Note: Descriptions are shown in the official language in which they were submitted.
2 ~ 95758
~ W0 96104905 ~ . P~
TITLE OF THE INVENTION
PHENOXYPHENYLACETIC ACID DERIVATIVES
RELATED APPLICATIONS
The present application is a continuation in part application
of Case 1~8g31B, U.S. Serial Number 08/287,374 iiled August ~, 1994
which is a continuation in part application of IJ.S. Serial Number
0~/197,467 filed February 24, 1994, which is a continuation in part
application of U.S. Serial Number 0~31034,455 filed on March 19,
1 993(abandoned).
SUMh~ARY OF THE IN~rENTION
This invention is concemed with non-peptidic endotlleli
receptor antagonists represented by the compound of Fc rmula I.
pharmaceutical compositions containing these compound.s, as well as
combination therapies which inclucde a compoulId of the present
invention. The compounds of the present invelItion are therapeutic
agelIts particularly useful ior the treatment of asthma, hypertension,
pulmonary hypertension. arteriosclerosis. congestive heart failure. renal
failure, particularly post-ischemic renal failure, cyclosporin
nephrotoxicity, vasospasm, vascular restenosis. cerebral and cardiac
i.schemia and other i.schemic states, myocardial infarction, Raynaud's
disease, benign prostatic hyperplasia. infl~mm ltory bowel diseases,
including Crohn's disease and ulcerative colitis, as well as other
inil~mm~ltory diseases, or endoto~ic shock caused by C~l associated with
endc~tllelin.
This invention further constitutes a method for
antagonizilIg endothelin receptors in a mamulIal, including humans,
r which compri.ses administering to a mammal in need of such treatnlent
an ei'i'ective amount of a compoulld of l'ormula I
~ i 9 5 7 5 ~ P~
- 2 -
BACKGROUN~) DF THF. ~FENTI(:)N
F.n~lothf~lin is a 21-amino acid peptide produced by
endothelial cells ~The peptide is secreted not o:n]y by endothelia:l cell.s
but also by tracheal epithelial cells or from kidney cells. End(lthelin
(ET-1) has a potent vasoconstrictor effect. The vasocon.stricting effect
is cau.sed by the binding of endothelin to its receptor on the va.scular
smooth muscle ceDs.1-3
Endothelin~l ~ET-l) is one of three recently identified
potent ~asoconstricting peptides which also includes endothelm-2 (ET-2)
and endothelin-3 ~F,T-33 whose sequences dil'fer from ET-I by two and
six amino acids. respectively.4
Incre;~.sed levels of endothelin are folmd in the blood of'
patients with essential hypertension, acute myocardial infarction,
pulmonary hypertens:io:n, Raynaudts disease or atherosclerosis or in the
washing fluids of the ~ hdl~ly tract of patients with asthma compared
to normal levels.S~~
An experimental model of cerebral vasospa~ln and a seccllld
model of acute renal failure have led to the conclusion that endothelin is
one of the mediators causing cerebral va.sospasm following a
subarachnoid hemorrhage~ and renal failure.9-l(~
Endothelin was also found to control the release of many
physiological s-lh~ nces such a.s renin, atrial natriuretic peptide,
endothelium-derived relaxing factor IE~DRF~, thrclmboxalle A2,14-
prclstacyclin, norepinephrine, angiotensin Il and substance p.~
Further, endothelin causes contract:ion of the smoot.h muscle oiF tbe
gastrointestinal tract and the uterine smooth muscle.l~-T~) Endothelin
has also been shown to promote the growth of rat ~ascular smooth
muscle cells which would suggest a possible relev;mce to arterial
hypertrophy.20
F.nflolTlf~lin receptors are present in high concentration in
the peripheral tissues and also in the central nervous systeln, and
cerebral ~(I",i"i~ rion of endothelin has been shown to induce
behavioral changes in animals, suggesting that endothel:in may play an
important role in controlling neural functions.2~
~ wo96104g0S ~ /L~.
Endotoxin has been shown to promote the release of
endothelin. This finding has suggested that endothelill is an import~mt
., mediator for endotoxin-induced diseases.2~-23
A study has shown that cyclosporin added to a renal cell
culture. increased endothelin secretion.24 Another study has shown that
ad".illi~l~dlion of cyclosporin to rats, led to a decrease in the glomerular
filtration rate and all increase in the blood pressure, in association with
a remarkable increase in the circulating endothelin level. This
cyclosporin-induced renal failure can be suppressed by the
admillistration of anti-elldothelin antibody.25 These studies suggest that
endothelin is ~ig;nific:lntly involved in the pathogene.si.s of cyclosporin-
induced renal disease.
A recent study in patients with conge.stive heart failure
demonstrated a good correlation between the elevated levels of
endothelin in the plasma and the severity of the disease.26
Endothelin is an endogenous substance which direct:ly or
indirectly (through the controlled release of various other endogenous
substances) induce.s sustained contraction of vascular o r noll-vascular
smooth muscles. Its excess production or excess secretion is believed to
2 be one of the factors responsible for hypertensioll, pulmonary
hypertension, Raynaud's disease, bronchicll astllma. acute renal failure,
myocardia] infarction. angina pectoris, arteriosclerosis, cerebral
vasospasm and cerebral infarction. ~ A. M. Doherty, Endothelin: A
Nçw Challen~e. J. Med. Chem., 35, 1493-150~ (1992).
Subst~mces which specifically inhibit the binding of
endothelin to its receptor are believed to block the physiological effects
of endothelin and are useful in treating patiellts with endothelin related
disorders.
The novel compounds of the present inventioll are useful as
a non-peptidic endothelin antagonists, ~md have not been disclosed in any
,, issued patents or published patent applications. Among the published
patent applications disclosing linear ~md cyclic peptidic compounds as
endothelin antagonists are the following: Fujisawa in European Patent
Application EP-457,195 and Patent Cooperation Treat:y (PCT)
wO96/049r5 . ~>1~9~7s8 r~ 7 --
- 4 -
International Application No. WO 93/10144, Banyu in EP-436,189 and
460,679, Imrnunopharmaceutics Inc. in WO 93/~225580, W'arner
Lambert Co. WO g2/20706 and Takeda Chemical Ind. in EP-528,312
EP-543,425, EP-547,317 and WO 91/1308g .
Fujisawa has also disclosed hh~o nonpeptidic endothelin
antagonist compounds: anthraquinone derivatives produced hy a
ferrnentation process u.sing Streptomyces sp. No. 8gO09 in EP-405,421
and U.S. Patent No. 5,187,195; and a 4-phenoxyphel1ol derivative
produced by a fermentation process using Penicillium citreonigrum F-
12880 in a UK Patent Application GB 2259450. Shionogi and Co. has
also disclosed nonpeptidic endothelin antagonist triterpene compounds
whicl1 were produced by a f~rm~nt~ion process USillg Mvrica cerifera
in WC) 9V12991.
Arnon~ the non-peptidic endothelin antagonist compounds
which are known in the patent literature are: 1) a series of substituted
(1.4-quinolinoxy~l11ethylbiphenylcarboxylic acids disclosed by Roussel-
UcL~f in F P-498,723; 2) a series of of N-~4-pyrimidinyl)benzene-
sulfonamides with different substitution patterns from Hoffm~mn-l,a
Roche. published in EP-510,526, EP-526,708 and EP-601,386; 3) a
series of naphthalenesulfonamides and benzenesulfonarnides disclo~sed by
E.R. Sc~uibb & Sons in EP-558,2~ and EP-5~9,193, respectively; 4) a
series of compounds represented by 3-~3-indolylmethyl~-1,4-diaz.t-2,5-
dioxobicyclo[4.3.0]nonane-9-carboxylic acid irom
ImrnunoPh:lrm~l çlitins Inc. in WO 93i23401; 53 a series of fused
~1,2.4]thiadiazoles substituted with an irminosulfonyl bUb~titU~ frOnl
Takeda Chemical Ind. has been disclosed in EP-562,599; and 6~ a series
of indane and indene derivatives from SmithKline Beechanl Corp.
disclosed in WO 93fO~779; and a series of related phenylal'kyl
derivatives from ~rmithKline Beecb~rn disclosed in WO 94102474,
3~
~ W0 96t04905 . ~ ~ ~ i ' ' 2 l 9 ~ 7 5 ~ 5,~7
" REFERENCES
~- I Nature, 332, 411-415 (1988).
2 FEBS Letters, 231, 440-444 (]988~.
3 Biochem. Biophys. Res. Commun. 154, 86S-875 (1988~.
4 TiPS, 13, 103-108, March 1992.
5 Japan J. Hypenensioll 12, 79 (1989).
6 J. Vascular Mledicille Biology, ?, 207 (1990).
7 J. Arn. Med. Association, 264, 2868 (1990).
o 8 The Lancet, ii. 207 (1990) and The Lancet, ii, 747-748 (1989).
9 Japan. Soc. Cereb. Blood Flow & Metahol. 1, 73 (1989).
10 J. Clin. Invest., 83, 1762-1767 (1989).
I l Biochem. Biophys. Res. Comm. 157, 1164-1168 (1988).
12 Biochem. Biophys. Res; Comm. 155, 167-172 (1989).
13 Proc. Natl. Acad. Sci. USA, 85, 9797-9800 (1989).
14 J. Cardiovasc. Pharmacol., 13, 589-592 (1989).
Japam J. Hypertension 12, 76 (1989).
16 Neuroscience Letters. ]02? 179-]84 (1989).
17 FEBS Letters, 247, 337-340 (1989).
2 18 Fur. J. Pharmacol. 154, 227-228 (1988).
19 Biochern. Biophys. Res. Commun., 159, 317-323 (1989).
Atherosclerosis, Z~, 225-228 (1989).
21 Neuroscience Letter.s, 97, 276-279 (1989'~.
22 Biochem. Biophys. Res. Commull. 161, 1220-1227 (1989).
23 Acta. Physiol. Scand., 137, 317-318 (1989).
24 F.ur.J. Pharmaccl., ]80. 191-192 (1990).
Kidney lnt. 37, 1487-1491 (1990).
26 Mayo Clinic Proc., 67, 719-724 (1992).
2~ 9~7~8
W0 96~1)4905 ~ . r~ u,.,~
- 6 -
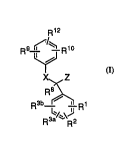
DETAILED DF~CCRIPTION OF Ti IE l:NVE:NTION. ~f
This in~ention is collcerlled with nove:l compounds of
structural formula 1:
~12
~/~
~
X~,Z
R 3~--~ R 1
or a pharm i~ellticsilly acc~ptable salt thereof~ wherein:
R 1, R2 R3a and R3b are independently:
(a~ H,
(b) F, Cl, Br, or 1,
(c) -N02.
(d) -NH2~
(e ) -NH(C I -C4)-alkyl,
(f) -N[(C I ~C4)-alkyl]2,
(~ -So2NHR7,
(h~ -CF3,
:30 ~i) (Cl-C6)-alkyl,
(.i ) -oR7,
(k) -s(o)n-(cl-c4)-alk
(1) -NHCO-~C I -C4)-alkyl,
(m ) -N~ ICO-O(C 1 -C~-alkyl,
(n) -CH~O-(Cl-C4)-alkyl,
. ! . ' ,! ;l i,
a-~7rQ
~ WOg6/04905 ~ L ~ 7 J ~ ~u r~ '7
(O) -o-(CH2)m-oR7
(P) -CoNR7R ] 1,
(q) -CooR7, or
(r) -phenyl;
K l and R2 on adjacent carboll atoms can he joined together to
fonn a ring structure:
~
;
A Ir~lt;,~ S:
a) -Y-C(R4)=C(R5)-.
b) -Y-C(R4)=N-,
c) -Y-N=C(R4)-,
d) -Y-l C(R6)(R6)1 s -Y-,
e) -Y-C(R6)(R6)-C(R6)(R6)-,
f) -C(R4)=C(R5)-Y-,
g) -N=C(R4)-Y-,
h) -C(R6)(R6j-C(R6)(R6) -Y-, or
i) -C(R4)=C(R5)-C(R4)=C~(R5)-;
n isO, I or2;
m is 2~ 3 or 4;
. I ".
s IS or,
Y is -O-, -S(O)n- and NR7;
wo s6~04sos
- 8 -
R4 and R5 are independently:
(a) H7
(b~ ~Cl-C6)-.llkyl or (C2-C6)-alkenyl each of which
iS unsubstituted or substituted with one or twcl
substituents selected from the group consisting of:
i) -OH,
ii ) -O-(C I -C4)-alkyl,
iii) -S(O)n-~C'l-C4)-alky!,
iv) -NR7-(CI-C4)-alkyl,
v) -NHR77
vi) -cooR7
vii) -CoNHR7~
viii) -oCoRI 1, or
ix) -CoNR7R l l ~
~c~) (C3-C7)-cycloalkyl,
(d~ F, Cl. Br, 1,
le) CF3,
(f) -CooR7,
(~) -CoNR7R 11,
(h) -NR7RI 1.
(i) -NR7CoNR7R 11,
(i) -NR7Coo:Rl I
(k) -S02NR7RI 1,
(1) -O-(CI-C4)-alkyl,
(m) -S(O)n-lCI-C4)-alkyl~ or
(n) -NHSO2Rl l;
R6 is:
(a) H~
(b~ (C]-C4~-alkyl unsubstituted or substitu~ed with
clle of the following substituents.
i) -OH,
ii) -NR7Rl I
iii) -cooR77
,
1 9~75~
~ ~VO 96104905 ~ .. .'C,. 7
iv) -CoNHR7, or
v) -CoNR7R1 1, or
(c) Cl, or F;
R7 is:
(a) H,
(b) (C I -C~ alkyl,
(c) phenyl,
o (d) (Cl-C6)-alkylp~henyl, or
(e) (C3-C7)-cycloalkyl;
R8 is:
(a) H,
(b) ~CI-C6)-alkyl, unsubstituted or substituted with a
subitituent .selected from the group consisting of:
phenyl,
(ii) -(C3-C7 j-cycloalkyl,
(iii) -NR7R1 1
(iv) -morpholin-4-yl,
(v~ -01 1.
(vi) -Co2R7, or
(vii~ -C'oN(R7)2,
(c~ phenyh unsubstit:uted or sub.stituted with a
substituent selected from the group consisting of:
;~ (Cl-C4)-a~
ii ~ -O-(C I -C4)-alkyl
iii) -CoNR7R1 1,
iv~ F, Cl, Br or 1, or
~ 30 v) -CooR7;
J R9 and R 10 are indepelldently:
(a~ H,
(b) (Cl-C6)-al~yl, unsubstituted or sllhsliluted with
(C3 -C7~-cycloalkyl or-CO2R,
7 ~ 9~ 75~
W096/04905 i~ r.,~l.. ,'.'{,~67
- ]O -
(c) (C2-C6)-alkenyl,
(d) (C2-C6)-alkynyl~
(e) Cl, Br, F, 1,
(f) (Cl-C6)-alkoxy,
(g) perfluoro-(CI-C6)-alkyl,
(h) (C3-C7~-cycloalkyl, unsubstituted or substituted
with (Cl-C6)-alkyl.
(i ) phenyl
(j) {cl-c6)-al}~yl-s(o)n-(cM2)n
lC (k) hydroxy-(C1-C6)-alkyl.
(I) -CF3,
(m) -Co2R7,
(n) -0~,
(~) -NR7RI 1~
(p) -I(Cl-C6)-alkyllNR7R1 1,
(q) -N02,
(r) -(cH2)n-so2-N~R7)2~
(.c) -NR7C'O-(CI-C4)-alkyl, c~r
2~ (t) -CoN(R7)2;
R9 and RIV 0n adjacent carbons can joic togethel to form a fu.sed
phenyl ring, unsubstituted or .substituted with a substituent
seleoted from the group consisting of: (Cl-C6)-alkyl, (Cl-
2s C~-alkoxy, (C3-C7)-cycloalkyl and ~CI-C6)-aLkyl-(C3-
C7)-eycloalkyl,
Rll is
(a) (C l -C6)-alkyl, unsubstituted or substituted with a
.substituent selected from the group consisting of:
i) -oR7, 4
ii ) -N[R7~2.
NH2.
iv ) -CooR7,
, ~j,7 ~ ~
, . ~ .
~ W0 96/04905 ~ ~ 2 1 9 5 7 5 ~ 'C,~67
v) -NLCH2CH2:]2
vi) -CF3. or
vii) -CoN(R7)2;
(b) aryl, wherein aryl is defined as phenyl or
naphthyl which is unsllb~lilut~d c~r substituted with
one or two substituents selected from the group
consisting of:
i) (Cl-C4)-alkyl,
ii) -O-(C I -C4)-alky h
iii) -Co[NR7]2~
iv) E~, Cl~ Br or 1,
v ~ -CooR7,
vi) -NE-12,
viil -NHL~CI -C4)-alkyl]~
viii) -NL(CI-C4)-aLkyl~2~ or
ix) -CON[CE 12cl 1212Q;
(c) -(Cl-C4)-alkylaryl~ wherein aryl is as defined
above,
(d) (C3-C7)-cycloalkyl,
R7
~,N~"N
(e~ N , or
(f) CF3,
R7 and Rl I on the same nitrogen atom they ccm join together to
form a ring .se}ected from the group consisting of:
morpholinyl, piperazinyl~ or pyrrolyl, or
QisO,Sor-NR7;
R12 iS
(a) H
W09610490~ ' ; 7'A 21~:~75~ F~ 7
- 12 -
(b) (Cl-C6)-alkyl, un~substituted or substituted with
one or two substituents selecte;l from the group
con~ in~ of
i) -OH,
ii) -O-(C I -C4)-alkyl,
iii) -O-(C I -C4)-cycloalkyl .
iv~ -SIO)n-(Cl -C4)-alkyl,
v) -NR7R
vi) -cooR7~
o vii) -CoN~R7-
viii) -OCOR
i~) -CoNR7Rl l ~
x) -NR7CoNR7R I 1 7
xi) -NR7cooRl 1~
xii) -C(R6)(oH)-C(R6)(R7)(oH),
xiii) -So2NR7R1 1, or
R~
N~N
xiv) N--N;
(c) (C3-C7)-cycloalkyl~
(d) -oR7,
(e) -CooR7
(f) -CONI 12
(g) -CONR160H,
(h) -CoNR7R 11,
(i) -CoNR7Co2R7,
(j ) -NH2,
(k) -NR7RI 1,
~1) -NR:7CoNR7RI I,
(m) -NR7C~ooR1 1,
(n~ -C(R6)(oH)-C(R6)(R7)(oH~,
(o) -S021~R7RI 1,
(p) -s(o)2NR7coRl 1,
t ~ I; 7 !~ n
W0 9611~4gO5 ~ 7 J ~ 'J ~ P~,lr~ 7
- 13 -
(q~ -S(0)2NR7Co2Rl I,
(r) -S(0)2NR7cONR7Rl 1
' (s) -NHS02RI 1~
(t) -NR7So2NR7Rl 1
(u) -CONHSO~RI 1~
(v) -CO-amino acid~ wherein amino acid is defined a~
an L-or D- amillo acid selected from the group
con.sisting of Ala~ lle. Phe~ Asp, Pro and Val and
which can be further substituted as a (Cl-C6~-
alkyl ester or an amide~ or
R7
N~N
~ N
xis
(a) -O,
(b) -S(~)n-
(c) -NR7 ~
(d) -CH2O-,
(e) -cH2s(o)n
(f~ -CH2NR7-~
(g) -ocH2
(h) -N(R7)CH2 ~
(i) -S(O)nCH2-, or
(j) -single bond;
Z is:
(a) -C~2~
(b) -CO2R 13.
