Note: Descriptions are shown in the official language in which they were submitted.
WO 96100308 PCT/US95I09199
r ' w : 'y
METHOD AND APPARATUS FOR EXTRACTING PRECIOUS METALS
FROM THEIR ORES AND THE PRODUCT THEREOF
STATEMENT AS TO RIGHTS IN INVENTIONS
MADE UNDER FEDERALLY-SPONSORED
RESEARCH AND DEVELOPMENT
This invention was made with U.S. Government support under Small Business
Innovation
Research Grant No. DI~tI-9461234 which was awarded by the National Science
Foundation
(NSF), an independent agency ofthe U.S. Government. The U.S. Government has
certain tights
in the invention.
TECHNICAL FIELD
This invention relates to a method and apparatus far extracting precious
metals from their
ores and the product thereof. In particular, it relates to the fbllowing: (1)
a biohydrometallurgical
process and apparatus for extraction and recovery of gold, silver and platinum
group elements
from their ores; (2} the products of that process and apparatus
BACKGROUND ART
2 0 Development of cost-effective techniques far extraction of precious metals
from their ores
has been a goal of metallurgists for hundreds of years. In recent years, the
addition of emzron-
mental costs to the total cost of products of precious metal miners has
encouraged a search for
environmentally acceptable options, especially for refractory ores.
In the prior art the first step in precious metal production from ore involves
preparing the
2 5 ore for precious metal extraMion. Preparation can take any one of a number
of courses depending
on the character of the are. Gold and silver ores ofren contain metallic
sulfides. Ores containing
platinum-group elements (PGE) typically also contain metallic sulfides. For
example, in the
Bushveld Igneous Complex in South Africa, platinum group element values occur
in solid solution
in the base-metal sulfides pyrrhotite (Fel_zS}, pentlandite (Fe,Ni)vSB, pyrite
(FeS2), and as the
30 discrete platinaid metal minerals cooperate (PtS), faurite (RuS:), braggite
(Pt,Pd,Ni)S and Pt-Fe
alloys or their intergrowths (see Guilbert, J.1~4. 8. Park, C.F , Jr. "The
aeolow~ of ore deposits."
New York: W.H. Freeman and Company, 1986}The 5tillwater Complex in Montana is
a similar
deposit Typically, refractory, non-oxidized (e.g., sulfide) Bald and silver
ores (especially those
CA 02194349 2004-03-O1
with a relatively high carbon content) are oxidized at elevated temperatures
and pressures in large
autoclaves (i.e., "roasted"), prior to the extraction of precious raetals by
means of cyanide
leaching, (see McQuiston, Jr., F.W., & Shoemaker, R.S., Gold and Silver
Cyanidation Plant
Pr ctice, Vol. II, Baltimore: Port City Press, 1980).
During the Last decade, heap leach processes for cost-effective bio-oxidation
of pyritic and
arsenopyritic sulfides in gold and silver ores have been developed to the poim
of commercial
application (see Totma, A.fi., Biotechnologyr: A Comprehensive Treatise in 8
Volumes, Deerfield
Beach, Florida: Verlag Chemie, I981). Recent improvements in the art are
disclosed by: Pooley
et al. in U.S. Patent No. 4,822,413, April 18, 1989; Hackl et a1. in U.S.
Patent No. 4,987,081,
January 32, 1991; Hunter in U.S. Patent No. 5,076,927, December 31, 1991;
Brierly et al. In U.S.
Patent No. 5,127,942, July 7, 1992; and Brierly and Hill in U.S. Patent No.
5,246,486, September
21, 1993.
A great variety of precious metal extraction processes have also been
developed
(see Gupta, C.K., & Mukherjee, T.K., I~ydrometallurav in Extraction Processes,
Vol. I, Boston:
CRC Press, 1990). Precious metal extraction processes are disclosed by: Pesic
in U.S. Patent
No. 4,778,519, October 18,.1988; Ball et al, in U.S. Patent No. 4,902,345,
February 20, 1990;
and Kandemir in UK Patent No. 2, I 80,829, published April 8, 1987.
The relatively
low economic cost ofcyanidation, however, has ensured its proliferation.
2 0 State-of the-art precious metal heap leach practice varies with the nature
of the ore. Bio-
oxidation process steps may include ore crushing, acid pretreatment,
inoculation with appropriate
sulfide-oxidizing bacteria, addition of nutrients, recirculating the
biolixiviant and cooling the heap
(for 3 to 8 days), and allowing the heap to "rest" (for 3 to 8 days). Precious
metal extraction by
means of cyanidation may include the process steps of washing the heap for an
extended period
(e.g., 14 days) to remove residual acidity or iron content, breaking the heap
apart in order to
agglomerate it with cement andlor lime to make a new heap, leaching it with an
alkaline cyanide
or thiosulfate solution for 30 to 40 days, and recovery of gold and silver
from the leach solution
by adsorption on activated carbon or zinc dust precipitation.
A significant amount of work in,the field of bio-oxidation and metals
extraction has been
accomplished by a variety of investigators. Tomizuka, N. & Yagisawa, M, in
"Optimum
conditions for leaching of uranium and oxidation of lead sulfide with
thiobaci>yus ferrooxidans and
recovery of metals from bacterial leaching solution with sulfated-reducing
bacteria," (in
et lu ' A Ii do o B a ' 1 Leac 'n d elat Mtcrob'o1 'cal om Murr,
2
W0 96100308 , PCTIUS95I09199
~' .; .' :, ,
L.E., Torma, A.E., & Brierly, J.A. (Eds.) New Yark: Academic Press, 1978),
describe a two-
step process for leaching of uranium and oxidation of lead sulfide where
recovery of metals is
accomplished by means of microbial sulfate reduction. Alper, J., in "Bacterial
methods may strike
it rich in refining metals, cleaning coal," (Hitfr Technolos;v. April, 1984,
pp. 32-35), describes the
bio-oxidation of gold-bearing arsenopyriteJpyrite and notes that production of
large amounts of
arsenic and sulfurous gases is avoided. Torma, A.E., in Biotechnoloev A
Comprehensive
Treatise in 8 Volumes, (Deerfield Beach, Fl: Verlag Chemie, 1988), reviewed
bioleaching
processes. Livesay-Goldblatt, E., in Fundamental and Applied
BiohvdrometallurQV, (Proc. 6th
International Symposium on Biohydrometallurgy, Vancouver, B.C. 89-96, 1986),
described a
i G process for gold recovery from arsenopyrite!pyrite ore by bacterial
leaching and cyarudation.
Torma, A.E., in "Biotechnala~~. A co>~rehensive treatise in 8 volumes,"
(Deerfield Beach, FL:
Verlag Chemie, 198x), reviews bio-oxidation of gold and silver ores. Hackl,
R.P., Wright, F., &
Bruynesteyn, A., in Proceedines of the Third .Annual General Meetinc of
Biominet, (August 20-
21, 71-90, 1986), described development of the BIOTANKL.EACH process for
leaching pyritic
materials from gold and silver ore. The results of bench-scale and pilot-scale
evaluations were
presented. Marchant, P.B., & Lawrence, R.W., in "Flowsheet design, process
control, and
operating strategies in the bio-oxidation of refractory gold ores,"
(Proceedings of the Third
Annual General Meeting ofBiominet, August 20-21, 39-51, 1986), listed
considerations in the
design of commercial bio-oxidation plants. Lawrence R.W , in "Biotreatment of
Gold,"
2 0 (Microbial Alineral Recovery New York: MCGraw-Hill edited by Ehrlich, H.L.
and Brierly, C.L,
1990), discussed biotreatment of gold ore. The benefits of using the BacTech
moderately
thermophilic cultures in bio-oxidation processes were discussed by Budden.
J.R., & Spencer, P.A.
in "Tolerance to temperature and water quality for bacterial oxidation: The
benefits of BacTech's
moderately thermophilic culture," (FEA1S Microbiolos3y Reviews, I I, 191-196,
1993). Chapman,
2 5 J.T., Marchant, P.B., Lawrence, R.W., & Knopp, R., in "Biooxidation of a
refractory gold
bearing high arsenic sulphide concentrate: A pilot study;' (FEMS hlicrobiolo;y
Reviews, I 1,
243-252, 1993), described a modular mobile bialeach pilot plant for bio-
oxidation of a refractory
gold-bearing high-arsenic sulfide concentrate. Moffat, A. S., in "Microbial
mining boosts the
environment," (Science 264, 778-779, 1994), disclosed bow bio-oxidation can
increase the
30 efficiency of mining.
Thermophilic versus mesophilic biolea.ching process performance was evaluated
by
Duarte, 7.C., Estrada, P.C., Pereira, P.C., & Beaumont, F1.P. in " FEMS
Microbiology Reviews,
11, 97-102, 1993. Two years of BIO?~ bio-oxidation pilot plant data were
analyzed by Hansford,
3
WO 96l0030S 4 ~ , ~ ~ PCTlUS95t09199
G. S., & bliiler, D.&I. in "Biooxidation of a sold-bearing pyrite-
arsenophyrite concentrate,"
FEMS Microbiolgs>v Reviews, 11, 175-182, 1993. Hoffman, W., ICatsikaros; N., &
Davs, G., in
"Design of a reactor bioleach process for refractory gold treatment," {FEMS
Microbiology
Reviews. 11, 221-230, 1994), described the design of a reactor bioleach
process For refractory
gold treatment Liu, X , Petersson, S., & Sandstrom, A., in "Evaluation of
process variables in
bench-scale bio-oxidation ofthe Olympias concentrate," (FEMS
Microbiolo~Reviews; I l, 207-
214, 1993), presented an evaluation of the effects of process variables on
pyritelarsenopyrite
oxidation and gold extraction. Maturana, H., Lagos, Ii., Flores, V., Gaeta,
~1., Comeja, I'., ~
Wicrtz, J.1' , in "Integrated biological process for the treatment of a
Chilean complex gold ore,''
{FEMS Microbiology Reviews, I l, 215-220, 1993), described an integrated
biological process for
treatment of a complex gold ore. Mineral sulfide oxidation by enrichment
cultures of a novel
thermoacidophilic bacteria were described by Norris, P.R. & Owen, J.P.
in"P4lineral sulphide
oxidation by enrichment cultures of novel thecmoacidophilic bacteria," FEMS
Microbiolouv
Reviews 11, >I-56, 1993). Rate controls on the bio-oxidation of heaps
ofpyritic material
I 5 imposed by bacterial upper temperature limits were described by Pantelis,
G. ~ Ritchie, A.LM. in
"Rate controls on the oxidation of heaps of pyritic material imposed by upper
temperature limits
an the bacterially catalysed process,' {FEMS Microbioloev Reviews, 1 I, 183-
190, 1993). Bio-
oxidation bacteria have been characterized in detail. Briefly, C.L., &
Brierly, J.A., in ".A
chemoautotrophic and thermophilic microorganism isolated from an acid hot
spring," (Canadian T'
Microhioloev, 19, 183-188, 1973), characterized a chemoautotrophic and
thermophilic (70°C)
microorganism isolated from an acid hot spring. De Rosa, M., Gambaeorta, A., &
Bullock, J.D.,
in "Extremely thermophilic acidophilic bacteria convergent wRh Sirlfolobus
acrdacaldarzrrs," (J.
General Microbioloev, 86, I >6-164, 1975), characterized the extremely
thermophilic (85°C),
acidaphilic {pH I .0) bacteria Szrlfvlobus aridocaldarius.
