Note: Descriptions are shown in the official language in which they were submitted.
WO 96/02525 PCT/FR95100975
1 .
Nouvelles pipérazides dérivées d'aryl pipérazine,
Ieurs procédés de préparation, leur utilisation à titre demédicament et les
compositions pharmaceutiques les comprenant.
La présente invention se rapporte à de nouvelles pipérazides dérivées d'aryl
pipérazine, ainsi qu'à leur procédé de préparation, les compositions
pharmaceutiques les
contenant et leur utiIisation comme médicaments.
La sérotonine ou 5-hydroxytryptamine (5-HT) est un neurotransmetteur et un
neuromodulateur du système nerveux central impliquée dans de nombreux
processus
physiologiques et pathologiques. La sérotonine joue un rôle important tant au
niveau, du
système nerveux qu'au niveau des systèmes cardiovasculaires et
gastrointestinaux. Au
niveau central, la sérotonine contrôle des fonctions aussi variées que le
sommeil, la
locomotion, la prise de nourriture, l'apprentissage et la mémoire, les
modulations
endocriniennes, le comportement sexuel, la thermorégulation. Dans la moelle,
la
sérotonine joue un rôle important dans les systèmes de contrôle des afférentes
nociceptives périphériques (cf. A. Moulignier, Rev. Neurol. (Paris), 150, 3-
15,1994).
La sérotonine peut jouer un rôle important dans divers types de conditions
pathologiques tels que certains désordres psychiatriques (anxiété, dépression,
aggressivité, attaques de panique, désordres compulsifs obsessionnels,
schizophrénie,
tendance au suicide), certains désordres neurodégénératifs (démence de type
Alzheimer,
Parkinsonisme, chorée de Huntington), l'anorexie, la boulimie, les troubles
liées à
l'alcoolisme, les accidents vasculaires cérébraux, la migraine ou encore les
céphalées
diverses (R. Glennon, Neurosci. Biobehavioral Reviews,l4, 35, 1990).
De nombreuses études pharmacologiques récentes ont mis en évidence la
diversité des récepteurs de la sérotonine ainsi que leur implication
respective dans ses
divers modes d'action (cf. E. Zifa, G. Fillion, Pharm Reviews, 44, 401, 1992 ;
S. Langer,
N. Brunello, G. Racagni, J. Mendlecvicz, "Serotonin receptor subtypes :
pharmacological significance and clinical implications", Karger Ed. (1992) ;
B.E.
Leonard, Int. Clin. Psycho-pharmacology, 1, 13-21 (1992) ; R.W. Fuller, J.
Clin.
Psychiatry, 51, 36-45 (1992) ; D.G. Grahame-Smith, Int. Clin.
Psychopharmacology, .6,
suppl. 4, 6-13, (1992). Ces récepteurs sont snbdivisés principalement en 4
grandes
classes (SHTI, 5HT2, 5HT3 et 5HT4) qui comportent elles-mêmes des sous-classes
WO 96102525 21 J 5.4 27 PCT/FR95/00975
2
telles que les récepteurs 5HTI qui sont divisés principalement en 5HT1A,
5HT1B,
5HT1D (cf G.R. Martin, P.A. Humphrey, Nettropharmacol., ââ, 261, 1994 ; P.R.
Saxena, Exp. Opin. Invest. Drugs, 3(5), 513, 1994). Les récepteurs 5HTID
renferment
eux-mêmes plusieurs sous-types de récepteurs ; c'est ainsi que les récepteurs
5HT1Da et '
SHTIDb ont été clonés puis identifiés chez l'homme (cf par exemple E. Hamel et
coll.,
Mol. Pharmacol., 44, 242, 1993 ; G.W. Rebeck et coil., Proc. Natl. Acad. Sci.
USA, 21,
3666, 1994). Par ailleurs, il a été démontré récemment que les auto-récepteurs
5HT1B
chez les rongeurs et 5HTID chez les autres espèces étaient capables de
contrôler la
libération de sérotonine dans les teminaisons nerveuses (cf M. Briley, C.
Moret, Cl.
Neuropharm. 16, 387, 1993 ; B.E. Léonard, Int. Clin. Psychopharmacol., 9, 7,
1994)
ainsi que la libération d'autres neurotransmetteurs tels que la
norepinéphrine, la
dopamine ou l'acétylcholine (M. Harrigton, J. Clin. Psychiatry, 53, 10, 1992).
Les composés ayant une activité antagoniste sélective au niveau des récepteurs
5HTID centraux tels que les composés nouveaux décrits dans la présente
invention
peuvent donc exercer un effet bénéfique sur des sujets souffrant de troubles
du système
nerveux central. En particulier, de tels composés trouvent leur utilité dans
le traitement
de la douleur, du cancer, des troubles de la locomotion, de la dépression, de
l'anxiété,
des attaques de panique, l'agoraphobie, les désordres compulsifs
obsessionnels, les
désordres de la mémoire incluant la démence, l'amnésie, et les troubles de
l'appétit, les
dysfonctionnements sexuels, la maladie d'Alzheimer, la maladie de Parkinson.
Les
antagonistes SHT1D trouvent également leur utilité dans le traitement des
désordres
endocriniens tels que l'hyperprolactinémie, le traitement des vasospasmes, de
l'hypertension et des désordres gastro-intestinaux dans lesquels interviennent
des
chanQements au niveau de la motilité et de la sécrétion.
Les éomposés selon la présente invention sont des antagonistes puissants et
sélectifs des récepteurs 5HTID et plus particulièrement des récepteurs
récemment
identifiés comme SHT1Da et 5HTIDR chez l'homme et de ce fait trouvent leur
utilité,
seuls ou en association avec d'autres molécules, comme médicaments et plus
particulièrement comme moyens thérapeutiques pour le traitement tant curatif
que
préventif de désordres liés à la sérotonine.
~
WO 96/02525 ' f i 3 k PCT/FR95/00975
2195427
3
L'état antérieur de la technique dans ce domaine est illustré notamment par
les
demandes de brevets européens 0533266, 0533267 et 0533268 qui décrivent de
nouveaux dérivés de benzanilides comme antagonistes 5HTID et les publications
récénies qui décrivent le GR 127,935 comme un antagoniste 5HT1D (cf. M.
Skingle et
coll., J. of Psychopharm. $(1), 14, 1994 ; S. Starkey, M. Skingle,
Neuropharmacol., 32,
393, I994).
Les dérivés de la présente invention se distinguent de l'art antérieur non
seulement par leur structure chimique originale qui les distingue sans
ambiguité des
dérivés précédemment décrits mais également par l'efficacité (rendement
global, nombre
d'étapes) de leur préparation et leur profil biologique original, en
particulier en ce qui
conceme leur sélectivité pour les sous-types de récepteurs de la sérotonine
(5HTID a et
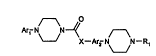
R)La présente invention concerne des dérivés de formule générale (I)
~--~ o
~ N \N --,\/ (0 )
X-Arz N.N-R,
dans laquelle
Arl représente un résidu aromatique tel qu'un phényle, un naphtyle, ou un
pyridyle, ou un résidu aromatique substitué par un ou plusieurs groupes
choisis parmi
les résidus alkyle linéaire ou ramifié comprenant de 1 à 6 atomes de carbone,
trifluorométhyle, 2,2,2 trifluoroéthyle, phényle, benzyle, cycloalkyle,
hydroxyl (OH),
thiol (SH), éther (OR'2), thioéther (SR'2), ester (OCOR' , carbamate
(OCONHR2),
carbonate (OCO2R'2), carbonyles (COR2, COOR'y CONHR2), halogènes (fluor,
chlore, brome ou iode), amine (NR2R3), nitro (NO2), nitrile (CN),
aminocarbonyle
(NHCOR'2, NHC02R'2, NHCONR2R3), aminosûlfonyles (NHSO2R'2, N(S02R'2)2,
NHSO2OR'2, NHSO2NR2R3), sulfonyles (S02R'2, S02NR2R3) et les hétérocycles
pouvant éventuellement être diversement substitués tels qu'un hétérocycle à 5
membres
pouvant contenir de 1 à 4 hétéroatomes tels que l'oxygène, l'azote ou le
soufre, ou deux
~
WO 96/02525
PCT/FR95/00975
4
substituants sur des carbones voisins pouvant former un cycle avec le résidu
aromatique
auquel ils sont attachés,
X représente O, NH, CH2O ou CH2NH,
Ar2 représente un radical aromatique tel qu'un phényle ou un naphtyle auquel
X et la pipérazine sont attachés sur des carbones différents et pouvant être
lui-même
diversement substitué par un radical alkyle ramifié ou linéaire comprenant de
1 à 6
atomes de carbone, un alcoxy (OR4), ou un halogène (chlore, fluor, iode ou
brome),
RI, R2, R3 et R4, identiques ou différents représentent un hydrogène, une
chaine alkyle linéaire ou ramifiée comprenant de 1 à 6 atomes de carbone,
R'2 représente une chaine alkyle linéaire ou ramifiée comprenant de 1 à 6
atomes de carbone,
et leurs sels, hydrates, solvates et bioprécurseurs physiologiquement
acceptables pour leur usage thérapeutique.
Les isomères géométriques et optiques des composés de formule générale (I)
font également partie de la présente invention ainsi que leurs mélanges en
toutes
proportions et sous forme racémique.
Parmi les sels physiologiquement acceptables des composés de formule
générale (I) sont inclus les sels obtenus par addition d'acides organiques ou
inorganiques
tels que les chlorohydrates, bromhydrates, sulfates, phosphates, benzoates,
acétates,
naphtoates, p-toluènesulfonates, méthanesulfonates, sulphamates, ascorbates,
tartrates,
citrates, oxaIates, maléates, salicylates, fumarates, succinates, lactates,
glutarates,
glutaconates.
L'expression "bioprécurseurs" telle qu'elle est utilisée dans la présente
invention s'applique à des composés dont la structure differe de celle des
composés de
formule (I) mais qui, administrés à un animal ou à un être humain sont
convertis dans
l'organisme en un composé de formule (I).
Une classe particulièrement appréciée de composés de formule (I) correspond
aux composés pour lesquels Ar2 représente un phényle pouvant être diversement
substitué, en particulier les composés de formule (la) 30
~ 2~9542i
WO 96/02525 PCT/FI295/00975
iRi
O
r-~ Ari N\IN--/< ( la )
XRS
dans laquelle Arl, X et RI sont définis comme dans la formule (I) et R5
représente un atome d'hydrogène, un radical alkyle ramifié ou linéaire
comprenant de 1
à 6 atomes de carbone, un alcoxy (OR4), ou un halogène (chlore, fluor, iode ou
brome),
R4 étant défini plus haut.
Une autre classe particulièrement appréciée de composés de formule (I)
correspond aux composés pour lesquels Ar2 représente un naphtyle, en
particulier les
composés de formule (Ib) :
Ri
1
Ari'N N
N
LD ~ ~ -rX / / ~ `Ib)
O ~ ~
dans laquelle Arl, X et RI sont définis conune dans la formule générale (I).
Lorsque Arl représente un aryle dont deux substituants sur des carbones
voisins forment"ün c cle avec le résidu aromatique, il s'agit avantageusement
d'un
phényle dont deux substituants sur des carbones voisins forment avec le
phényle auquel
ils sont attachés un cycle à 5 ou 6 éléments, pouvant comprendre 1 ou 2
hétéroatomes
(oxygène, azote ou soufre).
Les coüiposés de la présente invention peuvent être préparés par différentes
méthodes qui seront notamment dépendantes de la nature des substituants Arl,
Ar2, X
etR1
WO 96/02525 PCT/FR95/00975
6
On comprendra que dans certaines réactions ou suites de réactions chimiques
qui conduisent à la préparation de composés de formule générale (I) il soit
nécessaire ou
souhaitable de protéger des groupes sensibles éventuels dans les
intermédiaires de
synthèse afin d'éviter des réactions secondaires indésirables. Ceci peut être
réalisé par
l'utilisation (introduction et déprotection) des groupes protecteurs
conventionnels tels
que ceux décrits dans "Protective groups in Organic Synthesis", T.W. Greene,
John
Wiley & Sons, 1981 et "Protecting Groups", P.J. Kocienski, Thieme Verlag,
1994. Les
groupes protecteurs appropriés seront donc introduits et enlevés lors de
l'étape la plus
appropriée pour ce faire et en utilisant les méthodes et techniques décrites
dans les
références citées précédemment.
Les composés de formule générale (I) dans laquelle Arl, Ar2 et Rl sont décrits
comme précédemment et X représente -CH2O- ou -CH2NH- sont préparés par
condensation d'un intermédiaire de formule (II) :
O
ArF N\--IN ---- ~ I I ) -
Y - -
dans laquelle Arl est défini comme précédemment et Y représente un groupe
partant tel qu'un halogène (chlore, brome ou iode) un toxylate, un mésylate ou
un triflate
avec une aryl pipérazine de formule générale (III) :
HX' Arz N N-Ri (111)
~~ -
dans laquelle X' représente O ou NH, Ar2 représente un noyau aromatique tel
qu'un phényle ou un naphtyle sur lequel X' et le noyau pipérazine sont
attachés en des
positions différentes et RI est décrit comme précédemment. La condensation des
arylpipérazines de formule (III) avec les électrophiles de formule (II) est
réalisée en
présence d'une base organique ou inorganique telle que NaH, KH, DiPEA, 'DBU,
WO 96/02525 PCTIFR95100975
7
pyridine, DMAP, K2C03, CaC03, Cs2CO3, en présénce événtuelIément d'un iodure
tel
que NaI, KI, Bu4NI, dans un solvant anhydre polaire tel que le THF, le DME, le
n-
butanol, le t-butanol, le DMF, le DMSO, la méthyléthylcétone, à une
température
comprise entre 20 et 80 . Les intermédiaires de formule générale (II) sont
aisément
préparés par condensation d'une aryl pipérazine de formule générale (IV)
Ari- NNH ( IV )
dans laquelle Arl est défini comme précédemment et un chlorure d'acide de
formule générale (V) :
Y-CH2-C(0)Cl (V)
dans laquelle Y est décrit comme précédemment en présence d'une base
organique ou inorganique telle que la pyridine, la DiPEA, la DMAP, le DBU,
K2C03,
Cs2C0, ou CaC03 dans un solvant anhydre aprotique polaire tel que le =, le
DMF,
le DnIE, le DMSO ou la méthyléthylcétone à une température comprise entre - 10
C et
80 C.
Les intermédiaires de formules générales (III) et (IV) sont préparés par
diverses
méthodes et techniques bien connues de l'homme de métier pour la préparation
des aryl
pipérazines et dont le choix est dépendant de la nature de X', de Ar2, de Arl
et de RI.
C'est ainsi que, dans le cas particulier où X' est un oxygène, les
intermédiaires de
forniule (III) sont accessibles par condensation d'une arylamine de formule
(VI) :
- Ho-Ar2-NH2 (VI)
dans laquelle Ar2 est défini comme précédemment avec un dérivé d'amine de
formule (VII) :
R' 1-N-(CH2CH2Y)2 - (VII)
WO 96/02525 219-5.4 2 7 PCT/FR95/00975
8
dans laquelle R' I est équivalent à Rl tel que décrit précédemment, où R' I
représente un groupe protecteur tel que t-butoxycarbonyl ou un tosyle qui sera
transformé en RI ultérieurement, et Y représente un chlore, un brome, un iode,
un
tosylate ou un mésylate. Cette réaction est réalisée préférentiellement dans
un solvant
anhydre polaire tel que le DMF, l'acétonitrile, le THF, le n-butanol, le t-
butanol ou le
DMS0, généralement à température de reflux du solvant utilisé, en présence
d'une base
organique ou inorganique généralement utilisée pour ce type de réaction, telle
qu'un
carbonate de potassium, de sodium ou de calcium.
Dans certains cas particuliers, les dérivés de formule (III) dans lesquels X'
est
un oxygène sont préférentiellement préparés par réaction d'une lithio-
pipérazine de
formule VIII dans laquelle RI est défini comme précédemment :
R1 -NN-Li ( VIII )
avec un bis-methoxy aromatique symétrique de formule générale (IX)
MeO-Ar2-OMe (IX)
dans laquelle A172 représente un phényle ou un naphtyle,
dans les conditions décrites préalablement (cf : J. Org. Chem. M, 5101, 1993)
suivie de la déméthylation du groupement aryl-methoxy avec un réactif
approprié tel
que BBr3 dans le dichlorométhane.
Les composés de formule générale (III) dans lesquels X' représente NH sont
préparés par condensation d'une amine aromatique de formule générale (X)
X"-Ar2-NH2 (X)
dans laquelle Ar2 est défini comme précédemment et X" représente une
fonction qui pourra ultérieurement être transformée en amine (telle que par
exemple un
W O 96/02525 PCTIFR95/00975
9
groupe nitro) soit avec un dérivé de bis-(halogéno-éthyl)-amine de formule
(VII) dans
les conditions décrites précédemment pour ce type de réaction, soit avec un
aniino-acide
de formule générale (XI)
~--CO2H
Ri-N \-( XI
- COzH -
dans laquelle Rl est défini comme précédemment, en présence d'anhydride
acétique, suivi de la réduction de la dicétopipérazine intermédiaire ainsi
formée avec par
exemple un borane. Dans les deux cas, le dérivé de formule (III) sera
finalement obtenu
après Lransformation du groupe représenté par X" en amine. S'il s'agit d'un
groupe nitro,
cette transformation sera effectuée selon les méthodes et techniques bien
connues de
l'homme de métier pour transformer un nitroaromatique en un dérivé d'aniline
telles que
par exemple l'emploi de Nickel de Raney ou de catalyseur au rhodium en
présence
d'hydrazine, l'hydrogénation sur charbon-palladium à pression atmosphérique,
ou encore
l'utilisation de SnC12 ou de zinc.
Les composés de formule générale (I) dans laquelle Arl, Ar2 et RI sont décrits
comme précëdemment et X représente 0 ou NH sont préparés par condensation d'un
intermédiaire de formule générale (III) dans laquelle X' représente O ou NH,
Ar2 et RI
sont définis connne précédemment, et d'une arylpipérazine de formule (IV) dans
laquelle Arl est défini comme précédemment, avec un dérivé de formule générale
(XII):
t
x,
X2 (xu )
dans laquelle XI et X2, identiques ou différents représentent chacun un groupe
partant tel qu'un halogène (en particulier le chlore), un groupe 0-alkyle (en
particulier le
groupe OCCIg), un groupe succinimyle, phtalyle ou imidazolyle. La méthode de
la
présente invention comprend également l'utilisation de précurseurs ou
analogues bien
connus des réactifs de formule générale (XII). C'est ainsi et à titre
d'exemple que la
ry( ~
W O 96102525 /r 1~~ 4 2, 7 PCT/FR95100975
condensation des intermédiaires (III) et (IV) avec le phosgène peut être
avantageusement effectuée à l'aide de diphosgène ou de triphosgène selon une
procédure
bien connue de l'homme de l'art.
Les méthodes et techniques choisies pour la mise en muvre de la préparation
5 des composés de formule (I) dans laquelle X représente O ou NH par
condensation des
dérivés de formule (III) dans laquelle X' représente O ou NH et de dérivés de
formule
(IV) avec un réactif de formule (XII) telles que le choix de l'ordre des
réactifs, les temps
de réaction, l'isolation et/ou la purification des intermédiaires, la
température de la
réaction à différentes étapes de la condensation, la nature du ou des
solvants, la présence
10 de co-réactifs (tels qu'une base organique ou inorganique; par exérnple une
amine
tertiaire) ou de catalyseurs et le choix du réactif (XII) (choix de XI et X2)
seront
déterminés par la nature de Arl, Ar2, X(0 ou NH) et RI.
C'est ainsi qu'une méthode particulièrement appréciée pour la préparation de
dérivés de formule (I) dans laquelle X = NH et Arl, Ar2, RI sont définis comme
précédemment, consiste à faire réagir un intermédiaire de formule (III) dans
laquelle X'
représente NH, Arz et RI sont définis comme précédemment avec du triphosgène
en
présence d'une base telle que la triéthylamine dans un solvant anhydre tel que
le
dichlorométhane et d'ajouter ensuite un composé de formule (IV) dans laquelle
Arl est
défini comme précédemment en présence d'une base telle qu'une amine tertiaire.
Dans le cas de la préparation de dérivés de formule générale (I) dans laquelle
Arl, Ar2 et RI sont définis comme précédemment et X représente un oxygène, une
méthode particulièrement appréciée consiste à condenser tout d'abord une
arylpipérazine
de formule (IV) avec du triphosgène en présence de triéthylamine dans un
solvant
anhydre tel que le dichlorométhane et d'isoler l'intermédiaire de formule
générale (XIII)
ainsi formé
O
Ar N N ( cI XIII )
,.
21954,27
WO 96/02525 PCTIFR95/00975
11
avant de le condenser avec un nucléophile de formule générale (III) dans
laquelle X' représente un oxygène, en présence d'une base organique ou
inorganique
telle que NaH, KH, t-BuOK dans un solvant aprotique polaire tel que le THF ou
le
DMF.
Doivent également être considérées comme faisant partie de la présente
invention les méthodes qui permettent de préparer les produits de formule (I)
dans
laquelle X représente O ou NH par condensation d'une pipérazine aromatique de
formule (IV) avec un dérivé de formule générale (3üV) :
O /-,
y---X-Ar2 N N-Ri ( XIV )
Xi ~~ -
dans laquelle Xl, Ar2, R, sont définis comme précédemment et X représente 0
ou RZI, en présence d'une base organique ou inorganique dans un solvant
polaire
aprotique à une température comprise entre 20 et 1 00 C.