(c) -CONH-(tetrazol-5 -yl)~
(d) -CONHSO2OR 11
(e) -CoNHSo2NR7R 11
W0$16~04905 ~ ~2t 95758 F._~ s~ l ~
- 14 -
(f) -CONHSO2-a~l, wherein aryl is defined as
phenyl or naphthyl which is unsubsfituted or
substituted with one. two or three sllhititnl~n
selected frorn the group consisting of:
i) (Cl -C4)-alkyl,
ii) -O-(C I -C4)-alkyl,
iii) -CoNR7Rl I
iv) F, Cl, Br or 1,
) -cool~7,
v j) -NH2.
vii) -NHI(CI -C4)-alkyl'l,
vii i) -N [(C 1 -C4)-alkyl] 2,
ix) -phenyl,
x ) -OH,
~ OCH2CH20H,
xii) -CF3;
(g) -CONHS02-(CI-C~)-alkyl, wherein the alkyl
group is unsubstituted or ~ilhstitllt.od as defined in
K4(b),
(tl) -CONE-lS02-~Cl-C4)-perfluoroalk~
(i) -tetrazol-5-yl,
(.1~ -CONHSO2-heterc)aryl, wtlereill heteroaryl is
defined as carbazolyl, furyl. thiellyl, pyrrolyl,
isothiazolyl, imidazolyl, isox~olyl. thiazolyl.
oxazolyl, pyrazolyl, pyrdzinyl, pyridyl,
pyrimidyl, purillyl or quinolillyl, which is
nn~llhititllt~d or substituted Wittl on~, two clr three
~nh~tifll.ont~ selected from the ~sroup consi~fing of:
i) (Cl -C4)-alkyl,
ii) -O-(C I -C4)-alkyl,
iii) -CoNR7R~
iv~ F, Cl, Br or 1,
v) -CooR7,
vi ) -NR7CoNR7R 11 , and
~ WO 96/D4905 ~ 9 5 7 5 8 PCT~US95/09967
~ vii~ -NR7CooR I l;
(k) -S02NHCO-aryl. wherein aryl is defined in Z(dl
above,
(I) -SO2NHCO-(CI-Cg~-alkyl, wherein the alkyl
group is unsub.stituted or substituted as defined in
R4(b).
(m~ -SO2NHCO-(CI -C4)-perfluoroalkyl,
(n~ -SO2NI ICO-heteroaryl, wherein heteroaryl is as
defined in Z(g) above,
(o) -SO2NHCON(R1 1)2 wherein the Rl I groups are
the same or different,
(p) -Po(oR7)2, wherein the R7groups are the same
or different, or
(q~ -PO(R l I )oR7;
R13 is:
i ~) (C I -C4)-a Ikyl,
(b) CHR I 4-o-coR 15,
(c) Cl l2cH2-N~(c] -c2)-alkyl]
2Q (d) CH2cH2-N[cll2c}l2l2o
(e~ (CH2CI-12O~y-o-l(cl-c~)-alkyll~ whelein y is I or 2
(~) phenyl, naphthyl, CH2-phenvl or C}12-naphthyl,
where phenyl or naphthyl is substituted or
un.substituted with CO2-(CI-C4~-alk
.. ~ 2~ 957~8
wo sclo4sos ~ ru.~ 7
- 16-
-CH2 CH3
'g' 0)=(0 ~ ,
0 0
~h) ~o,
(j) 5sS~3, or
(j) -CH2
~ O; and
i
R 14 and R IS independently are ~Cl -C6)-alkyl or phenyl: ~md
R 16 i~ H, ~Cl-C6)-alkyl or (Cl-C6)-alkylphenyl.
r
~ wO 96/049(l5 ~ ? ~i 8 ~ 7
~ An embodiment of the invention is the compound of structural formula II:
R12
R9--~--R lo
X.~Z
R3 \
R3b ~ R
R3a ~ R2
or a pharmaceutically acceptable salt thereof, wherein:
Rl, R2, R3a and R3b are independently:
(a) 1-1,
(b) F, Cl, Br, or 1,
(c) -NO2,
(d) -NH2,
(e) -Nl l(Cl -C4)-alkyl,
(f} -N[(C I -C4)-alkYl]2
(g) -S02NHR7,
(h) -CF3,
(i) (Cl-C6)-alkyl,
~i ) -oR7,
(k) -S(O)Il-(Cl -C4~-alkyl,
3 o (1) -NHCO-(C I -C4 )-alkyl !
(m) -NHCO-O(CI -C4)-alkyl,
(n~ -CH~O-(C I -C4)-alkyl,
(O) -o-(CH2)rn-oR7
(P) -CON R7R I I . or
(q) -CooR7;
WO 961049Ol . ~ = , 2 ~ ~ 5 7 5 8 T ~
- 18 -
R l and R2 on adjacent carbon atoms can be joined together to
i:'orrn a ring structure-
~
~ ;
A represents:
a) -Y-C(R4~=C(R5)-,
b) -Y-C(R4~=N~,
c) -Y -N=C(R4~-,
d) -~r-LC~R6)(R6)]S-Y-,
e) -Y-C(R6)(R6)-C(R6)(R6)-,
f) -C~(R4')=C(R5~ Y,
g) -N=c(R4)-y-~
h) ~ R6)(R6)-C(R6)~R6) -Y-, or
i) -C(R4)=CIR5)-C(R4)=C(R5)-;
m is 2, 3 or 4,
n is0, 1 o:r2,
sis I or2,
Y is -O~ (~)n- and NR7;
R4 and R5 are independently:
(a) H,
~ WO 96/04~05 ~ 2 ~ 9 5 7 5 8
- 19 -
~ (b) (Cl-C6)-alkyl or (c2-c6)-alkenyl each of which
is unsubstituted or substituted with one or two
substituents selected from the group consisting of:
i) -OI~I,
ii) -0-(C1 -C4)-alkyl,
iii) -S(O)n-(C I -C4)-alkyl,
iv) -NR7-(C'1 -C4)-alkyl,
v) -Nl IR7,
vi) -cooR7
o vii ) -CoNHR7~
viii) -oCOR 11, or
i~) -CoNR7R 11,
(c) (C3-C7)-cycloalkyl.
(d) F, Cl, Br, I,
(e) CF3,
(f) -CooR7,
(g) -CoNR7R] 1,
(Il) -NlR7R1 1,
(i) -NR7CoNR7Rl 1
(J) -NR7CooR1 I
(k) -S02NR7Rl 1,
(I) -0-(C1 -C4)-alkyl,
(m) -S(O)n-(Cl-C4)-alkyl, or
(n) -NHS02Rl l;
R6 is:
(a) H,
(b) (Cl-C4)-alkyl un.substituted clr substituted with
one or two substituents selected from the group
consisting of:
i) -01 ~,
ii) -NR7RI 1,
iii) -cooR7 ~
iv) -CoNHR7~ or
W0 g6104905 ~ 2 1 ~ 5 7 5 ~
- 20 -
v) -CoNR7R 1 l, or
(c) Cl, or F;
5R7 is:
(a) E~,
(b) (Cl-C6)-alkyl,
(c) phenyl,
(d) ~C I -C6)-alkylphenyl, or
(e) (C3-C7)-cycloalkyl;
R2 is:
(a) H,
(b) (Cl-C6~-alkyl, unsubstituted or substituted with
one or two sub.stituents selected from the group
consisting of:
(i) -phenyl.
(ii) -(C3-C7)-cycloalkyl.
(iii) -NR7R 1 l,
(iv) -morpholin-4-yl,
(v~ -OH,
(vi) -Co2R7, or
(vii) -CoN(R7)2, or
(c) phenyl;
R9 ancl R 10 are independently:
~a) I 1,
(b) (Cl-C6)-alkyl, unsubstituted or substituted with
(C3 -C7)-cycloalkyl or -C02R7,
(c) (C2-C6)-alkenyl,
(d) (C2-C6)-alkynyl,
(e) Cl, Br, F, 1,
(f) (C I -C6)-alko~y,
(g) perfluoro-(Cl-C6)-alkyl,
~ WO96/0490!; ~ t' r~'. 2 1 9 5 7 5 ~} r~ ~/ L.~
(h) (C3-C7)-cycloalkyl, unsubstituted or substituted
v~ith (Cl-C6~-alkyl,
(i3 phenyl,
(j) (cl-c6)-alkyl-s(o)n-(cH2)
(k) hydroxy-(Cl-C6)-alkyl,
(I ) -CF3,
(In) -Co2R7,
(Il) -OH,
(o) -NR7Rl I,
(p) -[(Cl-C6)-alkyl]NR R,
(q) -N02.
(r) -(CH2)1l-So2-N(R7)2~
(s) -NR7CO-(CI-C4)-alkyl, or
(t) -CoN(R7)2;
R9 and R10 on adjacent carbons can join together to i'olm a fu.sed
phenyl ring, unsubstituted or substituted with a ,substituent
selected from the group consistillg of: (Cl-C6)-alkyl, (Cl-
C6)-alkoxy~ (C3-C7)-cycloalkyl cmd (Cl-C6)-alkyl-(C3-
C7)-cycloalkyl,
Rll iS
(a) (Cl -C6)-alkyl, unsubstituted c r substituted Wit}l a
subst:ituent selected from the group consisting of:
i) -oR7,
ii) -N[:R7 12,
iii) -NH2,
iv) -CooR7,
3() v) -N[CH2CH212Q,
vi) -CF3, or
vi i) -CCIN(R7)2;
(b) aryl, whereill aryl is defined as phenyl or
naphthyl which is unsubstituted or substituted Wittl
wo s6/0490~ 7 ~ 7 ~ 9 ~ s7 5 8 F~ u.......... .'~
- 2~ -
one or tw{) substituents select:ed frum the group
~ consisting of:
i) (Cl-C4)-alkyl,
ii) -O-(C 1 -C4)-alkyl,
S iii) -Co[NR7 12.
iv) P, Cl, Br or 1,
v) -CooR7.
vi) -NH~,
vii) -NHI(Cl-C4)-aLkyl],
viii) -N[(C;-C4)-aL~cyll2,
ix~ -CONrCH2CH21~Q;
(c) -(C-l-C4)-alkylaryl, wherein aryl is as de~med
above,
(d) (C3-C7)-cycloalkyl,
R7
N~N
(e) N- N, or
(f) CF3;
R7 and Rl I on the same nitrogen atom they can Join together to
form a ring selected from the group consisting of:
morpholinyl, piperazinyl, or pyrrol~yl, or
Q is O, S or -NR7;
R12 is
(a) H
(b) (Cl -C6)-alkyl, unsub.stituted or substituted ~,vith
one or two sub~stituent~. selected fronl the group
eonsisting of:
i) -OH7
ii) -O-(C I -C4)-alkyl,
iii) -O-(C I -C4)-cycloalkyl,
iv) -S(O)n-{Cl -C4)-alkyl,
~ wo 96~90~ 1 9 5 7 5 ~ I ~ " ~ ~ 9~ 57
v) -NR7R 11
vi) -cooR7
vi~ CoNHR7
viii) -OCORI l~
ix) -CoNR7RI 1,
x) NR7CoNR7RI 1
xi~ -NR7CooRl 1~
xii) -C(R6)(oH)-C~R6)(R7)(oH),
xiii) -So2NR7Rl 1, or
R7
~N'"N
xiv) N--N;
(c) (C3-C7)-cycloalkyl,
(d) -oR7,
(e) -CooR7
(f) -CONH2
(g) -CONR 16OH~
(h) -CoNR7RI 1
(i) -CoNR7Co2R7
(j) -Nl 12~
(k) -NR7Rl 1,
(I) -NR7CoNR7R1 1~
(m) -NR7CooRl 1,
(n) -C(R6)(oH)-C(R6)(R7)(oH),
(o) -So2NR7Rl I,
(p~ -S(0)2NR7CoRI I,
(~I) -S(0)2NR7Co2R I I,
3~ (r) -S(0)2NR7CoNR7RI 1,
(s) -NHS02RI 1,
(t) -NR7So2NR7R~ 1,
(u) -CONHS02R1 1,
W0 9611)4905 ~ 2 1 ~ ~ 7 5 ~ J --
- 24 -
(v) -CO-amino acid, wherein amino acid i.5 defined as
an L-or D- arnino acid selected from the group
consisting of Ala, lle, Phe, Asp, Pro ~md Val and
whicll can be further substituted as a (C'l-C6)-
alkyl ester or an amide, or
R7
o (w) N-N;
X is
(a) o,
(b~ -S(O)n,
(c~ -NR7-,
(d) -CH2O,
(e) -cH2s(o)n
~f) -CH2NR7-,
(g) -ocH2
(h) -N(R7)CH2-~
(i) -S(O)nCH2-, or
~') -single bond;
Z is:
(a) -CO2H,
(b) -Co2R 1 3,
(c) -CONH-(tetrazol-S-yl),
(d) -CONHSO2ORI I
(e) -CoNHSo2NR7R1 1
(:f) -CONH~O2-aryl. wherein a~l is deit'ined as
phenyl or naphthyi which is l",~"I,.~ lt d or
substituted with one, two or three substituents
selected from the group cc nsistillg ol':
i) (Cl-C4)-alkyl,
ii) -O-{CI -C4)-alkyl,
t, ,~ ~
W0 96/04905 '~ 2 t 9 5 7 5 8
- 25 -
iii) -CoNR7R 11,
iv) F, Cl, Br or 1,
v) -CooR7,
vi) -NH2,
vii) -NH[(CI -C4)-alkyl]~
viii) -N[(C I -C4)-alkyl]2,
ix) -phenyl,
x) -OH,
xi) -OCH2CH2OH,
xii) -CF3;
~g) -CONHSO2-(Cl-Cg)-alkyl, wherein the alkyl
group is unsubstituted or substituted as defined in
R4(b),
(h) -CONllS02-~Cl-C4)-perfluoroalkyl,
(i) -tetrazc~ -yl,
(j) -CONHS02-heteroaryl, wherein heteroaryl is
defined as c~rb~olyl, furyl, thienyl, pyrrolyl,
isothiazolyl, imidazolyl~ isoxazolyl, thiazolyl,
oxa~olyl. pyrazolyl, pyrazinyl, pyridyl,
pyrimidyl, purinyl or quinolinyl, which is
unsubstituted or substituted v~ith one, tu,o or three
substituents selected from the grc)up cclllsisting of:
i) (Cl -C4)-alkyl,
ii) -O-(C I -C4)-alkyl,
2s iii) -CoNR7RI 1,
iv) F, Cl, Br or 1,
v) -CooR7,
vi) -NR7CoNR7R l l, and
vii) -NR7CooR~ I;
(k) -SO2Nl~CO-aryl, wherein aryl is defined in Z(d)
above,
(I) -SO2NHCO-(C;l-C'~)-alkyl, wherein the alkyl
group i.s lln~llh~titl1t~d or substituted as defined in
R4(b),
~0 ~6/04sos ~ 1 9 5 7 5 8 r~ 67--
- 26 -
(m) -SO2NHco-(c I -C4)-perfluoroalkyl,
(n) -SO2NHCO-heteroaryl. wherein heteroaryl is a.s
defined in Z~g) above,
(o) -SO2NI ICON(R I 1 )2 wherein the R I I groups are
the same or different,
(p) -Po(oR7)2~ ~rherein the R7groups are the 6ame
or different? or
(q) -PO(R I ] )oR7;
R13 is:
(a) ~cl-c4)-alkyl~
(b) Cl IR14-O-COR] ~,
(c) CH2CH2-N[(CI-C2)-alkyll2,
(d~ CH2CH2-N[cH2cH2]2O~
(e) (CH2CH2Ojy~O-[(CI-C4)-alkyl:], wherein y is I or 2,
(f) phenyl, naphthyl, CH2-phenyl or CH2-naphthyl,
where phenyl or naphthyl i.s .substituted or
ull~u~;.LiLuL~d with
C O2-(C'I-C4)-alk
-CH~ ~CH ~
(9) o~fO '
0 0
(h) ~o,
(i~ ~3 . or
U) -CH2
O~o; and
\
~ W0 96/04905 ~ ! 2 1 q 5 7 5 8
R 14 and R 15 independently are (Cl -C6)-alkyl or phenyl; and
R16 is H, (Cl-C6)-alkyl, or (Cl-C~)-alkylphenyl.
An embodiment of the compounds of Forrnula II are the
compounds of Forrnula 111:
R12
X Z
R3b--a~
R3 R
R2
lll
or a pharmaceutically acceptable salt theleof, wherein:
Rl~ R2, R3a and R3b are independently:
(a) H,
(b) F, Cl, Br~ or 1,
2~ (C) -N02,
(d) (C I -C~,)-alkyl,
(e) -oR7,
(f) -NHC0-(C I -C~)-alkyl,
(g) -NHC0-O(C I -C4)-alkyl,
o (h) -C)-(CH2)m-OR7,
(i) CoNR7Rl 1, or
Ci) -C001~7;
R1 and R2 on adjacent carbon atoms can be joined together to
form a ring structure:
2~gr758
~o s6~04sos ~ 7
- 28 -
A l~plese~
a~ -Y-C(R4)=C(R5)-,
b) -Y-C~R4)=N -~
c) -Y-N=C(R4)-,
d) -Y-[C~R6)(R6)]s -Y-,
e) -~-C'(R6)(R6)-C'(R6)(R6)-,
f) -C~R4~=C(R5)-Y-~
g) -N=C(R4)-Y-,
h) -~:'(R6)(R6)-C(R6)(R6) -Y-, or
i) -C(R4)=C'(R5)-C(R4~=C(R5)-;
m is 2, 3 or 4,
n is1), 1 or2,
sis I or2,
2s
Y is -O-, -S- and NR7
R4 and R5 are independently:
(a) H,
(b): (Cl-C6)-alkyl,
(c) (C3-C7)-cyc]oalkyl,
(d) F, Cl, Br, 1,
(e) -NR7CooR I I .