2 5 While prior art has extensively studied and developed the bio-oxidation
process for
oxidizing metal sulfides present in gold and silver ore to expose or mobilize
precious metal values,
little attention has been given to biotechnologies for extracting
(solubilizing) and recovering
those values. Torma, A.E" in "Biotechnology: A Comprehensive Treatise in 8
Volumes,"
(Deerfield Beach, FL: Verlag Chemie, 1988), reviewed dissolution of gold by
microorganisms.
Olson, G.J., in "Microbial oxidation ofgold ores and gold bioleaching," {FENIS
Microbialouv
etters,119, I-6, 1994), reviewed microbial oxidation of gold ores arid gold
bioleaehing. He
described cyanogeruc microorganisms such as Chromo6acterirrnz violacerrm, and
he noted that
gold forms soluble sulfide and polysulfide complexes and suggested that
biogenesis of partially-
4
WU 9GI00308 t ~ ~ ~ PCT/I1S95109199
oxidized sulfur compounds may be a mechanism of gold dissolution. The U.S.
Bureau afMines,
(office of Technology Transfer in U. S. Bureau of Mines Cooperative Research
Opportunities,
Washington, DC: U.S. Bureau ofMines, 1995), disclosed that the solubility
ofgold in dilute
polysulfide solutions at elevated temperatures and pressures and neutral pH
levels is comparable
to the solubility of gold in cyanide solutions.
Investigators have hypothesized natural processes far gold solubilization
involving specific
sulfide-oxidizing bacteria such as Thiohcreillus ferroxidans and THiolxrcillus
denitrifrcarrs (see
Lyalikova, N.N. & Mokeicheva, L. Y. in "The role of bacteria in gold migration
in deposits,"
Microbiolo~v, 38: 805-810, 1969; Kulibakin, V.G., Raslyakow, N.A., Tsimbalist,
V G.,
Mel'nikova, R.D., and Nepeina, L.A., in "Role of sulfirr bacteria in supergene
migration and
concentration ofgold," Trans. Inst. Geol. Geofiz. Akad. SSSR Sib. Otd., 370:
75-86, 1977 (in
Russian)) and sulfate-reducing bacteria such as De.sulfovibrio (see Meyers,
W.B. "An hypothesis
ofthe chemical environment ofthe Rand Goldfceld, South Afiica," U.S.
Geological Sun~ey Open-
File Report 1389, 1970; Meyers W.B. "Precambrian pyritic gold-and uranium-
bearing
conglomerates," Geolos_zical Society of America, Abstracts with Programs, 3:
656-657, I971;
Myers, W.B.,"Genesis of Uranium and Gold-Bearing Precambrian Quartz-Pebble
Conglomerates," Geological Satrev Professional Paper 1161-Arl, 1981; and
Mossman, D.J. &
Dyer, B.D., "The geochemistry of Witwatersrand-type gold deposits and the
possible influence of
ancient prokaryotic communities on gold dissolution and precipitation,"
Precambrian Res. earth,
30: 303-319, 1985).
Speculation concerning the impact of biological organisms on the
solubilization of gold
and silver by bisulfide complexes is based on the findings of those who have
studied the formation
of precious metal-bisulfide complexes abiotically. Krauskopf, K.B., in "The
solubility of gold,"
(Eceyomic Geoloev, 46, 858-870, 1951 ), disclosed that "gcdd may be
transported in alkaline
sulfide solutions, even in dilute solutions near the neutral paint" and
"experimentally, one ofthe
most perplexing facts about the chemistry of gold is its ability to dissolve
in solutions of HS- of
moderate concentration even at room temperature, whereas it dissolves in S'=
(i.e., more alkaline
solutions) only in concentrated solutions at high temperature." Barnes, H.L.
in " ,~ochemistrv of
hvdrothermal ore deposits." (New York: Hold, Rinehart & Winston, Inc., 1967),
disclosed that
3 0 "gold is known to be soluble in alkaline solutions containing bisulfide ar
sulfide ion" and that
"when the pH is increased to 7.0 at constant ~;5, HS' increases relative to
H2S and the solubilities
(of silver) summarized by Anderson rise abruptly by a factor of 20." (See
Anderson, G.M., "The
solubility ofPbS in H,S-water solutions,'' Economic GeoIo~,v. 57, 809-829,
1962). Weissberg,
S
WO 96100308 ~ ~ ~ ~ ~ ~ ~ 1'CTIUS95709199
B.G., in "Solubility of sold in hydrothermal alkaline sulfide solutions,"
(Economic Geolostv, 65,
551-556, 1970), disclosed that "in less alkaline solutions, where the HS' ion
predominates, the
experimentally determined solubility of gold ranges from 100 to 200 ppm Au in
solutions
containing from 0.2 to 0.3 moles NaHS/ICg solution at temperatures between I
50 and 250"C...."
and that "the present results are in good agreement with results given by
Ggrvzlo and by Lindner
and Gruner and substantiate the high solubility of gold in near neutral pH
bisulfide solutions."
(See Ogryzlo, S.P., "Flydrothermal experiments with gold," Economic Geology,
30, 400-424,
1935; and Linden l.L & Gruner, Lid'., "Action of alkali sulphide solutions on
minerals at
elevated temperatures," Economic Geology, 34, 537-560, 1939). Seward, T.hL, in
"Thio
complexes of gold and the transport of gold in hydrothermal ore solution,"
(Cxeochimica et
cosmochimica Acta, 37, 379-399, 1973), disclosed that "...an increase in the
bisulfide ion
concentration at constant pH (or HSYH=S ratio) leads to higher gold
soiubilities" and that
"considerable quantities of gold may be transported in hydrothermal ore
solutions as thio
complexes, particularly in the near neutral pH region where the
Au(HS)~° complex predominates."
Seward, T.M., in "The stability of chloride complexes of silver in
hydrothermal solutions up to
350°C," (Geochimica et Cosmochimica Acts, 40, 1329-1341, 1976),
disclosed that "the solubility
data {up to 180°C) ofMelenfyev et al, suggest that Ag(HS); will
probably be important in near
neutral hydrothermal solutions...." {See hlelent'yev, B.N., Ivanenko, V.V.,
and Pamfilova, L.A.
"Solubility of some ore-forming sulfides under hydrothermai conditions,"
Rastvorimost'
nekotorykh tudoobrazuyushkikh sul'fidov v g~drotermal' nvkh uslovivakh'
Moskva, 27-102,
1968). Mountain, B.W. & Wood, S,A., in "Chemical controls on the solubility,
transport, and
depostion of platinum and palladium in hydrothermal solutions: A thermodynamic
approach,"
(~onomic Geology, 83, 492-510, 1988), disclosed that "Westland states that Pt
is soluble in
alkaline sulfide solutions, possibly a [Pt{HS),(OH)~~j- complexes. Recent
experiments ... have
2 5 yielded Pt concentrations of about 1 ppm after one month for Pt metal in
contact with a 1.0 m
Na_5 solution." (See Westland, A.D., "Inorganic chemistry ofthe platinum-group
elements,"
Canadian Inst Mining Metallurgyr_Snec L'ol. 23, 7-18). Gammons, C.H. & Barnes,
FLL., in "The
solubilitg~ of Ag=S in near-neutral aqueous sulfide solutions at 25 to
300°C," (Geochimica et
Cosmochimica Acta, 53, 279-290, 1989), disclosed that "Ag(HS): is the dominant
silver species
3 0 in hvdrothermal fluids with near-neutral to alkaline pH, relatively low
oxidation state, high total
sulfide, and T - 300°C'...." Shenberger, D,M. & Barnes, H.L., in
°Solubility of gold in aqueous
sulfide solutions from 150 to 350°C," {Geochimica et Cosmochimica Acta.
53, 269-278, 1989),
disclosed that "the fact that gold is soluble in alkaline sulfide solutions
has been known since at
6
CA 02194349 2004-03-O1
least the 17th century..." and that "the high stability of Au(HS~' indicates
that geologically
significant quantities of gold can be transported in typical hydrothermal
solutions." Wood, S.A.
& Mountain, B.W., in "Thermodynamic constraints vn the solubility ofplatinum
and paDadium in
hydrothennal solutions: Reassessmert of hydroxide, bisulinde, and ammonia
complexing,"
(E~,onomic Geoloav, 84, 2020-2028, 1989), disclosed "... Pt solubilities on
the order of 10 to 100
ppb are attainable in bisulfide solutions at alkaline pH at 25 degrees C" and
that "Pt and Pd
bisulfide complexes show a strong similarity to Ag and Au bisulfide complexes,
where the
Au(HS)Z complex predominates over a wide range of conditions." Gammons, C.H.,
Bloom,
M. S., do Yu, Y., in "Experimental imrestigation of the hydrothermal
geochemistry of platinum and
palladium: I. Solubility of platinum and palladium sulfide minerals in
NaCIIH=SO, solutions at
300°C," (G~ochi~nc~ et Cosmochimica Acta, 56, 3881-3894, 1992),
disclosed "... a broad
similarity in the chemical behavior of Au and the PGE elements."
In evaluating the potential for transport of precious metals in natural
systems as bisulfide
complexes in hydrothermal fluids, investigators have assumed the hydrogen
fugacity in their
abiotic.systems is set by the mineral assemblages through which the fluids
would move in nature.
For example, Weissberg, B.G., in "Solubility of gold in hydrothermal alkaline
sulfide solutions."
(Economic GeoI~r, 65, 551-556, 1970), disclosed that "... the solubility of
gall in natural
systems depends on the hydrogen fugacity, which is conuolled principally by
equilibria between
the minerals pyrrhotite, pyrite, magnetite and hematite." Seward, T.M., in
"Thin complexes of
, gold and the transport ofgold in hydrothermal ore solutions," (Geochimica et
Cosmochimica
Acta, 37, 379-399, 1973), disclosed that "since the dissolution ofgold is a
function of hydrogen
fugacity (see, for example, Raymahashay, B.C. & Holland, H.D., "Redox
reactions accompanying
hydrothermal wall rock alteration," Economic Geolo~v, 64, 291-305, 1969), a
pyrite-pyrrhotite
'redox' buffer was present in all experiments in order that the fm was
maintained at a known
2 S value."
In the field of biological waste degradation, investigators have long
understood and
utilized the bioprocessing opportunities preserted by "interspecies hydrogen
transfer" betroveen
hydrogen-producing and hydrogen-consuming anaerobic microorganisms. A
biohydrometallurgical application of this knowledge was disclosed by Hunter,
R.M., in
"Biocata~rzed Pa ' Demineralization of Acidic Metal-Sulfate Solutions," (Ph.D.
