solidified (0.669).
Compounds 199 and 201 may be prepared by methods similar to that
described in Example 5 but using an intermediate 11 as starting material in
place of the hydroxy compound. Compound 200 may be prepared by a method
similar to that described in Example 27 but again using intermediate Il.
The intermediate 11 is 3-(N-allyl)amino-1-3-b" I' yl-2-pyrrolidinone, a
compound of general formula II in which A is 3-b., ,' yl, X is CH2 and
R20 is NH-CH2-CH=CH2. For compounds 199 and 201 Compound Il is reacted
with the appropriate acyl chloride and for compound 200 it is reacted with
t-butyl i socyanate .
Compound 202, 203 and 204 may all be prepared using methods similar to
that described in Example 72 or Example 73. Compound I2, a compound of
general formula III in which A is 3-(trifluoromethyl)phenyl, X is CH2 and
R20 is methane sulfonyloxy may be used as an intermediate for compound 204
and a similar intermediate may be used for Compounds 202 and 203.
Compounds 205 to 20~3 may be prepared by methods similar to those
described in Examples 5, and 10 or 27 using as intermediates Compounds I3
(formula III, A is 3-chloro-4-fluorophenyl, X is CH2 and R15 is methane
sulfonyloxy), I4 (formula II, A is 3-chloro-4-fluorophenyl, X is CH2 and
R15 is NH2) and I5 (formula II, A is 3-chloro-4-fluorophenyl, X is CH2 and
R15 i s NHMe) .
Compounds 209 and 210 may be prepared by methods similar to those
described in Examples 27 and 5 respectively from intermediate I6 (formula
II, (formula II, A is 3-chloro-4-fluorophenyl, X is CH2 and R15 is
NH-CH2-CH=CH2) -
Compounds 211 and 212 may be prepared by methods similar to those
described in Example S but starting from intermediates I4 and I5
respecti vely .
WO 95133719 2 1 9 0 9 7 9 pCT,GB95,0l22~
- 163 -
Compounds 213, 214 and 217 to 219, 221, 222, 224 to 231 and 233 may
all be prepared by methods similar to those of Example 5. Compounds 220
and 232 may be prepared by methods similar to those of Example 20 and
Compounds 215, 216 and 223 by methods similar to that of Example 10.
lntermediates in the synthesis of these compounds include:
17 (formula III, A is 3-(trifluo,, ~I""ty)phenyl, X is CH2 and R20 is
methane su1fony10xy);
I8 (formula II, A is 3-(trifluoromethoxy)phenyl, X is CH2 and RlS is
NH-CH0);
19 (formula II, A is 3-(trifluoromethyl)phenyl, X is CH2 and RlS is
NH-CH0);
I10 (formula II, A is 3-(trifluoromethoxy)pheny1, X is CHz and R15 is
NH-Me);
111 (formu1a llI, A is 3-bromophenyl, X is CH2 and R20 is methane
sul fonyl oxy);
112 (formula 11, A is 3-b(. ,~ ,~1, X is CH2 and R15 is SH);
I13 (formula Il, A is 3-bl~ ,' y1, X is CH2 and R1S is NMe); and
114 (formu1a II, A is 3-b~ "l, X is CH2 and R15 is NH2).
The pyrrolidinone compound 282 was prepared from
3-(methylamino)-1-[3- (trif1u~,. lh~xy)phenyl]pyrrolidin-2-one (prepared
in analogy to the procedure for the preparation of
3-(methy1amino)-1-[3-(trifluoromethy1) pheny1]pyrrolidin-2-one described in
Examp1e 10 and trimethylsily1acetyl chloride and pyridine in
d i ch l oromethane .
lntermediates used in the preparation of Compounds 26, 271 and 271
include the compounds listed below.
2- (methyl thi o) -N- (3 -tri f 1 _ . L' J, ' .~ l ) acetami de
Prepared by a similar method to that described in Example 91, Step 1.
NMR(CDC13): ~ 2.20(3H,s); 3.35(2H,s); 7.0(1H,d); 7.35(1H,t); 7.45(1H,d~;
7.62(1H,s); 8.80(1H,bs).
MPt: 43.5-45C.
N-(~ . Lh~l)-N-(3-trifl , i' ~,' ,1)-2-(methylthio)acetamide
Prepared by a similar method to that described in Example 91, Step 2.
2 1 9 0 9 7 9 CT/G3395/0122.1
WO 95/33719 P
- 164 -
NMR(CDCl3): -dl 1.22(3H,t); 2.22(3H,s); 3.02(2H,bs); 3.68(2H,m);
5.10(2H,s); 7.18-7.29(3H,m); 7.47(1H,t).
N-(~:' .t Lh.~l)-N-(3-trifl~ phenyl)-2-(methylsulphinyl)acetamide
Prepared by a similar method to that described in Example 91, Step 3.
NMR(CDCl3); ~ 1.24(3H,t); 2.76(3H,s); 3.59(2H,d); 3.67(2H,q); 5.11(2H,d);
7.12(1H,s); 7.22-7.36(2H,m); 7.51(1H,t).
5-hydroxy-3-(3-trifl ~ phenyl)oxazolidin-4-one
Prepared by a similar method to that described in Example 91, Step 4.
NMR(CDCl3): ~ 5.44(1H,s); 5.65(2H,s); 7.06-7.16(1H,m);7.37-7.45(2H,m);
7 . 60 ( lH , s I ) .
5-[methylamino~-3-(3-trifl .1 '~ ~ phenyl)oxazolidin-4-one
Prepared by a similar method to that described in Example 91, Step 6
(used for Compound 269);
NMR(CDC~3): ~ 22.57(3H,s); 5.18(1H,s); 5.41(1H,m); 5.47(1H,m);
7.05-7.13(1H,m); 7.13-7.33(2H,m); 7.61(1H,s).
5-amino-3-(3-trifl .. ' y phenyl)ox~zo~idin-4-one
Prepared by a similar method to that described in Example 91, Step 6
(used for Compound 271);
NMR(CDCl3): ~ 1.62(2H,bs); 5.20(1H,bs); 5.37(1Hld); 5.45(1H,d);
7.03-7.13(1H,m); 7.32-7.47(2H,m); 7.60(1H,s).
5-(N-ethylamino)-3-(3-trifl .. i' ~,' ,l)oxazolidin-4-one
Prepared by a similar method to that described in Example 91, Step 6
(used for Compound 270);
NMR(CDCl3): ~ 1.25(3H,t); 2.90(2H,q); 4.27(1H,bs); 5.20(1H,s); 5.40(1H,m);
5.46(1H,m); 7.04-7.11(1H,m); 7.38-7.50(2H,m); 7.60(1H,s).
Intermediates in the preparation of Compounds 259, 261, 263, 264 and
268 are detailed below.
5-methylenechlorocarbonyl)-3-(3-trifluoromethylphenyl)oxazolidine
Prepared by a method similar to that described in Example 96, Step 8.
WO 95133719 , 2 1 9 0 9 7 9 PCT/GBg~10l22~
165 -
NMR(CDC13): ~ 3.52(2h,m); 4.79(1H,m); 5.52(2H,s); 7.50(1H,d); 7.55(1H,t);
7.75(1H,d); 7.80(1H,s).
l-methyl cycl opentyl -l-acet~mi de
Prepared by a method similar to that described in Example 96, Step 6.
NMR (CDCls): ~ 11.40(3H,s); 1.56-1.78(8H,m); 1.93(3H,s); 5.39(1H,bs).
1 -methyl cycl opentyl -l-dmi ne
Prepared by a method similar to that described in Example 96, Step 7
(used for the preparation of Compound 259)
NMR(CDC13): ~ 1.27(3H,s); 1.40-1.82(10H,bm).
1 -ethy 1-1- cyc 1 opropy 1 acetami de
Prepared by a method similar to that described in Example 96, Step 6
(used for the preparation of Compound 261)
NMR(CDC13): ~ 0-0.34(4H,m); 0.60-0.75(1H,m); 0.97(3H,d); 1.74(3H,s);
2.03-2.13(1H,m) .
l-ethyl-l-cyclopropyl Aminochloride
Prepared by a method similar to that described in Example 96, Step 7
(used in the preparation of Compound 261).
NMR(CDC13): ~ 0-0.6(1H,m); 0.12-0.30(3H,m); 0.51-0.72(1H,m); 0.97(3H,d);
2.13-2.21(1H,m); 7.84(3H,bs).
MPt: 130-155C.
1-methyl cycl ohexyl -l-acet,lmi de
Prepared by a method similar to that described in Example 96, Step 6
(used in the preparation of Compound 263).
NMR(CDC13): ~ 1.37(3H,s); 1.23-1.55(8H,m); 1.92-2.02(ZH,m); 1.95(3H,s);
5. 18(1H,bs) .
MPt: B3.5-86C.
l-methylcyclohexyl -1-aminochloride
Prepared by a method similar to that described in Example 96, Step 7
(used in the preparation of Compound 263).
NMR(CDC13): ~ 1.16(3H,s); 1.23-1.44(4H,m); 1.44-1.60(6H,m); 7.57(3H,bs).
_ _ _ . ... . _ _ _ . . . _ . . . .
WO 95/33719 2 1 9 0 9 7 9 PCTIGB95/0122.1
- 166 -
(1-methylcyclopropyl)-1-t-butoxy carbamate (used in the preparation of
1-methylcyclopropyl ) -l-aminochloride) .
Diphenylphosphonyl azide (14.839) was added in one go to a solution of
1-methylcyclo propane carboxzylic acid (5.009) in tert-butanol (150ml).
After twenty minutes triethylamine (8.4ml) was added to the resultant
mixture which was then heated under reflux under N2 for five hours. The
resultant mixture was quenched with water and extracted with ther. The
extracts were washed with water and brine dried over magnesium sulphate and
evaporated under reduced pressure. Ether was added to the residue by
filtration. The filtrate was dried over magnesium sulphate and evaporated
under reduced pressure to give the title compound (4.859) as a white solid.
NMR(CDCl3): ~ 0.52-0.60(2H,m); 0.70-0.76(2H,m); 1.33(3H,s); 1.44(9H,s).
MPt: 69.5-77C.
1-methyl cyclopropyl-1-~mino chloride
(Used in the preparation of Compound 268).
Hydrogen chloride was bubbled through a solution of (1-methyl
cyclopropyl)-1-t-butoxy carbamate (4.499 prepared as described) in ethanol.
The resultant mixture weas evaporated under reduced pressure. Ether was
added to the residue, this was then filtered to give the title compound
(1.569) as a white solid.
NMR(CDCl3): ~ 0.60(2H,t); 0.90(2H,t); 1.34(3h,s); 8.43(3H,bs)
MPt : 193-206C (dec) .
5tructural details and characterising data for the intermediates IS9
and I60 are given in Tables II and III. These intermediates are used in
the synthesis of Compound 329.
The intermediate I61 is a compound of general formula VI in which A is
2-trifluoromethylbenzoxazol-6-yl. It is used in the preparation of
Compound 332. Other intermediates in the synthesis of Compound 332 include
intermediates I62 and 163, details of which are given in Tables II and III.
Intermediates I64 and 165 are used in the preparation of Compound 331.
Further details of these intermediates are given in Tables II and III.
WO 95/33719 PCT/GB9~/0122 1
- 167 -
The intermediate 166 is used in the preparation of Compound 33g. 166
is a compound of general formula XXVII in which A is
2,2-bis(difl " ~1~u~y)pyrimidin-4-yl, RZ, R3, R4 and R5 are all hydrogen,
R1 jS t-butyl and R25 jS 1. This compound is prepared from the
intermediate 4-aminû-2,6-bis(difluoromethoxy)pyrimidine (m.p. 102-103C)
which, in turn, is prepared by treating 4-amino-Z,6-dihydroxypyrimidine
with chlorofluoromethane in aqueous dioxane in the present of sodium
hydroxide in a manner similar to that described in Example 66, for the
intermediate to Compound 181.
NMR (CDCl3): ~ 1.35(9H,s); Z.4(ZH,m)j 3.Z5(2H,t); S.û5(1H,bs);
5.25(1H,dd); 7.25(1H,t); 7.35(1H,t); 7.5(1H,s); 9.0(1H,bs). MS: MH 539.
The intermediate I67 is used in the preparation of Compound 332. 167
is a compound of general formula XXVII in which A is S-methoxycarbonyl-
thiazol-2-yl, R2, R3, R4 and RS are all hydrogen, R1 is t-butyl and R25 jS
I.
NMR and melting point details are given in Table III.
The intermediate I68 is used in the preparation of Compound 333. I68
is a compound of general formula XXVII in which A is
S-thiocyanato-thiazol-2-yl, R2, R3, R4 and R~ are all hydrogen, R1 is
t-butyl and R25 is I. NMR and melting point details are given in Table
III .
Intermediate I69 is used in the preparation of Compounds 273-277.
Intermediates I70, I72 and I73 are used in the preparation of Compounds
283-290; 291-292; and 295-296 respectively.
Structural details for intermediates are given in Table II and
characterising data for Compounds 199 to 233 and intermediates 11 to 168
are given in Table III. All of the compounds of Table II are of either
general formula II, III or IV.
WO 95133719 2 1 9 0 9 7 9 PCT/GB9~10122.1
-- 168 --
TASLE I I ~ ~ ~
Int Nc~. Pre,p. A X Rl~i/}120
Il i m-OSr Ph CH2 NHCH2CH=CH2
I2 ! m-CF3 Ph Ph CH2 OS02Me
I3 m-Cl Ph, p-~ Ph CH2 0502Me
I4 m-Cl Ph, p-F Ph CH2 NH2
I~ m-Cl Ph, p-F Ph CH2 ' NHMe
~ ~ m-Cl '-, -F Ph r NHC ~2r =CH2
.' O~ 3 h O ~ ~e
0~ 3 h , ' ~ O
~_ ~ O
~ ~ OC 3 1 : e
._. m- r I O ~ Me
._ m- r I
.. m- r I ~ N-.