Dans le cas particulier des composés de formule générale (I) dans laquelle RI
représente un hydrogène, il est préférable de mettre en oeuvre, pour certaines
réactions
qui le nécessitent, des intermédiaires réactionnels dans lesquels RI
représente un groupe
protecteur tel que par exemple un t-butoxycarbonyl (BOC) qui sera introduit
préalablement par condensation de l'intermédiaire approprié dans lequel RI = H
avec un
réactif adéquat tel que (BOC)20, BOC - ON = C (CN)-Ph, BOC - ONH2. Ceci
permettra de préparer, selon les méthodes et techniques présentées
préalablement, des
intermédiaires de formule générale (I) dans lesquels RI = BOC et de
transformer ces
intermédiaires en produits finaux de formule générale (I) dans lesquels RI = H
après
déprotection du t-butylcarbonate selon les méthodes et techniques bien connues
pour ce
type de transformation telle que l'utilisation d'acide (HCI, CF3CO2H, H2S04)
en milieu
organique.
Doivent également être considérées comme faisant partie intégrale de la
présente invention toutes les méthodes qui permettent de transformer un dérivé
de
formule (I) en un autre dérivé de formule (I) dans laquelle au moins un des
substituants
Arl, X, Ar2 ou RI sont différents, par les méthodes et techniques bien connues
de
- --
2495:~27 0
WO 96l02525 - PCTIFR95/00975
12
l'homme de l'art. C'est ainsi, et à titre d'exemple, que les dérivés de
formule générale (I)
dans lesquels Arl représente un phényle substitué par un groupe N02 peuvent
être
transformés en dérivés de formule (I) dans lesquels Arl représente un phényl
substitué
en même position par un groupe NH2 (par les méthodes et techniques bien
connues pour
ce type de réduction telles que décrites par exemple dans "Compréhensive
Organic
transformation", p. 412 ; (R. C. Larock, VCH, 1989) parmi lesquelles on peut
citer
l'hydrogénation atmosphérique catalysée au palladium sur charbon,
l'utilisation du
SnC12 ou de zinc ou encore de catalyseur au rhodium en présence d'hydrazine.
Les
composés de formule générale (I) dans lequels Arl représente un aromatique
substitué
par un groupement NH2 peuvent eux aussi être transformés en de nombreux autres
dérivés de formule (I) tels que des dérivés dans lesquels Arl représente un
aromatique
substitué par NR2R3, NHCOR2, NHC02R2; NHCOR2R3, NHS02R2, NHS020R2,
NHS02OR2, NHS02NR2R3 par les méthodes et techniques bien connues pour
transformer une amine aromatique en amide, carbonate, urée, sulfonamide,
sulfonate ou
sulfonylurée.
Lorsque l'on désire isoler un composé selon l'invention à l'état de sel, par
exemple de sel par addition avec un acide, on peut y parvenir en traitant la
base libre de
formule générale (I) par un acide approprié de préférence en quantité
équivalente, ou par
le sulfate de créatinine dans un solvant approprié.
Lorsque les procédés décrits ci-dessus pour préparer les composés de
l'invention donnent des mélanges de stéréoisomères, ces isomères peuvent être
séparés
par des méthodes conventionnelles telles que la chromatographie préparative.
Lorsque les nouveaux composés de formule générale (I) possèdent un ou
plusieurs centres asymétriques, ils peuvent être préparés sous forme de
mélange
racémique ou sous forme d'énantiomères que ce soit par synthèse
énantionsélective ou
par résolution. Les composés de formule (I) possédant au moins un centre
asymétrique
peuvent par exemple être séparés en leurs énantiomères par les techniques
habituelles
telles que la formation de paires diastéréomériques par formation d'un sel
avec un acide
optiqüement actif tel que l'acide (-)-di-p-toluoyl-l-tartrique, l'acide (+)-di-
p-toluoyl-l-
tartrique, l'acide (+)- camphorsulfonique, l'acide (-)-camphorsulfonique ,
l'acide (+)-
phénylpropionique, l'acide (-)-phénylpropionique, suivie par cristallisation
fractionnée
~ 2195'427
WO 96/02525 PCT/FR95100975
õ ,..
13
et régénération de la base libre. Les composés de formule (I) dans lesquels RI
est un
hydrogène comprenant au moins un centre asymétrique peuvent également être
résolus
par formation d'amides diastéréomériques qui sont séparés par chromatographie
et
hydrolysés pour libérer l'auxiliaire chiral.
Les exemples qui suivent illustrent l'invention sans toutefois en limiter la
portée.
Les spectres RMN du proton ont été'enregistrés sur un appareil Brilcker AC
200. Les déplacements chimiques sont exprimés en ppm et les abréviations
suivantes
ont été utilisées :"s" pour singulet ;"se" ou "brs" pour singulet élargi, "d"
pour doublet,
"dd" pour doublet de doublet, "t" pour triplet, "q" pour quadruplet, "sx" pour
sextuplet,
"m" pour multiplet, "M" pour massif.
Les spectres infra-rouge ont été enregistrés sur un appareil Nicolet 510P. Les
bandes d'absorbtion sont données en cm 1.
Les analyses élémentaires ont été réalisées sur un appareil Fisons EA 1108.
EXEMPLE 1 Furuaratede N-[4-méthoxy-3-(4-méthylpipérazin-1-yl)phényl]-4-(o-
tolyl pipérazin-1-ylamide
çj
/-\ NN~N O\
1
Une solution de triphosgène (320 mg, 1.08 mmol) dans le dichlorométhane (10
ml) est cannulée sur une solution de 4-méthoxy-3-(4-méthyl pipérazin-l-yl)
aniline
pouvant être préparée selon la méthode décrite dans le brevet européen 0 533
266-Al
(714 mg, 3.23 mmol) et de triéthylamine (450 l, 3.25 mmol) dans le
dichlorométhane
(10 ml) sous atmosphère d'azote. Pendant cette opération, le mélange
réactionnel est
WO 96/02525 219`5 412 i
Pca/FR95t00975
14
refroidi avec un bain de glace. Il est ensuite ramené à température ambiante
pendant V2h.
Après ce temps, la N-o-tolylpipérazine (570 mg, 3.23 mmol) et la triéthylamine
(450 l,
3.25 mmol) diluées dans le dichlorométhane (10 ml) sont ajoutées. Le mélange
réactionnel est agité 12 h à température ambiante puis il est dilué avec de
l'eau et du
dichlorométhane. Les phases sont séparées et la phase organique séchée sur
sulfate de
sodium. Le brut obtenu est purifié par chromatographie-éclair avec un mélange
(90/9/1)
de dichlorométhane/méthanol/ammoniaque.
Masse obtenue : 356 mg (Rdt 26 %).
Ce composé est dissous dans le méthanol et traité avec de l'acide fumarique
pour donner le fumarate correspondant. Celui-ci est cristallisé dans l'éther.
Analyse Elémentaire pour : C24H33N502-1.5C4F-I404
Calculées, C, 60.29 ; H, 6.58 ; N, 11.72 ; Expérimentales, C, 59.86 ; H, 6.77
;
N, 11.48
Masse : 424 (MH+), 248, 177
IR (Kbr) : 3416, 2924, 1707, 1637, 1500
RMN 1H (DMSO) : 2.27 (s, 3H) ; 2.39 (s, 3H) ; 2.72 (M; 4H) ; 2.82 (M, 4H) ;
3.00 (M, 4H) ; 3.56 (M, 4H) ;'3.72 (s, 3H) ; 6.57 (s, 3H) ; 6.78-7.19 (m, 4H)
; 8.36 (s,
1H).
Point de fusion : 110 C
EIEMPLE 2 Fumarate du N-[4-méthoxy-3-(4-méthylpipérazin-1-yl)phényl]-4-(0-
méthoxy phényl)pipérazin-1-ylamide
N
OCHj N
~-~ NN N \ / O
\ _
O
2
Le composé 2 est préparé suivant la procédure décrite dans l'exemple I à
partir
des réactifs suivants : triphosgène (273 mg, 0.92 mmol) ; 4-méthoxy-3-(4-
méthyl
~
WO 96/02525 219 5 4 2 7 pCTIFR95/00975
pipérazin-1-yI) aniline (609 mg, 2.76 mmol) ; triéthylamine (2 x 382 jil, 5.52
mmol) ; 1-
(o-méthoxyphényl) pipérazine (530 mg, 2.76 nunol) ; dichlorométhane (25 ml).
Le brut réactionnel est purifié par chromatographie-éclair avec un mélange
(95/5/1) de dichlorométhane/méthanol/ammoniaque.
5 Masse obtenue : 358 mg (Rdt 30 %)
Analyse Elémentaire pour : C24H33N503-C4H404
Calculées, C, 60.53 ; H, 6.71 ; N,12.60 ; Expérimentales, C, 59.61 ; H, 6.90
N, 11.63
Masse : 440 (MH+), 280, 248, 193
10 IR (Kbr) : 3420, 1701, 1638, 1508
RMN1H(DMSO):2.35(s,3H);2.67(M,4H);3.55(M,4H);3.72(s,3H);
3.79 (s, 3H) ; 6.57 (s, 2H) ; 6.78-7.10 (m, 7H) ; 8.3 (s, 1H).
Point de fusion : 128 C
FXFViPT.F. 3 Fumarate du N-14-méthoxy-3-(4-méthylpipérazin-1-yl)phényl]-4-(p-
15 méthoxy phényl) pipérazin-1-ylamide
çj
vN" ~-/ \
0
3
Le composé 3 est préparé suivant la procédure décrite dans l'exemple 1 à
partir
des réactifs suivants : triphosgène (240 mg, 0.81 mmol) ; 4-méthoxy-3-(4-
méthyl
pipérazin-1-yl) aniline (490 mg, 2.22 mmol) ; triéthylamine (2 x 340 l, 4.86
mmol) ; 1-
(p-méthoxy phényl) pipérazine (523 mg, 2.72 mmol) ; dioxane (25 ml).
En fm de réaction, le mélange est dilué avec de l'eau puis extrait trois fois
avec
de l'acétate d'éthyle. Les phases organiques sont rassemblées, lavées avec une
solution
saturée en chIorure de sodium, séchées sur sulfate de sodium et concentrées.
2195427 +
WO 9G/02525 PCT/FR95/00975
~ X ',a = .
16
Le brut est purifié par chromatographie-éclair avec un mélange (95/5/1) puis
(90/9/1) de dichlorométhane/méthanol/ammoniaque.
Masse obtenue : 464 mg (Rdt 48 %)
Analyse Elémentaire pour : C24H33N50g-C4H404
Calculées, C, 60.53 ; H, 6.71 ; N, 12.60 ; Expérimentales, C, 60.02 ; H, 6.72
N, 12.45
Masse : 440 (M'rl+), 248, 193
IR (Kbr) : 3414, 1717, 1655, 1512
RMNIH(DMSD):2.39(s,3H);2.70(M,4H);3.02(M,8H);3.57(M,4H);
3.71 (s, 3H) ; 3.75 (s, 3H) ; 6.59 (s, 2H) ; 6.81-7.13 (m, 7H) ; 8.40 (s, 1H).
Point defusion : 215 C
EXEVIPLE 4 Fumarate du N-[4-méthoxy-3-(4-méthylpipérazin-1-yl)phényl]-4-
(m-méthoxy phényl) pipérazin-1-ylamide
N
O cN_
NN N~O
0
4
Le composé 4 est préparé suivant la procédure décrite dans l'exemple 3 à
partir
des réactifs suivants : triphosgène (262 mg, 0.88 mmol) ; 4-méthoxy-3-(4-
méthyl
pipérazin-1-yl) aniline (531 mg, 2.40 mmol) ; triéthylamine (4 x 365 l, 10.55
mmol) ;
dichlorhydrate de la 1-(m-méthoxy phényl) pipérazine (636 mg, 2.4 mmol) ;
dioxane
(25 ml).
Le brut est purifié par chromatographie-éclair avec un mélange (90/9/1) de
dichlorométhane/méthanol/ammoniaque.
Masse obtenue : 390 mg (Rdt 37 %)
Analyse Elémentaire pour : C24H33N503-1.5C4H404
~ 2195427
WO 96/02525 PCT/FR95/00975
17
Calculées, C, 58.72 ; H, 6.41 ; N, 11.41 ; Expérimentales, C, 57.19 ; H, 6.66
;
N, 11.70
RMNIH(DMSO): 2.41 (s, 3H); 2.74 (M, 4H); 3.02 (M, 4H); 3.15 (M, 4H);
3.57 (M, 4H) ; 3.74 (s, 3H) ; 6.39-6.58 (m, 6H) ; 6.83 (d, IH) ; 7.07-7.18 (m,
3H) ; 8.41
(s, 1H).
EXEMPLE 5 Fumarate du N-[4-méthoxy-3-(4-méthylpipérazin-1-yl)phényl]-4-
(2,3-diméthyl phényl) pipérazin-1-ylamide
çj
f-\ CN1
5
Le composé 5 est préparé suivant la procédure décrite dans l'exemple 3 à
partir
des réactifs suivants triphosgène (282 mg, 0.95 mmol) ; 4-méthoxy-3-(4-méthyl
pipérazin-l-yl) aniline (567 mg, 2.57 mmol) ; triéthylamine (2 x 395 l, 5.70
mmol) ; 1-
(2,3-diméthylphényI) pipérazine (598 mg, 3.14 mmol) ; dioxane (25 ml).
Le brut est purifié par chromatographie-éclair avec un mélange (95/5/1) puis
(90/9/1) de dichlorométhane/méthanol/ammoniaque.
Masse obtenue : 405 mg (Rdt 36 %)
Analyse Elémentaire pour : C25H35N502-C4H404
Calculées, C, 62.91 ; H, 7.10 ; N, 12.65 ; Expérimentales C, 62.44 ; H, 7.45
N, 12.26
Masse : 438 (MH+), 248, 191
IR (Kbr) : 3407, 2949, 1707, 1638, 1500
WO 96/02525 PCTIFR95/00975
2195427
18
RMN1H(DMSO):2.20(s,3H);2.22(s,3H);2.36(s,3H);2.67(M,4H)
2.79 (M, 4H) ; 3.00 (M, 4H) ; 3.58 (M, 4H) ; 3.73 (s, 3H) ; 6.58 (s, 2H) ;
6.79-7.13 (m,
6H) ; 8.37 (s, 1H).
Point de fusion : 133 C
EXEMPLE 6 Fumarate du N-[4-méthoxy-3-(4-méthylpipérazin-1-yl)phényl]-4-(p-
nitrophényl)-pipérazin-1-ylamide
d
0 \ N NN N H
O
p
O
6
Le composé ¾ est préparé suivant la procédure décrite dans l'exemple 3 à
partir
des réactifs suivants : triphosgène (250 mg, 0.85 mmol) ; 4-méthoxy-3-(4-
méthyl
pipérazin-1-yl) aniline (510 mg, 2.31 mmol) ; triéthylamine (2 x 353 l, 5.10
mmol) ; 1-
(p-nitrophényl) pipérazine (480 mg, 2.31 mmol) ; dioxane (25 ml).
Le brut est purifié par chromatographie-éclair avec un mélange (85/14/1) de
dichlorométhane/méthanol/ammoniaque.
Masse obtenue : 437 mg (Rdt 42 %)
Analyse Elémentaire pour : C23H30N604-C4H404
Calculées, C, 56.83 ; H, 6.01 ; N, 14.73 ; Expérimentales, C, 56.47 ; H, 5.96
N, 14.50
Masse : 455 (MH+), 248, 208, 178
IR (KBf): 3368, 1692, 1664, 1597, 1510
RMN 1 H(DMSO) : 2.33 (s, 3H) ; 2.63 (M, 4H) ; 2.96 (Iv1, 4H) ; 3.15 (M, 4H)
3.54 (M, 4H) ; 3.71 (s, 3H) ; 6.55 (s, 2H) ; 6.79 (d, 1H) ; 7.01-7.09 (m, 4H)
; 8.05 (d,
2H) ; 8.39 (s, 1H).
Point defusion : 215 C
~ 219542~
WO 96/02525 PCT/FR95100975
19
EXÉIIPLE 7 _Difunzarate du N-[4-méthoxy-3-(4-méthylpipérazin-1-yI)phényl]-4-
(3,4-méthylène dioxy)phényi-pipérazin-1-ylamide
N
H
N\--/ NN
O O
7
Le composé Z est préparé suivant la procédure décrite dans l'exemple 3 à
partir
des réactifs suivants triphosg8ne (250 mg, 0.83 mmoI) ; 4-méthoxy-3-(4-méthyl
pipérazin-1-yl) aniline (510 mg, 2.31 mmol) ; triéthylamine (2 x 353 l, 5.10
mmol) ; I-
(3,4-méthylènedioxyphényl)pipérazine (420 mg, 2.04 mmol) ; dioxane (25 ml).
Le brut est purifié par chromatographie-éclair avec un mélange (94/6/1) de
dichlorométhane/méthanol/ammoniaque.
Masse obtenue : 461 mg (Rdt 50 %)
Analyse Elémentaire pour : C24H31N504-2C4H404
Calculées, C, 56.05 ; H, 5.73 ; N, 10.21 ; Expérimentales, C, 55.46 ; H, 5.93
;
N, 9.94
Masse : 454 (MH+), 248, 207, 136
IR (Kbr) : 3406, 2907, 1707, 1638, 1500
RMN 'H (DMSO) : 2.47 (s, 3H) ; 2.84 (M, 4H) ; 3.02 (M, 8H) ; 3.54 (M, 4H) ;
3.72 is, 3H) ; 5.91 (s, 2H) ; 6.38 (dd, 1H) ; 6.58 (s, 4H) ; 6.72-6.83 (m, 3H)
; 7.06-7.12
(m, 2H) ; 8.38 (s, IH).
Point de fusion : 109 C
EXE`IPLE 8_ Fumarate de N-[4-méthoxy-3-(4-méthylpipérazin-1-yl)phényl]-4-(o-
cyano phényl)pipérazin-1-ylamide
2195427 =
WO 96/02525 PCT1FR95/00975
/
o
N vN N O
~ ~ \
8
Une solution de 4-méthoxy-3-(4-méthyl pipérazin-l-yl) aniline (544 mg,
2.46 mmol) et de triéthylamine (374 l, 2.70 mmol) dans le dioxane (5 ml) est
cannulée
5 lentement sur une solution de triphosgène (268 mg, 0.90 mmol) dans le
dioxane (5 ml)
sous atinosphère d'azote. Pendant cette opération, le mélange réactionnel est
refroidi
avec un bain de glace. Il est ensuite ramené à température ambiante pendant 20
mn
avant l'ajout de la 1-(o-cyanophényl) pipérazine (460 mg, 2.46 mmol) et la
triéthylamine
(374 l, 2.70 mmol) diluées dans le dioxane (10 ml). Après 2 h à température
ambiante,
10 le mélange est dilué avec de l'eau puis extrait trois fois avec de
l'acétate d'éthyle. Les
phases organiques sont rassemblées, lavées une fois avec une solution saturée
en
chlorure de sodium, séchées sur sulfate de magnésium et concentrées.
Le brut est purifié par chromatographie-éclair avec un mélange (90/9/1) de
dichlorométhane/méthanol/ammoniaque.
15 Masse obtenue : 665 mg (Rdt 70 %)
Analyse Elémentaire pour : C24H30N602-C4H404
Calculées, C, 61.08 ; H, 6.22 ; N, 15.26 ; Expérimentalés, C, 60.06 ; H, 6.62
;
N, 15.14
Masse : 435 (MH+), 248, 188 -
20 IR (Kbr) : 3570, 3389, 1674, 1595
RMN 1H (DMSO) : 2.35 (s, 3H) ; 2.67 (M, 4H) ; 2.97 (M, 4H) ; 3.14 (M, 4H) ;
3.59 (M, 4H) ; 3.71 (s, 3H) ; 6.54 (s, 2H) ; 6.79 (d, 1H) ; 7.04-7.20 (m, 4H)
; 7.56-7.72
(m, 2H) ; 8.38 (s, 1H).
Point de fusion : 141-143 C
~ 2195427
WO 96/02525 PCT1FR95100975
21
EXEMPLE 9 Difumarate du N-[4-méthoxy-3-(4-méthylpipérazin-1-yl)phénylj-4-
(p-amino phényl)pipérazin-1-ylamide
No
H2 ~
N NN
O
Le composé ¾(392 mg, 0.86 mmol), dissous dans le méthanol (25 ml) est agité
pendant 4 h à température ambiante sous atmosphère d'hydrogène et en présence
d'une
quantité catalytique de Pd/C. Le mélange réactionnel est ensuite filtré sur
célite,
concentré et purifié par chromatographie-éclair avec un mélange (90/9/1) puis
(85/15/1)
de dichlorométhane/méthanol/ammoniaque.
Masse obtenue : 284 mg (Rdt 78 %)
Analyse Elémentaire pour : C23H32N602-2C4H404
Calculées, C, 56.70 ; H, 6.17 ; N, 12.80 ; Expérimentales, C, 56.33 ; H, 6.40
;
N, 12.48
IR (Kbr) : 3418, 1637, 1510
RMrV 1H (DMSO) : 2.48 (s, 3H) ; 2.88 (M, 8H) ; 3.05 (M, 4H) ; 3.54 (M, 4H) ;
3.74 (s, 3H) ; 6.50-7.14 (m, 11H) ; 8.37 (s, IH).