Thesis,
Montana State University, 1989) and by Hunter, R.M. in "BiocataI3rzed Partial
Demineralization
of Acidic Metal-Sulfate Solutions," (U.S. Patent No. 5,076,927, December 31,
1991),
Microbial hydrogen
7
:,- i ;
R'O 9GJ00308 ~ ~ ~ ~ ~ ~ ~ ~ ~ PCTlL1S95109199
management techniques are disclosed by Harper, S.R. & Pohland, F G. in'"Recent
developments
in hydrogen management during anaerobic biological wastewater treatment,'"
(Biotechnola and
Bioen insT~g, 28, 585-602, 1986). The following reactions illustrate the
consumption of
hydrogen by acetogens (ACET), methanogens (METH), sulfate-reducing bacteria
I;SRB) and
nitrate-reducing bacteria that produce ammonium (.AMM) and nitrogen (NRB):
Acetogens (acetogenic bacteria):
2HC0; + 4H, + H* --> CH3C00' + 4H,0 aGn = -104.6 kJ
Methanogens (methanogenic bacteria):
HCO,~+4H_+II' -->CH,+3H,0 aGa =-135.6 kJ
Sulfate-reducing bacteria:
SOi a + 4H$ + H' --> HS' + 4H_O aGa = -1 S 1.9 kJ
Nitrate-reducing bacteria that produce ammonium:
NOJ + 4H=+ 2H'--% NH; + 3H;0 aG,; _ -599.6 kJ
Nitrate-reducing bacteria that produce nitrogen gas:
2N0,-+SHr+2H"-> tv=+6H=O aGa =-1,120.5 kJ
The negative free energies of these reactions at pH 7.0 (aGo ) indicate it is
thermodynamically
feasible for oxyanion-reducing, hydrogen-consuming bacteria to reduce hydrogen
gas fiigacities in
reactor environments to very low levels under anaerobic conditions.
Na single prior art reference or combination of references have suggested
combining the
2 0 knowledge of the above lines of inquiry: the art of solubilization and
transport of precious metals
in hydrothermal fluids, and the arts of aerobic and anaerobic biopracessing.
The prior art does
not teach the use of bio-oxidation to liberate (mobiliae) platinum-group
elements from their ores.
The prior art does not teach the use ofsulfate-reducing bacteria to increase
the fugacity of
hydrogen sulfide gas and the activity of bisulfide ions in a reactor in order
to increase the
2 5 solubility of precious metal-bisulfide complexes. Neither does the prior
art teach the use of
microorganisms capable of biological reduction of an oxyanion to lower the
hydrogen gas Fugacity
in a reactor in order to increase the solubility of precious metal-bisulfide
complexes in the reactor.
In fact, the prior art teaches away from the present invention toward aerobic
processes far -
teaching of precious metals from their ores. Such aerobic processes are
disclosed in the following
30 recently published books on the subject: Ehrlich, H.L. ( 1990), Microbial
Mineral Recovery,
New Fork: McGraw-Hilt; Gupta, C.K., & Mukherjee, T.K. (1990), Hvdrometallurgy
in
Extraction Processes. Vols. I and II, Boston: CRC Press; Yannopoulos, J.C.
(1991), The
8
w0 96!00308 ~ ~ ~ ~ ~ ~ ~ PCf/US95109199
Extractive Metallur of Gold, New York: Van Nostrand Reinhold; Marsden, J. &
House, I.
( 1993), The Chemistry of Gold Extraction, New York: Ellis Horwood.
DISCLOSURE OF INVENTION
For the purposes of this disclosure, the term "ore" refers to a composition
that comprises
precious metal values. Thus, ore may be a mineral assemblage that is being
mined in-situ (in
place) or that has been mined conventionally; or it may be a waste product,
such as obsolete or
damaged electronic components. The term "precious metals" refers to gold(Au),
silver(Ag)
and/or platinum-group elements (PGE). The term "platinum-group elements"
refers to platinum
I O (Pt), palladium (Pd), rhodium (Rh), ruthenium (Ru), osmium (Rh) and
iridium (Ir). The term
"bisufide lixiviant" refers to an aqueous solution comprising HS° ions,
and may also comprise
dissolved H,S gas (HzS~,q,). The term "bisulfide complex" refers to a complex
comprising a
precious metal and bisulfide.
The present invention provides method and apparatus for leaching of precious
metals from
15 their ores by means of a leaching solution comprising a sulfide ion and
having a low fugacity of
hydrogen gas. Leaching is accomplished by formation of precious metal
complexes. For
example, in gold leaching at neutral pH's, the complex Au(HS)_ predominates.
At a pH above
about 10, the solubility of gold is increased by the formation of the complex
Au,S_°'-. Below a pH
of about 3, the solubility of gold is increased by the formation of the
complex AuHS° Thus,
20 formation of a variety of precious metal-sulfide complexes is possible.
The invention may be practiced on oxidized ore, suflide ore, or otherwise
refractory ore in
a tank reactor or heap (each operation. Preferably, a bio-oxidation step for
removing base-metal
sulfides from precious metal ores is coupled with a bisulfide precious metal
leaching step, but
conventional roasting may also be used to remove base-metal sulfides and
produce an acidic,
2 5 sulfate stream. Preferably, the leaching solution is essentially neutral
or allcaline. In a preferred
embodiment, the process ofproducing the leaching solution is biocatalyzed.
In a preferred embodiment, a first process step of bio-oxidation of are
particles is
accomplished to free (liberate) precious metals dispersed or occluded within
the ore. A portion of
the acidic, base-metal sulfate leach solution produced by the bio-oxidation
step is introduced to an
30 anaerobic reactor. In a heap leach embodiment ofthe process, the anaerobic
reactor is a side-
stream reactor or a series of such reactors in series. In one alternative
slurry (e.g., vat or tank)
leaching embodiment, the anaerobic process may occur on-line. One or more
preferably non-
toxic electron donors (such as hydrogen gas, formate, acetate andJor methanol--
which does not
WO 96109)308 ; +° '~ ~ ~ ~ l(. ~ j~, ~ PCTlU895/09199
bind effectively to activated carbon) and growth requirements (such as
vitamins and!'or salts), are
added to the anaerobic reactor to enrich within it a culture of at least one
oxyanion-reducing
bacterium (e.g., a sulfate-reducing bacterium). In an alternative embodiment,
the electron donors
andlor growth requiremems are derived from organic material deposited on the
ore by sulfide-
s oxidizing bacteria during the bio-oxidation step. The hydrogen fugacity in
the reactor, or at least
in the last reactor in a series of such reactors, is maintained at a low level
by at least one
hydrogen-consuming bacterium. The anaerobic reactor may be operated in a pH-
slat mode by
adding sufricient acidic sulfate solution to maintain a neutral pH in the
reactor (see Hunter, R.M.,
$i~ocatalvzed Partial Demineralization of Acidic Metal-Sulfate Solutions, Ph
D. Thesis, Montana
1 c) State University, 1989). In an alternative embodiment, the anaerobic
reactor may be operated in a
sulfide-slat mode by adding sufficient sulfate solution to maintain a constant
dissoh~ed sulfide
concentration in the reactor in response to sistnals from a sulfide sensor
(e.g., sulfide ion selective
electrode), Aase metals are preferably precipitated and removed and a portion
of the hydrogen
sulfide gas (H=S) produced in the anaerobic reactor is preferably removed. In
this way, oxyanian-
i 5 reducing bacteria are used to create an essentially neutral leaching
solution comprising a relatively
high concentration bisulfide ions, a high fugacity of hydrogen sulfide gas, a
low concentration of
dissolved base metals and a low firgacity of hydrogen gas.
In an alternative embodiment, the precious metal leaching solution is produced
in an
anaerobic environment by contacting a stream of gas comprising hydrogen
sulfide gas and
2 Q essentially no hydrogen gas with the solution until the environment has an
appropriately' high
concentration of hydrogen sulfide gas and an appropriately tow firgacity of
hydrogen gas. Tlre
gas may be produced biotically by a culture of sulfate-reducing; bacteria, or
it may be produced
abioticaliy by purifying HxS gas to remove Ha gas.
In a second process step, the oxidized ore (possibly in a heap that is covered
and
2 5 submerged to exclude oxygen) is leached (by recirculating the neutral or
alkaline bisulfide tixiviant
comprising, or saturated with, H1S) in a leaching reactor. In one embodiment,
the HrS partial
pressure is increased by introducing the lixiviant under pressure at the
bottom of a heap
submerged in water, causing ion concentrations to increase in direct
proportion to the increase in
H=S partial pressure.
30 In a preferred embodiment, the anaerobic reactor and the leaching reactor
are operated
together as a single, essentially completely-mixed reactor. A completely mixed
reactor is one that
produces an ett7uent concentration of a conservative tracer (e g., a non-
reactive dye) equal to 37
+3 percent ofthe initial tracer concentration (i.e., tracer mass divided by
liquid volume) one
WO96/00308 .. 1 ~':} ~ ~ ~ ~ ~ ~ Oj PCT~S951(19199
detention time (i.e., liquid volume divided by liquid volumetric flow rate)
afrer an impulse input
(i.e., slug addition) of the tracer
The complexed precious metal (e.g., gold and silver) is recovered (preferably
continuously) from the lixiv~iant solution. Recovery may be accomplished in a
conventional
manner by adsorption on activated carbon or by modifying either the solution
pFL, hydrogen
fugacity, or oxidation-reduction potential (ORP).
Recovered precious metals are converted into products. TMs may include the
operations
of separating, smelting and casting of each precious metal into bars, bullion
or other forms.
The present invention offers a variety of advantages not provided by the prior
art. One
object of the invention is to lower the monetary cost of gold, silver and
platinum-group element
production. By utilizing a waste product (excess sulfuric acid from a roasting
or bio-oxidation
pretreatment step) as the starting material far preparation of a bisulflde
lixiviant, the lixiviant (a
neutral bisulfide solution) would be produced biologically instead of being
purchased. Another
object of the invention is to use bath inorganic (salts) and organic (biofllm
carbonaceous
compounds} byproducts of biaxidization as inputs to a precious-metal
solubilization process.
Another object ofthe invention is to lower the environmental risk of precious
metal mining. TMs
is the case because the actual and perceived environmental risk of maintaining
a large inventory of
a neutral bisulflde solution is much lower than that associated with
maintaining an equivalent
volume of caustic cyanide solution. Another object of the invention is to
provide a method and
2 0 apparatus for both in-situ or ex-situ (conventional) mining. Further
objects and advantages of the
invention will become apparent from consideration of the drawings and the
ensuing description.
BRIEF DESCRIPTION OF DRAWINGS
The features of the invention will be batter understood by referring to the
accompanying
2 5 drawings which illustrate presently preferred embodiments of the
invention.
In the drawings:
Fig. 1 is a highly schematic block diagram illustrating a first representative
embodiment of
the present invention.
Fig. 2 is a Mghly schematic block diagram illustrating a second representative
embodiment
3 0 of the present invention.
Fig. 3 is a Mghly schematic block diagram illustrating a third representative
embodiment of
the present invention.
WO 96100308 s. ; ~;i ~ ~ ~ ~ ~ ~ ~ PCTfU895109199
Fig. 4 is a highly schematic block diagram illustrating a fourth represemative
embodiment
of the
present
invention.