__4 m- r ~ ~ ~
I15ex 99, m-SCF3 Ph CH2 Cl
I16 ex 10, m-CF3 Ph CH2 Br
I17 step ;b m-OCF3 Ph CH2 8r
I18 I step 1 m-OCHF2 Ph CH2 13r
Il9 ' step 1 m-Cl Ph CH2 i3r
I20 step 1 m-OCF3 Ph CH2
I21 la m-OCF3 Ph CH2 OH
~x 9, step CH2 OH
I22 1 m-OCHF2 Ph
I23 la m-SCF3 Ph CH2 OH
ex 1, step
I24 3 m-CF3 Ph CH2 N3
lex 1, step
I251 3 m-OCF3 Ph CH2 N3
ex 1, step }
I26 3 m-OCHF2 Ph CH2 I N3
ex 1, step
I27 4 m-CF3 Ph CH2 NH2
wo 9~,3371g 2 1 9 0 9 7 9 pcrlG~9slol22~
~ 169 --
Int 2~o. ! Prep. A X ! R15~R20
j ex 1, s tep
I28~ 4 m-OCF3 Ph CH2 NH2
I ex 1, step
I29 1 4 m-OCHF2 Ph CH2 NH2
ex 10,
I30 step 2 m-CF3 Ph CH2 NBMe
ex 10,
I31 seep 2 m-oCF3 Ph CH2 NHMe
ex 10,
I32 step 2 m-OCHF2 Ph CH2 NHMe
ex 99,
I33 step 2 m-SCF3 Ph ! CH2 NHMe
ex 27,
I34 step 1 m-OCF3 Ph CH2 NHrt
ex 27,
I35 I step 1 m-OCHF2 Ph CH2 NH~t
ex 27,
I36 I step 1 m-OCF3 Ph CH2 NHPr
ex 27,
I37 step 1 m-CF3 Ph CH2 NHCH~CH3)2
ex 27,
I38 step 1 m-CF3 Ph CH2 NHCH2CH20Me
ex 27,
I39step 1 m-OCF3 Ph CH2 Nn~s
ex 27,
I40step 1 m-CF3 Ph CH2 NH-cyclopropyl
ex 27,
Iqlstep 1 m-OCF3 Ph CH2 NHCH2CH2N ~Me~ 2
ex 27,
I42stèp 1 m-OCF3 Ph CH2 I~IH~ JI-I
ex 27,
I43step 1 m-OCF3 Ph CH2 NHCH2CCH
ex 27,
I44step 1 m-CF3 Ph CH2 NCH2CH=CH2
ex 27,
I45 step 1 m-OCF3 Ph CH2 NCH2CH=CH2
ex 27,
I46 step 1 m-OCF3 Ph CH2 NHCH2CH~OMe)2
ex 27,
I47 step 1 m-CF3 Ph CH2 NHCH2Ph
ex 27,
I48 step 1 m-CF3 Ph CH2 NHPh
ex 9~,
I49 step 2 m-OCF3 Ph CH2 NHCH2CF3
ex 24,
I50 step 1 m-CF3 Ph CH2 NHOMe
21 90979
WO 95/33719 PCIIGB95/0122
- 170 -
Int No. Prep. A I Y ' EU5tRZ0
ex 23, 1 1
ISls-ep 1 m-CF3 Ph CH2 I NHCH2CO2Et
ex 23,
IS2step l m-OCF3 Ph CH2 NHCHCO2~qe
ex 23,
IS3 step l m-OCF3 Ph CH2 NHCH2CO2C(CH3)3
~x 23,
IS4 step 1 m-OCF3 Ph CH2 NHCH2COC(CH3)3
ex 23,
ISS ~tep 1 m-OCF3 Ph CH2 NHCH2CN
ex 20,
IS6 step 1 m-CF3 Ph CH2 SAc
ex 20,
IS7 I step 1 m-OCF3 Ph CH2 SAc
ex 20,
IS8 I step 1 m-OCHF2 Ph CH2 SAc
2-triiluoromethyl
IS9 }~.~n-nYA.~1_5_yl H
I60 2-trifluoromethyl S OH
b~n7nv~7nl -S-yl
I62 2-trifluoromethyl 5 H
~.n7nY~7nl--6--yl
I63 2-~rifl.. n L11yl 5 OH
n7nY~7nl--6--yl
I641, 3-~n7n~ Ynl _5 H
yl
I651,3-3:1enzodioxol-S 5 OH
. 69 m-OCHF2 Ph O C~ 02H
.70 m-CF3 Ph Nn~ en3
_72 m-CF3 Ph ~ t
_73 I m-CF3 Ph NH-C ~ tH=CH2
WO 95133719 2 1 9 0 9 7 9 PCTIGB95~0122-1
-- 171 --
Ti~T~T~T III
No. Prep mp~ nmr
1.63 (6H, dl, 1.90-2.0B (lH, m~, 2.32 (lH, s),
B 2.42 (lH, dd), 2.40-2.59 (lH, m~, 2.65 (lH,
16 ex a 129 dd), 3.0-3.12 (lH, m), 3.76-3.93 (2H, m), 6-54
12 (lH, br 5), 7.51 (lH, d), 7.60 (lH, t), 7.85
(lH, d~, 7.89 (lH, br s)
0.84 (3H, t~, 1.29 (6H, s), 1.71 (2H, q), 1.90-
2.08 (lH, m), 2.39 (lX, dd), 2.44-2.57 (lH, m),
17 ex 16 117 2.72 (lH, dd), 2.95-3.08 (lH, m), 3.76-3.90
(3H, m), 5.86 (lH, br s), 7.40 (lH, d), 7.50
(lH, t), 7.85-7.81 (2H, m)
1.42 (6H, s), 1.90-2.08 (lH, m), 2.40 (lH, dd),
2.44-2.57 (lH, m), 2.74 (lH, dd), 2.99-3.12
18 ex 16 111 ( lH, m), 3 . 75-3 . 91 (2H, m), S . 02 ( lH, d), S .10
(lH, d), 6.01 (lH, dd), 6.15 (lH, br s), 7.40
(lH, br d), 7.50 (lH, t), 7.B2-7.91 (2H, m)
1.2B (3H, t), 1.38 (9H, s), 2.25-2.53 (2H, m),
112- 3.11-3.38 (2H, m), 3.71-3.95 (2H, m), 4.41 (lH,
19 ex 27 114 br s), 4.65 (lH, dd), 7.37 (lH, d), 7.47 (lH,
t), 7 . 87-? . 9S (2H, m)
1.67 (6H, 2s), 2.08-2.25 (lH, m~, 2.43-2.56
146- (lH, m), 2.88 (3H, s), 3.74-3.90 (2H, m~, 4.73
147 (lH, br s~, 5.22 (lH, dd), 7.41 (lH, br d),
7.49 (lH, t~, 7.86-7.95 (2H, m~
1.95-2.11 (lH, m), 2.50-2.70 (2H, m), 2.96 (lH,
137- dd), 3.10-3.25 (lH, m), 3.79-3.98 (2H, m), 7.09
22 ex 8 138 (lH, t~, 7.30 (2H, t), 7.40-7.56 (4H, m), 7.82-
7.93 (2H, m), 8.70 (lH, br s)
l.lS (3H, t), 1.90-2.08 (lH, m), 2.39-2.58 (2H,
m), 2.78 (lH, dd), 3.0-3.15 (lH, m), 3.25-3.38
23 ex 8 110 (2H, m), 3.76-3.95 (2H, m), 6.24 (lH, br s),
7.42 (lH, br d), 7.50 (lH, br t), 7.86 (lH, br
d), 7.92 (lH, br s)
-
1.60-2.05 (SH, m), 2.25-2.58 (3H, m), 2.74 (lH,
146- dd); 2.97-3.12 (lH, m), 3.75-3.82 (2H, m), 4.28
147 4.43 (lH, m), 6.40 (lH, br m), 7.41 (lH, d),
7.50 (lH, t), 7.85 (lH, br d), 7.90 (lH, br s)
.. .. . _ . . _ .. _ . . _ . .
21 90979
WO 95133719 PCT/GB95/0122.1
-- 172 -
No. Prep mpt nmr
1.25 (3Hr t), 1.53 (6H, 2s), 1.90-2.10 ~lH, m),
2.45 (lH, dd), 2.42-2.56 (lH, m), 2.78 (lH,
ex lB gg dd), 3.00-3.14 (lH, m), 3.76-3.91 (2H, m), 4.18
(2H, q), 6.75 (lH, br s~, 7.40 (lH, br d~, 7.49
(lH, t), 7.85 (lH, d), 7.90 (lH, br s)
1.90-2.08 (lH, m), 2.50-2.66 (2H, m), 3.06 (lH,
128- dd), 3.12-3.27 (lH, m), 3.79-3.95 (2H, m~, 6.90
26 ex 12 131 (lH, d), 7.40 (lH, d), 7.49 (lH, t), 7.58 (lH,
t), 7.86-7.98 (2X, m), 8.31 (lH, br s)
1.87-2.03 (lH, m), 2.49 (lH, t), 2.49-2.62 (lH,
m), 2.63 (lH, dd), 3.04 (lH, dd), 3.05-3.18
27 ex 12 oll (lH m) 3 80-3.94 (2H, m), 4.74 (2H, d), 7.40
(lH, d), 7.48 (lH, t), 7.85-7.90 (2H, m)
1.31 (9H, s), 1.96-2.15 (lH, m), 2.75-2.90 (lH,
175- m), 3.75-3.90 (2H, m), 4.40-4.55 (lH, m~, 5.20
28 ex 1 176 (lH, br s), 5.50 (lH, br d), 7.41 (lH, d), 7.47
(lH, t), 7.79 (lH, d), 7.92 (lH, br 5)
1.08 (9H, s), 1.90-2.08 (lH, m), 2.16 (2H, s),
138- 2.85-2.97 (lH, m), 3.79-3.93 (2H, m), 4.52-4.64
29 ex 5 139 (lH, m), 6.18 (lH, br d), 7.42 (lH, d), 7.50
(lH, t), 7.85 (lH, d), 7.93 (lH, br s)
1.38 (9H, s), 1.87-2.06 (lH, m), 2.46-2.60 (lH,
m), 2.91 (lH, dt), 3.10-3.35 ~2H, m), 3.80-3.95
30 ex 12 o11 (2H m) 7 40 (lH, d), 7.49 (lH, t), 7.82-7.92
(2H, m)
0.95-1.02 (6H, m~, 1.72-1.90 (2H, m), 1.90-2.20
(3H, m), 2.45 (lH, dd~, 2.43-2.56 (lH, m), 2.76
31 ex 8 87-90 (lH, dd), 3.00-3.11 (lH, m), 3.76-3.91 (2H, m),
6.24 (lH, br s), 7.40 (lH, d), 7.49 (lH, t),
7.82-7.91 (2H, m)
1.15 (3H, d), 1.37 (9H, s), 2.02-2.10 (lH, m),
2.26-2.38 (lH, m), 2.66-2.77 (lH, m), 2.99-3.09
32 ex 19 (lH, m), 3.75-3.84 (2H, m), 5.66 (lH, br 5),
111 7.38 (lH, br d), 7.48 (lH, t), 7.83-7.90 (2H,
m)
0.77 (6H, t), 1.20 (3H, s), 1.48-1.80 (6H, m~,
1. 9û-2 . 05 ( lH, m), 2 . 50-2 . 70 (lH, m), 3 . 80-3 . 95
33 ex 12 o-l (3H m) 5.50 (lH, s), 7.36-7.55 (2H, m), 7.81-
7.95 (2H, m)
WO95133719 2 1 9~979 PCT/GBgS10122l
-- 173 --
No. Prep mpt
0.89 (3H, t~, 1.32 (6H, s), 1.74 (2H, q), 2.07-
2.23 (lH, m), 2.42-2.55 (lH, m), 2.87 ~3H, s),
34 ex 16 5 3.73-3.90 (2H, m), 4.40 (lH, br s), 5.22 (lH,
dd), 7.40 (lH, br d), 7.49 (lH, t), 7.8B (lH,
br d), 7.96 (lH, br s)
1.46 (6H, s), 2.05-2.24 (lH, m), 2.42-2.55 (lH,
m), 2.89 (3H, s), 3.73-3.90 (2H, m), 4.63 (lH,
ex 16 171 br s), 5.04 (lH, d), 5.12 (lH, d), 5.21 (lH,
dd), 6.07 (lH, dd), 7.40 (lH, d), 7.49 (lH, t),
7.88 (lH, ~r d), 7.95 (lH, br s)
1.37 (9H, s), 2.04-2.21 (lH, m), 2.41-2.55 (lH,
132- m), 2.8~ (3H, s), 3.70-3.86 (2H, m), 4.45 (lH,
133 br s), 5.22 (lH, dd), 6.99-7.04 (lH, m), 7.38
(lH, t), 7.56 (lH, dd), 7.70 (lH, br s)
2.17-2.34 (lH, m), 2.45-2.58 (lH, m), 3.07 (3H,
s), 3.75-3.93 (2H, m), 5.08 (lH, dd), 7.01 (lH,
37 ex 10 t), 7.12 (lH, s), 7.22-7.31 (2H, m), 7.42 (3H,
169 dd), 7.50 (lH, t), 7.87 (lH, br d), 7.96 (lH br
s)
1.24 (3H, d), 1.31 t3H, d), 1.33 (9H, s), 2.22-
2.37 (lH, m), 2.51-2.69 (lH, m), 3.70-3.84 (2H,
38 ex 27 158 m) , 3.89-4.00 (2H, m) , 4.30 (lH, br s) , 7.35
(lH, br d), 7.45 (lH, t), 7.90 (lH, br d), 7.95
( lH, br s )
1.38 (9H, s), 2.16-2.32 (lH, m), 2.69-2.72 (lH,
104- m), 3.81-3.92 (2H, m), 4.35 (lH, t), 5.63 (lH,
41 ex 20 106 br s), 7.00-7.07 (lH, m), 7.39 (lH, t), 7.55
(lH, dd), 7.64 (lH, br s)
1.38 (9H, s), 2.18-2.34 (lH, m), 2.71-2.85 (lH,
119- m), 3.85-3.95 (lH, m), 4.35 (lH, t), 5.62 (lH,
42 ex 20 120 br 5), 7.42 (lH, d), 7.50 (lH, t), 7.85
(2H, m)
1.66 (6H, 2s), 2.06-2.34 (lH, m), 2.34 (lH, s),
127- 2.43-2.56 (lH, m), 2.88 (2H, m), 4.72 (lH, br
128 s), 5.22 (lH, dd), 7.40 (2H, m), 7.84-7.93 (2H,
m)
WO 95~33719 2 1 9`0 9 7 9 PCT/GB9S)~122~
- 174 -
~lo. Prep mp~ nmr
1.37 ~9H, s), 2.05-2.21 (lX, m), 2.41-2.55(1H,
136- m), 2.85 ~3H, s), 3.71-3.88 (2H, m), 4.45 (lH,
137 br s), 5.21 (1~, dd), 7.38-7.46 (2H, m), 7.84-
7.94 (2H, m)
10:1 ~ot~er mixture: 1.10 ~9H, s), 2.10-2.53
66.5- ~4~l, m), 2.8B and 3.~8 (3~, 2s), 3.~2-3.93 ~2H,
Sl ex S 68.5 m), 4.88 ~nd 5.23 (lH, 2t), 6.99-7.10 (lE, m),
7.38 (lH, t), 7.54-7.70 12H, m)
1.66 (6E~, d), 2.06-2.23 (lH, m), 2.34 (lH, s),
2.42-2.56 (lH, m), 2.87 (3H, Sl, 3.70-3.88 (2X,
52 ex 10 m), 4.72 (lH, br s), 5.22 (lH, dd), 6.97-7.05
136-5 (lH, m), 7.39 (lH, t), 7.55 (lH, dd), 7.68 (lH,
br s)
1.33 (9H, s), 1.91-2.~8 (lH, m), 2.78-2.90 (111,
175- m), 3.B0 ~2b, dd), 4.40-4.50 (1~, m), 4.86 ~
53 ex 176.5 br s), S.20 (lH, br d), 7.00-7.0a ~1~, m), 7.39
(lH, t), 7.48-7.53 (lH, m), 7.66 (lH br s)
1.06 (9H, s), 1.87-2.05 (lH, m), 2.15 ~2H, s~.
2.e4-2.97 (lH, m), 3.82 (lH, dd), 4.50-4.61
54 ex 5 36 (lH, m), 6.12 (lH, br d), 7.00-7.08 ~lH, m),
7.39 ~lH, t), 7.49-7.SS ~lH, m), 7.69 (lH, br
s)
1.62 (6E, 2s), 1.95-2.13 (lH, m), 2.44 (lH, s),
2.al-2.94 (lH, m), 3.75-3.8S (2~, m), 4.45-4.57
5sex 1 157 (lH, m), 5.31 (lH, br s), S.99 (lH, br d), 7.00
7.06 ~lH, m), 7.39 (lH, t~, 7.50 (lH, dd), 7.68
(lH, br s~
0.37 (3H, t), 1.31 (6H, s), 1.73 (2H, q), 2.04-
2.21 (lH, m), 2.40-2.53 ~lH, m), 2.85 (3X, 5),
56 ex 16 98-99 3.68-3.87 (2~, m), 4.36 ~lH, br s), 5.21
dd), 6.97-7.05 ~IH, m), 7.38 ~lH, t), 7.56
ddl, 7 . 6g (lH, br s )
0.87 (3H, t), 1.29 (6H, d), 1.69 (2H, q), 1.91-
2.10 (lH, m), 2.78-2.90 (1~, m), 3.ao (2H, dd),
57 ~a 16 4.38-g.48 (lH, m), 4.75 (lH, br s), S.l9 (lH,
172 br d), 7.~0-7.08 (lH, m), 7-3g ~lH, t), 7.52
(lH, dd), 7 . 67 (lH, br s)
,
_
WO 95133719 2 1 9 0 9 7 9 PCT/GB9510122.1
- 175 --
No. Prep mpt nmr
3:2 F~otamer mixture: 1.43 and 1.49 (9H, 2s),
2.11-2.35 llH, m), 2.35-2.54 (lH, m), 2.89 and
SE ex 21 2.96 (3H, 2s), 3.69-3.89 (2H, m), 4.53 and 4.99
107 (lH, 2 br t), 7.02 (lH d), 7.39 (lH, t), 7.58
(lH, d), 7.68 (lH, br s)
1.47 (9H, s), 1.94-2.12 (lH, m), 2.73-2.90 (lH,
S9 21 136- m), 3.75-3.85 (2H, m), 4.30-4.44 (lH, m), 5.14-
ex 137 5.30 (lH, v br 5), 7.00-7.08 (lH, m), 7.40 ~lH,
t), 7.55 (lH, dd), 7.65 (lH, br s)
1.36 (9H, s), 2.10 (lH, m), 2.46 (lH, m), 2.83
134- (3H, s), 3.77 (2H, m), q.43 (lH, br s), 5.21
ex 135 (lH, dd), 7.12 (lH, m), 7.29 (lH, t), 7.57 (lH,
t ), 7 . 72 ( lH, m)
5:4 Rotamer mixture: 1.44 and l.S0 (9H, 2s),
2.1q-2.37 (lH, m), 2.40-2.55 (lH, m), 2.89 and
61 ex 21 136 2.96 (3H, 2s), 3.82-3.90 (2H, m), 4.55 and 4.98
(lH, 2t), 7.41 (lH, d), 7.50 (lH, br t), 7.82-
8. 00 ~2H, m)
10:1 Rotamer miYture: 1.11 (9H, s), 2.12-2.30
(lH, m), 2.29 (lH, d), 2.38 (lH, d), 2.40-2.55
63 ex S gum (lH, m), 3.08 (3H, s), 3.76-3.96 (2H m), 5.20
(lH, dd), 7.40 (lH, d), 7.58 (lH, t) 7.86-7.94
(2H, m)
.
