Note: Descriptions are shown in the official language in which they were submitted.
,93178
RAN 4070/ 106
The present invention relates to compounds of formula I and
their pharmaceutically acceptable salts and esters thereof, that inhibit
matrix metalloproteases, particularly interstitial collagenases, and are
therefore useful in the treatment of mammals having disease states
alleviated by the inhibition of such matrix metalloproteases.
Matrix metalloproteases ("MMPs") are a family of proteases
(enzymes) involved in the degradation and remodeling of connective
tissues. Members of this family of endopeptidase enzymes are present
in various cell types that reside in or are associated with connective
tissue, such as fibroblasts, monocytes, macrophages, endothelial cells,
and invasive or metastatic tumor cells. MMP expression is stimulated
by growth factors and cytokines in the local tissue environment,
where these enzymes act to specifically degrade protein components
of the extracellular matrix, such as collagen, proteoglycans (protein
core), fibronectin and laminin. These ubiquitous extracellular matrix
components ara .present in the linings of joints, interstitial connective
tissues, basement membranes, and cartilage. Excessive degradation of
extracellular matrix by MMPs is implicated in the pathogenesis of
2 0 many diseases, including rheumatoid arthritis, osteoarthritis, multiple
sclerosis, chronic obstructive pulmonary disease, cerebral
hemorrhaging associated with stroke, periodontal disease, aberrant
angiogenesis, tumor invasion and metastasis, corneal ulceration, and
in complications of diabetes. MMP inhibition is, therefore, recognized
2 5 as a good target for therapeutic intervention.
The MMPs share a number of properties, including zinc and
calcium dependence, secretion as zymogens, and 40-50% amino acid
sequence homology. The MMP family currently consists of at least
3 0 eleven enzymes, and includes collagenases, stromelysins, gelatinases,
matrilysin, metalloelastase, and membrane-type MMP, as discussed in
greater detail below.
Lo/So 4.11.96
~~3~7g
_2_
Interstitial collagenases catalyze the initial and rate-limiting
cleavage of native collagen types I, II, and III. Collagen, the major
structural protein of mammals, is an essential component of the
matrix of many tissues, for example, cartilage, bone, tendon and skin.
Interstitial collagenases are very specific matrix metalloproteases
which cleave these collagens to give two fragments which
spontaneously denature at physiological temperatures and therefore
become susceptible to cleavage by less specific enzymes. Cleavage by
the collagenases results in the loss of structural integrity of the target
tissue, essentially an irreversible process. There are currently three
known human collagenases. The first is human fibroblast-type
collagenase (HFC, MMP-1, or collagenase-1) that is produced by a
wide variety of cells including fibroblasts and macrophages. The
second is human neutrophil-type collagenase {HNC, MMP-8, or
collagenase-2) that has so far only been demonstrated to be produced
by neutrophils. The most recently discovered member of this group of
MMPs is human collagenase-3 (MMP-13) which was originally found
in breast carcinomas, but has since shown to be produced by
2 0 chondrocytes. The only collagenase known to exist in rodents is the
homolog of human collagenase-3.
The gelatinises include two distinct, but highly related, enzymes:
a 72-kD enzyme (gelatinise A, HFG, MMP-2) secreted by fibroblasts
2 5 and a wide variety of other cell types, and a 92-kD enzyme
(gelatinise B, HNG, MMP-9) released by mononuclear phagocytes,
neutrophils, corneal epithelial cells, tumor cells, cytotrophoblasts and
keratinocytes. These gelatinises have been shown to degrade gelatins
(de.natured collagens), collagen types IV (basement membrane) and
3 0 V, fibronectin and insoluble elastin.
Stromelysins 1 and 2 have been shown to cleave a broad range of
matrix substrates, including laminin, fibronectin, proteoglycans, and
collagen types IV and IX in their non-helical domains.
~1~317~
-3-
Matrilysin (MMP-7, PUMP-1) has been shown to degrade a wide
range of matrix substrates including proteoglycans, gelatins,
fibronectin, elastin, and laminin. Its expression has been documented
in mononuclear phagocytes, rat uterine explants and sporadically in
tumors. Other less characterized MMPs include macrophage
metalloelastase (MME, MMP-12), membrane type MMP (MMP-14),
and stromelysin-3 (MMP-11 ).
Inhibitors of MMPs provide useful treatments for diseases
associated with the excessive degradation of extracellular matrix, such
as arthritic diseases (rheumatoid arthritis and osteoarthritis), multiple
sclerosis, bone resorptive diseases (such as osteoporosis), the
enhanced collagen destruction associated with diabetes, chronic
obstructive pulmonary disease, cerebral hemorrhaging associated
with stroke, periodontal disease, corneal or gastric ulceration,
ulceration of the skin, tumor invasion and metastasis, and aberrant
angiogenesis. The involvement of individual collagenases in the
degradation of tissue collagens probably depends markedly on the
tissue. The tissue distribution of human collagenases suggests that
2 0 collagenase-3 is the major participant in the degradation of the
collagen matrix of cartilage, while collagenase-1 is more likely to be
involved in tissue remodeling of skin and other soft tissues. Thus,
inhibitors selective for collagenase-3 over collagenase-1 are preferred
for treatment of diseases associated with cartilage erosion, such as
2 5 arthritis, etc.
Inhibitors of MMP also are known to substantially inhibit the
release of tumor necrosis factor (TNF) from cells, and which therefore
may be used in the treatment of conditions mediated by TNF. Such
3 0 uses include, but are not limited to, the treatment of inflammation,
fever, cardiovascular effects, hemorrhage, coagulation and acute
phase response, cachexia and anorexia, acute infections, shock states,
restinosis, aneurysmal disease, graft versus host reactions and
autoimmune disease.
,~~~~1~g
-4-
In addition to these effects on the release of TNF from cells, MMP
inhibitors have also been shown to inhibit the release of other
biologically active molecules from cells, including soluble receptors
(CI)30 and receptors for TNF (p55 and p75), IL-6, IL-1 and TSH),
adhesion molecules (e.g., L-selection, ICAM-l, fibronectin) and other
growth factors and cytokines, including Fas ligand, TGF-a, EGF, HB-EGF,
SC:F and M-CSF. Inhibition of the release or shedding of such proteins
may be of benefit in a number of disease states, including rheumatoid
arthritis, multiple sclerosis, vascular disease, Type II diabetes, HIV,
cachexia, psoriasis, allergy, hepatitis, inflammatory bowel disease, and
cancer.
Since non-specific inhibition of the shedding enzymes (sheddases)
may have opposite pharmacological effects, selectivity will be a
particular advantage, e.g., the inhibition of TNF release without the
concurrent inhibition of TNF receptor release.
The design and uses of MMP inhibitors is described, for example,
in .1. Enzyme Inhibition, 2, 1-22 (1987); Drug News & Prospectives,
2 0 3(8), 453-458 (1990); Arthritis and Rheumatism, 36(2), 181-189
(1993); Arthritis and Rheumatism, 34(9), 1073-1075 (1991);
Serninars in Arthritis and Rheumatism, 19(4), Supplement 1
(February), 16-20 (1990); Drugs of the Future, 15(5), 495-508
(1990); and J. Enzyme Inhibition, 2, 1-22 (1987). MMP inhibitors are
2 5 also the subject of various patents and patent applications, for
example, U.S. Patent Nos. 5,189,178 and 5,183,900, European
Published Patent Applications 438 223, 606 426, and 276 436, and
published Patent Cooperation Treaty International Applications
WO 92/21360, WO 92/06966, WO 92/09563, and WO 94/25434.
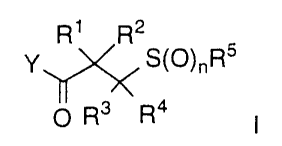
One aspect of the invention concerns compounds represented by
Formula I:
R1 R2
Y\~1~%~ S(O)nR5 I
O R3 R4
1931'8
-s-
wherein:
n is 0, 1 or 2;
Y is hydroxy or XONH-, where X is hydrogen or lower alkyl;
s R 1 is hydrogen or lower alkyl;
R2 is hydrogen, lower alkyl, heteroalkyl, aryl, aralkyl,
arylheteroalkyl, cycloalkyl, cycloalkylalkyl, heteroaryl,
heteroaralkyl, heteroarylheteroalkyl, heterocyclo,
heterocylo-lower alkyl, heterocyclo-lower heteroalkyl
or
-NR6R~, wherein:
R6 is hydrogen, lower alkyl, cycloalkyl or cycloalkyl
alkyl, aryl, heteroaryl and heteroaralkyl;
R~ is hydrogen, lower alkyl, cycloalkyl or cycloalkyl
alkyl, aryl, aralkyl, heteroaryl, heteroaralkyl, -C(O)R8,
1 5 -C(O)NRgR9, -S02NRgR9, -S02R10, aryloxycarbonyl, or
alkoxycarbonyl; or
R 6 and R~ together with the nitrogen atom to which
they are attached represent a heterocyclo group;
wherein
2 0 R g and R9 are independently hydrogen, lower
alkyl, cycloalkyl, cycloalkylalkyl, aryl, aralkyl,
heteroaryl, heteroaralkyl or heteroalkyl; and
R ~ 0 is lower alkyl, cycloalkyl, cycloalkylalkyl,
aryl, aralkyl, heteroaryl, heteroaralkyl,
2 s heteroalkyl or heterocyclo; or
R 1 and R2 together with the carbon atom to which they are
attached
represent a cycloalkyl or heterocyclo group;
R3 is hydrogen, lower alkyl, cycloalkyl, cycloalkylalkyl,
aryl,
aralkyl, heteroaryl, heteroaralkyl, heteroalkyl or
lower
3 0 alkoxy;
R4 is hydrogen, lower alkyl, cycloalkyl or cycloalkylalkyl;
or
R2 and R3 together with the carbons to which they are attached
represent a cycloalkyl or heterocyclo group; or
R 3 and R4 together with the carbon to which they are attached
3 s represent a cycloalkyl or heterocyclo group; and
i
-6-
RS is lower alkyl, cycloalkyl, cycloalkylalkyl, aryl, aralkyl,
heteroaryl; or heteroaralkyl;
or a pharmaceutically acceptable salt or ester thereof.
A second aspect of this invention relates to pharmaceutical
compositions containing a therapeutically effective amount of a
compound of Formula I or a pharmaceutically acceptable salt or ester
thereof admixed with at least one pharmaceutically acceptable
excipient.
A third aspect of this invention relates to methods for treating
mammals having a disease state alleviated by the inhibition of matrix
metalloproteases, by administering an effective amount of a
compound of Formula I, or a pharmaceutical composition thereof, to
the mammal. Such disease states include arthritic diseases, multiple
sclerosis, bone resorption disease (such as osteoporosis), the enhanced
collagen destruction associated with diabetes, chronic obstructive
pulmonary disease, cerebral hemorrhaging associated with stroke,
periodontal disease, corneal or gastric ulceration, ulceration of the
2 0 skin, and tumor metastasis.
A fourth aspect of this invention relates to methods for preparing
compounds of Formula I.
2 5 Among the family of compounds of the present invention as
defined above, a particular family of compounds of formula I consists
of n is 0, 1 or 2; Y is hydroxy or XONH-, where X is hydrogen or lower
alkyl; R1 is hydrogen or lower alkyl; R2 is hydrogen, lower alkyl,
aralkyl, cycloalkyl, cycloalkylalkyl, heterocyclo, or -NR6R~; or R1 and
3 0 R 2 together with the carbon atom to which they are attached
represent a cycloalkyl or heterocyclo group; in which R6 is hydrogen,
lower alkyl, or phenyl; and R~ is hydrogen, lower alkyl, benzyl,
-C(O)Rg, -C(O)NRgR9, -S02NRgR9, -S02R10, benzyloxycarbonyl, or
alkoxycarbonyl; or R6 and R~ together with the nitrogen atom to
3 5 which they are attached represent a heterocyclo group; wherein Rg
~9~~~'g
and R9 are independently hydrogen or lower alkyl; and R10 is lower
alkyl, aryl, heteroaryl, or heterocyclo; R3 is hydrogen, lower alkyl,
cycloalkyl, cycloalkylalkyl, aralkyl, heteroaralkyl, or lower alkoxy; R4
is hydrogen or lower alkyl; or R2 and R3 together with the carbons to
which they are attached represent a cycloalkyl or heterocyclo group;
or R3 and R4 together with the carbon to which they are attached
represent a cycloalkyl or heterocyclo group; and RS is lower alkyl,
aryl, aralkyl, heteroaryl, or heteroaralkyl.
Within these families a preferred category includes compounds
where n is 2 and Y is -NHOH.
Within this category, one preferred group includes the
compounds where R1 is hydrogen and RS is aryl. One preferred
1 5 subgroup within this group includes the compounds where R2 is
hydrogen and R3 is aralkyl, especially benzyl, and R4 is hydrogen and
RS is optionally substituted phenyl or naphthyl, more especially
where RS is 4-methoxyphenyl, phenylthiophenyl, phenoxyphenyl, or
biphenyl.
Another preferred subgroup within this group includes the
compounds where R3 and R'1 together with the carbon to which they
are attached form a cycloalkyl group, especially cyclopentyl and
cyclcohexyl, more especially in combination where RS is 4-methoxy-
2 5 phenyl or 4-phenoxyphenyl.
Yet another preferred subgroup within this group includes the
compounds where R3 and R4 together with the carbon to which they
are attached form a heterocyclo group, in particular optionally
3 0 substituted piperidinyl or tetrahydropyranyl, especially piperidin-4-
yl, 1-methylpiperidin-4-yl, 1-(cyclopropylmethyl)piperidin-4-yl, or
tetrahydropyranyl, more especially in combination where RS is 4-
phenoxyphenyl, 4-(4-chlorophenoxy)phenyl, 4-bromophenoxy)-
phenyl, or 4-(4-fluorophenoxy)phenyl.
~93~~8
_g_
Another preferred group within this category includes the
compounds where R2 is -NR6R~, R1, R3 and R4 are hydrogen, and RS
is aryl. One preferred subgroup within this group includes the
compounds where R$ is 4-phenoxyphenyl, 4-(4-chlorophenoxy)-
phenyl, or 4-(4-fluorophenoxy)phenyl, especially where R6 is
hydrogen and R~ is CBZ-valinamido, valinamido or dimethylamino-
sulfonyl.
Another preferred group within this category includes the
1 0 compounds where R1 and R2 together with the carbon to which they
are attached form a heterocyclo group. A preferred subgroup within
the group includes compounds where R3 and R4 are hydrogen and R1
and R2 together with the carbon to which they are attached form a
heterocyclo group, in particular optionally substituted piperidinyl or
tetrahydropyranyl, especially piperidin-4-yl, 1-methylpiperidin-4-yl,
1-(cyclopropylmethyl)piperidin-4-yl, or most preferably tetrahydro-
pyranyl, more especially in combination where RS is 4-phenoxy-
phenyl, 4-(4-chlorophenoxy)phenyl, 4-(4-bromophenoxy)phenyl,
4-(4-fluorophenoxy)phenyl, 4-(thiophen-2-yl)phenoxy)phenyl,
2 0 4-(thiophen-3-yl)phenoxy)phenyl, 4-(thiazol-2-yl)phenoxy)phenyl,
4-(2-pyridyloxy)phenyl, or 4-(5-chloro-2-pyridyloxy)phenyl.
Another preferred group within this category includes
compounds wherein Rl and R2 are both alkyl, R3 and R4 are hydrogen.
2 5 One preferred subgroup includes compounds wherein RS is 4-
phenoxyphenyl, 4-(4-bromophenoxy )phenyl, 4-(4-
chlorophenoxy)phenyl, or 4-(4-fluorophenoxy)phenyl.
Another group within this category includes compounds wherein
3 0 R2 and R3 together with the carbons to which they are attached form
a c:ycloalkyl group and RS is aryl. Preferably, the cycloalkyl group is
cyclopentyl or cyclohexyl and R~ is 4-phenoxyphenyl, 4-(4-bromo-
phenoxy)phenyl, 4-(4-chlorophenoxy)phenyl, or 4-(4-
fluorophenoxy)phenyl.
~g378
-9-
Preferred compounds are:
N-hydroxy-2-[4-(4-phenoxyphenylsulfonyl)-tetrahydropyran-4-
yl]-acetamide;
2- { 4-[4-(4-chlorophenoxy)-phenylsulfonyl]-tetrahydropyran-4-
yl}-N-hydroxyacetamide;
2-{ 4-[4-(4-fluorophenoxy)-phenylsulfonyl]-tetrahydropyran-4-
yl}-N-hydroxyacetamide;
N-hydroxy-2-[4-(4-phenoxyphenylsulfonyl)-piperidin-4-yl]-
ace.tamide;
2- { 4-[4-(4-chlorophenoxy)-phenylsulfonyl]-piperidin-4-yl } -
N-hydroxyacetamide;
2-{ 4-[4-(4-fluorophenoxy)-phenylsulfonyl]-piperidin-4-yl }-
N-hydroxyacetamide;
N-hydroxy-2-[ 1-methyl-4-(4-phenoxyphenylsulfonyl)-piperidin-
4-yl]-acetamide;
N-hydroxy-2-{ 1-methyl-4-[4-(4-chlorophenoxy)-
phenylsulfonyl]-piperidin-4-yl }-acetamide;
2 0 N-hydroxy-2-{ 1-methyl-4-[4-(4-fluorophenoxy)-phenylsulfonyl]-
piperidin-4-yl }-acetamide;
2-[ 1-cyclopropylmethyl-4-(4-phenoxyphenylsulfonyl)-piperidin-
4-yl]-N-hydroxyacetamide;
2-{ 1-cyclopropylmethyl-4-[4-(4-chlorophenoxy)-phenylsulfonyl]-
2 5 piperidin-4-yl }-N-hydroxyacetamide;
2-{ 1-cyclopropylmethyl-4-[4-(4-fluorophenoxy)-phenylsulfonyl]-
piperidin-4-yl }-N-hydroxyacetamide;
N-hydroxy-2-[4-(4-phenoxyphenylsulfinyl)-tetrahydropyran-4-
yl]-acetamide;
30 2-{4-[4-(4-chlorophenoxy)-phenylsulfinyl]-tetrahydropyran-4-
yl}-N-hydroxyacetamide;
2-{ 4-[4-(4-fluorophenoxy)-phenylsulfinyl]-tetrahydropyran-4-
yl } -N-hydroxyacetamide;
N-hydroxy-2-[4-(4-phenoxyphenylthio)-tetrahydropyran-4-yl]-
3 5 acetamide;
- 10 -
2- { 4-[4-(4-chlorophenoxy)-phenylthio]-tetrahydropyran-4-yl } -
N-hydroxyacetamide;
2-{ 4-[4-(4-fluorophenoxy)-phenylthio]-tetrahydropyran-4-yl }-
N-hydroxyacetamide;
4-[4-(4-chlorophenoxy)phenylsulfonylmethyl]-tetrahydropyran-
4-(.V-hydroxycarboxamide);
4-[4-(4-bromophenoxy)phenylsulfonylmethyl]-tetrahydropyran-
4-(N-hydroxycarboxamide);
4-[4-(4-fluorophenoxy)-phenylsulfonylmethyl]-tetrahydropyran-
4-(.N-hydroxycarboxamide);
3-[4-(4-chlorophenoxy)phenylsulfonyl]-2,2-dimethyl-N-hydroxy-
propionamide;
4-[4-(4-chlorophenoxy)phenylsulfonylmethyl]-1-(cyclopropyl-
methyl)piperidine-4-(N-hydroxycarboxamide);
4-[4-(4-chlorophenoxy)phenylsulfonylmethyl]-1-(nicotinoyl)-
piperidine-4-(N-hydroxycarboxamide);
4-[4-(phenoxy)phenylsulfonylmethyl]-tetrahydropyran-
4-(N-hydroxycarboxamide);
4-[4-(4-(thiaphen-2-yl)-phenoxy)phenylsulfonylmethyl]-
2 0 tetrahydropyran-4-(N-hydroxycarboxamide);
4-[4-(4-(thiophen-3-yl)-phenoxy)phenylsulfonylmethyl]-
tetrahydropyran-4-(N-hydroxycarboxamide);
4-[4-(4-(furan-2-yl)-phenoxy)phenylsulfonylmethyl]-
tetrahydropyran-4-(N-hydroxycarboxamide);
2 5 4-[4-(4-(benzofuran-2-yl)-phenoxy)phenylsulfonylmethyl]-
tetrahydropyran-4-(N-hydroxyc arboxamide);
4-[4-(4-(thiazol-2-yl)-phenoxy)phenylsulfonylmethyl]-
tetrahydropyran-4-(N-hydroxycarboxamide);
4-[4-(4-(thiazol-4-yl )-phenoxy)phenylsulfonylmethyl]-
3 0 tetrahydropyran-4-(N-hydroxycarboxamide);
4-[4-(4-(thiazol-5-yl)-phenoxy)phenylsulfonylmethyl]-
tetrahydropyran-4-(N-hydroxycarboxam.ide);
4-[4-(4-(imidazol-1-yl)-phenoxy)phenylsulfonylmethyl]-
tetrahydropyran-4-(N-hydroxycarboxamide);
3 5 4-[4-(4-(imidazol-2-yl)-phenoxy)phenylsulfonylmethyl]-
19378
- 11 -
tetrahydropyran-4-(N-hydroxycarboxamide);
4-[4-(5-chloro-2-pyridyloxy)phenylsulfonylmethyl]-
tetrah; dropyran-4-(N-hydroxycarboxamide);
3-[4-(5-chloro-2-pyridyloxy)phenylsulfonyl]-2,2-dimethyl-
N-hydroxypropionamide;
(R)-2-(CBZ-valinamido)-N-hydroxy-3-(4-phenoxyphenyl-
sulfonyl)propionamide;
(R)-N-hydroxy-2-valinamido-3-(4-phenoxyphenylsulfonyl)-
propionamide;
(R)-2-dimethylamino-N-hydroxy-3-(4-phenoxyphenylsulfonyl)-
propionamide;
(R)-2-dimethylaminosulfonGmido-N-hydroxy-3-(4-phenoxy-
phenylsulfonyl)-propionamide
and pharmaceutically acceptable salts thereof.
Definitions
The following definitions are set forth to illustrate and define the
meaning and scope of the various terms used to describe the
invention herein.
"Alkyl" means a branched or unbranched saturated hydrocarbon
chain containing 1 to 8 carbon atoms, such as methyl, ethyl, propyl,
tert-butyl, n-hexyl, n-octyl and the like.
2 5 "Lower alkyl" means a branched or unbranched saturated
hydrocarbon chain containing 1 to 6 carbon atoms, such as methyl,
ethyl, propyl, isopropyl, tert-butyl, n-butyl, n-hexyl and the like,
unless otherwise indicated.
3 0 The term "heteroalkyl" refers to a branched or unbranched, cyclic
or acyclic saturated organic radical containing carbon, hydrogen and
one or more heteroatom containing substituents independently
selected from ORa, NRaRb, and S(O)nRa (where n is 0, 1 or 2) and Ra is
hydrogen, alkyl, cycloalkyl, aryl, aralkyl, heteroaryl, heteroaralkyl or
3 5 acyl, Rb is hydrogen, alkyl, cycloalkyl, aryl, aralkyl, acyl,
X93178
- 12 -
alkylsulfonyl, carboxamido, or mono- or di-alkylcarbamoyl.
Representative examples include hydroxyalkyl, aminoalkyl,
alkoxyalkyl, aryloxymethyl, N-acylaminoalkyl, thienylthiomethyl and
the like.
"Acyl" refers to the group -C(O)-R', where R' is lower alkyl.
"Alkylene" refers to a straight chain or branched chain divalent
radical consisting solely of carbon and hydrogen, containing no
unsaturation and having from one to six carbon atoms, e.g.,
methylene, ethylene, propylene, 2-methylpropylene, butylene, 2-
ethylbutylene, hexylene, and the like.
"Lower alkoxy" means the group -O-R', where R' is lower alkyl.
"Alkoxycarbonyl" means the group RO-C(O)- where R is alkyl as
herein defined.
"Alkoxycarbonylalkyl" means the group ROC(O)(CH2)n- where R is
2 0 alkyl as herein defined and n is 1, 2 or 3.
"Aryl" refers to a monovalent aromatic carbocyclic radical having
a single ring (e.g., phenyl) or two condensed rings (e.g., naphthyl),
which can optionally be mono-, di- or tri-substituted, independently,
2 5 with hydroxy, carboxy, lower alkyl, cycloalkyl, cycloalkyloxy, lower
alkoxy, chloro, fluoro, trifluoromethyl and/or cyano. The rings) can
alternatively be optionally monosubstituted with the group Ra-Z-,
where Z is oxygen, sulfur, -CH=CH-, -CH2, carbonyl, a covalent bond, or
nitrogen optionally substituted with lower alkyl, and Ra is a
3 0 monovalent aromatic carbocyclic, heteroaryl or heterocyclo radical, or
a combination thereof, having 1 or 2 rings, for example phenyl,
pyridyl, thienyl, imidazolyl, furanyl, pyrimidinyl, benzothiophene,
azanaphthalene, indolyl, phenyl-(furan-2-yl), phenyl-(thien-2-yl),
phenyl-(thien-3-yl), phenyl-(imidazol-2-yl), phenyl-(thiazol-2-yl),
3 5 phenyl-(morpholin-2-yl), and phenyl-(oxazol-2-yl), (the rings)
;93~7~
- 13 -
represented by Ra being optionally mono-or disubstituted by
hydroxy, carboxy, lower alkyl, lower alkoxy, halo, trifluoromethyl
and/or cyano). Examples of aryl substituted by Ra-Z- are benzoyl,
diphenylmethane, biphenyl, 6-methoxybiphenyl, 4-(4-methyl-
phenoxy)phenyl, 4-phenoxyphenyl, 2-thiophenoxyphenyl,
4-pyridethenylphenyl, 4-(thiophen-2-yl)phenoxyphenyl, 4-
(thiophen-3-yl)phenoxyphenyl, 4-(2-pyridyloxy)phenyl, 4-(5-chloro-
2-pyridyloxy)phenyl, 4-(thiazol-5-yl)phenoxyphenyl, 4-(imidazol-
2-yl)phenoxyphenyl, and the like.
"Heteroaryl" refers to a monovalent aromatic carbocyclic radical
having one or two rings incorporating one, two or three heteroatoms
(chosen from N, O or S) within the ring(s), such as thiazole, oxazole,
imidazole, thiophene, quinolyl, benzofuranyl, pyridyl, and indolyl,
which can optionally be mono-, di- or tri-substituted, independently,
with OH, COOH, lower alkyl, lower alkoxy, halo, trifluoromethyl and/or
cyano.
"Aralkyl" refers to a radical of the formula Rb-Rc-, wherein Rb is
2 0 aryl as defined above and Rc is alkylene as defined above, for
example benzyl, phenylethylene, 3-phenylpropyl, biphenylpropyl.
"Benzyloxycarbonyl" refers to a radical of the formula
RdCH20C(O)-, where Rd is phenyl. "Benzyloxycarbonylamino" refers to
2 5 a radical of the formula RdCH20C(O)NH-, where Rd is phenyl.
"Cycloalkyl" means a saturated monovalent monocyclic
hydrocarbon radical containing 3-8 carbon atoms, such as cyclopropyl,
cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl and cyclooctyl.
"Cycloalkylalkyl" means cycloalkyl as defined above attached to
an alkylene radical as defined above.
"Halo" refers to bromo, chloro or fluoro.
'~g3178
- 14 -
"Heteroaralkyl" refers to a radical of the formula Relic-, where Re
is heteroaryl as defined above and Rc is alkylene as defined above.
"Heterocyclo" refers to a monovalent saturated carbocyclic radical,
consisting of either a 5 to 7 membered monocyclic ring or a 9 to 14
membered bicyclic ring, substituted by one, two or three heteroatoms
chosen from N, O, or S, optionally fused to a substituted or unsubsti-
tuted benzene ring. Examples of heterocyclo radicals are morpholino,
piperazinyl, piperidinyl, pyrrolidinyl, tetrahydrothiopyranyl,
tetrahydrothiopyranyl-l,l-dioxide, tetrahydropyranyl, and the like,
which can be optionally substituted by one or more substituents
independently selected from lower alkyl, lower alkoxy, alkylamino,
alkylaminoalkyl, acyl valyl, alkylsulfonyl, dialkylamino, heteroaroyl,
alkoxycarbonylalkyl, and an amino protecting group where
appropriate (e.g. CBZ, for example, 1-CBZ-piperidin-4-yl). However,
the definition "R6 and R7 together with the nitrogen to which they are
attached represent a heterocyclo group" clearly can refer only to a
heterocyclo group containing at least one nitrogen atom.
2 0 "Hydroxylamino" refers to the group -NHOH.
"BOC" refers to tent-butoxycarbonyl.
"CBZ" refers to benzyloxycarbonyl.
"DCC" refers to 1,3-dicyclohexylcarbodiimide.
"Valine amide" refers to the radical (CH3)2CHCH(NH2)C(O)NH-.
3 0 "Optional" or "optionally" means that the subsequently described
evE;nt of circumstances may or may not occur, and that the
description includes instances where said event or circumstance
occurs and instances in which it does not. For example, "optionally
substituted phenyl or aryl" means that the phenyl or aryl moiety may
3 5 or may not be substituted and that the description includes both
~93~78
- 15 -
substituted and unsubstituted phenyl. The phrase "optional
pharmaceutical excipients" indicates that a composition or dosage
form so described may or may not include pharmaceutical excipients
other than those specifically stated to be present, and that the
formulation or dosage form so described includes instances in which
optional excipients are present and instances in which they are not.
"Amino-protecting group" as used herein refers to those organic
groups intended to protect nitrogen atoms against undesirable
reactions during synthetic procedures, and includes, but is not limited
to, benzyl, acyl, benzyloxycarbonyl (carbobenzyloxy), p-methoxy-
benzyloxy-carbonyl, p-nitrobenzyloxycarbonyl, tert-butoxycarbonyl,
trifluoroacetyl, and the like.
"Base" as used here includes both strong inorganic bases such as
sodium hydroxide, lithium hydroxide, ammonium hydroxide,
potassium carbonate and the like, and organic bases such as pyridine,
diisopropylethylamine, 4-methylmorpholine, triethylamine,
dirnethylaminopyridine and the like.
"Pharmaceutically acceptable salt" refers to those salts which
retain the biological effectiveness and properties of the free bases or
fret; acids and which are not biologically or otherwise undesirable. If
the compound exists as a free base, the desired acid salt may be
2 5 prepared by methods known to those of ordinary skill in the art, such
as treatment of the compound with an inorganic acids such as
hydrochloric acid, hydrobromic acid, sulfuric acid, nitric acid,
phosphoric acid and the like; or with an organic acids such as acetic
acid, propionic acid, glycolic acid, pyruvic acid, oxalic acid, malefic acid,
3 0 malonic acid, succinic acid, fumaric acid, tartaric acid, citric acid,
benzoic acid, cinnamic acid, mandelic acid, methanesulfonic acid,
ethanesulfonic acid, p-toluenesulfonic acid, salicylic acid, and the like.
If the compound exists as a free acid, the desired base salt may also
be prepared by methods known to those of ordinary skill in the art,
3 5 such as the treatment of the compound with an inorganic base or an
- 16 -
organic base. Salts derived from inorganic bases include, but are not
limited to, the sodium, potassium, lithium, ammonium, calcium,
magnesium, iron, zinc, copper, manganese, aluminum salts and the
like. Salts derived from organic bases include, but are not limited to,
salts of primary, secondary, and tertiary amines, substituted amines
including naturally occurring substituted amines, cyclic amines and
basic ion exchange resins, such as isopropylamine, trimethylamine,
diethylamine, triethylamine, tripropylamine, ethanolamine,
2-dimethylaminoethanol, 2-diethylaminoethanol, trimethamine,
dicyclohexylamine, lysine, arginine, histidine, caffeine, procaine,
hydrabamine, choline, betaine, ethylenediamine, glucosamine,
methylglucamine, theobromine, purines, piperazine, piperidine,
N-e;thylpiperidine, polyamine resins and the like.
"Pharmaceutically acceptable ester" as used herein refers for
example to those non-toxic esters of a compound of Formula I where
R 1 is hydroxy, and are formed by reaction of such compounds, by
means well known in the art, with an appropriate alkanol of 1-8
carbon atoms, for example methanol, ethanol, n-propanol, isopropanol,
2 0 n-butanol, tert-butanol, i-butanol (or 2-methylpropanol), n-pentanol,
n-hexanol, and the like.
The terms "inert organic solvent" or °'inert solvent" mean a
solvent inert under the conditions of the reaction being described in
2 5 conjunction therewith, including, for example, benzene, toluene,
acetonitrile, tetrahydrofuran ("THF"), N,N-dimethylformamide
("rrMF" ), chloroform ("CHC13 "), methylene chloride (or
dichloromethane or "CHZC12"), diethyl ether, ethyl acetate, acetone,
methylethyl ketone, methanol, ethanol, propanol, isopropanol, tert-
3 0 butanol, dioxane, pyridine, and the like. Unless specified to the
contrary, the solvents used in the reactions of the present invention
are inert solvents.
The compounds of this invention may possess one or more
3 5 asymmetric centers; such compounds can therefore be produced as
19378
- 17 -
mixtures of stereoisomers or as individual (R)- or (S)- stereoisomers.
The: individual enantiomers may be obtained by resolving a racemic
or non-racemic mixture of an intermediate at some appropriate stage
of the synthesis. It is understood that the individual (R)- or (S)-
stereoisomers as well as racemic mixtures and other mixtures of
stereoisomers are encompassed within the scope of the present
invention.
The use of the symbol "(R)" or "(S)" preceding a substituent
designates the absolute stereochemistry of that substituent according
to the Cahn-Ingold-Prelog rules [see Cahn et al., Angew. Chem. Inter.
Edzt., 5, 385 (1966), errata p. 511; Cahn et al., Angew. Chem., 78, 413
( 1966); Cahn and Ingold, J. Chem. Soc., (London), 612 ( 1951 ); Cahn et
al., Experientia, 12, 81 ( 1956); Cahn J., Chem. Educ., 41, 116 (1964)].
Because of the interrelation of the designated substituent with the
other substituents in a compound having a or 13 prefixes, the
designation of the absolute configuration of one substituent fixes the
absolute configuration of all substituents in the compound and thus
the absolute configuration of the compound as a whole.
"Stereoisomers" are isomers that differ only in the way the atoms
are arranged in space.
"Enantiomers" are a pair of stereoisomers that are
2 ~ non-superimposable mirror images of each other. Enantiomers rotate
the plane of polarized light in opposite directions. The enantiomer
that rotates the plane to the left is called the levo isomer, and is
designated (-). The enantiomer that rotates the plane to the right is
called the dextro isomer, and is designated (+).
"Diastereoisomers" are stereoisomers which are not
mirror-images of each other.
"Racemic mixture" means a mixture containing equal parts of
3 5 individual enantiomers. "Non-racemic mixture" is a mixture
193'18
- 18 -
containing unequal parts of individual enantiomers.
"Mammal" includes humans and all domestic and wild animals,
including, without limitation, cattle, horses, swine, sheep, goats, dogs,
cats, and the like.
"Treating" or "treatment" as used herein cover the treatment of a
disease-state in a mammal, particularly in a human, and include:
(i) preventing the disease-state from occurring in a mammal, in
particular, when such mammal is predisposed to the disease-state but
has not yet been diagnosed as having it;
(ii) inhibiting the disease-state, i.e., arresting its development; or
(iii) relieving the disease-state, i.e., causing regression of the
disease-state.
The term "therapeutically effective amount" refers to that amount
of a compound of Formula I that is sufficient to effect treatment, as
defined above, when administered to a mammal in need of such
2 0 treatment. The therapeutically effective amount will vary depending
on the subject and disease state being treated, the severity of the
affliction and the manner of administration, and may be determined
routinely by one of ordinary skill in the art.
2 5 Nomenclature
The compounds of Formula I, illustrated below, will be named
using the indicated numbering system:
R' R2
Y t 2 s S(O)nR5
~ R3 Ra
A compound of Formula I wherein is Y is N-hydroxylamino; R 1
and R2 are hydrogen; R3 is benzyl; R4 is hydrogen; RS is 4-methoxy-
phf;nyl; and n is 2, is named 3-benzyl-3-(4-methoxyphenylsulfonyl)-
~93~~8
- 19 -
N-hydroxypropionamide.
A compound of Formula I wherein Y is N-hydroxylamino; R1 and
R 2 are hydrogen; R3 and R4 together with the carbon to which they
are attached represent tetrahydropyran-4-yl; R5 is 4-(4-fluoro-
phenoxy)phenyl; and n is 2, is named as an acetic acid derivative,
i.e.., 2-{4-[4-(4-fluorophenoxy)-phenylsulfonyl]-tetrahydropyran-4-
yl } ~-N-hydroxy-acetamide.
A compound of Formula I wherein Y is hydroxy; R1 is hydrogen;
R2 is methyl; R3 and R4 together with the carbon to which they are
attached represent 1-methylpiperidin-4-yl; R5 is biphenyl; and n is 1,
is named 2-[4-(biphenyl-4-sulfinyl)-1-methylpiperidin-4-yl]-
propionic acid.
A compound of Formula I wherein Y is N-hydroxylamino; R1 and
R2 together with the carbon to which they are attached represent
tetrahydropyran-4-yl, R3 and R4 are hydrogen, RS is 4-(4-chloro-
phenoxy)-phenyl; and n is 2, is named 4-[4-(4-chlorophenoxy)-
2 0 phenylsulfonylmethyl]-tetrahydropyran-4-(N-hydroxycarboxamide).
Synthetic Reaction Parameters
Unless specified to the contrary, the reactions described herein
take place at atmospheric pressure within a temperature range from
2 5 5°C: to 100°C (preferably from 10°C to 50°C;
most preferably at "room"
or "ambient" temperature, e.g., 20°C). Further, unless otherwise
specified, the reaction times and conditions are intended to be
approximate, e.g., taking place at about atmospheric pressure within a
terr~perature range of about 5°C to about 100°C (preferably from
3 0 about 10°C to about 50°C; most preferably about 20°C)
over a period
of about 1 to about 10 hours (preferably about 5 hours). Parameters
given in the Examples are intended to be specific, not approximate.
Amide couplings used to form the compounds of Formula I are
3 5 generally performed by the carbodiimide method with reagents such
19~~~g
- 20 -
as 1,3-dicyclohexylcarbodiimide or N'-ethyl-N'-(3-dimethylamino-
propyl)-carbodiimide hydrochloride or alternatively 1-(3-dimethyl-
aminopropyl)-3-ethylcarbodiimide hydrochloride (EDCI), in the
presence of 1-hydroxybenzotriazole hydrate (HOBT) in an inert
solvent such as N, N-dimethylformamide (DMF) or methylene chloride
(CH2C12). Other methods of forming the amide or peptide bond
include, but are not limited to, synthetic routes via an acid chloride,
acyl azide, mixed anhydride or activated ester such as a p-
nitrophenyl ester. Typically, solution phase amide couplings with or
without peptide fragments are performed.
The selection of amino protecting groups used in the preparation
of compounds of Formula I is dictated in part by the particular amide
coupling conditions, and in part by the components involved in the
coupling. Amino-protecting groups commonly used include those
which are well-known in the art, for example, benzyloxycarbonyl
(carbobenzyloxy) (CBZ), p-methoxybenzyloxycarbonyl, p-nitro-
benzyloxycarbonyl, N-tort-butoxycarbonyl (BOC), and the like. It is
preferred to use either BOC or CBZ as the protecting group for the a-
2 0 amino group because of the relative ease of removal by mild acids in
the case of BOC, e.g., by trifluoroacetic acid (TFA) or hydrochloric acid
in ethyl acetate; or removal by catalytic hydrogenation in the case of
CB'I
2 5 PREPARATION OF COMPOUNDS OF FORMULA I
One method for preparing a compound of the Formula I, in
particular wherein n is 1 or 2; Y is hydroxy or XONH-, where X is
hydrogen or lower alkyl; R1 is hydrogen or lower alkyl; R2 is
3 0 hydrogen, lower alkyl, aralkyl, cycloalkyl, cycloalkylalkyl, or
heterocyclo; or R1 and R2 together with the carbon atom to which
they are attached represent a cycloalkyl or heterocyclo group; R3 is
hydrogen, lower alkyl, cycloalkyl, cycloalkylalkyl, aralkyl,
het:eroaralkyl, or lower alkoxy; R4 is hydrogen or lower alkyl; or R2
3 5 and R3 together with the carbons to which they are attached
193~'1~
- ~1 -
10
represent a cycloalkyl or heterocyclo group; or R3 and R4 together
with the carbon to which they are attached represent a cycloalkyl or
heterocyclo group; and RS is lower alkyl, aryl, aralkyl, heteroaryl, or
heteroaralkyl; comprises contacting a compound of the Formula:
R1 R2
Y\~~~~ SR5
O R3 R4
with an oxidizing agent. Suitable oxidation conditions are outlined in
the description of reaction scheme VIII below.