The following
reference
numerals
are used
to indicate
the parts
of the
insrention
on the
drawings:
2 are
4 bio-oxidation reactor
6 sulfate ions
7 electron donor
8 sulfate reduction reactor
10 bisulfide lixiviant
oxidized ore
22 bisulfide leaching reactor
24 pregnant solution
26 precious metals recovery reactor
15 28 leached ore
ore
32 crushing
34 crushed are
36 acid leaching
20 37 aerobic reactor
38 air
acid-leach solution
42 pump
44 acid-leached ore
25 46 bisulfide leaching
47 essentially completely-mixed, anaerobic reactor
48 bisulfide lixiviant
pump
pH controller
3 62 valve
0
64 valve
66 pregnant bisulfide lixiviant
68 gold and silver recovery
12
W096100308~ (j ~ ~ ~ ~ PCTIUS95109199
70 spent Lixiviant
76 bisulfide lixiviant recirculation loop
78 activated carbon column
80 leached ore
82 sensarlcontroller
84 electron donor
90 dewatering
92 contained bisulfide lixiviant
94 waste ore
96 acid-leach solution portion
98 base metal removal
100 base metal removal reactor
102 acid leach solution portion
104 iron and other base metals
I 10 excess hydrogen sulfide gas
112 excess hydrogen sulfide gas portion
I 14 sulfur recovery
116 sulfur recovery reactor
120 elemental sulfur
2 200 heap
0
202 heap, second heap
204 crushed ore
205 crushed ore, oxidized ore
206 air
208 plenum
-
210 acidic"base-metal sulfate leach solution
212 pump
214 portion
216 distributor
3 220 portion
0
230 anaerobic, sulfate-reduction reactor
232 pH controller
234 valve
13
WO 96!00308~ ! ~ ~ ,~ 4 ~j PCTlU595l09199
238 hisuifide leach solution
240 non-toxic eiectron donor
244 base metals
250 settling tank
252 portion
2S4 bisulfide lixiviant
260 headspace
262 headspace
264 conduit
266 excess hydrogen sulfide gas
270 sulfurrecovery
272 elemental sulfiu
282 bisulfide lixiviant
284 plenum
286 pump
290 distributor
292 portion
294 portion
300 pregnant portion
302 reactor
306 barren lixiviant solution
312 aerobic, stirred batch reactor
314 air pump
318 pH monitor
320 liquid supernatant
322 ore
324 portion of dried, bio-oxidized ore
326 continuously stirred tank reactor
328 media reservoir
330 pump
332 pump
334 H,S canister
3 36 pH monitor/controller
14
W096100308 ' : PCT/US95/09199
19~..~~9
338 acidic supernatant
340 pump
342 efrluent storage
container
344 pump
346 pump
348 160-ml serum bottles
MODES) FOR CARRYING OUT TIgE INVENTION
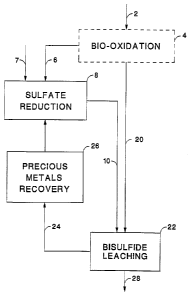
Reference is now made to Fig. 1 which is a schematic block diagram
illustrating a
preferred embodiment of the invention, with the dashed lines representing
possible variations in
the process and apparatus. Ore 2 is the input to the process and, under
certain conditions, may be
the only input to the process. In a preferred embodiment, ore 2 is crushed and
may be otherwise
treated to optimize bio-oxidation. In bio-oxidation reactor 4, oxidation of
metal sulfides is
accomplished to free or mobilized precious metals dispersed or occluded within
metallic sulfides
in ore 2.
Bio-oxidation reactor 4 produces a sidestream comprising sulfate ions 6 and
acidity. In
some instances, the sidestream also comprises biofilm carbonaceous compounds.
In an alternative
embodiment, bio-oxidation does not occur and sulfate ions 6 are an input to
the process Sulfate
ions 6 may be a component of a waste stream, such as acid none drainage, or by-
product of ore
2 0 roasting.
In a preferred embodiment, electron donor 7 is added to sulfate reduction
reactor 8 so that
sulfate ions 6 are biologically reduced therein. In a preferred embodiment,
sulfate reduction
reactor 8 is operated at a mean cell residence time low enough to cause
essentially-complete (99+
percent) utilization of electron donor 7. In a preferred embodiment, sulfate
reduction reactor 8 is
operated in a pH-stat mode so as to maintain an essentially Constant pH (~ 0.1
pH unit) in reactor
8 and in bisulfide lixiviant 10 that it produces.
Oxidized ore 20 is introduced to bisulfide leaching reactor 22. In reactor 22,
precious
metal values in oxidized ore 20 are dissolved and complexed by means of
bisul6de lixiviant 10.
Pregnant solution 24 comprising precious metal values is introduced to
precious metals recovery
reactor 26 fur precious metals recovery in a conventional manner by adsorption
on activated
carbon; or by modifying either the solution pH, hydrogen fiagacity, or
oxidation-reduction
potential (ORP). A product (e.g., gold bullion) is formed from said precious
metal by smelting
and casting. In one embodiment, leached ore 28 is disposed ofin a conventional
manner (e.g.,
IS
WO96100308 ~._ ' ; .. ~ ~ ,!~ ~ PCT/t1S95/09199
permanent storage) and need not be treated for removal of lixiviant. In a
preferred embodiment,
leached ore 28 is washed andlar dewatered to remove residual Iixiviant 10
prior to disposal.
Lixiviant 10 removed frora leached ore 28 is used to wet andJar neutralize the
acidic pH of
incoming oxidized ore 20 andlor it is returned to leaching reactor 22.
It is well known in the art that the composition of ores varies widely,
requiring
optimization of the leaching step based on ore compositions and other local
conditions. For this
reason, design and operation of reactors 8 and 26 are preferably optimized for
precious metal
dissolution and complex formation. Depending on ore composition, design and/or
operation are
varied to achieve the followinst conditions in the reactor environment:
1. Maximize dissolved bisulfide concentration and dissolved HZS~,st
concentration at
the precious metal surface;
2. Optimize pH;
3. Minimize hydrogen fugaciy at the precious metal surface;
4. Maximize pressure;
5. Maximize temperature.
For gold andlor silver leaching, information on the aqueous chemistry of gold
and silver bisulfide
complexes and other chemical species likely to be present in a bisulfide
lixiviant is used (see
Barnes, H.L. led.), C~~ehemistryfHvdrothermal Dre Deposits, 2nd ed., New York:
John
Wiley & Sons, 1979). Published information on the aqueous chemistry of gold
and silver bisulfide
2 0 complexes and other chemical species likely to be present in a bisulfide
lixi4iant are used to
produce a mathematical model of the salubilization step. The model
incorporates the data
presented in Tables 1 and Z. In the model, stability and equilibrium constants
are used to predict
the direction of a reversible chemical reaction under certain standard
conditions and under other
conditions. The standard conditions are 1.0 molar (h~ concentrations of
dissolved reactants and
2 5 products and I .0 atmosphere (.4tm) pressure of gaseous reactants and
products. The temperature
is usually taken as 25°C (298°K), but stability and equilibrium
constants are reported at other
temperatures as well.
16
W096/00308 9 ~ , '' ~ ~ ,~ ~ ~ ~ ~ PC1YUS95l09199
Table 1. Equilibrium Constants of Metal Sulfides and Sulfide Complexes
Metalt'reaction Temperature, Ionic strengthLog K
C
Cadmium
CDs + H=Siq, -~ Cd(HS)x 25 1.0 -4.57
CDs + HzS~,~~ + HS-- Cd(HS); 25 l .0 -2.69
CDs + HZS~,~~ + 2HS~-~Cd(HS); 25 l.0 -0.33
=
Copper
Cu=S + SHS~ + H* - 2Cu(HS)3 - 22 2.1 to 4.4 +2.020.26
Gold
Au~,~ + HS- -~ AuS- + O. SH2~R~ 25 -5.6'
Au~,~ + H~S - .AuHS + O.SHa~~ 25 - -11.1410.2
Au,a, + H:S~,y~ + HS' --~ Au(HS)~20 - -6.1
+ O. SH~R~
2Au + H=S~,q~ + 2HS-~ Au~S(HS=)~175 0.50 -2.14
+ H=~~
2Au + S=' ~ 2AuS' 25 -2.02
2Au + 2HS~ + 0.502 --~ 2AuS- 25 - +30.35
+ H=O
2Au + 2HS- ~ Au2S + HiiRa + SR 25 - -11.25
Lead
PbS + HAS"q, + HS -~ Pb(HS); 25 0 -5.62 f
0.2
PbS + H:Si,~~ ~ Pb(HS)_~,y, 25 0 -7.6'
PbS + 2H~S"q~ -~ PbS(H=S)z~,9, 200 0 -4 88
Mercury
HgS + 2H=S"~~ - HgS(HxS)uwr 20 1.0 -4.25"
HgS + H3S~ 20 1.0 -3.50
~ + HS' --~ Hg(HS);
w
HgS+2HS---r HgS(HS=)' 20 1.0 -3.51"
HgS + HS' + OH' - HgSi = + H,Of,~,25 0 0.31
Silver
Ag"~ + HZS~,y~+ HS' -~ Ag(HS)= 25 - -2.72f0.10
+ O,SH_~;~
Ag*+HSwAgHSi,q, 20 1.0 +(3.30
Ag* + 2HSw-r Ag(HS)= 20 1.0 +3,87
2Ag(HS), -~ Ag=S(HS),=+H,S~,~, 25 1.0 +3.2
17
rY0 96100308 ' , ~ ~',, ~ ~ ~ .~f ~ j~' ~ PCTlC~S95/09199
Table 1. Equilibrium Constants of Metal Sulfides and Sulf de Complexes (cont.
j
Metafreaction Temperature, °C Ionic strength Lo K
Silver (coot. )
Ag~S + HAS -~ 2AgSH 20 1.U -15.78
Ag=S + HjS + 2HS- --~ 2Ag(SH): 20 l .0 -8.05
Zinc
ZnS + 2HZS~,sy -1 ZnS{H2S)u,qy 80 - -2.24
ZnS + HsSt,~y + HS' ~ Zn(HS); 25 I .0 -3.0 t 0 4
ZnS + H,Sy-,~, + 2HS- ~ Zn(HS,)~' 25 1 0 -2 6
' Staichiometry unproven.
b'fotal sulfide is O.SM, and pressure is 1,000 bar.
'Less certain
d Solid phase is metacinnabar
' Pressure is 1 bar
Table 2. Stability (Formation) Constants
Log stability constant by Metal'
Liguid Gold, !3, Silver, (3, Platinum 13, Palladium 13,
Chloride (Ch) 9" 5.4' 13.99 11.54
Bromide (Bc ) I2" 7.1' 15.4 14.9
Nitrite {NO-~) - - 24.1 21.0
Iodide {I -) 19.6' - 29.6 24,9
Thiocyanate (SCN-) 17.1" - 33.6 25.6
Thiourea CS (NH,)~ 23.3° 13.10' - -
Thiosulfate (SOS z) 28.7a 13.3' 43.7 35.0
Bisulfide (HS-) 30.1' 17.43' ,..51 ...41
Cyanide (CN-) 38.3' 18,7' ,..78 63
' Far bivalent-ion complexes at 25°C
36 " Source: Hancock, R:D., Finkelstein, N.P., & Evers, A. (1977). A linear
free-energy
relation involving the formation constants of pailadium(Il) and platinum{II).
J.ino~.nucl.Chem. 39. 1031-1034.
Source' Kotz, J.C. & Purcell, K.F. (199I). Chemistry & Chemical Reactivity
Philadelphia, P.A: Sounders Col3ege Publishing.
° Source: Marsden, J. & House, I. (1993). The chemisnv of bald
extraGtipn. New Yark:
Ellis Hanvood.
' Source: Renders, PJ. & Seward, T.hl. (1989). The adsorption ofthia gald(Ij
complexes
by amorphous As7S3 and Sb,S, at 25 and 90°C. Geochimica et Cosmochimica
Acta. 53. 255-26'7.
18
WO 96!00308 b ''. ' ~ ~ (j j~ ~ ~ i~ PCT/US95/09199
Equilibrium constants can be derived in a number of ways. The stability
constant for a
reaction is related to the standard free energy change the reaction as
follows:
nG° _ -RT In K
Ln K = -eG°/RT
logK =-nG°/2.30*RT
where oG° = free energy change under standard conditions, kJ
R = gas law constant = 0.0083 l4 kJl(mol*°K)
T = absolute temperature in decrees Kelvin (°K)
2.30 = In 10
The value of the product (2.3*RT) is 4.1840* 1.3636 K calireaction = 5.705
kJlreaction at 25° C
and 5.935 kJ/reaction at 37°C.