(f~ -So2NR7Rl I
WO 96/04905 ' 2 ~ ~ r 7 ~ o PCT/US95/09967
- 29 -
- (g) -O-(C I -C4)-alkyl,
(h) -S(O)n-(C1-C4)-alkyl, or
(i) -NHS02R l l,
R6 is:
(a) H, or
(b) (C I -C4)-alkyl. or
(c) Cl, or F:
oR7 is:
(a) H,
(b) (C I -C6)-alkyl,
(c) phenyl, or
(d) benzyl;
R8 is:
(a) H,
(b) (C I -C6)-alkyl, or
(c) phenyl;
R9 and R10 are independently:
(a) H,
(b) (Cl-C6)-alkyl, unsubstituted or substituted with
(C3 -C7)-cycloalkyl,
(c) Cl, Br, F, 1,
(d~ (C I -C6)-alkoxy, or
(e) hydroxy-(CI-C6)-alkyl;
Rll is
(a) (Cl -C6)-alkyl, unsubstituted or sub.stituted with a
.substituent selected from the group consisting of:
i) -oR7,
-N[R7]2,
iii j -NH2,
W0 96/~W905 ~ 9 5 7 5 8 . ~ r ~ _7 --
- 30 -
iY) -CooR7.
v) -NICH2cH2]2Q,
vi) -CF3. or
vii) -CoNiR7)2;
(b) aryl, wherein aryl is defined as phenyl or
naphthyl whic:h is un.substitllted or s~ fitlltt~d vvith
one or two snhsrjtllenls selected from the group
g of:
i) ~Cl-C4~-alkyl,
o ii) -O-(CI-C4)-alkyh
iii ) -Co[NR7]2.
iv) P, Cl, Br or 1,
v ) -CooR7,
~/ i) -NH2~
Vii) -NHI(Cl-C4)-alkyl],
viii) -N[(cl-c4l-alkyl]2~
ix) -CON[CH2CH2]2Q,or
~c) -(Cl-C4)-aL~ylaryl, wherein aryl is a.s defined
above,
~d) (C3-C7)-cycloalkyl,
R7
N~N
~11
~e) N , or
(f) CF3;
R7 and R 1 l on the sarne nitrogen atom they cun jOill together to
forrn a ring selected from the group consistulg of.
morplloli:nyl, piperazinyk or ~pyrrolyl, or
QisO,Sor-NR7;
12 is
(~1) H,
. , .
~ W096/04905 . . i~ 2! 9 5 758 r~ 7
~1
~ (b) (Cl-C6)-alkyl. whereill alkyl is defined as
unsubstituted or substituted with one or two
substituent.s selected from the group consisting of:
i) -OH,
ii) -O-(C I -C4)-alkyl,
iii) -O-(CI -C4)-cycloalkyl,
iv) -S(O)n-(CI -C4)-alkyl,
iv) -NR7-(C I -C4)-alkyl,
v) -NR7R 11,
Vi) -cooK7
vii) -CoNHR7
viii) -OCORI 1,
ix) -CoNR7R l l,
~) -NR7coNR7R 11,
xi) -NR7cooRl 1,
xii) -C'(R6)(oH)-C(R6)(R7)(oH),
xiii) -So2NR7Rl 1, or
R7
~N~N
xiv) N--N,
(C) -CooR7
(d) -CONH2~
(e) -CONR 1 6OH,
2 5 (f) -CoNR7Rll~
(g) -CoNR7Co2R7,
(h) -C(R6)(OH)-C(R6)(R7)(OII), or
(i) -CONHS02RI 1,
(j) -S02NR7R1 1,
(k) -NR7So2NR7Rl 1,
(I) -CO-amino acid, wherein amino acid is de:t'ined as
an L-or D- amino acid selected from the group
consi.sting of Ala, lle, Phe, Asp, Pro and Val and
which can be turther substituted as a (Cl-C6)-
alkyl ester or an amide, or
w0 96/0490~ ? 1 ~ ~; 7 5 8 ~ f~
R7
(m ~ N- N ;
is
(a) -O,
(b) -NR7-, or
(c) -single bond;
Z is:
(a) -CO2H,
(b) -CO2R 13 ~
(C) -CONI-l(tetrazol-5-yl),
(d) -CoNHSo2NR7R 11,
(e) -CONIlS02-aryl. wherein aryl is defined a~
phenyl or naphthyl which is unsubstituted or
substituted with one, two or three substituents
selected from rhe group consisting of:
i) (Cl-C4)-alkyl,
ii~ -O-(CI-C4)-alkyl.
iii) -CoNR7RI I,
iv) F, Cl, Br or 1,
v) -CooR7,
vi) -NH2,
vii) -NH[(CI -C4)-alkyl],
viii) -N[(cl-c4)-alkyl]2
ix) -phenyl;
(f~f -CONHSO2-(CI-Cg)-alkyl, wherein alkyl is
unsubstituted or substituted as defined in R4(b~,
(g~f -CONHSO2-heteroaryh wherein heteroaryl is
defined as carbazolyl, furyl, thienyl, pyrrolyl,
isot~hiazolyl~ imidazolyl, isoxazolyl, thiazolyl,
oxazolyl. pyrazolyl, pyrazinyl, pyridyl,
~ WO 96/04905 ~ , 2 ~ 9 ~ 7 5 ~
~ pyrimidyl, purinyk or quinolinyl~ wh:ich is
unsubstituted or .substituted with one, two or three
b~iiiue~ki selected from the group consisting of:
i) (C l -C4)-alkyl,
ii~ -O-(Cl-C4)-alkyl~
iii) -CoNR7RI 1,
iv) F, Cl. Br or 1,
V) -CooR7,
vi) -NR7CoNR7R l l, and
Vii) -NR7CooRI l;
~h) -tet:razol-5-yl;
R13 is (Cl-C4)-alkyl; and
Rl6 is H, (C1-C6)-alkyl, or (Cl-C6)-alkylphenyl.
A subclas.s of the compounds of Formula III are the
compounds of Formula IV:
R12
Rg~
Xl,
R3rd J~l R
IV
or a ph~nn u-ellti~,llly acceptable salt thereof, wherein:
R l and R2 taken together form the ring structure:
2 7 ~ 5~ ~ --
W0 9610'19aS ~ ,.,. C,,.~7
;
- 34 -
A
~ ; ~
A repre.sents:
a) -Y-lC(R6)(R6)],s -Y-. or
b) -C(R4)=C(R5)-C~R43=C(R5)-;
sis l or2;
Y i~ -0-;
R3a is:
(a) H.
(b~ F, C1, Br, or I,
(c) (C'l-C'6)-alkyl,
(d) -oR7~
(e) -0-(Cl-12)m-OR7,
(f) -CoNR7Rl 1, or
(g) -CooR7;
mis2,30r4;
R4 and RS are independently:
(a) H,
(b) (Cl-c6)-alkyl~
(cl (C3-C7)-cycloalkyl,
(d) F, Cl, Br, I,
(e) -NR7CooR1 1,
(f) -S02NR7R l 1
(g) -0-(C l -C4)-alkyl,
(h) -S(O)n-(Cl-C4)-alkyl, or
(i ) -NHS02R 11;
~ W0 96/0490~ 2 1 9 5 7 5 ~ '05 :7
nisO, 1 or2,
R6 is:
(a) H, or
(b~ (Cl-C4)-all~yl, or
(c) Cl, or F;
R7 is:
(a) H,
o (b) (C'l-C6)-alkyl,
(c) phenyl, or
(d) benzyl;
R~ is:
~a) H.
(b) (Cl-C6)-alkyl, or
(c) phenyl;
R9 is:
(a) H,
(b) (Cl-C6)-alkyl, unsubstit:utecl Ol substituted witl
(C3 -C7)-cycloalkyl,
(c) Cl, Br, F, I,
(d) (C I -C6)-alkoxy, or
(ei hydroxy-(CI-C6)-alkyl;
Rll is
(a) (Cl-C6)-alkyl, unsubstituted or substitwted with a
- substituent select:ed from the group consisting of:
3 o i) -oR7~
ii) -N[R7]2,
iii~ -NH2,
iv) -CooR7,
v) -N[CI{2CH2]2Q.
wo ~go~ ~ ~ 2 ~ 9 ~ 7 5 ~
- 36 -
vi) -CF3, or
vii) -CoN(R7)2;
~b) aryl, wherein aryl is defined as phenyl or
naphthyl which is unsubstituted or substituted with
one or two substituents selected from the group
Coll.si.st~ g of:
i) ~CI-C4)-aLkyl,
ii) -O-(C I -C4)-alkyl,
iii) -CO[NR7 12.
iv) F. C]~ Br or 1,
V) -CooR7,
vi) -NH2,
vii) -NH[(Cl -C4)-alkyl],
~viii) -N[(CI-C4)-alkyl]2, or
ix) -CON~CI ~2CE-12]2Q;
(c3 -~CI-C~)-alkylalyl, wherein a:ryl is as det'ined
ahove,
(d) (C'3-C7)~cycloalkyl,
N
'"N
~e) N , or
(f) CF3;
R7 and R I I on the s~ne nitrogen atom they c.m jo;n tc~gether to.
form a ring selected from the group consisting of:
morpholinyl7 piperazinyl, or pyrrolyl, or
Q is O, S or -NR7;
R12 is
~a~ El,
(b) (C1-C6)-alkyl, wherein alkyl is defined as
unsubstituted or substituted with one or two
substituents selected from the group collsisting of:
~ w096~04go5 ;; ; ~ 2i 9575~ P~ 7
~ i) -OH,
ii) -O-(C 1 -C4)-alkyl,
iii) -O-(CI -C4)-cycloalkyl,
iv) -S(O )n-(C I-C 4)-alkyl,
iv) -NR7-(Cl-C4)-alkyl,
v) -NR7Rl 1,
vi) -cooR7 ~
vii) -CoNHR7
viii) -oCORl 1
iX) -CoNR7R1 1
x) -N-R7coN-R7R 1
xi) -NR7CooR 11~
xii) -C(R6)(oH)-C(R6)(R7)(oH), or
xiii) -So2NR7Rl 1,
R7
N'N
xiv) N--N
(C) -cooR7
(d) -CONH2~
(e) -CONR I 6OH,
(f) -CoNR7R1 1,
(g) -CoNR7Co2R7,
(h) -C'(R6)(oH)-C(R6)(R7)(oH), or
Z5 (i) -CONllSO,RI 1,
(j) -so2NR7Rl 1,
(k) -NR7So2NR7Rl I,
(I) -CO-amino acid, wherein amino acid is defined as
an L-or D- amillcl acid selecled from the group
consisting of Ala, lle, Phe. Asp. Pro and Val and
whicll can be furthel substitut:ed as a (Cl-C6)-
alkyl ester or an amide, or
wo s6~04sos ~ t Y ~ 7 ~ ~ r~
- 38 -
R7
N~N
(ro ~ N
xi~
(a) -O-,
(b) -NR7-~ or
(c~ -single bond;
Z is:
(al -C'02H,
(b) -Co2R13,
(c) -CONH-(tetrazol-~ -yl j,
(d~ -CONllSO2NR7Rl l,
(e) -CONl-ISO2-aryh wherein aryl is def~:ned as
phenyl or naphthyl which is u~ b~liluled or
substituted v~ith one, two or three .substituent.s
selected from the group consisting of:
i ~ (C I -C4)-alkyl,
ii) -O-(C l -C4)-alkyl,
iii) -CoNR7R l l,
iv) F~ Cl, Br or I~
v ) -CooR7,
vi) -NH2~
C I -C4)-~lkyl],
viii) -N~Cl -C4)-alkyl]2,
ix) -phenyl;
(f) -CONHSO~-(C1-Cgj-alkyl, wherein alkyl is
unsubstituted or substituted as defined ill R4~b),
(gi -CONHSO~-heteroaryl, wherein heteroaryl is
defined as carbazolyl, furyl, thienyl, pyrrolyl,
isothiazolyl, imidazolyl, isoxazolylt thiazolyl,
oxazolyl, pyrazolyh pyrazinyl, pyridylt
~ wo 96io490~ 2 1 9 r 7 c ~3
- 39 -
~ pyr:imidyl~ purinyl, or quinolinyl, ~hich is
Imsllhstitl~t~d or substituted with one, two, or
three substituents selected from the group
consisting of:
i) (Cl-C4)-alkyl,
ii) -O-(C I -C4)-alkyl,
iii) -CoNR7R1 1,
iv) F, Cl, Br or 1,
v ) -CooR7 .
vi) ~1R7CoNR7RI 1, and
vii) -NR7CooRI l;
(h) -tetrazol-5-y l;
R13 is (Cl-C4)-alkyl; and
R16 is H~ (Cl-C~)-all;yl, or (Cl-C6)-alkylphenyl.
A second embodiment of the cc~mpounds of Formula n
are the compounds of Forrnula V:
R12
R~ Rl~
X~z
R3b--~--R3a
R2 R1
or a pharmaceutically acceptable salt thereof, wherein:
Rl, R~, R3a and R3b are independently:
(a) H,
q ~ 7 5 ~
W09~v~04905 ~ C3~vl
-40 -
(b) F, Cl, Br, or 1,
(c) -NO2,
(d) (cl-c6)-alk
(e) -oR7,
(f) -NHCO-(C I -C4)-alkyl,
(g) -NHCO-O(C 1 -C4)-alkyl,
(h) -(:~ -(CH2)m-oR7,
(i) CoNR7Rl 1, or
(j) -CooR7;
m is 2, 3 or4,
R4 and R5 are independently:
(a) H,
(b) (Cl-C6)-alkyl,
(c~ (C3-C7)-cycloalkyl,
(d) F. Cl, Br, 1,
(e) -~R7CooRl I,
(f3 -So2NR7Rl 1,
2 0 (g) -0-(C I -C4)-alkyl,
(h) -S(O)n-(C1-C43-aLiyl, or
(i) -NHSO~RI l;
n is (), I or~,
2~
R~ is:
(a3 H,
(b) (C I -C4~-alkyl,or
(c i Cl or F,
~ W096/04905 ~ 9~758 1_1lu.. '~,3,_
- 41 -
- R7 is:
(a) H,
(b) (C I -C6)-alkyl,
(c) phenyl, or
(d) benzyl;
R~ is:
(a) I 1,
(b) (C I -C6)-alkyl, or
(c) phenyl;
R9 and RIQ are indepelldently:
(a) H,
(b) (Cl-C6)-alkyl, unsubstituted or substituted with
(C3-C7)-cyClOalkyl~
(c) Cl, Br, F, 1,
(d) (C 1 -C6)-alkoxy, or
(e) hydroxy-(Cl-C6)-alkyl;
R 11 i~i
(a) (Cl-C6)-alkyl, unsubstituted or suhstituted with a
substituent selected from the group consisting of:
i) -oR7,
ii) -Nl R7]2,
2s iii) -NH2,
i', ) -CooR7,
v) -N[ CH2CH2] 2Q.
vi) -CF3. or
vii) -CoN(R7)2;
(b) aryl, wherein aryl is defïned as phenyl or
naphthyl which is unsubstituted or substituted with
one or two .substituents selected from the group
consisting of:
i ) (C 1 -C ~ )-alkyl,
WO 96/r4905 ' ~ '. 2 ~ q 5 ? 5 8 r~ 3 l--
- 42 -
ii) -O-(C I -C4)-alliyl.
iii) -CO[NR712,
iv) F. Cl, Br or I~
v) -CooR7,
vi) -NH2
vii) -NH[(C 1 -C4)-alkyll,
viii) -N[~CI-C4)-alkyl]2~ or
ix) -CONl CH2CH212Q;
(c) -(C1-C4~-alkylaryl, wherein aryl is as defined
o above,
(d) (C3-C7)-cycloalkyl,
R7
~N~N
(e) N , or
(f~ CF3,
R7 and R11 on the same nitrogen atom they can join together to
forrrl a ring selected from the group consisting of
morphc linyl, pipera~ yl. or pyrrolyl, or
Q is O, S or -NR7;
R12 is
2s (a) H,
(b) (Cl-C~)-alkyl, wherein alkyl is defmed ias
unsubstihuted or~substituted with one or two
sllb~tihlents selected from the group consisting of:
i) -OH,
ii) -O-(Cl-C4)-alkyl,
iii) -O-(C 1 -C4)-cycloalkyl,
i~i ) -S~O)n-(C I -C4)-alkyl,
iv) -NR7-(CI -C4)-alkyl,
v) -NR7R
vi ) -cooR7
~ WO g6104905 . ! ~ ~ ~ 2 ~ 9 ~ 7 5 8 ~ S ~
-43 -
vii) -CoNHR7
viii) -OCOR 11,
ix) -CoNR7R 11
x) -NR7CoNR7R
s xi) -NR7CooRl 1~
xii) -C(R6)(oH)-C'(R6~(R7)(oHj, or
xiii) -So2NR7RI I, or
R7
<N'N
xiv) N-N;
(C) -cooR7
(d) -CONH2~
(e) -CONR I 6OH,
(f) -CoNR7R 11,
(g) -CoNR7Co2R7,
(h) -C(R6)(OII)-ClR6)(R7)(O}-I), or
(i) -CONHS02RI 1,
(j) -so2NR7R1 1,
(k) -NR7So2NR7Rl 1,
(I) -CO-amino acid, wherein amino acid i~ defined as
~Ul L-or D- amino acid selected from the group
consi.sting of Ala. Ile, Phe, Asp, Pro and Val and
which can be further substituted as a (Cl-C6)-
alkyl ester or an amide, or
R7
~,N'"N
- (m) N-N;
xis
(a) -O,
~b) -NR7-, or
(c) -single bond;
W096f04905 ~ 2195758 r~ o. I--
-44 -
Z is:
C02H,
(b) -CO2R13,
(c) -CONH-(tetrazol-5-yl),
(d) -CoNHSo2NR7R I I
(e) -CONHSO2-aryl, wherein aryl is defined as
phenyl or naphthyl which is unsubstituted or
.substituted with one. two or three substiluents
.selected from the group consistillg of:
i) (Cl -c4~-alky~l~
ii) -O-(CI -C4)-alkyl,
iii) -CoNR7R1 1,
iv) F, Ch Br or 1,
v) -Coo'R7,
vi) -NH2,
vii) -NH[~CI -C4)-alkyl j,
viii) -N[(C I -C4)-alkyll2,
ix) -phenyl:
(f) -CONllSO2-(Cl-C~)-alkyl, wherein alkyl is
un.substituted or substituted as defined in R4~b),
(g) -CONHS02-heteroaryl, wherein heteroaryl is
deFuled as carbazolyl, furyl, thienyl, pyrrolyl,
isothiazolyl, imidazolyl, iso~:azolyl, th:iazolyl,
oxazolyl, pyrazolyl, pyrazinyl, pyridylt
pyrimidyl, purinyl, or quinolinyl, which is
msllhstitllted or substituted with one, two or three
suhstihl~nts selected from the group consisting of:
i) (Cl-C4)-alkyl,
ii) -O-~CI -C4)-alkyl,
iii) -CoNR7R I I ~ r
iv) F, Cl, Br or I,
v ) -CooR7,
vi) -NR7CoNR7Rl 1, and
~ W096104905 .: : ,,'; 2t95758 1~1,1 ,~,
- 45 -
vii) -NR7CooRI l;
~h) -tetrazol-5-yl;
R l 3 is: (C l -C4~-alkyl; and
Rl6 is H, (Cl-C6)-alkyl, or (C'l-C'6)-alkylphenyl.