One method of preparing compounds of Formula I where n is 0,
R1 is hydrogen and R2 is not -NR6R~ is from the corresponding
unsaturated acid of Formula (4), the preparation of which is shown
below in Reaction Scheme I:
REACTION SCHEME I
O~R3 R2 step 1 R\ Rs
+ ~PPh I~3
R4 t BuO2C t BuO2C R4
(1) (2) (3)
step 2 R2 Rs
(3)
H02C Ra
(4)
2 0 Starting- Materials
Aldehydes and ketones of Formula ( 1 ) are commercially
available, for example from Aldrich Chemical Co., or may be prepared
as shown below, or prepared according to methods well known to
those skilled in the art. The ylides of Formula (2) are commercially
'~93~78
- 22 -
available, for example, (tert-butoxycarbonylmethylene)triphenyl-
phosphorane is available from Aldrich, or may be prepared by
standard methods known to those skilled in the art, for example by
reacting the appropriate bromo derivative of formula R2CHBrC02-
(tert-butyl) with triphenylphosphine, and reacting the resulting
triphenylphosphonium bromide derivative with a strong base.
Step 1 - Preparation of Compounds of Formula (3)
In general, a solution of an aldehyde or ketone compound of
1 0 Formula ( 1 ) is reacted in an inert organic solvent, for example
benzene, with a compound of Formula (2) (or alternatively, the
corresponding phosphonate, for example trimethyl phosphonoacetate)
for a period of 8 to 48 hours at 15°C to 30°C (aldehydes),
preferably
20"C, or 70°C to 90°C (ketones), preferably 80°C, until
starting
1 ~ material is consumed. The reaction product, an enoic ester of Formula
(3), is isolated and purified by conventional means.
S tep 2 - Preparation of Compounds of Formula l4)
The compound of Formula (3) is then hydrolyzed under acidic
2 0 conditions, optionally in the presence of an inert solvent, e.g.,
treatment with trifluoroacetic acid in methylene chloride for about 20
minutes to 3 hours. The reaction is carried out at a temperature
range from about 0°C to 40°C, preferably at about room
temperature.
In the case where trimethyl phosphonoacetate is used in Step 1, a
2 5 methyl ester is produced which may be hydrolyzed conventionally
under basic conditions, for example sodium hydroxide in aqueous
methanol or ethanol. The reaction product, an enoic acid of Formula
(4), is isolated and purified by conventional means.
3 0 Preparation of Compounds of Formula (4) where R3 and R4 together
with the Carbon to which they are attached represent a Piperidine
Derivative
The preparation of compounds of Formula (4) where R3 and R4
3 5 together with the carbon to which they are attached represent a
~~9317g
- 23 -
piperidine derivative, represented below as a compound of Formula
(4a~, in general requires the protection of the NH group. An example
is shown below in Reaction Scheme II.
REACTION SCHEME II
H CBZ CBZ
step 1 N step 2
OH OH O
(a) (b) (1 a)
CBZ CBZ
step 1 N step 2
(1 a) + (2)
R2 CO2t-BU R2 CO2H
(3a) (4a)
Step 1 - Preparation of Compounds of Formula (b)
In general, a solution of a hydroxypiperidine compound of
Formula (a) is protected by reaction of (a) in an inert organic solvent,
for example tetrahydrofuran, in the presence of an excess of a
tertiary base, for example triethylamine, with an equimolar amount
of benzyl chloroformate. The reaction is carried out in the
temperature range from about 0°C to 40°C, preferably at about
25°C,
for about 10 to 30 hours, preferably about 18 hours. The reaction
product of Formula (b) is isolated and purified by conventional
means.
~193:~7~
- 24 -
Step 2 - Preparation of Compounds of Formula ( 1 a)
A compound of Formula (la) is a compound of Formula (1) where
R3 and R4 together with the carbon to which they are attached
represent a protected piperidine derivative.
In general, a solution of a compound of Formula (b) is oxidized to
a l~etone of Formula (la) by reaction of (b) in an inert organic solvent,
for' example methylene chloride, with an oxidizing agent, for example
pyridinium chlorochromate, preferably in the presence of an inert
1 0 support, for example Celite.* The reaction is carried out in the
temperature range from about 0°C to 40°C, preferably at about
25°C,
for about 10 to 30 hours, preferably about 18 hours. The reaction
product of Formula ( 1 a) is isolated and purified by conventional
mE;ans.
20
Alternatively, reaction of commercially available 4-piperidone
monohydrate hydrochloride with benzyl chloroformate under
Schotten-Baumann conditions gives a compound of Formula (la) in a
single step.
Prf~paration of Compounds of Formula (4) where R3 and R4 Together
with the Carbon to which they areattached Represent a Piperidine
_De;rivative
A compound of Formula (4) where R3 and R4 together with the
2 5 carbon to which they are attached represent a piperidine derivative is
represented as a compound of Formula (4a).
The protected piperidine ketone of Formula ( 1 a) is converted to
(3;~), which is hydrolyzed to (4a) as described in Reaction Scheme I,
3 0 Steps 1 and 2. The compound of Formula (4a) is then converted to a
compound of Formula I where n is 0 as described in Reaction Scheme
III below. The benzyloxycarbonyl (CBZ) protecting group is removed
by catalytic hydrogenation, to give a compound of Formula I where
R-3 and R4 together with the carbon to which they are attached
3 S represent piperidine.
'~ rCrademark
%4
~93~78
- 25 -
Preparation of Compounds of Formula (4) where R3 and R4 To eg ther
with the Carbon to which thev are attached Represent a P.
Derivative
Compounds of Formula (4) where R3 and R4 together with the
carbon to which they are attached represent a tetrahydropyran
derivative, represented as Formula (4b), are prepared similarly to the
procedure shown above, starting from the corresponding 4-oxotetra-
hydropyran. The reaction is shown below in Reaction Scheme III and
described in Example 3.
REACTION SCHEME III
R2
5
SR
of of of
(1 b) (4b) la
The tetrahydropyran derivative of Formula (4b) is then
converted to the corresponding compound of Formula I, i. e., a
compound of Formula I where n is 0, as described in Reaction Scheme
VII.
Preparation of Compounds of Formula (4) where R3 and R4 To eg_ ther
with the Carbon to which they are Attached represent a Tetrah,
thiopyran-1,1-dioxide Derivative
2 5 Compounds of Formula (4) where R3 and R4 together with the
carbon to which they are attached represent a tetrahydrothiopyran-
1,1-dioxide derivative are prepared similarly to the procedure shown
above, starting from the corresponding 4-oxotetrahydrothiopyran.
E~~93~~$
- 26 -
The tetrahydrothiopyran-1,1-dioxide derivative of Formula (4) is
then converted to the corresponding compound of Formula I where n
is 0 as described in Reaction Scheme III.
Alternative Preparation of Compounds of Formula I
Another method of preparing compounds of Formula I where R2
is not -NR6R~ and R3 and R'l are both hydrogen is from the
corresponding lactone of Formula ( 10), the preparation of which is
shown below in Reaction Scheme IV.
REACTION SCHEME IV
Ri R2 R1 R2 Ri R2
Et0 OEt step 1 Et0 OH step 2 HO OH
O O O O
(7) ($) (9)
step 3
(9) Ri R2
O==
O
( 10)
Step 1 - Preparation of Compounds of Formula ($)
The starting compounds of Formula (7) are commercially
available, or may be prepared by means well known in the art
starting from diethyl malonate, e.g., Gibson and Johnson, J. Chem. Soc.,
2 0 p2525 (1930), (other diesters may be employed in place of the
diethyl ester if desired). In general, a solution of a compound of
Formula (7) is dissolved in an inert aromatic solvent, preferably
benzene or toluene, and cooled to about -40° to -20°C,
preferably
about -30°C. To this cold solution is added a suitable hindered
2 5 reducing agent, preferably diisobutylaluminum hydride in an inert
~9~1~8
- 27 -
aromatic solvent, maintaining the temperature at no higher than
abc>ut 25°C. After the addition is complete, the reaction is maintained
at about 15°C until all the starting material is consumed. After about
minutes the reaction is quenched by addition of a protic solvent,
5 preferably ethanol, maintaining the temperature at no higher than
about -15°C. Sodium borohydride is optionally added, but preferably
the reaction is simply allowed to warm to about room temperature.
The reaction product of Formula (8) is isolated and purified by
conventional means.
Step 2 - Preparation of Compounds of Formula (91
In general, the compound of Formula (8) is hydrolysed with a
base to form the hydroxymethyl acid of Formula (9).
The compound of Formula (8) is dissolved in an aqueous protic
solvent, preferably aqueous methanol, and reacted with about 3
molar equivalents of a base, for example potassium hydroxide or
lithium iodide, followed by sodium cyanide. The reaction is carried
out in the temperature range from about 80°C to 120°C,
preferably at
2 0 about the reflux temperature of the solvent mixture, for about 8
hours. The reaction product of Formula (9) is isolated and purified by
conventional means.
Step 3 - Preparation of Compounds of Formula X10)
2 5 In general, the compound of Formula (9) is dehydrated to form a
lactone of Formula ( 10).
To a mixture of the compound of Formula (9) and about 2 molar
equivalents of a tertiary base, preferably triethylamine, optionally in
3 0 the presence of 4-dimethylaminopyridine, in an inert solvent, for
example, diethyl ether or dichloromethane, at about -20°C, is added
about 1 molar equivalent of a dehydrating agent, for example
trifluoromethanesulfonic anhydride, methanesulfonic anhydride,
met:hanesulfonyl chloride, p-toluenesulfonyl chloride, benzenesulfonyl
3 5 chloride, preferably benzenesulfonyl chloride. The reaction is carried
293178
- 28 -
out at about -10°C, for about 10 minutes to 4 hours, preferably about
30 minutes. The reaction product of Formula { 10) is isolated by
conventional means synthesis without further purification.
Preparation of Compounds of Formula ~10~ where R1 and R2 to eg ther
with the Carbon to which they are attached Represent a Tetrah,
~yran Derivative
To give a specific example, the preparation of a compound of
1 0 Formula ( 10) where R 1 and R2 together with the carbon to which
they are attached represent a tetrahydropyran derivative
(represented as Formula ( l0a)) is shown below in Reaction Scheme V,
and. described in Example 5.
1 5 REACTION SCHEME V
O O O
Et0 OEt step 1 Et0 OH step 2 HO OH
O O O O
(7a) (8a) (9a)
step 3 O
(9a)
O
O
(1 Oa)
2 0 The starting compound of Formula (7a) is either commercially
available or may be prepared as shown in Example 31A. Steps 1-3 are
carried out in the same manner as shown in Reaction Scheme IV.
193~7~
- 29 -
Preparation of Compounds of Formula ( 10) where R3 and R4 are as
Defined in the compounds of formula I
The preparation of a compound of Formula (10) where R3 and R4
are as defined in the compounds of formula I, represented as Formula
( l Ob), is shown below in Reaction Scheme VI, and described in
Example 5.
REACTION SCHEME VI
R~ R2 R1
EtO~~~~OEt step 1 RO
'R
O O O
(11)
O R1 R2
(11) + R3~R4 step 2 RO OH
O Rs .Ra
(9b)
(9b) step 3 R~ RRs
O
4
R
1 0 (10b)
Step 1 - Preparation of Compounds of Formula ( 11 ~
The compound of Formula ( 11 ), where R is Et, may be prepared
from the compound of Formula (7) by decarboxylation. In general, the
1 5 diester is reacted with a mixture of lithium iodide and sodium
cyanide at about 130° to 140°C in a suitable solvent, for
example N, N-
dimethylformamide, for about 24 hours.
Step 2 - Preparation of Compounds of Formula (9b~
2 0 In general, an anion of a compound of Formula ( 11 ), where R is H
or lower alkyl, is reacted with a compound of the formula R3R4C=O to
- 30 -
form a hydroxy acid or hydroxy ester, respectively, of Formula (9b).
A solution of the compound of Formula ( 11 ) in an anhydrous
ethereal solvent, preferably tetrahydrofuran, is added to about 1.1
molar equivalent (when R is lower alkyl) or about 2 molar
equivalents (when R is hydrogen) of a hindered base, preferably
lithium diisopropylamide, in an anhydrous ethereal solvent,
preferably tetrahydrofuran, at about 0°C. When the addition is
complete, a small quantity of a polar solvent is optionally added,
preferably hexamethylphosphoramide. To this mixture is added an
excess of a compound of the formula R3R4C=O. The addition is carried
out at a temperature range of about -78 to 10°C, preferably at about
-78"C when R3 and R4 are hydrogen, or preferably 0°C for ketones,
followed by reaction at room temperature for about 2-24 hours,
1 5 preferably about 10 hours. Where R in the starting material of
Formula ( 11 ) is hydrogen, the reaction product of Formula (9b) is
isolated and purified by conventional means. Where R in the starting
material of Formula (11) is lower alkyl, the reaction product of
Formula (9b), where R = H, is obtained by hydrolyzing the ester
2 0 product using a base, preferably lithium hydroxide, as described
above, then isolating and purifying (9b) by conventional means.
Step 3 - Preparation of Compounds of Formula (lOb~
The compound of Formula (9b) is then converted to a compound
2 5 of Formula ( l Ob) in the same manner as described in Reaction Scheme
IV.
The method of Reaction Scheme VI can be used, for example, to
prepare compounds of Formula (10) where R1 and R2 taken together
3 0 with the carbon to which they are attached is tetrahydropyran-4-yl,
by starting with 4-carboxytetrahydropyran or an ester thereof, for
example, the ethyl ester. Similarly, compounds of Formula ( 10) where
R 1 and R2 taken together with the carbon to which they are attached
is piperidin-4-yl or derivatives thereof, may be prepared by starting
3 5 with 1-benzyloxycarbonyl-4-carboxypiperidine, N-(tent-butoxy-
r X1931?~
- 31 -
carbonyl)-4-carboxypiperidine, or an ester thereof, for example, the
ethyl ester.
Alternative Preparation of Compounds of Formula I
Compounds of Formula I can also be prepared from compounds of
Formula (13), the preparation of which is shown below in Reaction
Scheme VIa, and described in Example SA.
REACTION SCHEME VIA
R1 R3 R4 Ri R2
RO R2 + ~ step 1 RO R3
O X X
O X R4
(11) (13)
where R is hydrogen or lower alkyl, and X is halo or -p-tosyl.
I S Step 1 - Preparation of Compounds of Formula (13) from 11)
The starting compounds of Formula (13) are commercially
available, for example, an ester of commercially available
chloropivalic acid may be prepared conventionally, or compounds of
Formula (13) may be prepared by means well known in the art, for
2 0 example, Gibson and Johnson, J. Chem. Soc., p2525 (1930). In general,
an anion of a compound of Formula ( 11 ) is reacted with an alkyl
dihalide to form a halo-substituted hydroxy acid ester of Formula
(13).
2 5 A solution of the compound of Formula ( 11 ) in an anhydrous
ethereal solvent, preferably tetrahydrofuran, is added to about 1.1
molar equivalent (when R is lower alkyl) or about 2 molar
equivalents (when R is hydrogen) of a hindered base, preferably
lithium diisopropylamide, in an anhydrous ethereal solvent,
3 0 preferably tetrahydrofuran, at about -100 to 0°C, preferably at
about
-78°C. To this mixture is added an excess of an alkyl dihalide,
1~3~7$
- 32 -
preferably diiodomethane. The addition is carried out a temperature
range of about -5° to 50°C for about 1-5 hours. The reaction
product
of Formula ( 13) is isolated by conventional means, and preferably
used in the next step of the synthesis without further purification.
It should be noted that a compounds of Formula ( 13) where X is
p-tosyl, are obtained by tosylation by conventional means of
compounds of Formula 1;8) or (9b).
Preparation of Compounds of Formula I
The intermediates of Formulae (4), ( 10), and ( 13) may be
converted to compounds of Formula I where Y is hydroxy and n is 0,
designated as compounds of Formula Ia, as shown in Reaction Scheme
VII below.
REACTION SCHEME VII
R' R2
step 1 RO SR5
[(4) or (10) or (13)] + RSSH
O R3 Ra
la
where R is hydrogen or lower alkyl.
Compounds of Formula (4) are either commercially available, for
example from Aldrich, or may be prepared according to methods
known to those skilled in the art, for example, as described by
Mannich and Rister, Chem. Ber., 57, 1116 (1924) for acids where R3
2 5 and R4 are each hydrogen, or may be prepared as described above, or
as described in Example 3. Compounds of Formula (5) are
corr~mercially available, for example from Aldrich, Fluka, etc.), or may
be prepared according to methods known to those skilled in the art,
e.g., as described below in Example 4.
Step 1 - Preparation of Compounds of Formula Ia from ~4)
Compounds of Formula I where n is 0 and Y is hydroxy,
- 33 -
designated as compounds of Formula Ia, may be prepared by heating
an enoic acid of Formula {4) with an equimolar amount of a thiol of
Formula (5) in the presence of an approximately equimolar amount of
a secondary amine, preferably piperidine. The reaction is carried out
in the temperature range from about 70°C to 120°C, preferably at
about 100°C, for about 1 to 24 hours, preferably about 3 hours. The
sulfide reaction product, a compound of Formula Ia, is isolated and
purified by conventional means.
1 0 Step 1 - Preparation of Compounds of Formula Ia from X10),
Compounds of Formula I where n is 0 and Y is hydroxy,
designated as compounds of Formula Ia, may be prepared by reacting
a lactone of Formula ( 10) with about 1.1 molar equivalents of an
anion of a thiol of Formula (5) (generated by reaction of (5) with an
alkaline metal hydride, preferably sodium hydride in a polar solvent,
preferably N,N-dimethylformamide). The reaction is carried out in a
polar solvent, preferably N, N-dimethylformamide, at a temperature
range of about 0°C to 70°C, preferably at about 0° to
25°C. The sulfide
reaction product, a compound of Formula Ia, is isolated and purified
2 0 by conventional means.
Step 1 - Preparation of Compounds of Formula Ia from (13)
Compounds of Formula I where n is 0 and Y is hydroxy or lower
alkoxy, designated as compounds of Formula Ia, may be prepared by
2 5 reacting an enoic acid ester of Formula ( 13) with about 1.1 molar
equivalents of an anion of a thiol of Formula (5) (generated by
reaction of (5) with an alkaline metal hydride, preferably sodium
hydride in a polar solvent, preferably N, N-dimethylformamide). The
reaction is carried out in a polar solvent, preferably N, N-dimethyl-
3 0 formamide, at a temperature range of about 30°C to 120°C,
preferably
at about 80°C, for about 10 minutes. The sulfide reaction product, a
compound of Formula Ia, is isolated and purified by conventional
me ans.
-, ~93~T$
- 34 -
Conversion of Compounds of Formula Ia to other Compounds of
Formula I
One method of converting compounds of Formula Ia to other
compounds of Formula I is shown below in Reaction Scheme VIII.
REACTION SCHEME VIII
R1 R2
step 1 t BuOHN SR5
la + t BuONH2~HCi
p R3 Ra
Ib
R' R2
step 2 t BuOHN S(O)~R5
Ib
O R3 R4
Ic
R1 R2
step 3 HOHN S(O)"R5
Ic
O R3 Ra
Id
Step 1 - Preparation of Compounds of Formula Ib
In general, compounds of Formula I where n is 0 and Y is tert-
BuONH-, designated as compounds of Formula Ib, are prepared by
reacting a compound of Formula Ia with an excess of a O-(tert-butyl)-
hydroxylamine hydrochloride and N-ethyl-N'-(3-dimethylamino-
propyl)-carbodiimide hydrochloride (or other carbodiimide
derivatives, for example 1,3-dicyclohexylcarbodiimide), in the
presence of 1-hydroxybenzotriazole hydrate and a tertiary base, for
example dimethylaminopyridine, triethylamine, 4-methylmorpholine,
2 0 pyridine, or a mixture of such bases. The reaction is carried out in an
inert solvent, preferably methylene chloride, in the temperature
19378
- 35 -
range from about 0°C to 40°C, preferably at about 25°C,
for about 10
to 30 hours, preferably about 18 hours. The N-tert-butoxy reaction
product, a compound of Formula Ib, is isolated and purified by
conventional means.
Step 2 - Preparation of Compounds of Formula Ic where n is 1
In general, compounds of Formula I where n is 1 and Y is tert-
Bu(JNH-, (i. e., sulfoxides), designated as compounds of Formula Ic, are
prepared from campounds of Formula Ib by reaction with a mild
1 0 oxidizing agent, for example sodium periodate or one equivalent of
"OXONE"TM (potassium peroxymonosulfate, Aldrich Chemical Co.),
until starting material can no longer be detected. The reaction is
carried out in an inert solvent, preferably aqueous acetone, in the
temperature range from about 0°C to 40°C, preferably at about
25°C,
for about 10 minutes to 4 hours, preferably about 30 minutes. The
sulfoxide product, a compound of Formula Ic where n is 1, is isolated
and purified by conventional means.
Step 2 - Preparation of Compounds of Formula Ic where n is 2
2 0 In general, compounds of Formula I where n is 2, Y is tert-
BuONH-, and R1 is hydrogen (i.e., sulfones), designated as compounds
of Formula Ic, are prepared from compounds of Formula Ib by
reaction with about 1-3 molar equivalents, preferably about 1.5
molar equivalents, of a strong oxidizing agent, for example,
2 5 m-c:hloroperbenzoic acid or OXONE. The reaction is carried out in an
inert solvent, preferably a protic solvent, preferably aqueous
methanol, in the temperature range from about 0°C to 40°C,
preferably at about 25°C, for about 10 minutes to 4 hours, preferably
about 2 hours. The sulfone product, a compound of Formula Ic where
3 0 n is 2, is isolated and purified by conventional means.
Step 3 - Preparation of Compounds of Formula Id
In general, compounds of Formula I where Y is HONH-, designated
as compounds of Formula Id, are prepared by hydrolysing an N-tert-
3 5 butaxy compound of Formula Ib or Ic under acid conditions under
- 36 -
conditions similar to that shown for the preparation of compounds of
Formula (4) above, or using hydrochloric acid gas in a sealed tube in
an inert solvent, for example, 1,2-dichloroethane. The hydroxyamino
reaction product, a compound of Formula Id where Y is HONH-, is
isolated and purified by conventional means.
Alternative Method of Introduction of R3 and R4 into Compounds of
Formula I
An alternative method of introducing the groups R3 and R4 into
compounds of Formula I is shown below in Reaction Scheme VIIIA.
REACTION SCHEME VIVA
R1 R2 Ri R2
RO~~~~~S02R5 step 1 RO S02R5
O ~ R3 ~R4
Ib IW
where R is hydrogen or lower alkyl.
Step 1- Preparation of Compounds of Formula I where n is 2, and
R 3 is as defined in the compounds of formula I but is other than
2 0 Hydrogen
The compounds of Formula I where n is 2, Y is hydroxy or alkoxy,
R 3 is as defined in the compounds of formula I other than hydrogen,
and R1, R2, and R4 are defined in the compounds of formula I,
designated as compounds of Formula Iw are prepared by the
2 5 alkylation of compounds of Formula I where both R3 and R4 are
hydrogen.
A solution of the compound of Formula Iw in an anhydrous
ethereal solvent, preferably tetrahydrofuran, is added to a hindered
3 0 base, preferably lithium diisopropylamide, in a manner similar shown
above in Reaction Scheme VIA. To this mixture is added about 1
~2193~?8
- 37 -
molar equivalent of an alkyl or aralkyl halide. The reaction addition is
stirred for about 1-3 hours, then stirred stirred for an additional
1-5 hours, preferably 3 hours, at about room temperature. The
reaction product is isolated and purified by conventional means.
R 4 may be introduced in the same manner as shown above.
Compounds of Formula Iw can be converted to other compounds
of Formula I as shown previously.
Preferred Procedure for Preparing Compounds of Formula Id from
Corn~ounds of Formula Ia
A preferred method of converting compounds of Formula Ia to
1 5 other compounds of Formula I is shown below in Reaction Scheme IX.
REACTION SCHEME IX
R' R2 R1 R2
HO SR5 step 1 CI SR5
O Rs R4 O Rs R4
la (12)
R~ R2
step 2 HOHN SR5
(12)
O R3 R4
Iba
R1 R2
step 3 HOHN S(O)"R5
Iba
O R3 Ra
Id
~ g ,~ ~ 7g
- 38 -
Stets 1 - Preparation of Compounds of Formula Iba
In general, an acid halide of a compound of Formula Ia,
designated as compounds of Formula ( 12), is prepared by reacting a
compound of Formula Ia with a halogenating agent.
The compound of Formula Ia is reacted with an excess of a
halogenating agent, for example oxalyl chloride, oxalyl bromide,
phosphorous oxychoride, phosphorous trichloride, phosphorous
pentachloride, thionyl chloride, preferably oxalyl chloride in the
1 0 presence of a small amount of N, N-dimethylformamide as a catalyst.
The; reaction is carried out in an inert solvent, preferably methylene
chloride, in the temperature range from about 0°C to 40°C,
preferably
at about 25°C, for about 10 to 30 hours, preferably about 18 hours.
The. acid halide reaction product, a compound of Formula (12), is
isolated by conventional means.
Step 2 - Preparation of Compounds of Formula Iba
Compounds of Formula I where n is 0 and Y is HONH-, designated
as compounds of Formula Iba, may be prepared by reacting a
2 0 compound of Formula ( 12) with about 1-5 molar equivalents,
preferably about 3.5 molar equivalents, of N, O-bis(trimethylsilyl)-
hydroxylamine, or more preferably aqueous hydroxylamine dissolved
in a suitable solvent, for example a mixture of tert-butanol/tetra-
hydrofuran. The reaction is carried out in an inert solvent, preferably
2 5 methylene chloride, in the temperature range from about 0°C to
25°C,
preferably at about 25°C, for about 1-10 hours, preferably about 3
hours for N, O-bis(trimethylsilyl)hydroxylamine, or about 1.5 hours for
aqueous hydroxylamine. The N-hydroxamic acid product, a compound
of Formula Iba, is isolated and purified by conventional means.
Step 3 - Preparation of Compounds of Formula Id
The compound of Formula Iba is converted to a compound of
Formula Id where n is 1 or 2 in the same manner as shown in
Reaction Scheme VIII, steps 2 or 3, above.
:35
z~9~1r~
- 39 -
Alternative Preparation of Formula I
of Compounds
It should be noted that the of the steps in the above
sequence
Reaction Schemes for the preparationof compounds of Formula
Id
may be changed. That is, a compoundof Formula Ia may be oxidized
first to a sulfone,followed by conversionof the carboxy group
to
hydroxyamino as shown above, if desired.
so
Preparation of Compounds of Formula I where RS is Biphen
Compounds of Formula I where RS is optionally substituted
biphenyl are preferably prepared from compounds of Formula Ia
where RS is optionally substituted bromophenyl. For example,
compounds where RS is 4-biphenyl can be prepared from compounds
of Formula Ia where RS is 4-bromophenyl, represented below as a
compound of Formula Iaa, as shown below in Reaction Scheme X.
REACTION SCHEME X
Br / Br
/
R1 R2 ~ ~ R~ R2 W
HO S step 1 HO S02
O Rs Ra O Rs Ra
/
laa le
/
R' R2
/ B(OH)2 step 2 HO S02
le +
O R3 R4
If
/
R1 R2
step 3 HOHN S02
If
O R3 Ra
Ih
as~~~~
- 40 -
Sten 1 - Preparation of Compounds of Formula Ie
In general, compounds of Formula I where n is 2, Y is hydroxy, RS
is 4-bromophenyl, and Rl, R2, R3, and R4 are as defined in the
compounds of formula I, designated as compounds of Formula Ie, are
prepared from compounds of Formula Iaa by reaction with a strong
oxidizing agent in the same manner as shown above in Reaction
Scheme VIII, Step 2.
Sten 2 - Preparation of Compounds of Formula If
In general, compounds of Formula I where n is 2, Y is hydroxy, R5
is biphenyl, and R l , R~ , R3 , and R4 are as defined in the compounds of
formula I, designated as compounds of Formula If, are prepared by
reacting a compound of Formula Ie with phenylboronic acid and
zero-valent palladium catalysts, preferably tetrakis(triphenylphos-
phine)palladium. The reaction is carried out in a protic solvent,
preferably a mixture of ethanol and benzene, in the temperature
range from about 30°C to 100°C, preferably at about 80°C.
When the
desired temperature is reached, aqueous 2M sodium carbonate is
2 0 added, and refluxing continued for about 1-8 hours, preferably about
2 flOUrS. The reaction product, a compound of Formula If, is isolated
by conventional means and preferably purified using preparative
TLC.
2 5 Step 3 - Preparation of Compounds of Formula Ih
In general, compounds of Formula I where n is 2, Y is HONH-, R5
is biphenyl, and R l , R2 , R3 , and R4 are as defined in the compounds of
formula I, designated as compounds of Formula Ih, may be prepared
from the corresponding compounds of Formula If in the same manner
3 0 as shown above in Reaction Scheme VIII, or preferably as shown in
Reaction Scheme IX or X.
To prepare compounds of Formula I where RS is substituted
biphenyl, a compound of Formula Iaa optionally substituted on the 4
3 5 bromophenyl ring is reacted with an optionally substituted boronic
~93~78
I'
- 41 -
acid in the same manner as shown above,
Preparation of Compounds of Formula I where RS is Diphenylsulfide
Compounds of Formula I where RS is optionally substituted
diphenylsulfide are preferably prepared from the corresponding
compounds of Formula Ie, i. e., compounds of Formula I in which RS is
optionally substituted , '~romophenyl, prepared as in Reaction
Scheme X. For example, compounds where R5 is 4-diphenylsulfide can
be prepared from co_rt-.pounds of Formula Ie as shown below in
Reaction Scheme XI.
REACTION SCHEME XI
S
R' R2
step 1 HO S02
le
O R3 Ra
S
R1 RZ W
step 2 HOHN~~~~~S02
l ,~i
O R3 R4
Step 1 - Preparation of Compounds of Formula Ii
In general, compounds of Formula I where n is 2, Y is hydroxy, RS
is ~~-diphenylsulfide, and R1, R2, R3, and R4 are as defined in the
compounds of formula I, designated as compounds of Formula Ii, are
2 0 prepared from compounds of Formula Ie by heating an anion of
thiophenol (preferably prepared in situ, for example, by treatment of
!~~~~78
- 42 -
thiophenol with sodium or potassium hydride, preferably potassium
hydride, in a polar solvent, preferably N,N-dimethylformamide. The
reaction is carried out in a polar solvent, preferably N, N-dimethyl-
formamide, in the temperature range from about 30°C to 100°C,
preferably at about 75°C, for about 4-48 hours, preferably about 18
hours. The reaction product, a compound of Formula Ii, is isolated by
conventional means and preferably purified using preparative TLC.
Step 2 - Preparation of Compounds of Formula I_i
1 0 In general, compounds of Formula I where n is 2, Y is HONH-, RS
is 4-diphenylsulfide, and R1, R2, R3, and R4 are as defined in the
compounds of formula I, designated as compounds of Formula Ij, are
prepared from the corresponding compounds of Formula Ii in the
same manner as shown above in Reaction Scheme VIII, or preferably
1 5 as shown in Reaction Scheme IX or X.
To prepare compounds of Formula I where RS is substituted
4-diphenylsulfide, a compound of Formula Ie optionally substituted
on the 4-bromophenyl ring is reacted with an optionally substituted
2 0 anion of thiophenol in the same manner as shown above.
Preparation of Compounds of Formula I where R5. is 4-f4-(thiot~hen-
2-~l)phenox,~phenyl
Compounds of Formula I where RS is optionally substituted
2 5 4-[4-(4-thiophen-2-yl)phenoxy]phenyl are prepared from the
corresponding compounds of Formula I where RS is optionally
substituted 4-(4-bromophenoxy)phenyl. This reaction is shown in
Reaction Scheme XIA.
19~~78
- 43 -
SCHEME XIA
!~~
s
o ~ o
R~ R2 ~ R' R2
HO\ / S02 step 1 HO S02
O~ Rs Ra O Rs Ra
Ifa Ifb
Preparation of Compounds of Formula Ifb
The 4-bromo group of the compound of Formula (Ifa), which may
be prepared by methods analogous to those previously shown, or as
described in Example 16D, is displaced to give a compound of Formula
Ifb, using the same procedure as described in Reaction Scheme X, step
1 0 2.
The compound of Formula (Ifa) is reacted similarly in order to
introduce other aryl or heteroaryl groups.
1 5 Reduction of a compound of Formula Ifa with palladium and
hydrogen replaces the bromo group by hydrogen.
Preparation of Compounds of Formula I where R5 is 1.2-
Diphenylethene
2 0 Compounds of Formula I where RS is optionally substituted
1,2~-diphenylethene are preferably prepared from the corresponding
compounds of Formula I where RS is optionally substituted 4-
bro:mophenyl, as prepared in Reaction Scheme X. For example,
compounds where RS is 4-diphenylethene can be prepared from
2 5 compounds of Formula Ie as shown below in Reaction Scheme XII.
1~~~78
- 44 -
REACTION SCHEME XII
step 1 HO S02
le
O R3 Ra
Ik
Step 1 - Preparation of Compounds of Formula Ik
In general, compounds of Formula I where Y is hydroxy, RS is
4-( 1,2-diphenylethene), and R l , R2, R3, and R4 are as defined in the
compounds of formula I, designated as compounds of Formula Ik, are
prepared by reacting a compound of Formula Ie with an optionally
substituted styrene in the presence of a hindered tertiary organic
base, for example diisopropylethylamine, and palladium diacetate,
and trimethylphenylphosphine or other triphenylphosphine
derivatives> preferably trimethylphenylphosphine or tetrakis(tri-
phenylphosphine)-palladium(0). The reaction is carried out in the
absence of solvent, in the temperature range from about 30°C to
100°C, preferably at about 80°C, for about 4-48 hours,
preferably
about 16 hours. The reaction product, a compound of Formula Ik, is
isolated by conventional means and preferably purified using
preparative TLC.
Conversion of the carboxylic acid of Formula Ik to its
hydroxyamino equivalent is carried out in the same manner as shown
above in Reaction Scheme VIII, or preferably as shown in Reaction
Scheme IX or X.
Preparation of Compounds of Formula I where R3 and R4 to eg ther
with the Carbon to which they are attached ret~resent an N-
Substituted Piperidine Derivative
The preparation of compounds of Formula I where Rl and R2 or
~~93~~8
- 45 -
R 3 and R4 together with the carbon to which they are attached
represent an N-substituted piperidine derivative are prepared from
the corresponding unsubstituted piperidine derivative. This
procedure is exemplified by reference to a compound of Formula I
where R3 and R4 together with the carbon to which they are attached
represent an N-substituted piperidine derivative, designated as
compounds of Formula Il, as shown below in Reaction Scheme XIII.
REACTION SCHEME XIII
R1 ~ R i R2
t BuOHN S(O)"R5 step 1 t BuOHN S(O)~R5
O J o ~N~
N
H R
II Im
R ~ R2
step 2 HOHN S(O)ARS
Im
J
N
R
In
Step 1 - Preparation of Compoundsof Formula Im
Compounds t-BuONH-, R1 and R2 are
of Formula as
I where Y
is
1 5 defined the compounds of formula and R~ and R4 together
in I, with
the carbon to which they are attachedrepresent N-substituted
an
pipf:ridine derivative, are designatedcompounds of Formula
as Im.
In general, compounds of Formula Im are prepared by reacting a
2 0 compound of Formula Il with a compound of the formula RX, where R
is lower alkyl, cycloalkylalkyl, acyl, alkoxycarbonylalkyl, picolyl,
-SG2Ra, where Ra is lower alkyl or -NRbRc, where Rb and Rc are
l~~~is
- 46 -
independently hydrogen or lower alkyl; and the like, and X is chloro,
bromo or iodo; for example, RX may be methyl iodide, cyclopropyl-
methyl bromide, 3-picolyl chloride, ethyl bromoacetate, bromo-
acetamide, acetyl chloride, dimethylaminosulfonyl chloride, in the
presence of a base, for example triethylamine or potassium carbonate.
The reaction is carried out in a polar solvent, preferably
N, N-dimethylformamide, in the temperature range from about 0°C to
50"C, preferably at about 25°C, for about 4 to 48 hours, preferably
about 16 hours. The reaction product, a compound of Formula Im, is
isolated by conventional means and preferably used with no further
purification.
Alternatively, a reductive alkylation may be carried out on a
compound of Formula Il to give a compound of Formula Im. For
1 5 example, reducing a compound of Formula Il in acetone in the
presence of a catalyst, for example palladium on carbon, under
hydrogen gives an N-isopropyl derivative of Formula Im.
Step 2 - Preparation of Compoundsof FormulaIn
2 0 Compounds of Formul a I where and R2 are as
Y is HONH-,
R1
defined in the compounds of formula and R~ R4 together
I, and with
the carbon to which they are attachedrepresent N-substituted
an
piperidine derivative, designated compounds of Formula In.
are as
2 5 In general, compounds of Formula In are prepared from a
compound of Formula Im by reaction with a strong acid, preferably
hydrochloric acid. The reaction is carried out in a sealed tube in an
inert solvent, preferably 1,2-dichloroethane, in the temperature range
from about 0°C to 45°C, preferably at about 20°C, for
about 10 to 72
3 0 hours, preferably about 48 hours. The reaction product, a compound
of Formula In, is isolated and purified by conventional means,
preferably by chromatography.
Preparation of Compounds of Formula I where R2 is -NR6R7
3 5 Compounds of Formula I where R2 is -NR6R7, in which R6 is
~~~~78
I
- 47 -
hydrogen and R7 is CBZ, where CBZ represents benzyloxycarbonyl, and
R 1, R3 and R4 are hydrogen, shown below, for example, as Formulae
Ip and Iq, are prepared by a different route, as shown in Reaction
Schemes XIV, XV, and XVI. This route provides compounds of
Formula Iab, optically pure or as racemic mixtures, depending upon
the chirality of the starting lactone.
REACTION SCHEME XIV
NHCBZ NHCBZ
step 1 HO SR5
O + RSSH
O O
1 0 (6) (5) lab
Step 1 - Preparation of Compounds of Formula Iab
In general, compounds of Formula Ia where Y is hydroxy, R2 is
-NR6R7, in which R6 is hydrogen and R7 is CBZ, where CBZ represents
1 5 benzyloxycarbonyl, and R 1, R3 and R4 are hydrogen, designated as
compounds of Formula Iab, are prepared by treating an anion of a
thiol of Formula (5) (preferably prepared in situ, for example, by
treatment of Formula (5) with sodium or potassium hydride,
preferably potassium hydride, in a polar solvent, preferably N, N-
2 0 dirnethylformamide) with a lactone of Formula (6). The reaction is
carried out in a polar solvent, preferably N, N-dimethylformamide, in
the temperature range from about 0°C to 40°C, preferably at
about
25°C, for about 5 minutes to 10 hours, preferably about 30 minutes to
6 hours. The sulfide reaction product, a compound of Formula Iab, is
2 5 isolated by conventional means and preferably used directly in the
next step.
Preparation of Compounds of Formula I where R2 is -NR6R7
Compounds of Formula I where R2 is -NR6R7, in which R6 is
3 0 hydrogen and R7 is CBZ, where CBZ represents benzyloxycarbonyl, and
R 1, R3 and R4 are hydrogen, are prepared from compounds of
~~~~7s
- 48 -
Formula Iab as shown below in Reaction Scheme XV.
REACTION SCHEME XV
NHCBZ
step 1 t BuOHN SR5
lab
O
to
NHCBZ
step 2 t BuOHN S(O)"R5
to
O
Ip
NHCBZ
step 3 HOHN S(O)~R5
Ip
O
Step 1 - Preparation of Compounds of Formula Io
Compounds of Formula I where Y is tent-BuONH-, R2 is -NHCBZ
whf:re CBZ represents benzyloxycarbonyl, and Rl, R3 and R4 are
hydrogen, designated as compounds of Formula Io, are prepared as
shown in the same manner as shown in Reaction Scheme VIII, or
preferably as shown in Reaction Scheme IX or X.
Step 2 - Preparation of Compounds of Formula In
1 5 Compounds of Formula Ip where n is 2, Y is tert-BuONH-, R2 is
-NHCBZ where CBZ represents benzyloxycarbonyl, and Rl, R3 and R4
are hydrogen, designated as compounds of the Formula Ip, are
prepared in 'the same manner as shown in Reaction Scheme VIII, or
preferably as shown in Reaction Scheme IX or X.
~193~ 78
- 49 -
Step 3 - Preparation of Compounds of Formula Icy
Compounds of Formula I where n is 2, Y is HONH-, R2 is -NHCBZ
where CBZ represents benzyloxycarbonyl, and Rl, R3 and R4 are as
defined in the compounds of formula I, designated as compounds of
the Formula Iq, are prepared by hydrolyzing a compound of
Formula Ip in the same manner as shown above in Reaction Scheme
VIII, or preferably as shown in Reaction Scheme IX or X.
Preparation of Compounds of Formula I where R2 is -NR6R~
1 0 Compounds of Formula I where R2 is -NR6R~, in which R6 and R~
are both hydrogen, and Rl, R3 and R4 are hydrogen, are prepared
from compounds of Formula Ip as shown below in Reaction Scheme
XVI.
1 5 REACTION SCHEME XVI
NH2
step 1 t BuOHN S(O)"R5
Ip
O
Ir
N H2
step 2 HOHN S(O)~R5
Ir
O
Is
Step 1 - Preparation of Compounds of Formula Ir
2 0 In general, compounds of Formula I where n is 2, Y is tert-
BuONH-, R2 is -NH2, and Rl , R3 and R4 are hydrogen, designated as
compounds of Formula Ir, are prepared by reducing a compound of
Formula Ip using a metal catalyst, preferably palladium on carbon.