An opportunity for the solubilization ofprecious metal (e.g., gold, silver,
platinum and
palladium) is created by the complex-forming reactions:
Au~x>+ HaSc.v + HS'-_> Au(HS)=-+ 0.5 H=cap
Ag,x~ + H=S~,q, + HS' --> Ag(HS)= + O.SH~~a~
pt"~+ 2H_S~ay~ + 2HS- __> pt(HS), z + Hya>
Pd~" + 2HzS~,q, + 2HS --> Pd(HS), _ + Hnai
The equilibrium and stability constants for the gold and silver reactions have
been
determined experimentally and are dependent on the temperature at which they
occur.
Shenberger, D.M. & Barnes, H.L., in "Solubility of gold in aqueous sulfide
solutions from 150 to
350°C," (Geochimica et Cosmochimica Acta, 53, 269-278, 1989), have
derived the following
equation for the temperature dependence of the first reaction (between
150° to 350°C):
log Krumsy = -9.383 * 10'/T= + 2170.4iT - 2.28I6
where T = temperature in degrees Kelvin
The following equation applies between 25°C and 150°C:
log Ka"n,su = 3.32 + (_2,420/T}
Similarly, Gammons, C.H. & Barnes, H.L., in "The solubility of Ag2S in near-
neutral aqueous
sulfide solutions at 25 to 300°C," (Geochimica et Cosmochimica Acta,
53, 279-290, 1989), have
derived the following equation for the temperature dependence of the second
reaction:
log Kaa~sk = 0.439 - 943/T
The equilibrium and stability constants for the platinum group element
reactions can be
estimated using the methods disclosed by Hancock, R.D., )!inkelstein, N.P., &
Evers, A., in "A
linear free-energry relation involving the formation constants of palladium
(II) and platinum (II),"
19
WO 9b100308 ,' y ,,.: ,; ,,~J ~ ~ E~." (~ J ~ ~ PCT7US95/09199
r rrn,rnal of Inors~uvc and Nuclear Chemistry, 39, 1031-1034, 1977} and
Mountain, B. W. &
Wood, S.A., in "Chemical controls on the solubility, transport, and deposition
of platinum and
palladium in hydrothermal solutions: A thermodynamic approach," {Economic
Geolo~y, $3, 492-
510, 1988}, They have demonstrated that, for metals in the group Au, Ag, Pt
and Pd, plots of the
logarithms of the stability constants of one metal versus another are linear
for a variety of Ggands.
These equilibrium and stability constants may be used to determine fhe
equilibrium molar
concentration of each complex using the following equations:
[.4u(HS),~]=K,~su * [HaScw>] * [HS~If[H3i~I]°.
[~~$(HS)x ~ = K~q<ttsu * ~zS<.ar] * [HS~]~[Hnza~
[Pt(HS); x] = K~sn * ~xsan~]x * ~S ]xl[Hxcs~]
[Pd(HS).s x] = IiParns~s * [HxS<.a>]= * ~S ]1~[Hxcx~]
The above equations are used to estimate the hydrogen fugacity required to
solubilize gold
and silver at various temperatures and at two reactant (H,S + HS-}
concentrations. Those
estimates far gold and silver are presented in Table 3. In preparing the
estimates presented in
Z 5 Table 3, the following activity and fugacity coefficients are used: Au
(HS)3 = 0.7,
Ag (HS). = 0.7, HxS = 0 97, HS' = 0.7, and H3 = 1Ø
Since anaerobic digesters must be operated at Hxu~ fugacities below about 10'
atmosphere
(atm), achievement of the firgacities indicated in Table 3 are feasible. This
would produce gold
and silver concentrations in a pregnant bisulfide lixiviant of 0.1 to I.0 mgft
and 1.0 to 10 mgll,
respectively, which are the same concentrations that occur in pregnant cyanide
heap leach
solutions during conventional cyanidation,
Table 3. Hydrogen Gas Fugacities Required to Soiubilize Gold and Silver
2 5 Log of hydrogen gas (I-Ix~e~) fugacity, atm, required at indicated
temperature, °C, 2o achieve indicated metal concentration.
Metal concentration f ligand concentration° 25 35 65
Gold, U. I mgR Au
1,400 mg/I Fi:S,,q~+HS- -3.84 -3.32 -1.92
2,700 mgll H=S~,s~+I-IS~ -2.63 -2.11 -0.71
Gold, 1.0 mgll Au
1,400 mg!( HZS~,v~+HS- -5.84 -5.32 -3.92
2 700 m~/1 H S +HS- -4 63 -4.11 -2.71
WO 96100308 c~ , ~ ~ t~ ~ ,~ tt ~ PCT/US95109199
Table 3 (Cont). Hydrogen Gas Fugacities Required to Solubilize Gold and Silver
Log of hydrogen gas (H~~g,)fugacity, atm, required at indicated
temperature, °C, to achieve indicated metal concentration
Metal concentration / liuand concentration'25 35 65
Silver, 1.0 mg/1 Ag
1,400 mg/1 HzS~,q~+HS- -2.22 -2.00 -1.4b
2,700 mg/1 H=Sy+HS- -I .O1 -0.79 -0.25
Silver, 10 mg/1 Ag
1,400 mgll H=5~,~~+HS- -4.22 -4.00 -3.46
2,700 mgJl H=S~,~~+HS~ -3.01 -2.79 -2.25
' Concentration of each reactant (HZS or HS-) is about half of the indicated
total at pH 7Ø
In a preferred embodiment, bisulfide ions are generated biologically (by
naturally-
occurring sulfate-reducing bacteria) at very low cost using an acidic waste
product (bio-
oxidation heap leach effluent) as the sulfate source. For example, with
formate ion as the
electron donor, the following reaction occurs:
8HCOO-+ 2SOs Z + 3H' -~ 8HC0; + H,5 + HS'
With acetate ion as the electron donor, the follwving reaction occurs:
CH,COO- + SO; - -> 2HC0; -~ HS
A mass balance on typical heap leach et3luents has shown that biological
reduction of contained
sulfate ions produces bisulfide and sulfide ions in excess of the
concentration required for
2 0 essentially complete base metals precipitation (see Hunter thesis, 1989).
Thus, production of a
high-concentration bisulfide Iixiviant is passible. Moreover, H=S gas can be
recovered from
spent Iixiviant andlor leached ore by reducing the H=S gas partial pressure in
the gas mixture in
contact with said spent lixiviant andlor leached ore using a vacuum pump. More
complete H,S
gas recovery can be achieved by acidifying the spent lixiviant and/or leached
ore to a pH below
7.0 and/or by increasing gasiliquid interfacial area, (e.g., by forming the
liquid into droplets).
A variety of techniques may be used to macimize bisulfide concentrations in
the lixiviant.
The solubility of hydrogen sulfides in water decreases with increasing
temperature (from about
7,100 mgll at 0°C to 3,000 mgll at 30°C under a partial pressure
of one atmosphere),
(Environmental Protection Agency, Process Design n4anual for Sulfide Control
in Sanitarv
Sewera_ee Systems, EP.A 625ii-74-005,October 1974). In accordance with Henry's
law, the
21
W096100308 a .f ' y PCTlI3S95109199
saturation concentration of H=S in water is directly proportional to the
partial pressure of the
gas in the atmosphere in contact with the liquid. Removal of HZS increases the
pH of the
solution (by removing protons). Moreover, the proportion of bisulfide ion
(relative to H=S)
increases with pH over at least the range pH 5-9.
The optimal pH for the bisulfide lixiviant solution for precious metal
recovery is the pH
that maximizes the solubility of target precious metal compounds and the
stability of their
complexes. For example, Krauskopf, K.B., in "The solubility of gold" Economic
Geolosay, (46,
858-870, 1951 ), noted that "one of the most perplexing facts about the
chemistry of gold is its
ability to dissolve in solutions of HS- of moderate concentration even at room
temperature,
whereas it dissolves in Sn (i.e., more alkaline solutions) only in
concentrated solutions at high
temperature." Schwarzenbach, von G. & Widmer, M., in "Die loslichkeit von
MetaIlsuIfiden,"
(Helvetica Chimica Acta, 49, 11 I-I23, 1966), found that the solubility of
silver was greatest at
pH 7 at a temperature of 20°C in the presence of excess sulfide in the
form o.f HxS, HS-, and S''.
l~lelent'yev, B:N., ivanenka, V.V., and Pamfilova, L.A., in "Solubility ofsome
ore-forming
sulfides under hydrothermaI conditions," (Rastvorimosf nekotorvkh
rudoobrazuvushkikh
suffidov v gidrotermal' n~h usloyvakh' Moskva, 27-102, 1968), found that the
solubility of
Ag=S increases with pH in the range pH 4-8 in the temperature range 100-
300°C. Sewsrd
reported that for gold in solutions of reduced sulfur "a pronounced solubility
maximum occurs
in the region of pH about 7." ( Seward, T.M., "Thio complexes of gold and the
transport of
2 0 gold in hydrotherntal are solutions," Geochimica et cosmochimica Acta. 37,
379-399, 1973).
Options for reducing hydrogea fugaciry include bioprocessing hydrogen-
management
techniques Hydrogen-consuming bacteria (hydrogenotrophs) include such anion-
reducing
bacteria as acetogens, methanogens, sulfate-reducing bacteria and denitrifying
(nitrate-reducing)
bacteria in natural ecosystems, these bacteria participate in "interspecies
hydrogen transfer."
2 5 . Examples of acetogens include Acetobacterium u.~oocti (ATCC 29683, DSM
1030, or DSM
2396) and Clostridium aceticum (ATCC 35044 or DSM 1496). Examples of hydrogen-
consuming methanogens are numerous and include the mesophiles
Methcurobrevibacter
rumiuaratium (ATCC 35063 or DSM 1093) and Methanorarcina barkeri (ATCC 29786
or
DSM 805), and the thermophife aL9etlwraobarterium thermcxrutomophicum (ATCC
29096 or
3 0 DSM 1053). Examples of hydrogen-consuming sulfate-reducing bacteria are
shown in Table 4.
22
W0 96t0030ti , , ~ , ~ ~ t.~ P ~ PCT/U595I04199
~ ~
Table 4. Examples of Hydrogen-Consuming
Sulfate-Reducing Bacteria'
Selected other Optimum Optimum
electron donors(D)pH range, temp., Growth
Genus/species and acceptors(A)units C requirements
S Desuifobact~r
genus Acetate(D) 6.0-7.0 20-33 Vitamins,Salts''
cuwatus (DSM 3379) Acetate(D) 6.8-7.2
hvrosenophilus {DSM 6.6-7.0
3380)
14 Desulfobacterium
genus 6.6-7.6 20-30
or
30-35
anilini (DSM 4660}
autotrophicum (DSM 3382) Acetate(D}, formate(D)6.7 25-28 Vitamins",
salts'
catechoiicum (DSlli 3882) Acetate{D), formate{D)6.9-7.1 28 Vitamins,
Nitrate'{A) dithionite
15 ma tii (DSM 4194)
Desutfobulb~s
genus Propionate(D}, 6.6-7.5 25-40 Vitamin',acetate
ethanol(Dj, Nitrate'(A) as carbon
eloneatus (DSM 2908)
pro~ionicus (DSM 2032)
20 Desulfacoccus
genus 6.6-7.6 28-35 Salts',~7tamins
niacini (DSM 2650) Formate(D), ethanol{D)
Desulfomicrobium
genus 6.6-7.5 25-40 Acetate
as
carbon
2 5 aDSheronum (DSA4 5918)Ethanol(D) 25-30 Reduc.agents
baculatum
Desulfomona~
Qiera (ATCC 29098} Ethanol(D) 6.6-7.5 30-40 Acetate
as
carbon
23
W0 96f0030R i';. all ~ i '~x. ~ '~ ~ ~ ~ ~ '~' FG"flU895109199
Table 4 Examples of Hydrogen-Consuming Sulfate-Reducing Bacteria' (cont.)