A third embodiment of compounds of Forrnula II are the
compounds of Fonnula VI
R12
R9J¢l R10
X~Z
R3~ R2
v
or a pharmaceuticall~ acceptable salt thereof. wherein:
Rl and R2 are represented by the iollowing ring structure:
~-
~;
A represents:
a) -Y-LC(R6)(R6)]~ -Y-. or
b) -C(R4)=C(R5)-C(R4)=C(R5)-;
sis l or2,
2 ~ 9 5 7 5 8 ~
WO9C104905 ~,II~Jw.,.~, 7
- 46 -
Y is -O-, -S- and NR7;
R3~1 and R3b are independentlS~:
(a) H,
(b) P, Cl, Br, or 1,
(C) -N02,
(d) I C I -C,~)-alkyl,
(e) -oR7,
(f) -NHCO-(Cl-C4)-alkyl,
(g) -NHC'O-O(C1-C4)-alkyl,
~h) -o-(cH2im-oR7
(i) c'oNR7Rll~ or
(i ) -CooR7;
m i~ 2, 3 or4,
R4 and R5 are independently:
(a) 1-1,
(b) (C1-C~)-alkyl,
(c) (C3-C7)-cycloalkyl,
(d) F, Cl, Br, 1,
(e) -NR7cooRl I,
(f) -S02NR7R 11,
(g) -O-(C l-C4)-alkyl,
(h) -S(O~n-(CI -C4)-alkyl, or
(i) -NHSO2RI l;
nis0, 1 or2,
33 R6 is
(a) H, or
(b) (Cl-C4)-alkyl, or
(c) Cl or F;
9 5 7 5 ~
WO 96/0490S l _ " "~, -/,.. ,
- 47 -
R7 is:
(a) H,
(b) (Cl-C6)-alkyl,
(c) phenyl, or
(d) benzyl;
R8 i.s:
(a) H,
(b) (C I -C6)-alkyl, or
1 o (c) phen~T l;
R9 and R10 are independently:
(a) H,
(b) (Cl-C6)-alkyl, unsubstituled or substituted with
(C3 -C7 i-cycloalkyl,
(c) Cl, Br, F, 1,
(d) (C I -C6)-alkoxy, or
(e) hydroxy-(CI-C6)-alkyl:
Rl I is
(a) (C I -C~)-alkyl, unsubstituted or substituted with a
subslituent selectecl from the group consisting of:
i) -oR7,
ii) -Nl R7 12.
iii) -NH2.
iv) -CooR7,
v) -N[CH2CH2 12Q.
vi) -CF3, or
vii) -CoN(R7)2;
(b) aryl, wherein aryl is defined as phenyl or
~ naphthyl which is llncilb~titlltt~-l or substituted with
one or two ~ub~lilu~ selected from the group
consisting of:
i) (C I -C4)-alkyl,
2 1 ~ 5 7 5~ --
W0 96/1~4905 1
- 48 -
ii) -O-(C l-C~-alkyl~
iii) -Co~NR7]2
iv) F, Cl, Br or 1,
v ) -CooR7,
vi) -NH2,
vii) -2~H[(CI -C4)-alkyl],
viii) -N[(Cl-C4)-alkyl~2, or
ix) -CON[CH2CH2]2Q;
(c) -(Cl -C4)-alkylaryl, wherein aryl is as de.fined
above,
(d) (C3-C7)-cycloalkyl,
R7
~N~"N
~e) N--N or
(f) CF3;
K7 and Rl 1 on the .sarme nitrogen atom they can join together ~o
i'c~rm a ring selected from the group consist:ing of:
morpholinyl, piperazinyl, or pyrro:lyl, or
Q is O, S ar -NR7;
R12 is
2s (~) H,
(b) (C'l-C6)-alkyl, whereill alkyl is defined a.s
unsubstituted or substituted with one or t vo
substituents selected from the group consisting of:
i) -OH,
ii) -O-(CI-C4)-alkyl,
ii i) -O-(C I -C4)-cycloalkyl,
-S(OJn-(cl -C4)-alkyl,
iv) -NR7-(C'1 -C4)-alkyl,
v) -NR7R 11,
vi) -cooR7 ~
2 ~ 9575B
W0 96104905 ~ F~ ,. C7
- 49 -
vii ) -CoNHR7
viii) -OCOR] 1,
ix) -CoNR7RI 1~
x) -NR7CoNR7R1 1,
S xi) -NR7CooK l l ~
xii) -C(R6)(oH)-C(R6)(R7)(oH),
xiii) -So2NR7Rll~ or
R7
1 0 ~N~N
xiv) N-;
(C) -cooR7
(d) -CONH2~
(e) -CONR 16OH,
(f) -CoNR7R1 1,
(g) -CoNR7Co2R7,
(h) -C(R6)(oH)-C(R6)(R7~(oH), or
(i) -CONHS02RI 1,
(j) -SC)2NR7R1 1,
(k) -NR7So2NR7Rl 1,
(I) -CO-amino acid, wherein amino acid is defined as
an L-or D- amino acid selected from the group
consisting of Ala, lle, Phe, Asp, Pro and Val and
which can be filrther substituted as a (C I -C6)-
2s alkyl ester or an amide, or
R7
N~N
~ N
xis
(a) -O,
(b) -NR7-, or
(c) -single bond;
WO 9G/04905 ~ 9 :~ ~ 5 8 ~ 7--
Z is:
(a) -CO2H,
(b) -CO2R 13,
(c) -CONH-~tetraz.ol-~-yl),
(d) -CoNHSo2NR7R 11,
(e) -CONHSO~-aryl, wherein aryl is defined as
phenyl or naphthyl which is unsubstituted or
substituted witll one, two or tllree substituen~s
selected from the group consisting of:
i ~ (C l -C4)-alkyl,
ii) -O-(C I -C4)-alkyl,
iii) -CoNR7R 11,
iv) F, Cl. Br or 1,
v) -CooR7,
v i ) -NH2,
vii) -NH[(C I -C4)-alkyl],
viii) -Nl(cl -C4)-alkyl]2,
ix) -phenyl,
(f) -CONHSO2-(CI-C~)-alkyl, whereill alkyl is
unsubstituted or substituted as defined in R4(b),
(g i -CONHSO2-heteroaryl, whereill heteroaryl is
defined as carbazolyl, furyh thienyl, pyrrolyl,
isothiazolyl, imidazolyl, isoxazolyl, thiazolyl,
o~;azolyl, pyrazolyl, pyrazinyl, pyridyl,
pyrim:idyl, purinyl, or quinolinyl, which is
unsubstituted or sub.stituted with one, two or three
substituents selected from the group consisting of:
i) (C1 -C4)-alkyl,
3 ~ -O-(C l ~C4)-alkyl,
iii) -CoNR7Rl I
iv~ F~Cl,Brorl,
V) -CooR7,
vi) -NR7CoNR7R 1 l, and
~ wo96/0490~ ; 2 t 957S 8 ~IIL~ ' 7
- 51 -
vii) -NR7CooR 11;
(h) -tetrazol-5-yl;
R13 is (Cl-C4)-alkyl; and
R16 is H~ (Cl-C6)-alkyl, or (Cl-C6)-all;ylphenyl.
An embodiment of the compoullds of Forrnula I are:
2-1(2~6-dipropyl-4-hydroxymethyl)pilenoxy]-2-(3-methyl!lhenyl)acetic
acid;
2-[(2~6-dipropyl-4-hydroxymethyl)pherloxy]-2-(4-pllenoxypllenyl)-
acetic acid;
2-[(2,6-dipropyl-4-hydroxymethyl)p}lenoxyl-2-(4-phenylpllenyl)acetic
acid;
2-[(2,6-dipropyl-4-hydroxymethyl)phenoxyl-2-(3-carboxyphenyl)acetic
20 acid;
2-[(2,6-dipropyl-4-hydroxymethyl)pllenoxy j-2-(3,4-ethylenedioxy
phenyl)acetic acid;
2-[(2,6-dipropyl-4-hydroxymethyl)phenoxy]-2-(3,4,5-trimethoxy-
25 phenyl)acetic acid;
2-~(2,6-dipropyl-4-llydroxymethyl)phenoxy]-2-(3~4-methylelledioxy-
phenyl)acetic acid;
30 2-[(2,6-dipropyl-4-hydroxymethyl)phenoxy~-2-(3,4-dimethoxy-
phenyl)acetic acid;
2-[ (2~6-dipropyl-4-hydroxymethyl)phenoxy] -2-(3 ~5-dimethoxy-
phenyl)acetic acid;
... . . .. . . .
W0 ~6/04905 ~ 9 5 7 5 ~ r~ , 7--
- 52 -
2-((2,6-dipropyl-4-tetrazol-5-yl)phenoxy)-2-(3-bromophenyl):lcetic acid
~-1 (2,6-dipropyl-4-hydroxymethyl)phenoxyl -2-(3-bromophenyl ~acetic
acid;
2-L(2,6-dipropyl-4-hydroxymethyllphenoxyl-2-~2-napllthyl)acetic acid;
2-[(2,6-dipropyl-4-~2-hydrnx~ethyl)phenoxy~-2-(2-naphthyl~acetic
acid;
2-1 (2,6-dipropyl-4-(2-hydroxyethyl)phenoxy]-2-(3,4-1nethylelleclioxy-
phenyl)acetic acid;
2-[(2,6-dipropyl-4-(2-hydroxyethyl)phenoxy]-2-(3-methox~phenyl~-
15 acetic acid;
2-L(2,6-dipropyl-4~1 .2-dihydroxyethyl)phenoxy)l-2-~-naphthyl).~cetic
acid;
2o 2-~(2~6-dipropyl-4-(1-hydroxypentyl)phenoxy]-2-( -naplltllyllacetic
acid;
2-1~4-carboxy-2~6-dipropyl)phelloxy~-2-phenylacetic acid;
2-[(4-carboxy-2,6-dipropyl)phenoxyJ-2-(3,4-dichlurophenyl)acetic acid;
2-[(4-carhoxy-2,6-dipropyl)phenoxy]-2-(3-bromophenyl)acetic acid:
2-1(4-carboxy-2~6-dipropyl)phenoxyl-2-~3,4-methylenedioxyphenyl]
30 acetic acid;
2-[i4-carboxy-2,6-dipropyl)phenoxy]-2-(3-methoxyphenyl)acetic acid;
(N-ben~enesulfony 1~-2-[(4-(N -b~n7rnesl 1lfonyl~carboxam:ido-2,6-
dipropylphenoxyl-2-(3-bromophellyl)acetamide;
~ W096/0~9US ~ ~ 2 ~ 95758
- 53 -
(N-4-t-butylbenze[lesulfonyl)-2-(4-methoxycarbonyl-2-propylphelloxy)-
2-(3~4-methylenedioxyphenyl)acetamide;
5N-(benzenesulfonyl)-2-(4-methoxycarbonyl-2-propylphenoxy)-2-(3,4-
methylene,dioxyphenyl),-rel~ltni~P;
N-(4-phenylbenzenesulfonyl)-2-(4-methoxyc.lrbonyl-2-propyl-
phenoxy)-2-(3,4-methylenedioxyphenyl)acetamide;
N-(4-chlorobenzenesulfonyl)-2-(4-methoxycarbonyl-2-propyl-
phenoxy)-2-(3 ,4-methylenedioxyphenyl)acetamide;
N-(4-methylbenzenesulfonyl)-2-(4-methoxycarbonyl-2-propyl-
5phenoxy)-2-(3 ,4-methylenedioxyphenyl~;tcer tmi~iP
N-(5-iso-butylthien-2-ylsulfonyl)-2-(4-methoxycarbonyl -2-
propylphenoxy)-2-(3 .4-methylenedioxyphenyl),~ret~mirl,~;
20N-(4-methoxybenzenesulfonyl)-2-(4-methoxycarbcnyl-2-
propylphenoxy)-2-(3 ,4-methylenedioxyphenyl)acetamide;
N-(4-dimethylan~ obenzenesulfonyl)-2-(4-methoxycclrbonyl-2-
propylpllenoxy)-2-(3 ,4-methylenellioxypllenyl)acetamide;
N-(2-methylbenzenesulfonyl)-2-(4-methoxycarbonyl-2-
propylphenoxy)-2-(3,4-methylenedioxyphen) I)~tf et -rni~le;
N-(2-methoxycarbc~l~ylbel~zenesulf nyl)-2-(4-methoxycarbollvl-2-
30propylphenoxy)-2-(3,4-methylenedioxyphenyl)acetalnide;
N-(2-chlorobPnzP,IlP~nlfollyl)-2-(4-methoxyc~lrbonyl-2-
propylphenoxy)-2-(3,4-metllylenedioxyphenyl),l~et llni(l~,
W0 96/04905 ~ 7 !~ 9 5 7 5 3 r ~
- 54 -
N-(3-chloroben~.eneslllt'~ nyl)-2-(4-mett~ ycarb~ yl-2-
propylphenoxy)-2-(3 ,4-methylenedioxyphenyl)acetamide;
N-~phenylme,tl-~lnP,slllfonyl)-2-(4-methoxycarbonyl-2-p:ropylphelloxy)-
5 2-(3,4-methylenedioxypllenyl~acetarrlide;
N-(dansylsulfonyl)-2-14-methoxycarbonyl-2-propylphenoxy 1-2-(3,4-
methylelledioxypllellyl')acetanlide~
N-(8-quinolinesulfonyl)-2-(4-methoxycnrbonyl-2-propylp}lelloxy)-2-
(3,4-methylenediox~phenylacetamide;
N -(4-t-butylbenzerlesulfonyl)-2-(4-carboxy-2-propylphelloxy)-2-(3,4-
methylenedioxyphenyl)~rel:~mi-~P
N-(benzenesulfonyl)-2-(4-carboxy-2-propylphenoxy)-2-(3,4-
methylelledioxyphenyl)acetLqmide,
N -(4-phenylbenzcllesulfonyl)-2-(4-carboxy-2-propylphenoxy~-7-(3 ,4-
20 methylenedioxyphenyl)acetamide;
N-~4-chlorobenzenesulfonyl7-2-(4-carboxy-2-propylphenoxy)-2-(3,4-
methylenedioxyphenyl)acetamide;
N-(4-methylh~n7!~nP~ fonyl)-2-(4-carboxy-2-propylphenoxy)-2-(374-
metilylenedioxyphenyl)a~etamide,
N-(5-isobutylthien-2-ylsulfonyl)-2-(4-carboxy-2-propylphenoxy7-2-
(3 ,4-methylenedioxyphenyl)acetamide;
3~
N-(4-methoxybenzellesulfonyl)-2-(4-carboxy-2-propylphenoxy)-2-( 3 74-
methylenedioxyphenyl)~lcet~mi~P;
N-(4 -dimethylaminobe:nzenesulfonyl)-2-(4-carboxy-2-propylphenoxyJ-
3 ,4-methylene.dioxyphenyl)acetamide;
~ W0 96/04905 ' ; r~ . 67
2t,~575~
N-(2-meth~ Ibcll~cllcsulfonyl)-2-(4-carboxy-2-propylphelloxy)-2-(3,4-
met:hylenedillxyphenyl).lcetamide;
N-(2-methoxycarbonylbenzenesulfonyl)-2-(4-carboxy-2-
propylphenoxy)-2-(3,4-methylenedioxypllenyl)acetamide;
N-(2-chlorobc.lzcl.esulfonyl)-2-(4-carboxy-2-propylphelloxy)-2-(3,4-
methylenedioxyphenyl)acetamide;
N-(3-chlorobenzenesulfonyl)-2-(4-carboxy-2-propylphenc)xy)-2-(3 ,4-
methylenedioxyphellyl)acetamide;
N-(phenylmethanesulfonyl)-2-(4-carboxy-2-propylphenoxy)-2-(3 ,4-
methylenedioxyphenyl)acetamide;
N-(dansyl~ulfonyl)-2-(4-carboxy-2-propylphenoxy)-3,4-methylene-
dioxyphenyl)acetamide;
N-(~-quinolinesulfonyl ~-2-(4-carhoxy-2-propylphenoxy )-2-(3 ,4-
methylenedioxyphenyklcetamide;
N-(~-quinolinesull'(myl)-2-(4-carboxamidc)-2-propylphenoxy)-2-(3,4-
methylenedioxyphenyl)acetamide;
a-(4-carbomethoxy-2-n-propylphenoxy)-3,4-methylenedioxy-
phenylacetic acid;
N-(4-iso-propylbenzenesulfollyl)-a-(4-carbomethoxy-2-/l-propyl-
phenoxy)-3~4-methylenedioxyphellylacetamide;
N-(4-i.~o-propylbenzenesLIlfonyl)-a-(4-carboxy-2-n-propylphen(lxy)-
3,4-methylenedioxyphenylacetamide dipotassium salt;
- 2~9575û
W0 96/04905 r ~ u., a~,57--
- 56 -
a-(2-iso-butyl-4-carbomethoxyphenox!, )-3~4-methylenedioxy-
phenylacetic acid;
/\'-(4-iso-propylbenzenesulforlyl}-a-(2-iso-butyl-4-carbomethoxy-
5 phenoxy)-3,4-methylenedioxyphenylacetamide;
/~,'-(4-i.s~o-propylbell7en~sll1fo1lyl)-a-(2-iso-butyl-4-carboxypheno.Yy)-
3,4-methylenedioxyphe,nyklcet,lmide;
~ N-(4-iso-propylbenzenesulfonyl~-a-~2-/t-propyl-4-methoxycarbonyl-
phenoxy)-a-met~lyl-3~4-rlletllylenedioxyphellyl ~ret/mli~ir.;
N-(4-is(~-propylbrn7~nf~slilfonyl)-a-(2-n-propyl-4-carboxyphenoxy)-a-
rmethyl-3~4-metllylenedioxyphenyl~retslmi-lr dipotassium salt;
(4-i~o-propylb~n7l~nt~lllfonyl~-a-(2-n-prop~l-4-carhoxamid
phenoxy)-3.4-methylenedioxyphe.nylacet;lmide;
IV-(4-lso-propylber~enesulfonyl~-a-(2-ll-propyl-4-hydroxymethyl-
20 pllenoxy)-3,4-meth~ enedioxyphenyl~ t:~mi(l~,
N-(4-isd-propylken7PnP~Il If onyl)-a-(4-forrnyl-~-n-propylphenoxy)-3,4-
methylenedioxyphenyl~ret~mide,
25 a-(4-acetyl-2-n-propylphenoxy)-3,4-methylenedioxyphenylacetic acid;
N-(4-isv-propylbenzenesulfonyl~-oc-(4-acetyl-2-/l-propylphenoxy ~-3 ,4-
methyleneclioxyphenyl:~retslmitlr~;
3~ a-(2-n-propylphenoxy)-3,4-methylenedioxyphenylacetic acid
N-(4-iso-propyJben7rnrs-llfonyl)-a-(2-n-propylphenoxy)-3,4-
n~etllylelledioxypheny]acetamide;
~ wog~gos ~ 2~9~758 ~ .r..; 7
(3-methoxyphenoxy)-3,4-methylenedioxyphenylacetic acid:
a-(2-(2-hydroxyethyl)phenoxy)-3,4-methylenedioxyphenylacetic acid;
5 a,-(2-(2-carbomethoxyethyl)phenoxy)-3,4-methylenedioxyphenylacetic
acid;
a -(4-hydroxymethyl-2-n-propylphenoxy)-3 ~4-methylenedioxyphenyl-
acetic acid;
a-(4-(2-hydroxyethyl )-2-n-propylpllelloxy)-3 ,4-methylenedioxyphenyl -
acetic acid;
N-(4-i,so-propylbenzenesulfonyl)-a.-(2-(2-carbomethoxyethyl)phenoxy)-
3,4-rmethylenedioxyphenyl ~ret,lmi~l.o;
N-(4-isv-propylbenzenesulfonyl)-a-(2-(2-carboxyethyl)phenoxy)-3 .4-
methylenedic~xyphenylacet~mide;
20 o-(2-(2-carboxyetllyl)phenoxy)-3,4-methylelledioxyphenylacetic acid;
N-(4-isO-propylbenzenesulfonyl)-2-(4-carbomethoxy-2-n-propyl-
phenoxy)-2-(5-methoxy-3,4-methylenedioxyphenyl)~cet~mi~;
25 N-(4-isO-propylbenzenesulfonyl)-2-(4-carboxy-2-propylphenoxy)-2-(5-
methoxy-3 ,4-methylenedioxypllellyl)acetalllide;
N-(4-is0-propylbenzenesult'onyl)-2-(4-(N-(4-isv-propylbel~ene-
sulfonyl)carboxamido)-2-propylphenoxy)-2-(5-methoxy-3,4-1nethylene-
3~) dioxyphenyl)acetamide;
N-(4-iso-propylbenzenesull'onyl)-2-(4-carboxamido-2-propylphenoxy)-