The reaction is carried out under hydrogen at about 1 atmosphere, in
2 5 a p~rotic solvent, preferably ethanol, in the temperature range from
about 0°C to 40°C, preferably at about 25°C, for about 4
to 48 hours,
~~~317~
-so
preferably about 18 hours. The N-tert-butoxy reaction product, a
compound of Formula Ir, is isolated and purified by conventional
means.
s Stets 2 - Preparation of Compounds of Formula Is
In general, compounds of Formula I where n is 2, Y is HONH-,
R2 is -NH2, and R1, R3 and R4 are hydrogen, designated as
compounds of Formula Is, are prepared by reacting a compound of
Formula Ir with a strong acid, preferably hydrochloric acid. The
reaction is carried out in a sealed tube in an inert solvent, preferably
1,2-dichloroethane, in the temperature range from about -10°C to
40°C, preferably at about 2s°C, for about 4 to 48 hours,
preferably
about 18 hours. The hydroxyamino reaction product, a compound of
Formula Is, is isolated and purified by conventional means, preferably
1 s as its hydrochloride salt.
Preparation of Compounds of Formula I where R2 is -NR6R~
Alternatively, the compound of Formula Ir can be used to
produce other compounds of Formula I where R6 and/or R~ are as
2 0 defined in the Summary of the invention, but not both hydrogen. For
example, the preparation of a compound of Formula I where R2 is
vali.ne amide is shown below in Reaction Scheme XVII.
1931~g
- 51 -
REACTION SCHEME XVII
CBZHN
O NH
step 1 t BuOHN S(O)nR5
Ir
O
It
H2N
O~NH
step 2 t BuOHN S(O)nR5
It
O
lu
H2N
O~NH
step 3 HOHN S(O)nR5
lu
O
Iv
Step 1 - Preparation of Compounds of Formula It
In general, compounds of Formula I where n is 2, Y is tert-
BuONH-, R2 is 2-(S)-CBZ-valine amide, i. e., where R6 is hydrogen and
R~ is 2-(S)-CBZ-3-methyl-1-butanoyl, where CBZ represents
benzyloxycarbonyl, and Rl , R3 and R4 are hydrogen, designated as
compounds of Formula It, are prepared by reacting a compound of
Formula Ir with CBZ-(S)-valine in the presence of N-ethyl-N'-
(3-dimethylaminopropyl;)-carbodiimide and 1-hydroxybenzotriazole
and a slight excess of a tertiary amine, preferably triethylamine. The
reaction is carried out in an inert solvent, preferably methylene
y 19~1~8
- 52 -
chloride, in the temperature range from about 0°C to 40°C,
preferably
at about 25°C, for about 6-48 hours, preferably about 16 hours. The
reaction product, a compound of Formula It, is isolated by
conventional means, and is preferably used in the next step without
further purification.
Step 2 - Preparation of Compounds of Formula Iu
In general, compounds of Formula I where n is 2, Y is tert-
BuONH-, R2 is 2-(S)-amino-valine amide, i.e., where R6 is hydrogen
1 0 and R~ is 2-(S)-amino-3-methyl-1-butanoyl, and R 1, R3 and R4 are
hydrogen, designated as compounds of Formula It, are prepared by
reducing a compound of Formula It using a metal catalyst, preferably
palladium on carbon. The reaction is carried out under hydrogen at
about 1 atmosphere, in a protic solvent, preferably a mixture of
methanol and ethanol, in the temperature range from about 0°C to
40''C, preferably at about 25°C, for about 1 to 8 hours, preferably
about 3 hours. The reaction product, a compound of Formula Iu, is
isolated and purified by conventional means, preferably
chromatography.
Step 3 - Preparation of Compounds of Formula Iv
In general, compounds of Formula I where n is 2, Y is HONH-,
R2 is 2-(S)-amino-valine amide, i.e., where R6 is hydrogen and R~ is
2-(S)-amino-3-methyl-1-butanoyl, and R1, R3 and R4 are hydrogen,
2 5 designated as compounds of Formula Iv, are prepared by reacting a
compound of Formula Iu with a strong acid, preferably hydrochloric
acid. The reaction is carried out in a sealed tube in an inert solvent,
preferably 1,2-dichloroethane, in the. temperature range from about
-20°C to 40°C, preferably at about 25°C, for about 4 to
48 hours,
3 0 preferably about 24 hours. The hydroxyamine reaction product, a
compound of Formula Iv, is isolated and purified by conventional
means, preferably as its hydrochloride salt.
Preparation of Compounds of Formula I where R2 is -NR6R~
3 5 In a manner similar to that shown above, compounds of Formula
,; ~~93~~8
- 53 -
I where R2 is -NR6R~, in which R6 and R~ are both methyl, are
prepared by reacting a compound of Formula Ir in a polar solvent,
preferably N, N-dimethylformamide, with about two equivalents of
methyl iodide in the presence of a base, preferably potassium
carbonate, then treating the product with hydrochloric acid gas as
shown in Step 3 above.
Preparation of Compounds of Formula I where R2 is -NR6R~
In a manner similar to that shown above, compounds of Formula
1 0 I where where R2 is -NR6R~, in which R6 is hydrogen and R~ is
-NHS02N(CH3)2, are prepared by reacting a compound of Formula Ir
with about one equivalent of dimethylsulfamoyl chloride in an inert
solvent, preferably methylene chloride, in the presence of a base,
preferably pyridine, then treating the product with hydrochloric acid
1 5 gas as shown in Step 3 above.
Similarly, the compound of Formula Ir can be used to produce
other compounds of Formula I where R6 and/or R~ are as defined in
the: Summary of the invention, but not both hydrogen, in the same
2 0 manner as shown in Reaction Scheme XVII above.
Isolation and Purification of the Compounds
Isolation and purification of the compounds and intermediates
de scribed herein can be effected, if desired, by any suitable
2 5 separation or purification procedure such as, for example, filtration,
extraction, crystallization, column chromatography, thin-layer
chromatography, thick-layer chromatography, preparative low or
high-pressure liquid chromatography or a combination of these
procedures. Specific illustrations of suitable separation and isolation
3 0 procedures can be had by reference to the Examples hereinbelow.
However, other equivalent separation or isolation procedures could, of
course, also be used.
Salts of Compounds of Formula I
3 5 Some of the compounds of Formula I may be converted to a
'~9~~~g
- 54 -
corresponding acid addition salt by virtue of the presence of basic
nitrogen atoms. The conversion is accomplished by treatment with at
least a stoichiometric amount of an appropriate acid, such as
hydrochloric acid, hydrobromic acid, sulfuric acid, nitric acid,
phosphoric acid and the like, and organic acids such as acetic acid,
propionic acid, glycolic acid, pyruvic acid, oxalic acid, malic acid,
ma:lonic acid, succinic acid, malefic acid, fumaric acid, tartaric acid,
citric acid, benzoic acid, cinnamic acid, mandelic acid, methanesulfonic
acid, ethanesulfonic acid, p-toluenesulfonic acid, salicylic acid and the
like. Typically, the free base is dissolved in an inert organic solvent
such as diethyl ether, ethyl acetate, chloroform, ethanol or methanol
and. the like, and the acid added in a similar solvent. The temperature
is maintained at 0° to 50°C. The resulting salt precipitates
spontaneously or may be brought out of solution with a less polar
solvent.
In summary, the compounds of the present invention are made
by the procedures outlined below:
2 0 1. A process for preparing compounds of Formula I where Rl
is hydrogen comprises:
reacting a compound of the formula:
R2 R3
H02C R4
(4)
where R2, R3 and R4 are as defined in the compounds of formula
I, except that R2 cannot be -NR6R~;
with a compound of the formula RS SH, where RS is as defined in the
compounds of formula I, in the presence of a secondary base.
2. Alternatively, a process for preparing compounds of
193°~~8
- 55 -
Formula I comprises:
reacting a compound of the formula:
R1 R2
t BuOHN ~~~>~~ SR5
O R3 Ra
where R1, R2, R3, R4 and RS are as defined in the compounds of
formula I,
with a mild oxidizing agent, for example, sodium periodate.
3. Alternatively, a process for preparing compounds of
Formula I comprises:
reacting a compound of the formula:
R~ R2
r-BuOHN ~~~~~ SR5
O R3 R4
where R1, R2, R3, R4 and RS are as defined in the compounds of
formula I,
with a strong oxidizing agent, for example, OXONE or m-
chloroperbenzoic acid.
4. Alternatively, a process for preparing compounds of
Formula I where n is 2 comprises:
reacting a compound of the formula:
R1 R2
t-BuOHN S(O)~R5
2 5 O R3 R4
where R 1, R2, R3 , R4 and RS are. as defined in the compounds of
formula I,
~~~3~~8
- 56 -
wit:h a strong oxidizing agent, for example, OXONE or m-chloro-
perbenzoic acid.
5. Alternatively, a process for preparing compounds of
Formula I comprises:
reacting a compound of the formula:
R1 R2
HO ~~~<~ S(O)~R5
O R3 Ra
where n, R1, R2, R~, R4 and R5 are as defined in the compounds
of formula I,
with O-(tert-butyl)hydroxylamine hydrochloride in the presence of a
carbod.iimide, for example, N-ethyl-N'-(3-dimethylaminopropyl)-
carbodiimide hydrochloride, and a tertiary amine.
6. Alternatively, a process for preparing compounds of
Formula I comprises:
reacting a compound of the formula:
R1 R2
CI S(O)~R5
2 0 O R3 R4
where n, R1, R2, R3, R4 and R5 are as defined in the compounds of
formula I,
with hydroxylamine or N, O-bistrimethylsilyl hydroxylamine.
7. Alternatively, a process for preparing compounds of
Formula I comprises:
hydrolysing a compound of the formula:
~93~~8
,,
- 57 -
R1 R2
t BuOHN ~~~~~~ S{O)~R5
O R3 R4
where n, R1, R2, R3, R4 and RS are as defined in the compounds of
formula I,
under acid conditions, for example, with hydrochloric acid or
trifluoroacetic acid.
8. Alternatively, a process for preparing compounds of
Formula I comprises:
reacting a compound of the formula:
R' R2
t BuOHN S(O)"R5
J
N
H
where n, R 1, R2 and RS are as defined in the compounds of
1 5 formula I, except that R2 cannot be -NR6R7;
with a compound of the formula RX, where R is lower alkyl,
cycloalkylalkyl, acyl, alkoxycarbonylalkyl, acetamido, picolyl, -S02Ra,
where Ra is lower alkyl or -NRbRc, where Rb and Rc are
independently hydrogen or lower alkyl; and X is chloro, bromo or
2 0 iodo.
9. Alternatively, a process for preparing compounds of
Formula I comprises:
reacaing a compound of the formula:
,,
~~931~8
-sg-
R1 R2
t BuOHN S(O)~R5
J
N
H
where n, R 1, R2 and RS are as defined in the compounds of
formula I, except that R2 cannot be -NR6R~;
with acetone under hydrogen in the presence of a catalyst, for
example, palladium on carbon, to give the N-isopropyl derivative.
10. Alternatively, a process for preparing compounds of
Formula I comprises:
reacting a compound of the formula:
NHCBZ
O ,7
O
with an anion of a compound of the formula RSSH, where RS is as
defined in the compounds of formula I.
11. Alternatively, a process for preparing compounds of
Formula I comprises:
reacting a compound of the formula:
NH2
t BuOHN ~ S(O)nR5
O
where RS is as defined in the compounds of formula I, with an
acylating agent, for example CBZ-(S)-valine in the presence of N-
2 5 ethyl-N'-(3-dimethylamiisopropyl)-carbodiimide and 1-hydroxy-
ben.zotriazole and a tertiary amine, or an alkylating agent, for
,t
~~317g
- 59 -
example, methyl iodide in the presence of a base or a sulfamoyl
halide, such as dimethylsulfamoyl chloride in the presence of a base.
12. Alternatively, a process for preparing compounds of
Formula I comprises:
reacting a compound of the formula:
R1 R2
R3
O
O R4
1 0 where R l , R2, R3 and R4 are as defined in the compounds of
formula I, except that R2 cannot be -NR6R~;
with a compound of the formula RSSH, where RS is as defined in the
cornpounds of formula I, in the presence of a secondary base.
13. Alternatively, a process for preparing compounds of
Formula I comprises:
reacting a compound of the formula:
R' R2
RO X
O R3 R4
with an anion of a compound of the formula RSSH, where RS is as
defined in the compounds of formula I.
14. Alternatively, a process for preparing compounds of
2 5 Formula I comprises:
reacting a compound of the formula:
R' R2
RO~~~~ S02R5
O
- 60 -
with an alkyl or aralkyl halide in the presence of a hindered base.
15. Alternatively, a process for preparing compounds of
S Formula I comprises:
reacting a compound of the formula:
Br
O
R1 R2
HO S02
O R3 Ra
1 0 with a compound of the formula R"B(OH)z or R"SnMe3, where R" is aryl
or heteroaryl, in the presence of tetrakis(triphenylphosphine)palladium(0).
The compounds of Formula I inhibit mammalian matrix
15 metalloproteases, such as the stromelysins, gelatinases, matrilysin and
collagenases, and are therefore useful as therapeutically active
substances, especially for treating diseases associated with the MMP-
induced excessive degradation of matrix and connective tissue within
the mammal, for example, arthritic diseases (rheumatoid arthritis and
:Z 0 oste,oarthritis), multiple sclerosis, bone resorptive diseases (such as
osteoporosis), the enhanced collagen destruction associated with
diabetes, chronic obstructive pulmonary disease, cerebral
hemorrhaging associated with stroke, periodontal disease, corneal
ulceration, ulceration of the skin, tumor invasion and metastasis, and
:Z 5 aberrant angiogenesis.
The compounds of Formula I substantially inhibit the release of
tumor necrosis factor (TNF) from cells, and are therefore useful for
the treatment of conditions mediated by TNF, for example
~931~'8
- 61 -
inflammation, fever, cardiovascular effects, hemorrhage, coagulation
and acute phase response, cachexia and anorexia, acute infections,
shack states, restinosis, aneurysmal disease, graft versus host
reactions and autoimmune disease.
The compounds of Formula I also inhibit the release of other
biologically active molecules from cells, including soluble receptors
(CD30 and receptors for TNF (p55 and p75), IL-6, IL-1 and TSH),
adhesion molecules (e.,g., L-selection, ICAM-l, fibronectin) and other
1 0 growth factors and cytokines, including Fas ligand, TGF-a, EGF, HB-
EGF, SCF and M-CSF. Inhibition of the release or shedding of such
proteins, and are therefore useful for treating a number of disease
states, for example rheumatoid arthritis, multiple sclerosis, vascular
disease, Type II diabetes, HIV, cachexia, psoriasis, allergy, hepatitis,
inflammatory bowel disease, and cancer.
The ability of the compounds of Formula I to inhibit matrix
metalloprotease activity, such as the activity of collagenase-1, -2 and
-3, stromelysin-l, gelatinases A and B, and matrilysin may be
2 0 demonstrated by a variety of in vitro assays known to those of
ordinary skill in the art, such as the assay described in the MMP
Enzymatic Assay described in FEBS, 296, 263 (1992) or modifications
thereof. The ability of the compounds of Formula I to inhibit MMP
mediated processes in vivo may be tested using the interleukin-1
2 5 stimulated cartilage explant assay and cartilage plug implantation
assay.
The ability of the compounds of Formula I to inhibit the release of
TNF as shown in Examples 45 to 47.
The present invention also relates to a pharmaceutical
composition comprising a pharmaceutically acceptable non-toxic
excipient and a therapeutically effective amount of a compound of
formula I.
- 62 -
Administration of the compounds of Formula I or their
pharmaceutically acceptable salts, in pure form or in an appropriate
pharmaceutical composition, can be carried out via any of the
accepted modes of administration or agents for serving similar
utilities. Thus, administration can be, for example, orally, nasally,
parenterally, topically, transdermally, or rectally, in the form of solid,
semi-solid, lyophilized powder, or liquid dosage forms, such as for
example, tablets, suppositories, pills, soft elastic and hard gelatin
capsules, powders, solutions, suspensions, or aerosols, or the like,
preferably in unit dosage forms suitable for simple administration of
precise dosages. The compositions will include a conventional
pharmaceutical carrier or excipient and a compound of Formula I as
the/an active agent, and, in addition, may include other medicinal
agents, pharmaceutical agents, carriers, adjuvants, etc.
Generally, depending on the intended mode of administration, the
pharmaceutically acceptable compositions will contain about 1 % to
about 99% by weight of a compounds) of Formula I, or a
pharmaceutically acceptable salt thereof, and 99% to 1 % by weight of
2 0 a suitable pharmaceutical excipient. Preferably, the composition will
be about 5% to 75% by weight of a compounds) of Formula I, or a
pharmaceutically acceptable salt thereof, with the rest being suitable
pharmaceutical excipients.
2 5 The preferred route of administration is oral, using a convenient
daily dosage regimen which can be adjusted according to the degree
of severity of the disease-state to be treated. For such oral
administration, a pharmaceutically acceptable composition containing
a compounds) of Formula I, or a pharmaceutically acceptable salt
3 0 thereof, is formed by the incorporation of any of the normally
employed excipients, such as for example, pharmaceutical grades of
mannitol, lactose, starch, pregelatinized starch, magnesium stearate,
sodium saccharine, talcum, cellulose ether derivatives, glucose,
gelatin, sucrose, citrate, propyl gallate, and the like. Such compositions
3 5 take the form of solutions, suspensions, tablets, pills, capsules,
2~931'~8
- 63 -
powders, sustained release formulations, and the like.
Preferably such compositions will take the form of capsule, caplet
or tablet and therefore will also contain a diluent such as lactose,
sucrose, dicalcium phosphate, and the like; a disintegrant, such as
croscarmellose sodium or derivatives thereof; a lubricant such as
magnesium stearate and the like; and a binder such as a starch, gum
acacia, polyvinylpyrrolidone, gelatin, cellulose ether derivatives, and
the like.
The compounds of Formula I, or their pharmaceutically
acceptable salts, may also be formulated into a suppository using, for
example, about 0.5% to about 50% active ingredient disposed in a
carrier that slowly dissolves within the body, e. g. , polyoxyethylene
1 5 glycols and polyethylene glycols (PEG), e.g., PEG 1000 (96%) and PEG
40C)0 (4%).
Liquid pharmaceutically administrable compositions can, for
example, be prepared by dissolving, dispersing, etc., a compounds) of
2 0 Formula I (about 0.5% to about 20%), or a pharmaceutically acceptable
salt thereof, and optional pharmaceutical adjuvants in a carrier, such
as, for example, water, saline, aqueous dextrose, glycerol, ethanol and
the like, to thereby form a solution or suspension.
2 5 If desired, a pharmaceutical composition of the invention may
also contain minor amounts of auxiliary substances such as wetting or
emulsifying agents, pH buffering agents, antioxidants, and the like,
such as, for example, citric acid, sorbitan monolaurate, triethanol-
amine oleate, butylated hydroxytoluene, etc.
Actual methods of preparing such dosage forms are known, or
will be apparent, to those skilled in this art; for example, see
Rernington's Phcarmacezctical Sciences, 18th Edition, Mack Publishing
Company, Easton, Pennsylvania ( 1990). The composition to be
3 5 administered will, in any event, contain a therapeutically effective
X93178
- 64 -
amount of a compound of Formula I or a pharmaceutically acceptable
salt thereof, for treatment of a disease-state alleviated by the
inhibition of matrix metalloprotease activity in accordance with the
teachings of this invention.
The compounds of Formula I or their pharmaceutically acceptable
salts, are administered in a therapeutically effective amount which
will vary depending upon a variety of factors including the activity of
the specific compound employed, the metabolic stability and length of
action of the compound, the age, body weight, general health, sex,
diet, mode and time of administration, rate of excretion, drug
combination, the severity of the particular disease-state, and the host
undergoing therapy. Generally, a therapeutically effective daily dose
is from about 0.014 mg to about 14.3 mg/kg of body weight per day
of a compound of Formula I or a pharmaceutically acceptable salt
thereof; preferably, from about 0.07 mg to about 5 mg/kg of body
weight per day; and most preferably, from about 0.14 mg to about
1.4 mg/kg of body weight per day. For example, for administration to
a 70 kg person, the dosage range would be from about 1 mg to about
2 0 1.0 gram per day of a compound of Formula I or a pharmaceutically
acceptable salt thereof, preferably from about 5 mg to about 300 mg
per day, and most preferably from about 10 mg to about 100 mg per
dav.
~g~~~8
- 65 -
EXAMPLES
The following preparations and examples are given to enable
those skilled in the art to more clearly understand and to practice the
present invention. They should not be considered as limiting the
scope of the invention, but merely as being illustrative and
representative thereof.
EXAMPLE 1
1 0 Preparation of Compounds of Formula ( 1 )
1 A. Preparation of ( 1 ) where R3 and R4 when taken together with the
Carbon to which they are attached represent N-CBZ-piperidine
1. A solution of benzyl chloroformate (35 ml, 247 mmol) in
tetrahydrofuran (70 ml) was added to an ice-cold solution of
4-hydroxypiperidine (25 g, 247 mmol) and triethylamine (45 ml, 321
mmol) in tetrahydrofuran (350 ml). The mixture was stirred
overnight at room temperature and the solvent removed under
reduced pressure. The residue was partitioned between 5%
2 0 hydrochloric acid and ethyl acetate, and the organic layer washed
with brine, dried over magnesium sulfate, and the solvent removed
under reduced pressure to give 4-hydroxy-N-CBZ-piperidine as a pale
yellow oil.
2 5 2. Celite (66 g) was added to a solution of 4-hydroxy-N-CBZ-
piperidine ( 18 g, 76.5 mmol) in methylene chloride (500 ml), followed
by pyridinium chlorochromate (33 g, 153 mmol). The mixture was
stirred overnight, and then isopropyl alcohol ( 12 ml) was added over
a period of 3 hours. The reaction mixture was filtered through silica
3 0 gel and the filter cake was repeatedly rinsed with methylene chloride
and ethyl acetate. The combined filtrates were evaporated under
reduced pressure. Silica gel chromatography using 50% ethyl
acetate/hexane, gave 4-oxo-N-CBZ-piperidine as a yellow oil.
~~3~7g
- 66 -
EXAMPLE 2
Preparation of Compounds of Formula (3)
2A. Preparation of (3) where R2 is H,~gen, and R3 and R4 when
taken together with the Carbon to which then are attached represent
N-CBZ-piperidine
tert-(Butoxycarbonylmethylene)triphenylphosphorane (28 g, 74.4
mmol) was added to 4-oxo-N-CBZ-piperidine ( 14.2 g, 61.3 mmol) in
benzene ( 150 ml), and the solution was stirred at reflux overnight.
The solution was concentrated, and the residue triturated with hexane
(500 ml). Filtration and concentration of the filtrate gave 4-tert-
butoxycarbonyl-methylene-N-CBZ-piperidine as a colorless oil.
2B. Preparation of (3), varying-R2 R3, and R4
1 5 Similarly, following the procedures of Example 2A above, but
replacing 4-oxo-N-CBZ-piperidine with:
formaldehyde;
acetone;
2 0 propionaldehyde;
cyclopentanone;
cyclohexanone;
1,4-cyclohexanedione mono-ethylene ketal;
4-methylcyclohexanone;
2 5 phenylacetaldehyde;
4-(biphen-4-yl)butyraldehyde;
cyclopentylacetaldehyde;
tetrahydropyranone; and
tetrahydrothiopyran;
3 0 and optionally replacing tert-(butoxycarbonylmethylene)triphenyl-
phasphorane with:
tert-butyl-3-phenylpropionate-2-triphenylphosphorane;
tert-butyl-propionate-2-triphenylphosphorane; and
tert-butyl-3-methylpropionate-2-triphenylphosphorane;
3 5 the following compounds of Formula (3) were prepared:
~~3~7g
- 67 -
1-(tert-butoxycarbonyl)-1-benzylethene;
1-(tert-butoxycarbonyl)-2,2-dimethylethene;
1-(tert-butoxycarbonyl)-1-methyl-2-ethylethene;
tert-butoxycarbonylmethylenecyclopentane;
tert-butoxycarbonylmethylenecyclohexane;
tert-butoxycarbonylmethylene-4-methylcyclohexane;
1-(tert-butoxycarbonyl)-2-benzylethene;
1-(tert-butoxycarbonyl)-1-isopropyl-2-benzylethene;
1-(tert-butoxycarbonyl)-2-[3-(biphen-4-yl)]propylethene;
1 0 1-(tert-butoxycarbonyl)-2-cyclopentylmethylethene;
4-(tert-butoxycarbonylmethylene)-tetrahydropyran; and
4-(tert-butoxycarbonylmethylene)-tetrahydrothiopyran.
2C. Preparation of ~3), var,~g R2 R3, and R4
1 5 Similarly, following the procedures of Example 2A above, but
optionally replacing 4-oxo-N-CBZ-piperidine with other compounds of
Formula (1), and optionally replacing (tert-butoxycarbonyl-
methylene)triphenyl-phosphorane with other compounds of Formula
(2), other compounds of Formula (3) are prepared.
EXAMPLE 3
Preparation of Compounds of Formula ~4)
3A. Preparation of (4) where R2 is HXdrogen, and R3 and R4 when
taken together with the Carbon to which they are attached represent
2 5 N-CBZ-piperidine, a Compound of Formula (4a)
Trifluoroacetic acid (10 ml) was added to 4-tert-butoxycarbonyl-
methylene-N-CBZ-piperidine (20 g, 60.3 mmol) in methylene chloride
(30 ml) and the solution was stirred at room temperature for 1.5
3 0 hours. After evaporation of the solvent, the residue was triturated
with diethyl ether to give 4-carboxymethylene-N-CBZ-piperidine as a
crystalline white solid.
~93"~78
- 68 -
3B. Preparation of ~4) where R2 is H~gen and R3 and R4 when
taken together with the Carbon to which their are attached represent
Tetrah~pyran a Compound of Formula (4b~
Methanol (204 ml) was slowly added to a suspension of sodium
hydride (5.48 g, 228.2 mmol) in tetrahydrofuran (204 ml) at 0°C.
When addition was complete, trimethylphosphonoacetate (34.22 ml,
211..4 mmol) was added to the mixture at such a rate as to maintain
the temperature below 12°C. Stirring was continued for a further 10
minutes. To this reaction mixture was added a solution of 2,3,5,6-
tetrahydropyran-4-one ( 16.28 g, 163.0 mmol) in tetrahydrofuran (20
ml), keeping the temperature below 30°C. After the addition was
complete, stirring was continued for 30 minutes at room temperature,
then methanol ( 100 ml) and 2M sodium hydroxide (326 ml) was
added, and the mixture stirred overnight at room temperature. The
resulting solution was concentrated to one half of the original volume,
and acidified to pH 1.2 with 6M hydrochloric acid ( 108 ml). The
reaction mixture was partitioned between ethyl acetate and water,
the combined organic extracts dried over magnesium sulfate, and
2 0 solvent removed under reduced pressure to give 4-(carboxy-
methylene)-2,3,5,6-tetrahydropyran (22.62 g), which was used with
no further purification.
3C. Preparation of (4), varvin~R,2 R3, and R4
Similarly, following the procedures of Example 3A above, but
replacing 4-(tert-butoxycarbonylmethylene)-N-CBZ-piperidine with
other compounds of Formula (3), the following compounds of Formula
(4) were prepared:
1-benzyl-1-carboxyethene;
1-carboxy-2,2-dimethylethene;
1-carboxy-2-ethyl-1-methylethene;
carboxymethylenecyclopentane;
3 5 carboxymethylenecyclohexane;
~93~~"g
- 69 -
carboxymethylene-(4-methylcyclohexane);
4-carboxymethylenecyclohexanone mono-ethylene ketal;
2-benzyl-1-carbox yethene;
2-[3-(biphen-4-yl)propyl]-1-carboxyethene;
2-benzyl-1-carboxy-1-isopropylethene;
1-carboxy-2-cyclopentylmethylethene;
4-carboxymethylene-tetrahydrothiopyran; and
4-carboxymethylene-(tetrahydrothiopyran-1,1-dioxide).
1 0 3D. Preparation of (4), var, ing_ R2 R3, and R4
Similarly, following the procedures of Example 3A above, but
replacing 4-(tert-butoxycarbonylmethylene)-N-CBZ-piperidine with
other compounds of Formula (3), other compounds of Formula (4) are
prepared, or may be prepared by means well known to those skilled
in the art. Alternatively, they are commercially available, for
example, 1-cyclopentene carboxylic acid and 1-cyclohexene carboxylic
acid are available from Lancaster Synthesis Inc.
2 0 EXAMPLE 4
Preparation of Compounds of Formula ~5)
4A. Preparation of (5) where RS is 4-Phenox py= hen~l
A solution of sodium thiomethoxide (25 g) and 4-bromodiphenyl
2 5 ether (25 g) in N, N-dimethylformamide (DMF) ( 150 ml) was refluxed
overnight. The mixture was cooled and added to dilute aqueous
sodium hydroxide. The water layer was washed with ether to remove
by-products and acidified with hydrochloric acid. The product, 4-
(phenoxy)thiophenol, was extracted with ether, and the ether layer
3 0 dried and evaporated to give 4-(phenoxy)thio-phenol ( 19-20 g) as a
red oil. This material can be used without further purification.
-70-
4B. Alternative Preparation of ~5) where RS is 4-(4-Bromo-
phenox~)phen~
A solution of 4-bromodiphenyl ether (50 g, 200.7 mmol) in
methylene chloride ( 118 ml) was cooled to 0°C and chlorosulfonic acid
( 14.7 ml, 220.8 mmol) was added dropwise over a 20 minute period.
The solution was stirred an additional 10 minutes, warmed to room
temperature and stirred an additional 1 hour. To this mixture was
added oxalyl chloride (23.6 ml, 270.9 mmol), followed by N, N-
1 0 dimethylformamide ( 1.5 ml) as a catalyst, and the mixture refluxed
for 2 hours. The mixture was cooled to room temperature, and
additional oxalyl chloride (23.6 ml, 270.9 mmol) was added, the
mixture refluxed for 3 hours, cooled to room temperature and stirred
12 hours more. The solution was concentrated to an oil, azeotroped
several times using methylene chloride and put under high vacuum
( 1 torr) for several hours until the mixture had completely solidified.
This mixture was immediately dissolved in methylene chloride ( 160
ml) which was added dropwise to a solution of triphenylphosphine
( 157.0 g, 602 mmol) in methylene chloride ( 160 ml) containing N, N-
2 0 dimethylformamide (4 ml, 52.2 mmol). The mixture was stirred 2
hours, diluted with 1M aqueous hydrochloric acid (300 ml) and
stirred for 1 hour. The aqueous layer was separated, extracted with
methylene chloride (200 ml), and the organic layers were combined,
washed with 200 ml of brine, dried (MgS04) and concentrated in
2 5 vacuo. The resulting solid was further purified through trituration
with 750 ml of hexane. The solid was then dissolved in 750 ml of
diethyl ether, extracted with 2M aqueous sodium hydroxide (2 x 350
ml), and the basic aqueous layer back extracted using diethyl ether
(2 x 400 ml). The aqueous layer was adjusted to pH 2, extracted
3 0 with diethyl ether (3 x 200 ml) and the combined organic layers dried
(MgS04) and concentrated to afford 4-(4-bromophenoxy)thiophenol
(45.6 g, 81%). 1HNMR (CDC13) 8 3.43 (s, 1H), 6.86 (d, J = 8.9 Hz, 2H),
6.89 (d, J = 8.6 Hz, 2H), 7.28 (d, J = 8.6 Hz, 2H), 7.43 (d, J = 8.9 Hz, 2H).
~'~9~~~8
- 71 -
The corresponding 4-chloro and 4-fluoro analogues were obtained
in similar fashion from the corresponding commercially available
4-halodiphenylethers, respectively.
4-(4-chlorophenoxy)thiophenol: 1 HNMR (CDC13) S 3.43 (s, 1H),
6.90 (mc, 4H), 7.27 (mc, 4H).
4-(4-fluorophenoxy)thiophenol: 1HNMR (CDC13) 8 3.41 (s, 1H),
6.85 (d, J = 8.7 Hz, 2H), 7.00 (mc, 4H), 7.26 (d, J = 8.7 Hz, 2H).
4-(4-pyridyloxy)thiophenol: 1 HNMR (CDC13 ) b 7.05 (d, J =9.0 Hz,
2H), 7.29 (d, J = 7.3 Hz, 2H), 7.44 (d, J = 8.8 Hz, 2H), 8.70 (d, J = 7.3 Hz,
1 0 2H); EIMS (M+): 203.
4-(5-chloro-2-pyridyloxy)thiophenol: 1HNMR (CDC13) 8 6.87 (d, J
=8.5 Hz, 1H), 7.01 (d, J = 8.7 Hz, 2H), 7.32 (d, J = 8.7 Hz, 2H), 7.63 (d,
J = 8.6 Hz, 1 H), 8.15 (d, J = 2.8 Hz, 1 H).
1 5 EXAMPLE 5
Preparation of Compounds of Formula ( 10)
SA. Preparation of a Compound of Formula (8) where R1 and R2 taken
together with the Carbon to which they are attached represent
2 0 Tetrah~pyran, a Compound of Formula (8a)
A solution of 1.SM diisobutylaluminum hydride (DIBAL-H) (419
ml, 629 mmol) in toluene was added to a 3-L Morton flask equipped
with a nitrogen gas inlet, mechanical stirrer, low temperature
2 5 thermometer, 500 ml pressure equalizing funnel, and containing
tetrahydropyran-4,4-dicarboxylic acid diethyl ester (70.78 g, 307.4
mmol) in toluene (600 ml) at -40°C, at a rate to maintain an internal
temperature no higher than -25°C. The mixture was stirred an
additional 10 minutes and anhydrous ethanol (595 ml) was added
3 0 dropwise over 20 minutes maintaining an internal temperature no
higher than -15°C. Solid sodium borohydride (11.6 g, 307.4 mmol)
was added in three portions over a 15 minute period, the cooling bath
wa<.> removed, the mixture allowed to warm to room temperature over
1 hour, and saturated aqueous sodium sulfate (325 ml) added over 15
3 5 minutes. The mixture was cooled to -15°C, ethyl acetate (250 ml)
was
~193~7'8
- 72 -
added, and the flocculent white precipitate filtered over a pad of
celite. The celite pad was washed with ethyl acetate (7 x 450 ml), the
filtrate washed with brine (200 ml), dried over magnesium sulfate,
and concentrated in vacuo. The residue was dissolved in the minimum
amount of ethyl acetate, filtered through a sintered glass funnel
containing silica gel (40 g), eluting with ethyl acetate, and the filtrate
concentrated in vacuo to afford the hydroxyester, 4-(hydroxy-
methyl)tetrahydropyran-4-carboxylic acid ethyl ester, as a pale
yellow oil (48.5 g, 84%).
SB. Alternative Preparation of a Compound of Formula (8) where R1
and R2 taken together with the Carbon to which then are attached
represent Tetrahvdrop
1. To a solution of tetrahydropyran-4,4-dicarboxylic acid
diethyl ester (400 mg, 1.74 mmol) in N, N-dimethylformamide (4 ml),
was added lithium iodide ( 1.16 g, 8.66 mmol), followed by sodium
cyanide (94 mg, 1.91 mmol). The mixture was heated at 130°C for 7
hours, 140°C for 25 hours, after which GC analysis indicated the
2 0 reaction to be >95% complete. The mixture was partitioned between
33°o diethyl ether/hexanes (100 ml) and brine (25 ml). The organic
layer was washed with additional brine (25 ml), dried (MgS04) and
concentrated in vacaio to afford the tetrahydropyran-4-carboxylic
acid ethyl ester (253 mg, 92%). Note: Substitution of 2 equivalents of
2 5 sodium acetate for 1.1 equivalents of sodium cyanide in this reaction
and heating 12 hours longer provides identical results.
2. Lithium diisopropylamide was prepared by the addition of
2.SM N-butyl lithium (30.3 ml, 75.6 mmol) in hexanes to a solution of
3 0 diisopropylamine ( 10.6 ml, 75.6 mmmol) in tetrahydrofuran (244 ml)
at 0°C and stirring for 20 minutes. Then a solution of tetrahydro-
pyran-4-carboxylic acid ethyl ester ( 10 g, 63.2 mmol) in tetrahydro-
furan (50 ml) was added to the solution of lithium diisopropylamide
over 15 minutes at -78°C. The resulting solution was stirred an
3 5 additional 50 minutes, and solid paraformaldehyde (10 g) was added
~193~78
- 73 -
in one portion. The mixture was slowly allowed to warm to room
temperature over 9 hours, diluted with 2M aqueous hydrochloric
acid ( 100 ml), and filtered over a pad of celite pad which was washed
with diethyl ether (2 x 200 ml). The aqueous layer of the filtrate was
washed with additional portions of diethyl ether (2 x 200 ml). The
combined organic layers were washed once with 2M aqueous
hydrochloric acid ( 100 ml), saturated aqueous sodium bicarbonate
( 100 ml), dried over magnesium sulfate, and concentrated in vacuo to
afford a slightly impure product 4-(hydroxymethyl)tetrahydropyran
4-carboxylic acid ethyl ester (11.5 g, 97°Io), which was taken into the
next reaction without further purification. IR (neat) 3433 (br), 1726
cmw 1; 1 HNMR (CDC13) b 1.30 (t, J = 7.1 Hz, 3H), 1.57 (ddd, J = 13.8,
10.1, 4.4 Hz, 2H), 2.07 (dm, J = 13.8 Hz, 2H), 2.30-2.45 (br s, 1H), 3.56
(ddd, J = 11.9, 10.3, 2.7 Hz, 2H), 3.66 (s, 2H), 3.82 (dt, J = 11.9, 4.2 Hz,
1 5 2H), 4.24 (q, J = 7.2 Hz, 2H); 13CNMR (CDC13) 8 14.25 (q), 30.54 (t),
46.63 (s), 61.04 (t), 64.79 (t), 69.02 (t), 175.24 (s); HRMS Calcd for
C9H 1604: 188.1049. Found: 188.1053.
SC. Preparation of a Compound of Formula (8) where R1 and R2 taken
2 0 together with the Carbon to which then are attached represent
Phi eridine a Compound of Formula (8)
Lithium diisopropylamide was prepared by the addition of 1.6M
N-butyl lithium (29.1 ml, 46.6 mmol) in hexanes to a solution
2 5 diisopropylamine (6.5 ml, 46.6 mmmol) in tetrahydrofuran ( 150 ml)
at ~°C with stirring for 20 minutes at -78°C. Then a solution of
neat N-
(tort-butoxycarbonyl)-piperidine-4-carboxylic acid ethyl ester (10 g,
38.9 mmol) was added over 5 minutes, and the resulting solution
was stirred an additional 50 minutes. Solid paraformaldehyde (13.5 g.
3 0 155.4 mmol) was added in one portion, and the mixture slowly
allowed to warm to room temperature over 9 hours. The mixture was
diluted with 2M aqueous hydrochloric acid ( 100 ml), filtered over a
pa<i of celite, washed with diethyl ether (2 x 200 ml). The combined
organic layers were washed once with 2M aqueous hydrochloric acid
3 5 ( 100 ml), saturated aqueous sodium bicarbonate ( 100 ml), dried over
~~3~'~
- 74 -
magnesium sulfate, and concentrated in vacuo. Chromatography on
silica gel, and eluting with 50% ethyl acetate/hexanes, yielded slightly
impure N-(tert-butoxycarbonyl)-4-(hydroxymethyl)piperidine-4-
carboxylic acid ethyl ester (10.57 g, 95%) as a pale yellow oil which
was taken immediately into the hydrolysis reaction (LiOH): 1 H NMR
(CI)C13) b 1.26 (t, J = 7.4 Hz, 3H), 1.40-1.53 (m, 2H), 1.46 (s, 9H), 2.00-
2.12 (m, 2H), 3.05-3.16 (m, 2H), 3.65 (s, 2H), 3.70-3.83 (m, 2H), 4.23
(q, .1 = 7.2 Hz, 2H).
1 0 SD. Preparation of a Compound of Formula (9) where R1 and R2 taken
together with the Carbon to which they are attached represent
TetrahXdropyran a Compound of Formula (9a)
Lithium hydroxide monohydrate ( 16.7 g, 398.5 mmol) was added
to a solution of 4-(hydroxymethyl)tetrahydropyran-4-carboxylic acid
ethyl ester (25.0 g, 132.8 mmol) in 4.5:1 methanol/water (220 ml).
ThE; mixture was heated to reflux for 40 minutes and the methanol
removed in vacuo by concentration using a bath temperature no
higher than 45°C. The aqueous layer was then extracted into diethyl
2 0 ether (4 x 100 ml) and the combined ether layers washed twice with
2M sodium hydroxide ( 15 ml). The combined aqueous base layers
were cooled to 0°C, acidified to pH 3.0 with 8M aqueous hydrochloric
acid, saturated with solid sodium chloride and extracted with ethyl
acetate (8 x 250 ml). The combined organic layers were dried over
2 5 magnesium sulfate, concentrated in vacuo. The white fluffy powder
residue was recrystallized from the minimum amount of methylene
chloride/hexanes to afford pure 4-(hydroxymethyl)tetrahydropyran-
4-carboxylic acid (17.05 g, 80%).