Point de fusion : 125 C
EXEMPLE 10 N-14-méthoxy-3-(4-méthylpipérazin-1-yl)phényl]-4-(p-méthyl-
sulfonylamino phényl) pipérazin-1-ylamide
WO 96/02525 PCT/FR95100975
22
/
~N
O- N~/
0=S
NH O N\_/ NN \
O
Le composé 9 (619 mg, 1.46 mmol) est dissous dans le dichlorométhane
(10 ml) en présence de triéthylamine (300 l, 2.19 mmol) et refroidi à 0 C. Le
chlorure
5 de mésyle (135 l, 0.75 mmol) est additionné puis le mélange réactionnel est
agité 30
minutes à 0 C et 4 h à température ambiante. Après ce temps, du chlorure de
mésyle
(135 l) et de la triéthylamine (300 l, 2.19 mmol) sont rajoutés. Le mélange
réactionnel est encore laissé 30 minutes à température ambiante avant d'être
dilué avec
de l'eau. Les phases sont séparées et la phase aqueuse extraite deux fois avec
du
10 dichlorométhane. Les phases organiques rassemblées sont lavées avec une
solution
saturée en chlorure de sodium, séchées sur sulfate de magnésium, filtrées et
concentrées.
Le brut est purifié par chromatographie-éclair avec un mélange (95/5/1) puis
(90/9/1) de dichlorométhane/méthanol/ammoniaque. Deux composés sont isolés.
Composé le moins polaire : produit de dimésylation ; 166 mg (Rdt 19 %)
Description complète dans l'exemple 11
Composé le plus polaire : produit attendu ; 281 mg (Rdt 38 %)
RMN 1H (DMSO) : 2.23 (s, 3H) ; 2.46 (M, 4H) ; 2.89 (s, 3H) ; 2.95 (M, 4H) ;
3.13 (M, 4H) ; 3.58 (M, 4H) ; 3.74 (s, 3H) ; 6.81 (d, 1H) ; 6.97-7.15 (m, 6H)
; 8.39 (s,
1H) ; 9.29 (s, 1H).
EXEMPLE 11 N-[4-méthoxy-3-(4-méthylpipérazin-1-yl)phényl]-4-[(p-N,N-bis-
(méthyl sulfonyl) amino phényl] pipérazin-1-ylamide
~ ~ .,.
WO 96102525 2195/j27 PCT/FR95/00975
23 Y
~
N
O- N
0=S
O-S~N
O 0
11
Le composé 11 est obtenu comme produit secondaire lors de la préparation du
coniposé IQ. Le fumarate est-préparé selon la méthode décrite précédemment
(dans
l'exemple 1 par exemple).
Analyse Elémentaire pour : C25H36N606S2-0.5C4H404-IH20
Calculées, C, 49.38 ; H, 6.14 ; N, 12.80 ; Expérimentales, C, 48.44 ; H, 6.10
N, 12.06
IR (Kbr) : 3418, 1676, 1601, 1510
RMN 1H (DMSO) : 2.30 (s, 3H) ; 2.58 (M, 4H) ; 2.97 (M, 4H) ; 3.16 (s, 2H,
H20);3.26(M,4H);3.46(s,6H);3.57(M,4H);3.72(3H,s);6.57(s, 1H);6.80(d,
1H) ; 6:98-7.10 (m, 4H) ; 7.30 (d, 2H) ; 8.39 (s, IH).
Point de fusion : 160 C
F.XE`PL.12 Funmarate .de_ la _2-[4-méthoxy-3-(4-méthylpipérazin-1-yl)
phénylamino]-1-(4-o-tolyl pipérazin-1-yl) éthanone
~
ON
O
/-\ N\_/N-~N / \ O
- \
12
llâ : 2-chloro-1-(4-o-tolyl pipérazin-1-yl) éthanone
WO 96/02525 - - PCT/FR95100975
24
Le chlorure de chloroacétyle (4.5 ml, 56.6 rnmol) est ajouté goutte à goutte à
une solution de 1-o-tolylpipérazine (8.31, 47.2 mmol) et de carbonate de
calcium
(14.2 g, 142 mmol) dans la méthyléthylcétone (100 ml) refroidie à 0 C. Le
mélange
réactionnel est agité à cette température pendant 1 h 30 puis il est filtré
sur célite. La
célite est rincée plusieurs fois avec de l'acétate d'éthyle et une solution de
soude 3M. Les
deux phases du filtrat sont ensuite séparées et la phase organique est séchée
sur sulfate
de magnésium, filtrée et concentrée pour donner le produit attendu sous la
forme d'un
solide brun.
Masse obtenue : 9.9 g(itdt 83 %)
IR (Kbr) : 2999, 2945, 1657, 1491, 1433, 1223
RMN 1H (DMSO) : 2.32 (s, 3H) ; 2.93 (m, 4H) ; 3.66 (m, 2H) ; 3.77 (m, 2H) ;
4.11 (s, 2H) ; 6.97-7.25 (m, 4H).
12 Une solution de chlorure 12,t1 (806 mg ; 3.27 mmol) de 4-méthoxy-3-(4-
méthyl-pipérazin-1-yl) aniline (602 mg, 2.72 mmol), de carbonate de sodium
(520 mg,
4.9 mmol) et de pyridine (25 1, 0.3 nnnol) dans le butan-l-ol (50 ml) est
portée au
reflux. Après 6 h, le mélange réactionnel est dilué avec de l'eau et de
l'acétate d'éthyle.
Les phases sont séparées et la phase aqueuse est extraite deux fois avec de
l'acétate
d'éthyle. Les phases organiques sont alors rassemblées, lavées avec une
solution saturée
en chlorure de sodium, séchées sur sulfate de magnésium, filtrées et
concentrées.
Le brut est purifié par chromatographie-éclair avec un mélange (97/3/1) puis
(95/5/1) de dichlorométhane/ méthanol/ammoniaque.
Masse obtenue : 253 mg (Rdt 21 %)
Le fumarate correspondant est préparé par addition de 0.9 équivalents d'acide
fumarique à une solution du composé dans le méthanol chaud. Le méthanol est
évaporé
et l'huile obtenue est cristallisée dans l'éther éthylique.
Analyse Elémentaire pour : C25H35N502-C4H404
Calculées, C, 62.91 ; H, 7.10 ; N, 12.65 ; Expérimentales, C, 61.89 ; H, 7.09
N, 12.26
Masse : 438 (MH+), 222, 177
IR (Kbr) : 3372, 2922, 2816, 1707, 1655, 1612, 1508
W O 96/02525 PCT/FR95100975
~
RMN 1H (DMSO) 2.26 (s, 3H) ; 2.35 (s, 3H) ; 2.64 (M, 4H) ; 2.84 (M, 4H) ;
2.99-(M, 4H) ; 3.65 (M, 7H) ; 3.87 (s, 2H) ; 6.19 (d, 1H) ; 6.31 (s, 1H) ;
6.57 (s, 2H) ;
6.68 (d, 1H) ; 6.96-7.18 (m, 5H).
Point de fu' sion : 179 C (déc.)
5 EXEMPLE 13 Dichlorhydrate de la 2 j3-(4-méthylpipérazin-1-yl)-phénoxy-l-(4-
o-tolyl pipérazin-1-yl) éthanone
/
K-:>
O
/-\ NN~ -- /_\
13
10 .13A : 3-(4-méthyl pipérazin-1-yl)phénol
Le 3-aminophénol (2 g, 18 mmol) est porté au reflux du butan-1-ol (30 ml) en
présence du chlorhydrate de la 2-chloro-N-(chloroéthyl)-N-méthyléthanamine
(3.5 g,
18 mmol) et de carbonate de sodium (95 mg, 9 mmol). Après 48 h, le mélange
réactionnel est concentré et adsorbé sur silice avant d'être purifié par
chromatographie-
15 éclair avec un mélange (90/10) de dichlorométhane/méthanol.
Masse obtenue : 574 mg (Rdt 17 %)
RMN 1 H(DMSO) : 2.28 (s, 3H) ; 2.65 (M, 4H) ; 3.14 (M, 4H) ; 6.20-6.42 (m,
3H) ; 6.96 (dd, 1H) ; 9.22 (M, 1H).
13 : Les composés IZA (529 mg, 2.75 mmol) et UA (1.04 g, 4.13 mmol) sont
20 portés au reflux de la méthyléthylcétone (20 ml) en présence de carbonate
de potassium
(1.26 g, 6.9 mmol) et d'iodure de potassium (55 mg, 0.33 mmol). Après 4 h, le
mélange
réactionnel est dilué avec de l'eau et de l'acétate d'éthyle. Les phases sont
séparées puis
la phase aqueuse est extraite trois fois avec de l'acétate d'éthyle. Les
phases organiques
sont rassemblées, lavées avec une solution saturée en chlorure de sodium,
séchées sur
25 sulfate de magnésium et concentrées. Le brut obtenu est purifié par
chromatographie-
WO 96/02525 !' - PCT/FR95/00975
26 2195427
éclair avec un mélange (97/3/1) puis (95/5/1) de dichlorométhane / méthanol /
ammoniaque.
Masse obtenue : 529 mg (Rdt 47 %)
Le dichlorhydrate est obtenu par ajout d'une solution de chlorure d'hydrogène
dans l'éther éthylique sur une solution du composé dans le dichlorométhane.
Analyse Elémentarre pour : C24H32N402-2HCI-0.6H20
Calculées, C, 58.56 ; H, 7.21 ; N, 11.38 ; CI 14.40 Expérimentales, C, 58.64
H,7.47;N, 11.16; Cl 12.53
Masse : 409 (MH+), 219, 193 -
IR (Kbr) : 3429, 2928, 2698, 1655, 1603
RMNIH(DMSO): 2.32(s,3H);2.79(d,3H);2.88(M,4H)3.13(M,4H);
3.46 (M, 2H) ; 3.67 (M, 4H) ; 3.82 (M, 4H) ; 4.84 (s, 2H) ; 6.45-6.63 (m, 4H)
; 6.99-
7.23 (m, 5H) ; 11.22 (M, 1H).
Point de fusion : 143 C
EXEMPLE 14 2-[4 - chloro - 3 - (4 - méthyl pipérazin - 1 - yl) phénoxy] - 1-[4-
(o- tolylpipérazin - 1 - yl) éthanone
/
K__5-
ï
14
JIA: 4-chloro-3-aminophénol
Une solution de 4-chloro-3-nitrophénol (2.67 g, 15.4 mmol) dans le méthanol
(30 ml) est agitée pendant 24 h sous atmosphère d'hydrogène en présence d'une
quantité
catalytique de Pd/C. Après ce temps, le mélange réactionnel est filtré sur
célite puis
concentré. Il est purifié par chromatographie-éclair avec un mélange (95/5/1)
puis
(90/9/1) de dichlorométhane/méthanol/ammoniaque.
- - - - - - - - - -
WO 96/02525 PCT/FR95/00975
27
Masse obtenue : 1.1 g (Rdt 66 %)
RMN 1H (DMSO) : 5.16 (M, 2H) ; 5.97 (dd, IH) ; 6.24 (d, 1H) ; 6.93 (d, 1H) ;
9.15 (s, 1H).
14R: 4-chloro-3-(4-méthylpipérazin-1-yl)phénol
Le composé ]4R est préparé suivant la même procédure que IIA à partir des
réactifs suivants : 14A (1.1 g, 10.1 mmol) ; chlorhydrate de la 2-chloro-N-(2-
chloroéthyl)-N-méthyléthanamine (1.96 g, 10.1 mmol) ; carbonate de sodium (535
mg,
5.05 mmol) dans le butan-1-ol (40m1).
Le brut obtenu est purifié par chromatographie-éclair avec un mélange (95/5/1)
puis (90/9/1) de dichlorométhane/méthanol/ammoniaque.
Masse obtenue : 1.1 g (Rdt 48 %)
BMN 1H (DMSO) : 2.23 (s, 3H) ; 2.47 (M, 4H) ; 2.93 (M, 4H) ; 6.45 (dd, 1H) ;
6.54 (d, 1H) ; 7.15 (d, 1H) ; 9.53 (M, 1H).
14 Le composé 14 est préparé suivant la même procédure que 'u à partir des
réactifs suivants : composé 12A (660 mg, 2.61 mmol) ; composé j4n (392 mg,
1.73 mmol) ; carbonate de potassium (600 mg, 4.33 mmol) ; iodure de potassium
(30 mg, 0.19 mmol) dans la méthyléthylcétone (20 ml).
Dans ce cas, l'extraction se fait avec le dichlorométhane au lieu de l'acétate
d'éthyle. Le brut est purifié par chromatographie-éclair avec un mélange
(97/3/1) de
dichlorométhane/méthanol/ammoniaque.
Masse obtenue : 424 mg (Rdt = 55 %)
RMN 1H (CDC13) : 2.31 (s, 3H) ; 2.35 (s, 3H) ; 2.62 (M, 4H) ; 2.88 (M, 4H) ;
3.08 (M, 4H) ; 3.71 (M, 4H) ; 4.70 (s, 2H) ; 6.55 (dd, 1H) ; 6.69 (d, 1H) ;
6.93-7.27 (m,
5H).
EXEMPLE 15 4-o-tolylpipérazin-1-yloate de [4-chloro-3-(4-méthylpipérazin-1-yl]
phén-1-yle
WO 96/02525 PCT/FR95100975
28
o
c N N CI 0
MA: 1-chloroformyl-4-o-tolylpipérazine
Une solution de o-tolylpipérazine (1.12 g, 6.4 mmol) et de triéthylamine
5 (885 1, 6.4 mmol) dans le dichlorométhane (20 ml) est cannulée sur une
solution de
triphosgène (700 mg, 2.35 mmol) dans le dichlorométhane à 0 C et sous
atmosphère
d'azote. Le mélange réactionnel est ramené à température ambiante et agité
pendant 2 h
avant d'être dilué avec de l'eau. Les phases sont séparées et la phase aqueuse
extraite
deux fois avec du dichlorométhane. Les phases organiques réunies sont lavées
avec une
10 solution saturée en chlorure de sodium, séchées sur sulfate de magnésium,
filtrées et
concentrées.
Le brut est purifié par chromatographie-éclair avec un mélange (96/4) d'éther
de pétrole/acétate d'éthyle.
Masse obtenue : 738 mg (Rdt : 48 %)
15 IR üzlm) : 3019, 2920, 2818, 1736
RMN 1H (CDC13) : 2.36 (s, 3H) ; 2.95 (M, 4H) ; 3.87 (M, 4H) ; 6.66-7.61 (m,
4H)
15 : Une solution de 1.4B (190 mg, 0.84 mmol) dans le tétrahydrofurane (5 ml)
est cannulée sur une suspension d'hydrure de sodium (66 %, 37 mg, 0.92 mmol)
dans le
tétrahydrofurane (5 mI) à 0 C et sous atmosphère d'azote. Après 15 mn, une
solution de
,l. à dans le tétrahydrofurane (10 ml) est cannulée sur le mélange
réactionnel. Celui-ci
est ramené à température ambiante et laissé 2 h sous agitation. Après ce
temps; il est
dilué avec de l'eau et extrait trois fois avec de l'acétate d'éthyle. Les
phases organiques
rassemblées sont alors lavées avec une solution saturée en chlorure de sodium,
séchées
sur sulfate de magnésium, filtrées et concentrées.
219a ~ ~2.7
_.
R'O 96/02525 PCT/FR95/00975
29
Le brut est purifié par chromatographie-éclair avec un mélange (95/5/1) de
dichlorométhane/méthanol/ammoniaque.
Masse obtenue : 328 mg (Rdt : 91 %)
RMN 1H (CDC13) : 2.33 (s, 3H) ; 235 (s, 3H) ; 2.60 (M, 4H) ; 295 (M, 4H) ;
3.09 (M, 4H) ; 3.75 (M, 41-i) ; 6.73-6.92 (m, 2H) ; 6.99-7.05 (m, 2H) ; 7.15-
7.34 (m,
3H).
.XF.MP . .16 2 - [8 - (4 - méthylpipérazin - 1 - yl) naphtalèn - 2 - yloxyl -
1 - (4-
o-tolylpipérazin - 1 - yl) - éthanone
N
O û
/-~ N \--j N_I\', O
16
1-6A : 8-(4-méthyl pipérazin-1-yl)naphtalèn-2-ol
Le composé 1fi est préparé suivant la même procédure que L2â à partir des
réactifs suivants : 8-aminonaphtalèn-2-ol (1.6 g, 10 mmol), chlorohydrate de
la 2-
chloro-N-(2-chloroéthyl)-N-méthyléthananine (2 g, 10.4 mmol), carbonate de
sodium
(0.54 g, 5 mmol), butan-1-ol (20 ml).
Le brut réactionnel est purifié par chromatographie-éclair avec un mélange
(95/4/1) puis (90/9/1) de dichlorométhane/méthanol/ammoniaque.
Masse obtenue : 1.17 g (Rdt 48 %)
IR (KBr) :3200, 3600, 3073,2946, 2812, 1622, 1597, 1448
RMN IH (DMSO) : 2.28 (s, 3H) ; 2.58 (M, 4H) ; 2.96 (M, 4H) ; 6.98-7.72 (m,
6H) ; 9,66 (s, 1H).
M : Le composé M est préparé suivant la même procédure que décrite
précédemment pour la préparation de l'exemple 1â à partir des intermédiaires
12A
WO 96/02525 219 5 4 27 PCT/FR95/00975
(640 mg, 2.54 mmol), 1fi (512 mg ; 2.11 mmol), du carbonate de potassium (35
mg,
0.21 mmol) dans la méthyléthylcétone (60 ml).
Le brut réactionnel est purifié par chromatographie-éclair avec un mélange
(90/9/1) de dichlorométhane/méthanol/ammoniaque.
5 Masse obtenue : 694 mg (Rdt : 72 %)
Analyse élémentaire pour C28H34N402
Calculées : C, 73,33 ; H, 7,47 ; N, 12,22 ; Expérimentales : C, 72,83 ; H,
7,44 ;
N, 12,07
IR (KBr) : 2938, 2835, 2787, 1668, 1657, 1448
10 RMN 1H (CDC13) : 2.29 (s, 3H) ; 2.46 (s, 3H) ; 2.88 (M, 8H) ; 3.13 (M, 4H)
3.75 (M, 4H) ; 4.87 (s, 2H) ; 6.89-7.77 (m, 10H).
FXEM PL.E 17 2-[8-(4-méthylpipérazin-1-yl)-naphtalèn-2-yloxy]-1-[4-(2-
cyanophényl)-pipérazin-1-yl] éthanone
CND
O
N \-J N - 0 N
15 17
1_7A: 2-chloro-I-[4-(2-cyanophényl)pipérazin-1-yl]éthanone
Le composé 12à est préparé suivant la même procédure que ]M à partir
des réactifs suivants : chlorure de chloroacétyle (670 I, 8.4 mmol), 1-(2-
20 cyanophényl)pipérazine (1.32 g, 7 mmol), carbonate de calcium (2.1 g, 21
mmol),
méth~,léthylcétone (30 ml).
Masse obtenue : 1.8 g (Rdt quantitatif)
RMN IH (DMSO) : 3.14 (M, 4H) ; 3.64 (M, 4H) ; 4.44 (s,s 2H) ; 7.09-7.27 (m,
2H) ; 7.57-7.74 (m, 2H).
~ 2195427
W 0 96102525 PCT/FR95/00975
31
le composé 11 est préparé suivant la procédure décrite précédemment pour
la préparation de 12 à partir des intermédiaires suivants :17A (1.04 g, 3.95
mmol), 16A
(1.01 g, 4.17 mmol), carbonate de potassium (1.36 g, 9.88 mmol), iodure de
potassium
(68 mg, 0,41 mmol), dans la méthyléthylcétone (50 ml).
Le brut réactionnel est purifié par chromatographie-éclair avec un gradient de
(97/3/1) jusqu'à (90/9/1) de dichlorométhane/ méthanol/ ammoniaque.
Masse obtenue : 925 mg (Rdt 50 %)
RMN I H(CDC13) : 2.45 (s, 3H) ; 2.76 (M, 4H) ; 3.21 (Ivl, 8H) ; 3.87 (M, 4H) ;
4.90 (s, 2H) ; 6.95-7.79 (m, 10H).
EXEMPLE 18 2-[8-(4-méthylpipérazin-1-yl)-naphtalèn-2-yloxy]-1-14-(4-
nitrophényl)-pipérazin-1-yl]éthanone.
O N+ ,-, NN ~ N
O
O / /
18
j$A : 2-chloro-l-[4-(4-nitrophényl)pipérazin-1-yl]éthanone.
Le composé 1$A est préparé suivant la même procédure que IZA à partir des
réactifs suivants : chlorure de chloroacétyle (4.2 ml, 53 mmol), 4-
nitrophénylpipérazine
(9.18 g, 44.3 mmol), carbonate de calcium (13.3 g, 133 mmol),
méthyléthylcétone
(100 ml).
Dans ce cas, le brut est purifié par chromatographie-éclair avec un mélange
(97 3) puis (95/5) de dichlorométhane/méthanol.
Fraction la moins polaire : 3.8 g (Rdt 30 %)
RMN 1H (DMSO) : 3.35-3.62 (M, 8H) ; 4.46 (s, 2H) ; 7.03 (d, 2H) ; 8.07 (d,
2H).
Fraction la plus polaire : 2.12 g
2195~27 ~
WO 96/02525 ,; PCT/FR95100975
32 -
Produit de double alkylation (10) 1,2-bis-(4-(4-nitrophényl)pipérazin-1-yl]
éthanone.
Analyse Elémentaire pour C22H26N605
Calculées : C, 58.14 ; H, 5.77 ; N, 18.49 ; Expérimentales : C, 57.94 ; H,
5.72 ;
N, 18.04
Masse : 455 (MH+)
IR (KBr) : 2837, 1647, 1597, 1323
RMN IH (CDC13) :2.71 (m, 4H) ; 3.33 (s, 2H) ; 3.46 (M, 8H) ; 3.83 (M, 4H) ;
6.82 (dd, 4H) ; 8.13 (m, 4H).