2-(5 -methoxy-3,4-methylenedioxyphenyl)acetamide;
~: ~ ? ~ ~ ~ O ~ --
WO 96/04905 ~ 7 7 5 8 . ~ ~ " . ~
- 5X -
N-(4-iso-propylbenzenesulfonyl~-2-(4-(N-methylcarbox~nido)-2-
propylphe,noxy)-2-(5-methclxy-3 ,4-methylenedioxyphenyl~ace.~slmi~
N-(4-iso-propylbenzellesulfonyl)-2-~4-(N-2-hydroxyethylcarhl)x.~ ido)-
2-propvlplle:noxy~-2-(5-methoxy-3.4-methylelledioxyphenyl)acetan1ide;
N-(4-iso-propylb ~n7e.n~sll1fonyl~-2-(4-(N-morpholinylcarhclxam jdo)-2
propylphenoxy)-2-(5-methoxy-3,4-methylenedioxyphenyl)acetamide;
N-(4-i.so-propylben7enesulfonyl)-2-(4-(N-3-methylbutylc.lrbo.~amido)-
2-propylphenoxy~-2-(5-methoxy-3 ,4-methylenedioxyphenyl)acelarl-ide:
:N-(4-i.so-propyl:benzenesulfonyl)-2-~4-(N-carboxymethylcarboxamido)-
2-propylphenoxy)-2-(5-1llethoxy-3 ,4-methylenedioAYyphellyl)acetamide;
N-(4-iso-propyl~en7f,n~ l1fonyl)-2-(4-~N-(L-Ala-OEt)carboxamido)-2-
propylphenoxy~-2-(~i-methoxy-3,4-methylenedioxyphenyl)ac.etamide;
N -(4-i.~o-propylbenzenesulfonyl)-2-~4-(N-2-ethoxycarbonylethyl-
carboxamido)-2-propylphenoxy~-2-(5-methoxy-3,4-methylelledioxy-
phenyl )acetamide;
N-(4-isv-propylbenzenesulfonyl)-2-(4-(N-~L-Ala)carbox.lmido)-2-
propylphenoxy)-2-(5-methoxy-3.4-methylenedioxypllenyl )acetamide:
N-(4-iso-propylbenzenesulfonyl)-2-~4-(N-2-carboxyethylcarboxamido)-
2-propylphenoxy)-2-(5-methoxy-3 ,4-methylenedivxyphenyl)acet~lnli(le:
N-(4-isv-propylbPn7.~n~sll1fonyl)-2-(4-(N-3-hydroxyprclpyl-
3~ carb{)x~ id{))-2-propylphenoxy)-2-(5-metho~xy-3.4-methylenedioxy-
phenyl)a cetamide;
~ W096/04905 ~ 219~75~3 r~ .J.,S/~ ~
- 59 -
- N-(4-iso-propylbenzenesulfonyl)-2-(4-(N-t:etra7.ol-S-ylcarbox,lmido)-2-
propylphenoxy)-2-(5-metho~y-3,4-methylenedioxyphellyl)acetamide;
N-(4-iso-propylben7.l~nP.slllfonyl)-2-(4-(N-3-(morpholin-4-yl)propyl-
carboxamido)-2-propylphenoxy)-2-(5-metlloxy-3.4-1llethylenedioxy-
phenyl)acetamide;
N-(4-i.so-propylbenzenesulfonyl)-2-(4-~N-(D-Ala-O~Ie)carboxamido)-
2-propylphenoxy)-2-(5 -methoxy-3 .4-methylenedioxyphenyl).lcetamide;
N-(4-isf.~-propylbenzenesulfonyl)-2-(4-(N-(D-Ala)carboxamido)-2-
propylphenoxy)-2-(5 -methoxy-3 ,4-methylened ioxyphenyl)acetamide;
N-(4-iso-propylbenzenesulfonyl)-2-(4-(N-(3-carboxymetllylpropyl)-
CarbOX.lmidO)-2-prOpylphenOXy)-2-(5-nlettlOXy-3~4-methylenedi
phenyl)acetamide;
N-(4-i.so-propylbenzerlesuli'onyl)-2-(4-(N-(3-carboxypropyl)-
carboxamido)-2-7t-propylphenoxy)-2-15-methoxy-3 .4-methylenedioxy-
phellyl)acetarllide;
N-(4-isz~-propylbenzenesulfonyl)-2-(4-(N-iso-propylcarballloyl).lmillo-
2-n-propylphenoxy)-2-(3~4-nletbylenedioxypilenyl)a(~e~mir1
a-(2-)1-prOpyl-4-nlethylanlillOSUlfOllylphenoxy)-374-methylenedi
phenylacetic acid;
N-(4-iso-propylbenzenesuli'onyl)-a-(2-n-propyl-4-methylamino-
suli'onylphenoxy)-3,4-methylenedioxyphenylacetamide pc tassium salt;
3~
N-(4-iso-propylbellzenesulf onyl)-~-[4-('cyanomethyl)-2-/1-
propylphenoxy)~l-3 ,4-methylenedioxyphenylacetamide,;
N-(4-i.so-propylbPn7.~.nPslllfonyl)-cY.-[4-(tetrllz.ol-5-ylmethyl)-2-n-
propylphenoxy)]-3 ,4-methyle,nedioxyphenylacetamide;
~1957
Wo g6/0490s ~ r ~- 5 ~ u~
- 60 -
N-(4-i.fo-propylben~enesulfollyl)~ N-(4-carbomethoxyphenylamillo)l-
3 ,4-methylenedioxyphenylacet:amide;
5 N-(4-i.so-propyl'ben~enesulfonyl)-G~-[N-(4-carboxyp}lenylclmillt))]-
3,4-methyle.nedioxyphenylllcP~:Imi~1P;
N -(3-pyridi:nesul~onyl)-2-(4-carboxy-~-propylphenoxy)-2-(3 .4-
methylenedioxypllenyl)acetatmide.;
N-(2-metllyl-3-quinolineslllfc~nyl)-2-(4-carboxy-2-propylphelloxy)-2-
(3 74-methylenedioxyphenyl)acet:amide;
N-(3 -quinoline~ulfonyl)-2-(4-carboxy-2-propylphenoxy)-2-(3 ,4-
15 methylenedioxyphenyl)~( et~mi~
N-~4-bydro~;y-~ pyridinesulfonyl)-2-(4-cLIrboxy-2-plopylphetloxy)-2-
(3,4-tnetllylelledioxyphenyl)acetamide;
20 N-(4-ethoxybPn7~l-Pslllfonyl)-2-(4-carboxy-'~-propylpllel:loxy)-2-l3,4-
methylenedioxyp}lenyl~ Pf:,mirl~;
N -(4-carboxamidobenzenesulfonyl)-2-(4-carboxy-2-propylpheIloxy)-2-
(3 ,4-methylenedioxyphenyl)acetamide;
5 N-14-(N,:N-dilrletllylcarboxamido)benzenesulfollyl]-2-(4-carboxy-2-
propylphenoxy3-2-(3 .4-methylenedioxyphenyl,l~cet~mifl.o;
N-(4-ethyltlli~:~-3-pyridinesulfonyl)-2-(4-carboxy-2-propylphenoxy)-2-
13 ,4-methylenedioxypllenyl)acetamide:
N-(4-ethoxy-3-pyrid:inesulfonyl)-2-(4-carboxy-2-propylphenoxy)-2-
(3 ,4-methylenedioxyphenyl~acetarnide;
N-1 (4-amino-2,5-dimethoxy)ben~.enesulf'ollyl]-2-(4-carboxy-2-
propylphenoxy)-2-(3 ,4-methylenedioxyphenyl)acetLImide;
21 9~75~
W0 96/04905 ' ~ ; F~ JO7~ ~1S~S7
- 61 ~
N-[(2.5-dimethoxy)benzenesulfonyl] -2-(4-carboxy-2-propylphenoxy)-2-
(3 ,4-methylenedioxyphenyl)acetamide;
N-[(3,4-dimethoxy)benzenesulfonyl]-2-(4-carboxy-2-propylphenoxy)-2-
(3 .4-methylenedioxyphenyl)acetamide;
N-[2-1'5-(morpholin-4-yl)benzothiophene]sulfonyl]-2-(4-carboxy-2-
propylphenoxy)-2-(3 .4-methylened ioxyphenyl )acetamide;
1~
N-[[2-(4-methoxy)benzothiophenel.sult'ollyl]-2-(4-carboxy-2-
propylphenoxy)-2-(3?4-methylenediox~phenyl)"~et,lmirl~:
N-[4-[2-(benzyloxycarbonylamino)ethyl]benzenesulfonyl ] -2-(4-carboxy-
2-propylphenoxy)-2-(3,4-methylenedioxyphellyl)acetamide;
N-[12,5-dimethoxy-4-((N-i.~o-propylcarbamoyl)amirlo)]benzene-
sulfonyl] -2-(4-carboxy-2-propylphenoxy)-2-(3 ,4-methylenedioxy-
phenyl)acetamide,
N-1~(2.4-dimethoxy)benzene.sulfonyl]-2-(4-carboxy-2-propylphenoxy)-2-
(3 ,4-methylenedioxyphellyl)acetami(ie;
N-[(2,4,6-trimethoxy)benzenesulf(myl l-2-(4-carboxy-2-propylphenoxy)-
2-(3 ,4-methylenedioxyphenyl )acetamide;
N-(8-~uinolinesulfonyl)-2-(4-carboxy-2-propylphelloxy)-2-(3,4-
methylenedioxyphenyl~acetamide;
N-(3-quinolinesulfonyl)-2-(4-carboxy-2-propyll)helloxy)-2-(3.4-
3 ~ methylenedioxyphenyl)acetamide;
N-(8-4uinolinesulfonyl)-2-(4-carboxamido-2-propylphenoxy)-2-(5-
methoxy-3,4-n~ethylenedioxyphenyl): ce~mi~
WO 96/04905 2 ~ 9 5 7 5 ~
N-(4-rerf-hutylb~nzenesulfonyl)-2-~4-carboxamido-2-1-ropylp}lello~y)-
2-(5-methoxy-374-methylenediox!,phenyl)acetamide;
N-(4-amino-2,5-dimetho~yl enz~.lesulfonyl3-2-(4-carboxamido-2-
propylphenoxy 1-2-(~-methoxy-3,4-methylenediox$~phellyl)acetamide:
N-[4-r.sci-propylhen7P,nf~.slllfonyl]-2-(4-carboxy-2-propylphenoxy)-2-
(3 ~4-methylenedioxyphenyl)acelamide;
N-(4-iso-propylber~ nP.~Illfonyl)-2-[l4-[N-[2-(c~arbethox$')ethyl 1-
carbamoyll~-2-propylphenoxy]-2-(3.4-methylenedioxypi~ellyl)acetamide,
N-(4-iso-propylbe:nzeneslllfollyl)-2-[ L4-[N-(2-ca:rboxyethyl )carbamoyl] 1-
2-propylphenoxy] -2-(3 ,4-methylenedioxyptlenyl)acet.tlnide;
N-(4-iso-propylbenzenesulfonyl)-2-[[4-[N-(2-carbamoylethyl~-
carbamoyll j-2-prop$~1phenoxyI-2-t3,4-methylenedioxyphenyl)acetamide;
N-(4-;s~)-propylbell7,ellesulfonyl3-2-[4-lN-(2,2,2-trifluoroethyl)-
20 carbamoyl]-2-pr~ 1~ylphe~ y~-2-(3~4-methyletledioxyl~tle~ acetalllide;
A perferred embodiment of the compounds of this
in~ention are:
25 N-(4-isO-prOpylbelll:elleSUlfOllyl)-o(-(4-carboxy-2-n-prop~rlphenoxy)-
3,4-methylenedioxyphe:nylacetamide dipotassium salt;
N -(~-quinolinesulfc~rlyl~-2-(4-carboxy-2-propylphenoxy)-2-(3 ,4-
methylenedioxy~ phenyl)acetamicle;
N-(4-dimethylaminobenzenesulfonyl~-2-~4-carboxy-2-n-p}opyl-
phenoxy)-2-(3 ,4-methylenedioxyphenyl)acetamide .
~ ~ 219~758
~ WO 96/0490~ J5,_7
-63 -
The alkyl substituents recited above denote straight and
branched chain hydrocarbon.s of the length specified such as methyl,
ethyl, isopropyl, isobutyl, neopentyl, isopentyl, etc.
The alkenyl-substituents denote al~yl groups as
5 described above which are modified so that each contains a carbon to
carbon double bond such as vinyl, allyl and 2-butenyl.
Cycloalkyl denotes rings Goml70sed of 3 to 8 methylene
groups, each of which may be substituted or unsubstituted with other
hydrocarbon sub.stituents. and include for e~ample cyclopropyl,
o cyclopentyl, cyclohexyl and 4-methylcyclohexyl.
The alkoxy substituent represents an alkyl group as
described above attached through an oxygen bridge.
The heteroaryl is defined as carbazolyl, furyl, thienyl,
pyrrolyl, isothiazolyl, imidazolyl, isoxazolyl, thiazolyl, o~azolyl,
5 pyrazolyl, pyrazinyl, pyridyl, pyrimidyl, purinyl or quinolinyl.
Although the reaction schemes described below are
reasonably general, it will be understood by those .skilled in the art
of organic synthesis that one or more functional groups present in a
given compound of FormuLI I may render the molecule
2Q incompatible ~vith a particular synthetic sequence. ln such a case an
alternative synthetic route, an altered order of steps, or a strategy of
protection and deprotection may be employed. In all cases the
particular reaction conditions, including reagellts, solvent,
U~Id~ and time, should be chosen so that tlley are consistent
25 witll the nature of the functionality present in the molecule.
The compounds of Formula I and specifically
compounds c f Formula III can be synthesized using the reactions
and techniques de.scribed t'or the synthesis of the non-heterocyclic
components in the patent application W091/11999 ~Merck & Co.;
30 published on August 22,1991 under the Patent Cooperation Treaty),
US Patent 5,177.095 (Merck & Co.; January 5, 1993), and also US
Patent 5,240,938 (Merck & Co.: August 31, 1993).
The reaction .schemes desclibed below have been
generalized for simplicity. It is further to be understood that in the
2 1 ~ 5 7 ~ 8
W0 96/04905
- 64 -
generalized schemes below, unless specified more narrowly in the
text~ the alkyl and aryl groups rrpresent unfamctionalized or
functionalized derivatives as described before. The leaving group Q
present in lhe alkyiating agents is either chloro, bromo, iodc),
5 methanesulfonate, p-toluenesulfonate or triflate.
Scheme I
R12
~S Q R9--~, Rto
R9-- _ R10 + 1 zl Base.
XH Ar
3L
~r ~r
Ar = R -~--R1 R3b_~ or
~,~,~ R12
R3a R2 R a
~ Rl3--~ Rl~
2 5 R3b_ ~3a X~ Z
Q = Cl, Br, 1, OMs, OTs or OTf Ar
~, Y or Vl
Zl = a precursor IO Z
;. 2 1 q J 7 ~ ~
WO 9~/04'70S P~, I Iv~., '0~ _ /
- 65 -
More specifically, the compounds of Formula 111, V or ~,'1
(where X is oxygen. sulphur or appropriately substituted nitrogen) Catl
be synthesized as outlined in Scheme I . The substituted compound 1
may be reacted with the alkylating agent ~ in an d~ riate solvent
5 such ~1S alcohols (methanoh ethanol, isopropanol and liice),
dimethylformamide ~DMF), dimethylsulfoxide (DMSO),
tetrahydrofuran (THF) ~md acetone in the presence of an alkali metal
salt such as alkoxides. carbonates, hydroxides and hydrides, or organic
bases such as trialkylamines or alk;yl lithiums to provide comp(7und ~.
The zl group present in compound 3 may then be further transt'ormed
to provide the appropriate compounds of Fonnula 111, ~/' or Vl.
In general, the alicylating agent ,~ can be prepared using
methods and fP-hni(lllPs outlined in US Patent 5,177.09~. More
specifically, compound 2 (where Zl is COOR and Q is Br) can be
syntilesized from the substituted arylacetic acids 4 as outlined in Scheme
The substituted arylacetic acid 4 is com~erted to the correspollding
ester either by refluxing the acid in an appropliate alcohol in the
presence of ~1 catalytic amount of conc. sulfuric acid, or using other
conventional methods of esterificatiotl. 'I'he resulting ester is then
20 refluxed in carbon tetrachloride ~vith N-bromosuccinimide and a
catalytic amount of a radical initiator (e.g., AII3N or ben7,0yll7eroxide~
to provide the 2-bromo-arylacetic acid ester ~.
Sclleme 2
1. ROH, H~ Br
2. NBS, AIBN
Ar/~COOH CC4 > Ar COOR
- Alternatively, the ester ~ may alsc be prepared from
appropriate aryl aldehydes (Scheme 3). T-he aldehyde 6 can be reacted
with trimethylsilyl cy~mide and catalytic amounts of KCN and 18-
crown-6 to provide the corresponding trimethylsilyl cyanohydrin 7,
i i 2 1 ~5~58 1~ 7--
- 66 -
which upon further treatment with the gaseous llCl and alcotlol affords
the, 2-hydroxy e~ster 3~. The ester 8 is treated with triphenylphc~spllirle
and carbon tetrabrorllide in methylene chloride to give the 2-
bromoarylacetate derivatives 5 .
Scheme 3
a OTMS b OH c Br
Ar--CHO 1 1 1
0 Ar CNAr COOEt Ar COOEt
6 7 8
a. TMSCN, Clat. KCN, CH2CI~, IS-Crown-6; b. HCl(g), F,tOH,
5 c. CBr4, Ph3P, CH2cl2
Scheme 4 illustrate~ a typical synthesi~ of an alk~lating
agent ~ where Ar ,~ ,..t~ a heterocycle SUCtl as ~all indole). The
appropriately su~stituted cyanoindole ~ (for ~ gener.ll ~ynthesis of
20 suh~tituted indoles refer to, R. K. Brown, Indole~u Part Orle. Ed. W. J.