3 0 SE. Alternative Preparation of a Compound of Formula (91 where R1
anti R2 taken together with the Carbon to which thev are attached
represent Tetrah~drop
Lithium diisopropylamide was prepared by the addition of 2.45M
3 5 N-butyl lithium ( 16.5 ml) in hexanes to a solution diisopropylamine
X193178
- 75 -
(5.80 ml, 41.4 mmmol) in tetrahydrofuran (40 ml) at 0°C with
stirring for 20 minutes. Then a solution of tetrahydropyran-4-
carboxylic acid (2.5 g, 19.2 mmol) in tetrahydrofuran ( 10 ml) was
added to the solution of lithium diisopropylamide over 15 minutes to
form a slurry, followed by hexamethylphosphoramide (2 ml). The
resulting solution was stirred for 25 minutes, then immediately
warmed to room temperature after a stream of gaseous formaldehyde
(prepared by heating 4 g of paraformaldehyde at 175-200°C over 5-
minutes) was passed through the solution. The slurry was
10 carefully concentrated at ambient temperature, acidified to pH 3 with
8M hydrochloric acid, saturated with solid sodium chloride, and
extracted with ethyl acetate (8 x 100 ml). The combined organic
layers were dried over magnesium sulfate, concentrated in vacuo.
Chromatography over silica gel (80 g), and eluting with 10%
methanol/methylene chloride, yielded 4-(hydroxymethyl)tetrahydro-
pyran-4-carboxylic acid as a white solid (1.80 g, 58%). mp 113.7-
115°C; IR (KBr) 3420 (br), 1724 cm-1, 1HNMR (DMSO-d6) 8 1.43 (ddd,
J =: 13.5, 11.0, 4.4 Hz, 2H), 1.85 (dm, J = 13.4 Hz, 2H), 3.37 (td, J =
11.3, 3.0 Hz, 2H), 3.43 (s, 2H), 3.71 (dt, J = 11.6, 3.9 Hz, 2H), 4.81 (br, s,
2 0 1H); 12.24 (s, 1H); 13CNMR (DMSO-d6) 8 30.42 (t), 46.38 (s), 64.35 (t),
68.15 (t), 69.02 (t), 176.08 (s); HRMS Calcd. for C7H 1204: 160.0735.
Found: 160.0731. Anal. Calcd. for C7H 1203: C, 52.49; H, 7.55. Found: C,
52.50; H, 7.62.
2 5 5F. Preparation of a Compound of Formula (9) where R1 and R2 taken
together with the Carbon to which they are attached represent
Pi~eridine a Compound of Formula f 9b)
Lithium hydroxide monohydrate (6.95 g, 165.6 mmol) was added
3 0 to solution of N-(tert-butoxycarbonyl)-4-(hydroxymethyl)piperidine-
4-carboxylic acid ethyl ester (9.52 g, 33.1 mmol) in 2:1 methanol/
water (100 ml). The mixture was heated to reflux for 30 minutes, the
methanol removed in vacuo by concentration using a bath
temperature no higher than 45°C. The aqueous layer was cooled to
3 5 0°C, acidified to pH 3.0 using 6M aqueous hydrochloric acid, and
~1g~~78
- 76 -
extracted with ethyl acetate (4 x 75 ml). The combined organic layers
were dried over magnesium sultate, and concentrated in vacuo, and
recrystallized from dichloromethane/hexanes to afford N-(tert-
butoxycarbonyl)-4-(hydroxymethyl)piperidine-4-carboxylic acid
(8.59 g, 100%).
5G. Alternative Preparation of a Compound of Formula (9) where R1
and R2 taken together with the Carbon to which they are attached
represent Piperidine
Lithium diisopropylamide was prepared by the addition of 2.45M
N-butyllithium (69 ml, 168.8 mmol) in hexanes to a solution
diisopropylamine (24 ml, 171.2 mmmol) in tetrahydrofuran (40 ml)
at 0°C with stirring for 20 minutes. Then a solution of N-(tert-
1 5 butoxycarbonyl)-piperidine-4-carboxylic acid ( 18 g, 78.5 mmol) in
tetrahydrofuran (35 ml) was added to the solution of lithium
diisopropylamide over 15 minutes to form a slurry, followed by
hexamethylphosphoramide (2 ml). The resulting solution was stirred
for 25 minutes, then stream of gaseous formaldehyde (prepared by
2 0 heating paraformaldehyde (16.4 g, 189 mmol) at 175-200°C over 5-
10 minutes) was passed through the solution, which was allowed to
immediately warm to room temperature. The slurry was concentrated
at ambient temperature, acidified to pH 4 with 6M hydrochloric acid,
saturated with solid sodium chloride, and extracted with ethyl acetate
2 5 (8 x 100 ml). The combined organic layers were dried over
magnesium sulfate, concentrated in vacuo. Chromatography over silica
gel, and eluting with 1 % methanol/ methylene chloride, afforded
N-(tert-butoxycarbonyl)-4-(hydroxymethyl)piperidine-4-carboxylic
acid as a white solid (4 g, 20%). mp 156.6-157.3 °C; 1HNMR (DMSO-
3 0 d6) 8 1.25-1.37 (m, 2H), 1.38 (s, 9H), 1.85 (dm, J = 13.7 Hz, 2H), 2.78-
2.94 (br m, 2H), 3.41 (s, 1H), 3.70 (dm, J = 12.8 Hz, 2H), 4.87 (br s,
1H), 12.34 (s, 1H); Anal. Calcd. for C12H21N05~ C, 55.58; H, 8.16; N,
5.40. Found: C, 55.72; H, 8.10; N, 5.53.
~93~78
_ 77 _
SH. Preparation of ( 10) where R 1 and R2 taken together with the
Carbon to which they are attached represent Tetrah~pyran, a
Compound of Formula ( 10a)
Trifluoromethanesulfonic anhydride ( 11.1 ml, 66.2 mmol),
followed by triethylamine ( 17.8 ml, 127.4 mmol) was added to a
slurry of 4-(hydroxymethyl)tetrahydropyran-4-carboxylic acid
( 10.20 g, 63.68 mmol) in anhydrous diethyl ether cooled to 0°C ( 115
ml). The biphasic solution was stirred for 20 hours, warmed to room
1 0 temperature, stirred an additional 2 hours. The layers were
separated by decantation, and the lower layer diluted with 2%
aqueous sodium bicarbonate solution (50 ml) and extracted with
met:hylene chloride (4 x 200 ml). The combined organic extracts were
washed with additional 2% aqueous sodium bicarbonate ( 100 ml),
dried over magnesium sulfate, and concentrated in vacuo to afford
2,7~-dioxa-spiro[3.5]nonane-1-one as a pale yellow oil (10.8 g). IR
(KI3r) 1821 cm-l; 1HNMR (CD3C13) b 1.92 (ddd, J = 13.4, 8.1, 4.0 Hz,
2H;1, 2.10 (dddd, J = 13.4, 6.1,3.4, 0.8 Hz, 2H), 3.70 (ddd, J = 11.8, 6.3,
3.9 Hz, 2H), 3.92 (ddd, J = 11.8, 7.9, 3.4 Hz, 2H), 4.15 (s, 2H); 13 C N M R
2 0 (CD3C13) b 30.78 (t), 55.78 (s), 64,46 (t), 71.50 (t), 173.42 (s), MS(EI)
m/E:=142. MS(CI) M+ =H m/e=143, M+ +HNH4 m/e=160.
5 I . Preparation of a Compound of Formula ( 10~ where R 1 and R2
taken together with the Carbon to which then are attached represent
2 5 Piperidine a Compound of Formula ( l Ob)
Trifluoromethanesulfonic anhydride (2.60 ml, 15.39 mmol),
followed by triethylamine (4.30 ml, 30.78 mmol) was added to a
slurry of N-(tert-butoxycarbonyl)-4-hydroxymethylpiperidine-4-
3 0 carboxylic acid (3.80 g, 14.65 mmol) in anhydrous diethyl ether (27
ml;~ cooled to 0°C. The biphasic solution was stirred for 23 hours,
warmed to room temperature, stirred an addition 1 hour, and the
upper diethyl ether layer separated by decantation. The lower was
extracted with additional portions of diethyl ether (2 x 100 ml), and
3 5 the combined organic extracts washed with aqueous sodium
~1~.~~Tg
_ 78 _
bicarbonate solution (2 x 50 ml), dried over magnesium sulfate, and
concentrated in vacuo to afford 7-(butoxycarbonyl)-2-oxa-
7-araspiro[3.5]nonan-1-one as a pale yellow oil (2.88 g, 82%). 1HNMR
(CI)C13) 8 1.48 (s, 9H), 1.79-1.89 (m, 2H), 2.02-2.10 (m, 2H), 3.48-3.66
(m, 4H), 4.13 (s, 2H).
EXAMPLE 6
Preparation of a Compound of Formula (13~
1 0 6A. Preparation of (13) where Rl and R2 taken together with the
Carbon to which they are attached represent Tetrah~pyran, and X
is Iodo
Lithium diisopropylamide was prepared by the addition of 2.SM
1 5 N-butyl lithium (5.6 ml, 13.9 mmol) in hexanes to a solution of
diisopropylamine ( 1.95 ml, 13.9 mmmol) in tetrahydrofuran (30 ml)
at 0°C with stirring for 20 minutes. Then a solution of tetrahydro-
pyran-4-carboxylic acid ethyl ester (2 g, 12.7 mmol) in tetrahydro-
furan (8 ml) was added to the solution of lithium diisopropylamide at
2 0 a temperature of -78°C over 15 minutes. The resulting solution was
stirred an additional 50 minutes, and diiodomethane ( 1. l4ml, 14.2
mmol) was added. The resulting mixture was stirred an additional 50
minutes, warmed to room temperature over 30 minutes, then
recooled to 0°C. The mixture was diluted with 1M aqueous
2 5 hydrochloric acid (25 ml), extracted with diethyl ether (2 x 100 ml),
and washed with additional portions of diethyl ether (2 x 50 ml).
The combined organic layers were washed once with 1M aqueous
hydrochloric acid ( 100 ml), saturated aqueous sodium bisulfite ( 100
ml;l, saturated aqueous sodium bicarbonate ( 100 ml), and dried over
3 0 magnesium sulfate, and concentrated in vacuo. The residue was
filtered over a plug of silica gel, eluting successively .with hexanes and
ethyl acetate, removing excess alkylating agent with the hexane wash,
to afford pure 4-(iodomethyl)tetrahydropyran-4-carboxylic acid ethyl
ester as a pale yellow oil which was taken directly into the next
3 5 reaction without further purification (3.20 g, 85%). IR (KBr) 1732
19~~~~
- 79 -
cmw l ; 1 HNMR (CDC13 ) 1.31 (q, J = 7.3 Hz, 3H), 1.56 (ddd, J = 14.6, 10.9,
4.5, 2H), 2.17 (ddd, J = 14.6, 5.7, 3.3, 2H), 3.31 (s, 2H), 3.51 (ddd, J =
11.'7, 11.1, 2.5 Hz, 2H), 3.51 (td, J = 11.7, 4.3 Hz, 2H), 4.24 (q, J = 7.1
Hz, 2H); 13CNMR (CDC13) b 14.33 (q), 15.04 (t), 34.70 (t), 45.26 (s),
61.:34 (t), 65.22 (t), 172.89 (s); EIHRMS Calcd. for C9H 15I03 (M+):
298.0066. Found: 298.0066. Anal. Calcd. for C9H 15I03: C, 36.26; H,
5.0'7. Found: C, 36.56; H, 5.09.
6B. Preparation of (137 where Rl and R2 taken together with the
Carbon to which they are attached represent Tetrah~ropyran, and
Var iy ng XX
Similarly, replacing diiodomethane with dibromomethane or
bromochloromethane, the following compounds of Formula (13) were
prepared:
4-(bromomethyl)tetrahydropyran-4-carboxylic
acid ethyl ester:
IR (neat) 1732 cm-1; 1 HNMR (CDC13 ) 1.30 (q, J = 7.1 Hz, 3H),
1.59
(ddd, J = 14.6, 10.9, 4.5, 2.17 (dm, J = 14.7, 2H), 3.48 (s,
2H), 2H), 3.53
2 0 (dt" J = 11.9, 4.5 Hz, (dt, J = 11.9, 4.5 Hz, 2H), 4.23
2H), 3.84 (q, J = 7.1
Hz, 2H); 13CNMR (CDC13) 8 14.27 (q), 33.17 (t), 40.16 (t),
46.05 (s),
61.29 (t), 64.97 (t), 172.91 CIMS (M+ + H): 251, (M+ + NH4+)
(s); 268.
4-(chloromethyl)tetrahydropyran-4-carboxylic
acid ethyl ester:
IR (neat) 1734 cm- l ; 1 HNMR (CDC13 ) 1.30 (q, J = 7.1 Hz, 3H),
1.59
2 5 (ddd, J = 14.6, 10.9, 4.5,2.16 (dm, J = 14.7, 2H), 3.53 (dt,
2H), J = 11.9,
4.5 Hz, 2H), 3.61 (s, 2H), (dt, J = 11.7, 4.3 Hz, 2H), 4.24
3.84 (q, J = 7.1
Hz, 2H); 13CNMR (CDC'.13) b 14.24 (q), 32.14 (t), 46.69 (s),
51.40 (t),
61.29 (t), 64.85 (t), 173.01 CIMS (M+ + H): 207. Anal. Calcd.
(s); for
C9H15C1O3: C, 52.31; H, 7.32 . Found: C, 52.51; H, 7.30.
6C. Alternative Preparation of a Compound of Formula (13) where Rl
and R2 taken together with the Carbon to which they are attached
re~>resent Tetrahydropyran, and X is p-Tosyl
~19~~~8
-so-
To a solution of tetrahydropyran-4-carboxylic acid ethyl ester
(820 mg, 4.356 mmol) in pyridine ( 10 ml) at 0°C, was added
p-toluenesulfonyl chloride (997 mg, 5.23 mmol), and the mixture
allowed to warm to room temperature over 1 hour period. The
mixture was stirred 36 hours and partitioned between methylene
chloride (150 ml) and 3N aqueous hydrochloric acid (50 ml). The
organic layer was washed with 25 ml of saturated aqueous sodium
bicarbonate, dried (MgS04), concentrated and the residue
chromatographed over 45 g of silica gel, eluting with 30% ethyl
1 0 acetate/hexanes, to afford the tosylate as a white solid ( 1.03 g, 69%).
mp 87.7-88.6 °C; IR (KBr) 1717 cm-1; 1 NMR (CDC13 ) 8 1.21 (q,
J = 17.1 Hz, 3H), 1.52 (ddd, J = 13.4, 10.6, 4.1 Hz, 2H), 2.00 (dm,
J = 13.4 Hz, 2H), 2.46 (s, 3H), 3.49 (ddd, J = 11.7, 10.6, 2.5 Hz, 2H),
3.76 (dt, J = 11.9, 4.1 Hz, 2H), 4.03 (s, 2H), 4.13 (q, J = 7.1 Hz, 2H), 7.35;
1 5 13C NMR (CDC13) b 14.10 (q), 21.67 (q), 30.43 (t), 44.93 (s), 61.37 (t),
64.43 (t), 74.65 (t), 127.95 (d), 129.89 (d), 132.67 (s), 145.05 (s),
172.57 (s); HRMS Calcd for C16H2206~ 343.1215. Found: 343.1217.
Anal. Calcd. for C16H22(J6: C, 56.12; H, 6.48. Found: C, 56.22; H, 6.46.
2 0 EXAMPLE 7
Preparation of Compounds of Formula Ia
7A. Preparation of Ia where R1 and R2 are Hero eg nR3 and R4
when taken together with the Carbon to which they are attached
2 5 represent Piperidine, and RS is Diphenylether, from a Compound of
Formula (4)
1. 4-Phenoxythiophenol (7.4 g, 36.3 mmol), 4-carboxy
methylene-N-CBZ-piperidine (10 g, 36.3 mmol) and piperidine (1.8
3 0 ml, 36.3 mmol) were stirred overnight at 100-110°C in a sealed
flask.
After cooling, the crude reaction mixture was partitioned between
ethyl acetate and 1 N hydrochloric acid, the organic layer was washed
with brine, dried over magnesium sulfate, filtered, and concentrated
in vacuo to give a yellow solid. The solid was triturated in l:l (v/v)
3 5 ethyl ether/hexane (500 ml) to give 2-[4-(4-phenoxyphenylthio)-N-
~~.~93~7~
- sl -
CBZ-piperidin-4-yl]-acetic acid as a white solid.
2. A solution of 2-[4-(4-phenoxyphenylthio)-N-CBZ-piperidin-
4-yl)]-acetic acid (150 mg, 0.29 mmole) in dry 1,2 dichloroethane (3
ml) under nitrogen was cooled to -10°C and saturated with
hydrochloric acid gas for 15 minutes. The reaction vessel was then
sealed and the solution stirred for two days at 25°C. The tube was
cooled to -10°C prior to opening to release gaseous hydrochloric acid,
and then allowed to warm to 25°C. The solvent was removed in vacuo
and the product triturated with ethyl acetate to give 2-[4-(4-
phenoxyphenylthio)-piperidin-4-yl)]-acetic acid hydrochloride as a
white powder. 1HNMR (CD30D): 7.93 (d,2H); 7.45 (t,2H); 7.27 (t,lH),
7.14 (t,4H); 3.52 (m,2H); 3.25 (m,2H); 2.70 (s,2H), 2.35 (m,4H).
1 5 7B. Preparation of Ia where R1 and R2 are Hydrogen. R3 and R4
when taken together with the Carbon to which they are attached
represent Cyclo~entyl, and RS is Di~henXlether. from a Compound of
Formula (4)
2 0 A mixture of cyclopentylideneacetic acid (2 mmol) and
p-(phenoxy)-thiophenol (2 mmol) was heated at 110°C under nitrogen
in the presence of piperidine ( 100 p.L) for 24 hours. The residue was
dissolved in ethyl acetate and washed with dilute hydrochloric acid.
The organic layer was separated, dried and evaporated under reduced
2 5 pressure to give crude 2-[1-(4-phenoxyphenylthio)-cyclopent-1-yl]-
acetic acid, which can be used in the next reaction without further
purification.
7C. Preparation of Ia where R1 R2 and R3 are Hydrogen. R4 is
3 0 Benzyl, and RS is 4-Bromophenyl
A mixture of E-2-benzylacrylic acid ( 1 g) and p-bromothiophenol
( 1.12 g) were stirred overnight at 110°C in the presence of piperidine
(300 ~L). The residue was partitioned between ethyl acetate and
3 5 dilute hydrochloric acid. The organic layer was separated, dried and
~'~93178
- 82 -
evaporated under reduced pressure to give 3-benzyl-3-(4-
bromophenylthio)-propionic acid (Iaa), which was used in the next
reaction with no further purification.
7D. Preparation of Ia where R1 and R2 when taken together with the
Carbon to which then are attached represent Tetrah~pyran, R3
and R4 are HXdrogen, and RS is 4-(4-Chlorophenoxy~phenyl, from a
Cornnound of Formula ( 10)
2,7-dioxa-spiro[3.5]nonane-1-one (10.8 g), obtained as described
in lJxample 5H, was immediately dissolved in N,N-dimethylformamide
(95 ml) and slowly added to a solution containing the sodium salt of
4-(4-chlorophenoxy)thiophenol (generated by the addition of sodium
hydride powder (2.14 g, 89.2 mmol) to a solution of 4-(4-chloro-
phenoxy)thiophenol (15.83 g, 66.8 mmol) in N,N-dimethylformamide
(19. ml) at 0°C and stirring for 30 minutes) over a 10-15 minute
period, and then stirred an additional 15 minutes. The resulting
slurry was heated to 40°C, stirred for 5 minutes, tert-butanol (2 ml)
wa<,> added, and the mixture cooled to room temperature over 20
2 0 minutes. The majority of the N, N-dimethylformamide was removed i n
vac. aco, the pH adjusted to 9.2, the resultant slurry diluted with 30%
diethyl ether-hexanes ( 120 ml) and filtered. The filter cake was
washed with additional portions of ether (3 x 70 ml), acidified to pH
3.5 with 2N aqueous hydrochloric acid, and extracted into methylene
2 5 chloride (4 x 350 ml). The combined organic layers were dried over
magnesium sulfate, concentrated in vacuo. The solid residue was
recrystallized from the minimum amount of methylene chloride-
he~;anes to afford pure 4-[4-(4-chlorophenoxy)phenylthiomethyl]-
tetrahydropyran-4-carboxylic acid as a white crystalline solid (19.50
3 0 g). mp 140.6-141.9°C; IR (KBr) 3429 (br), 1732 cm-1; 1HNMR (DMSO-
d6;1 8 1.54 (ddd, J = 14.2, 10.0, 4.2 Hz, 2H), 1.95 (dm, J = 14.2 Hz, 2H),
3.19 (s, 2H), 3.56 (ddd, J = 11.8, 10.0, 4.2 Hz, 2H), 3.70 (dt, J = 11.8, 4.2
Hz, 2H), 6.98 (d, J = 8.8 Hz, 2H), 7.02 (d, J = 8.9 Hz, 2H), 7.02 (d, J = 8.9
Hz, 2H), 7.42 (d, J = 9.0 Hz, 4H), 12.66 (s, 1H); 13CNMR (DMSO-d6) 8
3 5 33.06 (t), 43.56 (t), 45.03 (s), 64.13 (t), 119.43 (d), 120.11 (d), 110.43
193~~'8
- 83 -
(d), 127.35 (s), 129.80 (d), 131.09 (s), 131.59 (d), 154.90 (s), 155.50
(s), 175.25 (s); HRMS Cald. for C19H19S04C1: 378.0693. Found:
378.0685. Anal. Calcd. for C19H19SO4C1Ø25 H20: C,59.53; H, 513.
Found: C, 59.53; H, 5.07.
Similarly, replacing 4-(4-chlorophenoxy)thiophenol with
4-(~4-bromophenoxy)thiophenol and 4-(4-fluorophenoxy)thiophenol,
the following compounds were prepared:
4-(4-(4-bromophenoxy)phenylthiomethyl]tetrahydropyran-4-
carboxylic acid: mp 143.7-144.5 °C; IR (KBr) 3434 (br), 1732 cm-1;
1H NMR (DMSO-d6) 8 I.54 (ddd, J = 13.8, 10.1, 4.3 Hz, 2H), 1.94 (dm, J
- 13.5 Hz, 2H), 3.19 (s, 2H), 3.37 (ddd, J = 11.8, 10.1, 2.5 Hz, 2H), 3.70
(dt, J = 11.8 Hz, 4.0 Hz, 2H), 6.96 (d, J = 9.2 Hz, 2H), 6.98 (d, J = 8.8 Hz,
1 5 2H), 7.41 (d, J = 8.8 Hz, 2H), 7.55 (d, J = 9.0 Hz, 2H), 12.68 (s, 1H);
13C
NMR (DMSO-d6) 8 33.04 (t),43.34 (t), 45.00 (s), 64.10 (t), 115.14 (s),
119.59 (d), 120.53 (d), 131.15 (s), 131.51 (d), 132.77 (s), 154.71 (s),
15f>.06 (s), 175.28 (s); ELMS (M+): 424. Anal. Calcd. for C19H19S04Br:
C, 53.91; H, 4.52. Found: C, 53.53; H, 4.54;
4-[4-(4-fluorophenoxy)phenylthiomethyl]tetrahydropyran-4-
carboxylic acid: mp 143.0-143.4 °C; IR (KBr) 3436 (br), 1721 cm- l ; 1
H
NMR (DMSO-d6) 8 1.54 (ddd, J = 13.5, 10.1, 4.0 Hz, 2H), 1.94 (dm, J =
13.5 Hz, 2H), 3.17 (s, 2H), 3.38 (td, J = 11.8, 2.5 Hz, 2H), 3.70 (dt, J =
2 5 11.8 Hz, 4.0 Hz, 2H), 6.93 (d, J = 8.8 Hz, 2H), 7.05 (dd, J = 9.2. 4.6 Hz,
2H), 7.21 (dd, J = 9.1, 8.4 Hz, 2H), 7.40 (d, J = 8.8 Hz, 2H), 12.65 (s,
1H); 13C NMR (CDC13) 8 33.05 (t), 43.65 (t), 45.49 (s), 64.12 (t), 116.53
(dd, JC-F = 23.2 Hz), 118.71 (d), 120.63 (dd, JC_F = 8.5 Hz), 130.31 (s),
131.69 (d), 152.38 (s), 155.85 (s), 158.29 {d, JC_F = 239.9 Hz), 175.28
3 0 (s); EIMS (M+): 362. Anal. Calcd. for C I9H 19SO4F: C, 62.97; H, 5.28.
Found: C, 62.79; H, 5.26.
7E. Alternative Preparation of Ia where R 1 and R2 are both Methyl,
R=' and R4 are Hydrogen, and RS is 4-(4-Chloro~henoxY)phenvl
X193178
- 84 -
Sodium hydride powder (0.86 g, 35.8 mmol) was added to a
mixture of 4-(4-chlorophenoxy)thiophenol (3.55 g, 15 mmol) in N,N-
dim.ethylformamide ( 12 ml) at 0°C. The mixture was warmed to room
temperature over 5 minutes, stirred for an additional 20 minutes, and
solid chloropivalic acid ( 1.64 g, 12.0 mmol) was added in one portion.
This mixture was heated to 80°C for 18 hours, cooled to room
temperature, and water { 1 ml) added. The residue was partitioned
between methylene chloride (50 ml) and 2N hydrochloric acid (25
ml). The aqueous layer was separated and washed with additional
methylene chloride (2 x 25 ml). The combined organic extracts were
dried over magnesium sulfate, concentrated in vacuo.
Chromatography over silica gel, and eluting with 5% methanol/
methylene chloride, gave slightly impure 3-[4-(4-chlorophenoxy)-
phenylthio]-2,2-dimethyl propionic acid (4 g, 99%). This material was
recrystallized from the minimum amount of diethyl ether/hexanes to
afford analytically pure acid as a white solid (3.20 g, 80%). mp 84.4-
84.!~°C; IR (KBr) 3433 (br), 1732 cm-1; 1 HNMR (DMSO-d6) 8 1.19 (s,
6H;), 3.14 (s, 2H), 6.97 (d, J = 8.7 Hz, 2H), 7.01 (d, J = 8.9, 2H), 7.40 (d,
J = 8.8 Hz, 2H), 12.36 {br s, 1H). EIMS(M+): 378. Anal. Calcd. for
2 0 C 1-7H 17S03C1: C, 60.62; H, 5.09. Found: C, 60.31; H, 4.96.
7F. Preparation of Ia where R1 and R2 when taken together with the
Carbon to which theX are attached represent N-BOC-Piperidine, R3
and R4 are H~dro g-een, and R5 is 4- 4-Chlorophenox~phenyl, from a
2 5 Co mpound of Formula ( 1 Ob)
7-(tert-Butoxycarbonyl)-2-oxa-7-azaspiro[3.5]nonan-1-one
obtained in Example 5I above, was immediately dissolved in N, N-
dimethylformamide (4 ml), slowly added to a solution containing the
3 0 sodium salt of 4-(4-chlorophenoxy)thiophenol (generated by the
addition of sodium hydride power (340 mg, 14.17 mmol) to a solution
of 4-(4-chlorophenoxy)thiophenol (3.00 g, 12.7 mmol) in
N,,?V-dimethylformamide (19 ml), at 0°C and stirred for 30
minutes)
over a 10-15 minute period, and was stirred an additional 15
3 5 minutes. The resulting slurry was heated to 80°C, stirred for
2193~~8
-8s-
s minutes, tert-butanol (2 ml) added, and the mixture cooled to
room temperature over 20 minutes. The majority of the N,N-
dimethylformamide was removed in vacuo, the pH adjusted to 3.s
using 2M aqueous hydrochloric acid and extracted into ethyl acetate
s (4 x 1 s0 ml). The combined organic layers were dried over
magnesium sulfate, concentrated in vacuo and the residue
chromatographed over silica gel, eluting with 1 % to 10%
methanol/methylene chloride, to afford the piperidine acid,
4-[4-(4-chlorophenoxy)phenylthiomethyl]-N-(tert-butoxycarbonyl)-
1 0 piperidin-4-yl carboxylic acid as a pale yellow oil (s g, 89%). 1 H N M R
(OH not observed; CDCl3) 8 1.37 (s, 9H), l.ss (mc, 2H), 2.10 (mc, 2H),
3.0:5 (mc, 2H), 3.06 (s, 2H), 3.72 (mc, 2H), 6.81 (d, J = 8.8 Hz, 2H), 6.8s
(d, J = 8.9 Hz, 2H), 7.21 (d, J = 8.9 Hz, 2H), 7.30 (d, J = 8.7 Hz, 4H).
1 s 7G. Preparation of Ia where R 1 and R2 when taken together with the
Carbon to which they are attached represent Tetrahydropyran, R3
and R4 are Hydr~en, RS is 4-(4-Chlorophenoxy)phenyl, from a
Coyound of Formula Ia where R is Ethyl
2 0 To a solution of 4-[4-(4-chlorophenoxy)phenylthiomethyl]-
tetrahydropyran-4-carboxylic acid ethyl ester (70 mg, 0.17 mmol) in
ethanol (2 ml) containing two drops of water, was added potassium
hydroxide (s8.3 mg, 1.04 mmol). The mixture was refluxed for 13
hours, cooled to room temperature, acidified to pH 4, and extracted
2 s with ethyl acetate (4 x s0 ml). The combined organic layers were
dried over magnesium sulfate, and concentrated to afford 4-[4-(4-
chl.orophenoxy)-phenylthiomethyl]-tetrahydropyran-4-carboxylic
acid (66 mg, 100%), which is spectroscopically identical to that
isolated from the prior procedure of Example 7D.
7H. Preparation of Ia where R1 and R2 when taken together with the
Carbon to which they are attached represent TetrahxdropYran, R3
and R4 are H,~!drog_en, RS is 4-(4-Bromo~henox~)phenyl, from a
Compound of Formula Ia where R is Ethyl
3s
219378
- 86 -
10
Similarly, following the procedure of Example 7G above,
4-[4-(4-bromophenoxy)phenylthiomethyl]-tetrahydropyran-4-
carboxylic acid and 4-[4-(4-fluorophenoxy)phenylthiomethyl]-
tetrahydropyran-4-carboxylic acid were prepared.
7I. Preparation of Ia where R1 and R2 when taken together with the
Carbon to which they are attached represent Tetrah~pyran, R3
and R4 are Hydro a°~ n~RS is 4-(4-Chlorophenox~)phen~l, and R is
Methyl from the Corresponding Carboxylic Acid
To a solution of 4-[4-(4-chlorophenoxy)phenylthiomethyl]-
tetrahydropyran-4-carboxylic acid (580 mg, 1.53 mmol) and
N,N-dimethylformamide catalyst (22 ~,L) in methylene chloride (15
ml) at 0°C was added oxalyl chloride (0.33 ml, 3.83 mmol) dropwise
over 10 minutes. The mixture was warmed to room temperature over
1 hour, the partial slurry stirred an additional 12 hours, and
concentrated in vacuo until the theoretical mass of the acid chloride
was obtained. The residue was suspended in tetrahydrofuran (7.5 ml),
and. methanol (0.19 ml, 4.59 mmol), followed by triethylamine (0.64
2 0 ml, 4.59 mmol) was added. The mixture was heated to reflux for 14
hours, concentrated, and the resulting residue partitioned between
methylene chloride ( 150 ml) and 1 M aqueous hydrochloric acid (50
ml). The aqueous layer was back extracted with additional portions of
methylene chloride (2 x 30 ml), the combined extracts dried over
2 5 magnesium sulfate, and concentrated to afford crude
4-[4-(4-chlorophenoxy )phenylthiomethyl]-tetrahydropyran-4-
carboxylic acid methyl ester, which was taken directly into the next
reaction without further purification. 1 HNMR (CDC13) b 1.62 (mc, 2H),
2.15 (dm, J = 13.6 Hz, 2H), 3.13 (s, 2H), 3.47 (td, J = 11.9, 2.4 Hz, 2H),
3 0 3.59 (s, 3H), 3.81 (dt, J = 12.0, 4.1 Hz, 2H), 6.92 (d, J = 8.9 Hz, 2H),
7.29
(d, J = 8.8 Hz, 2H), 7.36 (d, J = 8.8 Hz, 2H).
7J. Preparation of Ia where R1 and R2 taken together with the
Carbon to which they! are attached represent Tetrah~ropvran, R3
3 5 and R4 are Hydro,gen, RS is 4-(4-Chloro~henoxY~phenyl, and R is
~~9~1~8
_ 87 -
Eth~l from a Compound of Formula ( 13)
4-(Iodomethyl)tetrahydropyran-4-carboxylic acid ethyl ester
(300 mg, 1 mmol) was added to a solution containing the sodium salt
of 4-(4-chlorophenoxy)thiophenol (generated by the addition of
sodium hydride powder (36 mg, 1.5 mmol) to a solution of
4-(4-chlorophenoxy)thiophenol (262 mg, 1.1 mmol) in N,N-dimethyl-
formamide (2 ml) at 0°C and stirring for 30 minutes). The mixture
was warmed to room temperature over 5 minutes, stirred for an
additional 20 minutes, cooled to room temperature, and 1M aqueous
hydrochloric acid (5 ml) added. The mixture was then partitioned
between ethyl acetate ( 100 ml) and 2M hydrochloric acid (25 ml). The
aqueous layer was separated and washed with additional ethyl
acetate (2 x 50 ml). The organic extracts were combined, washed with
1 5 1 M sodium hydroxide (2 x 30 ml), dried over magnesium sulfate,
concentrated in vacuo. Chromatography over silica gel, and eluting
with 20% ethylacetate/ hexanes, yielded pure 4-[4-{4-chloro-
phenoxy)phenylthiomethyl]-tetrahydropyran-4-carboxylic acid ethyl
ester (370 mg, 91 %), followed by impure 4-[4-(4-chlorophenoxy)-
2 0 phenylthiomethyl]tetrahydropyran-4-carboxylic acid ethyl ester (40
mg). IR (KBr) 1728 cm'l; 1HNMR (CDC13) 1.23 (q, J=7.1 Hz, 3H),
1.56 (ddd, J = 14.6, 10.9, 4.4, 2H), 1.63 (ddd, J = 14.6, 5.7, 3.3, 2H),
3.13 (s, 2H), 3.51 (ddd, J = 11.8, 11.1, 2.4 Hz, 2H), 3.80 (dt, J = 11.8,
4.1 Hz, 2H), 4.07 (q, J = 7.1 Hz, 2H), 6.91 (d, J = 8.9 Hz, 2H), 6.92 (d, J =
2 5 8.9 Hz, 2H), 7.29 (d, J = 9.0 Hz, 2H), 7.39 (d, J = 8.9 Hz, 2H); 13C NMR
(CDC13) 8 14.20 (q), 33.72 (t), 45.72 (t), 46.07 (s), 60.92 (t), 65.06 (t),
119.29 (d), 120.20 (d), 128.43 (s), 129.85 (d), 130.57 (s), 133.05 (s),
155.40 (s), 156.21 (s), 174.02 (s); EIHRMS Calcd. for C21H23S~4C1
(M+): 406.1006. Found: 406.1008. Anal. Calcd. for C21H23S~4C1: C,
3 0 61.98; H, 5.70. Found: C, 61.86; H, 5.68.
7K. Preparation of Ia where R1 and R2 when taken together with the
Carbon to which they are attached represent Tetrahydropyran, R3
and R4 are Hydro~e~ n-R5 is 4-(4-Bromophenox~)phenYl and R is
3 5 Ethyl from a Compound of Formula ( 13)
2193178
_ss_
Similarly, replacing 4-(4-chlorophenoxy)thiophenol with
4-(4-bromophenoxy)thiophenol, and following the procedures of
Example 7J above, 4-[4-(4-bromophenoxy)phenylthiomethyl]-
tetrahydropyran-4-carboxylic acid ethyl ester was prepared (2.10 g,
93 io). IR (KBr) 1728 cm-1; 1 HNMR (CDC13 ) 8 1.22 (q, J = 7.1 Hz, 3H),
1.6() (ddd, J = 14.6, 10.9, 4.5, 2H), 2.14 (ddd, J = 14.6, 5.7, 3.3, 2H),
3.13 (s, 2H), 3.81 (ddd, J = 11.8, 11.1, 2.4 Hz, 2H), 4.07 (q, J = 7.1 Hz,
2H), 6.87 (d, J = 9.0 Hz, 2H), 6.92 (d, J = 8.8 Hz, 2H), 7.37 (d, J = 8.8 Hz,
1 0 2H), 7.43 (d, J = 9.0 Hz, 2H); 13CNMR (CI>C13) b 14.20 (q), 33.71 (t),
45.69 (t), 46.05 (s), 60.92 (t), 65.05 (t), 116.06 (s), 119.40 (d), 120.59
(d), 130.69 (s), 132.81 (d), 133.03 (s), 156.04 (s), 156.16 (s), 174.01
(s); EIHRMS Calcd. for C21H23S04Br (M+): 450.0500. Found: 450.0505.
Anal. Calcd. for C21H23S~4C1: C, 55.88; H, 5.14. Found: C, 55.52; H,
1 5 5.09.
Similar reactions were carried out, starting from compounds of
Formula ( 13) where X is iodo, bromo, and chloro, and moderate to
good yields were obtained in all cases.
7L. Preparation of Ia, varying R1 R2 R3, R4, and RS
Similarly, optionally replacing 4-carboxymethylene-N-CBZ-
piperidine with other N-protected compounds of Formula (4) and
2 5 following the procedures of Example 7A ( 1 ) and (2) above, or
optionally replacing cyclopentylideneacetic acid with other
compounds of Formula (4) and following the procedures of Example
7B above, and optionally replacing p-phenoxythiophenol with other
compounds of Formula (5), the following compounds of Formula Ia
3 0 were prepared:
2-[4-(4-methoxyphenylthio)-N-CBZ-:piperidin-4-yl-]-acetic acid;
2-[4-(4-methoxyphenylthio)-piperidin-4-yl)]-acetic acid;
2-benzyl-3-(3-methoxyphenylthio)-propionic acid;
2-benzyl-3-(4-methoxyphenylthio)-propionic acid;
3 5 3-benzyl-3-(4-methoxyphenylthio)-propionic acid;
~~93~7~
- 89 -
3,3-dimethyl-3-[(4-chlorophenoxy)phenylthio]-propionic acid;
2-{4-[4-(4-fluorophenoxy)phenylthio]-piperidin-4-yl}-acetic acid;
2-{ 4-[4-(4-fluorophenoxy)phenylthio]-N-CBZ-piperidin-4-yl }-
acetic acid;
3-benzyl-3-[(4-phenylthiophenyl)thio]-propionic acid;
3-benzyl-3-(phenylthio)-propionic acid;
3-benzyl-3-(4-phenoxphenylthio)-propionic acid;
3-benzyl-3-[(4-biphenyl)thio)-propionic acid;
3-benzyl-3-(2-naphthylthio)-propionic acid;
3-benzyl-3-(4-methoxystyrylphenylthio)-propionic acid;
3-cyclopentylmethyl-3-(4-methoxyphenylthio)-propionic acid;
3-cyclopentylmethyl-2-isopropyl-3-(4-methoxyphenylthio)-
propionic acid;
3-ethyl-2-methyl-3-(4-methoxyphenylthio)-propionic acid;
3,3-dimethyl-(4-methoxyphenylthio)-propionic acid;
2-[1-(4-methoxyphenylthio)-cyclopent-1-yl]-acetic acid;
2-(4-(4-methoxyphenylthio)-cyclohexanone-4-yl]-acetic acid
ethylene ketal;
2-[1-(4-methoxyphenylthio)-(4-methylcyclohex-1-yl]-acetic acid;
2 0 2-[1-(4-phenoxyphenylthio)-cyclohex-1-yl]-acetic acid;
2-[4-(4-phenoxyphenylthio)-tetrahydropyran-4-yl]-acetic acid;
{ 4-[4-(4-benzo[b]thiophen-2-yl-phenoxy)phenylthio)-
tetrahydropyran-4-yl]-acetic acid;
2- { 4-[4-(phenylmethyl)phenylthio]-tetrahydropyran-4-yl }-acetic
2 5 acid;
2-{4-[4-(4-fluorophenoxy)phenylthio]-tetrahydropyran-4-yl }-
acetic acid;
2-{4-[4-(4-chlorophenoxy)phenylthio]-tetrahydropyran-4-yl}-
acetic acid: mp 138.5-138.8 °C; 1HNMR (CDC13, OH not seen) 8 1.73 (d,
3 0 J == 14.7, 2H), 1.91 (ddd, J = 14.7, 10. l, 4.3 Hz, 2H), 2.58 (s, 2H),
3.76
(dt., J = 11.8, 4.1 Hz, 2H), 4.02 (dt, J = 11.8, 2.6 Hz, 2H), 6.94 (d, J = 8.8
Hz., 2H), 6.98 (d, J = 8.9 Hz, 2H), 7.33 (d, J = 8.9 Hz, 2H), 7.53 (d, J = 8.8
Hz, 4H); FABMS (M+): 379.2. Anal. Calcd. for C 19H 19SO4C1: C, 60.23; H,
5.05. Found: C, 60.39; H, 5.01;
35 2-{4-[4-(4-chlorophenoxy)phenylthio]-tetrahydropyran-4-yl}-
2~9317~
- 90 -
acetic acid;
2- { 4-[4-(4-bromophenoxy)phenylthio]-tetrahydropyran-4-yl }-
acetic acid;
2-[4-(4-phenoxyphenylthio)-tetrahydrothiopyran-1,1-dioxide-4-
yl]-acetic acid;
traps-2-(4-methoxyphenylthio)-cyclopentanecarboxylic acid; and
2-(4-methoxyphenylthio)-cyclohexanecarboxylic acid.