1$ : le composé 18 est préparé suivant la procédure décrite précédemment pour
la préparation 12 à partir des intermédiaires jIA (1.75 g, 6 mmol) et 1.6à
(1.21 g,
5 mmol), de carbonate de potassium (1.7 g, 12.5 mmol) et d'iodure de potassium
(83 mg, 0.5 mmol) dans la méthyléthylcétone (150 ml).
Le brut réactionnel est adsorbé sur silice puis purifié par chromatographie-
éclair avec un mélange (97/3/1) de dichlorométhane/méthanoUammoniaque.
Masse obtenue : 111 mg (Rdt 4 %)
RMN IH (CDC13) : 2.43 (s, 3H) ; 2.76 (M, 4H) ; 3.12 (M, 4H) ; 3.45 (M, 4H) ;
3.84 (m, 4H) ; 4.90 (s, 2H) ; 6.80 (d, 2H) ; 7.11-7.78 (m, 6H) ; 8.13 (d, 2H).
EXENZPLE 19 4 - phénylpipérazin - 1 - yloate de 8 - (4 - méthyl pipérazin - 1-
yl)
naphtalèn - 2 - yle
ô
N
OCç55
19
19A : 1-chloroformyl-4-phényl pipérazine
W O 96102525 PCT/FR95100975
33
Une solution de 1-phénylpipérazine (237 l, 1.55 mmol) et de triéthylamine
(215 l, 1.55 mmol) dans le tétrahydrofurane (10 ml) est ajoutée à une
solution de
triphosgène (155 mg, 0.52 mmol) dans le tétrahydrofurane (10 ml) à 0 C et sous
atmosphère d'azote.
Le mélange réactionnel est ramené à température ambiante. Après 30 minutes,
il est dilué avec de l'eau et de l'acétate d'éthyle. Les phases sont séparées
et la phase
aqueuse est extrâite trois fois avec de l'acétate d'éthyle. Les phases
organiques sont alors
rassemblées, lavées avec une solution saturée en chlorure de sodium, séchées
sur sulfate
de magnésium, filtrées et concentrées.
Le brut réactionnel est purifié par chromatographie-éclair avec un mélange
(97/3/1) de dichlorométhane/méthanollammoniaque.
Masse obtenue : 336 mg (Rdt 96 %)
Analyse Elémentaire pour C 11 H 14C1N2O
Calculées : C, 58.80 ; H, 5.83 ; N, 12.47 ; Expérimentales : C, 58.90 ; H,
5.91 ;
N, 12.30
Masse : 225 (MH+), 189, 163
IR (KBr) : 2826, 1728, 1599, 1496, 1410
RMN 1H (CDC13) : 3.23 (M, 4H) ; 3.84 (m, 4H) ; 6.92-6.99 (m, 3H) ; 7.27-
7.37 (m, 2H).
1Q : Une sôlution de 1{A (205 mg ; 0.85 mmol) dans le tétrahydrofurane (5 ml)
est cannulée sur une suspension d'hydrure de sodium (60 %, 37 mg, 0.92 mmol)
dans le
tétrahydrofurane (5 ml) à 0 C et sous atmosphère d'azote. Après 15 minutes, le
mélange
réactionnel est cannulé sur une solution de 12A (190 mg, 0.85 mmol) dans le
tétrahvdrofurance (5 ml) puis il est ramené à température ambiante et agité 15
niinutes.
La solution est alors diluée avec de l'eau puis extraite trois fois avec de
l'acétate d'éthyle.
Les phases organiques sont rassemblées, lavées avec une solution saturée en
chlorure de
sodium, séchées sur sulfate de sodium, filtrées, concentrées. Le brut est
purifié par
chromatographie-éclair avec un mélange (95/5/1) de dichlorométhane / méthanol
/
ammoniaque.
_Masse obtenue : 301 mg (Rdt 70 %)
WO 96/02525 PCT/FR95/00975
34
Le finnarate est préparé par addition de 0.9 équivalent d'acide fumarique sur
une solution de la base dans le méthanol. Il est ensuite cristallisé dans
l'èther éthylique.
Analyse Elémentaire pour C26H30N402-1.3xC4H404
Calculées : C, 62.51 ; H, 6.26 ; N, 9.35 ; Expérimentales : C, 64.95 ; H, 6.06
N, 9.72
Masse : 431 (MH+), 243, 191, 163, 136
IR(KBr) : 3439, 1717, 1597, 1414, 1225
RMN 1H (DMSO) : 2.39 (s, 3H) ; 2.78 (M, 4H) ; 3.05 (M, 4H) ; 3.24 (M,4H)
3.62 (M, 2H) ; 3.79 (M, 2H) ; 6.58 (s, 2H) ; 6.79.7.94 (m, 11H).
Point de fusion : 206 C
EXEMPLE 20 4-o-totylpipérazin - 1 - yloate de 8-(4 - méthyl pipérazin - 1-y1)
naphtalèn - 2 - yle.
CND
N
N/-\N O / /
- v,-~ ~ ~ ~
15 Le composé 1,.Q est préparé suivant la procédure décrite précédemment pour
la
préparation de 15 à partir de Mà (430 mg, 1.8 mmol), 16à (436 mg, 1.8 mmol),
d'hydrure de sodium (60 %, 80 mg, 2 mmol), dans le tétrahydrofurane (30 ml).
Le brut réactionnel est purifié par chromatographie-ëclair avec un mélange
(95/5/1) de dichlorométhane/méthanol/ammoniaque.
20 Masse obtenue : 664 mg (Rdt 83 %)
RMV 1H (CDC13) : 2.36 (s, 3H) ; 2.42 (s, 3H) ; 2.72 (M, 4H) ; 2.98 (M, 4H) ;
3.14 (M,
4H) ; 3.80 (M, 4H) ; 7.00-7.41 (m, 7H) ; 7.55 (d, IH) ; 7.82 (d, 1H)
7.89 (d, IH).
~ _ ..
WO 96/02525 219 5~~~~ PCT/FiL95/00975
EXEMPLE 21 Fumarate du N-[4-méthoxy-3-(4-méthylpipérazin-1-y1)phényl]-4-
phényIpipérazin-1-ylamide
. ~~
r_\
N N O
5 2.~
Le composé 21 est préparé suivant la procédure décrite dans l'exemple 3 à
partir des réactifs suivants : triphosgène (200 mg, 0.68 mmol) ; 4-méthoxy-3-
(4-
méthylpipérazin-1-yl)aniline (447 mg, 2.02 mmol) ; triéthylamine (820 l, 5.90
mmol) ;
10 1-phénylpipéraiine (310 ml, 2.02 mmol) ; tétrahydrofurane (15 ml).
Le brut est purifié par chromatographie-éclair avec un gradient de (98/2/1) à
(90/9,11) de dichlorométhane/méthanol/ammoniaque.
Masse obtenue : 92 mg (Rdt 12 %)
Ce composé est dissous dans le méthanol et traité avec de l'acide fumarique
15 pour donner le fumarate correspondant. Celui-ci est cristallisé dans
l'éther.
Analyse Elémentaire pour: C23H31N502-C4H404-H20
Calculées, C, 59.66-; H, 6.86 ; N; 12.88 ; Expérimentales, C, 59.62 ; H, 6.79
N, 12.89
Masse (DCI/NH3) : 410 (MH+), 248, 163
20 IR (Kbr) : 3395,2955,2835,1707,1637,1599,1500.
RMN'H. (DMSO) : 2.35 (s, 3H) ; 2.65 (M, 411) ; 2.99 (M, 4H) ; 3.14 (M, 4H) ;
3.57 (M, 4H) ; 3.72 (s, 3H) ; 6.58 (s, 2H) ; 6.79-7.27 (m, 8H) ; 8.39 (s,1H).
Point de fusiôn : 112 C
EXEMPLE 22- _Fumarate- du Nr[4-méthoxy-3-(4-méthylpipérazin-1-yl)phényl]-4-
25 (1-naphtyl)pipérazin-1-ylamide
;2~.95427 =
WO 96/02525 PCT/FR95/00975
36
-/
/-t
N~~
O
22
Une solution de 4-méthoxy-3-(4-méthylpipérazin-l-yl)aniline (520 mg,
2.35 mmol) et de triéthylarnine (325 l, 2.35 mmol) dans le dichlorométhane
(10 ml) est
cannulée lentement sur une solution de triphosgène (235 mg, 0.78 mmol) dans le
dichlorométhane (10 ml) sous atmosphère d'azote. Pendant cette opération, le
mélange
réactionnel est refroidi avec un bain de glace. Il est ensuite ramené à
température
ambiante pendant 20 mn avant d'être cannulé sur une suspension de 1-(I-
naphtyl)pipérazine (500 mg, 2.35 mmol) et triéthylamine (235 l, 2.35 mmol)
dans le
dichlorométhane (10 ml). Après 2 h à température ambiante, le mélange est
dilué avec
de l'eau puis extrait trois fois avec de l'acétate d'éthyle. Les phases
organiques sont
rassemblées, lavées une fois avec une solution saturée en chlorure de sodium,
séchées
sur sulfate de magnésium et concentrées.
Le brut est purifié par chromatographie-éclair avec un mélange (95/5/1) puis
(90/911) de dichlorométhane/méthanol/ammoniaque.
Masse obtenue : 910 mg (Rdt 84 %)
Ce composé est dissous dans le méthanol et traité avec de l'acide fumarique
pour donner le fumarate correspondant. Celui-ci est cristallisé dans l'éther.
Analyse Elémentaire pour : C27H33N502-C4H404-0.6H20
Calculées, C, 63.49 ; H, 6.56 ; N, 11.94 ; Expérimentales, C, 63.16 ; H, 6.65
N, 11.58 Masse (DCI/NH3) : 460 (MH+), 248, 213.
IR (Kbr) : 3375,1686,1608,1500.
WO 96/02525 2195427 PCT/FR95/00975
37
RMN 1H (DMSO) : 2.37 (s, 3H) ; 2.68 (M, 4H) ; 3.03 (M, 8H) ; 3.75 (M,
7M) ; 6.59 (s, 2H) ; 6.82 (d, 8.7Hz,1H) ; 6.85-7.19 (m, 3H) ; 7.41-7.66 (m,
4H) ; 7.91
(m, 1 H) ; 8.20 (m, 1 H) ; 8.43 (s, 1 H).
Point de fusion : 130 C
EXEMPLE 23 Fumarate du N-[4-méthoxy-3-(4-méthylpipérazin-1-yl)phényl]-4-
(pyridin-4-yl)pipérazin-1-ylamide
N
- --
N // N\_1N H O
O
22
Une solution de 4-méthoxy-3-(4-méthylpipérazin-1-yl)aniline (546 mg,
2_47 mmol) et de triéthylamine (345 l, 2.47 mmol) dans le dichlorométhane (10
ml) est
cannulée lentement sur une solution de triphosgène (245 mg, 0.82 mmol) dans le
dichlorométhane (20 ml) sous atmosphère d'azote. Pendant cette opération, le
mélange
réactionnel est refroidi avec un bain de glace. Il est ensuite ramené à
température
ambiante pendant 20 mn avant l'ajout de la 1-(pyridin-4-yl)pipérazine (403 mg,
2.47 mmol) et la triéthylamine (345 l, 2.47 mmol) diluées dans le
dichlorométhane
(10 ml). Après 2 h à température ambiante, le mélange est dilué avec de l'eau
et la phase
organique est lavée avec une solution saturée en chlorure de sodium, séchée
sur sulfate
de magnésium et concentrée.
Le brut est purifié par chromatographie-éclair avec un mélange (90/9/1) de
dichlorométhane/méthanol/ammoniaque.
Masse obtenue : 762 mg (Rdt 77 %)
Ce composé est dissous dans le méthanol et traité avec de l'acide fumarique
pour donner le fumarate correspondant. Celui-ci est cristallisé dans l'éther.
219542~
WO 96/02525 PCT/FR95/00975
38
Analyse Elémentaire pour : C22H30N602-C4H404-H20
Calculées, C, 57.82 ; H, 7.63 ; N, 15.56 ; Expérimentales, C, 57.32 ; H, 7.06
N, 15.30
Masse (DCI/NH3) : 411 (MH+), 248, 222, 164.
IR (Kbr) : 3306,1664,1641,1535,1514,1348,1250.
RMN 1H (DMSO) : 2.33 (s, 3H) ; 2.63 (M, 4H) ; 2.98 (M, 4H) ; 3.43 (M, 4H) ;
3.58 (M, 4H) ; 3.73 (s, 3H) ; 6.57 (s, 2H) ; 6.79-7.12 (m, 5H) ; 8.20 (d,
6.2Hz, 2H)
8.72 (s, 1 H).
Point de fusion : 199 C
EXEMPLE 24 Fumarate du N-[4-méthoxy-3-(4-méthylpipérazin-1-y1)phényl]-4-
(pyridin-2-yl)pipérazin-1-ylamide
O
N
C N NNO
H
24
Le composé Z4 est préparé suivant la procédure décrite dans l'exemple 23 à
partir des réactifs suivants : triphosgène (210 mg, 0.71 mmol) ; 4-méthoxy-3-
(4-
méthylpipérazin-l-yl)aniline (481 mg, 2.17 nunol) ; triéthylamine (2 x 300
jil,
4.34 mmol) ; 1-(pyridin-2-yl)pipérazine (330 ml, 2.17 mmol) ; dichlorométhane
(40 ml).
Le brut est purifié par chromatographie-éclair avec un mélange (95/5/1) puis
(90/911) de dichlorométhane/méthanol/ammoniaque.
Masse obtenue : 743 mg (Rdt 83 %)
Ce composé est dissous dans le méthanol et traité avec de l'acide fumarique
pour donner le fumarate correspondant. Celui-ci est cristallisé dans l'éther.
Analyse Elémentaire pour : C25H35N502-C4H404-0.37H2O _
,~P=.
WO 96/02525 2195427 PCT/FR95/00975
39
Calculées, C, 58.45 ; H, 6.74 ; N, 15.73 ; Expérimentales, C, 58.58 ; H, 6.82;
N, 15.24
Masse (DCI/NH3) : 411 (MH+),248,164.
IR (Kbr) : 3366,2837,1707,1639,1595;1510.
RMN 1H (DMSO) : 2.36 (s, 3H) ; 2.67 (M, 4H) ; 2.99 (M, 4H) ; 3.52 (M,
8H) ; 3.73 (s, 3H) ; 6.58 (s,2H) ; 6.67 (dd, 5.0 et 6.9 Hz, I H) ; 6.83 (m,
2H) ; 7.07 (m,
,
2H) ; 7.56 (m, IH) ; 8.12 (m, 1H) ; 8.39 (s,1H).
Point de fusion : 110 C
F=X NiP ,. 5 Fumarate du N-[4-méthoxy-3-(4-méthylpipérazin-1-yl)phényl]-4-
(2,6-diméthylphényl)pipérazin-1-ylamide
\NJ
f -~ N~-%\ ~N \
O
Le composé M est préparé suivant la procédure décrite dans l'exemple 22 à
partir des réactifs suivants : triphosgène (235 mg, 0.79 mmol) ; 4-méthoxy-3-
(4-
méth~IIpipérazin-1-yl)aniline (525 mg, 2.38 mmol) ; triéthylamine (2 x 367 1,
5.24 mmol) ; 1-(2,6-diméthylphényl)pipérazine (453 mg, 2.38 mmol) ;
dichlorométhane
(40 ml).
Le brut est pnrifié par chromatographie-éclair avec un mélange (95/5/1) puis
(90~9'I) de dichlorométhane/méthanol/ammoniaque.
Masse obtenue : 656 mg (Rdt 63 %)
Ce composé est dissous dans le méthanol et traité avec de l'acide fumarique
pour donner le fumarate correspondant. Celui-ci est cristallisé dans l'éther.
Analyse Elémentaire pour : C25H35N502-C4H404
2~95~427.::,;
WO 96/02525 PCT/FR95/00975
Calculées, C, 62 91 ; H, 7.10 ; N, 12.65 ; Expérimentales, C, 62.74 ; H, 7.31
N, 12.86
Masse : (DCI/NH3) : 438 (MH+), 294,248,191.
IR (Kbr) : 3366,2949,2829,1701,1637,1585,1510.
5 RMN 1H (DMSO) : 2.26 (s, 6H) ; 2.31 (s, 3H) ; 2.61 (M, 4H) ; 2.98 (M, 8H)
3.50 (M, 4H) ; 3.71 (s, 3H) ; 6.55 (s, 2H) ; 6.78 (d, 8.6Hz, 1H) ; 6.91-7.10
(m, 5H)
8.31 (s, 1H).
Point de fusion : 172 C
EXEMPLE 26 Fumarâte du N-[4-méthoxy-3-(4-méthylpipérazin-1-yl)phényl]-4-
10 (5,6,7,8-tétrahydronaphtalèn-1-yl)pipérazin-1-ylamide
d
_\
N NYH --~ / ~
ZS
15 2_~à: 1-(5,6,7,8-tétrahydronaphtalèn-1-yl)pipérazine
La 5,6,7,8-tétrahydronaphtalèn-1-ylamine (1.4 ml, 15 mmol) est portée au
reflux du butan-1-ol (50 ml) en présence du chlorhydrate de la bis(2-
chloroéthyl)amine
(2.68 g, 15 mmol) et de carbonate de sodium (800 mg, 7.5 mmol). Après 48 h, le
mélange réactionnel est concentré et imprégné sur silice avant d'être purifié
par
20 chromatographie-éclair avec un mélange (95/5/1) puis (90/9/1) de
dichlorométhane /
méthanol / ammoniaque.
Masse obtenue : 745 mg (Rdt 22 %)
RMN 1H (DMSO) : 1.68 (t, 3.2 Hz, 4H) ; 2.67 (t, 6.6 Hz, 4H) ; 2.81 (t, 4.5 Hz,
4H) ; 2.97 (t, 3.7 Hz, 4H) ; 5.33 (se, 2H) ; 6.83 (m, 2H) ; 7.04 (m,1H).
~ 2195427
WO 96/02525 PCT/FTt95/00975
. t : ~ i ..
41
le composé 2¾ est préparé suivant la procédure décrite dans l'exemple 23 à
partir des réactifs suivants : triphosgène (181 mg, 0.61 mmol) ; 4-méthoxy-3-
(4-
méthylpipérazin-I-yl)aniline (405 mg, 1.83 mmol) ; triéthylamine (2 x 253 l,
3.66 mmol) ; 1-(5,6,7,8-tétrahydronaphtalèn-1-yl)pipérazine (j,fià) (418 mg,
1.83 mmol) ; dichlorométhane (40 ml).
Le brut est purifié par chromatographie-éclair avec un mélange (95/5/1) de
dichlorométhane/méthanol/ammoniaque.
Masse obtenue : 799 mg (Rdt 94 %)
Ce composé est dissous dans le méthanol et traité avec de l'acide fumarique
pour donner le fumarate correspondant. Celui-ci est cristallisé dans l'éther.
Analyse Elémentaire pour : C27H37N502-C4H404-0.4H20
Calculées, C, 63.44 ; H, 7.18 ; N, 11.93 ; Expérimentales, C, 63.54 ; H, 7.22
N, 12.13
Masse (DCI/NH3) : 464(MH+), 294, 248, 217.
IR (Kbr) : 3414, 2934, 2837, 1637, 1604, 1508.
itMN 1H (DMSO) : 1.71 (M, 4H) ; 2.35 (s, 3H) ; 2.72 (M, 12H) ; 2.99 (M,
4H) ; 3.56 (M, 4H) ; 3.73 (s, 3H) ; 6.58 (s, 2H) ; 6.83 (m, 3H) ; 7.06 (m, 3H)
; 8.36 (s,
1H).
Point de fusion : 95 C
EXEMPLE 27 Fumarate du N-[4-méthoxy-3-(4-méthylpipérazin-1-yl)phényl]-4-
(1,2,3,4,5,6-h examéthylphényl)pipérazin-1-ylamide
/
0
H
2'I
WO 96/02525 2195 42 7 PCT/FR95/00975
42
Le composé 21 est-préparé suivant la procédure utilisée pour le composé 23 à
partir de la 4-méthoxy-3-(4-méthylpipérazin-l-yl)aniline et de la 1-
(1,2,3,4,5,6-
hexaméthylphényl)pipérazine.
FX_F._14Pi.E 28 Fumarate du N-[4-méthoxy-3-(4-méthylpipérazin-1-y1)phényl]-4-
(2,4-diméthylphényl)pipérazin-1-ylamide
N
N
O
2$
Le composé M est préparé suivant la procédure décrite dans l'exemple 22 à
partir des réactifs suivants : triphosgène (226 mg, 0.76 mmol) ; 4-méthoxy-3-
(4-
méth}.lpipérazin-l-yl)aniline (504 mg, 2.28 mmol) ; triéthylamine (2 x 315 l,
4.56 mmol) ; 1-(2,4-diméthylphényl)pipérazine j433 mg, 2.28 mmol) ;
dichlorométhane
(40 ml).
Le brut est purifié par chromatographie-éclair avec un mélange (95/5/1) de
dichlorométhane/méthanol/ammoniaque.
Masse obtenue : 230 mg (Rdt 23 %)
Ce composé est dissous dans le méthanol et traité avec de l'acide fumarique
pour donner le fumarate correspondant. Celui-ci est cristallisé dans l'éther.
Analyse Elémentaire pour : C251-135N502-C4H404-0.I 8H20
Calculées, C, 62.55 ; H, 7.12 ; N, 12.58 ; Expérimentales, C, 62.22 ; H, 7.18
;
N, 13.35
Masse (DCI/NH3) : 438 (MH+), 248, 191.
IR (Kbr) : 3339, 2945, 2847, 2810, 1662, 1606, 1510, 1246.