Houli}~an~ ~fol. ?57 Chapter Il~ ~iley-lnterscience, New York. 1972) is
reduced witll DIBAL-I I to provide the corresponding aldetlyde, whictl i~
then converte,d into the N-Boc derivative l~ . Reaction of l0 with the
trichlorometllide anion Igenerated from KOH and CHCl3: J. ~1. Wyvratt
25 et. al., J. Org. Chem.. ~, 944-94S ~l9~7)~1 followed hy t,reatmellt with
aqueous NaOH ;n D~F provides the alcohol LL Treatment of ~, with
diazomethane followed by the reaction with CBr4/Ph3P yields the
alkylating agent 12.
wo 96/0490~ 2 1 ~ ~ 7 5 ~ P~,lrL ,~7
- 67 -
Scheme 4
R3b R3b
~CN a ¢~,~3~CHO b
N N
H R3a ' R3a
2 Boc
R3b OH R3b Br
~\~COOH c <~-\~COOMe
N~\ 3 NJ~\R3a
Boc 11 Boc
12
a. (i~ DIBALH, Toluene; (ii) Boc2O, DMAP, CH2CI2
b. (i) CHCb, KOH, DMF, 0~C; (ii) NaOH, DME / H2O
c. (il CH2N2; (ii) CBr4/Ph3P. CH2CI2
A t~pical .synthe.sis of alkylatin~ agents bearing a substituted
20 benzoxazole or b~n7thi~7l)1e ring is outlined in Scheme 5. The
substituted benzoxazole 14 is prepared from the corresponding o-
aminophellol 13 by the reaction of an appropriate orthoester under
refluxing conditions (for other methods of synthesis of benzoxazoles
see, S. A. Lang and Y. Lin, Comprehensive lleterocyclic Chemistry,
2s Vol. G, 1-130, Ed. C. \~'. Rees; and references cited therein). Reduction
of 14 with NaBH4 provides the alcohol 15 which is then subjected to
pyridinium dichromate (PDC) oxidation to yield the corlesponding
aldehyde 16. Further elaboration of 16 as outlined provides the key
intermediate 17. Similarly~ the benzothia7.01e 19 can also be prepared
30 form the appropriately substituted o-amincthiophenol 18.
W0 96104905 ' ~ . 2 1 9 ~; 7 5 ~ r~In . n )s.. ~--
- 68 -
R3b R3b
NH2 ~ ,COOH < ~
H O ~ \ 3~ R3a
13 14
R3b
</ ~,~ C <~
N~-,\s~GOOMe
a. C H(O Et~3. EtO H.reflux < ~ ~
~. (i) ClC O OEt, Et3N, THF;~ii) NaBH4, THF-H2 ~ ~R3a
c. Pyridiniurn dichrom ate, C H2C12 17
d. (i) C H Cl3, KO H, D M F,0~C;(ii) NaO H, D M E / H20:
(iii1 H Cli MeO H;(iv~ C Br~P~3P, C H2C12
R3b R3b Br
NH2 ~\~COOH <N ~ COOll,Ae
H S ~ \R3a S R~a
18 1~2
~ ~; 2
~ W0 96/04905 ~ 9 5 7 5 g r~
- 69 -
Scheme 6 illustrates the synthesis of benzofuran and
dihydrobenzofuran alkylating agents ~ and 2~. The benzofuran ~.L can
be prepared from the (x-phenoxy carbonyl compound 20 via a ring
closure reaction [Stoermer and Wehtn, Chem. Ber., 35, 3549 (190~)
5 (for general methods of synthesis of benzofurans and
dihyd-u~ ufuld~ls see, R. C. Elderfield and V. B. Meyer, Heterocyclic
Compounds. Vol. 2, Chapter 1, Ed. R. C. Elderfield. Wiley; and
references cited therein). The ester ~ is reduced to provide the
aldehyde 22 which is then transfonned into the corresponding
alkylating agent 23. The dihydrobenzofurall ester 2~, obtained by
catalytic reduction of 21, can also be tr~msfomled into the
corresponding atkylating agelIt 2~ using the sequence of reactions
outlined in Scheme 6.
Benzothiophene ~ may be s~nthesized from the
5 corresponding aldehyde ~ in a manner similar to that outlined in
Scheme 6 for benzofuran ~. Benzothiophene ~k can be prepared by
the oxidative cyclization (using an alkaline solution of potassium
ferricyanide) of a~l,.u~-ialely substituted o-mercaptocinnamic acid ~~~
~C. Ctlmelewsliy and P. Friedlander, Chem. Ber., 46, 1903 (1913)1.
20 (For general methods of synthesis of benzothiophene, ~ E.
Champaigne in Comprehensive Heterocyclic Chemistry. vol. 4~ Chapter
3-]5~ Eds. A. Katritzky and C.W. Rees.)
Scheme 7 outlines a typical synthesis of oc-bromoaryl-
acetates, 30 and 32,bearing appropriately substituted methylenedioxy or
25 1~4-dioxane rings. The substituted catechol derivative ~ is treated with
~m appropriate dibromide (wllere m is I or 2) in the presence of cesium
carbonate in dimethyli'ormamide to provide 28. Treatment of 28 witl
DIBALH yields the aldehyde ~2 which is then transformed into the
desired alkyl bromide as described.
2 ~ 9 ' 75 8
W0 96/~490~ 7
- 70 -
Scheme 6
(EtO~2HC~0~,,;3~COOMe
R3a 20
a
o R COOMer~ R3b
~'~R3a R3a
21 22
S ¦ d R3b I c Br
o R~COOMe ~,~ COOMe
R3a R3a 2;~
2~
¦ b,c a. ZnCi2
b. DIBALH, toluene
Br c. (i) CHCI3, KOH, DMF, 0~C;
R3b 1 (li) NaOH, DME / H20,
\s~COOMe ~iii) HCI / MeOH;
\~l ~J j~ (Iv) CBr4~h3P. CH~C12;
R3a d. Ra-NI I H2
R3b ~ COOMe R3b Br
HOOC~R ~6~3~L
f t 8
~ Wo s6/04905 ~ ~ ~ 9 ~:) 7 ~ P~ 7
- 71 -
Scheme 7
R3b 0 R3b COOM
a (CH2~mJ~"J b
27 28
R3b
(CH2)m~f ~
~0~\ 3a
29
Ic
p R3bCH2COOMe R3b Br
15(CH2)m~f ~ d O~,\s~COOMe
~ R3a oJ~\R3a
31 ~Q
a. Br-(CH2)m-Br, Cs2CO3, DMF
b. DIBALH, toluene
c. (i) CHC13~ KOH. DMF, O~C; (ii) NaOH, DME I H20;
(iii) HCI / MeOH; (iv) CBr4/Ph3P, CH2C12;
d. NBS, AIBN, CC14
Br
X ~r ~COOMe
R12 0~\R3a 32
W096~04905 ~ 9 5 ~5~ 7--
- 72 -
Fcolk~ing the synthetic route outlined in Scheme ~ below
N-(4-iso-propylhPn~PnPslllfollyl)-a-(4-carhoxy-2-n-propylphenoxy~-
3,4-methylenedioxyphenylacetamide dipotassium salt, 4~ i.s prepared.
Scheme 8
Methyl 4-hydroxy-3-1,1u~)yllJe-~ùal~. 36:
CO2CH3 CO2CH3
Br~ ~ 1 60-5~C
~/ K2CO3, acetone ~ ~,CI
OH reflux 0 ~ W'CI
33 3~
CO2CH3 co2CH3
1~1 H2,PdiC
MeOH
OH OH
~5 36
~ W0 96104905 ~ 7 5 ~ u~ . ~
Ethyl a-bromo-3,4-methylenedioxyphenylacetate, 39:
OH
5 <o~CHO 1)TMSCN <O ~CO2C2Hs
O 2) EtOH o 38
10PBr3, Et20, <~~CO2C2Hs
3g
4-is~l-propylbenzenesull'onate, 41:
SO2CI ~, SO2NH2
~ NH~OH
H3C~ ' H3C~/ \~
CH3 40 3 4
WO 9v/~v~4905 ~ 5 7 5 ~ l/L~
- 74 -
N-~4-iso-propyll~ ,..1lfonyl)-a-(4-carboxy-2-n-propyl-
phenoxy)-3.4-methylenedioxypherlylacetamide dipotassium salt. 45:
H3CO2C~ <O~CO2CH2CH3 aC2et~ e
oJ~ref~ux
36 39
H3CO2C~¢~ H3CC~2C~
N~OH.
15~f CO2C2Hs ~1eOH~,~~~3--CO2H
42 43
H3CO2C~ ~ CH3
20cr)l. DBU. THF ~oK+ ~CH3
Ctl3<~ ~Jb- N~S ~J
~CH3 O~ ~ ~ ~
H2N~S~ 41
O O 44Crystalline
K 02C ~¢~[~~ CH3
I) NaOH, MeOI~, ~ K~ ~CH3
O /~
2) KO~I < ~ 1I S~
CrYstalline, pK Il = 5.4, pK~ .8, MW = 615.~,
logP 2.7, Aqueou.s solubilitv > 1;0 mg/mL
21 ~75~
~ W0 91;104905 . ..... , ,
- 75 -
Similarly, compounds of Fomlula IV wherein -A- is
represented by -OCH2- and X is ~ d by -NH- can be prel~ared
using the route described below
~ ~ } ~ 1 ~7~&
WOg6/04g05 . ~,9JI J r.l.
- 76 -
N-(4-iso-propylben~;enesulfonyl~ -[N-(4-carboxyanilinyl) 1-
3,4-methylenedioxyphenyl~(~et:lmi~ dipotassillm salt, 50:
Br
S H3CO2C~3~ + < ~ X5~C
46 ~AIdrich) 39
H3CO2C~'NH H3C02C~3'NH
KOH ~ I
<~ ~3--CO2C2H5 MeOH' ~~ ~ CO2H
47
H3CO2C~ CH3
DMAP. EDC~ ~ , NH ~, ~CH3
- CH3 o~,l~,
2 S~ ~CH3 <o~J ~ ~ ~
2 5 O O 49
HO2C ~3~ '~H3
I) KOH. I leOH , I H
reflux <~~~ "S~
o~ ~
5Q
, ,,,:,3< ~9 7 ~
~ ~0 96~0490~ J ~ .,u~ .9,67
- 77 -
HO~\/ CH3
H l~CH3
o~ N ~S ~J
¦ PBr3/Et20/0~C
0 Br/\~/ CH3
~ H ~ CH3
< ~ ~,5~'
¦ KCN/DMSO
I room temperature
NC ~¢~/ CH3
H ~CH3
o~ N 'S~)
(cH3)3snN3
Toluene,120~C
N,N~ CH3
- H O ~ C
3Q I H l ll H3
o~ N 'S J~
W09610490~ ~ 2 ~ 95;~58 1~ 7--
- 78 -
Scheme 8 illustrates the, synthetic route to cc~mpoullds
bearing N-acylsulfamides (~) (Z=-CONHS02NH-R). The c.lrboxylic
acid 33 tZI=-C~OOH: Scheme I ) is reacted with 1,1'-
carbonyl-liimi-1~7c-1e (CDI) to provide the acylimidazole which is thell
5 reacted with an appropriate sulfamide (~L) in the prexeuce c~f DBU. 'I'he
sulfamides (R-NI-ISO2N112) can be prepared from ~ .,u~ primary
amilles using the literature procedures [W. L. Matie,r~ ~hh T. Comer and
D. Deitchman, J. Med. Chem.. 15, 538-~;41(1972); J. D. Calt and W. 1,.
Maiter, J. Or~. Chelll.. 3c3, 566-568(19~4)l.
Scheme ~
R12 R12
R9--~ R1o 1)Cd~ y'" ~ ul~ R9 ~ 0
2) R-NHSO2NH2 (34), DBU ~
X~,COOH X~CONHSO2NH-R
Ar Ar
33 35
2~
~ W0 96/04905 ' ~ 2 J 9 5 7 5 ~ P~
- 79 -
The reactions are performed in a solvent appropriate to the
reagents and materials employed and suitable for the tran.sformatiorl
being eft'ected. It is under.stood by those skilied in the art of organic
synthesis that the functionality present on the heterocycle and in the
reactants being employed should be consistent with the chemical
transformations being conducted. L)epending upon the reactions and
techni4ues employed, optimal yields may require changing the order of
synthetic steps or use of protecth1g groups follo~ved by deprotection.
The compounds useful in the novel method treatment of
this invention forl11 salts with various inorganic and organic acids and
ba.ses whictl are .tlso withirl the scope of the invel1tion. Such salts
include ammonium salts, alkali metal salts like sodium and potassium
.salts, alkaline earth metal salts like the calcium and magnesium salts,
salts with organic base.s; e.g., dicyclohexylamine .salts, N-rnethyl-D-
ghlcamine salts, salts with amino acids like arginine~ Iysine, and the like.
Also, salt.s with organic and inorganic acids may be prepared; e.g.~ HCI,
I lBr, i-12SO4, H3PO4, methanesulfonic, toluenesulfonic, maleic,
fumaric, camphorsulfonic.
The salts can be fonned by convel1tion.tl means, such as by
reacting the free acid or free ba~e forms oi' the product with one or
more equivalel1ts of the ;~ u~liale base or acid in a .solvent or medium
in which the salt is iusoluble, or in a solvel1t such as water wtlicrl is then
removed i~l uaclu~ or by freeze-drying or by exch~nging the cations of
an existing salt for another cation on a suitable ion exchange resin.
It will be ;~ .;ial~d that the compounds of general
Formula I in this invelltiol1 may be derivatised at functional group.s to
provide prodrug derivatives which are capable of conversion b~ck to
the parent compounds i~ vi~o. The concept of prodrug administration
has been extensively reviewed (e.g. A.A. Sinkula h1 Annual Reports in
Medicil1al Chemistry. Vol 10, K.V. Heinzelman, Ed., Academic Press,
New York London, 1975, Ch. 31, pp- 306-326, H. Ferres, Dmgs of
TodaY, Vol 19, 499-538 (1983) .md J. Med. Chem.~ 18, 172 (1975)).
Examples of such prodnugs include the physiologically acceptable and
metabolically labile ester deriv.ttives~ such as lower alkyl (e.g. methyl
Wo 9h/~l4gl~5 :~ ~ ; 2 . 9 5 7 5 8 F.~
- 80 -
or ethyl e.sters), aryl (e.g. ~-indanyl esters~. alkenyl (e.g. vinyl ester.s),
alkoxyalkyl (e.g. methoxymethyl esters), alkylthioalkyl ~e.g.
methylthiomethyl esters~, alkanoyloxyaLkyl (e.g. pivaloyloxymetllyl
esters), and sub.stituted or lln.illh~tihl~d aminoethyl esters (e.g. 2-
dimethyl:~mino~thyl esters). Additionally, any physiologically
acceptable equivalent.s of the compounds of general Formula 1, similar
to the metabolically labile esters, which are capable of produchlg the
paretlt compclullcls of general Fonnula I in vil!o, are within the scope clf
this invention.
It will be t'urther appreciated that the majority of
compounds of general Formula I claimed herein are a.syrrmletlic and are
produced as racemic mixtures of enalltiomer~s and that bc~th the racenlic
compo~mds and the resolved indivi(lual enantiomers are considered to be
witllirl the scope of this invention. The racemic compounds of this
invelltion may be resolved to pro-dde individual enantiomers utilizing
methods known to those skilled in the art of organic synthesis. For
example, diastereoisonleric salts, e.sters or imides may be obtained from
a racemic compound of general Formula I and a suitable optically active
amine, amino acid, alcohol or the like. The dia.stereoisomeric .salts,
ester.s or imides are separated and purified, the optically active
enantiomers are regenerated and the preferred enantiolller is the rmore
pc~tellt isomer. ~'he resolved en~ln~ m~rs c f the coml-ounds of general
Formula 1, their ph~rmSIrellrir,llly acceptable salts and their prodrug
forms are also included within the scope of this invention.
2s Endothelin (ET-I~, and two closely related bioactive
peptides, E~7'-2 ~und E'I'-3, are widely distributed in mSImm~ n tissues.
and they can induce numerous biologioal respon.ses in non-vascular as
well as vascular tissues by bindnng to at least two distinct endothelhl
receptor subtypes. In addition to cardiovascular smooth muscle, neural
30 and atrial sites~ endothelin receptors may also be iiound in brain,
gastroilltestinal, kidney, Iung, urogenital, uteral and placental tissues.
Endothelin is a potent ~asoconstrictor peptide and thus
prays a role in vivo in arterial pressure-volume homeostasis. Not only
peripheral, bul c-oronary vascular resistance as well, is increased by
_ . . . . . . . . . . . . .. . . . ...
~ W0 96/04905 , ~ 9 ~ 7 5 ~ r~ ,3~s7
.
- 81 -
endotheiin; cardiac output is decreased, while plasrna renin activity i.s
increased. There is a reduction in renal blood flow and glomerular
filtration rate, while levels of atrial natriuretic factor, vasopressin! and
aldosterone become elevated.
It is also considered, in accordance with the present
invention, that antagoni.st.s for the endothelin receptor may be useful in
preventing or reducing restenosis subsequent to denudation following
angioplasty. Such denudation results in myointimal thickening
following angioplasty~ due to increased endothelin release. Endothelin
o acts as a growth factor with respect to smooth muscle and fibroblastic
cells, and possibly other types of cells, as well.
F,ndothelin is also a neuropeptide, acting on the posterior
pituitary, where it modulates the release of the neurosecretory
hormones vasopressin and oxytocin. Endothelin released from the
posterior pituitary al.so acts as a circulating hormone, ha~ing a wide
range of actions as discussed further above. This includes effects on the
endocrine system, especially the adrenal glands. Endothelin increases
plasma levels c f epinephrine.
Consequently, the novel compounds of the present
invention, which are receptor antagonists of endothelin, have
therapeutic usefulness in preventing, decreasing or modulatillg the
various physiological effects of endothelin discus.sed above, bv wholly
or partially blocking access of endothelin to its receptor.
Endothelin Receptor Bindin~ Assays
The binding of the novel compounds of this invention tv the
endotllelin receptor was detertnined in accord~unce with the assay
describecl in detail immef~ tt~ly below. It is similar to the assay
described in .~mbar et al. (1989) Biochem. Biopllys, Res. Commun.
158, 195-201; and Khoog et al. (1989) FEBS Letters~ ~53, 199-202.
The endothelins (ETs) ha\le a number of potent effects on a
variety of cells, and exert their action by interacting with specific
receptors present on cell membranes. The compounds described in the
present invention act as antagonists of ET at the receptors. In order to
wO 96rO490S ~ ~ - 2 1 9 5 7 5 8
- 82 -
identif~ ET antagonist.s and (1etl~rmin~. their eftïcacy irl ~r'tr v, the
fclllowirlg three llgand receptor assays were established.
Receptor bindin.~ ~ssay usin~ cow aorta membrane preparatio:n:
Thoracic aortae u~ere obtained from freshl~ slallg.htered
calves and brought to the lab on wet ice. The adventitia were removed,
and the aorta was opened up lengthwise. The lumenal surface of the
tissue was scrubbed with cheesecloth to remove the endothelial layer.