7M. Preparation of Ia, varying R1 R2 R3 R4, and RS
Similarly, optionally replacing 2,7-dioxa-spiro[3.5]nonane-1-one
with other compounds of Formula ( 10) and following the procedures
of Example 7D above, and optionally replacing 4-(4-chlorophenoxy)-
thiophenol with other compounds of Formula (5), the following
compounds of Formula Ia were prepared:
4-[4-(4-fluorophenoxy)phenylthiomethyl]tetrahydropyran-4-
car~boxylic acid;
4-[4-(4-bromophenoxy)phenylthiomethyl]tetrahydropyran-4-
carboxylic acid;
2 0 3-(4-benzoylphenylthio)-2,2-dimethyl propionic acid;
3-[4-(4-chlorophenoxy)phenylthio]-2,2-dimethyl propionic acid;
4-[(4-phenoxypyrid-4-yl)thiomethyl]tetrahydropyran-4-
carboxylic acid: 1 HNMR (OH not observed; CDC13) 8 1.65 (mc, ZH), 2.16
(dm, J = 14.2 Hz, 2H), 3.20 (s, 2H), 3.57 (tm, J = 11.4 Hz, 2H), 3.84 (dm,
2 5 J =: 12.0 Hz, 2H), 6.87 (d, J =6.2 Hz, 2H), 7.00 (d, J = 8.6 Hz, 2H), 7.47
(d,
J = 8.9 Hz, 2H), 8.43 (d, J = 6.0 Hz, 2H).
7N. Preparation of Ia, varying R1 R2 R3,_R4. and R5
3 0 Similarly, following the procedures of Example 7 above, other
compounds of Formula Ia are prepared.
~'~93178
- 91 -
EXAMPLE 8
Preparation of Compounds of Formula Iba
8A. Preparation of Iba where Rl and R2 when taken together with
the Carbon to which they are attached represent Tetrah, dropyran R3
and R4 are Hydro~en, and R5 is 4-f'4-ChlorophenoxY,~phen~
Oxalyl chloride (37.5 ml, 429.5 mmol) was added dropwise over
minutes to a suspension of 4-[4-(4-chlorophenoxy)phenylthio-
10 methyl]-tetrahydropyran-4-carboxylic acid (65.1 g, 171.8 mmol) and
N, N-dimethylformamide catalyst (2 ml) in methylene chloride ( 1 litre)
at 0°C. The mixture was warmed to room temperature over 1 hour
and the resultant partial slurry stirred an additional 20 hours,
concentrated under reduced pressure until the theoretical mass of the
acid chloride was obtained. This mixture was dissolved in methylene
chloride (600 ml), cooled to 0°C, and N,O-bis(trimethylsilyl)hydroxyl-
amine ( 109.1 ml, 510.45 mmol) added dropwise over 10 minutes.
The mixture was immediately warmed to room temperature, stirred 3
hours, and recooled to 0°C. Aqueous 2.4M hydrochloric acid solution
2 0 (400 ml, 960 mmol) was added to the solution, causing precipitation
of the hydroxamic acid product within several minutes after the
addition. The slurry was stirred an additional 30 minutes and filtered.
Thf: filter cake was washed with water (3 x 30 ml) and 50% diethyl
ether-hexanes (2 x 25 ml) and dried at 70°C to afford
2 5 4-[4-(4-chlorophenoxy)phenylthiomethyl]-tetrahydropyran-
4-(N-hydroxycarboxamide) (61.8 g, 92%). mp 146.6-148.0 °C; IR (KBr)
3426 (br), 1636 cm-l; lHNMR (DMSO-d6) 8 1.54 (ddd, J = 13.8, 10.2,
4.0 Hz, 2H), 2.00 (dm, J = 13.8 Hz, 2H), 3.16 (s, 2H), 3.39 (m, 2H), 3.66
(dt, J = 11.7, 3.8 Hz, 2H), 6.98 (d, J = 8.8 Hz, 2H), 7.02 (d, J = 9.0 Hz,
3 0 2H), 7.40 (d, J = 8.8 Hz, 2H), 7.41 (d, J = 8.9 Hz, 2H), 8.78 (s, 1H),
10.63
(s, 1H); 13CNMR (CDC13) 8 32.79 (t), 43.60 (s), 43.70 (t), 63.93 (t),
119.56 (d), 120.07 (d), 127.19 (s), 129.85 (d), 131.24 (d), 131.34 (s),
154.62 (s), 155.59 (s), 169.69 (s); FABHRMS Calcd. for C19H21NS04C1
(M+ + H): 394.0880. Found: 378.0872. Anal. Calcd. for C19H2pNS04C1:
3 5 C, 57.94; H, 5.12; N, 3.56. Found: C, 57.98; H, 5.04; N, 3.68.
X193178
- 92 -
8B. Alternative Preparation of Iba where Rl and R2 when taken
together with the Carbon to which they are attached represent
Tetrahydro~yran, R3 and R4 are H~g_en, and R5 is 4- 4-
ChlorophenoxX)phenyl
Oxalyl chloride (37.5 ml, 429.5 mmol) was added dropwise over
minutes to a solution of 4-[4-(4-chlorophenoxy)phenylthio-
methyl]-tetrahydropyran-4-carboxylic acid (65.1 g, 171.8 mmol) and
1 0 N, N-dimethylformamide catalyst (2 ml) in methylene chloride ( 1 litre)
at 1)°C. The mixture was warmed to room temperature over 1 hour,
and the resultant partial slurry stirred an additional 20 hours and
concentrated in vacuo until the theoretical mass of the acid chloride
was obtained. A solution of the acid chloride mixture (650 mg, 1.68
1 5 mmol) in methylene chloride (3.4 ml) was added dropwise over 2
minutes to a solution of 50% aqueous hydroxylamine (556 mg) in 2:1
tetrahydrofuran/tert-butanol (5.1 ml). The mixture was stirred 1.5
hours and concentrated until approximately 1 ml of aqueous solution
was remaining. The slurry was filtered, washed with 1:1 diethyl
2 0 ether-hexanes (3 X 15 ml) and the solid dried overnite at 70°C in a
vacuum oven, to afford 4-[4-(4-chlorophenoxy)phenylthiomethyl]-
tetrahydropyran-4-(N-hydroxycarboxamide) (584 mg, 91 %). mp
146.6-148.0 °C; IR (KBr) 3426 (br), 1636 cm- l ; 1 HNMR (DMSO-d6) 8
1.54 (ddd, J = 13.8, 10.2, 4.0 Hz, 2H), 2.00 (dm, J = 13.8 Hz, 2H), 3.16
2 5 (s, 2H), 3.39 (m, 2H), 3.66 (dt, J = 11.7, 3.8 Hz, 2H), 6.98 (d, J = 8.8
Hz,
2H), 7.02 (d, J = 9.0 Hz, 2H), 7.40 (d, J = 8.8 Hz, 2H), 7.41 (d, J = 8.9 Hz,
2H), 8.78 (s, 1H), 10.63 (s, 1H); 13C NMR (CDC13) b 32.79 (t), 43.60 (s),
43.'70 (t), 63.93 (t), 119.56 (d), 120.07 (d), 127.19 (s), 129.85 (d),
131.24 (d), 131.34 (s), 154.62 (s), 155.59 (s), 169.69 (s); FABHRMS
3 0 Calcd. for C19H21NS04C1 (M+ + H): 394.0880. Found: 378.0872. Anal.
Calcd. for C19H20NS04C1: C, 57.94; H, 5.12; N, 3.56. Found: C, 57.98; H,
5.04; N, 3.68.
- 93 -
8C. Preparation of Iba, varying Rl R2 R3 R4, and RS
Similarly, replacing 4-[4-(4-chlorophenoxy)phenyl-thiomethyl]-
tetrahydropyran-4-carboxylic acid with other compounds of Formula
Ia and following the procedures of Example 8A above, the following
compounds of Formula Iba were prepared:
4-[4-(4-fluorophenoxy)phenylthiomethyl]tetrahydropyran-
4-(N-hydroxycarboxamide): mp 146.2-146.5 °C; IR (KBr) 3431 (br),
162 8 cm- l ; 1 HNMR (CDC13 ; NH and OH not observed) 8 1.35 (ddd, J =
1 0 13.8, 10.2, 4.0 Hz, 2H), 1.83 (dm, J = 13.8 Hz, 2H), 2.85 (s, 2H), 3.23
(m,
2H), 3.46 (dt, J = 11.9, 3.9 Hz, 2H), 6.58 (d, J = 8.8 Hz, 2H), 6.57 (d, J =
8.8 Hz, 2H), 6.65-6.78 (m, 4H),7.06 (d, J = 8.8 Hz, 2H); 13C NMR (CDC13)
8 ?~2.99 (t), 44.27 (s), 45.49 (t), 64.63 (t), 116.28 (dd, JC-F = 23.2 Hz),
118.64 (d), 120.49 (dd, JC_F = 8.5 Hz), 130.41 (s), 132.49 (d), 152.46
1 5 (s), 156.49 (s), 160.29 (d, JC_F = 241.9 Hz), 170.23 (s); FABMS (M+ +
H): 378. Anal. Calcd. for C19H2pNS04F: C, 60.46; H, 5.34; N, 3.71.
Found: C, 60.08; H, 5.29; N, 3.65.
4-[4-(4-bromophenoxy)phenylthiomethyl]tetrahydropyran-
4-N-hydroxycarboxamide: mp 153.1-154.0 °C; IR (KBr) 3434 (br),
2 0 16;14 cm-1; 1 HNMR (CDC13 ; NH and OH not observed) 8 1.68 (ddd, J =
14.0, 10.0, 4.0 Hz, 2H), 2.13 (dm, J = 14.0 Hz, 2H), 3.15 (s, 2H), 3.55
(ddd, J = 12.0, 10.2, 2.5 Hz, 2H), 3.76 (dt, J = 12.0 Hz, 4.1 Hz, 2H), 6.87
(d, J = 9.0 Hz, 2H), 6.90 (d, J = 8.8 Hz, 2H), 7.37 (d, J = 8.8 Hz, 2H), 7.43
(d, J = 9.0 Hz, 2H); 13CNMR (CDC13) 8 33.01 (t), 44.32 (s), 45.40 (t),
2 5 64.65 (t), 115.95 (s), 119.50 (d), 120.53 (d), 130.67 (s), 132.76 (d),
132.80 (d), 155.92 (s), 156.16 (s), 170.60 (s); FABMS (M+ + H): 438.
Anal. Calcd. for C19H20NS04Br: C, 52.06; H, 4.60; N, 3.20. Found: C,
51.84; H, 4.52; N, 3.54.
3-(4-benzoylphenylthio)-2,2-dimethyl-N-hydroxypropionamide;
3 0 3-[4-(4-chlorophenoxy)phenylthio]-2,2-dimethyl-N-hydroxy-
propionamide: mp 114.7-115.3 °C; 1HNMR (CDC13) 8 1.30 (s, 6H), 3.14
(s, 2H), 6.90 (d, J = 8.8 Hz, 2H), 6.92 (d, J = 8.8 Hz, 2H), 7.29 (d, J = 8.9
Hz, 2H), 7.37 (d, J = 8.8 Hz, 1H); FABHRMS Calcd. for C17H18NS03C1
(M+ + H): 352.0772. Found: 352.0774. Anal. Calcd. for C17H1gNS03Cl:
~~g3178
- 94 -
C, 58.03; H, 5.16; N, 3.98. Found: C, 57.85; H, 5.10; N, 4.12.
3,3-dimethyl-3-[(4-chlorophenoxy)phenylthio]-N-hydroxy-
propionamide;
{ 4-[4-(4-benzo[b]thiophen-2-yl-phenoxy)phenylthio)-
tetrahydropyran-4-yl]-N-hydroxyacetamide;
2- { 4-[4-(phenylmethyl)phenylthio]-tetrahydropyran-4-yl }-
N-hydroxyacetamide;
2-{4-[4-(4-chlorophenoxy)phenylthio]-tetrahydropyran-4-yl}-
N-hydroxyacetamide; and
2-{4-[4-(4-bromophenoxy)phenylthio]-tetrahydropyran-4-yl}-
N-hydroxyacetamide.
8D. Preparation of Iba. var~~ R1 R2 R3 R4, and RS
Similarly, replacing 4-[4-(4-chlorophenoxy)phenylthiomethyl]-
tetrahydropyran-4-carboxylic acid with other compounds of Formula
Ia and following the procedures of Example 8A above, other
compounds of Formula Iba are prepared, for example:
4-(4-phenoxyphenylthiomethyl)tetrahydropyran-4-(N-hydroxy-
2 0 carboxamide);
4-[4-(4-fluorophenoxy)phenylthiomethyl] tetrahydropyran-
4-(N-hydroxycarboxamide);
4-[4-(4-chlorophenoxy)phenylthiomethyl]piperidine-
4-(N-hydroxycarboxamide);
4-[4-(4-chlorophenoxy)phenylthiomethyl]-1-methylpiperidine-
4-(N-hydroxycarboxamide);
4-[4-(4-chlorophenoxy)phenylthiomethyl]-1-(cyclopropyl-
methyl)piperidine-4-(N-hydroxycarboxamide;);
4- [4-(4-chlorophenoxy)phenylthiomethyl]-1-acetylpiperidine-
3 0 4-(N-hydroxycarboxamide);
4-[4-(4-chlorophenoxy)phenylthiomethyl]-1-(3-pyridyl)-
piperidine-4-(N-hydroxycarboxamide);
4-[4-(4-chlorophenoxy)phenylthiomethyl]-1-(3-pyridoyl)-
piperidine-4-(N-hydroxycarboxamide);
3 5 2-[4-(4-methoxyphenylthio)-N-CBZ-piperidin-4-yl-]-N-
~~~'~'18
- 95 -
hydroxyacetamide;
2-[4-(4-methoxyphenylthio)-piperidin-4-yl)]-N-hydroxy-
acetamide;
2-benzyl-3-(3-methoxyphenylthio)-N-hydroxypropionamide;
2-benzyl-3-(4-methoxyphenylthio)-N-hydroxypropionamide;
3-benzyl-3-(4-methoxyphenylthio)-N-hydroxypropionamide;
2- { 4-[4-(4-fluorophenoxy)phenylthio]-piperidin-4-yl } -
N-hydroxyacetamide;
2- { 4- [4-(4-fluorophenoxy)phenylthio]-N-CBZ-piperidin-4-yl } -
N-hydroxyacetamide;
3-benzyl-3-[(4-phenylthiophenyl)thio]-N-hydroxypropionamide;
3-benzyl-3-(phenylthio)-N-hydroxypropionamide;
3-benzyl-3-(4-phenoxphenylthio)-N-hydroxypropionamide;
3-benzyl-3-[(4-biphenyl)thio]-N-hydroxypropionamide;
3-benzyl-3-(2-naphthylthio)-N-hydroxypropionamide;
3-benzyl-3-(4-methoxystyrylphenylthio)-N-hydroxy-
propionamide;
3-cyclopentylmethyl-3-(4-methoxyphenylthio)-N-hydroxy-
propionamide;
2 0 3-cyclopentylmethyl-2-isopropyl-3-(4-methoxyphenylthio)-
N-hydroxypropionamide;
3-ethyl-2-methyl-3-(4-methoxyphenylthio)-N-hydroxy-
propionamide;
3, 3-dimethyl-(4-methoxyphenylthio)-N-hydroxypropionamide;
2-[1-(4-methoxyphenylthio)-cyclopent-1-yl]-N-hydroxy-
ace:tamide;
2-[4-(4-methoxyphenylthio)-cyclohexanone-4-yl]-N-hydroxy-
acetamide ethylene ketal;
2-[ 1-(4-methoxyphenylthio)-(4-methylcyclohex-1-yl]-
3 0 N-hydroxyacetamide;
2-[1-(4-phenoxyphenylthio)-cyclohex-1-yl]-N-hydroxyacetamide;
2-[4-(4-phenoxyphenylthio)-tetrahydropyran-4-yl]-N-
hydroxyacetamide;
2- { 4-[4-(4-fluorophenoxy)phenylthio]-tetrahydropyran-4-yl } -
3 5 N-hydroxyacetamide;
. ~~9~.;~~~
- 96 -
2- [4-(4-phenoxyphenylthio)-tetrahydrothiopyran-1,1-dioxide-4-
yl]-N-hydroxyacetamide;
traps-2-(4-methoxyphenylthio)-cyclopentanecarboxylic acid; and
2-(4-methoxyphenylthio)-cyclohexanecarboxylic acid.
EXAMPLE 9
Preparation of ComRounds of Formula Ib
9A.. Preparation of Ib where R1 and R2 are Hydrogen. R3 and R4
when taken together with the Carbon to which they are attached are
Cyc;lopent~, and RS is 4-Phenox~phenyl
The 2-[1-(4-phenoxyphenylthio)-cyclopent-1-yl]-acetic acid
obtained in Example 5 was dissolved in methylene chloride (8 ml) and
treated with 4-dimethylaminopyridine (180 mg), O-(tert-butyl)-
hydroxylamine hydrochloride (360 mg), triethylamine (540 p,L),
pyridine (400 p.L), and 1-(3-dimethylaminopropyl)-3-ethylcarbodi-
imide hydrochloride (750 mg). After stirring overnight the reaction
mixture was partitioned between ethyl acetate and water, the organic
2 0 layer separated, and the solvent removed under reduced pressure.
Preparative TLC of the residue and elution with 2:1 hexane/ethyl
acetate gave N-(tert-butoxy)-2-[ 1-(4-phenoxyphenylthio)-cyclopent-
1-yl]-acetamide (270 mg) as a white foam, which can be used in the
nexa reaction without further purification.
9B. Preparation of Ib where R1 and R2 are H,ydro~en, R3 and R4
when taken together with the Carbon to which then are attached are
Tet:rahydro~yran, and RS is 4-PhenoxYphen,~l
3 0 O-(tert-Butyl)hydroxylamine hydrochloride (9.57 g),
4-methylmorpholine ( 15.64 ml), hydroxybenzotriazole (6.87 g), and
1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (19.5
g) was added to a solution of 2-[4-(4-phenoxyphenylthio)-
tetrahydropyran-4-yl]-acetic acid (17.5 g) in methylene chloride (200
3 5 ml;). After stirring for 3 hours at room temperature, 0.5 M
X193 ~7~
- 97 -
hydrochloric acid (200 ml) was added to the mixture, and the mixture
extracted with methylene chloride. The solvent was removed from
the combined extracts under reduced pressure. Silica gel
chromatography of the residue and elution with 35%-80% ethyl
acetate/hexane gave N-tert-butoxy-2-[4-(4-phenoxyphenylthio)-
tetrahydropyran-4-yl]-acetamide ( 15.3 g) as an oil, which can be used
in the next reaction without further purification.
9C. Preparation of Ib where R3 and R4 are Hydro eg nR1 and R2
when taken together with the Carbon to which then are attached are
N-BOC-Piperidine, and R5 is 4- 4-Chlorophenoxx.l~henyl
4-Methylmorpholine (2.60 ml, 23.68 mmol) was added dropwise
to a solution of 2-{4-[4-(4-chlorophenoxy)phenylthiomethyl]-N-BOC-
1 5 piperidin-4-yl }-carboxylic acid obtained in Example 6 (2.83 g, 5.92
mmol), O-(tent-butyl)hydroxylamine hydrochloride (2.23 g, 17.76
mmol), and 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide
hydrochloride (2.27 g, 11.84 mmol) in anhydrous methylene chloride
(25 ml) cooled to 0°C. After the resulting mixture was allowed to
2 0 warm to room temperature over 1 hour and stirred for an additional
12 hours, the mixture was partitioned between diethyl ether/1 N
aqueous hydrochloric acid (300 ml). The acid layer was back
extracted using diethyl ether (2 x 100 ml), and the combined ether
extracts dried over magnesium sulfate and concentrated.
2 5 Chromatography over silica gel, and eluting with 25% ethyl
acetate/hexanes, gave N-(tert-butoxy)-2-{4-[4-(4-
chlorophenoxy)phenylthiomethyl]-N-BOC-piperidin-4-yl }-
carboxamide (2.88 g, 89%). 1HNMR (CDC13) 8 1.31 (s, 9H), 1.45 (s, 9H),
1.58 (mc, 2H), 2.10 (br d, J = 14.2 Hz, 2H), 3.13 (s, 2H), 3.19 (mc, 2H),
3 0 3.73 (mc, 2H), 6.93 (d, J = 8.8 Hz, 2H), 6.95 (d, J = 8.9 Hz, 2H), 7.30
(d, J
= 8.9 Hz, 2H), 7.38 (d, J = 8.7 Hz, 2H), 8.15 (br s, 1H).
9D. Preparation of Ib, varying R1 R2 R3~R4. and R5
3 5 Similarly, following the procedures of Example 9A above, but
~~1 ~~v ~~
- 98 -
replacing 2-[1-(4-phenoxyphenylthio)-cyclopent-1-yl]-acetic acid
with other compounds of Formula Ia, the following compounds of
Formula Ib were prepared:
N-tert-butoxy-2-[4-(4-phenoxyphenylthio)-N-CBZ-piperidin-4-
yl)]-acetamide;
N-tert-butoxy-2-[4-(4-methoxyphenylthio)-N-CBZ-piperidin-4-
yl)]-acetamide;
N-tert-butoxy-2- { 4- [4-(4-fluorophenoxy)phenylthio]-N-CB Z-
piperidin-4-yl}-acetamide;
1 0 N-tert-butoxy-2-{ 4-[4-(4-fluorophenoxy)phenylthio]-piperidin-
4-yl }-acetamide;
N-tent-butoxy-2-[4-(4-phenoxyphenylthio)-piperidin-4-yl)]-
acetamide;
N-tert-butoxy-2-[4-(3-methoxyphenylthio)-piperidin-4-yl)]-
acetamide;
N-tert-butoxy-2-[4-(4-methoxyphenylthio)-piperidin-4-yl)]-
acetamide;
N-tert-butoxy-2-benzyl-3-(phenylthio)-propionamide;
N-tert-butoxy-3-benzyl-3-(phenylthio)-propionamide;
2 0 N-tert-butoxy-3-benzyl-3-(4-methoxyphenylthio)-propionamide;
N-tert-butoxy-3-benzyl-3-[(4-phenylthiophenyl)thio]-
propionamide;
N-tert-butoxy-3-benzyl-3-(4-phenoxyphenylthio)-propionamide;
N-tert-butoxy-3-benzyl-3-[(4-biphenyl)thio]-propionamide;
2 5 N-tert-butoxy-3-benzyl-3-(2-naphthylthio)-propionamide;
N-tert-butoxy-3-benzyl-3-(4-methoxystyrylphenylthio)-
propionamide;
N-tert-butoxy-3-cyclopentylmethyl-3-(4-methoxyphenylthio)-
propionamide;
3 0 N-tert-butoxy-3-cyclopentylmethyl-2-isopropyl-
3-(4-methoxyphenylthio)-propionamide;
N-tert-butoxy-3-ethyl-2-methyl-3-(4-methoxyphenylthio)-
propionamide;
N-tert-butoxy-3,3-dimethyl-(4-methoxyphenylthio)-
3 5 prapionamide;
- 99 -
N-tert-butoxy-2-[1-(4-methoxyphenylthio)-cyclopent-1-yl]-
acetamide;
N-tert-butoxy-2-[ 1-(4-methoxyphenylthio)-(4-methylcyclohex-
1-yl]-acetamide;
N-tert-butoxy-2-[4-(4-phenoxyphenylthio)-cyclohexanone-4-yl]-
acetamide ethylene ketal;
N-tert-butoxy-2-[1-(4-phenoxyphenylthio)-cyclohex-1-yl]-
acetamide;
N-tert-butoxy-2-[4-(4-methoxyphenylthio)-N-CBZ-piperidin-4-
yl)]-acetamide;
N-tert-butoxy-2-[4-(4-methoxyphenylthio)-piperidin-4-yl)]-
acetamide.
N-tert-butoxy-2-{ 4-[4-(4-fluorophenoxy)phenylthio]-
tetrahydropyran-4-yl }-acetamide;
N-tert-butoxy-2-{4-[4-(4-chlorophenoxy)phenylthio]-
tetrahydropyran-4-yl }-acetamide;
N-tert-butoxy-2-[4-(4-phenoxyphenylthio)-tetrahydrothiopyran-
l , l -dioxide-4-yl]-acetamide;
N-tert-butoxy-4-[4-{4-pyridyloxy)phenylthiomethyl]-
tetrahydropyran-carboxamide: 1HNMR (CDC13) 8 1.31 (s, 9H), 1.70
(m~,, 2H), 2.14 (dm, J = 11.8 Hz, 2H), 3.21 (s, 2H), 3.63 (mc, 2H), 3.82
(m~;, 2H), 6.84 (d, J =6.4 Hz, 2H), 7.03 (d, J = 8.6 Hz, 2H), 7.44 (d, J =
8.4 Hz, 2H), 8.20 (s, 1 H), 8.48 (d, J = 5.8 Hz, 2H).
N-tert-butoxy-4-[4-(5-chloro-2-pyridyloxy)phenylthiomethyl]-
2 5 tetrahydropyran-carboxamide: mp 100.5-102.7 °C; IR (KBr) 3438 (br),
167 cm-1; 1HNMR (DMSO-d6) 1.19 (s, 9H), 1.57 (ddd, J =13.5, 10.1,
4.0 Hz, 2H), 2.05 {dm, J =13.5 Hz, 2H), 3.34 (s, 2H), 3.42 (mc, 2H), 3.65
(dm, J = 11.6 Hz, 2H), 7.09 (d, J = 8.8 Hz, 2H), 7.10 (d, J = 8.8 Hz, 2H),
7.41 (d, J = 8.7 Hz, 2H), 7.95 (dd, J = 8.8, 2.7 Hz, 1 H), 8.19 (d, J = 2.7
Hz,
3 0 1H;1, 10.37 (s, 1H); 13CNMR (DMSO-d6) ~ 26.66 (q), 33.03 (t), 43.20 (t),
44.25 (s), 64.10 (t), 80.78 (s), 113.00 (d), 121.88 (d), 124.88 (s),
130.43 (d), 132.67 (s), 139.93 (d), 145.51 (d), 151.89 (s), 161.58 (s),
171.64 (s); FABHRMS Calcd. for C22H28N2S04C1 (M+ + H): 451.1458.
Found: 451.1461. Anal. Calcd. for C22H27N2S04C1: C, 58.59; H, 6.03; N,
3 5 6.21. Found: C, 58.70; H, 6.05; N, 6.43.
;~9~~~'g
- 100 -
N-tert-butoxy-3-[4-(5-chloro-2-pyridyloxy)phenylthio]-2,2-
dimethyl-N-hydroxypropionamide: mp 90.8-91.9°C; IR (KBr) 3438
(br), 1651 cm-1; 1 HNMR (DMSO-d6) 8 1.18 (s, 9H), 1.21 (s, 6H), 3.20
(s, 2H), 7.08 (mc, 3H), 7.40 (d, J = 8.7 Hz, 2H), 7.93 (dd, J = 8.7, 2.7 Hz,
1H), 8.17 (d, J = 2.7 Hz, 1H), 10.17 (s, 1H); 13C NMR (DMSO-d6) 8 24.67
(q), 26.48 (q), 42.54 (s), 44.31 (t), 80.62 (s), 112.95 (d), 121.79 (d),
125.28 (s), 130.32 (d), 133.31 (s), 139.86 (d), 145.48 (d), 151.77 (s),
161.58 (s), 173.77 (s); FABHRMS Calcd. for C2pH26N2SO3C1 (M+ + H):
409.1353. Found: 409.1354. Anal. Calcd. for C2pH25N2SO3C1: C, 58.74;
1 0 H, 6.16; N, 6.85. Found: C, 58.91; H, 6.13; N, 7.07.
N-tert-butoxy-2-(4-methoxyphenylmercapto)-cyclohexane-
carboxamide; and
N-tert-butoxy-trans-2-(4-methoxyphenylmercapto)-
cyclopentanecarboxamide.
9E. Preparation of Ib, varying R2 R3 R4~ and RS
Similarly, following the procedures of Example 9A above, but
replacing 2-[1-(4-phenoxyphenylthio)-cyclopent-1-yl]-acetic acid
2 0 with other compounds of Formula Ia, other compounds of Formula Ib
are prepared.
EXAMPLE 10
Preparation of Compounds of Formula Id
10A. Preparation Id where 0, R1 R2 are H.~ en,
of n is and R3
and when taken ether with Carbon which they are
R4 tog the to
attachedare Cyclopentyl, and RS -Phenox~phenyl
is 4
3 0 The N-tert-butoxy-2-[1-(4-phenoxyphenylthio)-cyclopent-1-yl]-
acetamide was dissolved in trifluoroacetic acid (6 ml) and allowed to
stand for 24 hours. The acid was evaporated off under reduced
pressure and the product purified by preparative TLC, eluting with
6.5% methanol/methylene chloride gave N-hydroxy-2-[1-(4-
3 5 phenoxyphenylthio)-cyclopent-1-yl]-acetamide (100 mg).
X193178
- 101 -
lOB. Preparation of Id where n is 0, var ping R1 R2 R3~R4, and RS
Similarly, following the procedures of Example l0A above, but
replacing N-tert-butoxy-2-[1-(4-phenoxyphenylthio)-cyclopent-1-yl]-
acetamide with other compounds of Formula Ib, the following
compounds of Formula Id where n is 0 are prepared:
N-hydroxy-2-[4-(4-phenoxyphenylthio)-N-CBZ-piperidin-4-yl)]-
acetamide;
N-hydroxy-2-[4-(4-methoxyphenylthio)-N-CBZ-piperidin-4-yl)]-
acetamide;
2-{ 4-[4-(4-fluorophenoxy)phenylthio]-N-CBZ-piperidin-4-yl }-N-
hydroxy-acetamide;
2-{ 4-[4-(4-fluorophenoxy)phenylthio]-piperidin-4-yl }-N-
hydroxy-acetamide;
3-benzyl-N-hydroxy-3-(3-methoxyphenylthio)-propionamide;
N-hydroxy-2-[4-(4-phenoxyphenylthio)-piperidin-4-yl)]-
acetamide;
N-hydroxy-2-[4-(4-methoxyphenylthio)-piperidin-4-yl)]-
2 0 acetamide;
2-benzyl-N-hydroxy-3-(phenylthio)-propionamide;
3-benzyl-N-hydroxy-3-(phenylthio)-propionamide;
3-benzyl-N-hydroxy-3-(4-methoxyphenylthio)-propionamide;
3-benzyl-N-hydroxy-3-[(4-phenylthiophenyl)thio]-propionamide;
2 5 3-benzyl-N-hydro:cy-3-(4-phenoxyphenylthio)-propionamide;
3-benzyl-N-hydroxy-3-[(4-biphenyl)thio]-propionamide;
3-benzyl-N-hydroxy-3-(2-naphthylthio)-propionamide;
3-benzyl-N-hydroxy-3-(4-methoxystyrylphenylthio)-
propionamide;
3 0 3-cyclopentylmethyl-N-hydroxy-3-(4-methoxyphenylthio)-
propionamide;
3-cyclopentylmethyl-N-hydroxy-2-isopropyl-3-(4-
methoxyphenylthio)-propionamide;
3-ethyl-N-hydroxy-2-methyl-3-(4-rnethoxyphenylthio)-
3 5 propionamide;
X93178
- 102 -
3,3-dimethyl-N-hydroxy-(4-methoxyphenylthio)-propionamide;
N-hydroxy-2-[1-(4-methoxyphenylthio)-cyclopent-1-yl]-
acetamide;
N-hydroxy-2- [ 1-(4-methoxyphenylthio)-(4-methylcyclohex-1-
yl]-acetamide;
N-hydroxy-2- [ 1-(4-phenoxyphenylthio)-cyclohex-1-yl]-
acetamide;
N-hydroxy-2-[4-(4-methoxyphenylthio)-N-CBZ-piperidin-4-yl)]-
acetamide;
N-hydroxy-2-[4-(4-methoxyphenylthio)-piperidin-4-yl)]-
acetamide;
N-hydroxy-2-[4-(4-phenoxyphenylthio)-tetrahydropyran-4-yl]-
acetamide; 2-{4-[4-(4-chlorophenoxy)-phenylthio]-
tetrahydropyran-4-yl }-N-hydroxy-acetamide;
2-{4-[4-(4-fluorophenoxy)phenylthio]-tetrahydropyran-4-yl}-N-
hydroxy-acetamide, m.p. 50-55°C; and
N-hydroxy-2-[4-(4-phenoxyphenylthio)-tetrahydrothiopyran-
1,1-dioxide-4-yl]-acetamide.
2 0 1 OC. Preparation of Id where n is 0, varying R 1 L R21 R3 R4, and R5
Similarly, following the procedures of Example l0A above, but
replacing N-tert-butoxy-2-[1-(4-phenoxyphenylthio)-cyclopent-1-yl]-
acetamide with other compounds of Formula Ib, other compounds of
2 5 Formula Id where n is 0 are prepared.
EXAMPLE 11
Preparation of Compounds of Formula Id
3 0 1 1 Preparation Id where 1. R1 and R2 are H.Ydrogen,
A. of n is R3
and R4 when taken ether with Carbon which the are
tog the to
attached are C,~pentX l, and RS 4-Phenox~ hen~
is
A solution of N-hydroxy-2-[ 1-(4-phenoxyphenylthio)-cyclopent-
3 5 1-yl]-acetamide (45 mg) in acetone (4 ml) was treated with sodium
~~93~~'8
- 103 -
periodate (260 mg) in water (2 ml). Over the course of 24 hours, two
additional portions of sodium periodate (260 mg) were added. After
complete disappearance of starting material the solution was diluted
with methylene chloride, filtered, dried, and the solvent evaporated
under reduced pressure. Preparative TLC on silica gel and elution
with 10% methanol/methylene chloride gave N-hydroxy-
2-[1-(4-phenoxyphenylsulfinyl)-cyclopent-1-yl]-acetamide (15 mg),
1H NMR (CDCl3) 7.64 (d,2H), 7.44 (t,2H), 7.30-7.05 (m,SH), 2.97
(d, l H), 2.53 (d,1H), 2.15-1.65 (m,BH).
11 B . Preparation of Id where n is 1, R1 and R2 are Hydro eg-n-R3
and R4 when taken together with the Carbon to which they are
attached are Tetrah_ydropyran-4-yl, and RS is 4- 4-Fluorophenoxx)-
~'.nyl
2- { 4-[4-(4-Fluorophenoxy)phenylthio]-tetrahydropyran-4-yl }-
N-hydroxyacetamide (500 mg) was dissolved in methanol (25 ml).
OXONE (400 mg) in water (5 ml) was added. After stirring for 30
minutes, the mixture was partitioned between methylene chloride
2 0 and water. Preparative TLC on silica gel and elution with 10%
methanol/methylene chloride gave 2-{4-[4-(4-fluorophenoxy)phenyl-
sul.finyl]-tetrahydropyran-4-yl } -N-hydroxyacetamide (402 mg, m.p.
120°C).
2 5 11 C. Preparation of Id where n is 1, varying R 1 R2 R3 R4, and RS
Similarly, following the procedures of Example 11A or 11B above,
but replacing N-hydroxy-2-[1-(4-phenoxyphenylthio)-cyclopent-1-
yl]-acetamide with other compounds of Formula Id where n is 0,
3 0 other compounds of Formula Id where n is 1 are prepared, for
example;
N-hydroxy-2-[4-(4-phenoxyphenylsulfinyl)-N-CBZ-piperidin-4-
yl)]-acetamide;
N-hydroxy-2-[4-(4-phenoxyphenylsulfinyl)-piperidin-4-yl)]-
3 5 acetamide;
19~~~~
- 104 -
N-hydroxy-2-[4-(4-methoxyphenylsulfinyl)-N-CBZ-piperidin-4-
yl)]-acetamide;
2-{ 4-[4-(4-fluorophenoxy)phenylsulfinyl]-piperidin-4-yl }-
N-hydroxyacetamide;
N-hydroxy-2-[4-(4-methoxyphenylsulfinyl)-piperidin-4-yl)]-
acetamide;
2-benzyl-N-hydroxy-3-(4-methoxyphenylsulfinyl)-propionamide;
3-benzyl-N-hydroxy-3-(3-methoxyphenylsulfinyl)-propionamide;
3-benzyl-N-hydroxy-3-(4-methoxyphenylsulfinyl)-propionamide;
3-benzyl-N-hydroxy-3-[(4-phenylthiophenyl)sulfinyl]-
propionamide;
3-benzyl-N-hydroxy-3-(4-phenoxyphenylsulfinyl)-propionamide;
3-benzyl-N-hydroxy-3-[(4-biphenyl)sulfinyl]-propionamide;
3-benzyl-N-hydroxy-3-(2-naphthylsulfinyl)-propionamide;
3-benzyl-N-hydroxy-3-(4-methoxystyrylphenylsulfinyl)-
propionamide;
3-cyclopentylmethyl-N-hydroxy-3-(4-methoxyphenylsulfinyl)-
propionamide;
3-cyclopentylmethyl-N-hydroxy-2-isopropyl-
2 0 3-(4-methoxyphenylsulfinyl)-propionamide;
3-ethyl-N-hydroxy-2-methyl-3-(4-methoxyphenylsulfinyl)-
propionamide;
3,3-dimethyl-N-hydroxy-(4-methoxyphenylsulfinyl)-
propionamide;
N-hydroxy-2-[1-(4-methoxyphenylsulfinyl)-cyclopent-1-yl]-
acetamide;
N-hydroxy-2-[ 1-(4-methoxyphenylsulfinyl)-(4-methylcyclohex-
1-yl]-acetamide;
N-hydroxy-2- [ 1-(4-phenoxyphenylsulfinyl)-cyclohex-1-yl]-
3 0 acetamide;
N-hydroxy-2-[4-(4-methoxyphenylsulfinyl)-N-CBZ-piperidin-4-
yl)]-acetamide; and
N-hydroxy-2-[4-(4-methoxyphenylsulfinyl)-piperidin-4-yl)]-
acetamide.
3 5 N-hydroxy-2-[4-(4-phenoxyphenylsulfinyl)-tetrahydropyran-4-
1g3~7~
- 105 -
yl]-acetamide;
4-[4-(4-chlorophenoxy)phenylsulfinylmethyl]-tetrahydropyran-
4-(,?V-hydroxycarboxamide): mp 141.3-142.1 °C; IR (KBr) 3436 (br),
1649 cm-1; 1 H NMR (DMSO-d6) b 1.67 (dm, J = 13.9 Hz, 1 H), 1.79 (dm,
J = 13.9 Hz, 1 H), 1.97 (dm, J = 13.9 Hz, 1 H), 2.24 (dm, J = 13.9 Hz, 1 H),
2.9'7 (d, J = 13.7 Hz, 1H), 3.07 (d, J = 13.7 Hz, 1H), 3.33-3.54 (mc, 2H),
3.69 (mc, 2H), 7.12 (d, J = 8.9 Hz, 2H), 7.21 (d, J = 8.8 Hz, 2H), 7.48 (d, J
= 8.9 Hz, 2H), 7.66 (d, J = 8.8 Hz, 2H), 8.87 (br s, 1 H), 10.76 (s, 1 H),
13~JNMR (DMSO-d6) x32.43 (t), 33.71 (t), 42.69 (s), 63.65 (t), 67.12
(t), 118.90 (d), 121.07 (d), 126.11 (d), 128.19 (s), 130.07 (d), 139.51
(s), 154.62 (s), 158.72 (s), 169.68 (s); FABHRMS Calcd. for
C 1 c~H21 NS 05C1 (M+ + H): 410.0829 Found: 426.0825. Anal. Calcd. for
ClgH2pNS05Cl: C, 55.68; H, 4.92; N, 3.42. Found: C, 55.70; H, 4.93; N,
3.64.
2-{4-[4-(4-chlorophenoxy)-phenylsulfinyl]-tetrahydropyran-4-
yl } -N-hydroxyacetamide; and
N-hydroxy-2-[4-(4-phenoxyphenylsulfinyl)-
tetrahydrothiopyran- l , l -dioxide-4-yl]-acetamide.
2 0 EXAMPLE 12
Preparation of Compounds of Formula Id
12A. Preparation Id where n 2, R1 R2 are Hydrogen,
of is and R3
and R4 when taken ether with Carbon which they are
tog the to
2 5 attachedare Cyclopentyl. and R5 4-Phenoxy~hen~
is
A solution of N-hydroxy-2-[ 1-(4-phenoxyphenylthio)-cyclopent-
1-yl]-acetamide (45 mg) in methanol (4 ml) was treated with a
solution of OXONE (260 mg) in water (2 ml). The mixture was stirred
3 0 for 1 hour, then partitioned between methylene chloride and water.
The. organic layer was separated, and the solvent removed under
reduced pressure. Preparative TLC on silica gel and elution with 10%
methanol/methylene chloride gave N-hydroxy-2-[1-(4-
phenoxyphenylsulfonyl)-cyclopent-1-yl]-acetamide (20 mg), m/e -
3 5 393 (MNH4+, CIMS).