~ 2195427
WO 96/02525
- =.' =' :, :,. - . PCTYF7t95/00975
43
RIvIIV 1H (DIVISO) : 2.20 (s, 3H) ; 2.23 (s, 3H) ; 2.33 (s, 3H) ; 2.63 (M, 4H)
;
2.77 (M, 4H) ; 2.97 (M, 4H) ; 3.54 (M, 4H) ; 3.71 (s, 3H) ; 6.56 (s, 2H) ;
6.79 (d, 8.7Hz,
IH) ; 6.92-7.10 (m, 5H) ; 8.34 (s, IH).
Point de fusion : 200 C
EXEMPLE 29 Fumarate du N-[4-méthoxy-3-(4-méthylpipérazin-1-y1)phényl]-4-
1 (3,4-diméthoxyphényl)pipérazin-1-ylamide
N
-O O- N
~- -a i o
N- - \
~ ~ N ~/ ~
O
Z2
2M: 1-(2,3-diméthoxyphényl)pipérazine :
Une solution de Butyllithium (26 ml, 1.6 M dans l'hexane, 42 mmol) est
ajoutée goutte à goutte sur une solution de pipérazine (3.446 g, 40 mmol) dans
le
tétrahvdrofurane (50 ml) maintenue à 0 C. Le mélange réactionnel est ensuite
ramené à
température ambiante et agité pendant 2 h . Une solution de 1,2,3-
triméthoxybenzène
(6.72 g, 40 mmol) dans le tétrahydrofurane (20 ml) est ensuite additionnée
puis le
mélange est porté à reflux pendant 12 h. Après avoir été refroidi à
température
ambiante, il est finalement versé sur une solution IN d'acide chlorhydrique et
extrait
trois fois avec de l'acétate d'éthyle. La phase aqueuse est ensuite basifiée
avec une
solution JN d'hydroxyde de soude et extraite trois fois avec de l'acétate
d'éthyle. Ces
dernières phases organiques sont rassemblées, lavées une fois avec une
solution saturée
en cblorure de sodium, séchées sur sulfate de magnésium et concentrées. La
purification
finale se fait par chromatographie éclair avec un mélange (90/9/1) de
dichlorométhane /
méthanol / ammoniaque.
Masse obtenue : 1.63 g (Rdt 18 %)
f ) 0
WO 96f02525 21954b PCT/FR95/00975
44
RMN 1H (CDC13) : 2.02 (s, 1H) ; 3.03 (M, 8H) ; 3.82 (s, 3H) ; 3.83 (s, 3H)
6.57 (m, 2H) ; 6.94 (t, 4.0 Hz, 1H).
22 : Le composé 22 est préparé suivant la procédure décrite dans l'exemple
à partir des réactifs suivants : triphosgène (240 mg, 0.81 mmol) ; 4-méthoxy-3-
(4-
méthylpipérazin-1-yl)aniline (534 mg, 2.41 mmol) ; triéthylamine (2 x 333 l,
4.82 mmol) ; 1-(2,3-diméthoxyphényl)pipérazine (2M) (538 mg, 2.41 mmol)
dichlorométhane (40 ml).
Le brut est purifié par chromatographie-éclair avec un mélange (92/8/1) de
dichlorométhane/méthanol/ammoniaque.
Masse obtenue : 879 mg (Rdt 78 %) -
Ce composé est dissous dans le méthanol et traité avec de l'acide fumarique
pour donner le fumarate correspondant. Celui-ci est cristallisé dans l'éther.
Analyse Elémentaire pour : C25H35N504-C4H404
Calculées, C, 59.48 ; H, 6.71 ; N, 11.96 ; Expérimentales, C, 59.49 ; H, 6.85
N, 12.12
Masse (DCI/NH3) : 470 (MH+), 248, 223. - -
IR (Kbr) : 3354, 2932, 2849, 1672, 1510, 1234.
RMN 1H (DMSO) : 2.32 (s, 3H) ; 2.63 (Ivi, 4H) ; 2.96 (M, 8H) ; 3.54 (M, 4H) ;
3.70 (s, 6H) ; 3.73 (s, 3H) ; 6.54 (s, 2H) ; 6.57 (d, 6.6Hz, 1H), 6.66-7.09
(m, 5H) ; 8.33
(s, IH).
Point de fusion : 184 C
EXEMPLE 30 Difumarate du N-[4-méthoay-3-(4-méthylpipérazin-1-yl)phényl]-
4-(benzodioxan-5-yl)pipérazin-1-ylamide
N~
O
QrTT6 _
o
~O
~!!
~ 2195427:
WO 96/02525 PCTIFR95100975
Le composé 3Q est préparé suivant la procédure décrite dans l'exemple 7â à
partir des réactifs suivants : triphosgène (164 mg, 0.55 mmol) ; 4-méthoxy-3-
(4-
méthylpipérazin-1-yl)aniline (370 mg, 1.66 mmol) ; triéthylamine (2 x 230 i,
3.32 mmol) ; 1-(benzodioxan-5-yl)pipérazine (EP-0 574 313) (366 mg, 1.66 mmol)
5 dichlorométhane (40 ml).
Le brut est purifié par chromatographie-éclair avec un mélange (92/8/1) puis
(90/9/1) de dichlorométhane/méthanol/ammoniaque.
Masse obtenue : 613 mg (Rdt 79 %)
Ce composé est dissous dans le méthanol et traité avec de l'acide fumarique
10 pour donner le fumarate correspondant. Celui-ci est cristallisé dans
l'éther.
Analyse Elémentaire pour : C25H33N504-C4H404-0.6H20
Calculées, C, 58.60 ; H, 6.48 ; N, 11.78 ; Expérimentales, C, 58.82 ; H, 6.72
;
N, 11.29
Masse (DCI/NH3) : 468 (MH+), 248, 221, 108.
15 IR (Kbr) : 3414, 1637, 1599, 1510 .
RMN 1H (DMSO) : 2.36 (s, 3H) ; 2.67 (M, 4H) ; 2.97 (M, 8H) ; 3.54 (M, 4H) ;
3.73 (s, 3H) ; 4.24 (M, 4H) ; 6.48-6.58 (m, 4H) ; 6.69-7.11 (ru, 2H); 7.06-
7.11 (m, 2H) ;
8.35 (s, IH).
Point de fusion : 120-121 C
20 EXEMPLE 31 Fumarate du N-[4-méthoxy-3-(4-méthylpipérazin-1-yl)phényl]-4-
(2,4,6-triméthylphényl)pipérazin-1-ylamide
~
'N J
, -- - -
\ - -: . .
N /-- N H
O
25 LA: 1-(2,4,6-triméthylphényl)pipérazine
21954?7 , ,, . =
WO 96102525 PCT/FR95/00975
46
Le composé UA est préparé suivant la procédure décrite pour le composé MA
à partir des réactifs suivants : 2,4,6-triméthylaniline (4.8 ml, 34 mmol) ;
chlorhydrate de
la bis(2-chloroéthyl)amine (6.05 g, 34 mmol) ; carbonate de sodium (1.8 g, 17
mmol)
butan-1-ol (75 ml). Le brut réactionnel, après avoir été imprégné sur silice,
est purifié
par chromatographie-éclair avec un mélange (95/5/1) puis (90/9/1) de
dichlorométhane /
méthanol / ammoniaque.
Masse obtenue : 3.79 g (Rdt 55 %)
RMN 1H (CDC13) : 2.23 (s, 3H) ; 2.29 (s, 6H) ; 3.01 (m, 8H) ; 3.33 (se, 1H)
6.81 (s, 2H).
31 : Le composé 31 est préparé suivant la procédure décrite dans l'exemple 21
à partir des réactifs suivants : triphosgène (218 mg, 0.74 mmol) ; 4-méthoxy-3-
(4-
méthylpipérazin-1-yl)aniline (490 mg, 2.21 mmol) ; triéthylamine (2 x 305 l,
4.41 mmol) ; 1-(2,4,6-triméthylphényl)pipérazine (31 ) (450 mg, 2.21 mmol)
dichlorométhane (40 ml).
Le brut est purifié par chromatographie-éclair avec un mélange (95/5/1) puis
(90/9/1) de dichlorométhane/méthanol/azmnoniaque.
Masse obtenue : 622 mg (Rdt 63 %)
Ce composé est dissous dans le méthanol et traité avec de l'acide fumarique
pour donner le fumarate correspondant. Celui-ci est cristallisé dans l'éther.
Analyse Elémentaire pour : C26H37N502-C4H404
Calculées, C, 63.47 ; H, 7.28 ; N, 12.34 ; Expérimentales, C, 63.24 ; H, 7.53
N, 12.52
Masse (DCI/NH3) : 452 (MH+), 248, 205.
IR (Kbr) : 3356, 2949, 2829, 1637, 1518, 1250, 1215.
RMN 1H (DMSO) : 2.17 (s, 3H) ; 2.24 (s, 6H) ; 2.33 (s, 3H) ; 2.62 (M,4 H)
2.98 (M, 8H) ; 3.51 (M, 4H) ; 3.73 (s, 3H) ; 6.58 (s, 2H) ; 6.80 (m, 3H) ;
7.04-7.13 (m,
2H) ; 8.33 (s, 1H).
Point de fusion : 171 C
EXEMPLE 32 Fumarate du N-[4-méthoxy-3-(4-méthy_lpipérazin-1-yl)phényl]-4-
(p-acétylaminophényl)pipérazin-1-ylamide
WO 96/02525 PCT/FR95100975
47
N
.__/
NH /-\ ~N~N
O li
L'anhydride acétique ( 250 ml, 2.65 mmol) est ajouté à une solution du
composé 2(634 mg, 1.49 mmol) dans la pyridine (20 ml) à 0 C. Le mélange
réactionnnel est ensuite ramené à température ambiante et agité pendant 12 h.
La
pyridine est évaporée puis le brut est dilué dans le toluène et évaporé à
nouveau avant
d'être imprégné sur silice et purifié par chromatographie-éclair avec un
mélange (95/5/1)
de dichlorométhane/méthanol/ammoniaque.
Masse obtenue : 538 mg (Rdt 77 %)
Analyse Elémentaire pour : C25H34N603-C4H404-H20
Calculées, C, 57.99 ; H, 6.71 ; N, I3.99 ; Expérimentales, C, 58.01 ; H, 6.84
N, 13.88
Masse (DCI/NH3) : 467(MH+), 248, 220, 177.
IR (Kbr) :3364, 1643, 1604, 1514.
RMN 1H (DMSO) : 2.01 (s, 3H) ; 2.38 (s, 3H) ; 2.69 (M, 4H), 3.05 (M, 4H)
3.18 (s, H20) ; 3.57 (M, 4H) ; 3.74 (s, 3H) ; 6.59 (s, 2H) ; 6.80-7.14 (m, 5H)
; 7.45 (d,
8.9Hz, 2H) ; 8.40 (s;1H) ; 9.73 (s,1H).
Point de fusion : 178 C
E}.EMPLE 33 Hémifumarate du N-[4-méthoxy-3-(pipérazin-1-yl)phényl]-4-(2,3-
dim éthylphényl)pip érazin-1-ylamide
WO 96/02525 " PCT/FR95/00975
48
&'NN N
IOI \
O
3.îA : La 2-(pipérazin-1-yl)-4-nitroanisole
La 2-(pipérazin-1-yl)anisole (5,1 g ; 26 mmol) est lentement acidifiée avec
6 ml d'acide sulfitrique (5N) puis déshydraté par évaporation sous pression
réduite.
L'acide sulfurique concentré est ensuite ajouté (22 ml) très lentement (25
min) et le
milieu réactionnel est agité jusqu'à obtention d'un mélange homogène. Le
nitrate de
potassium (3.1 g ; 31 mmol) est ajouté sur 35 minutes par petites portions. La
réaction
est agitée 4 heures. La solution est ensuite versée sur de la glace et
neutralisée par ajout
de carbonate de sodium (jusqu'à pH-7-8). Le dérivé nitré est extrait de la
phase aqueuse
avec de l'acétate d'éthyle (4 x 80 ml), séché sur sulfate de magnésium et
purifié par
chromatographie-éclair avec un gradient d'éluant (2 à 5 % de méthanol dans du
dichlorométhane en présence de 0.5 % d'anitnoniaque).
Masse obtenue : 5,7 g(93 %)
1Fj-Elib[ (200MHz, dmso-d6) d: 7,90 (dd, 1H, 2.8 et 9Hz) ; 7,62 (d, 1H,
2.8Hz) ; 7,14 (d, 1H, 9Hz) ; 3,93 (s, 3H) ; 2,93-(brs, 4H) ; 2,83 (brs, 4H).
3M: La 2-(N-tert-butoxycarbonylpipérazin-1-yl)-4-nitroanisole
L'amine 33A (5,7 g ; 24 mmol) est dissoute dans du dichlorométhane (48 ml)
sous atmosphère inerte d'azote à température ambiante en présence de
triéthylamine
(3,7 ml ; 26 mmol). Le di-tert-butyldicarbonate (5,7 g ; 26 mmol) est ajouté
lentement,
dilué dans du dichlorométhane. La réaction est immédiate. Le mélange
réactionnel est
dilué dans du dichlorométhane, lavé avec une solution de chlorure d'ammonium
saturée
et séché sur sulfate de magnésium. Le composé MB est purifié par filtration
sur silice en
éluant abondamment avec du dichlorométhane pur (pour enlever le di-tert-
WO 96/02525 PCTlFR95/00975
49
butyldicarbonate résiduel) puis avec une solution 3 % de méthanol dans le
dichlorométhane.
Masse obtenu_e : 5,1 g (60 %)
la-IM (200MHz, dmso-d6) d : 7,93 (dd, 1H, 2.8 et 9Hz) ; 7,62 (d, 1H,
2.8Hz) ; 7,15 (d, 1H, 9Hz) ; 3,93 (s, 3H) ; 3,43 (brs, 4H) ; 2,97 (brs, 4H) ;
1,41 (s, 9H).
La 2-(N-tert-butoxycarbonylpipérazin-1-yl)-4-aminoanisole
Le composé 3M (5,1 g ; 15 mmol) est dissous dans de l'éthanol (48 ml) sous
atmosphère inerte d'azote en présence d'une quantité catalytique de nickel de
Raney.
L'hydrazine (3,6 ml) est ajouté lentement pré-diluée dans un peu d'éthanol. La
réaction
est chauffée à 50 C pendant 3 heures et le solvant est évaporé sous pression
réduite.
L'aniline est purifiée par chromatographie-éclair avec un mélange d'éluants
(ammoniaque-mëthanol-dichlorométhane= 1-5-95).
Masse ohtenue: 3,9 g (83 %)
1H-RMN (200MHz, dmso-d6) d 6,62 (d, 1H, 8Hz) ; 6,15-6,10 (m, 2H) ; 4,56
(s, 2H, NH2) ; 3,64 (s, 3H) ; 3,42 (brs, 4H) ; 2,83 (brs, 4H) ; 1,41 (s, 9H).
le N-[4-méthoxy-3-(4-tert-butoxycarbonylpipérazin-1-yl)phényl]-4-(2,3-
diméthylphényl)pipérazin-1-ylamide
Le dérivé 3M est préparé selon la même procédure décrite pour 23 à partir des
réactifs suivants: MC (1 g ; 3,2 mmol) ; 1-(2,3-xylyl)pipérazine (912 mg ; 4,8
mmol)
triéthylamine (0,9 ml ; 6,5 mmol) ; triphosgène (318 mg ; 1 mmol) ;
dichlorométhane
(25 ml).
Masse obtenue : 1,1 g (65 %)
1H_$~T (200MHz, dmso-d6) d : 8,34 (s, 1H, NH) ; 7,15-6,7 (m, 6H) ; 3,71
(s, 3H) ; 3,54 (brs, 4H) ; 3,42 (brs, 4H) ; 2,84 (brs, 4H) ; 2,76 (brs, 4H) ;
2,19 (s, 3H)
2,17 (s, 3H) ; 1,35 (s, 9H).
M Le dérivé 3M (1,1 g ; 2,1 mmol) est dissous dans 60 ml de
dichlorométhane sous atmosphère inerte d'azote à température ambiante et
l'acide
trifluoroacétique (10,2 ml) est ajouté. La réaction est agitée 1 heure puis
basifiée avec de
la soude 0,5M. La phase organique est lavée avec une solution saturée en
chlorure de
sodinin, séchéé sur sulfate de magnésium et évaporée sous pression réduite. Le
produit
~195~~~ r!
WO 96/02525 PCT/FR95/00975
final est purifié par chromatographie-éclair avec un gradient d'éluants
(dichlorométhane-
méthanol-ammoniaque de 98-2-0,5 à 96-4-0,5).
M sc.ob nu :641mg (71%)
1H=$LE (200MHz, dmso-d6) d: 8,36 (s, 1H, NH) ; 7,15-6,7 (m, 6H) ; 3,73
5 (s, 3H) ; 3,58 (brs, 4H) ; 3,82 (brs, 12H) ; 2,23 (s, 3H) ; 2,21 (s, 3H).
Ce composé est dissous dans le méthanol et traité avec de l'acide fumarique
pour donner le fumarate correspondant.
lH-$pJ~j, (200M_Idz, dmso-d6) d: 8,39 (s, 1H, NH) ; 7,15-6,7 (m, 6H) ; 6,42
(s, fumarate) ; 3,73 (s, 3H) ; 3,58 (brs, 4H) ; 3,00 (brs, 8H) ; 2,79 (brs,
4H) ; 2,22 (s,
10 3H) ; 2,20 (s, 3H).
Analyse élémentaire pour : Ç24 H33 N5 02 ; 0,5 Cq H4 04
Calculée C=64.84 ; H=7.32 ; N=14.54
Trouvée C=64.52 ; H=7.31 ; N=14.43
I$(KBr) : 3269, 1633, 1604, 1533, 1510, 1473,1200.
15 DCI (Nli3) : 424 (MH+), 191 (base).
$f : 0.2 (dichlorométhane-méthanol-ammoniaque = 85-15-1)
EXEIIPLE34 Fumarate du._..N-[4-méthoay-3-(4-éthylpipérazin-1-yl)phényl]-4-
(2,3-diméthylph ényl)pipérazin-1-ylamide
/
~
~N H
/1I I
N N N
O
34
L'amine 23 ( 613 mg, 1,44 mmol) est dissoute dans du DMF (10 ml) et le
bromoéthane (0,11 ml ; 1,44 mmol) est ajouté. Du carbonate de césium (470 mg ;
1,44 mmol) est introduit et l'agitation est pousuivie à température ambiante
pendant
WO 96102525 -- - PCT/FR95100975
_ ~: =-. s .
51
1 jour. Le solvant est évaporé sous pression réduite (<1 rnmHg), le résidu est
repris au
dichlorométhane,lavé avec une solution saturée de bicarbonate de sodium et
séché sur
sulfate de magnésium. Le produit est purifié par chromatographie-écl.air avec
un
mélange de méthanol (8 %) et d'ammoniaque (0,5 %) dans le dichlorométhane.
Masse obtenue : 427 mg (65 %)
Ce composé est dissous dans le méthanol et traité avec de l'acide fumarique
pour donner le fumarate correspondant.
113$MN (200MHz, dmso-d6) d 8,35 (s, 1H, NH) ; 7,15-6,7 (m, 6H) ; 6,54
(s, fumarate) ; 3,71 (s, 3H) ; 3,56 (brs, 4H) ; 2,98 (brs,4H) ; 2,78 (brs, 4H)
; 2,63 (brs,
4H); 2,55-2,4 (m, 2H) ; 2,20 (s, 3H) ; 2,19 (s, 3H) ; 1,05 (t, 3H, 7Hz).
Analyse élémentaire nour : C26 H37 N5 02 ; 0,5 C4 H4 04 ; 0,75 H2 O
Calculée C=64.28 ; H=7.80 ; N=13.39
Trouvée C=64.38 ; H=7.59 ; N=13.29
IB(KBi) :3431, 1637, 1580, 1541, 1518, 1234.
DCI (~3) : 452 (IvlfiF, base).
$f : 0.65 (dichlorométhane-méthanol-ammoniaque = 86-14-1)
EXEMPLE 35 Iiémïfumarate du N- [4- méthoxy - 3 - (4- propylpipérazin - 1 - yl)
phényl] - 4 - (2,3- diméthylphényl) pipérazin - 1 - ylamide
H
NI/N / N
Olf \ I
Le dérivé M est préparé selon la même procédure décrite pour 34 à partir des
réactifs suivants: M (800 mg ; 1,8 mmol) ; 1-bromopropane (0,16 ml g ; 1,8
mmol)
carbonate de césium (586 mg ; 1,8 mmol) ; DMF (13 ml).
.. 1' i 7 çt ~
WO 96102525 PCTIFR95100975
52
Masse obtenue :547 mg (65 %)
Ce composé est dissous dans le méthanol et traité avec de l'acide fumarique
pour donner le fumarate correspondant.
lH:$~ (200MHz, dmso-d6) d : 8,37 (s, 1H, NH) ; 7,2-6,75 (m, 6H) ; 6,57
(s, fumarate) ; 3,73 (s, 3H) ; 3,57 (brs, 4H) ; 2,97 (brs,4H) ; 2,79 (brs, 4H)
; 2,60 (brs,
4H) ; 2,38 (t, 2H, 7.2Hz) ; 2,22 (s, 3H) ; 2,20 (s, 3H) ; 1,49 (q, 2H, 7.2Hz)
; 0.88 (t, 3H,
7.2Hz).
Analyse élémentaire pour : C27 H39 N5 02 ; 0,5 C4 H4 04 ; 0,5 H2 O
Calculée C=65.39 ; H=7.95 ; N=13.15
Trouvée C=65.53 ; H=7.87 ; N=13.05
jg(KBr): 3300, 2964, 1833, 1643, 1604, 1539, 1508, 1234.