The tissue was ~round in a meat grinder, and .suspended in ice-cc~ld ().2~5
M .sucrose, 5 mll1 tris-~CI, pH 7.4, containing 0.5 mglmL leupeptin and
7 mg/mL pepstatin A. Tissue was homogenized twice and then
centrifuged for lO minutes at 750 x g at 4~C. The supernatant was
filtered through cheesecloth and centrifuged again for 30 minutes at
4~,000 x g at 4"C. The pellet thus obtained was resuspended in the
buffer solution described above (i:ncluding the protea.se inhibitors). and
aliquots were quick-frozen and stored at -70~C until use. Membranes
wele d:iluted into 50 rnM pota.ssium phosphate (K:Pi), 5 m:M EDTA E~H
7.5 containing 0.01 q~c human serum albumin. Assays were done in
triplicate. Test compound.s and lOOpM [1251]-endothelin-1 (2(:)0(:)-22(:)0
2(1 Ci/mmole, obtained from New Englalld Nuclear or .Amersham) were
placed in a tube containing this buffer, and the membranes prepared
above were added la.st. The .sarnple.s were incubated for 60 rmin at 37'~C.
At Ihe end of th:i.s incubation~ sarnples were filtered onto prewetted
(with Z~; BSA in water) glass fiber filter pads and wa.shed wit:h 150 rnA~/
2~ NaCI, O.l~o BS~. The filter.s were assayed for IZSI radioactivity in a
gamma counter. Nondisplaceable binding of [l251]-endothelin-1 is
measured in the presence of 100 nM unlabelled endothelin-l
[Endothelin-l ~ET-I~ was purchased from Peptide.s Intematiollal
(Louisville, KYl. 125T-ET-1 (2000 Ci/m~lol~ w~s pur~hased from
Amersham ~Arlington lleights, L)l. Specific binding is total binding
mhlus nondisplaceable binding. The inhibitory c.oncentration (ICso)
YY'hiCh gives 509to displacement of the total specifically bound [ I ~Sl~-
endothelin-l was presented as a measure of the efficacy of such
compounds as ET antagonists.
~ W0 96/04905
- R3 -
Receptor bindin~ assay usin~ rat hippocampal membrane preparation:
Rat hippocampi were obtained from freshly sacrificed male
Sprague-Dawley rats and placed in ice cold 0.25 M sucrose, 5 mM tris-
HCI, pH 7.4 containing 0.5 mg/mL leupeptin, ~ mg/mL pepstatin A.
5 lIippocampi were weighed and placed in a Dounce homogenizer with 25
volumes (wet weight to volume) ice-cold sucrose buffer in the presence
of protease inhibitors. I~ippocampi were homogenized using a Dounce
(glass-glass) homogenizer with t~Tpe A pestle, with homogenizer in ice.
Tissue homoge.nate was centrifuged at 750 x ,~,~ for l O min at 4~C.
o Supennatant was filtered through dampened cheesecloth, and centrifuged
again at 48,000 x g l'or 30 min at 4~C. Pellets were resuspended in
sucrose buft'er with protease inhibitors. Aliquots of this preparation
wele quick frozen and stored at -70~C ulltil use. Membranes were
diluted into 50 rrlll~ KPi, 5 mM EDTA pH 7.5 containing 0.01% human
5 serum albumin. Assays were done in triplicate. Test compounds and 25
pM [1251]-endothelin-l (200()-2200 Ci/mmole, obtained from New
England Nuclear or Amersham) were placed in a tube containing this
buft'er, and the membranes prepared above were added last. The
samples were incubated for 60 min at 37~C. At the end of this
20 incubatiorl, samples were filtered onto prewetted (Witll 2~o BSA in
watel ) glass fiber itilter pads and washed with 150 m,P!~ NaCl, 0. l ~o
BSA. l'he filters were assayed ~or 1~51 radioactivity in a gamma
counter. Nondisplaceable binding of [l251]-endothelin-l is measured in
the presence of l O0 ~?M unlabelled endothelin- I [Endothelin- l (ET- l )
25 was purchased from Peptides lntemational (Louisville, KY). 1251-ET-
l (2000 Ci/mMol) was purchased t'rom Amersham (Arlingtoll Heights,
IL,~]. Specific binding is total binding minus nondisplaceable binding.
The inhibitory concentration (IC~ol WlliCIl gi~,es 50% displacement of
the total specifically bound 1~l~5ll-elldothelill-l was presented as a
30 measure of the efficacy of such compounds as endothelin antagonists.
Receptor b:inding assay using cloned hum~m ET receptors expressed in
Chinese Hamste.r Ovarv Cells:
Both endc)the]ill receptor subtypes were cloned from a
human cDNA library and were individually expresseci in Chinese
_ . .. _ . . . _ . . .. . ... . ... ... . . .. .. . ..
wo s6/049~5 ~ ~ 2 1 ~ ~ 7 5 ~
- 84 -
Halnster Ovary cell.s. Cells were harvested hy addition of 126 mM
NaCI, 5 mll1 KCI, 2 ml~ EDTA, I mM NaH2PO~, 15 mM gluco.se~ 10
171Aq tris/HEPES pH 7.4 Cells were centrifuged at 250 .~ g for 5
minutes. The ~u~e~ ~nt was aspirated off, and the cell.s were
resuspended in the 50 mM KPi, 5 mM EDTA pH 7.5 containing ().01~c.
human serurn albumin. Assays were done in triplicate. Test compounds
and 25-100 pM 1 1 25ll-endothelin- 1 (2000-2200 Cilmmole, obtained
from New England Nuclear or Amersham) were placed in a tube
co:ntaining 50 mM KPi, 5 mM EDTA pH 7.5 containing 0,01~! human
serum albumin, and the cells prepared above were added last The
.samples were incubated for 60 m:in at 37~C. At the end of thi.s
incubatiol1, sarnples were filtered onto prewetted (witl~ 2~o 13SA in
water) glass fiber t'ilter pads and wa.shed with 150 mM NaCI, 0.19'~
BSA.
The filters were assayed for 1251 radioactivity in d gamma
counter. Nondisplaceable binding of [1251]-endothelin-1 is measLIred in
the presence of 100 nM unlabelled endothelin-] [:Endottlelir~ ET-I)
was purchased frorn Peptides Interllational ~Louisville, KY~. 125I-ET-
I (2(:)00 Ci/mMol) was purchased from Amersham (Arlington l-leights,
IL)~. Specific binding is total binding minus nondisplaceable bi:nding.
The inhibitory c.oncentration (ICso) which gives 50~ di.splacernellt of
the total specifically ~und ~1251]-endothelin-1 wa.s presented as a
measure of the efficacy af such compounds as endothelin antagonists.
The binding assays described above were used to evaluate
the pote:ncy of interaction of ~ s~l.L~ltive compounds of the invention
with enduthelin receptors. To fl~t~rrnin~- ~vhether these cormpoLInds
were endothelin antagonists, assays which measure the ability of the.
compounds to inhibit endothelin-~tim~ tpd phosphatidylinositol
hydrolysis were established. Rat uterus contains predominantly one of
the knowll end(lt.heli:n receptor subt~pes (F,l'A).
Phospl]atidylinositol hydrolysis assays using rat uterine slices:
Diethylstilbestrol primed femlle Sprague-Dawley rats were
.sacrificed and their uteri were collected, dissected of fat and colmecti~e
ti.ssue and minced. ~1ince-d tissue was added to oxygenated (95~o ~2~
2 ~ 9575~
~W0 96/04905 ~ 5, _ /
5'io CO2) 127 mM NaCI, 25 mll1 NallC03, 10 mM Glucose, 2.5 ntM
KCI, 1.2 mM KH2PO4, ~ .2 mM MgSO4, ] .~ mM CaC12. To the tissue
~ mince, 1.2 mM myo-[3H]~ ositol (.D.mersham) was added. 'I'he mincewas incubated 90 min at 37~C, with constant oxygenation. After
incubation, the loaded tissue mince was washed five times with t:he same
oxygenated buffer to remove excess radiolabelled inositol. The tissue
mince was resuspended in the aboie buffer, containing 10 m~l LiCI,
aliquotted into tubes, and 3 nM endothelin-l wit:h and without test
compounds was added to start the assay. Assays were done in
quadruplicate. Samples were incubated at 37~C under blowin~ ~2 in a
hooded water bath for 30 minutes. Reaction ~as stopped by addition of
trichloroacetic acid to 6~o concentration. Samples were sonicated for
10 min, centrifuged .'~.0 min, then trichloroacetic acid was extracted with
5 water-saturated ethyl ether. An ali~uot of eacll sample was neutralized
and diluted by addition of 50 mM tris-HCI pH 7.4. A 100 mL aliquot of
this solution was assayed for radioactivity in .l beta counter. The diluted
neutralized sample was applied to Dowex I x ~-formate columns,
washed with water, then washed with 60 mM ammonium formate, 5
20 mM sodium tetraborate. Samples were eluted with 200 mlll ammonium
formate, 5 mM sodium tetraborate. The radioactivity of each eluted
.sample was measured in a beta counter. Radioactivity was normalized
by dividing r~ldioactivity in post column sample by radioactivity in
precolumn sample. Control values (100% stim~ t~d) are values in the
presence of endt t~ minus the values in the absence of endothelin
(basal). Test sample values are the values in the presence of endothelin
and test sample minus basal. Inhibitory concentrLItion ~ICso) is the
concentration of test compound required to give a sample activity of
50~c, of control value.
~Sarafotl)xin S6c is a member of the endothelin family
which bhlds preferentially to one of the known endothelin receptor
subtypes (ETg).
Phosphatidvlinositol hydrolysis a.s.says usin~ rat lun~ slices:
Male Sprague-Dawley rat:s were sacrificed and their lungs
were collected. dis.sected of fat and connective tissue and minced.
WO 96~04905 ' ~
- 86 -
Minced tissue was added to oxygenated (95% O~, 5~ CO2) 1~7 r
NaCI~ 25 mA1 NaHCO3, lû mM glucose, 2.5 mM KCI, l.. mM
KH2PO47 1.2 rflM MgSO4, 1.8 mM CaC12. To the tissue mince, 1.2
,~1 myo-~3H]-inositol wa.s added. The mince vdas incubated 60 min ,It
37~C, with con~tant oxygenation. After incubation, loaded tissue mince
was wa.shed five times with the salne oxygenated buffer to remove
excess radiolabelled illOSitOh Tissue mince was resuspended in the
above buffer, colltaining 10 m~M LiC1, aliquotted into tubes, ancl 3 n~l
.sarafotoxin S6c with and without test compounds was added to start the
assay. As.says were done in quadruplicate. Sarmples were. incubated at
37~C under blov~ing ~2 in a hooded water bath for 30 minutes.
Reaction was stopped by addition of 0.5 rmL 18~ trichloroacetic acid to
69to concentration. Samples were sonicated for lO min, centrifuged 20
minl then trichloroacetic acid was extracted with water-saturated ethyl
ether. An aliquot of each sample was neutralized and diluted by
addition of 50 rnM tris-HCI pH 7.4. A 100 mL aliquot of this solution
wa.s assayed for radic7activity in a beta counter. The diluted neutra]i~ed
sample was applied to Dowex l x 8-formate columns, washed wit~h
water, then washed with 60 mM ammonium formate, 5 mlll sodhlm
tetraborate. Samples were eluted witll 200 mM amJnonium forrnate. 5
mM sodium tetraborate. The radic~activity of each eluted sample was
measllred in a beta counter. Radioactivity was normalized by dividing
radioacti~ity in post column sample by radioactiYity in precolumn
sample. Control values (lûO% stimulated~ are values in the presence of
sarafotoxin minus the values in the absence of sarafotoxin (hasal). l'est
sample values are the values in the presence of sarLIfotoxin and test
sample minus basal. Inhibitory concentration (ICso~ is the
concentration of test compound required to give a sample activity of
50~7c of control value.
Phosphatidylinositol hydrolysis assayx using cloned hum~m endothelin
receptors expressed in Chinese Hal~ster Ovary cells:
Endotllelin receptors of both receptor subtype.s were cloned
from a human cDNA library and were indii/idually expressed in
~ W09~049~ qrJ75~ r .IIU~
- 87 -
Chinese Hamster Ovary cells. (~ells were loaded ovennight by the
addition of 1.2 ~M myo-l3~1]-ino.sitol to their growtl1 medium. Cells
were harvested by addition of 126 mM NaCI, ~5 mM KCI, 2 fnM EDTA,
I mM NaH2P04, 15 mM glucose, 10 rnM tris,/HEPES pH 7.4. Cells
were washed five times by centrifugation at 250 x g for 5 minutes to
remove excess radiolabelled inositol. The supernatant was aspirated off,
and the cells were resuspended in the same o~ygenated (95% ~2~ 5%
CO2) buffer containing 10 mM l iCI. aliquotted into tubes, and 0.3 nM
endothelin-l with and without test compounds was added to start the
a.ssay. Assays were done in quadruplicate. ~amples were incubated at
37~C under blowing ~2 in a hooded water bath tor 30 minute.s~
Reaction was stopped by addition of 0.5 mL 18~o trichloroacetic ;~cid to
6% concentration. Samples were sonicated for 10 min, centril'uged 20
min, then trichloroacetic acid was extracted with water-saturated ethyl
ether. An aliquot of each .sarnple was neutralized and diluted by
addition of 50 l7~M tris-~lCI pll 7.4. A 100 mL aliqllot of this solution
was assayed for radioactivity in a beta counter. The diluted neutralized
.sample was applied to Dowe~ X-formate colunltls, washed with
water, then washe(l with 60 ~nM .Immnninm formate, 5 ~7lM sodium
tetraborate. Samples were eluted with 2()0 ~nM ammonium f'onmate, 5
n~JM sodium tetraborate. The radioactivity of each eluted sample was
measured in a beta counter. Radioactivity ~as nclntlalized by dividing
radioactivity in poit column sample by radioactivity in precolumn
sample. Control values (100~,'o stimulated) are values in the presence of
endothelin mil1u.s the values in the absence of endothelin ~basal). Test
sample vahles are the values hl the presence of endothelill and test
sample minus basal. Inhibitory concentration (ICso) is the
concentration of test compound required to give a sample activity of
50% of control value.
Using the methodology described above, representative
compounds of the inventio~ ere evaluated and found to exhibit ICso
values of at least <50 ~1 thereby demonstrating and confirming the
utility of the compounds of the inventiol1 as effective endothelin
~untagonists.
WO 961049115 ~ 19 5 75 8
Intravenuus Effect of Endothel:in-l Antagonist, IV-(4-iso-proT-ylbellzerle~
sulfonyl~-a-(4-carbc~xy-2-n-prol3ylphenox,y)-3~4-illethylenedioxyphenyl-
~3~et:ut~ dipotassiunl .sult [Example 58~ on Endothelin l -lnducc d
(:~hanges i:n Diastol;c and Urethral Pre.ssures in the Anesthetized Male
Dog
Methodology for rlpfl~rminin~ whether an ET-1 .selective antagonist
could inhibit the ET-I mediated prostatic urethral contractions in a
mongrel dog nlodel:
On separate days, two f:dsted male mongrel dog.s (HRP.
Inc.) weighing 11.0 and 12.4 kg, were anestheti~ed with Sodium
Pentobarbital ~Steri.s Laboratorie.s, Inc.) at 35 mg~kg (i.-iu) to effect,
followed by 4 mg/kg/hr ~i.v.) infusioll. A cuffed endotracheal tube was
5 inserted and each animal ~h~a.s ventilated with room air using ~ positive
displacement large animal ventilator (Harvard Apparatus) at a rate of
1~ breaths/minute and an average tidal volume. of 18 mlJkg body
weight. Body temperature was ma~ntained with a heatinsg pad and heat
k!mp using a te~nlperature controller (YSI) and esophageal probe. Tu,o
catheters (PE 2~31)) were placed in the aorta via the femor.ll arteries
(one in each artery) for admini.stration of endothelirl or phenylephrille
iand for continuous direct monitoring of blood pressure and heart rate
u.sing a Statham blood pressure transducer ~Spectrarned) and a compllter
system ~Modular In.strument~s, Inc.). Two other catheters (PE 260~
25 were placed in the vena cava via the femoral veins (one catheter in each
vein) for administration of pentobarbital and N-~4-isc3-propylberlzene-
sulionyl)-a-(4-carboxy-2-rl-propylptlenoxy)-3,4-1rlethylelledic)xyphellyl-
acetamide dipotassium salt ['Exarnple 5X] . A supra-pubic ;ncision
approxirnately one-half inch lateral to the penis was made to expose the
30 ureters, urinary bladder, pro.state, and urethra. The dome of the
bladder was retracted to facilitate dissec.tion of the ureters. The ureters
were cannulated with PE 90 and tied off to the bladder. Umbilical tape
was passed beneath the uret:hra at the bladder neck and another piece of
tape was placed approximately l-2 cm. distal to the prostate. l'he
~ wos6l04so~ r~.,.a,,...~ 7
7 5 8
- 89 -
bladder dome was incised and a l~icro-tip~ catheter transducer (Millar
In~lrullle~ , Inc.) was advanced into the urethra. The neck of the
bladder was ligated with the umbilical tape to hold the trall.sducer. The
bladder incision was sutured with 3-0 sili~ (purse string suture). The
transducer was withdrawn until it was positioned in the prostatic
urethra. The position of the ~licro-tip~ catheter was verified by gently
squeezing the prostate and noting the large change in urethral pressure
prior to ligating the distal urethra.
o Experimental Protocol:
Phenylephrine (PE) ~l0 llg/kg, intra-atterial) was
administered and pressor effects on diastolic blood pressure (DBP) and
intra-urethral pressure (IUP) were noted. When blood pressure
returned to baseline, endothelin-l (ET-I) (I nmole/kg, intra-arterial)
was ~(lmini~t~red. Changes in DBP and IUP were monitored for one
hour and an ET-1 selective endothelin antagonist~ such as the compound
of Example 58, N-(4-iso-plopylbenzene-sulfonyl)-o~-(4-carboxy-2-n-
propylphenoxy)-3,4-methyienedioxyphenyl-acetamide dipotassium salt
(30 mg/kg, intra-venous) was admil1istered. l'en to fifteen minutes
later when blood pressure had stabilized, ET-I was administered again.
and inhibition of ET-I induced effects were noted. PE was
administered at the end of the experiment to verify specificity for E'l'-l
blockade. The dogs were e-ltll~li7.~d with .LIl overdose of pentobalbit,ll
followed by saturated KCI.
The drugs utilized in the experiment described above were:
I ) Phenylephrine, HCI (PE) (Sigma Chemical, Co.) was given
at a volume of 0.05 ml./kg;
2) Endothelin-l (ET-I) (:Eluman, Porcine, Canine, Rat, Mouse,
Bovine) (Peninsula Laboratories, iilC.) was given at a volllme of
3 c 0 05 mLllcg;
3) ET-l selective antagonist, SUCil as the compound of
Example 58, N-(4-iso-propylbellzene-sulfonyl)-~-(4-carboxy-2-
n-propylphenoxy)-3 ,4-methylenedioxyphellylacetamide
dipotassium salt, was given at a volume of 0.3 ml,lkg.
All drugs were dissolved in isotonic saline solution.