1~3~78
- 106 -
128. Preparation of Id where n is 2, R1 and R2 when taken
together with the Carbon to which they are attached represent
Tetrahydro~yran, R3 and R4 are Hydrogen, and RS is 4- 4-
Chloro~henox~)phen~
To a mechanically stirred suspension of 4-[4-(4-chlorophenoxy)-
phe.nylthiomethyl]tetrahydropyran-4-(N-hydroxycarboxamide) (59.8
g, 151.8 mmol) in 20% tetrahydrofuran-methanol ( 1570 ml) cooled to
1 0 5°C' was added dropwise a solution of OXONE (152 g, 247 mmol) in
water ( 1 litre), maintaining an internal temperature of 15-20°C. The
mixture was stirred for 5.5 hours, and the mixture then partitioned
between 30% ethyl acetate/water (3 litres). The aqueous layer was
washed with ethyl acetate (2 x 300 ml), the combined ethyl acetate
layers dried over magnesium sulfate, concentrated under reduced
pressure, and the residue crystallized from the minimum amount of
met:hylene chloride/hexanes, to afford analytically pure 4-[4-(4-
chlorophenoxy)phenylsulfonylmethyl]-tetrahydropyran-4-(N-
hydroxycarboxamide) as a white powder (54.2 g, 84%). mp 147.7-
148.9 °C; IR (KBr) 3429 (br), 1636 cm-1; 1HNMR (DMSO-d6) 8 1.70
(dire, J = 13.9, 2H), 1.96 (dm, J = 13.9 Hz, 2H), 3.38-3.48 (m, 2H), 3.58-
3.68 (m, 2H), 3.58-3.68 (m, 2H), 3.66 (s, 2H), 7.19 (d, J = 8.9 Hz, 2H),
7.19 (d, J = 8.9 Hz, 2H), 7.52 (d, J = 8.9 Hz, 2H), 7.85 (d, J = 8.9 Hz, 2H),
8.68 (d, J = 2.0 Hz, 1H), 10.54 (d, J = 2.0 Hz, 1H), 13CNMR (DMSO-d6) 8
2 5 32.83 (t), 41.70 (s), 61.02 (t), 63.19 (t), 118.01 (d), 121.71 (d), 128.73
(s), 130.08 (d), 130.19 (d), 135.20 (s), 153.83 (s), 160.86 (s), 168.96
(s); FABHRMS Calcd. for C19H2pNS06C1: 426.0778. Found: 426.0774.
Anal. Calcd. for C19H2pNS06C1: C, 53.59; H, 4.73; N, 3.29. Found: C,
53.58; H, 4.70; N, 3.40.
12C. Preparation of Id where n is 2, R1 and R2 when taken
together with the Carbon to which then are attached represent
Tetrah~drop,Yran, R3 is hXdro~en, R4 is Benzxl, and RS is 4- 4-
Chloro~phenox~)phen,~
~~317~
- 107 -
To a solution of 3-benzyl-4-[4-(4-chlorophenoxy)-
phenylsulfonylmethyl]-tetrahydropyran-4-carboxylic acid (316 mg,
0.63 mmol) and N, N-dimethylformamide catalyst ( 10 p.L) in
methylene chloride (6 ml;j at 0°C was added oxalyl chloride (200 ~L,
2.2CI mmol) dropwise over 10 minutes. The mixture was warmed to
room temperature over 1 hour, the partial slurry stirred an additional
8 hours, and concentrated in vacuo until the theoretical mass of the
acid. chloride was obtained. This mixture was dissolved in methylene
chloride (8 ml), cooled to 0°C, and a neat solution of N,O-
bis(trimethylsilyl)hydroxylamine (0.56 g, 3.15 mmol) added dropwise
over 5 minutes. The mixture was immediately warmed to room
temperature, stirred for 48 hours, and recooled to 0°C. To this
solution
was added aqueous 1M hydrochloric acid (5 ml, 150 mmol), and the
solution stirred for an additional 30 minutes, partitioned between
1 5 ethyl acetae ( 150 ml) and brine (50 ml). The organic layer was dried
over magnesium sulfate, concentrated in vacuo, chromatographed
over silica gel, eluted with 4% methanol/methylene chloride) to
afford 280 mg (86%) of 3-benzyl-4-[4-(4-chlorophenoxy)-
phenylsulfonylmethyl]-tetrahydropyran-4-(N-hydroxycarbamide)
hydroxamic acid. mp 108-113°C; IR (KBr) 3422 (br), 1653 cm-1;
1 HNMR (CDC13 ) 8 1.76-1.86 (m, 1 H), 2.08-2.27 (m, 2H), 2.34 (dm, J =
13.8 Hz, 1H), 2.91 (dd, J = 16.5, 7.2 Hz, 1H), 3.17 (dd, J = 16.4, 4.0 Hz,
~1 H), 3.19-3.23 (tm, J = 9.0 Hz, 1 H), 3.43 (td, J = 11.9, 2.4 Hz, 2H), 6.65-
6.7:? (m, 2H), 6.76 (d, J = 8.9 Hz, 2H), 6.88 (d, J = 8.8 Hz, 2H), 6.98-7.04
2 5 (m, 3H), 7.30 (d, J = 8.9 Hz, 2H), 7.49 (d, J = 8.8 Hz, 2H); 13CNMR
(CDC13) b 31.76 (t), 34.23 (tj, 47.30 (s), 64.07 (tj, 64.66 (t), 72.68 (d),
117.50 (d), 121.64 (d), 126.47 (d j, 127.96 (d), 128.53 (d), 130.31 (d),
136.69 (d), 132.91 (s), 137.83 (s), 153.34 (s), 162.12 (s), 171.30 (s);
FA:BMS (M+ +H): 516; Anal. Calcd. for C26H26NS06Cl: C, 60.52; H, 5.08;
3 0 N, 2.71. Found: C, 60.45; H, 5.10; N, 2.55.
12D. Preparation of Id where n is 2, var~g R1 R2 R3 R4, and RS
Similarly, following the procedures of Example 12C above, but
3 5 replacing 4-[4-(4-chlorophenoxy)phenylthiomethyl]-tetrahydro-
~9317~
- 108 -
pyran-4-(N-hydroxycarboxamide) with other compounds of Formula
Iba, the following compounds of Formula Id where n is 2 were
prepared:
4-[4-(4-fluorophenoxy)phenylsulfonylmethyl]tetrahydropyran-
4-(N-hydroxycarboxamide): mp 153.1-153.9 °C; IR (KBr) 3434 (br),
1636 cm-1; 1 HNMR (CDC13 ) b 1.87 (ddd, J = 13.6, 8.8, 4.0 Hz, 2H),
2.22 (dm, J = 13.6 Hz, 2H), 3.52-3.78 (m, 4H), 7.00-7.16 (m, 6H), 7.84
(d, .1 = 8.9 Hz, 2H); 13CNMR (CDCl3) 8 33.12 (t), 42.19 (s), 62.52 (t),
63.96 (t), 116.88 (dd, JC_F = 21.3 Hz), 117.30 (d), 121.97 (dd, JC-F =
1 0 8.4 Hz), 130.18 (s), 134.21 (d), 150.66 (d, JC_F = 2.6 Hz), 159.73 (d, JC-
F =: 243.8 Hz), 162.61 (s), 169.73 (s); FABMS (M+ + H): 410. Anal. Calcd.
for C19H2pNS06F: C, 55.74; H, 4.92; N, 3.42. Found: C, 55.45; H, 4.91;
N, 3.38.
4-[4-(4-bromophenoxy)phenylsulfonylmethyl] tetrahydropyran-
4-(,?V-hydroxycarboxamide): mp 150.1-151.0 °C; IR (KBr) 3432 (br),
1636 cm- l ; 1 HNMR (CDC13 ; NH and OH not observed) 7 1.87 (ddd, J =
13.6, 8.7, 3.9 Hz, 2H), 2.12 (dm, J = 13.6 Hz, 2H), 3.52 (s, 2H), 3.62-3.80
(m, 4H), 6.97 (d, J = 8.8 Hz, 2H), 7.06 (d, J = 8.8 Hz, 2H), 7.52 (d, J = 8.8
Hz, 2H), 7.85 (d, J = 8.8 Hz, 2H); 13CNMR (CDC13) 8 33.10 (t), 42.16 (s),
2 0 62.49 (t), 63.93 (t), 117.66 (s), 117.83 (d), 121.93 (d), 130.20 (d),
13 3.17 (d), 134.61 (s), 154.13 (s), 161.79 (s), 169.53 (s); FABHRMS
Calcd. for C19H2pNS06Br (M+ + H): 470.0273. Found: 470.0268. Anal.
Calcd. for C19H20NS06Br: C, 48.51; H, 4.28; N, 2 .98. Found: C, 48.29; H,
4.02; N, 2.94.
2 5 3-(4-benzoylphenylsulfonyl)-2,2-dimethyl-N-hydroxy-
propionamide;
3-[4-(4-chlorophenoxy)phenylsulfonyl]-2,2-dimethyl-
N-hydroxypropionamide: mp 154.9-156.1 °C; 1HNMR (CDC13) 8 1.45 (s,
6H), 3.48 (s, 2H), 7.02 (d, J = 8.9 Hz, 2H), 7.04 (d, J = 8.9 Hz, 2H), 7.38
3 0 (d, J = 8.9 Hz, 2H), 7.85 (d, J = 8.9 Hz, 2H); FABMS (M+ +H): 384Ø Anal.
Calcd. for C17H18NS05Cl: C, 53.19; H, 4.73; N, 3.65. Found: C, 52.98; H,
4.69; N, 3.73.
4-(4-phenoxyphenylsulfonylmethyl)-tetrahydropyran-
4-(N-hydroxycarboxamide): mp 141.8-142.9 °C; IR (KBr) 3432 (br),
~19~1~g
- 109 -
1636 cm-1; 1 H NMR (DMSO-d6) 8 1.74 (ddd, J = 13.8, 10.0, 3.9 Hz, 2H),
1.98 (dm, J = 13.8 Hz, 2H), 3.45 (mc, 2H), 3.64 (mc, 2H), 3.65 (s, 2H),
7.15 (d, J = 8.8 Hz, 2H), 7.26 (d, J = 7.5 Hz, 2H), 7..47 (t, J = 7.5 Hz, 1H),
7.85 (d, J = 8.8 Hz, 2H), 8.68 (s, 1H), 10.52 (;s, 1H); 13C NMR (DMSO-d6)
8 32.87 (t), 41.76 (s), 61.19 (t), 63.28 (t), 117.71 (d), 119.99 (d),
124.91 (d), 130.04 (d), 130.34 (d), 134.85 (s), 154.85 (s), 161.39 (s),
168.97 (s); FABHRMS Calcd. for C19H22NS06 (M+ + H): 392.1168.
Found: 392.1162. Anal. Calcd. for C19H21NS06Ø5H20: C, 56.99; H,
5.54; N, 3.50. Found: C, 57.06; H, 5.35; N, 3.93.
4-[4-(4-thiophen-2-yl)phenoxyphenylsulfonylmethyl]-
tetrahydropyran-4-(N-hydroxycarboxamide): mp 172.2-176.5 °C; IR
(KBr) 3428 (br), 1636 cm-1; 1 H NMR (DMSO-d6) b 1.72 (dm, J = 14.5
Hz, 2H), 1.99 (dm, J = 14.5 Hz, 2H), 3.46 (mc, 2H), 3.65 (mc, 2H), 3.66
(s, 'ZH), 7.14 (dd, J = 4.9, 3.6 Hz, l H), 7.19 (d, J = 8.7 Hz, 2H), 7.20 (d,
J =
1 5 8.9 Hz, 2H), 7.48 (dd, J = 3.6, 1.2 Hz, 1H), 7.52 (dd, J = 4.9, 1.2 Hz,
1H),
7.73 (d, J = 8.8 Hz, 2H), 7.86 (d, J = 8.8 Hz, 2H), 8.68 (s, 1H), 12.58 (s,
1H); 13CNMR (DMSO-d6) 8 32.89 (t), 41.78 (s;), 61.20 (t), 63.28 (t),
117.88 (d), 120.55 (d), 123.66 (d), 125.56 (d), 127.34 (d), 128.45 (d),
130.07 (d), 130.62 (s), 135.04 (s), 142.45 (s), 154.30 (s), 161.16 (s),
2 0 169.03 (s); FABHRMS Calcd. for C23H24NS206 (M+ + H): 474.1045.
Found: 474.1050. Anal. Calcd. for C23H23NS206: C, 58.33; H, 4.90; N,
3.00. Found: C, 58.18; H, 4.84; N, 3.19.
4-[4-(4-thiophen-3-yl)phenoxyphenylsulfonylmethyl]-
tetrahydropyran-4-(N-hydroxycarboxamide): mp 183.5-184.4 °C; IR
2 5 (KBr) 3432 (br), 1636 cm-1; 1 H NMR (DMSO-d6) 8 1.72 (mc, 2H), 1.98
(m,~, 2H), 3.48 (mc, 2H), 3.65 (mc, 4H), 7.18 (mc, 4H), 7.55 (dd, J = 5.1
Hz, 1H), 7.62 (d, .7 = 4.9, 3.7 Hz, 2H), 7.80 (d, J = 8.6 Hz, 2H), 7.86 (mc,
3H), 8.69 (s, 1H), 10.58 (s, 1H); 13C NMR (DMSO-d6) 8 32.88 (t), 41.79
(s), 61.19 (t), 63.28 (t), 117.71 (d), 120.42 (d), 120.81 (d), 126.09 (d),
3 0 127.10 (d), 127.97 (d), 130.06 (d), 132.10 (s), 134.89 (s), 140.54 (s),
153.86 (s), 168.85 (s); FABHRMS Calcd. for C23H24NS206 (M+ + H):
474.1045. Found: 474.1049. Anal. Calcd. for C23H23NS206Ø75H20: C,
56.72; H, 5.07; N, 2.88. Found: C, 56.74; H, 4.78; N, 3.22.
3,3-dimethyl-3-[(4-chlorophenoxy)phenylsulfonyl]-N-hydroxy-
X193178
- 110 -
propionamide;
{ 4-[4-(4-benzo[b]thiophen-2-yl-phenoxy)phenylsulfonyl)-
tetrahydropyran-4-yl]-N-hydroxyacetamide;
2- { 4-[4-(phenylmethyl)phenylsulfonyl]-tetrahydropyran-4-yl }-
N-hydroxyacetamide;
2- { 4-[4-(4-chlorophenoxy)phenylsulfonyl]-tetrahydropyran-4-
yl } ~-N-hydroxyacetamide; and
2- { 4-[4-(4-bromophenoxy)phenylsulfonyl]-tetrahydropyran-4-
yl } ~-N-hydroxyacetamide.
12E. Preparation of Id where n is 2, var~ng R1 R2 R3 R4. and RS
Similarly, following the procedures of Example 12A or 12B above,
but replacing N-hydroxy-2-[1-(4-phenoxyphenylthio)-cyclopent-1-
yl]-acetamide with other compounds of Formula Id where n is 0, the
following compounds of Formula Id where n is 2 are prepared, for
example;
4-(4-phenoxyphenylsulfonylmethyl)tetrahydropyran-
4-(.N-hydroxycarboxamide);
2 0 4-[4-(4-fluorophenoxy)phenylsulfonylmethyl]tetrahydropyran-
4-(.N-hydroxycarboxamide);
4-[4-(4-chlorophenoxy)phenylsulfonylmethyl]piperidine-
4-(N-hydroxycarboxamide);
4-[4-(4-chlorophenoxy)phenylsulfonylmethyl]-1-methyl-
2 5 piperidine-4-(N-hydroxycarboxamide);
4-[4-(4-chlorophenoxy)phenylsulfonylmethyl]-1-cyclopropyl-
methylpiperidine-4-(N-hydroxycarboxamide);
4-[4-(4-chlorophenoxy)phenylsulfonylmethyl]-1-acetyl-
piperidine-4-(N-hydroxycarboxamide);
3 0 4-[4-(4-chlorophenoxy)phenylsulfonylmethyl]-1-(3-pyridyl)-
piperidine-4-(N-hydroxycarboxamide);
4-[4-(4-chlorophenoxy)phenylsulfonylmethyl]-1-(3-pyridoyl)-
piperidine-4-(N-hydroxycarboxamide);
N-hydroxy-2-[4-(4-phenoxyphenylsulfonyl)-N-CBZ-piperidin-4-
3 5 yl)]-acetamide;
~ 93178
- 111 -
N-hydroxy-2-[4-(4-methoxyphenylsulfonyl)-N-CBZ-piperidin-4-
yl)]-acetamide;
2-{4-[4-(4-fluorophenoxy)phenylsulfonyl]-N-CBZ-piperidin-4-yl}-
N-hydroxyacetamide;
2-{4-[4-(4-fluorophenoxy)phenylsulfonyl]-piperidin-4-yl}-
N-hydroxyacetamide;
N-hydroxy-2-[4-(4-methoxyphenylsulfonyl)-piperidin-4-yl)]-
acetamide;
N-hydroxy-2-[4-(4-phenoxyphenylsulfonyl)-piperidin-4-yl)]-
acetamide;
2-benzyl-N-hydroxy-3-(4-methoxyphenylsulfonyl)-
propionamide;
3-benzyl-N-hydroxy-3-(3-methoxyphenylsulfonyl)-
propionamide;
3-benzyl-N-hydroxy-3-(4-methoxyphenylsulfonyl)-
propionamide;
3-benzyl-N-hydroxy-3-[(4-phenylthiophenyl)sulfonyl]-
propionamide;
3-benzyl-N-hydroxy-3-(phenylsulfonyl)-propionamide;
2 0 3-benzyl-N-hydroxy-3-(4-phenoxyphenylsulfonyl)-
propionamide;
3-benzyl-3-[(4-biphenyl)sulfonyl]-N-hydroxypropionamide;
3-benzyl-N-hydroxy-3-(2-naphthylsulfonyl)-propionamide;
3-benzyl-N-hydroxy-3-(4-methoxystyrylphenylsulfonyl)-
2 5 propionamide;
3-(cyclopentylmethyl)-N-hydroxy-3-(4-methoxyphenylsulfonyl)-
propionamide;
3-(cyclopentylmethyl)-N-hydroxy-2-isopropyl-3-(4-
methoxyphenyl-sulfonyl)-propionamide;
3 0 3-ethyl-N-hydroxy-3-(4-methoxyphenylsulfonyl)-2-
methylpropionamide;
3,3-dimethyl-N-hydroxy-(4-methoxyphenylsulfonyl)-
propionamide;
N-hydroxy-2-[1-(4-methoxyphenylsulfonyl)-cyclopent-1-yl]-
3 5 acf;tamide;
19~~7'8
- 112 -
N-hydroxy-2-[ 1-(4-methoxyphenylsulfonyl)-(4-methylcyclohex-
1-yl]-acetamide;
N-hydroxy-2-[ 1-(4-phenoxyphenylsulfonyl)-cyclohex-1-yl]-
acetamide;
N-hydroxy-2-[4-(4-phenoxyphenylsulfonyl)-tetrahydropyran-4-
yl] ~-acetamide;
2- { 4-[4-(4-chlorophenoxy)phenylsulfonyl]-tetrahydropyran-4-
yl }-N-hydroxyacetamide;
2-{ 4-[4-(4-fluorophenoxy)phenylsulfonyl]-tetrahydropyran-4-
yl}-N-hydroxyacetamide; and
N-hydroxy-2-[4-(4-phenoxyphenylsulfonyl)-tetrahydrothio-
pyran-1,1-dioxide-4-yl]-acetamide.
12F. Preparation of Id where n is 2, varying R1~,R2 R3 R4, and RS
Similarly, following the procedures of Example 12A above, but
replacing N-hydroxy-2-[1-(4-phenoxyphenylthio)-cyclopent-1-yl]-
acetamide with other compounds of Formula Id where n is 0, other
compounds of Formula Id where n is 2 are prepared.
EXAMPLE 13
Preparation of Compounds of Formula I where Y is tert-BuONH-
13.A. Preparationof Ic where is 2, R1 and R2 are H,~rogen,
n R3
2 5 and R4 when takentogether the Carbon to which they are
with
attached are Tetrahydropyran, RS is 4-PhenoxYphenyl
and
To a cooled solution of N-tert-butoxy-2-[4-(4-phenoxyphenyl-
thio)-tetrahydropyran-4-yl]-acetamide ( 14.1 g, 33.9 mmol) in
3 0 methanol (340 ml) was added a solution of OXONE (33.9 g) in water
(1 i'0 ml). The reaction mixture was stirred for 5 hours at room
temperature, concentrated to half the original volume under reduced
pressure, and the residue then partitioned between ethyl acetate and
water. The solvent was removed from the ethyl acetate extracts
3 5 under reduced pressure. The residue chromatographed on silica gel,
~~~1~'8
- 113 -
eluting with 10% methanol/methylene chloride, to give N-tert-
butoxy-2-[4-(4-phenoxyphenylsulfonyl)-tetrahydropyran-4-yl]-
acetamide as a white foam.
13B. Preparation Ic where n is 2, R3 and R4 H.~gen, R1
of are
and R2 when taken ether with the Carbon to they are
tog which
attached are N-BOC-Pi peridine, and RS is 4-(4-ChlorophenoxX)phenXl
To a solution of N-tert-butoxy-2-[4-(4-phenoxyphenylthio-
methyl)-N-BOC-piperidin-4-yl]-carboxamide (4.96 g, 9.03 mmol) in
anhydrous methylene chloride (70 ml) cooled to 0°C, was added 60%
3-chloroperoxybenzoic acid (4.96 g). After the resulting mixture was
allowed to warm to room temperature over 30 minutes and stirred
for 5 minutes, 13.6M aqueous methyl sulfide ( 1 ml, 13.62 mmol) was
1 S added in one portion. The mixture was stirred 10 minutes, partitioned
with saturated aqueous sodium bicarbonate (2 x 50 ml), dried over
magnesium sulfate, and concentrated in vacuo. Chromatography over
silica gel, and eluting with 25% ethyl acetate/hexanes, gave
N-,tert-butoxy-2-[4-(4-phenoxyphenylsulfonylmethyl)-
2 0 N-BOC-piperidin-4-yl]-carboxamide as a white foam (4.70 g, 90%).
1HNMR (CDC13) 8 1.31 (s, 9H), 1.46 (s, 9H), 1.59 (mc, 2H), 2.18 (mc,
2H), 3.42 (mc, 2H), 3.45 (s, 2H), 3.62 (mc, 2H), 7.01 (d, J = 8.9 Hz, 2H),
7.04 (d, J = 8.8 Hz, 2H), 7.38 (d, J = 8.8 Hz, 2H), 7.84 (d, J = 8.8 Hz, 2H),
8.44 (br s, 1H).
13C. Preparation of Ic where n is 2 and Y is tert-BuONH-, varying
R1 R2 R3 R4~and RS
Similarly, following the procedures of Example 13B above, but
3 0 reI>lacing N-tert-butoxy-2-[4-(4-phenoxyphenylthiomethyl)-
N-BOC-piperidin-4-yl]-carboxamide with other compounds of Formula
Ib, the following compound of Formula Ic where n is 2 and Y is tert-
BuONH- was prepared:
N-tert-butoxy-4-[4-(4-pyridyloxy)phenylsulfonylmethyl]-
3 5 tetrahydropyran-carboxamide: IR (KBr) 3434, 1684 cm- I ; 1 H N M R
193~~'8
- 114 -
(CDC13) 8 1.33 (s, 9H), 2.01 (mc, 2H), 2.24 (mc, 2H), 3.55 (s, 2H), 3.79
(m~;, 4H), 6.93 (d, J =6.3 Hz, 2H), 7.22 (d, J = 8.8 Hz, 2H), 7.96 (d, J = 8.8
Hz, 2H), 8.38 (s, 1 H), 8.57 (d, J = 6.3 Hz, 2H); FABHRMS Calcd. for
C2pH28N2S06 (M+ + H) 449.1746. Found: 449.1757.
N-tert-butoxy-4-[4-(5-chloro-2-pyridyloxy)phenylsulfonyl-
met.hyl]-tetrahydropyran-carboxamide: mp (broad) 100.8-135.8 °C; IR
(KBr) 3436 (br), 1684 cm-1; 1HNMR (DMSO-d6) 8 1.20 (s, 9H), 1.72
(m~,, 2H), 2.03 (mc, 2H), 3.48 (mc, 2H), 3.67 (mc, 2H), 3.76 (s, 2H), 7.23
(dd, J = 8.8, 0.5 Hz, 1H), 7.41 (d, J = 8.8 Hz, 2H), 7.91 (d, J = 8.8 Hz, 2H),
1 0 8.03 (dd, J = 8.8, 2.7 Hz, 1H), 8.25 (dd, J = 2.7, 0.5 Hz, 1H), 8.30 (s,
1H),
10.:32 (s, 1H); 13CNMR (DMSO-d6) b 26.66 (q), 33.09 (t), 42.37 (s),
61.()3 (t), 63.36 (t), 80.64 (s), 113.89 (d), 121.38 (d), 126.33 (s),
129.53 (d), 137.00 (s), 140.34 (d), 145.74 (d), 157.87 (s), 160.66 (s),
171.25 (s); FABHRMS Calcd. for C22H28N2S06C1 (M+ + H): 483.1357.
1 5 Found: 483.1354. Anal. Calcd. for C22H27N2S06C1: C, 54.71; H, 5.63; N,
5.8(). Found: C, 54.46; H, 5.60; N, 5.98.
N-tert-butoxy-3-[4-(5-c,hloro-2-pyridyloxy)phenylsulfonyl]-
2,2~-dimethyl-propionamide: mp (broad) 64.5-70.5 °C; 1HNMR (DMSO-
d6) 8 1.19 (s, 9H), 1.29 (s, 6H), 3.65 (s, 2H), 7.24 (d, J = 8.7 Hz, 1H),
2 0 7.41 (d, J = 8.8 Hz, 2H), 7.91 (d, J = 8.8 Hz, 2H), 8.04 (dd, J = 8.8, 2.7
Hz,
1H), 8.26 (d, J = 2.7 Hz, 1H), 10.17 (s, 1H); 13C NMR (DMSO-d6) 8 25.01
(q), 26.47 (q), 40.74 (s), 63.03 (t), 80.79 (s), 113.91 (d), 121.38 (d),
126.32 (s), 129.35 (d), 130.66 (s), 140.36 (d), 145.75 (d), 157.72 (s),
160.68 (s), 173.14 (s); FABHRMS Calcd. for C20H26N2SO5Cl (M+ + H):
2 5 441.1251. Found: 441.1248. Anal. Calcd. for C2pH25N2S05C1: C, 54.48;
H, 5.71; N, 6.35. Found: C, 54.37; H, 5.69; N, 6.57.
13D. Preparation of Ic where n is 2 and Y is tert-BuONH-, vary
R1, R2 R3 R4, and R5
Similarly, following the procedures of Example 13A above, but
replacing N-tert-butoxy-2-[4-(4-phenoxyphenylthio)-tetrahydro-
pyran-4-yl]-acetamide with other compounds of Formula Ib, the
following compounds of Formula Ic where n is 2 and Y is tert-BuONH-
~1~31~8
- 115 -
were prepared;
N-tert-butoxy-2-[4-(4-phenoxyphenylsulfonyl)-N-CBZ-piperidin-
4-yl)]-acetamide;
N-tert-butoxy-2-[4-(4-methoxyphenylsulfonyl)-N-CBZ-piperidin-
4-yl)]-acetamide;
N-tert-butoxy-2-{ 4-[4-(4-fluorophenoxy)phenylsulfonyl]-
piperidin-4-yl }-acetamide;
N-tert-butoxy-2-[4-(4-methoxyphenylsulfonyl)-piperidin-4-yl)]-
acetamide;
N-tert-butoxy-2-[4-(4-phenoxyphenylsulfonyl)-piperidin-4-yl)]-
acetamide;
2-benzyl-N-tert-butoxy-3-(4-methoxyphenylsulfonyl)-
propionamide;
3-benzyl-N-tert-butoxy-3-(3-methoxyphenylsulfonyl)-
propionamide;
3-benzyl-N-tert-butoxy-3-(4-methoxyphenylsulfonyl)-
propionamide;
3-benzyl-N-tert-butoxy-3-[(4-phenylthiophenyl)sulfonyl]-
propionamide;
2 0 3-benzyl-N-tert-butoxy-3-(phenylsulfonyl)-propionamide;
3-benzyl-N-tert-butoxy-3-(4-phenoxyphenylsulfonyl)-
propionamide;
3-benzyl-N-tert-butoxy-3-[(4-biphenyl)sulfonyl]-propionamide;
3-benzyl-N-tert-butoxy-3-(2-naphthylsulfonyl)-propionamide;
2 5 3-benzyl-N-tert-butoxy-3-(4-methoxystyrylphenylsulfonyl)-
propionamide;
N-tert-butoxy-3-(cyclopentylmethyl)-3-(4-methoxyphenyl-
sulfonyl)-propionamide;
N-tert-butoxy-3-(cyclopentylmethyl)-2-isopropyl-
3 0 3-(4-methoxyphenylsulfonyl)-propionamide;
N-tert-butoxy-3-ethyl-2-methyl-3-(4-methoxyphenylsulfonyl)-
propionamide;
N-tert-butoxy-3,3-dimethyl-(4-methoxyphenylsulfonyl)-
propionamide;
3 5 N-tert-butoxy-2-[1-(4-methoxyphenylsulfonyl)-cyclopent-1-yl]-
~1931~'8
- 116 -
acetamide;
N-tert-butoxy-2-[ 1-(4-methoxyphenylsulfonyl)-(4-
methylcyclohex-1-yl]-acetamide;
N-tert-butoxy-2-[4-(4-phenoxyphenylsulfonyl)-cyclohexanone-4-
yl]-~acetamide ethylene ketal;
N-tert-butoxy-2-[ 1-(4-phenoxyphenylsulfonyl)-cyclohex-1-yl]-
acetamide;
N-tert-butoxy-2-[4-(4-phenoxyphenylsulfonyl)-tetrahydropyran-
4-yl]-acetamide;
N-tert-butoxy-2-{4-[4-(4-chlorophenoxy)phenylsulfonyl]-
tetrahydropyran-4-yl }-acetamide;
N-tert-butoxy-2- { 4-[4-(4-fluorophenoxy)phenylsulfonyl]-
tetrahydropyran-4-yl } -acetamide;
N-tert-butoxy-2-[4-(4-phenoxyphenylsulfonyl)-tetrahydrothio-
pyran-l,l-dioxide-4-yl]-acetamide;
N-tert-butoxy-2-(4-methoxyphenylsulfonyl)-
cyclohexanecarboxamide; and
N-tert-butoxy-traps-2-(4-methoxyphenylsulfonyl)-
cyc;lopentanecarboxamide.
13E. Preparation of Ic where n is 2, varying-R1 R2 R3 R4, and RS
Similarly, following the procedures of Example 13A above, but
replacing N-tert-butoxy-2-[4-(4-phenoxyphenylthio)-N-CBZ-
2 5 piperidin-4-yl)]-acetamide with other compounds of Formula Ib,
other compounds of Formula Ic where n is 2 and Y is tert-BuONH- are
prepared.
EXAMPLE 14
3 0 Preparation of Compounds of Formula Ic where Y is tert-BuONH-
14 A. Preparation of Ic where n is 2, R1 and R2 are H,~ro eg_ nR3
and R4 when taken together with the Carbon to which they
attached are Piperidine, and R5 is 4-Phenoxy~hen~
. - 21 9 3 '1 7 ~
- 117 -
To a solution of N-tert-butoxy-2-[4-(4-phenoxyphenylsulfonyl)-
N-CBZ-piperidin-4-yl)]-acetamide ( 1.2 g, 2.1 mmol) in ethanol (21 ml)
was added 10% palladium on carbon ( 1 g) and ammonium formate
(6.7 g), and the mixture refluxed for 1 hour. The mixture was filtered
through Celite~ the filter cake washed with ethanol ( 150 ml) followed
by 10% methanol in methylene chloride ( 150 ml). Solvent was
removed from the filtrate under reduced pressure and the residue
was dissolved in hot ethyl acetate. Filtration, concentration of the
filtrate, ~ followed by silica gel chromatography and elution with 10%
methanol/methylene chloride gave N-tert-butoxy-2-[4-(4-
phenoxyphenylsulfony()-piperidin-4-yl)]-acetamide as a colorless oil.
14B. Preparation of Ic where n is 2 var ing- R1 R2 R3 R4, and R5
Similarly, following the procedures of Example 14A above, but
replacing N-tert-butoxy-2-[4-(4-phenoxyphenylsulfonyl)-N-CBZ-
piperidin-4-y1)]-acetamide with other N-CBZ protected compounds of
Formula I, other compounds of Formula I where n is 2 and Y is tert-
BuONH- are prepared.
EXAMPLE 15
Preparation of Compounds of Formula Id where Y is HONH-
15 A. Preparation of Id where n is 2 R 1 and R2 are Hxdrogen, R3
2 5 and R4 when taken together with the Carbon to which they are
attached are Pi~eridine, and R5 is 4-PhenoxXphen~
A solution of N-teat-butoxy-2-[4-(4-phenoxyphenylsulfonyl)
piperid-4-yl)]-acetamide (27 mg, 0.05 mmol) in dichloroethane (2 ml)
3 0 was cooled to -2.0°C, and saturated with hydrochloric acid gas for
30
minutes. The reaction vessel was then sealed and the solution stirred
for two days at 25°C. Solvent was removed from the reaction mixture
under reduced pressure, and the residue dissolved in 50% methanol
in methylene chloride. Addition of hexane precipitated N-hydroxy-2-
3 5 [4-(4-phenoxyphenylsulfonyl)-piperidin-4-yl)]-acetamide, m/e - 391
* 'Trademark
1~3~~'$
- 118 -
(MH+, FAB).
158. Preparation of Id where n is 2, var,~ring R1 R2 R3 R4, and R5
Similarly, following the procedures of Example 15A above, but
replacing N-tert-butoxy-2-[4-(4-phenoxyphenylsulfonyl)-piperidin-
4-yl)]-acetamide with other compounds of Formula Ic where Y is tert-
BuONH-, the following compounds of Formula Id where n is 2 and Y is
HONH- were prepared:
N-hydroxy-2-[4-(4-phenoxyphenylsulfonyl)-N-CBZ-piperidin-4-
yl)]-acetamide, m/e = 525 (MH+) ;
N-hydroxy-2-[4-(4-methoxyphenylsulfonyl)-N-CBZ-piperidin-4-
yl)]-acetamide, m/e = 463 (MH+, FAB);
2-{ 4-[4-(4-fluorophenoxy)phenylsulfonyl]-piperidin-4-yl }-
N-hydroxyacetamide, m.p. 196-197°C;
2-{4-[4-(4-chlorophenoxy)phenylsulfonyl]-piperidin-4-yl}-
N-hydroxyacetamide, m.p. 200-201 °C;
2- { 4-[4-(4-chlorophenoxy)phenylsulfonyl]-tetrahydropyran-4-
yl}-N-hydroxyacetamide: mp 135.7-136.1 °C; 1HNMR (CDC13) b 1.60
2 0 (m~,, 2H), 1.83 (mc, 2H), 3.00 (s, 2H), 3.66 (mc, 2H), 3.88 (mc, 2H), 7.06
(d, J = 8.8 Hz, 2H), 7.09 (d, J = 8.8 Hz, 2H), 7.42 (d, J = 8.9 Hz, 2H), 7.79
(d, J = 8.9 Hz, 2H), 7.25 (s, 1H), 9.49 (s, 1H); FABHRMS Calcd. for
C 1 ~H2pNS 06C1 (M+ + H): 426.0778. Found: 426.0775. Anal. Calcd. for
C 1 yH2pNS06Cl: C, 53.59; H, 4.73; N, 3.29. Found: C, 53.30; H, 4.67; N,
2 5 3.35.
2-[4-(4-cyclohexyloxyphenylsulfonyl]-tetrahydropyran-4-yl }-
N-hydroxyacetamide: m.p. 77-78°C;
N-hydroxy-2-[4-(4-methoxyphenylsulfonyl)-piperidin-4-yl)]-
acetamide, m/e = 329 (MH+);
3 0 N-hydroxy-2-[4-(4-phenoxyphenylsulfonyl)-piperidin-4-yl)]-
acetamide, m/e = 391 (MH+);
2-benzyl-N-hydroxy-3-(4-methoxyphenylsulfonyl)-
prapionamide, m/e = 350.2 (MH+);
3-benzyl-N-hydroxy-3-(3-methoxyphenylsulfonyl)-
3 5 propionamide, m/e = 350.2 (MH+);
~93~~g
- 119 -
3-benzyl-N-hydroxy-3-(4-methoxyphenylsulfonyl)-
propionamide, m/e = 350.2 (MH+);
3-benzyl-N-hydroxy-3-[(4-phenylthiophenyl) sulfonyl]-
propionamide, m/e = 427 (MH+);
3-benzyl-N-hydroxy-3-(phenylsulfonyl)-propionamide,
m/e: = 320 (MH+);
3-benzyl-N-hydroxy-3-(4-phenoxyphenylsulfonyl)-
propionamide, m/e = 412.2 (MH+);
3-benzyl-N-hydroxy-3-[(4-biphenyl)sulfonyl]-propionamide;
1 0 m/e = 395 (MH+);
3-benzyl-N-hydroxy-3-(2-naphthylsulfonyl)-propionamide,
m/e: = 370.1 (MH+);
3-benzyl-N-hydroxy-3-[(4-methoxystyrylphenylsulfonyl]-
propionamide, m/e = 452.2 (MH+);
3-(cyclopentylmethyl)-N-hydroxy-3-(4-methoxyphenylsulfonyl)-
propionamide, m/e = 342 (MH+);
3-(cyclopentylmethyl)-N-hydroxy-2-isopropyl-3-(4-methoxy-
phenylsulfonyl)-propionamide;
3-ethyl-N-hydroxy-2-methyl-3-(4-methoxyphenylsulfonyl)-
2 0 propionamide, m/e = 301 (MH+);
3,3-dimethyl-3-(4-methoxyphenylsulfonyl)-N-hydroxy-
propionamide, elemental analysis: C 1 H 1 N ;
N-hydroxy-2-[4-(4-methoxyphenylsulfonyl)-cyclopent-1-yl]-
acetamide, m/e = 313 (MH+);
2 5 N-hydroxy-2-[4-(4-methoxyphenylsulfonyl)-(4-methylcyclohex-
1-yl]-acetamide, m/e = 341 (MH+);
N-hydroxy-2-[4-(4-phenoxyphenylsulfonyl)-cyclohex-1-yl]-
acetamide, m/e = 389 (MH+);
N-hydroxy-2-[4-(4-phenoxyphenylsulfonyl)-tetrahydropyran-4-
3 0 yl]-~acetamide, m.p. 88.5-90°C, m/e = 391 (MH+);
2- { 4-[4-(4-chlorophenoxy)phenylsulfonyl]-tetrahydropyran-4-
yl}-N-hydroxyacetamide;
2- { 4-[4-(4-fluorophenoxy)phenylsulfonyl]-tetrahydropyran-4-
yl}-N-hydroxyacetamide, m.p. 91-95°C;
3 5 N-hydroxy-2-[4-(4-phenoxyphenylsulfonyl)-tetrahydro-
:193178
- 120 -
thiopyran-l,l-dioxide-4-yl]-acetamide, m/e = 440.1 (MH+);
N-hydroxy-traps-2-(4-methoxyphenylsulfonyl)-
cyclopentanecarboxamide, m/e = 313 (MH+);
N-hydroxy-traps-2-(4-methoxyphenylsulfonyl)-
cyclohexanecarboxamide, m/e = 327 (MH+); and
2-benzyl-N-hydroxy-traps-2-(4-methoxyphenylsulfonyl)-
cyclopentane-carboxamide, m/e = 390 (MH+, FABMS).
15C'.. Preparation of Id where n is 2. varying R1 R2 R3 R4, and RS
Similarly, following the procedures of Example 15A above, but
rep acing N-tert-butoxy-2-[4-(4-phenoxyphenylsulfonyl)-piperidin-
4-yl)]-acetamide with other compounds of Formula Ic where Y is tert-
BuONH-, other compounds of Formula Id where n is 2 and Y is HONH-
are prepared, for example:
2- { 4-[4-(4-fluorophenoxy)phenylsulfonyl]-N-CBZ-piperidin-4-yl } -
N-hydroxyacetamide;
2-{ 1-methyl-4-[4-(4-chlorophenoxy)-phenylsulfonyl]-piperidin-
4-yl }-N-hydroxyacetamide;
N-hydroxy-2-{ 1-methyl-4-[4-(4-fluorophenoxy)-phenylsulfonyl]-
piperidin-4-yl }-acetamide; and
2-{ 4-[4-(4-bromophenoxy)-phenylsulfonyl]-tetrahydropyran-4-
yl}-N-hydroxyacetamide.
2 5 15I). Preparation of Id where n is 2. R1 and R2 are H.~~en, R3
and R4 when taken together with the Carbon to which they are
attached are Cyclohexanone, and RS is 4-PhenoxXphenyl
Following the procedure outlined in Example 15A, N-hydroxy-
3 0 2-[4-(4-phenoxyphenylsulfonyl)-cyclohexanone-4-yl]-acetamide
ethylene ketal (400 mg) was prepared from the corresponding N-
ter;!-butoxy precursor. The above product was dissolved in a 1:1
mixture of acetone and 1M hydrochloric acid (40 ml) and stirred at
room temperature for 18 hours. The reaction was concentrated under
3 5 reduced pressure and extracted with ethyl acetate. Silica gel
19378
- 121 -
S
chromatography using 10% methanol/methylene chloride gave
2-[4-(4-phenoxyphenylsulfonyl)cyclohexanone-4-yl]-
N-hydroxyacetamide as a white solid: m.p. 106°C (dec), m/e = 404
(MH+, FABMS).