1.28 (lH, t), 1.36 (9H, s), 2.22-2.50 (2H, m),
64 7 108.5- 3.11-3.37 (2H, m), 3.67-3.90 (2H, m), 4.41 (lH,
ex 2 110 br s), 4.68 (lH, dd), 6.96-7.04 (lH, m), 7.36
(lH, t), 7.58 (lH, dd), 7.69 (lH, br s)
1.30 (3H, t), 1.66 (6H, d), 2.30-2.54 (2H, m),
146- 2.33 (lH, s), 3.15-3.37 (2H, m), 3.68-3.91 (2H,
ex 27 148 m), 4.60-4.71 (2H, m), 6.97-7.03 (lH, m), 7.36
(lH, t), 7.56 (lH, dd), 7,68 (lH, br s)
1.46 (9H, s), 1.95-2.15 (lH, m), 2.72-2.88 (lH,
147- br m), 3.79-3.87 (2H, m), 4.30-4.45 (lH, br m),
69 ex 21 148 S 21 (lH, br s), 7.42 (lH, d), 7.50 (lH, t),
7 . 85-7 . 94 (2H, m)
WO 95133719 2 ~ 9 O q 7 9 PCT/GB9510122-J
~ 176 --
No. Prep mpt nmr
1.36 ~9H, s), 2.02-2.Z0 (lH, m), 2.41-2.54 llH,
m), 2.34 ( 3H, s), 3.70-3.86 (2H, m), 4.4~ (lH,
ex 10 5 br s), 5.18-5.27 (lH, dd), 6.54 (lH, t), 6.92
13 (lH, dd), 7.35 (lH, t), 7.45 (lH, dd), 7.60
(lH, t)
1. 67 ( 6H, 2s ), 2 . 05-2 . 22 ( lH, m~, 2 . 42-2 . SS
(lH, m), 2.87 (3H, s), 3.70-3.86 (2H, m), 4.73
71 ex 10 3 (lH, br s), 5.24 (lH, dd), 6.54 (lH, t), 6.92
12(lH, dd), 7.35 (lH, t), 7.45 (lH, dd), 7.60
(lH, t)
1.08 (9H, s), 2.08-2.28 (lH, m), 2.09 (lH, d),
2.36 (lH, d), 2.36-2.52 (lH, m), 3.06 (3H, s),
72 ex S gum 3.72-3.g2 (2H, m), 5.23 (lH, dd), 6.53 (lH, t),
6.92 (lH, dd), 7.35 (lH, t), 7.45 (lH, dd),
7.59 (lH, t)
5:4 Rotamer mixture: 1.44 and 1.48 (9H, 2s),
2.12-2.35 (lH, m), 2.38-2.53 (lH, m), 2.88 and
73 ex 21 140 2.96 (3H, 2s), 3.69-3.87 ~2H, m), 4.51 and 4.99
(lH, 2 br t), 6.54 (lH, t~, 6.92 (lH, br d),
7.35 (lH, t~, 7.41-7.52 (lH, br m), 7.60 (br s)
1.26 (3H, t), 1.33 (6H, s), 1.67-1.78 (2H, m),
2.01-2.20 (lH, m), 2.40-2.52 (lH, m), 2.85 (3H,
74 ex 16 77-81 s), 3.69-3.85 (2H, m), 4.36 (lH, br 5), 5.21
(lH, dd), 6.53 (lH, t), 6.91 (lH, dd), 7.35
(lH, t), 7.45 (lH, dd), 7.61 (lH, t)
1.66 (9H, s), 2.20-2.35 (lH, m), 2.39 (lH, s),
111- 2.72-2.86 (lH, m), 3.86-3.95 (2H, m), 4.40 (lH,
ex 20 112.5 t), 6.07 (lH, br 5), 7.39-7.54 (2H, m), 7.8g-
7 . 92 (2H, m)
1.66 (6H, s), 2.18-2.32 (lH, m), 2.40 (lH, s),
2.70-2.85 (lH, m), 3.81-3.92 (2H, m), 4.39 (lH,
76 ex 20 83-86 t) 6 09 (lH, br s), 7.00-7.08 (lH, m), 7.40
(lH, t), 7.50-7.58 (lH, m), 7.62-7.66 (lH, m)
1.52 (9H, s), 2.16-2.33 (lH, m), 2.64-2.76 (lH,
m), 3.75-3.93 (2H, m), 5.36 (lH, t), 7.05 ( lH,
77 ex 22 80-92 dt) 7 40 (lH, t), 7.58 (lH, dd), 7.65 (lH, br
s)
WO 95/33719 2 1 9 ~ 9 7 9 PCrIGB95/0122.1
-- ~177 --
N~. E'rep mpt nmr
0.96 (3H, t), 1.35 (9H, s), 1.55-1.81 (2H, m),
2.30-2:50 (2H, m), 3.02-3.22 (2H, m), 3.68-3.78
78 ex 27 111 5 (lH, m), 3.81-3.91 (lH, m~, 4,40 (lH, br s),
4.53 (lH, dd), 6.95-7.03 (lH, m~, 7.36 (lH, t),
7.58 (lH, dd), 7.68 (lH, br s)
0.90 (3H, t), 1.33 (6H, s), 1.72 (2H, q), 2.18-
2.35 (lH, m), 2.70-2.85 (lH, m), 3.85-3.94 (2H,
79 ex 16 gum m) 4 35 (lH, t), 5.50 (lH, br s), 7-42 (lH~
d), 7.50 (lH, t), 7.85-7.92 (2H, m)
0.90 (3H, t), 1.32 (6~, s), 1.72 (2H, q), 2.15-
2.34 (lH, m), 2.68-2.84 (lH, m), 3.82-3.90 (2H,
B0 ex 16 5 m), 4.35 (lH, t), 5.52 (lH, br s), 7.00-7.08
84. (lH, m), 7.39 (lH, t), 7.52-7.59 (lH, m), 7.65
(lH, br s)
1.53 (9H, s), 2.34-2.51 (2H, m), 3.78-3.88 (2H,
105- m), 3.82 (3H, s) 4.96 (lH, t), 7.41 (lH, br
81 ex 22
d)
1.31 (9H, s), 2.35-2.50 (lH, m), 2.54-2.71 (lH,
136.5- m), 3.16-3.29 (lH, m), 3.66-4.00 (4H, m), 4.24
82 ex 27 133.5 (lH, t), 4.63 (lH, br t), 5.06 (lH, br s), 6.99
7,06 (lH, m), 7.37 ~lH, t), 7.53-7.63 (2H, m)
1.25 ~9H, s), 2.27-2.50 (2H, m), 2.67-2.83 t2H,
83 ex 27 gl~m m), 4.15 ~lH, br s~, 5.04 ~lH t), 7.31-7.50
(7H, m), 7.86 (lH, d), 7.95 (lH, br s)
1.34 (9H, s), 2.14-2.30 (lH, m), 2.44-2.57 (lH,
111- m), 3.32-3.40 (2H, m), 3.44 (3H, s), 3.70-3.91
84 ex 27 112 (3H, m), 4.36 (lH, dd), 6.34 ( lH, br s), 7.38
(lH, br d), 7.47 (lH, t), 7.86-7.95 (2H, m)
0.79-0.91 (4H, m), 1.35 (9H, s), 2.29-2.41 (lH,
m), 2.48-2.66 (lH, m), 2.6;-2.76 (lH, m), 3.75
85 ex 27 138 (lH, q), 3.87-3.93 (lH, m), 5.31 (lH, br s),
7.35 (lH, d), 7.45 (lH, t), 7.90 (lH, d), 7.95
(lH, br s)
-
1.41 (9H, s), 2.26-2.39 (lH, m), 2.39-2.55 (lH,
127.5- m), 3.74-3.90 (2H, m), 3.78 (3H, s), 5.12 (lH,
6 128.5 dd), S.90 (lH, br s), 7.40 (lH, d), 7.48 (lH,
t), 7 . 87-7 . 97 (2H, m)
.
WO 9S/33719 2 1 9 0 9 7 9 PCT/GB9S/0122.1
-- 178 --
~o. ~r~p mpt nmr
1.10 (9H, 5), 2.12-2.23 (lH, m), 2.33-2.48 (lH,
llS- m), 3.58--3.78 (2H, m), 4.24-4.49 (3H, m), 4.85
87ex 27 116 4.97 (lH, m), 7.17-7.41 (7H, m), 7.75 (lH, br
d), 7.91 (lH, s)
1.41 (9H, s), 2.24-2.37 (lH, m), 2.39-2.51 (lH,
102- m), 3.68-3.87 (2H, m), 3.77 (3H, s), S.ll (lH,
104 dd), 5.89 (lH, br s), 6.52 (lH, t), 6.92 (lH,
dd), 7.34 (lH, t), 7.47 (lH, dt), 7.58 (lH, t)
1.10 (9H, 5), 2.40 (2H, s), 2.41-2.53 (2H, m),
3.78-3.88 (lH, m), 3.85 (3H, s), 3.90-4.00 (lH,
89ex S 72-73 m) S 04 (lH t), 7.41 (lH, br d), 7.49 (lH,
t), 7 . 89-7 . 9S (2H, m)
1.33 (9H, s), 2.11-2.29 (lH, m), 2.45-2.59 (lH,
m), 3.16-3.37 (2H, m), 3.49 (3H, s~, 3.53 (3H,
90 ex 27 96 5 s), 3.69-3.90 (2H, m), 4.6B (lH, t), 4.85 (lH,
dd), 6.25 (lH, br s), 6.97-7.05 (lH, m), 7.38
(lH, t), 7.56 (lH, dd~, 7.68 (lH, br s)
2.23-2.41 (lH, m~, 2.38 (lH, t), 2.48-2.60 (lH,
m), 3.70-3.90 (2H, m), 3.93 (lH, dd), 4.05 (lH,
91 ex 27 137 5 dd), 4.92 (lH, br s), 5.07 (lH, dd), 7.00-7-07
(lH, m), 1.39 (lH, t), 7.57 (lH, dd), 7.67 (lH,
br s)
1.36 (9H, s), 2.20-2.38 (lH, m), 2.40-2.59 (lH,
m), 2.6~ (6H, s), 3.10-3.22 (2H, m), 3.56-3.68
92 ex 27 (2H, m), 3.68-3.83 (lH, m), 3.83-3.96 (lH, m),
g~ 4.77 (lH, t), 6.62 (lH, v br s), 7.03 (lH, d),
7.39 (lH, t), 7.50 (lH, d), 7.68 (lH, s)
1.33 (9H, s), 2.10-2.26 (lH, m), 2.42-2.54 (lH,
m), 3.65-3.93 (4H, m), 4.62 (lH, br s), 5.07
93 ex 27 (lH, dd), 5.29 (lH, dd), 5.37 (lH, dd), S.91-
108 6.08 (lH, m), 6.98-7.05 (lH, m), 7.38 (lH, t),
7.57 (lH, dd), 7.69 (lH, br s)
1.58 (9H, s), 1.95-2.10 (lH, m), 2.63-2.75 (lH,
141- m), 3.00 (3H, s), 3.78-3.85 (2H, m), 5.63 (lH,
143 br ~), 6.54 (lH, dd), 6.94 (lH, br d), 7.36
(lH, t), 7.44 (lH, dt), 7.60 (lH, t)
WO 95133719 2 1 9 0 9 7 9 PCTIGB95/0122.1
-- 179 -
llo. Prep mpt nmr
l.S9 (9H, s), 1.97-2.14 (lH, m), 2.65-2.77 (lH,
141- m~, 3.82-3.91 (2H, m), 5.63 (lH, br s), 6.68
9S ex 26 144 (lH, dd), 7.42 (lH, br d), 7.51 (lH, t), 7.88
(lH, br d), 7.95 (lH, br s)
0.98 (3H, t), 1.07 (9H, s), 1.61-1.79 (2H, m),
15 2.27 (2H, s), 2.30-2.55 (2H, m), 3.30-3.50 (2H,
96 ex 5 m), 3.75 (lH, q), 3.91-4.01 (lH, m), 4.05 (lH,
116 t) 6 94-7.01 (lH, m), 7.34 (lH, t), 7.55-7.67
(2H, m)
1.08 (9H, s), 1.29 (3H, t), 2.28 (2H, s~, 2.34-
2.53 (2H, m), 3.39-3.61 ~2H, m), 3.76 (lH, q),
97 ex 5 87-88 3.91-4.01 (lH, m), 4.12 (lH, t), 6.95-7.02 (lH,
m), 7.36 (lH, tl, 7.60 (lH, dd), 7.66 (lH, br
s)
0.88 (3H, t), 1.29 (3H, t), 1.32 (6H, s), 1.82
1 5 (2H, q), 2.23-2.53 (2H, m), 3.12-3.35 (2H, m),
98 ex 16 3.67-3.90 (2H, m), 4.34 (lH, br s), 4.68 (lH,
112-5 dd) 6 96-7.04 (lH, m), 7.36 (lH, t), 7.58 (lH,
dd), 7.68 (lH, br s)
1.35 (9H, s), 2.14-2.31 (lH, m), 2.51-2.64 (lH,
99 23 144- m), 3.74-3.90 (2H, m), 3.95 (lH, d), 4.15-4.32
ex 146 (2H, m), 4.93 (lH, dd), 5.23 (lH, br s), 7.41
(lH, br d), 7.50 (lH, t), 7.85-7.94 (2H, m)
1.58 (9H, 5), 1.95-2.13 (lH, m), 2.63-2.76 (lH,
100 26 140- m), 3.01 (3H, s), 3.78-3.86 (2H, m), 5.62 (lH,
ex 142 br s), 6.65 (lH, dd), 7.01-7.08 (lH, m), 7.40
(lH, t), 7.55 (lH, dd), 7.70 (1 H, br s)
1.61(6H, 2s), 2.11-2.29 (lH, m), 2.31 (lH, 5),
2.47-2.61 (lH, m), 3.22 (lH, dd), 3.36 (lH,
101 27 111.5- dd), 3.50 (3H, s), 3.56 (3H, s), 3.70-3.90 (2H,
ex 112.5 m), 4.72 (lH, dd), 4.90 (lH, dd), 6.61 (lH, s),
6.98-7.05 (lH, m), 7.38 (lH, t), 7.55 (lH, dd),
7.68 (lH, br s)
1.08 (9H, s), 2.30-2.40 (3H, m), 2.40-2.57 (2H,
0 m), 3.71-3.98 (2H, m), 4.12-4.32 (2H, m), 4.88
1 2 ex 5 69 71 (lH t) 6.97-7.06 (lH, m), 7.37 (lH, t), 7.58
(lH, dd), 7.66 (lH, br s)
WO 95/33719 . 2 1 9 0 9 7 9 PCTlGB9510122f
-- 180 -- ~
No. Prep mpt nmr
1.07 (9H, s), 2.26 (2H, d), 2.3I-2.56 (2H, m),
3.74 (lH, q), 3.87-3.97 (lH, m~, 3.97-4.21 (2H,
103 ~x 5 91-93 m), 4.40 (lH, t), 5.28 (lH, dd), 5.36 (lH, dd),
5.81-5.97 (lH, m), 6.95-7.02 (lH, m), 7.35 (lH,
t), 7.54-7.68 (2H, m),
0.98 (3H, t), 2.07 (9H, s), 1.61-1.80 (2H, m),
2 27 (2H, s), 2 33-2.56 (2H m), 3 30-3 51