DCI (NI33) : 466 (MH+), 276 (base).
gf : 0.71 (dichlorométhane-méthanol-ammoniaque = 90-10-1)
EXEMPLE 36 Fumarate de la 2-[3-(4-méthylpipérazin-1-yl)naphtalèn-2-yloxy]-
1-[4-(2,3-diméthylphényl)pipérazin-1-yl]éthanone
\
0
-\ O
\ /. ~%~_
3_fià: 2-chloro-l-[4-(2,3-xylyl)pipérazin-I-yljéthanone
Le chlorure de chloroacétyle (1.47 ml, 18.5 mmol) est ajouté goutte à goutte à
une solution de 1-(2,3-xylyl)pipérazine (3.2 g, 16.8 mmol) et de carbonate de
calcium
(5g, 50 mmol) dans la méthyléthylcétone (40 ml) refroidie à 0 C. Le mélange
réactionnel est agité à cette température pendant 1 h 30 puis il est filtré
sur célite. La
célite est rincée plusieurs fois avec de l'acétate d'éthyle et une solution de
soude 3M. Les
i~i 2195~ WO 96102525 42
} Y '~
PCT/FR95100975
53
deux phases du filtrat sont ensuite séparées et la phase organique est séchée
sur sulfate
de magnésium; filtrée et concentrée.
Masse obtenue : 4.017g (Rdt 90 %)
RMN 1H (CDC13) : 2.24 (s, 3H) ; 2.27 (s, 3H) ; 2.87 (M, 4H) ; 3.67 (M, 4H) ;
4.11 (s, 2H) ; 6.75-7.11 (m, 3H).
M$ : 3-(4-méthylpipérazin-1-yl)naphtalèn-2-ol
Le 3-aminonaphtalèn-2-ol (5 g, 31.4 mmol) est porté au reflux du butan-l-
ol,(100 ml) en présence du chlorhydrate de la 2-chloro-N-(chloroéthyl)-N-
méthyléthanamine (6.05 g, 31.4 mmol) et de carbonate de sodium (1.7 g, 16
mmol).
Après 48 h, le mélange réactionnel est concentré et imprégné sur silice avant
d'être
purifié par chromatographie-éclair avec un mélange (93/7/1) de dichlorométhane
/
méthanol / ammoniaque.
Masse obtenue : 3.8 g (Rdt 50 %).
RMN 1H (DMSO) : 2.24 (s, 3H) ; 2.50 (M, 41I) ; 3.09 (M, 4H) ; 7.11 (s, 1H)
7.16-7.27 (M, 311) ; 7.57 (m,1 H) ; 7.66 (m,1 H) ; 9.65 (s, 1 H).
le composé 3.6à (624 mg, 12.75 mmol) et le composé 3a (556 mg
2.29 mmol) sont agités à température ambiante sous atmosphère d'azote dans le
diméthylformamide (15 ml) en présence de carbonate de césium (1.87 g,5.73
mmol)
pendant 12 h. Le mélange réactionnel est ensuite dilué avec de l'eau et
extrait trois fois
avec de l'acétate d'éthyle. Les phases organiques sont rassemblées et lavées
trois fois
avec une solution saturée en chlorure de sodium avant d'être séchées sur
sulfate de
magnésium et concentrées. Le brut réactionnel est purifié par chromatographie-
éclair
avec un mélange (95/5/1) puis (90/9/1) de dichlorométhane/méthanol/ammoniaque.
Masse obtenue : 827 mg (Rdt 77 %)
Analyse élémentaire pour C29H36N404-C4H404
Calculées : C, 67.33 ; H, 6.85 ;N, 9.52 ; Expérimentales : C, 67.01 ; H, 6.89
N. 9.42
IR (KBr) : 3433, 2910, 2855, 1710, 1677, 1657, 1479.
RMN IH (DMSO) 2.18 (s, 3H) ; 2.20 (s, 3H) ; 2.33 (s, 3H) ; 2.66 (M, 4H)
2.76 (M, 2H) ; 2.86 (M, 2H) ; 3.19 (M, 4H) ; 3.65 (M,4H) ; 4.98 (s, 2H) ; 6.56
(s, 211)
;
6.57 (m, 2H) ; 7.01 (d, 7.6Hz, IH) ; 7.27 (m, 4H) ; 7.67 (m, 2H).
WO 96/02525 2195 4 27 PCT/FR95100975
54
Point de fusion : 186 C
F.X.MP , .;7 Funmarate_ dela.__2-[2-(4-méthylpipérazin-1-yl)naphtalèn-1-yloxy]-
1-[4-(2,3-diméthylphényl)pipérazin-1-yl] éthanone
O
a ~\ ~0
v
C
31à: 2-(4-méthylpipérazin-1-yl)naphtalèn-1-ol
Le chlorhydrate du 2-aminonaphtalèn-1-ol (3 g, 15.3 mmol) est porté au reflux
du butan-1-ol (150 ml) en présence du chlorhydrate de la 2-chloro-N-
(chloroéthyl)-N-
méthyléthanamine (2.95 g, 15.3 mmol) et de carbonate de sodium (2.44 g, 23
mmol).
Après 48 h, le mélange réactionnel est concentré et imprégné sur silice avant
d'être
purifié par chromatographie-éclair avec un mélange (95/5/1) puis (90/9/1) de
dichlorométhane/méthanol/ammoniaque
Masse obtenue : 1.12 g (Rdt 30 %).
RMN 1H (CDC13) : 2.38 (s, 3H) ; 2.67 (M, 4H) ; 3.02 (M, 4H) ; 7.20-7.76 (m,
5H) ; 8.28 (d, 8.3Hz, 1H).
31 : le composé 3fiA (396 mg, 1.49 mmol) et le composé 31à (308 mg ; 1.28
mmol) sont agités à température ambiante sous atmosphère d'azote dans le
diméthylformamide (15 ml) en présence de carbonate de césium (1.03 g, 3.2
mmol)
pendant 12 h. Le mélange réactionnel est ensuite dilué avec de l'eau et
extrait trois fois
avec de l'acétate d'éthyle. Les phases organiques sont rassemblées et lavées
trois fois
avec une solution saturée en chlorure de sodium avant d'être séchées sur
sulfate de
magnésium et concentrées. Le brut réactionnel est purifié par chromatographie-
éclair
avec un mélange (90/9/1) de dichlorométhane/méthanol/ammoniaque.
Masse obtenue : 392 mg (Rdt : 65 %)
WO 96/02525 . PCT/FIt95/00975
RMN 1H (CDC13) : 2.25 (s, 3H) ; 2.26 (s, 3H) ; 2.34 (s, 3H) ; 2.59 (M, 4H)
2.91 (M, 4H) ; 3.26 (Iv1, 4H) ; 3.67 (M, 2H) ; 3.88 (M, 2H) ; 4.99 (SAB, 2H) ;
6.86 (d,
7.8Hz, 1H) ; 6.92 (d, 7.1Hz, 1H) ; 7.05 (d, 7.7Hz, 1H) ; 7.11-7.47 (m, 4H) ;
8.33 (d,
8.3Hz, H).
5 FX .MP ..38 Fumarate de la 2-[1-(4-méthylpipérazin-1-yl)naphtalèn-2-yloxy]-
1-[4-(2,3-diméthylph ényl)ipérazin-1-yI) éthanone
~
O ~N~
N ;
3M: 1-(4-méthylpipérazm-1-yl)naphtalèn-2-ol
Le chlorhydrate du 1-aminonaphtalèn-2-ol (5 g, 25.6 mmol) est porté au reflux
du butan-1-ol (100 ml) en présence du chlorhydrate de la 2-chloro-N-
(chloroéthyl)-N-
méthyléthanamine (4.9 g, 25.6 mmol) et de carbonate de sodium (5.4 g,51 mmol).
Après
48 h, le mélange réactionnel est concentré et imprégné sur silice avant d'être
purifié par
chromatographie-éclair avec un mélange (93/7/1) de dichlorométhane / méthanol
/
ammoniaque.
Masse obtenue : 4.35 g (Rdt 70 %)
RMN IH (CDC13) : 2.29 (m, 2H) ; 2.41 (s, 3H) ; 2.91 (M, 4H) ; 3.86 (M, 2H)
7.20-7.42 (m, 3H) ; 7.62 (d, 8.8Hz, 1H) ; 7.76 (d, 8.1Hz, 1H) ; 8.03 (d,
8.2Hz, 1H).
M : le composé M (477 mg, 1.79 mmol) et le composé 3M (370 mg
1.52 mmol) sont agités à température ambiante sous atmosphère d'azote dans le
diméthylformamide (15 ml) en présence de carbonate de césium (1.25 g, 3.82
mmol)
pendant 12 h. Le mélange réactionnel est ensuite dilué avec de l'eau et
extrait trois fois
avec de l'acétate d'éthyle. Les phases organiques sont rassemblées et lavées
trois fois
avec une solution saturée en chlorure de sodium avant d'être séchées sur
sulfate de
2195427 ;; ~
WO 96/02525 PC'd'/FR95100975
56
magnésium et concentrées. Le brut réactionnel est purifié par chromatographie-
éclair
avec un mélange (90/9/1) de dichlorométhane/méthanol/ammoniaque.
Masse obtenue : 615 mg (Rdt : 86 %)
RMN 1H (CDC13) : 2.04 (M, 2H) ; 2.23 (s, 3H) ; 2.26 (s, 3H) ; 2.41 (s, 3H)
2.47 (M, 2H) ; 2.87 (M, 8H) ; 3.65 (M, 2H) ; 3.81 (M, 2H) ; 4.84 (s, 2H) ;
6.82 (d,
7.8Hz, 1H) ; 6.91 (d, 7.1Hz, 1H) ; 7.05 (t, 7.7Hz, 1H) ; 7.28-7.52 (m, 3H) ;
7.66 (d,
9.1Hz, 1H) ; 7.75 (d, 7.8Hz, 1H) ; 8.42 (d, 8.4Hz, 1H).
EXEMPLE Fumarat_e du_4-(o-méthoxyphényl)pipérazin-1-yloate de [4-chloro-
3-(4-méthylpipérazin-1-yl)]phényle
N
O
0 N~ -
N N--/
~ O c5 CI
3.2
39A : 1-chlorocarbonyl-4-(o-méthoxyphényl)pipérazine
Le composé 3M est préparé suivant la procédure décrite pour le composé MA
à partir des réactifs suivants : 1-(o-méthoxyphényl)pipérazine (441 mg, 2.3
mmol) ;
triphosgène (210 mg, 0.7 mmol) ; triéthylamine (315 ml, 2.3 mmol) ;
tétrahydrofurane
(30 ml).
Pour le traitement, après dilution avec de l'eau, le mélange réactionnel est
extrait trois fois avec de l'acétate d'éthyle. Les phases organiques sont
rassemblées,
lavées une fois avec une solution saturée en chlorure de sodium, séchées sur
sulfate de
magnésium et concentrées. Le brut est purifié par chromatographie-éclair avec
un
gradient de (98/2/1) à (90/9/1) de dichlorométhane/méthanol/ammoniaque.
Masse obtenue : 333 mg (Rdt 57 %)
Masse (DCI/NH3) : 255 (MH+).
~ 219542t
WO 96/02525 - YCT/FR95/00975
57
RMN 1H (CDCI3) : 3.06 (M, 4H) ; 3.79 (M, 4H) ; 3.86 (s, 3H) ; 6.86-7.08 (m,
4H),
32: Le composé 32 est préparé suivant la procédure décrite pour le composé 15
à partir des réactifs suivants : composé 14B (316 mg, 1.39 mmol) ; composé 22à
(333 mg, 1.39 mmol) ; hydrure de sodium (60 %, 67 mg, 1.67 mmol) ;
tétrahydrofurane
(35 ml).
Le brut est purifié par chromatographie-éclair avec un mélange (90/9/1) de
dichlorométhane/méthanol/ammoniaque.
Masse obtenue : 302 mg (Rdt 49 %)
Ce composé est dissous dans le méthanol et traité avec de l'acide fumarique
pour donner le fumarate correspondant. Celui-ci est cristallisé dans l'éther.
Analyse Elémentaire pour : C23H29CIN403-1.5C4H404-0.5H20
Calculées, C, 55.46 ; H, 5.78 ; N, 8.92; Expérimentales, C, 55.61 ; H, 5.68 ;
N,
8.99
Masse (DCUNH3) : 445(MH+), 411, 371, 221, 193, 136.
IR (Kbr) : 3433, 2837, 1714, 1595, 1242.
RMN 1H (DMSO) : 2.34 (s, 3H) ; 2.64 (M, 4H) ; 3.02 (M, 8H) ; 3.57 (M, 2H) ;
3,71 (M, 2H) ; 3.80 (s, 3H) ; 4.41 (très large - H20) ; 6.60 (s, 2H) ; 6.92
(m, 6H) ; 7.41
(d, 8.6 Hz, 1 H).
Point de fusion : 105 C (déc.)
ElEMPLE 40 Fumarate du. 4-(2,3-diméthylphényl)pipérazin-1-yloate de [4-
chloro-3-(4-méthylpipérazin-1-yl)] phényle
N
N
- - -
JN\-- Nr0 d Cl
4S2
40A ' 1-chlorocarbonyl-4-(2,3-diméthylphényl)pipérazine
4 :
WO 96/02525 g 195427 PCT/FR95/00975
+G 58
Le composé 4QA est préparé suivant la procédure décrite pour le composé j.$A
à partir des réactifs suivants : 1-(2,3-diméthylphényl)pipérazine (3.94 g,
20.7 mol) ;
triphosgène (2.05 g, 6.9 mmol) ; pyridine (1.68 ml, 21 mmol); dichlorométhane
(150 ml).
Le brut est purifié par chromatographie-éclair avec du dichlorométhane.
Masse obtenue : 4.067g (Rdt 78 %)
Analyse Elémentaire pour: C13HI7CIN2O
Calculées, C, 61.78 ; H, 6.78 ; N, 11.08; Cl, 14.03 ; Expérimentales, C, 61.76
;
H, 6.79 ; N, 10.96; Cl, 14.08.
IR (Kbr) : 2922, 2824, 1759, 1720, 1406.
RMN 1H (CDCL3) : 2.25 (s, 3H) ; 2.29 (s, 3H) ; 2.92 (M, 4H) ; 4.15 (M, 4H) ;
6.87-7.26 (m, 3H).
4Q : Le composé Q est préparé suivant la procédure décrite pour le composé ~
à partir des réactifs suivants : composé L4B (560 mg, 2.47 mmol) ; composé -UA
(624 mg, 2.47 mmol) ; hydrure de sodium (60 %, 110 mg, 2.72 mmol)
tétrahydrofurane (20 ml).
Le brut est purifié par chromatographie-éclair avec un mélange (95/5/1) de
dichlorométhane/méthanol/ammoniaque.
Masse obtenue : 1.03 g (Rdt 94 %)
Ce composé est dissous dans le méthanol et traité avec de l'acide fumarique
pour donner le fumarate correspondant. Celui-ci est cristallisé dans l'éther.
Analyse Elémentaire pour : C24H31 C1N402-1.2C4H404-0.15H20
Calculées, C, 59.13 ; H, 6.22 ; N, 9.58 ; Expérimentales, C, 59.25 ; H, 6.15 ;
N,
9.43
IR (KBr) : 3433, 2916, 1714, 1579
RMN 1H (DMSO) : 2.20 (s, 3H) ; 2.22 (s, 3H) ; 2.33 (s, 3H) ; 2.62 (M, 4H) ;
2.85 (M, 4H) ; 3.17 (M, 4H) ; 3.59 (M, 2H) ; 3.72 (M, 2H) ; 6.60 (s, 2H) ;
6.83-7.10 (m,
5H) ; 7.41 (d, 8.6Hz, 1H).
Point de fusion : 150 C
EXEMPLE 41 Fumarate du 4-(2-fluorophényl)pipérazin-1-yloate de [4-chloro-3-
(4-méthylpipérazin-1-yl)] phényle
i 21954,~~..
WO 96/02525 PCT/FR95/00975
59
F
N
O
TIQI(
CI
41
41A: 1-chlorocarbonyl-4-(o-fluorophényl)pipérazine
Le composé 41A est préparé suivant la procédure décrite pour le composé mà
à partir des réactifs suivants : 1-(2-fluorophényl)pipérazine (1.54 g, 8.06
mmol) ;
triphosgène (850 mg, 2.85 mmol) ; pyridine (0.69 ml, 8.6 mmol) ;
dichlorométhane
(60 mI).
Le brut est purifié par chromatographie-éclair avec un mélange (90/10) d'éther
de pétrole/acétate d'éthyle).
Masse obtenue : 1.83 g (Rdt 88 %)
Analyse Elémentaire pour : CI 1H12CIFN20
Calculées, C, 54.44 ; H, 4.98 ; N, 11.54; Expérimentales, C, 54.23 ; H, 4.96
N, 11.38.
IR (Kbr) : 3448, 2815, 1731, 1502, 1207.
RMN 1H (CDCI3) : 3.10 (M, 4H) ; 3.86 (M, 4H) ; 6.88-7.12 (m, 4H).
41: Le composé ,41. est préparé suivant la procédure décrite pour le composé ~
à partir des réactifs suivants : composé MR (934 mg, 4.12 nunol) ; composé 41â
(1.0 g,
4.12 mmol) ; hydrure de sodium (60 %, 218 mg, 4.54 mmol) ; tétrahydrofurane
(100 mI).
Le brut est purifié par chromatographie-éclair avec un mélange (95/5/1) de
dichlorométhane/méthanol/animoniaque.
Masse obtenue : 1.6 g(Rdt 90 %)
Ce composé est dissous dans le méthanol et traité avec de l'acide fumarique
pour donner le fumarate correspondant. Celui-ci est cristallisé dans l'éther.
2495427
WO 96/02525 PCT/FR95100975
~ ' .A .
Analyse Elémentaire pour : C22H26C1FN402-C4H404 -
Calculées, C, 56:88 ; H, 5.51 ; N, 10.21 ; Cl, 6.46 ; Expérimentales, C, 57.01
;
H, 6.65 ; N, 9.95; CI, 6.36 -
IR (KBr) : 2827, 1726, 1711, 1237.
5 RMN 1H (DMSO) : 2.31 (s, 3H) ; 2.59 (M, 4H) ; 3.00 (M, 8H) ; 3.59 (M; 2H) ;
3.72 (M, 2H) ; 6.59 (s, 2H) ; 6.85 (dd, 2.6 et 8.5Hz, IH) ; 6.95 (d, 2.6Hz,
1H) ; 7.00-
7.20 (m, 4H) ; 7.40 (d, 8.7Hz, 1H).
Point de fusion : 177 C
EXEMPLE 42 4 -_ (2 -.naphtyl) pipérazin - 1 - yloate de 14 - chloro - 3 - (4 -
10 méthylpipérazin - 1 - yl)] phényle
Nï
N
~ N y O
\ ~ ~ N uci
42
9ZA :1-(2-naphtyl)pipérazine
Le composé -42A est préparé suivant la procédure décrite pour le composé 13A
à partir des réactifs suivants : 2-naphtylamine (5 g, 35 mmol) ; chlorhydrate
de la bis(2-
chloroéthyl)amine (6.23 g, 35 mmol) ; carbonate de sodium (1.85 g, 17 mmol) ;
butan-
1-01 (100 ml).
Le brut obtenu est purifié par chromatographie-éclair avec un gradient de
(95/5/1) à (85/15/1) de dichlorométhane/méthanol/ammoniaque.
Masse obtenue : 4.2 g (Rdt 56 %)
Analyse Elémentaire pour : C14H16N2-0:8H20
Calculées, C, 74.17; H, 7.82 ; N, 12.36;_Expérimentales, C, 74.27 ; H, 7.68 ;
N,
12.42.
IR (Kbr) : 3410, 2816, 1620, 1597, 1219.
# ~495427
WO96/02525 PCT/FR95/00975
61
RMN 1H (DMSO) : 3.17 (M, 4H) ; 2.92 (M, 4H) ; 7.14-7.44 (m, 4H) ; 7.71-
7.77 (m, 3H).
42B: 1-chlorocarbonyl-4-(2-naphtyl)pipérazine
Le composé UR ést préparé suivant la procédure décrite pour le composë j$A
à partir des réactifs suivants : 1-(2-naphtyl)pipérazine (600 mg, 2.82 mmol) ;
triphosgène (298 mg, 0.94 mmol) ; pyridine (230 ml, 2.82 mmol) ;
dichlorométhane
(20 ml).
Le brut est purifié par chromatographie-éclair avecun mélange (94/6) d'éther
de pétrole/acétate d'éthyle.
Masse obtenue : 530 mg (Rdt 68 %)
Analyse Elémentaire pour : C15H15C1N20-0.45CH2CI2
Calculées, C, 59.29 ; H, 5.12 ; N, 8.95 ; Expérimentales, C, 59.03 ; H, 5.10 ;
N,
9.01.
RMN 1H (CDCL3) 3.34 (t, 5.3Hz, 4H) ; 3.94 (M, 4H); 7.16-7.49 (m, 4H)
7.70-7.80 (m, 3H). -
42: Le composé 42 est préparé suivant la procédure décrite pour le composé ~
à partir des réactifs suivants : composé 14R (415 mg, 1.83 mmol) ; composé
(503 mg, 1.83 mmol) ; hydrure de sodium (50 %, 97 mg, .01 nunol) ;
tétrahydrofurane
(45 ml).
Le brut est purifié par chromatographie-éclair avec un mélange (95/5/1) de
dichlorométhane/méthanol/ammoniaque.
Masse obtenue : 639 g (Rdt 75 %)
RMN 1H (CDC13) : 2.35 (s, 3H) ; 2.60 (M, 4H) ; 3.09 (M, 4H) ; 3.33 (t, 5.0Hz,
4H) ; 3.78 (M, 2H) ; 3.84 (M, 2H) ; 6.74-6.82 (m, 2H) ; 7.15-7.45 (m, 5H);
7.68-7.77
(m, 3H).