15E. Preparation Id where n is 2, R3 R4 are Hydrogen,
of and R1
and when taken ether with the Carbon which they
R2 tog to
attachedare Piperidine,and RS is 4-(4-Chloronhenox )Y ohen~
1 0 To a sealed tube containing the free base N-tert-butoxy-2- { 4-[4-
(4-phenoxy)phenylsulfonylmethyl]-piperidin-4-yl}-carboxamide (780
mg, 1.62 mmol) in 1,2-dichloroethane (35 ml) at -30°C, was bubbled
in gaseous hydrochloric acid until the saturation point was reached.
The reaction vessel was then sealed and the solution stirred for two
15 days. After the vessel was recooled to -30°C and opened, a stream of
nitrogen gas bubbled through the solution, which was then warmed to
room temperature. The mixture was concentrated to afford 2-{4-[4-
(4-chlorophenoxy)phenylsulfonylmethyl]-piperidin-4-yl }-N-
hydroxycarboxamide (747 mg, 100%). mp 166.7-176.2°C; 1HNMR
2 0 (CD30D) b 2.39 (mc, 2H), 3.12 (mc, 2H), 3.36 (mc, 2H), 3.63 (s, 2H),
7.12 (d, J = 8.9 Hz, 2H), 7.15 (d, J = 8.9 Hz, 2H), 7.44 (d, J = 9.0 Hz, 2H),
7.89 (d, J = 8.9 Hz, 2H); FABMS (M+ +H): 425.0; Anal. Calcd. for
C 19H21 N2SOSCl.HCl.1.5 H20: C, 46.73; H, 4.33; N, 5.74. Found: C, 46.83;
H, 4.66; N, 5.71.
15F. Preparation of Id where n is 2, var~g R1 R2~R3 R4, and RS
Similarly, following the procedures of Example 15E above, but
replacing N-tert-butoxy-2-{4-[4-(4-chlorophenoxy)phenylsulfonyl-
3 0 methyl]-piperidin-4-yl) }-carboxamide with other compounds of
Formula Ic where Y is tert-BuONH-, other compounds of Formula Id
where n is 2 and Y is HONH- were prepared, for example:
2-{ 4-[4-(4-chlorophenoxy)phenylsulfonylmethyl]-1-(cyclo-
propylmethyl)piperidin-4-yl}-N-hydroxycarboxamide hydrochloride
3 5 ( 1.30 g, 84%). mp 120.5-124.0 °C; IR (KBr) 3429 (br), 1582 cm-1;
X193178
- 122 -
1HNMR (CD30D) b 0.40-0.50 (m, 2H), 0.73-0.81 (m, 2H), 1.12 (mc, 1H),
2.18 (mc, 2H), 2.41 (d, J = 14.8 Hz, 2H), 2.63 (d, J = 14.3 Hz, 2H), 3.03
(mc, 2H), 3.10 (mc, 2H), 3.60 (mc, 3H), 7.13 (mc, 4H), 7.43 (d, J = 8.7
Hz, ZH), 7.89 (d, J = 8.8 Hz, 2H), 7.93 (d, J = 8.8 Hz, 2H); FABMS
(M+ +H): 479.1. Anal. Calcd. for C23H27N2S05Cl.HCl.H20: C, 51.77; H,
5.09; N, 5.25. Found: C, 51.90; H, 5.53; N, 5.26.
2- { 4-[4-(4-chlorophenoxy)phenylsulfonylmethyl]-N-hydroxy-
1-nicotinoylmethylpiperidin-4-yl}-carboxamide hydrochloride (590
mg, 89%). mp 160.5 °C (effervescence); IR (KBr) 3426 (br), 1638 cm-1;
1 0 1HNMR (CD30D) 8 1.97 (mc, 2H), 2.25 (mc, 2H), 3.55 (mc, 4H), 3.64 (s,
2H), 7.10 (d, J = 8.9 Hz, 2H), 7.13 (d, J = 8.7 Hz, 2H), 7.43 (d, J = 8.6
Hz, 2H), 8.12 (mc, 1H), 8.61 (d, J = 7.9 Hz, 2H), 8.92 (d, J = 5.5 Hz, 2H),
8.98 (br s, 1H); FABMS (M+ +H): 530Ø Anal. Calcd. for
C25H29N3S06C1.HC1Ø5H20: C, 51.38; H, 4.14; N, 7.19. Found: C, 51.80;
1 5 H, 4.46; N, 7.25.
2-{ 4-[4-(4-chlorophenoxy)phenylsulfonylmethyl]-N-hydroxy-
1-methansulfonylpiperidin-4-yl}-carboxamide hydrochloride (682
mg, 69%). mp 107.3-112.3 °C; 1 HNMR (CDC13 ) 8 1.95 (mc, 2H), 2.40
(mc, 2H), 2.79 (s, 3H), 3.12 (mc, 2H), 3.42 (s, 2H), 3.51 (mc, 2H), 7.01
2 0 (d, J = 8.9 Hz, 2H), 7.07 (d, J = 8.9 Hz, 2H), 7.39 (d, J = 8.9 Hz, 2H),
7.83
(d, J = 8.9 Hz, 2H); FABMS (M+ +H): 503.2. Anal. Calcd. for
C20H23N2S207C1: C, 47.76; H, 4.61; N, 5.57. Found: C, 47.32; H, 4.56;
N, 5.52.
4-[4-(4-pyridyloxy)phenylsulfonylmethyl]-tetrahydropyran-
2 5 4-(N-hydroxycarboxamide) hydrochloride: mp 188-197°C; IR (KBr)
3431, 1638 cm-1; 1HNMR (DMSO-d6) b 1.73 (mc, 2H), 2.01 (dm, J
=14.7 Hz, 2H), 3.43 (mc, 2H), 3.65 (mc, 2H), 3.78 (s, 2H), 7.56 (mc, 4H),
8.02 (d, J = 8.7 Hz, 2H), 8.82 (d, J = 6.6 Hz, 2H), 10.64 (s, 1 H); 13 C N M R
(DMSO-d6) 8 33.01 (t), 39.78 (t), 61.13 (s), 63.26 (t), 114.48 (d),
3 0 121.81 (d), 130.87 (d), 138.41 (s), 144.92 (d), 156.14 (s), 168.4 (s),
168.8 (s); Anal. Calcd. for C18H21N2S06C1.HC1Ø6 H20: C, 49.17; H,
5.09; N, 6.37. Found: C, 49.16; H, 5.03; N, 6.27.
4-[4-(5-chloro-2-pyridyloxy)phenylsulfonylmethyl]-
tetrahydropyran-4-(N-hydroxycarboxamide): mp 141.9-142.7°C; IR
~93°~~8
- 123 -
(KBr) 3432, 1636 cm-1; 1HNMR (DMSO-d6) b 1.73 (mc, 2H), 2.01 (dm,
J =14.7 Hz, 2H), 3.33 (s, 2H), 3.46 (mc, 2H), 3.64 (mc, 2H), 7.23 (dd, J =
8.7, 0.4 Hz, 2H), 7.40 (d, J = 8.8 Hz, 2H), 7.92 (d, J = 8.8 Hz, 2H), 8.03 (d,
J = 8.7, 2.7 Hz, 2H), 8.26 (dd, J = 2.7, 0.4 Hz, 1 H), 8.69 (s, 1 H), 10.62
(s,
1H); 13CNMR (DMSO-d6) 8 32.89 (t), 41.81 (s), 60.96 (t), 63.26 (t),
113.88 (d), 121.32 (d), 126.31 (s), 129.58 (d), 136.93 (s), 140.33 (s),
145.74 (d), 157.82 (s), 160.69 (s), 169.02 (s); FABHRMS Calcd. for
C18H19N2S06C1 (M+ + H): 427.0731. Found: 427.0726. Anal. Calcd. for
C18H19N2S06C1.O.SH20: C, 49.49; H, 4.61; N, 6.41. Found: C, 49.54; H,
1 0 4.35; N, 6.47.
3-[4-(5-chloro-2-pyridyloxy)phenylsulfonyl]-2,2-dimethyl-
N-hydroxypropionamide: mp 115.8-116.6 °C; IR (KBr) 3412 (br), 1644
cm-1; 1 HNMR (CD30D) 8 1.38 (s, 6H), 3.58 (s, 2H), 7.13 (d, J = 8.7 Hz,
1 H), 7.34 (d, J = 8.8 Hz, 2H), 7.89 (dd, J = 8.7, 2.7 Hz, 2H), 7.95 (d, J =
1 5 8.8 Hz, 1H), 8.15 (d, J = 2.5 Hz, 1H); 13C NMR (CD30D) 8 25.55 (q),
41.76 (s), 65.06 (t), 114.91 (d), 122.35 (d), 128.40 (s), 130.98 (d),
138.21 (s), 141.44 (d), 146.88 (d), 159.89 (s), 162.32 (s), 174.51 (s);
FABHRMS Calcd. for C16H18N2SOSC1 (M+ + H): 385.0625. Found:
383.0625. Anal. Calcd. for C16H17N2SOSC1: C, 49.94; H, 4.48; N, 7.28.
2 0 Found: C, 49.58; H, 4.42; N, 7.30.
15G. Preparation of Id where n is 2, R3 and R4 are H,~ eg nR1
and R2 when taken together with the Carbon to which they
attached are 1-Picol.~ni~eridine, and RS is 4~4-Chlorophenoxy)-
25 h
A solution containing N-tert-butoxy-2-{4-[4-(4-chlorophenoxy)-
phenylsulfonylmethyl]-1-picolylpiperidin-4-yl}-carboxamide
(324 mg, 0.566 mmol) in trifluoroacetic acid (5 ml) was heated to
3 0 30°C for 1.5 hours, cooled to room temperature, and concentrated i
n
vac.uo. The residue was dissolved in ethyl acetate (100 ml), washed
with saturated sodium bicarbonate (2 x 30 ml), dried over magnesium
sulfate, and concentrated in vacuo. Chromatography over silica gel,
eluting with 6% methanol/methylene chloride, yielded 2-{4-[4-(4-
1g3~78
- 124 -
chlorophenoxy)-phenylsulfonylmethyl]-1-picolylpiperidin-4-yl } -N-
hydroxycarboxamide hydrochloride: mp 222.5-223.9°C; IR (KBr) 3436
(br), 1645 cm-1; 1 HNMR (DMSO-d6) 8 2.15 (mc, 3H), 2.40 (mc, 2H),
3.32 (mc, 2H), 3.57 (mc, 2H), 3.97 (mc, 2H), 4.44 (mc, 2H), 4.51 (mc,
2H;), 7.19 (mc, 4H), 7.50 (d, J = 8.8 Hz, 2H), 7.87 (mc, 3H), 8.49 (mc,
1H), 8.85 (mc, 1H), 8.99 (br s, 1H); FABMS (M+ +H): 516.1. Anal. Calcd.
for C29H34N3S05C1.2HC1Ø5 H20: C, 50.22; H, 4.89; N, 7.03. Found: C,
50.17; H, 4.65; N, 7.00.
1 0 EXAMPLE 16
Preparation of Compounds of Formula Ih
16A. Preparation of Ie where R1 R2 and R3 are H.~gen, and R4
is Benzyl
To a cooled solution of 3-benzyl-3-(4-bromophenylthio)-
propionic acid in methanol (50 ml) was added a solution of OXONE (8
g) in water (50 ml). The reaction mixture was stirred for 2 hours at
room temperature, and then partitioned between methylene chloride
2 0 and water. The solvent was removed from the organic layer under
reduced pressure, to give 3-benzyl-3-(4-bromophenylsulfonyl)-
propionic acid, as a crystalline solid.
16B. Preparation of If where R1 R2 and R3 are H.ydrogen, and R4
2 5 is Benzyl
1. A solution of 3-(4-bromophenyl)sulfonyl-4-benzylpropionic
acid (200 mg, 0.52 mmol), phenylboronic acid ( 127 mg, 1.04 mmol),
and tetrakis(triphenylphospine)palladium(0) (24 mg, 0.021 mmol) in
3 0 a l: l mixture of ethanol and benzene (5 ml) was heated to reflux
temperature with stirring. A solution of 2M sodium carbonate ( 1 ml)
was added to the reaction mixture, and stirring continued at reflux for
approximately 2 hours. The mixture was cooled and then partitioned
between ethyl acetate and water. The solvent layer was washed with
3 5 brine, dried over magnesium sulfate, filtered, and solvent removed
19378
- 125 -
under reduced pressure. The residue was chromatographed, eluting
with 7% methanol/methylene chloride, to yield 3-(4-biphenyl)-
sulfonyl-4-benzylpropionic acid. 1HNMR (CDC13): 7.75 ppm (m, 14H);
3.42 ppm (dd, 1H); 2.82 ppm (dd, 1H); 2.77 ppm (dd, 1H); 2.51 ppm
(dd, 1H).
16C. Preparation of Ih where R1 R2, and R3 are H,~gen and R4
is Benzvl
The 3-(4-biphenyl)sulfonyl-4-benzylpropionic acid, prepared as
shown above, was then converted to 3-(4-biphenyl)sulfonyl-4-
benzyl-N-hydroxypropionamide, m.p. 65°C (shrinks with
decomposition) as described in Examples 10A.
1 5 16D. Preparation of Ifb where R1 and R2 Together with the
Carbon to which thex are attached represent Tetrah~pyran-4-
R3 and R4 are H,~rogen, RS is 4-(Thiophen-2-vl)nhenoxYphen~
1. To a mechanically stirred suspension of 4-[4-(4-
2 0 bromophenoxy)phenylthiomethyl]-tetrahydropyran-4-carboxylic acid
(5.50 g, 13.0 mmol) in 20% tetrahydrofuran/methanol (135 ml) cooled
to 15 °C, was added a solution of OXONE ( 13.0 g, 21.2 mmol) in water
(86 ml) dropwise, maintaining an internal temperature of 15-20°C.
The mixture was stirred for 12 hours and dissolved in 40% ethyl
2 5 acetate/water ( 1200 ml). The layers were partitioned, and the water
layer back extracted using ethyl acetate (2 x 300 ml). The combined
ethyl acetate layers were dried (MgS04), concentrated, and the
residue crystallized from the minimum amount of methylene
chloride/hexanes to afford 4-[4-(4-bromophenoxy)-phenyl-
3 0 sulfonylmethyl]-tetrahydropyran-4-carboxylic acid as a white
powder, which was used without further purification (5.00 g, 84%).
2. To a solution of 4-[4-(4-bromophenoxy)phenylsulfonyl
methyl]-tetrahydropyran-4-carboxylic acid ( 1.10 g, 2.42 mmol) of in
3 5 N, N-dimethylformamide ( 15 ml) was added tetrakis(triphenyl-
. ~ ~g3178
- 126 -
phosphine)-palladium(0) ( 108 mg), 2-thiophene boronic acid (857 mg,
6.70 mmol), followed by 2M aqueous sodium carbonate (2.7 ml, 5.4
mmol). The reaction was heated to reflux for 10 hours, cooled to room
temperature, and the mixture partitioned between methylene
chloride ( 100 ml) and 1 N aqueous hydrochloric acid (20 ml). The
aqueous layer was back extracted with methylene chloride ( 100 ml),
and the combined organic layers dried (MgS04), the residue
chromatographed over 100 g of silica gel (eluted with methylene
chloride to 10% methanol/methylene chloride), and the resulting foam
crystallized from the minimum amount of methylene
chloride/hexanes to afford 4-[4-(4-(thiophen-2-yl)phenoxy)phenyl-
sulfonylmethyl]-tetrahydropyran-4-carboxylic acid ( 1.04 g, 94%). mp
181.2-193.3°C; IR (KBr) 3432 (br), 1718.9 cm-1; 1H NMR (DMSO-d6) b
1.67 (ddd, J = 13.8, 9.4, 4.0 Hz, 2H), 1.95 (dm, J = 13.8 Hz, 2H), 3.47
1 5 (mc, 2H), 3.67 (mc, 2H), 3.68 (s, 2H), 7.14 (dd, J = 4.9, 3.6 Hz, 1H),
7.20
(d, J = 8.8 Hz, 2H), 7.22 (d, J = 8.9 Hz, 2H), 7.50 (dd, J = 3.6, 1.2 Hz, 1H),
7.54 (dd, J = 4.9, 1.2 Hz, 1 H), 7.74 (d, J = 8.8 Hz, 2H), 7.87 (d, J = 8.8
Hz,
2H), 12.80 (s, 1H); 13CNMR (DMSO-d6) b 32.92 (t), 42.25 (s), 61.73 (t),
63.26 (t), 117.82 (d), 123.75 (d), 125.66 (d), 127.39 (d), 128.50 (d),
2 0 130.08 (d), 130.74 (s), 134.90 (s), 142.42 (s), 154.13 (s), 161.33 (s),
174.39 (s); FABHRMS Calcd. for C23H24S206 (M+ + H): 459.0936.
Found: 459.0936. Anal. Calcd. for C23H23S206~ C, 60.24; H, 4.83.
Found: C, 60.57; H, 4.90.
2 5 16E. Preparation of Ifb where R 1 and R2 Together with the
Carbon to which they are attached represent Tetrahydro~Yran-4-yl,
R3 and R4 are Hydrogen, R5 is 4-~Thiophen-3 ,Yl,~phenoxXphenXl
Similarly, following the above procedure, other compounds of
3 0 Formula Ifb, were prepared, for example replacing 2-thiophene
boronic acid with 3-thiophene boronic acid, 4-[4-(4-(thiophen-
3-yl)phenoxy)-phenylsulfonylmethyl]-tetrahydropyran-4-carboxylic
acid was prepared: mp 206.6-212.4 °C; IR (KBr) 3430 (br), 1719 cm-
l ; 1. HNMR (DMSO-d6) 8 1.67 (mc, 2H), 1.95 (mc, 2H), 3.47 (mc, 2H),
3 5 3.66 (mc, 2H), 3.67 (s, 2H), 7.20 (mc, 4H), 7.56 (dd, J = 5.0, 1.4 Hz,
1H),
13178
- 127 -
7.64 (d, J = 5.0, 2.9 Hz, 2H), 7.81 (d, J = 8.7 Hz, 2H), 7.87 (mc, 2H), 7.96
(s, 1H), 12.77 (s, 1H); 13CNMR (DMSO-d6) 8 32.92 (t), 40.38 (s), 61.19
(t), 63.26 (t), 117.66 (d), 120.54 (d), 120.87 (d), 126.04 (d), 127.07
(d), 127.96 (d), 130.02 (d), 132.00 (s), 134.66 (s), 140.45 (s), 160.80
(s), 174.32 (s); FABHRMS Calcd. for C23H23S206 (M+ + H): 459.0936.
Found: 459.0934. Anal. Calcd. for C23H22S2O6Ø5H20: C, 59.08; H,
4.96. Found: C, 58.82; H, 4.69.
16F. Catalytic Reduction of 4-f4-(4-bromophenox,X)-
phenylsulfonylmethyll-tetrah~pyran-4-carboxylic acid
A solution of 660 mg ( 1.45 mmol) of 4-[4-(4-bromophenoxy)-
phenylsulfonylmethyl]-tetrahydropyran-4-carboxylic acid in 80%
ethanol/tetrahydropyran (40 ml) was hydrogenated at atmospheric
pressure for 14 hours using palladium on carbon catalyst, filtered
over a celite pad washing with methylene chloride and concentrated
in vacuo to afford 4-[4-phenoxyphenylsulfonylmethyl]-
tetrahydropyran-4-carboxylic acid as a light orange solid (546 mg,
100%), which was taken directly into the next reaction without
2 0 further purification: mp 162.5-165.3°C; IR (KBr) 3431 (br), 1727 cm-
1; 1 HNMR (DMSO-d6) 8 1.67 (ddd, J = 14.1, 10.0, 4.0 Hz, 2H), 1.95 (dm,
J = 14.1 Hz, 2H), 3.47 (mc, 2H), 3.65 (mc, 2H), 3.66 (s, 2H), 7.15 (d, J =
8.8 Hz, 2H), 7.27 (t, J = 7.4 Hz, 1H), 7.45 (t, J = 7.5 Hz, 2H), 7.86 (d, J =
7.9 Hz, 2H), 12.74 (s, 1H); 13C NMR (DMSO-d6) 8 32.88 (t), 42.26 (s),
2 5 61.75 (t), 63.26 (t), 117.64 (d), 120.11 (d;l, 125.03 (d), 130.04 (d),
130.39 (s), 134.69 (s), 154.69 (s), 161.53 (s), 174.39 (s); FABHRMS
Calcd for C19H21SO6 (M+ + H): 377.1059. Found: 378.1064. Anal.
Calcd. for C19H2pSO6Ø75H20: C, 58.52; H, 5.56. Found: C, 58.54; H,
5.19.
EXAMPLE 17
Pr~aration of Compounds of Formula Ii
17 A . Preparation of Ii where R 1 R2 and R3 are H,~gen, and R4
3 5 is Benzvl
~~~~:~'8
- 128 -
Thiophenol (80 mg) was stirred for 45 min with potassium
hydride (40 mg) in N, N-dimethylformamide ( 1 ml) to produce a
homogeneous solution of potassium thiophenolate. To this mixture
was added 3-benzyl-3-(4-bromophenylsulfonyl)-propionic acid ( 100
mg) dissolved in N, N-dimethylformamide ( 1 ml) at room temperature.
After stirring for 16 hours at 75°C the mixture was partitioned
between aqueous citric acid and water, giving a product which was
purified by preparative TLC to afford 3-benzyl-3-(4-phenyl-
1 0 thiophenylsulfonyl)-propionic acid (30 mg).
17B . Preparation of I~ where R 1 i R2 and R3 are Hxdrogen, and R4
is Benzyl
The 3-benzyl-3-(4-phenylthiophenylsulfonyl)-propionic acid,
prepared as shown above, was then converted to 3-benzyl-3-
(4-phenylthiophenylsulfonyl)-N-hydroxypropionamide as descibed in
Example 10A.
2 0 EXAMPLE 18
Preparation of Compounds of Formula Ik
18A. Preparation of Ik where R1 R2 and R3 are H~rogen, and R4
is Benzyl
A mixture of 3-benzyl-3-(4-bromophenylsulfonyl)-propionic acid
(250 mg), p-methoxystyrene (0.1 ml), diisopropylethylemine (0.25
ml;), palladium acetate (5 mg) and trio-methylphenyl)phosphine ( 16
mg) was stirred overnight at 80°C. The reaction mixture was dissolved
3 0 in methylene chloride and washed with aqueous citric acid. Solvent
was removed from the methylene chloride solution, and the residue
chromatographed on silica gel (preparative TLC, eluting with 10%
methanol/methylene chloride), to afford 3-benzyl-3-(4-
styrylphenylsulfonyl)-propionic acid (21 mg).
i~
~~~ g 3178
- 129 -
18B . Preparation of Ik where R 1 R2 and R3 are H,~drogen, and R4
is Benzvl
The 3-benzyl-3-(4-styrylphenylsulfonyl)-propionic acid,
prepared as shown above, was then converted to 3-benzyl-3-(4-
styrylphenylsulfonyl)-N-hydroxypropionamide, LSIMS m/e=452.2
(M+H)+, as descibed in Example 10A.
EXAMPLE 19
Pr~aration of Compounds of Formula Il
Preparation of Il where n is 2. R 1 and R2 together with the Carbon
to which they are attached are Piperidine, R2 and R3 are H,ydrogen,
and RS is 4-(4-ChlorophenoxXlphen~
1 5 Trifluoroacetic acid (4 ml) was added to a solution of N-tert-
butoxy-2-[4-(4-phenoxyphenylsulfonylmethyl)-N-BOC-piperidin-4-
yl]-carboxamide (2 g, 3.64 mmol) dissolved in methylene chloride (4
ml). The reaction mixture was stirred for 1.3 hours and concentrated
in vacuo. The crude salt residue was dissolved in ethyl acetate ( 150
2 0 ml), washed with saturated aqueous sodium bicarbonate (2 x 50 ml),
dried over magnesium sulfate, concentrated in vacuo, to afford the
free base, N-tert-butoxy-2-[4-(4-phenoxyphenylsulfonylmethyl)-
piperidin-4-yl]-carboxamide ( 1.57 g, 90%). 1 HNMR (CDC13) b 1.28 (s,
9H), 2.23 (mc, 2H), 2.56 (mc, 2H), 3.30 (mc, 2H), 3.44 (mc, 2H), 3.53
2 5 (mc, 2H), 7.00 (d, J = 8.9 Hz, 2H), 7.05 (d, .l = 8.8 Hz, 2H), 7.38 (d, J
=
8.8 Hz, 2H), 7.82 (d, J = 8.8 Hz, 2H), 8.25 (br s, 1 H), 8.48 (br s, 1 H).
EXAMPLE 20
Preparation of Compounds of Formula Im
20A. Preparation of Im where n is 2, R is Ethox~carbon, ly_methyl,
R1. and R2 are Hydrogen, and RS is 4-Phenox~phenyl
A solution of N-tert-butoxy-2-[4-(4-phenoxyphenylsulfonyl)-
3 S piperidin-4-yl)]-acetamide (750 mg) in N, N-dimethylformamide ( 10
19~~~8
- 130 -
ml) was treated with ethyl bromoacetate (0.2 ml) and potassium
carbonate (600 mg). The mixture was stirred overnight at room
temperature, and then partitioned between ethyl acetate and water.
After drying, solvent was removed from the organic layer under
S reduced pressure to yield N-tert-butoxy-2-[4-(4-phenoxyphenyl-
sulfonyl)-1-(ethoxycarbonylmethyl)piperidin-4-yl]-acetamide, which
was used in the next step without further purification.
20B. Preparation of Im where n is 2 R is Isonrop,~l and R2
1 0 are H.~gen, and R5 is 4-PhenoxYphen~l
To a solution of N-tert-butoxy-2-[4-(4-phenoxyphenylsulfonyl)-
piperidin-4-yl)]-acetamide (500 mg) in acetone (20 ml) was added
10% palladium on carbon (100 mg), and the mixture stirred under
15 hydrogen for three days. The catalyst was filtered off, and solvent
removed from the filtrate under reduced pressure. The residue was
chromatographed on silica gel, eluting with 10% methanol/methylene
chloride, to give N-t-butoxy-2-[4-(4-phenoxyphenylsulfonyl)-1-
(isopropyl)piperidin-4-yl)]-acetamide (300 mg).
20C. Preparation of Im where n is 2, var ing R
Similarly, following the procedures of Example 20A above, but
replacing ethyl bromoacetate with 3-picolyl chloride, N-tert-butoxy-
2-[4-(4-phenoxyphenylsulfonyl)-1-(3-picolyl)piperidin-4-yl]-
acetamide was prepared.
Similarly, following the procedures of Example 20A above, but
replacing N-tert-butoxy-2-[4-(4-phenoxyphenylsulfonyl)-piperidin-
4-yl)]-acetamide with N-tent-butoxy-2-{4-[4-(4-fluorophenoxy)-
phenylsulfonyl]-piperidin-4-yl }-acetamide, and replacing ethyl
bromoacetate with cyclopropylmethyl bromide, N-tert-butoxy-2- { 4-
[4-(4-fluorophenoxy)-phenylsulfonyl]-1-(cyclopropylmethyl)-
piperidin-4-yl}-acetamide was prepared.
i~
~1~3178
- 131 -
Similarly, N-tert-butoxy-2-[4-(4-phenoxyphenylsulfonyl)-
1-(acetamidocarbonylmethyl)piperidin-4-yl]-acetamide was
prepared.
20D. Preparation of Im where n is 2, varying_ R
Similarly, following the procedures of Example 20A above, but
optionally replacing N-tert-butoxy-2-[4-(4-phenoxyphenylsulfonyl)-
piperid-4-yl)]-acetamide with other compounds of Formula Iy, and
optionally replacing ethyl bromoacetate with other compounds of
formula RX, where R is lower alkyl, cycloalkylalkyl, acyl,
alkoxycarbonylalkyl, picoline, -S02Ra, where Ra is lower alkyl or
-NRbRc, where Rb and Rc are independently hydrogen or lower alkyl;
and the like, and X is chloro, bromo or iodo, other compounds of
Formula Im were prepared:
N-tert-butoxy-2-[ 1-ethyl-4-(4-phenoxyphenylsulfonyl)-
piperidin-4-yl]-acetamide;
N-tert-butoxy-2-[ l -methyl-4-(4-phenoxyphenylsulfonyl)-
piperidin-4-yl]-acetamide, m.p. 152-155°C;
N-tert-butoxy-2-[1-(2-methylpropyl)-4-(4-phenoxyphenyl-
sulfonyl)-piperidin-4-yl]-acetamide;
N-tert-butoxy-2-[ 1-cyclopropylmethyl-4-(4-phenoxyphenyl-
sulfonyl)-piperidin-4-yl]-acetamide;
N-tert-butoxy-2-[ 1-cyclopropylmethyl-4-[4-(4-chlorophenoxy)-
2 5 phenylsulfonyl]-piperidin-4-yl]-acetamide; and
N-tert-butoxy-2-[ l -acetyl-4-[4-(4-fluorophenoxy)phenyl-
sulfonyl]-piperidin-4-yl]-acetamide.
20E. Preparation of Ic where n is 2, R3 and R4 are H,~rdrogen, R1
3 0 and R2 when taken together with the Carbon to which the~are
attached is 1-C,ycloprop, ly methylPiperidine, and R5 is 4- 4-
Chlorophenox~)t~hen
To a solution of the free base N-tert-butoxy-2-[4-(4-
3 5 phenoxyphenylsulfonylmethyl)-piperidin-4-yl]-carboxamide ( 1.28 g,
n
~9~178
- 132 -
2.66 mmol) dissolved in N, N-dimethylformamide ( 17 ml), was added
cyclopropylmethyl bromide (0.26 ml, 2.66 mmol), followed by
potassium carbonate ( 1.84 g, 13.3 mmol). After the reaction mixture
was stirred for 20 hours, water was added ( 100 ml), and the aqueous
solution extracted with ethyl acetate (3 x 100 ml). The combined
organic extracts were washed with brine (2 x 50 ml), dried over
magnesium sulfate, concentrated in vacuo. Chromatography over silica
gel, and eluting with 25% ethyl ~ acetate/hexanes, gave N-tert-butoxy-
2-[4-(4-phenoxyphenylsulfonylmethyl)-l.-(cyclopropyl)piperidin-4-
1 0 yl]-carboxamide (1.30 g, 92%). 1HNMR (CDC13) b 0.10 (ddd,J = 5.6, 4.7,
4.6 Hz, 2H), 0.53 (ddd,J =8.7, 4.7, 4.5 Hz, 2H), 0.85 (mc, 1H), 1.31 (s,
3H;), 1.64 (mc, 2H), 2.06 (mc, 2H), 2.24 (mc, 2H), 2.28 (d, J = 6.5 Hz,
2H;), 2.67 (mc, 4H), 3.50 (mc, 2H), 7.01 (d, J = 8.8 Hz, 2H), 7.04 (d, J =
8.8 Hz, 2H), 7.37 (d, J = 8.8 Hz, 2H), 7.85 (d, J = 8.8 Hz, 2H), 8.33 (br s,
1 5 2H); FABMS (M+ +H): 535.2.
20F. Preparation of Ic where n is 2. R3 and R4 are Hvdro en. R1
and R2 when taken together with the Carbon to which they are
attached is 1-(3-Picolyl)~iperidine, and RS is 4-(4-ChlorophenoxX)-
20 h~ enyl
Similarly, following the procedures of Example 20E above, but
replacing cyclopropylmethyl bromide with 1.25 equivalents of 3-
picolyol chloride hydrochloride, N-tert-butoxy-2-[4-(4-phenoxy-
2 5 phenylsulfonylmethyl)-1-(3-picolyl)piperidin-4-yl]-carboxamide was
prepared: mp 83.3-93.8°C; IR (KBr) 3436, 1661 cm-1; 1HNMR (CDC13)
8 1.31 (s, 9H), 2.00 (mc, 2H), 2.24 (mc, 2H), 2.55 (mc, 4H), 3.48 (s, 2H),
3.53 (s, 2H), 7.01 (d, J = 8.9 Hz, 2H), 7.04 (d, J = 8.9 Hz, 2H), 7.25 (dd, J
= 7.6, 4.6 Hz, 2H), 7.38 (d, J = 8.8 Hz, 2H), 7.64 (brd, J = 7.8 Hz, 2H),
3 0 7.85 (d, J = 8.9 Hz, 2H), 8.36 (br s, 1H), 8.52 (m, 2H); FABMS (M+ +H):
572Ø Anal. Calcd. for C29H34N3S05C1Ø5 H2O: C, 59.03; H, 5.81; N,
7.12. Found: C, 59.37; H, 6.15; N, 7.98.
i~
- 133 -
20CJ. Preparation of Ic where n is 2, R3 and R4 are H.~gen, R1
and R2 when taken together with the Carbon to which they are
attached is 1-(NicotinoXl~Piperidine, and RS is 4- 4-Chloronhenox~l-
hp enyl
To a solution of the free base N-tert-butoxy-2-[4-(4-
phenoxyphenylsulfonylmethyl)-piperidin-4-yl]-carboxamide (491
mg, 1.02 mmol) and N, N-diisopropylethylamine (444 mg, 2.55 mmol)
in methylene chloride (2 ml) cooled to 0°C, was added nicotinyl
chloride hydrochloride (219 mg, 1.27 mmol) in one portion. After the
reaction mixture was stirred for 3 hours, water (30 ml) was added,
and the aqueous solution extracted with ethyl acetate (2 x 60 ml). The
combined organic extracts were washed with brine (2 x 50 ml), dried
over magnesium sulfate, concentrated in vacuo. Chromatography over
silica gel, and eluting with 6% methanol/methylene chloride, afforded
N-tert-butoxy-2-[4-(4-phenoxyphenylsulfonylmethyl)-
1-(nicotinoyl)piperidin-4-yl]-carboxamide (233 mg, 39%). 1HNMR
(CDC13) 8 1.33 (s, 9H), 1.95 (mc, 2H), 2.35 (mc, 2H), 3.45 (mc, 2H), 3.49
(s, 2H), 3.55 (mc, 4H), 7.01 (d, J = 8.8 Hz, '?H), 7.06 (d, J = 8.8 Hz, 2H),
2 0 7.39 (d, J = 8.8 Hz, 2H), 7.41 (mc, 2H), 7.79 (mc, 2H), 7.83 (d, J = 8.8
Hz,
2H), 8.69 (br s, 1H), 8.52 (mc, 2H).
20H. Preparation of Ic where n is 2, R3 and R4 are H,~rogen, R1
and R2 when taken together with the Carbon to which they are
2 5 attached is 1-lMethanesulfonvllPiperidine, and R5 is 4- 4-
Chlorophenox~~he girl
To a solution of the free base N-tert-butoxy-2-[4-(4-
phenoxyphenylsulfonylmethyl)-piperidin-4-yl]-carboxamide ( 1.57 g,
3 0 3.26 mmol) in 67% methylene chloride/pyridine (16.5 ml) cooled to
-78°C, was added a solution of methanesulfonyl chloride (0.51 ml,
6.53 mmol) in methylene chloride (2 ml). After the reaction mixture
was stirred for 4 hours, 3N aqueous hydrochloric acid (25 ml) was
added, and the aqueous solution extracted with ethyl acetate (2 x
3 5 60 ml). The combined organic extracts were washed with brine (2 x
- 134 -
50 ml), dried over magnesium sulfate, concentrated in vacuo.
Chromatography over silica gel, and eluting with 45% ethyl
acetate/hexanes, afforded N-tert-butoxy-2-[4-(4-phenoxy-
phenylsulfonylmethyl)-1-(methanesulfonyl)piperidin-4-yl]-
carboxamide (1.16 g, 64%). 1HNMR (CDC13) 8 1.33 (s, 9H), 2.05 (mc,
2H), 2.37 (mc, 2H), 2.79 (s, 3H), 3.23 (mc, 2H), 3.43 (s, 2H), 3.47 (mc,
2H), 7.01 (d, J = 8.9 Hz, 2H), 7.06 (d, J = 8.9 Hz, 2H), 7.39 (d, J = 8.9 Hz,
2H), 7.85 (d, J = 8.9 Hz, 2H); FABMS (M+ +H): 559.1.
1 0 EXAMPLE 21
Preparation of Compounds of Formula In
21 A. Preparation of In where n is 2, R is Ethoxycarbon ly meth,
R1 and R2 are Hydro~en, and R5 is 4-Phenox n~ hens
The product from Example 20A, N-tert-butoxy-2-[4-(4-
phenoxyphenylsulfonyl)-1-(ethoxycarbonylmethyl)piperidin-4-yl]-
acetamide, was dissolved in dichloroethane ( 10 ml), cooled to 0°C, and
saturated with hydrochloric acid gas. The reaction vessel was then
2 0 sealed and the solution stirred for two days at 25°C. Solvent was
removed from the reaction mixture under reduced pressure, and the
residue purified by preparative TLC, eluting with 10°Io methanol/
methylene chloride, to give N-hydroxy-2-[4-(4-phenoxyphenyl-
sulfonyl)-1-(ethoxycarbonylmethyl)piperidin-4-yl]-acetamide
2 5 (420 mg), m/e = 477.1 {MH+, FABMS).
21 B . Preparation of In where n is 2, R is Isopropyl, R1 and R2 are
HXdro,g;en, and RS is 4-Phenoxy~phenyl
3 0 The product from Example 20B, N-t-butoxy-2-[4-(4-
phenoxyphenylsulfonyl)-1-(isopropyl)piperidin-4-yl)]-acetamide, was
reacted with hydrochloric acid gas as described above, to yield N-
hydroxy-2-[4-(4-phenoxyphenylsulfonyl)-1-(isopropyl)piperidin-4-
yl)]-acetamide (155 mg), m.p. 128°C, m/e = 432 (MH+,EIMS).
- 135 -
21C. Preparation of In where n is 2, var ing R
Similarly, following the procedures of Example 21A above, but
replacing ethyl bromoacetate with 3-picolyl chloride, N-hydroxy-2-
[4-(4-phenoxyphenylsulfonyl)-1-(3-picolyl)piperidin-4-yl]-acetamide
was prepared, m.p. 185-192°C (dec).
Similarly, following the procedures of Example 19A above, but
replacing N-tert-butoxy-2-[4-(4-phenoxyphenylsulfonyl)-piperidin-
4-yl)]-acetamide with N-tert-butoxy-2-{4-[4-(4-fluorophenoxy)-
phenylsulfonyl]-piperidin-4-yl}-acetamide, and replacing ethyl
bromoacetate with cyclopropylmethyl bromide, N-hydroxy-2- { 4-[4-
(4-fluorophenoxy)phenylsulfonyl]-1-cyclopropylmethylpiperidin-4-
yl}-acetamide was prepared, m.p. 104-105°C.
Similarly, N-hydroxy-2-[4-(4-phenoxyphenylsulfonyl)-
1-acetamidocarbonylmethylpiperidin-4-yl]-acetamide was prepared.
21D. Preparation of In where n is 2, var i~n~ R
Similarly, following the procedures of Example 21A above, but
optionally replacing N-tert-butoxy-2-[4-(4-phenoxyphenylsulfonyl)-
piperid-4-yl)]-acetamide with other compounds of Formula Iy, and
optionally replacing ethyl bromoacetate with other compounds of
2 5 formula RX, where R is lower alkyl, cycloalkylalkyl, acyl,
alkoxycarbonylalkyl, picoline, -S02Ra, where Ra is lower alkyl or
-NRbRc, where Rb and Rc are independently hydrogen or lower alkyl;
and the like, and X is chloro, bromo or iodo, other compounds of
Formula In were prepared:
3 0 2-[ 1-ethyl-4-(4-phenoxyphenylsulfonyl)-piperidin-4-yl]-
N-hydroxyacetamide, m.p. 182-183°C;
N-hydroxy-2-[ 1-methyl-4-(4-phenoxyphenylsulfonyl)-piperidin-
4-yl]-acetamide, m.p. 152-155°C;
N-hydroxy-2-[ 1-(2-methylpropyl)-4-(4-phenoxyphenylsulfonyl)-
3 5 piperid-4-yl]-acetamide, m.p. 226-227°C;
- 136 -
2-[ 1-cyclopropylmethyl-4-(4-phenoxyphenylsulfonyl)-piperidin-
4-yl]-acetamide, m.p. 2 l0-211 °C;
2-[1-cyclopropylmethyl-4-[4-(4-chlorophenoxy)-phenylsulfonyl]-
piperidin-4-yl]-N-hydroxyacetamide, m.p. 110-112°C; and
2-[1-acetyl-4-[4-(4-fluorophenoxy)phenylsulfonyl]-piperidin-4-
yl]~-N-hydroxyacetamide, m/e = 450 (MH:+).
EXAMPLE 22
Preparation of Compounds of Formula Iab
Preparation of Iab where R5 is 4-phenox~phenyl
4-Phenoxythiophenol (4.8 g) was stirred for 45 min with
potassium hydride (0.98 g) in N, N-dimethylformamide ( 100 ml) to
produce a homogeneous solution of potassium 4-phenoxythio-
phenolate. The lactone, (S)-3-carbobenzyl-oxyamino-2-oxetanone (5.3
g) (Arnold, L.D. et al., J. Am. Chem. Soc., 107, 7105 ( 1985)), dissolved
in N, N-dimethylformamide (50 ml) was then added at room
temperature. After stirring for 30 minutes the mixture was poured
into water and extracted with ethyl acetate. The combined extracts
2 0 were dried over magnesium sulfate, and solvent removed under
reduced pressure to give (R)-2-(benzyloxycarbonylamino)-3-(4-
phenoxyphenylthio)-propionic acid (9.2 g). It can be used directly in
the next step.