104 ex 5 gum (2H,m), 3.78 (lH, q) 3.94-4.08 (2H, m); 7.35
(lH, br d), 7.45 (lH t~, 7.87-7.95 (2H, m)
0.97 (3H, t), 1.35 (9H, s), 1.55-1.83 (2H, m),
2.30-2.50 (2H, m), 3.02-3.24 (2H, m), 3.70-3.81
105 ex 27 (lH, m), 3.84-3.95 (lH, m), 4.40 (lH, br s),
13B.5 4 46-4.55 (lH t), 7.37 (lH, d), 7.43-7.40 (lH,
t), 7 . 88-7 . 95 (2H, m)
0.96 (3H, t), 1.35 (9H, s), 1.54-1.81 (2H, m),
2.27-2.49 (2H, m), 3.02-3.23 (2H, m), 3.65-3.76
106 ex 27 84-87 (lH, m), 3.80-3.90 (lH m), 4.41 (lH, br s),
4.55 (lH, t), 6.53 (lH, t), 6.89 (lH, dd), 7.33
(lH, t), 7.46 (lH, dd~, 7.61 (lH, t)
0.95 (3H, t), 1.58 (9H, s), 1.58-1.70 (lH, m),
1.80-1.98 (lH, m), 2.05-2.21 (lH, m), 2.58-2.70
100- (lH, m), 3.16-3.43 (2H, m), 3.74-3.89 (2H, m),
ex 2 107 5.76 (lH, b~ 5), 6.41 (lH, dd), 6.54 (lH, t),
6.94 (lH, dd), 7.36 (lH, t), 7.44 (lH, dd),
7.60 (lH, t)
1. 33 ( 9H, 5 ), 2 .11-2 . 29 ( lH, m), 2 . 42-2 . 56 ( lH,
107- m) 3.66-3.95 (4H m) 4.61 (lH, br s), 5.05
108 27
7.40 (lH, d), 7.49 (lH, t), 7.88-7.95 (2H, m)
1.53 ~9H, s), 1.95-2.15 (lH, m), 2.64-2.77 (lH,
m), 3.78-3.94 (3H, m), 4.02-4.15 (lH, m), 5.35
109 cx 26 150 (lH, d), 5.40 (lH, br s), 5.99-6.16 (2H, m),
6.71 (lH, dd), 7.41 (lH, d), 7.50 (lH, t), 7.88
(lH, d), 7.93 (lH, br 5)
wo gsl337l9 2 1 9 0 9 7 9 PCTIGB9S/0122.1
-- 181 -- ~ ~
No. Prep mpt nmr
1.06 (9H, s~, 2.23 (lH, d), 2.31 (lH, d), Z.33-
2.45 (2H, m), 3.74 (lH, q), 3.85-4.20 (3H, m),
110 ex S gum 4.41 ~lH, t), 5.23-5.40 (2H, m), 5.81-5.96 (lH,
m), 6.51 (lH, t), 6.39 (lH, dd), 7.32 (lH, t),
7.46 (lH, dd), 7.53 (lH, t~
1.32 (9H, s~, 2.05-2.25 (lH, m), 2.40-2.53 (lH,
m), 3.64-3.92 (3H, m), 4.61 (lH, br s), 5.07
111 ex 27 gum (lH, dd), 5.25-5.41 (2H, m), 5.91-6.06 (lH, m),
6.53 (lH, t), 6.90 (lH, dd~, 7.34 (lH, t~, 7.44
(lH, dt~, 7.60 (lH, t~
l.Sl (9H, s~, 1.93-2.11 (lH, m~, 2.62-2.75 (lH,
130- m), 3.73-4.14 (4H, m), 5.30-5.42 (2H, m), 6.00-
132 6.16 (2H, m), 6.70 (lH, dd), 7.01 (lH, dd),
7.38 (lH, t), 7.54 (lH, dd), 7.68 (lH, br s)
3:1 Rotamer mix~ure: 1.06 (9H, 2s), 2.20-2.60
(4H, m), 2.75-2.90 (7H, m), 3.15-3.30 (lH, m),
113 ex S 3.40-3.56 (lH, m), 3.72-3.8B (2H, m), 3.90-4.10
g (lH, m), 4.35 and 4.85 (lH, 2t), 6.99-7.10 (lH,
m), 7.31-7.67 (3H, m)
1.34 (3H, t), 1.60 (9H, s), 2.02-2.20 (lH, m),
2.60-2.74 (lH, m), 3.29-3.60 (2H, m), 3.74-3.88
114 ex 26 109 (2H, m), 5.74 (lH, br s), 6.53 (lH, dd), 6.98-
7.05 (lH, m), 7.49 (lH, t), 7.54 (lH, dd), 7.68
(lH, br s)
1.45 (9H, s), l.90-Z.10 (lH, m), 3.07-3.25 (lH,
142- m), 3.75-3.84 (2H, m), 4.90-5.06 (lH, m), 6.54
llS ex 26 143 (lH br d), 6.65 (lH, v br s), 7.05 (lH, dd),
7.40 (lH, t), 7.45-7.56 (lH, m), 7.70 (lH, 5)
0.97 (3H, t), 1.65 (6H, 2s), 1.65-1.81 (lH, m),
2.31 (lH, s), 2.32-2.48 (2H, m), 3.08-3.22 (2H,
116 ex 27 173 5 m), 3.74 (lH, q), 3.81-3.93 (lH, m), 4.52 (lH,
t), 4.68 (lH, br s), 6.95-7.04 (lH, m), 7.37
(lH, t), 7.57 (lH, dd), 7.67 (lH, s)
1.33 (9H, s), 2.10 (3H, s), 2.13-2.32 (lH, m),
2.40-2.55 (lH, m), 3.38 (2H, t), 3.67-3.90 (2H,
117 ex 28 m), 4.30 (2H, t), 4.81 (lH, dd), 5.43 (lH, 5),
131 6 98-7.05 (lH, m), 7.38 (lH, t), 7.57 (lH, dd)~
7.67 (lH, br s)
.
WO 95133719 2 ~ 9 0 9 7 9 PCT/GB951012~
-- 182 --
No. Prep mpt r~mr
10:1 Rotam~r mixture, major rotamer: 0.86 (3H,
t)/ O.9S (3H, t), 1.29 (6H, s), 1.51-1.82 (4H,
114.5- m), Z.27-2.50 (2H, m), 3.00-3.25 (2H, m), 3.65-
llB ex 16 116 3 91 (2H, m~, 4.32 (lH, br s), 4.53 (lH, t),
6.93-7.05 (lH, m), 7.36 (lH, tj, 7.50-7.71 (2H,
m)
1.35 (9H, s), 2.13-2.31 (lH, m), 2.49-2.63 (lH,
m), 3.70-3.90 (SH, m), 3.91 (lH, d), 4.02 (lH,
119 ex 23 dl, 4.90 (lH, dd), 5.20 (lH, s), 7.00-7.08 (lH,
132-5 m), 7.39 (lH, t), 7.55 (lH, dd), 7.64-7-68 (lH,
m)
0.97 (3H, t), 1.20-1.46 (llH, m), 1.45-1.78
(2H, m), 2.27-2.50 (ZH, m), 3.04-3.27 (2H, m),20 ex 27 107 _ 3.66-3.78 (lH, m), 3.82-3.91 (lH, m), 4.42 (lH,
s), 4.54 (lH, t), 6.95-7.02 (lH, m), 7.35 (lH,
t), 7.57 (lH, dd), 7.6B (lH, ~r s)
1.34 (9H, s), 2.10-2.26 (lH, m), 2.44-2.56 (lH,
m), 3.25-3.43 (2H, m), 3.43 ~3H, s), 3.54-3.62
121 ex 27 (lH, m), 3.66-3.85 (3H, m), 4.89 (lH, dd), 6.35
118-5 (lH, s), 6.96-7.04 (lH, m), 7.37 (lH~ t), 7.57
(lH, dd), 7.68 (lH, br 5)
1.36 (9H, s), 2.23 (3H, s), 2.28-2.54 (2H, m),
2.80 (lH, t), 3.35-3.54 (2H, m), 3.65-3.93 (2H,22 ex 27 107 5 m), 4.54 (lH, t), S.l9 (lH, s), 7.01 (lH, br
d), 7.37 (lH, t), 7.57 (lH, dd), 7.67 (lH, br
s)
1.62 (9H, s), 2.13-2.27 (lH, m), 2.S0-2.63 (lH,
128 ex 29 gum m), 3.75 (lH, d), 3.84-3.97 (3H, m), 5.03 (lH,
dd), 7.41-7.56 (2H, m), 7.85-7.94 (2H, m)
~ 7.9(1H,m); 7.6(1H,m); 7.2-7.3(2H m); S.9S-
6.1 (lH,m); S . 8-S . 9S (lH,m); S .2s-s.i (2H,m); 4 . 9-
199 S.OS(2H,m); 4.4(1H,t); 3.9-4.2(2H,m); 3.85-
3.9(1H,m); 3.7-3.5(1H,q); 2.3-2.5(4H,m);
1.2 (6H,d) .
7.9(1H,m); 7.6(1H,m); 7.2-7.3(2H,m); S.9-
6.1(1H,m); S.25-5.45(2H,m); S.l(lH,m);
200 4.55(1H,s); 3.7-3.9(4H,m); 2.4-2.5S(lH,m); 2.1-
2.3~1H,m); 1.3(9H,s).
~` 2190979 ~o
WO 95/33719 PCT/GB95 122-1
-- 183 -- :
No. Prep mpt nmr
7.9(1H,m); 7.6(1H,m); 7.2-7.3(ZH,m); 5.8-
6.0(1H,m); 5.2-5.4(2H,m); 4.4(1H,t); 3.95-
201 4.212H,m); 3.85-3.9511H,m); 3.7-3.8(1H,m); 2.35
2.45(2H,m); 2.25(2H,d); l.l(9H,s).
7.7(1H,s); 7.5~1H,m); 7.q(lH,t); 7.0(1H,m);
136.0- 5.05(1H,m); 4.4-4.5(1H,m); 3.812H,m);
202 138.0 2.9(3H,s)i 2.85-2.95(1H,m); 1.9-2.1 (lH,m)
1.4 (9H,s) .
7.85(2H,m)i 7.2-7.5(7H,m)i 5.0(1H,m)i
203 4.6(2H,s)i 4.5(1H,m)i 3.8(2H,m)i 2.75-
2.9(1H,m)i 1.85-2.0(1H,m)i l.5(9H,s) .
8.0(1H,s): 7.9(1H,m)i 7.4-7.6(2H,m)i 5.1(1H,m)
204 4.5(1H,m)i 3.8-4.0(4H,m); 2.9-3.0(1H,m)i 1.9-
2.1 (lH,m) i 1.3 (12H,m) .
l.O9(9H,s) i 2.0(1H,m) i 2.15(2H,s) i 2.9(1H,m) i
205 164 3.8(2H,m); 4.55(1H,m)i 6.1(1H,m)i 7.1(1H,t)
7.5(1H,m)i 7.8(1H,m).
1.3(9H,s)i 2.0(1H,m)i 2.8(1H,m)i 3.8~2H,m)
206 173 4.4(1H,m~i 4.85(1H,s)i 5.2(1H,d~i 7.1(1H,t)
7.45(1H,=)i 7.75(1H,m).
1.35(9H,s) i 2.1(1H,m) i 2.45(1H,m) i 2.83(3H,s) i
207 137 3.75(2H,m)i 4.42~1H,s)i 5.2(1H,m); 7.1(1H,t);
7.5(1H,m)i 7.8(1H,m).
l.l(9H,s) i 2.1-2.5(1H,m) i 2.3(2H,m) i 3.1(3H,s)
208 3.8(2H,m)i 7.1(1H,t)i 7.5(1H,m)i 7.8(1H,m).
1.3(9H,s)i 2.2(1H,m); 2.45(1H,m); 3.6-
209 103 3.9(4H,m); 4.6(1H,s); 5.0(1H,m); 5.3(2H,m)i
6.0(1H,m)i 7.1(1H,t)i 7.5(1H,m)i 7.8(1H,m).
.05(9H,s); 2.25(2H,s)i 2.4(2H,m)i 3.7(1H,~);
210 3.9(1H,m~i 4.1(2H,m~; 4.3(1H,t)i 5.3(2H,m);
5.9(1H,m); 7.3(1H,t)i 7.5(1H,m)i 7.8(1H,m).
1.15(3H,s)i 1.16(3H,s); l.9(1H,m)i 2.3(2H,s)i
2.85(1H,m); 3.8(2H,m)i 4.5(1H,m); 5.1(2H,m)
211 115 5.95(1H,m)i 6.2(1H,m)i 7.15(1H,t)i 7.5~1H,m
7 . 8 llH,m) .
WO 95/33719 2 1 9 0 9 7 9 PCT/GB9510122.1
-- 184 --
No. ~rep mpt nmr
1.2(6H,s); 2.2(1H,m): 2.4(3H,m); 3.05(3H,s)i
212 3.8(2H,m); 4.9-5.2(3H,m); 6.0(1H,m); 7.1(1P.,t);
7.5(1H,m); 7.8(1H,m) .
l.l~9H,s); 1.3(3H,t); 2.3~2H,s); 2.45~2H,m);
213 93 3.5~2H,m); 3.8~1H,q); 4.0~1H,m); 4.1~1H,t);
7 . 4 ~2H,m); 7 . 9 (2H,m) .
0.85~3H,t); 1.05~3H,s); 1.06~3H,s); 1.3(3H,t);
l . 4 (2H, q); 2 . 25 (2H, s ); 2 . 45 (2H,m); 3 . S (2H,m);
214 3.8(1H,q); 4.0(1H,m); 4.1(1H,t); 7.4(2H,m);
7 . 9 (2H,m) .
1.12~9H,s); 2.30-2.53~2H,m); 2.63~2H,s); 3.81-
215 4.02~2H,m); 5.39-5.47~1H,t); 7.00-7.04(1H,m);
7.35-7.41(1H,t); 7.55-7.62(2H,m); 9.28(1H,s).
.11(9H,s); 2.30-2.55~2H,m); 2.63(2H,s); 3.85-
216 4.05(2H,m); 5.40-5.48(1H,t); 7.40-7.52(2H,m);
7.84-7.94~2H,m); 9.28(1H,s).
0.84-0.91(3H,t); 1.01(6H,s); 1.34-1.44(2H,q);
1. 91-2. 07 (lH,m); 2 . 02 (2H,s); 2 . 88-2 . 99 (lH,m);
217 3.81-3.90(2H,m); 4.51-4.60(1H,m); 6.08(1H,s);
7 . 40-7 . 54 (2H,m); 7 . 37-7 . 93 (2H,m) .
0.84-0.90(3H,t); 1.03-1.04(6H,d); 1.40-
1. 48 (2H, q); 2 .13-2 .29 (lH,m); 2 . 30-2 . 32 (2H,d);
218 2.42-2.54(lH,m); 3.09(3H,s); 3.78-3.95(2H,m);
5.15-5.23(1H,t); 7.39-7.53(2H,m); 7.89-
7 . 93 (2H,m) .
0 . 84-0 . 90 (3H, t); 1. 03 ( 6H, d); 1. 39-1. 47 (2H, q);
2.10-2.27(lH,m); 2.29-2.34(2H,d); 2.40-
219 2.52(1H,m); 3.07(3H,s); 3.73-3.92(2H,m); 5.18-
5.25(1H,t); 7.00-7.04(1H,m); 7.34-7.41(1H,t);
7.54-7.60(1H,m); 7.67(1H,s).
2 1 9 0 9 CT~GB9C,0,22
WO95/33719 ~9 P
- 185 -
No. Prep mpt nmr
0.86-0.91(3H,t); 1.32(6H,s); 1.6B-1.76(2H,q);
2.16-2.29(1H,m); 2.6g-2.80(1H,m); 3.80-
220 3.86(2H,m); 4.30-4.36(1H,t); 5.54(1H,s); 7.20-
7.32(2H,m); 7.58-7.63(1H,m); 7.80-7.82(1H,m).