EXENIPLE 43 _4 - _ (2,3 - diméth lphényl) pipérazin - 1 - yloate de [2 - (4 -
méthylpipérazin - 1 - yl)] phényle
R'O96/02525 21 9 JC i.A' / :,
PCT/FR95/00975
# ! 62
CN
PNNYOb
~
43A : Le 2-(pipérazin- 1 -yl)phénol
Le 2-aminophénol (6 g ; 54 mmol) est dissous dans du 1-butanol (110 ml) en
présence de carbonate de sodium (2,9 g ; 27,5 mmol) et de l'hydrochlorure de
la N-
méth~.lbis(2-chloréthyl)amine (11,1 g ; 58 mmol). La suspension est chauffée à
123 C
pendant 3 jours. Le butanol est évaporé sous pression réduite et le composé
41A est
purifié par chromatographie-éclair avec les mélanges d'éluants suivants :
NH4OH-
acétone-CH2CI2 (0,5-20-80) puis NH4OH-méthanol-CH2CI2 (1-5-95).
Masse obtenue : 9,09 g (86 %)
1~ (200MHz, dmso-d6) d : 8.95 (brs, 1H, OH) ; 6.95-6.65 (ni; 4H)
,
2.95 (brs, 4H) ; 2.48 (brs, 4H) ; 2.24 (s, 3H).
_4,1 : Le dérivé -42 est préparé selon la même procédure décrite pour M à
partir
des réactifs suivants : 4QA (656 mg ; 2,6 mmol) ;-43A-(500 mg ; 2,6 mmol) ;
hydrure de
sodium (137 mg ; 2.8 mmol) ; THF (30 ml).
Masse obtenue :.741mg (70-"L )--
1H-$~T (200MHz, dmso-d6) d 7.25-6.85 (m, 7H) ; 3.74 (brs, 2H) ; 3.59
(brs, 2H) ; 2.87 (brs, 8H) ; 2.43 (brs, 4H) ; 2.21 (s, 3H) ; 2.19 (s, 6H).
EXEMPLE 44 Hémifum-arate de la 2-[2-(4-méthylpipérazin-1-y1)phénoty]-1-[4-
(2,3-diméthylphényl)pip érazin-1-yl] éthanone
WO 96102525 f. i a7 5~~ 1 PCT/FR95100975
63
O N
0
44
49A : La 2-bromo-l-[4-(2,3-diméthylphényl)pipérazin-1-yl]éthanone
L'hydrochlorure de la 1-(2,3-xylyl)pipérazine (5 g ; 25 mmol) est mise en
suspension dans du dichlorométhane et la triéthylamine (7.6 ml ; 54 mmol) est
ajoutée.
Le mélange est refroidi à 0 C puis le bromure de bromoacétyle (2,4 ml ; 27
mmol) est
ajouté goutte à goutte. La réaction est agitée 30 minutes puis diluée dans du
dichlorométhane, lavée avec une solution de bicarbonate de sodium demi-saturée
et
séchée sur sulfate de magnésium. Le produit final est purifié par
chromatographie-éclair
avec un gradient d'éluants (30-70 à 50-50; EtOAc-EDP).
Masse ob .n u : 2,5 g (32 %)
IH:$NIN (200MHz, dmso-d6) d: 7.06 (t, 1H, 7.6Hz) ; 7.0-6.85 (m, 2H) ;'4.44
(s, 2H) ; 3.61 (brs, 4H) ; 2.79 (brs, 4H) ; 2.23 (s, 3H) ; 2.20 (s, 3H).
_4_4: Le composé 41A (800 mg ; 4.1 mmol) est dissous dans le DMF (9 ml) en
présénce de carbonate de césium (4 g ; 12.5 mmol) et du composé 4M (1.4 g ;
4.5
mmol) à température ambiante. Aprés 7 heures d'agitation, le mélange
réactionnel est
dilué dans de l'acétate d'éthyle et lavé plusieurs fois à l'eau. Les phases
aqueuses sont
combinées et extraites une fois avec de l'acétate d'éthyle. Les phases
organiques sont
combinées et séchées sur sulfate de magnésium. Le composé 44 est purifié par
chromatographie-éclair avec un mélange d'éluants (1-4-96 = NH40H-MeOH-CH2C12).
Masce obtenue : 659 mg (37 %)
Ce composé est dissous dans le méthanol et traité avec de l'acide fumarique
pour donner le fumarate correspondant. -
2~9542`7.
R'O 96102525 . . PCT/FR95/00975
64
1H-RMN (200MHz, dmso-d6) d: 7.15-6.8 (m, 7H) ; 6.56 (s, fumarate) ; 4.87
(s, 2H) ; 3.62 (brs, 4H) ; 3.06 (brs, 4H) ; 2.83 (brs, 2H) ; 2.75 (brs, 2H) ;
2.59 (brs, 4H) ;
2.30 (s, 3H) ; 2.21 (s, 3H) ; 2.19 (s, 3H).
Analyse élémentaire nour : C25 H34 N4 02 ; 0,5 C4 H4 04 ; 0;5 H2 O
Calculée C=66.23 ; H=7.62 ; N=11.44
Trouvée C=66.36 ; H=7.53 ; N=11.44
j$(KBr): 3500, 1674, 1657, 1498.
DCI (NH3) : 423 (MH+, base)
Ef: 0.56 (dichlorométhane-méthanol-ammoniaque = 90-10-1)
EXEMPLE 45 Fumarate de la-2-[4-chloro-2-(4-méthylpipérazin-1-yl)phénoxyj-l-
[4-(2,3-d iméthylphényl)pipérazin-1-yl]éthanone
1
0 ~N~
N II O /
ci
MA: Le 2-(pipérazin-1-yl)-4-chlorophénol
Le dérivé MA est préparé selon la même procédure décrite pour -43A à partir
des réactifs suivants : 2-amino-4-chlorophénol (3 g ; 20,9 mmol) ;
l'hydrochlorure de la
N-méthylbis(2-chloréthyl)amine (4,2 g ; 22 mmol) ; carbonate de sodium (1,1 g
10,5 mmoI) ; 1-butanol (42 ml).
Masse obtenue : 2,8 g (59 %)
IH-$Yj~j. (200MHz, dmso-d6) d: 9,38 (brs, 1H, OH) ; 6,9-6,7(m, 3H)_; 2,95
(brs, 4H) ; 2,43 (brs, 4H) ; 2,20 (s, 3H).
45 : Le dérivé 45. est préparé selon la même procédure décrite pour 44 à
partir
des réactifs suivants: MA (982 mg ; 4,3 mmol) ; 44A (1.3 g ; 4.3 mmol) ;
carbonate de
césium (1,4 g; 4,3 mmol) ; DMF (15 ml).
~ 219542~,
..._~ .
WO 96/02525 PCT/FR95100975
Masse : 1,04 g (53 %)
Ce composë est dissous dans le méthanol et traité avec de l'acide fumarique
pour donner le fiimarate correspondant.
l11-$pN (200MHz, dmso-d6) d: 7.15-6.8 (m, 6H) ; 6.57 (s, fumarate) ; 4.90
5 (s, 2H) ; 3.59 (brs, 4H) ; 3.09 (brs, 4H) ; 2.9-2.7 (m, 4H) ; 2.64 (brs, 4H)
; 2.33 (s, 3H) ;
2.20 (s, 3H) ; 2.18 (s, 3H).
Analyse élémentaire pour : C25 H33 CI N4 02 ; 1 C4 H4 04 ; 0,25 H2 0
Calculée C=60.31 ; H=6.54 ; N=9.70
Trouvée C=60.-I_6 ; H=6.52 ; N=9.47
10 I$1KBr~ : 3450, 2916,1709, 1662, 1591, 1498, 1473, 1338, 1219.
D CI(LM3) : 457 (MH+, base)
$f : 0.42 (dichlorométhane-méthanol-ammoniaque = 90-10-0.8)
E.XENIPLE 46 Difumarate de la 2-[8-(4-méthylpipérazin-1-yl)naphtalèn-2-yloxy]-
1-[4-(4-aminaphényl)pipérazin-1-yl] éthanone.
CND
0 N
HZN NN~O
4~ - -
Le composé l.$ (1.40 g, 2.86 nunol) dissous dans le méthanol (30 ml) est agité
pendant 5j à température ambiante sous une pression de 1 atm. d'hydrogène en
présence
d'une spatule de Pd/C. Le mélange réactionnel est ensuite filtré sur célite,
concentré et
purifié par chromatographie-éclair avec un mélange (95/5/1) de dichlorométhane
/
méthanol / ammoniaque.
Masse obtenue : 834 mg (Rdt 63 %)
W 0 96102525 YCT/FR95100975
66
Ce composé est dissous dans le méthanol et traité avec de l'acide fumarique
pour donner le fumarate correspondant. Celui-ci est cristallisé dans l'éther.
Analyse Elémentaire pour : C27H33N502-2C4H404 -
Calculées, C, 60.77 ; H, 5.97 ; N, 10.12 ; Expérimentales, C, 61.07 ; H, 6.28
N,9.94
Masse (DCI/NH3) : 460 (MH+).
IR (Kbr) : 3427, 2922, 1701, 1637, 1516.
RMN 1 H (DMSO) : 2.41 (s, 3H) ; 2.86-3.18 (M, 12H) ; 3.63 (M, 4H) ; 5.05 (s,
2H) ; 6.50 (d, 8.7Hz, 2H) ; 6.62 (s, 2H) ; 6.73 (d, 8.71-1z, 2H) ; 7.09-7.33
(m, 4H) ; 7.55
(d, 8.0Hz, 1H) ; 7.83 (d, 8.9Hz, 1H).
Point de fusion : 101 C
EXEMPL=E 47 Eumarate de, la 2-[8-(4-méthylpipérazin-1-yl)naphtalèn-2-yloxyj-
1-[4-(4-méthylsulfonylaminophényl)pipérazin-1-yl] éthanone.
1
o CD
\\ S O N
41
Le composé M (482 mg, 1.05 mmoI) est agité dans la pyridine (10 ml) à
température ambiante et sous atmosphère d'azote en présence de chlorure de
mésyle
(I35 ml, 1.74 mmol) pendant 4 h. La pyridine est ensuite évaporée. Le brut est
dilué
dans le toluène et évaporé deux fois afin d'éliminer complètement la pyridine.
Il est
ensuite imprégné sur silice et purifié par chromatographie-éclair avec un
mélange
(95/5!1) de dichlorométhane/méthanol/ammoniaque.
Masse obtenue : 475 mg (Rdt 84 %)
WO 94/02525 PCT/F1195/00975
67
Ce composé est dissous dans le méthanol et traité avec de l'acide fumarique
pour donner le fumarate correspondant. Celui-ci est cristallisé dans l'éther.
Analyse Elémentaire pour : C28H35N504S-C4H404
Calculées, C, 58.79 ; H, 6.01 ; N, 10.71 ; Expérimentales, C, 58.39 ; H, 6.16
N, 10.89
Masse (DCI/NH3) : 538 (MH+).
IR (Kbr) : 3418, 3215, 2922, 2828, 1647, 1512.
RMN IH (DMSO) : 2.29 (s, 3H) ; 2.71 (M, 4H) ; 2.87 (s, 3H) ; 3.01-3.21 (M,
8H) ; 3.65 (M, 4H) ; 5.06 (s, 2H) ; 6.60 (s, 2H) ; 6.94-7.32 (m, 8H) ; 7.53
(d, 8.IHz,
IH);7.82(d,9.0Hz,IH);9.30(s,1H).
Point de fusion : 144-146 C
EXEMPLE 48 Fumarate de la 2-[8-(4-méthylpipérazin-1-yl)naphtalèn-2-yloxy]-
1-[4-(2,3-diméthylphényl)pipérazin-1-yl] éthanone
o N
0
~ i - N \--/N
4$
Le composé 4â est préparé suivant 1 a procédure décrite pour le composé 13 à
partir des intermédiaires 3-6A (1.07 g, 4.0 mmol), MA (800 mg ; 3.3 mmol), du
carbonate de potassium (1.1g, 8.3 nnnol), de l'iodure de potassium (55 mg,0.33
mmol)
dans la méthyléthylcétone (80 ml).
Lebrut réactionnel est purifié par chromatographie-éclair avec un mélange
(95/5!1) de dichlorométhane/méthanoUammoniaque.
Masse obtenue : 1.038 g (Rdt 67 %)
2195427 . ~
WO 96102525 PCT/PR95100975
, .
68
Ce composé est dissous dans le méthanol et traité avec de l'acide fiunarique
pour donner le fumarate correspondant. Celui-ci est cristallisé dans l'éther.
Analyse élémentaire pour C29H36N402-1.5C4H404
Calculées : C, 65.00 ; H, 6.55 ; N, 8.66 ; Expérimentales : C, 64.67 ; H, 6.57
N, 8.66 - -
Masse (DCI/NH3) : 473(MH+), 233, 136.
IR (KBr) : 3424, 2887, 2833, 1716, 1697, 1651.
RMN 1H (DMSO) : 2.18 (s, 3H) ; 2.21 (s, 3H) ; 2.40 (s, 3H) ; 2.80 (M, 8H)
3.03 (M, 4H) ; 3.69 (M, 4H) ; 5.05 (s, 2H) ; 6.59 (s, 3H) 6.83-7.32 (m, 7H) ;
7.53 (d,
9H7, 1H) ; 7.81 (d, 8Hz, 1H).
Point de fusion : 178 C
EXEMPLE 49 Fumarate de la 2-[8-(4-méthylpipérazin-1-yl)naphtalèn-2-yloxy]-
1-[4-(1-naphtyl)pipérazin-1-yl] éthanone
1
CN~
O N
N /~~ ~O
N _ \ \ I
IS
42 - -
4Q 1 : 2-chloro-l-[4-(1-naphtyl)pipérazin-1-yl]éthanone
Le composé 4M est préparé suivant la procédure décrite pour le composé 12A
à partir des réactifs suivants : chlorure de chloroacétyle (270 ml, 3.45 mmol)
; 1-(1-
naphtyl)pipérazine (610 mg, 2.88 mmol) ; carbonate de calcium (860 mg, 8.64
mmol)
méthyléthylcétone (20 ml).
Masse obtenue : 765 mg (Rdt 92 %)
~ +~=~ {~ q-ry ,
WO 96/02525 fa à ei ~. ~ l+. !__ PCTIFR95/00975
69
RMN 1H (CDCI3) : 3.12 (M, 4H) ; 3.75 (M, 4H) ; 4.14 (s, 2H) ; 7.04 (m, 1H) ;
7.27-7.63 (m, 4H) ; 7.85 (m, 1H) ; 8.22 (m, 1H).
Le composé 42 est préparé suivant la procédure décrite pour le composé 13 à
partir des intermédiaires 4QA (712 mg, 2.46 nunol), lbà (375 mg ; 1.55 mmol),
du
carbonate de potassium (536 mg, 3.88 mmol) et de iodure de potassium (25 mg,
0.15
mmol) dans la méthyléthylcétone (20 ml).
Le brut réactionnel est purifié par chromatographie-éclair avec un mélange
(97/3/1) de dichlorométhane/méthanol/ammoniaque.
Masse obtenue : 393 mg (Rdt 51 %)
Ce composé est dissous dans le méthanol et traité avec de l'acide fumarique
pour donner le fumarate correspondant. Celui-ci est cristallisé dans l'éther.
Analyse élémentaire pour C31H34N402-C4H404-H20
Calculées : C, 66.86 ; H, 6.41 ; N, 8.91 ; Expérimentales : C, 67.13 ; H, 6.30
;
N,8.85
Masse (DCI/NH3) : 495 (MH+), 255, 243.
IR (KBr) : 3429, 3051, 2953, 2826, 1701, 1637, 1448.
RMN 1H (DMSO) : 2.36 (s, 3H) ; 2.76 (M, 4H) ; 3.10 (M, 8H) ; 3.81 (M, 4H) ;
5.06 (s, 2H) ; 6.57 (s, 2H) ; 7.06-8.19 (m, 13H).
Point de fusion : 118 C
F.X EMP = = 0 2-[8-(4-méthylpipérazin-1-yl)naphtalèn-2-yloxy]-1-[4-(2,3-
diméthoxyphényl)pipérazin-1-yl]éthanone
CN~
O N
- ~~
N \--/N
-O O-
~4
PCT/FIt95l00975
WO 96/02525 21 9 J4 2 {-
2-chloro- 1 -[4-(2,3-diméthoxyphényl)pipérazin- 1 -yl]éthanone
Le composé fQA est préparé suivant la procédure décrite pour le composé nà
à partir des réactifs suivants : chlorure de chloroacétyle (150 ml,1.87 mmol)
; 1-(2,3-
diméthoxyphényl)pipérazine (?,2.A) (416 mg, 1.87mmol) ; carbonate de potassium
5 (645 mg, 4.67 mmol) ; méthyléthylcétone (20 ml).
Masse obtenue : 540 mg (Rdt 98 %)
M : le composé 5QA (540 mg, 1.8 mmol) et le composé 1fiA (360 mg
1.5 mmol) sont agités à température ambiante sous atmosphère d'azote dans le
diméthylformamide (20 ml) en présence de carbonate de césium (1.4 g, 4.5 mmol)
10 pendant 12 h. Le mélange réactionnel est ensuite dilué avec de l'eau et
extrait trois fois
avec de l'acétate d'éthyle. Les phases organiques sont rassemblées et lavées
trois fois
avec une solution saturée en chlorure de sodium avant d'être séchées sur
sulfate de
magnésium et concentrées. Le brut réactionnel est purifié par chromatographie-
éclair
avec un mélange (95/5/1) de dichlorométhane/méthanol/ammoniaque.
15 Masse obtenue : 379 mg (Rdt 50 %)
Analyse élémentaire pour C29H36N404-0.22H20
Calculées : C, 68.49; H, 7.22 ; N, 11.02 ; Expérimentales : C,b8.25 ; H, 7.17
;
N, 10.84
Masse (DCI/NH3) : 505 (MH+), 265:
20 IR (KBr) : 3431, 2957,2828, 2791, 1680, 1595.
RMN IH (CDC13) : 2.44 (s, 3H) ; 2.77 (M, 4H) ; 3.13 (M, 8H) ; 3.82
(M,lOH) ; 4.87 (s, 2H) ; 6.49 (d, 8.2Hz, IH) ; 6.63 (d, 8.1Hz, 1H) ; 6.92 -
7.33 (m, 4H) ;
7.49 (d, 8.0Hz, 1H) ; 7.57 (d, 2.4Hz, 1H) ; 7.74 (d, 8.9Hz, IH).
Point de fusion : 181 C -
25 EXEMPLE 51 . Fumarate de la 2-j8-(4-méthylpipérazin-1-yl)naphtalèn-2-yloxy]-
1-[4-(benzodioxan-5-yl)pipérazin-1-yl]éthanone
(~G.J
~ ~ 4.2 sy
i PCTlFR95/00975
WO 96/02525 ~7
`..
71
O N
,~\~O
N N
O O
~f ,- - - -;- `
M
51A : 2-chloro-l-[4-(benzodioxan-5-yl)pipérazin-1-yl]éthanone
Le composé 51A est préparé suivant la procédure décrite pour le composé 12A
à partir des réactifs suivants : chlorure de chloroacétyle (127 ml, 1.6 mmol)
; I-
(benzodioxan-5-yl)pipéra2ine (321 mg, 1.46 mmol) ; carbonate de potassium (504
mg,
3.6,5 mmol) ; méthyléthylcétone (15 ml).
Masse obtenue : 368 mg (Rdt 85 %)
Le composé 51 est préparé suivant la procédure décrite pour le composé 51à
partir des intermédiaires 51A (368 mg, 1.24 mmol), 1fiA (300 mg ; 1.24 mmol),
du
carbonate de césium (978 mg, 3.0 mmol) dans le diméthylformamide (20 mI).
Le brut réactionnel est purifié par chromatographie-éclair avec un mélange
(95/5/1) puis (90/9/1) de dichlorométhane/méthanoUammoniaque.
Masse obtenue : 385 mg (Rdt 62 %)
Ce composé est dissous dans le méthanol et traité avec de l'acide fumarique
pour donner le fumarate correspondant. Celui-ci est cristallisé dans l'éther.
Analyse élémentaire pour C29H34N404-1.4C4H404-0.25H20
-- --Calculées : C, 62.06 ; H, 6_04 ; N, 8.37 ; Expérimentales : C, 61.89 ; H,
6.04
N, 838
Masse (DCI/NH3) : 503 (MH+), 263.
IR (KBr) : 3431, 1697, 1653.
~
WO 96/02525 - PCTIFR95100975
72
RMN 1H (DMSO) : 2.33 (s, 3H) ; 2.72 (M, 4H) ; 3.00 (M, 8H) ; 3.60 (M, 4H) ;
4.24 (M, 4H) ; 5.01 (s, 2H) ; 6.42-6.74 (m, 5.8H) ; 7.04-7.29 (m, 4H) ; 7.51
(d, 8.1Hz,.
1H) ; 7.79 (d, 9.OHz,.1H).
Point de fusion 206-207 C
EXEMPLE 52 .Fumarate _de la 2-18-(4-méthylpipérazin-1-yl)naphtalèn-2-yloxyj-
1-[4-(2,4,6-triméthylphényl] pipérazin-1-yl)éthanone
CND
O N
N --, N ,~,O
fu
5ZA.: 2-çhloro-l-[4-(2,4,6-triméthylphényl)pipérazin-1-yl]éthanone
Le composé 52A est préparé suivant la procédure décrite pour le composé 17.9
à partir des réactifs suivants : chlorure de chloroacétyle (480 ml, 6.05 mmol)
; 1-(2,4,6-
triméthylphényl)pipérazine (3lA) (1.122 g, 5.5 mmol) ; carbonate de calcium
(1.65 g,
16.5 mmol) ; méthyléthylcétone (25 ml) et diméthylformamide (10 ml), pour
solubiliser
la 1-(2,4,6-triméthylphényl)pipérazine).