2 5 EXAMPLE 23
Preparation of Compounds of Formula Io
Preparation of Io where RS is 4 ~phenoxy~hen~
The above-prepared (R)-2-(benzyloxycarbonylamino)-3-(4-
3 0 phenoxyphenylthio)-propionic acid was dissolved in methylene
chloride (175 ml), cooled to 0°C, and treated with O-(tert-
butyl)hydroxylamine hydrochloride (7.7 g), 4-methylmorpholine (9.4
ml), 1-hydroxybenzotriazole (2.8 g), and N-ethyl-N'-(3-dimethyl-
aminopropyl)-carbodiimide (7.9 g). The mixture was allowed to warm
3 5 to room temperature, stirred for 1.5 hours, then partitioned between
~~93~7g _ 137-
methylene chloride and water. Solvent was removed from the organic
phase under reduced pressure, and the residue purified by flash
chromatography on silica gel, eluting with 0 to 50% ethyl
acetate/hexane, to provide (R)-2-(benzyloxycarbonylamino)-N-tert-
butoxy-3-(4-phenoxyphenylthio)-propionamide (7.4 g) as a white
foam.
EXAMPLE 24
Preparation of Compounds of Formula Ip
1 0 Preparation of Ip where n is 2 and R5 is 4-phenoxXphenyl
(R)-N-tert-butoxy-2-(benzyloxycarbonylamino)-3-(4
phenoxyphenylthio)-propionamide ( 1.5 mmol) was dissolved in
methanol ( 140 ml), and a solution of OXONE ( 15 g) in water (50 ml)
was added with vigorous stirring. The oxidation is usually complete
within 2 hours. The mixture is then partitioned between methylene
chloride and water. Solvent was removed from the dried organic
phase under reduced pressure, to afford (R)-2-(benzyloxycarbonyl-
amino)-N-tert-butoxy-3-(4-phenoxyphenylsulfonyl)-propionamide
2 0 (8.3 g) in near-quantitative yield.
EXAMPLE 25
Preparation of Compounds of Formula Ia
Preparation of Iq where n is 2, Rl is Hydro eg n~R2 is -NR6R~, in
2 5 which R6 is Hvdro~en and R~ i5Benzyloxycarbonvlamino, and R5 is
4-phenox,~phen
A solution of (R)-2-(benzyloxycarbonylamino)-N-tert-butoxy-
3-(4-phenoxyphenylsulfonyl)-propionamide ( 1.2 g) obtained from
3 0 Example 16 in methylene chloride (5 mi) was diluted with
trifluoroacetic acid (30 ml). The solution was allowed to stand
overnight, and solvent was removed under reduced pressure. This
residue was chromatographed on silica gel, eluting with 10%
methanol/methylene chloride to give (R)-2-(benzyloxycarbonyl-
3 5 amino)-N-hydroxy-3-(4-phenoxyphenylsulfonyl)-propionamide (400
~19~17$ - 13s-
mg), m.p. 195-202°C.
EXAMPLE 26
Preparation of Compounds of Formula Ir
Preparation of Ir where n is 2 and R~ is 4-Phenox~Phen~
(R)-2-(benzyloxycarbonylamino)-N-tert-butoxy-3-(4-phenoxy-
phenylsulfonyl)-propionamide (6.0 g) obtained from Example 17 was
dissolved in ethanol ( 100 ml) and hydrogenated at 1 atmosphere in
1 0 the presence of 10% palladium on carbon (6 g) for a period of 18
hours. The catalyst was filtered off and the solvent removed from the
filtrate under reduced pressure to give (R)-2-amino-N-tert-butoxy-
3-(4-phenoxyphenylsulfonyl)-propionamide as a glass.
1 5 EXAMPLE 27
Preparation of Compounds of Formula Is
Preparation of Is where n is 2, R1 is Hydrogen, R2 is -NR6R7, in
which R6 and R7 are both HXdrogen, and RS is 4-phenoxyphenyl
2 0 Similarly as in Example 25, (R)-2-amino-N-tert-butoxy-
3-(4-phenoxyphenylsulfonyl)-propionamide (6.U g) was dissolved in
1,2-dichloroethane (5 ml) and cooled to -20°C and bubbled for 20
minutes with hydrochloric acid gas in a pressure tube. The flask was
then sealed and the mixture stirred overnight. The tube was cooled,
2 5 vented, and allowed to warm. The solution was rinsed with methanol,
the solvent removed from the filtrate under reduced pressure,
triturated with 1:1 hexane/ethyl acetate (4 ml). The residue was
filtered and dried to give (R)-2-amino-N-hydroxy-3-(4-phenoxy-
phenylsulfonyl)-propionamide hydrochloride, m.p. 178-180°C (dec).
- 139
EXAMPLE 28
Preparation of Compounds of Formula It
Preparation of It where n is 2, R1 is Hydro eg~nR2 is -NR6R~, in
which R6 is H,~gen and R~ is CBZ-fS)-Valinamido, and RS is 4-
phenoxxphenXl
To a solution of (R)-2-amino-N-tert-butoxy-3-(4-phenoxyphenyl-
sulfonyl)-propionamide ( 1.9 g) in methylene chloride (30 ml) was
added CBZ-(S)-valine (1.6 g), 1-hydroxybenzotriazole (0.9 g),
triethylamine (1 ml), and N'-ethyl-N'-(3-dimethylaminopropyl)-
carbodiimide ( 1.3 g). After stirring overnight at room temperature,
the solution was partitioned between methylene chloride and water,
and after the organic layer was dried over magnesium sulfate, solvent
was removed under reduced pressure to give (R)-N-tert-butoxy-2-
(CBZ-valinamido)-3-(4-phenoxyphenylsulfonyl)-propionamide, which
was used without further purification.
EXAMPLE 29
Preparation of Compounds of Formula Iu
2 0 Preparation of Iu where n is 2 R1 is Hydrogen, R2 is -NR6R~, in
which R6 is Hydrogen and R~ is (S)-Valinamido, and RS is 4-
phenoxy_ hp enyl
A solution of (R)-N-tert-butoxy-2-(CBZ-valinamidoj-3-
2 5 (4-phenoxyphenylsulfonyl)-propionamide (prepared above) in a
mixture of methanol (300 ml) and ethanol ( 100 ml) was stirred under
hydrogen at 1 atmosphere with palladium on carbon catalyst ( 10%
Pd, 4 g) for 3 hours. The mixture was filtered, and the filtrate
evaporated under reduced pressure. The residue was
3 0 chromatographed on silica gel, eluting with 0-3% methanol in
methylene chloride, to give (R)-N-tert-butoxy-2-valinamido-3-(4-
phenoxyphenylsulfonyl)-propionamide ( 1.6 g).
~~9~~~a - 140-
EXAMPLE 30
Preparation of Compounds of Formula Iv
Preparation of Iv where n is 2. R1 is Hydrogen, R2 is -NR6R~, in
which R6 is Hydrogen and R~ is (S~-V alinamido, and RS is 4-
phenoxy~phenyl
A solution of (R)-N-tert-butoxy-2-valinamido-3-(4-phenoxy-
phenylsulfonyl)-propionamide (1.6 g) in 1,2-dichloroethane (50 ml)
was cooled to -20°C and bubbled for 15-20 minutes with hydrochloric
1 0 acid gas in a pressure tube. The flask was then sealed and the mixture
stirred for 24 hours. After cooling the tube was cautiously vented and
its contents evaporated to yield a gum, which upon trituration with
ethyl acetate gave a crude product as a white powder. This product
was stirred overnight with 10% methanol/methylene chloride (20 ml)
and filtered to remove impurities. This was repeated three times to
give (R)-N-hydroxy-2-valinamido-3-(4-phenoxyphenylsulfonyl)-
propionamide hydrochloride (760 mg), m.p. 214-217°C.
EXAMPLE 31
2 0 Preparation of Compounds of Formula Iw
Preparation of Iw where n is 2. Y is lZVdroxy or lower alkoxyRR1
and R2 when taken together with the carbon to which thev are
attached are Tetrahydropyan-4-Xl, R3 is h~gen, and R4 is Benzes
and RS is 4- 4-Chlorophenox~)ahen~l
1. To a solution of 4-[4-(4-chlorophenoxy)phenylthiomethyl]-
tetrahydropyran-4-carboxylic acid methyl ester in 20%
tetrahydrofuran-methanol (9.5 ml) was added dropwise a solution of
OX:ONE ( 1.53 g, 2.49 mmol) in water (8 ml) while maintaining an
3 0 internal temperature of 15-20°C. The mixture was stirred 2 hours
and
the mixture dissolved in 40% ethyl acetate/water (200 ml). The
layers were partitioned, and the water layer back extracted using
ethyl acetate (2 x 50 ml). The combined organic layers were dried
over magnesium sulfate, concentrated, and the residue purified by
3 5 preparative chromatography (20 x 40-1000 um plates), eluting with
- 141 -
50% ethyl acetate/hexanes) to afford 4-[4-(4-chlorophenoxy)phenyl-
sulfonylmethyl]-tetrahydropyran-4-carboxylic acid methyl ester (460
mg, 71%). 1HNMR (CDC13) b 1.71-1.82 (m, 2H), 2.23 (dm, J= 13.6 Hz,
2H), 3.47 (s, 2H), 3.58-3.67 (m, 2H), 3.59 (s, 3H), 3.73-3.81 (m, 2H),
6.97-7.10 (m, 4H), 7.39 (d, J = 8.7 Hz, 2H), 7.84 (d, J = 8.7 Hz, 2H).
2. Lithium diisopropylamide was prepared by the addition of
2. S M N-butyl lithium (610 ~ L, 1.53 mmol) in hexanes to a solution of
diisopropylamine (200 ~,L, 1.53 mmol) in tetrahydrofuran (3 ml) at
0°C'. and stirring for 20 minutes. Then a solution of 4-[4-(4-
chlorophenoxy)-phenylsulfonylmethyl]-tetrahydropyran-4-carboxylic
acid methyl ester (540 mg, 1.27 mmol) in tetrahydrofuran ( 1 ml) was
added to the solution of lithium diisopropylamide at -78°C, and
stirred for an additional 60 minutes. Benzyl bromide (181 ~.L, 1.53
mmol) of was added to the mixture, stirred for an 50 minutes,
warmed to room temperature over 30 minutes, and stirred for an
additional 3 hours. The mixture was then diluted with 0.1 M aqueous
hydrochloric acid (25 ml) and extracted with methylene chloride (2 x
50 ml). The combined organic layers were dried over magnesium
2 0 sulfate, concentrated in vacuo, chromatographed over silica gel, eluted
with 20% ethyl acetate/hexanes, to afford 3-benzyl-4-[4-(4-chloro-
phenoxy)phenylsulfonylmethyl]-tetrahydropyran-4-carboxylic acid
methyl ester (440 mg, 67%). IR (KBr) 1736 cm-1; 1HNMR (CDC13) 8
1.78 (dm, J = 13.5 Hz, 1H), 2.02-2.17 (m, 2H), 2.39 (dm, J = 13.5 Hz,
2 5 1 H), 3.19-3.23 (m, 2H), 3.37-3.45 (td, J = 11.9, 2.4 Hz, 2H), 3.77-3.85
(m, 1H), 3.84 (s, 3H), 3.88-3.98 (m, 2H), 4.07-4.17 (m, 2H), 6.83-6.90
(m, 4H), 6.94 (d, J = 8.7 Hz, 2H), 7.08-7.15 (m, 3H), 7.37 (d, J = 8.7 Hz,
2H), 7.62 (d, J = 8.7 Hz, 2H); FABMS (M+ +H): 515.
3 0 EXAMPLE 32
Preparation of Compounds of Formula Ix
Preparation of Ix where n is 2, Y is hXdroxy-RR1 and R2 when
taken together with the carbon to which they are attached are
TetrahvdroDVan-4-vl, R3 is hvdro~en and R4is Benzyl, and RS is
3 5 4-(4-Chlorophenoxy)phenyl
- 142 -
To a solution of 3-benzyl-4-[4-(4-chlorophenoxy)-phenyl-
sulfonylmethylJ-tetrahydropyran-4-carboxylic acid methyl ester
(410 mg, 0.80 mmol) in N,N-dimethylformamide (4 ml) was added
lithium iodide ( 1.06 g, 7.96 mmol), followed by sodium cyanide (78
mg, 1.59 mmol). The mixture was heated to 120°C for 8 hours, cooled
to room temperature, the N, N-dimethylformamide solvent removed
by heating under reduced pressure, and the residue partitioned
between ethyl acetate ( 150 ml) and saturated aqueous sodium
bisulfite (50 ml). The ethyl acetate layer was dried over magnesium
sulfate, concentrated in vacuo, purified by preparative
chromatography (20 x 40-1000 um plates), eluted with 8%
methanol/methylene chloride) to afford 3 L7 mg (80%) of 3-benzyl-
4-[4-(4-chlorophenoxy)-phenylsulfonylmethyl]-tetrahydropyran-4-
carboxylic acid 1HNMR (N,N-dimethylformamide contaminant, CDC13)
8 1.74 (dm, J = 13.5 Hz, 1 H), 2.05-2.18 (m, 2H), 2.42 (dm, J = 13.5 Hz,
1H), 3.22-3.26 (m, 2H), 3.48-3.58 (m, 2H), 3.78-4.18 (m, SH), 6.83-
6.88 (m, 4H), 6.93 (d, J = 8.5 Hz, 2H), 7.08-7.13 (m, 3H), 7.36 (d, J = 8.7
Hz, 2H), 7.62 (d, J = 8.7 Hz, 2H); CIMS (NH3, M+ + NH4+): 518.
EXAMPLE 33
Preparation of Compounds of Formula I
Preparation of I where n is 2, R2 is -NR6R7 in which R6 and R7
are both Methyl, and R5 is 4-phenoxyphenyl
2 ~
To a solution of (R)-2-amino-N-tert-butoxy-3-(4-phenoxyphenyl-
sulfonyl)-propionamide ( 1.6 g) in N, N-dimethylformamide (5 ml) was
added potassium carbonate (0.5 g) and methyl iodide (550 p,l). After
stirring for 2.5 hours, the mixture was partitioned between ethyl
3 0 acetate and water, and after the organic layer was dried over
magnesium sulfate, solvent was removed under reduced pressure.
The residue was chromatographed on silica gel, eluting with 50% ethyl
acetate/hexane to give (R)-N-tent-butoxy-2-dimethylamino-3-(4-
phenoxyphenylsulfonyl)-propionamide (0.6 g).
143 -
This compound, (R)-N-tert-butoxy-2-dimethylamino-
3-(4-phenoxyphenylsulfonyl)-propionamide, was dissolved in
1,2-dichloroethane (50 ml), cooled to -30°C and bubbled for 15-20
minutes with hydrochloric acid gas in a pressure tube. The flask was
then sealed and the mixture stirred overnight. After cooling the tube
was cautiously vented and its contents evaporated, to yield a gum,
which upon trituration with 2:1 hexane/ethyl acetate gave a white
powder, (R)-2-dimethylamino-N-hydroxy-3-(4-phenoxyphenyl-
sulfonyl)-propionamide hydrochloride (0.43 g), m.p. 65-70°C.
EXAMPLE 34
Preparation of Compounds of Formula I
Preparation of I where n is 2. R2 is -NR6R7 in which R6 is
HXdro~en and R7 is Dimethylaminosulfonyl, and RS is 4-
phenoxX,phenXl
To a solution of (R)-2-amino-N-tert-butoxy-3-(4-phenoxyphenyl-
sulfonyl)-propionamide ( 1.5 g) in methylene chloride (20 ml) and
pyridine ( 1.2 ml) was added dimethylsulfamoyl chloride ( 1 ml), and
2 0 the mixture stirred overnight at room temperature. The mixture was
partitioned between methylene chloride and water, and after the
organic layer was dried over magnesium sulfate, solvent was
removed under reduced pressure. The residue was chromatographed
on silica gel, eluting with 0-45°Io ethyl acetate/hexane, to give (R)-N-
2 5 tert-butoxy-2-dimethylaminosulfonamido-3-(4-
phenoxyphenylsulfonyl)-propionamide ( 1.6 g).
This compound, (R)-N-tert-butoxy-2-dimethylaminosulfonamido-
3-(4-phenoxyphenylsulfonyl)-propionamide, was dissolved in
3 0 , trifluoroacetic acid (30 ml) and the mixture stirred overnight at room
temperature. The trifluoroacetic acid was removed under reduced
pressure, and the residue chromatographed on silica gel, eluting with
l0~lo methanol/methylene chloride, to give (R)-2-dimethylamino-
sulfonamido-3-(4-phenoxyphenylsulfonyl)-N-hydroxypropionamide
3 5 hydrochloride (550 mg). 1 H NMR (d6-DMSO) 7.90 (d,2H), 7.47 (d,2H),
2193v~'8
- 144 -
7.25 (t, l H), 7.13 (m,4H), 3.95 (m, l H), 3.55 (m,2H), 2.6 (s,6H).
EXAMPLE 35
Example of Preparation of Compounds of Formula I on a Large Scale
Preparation of I where n is 2, R1 and R2 when taken together with
the Carbon to which they are attached represent Tetrah,~pyran R3
and R4 are H drogen, and RS is 4-(4-Chloro~henoxY)phenyl
1. Preparation of a Compound of Formula (7a)
1 0 To a mixture of N, N-dimethylformamide (56 Kg) and diethyl
malonate (22 Kg) was added a 21 % solution of sodium ethoxide in
ethanol (45 Kg), followed by 2-chloroethyl ether ( 19 Kg). The mixture
was heated to 85°C, causing ethanol to distil from the mixture. The
temperature was raised to 120°C until all the ethanol formed was
removed (3 hours), and then the mixture was allowed to cool to 25°C.
The mixture was then rewarmed to 120°C and a further 45 Kg of a
21 % solution of sodium ethoxide in ethanol added at such a rate as to
cause the ethanol formed to distil off. When the distillation was
complete, the mixture was cooled to 100°C, and after it was
2 0 determined that the reaction was complete then cooled to 25°C. The
mixture was partitioned between toluene (80 Kg) and water (216 Kg)
and solvent removed from the organic layer by distillation. The
product was used in the next step with no further purification.
2 5 2. Preparation of a Compound of Formula (bay where R1 and R2
when taken to~;ether with the Carbon Atom to which they are
attached represent Tetrahydro,pxran
A solution of diethyl tetrahydro-4H-pyran-4,4-dicarboxylate, the
compound of Formula (7a), ( 12 Kg) in toluene ( 104 Kg) was cooled to
3 0 between -30°C to -35°C, and diisobutylaluminum hydride (69
Kg) was
added at such a rate so as to maintain a reaction temperature of
-25°C. After the addition was complete, the temperature was raised to
15"C over 3 hours, and the reaction stirred until all starting material
was consumed. The mixture was then recooled to -15°C and allowed
3 5 to stand overnight. The product was partitioned between ethyl
X193178
- 145 -
acetate (54 Kg), ethanol (48 Kg), and saturated sodium sulfate solution
(60 litres), and the mixture stirred overnight at 25°C. The
precipitated
salts were filtered off, washed with tetrahydrofuran, and the filtrate
washed with brine and separated. The organic layer was dried over
magnesium sulfate and solvent removed under reduced pressure, to
give ethyl 4-hydroxymethyltetrahydropyran-4-carboxylate (3.8 Kg),
the. compound of Formula (8a).
3. Preparation of a Compound of Formula ~9a) where Rl and R2
when taken together with the Carbon Atom to which they are
attached represent Tetrahydrop, ran
To a solution of lithium hydroxide monohydrate (4.46 Kg) in
methanol (44 litres) and water ( 11 Kg) was added ethyl 4-
hydroxymethyl-tetrahydropyran-4-carboxylate (8.0 Kg). The mixture
1 5 was refluxed for 30 minutes, then solvent removed under reduced
pressure. The mixture was cooled to 20°C, methyl tert-butyl ether
( 14.8 Kg) added, stirred for 10 minutes, and allowed to settle. The top
organic layer was separated. This was repeated twice more, then the
remaining mixture cooled to -10°C, and a solution of 31% hydrochloric
2 0 acid (13 Kg) in water (3 Kg) added, maintaining the temperature
below 5°C. The mixture. was extracted several times with
tetrahydrofuran, and the combined organic phases dried over
magnesium sulfate. Approximately 90% of the tetrahydrofuran was
removed, and the remaining solution added to a mixture of hexane
2 5 (64.5 Kg) and methyl tert-butylether (23.'7 Kg) with stirring. The
precipitated solid material was filtered off and dried under reduced
pressure at 60°C, to give 4-hydroxymethyl-tetrahydropyran-4-
carboxylic acid (3.7 Kg), the compound of Formula (9a).
3 0 4. Preparation of a Compound of Formula Ia where R1 and R2 when
taken together with the Carbon Atom to which they are attached
represent Tetrahydropxran
To a mixture of 4-hydroxymethyl-tetrahydropyran-4-carboxylic
acid (3.84 Kg), 4-dimethylaminopyridine (0.6 Kg) in dichloromethane
3 5 (32 litres) was added triethylamine (4.88 Kg). The mixture was
2193178
- 146 -
cooled to -20°C, and a solution of benzenesulfonyl chloride (4.66 Kg)
in dichloromethane (5 litres) was added over a period of 35 minutes,
maintaining the temperature below -10°C. The mixture was stirred at
-10''C for 30 minutes, then 3N hydrochloric acid (10 litres) and water
( 10 litres) were added with stirring, then the layers allowed to
separate. The organic layer was separated, the aqueous layer washed
with dichloromethane ( 16 litres), the combined organics washed with
aqueous 5% sodium bicarbonate solution (121itres), then with water
(12 litres), and solvent removed under reduced pressure, to give 2,7-
1 0 dioxaspiro[3,5]nonane-1-one, a compound of Formula (l0a)
To a mixture of 60% sodium hydride (0.92 Kg) in tetrahydrofran
(261itres) at 0°C was added a solution of 4-(4-chlorophenoxy)thio-
phenol (4.37 Kg) in tetrahydrofuran ( 15 litres), maintaining the
temperature below 10°C. The mixture was allowed to warm to room
temperature for 30 minutes, then recooled to 0°C. The concentrated
solution of 2,7-dioxaspiro[3,5]nonane-1-one obtained above was then
added slowly to this mixture, maintaining the temperature below
10°C. The mixture was allowed to warm to room temperature, and
2 0 stirred for 30 minutes. The mixture was then treated with
3N hydrochloric acid ( 16 litres) and dichloromethane (30 litres). The
organic layer was separated and the aqueous layer extracted twice
with dichloromethane (20 litres). The combined organics were washed
with water (20 litres), filtered, and 1001itres of solvent removed
2 5 under atmospheric pressure. To the remaining reaction product was
added acetonitrile (60 litres) and after a further 601itres of solvent
were removed by distillation, acetonitrile (40 litres) was added and
the total volume of the remainder reduced to 301itres by distillation.
This mixture was then heated to mild reflux (80°C), and then
slowly
3 0 cooled to 0°C. The product was filtered off, washed with hexane,
and
dried to about 60°C under reduced pressure, to yield
4-[4-(4-chlorophenoxy)phenylthiomethyl]tetrahydropyran-4-
carboxylic acid (5.61 Kg).
~'~9~1~'8
- 147 -
5. Preparation of a Compound of Formula Iba where R1 and R2
when taken together with the Carbon Atom to which the, are
attached represent Tetrahydropyran
A solution of 4-[4-(4-chlorophenoxy)phenylthiomethyl]tetra-
hydropyran-4-carboxylic acid (5.5 Kg) and N,N-dimethylformamide
(27 ml) in dichloromethane (27.5 litres) was cooled to 5°C, and oxalyl
chloride ( 1.4 litres) added slowly with stirring. After addition was
complete, the mixture was allowed to warm to room temperature and
stirred for 2 hours, thus forming a compound of Formula ( 12). The
solution was then recooled to 10°C, and a mixture of 50% aqueous
hydroxylamine (5.4 litres), tert-butanol ( 12.1 litres) and
tetrahydrofuran (30.5 litres) was added slowly, maintaining the
temperature below 21 °C. The mixture was then allowed to warm to
room temperature until the reaction was complete. The solvent was
then evaporated under reduced pressure until 90% had been
removed, at which point acetonitrile (42.5 litres) was added and the
remaining dichloromethane removed by distillation under reduced
pressure. The remaining solution was heated under reflux, and water
( 126 Kg) added at such a rate so as to maintain reflux. The solution
2 0 was then cooled to 5°C for 12 hours, and the solid thus obtained
filtered off. This product was washed with water and dried under
vacuum at 50°C to yield 4-[4-(4-chlorophenoxy)phenylthiomethyl]-
tetrahydropyran-4-(N-hydroxycarboxamide) (5.06 Kg), a compound
of Formula Iba.
6. Preparation of a Compound of Formula Id where R1 and R2 when
taken together with the Carbon Atom to which then are attached
represent Tetrah,~p~ran
To a solution of 4-[4-(4-chlorophenoxy)phenylthiomethyl]-
3 0 tetrahydropyran-4-(N-hydroxycarboxamide) (5.06 Kg) in
tetrahydrofuran (28 litres) and methanol { 112 litres) at 15°C was
added a solution of OXONE ( 14.23 Kg) in water (72 litres) with stirring,
ensuring that the temperature did not exceed 16°C. After the addition
was complete, the temperature was raised to 20°C and the mixture
3 5 stirred for 3 hours, then poured into a cold mixture (5°C) of
toluene
~1931~8
- 148 -
(60 litres) and ethyl acetate (98 litres) with stirring. The resultant
mixture was filtered, the organic and aqueous layers thus obtained
separated, and the aqueous layer washed with a mixture of ethyl
acetate (25 litres) and toluene (10 litres). This wash was repeated
twice more. The combined extracts and organic layer was washed
twice with water (25 litres), and solvent removed under reduced
pressure to a volume of 30 litres. The solution was cooled to 5°C, and
the solid filtered off, washed with ethyl acetate/water and dried
under vacuum at 50°C, to yield 4-[4-(4-chlorophenoxy)phenylsul-
fonylmethyl]-tetrahydropyran-4-(N-hydroxycarboxamide) (4.3 Kg).
7. Similarly other Compounds of Formula I may be prepared.
EXAMPLE 36
This example illustrates the preparation of representative
pharmaceutical compositions for oral administration containing a
compound of Formula I, or a pharmaceutically acceptable salt
thereof, e.g., N-hydroxy-2-[4-(4-phenoxyphenylsulfonyl)-piperidin-4-
2 0 yl)]-acetamide:
A. Ingredients % wt./wt.
Compound of Formula I 20.0%
Lactose 79.5%
2 5 Magnesium stearate 0.5%
The above ingredients are mixed and dispensed into hard-shell
gelatin capsules containing 100 mg each, one capsule would
approximate a total daily dosage.
X193178
- 149 -
B. Ingredients% wt./wt.
Compound of Formula I 20.0%
Magnesium stearate 0.9%
Starch 8.6%
Lactose 79.6%
PVP (polyvinylpyrrolidine) 0.9%
The above ingredients with the exception of the magnesium
stearate are combined and granulated using water as a granulating
liquid. The formulation is then dried, mixed with the magnesium
stearate and formed into tablets with an appropriate tablet machine.
C Ingredients
Compound of Formula I 0.1 g
Propylene glycol 20.0 g
Polyethylene glycol 400 20.0 g
Polysorbate 80 1.0 g
Water q.s. 100 ml
2 0 The compound of Formula I is dissolved in propylene glycol,
polyethylene glycol 400 and polysorbate 80. A sufficient quantity of
water is then added with stirring to provide 100 ml of the solution
which is filtered and bottled.
2 5 D Ingredients % wt./wt.
Compound of Formula I 20.0%
Peanut Oil 78.0%
Span 60 2.0%
3 0 The above ingredients are melted, mixed and filled into soft
elastic capsules.
X19317$
- 150 -
EXAMPLE 37
This example illustrates the preparation of a representative
pharmaceutical formulation for parenteral administration containing a
compound of Formula I, or a pharmaceutically acceptable salt thereof,
e.g., N-hydroxy-2-[4-(4-phenoxyphenylsulfonyl)-piperidin-4-yl)]-
acetamide:
Ingredients
1 0 Compound of Formula I 0.02 g
Propylene glycol 20.0 g
Polyethylene glycol 400 20.0 g
Polysorbate 80 1.0 g
0.9% Saline solution q.s. 100 ml
The compound of Formula I is dissolved in propylene glycol,
polyethylene glycol 400 and polysorbate 80. A sufficient quantity of
0.9% saline solution is then added with stirring to provide 100 ml of
the I.V. solution which is filtered through a 0.2 p, membrane filter
2 0 and packaged under sterile conditions.
EXAMPLE 3 g
This example illustrates the preparation of a representative
2 5 pharmaceutical composition in suppository form containing a
compound of Formula L, or a pharmaceutically acceptable salt thereof,
e.g., N-hydroxy-2-[4-(4-phenoxyphenylsulfonyl)-piperidin-4-yl)J-
acetamide:
3 0 Ingredients % wt./wt.
Compound of Formula I 1.0%
Polyethylene glycol 1000 74.5%
Polyethylene glycol 4000 24.5%
3 5 The ingredients are melted together and mixed on a steam bath,
X193178
- 151 -
and poured into molds containing 2.5 g total weight.
EXAMPLE 39
This example illustrates the preparation of a representative
pharmaceutical formulation for insufflation containing a compound of
Formula I, or a pharmaceutically acceptable salt thereof, e. g. , N-
hydroxy-2-[4-(4-phenoxyphenylsulfonyl)-piperidin-4-yl)]-acetamide
Ingredients % wt./wt.
Micronized compound of Formula I 1.0%
Micronized lactose 99.0%
The ingredients are milled, mixed, and packaged in an
insufflator equipped with a dosing pump.
EXAMPLE 40
This example illustrates the preparation of a representative
pharmaceutical formulation in nebulized form containing a compound
2 0 of Formula I, or a pharmaceutically acceptable salt thereof, e.g., N-
hydroxy-2-[4-(4-phenoxyphenylsulfonyl)-piperidin-4-yl)]-acetamide:
Ingredients % wt./wt.
Compound of Formula I 0.005%
2 5 Water 89.995%
Ethanol 10.000%
The compound of Formula I is dissolved in ethanol and blended
with water. The formulation is then packaged in a nebulizer
3 0 equipped with a dosing pump.
EXAMPLE 41
This example illustrates the preparation of a representative
3 5 pharmaceutical formulation in aerosol form containing a compound of
~19311g
- 152 -
Formula I, or a pharmaceutically acceptable salt thereof, e.g., N-
hydroxy-2-[4-(4-phenoxyphenylsulfonyl)-piperidin-4-yl)]-acetamide:
Ingredients % wt./wt.
Compound of Formula I 0.10%
Propellant 11 / 12 98.90%
Oleic acid 1.00%
The compound of Formula I is dispersed in oleic acid and the
propellants. The resulting mixture is then poured into an aerosol
container fitted with a metering valve.
EXAMPLE 42
In Vitro Assay
42A. Isolation of MMPs for Assays
The catalytic domain of human collagenase-1 was expressed as a
fusion protein with ubiquitin in E. Coli (Gehring, E.R. et al., J. Biol.
Chem., 270, 22507, (1995)). After purification of the fusion protein,
2 0 the fibroblast collagenase-1 catalytic domain was released by
treatment with 1 mM of aminophenylmercuric acetate (APMA) for 1
hour at 37°C and purified by zinc chelate chromatography.
Human collagenase-2 and gelatinase B were isolated in active
2 5 form from huffy coats (Mookhtiar, K.A. et al., Biochemistry, 29,
lOfi20, (1990)).
The propeptide and catalytic domain portion of human
collagenase-3 was expressed in E. Coli as an N-terminal fusion protein
3 0 with ubiquitin. After purification, the catalytic domain was obtained
by treatment with 1 mM APMA for 1 hour at 37°C, and purified by
zinc chelate chromatography.
Rat collagenase-3 was purified in active form from the culture
3 5 media of uterine smooth muscle cells (Roswit, W.T. et al., Arch.
X193178
- 153 -
Biochem. Biophys., 225, 285-295 (1983)).
The catalytic and fibronectin-like portion of human progelatinase
A was expressed as a fusion protein with ubiquitin in E. Coli. Assays
were carried out on autolytically activated material. Rat progelatinase
A was purified from the culture media of interleukin-1 stimulated
keratinocytes and activated by treatment with 1 mM APMA for 1
hour at 37°C, and subsequently dialyzed to remove excess APMA.
Human prostromelysin-1 was purified from the culture medium
of synovial fibroblasts by affinity chromatography using an
immobilized monoclonal antibody. The zymogen was activated by
treatment with trypsin ( 1.5 p,g/ml) for 1 hour at 23°C to give a
mixture of 45 and 28 kD species. The catalytic domain of human
stromelysin was prepared by expression and purification of
prostromelysin-1 from E. Coli and activated with 1 mM APMA for 1
hour at 37°C, followed by dialysis. Rat prostromelysin-1 was
expressed in Chinese Hampster Ovary cells and purified from the
culture media. It was activated by 1 mM APMA for 1 hour at 37°C,
2 0 followed by dialysis.
Human promatrilysin was expressed and purified from Chinese
Hampster Ovary cells (Barnett, J. et al., Prot. Expres. Pur., 5, 27,
( 1994)). The zymogen was activated by treatment with 1 mM APMA
2 5 for 1 hour at 37°C, and purified by zinc chelate chromatography.
Compounds of Formula I exhibited the ability to inhibit the
collagenases when tested in this assay.
3 0 42B. In Vitro Assay Procedure
Assays were performed in assay buffer (50 mM Tricine pH 7.5,
200 mM sodium chloride, 10 mM calcium chloride, 0.005% Brij-35)
containing 2.5% methyl sulfoxide (DMSO) once the substrate and
3 5 inhibitor were diluted into it. Stock solutions of inhibitors were
~93~7g
- 154 -
prepared in 100% DMSO. Stock solutions of the substrate were
prepared in 100% DMSO at a concentration of 2 mM.
The assay method was based on the hydrolysis of MCA-Pro-Leu-
Gly-Leu-DPA-Ala-Arg-NH2 (Bachem, Inc.) at 37°C (Knight, C.G. et
al.,
FEBS, 296, 263-266 (1992)). The fluorescence changes were
monitored with a Perkin-Elmer LS-SOB fluorimeter using an
excitation wavelength of 328 nm and an emission wavelength of 393
nm. The substrate concentration used in the assays was 10 ,mole. The
inhibitor was diluted into the assays from a solution in 100% DMSO,
and controls substituted an equal volume of DMSO so that the final
DMSO concentration from inhibitor and substrate dilutions in all
assays was 2.5%. The inhibition results are expressed as the inhibitor
concentration that produced 50% inhibition (IC50) of the activity in
the control (non-inhibited) reaction.
EXAMPLE 43
In Vitro Assay
2 0 This assay determines the ability of the compounds of Formula I
to inhibit the degradation of the collagen matrix (as judged by release
of hydroxyproline), and proteoglycan (as judged by the release of
35S_labelled glycosaminoglycans) from cartilage explants.
2 5 Small cartilage explants (3 mm diameter) were prepared from
freshly sacrificed bovine knee joints and labeled with 3 5 S 04. 3 5 S _
labelled glycosaminoglycans (GAG'S) and collagen fragments are
released into the culture medium in response to the addition of rhIL-
1-alpha, which induces the expression of chondrocyte matrix
3 0 metalloproteases (MMP's), including stromelysin and collagenase. The
percent inhibition of hydroxyproline and GAG's released was
corrected for spontaneous release in the absence of rhlL-1-alpha.
Compounds of Formula I, when tested in this assay, displayed the
3 5 ability to inhibit the release of both collagen fragments and 3 S S -
~~9~178
- 155 -
labelled GAG's from cartilage explants.
EXAMPLE 44
In Vivo A s s a ~r
The cartilage plug implantation assay measures the destruction of
the collagen matrix of a cartilage plug implanted in a rat (Bishop, J. a t
al., J. Pharm. Tox. Methods, 30, 19, ( 1993)).
Previously frozen bovine nasal cartilage plugs weighing
approximately 20 mg were embedded in polyvinyl sponges
impregnated with Mycobacterium tuberculosis and implanted
subcutaneously in female Lewis rats. Dosing was begun 9 days after
implantation and the plugs were harvested about one week later. The
plugs were weighed, hydrolyzed, and the hydroxyproline content
measured. Efficaciousness was determined by the comparison of the
compound-treated groups with vehicle treated controls.
The compounds of Formula I exhibited the ability to inhibit the
2 0 degradation of the cartilage plugs in this assay.
EXAMPLE 45
In Vivo Assay Procedure
2 5 45A. Determination of TNF Production Followin~~ LPS Stimulation
Female Balb/c mice, 6-8 weeks old (Jackson Labs or Harlan) were
used. For each treatment group, 6-8 mice were used. Mice were
injected LP. with LPS {Sigma, 13129, 10-20 p.g/mouse) after
3 0 treatment with a compound of Formula I. The compound of Formula I
or vehicle was administered subcutaneously (S.C.) once, 30-60
minutes prior to LPS challenge. Control animals received CMC vehicle
alone or CMC + 2-5% DMSO. Animals were bled 1.5 hours after LPS
injection under anesthesia with metofane from the retro-orbital
3 5 plexus, using a Pasteur pipette. Blood was collected in a microtainer
~g~l7g
- 156 -
serum separator tube (Becton Dickinson #5960). The sera were
separated and either tested the next day or they were kept at -20°C
until ready to test for TNF-a.
458. ELISA Assay for Murine TNF-a
The Endogen (EM-TNFA kit) mouse tumor necrosis factor alpha
(m'TNF-a) kit is an in vitro enzyme-linked immunosorbent assay for
the quantitative measurement of mouse TNF-a (ordering code: EM-
TNEA; Endogen, 30 Commerce Way, Woburn, MA 01801-1059, USA).
Standards (lyophilized recombinant E. coli-derived mouse TNF-a) or
serum samples (50 p.l each) were added in duplicate to each well of
the precoated anti-mTNF-a plate. Biotinylated antibody (50 p.l) was
added, the plates were incubated for 2-3 hours at room temperature.
1 5 The wells were washed five times with wash buffer and 100 ~,1 of
diluted strepavidin HRP were added to each well and then were
incubated at room temperature for 30 minutes. After washing (SX j,
100 ~.l premixed TMB substrate solution were added to each well and
plates were developed at room temperature in the dark for
2 0 30 minutes. The reaction was stopped by adding 100 ~,1 of the stop
solution. Absorbance at 450-575 nm was measured in a plate reader
(ThermoMax, Molecular Devices). Results are calculated at pg/ml TNF-
a by comparison to the standard curve, using Immunofit Beckman
software. They are expressed as mean pg/ml of TNF-a, and as
2 5 percentage of inhibition compared to controls (animals injected with
LPS alone), considered 100% of TNF-a production.
The compounds of Formula I, when tested in this assay, exhibited
the ability to inhibit TNF-a production.
EXAMPLE 46
TNF Conjugate Immunoassay
Human Monomac 6 cells were cultured at 37°C in RPMI 1640
3 5 medium supplemented with 10% fetal calf serum to a density of 1 X
'~9W1~$ - 157-
105 cells/mL. All subsequent incubations were performed at 37°C.
23 0 ~ 1 of these cells were placed in each well of a 96-well tissue
culture plate and the cells incubated for 15 minutes. 10 p,l of desired
concentration of compounds of Formula I in the above mentioned
medium were added to the appropriate wells and incubated for an
additional 15 minutes. To each well was added 10 ~,l of an LPS/PMA
mixture which brings the final concentration of LPS to 10 ng/mL and
the final PMA concentration to 30 ng/mL. The cells were then
incubated for 2 hours after which the plate was centrifuged and the
medium removed and analyzed for TNF content. The analysis was
performed using an R & D Systems TNF Quantikine Immunoassay
and following the manufacturer's protocol (R & D. Systems, 614
Mckinley Place N.E., Minneapolis, MN 55413, USA; Catalog No. DTA50).
The IC50 was calculated from the percent inhibition of TNF released
into the medium.
The compounds of Formula I, when tested in this assay, exhibited
the ability to inhibit TNF production.
2 0 EXAMPLE 47
TNFR Shedding ImmunoassaX
Human Monomac 6 cells are cultured to a density of 1 X 106
cells/mL at 37°C in RPMI 1640 medium supplemented with 10% fetal
2 5 calf serum. All subsequent incubations are performed at 37°C.
230 ~.l of these cells are placed in each well of a 96-well tissue
culture plate and the cells are incubated for 15 minutes. 10 p.l of
desired concentration of compounds of Formula I in the above
mentioned medium are added to the appropriate wells and incubated
3 0 for an additional 15 minutes. To each well is added 10 p,l of PMA at a
final concentration of 30 ng/mL. The cells are then incubated for
16 hours after which the plate is centrifuged and the medium is
removed and analyzed for TNF receptor content. The analysis is
performed using the R & D Systems TNF receptor Quantikine
3 5 Immunoassay following the manufacturer's protocol. Measurements
~193'~78 - lsg-
of each TNF receptor (receptor I and receptor II) are performed in
this way. The IC50 is calculated from the percent inhibition of TNF
released into the medium.
s The compounds of Formula I, when tested in this assay, exhibited
the ability to selectively inhibit TNF production.
While the present invention has been described with respect to
specific embodiments thereof, it will be understood by those skilled in
the art that various changes may be made and equivalents may be
substituted without departing from the scope of the invention. All
such modifications are intended to be within the scope of the claims
appended hereto.