1.07(9H,s); 1.87-2.04(1X,m); 2.14(2H,s); 2.84-
2.95(1H,m); 3.77-3.83(2H,m); 4.50-4.60(1H,m);
221 6.05-6.11(1H,d); 7.21-7.33(2H,m)i 7.58-
7 . 62 (lH,m); 7 . 83-7 . 85 (lH,m) i
0.84-0.91(3H,t); 1.00-1.04(6H,d); 1.34-
1.43(2H,q); 1.8702.05(lH,m); 2.13(2H,sl; 2.85-
222 2 . 96 ( lH,m); 3 . 77-3 . 84 (2H,m); 4 . 48-4 . 57 (lH,m);
6.06(1H,d): 7.22-7.33~2H,m)i 7.58-7.62~1H,m);
7 . 83-7 . 86 (lH,m) .
0.84-0.90(3H,t); 1.28-1.30(6H,d); 1.65-
1.74(ZH,q); 1.90-2.04(1H,m); 2.78-2.90(1H,m);
223 3.74-3.82(2H,m); 4.35-4.44(1H,m); 4.58(1H,s);
4.94-4.98(1H,d); 7.21-7.33(2H,m); 7.57-
7 . 61 (lH,m); 7. 82-7 . 84 (lH,m) .
1.20(6H,s); 2.05-2.22(1H,m); 2.36-2.50~3H,m);
3.03(3H,s); 3.71-3.89(2H,m); 4.94-5.03(2H,m);
224 5.18-5.26(1H,t); 5.96-6.06(1H,m); 7.19-
7 . 31 (2H,m); 7 . 59-7 . 63 (lH,m); 7 . 84-7 . 86 (lH,m) .
1.16-1.18(6H,d); 1.85-2.01~1H,m); 2.29(2H,s);
2.83-2.93(1H,m); 3.77-3.82(2H,m); 4.48-
225 q .57 (lH,m); 5 . 03-S. 11 (2H,m); 5 . 90-6. 00 (lH,m);
6.13-6.18(lH,d); 7.22-7.33(2H,m); 7.58-
7 . 62 (lH,m); 7 . 83-7 . 85 (lH,m) .
1.16-1.19 (6H, d); 1. 91-2 . 04 (lH,m); 2 .29 (2H, ~ );
2.85-2.96(1H,m); 3.81-3.89(2H,m); 4.50-
226 4.60(1H,m); 5.03-5.11(2H,m); 5.90-6.00(1H,m);
6.18-6.22(1H,d); 7.41-7.54(2H,m); 7.86-
7 . 93 (2H,m) .
WO 95/33719 2 1 9 0 9 7 9 PCTlGB95/Olt2.1 ~
-- 186 --
No. Prep mpt Dmr
1.20~6H,s); 2.09-2.25~1H,m); 2.41-2.42~2H,d);
2.40-2.51~1H,m); 3.04~3H,s); 3.73-3.gl~2H,m);
227 4.93-5.03~2H,m); 5.17-5.24(1H,t); S.9S-
6.05(1H,m); 7.00-7.05(1H,m); 7.34-7.41(1H,t~;
7.54-7.59(1H,m); 7.66(1H,s).
l.lO~9H,s); 2.05-2.52 4(m) 3.06~s,3H)+2.8B (s);
228 3.73-3.90(2H,m); 5.20-5.28(1H,t~ i+4.83-4.91
(t); 7.59-7.63(lH,m); 7.85-7.87~lH,m) .
.38~9H,s); 2.02-2.17~1H,m); 2.41-2.52~1H,m];
2.a3~3H,s); 3.69-3.83~2H,m); 4.44~1H,s); 5.18-
229 S . 26 ~ lH,m); 7 . 20-7 . 30 (2H,m); 7 . 60-7 . 64 (lH,m);
7 . B4-7 . 86 (lH,m) .
0.84-0.91(3H,t); 1.03-1.05(6H,d); 1.40-
1.48(2H,q); 2.05-2.51(2H,m); 2.29-2.32(2H,d);
230 3.06(3H,s)+ 2.86(s); 3.72-3.90(2H,m~; S.l9-
5.26(1H,t)+4.84-4.92(tl; 7.20-7.30(2H,m); 7.59-
7 . 63 (lH,m); 7 . 84-7 . 87 (lH,m) .
1.33(9H,s); 1.90-2.09(1H,m); 2.78-2.90(1H,m);
3.74-3.81(2H,m); 4.36-4.46(1H,m); 4.70(1H,s);
231 4.97-S.OO(lH,m); 7.20-7.32(2H,m); 7.55-
7.60(1H,m); 7.82-7.84(1H,m).
.38(9H,s); 2.15-2,29(1H,m); 2.69-2.81(1H,m);
3.81-3.87(2H,m); 4.30-4.37(1H,t); 5.67(1H,s);
232 7.20-7.31(2H,m); 7.59-7.63(1H,m); 7.80-
7 . 82 (lH,m) .
2.11-2.25(1H,m); 2.42(3H,s); 2.69-2.81(1H,m);
233 3.84-3.90(2H,m); 4.38-4.44(1H,t); 7.21-
7.33(2H,m); 7.59-7.64(1H,m); 7.80-7.82(1H,m).
1.96 (3H, s), 1.90-2.10 (lH, m), 2.20 (3H, s),
2.85-2.98 (lH, m), 3.77-3.86 (2H, m), 4.55-4.65
305 ex S (lH, m), 5.65 (lH, t), 6.17 (lH, br d), 7.00-
127 7.08 (lH, m), 7.39 (lH, t), 7.53 (lH, dd), 7.68
(lH, br s)
WO 95~33719 2 ~ 9 0 9 7 9 PCTIGB9slol22~
- la7 -~
No. Prep mpt nmr
0.94-1.04 ~6H, m), l.a8-2.21 (4H, m), 2.85 2.98
163.5- ~lH, m), 3.77-3.88 (2H, m), 6.20 (lH, br d),
164.5 7.01-7.09 (lH, m), 7.40 (lH, t), 7.53 (lH, dd),
7.68 (lH, br s)
0.88 (3H, t), 1.01 (6H, s), 1.39 (2H, q), 1.87-
2.05 (lH, m), 2.1q (2H, s), 2.85-2.96 (lH, m),
307 ex S 3.82 (lH, dd), 4.50-4.60 (lH, m), 6.13 (lH, br
107-5 d) 7 00-7.08 (lH, m), 7.39 (lH, t), 7.50-7.57
(lH, m), 7.68 (lH, br s)
3:2 Rotamer mixture: 2.21-2.45 (lH, m), 2.46-
2.63 (lH, m), 3.08 (3H, s;, 3.80-3.95 (2H, m),
308 ex 90 gum 4 75_4 96 (lH, 2m~, 4.78 (2H, sl, 7.43 (lH~ d),
7.51 (lH, t~, 7.88-7.92 (2H, m)
1.38 (9H, s), 2.16-2.30 (lH, m), 2.68-2.83 (lH,
30 0 89_92 m)~ 3.79-3.90 (2H, m), 4.35 (lH, t), 5.67 (lH,
9 ex 2 br s), 6.53 (lH, t), 6.84 (lH, dd), 7.36 (lH,
t), 7.44 (lH, dt), 7.56 (lH, t)
3:1 Rotamer mixture: 2.19-2.38 (lH, m), 2.49-
2.64 (lH, m), 3.01 (0.75H, s), 3.24 (2.25H, t),
310 ex S gum 3.78-3.98 (2H, m), S.00-S.lS (lH, m), 7.02-7.10
~lH, m), 7.36-7.46 (lH, m), 7.52-7.60 (lH, m),
7, 66 (lH, br s)
1.37 (9H, s), 2.06-2.25 (lH, m), 2.35-2.50 (lH,
153- m), 3.22-3.51 (4H, m), 3.71-3.87 (2H, m), 4.84
311 ex 89 154 (lH, dd), 6.96-7.06 (lH, m), 7.38 (lH, t), 7.56
(lH, dd), 7.71 (lH, br s)
0.85 ~3H, t), 1.28 (6H, s), 1.68 (2H, q), 2.08-
2.25 (lH, m), 2.40-2.54 (lH, m), 3.66-3.95 (4H,
m), 4.53 (lH, br 5), 5.05 (lH, dd), 5.29 (lH,
312 ex 27 89 91 dd) S 38 (lH, dd), 5.91-6.08 (lH, m), 6.96-
7.04 (lH, m), 7.37 (lH, t), 7.57 (lH, dd), 7.6g
( lH, br s )
1.06 (9H, d), 1.86-2.03 (lH, m), 2.15 (2H, s),
117 2.85-2.96 (lH, m), 3.82 (lH, dd), 4.50-4.61
313 ex S 11 (lH, m), 6.10 (lH, br d), 6.54 (lH, t), 6.94
9 (lH, dd), 7.36 (lH, t), 7.44 (lH, dt), 7.59
(lH; t)
WO 95133719 2 1 9 0 9 7 q PCT/GB9510122.1
-- 188 -
No. E'rep mpt nmr
1,66 ~6H, s), 2.16-2.31 ~lH, ml, 2.38 ~lh, s),
2.70-2.85 ~lH, m), 3.B0-3.90 ~2H, m), 4.3a ~
314 ex 20 gum t) 6 19 ~lH, br s), 6.54 ~lH, t), 6.94 ~lH, br
d), 7.35 ~lH, t), 7.44 ~lH, dt), 7.55 ~lH, t)
3:1 Rotamer mixture: 1.96 ~6H, s), 2.17-2.38
~lH, m), 2.43-2.55 ~lH, m), 2.94 and 2.97 ~3H,
315 ex 90 84 ) 2s), 3-72-3-91 ~2H, m), 4.84-4-98 ~lH, m), 7-04
~lH, br d), 7.40 ~lH, t), 7.57 ~lH, dd), 7.68
~ lH, br s )
1.35 ~9H, s), 1.51 ~9H, s), 2.15-2.30 ~lH, m),
122- 2.47-2.60 ~lH, m), 3.70-3.85 ~4H, m), 4.96
123 dd), 5.14 ~lH, br s), 7.02 ~br d), 7.39 ~lN,
t), 7.55 ~lH, dd~, 7.71 ~ lH, br s)
1.22 ~9H, s~, 1.31 ~9H, s), 2.02-2.18 ~lH, m),
2.47-2.60 ~lH, m), 3.68-3.85 ~lH, m), 4.20 ~lH,
317 ~x 27 d), 4.38 ~lH, d), 4.66-4.78 ~2H, m), 6.99-7.06
151 ~lH, m), 7.39 ~lH, t), 7.52-7.57 ~lH, m), 7-67
~ lH, br s )
1.36 I9~, s), 2.30-2.48 ~lH, m), 2.64-2.77 llH,
m), 3.78-3.96 ~2~, m), 4.15 ~lH, d), 4.30 ~lH,
318 ex 27 114 5 d), 4.73 tlH, dd), 5.07 (1~, br s), 7.08 ~lH,
dd), 7.43 ~lH, t), 7.50-7.58 ~lH, m), 7.64 ~lH,
br s)
0.93 ~9H, s), 2.08-2.26 ~lH, m), 2.41-2.55 ~lH,
m), 2.92 (3H, s), 3.08 (2H, d), 3.71-3.90 (2H,
319 ex 20 m), 4.64 (lH, br t), 5.15 (lH, dd), 6.98-7.05
134 (lH, m), 7.38 (lH, t), 7.57 (lH, dd), 7.70 (lH,
br s)
1.07 (9H, s), 2.24 (lH, d), 2.31 (lH, d), 2.34-
2.47 (2H, m), 3.77 (lH, q), 3.90-4.04 (lH, m),
320 ex 20 oil 4.04-4.21 (2H, m), 4.37 (lH, t), 5.28 (lH, dd),
5.36 (lH, dd), 5.82-5.98 (lH, m), 7.37 ~1~, br
d), 7.46 ~lH, t), 7.96-7.95 ~lH, m)
3:2 F(otamer mixture: 2.18-2.43 ~lH, m), 2.43-
2.63 ~lH, m), 3.06 ~3H, s), 3.74-3.92 ~2H, m),21 ex 90 73 75 4 72-5 00 ~3H, m), 7.00-7.09 ~lH, m~, 7.40 ~lH,
t),.7.56 (lH, br t), 7.68 ~lH, br s)
WO 95/33719 ` 2 1 9 0 9 7 9 PCT/GB9S/0122.1
-- ~189 -- ~ ;
No. Prep mpt nmr
l.09 (9H, s), 1.29 (3H, t), 2.28 ~2H, s), 2.32-
2.52 (2H, m), 3.40-3.62 (2~, m), 3.75 (lH, q),
322 ex 5 128 3.90-4.01 (lH, m), 4.15 (lH, t), 6.52 (lH, t),
131 6.89 (lH, dd), 7.33 (lH, t), 7.48 (lH, dd),
7.60 (lH, t)
1.26 (3H, s), 1.36 (9H, s), 2.21-2.52 (2H, m),
3.10-3.36 (2H, m), 3.65-3.90 (2H, m), 4.44 (lH,
323 ex 20 7 br s) , 4.70 (lH, dd) , 6.54 (lH, t) , 6.90 (lH,
14 dd), 7.34 (lH, t), 7.42-7.48 (lH, m), 7.61 (lH,
t)
1.08 (9H, s), 2.20 (lH, d), 2.40 (lH, d), 2.50
324 S (2H, qO, 3.72-4.08 (gH, m), 4.30-4.47 (lH, m),
ex gum 6.97-7.04 (lH, m), 7.37 (lH, t), 7.55-7.65 (2H,
m)
10:1 Rotamer mixture: 2.10-2.30 (lH, m), 2.41-
2.57 (lH, m), 2.79-3.20 (7H, m), 3.75-3.94 (2H,
325 ex 5 gum m), 4.81 zmd 5.20 (lH, 2dd), 7.01-7.10 (lH, m),
7.40 (lH, t), 7.54-7.70 (2H, m)
5:2 Rotamer mixture: 2.19-2.39 (lH, m), 2.50-
2.65 (lH, m), 3.00 (6~7H, s), 3.24 (lS/7H, t),
326 ex 88 7 3.78-3.98 (2H, m), 5.0-5.16 (lH, m), 7.02-7.13
6 5 (lH, m), 7.36-7.47 (lH, m), 7.53-7.61 (lH, m),
7.65 (lH, br s)
1.34 (9H, s), 2.40-2.52 (lH, m), 3.70-3.94 (3H,
128- m), 4.05-4.23 (lH, m), 4.31 (lH, t), 4.77 (lH,
327 ex 20 130 br s), 6.98-7.05 (lH, m), 7.38 (lH, t), 7.56
(lH, t), 7.56 (lH, dd), 7.65 (lH, br s)
lO:l Rotamer mixture: 2.08-2.28 (lH, m), 2.40-
2.80 (SH, m), 2.91 and 3.05 (3H, 25), 3.73-3.93
328ex 87 gum (2H, m), 4.75 and 5.20 (lH, 2dd), 6.99-7.10
(lH, m), 7.35-7.45 (lH, m), 7.52-7.60 (lH, m),
7 . 66 ( lH, br s )
1.34 (9H,s), 4.72 (lH, d), 4.90 (lH, br s),
329 ex 35 109 5-08 (lH, dd), 6.21 (lH, d), 7.72 (2H, s+d),
7. 94 (lH, d)
1.34 (9H, s), 4.74 (lH, d), 4.88 (lH, br s),
330 ex 35 S.ll (lH, dd), 6.22 (lH, d), 7.55 (lH, dd),
153 7,92 (lH, d), 8.00 (lH, d)
WO 95/33719 2 1 9 0 9 7 9 PCTIGB9S/0122.1
-- 19(~ --
No. Prep mpt nmr
1.35 (9H, s), 4.59 (lH, d), 4.90 12H, dd +b~
331 ex 35 119 s), 6.01 (2H, s), 6.18 (lH, d), 6.83 (2H, m~,
6. 99 (lH, d)
1.35 (9H, s), 2.2(1H, m), 2.8 (lH, m), 3.9 (3H,
332 77-78 s), 4.0 (lH, m), 4.3 (lH, m), 4.9 (lH, br s),
62/67 5.5 (lH, t), 8.15 (lH, s)
1.35 (9H, s), 2.2 (lH, m), 2.8 (lH, m), 4.0
t), 7 8 (lH, s)
1.35(9H,s); 2.15(lH,m); 2.7(lH,m); 3.8(lH,m);
339 gum 4.2:1H,m); 4.95(1H,br s); 5.4(1H,t);
62/66 7.30(1H,t); 7.40(1H,t); 7.8(1H,s).