Selected other Optimum Optimum
electron donors(D)pH range,temp., Growth
Genuslspecies and acceptors(A) units C requirements
Desulfomonile
tierliei (ATCC 49306) Formate(D} 6,8-7.0 37 Vitamins,
reduc. agentsb
Desulfonema
limcola (DS(v12076) Acetate(D) 7.0-7.G 28-32 Salts',
sediment
Desulfosarcina
variabilis (DSM 2060} 7.2-7.6 33 Dari.ness
Desulfotomaculum
genus 6.6-7.4 Vitamins?
ueathermicum 50-60 Acetate
as
carbon?
kuznetsovni (DSM 6115}Acetate(D), formats{D)-7.0 60-65 None
ni ri is (ATCC 1999$) 50-60
orientis (ATCC 19365) 25-40
ruminis (ATCC 2393) 25-40
thermoaceto-oxidans Acetate(D), formate(D)6.5 55-6fl Vitamins
(DSM 5813)
Desulfovibrio
genus Ethanol(D) 6.6-7.5 25-40 Biotin
africanus (DSM 2603) 34-37
carbinolicus (DSM 3852)
desulfuricans (ATCC Nitrate{A), 34-37 Acetate
as
2774) nitrite{A) carbon
fructosovorans Formats {D} -7.0 -35 Acetate
as
(ATTC 49200) carbon
furfuralis Nitrate(A) -6.8 -38
.
gisanteus Formater{D) 7.5 35 Acetate
as
carbon;
saltR
aiaas (ATCC 19364) Nitrite(A) 34-37
24
W~ 96100308 , , ~ ~ ~ ~ ~ ~ ~ PCTIUS95I09199
Table 4. Examples of Hydrogen-Consuming Sulfate-Reducing Bacteria' (cont.}
Selected other Optimum Optimum
electron donors(D}pH range, temp., Growth
Genus/species and acceptors(A)units C req uirements
salexine~ ns (ATCC Formate(D), 34-37 Saltk
14822)
ethanol{D)
simplex (DSM 4141) Formate(D), -7 -37 Acetate
as
ethanol(D), carbon
Nitrate(A) Ni*z,
WO;
z
sulfodismu_tans
(ATCC 43913)
termitidis (DSM >308)
v_ulearis (.ATCC 29579) 34-37 Acetate as
carbon
Thermodesulfo-bacterium 6.6-7.5
commune (ATCC 33708) ~7.0 70 Acetate as
carbon
mobile (DSM 1276} Formate(D} 70
' Sources' Widdei and pfennig, 1984; Holt et aL, 1994
" Vitamins are nicotinamide, 1-4-napthoquinone and thiamine in a defined
mineral medium.
Reductants are 1 mW NazB~9HzO or 0.:5m144 NazS=O,,
' Denitrified to ammonium.
Vitamins are biotin and p-aminobenzoate
° Salts are 20811 NaCI and 3g/I MgCI= 6F1=O.
'Completely oxidized.
° Salt is 2 to 25 g/l NaCI.
" Salts are >7g/I NaCI and =~ 1 gll MgCI_~6H,0
' Vitamin is p-aminobenzoic acid.
2 0 ' Salts are > 1 Sg/( NaCI and >2gll MgCI= 6HzO.
'' Salt is 20glI NaCI.
°' Salts are 7-20g~1 NaCI and l-3gJ1 MgClz~6Hz0.
" Vitamins are thiamine, biotin and p-aminobenzoic acid.
° Vitamins are biotin, p-aminobenzoate and nicotinate.
As noted, some of the sulfate-reducing bacteria listed in Table 4 are also
nitrate-reducing
bacteria because they can also reduce nitrate to produce ammonium. In a
preferred,
embodimentrt leaching solution is produced in an anaerobic reactor by
culturing in the reactor
sulfate-reducing bacteria capable of using forntate or acetate, as well as
hydrogen as electron
donors, and both sulfate and nitrate as electron acceptors. Since anions, such
as sulfate and
2S
WO 96/00308 * ~~ ~,j ~ <,~ L f ~ ~ ~ t~ ~' PCTlUS95l09199
,:
nitrate, are reduced, such bacteria are oxyaruon-reducing bacteria. Examples
of such bacteria
include mesophihc, fresh-water species such as Desrrlfolxxcterinm
catechodicarm DSM 3882
(acetate and formate) and Desulfovi6rio simplex DSM 4141 (formate);
mesophilic, salt-water
species, such as Desxrlfovi8rio salextgems DSM 2638 (formate); and
thermophilic, fresh-water
species such as Destrtfomaculum krratetsovii DSM 61 I5 or VKM B-1805 (acetate
and
formate). Microorganisms with ATCC accession numbers can be obtained from the
American
Type Culture Collection. 12301 Parklaevn Drive, Rockville, Maryland 20852-
1776, tel i-800-
638-b597, fax 1-301-231-5826. Microorganisms with DSM accession numbers can be
obtained
from the Deutsche Sammlung van Mikroorganismen and Zellkulturen GmbH,
Mascheroder
Weg ib, D-38124 Braunschweig, Germany, tel O11-49 (0)531-2616-336, fax 01 I-49
(0)531-
2616-418. niicroarganisms with VICIvt accession numbers can be obtained from
the Institute of
Biochemistry and Physiology of Microorganisms of the Russian Academy of
Science,
Pushchino-na-Oke, I42292 Moscow Region, Russian Federation.
In an alternative embodiment, preferred especially for laboratory (process
optimization)
studies, additional hydrogen consumption is accomplished by purging the
headspace of bisulfide
leaching reactor 22 through a H=S-scrubbing means (e.g., a zinc acetate
solution "bubbler") into
a nitrate-fed reactor containing a culture of sulfate-reducing bacteria that
are also capable of
nitrate reduction, operated in parallel (side-stream) or in series with
reactor 22. In this way,
"hydrogen-scrubbed" headspace gas is recycled back to reactor 22.
Alternatively, a zinc acetate
2 0 bubbler would not be required if the H,S concentration in reactor 22 were
controlled
independently by a side-stream bubbler controlled by a sulfide ion-selective
electrode that would
turn an a H,S-scrubbing bubbler loop when a high H,S setpoint was reached.
In an alternative embodiment, the HtSrst concentration and the HS'
concentration may
be increased to an appropriate level, and the H= fugacity may be reduced to an
appropriate level
2 5 in the environment provided by reactor 22 by contacting the contents of
reactor 22 with a
stream of gas having an appropriate H=S fugacity and effectively no H,. This
stream of gas may
be produced biologically by a culture comprising sulfate-reducing bacteria or
it may be
produced abiotically using conventional means. An equilibrium will be reached
that partitions
the constituents of reactor 22 of limited solubility between the gas and
liquid phases in reactor
3 0 22. Henry's Law can be used to predict equilibrium and steady state
constituent Levels.
While gold, silver and platinum-group elements are soluble in bisulfide
solutions at
ambient (atmospheric) pressures and at room temperature, their solubilities
generally increase
with pressure and temperature (Krauskopf, K.B., Economic Geolo$v, 46, 858-870,
1951;
26
WO 9fi/00308 ' ~' ': ; ~' ~.' ~ ~ ~ ~ ~ ~ ~ PCTlU595109199
Weissberg, B.C., Economic Geolos.Tv, 65, 551-556, 1970). For ihis reason, in
an alternative
embodiment, sulfate-reduction reactor 8 is operated in the thermophilic (50-
100°C) and
barophilic (over one atmosphere) ranges (e.g., in a submerged, covered heap).
If sulfate-reduction reactor 8 is operated at steady-state at relatively high
total dissolved
sulfide (H2S ~,q~ + HS- + S-Z ) concentrations (say over about 1,000 mgll in
the liquid), then
sulfate-reducing bacteria will be enriched in reactor 8 that are relaiively
resistant to growth rate
inhibition by such total sulfide concentrations. Many investigators have
reported that common
sulfate-reducing bacteria can grow in media containing over 2,700 mgJl of
total sulfides (See
Miller, L.P. ( 1950). Formation of metal sulfides through the activities of
sulfate-reducing
l0 bacteria. Contributions from Boyce Thompson Institute. 16,. 85-89; Saleh,
A.M., MacPherson,
R., & Miller, J.D.A. (1964). The erect of inhibitors an sulphate reducing
bacteria: a
compilation, Journal of~olied Bacteriolo~,y ~7 281-293)
In a preferred embodiment, sulfate-reduction reactor 8 is operated at a
relatively low
fatal sulfide concentration (say less than about 1,000 mgll in the liquid) in
order to minimize
inhibition of the sulfate-reducing bacteria growing in it. This may be
achieved by using a
vacuum pump or purging gas stream to transfer H=S gas from the headspace of
reactor 8 to the
liquid in leaching reactor 22.
In one embodiment, a HZS gas pump is used to increase the H:S partial pressure
in the
transferred gas stream. R'ith this embodiment, the 1-i:S gas removed from
reactor 8 is absorbed
in a basic (pH >7) solution as dissolved HS ions during the intake portion
ofthe pumping cycle.
During a subsequent discharge portion of the pumping cycle, the solution
containing dissolved
HS' ions is acidified to convert the dissolved HS to H=S gas and the H=S is
pumped into
leaching reactor 22. In one embodiment, waste sulfuric acid produced by
oxidation of metal
sulfides is used to acidify the solution containing the dissolved HS- ions.
In an alternative embodiment, H:S gas pumping is accomplished by dissolving it
in a
liquid solution at a relatively low temperature (e.g., 10°C). The H:S
is then driven out of the
solution by heating the liquid to a relatively higher temperature (e.g.,
60°C). This form of HAS
pumping is made possible by the significant change in Henry's law coelTtcient
for H2S gas with
temperature.
Precious metals recovery options include adsorption on activated carbon;
adsorption on
ion-exchange resin; and modification of the solution pH, hydrogen fiagacity,
or oxidation-
reduction potential (ORP). In an alternative embodiment, precious metals are
adsorbed on the
cell walls of bacteria and the bacteria are separated from the liquid in which
they are suspended
27
WO96100308 ,., l: ~ ~ ~ ~ ~ ~ ~ PLTlUS9'Sf09199
:..- f
by settling andlor filtration of the liquid after sextling of the ore
particles. Options that do not
otherwise modify lixiviant solution chemistry are preferable. For this reason,
in preferred
embodiments, at least reactors 8 and 22, and preferably also reactor 26, are
operated together as
a single, essentially completely-mixed reactor.