Masse obtenue 1.49 g (Rdt 96 %)
Le composé 51 est préparé suivant la procédure décrite pour le composé 51à
partir des intermédiaires 5M (559 mg, 1.99 mmol), 1¾A (335 mg ; 1.38 mmôl), du
carbonate de césium (1.13 g, 3.46 mmol) dans le diméthylformamide (15 ml).
Le brut réactionnel est purifié par chromatographie-éclair avec un mélange
(95/5/1) de dichlorométhane/méthanol/ammoniaque.
Masse obtenue 234 mg (Rdt 35 %)
Ce composé est dissous dans le méthanol et traité avec de l'acide fumarique
pour donner le fumarate correspondant. Celui-ci est cristallisé dans l'éther.
~ 2195427
W0 96/02525 -- PCT/FR95/00975
73
Analyse élémentaire pour C3pH-8N402-C4H404-0.85H20
Calculées : C, 66.08 ; H, 7.13 ; N, 9.07 ; Expérimentales : C, 65.95 ; H, 7.03
N,9.09
Masse (DCI/NH3) : 487 (MH+), 290, 247, 136.
~ IR(KBr) : 3433, 2918, 1701, 1637, 1448, 1211.
RMN 1H (DMSO) : 2.16 (s, 3H) ; 2.19 (s, 6H) ; 2.40 (s, 3H); 2.75 (M, 4H) ;
3.03 (M, 8H) ; 3.57 (M, 4H) ; 5.04 (s, 2H) ; 6.59 (s, 2H) ; 6.78 (s, 2H) ;
7.07-7.34 (m,
4H) ; 7.54 (d, 8.0Hz, 1 H) ; 8.04 (d, 9.0Hz, 1 H).
Point de fixsion : 106 C
EXEMPLE 53 - Funmarate de la 2-[8-(4-méthylpipérazin-1-yl)naphtalèn-3-ylozyj-
1-(4-o-tolylp ip érazin-1-yl)éthanone
~
CN~
N
/ /
1-~ ~ ~p ~ ~ I
0
~
5M: 8-(4-méthylpipérazin-1-yl)naphtalén-3-ol
Le composé $,îA est préparé suivant la même procédure que jm à partir des
réactifs suivants : 8-aminonaphtalèn-3-ol (10 g, 62.8 mmol), chlorohydrate de
la 2-
chloro-N-(2-chloroéthyl)-N-méthyléthananine (11.8 g, 62.8 mmol), carbonate de
sodium (3.32 g, 31.4 mmol), butan- l -ol (200 ml).
Le brut réactionnel est purifié par chromatographie-éclair avec un gradient de
(98r2.'1) à (90/9/1) de dichlorométhane/méthanol/ammoniaque.
Masse obtenue : 5.63 g (Rdt 37 %)
t
f i.
WU 96/02525 219 54(2 i PCT/FR95/00975
74
RMN 1H (DMSO) : 2.25 (s, 3H) ; 2.55 (M, 4H) ; 2.97 (M, 4H) ; 6.84 (dd,
1.2Hz et 7.1 Hz, 1H) ; 7.07 (m, 2H) ; 7.30 (rn, 2H) ; 7.94 (d, 9.9Hz, IH) ;
9.68 (s, IH).
.U : Le composé 53 est préparé suivant la procédure décrite pour la
préparation
du composé 12 à partir des intermédiaires 5M (617 mg, 2.55 mmol), Ilà (773 mg
3.06 mmol), du carbonate de potassium (880 mg, 6.38 mmol) et d'iodure de
potassium
(60 mg, 0.37 mmol) dans la méthyléthylcétone (80 ml).
Le brut réactionnel est purifié par chromatographie-éclair avec un mélange
(90/91) de dichlorométhane/méthanol/ammoniaque.
Masse obtenue : 967 mg (Rdt 83 %)
Analyse élémentaire pour C28H34N402-C4H404
Calculées : C, 66.88 ; H, 6.66 ; N, 9.75 ; Expériméntales : C, 66.18 ; H, 6.78
N,9.52
Masse (DCI/NH3) : 459(MH+), 219, 177.
IR (KBr) : 3431, 2920, 2824, 1707, 1655.
RMN 1H (DMSO) : 2.29 (s, 3H) ; 2.43 (s, 3H) ; 2.82 (M, 8H) ; 3.07 (M, 4H) ;
3.66 (M, 4H) ; 4.99 (s, 2H) ; 6.60 (s, 2H) ; 7.00-7.51 (m, 9H) ; 8.04 (d,
9.2Hz, 1H).
Point de fusion : 124 C
EXE1iPLE 54 Fumarate du N-[2-méthoxy-3-(4-méthylpipérazin-l-yl)phénylJ-4-
(2,3-dim éthylph ényl)pipérazin-1-vlamide
-
N - - NH / - ~ - - _- ,
~i -
o \ -~
N
\
5-4 : 2-méthoxy-3-(4-méthylpipérazin-1-yl)aniline
2; Le composé 5-4A est préparé suivant la procédure décrite pour le composé uc
à partir des réactifs suivants : 2-méthoxy-3-(4-méthylpipérazin-1-yl)1-
nitrobenzène
WO 96/02525 2195427 PCTlF'R95/00975
(3.5 g) ; hydrazine hydrate (4 ml); éthanol (50 ml); nickel de Raney (une
spatule). Le
brut est purifié par chromatographie-éclair avec un mélange (90/9/1) de
dichlorométhane/méthanol/ammoniaque).
Masse obtenue : 2.6 g (Rdt 67 %)
5 RMN IH (CDC13) : 2.34 (s, 3H) ; 2.58 (M, 4H) ; 3.12 (M, 4H) ; 3.80 (se, 2H)
;
3.84 (s, 3H) ; 6.33 (dd, 1.4 et 8.0Hz, IH) ; 6.42 (dd, 1.4 et 8.0Hz, IH) ;
6.82 (t, 8.0Hz,
1H).
Le composé S est préparé suivant la procédure décrite dans l'exemple 2.3 à
partir des réactifs suivants : triphosgène (282 mg, 0.95 mmol) ; 2-méthoxy-3-
(4-
10 méthylpipérazin-1-yl)aniline (326 mg, 1.47 mmol) ; triéthylamine (2 x 206
l,
2.99 mmol) ; 1-(2,3-diméthylphényl)pipérazine (304 mg, 1.6 mmol) ;
dichlorométhane
(25ml).
Le brut est purifié par chromatographie-éclair avec un mélange (95/5/1) puis
(90/9/1) de dichlorométhane/méthanol/ammoniaque.
15 Masse obtenue : 396 mg (Rdt 63 %)
Ce composé est dissous dans le méthanol et traité avec de l'acide fumarique
pour donner le fumarate correspondant. Celui-ci est cristallisé dans l'éther.
Analyse Elémentaire pour : C25H35N502-C4H404
Calculées, C, 62.91 ; H, 7.10 ; N, 12.65 ; Expérimentales C, 62.12 ; H, 7.09
20 N, 12.49
IR (Kl3r) : 3439, 2822, 1677, 1603, 1529, 1477.
RMN 1H (DMSO) : 2.21 (s, 3H) ; 2.23 (s, 3H) ; 2.33 (s, 3H) ; 2.63 (M, 4H)
2.82 (M, 4H) ; 3.05 (M, 4H) ; 3.60 (M, 4H) ; 3.80 (s, 3H) ; 6.60 (s, 2H) ;
6.65 (dd, 1.2 et
8.0Hz, 1H) ; 6.89-7.10 (m, 4H) ; 7.38 (dd, 1.2 et 8.1Hz, IH) ; 8.10 (s, IH).
25 Point de fusion : 194 C
', -
~
WO 96/02525 PCTIFR95/00975
76
RESULTATS BIOLOGIQUES
Les récepteurs humains 5HTIDa et 5HTIDR ont été clonés selon les
séquences publiées par M. Hamblin et M. Metcalf, Mol. Pharmacol., 4Q, 143
(1991) et
Weinshenk et coll., Proc. Natl. Acad. Sci. $Q, 3630 (1992).
La transfection transitoire et la transfection permanente des gènes de ces
récepteurs a été réalisée dans des lignées cellulaires Cos-7 et CHO-KI en
utilisant un
électroporateur.
La lignée cellulaire HeLa HA7 exprimant le récepteur 5HTlp, humain a été
obtenue de Tulco (Duke Univ., Durham, N.C., USA) et cultivée selon la méthode
de
Fargin et coll., J. Biol. Chem. 2a 14848 (1989).
L'étude de la liaison des dérivés de la présente invention avec les récepteurs
5HT1Da> 5HT1DR et 5HTI,ç humains a été réalisée selon la méthode décrite par
P.
Pauwels et C. Palmier (Neuropharrnacology, 31, 67, 1994).
Les milieux d'incubation pour ces mesures de liaison comprennent 0.4 ml de
préparation de membrane cellulaire, 0.05 ml d'un ligand tritié [[3H]-5CT
(concentration
finale : 2 nM) pour les récepteurs 5HT1Da et 5HT1Dp et [3H]-80H-DPAT
(concentration finale : 1 nM) pour le récepteur 5HTIA] et 0.05 ml de la
molécule à
tester (concentrations finales de 0.1 nM à 1000 nM) ou 10 M (concentration
finale) de
sérétonine (SHT1Da et 5HT1DR) ou 1 gM (concentration finale) de spiroxatrine
(5HT1A)=
L'étude de l'inhibition de la formation d'AMP cyclique (stimulée par la
forskoline) médiée par le récepteur 5HTIDR humain a été réalisée dans les
cellules
CHO-KI transfectées par le récepteur selon la technique décrite préalablement
pour le
récepteur 5HTIB (P. Pauwéls et C. Palmier, Neuropharmacology, 31, 67, 1994).
WO 96102525 PCTIFR95/00975
77
RESULTATS OBTENUS
Exemple 5HT1Da 5HT1D(3 5HTl,e, 5HTID(3
Ki* Ki* Ki* EC**50
(nM) (nM) (nM)
(nM)
1 48 2.3 396 >1000
2 68 2 509 >1000
3 230 8_4 1000 >1000
42 1.2 557 >1000
6 114 5 457 >1000
7 140 8.2 786 >1000
8 120 11.2 714 >1000
12 420 28 3000 >1000
16 0.68 0.28 50 >1000
19 2.8 0.5 16 >1000
* Affinité pour les récepteurs concernés
5 Activité agoniste intrinsèque (inhibition de la formation d'AMP cyclique
induite par la forskoline dans les cellules CHO-KI)
Les nouveaux composés dérivés d'aryl pipérazines faisant partie de la présente
invention sont des antagonistes puissants et sélectifs des récepteurs 5HT1D
comme le
démontrent les exemples cités ci-dessus. De nombreux composés faisant partie
de la
présente invention présentent l'avantage d'être particulièrement sélectifs
pour les
récepteurs 5HT1D a et (3 humains en particulier par rapport aux récepteurs
5HT1A,
5HT1C, SHT2, al, a2 et D2.
Les dérivés de la présente invention sont en outre capables d'inhiber la
contraction induite par la 5-hydroxy-tryptamine dans les anneaux de veine
saphène de
lapin et'd'antagoniser l'inhibition induite par la 5-carboxamido-tryptamine
(5CT) au
niveau de la libération de sérotonine dans les tranches de cerveau de cobaye.
~
WO 96102525 21 9C 4 2 ry PCTIFR95/00975
J 0 78
Un autre aspect particulièrement intéressant de la présente invention comprend
la découverte d'antagonistes sélectifs pour le récepteur 5HTIDR. humain. En
éffet,
comme l'indiquent les résultats pharmacologiques décrits ci-dessus, de
nombreux
composés nouveaux répondant à la formule générale (I) ont une affmité
nettement
supérieure pour le récepteur SHTIDP vis-à-vis des autres récepteurs y compris
le
récepteur 5HT1Da= La plupart des composés ayant cette originalité qui les
distingue de
tous les dérivés de l'art antérieur sont plus précisément définis par une sous-
classe
particulièrement appréciée des produits de formule (1) et qui a été définie
comme
répondant à la formule (la).
Les composés ayant une action antagoniste sélective au niveau des récepteurs
"5HT1-liké" et/ou 5HTID tels que ceux décrits dans la présente invention
peuvent
exercer un effet bénéfique sur des patients souffrant de désordres au niveau
du système
ner-veux central. De ce fait, la présente invention comprend également une
méthode pour
traiter de tels patients, méthode qui met en oeuvre l'administration d'une
dose active d'un
composé répondant à la formule générale (I).
La présente invention a également pour objet les compositions
pharmaceutiques contenant comme principe actif un composé de formule générale
I ou
un de ses sels acceptables pour l'usage pharmaceutique, mélangé ou associé à
un
excipient approprié. Ces conipôsitions peuvént revêtir, par exemple, la forme
de
compositions solides, liquides, d'émulsions, lotions ou crèmes.
Comme compositions solides pour administration orale, peuvent être utilisés
des comprimés, des pilules, des poudres (capsules de gélatine, cachets) ou des
granulés.
Dans ces compositions, le princïpe àctif selon l'invention est mélangé à un ou
plusieurs
diluants inertes, tels que amidon, cellulose, saccharose, lactose ou silice,
sous courant
d'argon. Ces compositions peuvent également comprendre des substances autres
que les
diluants, par exemple un ou plusieurs lubrifiants tels que le stéarate de
magnésium ou le
talc, un colorant, un enrobage (dragées) ou un vernis.
Comme compositions liquides pour administration orale, on peut utiliser des
solutions, des suspensions, des émulsions, des sirops et des élixirs
pharmaceutiquement
acceptables contenant des diluants inertes tels que l'eau, l'éthanol, le
glycérol, les huiles
végétales ou l'huile de paraffine. Ces compositions peuvent comprendre des
substances
W O 96/02525 2195427 PCT/FR95/00975
79 /
autres que les diluants, par exemple des produits mouillants, édulcorants,
épaississants,
aromatisants ou stabilisants.
Les compositions stériles pour administration parentérale, peuvent être de
prëférence des solutions aqueuses ou non aqueuses, des suspensions ou des
émulsions.
Comme solvant ou véhicule, on peut employer l'eau, le propylèneglycol, un
polyéthylèneglycol, des huiles végétales, en particulier l'huile d'olive, des
esters
organiques injectables, par exemple l'oléate d'éthyle ou autres solvants
organiques
convenables. Ces compositions peuvent également contenir des adjuvants, en
particulier
des agents mouillants, isotonisants, émulsifiants, dispersants et
stabilisants. La
stérilisation peut se faire de plusieurs façons, par exemple par filtration
aseptisante, en
incorporant à la composition des agents stérilisants, par irradiation ou par
chauffage.
Elles peuvent également être préparées sous forme de compositions solides
stériles qui
petivent être dissoutes au moment de l'emploi dans de l'eau stérile ou tout
autre milieu
stérile injectable.
Les compositions pour administration rectale sont les suppositoires ou les
capsules rectales qui contiennent, outre le produit actif, des excipients tels
que le beurre
de cacao, des glycérides semi-synthétiques ou des polyéthylèneglycols.
Les compositions pour administration topique peuvent être par exemple des
crèmes, lotions, collyres, collutoires, gouttes nasales ou aérosols.
Les doses dépendent de l'effet recherché, de la durée du traitement et de la
voie
d'administration utilisée ; elles sont généralement comprises entre 0,001 g et
1 g (de
préférence comprises entre 0,005 g et 0,25 g) par jour de préférence par voie
orale pour
un adulte avec des doses unitaires allant de 0,1 mg à 500 mg de substance
active, de
préférence de 1 mg à 50 mg.
D'une façon générale, le médecin déterminera la posologie appropriée en
fonction de l'âge, du poids et de tous les autres facteurs propres au sujet à
traiter. Les
exeinples suivants illustrent des compositions'selon l'invention .
Dans les exemples de compositions ci-dessous, le terme "composant actif'
désigne un ou plusieurs (généralement un) des composés de formule (I) selon la
présente invention :
WO 96/02525 2195427 PCT/FR95100975
ComDrimés
On peut les préparer par compression directe ou en passant par une granulation
au mouillé. Le mode opératoire par compression directe est préféré mais il
peut ne pas
convenir dans tous les cas selon les doses et les propriétés physiques du
composant
5 actif.
A - Par compression directe
mg pour 1 comprimé
composant actif 10,0
cellulose microcristalline B.P.C. 89,5
10 stéarate de magnésium ~M
100,0
On paàse le composant actif au travers d'un tamis à ouverture de maille de
250 m de côté, on mélange avec les excipients et on comprime à l'aide de
poinçons de
15 6,0 mm. On peut préparer des comprimés présentant d'autres résistances
mécaniques en
modifiant le poids de compression avec utilisation de poinçons appropriés.
B - Granulation au mouillé
mg pour un comprimé
composant actif 10,0
20 lactose Codex 74,5
amidon Codex 10,0
amidon de maïs prégélatinisé Codex 5,0
stéarate de magnésium m
Poids à la compression 100,0 -
25 On fait pâsser le composant actif au travers d'un tamis à ouverture de
maille de
250 um et on mélange avec le lactose, l'amidon et l'amidon prégélatinisé. On
humidifie
les poudres mélangées par de l'eau purifiée, on met à l'état de granulés, on
sèche, on
tamise et on mélange avec le stéarate de magnésium. Les granulés lubrifiés
sont mis en
comprimés conune pour les formules par compression directe. On peut appliquer
sur les
30 comprimés une pellicule de revétement au moyen de matières filmogènes
appropriées,
~ 2195427
WO 96/02525 PCTlFR95/00975
81
par exemple la méthylcellulose ou l'hydroxy-propyl-méthyl-cellulose, selon des
techniqués classiques. On peut également revêtir les comprimés de sucre.
~
mg pour une capsule
composant actif 10,0
*amidon 1500 89,5
stéarate de magnésium Codex
Poids de remplissage 100,0
*une forme d'amidon directement compressible provenant de la firme Colorcon
Ltd, Orpington, Kent, Royaume Uni.
On fait passer Ie composant actif au travers d'un tamis à ouverture de maille
de
250 m et on mélange avec les autres substances. On introduit le mélange dans
des
capsules de gélatine dure N 2 sur une machine à remplir appropriée. On peut
préparer
d'autres unités de dosage en modifiant le poids de remplissage et, lorsque
c'est
nécessaire, en changeant la dimension de la capsule.
~it4R -
mg par dose de 5 ml
composant actif 10,0
saccharose Codex 2750,0
glycérine Codex 500,0
tampon )
arôme )
colorant ) q.s.
préservateur )
eau distillée 5,0
On dissout le composant actif, le tampon, l'arôme, le colorant et le
préservateur
dans une partie de l'eau et on ajoute la glycérine. On chauffe le restant de
l'eau à 80 C et
on y dissout le saccharose puis on refroidit. On combine les deux solutions,
on règle le
volume et on mélange. Le sirop obtenu est clarifié par filtration.
2195427':?':'
WO 96/02525 - PCT/FR95/00975
82
~ypnositoires _ _ _
Composant actif 10,0 mg
*Witepsol H15 complément à 1,0 g
*Marque commercialisée pour Adeps Solidus de la Pharmacopée Européenne.
On prépare une süspension du composant actif dans le Witepsol H15 et on
l'introduit dans une machine appropriée avec moules à suppositoires de 1 g.
Liquide pour administration par i'ection intraveineuse
g/1
composant actif 2,0
eau pour injection Codex complément à1000,0
On peut ajouter du chlorure de sodium pour régler la tonicité de la solution
et
régler le pH à la stabilité maximale et/ou pour faciliter la dissolution du
composant actif
au moyen d'un acide ou d'un alcali dilué ou en ajoutant des sels tampons
appropriés. On
prépare la solution, on la clarifie et on l'introduit dans des ampoules de
dimension
appropriée qu'on scelle par fusion du verre. On peut également stériliser le
liquide pour
injection par chauffage à l'autoclave selon l'un des cycles acceptables. On
peut
également stériliser la solution par filtration et introduire en ampoule
stérile dans des
conditions aseptiques. La solution peut être introduite dans les ampoules en
atmosphère
gazeuse.
rto u h a nour irihalation
g/cartouche
composant actif micronisé 1,0
lactose Codex 39,6
Le composant actif est micronisé dans un broyeur à énergie de fluide et mis à
l'état de fines particules avant mélange avec du lactose pour comprimés dans
un
mélangeur à haute énergie. Le mélange pulvérulent est introduit en capsules de
gélatine
dure N 3 sur une machine à encapsuler appropriée. Le contenu des cartouches
est
administré à l'aide d'un inhalateur à poudre.
WO 96/02525 2la5427 PCTIFR95/00975
83
Aérosol sous pression à valve doseuse
mg/dose pour 1 boite
composant actif micronisé 0,500 120 mg
acide oléique Codex 0,050 12 mg
trichIorofluorométhane pour usage
pharmaceutique 22,25 5,34 g
dichiorodifluorométhane pour usage
pharmaceutique 60,90 14,62 g
Le composant actif est micronisé dans un broyeur à énergie de fluide et mis à
l'état de fines particules. On mélange l'acide oléique avec le
trichlorofluorométhane à
une température de 10-15 C et on introduit dans la solution à l'aide d'un
mélangeur à
haut effet de cisaillement le médicament micronisë. La suspension est
introduite en
quantité mesurée dans des boîtes aérosol en aluminium sur lesquelles on fixe
des valves
doseuses appropriées délivrant une dose de 85 mg de la suspension ; le
dichlorodifluorométhane est introduit dans les boites par injection au travers
des valves.