0.15~9H,s); 2.06(2H,m); 2.14~1H,m);
92.6- 2.44(1H,m); 2.99(3H,s); 3.78(2H,m);
282 93.4 5.27(1H,dd); 6.98(1H,m); 7.35(1H,t);
7.55(1H,dd); 7.64(1H,s).
l.lO(9H,s); 1.31(3H,t); 2.30t2H,q); 3.58(2H,m);
291 98.55 4.63(1H,d); 5.23(1H,dd); 5.27(1H,s);
7.51(2P.,m); 7.70(1H,m); 7.76~1H,s).
1.29(3H,t); 1.35(9H,s); 3.31(2H,m); 4.56(1H,s);
292 104.7 4.71(1H,d); 4.99(1H,dd); 5.86(1H,s);
105.7 7.52(2H,m); 7.70(1H,m); 7.79(1H,s).
1.01(3H,t); 1.06(9H,s); 1.71(2H,sextet);
2.28(2H,q); 3.45(2H,dt); 4.61(1H,d);
293 oll 5.20(2H,m); 7.48-7.55(2H,m); 7.70(1H,m);
7.76(1H,s) .
0 . 96 (3H, t); 1. 31 (9H, s ); 1. 70 (2H, 3extet);
115.7- 3.19(2H,dd); 4.54(lH,s); 4.70(1H,d);
294 116.7 S.Ol(lH,dd); 5.70(1H,s); 7.50(2H,m);
7.69(1H,m); 7.79(1H,s) .
1.06(9H,s); 2.27(2H,dd); 4.26(2H,m);
62.0- 4.62(1H,d); 5.17(1H,dd); 5.31(1H,s); 5.32(1H,br
295 63.8 s); 5.43(1H,d); 5.89(1H,m); 7.50(2H,m);
7.67(1H,m); 7.75(1H,s).
1.30(9H,s); 3.88(2H,m); 4.73(2H,d);
121.5- 4 . 93 (lH,dd); 5.30 (lH,dd); 5.41 (lH,dd);
296 122.5 i.96(11~,m); 6.13(1H,s); 7.52(2H,m); 7.70(1H,m);
7.76(1H,s) .
WO 95133719 - 2 1 9 0 9 7 9 PCT/GB9510122.1
-- ~91 --
No. Prep mpt nmr
1.0819H,s); 2.36(2H,q); 2.45(1H,s);
297 91-92 4.30(2H,dq); 4.68(1H,d); 5.15(1H,d);
5.61(1H,s); 7.51(2H,m); 7.69(1H,m); 7.74(1H,s).
1.33(9H s);
298 141-5- 4 77(1H d); 4 96(1H dd;; 5 27(1H s)
142~-8 6.13(1H,s); 7.53(2H,m); 7.70(1H,m); 7.7a(1H,s).
1.08(9H,s); 2.30(1H,d); 2.41(1H,d); 3.84(3~.,s);
126 96 4.65(1H,d); 5.19(1H,dd); 5.80(1H, br s); 7.52-
7 . 73 (4H,m) .
128.5- 3.71(3H,s); 5.11(2H,s); 7.40(1H, br s); 7.49-
129.5 7.71(4H,s); 7.82(1H,s).
0.89(9H,s); 3.02(2H,d); 3.95(2H,m); 4.71(1H,d);
111.3- q.84(1H,t); 5 Ol(lH dd) S 34(1H m) -
3 111.8 S.50(1H,m); 5 92(1H s); 6.00(1H,m;; ;.50(2H,m);
7 .70 (lH,m); 7. 77 (lH,s) .
O.91(9H,s); 2.43(1H,t), 3.08(2H,m)-
1451- 4 .11 ( lH, dd); 4 . 24 ( lH, dd) 4 . 74 ( lH, d) -
301 145J5 5.00(1H,dd); 5.32(1H,t~- 5.99(1H ) -
7 . 50 (2H,m); 7 . 75 (2H,m) . , s,
1.43(6H,s); 2.39 t 7
(lH, ), 3. 9(2H,s),
302 96 98 4.97(1H,dd); 5.47(1H,s); 6.10(1H,s);
7.54 (2H,m); 7.72 (lH,m); 7.78 (lH,s) .
1.62(9H,s); 3.77(1H,d); 3.99(1H,d); 4.84(1H,d);
303 4.91(1H,dd); 6.17(1H,s), 7.56(2H,m)-
201 7.74(2H,m).
1.37(9H,s); 4.74(1H d) 5.01(1H dd) -
304 150 6 5 47(1H s), 5.58(1H s), 7.51_7.;2(3H,m);
l.lO(9H,s); 2.31(2H,s); 3.14(3H,s); 4.74(1H,d);
125 1167 5.04(1H,dd); 6.10(1H,br s); 7.55(2H,m);
7 . 75 (2H,m) .
1.37(9H,s); 2.88(3H,s); 4.55(1H,br s)
124 139 4.79(1H,d); 4.88(1H,dd) 6.38(1H,s) -
140 7.ss(2H,m); 7.72(1H m)- 7.80(1H m)-
1.60(3H,s); 1.63(3H s); 2.15(1H m); 2.50(1H,m);
115- 2.89(3H,s); 3.80(2H,m); 4.70(1H,br s);
281 116 5.13(1H,dd); 7.02(1H m)- 7.40(1H,br)-
7 . 56 (lH,dd); 7 . 68 (lH m)
.36~9H,s); 2.69(2H,dq); 4.79(1H,m); 5.46(2H,d);
273 91-93 5.65(1H,br s), 6.56(1H,t) 6.98(1H,m)-
~ 7.28(1H,m); 7.39(1H,t), 7 51(1H,t).
.
WO 9~/33719 2 1 9 0 9 7 9 PCT/GB9510122.1
-- 192 --
No. Prep mpt nmr
1.65~6H,s); 2.35(1H,s); 2.74(2H,dq);
104- 4.80(1H,m); 5.48(2H,d); 6.07(1H,br s);
277 106 6.55(1H,t); 6.98(1H,dd); 7.29(1H,m);
7.39~1N,t); 7.51~1H,t) .
1.35(9H,s); 2.71(2H,dq); 4.80(1H,m);
67 oil 5.48(2H,s); 5.71(1H,br s); 7.06(1H,m);
7.26(1H,m); 7.40(1H,t); 7.50(1H,s); 7.64~lH,t).
1.28(3H,t); 2.95(2H,dq); 4.19(2H,q);
66 gum 4.80~1H,m); 5.50(2H,s); 7.05(1H,m); 7.28~1H,m);
7.42~1H,t); 7.49(1H,s); 7.63(1H,t) .
1.34(3H,t); 2.95(2H,dq); 4.19(2H,q);
62 58-S9 4.79(1H,m); 5.48~2H,s); 6.56~1H,t); 6.97~1H,m);
7.29~1H,m); 7.39~1H,t); 7.55~1H,t).
0.92~9H,s); 2.82~2H,dq); 3.10~2H,??~;
4.80~lH,m); 5.49(2H,d); 5.95(1H,br s);
274 6.55~1H,t); 6.97~lH,dd); 7.28~lH,m);
7.39~1H,t); 7.53~1H,t).
0.86~3H,t); 1.3D~6H,s); 1.73~2H,q);
2.71~2H,dq); 4.79~1H,m); 5.47~2H,s); 5.54~1H,br
275 s); 6.55~1H,t); 6.97~1H,dd); 7.29(1H,m);
7.39~1H,t); 7.53~1H,t) .
1.45~6H,s); 2.72~2H,dq); 4.79~1H,m);
5.07~2H,m); 5.47~2H,d); 5.78~1H,br s);
276 6.02~1H,m); 6.56~1H,t); 6.98~1H,dd);
7.29~1H,m); 7.39~1H,t); 7.53~1H,t).
1.35(3H,d); 3.17(1H,m); 4.88(1H,q);
5 . 20 (2H,A3q); 5 . 46 (lH,m); 5 . 50 (lH,m);
278 g 7.34(5H,m); 7.50(2H,m); 7.76(1H,m); 7.80(1H,br
s) .
1.46 (3H,d); 3.24 (lH,m); 4 .54 (lH,m); 4.98 (lH,d);
279 5.13(1H,m); 5.24(1H,d); 5.39(1H,m); 7.18(5H,m);
7 . 47 (2H,m); 7 . 65 (2H,m) .
1.28(3H,d); 1.37(9H,s); 2.83(1H,dq);
280 99-101 4.75(1H,m); 5.51(2H,d); 5.73(1H,br 5);
7.50(2H,m); 7.76(1H,d); 7.85(1H,m).
1.08 (9H,s); 2.20-2.50 (4H,s+m); 3.72 (lH,m);
283 gum 3.91(1H,m); 4.23(1H,t); 4.67(1H,d); 4.79~1H,d);
7 .24 ~2H,m); 7 .41 (4H,m); 7 . 90 ~2H,m) .
WO 95~337l9 2 1 9 0 9 7 9 PCTIGB95/0122J
-- =193
No. Prep mpe nmr
l.l9(9H,s): 2.28(1H,m); 2.52(1H,m); 3.80(2H,m);
284 gum 4.30(1H,s); 4.38(1H,d); 4.54(1H,d)i 4.99(1H,m);
7.25(2H,d); 7.41(1H,t); 7.51(3H,m); 7.90(2H,m).
1. 08 (9H, s ); 2 . 30 (2H,A3q); 2 . 39 (2H,m);
3.78(1H,m); 3.90(1H,m); 4.37(1H,t); 4.68(1H,d);
285 gum 4.94(lH,d); 7.25(lH,m); 7.43(2H,m); 7.75(2H,m);
7.90(2H,m); 8.58(1H,m).
1.29 (9H,s); 2.34 (lH,m); 2.50 (lH,m); 3. 83 (2H,m);
147- 4.48(2H,Ai3q); 4.93(1H,m); 6.16(1H,s);
286 149 7.27(1H,m); 7.40(1H,m); 7.49(1H,m); 7.63(1H,m);
7.75(1H,dt); 7.92(2H,m); 8.55(1H,m).
l.O9(9H,s); 2.23-2.50(4H,s+m); 3.76(1H,m);
287 gum 3.93(1H,m); 4.24(1H,t); 4.77(2H,A3q);
7 . 40 (3H,m); 7 . 88 (3H,m); 8 . 55 (2H,m),
1.25 (9H,s); 2.29 (lH,m); 2.51 (lH,m); 3. 82 (2H,m);
a8 140- 4.38(1H,s); 4.42(1H,d); 4.57(1H,m); 4.94(1H,m);
143 7.31-7.52(3H,m); 7.88(2H,m); 7.94(lH,m:;
8 . 58 (2H,m) .
1.08 (9H,s); 2.24 (2H,A3q~; 2.39 (2H,m);
3.77(1H,m:; 3.91~1H,m); 4.33(1H,t); 4.62(1H,d);
289 g 4.82(1H,d~; 7.38(2H,m); 7.49(2H,m); 7.90(2H,m);
8.64 (2H,m) .
1.25(9H,s); 2.26(1H,m); 2.54(1H,m;: 3.82(2H,m);
290 153 4.30 (lh,S); 4.38(lH,m); 4.56(lH,m); 4.95llH,m);
7.37-7.54(4H,m); 7.89(2H,m); 8.65(2H,m).
1.35, (9H,s); 2.27 (lH,m); 2.77 (lH,m); 4.15
(lH,q); 4.36 (lH,dt); 4.70 (3H,s); 4.92 (lH,br
504 s); 5.41 (lH,t); 8.07 (lH,dd); 8.81 (lH,d);
9.14 (lH,d); 9.70 (lH,s).
7.8(1H,m); 7.6(1H,m); 7.2-7.3(2H,m); S.9(1H,m);
1~ }1 42 5 5 . l-S . 3 (2H,m); 3 . 7-3 . 8 (2H,m); 3. 6 (lH,m);
44 5 3.4(2H,m); 2.S(lH,m); l.9(2H,m).
2.4(1H,m); 2.8(1H,m); 3.3(3H,s); 3.9(2H,m);
I2 llS 5.4(1H,t); 7.5(2H,m); 7.9(2H,m)-
2.4(1H,m); 2.75(1H,m); 3.3(3H,s); 3.9(2H,m);
~3 120 5 35(1H t); 7.2(1H,t); 7.5(1H,m); 7-8(1H~m)-
. _ _ . _ _ . . . . . . _ _ . . . .
WO 9~/33719 2 1 9 0 9 7 9 PCT/GB95/0~22.1
-- 194 -
No. Prep mpt nmr
1.7~2H,s); l.9(1H,m); 2.6~1H,m); 3.75~2~.,m~;
I4 106 7.1~1H,t); 7.s~1H,m); 7-75(1H~m)-
2.0(1H,m)i 2.5(1H,m): 2.55(3H,s); 3.5(1H,m);
IS 9 3.75(2H,m); 7.1(lH,t); 7.55(lH,m); 7.75(lH,m) .
1.95(2H,m)i 2.5(1H,m); 3.4(2H,m); 3.6(1H,m);
I6 3.75(2H,m); 5.2(2H,m); S.9(1H,m); 7.1(1H,t);
7.5(1H,m); 7.75(1H,m).
2.36-2.50(1H,m); 2.70-2.82(1H,m); 3.32(3H,s);
3.79-4.00(2H,m); 5.34-5.40(lH,t); 7.07-
I7 7.12(lH,m); 7.40-7.46(1H,t); 7.50-7.54(lH,m);
7.66(1H,s) .
1. 96-2.12 (lH,m); 2 . 89-3. 00 ilH,m); 3 . 82-
3.88(2H,m); 4.57-4.67(1H,m); 6.35(1H,s); 7.02-
I8 7.09(lH,m); 7.38-7.43(lH,t); 7.51-7.57(lH,m);
7.68(1H,s); 8.32(1H,s~.
1.99-2.14~1H,m~; 2.87-2.99(1H,m); 3.83-
3.91(2H,m); 4.59-4.70(1H,m); 6.48(1H,s); 7.41-
7.54(2H,m); 7.82-7.87(1H,d); 7.90(1H,s);
8.32(1H,s) .
1.97-2.12(lH,m); 2.20-2.70(1 H,s); 2.45-
2.55(1H,m); 2.58(3H,s); 3.57-3.64(1H,m); 3.74-
I10 3.88(2H,m); 7.00-7.04(1H,m); 7.34-7.41(1H,t);
7 .53-7.59 (lH,m); 7.64 (lH,s) .
2.33-2.48(1H,m); 2.68-2.80(1H,m); 3.32(3H,s);
Ill 3.77-3.96(2H,m); S.32-5.38(1H,t); 7.24-
7.38(2H,m); 7.54-7.59(1H,m); 7.82-7.85(1H,m).
2.38-2.76(2H,m); 3.73-4.01(3H,m); 7.13-
I12 7.30(2H,m); 7.50-7.65(1H,m); 7.76-7.83(1H,m).
1.90-2.04(lH,m); 2.43-2.52(1H,m); 2.51(3H,s);
I13 3.47-3.5S~lH,m); 3.70-3.84(2H,m); 7.20-
7.30(2H,m); 7.60-7.65(1H,m); 7.80-7.82(1H,m) .
1.80-1.96(1H,m); 2.S3-2.64(1H,m); 3.66-
I14 3.78(3H,m); 7.21-7.31(2H,m); 7.62-7.66(1H,m);
7 . 82-7 . 84 (lH,m) .