In an alternative embodiment, pregnant solution 24 is degassed to reduce its
total
dissolved sulfide concentration before andlor concurrent with contacting it
with granular
activated carbon in precious metals recovery reactor 26. Degassing may be
accomplished by
pumping gas from the headspace of reactor 26 into the liquid in leaching
reactor 22. Precious
metals that have absorbed to the activated carbon are eluted into a
concentrated solution that is
a solvent for the precious metals. Precious metals are recovered from the
concentrated solution
by conventional means.
Recovered precious metals are converted into products. This may include the
operations
of separating, smelting and casting of each precious metal into bars or
bullion.
Reference is now made to Fig. Z which is a schematic diagram illustrating a
second
alternative representative embodiment of the invention, with dashed lines
representing possible
variations in the process and apparatus. In this embodiment, ore 30 preferably
undergoes
crushing 32 to facilitate exposure of precious metal values in the ore to
processing solutions.
Crushed ore 34 then undergoes acid leaching 36 in aerobic reactor 37. If
necessary, air 38
containing oxygen and carbon dioxide is added in the acid leaching step Acid-
leach solution 40
2 ~ is recirculated through the ore undergoing acid leaching by means of pump
42.
Acid-leached ore 44 then undergoes bisulfide leaching 4G in essentially
completely-
mixed, anaerobic reactor 47. Bisulfide lixiviant 48 is recircufated through
the ore undergoing
bisulfide leaching by means of pump 50. The pH of bisulfide fixiviant 48 is
established at an
optimum pH by pH controller 60 which controls the rate of addition of acid-
leached ore 44 and
2 5 acid-leach solution 40 to reactor 47 by means of valves 62 and 64. The
sulfate andlor the
sulfide concentration in bisulfide lixiviant recirculation loop 76 is
monitored by sensorlcontroller
82, which may comprise an ion-specific electrode. Sensorlcontroller 82 is
programmed to add
up to a stoichiometric amount of electron donor 84, which is a sulfate-
reducing bacteria growth
substrate such as formate, acetate or methanol, to bisulfide lixiviant
recirculation loop 76.
3t~ Pregnant bisulfide lixiviant 66, which contains precious metal values is
subjected to gold
and silver recovery 68. Recovered gold and silver is converted into products
(e.g., bars of
essentially pure metal). Spent lixiviant 70 is returned to bisulfide lixiviant
recirculation loop 76.
28
CA 02194349 2004-03-O1
In a preferred embodiment, gold and silver recovery 68 is accomplished by
passing pregnant
bisulfide lixiviant 66 through activated carbon coh~mn 78.
Leached ore 80 undergoes dewatering 90 by conventional means, such as settling
and/or
vacuum filtration. Contained bisulfrde lixiviant 92 is retetrned to bisul8de
lixiviant recirculation
loop 76. Waste ore 94 is disposed ofby using conventional means.
In an alternative embodiment, acid-leach solution portion 96 undergoes base
metal
removal 98 in base metal removal reactor 100. Excess hydrogen sulfide gas 110
removed from
anaerobic reactor 47 is introduced to base metal removal reactor 100 to
precipitate iron and
other base metals 104. Acid-leach solution portion 102 having a reduced base
metal content
may be returned to reactor 37, or optionally, to reactor 47.
In an alternative embodiment, excess hydrogen sulfide gas portion 112
undergoes sulfur
recovery I 14 in sulfur recovery reactor 116. Recovery of element sulfur I20
may be
accomplished by the conventional Claus process or by means ofthe process
disclosed in U.S.
Patent No. 4,666,852.
Reference is now made to Fig. 3 which is a schematic diagram illustrating a
third
alternative representative embodiment of the invention, with dashed lines
representing possible
variations in the process and apparatus. In this embodiment, sequential
processing of heaps 200
and 202 of crushed ore 204 and 205 is accomplished. In heap 200, conventional
bio-oxidation
of crushed ore particles 200 is accomplished to free precious metals dispersed
or occluded
2 0 within the ore. Air 206 may be introduced to heap 200 via plenum 208.
Acidic, base-metal
sulfate leach solution 2I0 is collected from the bottom of heap 200 through
plenum 208 by
means of pump 212. Portion 214 of leach solution is recirculated by means of
pump 212 and
distributor 2I6 to the top of heap 200.
As was noted above, bio-oxidation of heap 200 may include ore crushing, acid
2 5 . pretreatment, inoculation with appropriate sul$de-oxidizing bacteria,
addition of nutrients,
recircuIating the biolixiviant and cooling the heap (for 3 to 8 days), and
allowing the heap to
"rest" (for 3 to 8 days). Additional process steps may include washing heap
200 for an extended
period (e.g., 14 days) to remove residual acidity or iron content, and
breaking heap 200 apart in
order to agglomerate ore 202 with cement and/or lime to make a new heap, such
as heap 202.
3 0 Portion 220 of aadic, base-metal sulfate leach solution 210 produced by
the bio-
oxidation step is introduced to anaerobic, sulfate-reduction reactor 230. In
this embodiment of
the process, reactor 230 is a side-stream reactor. The rate of addition of
portion 220 to reactor
' 230 may be controlled by pH controller 232 which operates valve 234 to
create an optimum pH
29
wo 9GIOO308 ~. '~ .,.; ( ~ C~ ~ 3 ~ ~ PC1YU8~5109199
for precious metals leaching in bisulfide leach solution 238 produced by
reactor 230. Preferably,
non-toxic electron donor 240 (such as formate, acetic acid (e.g., vinegar),
acetate, or methanol--
which does not bind effectively to activated carbon), is added to anaerobic
reactor 230 to enrich
within reactor 230 a microbial culture comprising sulfate-reducing bacteria.
Arraerabic reactor
230 is preferably operated in a pH-stet mode by adding a sufficient pardon 220
of acidic sulfate
solution to maintain a neutral pH in reactor 230. In some embodiments, the
concentration of
dissolved sulfide (HxS, HSw, and S') in the anaerobic reactor is maintained
below about 2,500
mg/l to prevent inhibition of the microbial culture comprising sulfate-
reducing bacteria.
In a preferred embodiment, base metals 244 (such as iron) are precipitated in
downstream settling tank 250, and portion 252 of clarified bisulfide
lixic~iant 254 is recirculated
to reactor 230. The rate of recirculation of portion 252 is preferably chosen
so that reactor 230
and settling tank 250 are operated together as a single, essentially
completely-mixed reactor.
Headspace 260 of reactor 230 and headspace 262 of settling tank 250 are
preferably connected
by~ conduit 264. Excess hydrogen sulfide gas (H~S) 266 produced in anaerobic
reactor 230
(e.g., that amount over about Z. X00 mg9) and tank 250 is preferably removed.
In some
embodiments, excess hydrogen sulfide gas undergoes sulfur recovery 270 to
produce elemental
sulfitr 273. By means afreaMOr 230 and tank Z50, sulfate-reducing bacteria are
used to create
clarified, approximately neutral (pH 6 to 9) leaching solution 254 comprising
bisulfide tans and a
low concentration of dissolved and suspended base metals. Bisulfide lixiviant
254 and
2 0 headspace 260 comprise the reactor envirorunent of reactor 230.
In another preferred embodiment, excess H=S gas produced in reactor 230 is
removed
from headspace 260 andfor headspace 262 by means of a H=S gas pump (not shown)
and
transferred into clarified bisulfide iixiviant 254 downstream from settling
tank 250. In this was
the concentrations of H,S~,~~ and HS in the lixiviant are increased after mast
of base metals 244
are removed from it.
In a preferred embodiment, heap 200 is undergoing bio-oxidation while a second
heap
202, which has previously undergone bio-oxidation, undergoes leaching with
bisulfide lixiviant.
In a second process step, oxidised ore 205 is preferably covered with cover
208 and submerged
in bisulfide iixiviant 282 to exclude oxygen. Heap 202 is leached by
recirculating portion 292 of
3 0 neuttal bisulfide lixiviant 282 saturated with H=S through it by means of
plenum 284, pump 286,
and distributor 290. In an alternative embodiment, the H=S pattial pressure is
increased by
introducing the lixiviant [andlor HZS gas having a low concentration (less
than 1,000 parts per
million by volume) of H; gas] under pressure at the bottom of a heap via
plenum 284 which is
Vf~O 96100308 y , .- ~ f ~ ~ ~ ~ y PCTlUS95109199
submerged in lixiviant 282, causing H5- ion concentrations to increase in
direct proportion to
the increase in H2S partial pressure. This may increase the concentration of
dissolved sulfide
(H2S, HS-, and S =) in heap 202 above ?, 500 mg/l. In a preferred embodiment,
anaerobic reactor
230, settling tank 250, and heap 202 are operated together as a single,
essentially completely-
mixed reactor by recirculating portion 294, from heap 202, to reactor 230.
In an alternative embodiment, pressure sensors are placed at multiple paints
throughout
the system far safety reasons. This provides a warning system for users of the
system, since
releases of H,S~g~ can he toxic. Low pressures sound an alarm, indicating a
leak somewhere,
while high pressures indicate unsafe operation. The use of multiple gauges
pinpoints the source
1 G of the problem quickly. The pressure gauges are also used to monitor and
regulate the HzS,&~
pressures to optimize the solubility of the gold and silver.
Additionally, conductivity and total dissolved solids meters are placed in the
effluent
streams of the sulfate-reducing reactor in order to measure the ionic strensnh
of the solvent.
The meters are used to monitor the ionic strength of the solvent, which
controls the activity
coefficients of the gold and silver complexes, HZS~,q~, HS-, and H,~r,.
Control of the activities of
these compounds increases the efficiency of solubilizing the Bald and silver.
Complexed gold and silver in pregnant portion 300 of lixiviant 282 is
recovered
continuously from the lixiviant solution in reactor 302. Recovery may be
accomplished in a
conventional manner by adsorption on activated carbon or by precipitation on
zinc dust or by
modifying either the solution pH, hydrogen fugacity, or oxidation-reduction
potential (ORP).
R4etal that has been recovered from activated carbon eluent by electrowinning
or zinc dust may
be smelted to recover precious metal values as products such as jewelry or
electronic system
components. Barren lixiviant solution 306 is recycled to heap 202.
Working Example No. 1
A chemostat having a working (liquid) volume of 5 liters and a headspace
volume of 2.5
liters was operated at a dilution rte of O.OOfi per hour for over 6 hydraulic
detention times sa
that steady state conditions were achieved. A sulfate-reducing bacteria growth
medium
comprising farmate ions was pumped into the chemostat at a constant rate. The
pH of the
liquid in the chemostat was maintained at 7.0 by~ means of a pH controller
that added bio-
oxidation process efr7uent (acidic metal sulfate solution) to the reactor as
required.
Sufficient fomtate ions and sulfate ions were introduced to the chemostat to
produce a
headspace H=S partial pressure of about 1 atmosphere. Achievement of this
partial pressure of
31
W096J00308 :w r . .~ rz 's ~,, ~ PCTIUS95J09199
HzS was assured by purging the chemostat wish a gas contaitung 99.5+ percent
H=S at the
beginning of the experiment.
The concentration of H= gas in the chemostat headspace before it was purged
and in the
gas used to purge the chemostat was measured by means of a gas chromatograph
with a thermal
canductir~ity detector. The concentration of H~ in the headspace was about 300
parts per
tttilIion (ppm) by volume and the H: concentration in the purging gas was
about 200 ppm.
The liquid level and the liquid volume in the chemostat was kept constant by
withdrawing liquid and headspace gas from the chemostat at a greater rate than
liquid was
added to the chemostat. The chemostat effluent contained about 200 mg/1 of
formate. The
effluent was discharged to a reservoir, the headspace of which was connected
to the headspace
of the chemostat.