WO 95133719 2 1 9 0 9 7 9 PCT/GB9510122~
- 195
No. Prep mpt nmr
2.36-2.48 (lH, m), 2.67-2.81 (lH, m), 3.al-3.92
I15 oil (lH, m), 3.99-4.10 (lH, m), 4.56 (lH, dd), 7.42
7 . 51 (2H, m), 7 . 84-7 . 93 (2H, m)
2.41-2.55 (lH, m), 2.68-2.84 (lH, m), 3.80-3.92
(lH, m), 4.00-4.13 (lH, m), 4.60 (lH, dd), 7.43
Il6 (lH, d), 7.50 (lH, t), 7.87 (lH, dt), 7.94 (lH,
t)
2.41-2.55 (lH, m), 2.66-2.83 (lH, m), 3.76-3.90
(lH, m), 3.99-4.12 91H, m), 4.54-4.63 (lH, m),
Il7 ll 7.01-7.12 (lH, m), 7.40 (lH, t), 7.56 (lH~ dd)~
7.66 (lX, s)
2.41-2.52 (lH, m), 2.67-2.Bl ~lH, m), 3.79-3.88
(lH, m), 4.00-4.12 (lH, m), 4.60 (lH, dd), 6.55
I18 oll (lH, t), 6.94-7.00 (lH, m), 7.39 (lH, t), 7.45-
7.50 (lH, m), 7.57 (lH, t)
2.45 (lH, m), 2.75 (lH, m), 3.82 (lH, m), 4.16
Il9 86 5 (lH, m), 4.59 (lH, dd), 7.17 (lH, m), 7.31 (lH,
t), 7.58 (lH, m), 7.70 (lH, m)
2.35-2.46 (lH, m), 2.55-2.70 (lH, m), 3.70-3.81
(lH, m), 3.89-4.01 (lH, m), 4.73 (lH, dd), 7.02
I20 ll 7.11 (lH, m), 7.40 (lH, t), 7.59 (lH, t), 7.65
(lH, s)
2.05-2.20 (lH, m), 2.56-2.70 (~H, m), 3.37 (lH,
s), 3.71-3.88 (2H, m), 4.45-4.55 91H, m), 7.00-
I21 74 7 7.08 (lH, m), 7.39 (lH, t), 7.57 (lH, dd), 7.64
(lH, s)
2.03-2.19 (lH, m), 2.56-2.69 91H, m), 3.70-3.89
78.5- (2H, m), 4.49 (lH, dd), 6.54 (lH, t), 6.91-6.98
I22 80 (lH, m), 7.36 (lH, t), 7.47 (lH, dd), 7,57 (lH,
t)
2.05-2.22 (lH, m), 2.56-2.70 (lH, m), 3.68 (lH,
I23 1124 br s), 3.74-3.90 (2H, m), 4.52 (lH, t), 7.40-
7.51 (lH, m), 7.84-7.94 (2H, m)
1.97-2.11 (lH, m), 2.46-2.61 (lH, m), 3.76-3.94
I24 55-56 (2H, m), 4.35 (lH, t), 7.44 (lH, d), 7.51 (lH,
t), 7.86 (lH, d), 7.91 (lH, s)
1.96-2.10 (lH, m), 2.46-2.60 (lH, m), 3.73-3.91
I25 53 5 (2H, m), 4.34 (lH, t), 7.02-7.10 (lH, m), 7.41
(lH, t), 7.56 ~lH, dd), 7.65 (lH, ~r s)
_ _ _ . _
WO 95/33719 2 1 9 0 9 7 9 PCTIGB95/0122.1 ~
- 196 -
No. Prep mpt nmr
1.95-2.10 (lH, m), 2.45-2.59 (lH, m), 3.72-3.90
(2H, m), 4.35 (lH, t), 6.54 (lH, t), 6.92-7.0D
I26 ~7um (lH, m), 7.37 (lH, t), 7.42-7.48 (lH, m), 7.56
(lH, t)
1,69-1.93 (3H, m), 2.44-2.60 (lH, m), Z.56-Z.77
IZ7 69-70 (3H, m), 7.3Z (lH, d), 7.41 (lH, t), 7.31 (ZH,
s)
1.78 (ZH, s), 1.81-1.97 (lH, m), Z.54-Z.67 (lH,
I28 37-38 m), 3.67-3.81 (3H, m), 6.98-7.07 ~lH, m), 7.39
(lH, t), 7.52-7.60 (lH, m), 7.66 ~lH, Sr
1.78 (2H, s), 1.78-1.97 (lH, m), Z.52-2.66 (lH,
I29 67-69 m), 3.65-3.30 93H, m), 6.54 (lH, t), 6.9Z (lH,
dd), 7.35 (lH, t), 7.47 (lH, dd), 7.58 (lH, t)
1.90 (lH, br s), l.90-Z.08 (lH, m), Z.43-2.58
0 82 83 (lH, m) , 2.53 (3H, s) , 3.53 (lH, dd) , 3.75-3.90
I3 (2H, m), 7.40 (lH, d), 7.49 (lH, t), 7.67 (lH,
s), 7.9Z (lH, d)
1.90-2.08 (2H, m), 2.42-2.57 (lH, m), 2.53 (3H,
sl, 3.54 (lH, dd), 3.70-3.86 (2H, m), 6.98-7.05
I31 (lH, m), 7.3B (lH, t), 7.56 (lH, dd), 7.65 (lH,
br s)
1.90 (lH, br s), 1.90-2.06 (lH, m), 2.41-2.53 -
(lH, m), 2.53 (3H, s), 3.52 (lH, dd), 3.70-3.85
I32 (2H, m), 6.54 (lH, t), 6.91 (lH, dd), 7.35 (lH,
t), 7.43-7.49 (lH, m), 7.56 (lH, t)
1.90-2.07 (lH, m), 2.12 (lH, s), 2.43-2.54 (lH,
I33 _ m), 2.54 (3H, s), 3.54 (lH, dd), 3.74-3.89 (2H,
m), 7.37-7.48 (2H, m),7.83-7.94 (2H, m)
1.19 (3H, t), 1.71 (lH, v br s), 1.90-2.06 (lH,
_34 m), 2.44-2.57 (lH, m), 2.68-2.85 (2H, m), 3.61
(lH, dd), 3.72-3.86 (2H, m), 6.98-7.05 (lH, m),
7.38 (lH, t), 7.52-7.58 (lH, m), 7.63 (lH, s)
1.19 (3H, t), 1.78 (lH, br s), 1.90-2.05 (lH,
m), 2.43-2.56 (lH, m), 2.69-2.83 (2H, m), 3.61
I35 oil (lH, dd), 3.71-3.85 (2H, m), 6.54 (lH, t), 6.92
(lH, dd), 7.35 (lH, t), 7.46 (lH, dt), 7.55
(lH, t)
21 9û979
WO 9S/33719 PCTIGB9~/0122.1
-- 197 - ~
~o. Prep mpt nmr
- 0.96 ~3H, t), 1.56-1.71 ~2H, m), 2.01-2.22 ~
m), 2.43-2.59 ~lH, m), 2.70-3.00 r3H, m), 3.67-
I36 - 3.90 (3H, m), 6.97-7.06 (lH, m), 7.38 ~lH, t),
7.54 ~lH, dd), 7.64 ~lH, s)
1.13 ~6H, 2d), 1.87-2.09 ~2H, m), 2.47-2.60
~lH, m), 3.00 ~lH, hept), 3.64 ~lH, dd), 3.78
I37 oll ~lH, dd), 7.38 ~lH, d), 7.47 ~lH, t), 7.85 ~lH,
d), 7.93 ~lH, br s)
1.92-2.08 ~lH, m), 2.14 ~lH, v br s), 2.45-2.58
~lH, m), 2.81-2.92 ~lH, m), 2.96-3.06 ~lH, m),
I38 _ 3.39 ~3H, s), 3.43-3.60 r2H, m), 3.64 ~lH, dd),
3.75-3.87 (2H, m), 7.4G (lH, d), 7.49 ~lH, t),
7.77 ~lH, s), 7.81 ~lH, d)
1.90-2.08 (2H, m1, 2.15 (3H, s), 2.45-2.59 (lH,
m), 2.54-2.79 (2H, m), 2.85-3.08 (2H, m), 3.64
I39 oll (lH, dd), 3.72-3.87 (2H, m), 6.97-7.06 (lH, m),
7.39 (lH, t), 7.50-7.60 (lH, m), 7.65 (lH, s)
0.36-0.60 (4H, m), 2.00-2.19 (lH, m), 2.23-2.34
(lH, m), 2.41-2.56 (2H, m), 3.74 (lH, dd), 3.74
I40 3.89 (2H, m), 7.41 (lH, d), 7.50 (lH, t), 7.87
(lH, s), 7.94 (lH, d)
1.90-2.08 (2H, m), 2.25 (6H, 5), 2.40-2.56 (3H,
m), 2.70-2.81 (lH, m), 2.85-2.96 (lH, m), 3.61
I41 oll (lH, dd), 3.71-3.87 (2H, m), 6.96-7.06 (lH, m),
7.38 (lH, t), 7.56 (lH, dd), 7.64 (lH, s)
1.90-2.08 (lH, m), 2.26 (ZH, v br s), 2.47-2.61
(lH, m), 2.82-3.05 (2H, m), 3.56-3.84 (SH, m),
I42 3 7.02 (lH, dd), 7.39 (lH, t), 7.55 (lH, dd),
7.65 (lH, 5)
1.86-2.07 (lH, m), 2.26 (lH, t), 2.57-2.60 (lH,
m), 3.47 (lH, dd), 3.71 (lH, dd), 3.75-3.91
I43 oll (3H, m), 6.98-7.07 (lH, m), 7.39 (lH, t), 7.57
(lH, dd), 7.64 (lH, s)
1.82-2.06 (2H, m), 2.44-2.58 (lH, m), 3.31-3.48
(2H, m), 3.65 (lH, dd), 3.75-3.89 (2H, m), S.lS
I44 oil (lH~ dt), 5.26 (lH, dt), 5.85-6.01 (lH, m),
7.41 (lH, d), 7.50 (lH, t), 7.86 (lH, s), 7.92
( lH, d)
WO 95/33719 2 1 9 0 9 7 9 PCT~GB95/01221
-- 198 -
No. Prep mpt nmr
1.86-Z.07 (lH, m), 2.42-2.57 (lH, m), 3.31-3.47
(2H, m~, 3.64 (lH, dd), 3.71-3.86 (2H, m), 5.15
I45 oil (lH, dd), 5.26 (lH, dd), 5.93-6.01 (lH, m),
6.9B-7.07 (lH, m), 7.38 (lH, t), 7.56 (lH, dd),
7.64 (lH, s)
l.B4 (lH, v br s), 1.90-2.06 (lH, m), 2.44-Z.58
(lH, m), 1.81 (lH, dd), 1.95 (lH, dd), 3.42
I46 oil (6H, 2s), 3.64 (lH, dd), 3.73-3.85 (lH, m),
4.51 (lH, t), 6.98-7.06 (lH, m) 7.38 ~lH, t)
7.56 (lH, dd), 7.63-7.66 (lH, m;
2.00-2.16 (lH, m), 2.85-2.98 ~lH, m) 3.B5-4.00
(2H, m), 4.12-4.23 (lH, m), 4.56 (lH s), 6.73
I47 93 (2H, d), 6.81 (lH, t), 7.19-7.28 (2H, m), 7.45
(lH, d), 7.54 (lH, t), 7.90-8.00 (2H, m)
1.91-2.06 (lH, m), 2.14 (lH, v br s), 2.38-2.50
(lH, m), 3.63 (lH, dd), 3.70-3.86 (2X, m), 3.91
I48 (lH, d), 3.98 ~lH, d) 7.20-7.43 ~6H, m), 7.47
~lH, t), 7.82-7.95 ~2H, m)
1.91-2.07 (lH, m~, 2.28 (lH, v br s~, 2.50-2.64
(lH, m~, 3.23-3.57 (2H, m~, 3.66-3.85 (3H, m~,
I49 ll 6.99-7.09 (lH, m), 7.40 (lH, t), 7.55 (lH~ dd)~
7.64 ~lH, s)
2.28-2.58 ~2H, m), 3.60 ~3H, s), 3.76-3.93 ~3H,
I50 81-82 m), 8.44 ~lH, s), 7.41 (lH, d), 7.50 (lH, t),
7 . 86-7 . 98 (2H, m)
1.30 (3H, t), 1.41-1.59 (lH, m), 3.55-3.90 ~4H,
ISl m), 4.22 ~2H, q), 7.41 ~lH, d), 7.49 ~lH, t),
80.5 7 84-7-95 ~2H, m)
1.89-2.07 (lH, m~, 2.41 (lH, br s~, 2.44-2.57
I52 71-72 (lH, m~, 3.60-3.87 (8H, m), 6.99-7.07 (lH, m),
7.38 (lH, t~, 7.55 (lH, dd~, ~.64 (lH, s)
1.45 (9H, m), 1.90-2.08 (lH, m), 2.43-2.56 (lH,
81- m~, 3.47 (lH, d~, 3.55 (lH, d), 3.63 (lH, dd),
I53 82.5 3.70-3.86 (2H, m), 6.98-7.05 ~lH, m), 7.39 ~lH,
t), 7.50-7.58 ~lH, m), 7.65 ~lH, s)
1.18 (9H, s), 1.92-2.08 (lH, m), 3.54 ~lH, t)
I54 3.68-4.00 (4H, m), 6.97-7.05 (lH, m), 7.38 (lH
103 t), 7.50-7.58 (lH, m), 7.64 (lH, s)
WO 95133719 2 1 9 0 9 7 9 PCT~GB9S/0122.1
- i99 -
~lo. Prep mpt nmr
1.90-2.05 (lH, m), 2.2B (lH, v ~r s), 2.53-2.66
I55 66 5 (lH, m), 3.61-4.01 (SH, m), 7.00-7.08 (lH, m~,
7.41 (lH, t), 7.54 (lH, dd), 7.65 (lH, s)
2.22 (lH, m), 2.42 (3H, s), 2.77 (lH, m), 3.91
IS6 _ (2H, m), 4.41 (lH, t), 7.4-7.55 (2H, m), 7.89
(2H, m)
2.20 (lH, m), 2.42 (3H, s), 2.76 ~lH, m), 3.89
IS7 _ (2H, m), 4.43 (lH, t), 7.04 (lH, d), 7.40 (lH,
t), 7.56 (lH, d), 7.62 (lH, s)
2.10-2.28 (lH, m), 2.42 (3H, s), 2.66-2.81 (lH,
m), 3.84-3.93 (2H, 2d), 4.43 (lH, t), 6.14 ~lH,
IS8 gum t), 6.90-6.97 (lH, m), 7.36 (lH, t), 7.44 (lH,
dd), 7.56 (lH, t)
I S 9 lloo3
I60 114424-
I61 95-97
I62 llol9-
I63 113368-
I64 95-97
I65 114520-
1. 35 (9H, s ); 2 . 4 (2H,m); 3 . 25 (2H, t); S . OS (lH, br
I66 81-82 s); 5.25(1X,dd); 7.25(1H,t); 7.35(1H,t);
7.5(1H,s); 9.0(1H,br s).
1.35 (9H, s), 2.5 (2H, m), 3.2 (2H, t), 3.9
I67 67-69 (3H, s), S.0 (lb, br s), 5.35 (lH, dd), 8.15
(lH, s), 10.1 (lH, br s)
1.35 (9H, s), 2.5 (2H, m), 3.2 (2PI, t), S.0
I68 67-69 (lH, br s), 5.35 (lH, dd), 7.8 (lH, s), 10.0
(lH, br s)
3.00 (2H, m), 4.80(lH, m) 5.47 (2H, s): 6.55
I69 oil (lH, t); 6.99 (lH, dd); 7.28(1H, m);7.39 (lH,
t); 7.47 (lH, t); 10.35 (lH, s)
0.96(3H, t); l.S9 (2H, sextet); l.91(1H, br m);
I70 oil 2.70 (lH, m); 2.80 (lH, m) 4.80 (2H, s); 5.15
(lH, s); 7.49 (2H, m); 7.74 (2H, m)
.
21 90979
WO 95133719 ' PCTIGB9S1012~.1
- 200 -
No. Prep mpt ~mr
I72 62 6 1.20 (3H, t); 1;79(1H, s); 2 34 (2H, m)i 4 B0
2.10 (lH, br sl; 3.42 (2H, dt); 4.79 t2H, m);
I73 5.13 ~lH, m) 5.15 (lH, s); 5.2B (lH, m) i S.90
(lH, ~n); 7.49 12H, m); 7.69 (lH, m) i 7.77 (lH,
WO 95/33719 2 1 9 D ~ 7 9 PCT/GB95/0122J
- 201 -
Bio10qical Data
The herbicidal activity of the compounds was tested as follows. Each
chemical was formulated in one of two ways. Either the chemical was
dissolved in an appropriate amount of water, dependent on the amount of
solvent/surfactant blend required such that the total volume is Scm3. Then
a solvent sufficient blend comprised 78.2 g/litre of Tween Z0 and 21.8
g/litre of Span 80 adjusted to 1 litre using methylcyclohexanone was added
to the solution. Alternatively, the chemical was dissolved in water to the
required concentration and 0.1% Tween added. Tween 20 is a Trade Mark for
a surface-active agent comprising a condensate of 20 molar proportions of
ethylene oxide with sorbitan laurate. Span 80 is a Trade Mark for a
surface-active agent comprising sorbitan mono-laurate. If the chemical did
not dissolve, the volume was made up to 5cm3 with water, glass beads were
added and this mixture was then shaken to effect dissolution or suspension
of the chemical, after which the beads were removed. In all cases, the
mixture was then diluted to the required spray volume. If sprayed
inde~,~".l~lllly, volumes of 25cm3 and 30cm3 were required for post: y....e
tests; if sprayed together, 45cm3 was required. The sprayed aqueous
emulsion contained 4% of the initial solvent/surfactant mix and the test
chemical at an appropriate concentration.
The spray compositions so prepared were sprayed on to young pot plants
(post .y.,..e test) at a spray volume equivalent to 1000 litres per
hectare. Damage to plants was assessed 13 days after spraying by
comparison with untreated plants, on a scale of 0 to 9 where 0 is 0%
damage, 1 is 1-5% damage, 2 is 6-15% damage, 3 is 16-25% damage, 4 is
26-35% damage, 5 is 36-5g% damage, 6 is 60-69% damage, 7 is 70-79% damage,
8 is 80-89% damage and 9 is 90-100% damage.
In a test carried out to detect prc: y.nce herbicidal activity,
crop seeds were sown at 2 cm depth and weed seeds at 1 cm depth beneath
compost and sprayed with the compositions at the rate of 1000 litres per
hectare. 20 days after spraying, the seedlings in the sprayed plastic
trays were compared with the seedlings in unsprayed control trays, the
damage being assessed on the same scale of 0 to 9.
The results of the prc ~ tests are given in Table IV below.
21 90979
WO 95133719 _ 'Q2 PCTIGB9S10122-1 --
o 0 o:o.o N ~ N o.o.o o O O N 0 ~ 0 N 0 0 o:o~o:o:o O O
O O N 0 0 0 0: 0: 0 o O o 0n 0 0 0 ~ 0 0: 0 ul . o: o ~ o O: 0
~ o 0 ~ o u~ 0 ~ o 0 .o o o .o .o 0 0 0 0 N 0 0 0 ~O :~ ~O O O 0
n o 0 0 I 0 0 0 0~0,0 o ~r O o N 0 0 0 ~ 0 0 N O N~O,O o:0
m
r~ o o:0 0 o:0:0.0 o:o o 1~ 0 0:0 u~ 0:0 o:o:o o:o o 0
N~ ~ O O ~ 0 0 0~ O.o~o~o C ~ 0 1~ N.-n ~ o:O;o:O:O O N
~ o O o ' ' ' ' '' ' ' ' '
X
O 1~ 0 0.0 0~0:0:0.0 ~ O:O:o:~ 117 0 N N ~ 0:0~0:0:0:0 O ~
o:c N O:O:N~O.O:O:O ~n:o O:o ~0 N'0 N:0 0 O.N:O:O~O o~r
2 ~ 01
I o o
Ir. C10:0:0:~ 010'~q10:0:~:0:0:0:0:0:0'0i0:0~0:010'C'~0:0 o:0
O O ~
0:0.U~O:~!0101N:010:0 O:NIO 0:0:01Q:0 010:0:011--~0:0;0,0
~ ' 0 ! O ~ ' 0
.I: o O C" 0
O O O ~ '
O ~'N:O.O:O,O:OI~'~:O'N:0:0:0:N N~N O~N 0.0:010:0:N.N O:O
O:C.O:O.O -- C~1.0~0:0 N ~O,N:O ~ o,0:0 0:C:O~o:olO:O o:O
C~ C:o:o.o,o _ o;o:o ~r~N~O.O,O ~ u~,~ N~0~-r o:o~o~o:o:o:o
Z, C ~ C~
> ~ ~qlO ~ 01
. o o O ~ O ~ ~N o, O O, O . o, o . 8 o ' o ~ O ~ ~ o O ' O ~ O ~ ~ O ' o ' ' O
O ~ O ' N; N ' N N ' N N ' N ' æ 0 N ' N ' NO ' N Q _ ' _ 0 NO N N ION ' N ' æ; N N N
0: 5 ,5 '~ ~ S _ ~,_ '~ ~_ ~ ~ ,5 ,5, ~ '5 ~ ~ ,5 '_ ' ~ ,5
, _ ~ .
z ~ C . C t_ 0: Q _, _, _, _, r- _ ' _ N N '--N ' N ' N7 ' ~N I uN7 ! N ' ~N ' N
SUBSTITUTE SHEET (RULE 26)
... . = . , . _ _, _ _ _ . ... . ... ... . . . . . .
21 90979
W0 95133719 PCT/CB95/0122
- 03 -
CD o o tq o ~ O 0 t'~ O: O O U~ tO ~ N Iq O tD: O ' ~ N C:l 10 O 1` O
~7 0:0'0'0'0 0 0:0 0 0 0 0'0 0 0'0 ¢~:0:0 0'0'0 0:0:U~!0'0 Irl 0:0
~ 0'0:N 0 0 0 0 0:0'0 O O O 0 0 0 ~D 0 0 0'0 0:0:0 0 0.0'~0 0 0
m 0!0'0 0'0 ~ 0'0:0'0 O.N O'0 0 0 ~.D.0'0 0 Ct.0:0:0 1~0:0 ~q 0:0
~ 0:0't~'0 0 0 0:0'0:0'0.0'0 0 0 0 N 1-- 0 0.0:0:0 0:CO:010'~ 0'0
010:0 ~D 0 ~D 0 0:0 0 o:o o q 0:0 ~o;o N'O:O'~ 0 1~ N 0 O'0'0
- t~
0 , . --.
0 '
U_ 0' : ' ' ' ' " '
o :o )n 0 CD: 0 0 . 0 :0 '0: 0 0 ~D ~rt ~r 0 0 :0 Ir~ O :~ 0 1
, _ , _ . 0 0 0 fO ~D
~'0:0 ~ :~'~ 0 0 ~ O:O:O.~ ~ Cq'N:NlO'N:O:N' ' ' ' : llt~
0~0,~ r-- ~0Ø0
C ~
~ .
~ 010:0:0:0'0:0!0:0:0:0~0!0:0:0'010:0:0:0:t'~:010:010:0!0101010
'~ 010:0:0:0'010:0:0:0:0:0:0.0:0:010.0:0:0111~:010:010101010:010
0:
Q 0
~ ~r I
OO:O~O:O:O.OI~r o~ Olo:o:o:o:o:~Nlo:o:o N~o:o~o ollo~ :O
5: N10:0:~:0'~'U~10:1` 0:0:0:0'0'0:¢1:0:~''):0:11~:0:~DI~:0:1`'010:~:0:0
NlO!O'N'lr~ NI~D:I!I`~IIl:O:O'O ~ N:O~N~ O!O:O'O:O~ q'~r:lnlO:eO:O
Z: 0
>
= ' 0 0 ' 0 ' 0 ' O ' OO o uo~ o ~o~ ~,o~ o o o cot o ' ~ ~o ~ o oo oo oo ~ oo oo oo ~ o
c~ . N æ N N N ' N, N N, N N N ~ N ' N N, N ' N: N N ' ' ' ' ' ' '
o ~'_ 5'_'_ _ .5 .5'5 5 5 5,5 5 5 5 ~ 5' 5~ 5 5
.Q.Q.Q.Q.Q.Q.Q.Q.Q.Q.Q.Q.Q.Q.Q Q.Q.Q.Q.Q.Q.Q.Q.Q.Q.Q:Q:Q:Q Q:Q
SUaSTlTUTE SHEET (RULE 26~
2 1 90979
WO 95/33719 ,~ PCT/GB95/0122-1
- ~0
0 ' O ' tO ' 0~ O O 1:') N ' O .11 ID r~ U) N O O 0: O O O N O ~ N N N
0:0 1` CO 0 0 0 0 0 ~ 0~0'0:0 0:0'0'0 N 0 0 0 0 0 O.0:-~1 0 C:~ 0
C~ 0 10 '0 1~ 0 0 0 0 1`_ 0 ~0 '0 ~0~0 0 0 ~0 0 0 0 0 ' 0 0 1` 0 :0 !Ul:0 0 0
m 0 0 r-- ~ 0 0 ~ o N ~ 0 0 0 '0 0 0 U~ 0 ' 0 0:0 . 0 0 0 o 0 ~ 0 o,0
~ 0'011n'0 0 0'0 0 0 0 0'1~ 0'0 0:0:0'0'0 0 0' 0 0:0 O.0!~0:0 O:l 0
~ 0 ' ~ D: 0 0 u~ O O ' ~D: ~ ' ~ 0: 0 II~ D 0 0: 0 0 CD C'~ O ~ r~ 0
C
<~ 0'~ O:O'I~ 1-_ 0:0:0'N r~ l C.)'0:01N'~) O'~ L~ O U~O:~:O 0 O:0
NIO:N:O:~O ~ ~t'O:O: D:N 0'0'0:0:N~N O tDIN O'O'N:0:~'0:U~0:0
I .
UJ .
Q 010~010 0:0:0:0:~:11010:010:0~0:Cll0:0:N:010:0:0~010:01010.010
~ 010:C~1010'0:0:0:0'010:0'0:0'Q10:0~0:0:010:Q1010 0101010~010
Q
<1
o NlOiO:OlO.O O~O~O:O:~r~O:N:O:N O:O OIO.O'O'O.
~1~10:0:0:0 III:O:~'~D:0:0101~ ~.N 011r~ 1/'1 n ~'~)'~D'11~:0'1r):0
~5~ o o:o:o l` N O O:O _ ~n O:~D:N 1` ~:O:O O:~ O: o.o'c.~'o:lrl~O:~t:O'O~
m
>
O O O O ~O O o o O o O O ~O O O ~O O 0 :0 0 0 0 O O O ~O O O O jO
'1 N; N, N N ' N N N N N N N N I N N N N N N N N N N N N N, N N ' N N N
o 5--5~5~5 ~ '5 5'5 5 5 5~5 5 5 5 5 5 ~-~ 5 ~ 5 5'~
1~ 0 ~ o -- t') ~ n r- ~ 0 ~ o N tq ~ ~ U') ~ ~` ~ 0 ' C~ -- N r~
z
SU~STITUTE SUEET (Rl!LE 26)
2 1 90979
WO 95133'T19 PCT/G1~9~/0122.1
- 05 -
~ N ~r/ ' tD: O ~1 l`7 ~ n N N N O O N O O N tq n O O O ' O 1~ O N~ O O
0 ~ C ' Q ! 0 C 0 0 0 0 Q C ' 0 0 C C ~_ 1~ C ' C C ~ C C C ' 0 C ' 0: C ' 0 0~
3.0 0:0 C C 0 0 C 01C'0 1~ C 0 ~ 0'C:C:0:~:/ 0'C,C 0'0 1~ C O:C
C'C'C O:C~C 0 C 0 C C'0'~ C C ll-~'C 0 01C'0 C~IC 1~ O'C'~ 0:1:0'0
~ C10'010~0'0 0 0:0 C U~ C:o~0:0~'~r C'0 1~ 0 O:O~:0:0 O:C O:C'O'C
I: ~nl0:0!0:0'0:q:1:0 CIC:C~'0~N 0'C O:C:C 1~ C'O 0'C 1-- O~C:O ~ O:C
. . _ ~
X , ~
-- O:C~ lD:O:0!01C:N N'~'l`')'C'O:~ O'O:O:~'O'N,O:O:N O:O'N.O:O~IO:N
Ir~ Y~::~n C:0'1'~'N:N~ ='O:O'O:O C:l:O.O'O.O:O:O:O ~'O:O:olo
- : ~
0101010:01010!C~010!C:01C'C:010!CIC!0!0!0:0!0:010:0!1~.0!CO10
~ I
~, _
0 !0 !0 10 :C !C !C :0:0 :0 10 :0 10 10 !0 !0:0 !C !0 10 !0 10 10 IC 10:0 !0!0 !0 10
5~ : -
C . - .-- ~
-
~n
O . ._
U~ !O:II~:O'0'U'):Q:O'~rl0iO:¢III` O:C~:0i~:~0~10~11'):O:0:0.010:~
~5~ O:O.Ul!O:tD:~'lr~:~'N~ :O O:O:O:O'N!N'I~ O.O:O:O:O:O'O'O!O:O
Z
m~ ~ ~
~ ~ ~ o~ .~ o a o o o.o o jOO 'Oo. 'oo o OO' OO 'OO'O 'O U~ a o' o o 'o
~ N . N N . N . N N; N N N N N ' N ON . N ! N; N N N i N N ~ NO N ' N N N ON ' 2 ' NO ' N; O
o ~ ~ 5 5:5 ~'~:~} :'_''5:5:5:~:~:5 5 ~ 5 5 5 5 _ ~ 5 ~ 5 5 ~
o 01, C 0; C, C :0 0: 0 0 . 0 0 _ . _ _ ., ., . ._ ._ ._ _ ._ _ _, . . . .
,Z
SUBSTITUTE SHEET ~RULE 26)
. _ ... _ ... . _ . _ . _ _ . , . _ , .
WO 95/33719 2 t 9 0 9 7 9 PCTIGB95101221
- ~06 -
C~ CD,0 0 O O N N O O O:O:O:O:O O N
l C~: Q O ~D 0 ~D U'l O O . N O Q; Q 0: Q Q
: c) o ~ 0 1~ o ~ ' 0 ~ Cll Q C) :C~ ,
n. ~ ! C~: Q O ~ C~: 0 ~r O O CD N
.O . O O~ n O: N ~01~r ~ : Q ~
C~.01~ O O O~:N O O:O:O O.Q 0 Q:tD:Q
~1:
X -- _ .
r~ O:O ~ N~N O:O:O~O:O:O,O:O:N
Nltn:O~:O:O 1~ NIO:O~O~O O O
o
I .
0 1 C~ l N, ~ l O ~ Cl l Q
=
o 010 0: 0
CDI~IQ:I~ O.O~ i~'O:O:O:O:O:O:O10.0
0:0:0,0:0:a~ NIO.O:O:O:O-O:O:O:O:O
m
o
o'o o:g o o:o:O:o:o:o o:o 0:0 0:0
~ ' ' ' ' ~ ' N ,1~ N ~ N N N N: N; U~
o:o:o:o:O,o o~o o :O~O o o o:o o
" N:N:N:N N N NIN N ælN N N N N N.N
0 1 C~: 0 -- N ' r~ N: ~
O _ i, . _ ._ ., . _ ._ . ~_ . _ _, ~ . _ _, _ _ _
SUaSTlTUTE SHEET (RULE 26)
~ 2 1 9 ~ 9 7 9 PCT/GB9~/0l2~
WO 9~i/33719
- 207 -
TA3LE V
Abbreviations used for Test Plants in Table IV
BV - Sugar beet
GM - Soybean
ZM - Maize
OS - Rice
PA - Polvgonum aviculare
CA - ChenoDod i um a 1 bum
GA - Galium aDarine
AR - Amaranthus retroflexus
Ml - Mat ri c ari a i nodora
BP - Bidens ~Q~
EH - Euphorbi a heteroDhvl 1 a
IH - IDomoea hederacea
AT - Abutilon theoDhrasti
XT - Xanthium strumarium
AF - Avena fatua
AM - AloPecurus mvosuroides
LR - Lol; um ~i~Ym
SH - SorGhum haleDense
SV - Setaria viridis
PD - Panicum dichotomiflorum
EC - Echinochloa crus-Galli
CE - CvDerus esculentus
.
21 90979
WO 95/33719 - 2 0 8 - PCTIGB95101Z2
STRUCTURES
O O O
A--NJ~ 2~X R2
11 111 IV
y NH2 ~ ~ R
V Vl Vll Vlll IX
o
A--N~ J~ A`N~CH(OH)
XXl Xll
N CH(OR )2 N CH(oR19)2 Ho~cH(oR )2
~OR H
Xlll XIV XV
N ~S ~ R23 R `N~s ~R23
R'90~CH(oR19)2 --~OR2~ ~oR2-
XVI XVII XVIII
21 90979 ~
W09~5r33719 -- 209 - PCT/GB9~101221
~ 'l'~U~ 1 ~ES o
O X 11
A`N~S`R23 ~oR2. t~
XIX XX XXI
O O
AR2N J',o ~, R ~R'
XXII XXIII XXIV XXV
HR2N~Rs R~R~
XXVI XXVII XXVIII
~ HO~ R22O~S~R23
R2 R5 o A,NH~S
XXIX XXX XXXI
R1 RJ~C
XXXII XXXIII XXXIV
o . ~
XXXV XXXVI