A square of gold foil about 0.1 inch on a side and 0.025 inch thick was placed
in a 160-
milliliter (ml) serum bottle and a Teflon ~ septum stopper was crimped on the
bottle mouth.
The bottle was purged with oxygen-free nitrogen gas and 100 ml of chemostat
effluent was
I S transferred to the battle without exposing it to air.
The contents of the bottle were then purged three times with the afore
described HjS gas
mixture at about 3-day intervals. Within four hours of the itutial purging,
the liquid in the bottle
took on a bright yellow color. Testing of a six-ml sample of the liquid plus
two ml of aqua regia
revealed that the liquid contained about 0.3 mgll of gold.
2 0 A 10-ml sample of the liquid was withdrawn from the bottle and introduced
anaerobically to a similar serum bottle containing washed granular activated
carbon and the Ii:S
gas mixture. The liquid in contact with the activated carbon immediately
became colorless
indicating adsorption of the gold on the activated carbon granules. Assays of
the activated
carbon revealed that gold was adsorbed on the carbon.
Working Example No. 2
Reference is now made to Fig. 4 which is a schematic diagram illustrating a
fourth
alternative representative embodiment of the invention, with dashed lines
representing possible
variations in the process and apparatus. In this embodiment, an experiment was
conducted to
3 0 illustrate the disclosed method and apparatus on low-grade samples of gold
ore. Experimental
procedures and results are presented below.
Each I .5 kilogram ore sample was ground to a fine powder that could pass
through a
150-mesh sieve (sieve opening = 0.004 inch). The entire sample was split into
three
32
W~09G100308 , ~; 2 ~ ~ j~ ~ q ~ PC'TlUS95109199
representative samples of approximately 500 grams each. The first
representative aliquot of the
ore was assayed three times for gold and silver content, as well as for the
presence of trace
elements. The second and third representative aliquots were bio-oxidized to
oxidize (and
solubiIize} metal sulfides and mobilize gold and silver values.
Bio-oxidation was accomplished in aerobic, stirred, batch reactor 312 having a
working
volume of 5 liters. Batch reactor 312 was placed in a water bath (not shown)
having a
temperature of 35°C. About 1,000 grams of the ground ore was suspended
in about 5 liters of
an acidic :rhiohacillusferrnoxidarzs medium in the reactor. The acidic medium
as described in
ASTM Standard E 1357 contained the constituents shown in Table 5 and its pH
was adjusted to
pH 2 with concentrated sodium hydroxide (NaOH). Air and carbon dioxide were
introduced
into the suspension by pumping air into it at a relatively high rate with air
pump 314. The
suspension was inoculated with an active culture of T?riohacilhrs
ferrooxidafrs, ATCC 13661,
obtained from the American Type Culture Collection at the address given above.
The progress
of bio-oxidation was monitored by measuring pH (with first pH monitor 318) and
dissolved iron
concentrations in the acidic medium.
Table ~. Bio-Oxidation Medium
Compound Concentration, mg/I
Ammonium sulfate, (NH,)xSO, 300
Calcium nitrate,Ca(N0,)z 1
Magnesium sulfate, MgSO,7H=0 50
Potassium chloride, KCI 10
Potassium phosphate dibasic, K:HPO, 50
Iron sulfate, FeSO,7H,0 44.22
Sulfuric acid, H,SO,195-98%) 2.6 ml
After a period of ten to twelve days, when the rate of increase in iron
concentrations in
the acidic medium slowed and the pH stabili~aed, ore 322 was separated from
liquid supernatant
3 0 320 by settling and drying in an oven at 140°C. A first
representative portion of dried, bio-
oxidized ore 324 was subjected to conventional cyanide e~,~traction, and then
assayed For gold
content to provide a basis of comparison with the bisulfide extraction.
33
9 ~ ~ ~ PC'flUS95/09199
WO 9Gl00308 ~~
i 5 F. ~ ~
A leaching solution comprising dissolved hydrogen sulfide gas and bisulfide
ions was
produced in continuously stirred tank reactor (CSTR or chemostat} 32b having a
working
(liquid) volume of five liters and a headspace volume of 2.5 liters. Chemostat
326 was placed in
a water bath (not shown) having a temperature of 35°C. Chemostat 326
was started in a batch
mode by placing a sulfate-reducing bacteria medium in the chemostat,
inoculating the chemostat
with wild sulfate-reducing bacteria and allowing the culture to acclimate for
5-7 days.
After an acclimation period, sulfate-reduction medium 328 containing the
constituents
shown in Table 6 and formate as a carbon source was pumped into reactor 326 by
pump 330 at
a rate that produced a dilution rate of about 0,005 per hour. Liquid effluent
was removed from
the chemostat by pump 332 at the rate required to maintain the liquid level in
the chemostat at a
set level and discharged to efiiuent storage container 342. This dilution rate
produced a mean
cell residence time in the reactor that was much less than the maximum
specific growrth rate of
the sulfate-reducing bacteria used to inoculate it. Chemostat 326 was operated
in a pH-stat
mode at pH 7.0 by continuously monitoring the pH of the liquid in chemostat
326 with pH
monitoricontroller 336, and by intermittently pumping acidic supernatant 338
produced by the
bio-oxidation step into chemostat 326 with pump 340. Addition of acidic
supernatant 338 to
chemostat 326 increased the dilution rate to about 0.006 per hour. The
chemostat headspace
was periodically purged with hydrogen sulfide from canister 334 to maintain
positive pressure
within the reactor. After chemastat 326 had been operating for about three
hydraulic detention
times and had reached steady state, the effluent from the reactor was used as
solvent in leaching
experiments
Pyrex serum bottles 348 with a capacity of 160 ml were used as batch leaching
reactors.
The reactors had previously been washed with aqua regia because Pyrex is known
to adsorb
gold complexes under certain conditions. Representative four-gram portions of
the bio-oxidized
2 5 ore were added to the reactors. The reactors were augmented with 4 gram
portions of
prewashed 4-12 mesh activated carbon. Effluent collected from the chemostat
was dispensed in
I00 m1 aliquots into the leashing reactors by pump 346. The reactors were
immediately capped,
sealed, purged and pressurized to 1 atmosphere absolute with 99.5 percent pure
hydrogen
sulfide gas. The reactors were then placed in both 35 and 65 °C
incubators. The experiments
were mixed by hand about two times daily and purged and pressurized with
hydrogen sulfide
gas at least every 48 hours.
34
WO96l00308 " ' ~ , PCT/US95/09199
Table 6. Sulfate-Reduction Medium
Compound Concentration
Solution 1: Mineral Media mg/l
Ammonium chloride, NH,CI 300
Calcium chloride, CaCI: H,O 150
Magnesium chloride, MgCl2 6H=0 400
Potassium chloride, KCI 500
Potassium phosphate monobasic, ICH_PO~ 200
Sodium chloride, NeCI 1,200
Yeast extract 6.5
Solution 2: Trace Minerals' mgR
Boric acid, H,BO, 60
Cobalt chloride, CoCI. 6H=O 120
Copper chloride, CuCI= GH_O 15
Ferric chloride, FeCI4H,0 1500
2 0 Manganese chloride, MnCl2 4H~0 100
Nickel chloride, NiCI6HZ0 25
Sodium molybdate, NaMoO; 2H_O 25
Zinc chloride, ZnCI 70
Hydrochloric acid, HCI (25l0) 6.25 ml
?5
Solution 3: Vitaminsb mg/
Biotin 50
Calcium pantothenate 125
Thiamine 250
30 p-Aminobenzoic acid 250
Nicotinic acrid 500
Pyridoxamine 1,000
lotion 4: Sodium Bicarbonate Solution' mg/100 ml
35 Sodium bicarbonate, NaHCO, 8,500
Distilled water 100 ml
Solution 5: Selenium TunEstate Solutian° mgt!
Sodium hydroxide. NaOH 500
~i 5
WO l6l00308 ~ ~ g C~ ~ jt1 ~ PCTtUS95109I49
,. x .,. ,~, ; ~ f '~
,~W y. ,;. cW s
Table 6. Sulfate-Reduction Medium (coot.)
Compound Concentration
Sodium selenite, Na=SeO; SH;O 12
Sodium tungstate, Na=WO; 2H:0 16
Solution 6: Vitamin BI2' mg,'200 ml
Cyanocobalamia 20
Distilled water 200 ml
Add 1 ml per liter of Mineral Media
" Add 200 pl per liter of Mineral Media
' Add 30 ml per liter of Mineral Media
~ Add 250 pl per titer of &Iineral Media
'Add 200 lCl per liter of Mineral Media
Samples of the liquid phase from the reactors were taken several times
throughout the
experiment. The 6 ml liquid samples were taken from well settled reactors and
filtered with a
0.2 pm millipore filter into IO ml serum bottles. The samples were preserved
in 2 ml aqua regia
and analyzed for gold concentrations by inductively-coupled plasma atomic
emission
spectroscopy (ICP) analysis.
Upon completion of the experiments, all the components of the experiments were
aaalyzed for gold and silver concentrations. Liquid samples were taken from
the reactors, then
2 0 the bottles were opened and the components were separated. The reactors
were shaken and
poured through a small mesh sieve (to collect the activated carbon) into a
vacuum filter funnel
containing a Whatman glass fiber filter. The liquid passed through and the ore
portion was
collected on the filter. The activated carbon was washed with distilled water,
blotted dry with
filter paper and dispensed into serum bottles. The ore and the filter were
removed from the
2 5 apparatus and inserted into serum bottles. Both the carbon and the ore
samples, were analyzed
by fire assay for gold and silver concentrations. The liquid samples were
prepared as described
above and analyzed for gold by ICP analysis.
Bisulfide leaching results are presented in Table 7. The relatively low
recovery
percentages were attributed to infrequent mixing of serum bottle contents and
infrequent
3 0 purging with HxS gas. Because the rates of metal dissolution reactions are
controlled by the
rates at which reactants can reach and products can leave the metal surface as
well as by
reactant and product concentrations at the metal surface, subsequent
experimental designs
addressed these factors.
36
W096100308 ' ' ' ) ~ PCT/U595109199
~ - l; ~t~.S~~
Table 7. Bisulfide Leaching Results
Placer PlacerPlacer Placer
Mine Suurcc BatrickBarrickBarnckBarrickdrnne dome dome dcmte
Leaching temp. 35 36 65 66 35 35 65 65
~~
Solventtrcatmeat non- non- non- non-
(tiltratiott) filteredtilten-
~1filteredfilteredIileemdfilteredfilteredfiltered
Gold wncentrations
Bio-oxidizedorc 0.10 0.105 U.10~ 0.10 O.Ui2 0.052 0.052 0.052
(ozhon)
Bisultide LeachedO.t)82O.U72 0,076 0.078 0.039 U.035 O.U36 U.037
ore (oz/ton)
Crold recoven~ 22 31 28 25 25 34 32 U
3
percent
INDUSTRIAL APPLICABILITY
The invention has utility as a means of extracting precious metals from ore
that is being
mined in situ or ex situ. The invention can also be used to recover precious
metals from scrap.
Many variations in configurations have been discussed and others will occur to
those
skilled in the art. Some variations within the scope ofthe claims include
biotic and abiotic
means far producing the bisuifide lixiviant and other 1-I=S gas pumping
schemes. All such
variations within the scope of the claims are intended to be within the scope
and spirit of the
present invention
37