W0 96/U~05 2 sl 9 5 7 5 8 r~ J--
- 90 -
Re.sults:
ET-I elicited an initial depre.ssor effect followed by a
longer pressor effect. In one dog, the pressor effect wa.s bipllasic. The
decrease :in DBP in hot'h dog.s aver.iged 13 rmnHg, while the peali
5 pressor efl'ect averaged 26 mrnHg. 'l~e average ET-I induced increa.se
in IUP was 15 mmHg. Ten to 14 minutes after administration of hl'-(4-
isa-propylbenzene-sulfonyl3-t.lG-(4-carboxy-2-n-propylpllenoxy~)-3,4-
methylenedioxy{3henyl-Sl~elSlrni-lP dipotassium salt IExarnple 5~. the
dogs we.re challenged with ET-I again and the depressor and pressor
effec.ts on DBP were. inhibited 69% and ~6%, respectively. The pressor
effect on IUP s~as inhibited 930i~G (T-able I ~. Intra-arterial :PP.i-induced
increases in DBP and IUP did not change ~ignitlrs~ntly after
adm:illistration C13' N-(4-is~3-propyll3enzene-.sullonyl i-o~-(4-carboxy-2-n-
propylphenoxy)-3.4-methylenedio~yphenyl-S~netSImide dipotassilun s alt
[Exarllple 5~] in the one dog studied. Increases in DBP and IU:P were
inhibited 35 and 13~.ro, re.spectively ~Table 2).
~ W09C1049~5 ~ 9~75,~ P~l/~J~. 5~1,rc7
- 91 -
Table 1. Eff'ects of ET-1 Antagonist, N-(4-i.so-propylbenzene-
sulfonyl)~ 4-carboxy-2-n-prop~!lphenoxy)-374-methylenedioxyphenyl-
acetamide dipotassium salt [Example 581, on ET-I Induced Changes in
DBP and IUP in Anestheti~.ed Male Do~ (n=2)
CHANGE
Cl-IANGE IN DBP IN IUP
(mmHg) (mmH~)
DOG # DEPRF,SSOR ¦ PRESSOR PRESSOR
ET- I (i.a.)
HG FMJC -10 19 18
IIGFMHK -15 33 11
MEAN -13 26 15
SEh/l 3 7 4
ET-I + Example 58
HG F'MJC -3 8
HG FMHK -5 2
MEAN -4 5
2 5 SEM I 3 0
~) INH131TION
HG FMJC 70 58 94
~ HG FMHK 67 g4 91
MEAN 69 76 93
SEM 2 18 2 ,
W096104905 ~ ' a ~95758 r~ sn--
_ 9~ _
Table 2. Effects of ET-I Antagonist, N-~4-iso-propylbenzene-
sulfonyl)-o~-(4-carboxy-2-n-propylphenoxy)-3 ,4-
methylenedioxyphenylacetamide dipotassiunl salt [Exarmple
5~1, On PE-lnduced Changes ;n Di3~P and I~TP iu
An~Clllp~i7~ Male Dog # H~ FMJC~
TREA1'MENT INCREASE IN DBP INCREASE 11~ IUP
(mm~
Phenylellhrille 17 31
Phenylephrine 1 i 27
Example 5~
% inhibitio:n of Control 3~ ]3
Conclusions:
ET- I causes constriction of the prostatic urethra, a.s well as
a complex hemodynarriic re,sponse comprised of an initial depre.s.sor and
20 subsequent pressor response in anestheti~ed dog.s. rhe hemodynamic
~nd prostatic urethrai respons~ to ET-I were .speciilcall~, inhibited b~
IV-~ 4-i~<~-propylb~n~ne~ulf'onyl)-a-~4-carboxy-~ prop~ lphello~y )-
3,4-methylelled:ioxyphe:nylacetam:ide dipotassium salt. The efficacy o~'
the N-(4-iso-propylbellzenesulfonyl)-o~-~4-carboxy-2-n-propyl-
25 phenoxy)-3,4-methylenedioxyphenyl~iret~mi;e dipotassium .salt Ul
inhibiting the prostat:ic urethral pressor effect of ET- I suggests that
seiective antagonists of E1'-1 will be usefui in the treatment of urin~
ob.struction in benign prostatic hyperplasia.
30 1~ Sifli Rat Pro,~tate:
Male Sprague-Dawley rats (Taconic Farms) weighin~ 300-
400 grams were anesthetized with urethane (1.75 gll~g, ip). a tractleal
canmlla was inserted, and the femoral arter~,~ was cslrin~ t~ri Core
body temperature ~h'a.S maintained at 37 + 0.5 ~CI. A 4-5 cm midline
,lhdolnin ll incision was made to expose the bladder and prostate. Ttle
1 9~75~
~ WO 96104905 ' r~
- g3 -
prostate was .separated from the bladder and surrounding capsule by
blunt dissection with a iorcep. A length of surgical silk was gently
secured around the anterior tips of the prostate lobes. A second length
of surgical silk attached to an atraumatic needle was passed through and
tied to the base of the prostate approxilIlately 10-] 2 mm posterior to the
first tie. The posterior ligature was secured to an anchor post whereas
the anterior ligature was connected to a Grass FT03 tran.sducer ~Grass
Instruments, Quincy, MA) and m~int~in~d at a tension of I g. Signals
from the transducer were amplified and recorded on a polygraph
(Hewlett-Packard ~0:i13 amplifiers and 775~A recorder, Palo Alto.
CA). After equilibrating for approximately 15 min, the rats were
administered pretreatment drugs (atropine I mg/kg, (+) propranolol I
mg/kg) lO min apart through the intra-arterial (IA) cannula. Thirty
minutes later, ET-I (0.3 nmoles/kg) was injected intra-arterial every
thirty minutes for a total of three times. Five minutes before the third
injection of ET-I, vehicle ~ith or without an endothelin antagonist was
injected IA. The response of the prostate to ET-l was qu.mtified by
mea.suring the change (~) from baseline tension to the peak of the
response during the 5-minute period after the third ET- I injection.
The in .Sitll rat postate protocol has been utilized to
deterrnine the antagonist activity and potency of compounds of this
invention to block the direct contractile effects of ET-I on the rat
prostate in vivo. in this protocol, N-(4-is~i-propylbenzene-sulfollyl)-o~-
(4-carboxy-2-n-propylphenoxy)-3,4-methylenedioxyptlenylacetamide
dipotassium salt was dernonstrdted to cause a specific inllibitioll of F,T-I
to contract the prostate and will be useful in the treatment of urinary
obstruction itl benign prostatic hyperplasia.
Accordingly the novel compounds of the present invention
are useful in humall therapy for treating asthma. hypertension, renal
failure particularly post-ischemic renal ~:ailure, cyclospcrin
nephrotoxicity, vasospasm, cerebral and cardiac ischemia, myocardial
infarction, or endotoxin shock caused by or associated ~vith endothelin,
. . 2 ~ 9 5 7 5 ~
WO 9611~4905 , ~ "
- 94 -
hy admin.stration to a patient in need of such treatmelIt of a
therapeutically ei'fective amount thereof
In the manageme:nt of hypertension and the cl:inical
conclitiolls noted above, the cc~rllpoll:nds of this invent:ion may be utilized
in compositions such as tablets, capsules or elixirs for oral
administration, suppositories for rectal administration, sterile solutions
or suspensions for parenteral or intramuscular administration, and the
like. The compounds of this invention can be ~1mini~ red to patients
(an:imals and human) in need of such treatment in dosages that will
provide optimal ph~rm ~ ellti-~l efficacy. Although the dose will vary
from patient to patient depending upon the nature and severity of
disease, the patient's weight, special diet.s then be:ing followed by a
patiellt, concurrellt medic.ltion, and other factc rs which those skilled in
the art will recognize7 the dosage r~mge will gene.rally be. about 0.~ mg
5 to l.0 ~. per patient per day which can be sl~mini~t~red in single or
multiple doses. :Peri'erably, the dosage range will be about 0.5 mg to
500 mg. per patient per day; l:nc~re preferably at~out 0.5 mg t(J 20() m~.
per patient per day.
The compound.s of this i:nvention can also be adlIlinistered
20 ill combination ~,vi~h A2-adrenosine receptoragonists, a-adrenergic
antagonists, angioten.sin II antagonists7 angiotensin converting enzyme
inhibitors, ~-adrenergic antagonists, atriopeptidase inhibitors~alone or
with ANP), calcium channel blockers7 diuretics, potassium chamlel
agonists7 renin inhibitors, sertonin antagonists, sympathol~tic ~gents7 as
25 well as otber antihypertensive agents. For example7 the compc~unds oi
this invention c.~m be given in combination with such compound.s as A-
69729, FK 906, FK 744, UK-739007 CSG 22492C7 amiloride, atenolol~
atriopeptin. bendroflumetlliazide7 chlorothalidone, chlorc~tlliazide,
clonidine, cromakalin, cryptenamine acetates and cryptenamine talmates,
30 deserpidine, diazox:ide, doxazosin7 guanabenz, gua:nethidine,
guanethidine sulfate, h~dral,lzine hydroctlloride7 hydrochlorothiazide,
isradipirle7 ketan.serin7 losartan7 metolazone, metoprolol7 met:oprolol
tartate, rmetllyc:lott1iazide, methyldopa. methyldopate hydrochlor:ide,
minoxidil, nadolol, pargyline hydroclllc ride, pinacidil, pindolol7
2 t ~ 5 7 ~ Q
wo 96/0490~ -~ V I ~ l/lJ". ...,v,
- 95 ~
polythiazide, prazosim propranolol, rauwolfia serpentina. rescilmamine,
reserpine, sodium nitroprusside, spironol.lctcme, terazosin, timolol
maleate, trichlormethiazide, trimethophan cam.sylate, verapamil.
benzthiazide, quinethazone, ticrynafan, triamterene, acetazolamide,
S aminophylline, cyclothiazide, ethacrynic acid, furosemide,
merethoxylline procaine, sodium ethacrynate, captopril, delapril
hydrochloride, enalapril~ enalaprilat, fosinopril sodium, lisinopril,
pentopril, quinapril, quinapril hydrochloride, ramapril~ teprot:ide,
zofenopril, zofenopril calcium, diflusinal, diltiazem, felodipine,
nicardipine, nifedipine, niludipine, nilïlodipine, nisoldipine~
nitrendipine, and the like, a.s well as admixtures and combinations
thereof. Combinations useful in the management of congestive heart
failure include, in addition, compounds of this invention with cardiac
stimulants such as dohllt~mi~ and xamoterol and phosphodiesterase
5 inhibitors including amrinone and milrinone.
Typically, the individu.ll daily dosages for the.se
combinations can range from about one-fifth of the minimllm
recnmmPn(~Pd clinical dosages to the maximum recommended levels for
those entities given singly. To illustrate these c.ombinations, one of the
20 endotheliIl antagonists of this invention effective clinically at a given
dail~ dose range can be effectively combined, at levels wtlicll are less
than that daily dose range, with the following compounds at the
indicated per day dose range: hydrochlorothiazide (6-lO0 mg),
chlorothiazide (125-S()0 mg), furclsemide(S-80 mg), ethacrynic acid (5-
25 200 mg), amiloride (5-20 mg), diltiazem(30-540 mg), felodipine( l -20
mg), nifedipine(5- 120 mg), nitrendipine(S-60 mg), timolol maleate (1-
20 mg), propanolol (10-480 mg), and methyldopa(l25-2000 mg). In
addition triple drug combinations of hydrochlorothiazide(6-100 mg)
plus amiloride (S-20 mg) plus endothelin antagonists of this invention,
30 or hydrochlorothiazide(6-lO0 mg) plus timolol maleate (I-20 mg) plus
- endothelin antagonists of this inventiol, or hydrochlorothiazide(6-lO0
mg) plus nifedipine (5-60 mg) plus endothelin ant.lgonists of this
invention are effective combilIati- ns to control blood pressure in
hypertensive patients. Naturally, these dose ranges can be adjusted on a
2 1 ~75~
WO 46~04905 i r~.,. . Y.. _7--
- 96 -
unit basi.s a.s necessary to permit divided daily dosage and the dose will
vary depending on the nature and severity of thc disease. weigllt of the
patient. special diets and other factors.
The present invention also relates to pharmaceutical
5 compositions t'or treating asthma, hypertension, renal failure,
particularly post-ischemic renal failure, cyclosporin nephroto~icity,
vasospasm, cerebral and cardiac ischem:ia, benign prostatic hyperplasia,
myocardial infarct:ion, or endotoxin shock caused by or as~sociated with
endothelin, comprising a therapeutically effective amount of the novel
compound of this invention together with a ph~m~ elltic ~lly acceptahle
carrier therefor.
About 0.~ mg to 1.0 g. of compound or mixture of
compounds of Formula I or a physiologically acceptable salt is
compounded with a physiologically acceptable Yehicle, carrier,
5 excipient, binder, pre.servative, stabilizer, flavor, etc., in a Ullit dosage
form as called for by accepted pharrnaceutical practice. The arnoullt of
acti~e substance in these cc~mpositiolls or preparations i.s such that a
suitable dosage in the range indicated :i.s obtained.
Illu.strative of the adju~ants whicll can be incorporated in
20 ta:blets, capsules and the like are the following: a binder .such as gum
tragacanth, acacia, corn starch or gelatin: an excipient such as
microcry.stalline cellulose; a disintegratillg agent such as com starch,
pregelatini~ed starch, alginic acid and the like; a lubricant such as
magnesium stearate; a sweetening agent such as sucrose, lactose or
25 saccharin; a flavoring agent such as p~,pe,..lil.~, oil of wintergreen or
cherry. Whe.n the dosage unitform is a capsule, it may contain, in
addition to materia:ls of the above type, a liquid carrier such a5 fatty oil.
Variolls other rnaterials may be present as coatings or to otherwise
modify the physical form of the dosage unit. Fclr inst~mce, tablets mày
30 be coated with .shellac, .sugar or both. A .syrup or elixir may cc~ntain the
active compound, sucrose as a sweetening agent, methyl and propyl
parabens as pre.servatives, a dye and a flavoring such as cherry or
orange flavor.
~ wos6/04sos ~ " ' ' 2 ~ 35 75~ P~
- 97 -
Sterile compositions for injection can be fonmulated
according to conventional pharmaceutical practice by dissolving or
suspending the active substance in a vehicle such as water for injection,
a naturally occurring vegetable oil like sesame oil, coconut oil, peanut
5 oil, cottonseed oil, etc.. or a synthetic fatty vehicle like ethyl oleate or
the lilce. Buffers, preservatives, antioxidants and the like can be
incorporated as required.
The following examples illustrate the preparation of the
compounds of Formula I and their incorpor~ltion into pharmaceutical
o compositions and as such are not to be considered as limiting the
invention set forth in the claims appended hereto.
EXAMPLE l
5 2-(2,6-Dipropyl-4-hydroxymethyl)phelloxyphenylacetic acids
Step A: Preparation of Alliyl 2-bromo-2-phenvlacetate.s
Method A:
Substituted phenylacetic acid is convered to the
20 corresponding methyl ester by refluxing the acid in methanol in the
presence of a catalytic amount of conc. sulfuric acid. The e.ster thu~s
obtained is then refluxed in carbon tetrachloride with N-
bromosuccinimide (1.1 equiv) and AIBN (0.0~-O.l equiv). Upon
completion of the reaction, the resulting product is purified by flash
25 column chromatography using silica gel and ethyl acetate in hexane
as eluent to provide the desired al~yl bronlide.
Method B:
An arylaldeh~de is reacted ovemight with trimethylsilyl
cyanide in the presence of catalytic amounts of KCN and l~-croivn-6
30 in methylene chloride. The reaction mixture is quenched with water
and extracted with CH2Cl2f ethyl acetate/ether (l/2/2) mixture. The
organic phase is washed with saturated aq. NaHC03 solutioll. After
dryhlg and concentration of the organic phase, the resulting
trimethyisilyl cyanohydrin is hydrolyzed to give the corresponding
hydroxy acid. 1'reatment wit}l gaseous l lCl in methanol or ethanol at
WO 96104905 r / ~ ~; 2 1 ~ 5 7 5 ~ ~ " Ll ~ i. c ~ hC7
~ 98 ~
O"C for 0.5 h and then overrLight at room t~mrerltl~re alfords the
crude 2-hydroxy ester. The ester is then treated with
triphellylphosplline and carbon tetrabromide in methylene chloride at
O'~'C o~ernight. Methylene chloride is removed and flash column
5 chromatography of the crude product using silica gel and ethyl
acetate/lLexane as eluent gives the desired 2-bromophenylacetates.
StepB: Alkykltion of the ptlenol
(2j6-Dipropyl-4-hydroxymethyl)phenol (prepared as
described in patent application WO 91/11999) is alkylated with 2-
bromo-2-aryl e.iters in DMF u.sing either cesium carbonate
(Cs2C03), or polassium carbonate (K2C03). or sodium hydride
(NaH~. The aL~ylated product is purified by flash column
chromatograph~ using silica gel and ethyl acetate/hexane mixtlJre as
5 eluent to provide the desired sl-hstih-l~d 2-phenoxy-2-phenylacetic
',1 ~
acl~l es.ers.
Step C: (~ener~l pro~P~ re for ester hvdrolv.si.~
Th~ product of Step C is dissolved in methanol or e~Tlanol
20 and reacted with aqueous l~aOH or L.iOH, or E~OH solution at room
temperature for 1- 6 hours, neutralized to pH 7 with I N HCI and then
c.-nni~ntr~tPd in vacuo. The residue i.s purified 011 'd silica gel i1ash
chromatography column to afford the corresponding carbo~ylic acid.
l'he follc~wil1g phenoxyphenylacetic acid derivatives
were prepared using the general procedures mltlil1ed in Example 1.
EXAMPLE 2
2-[~2,6-Dipropyl-4-hydroxymethyl)phenoxy]-2-(3-methylphenyl)acetic
acid
IH NMR (400 Ml-lz, CD30D, ppm): o 7.15 - 6.95 (m, 4H), 6.86 (s,
2H), 4.92 (br s. IH), 4.5 (~s, 2H), 2.3-2.1 ~m, 4H3, 2.2 (s. 3lr)~ 1.5-
1.35 (m. 2H), 1.32-1.lX (m, 2H), 0.7 (t, GH).
~ W0961049(15 ~ ' 2 ~ 9 57 5~ T.,I/L . 3 .................... ~7
_ 99 _
EXAMPLE 3
2-[(2,6-Dipropyl-4-hydroxymethyl)phenoxyl-2-(4-p}lenoxyphenyl)-
5 acetic acid
IH NMR (400 MlIz, CD30D,ppm):o7.42 (d~ 2H,J=8.4 Hz),7.33
(dd,2H,J=7.4 Hz.,8.5),7.09(t,1H,J=7.411z),6.97-6.g5(m,4H),
6.91 (d, 2H,J=8.4 Hz),4.85(s,IH),4.47(s,2H),2.38(t~4H,J=8.0
o Hz),1.56(sx.2H,J=7.11Iz),1.42(sx,2H,J=7.1 Hz),0.85(t,6H,
J=7.3 H7)-
FAB-MS m/e = 435 (M+ I )
EXAMPLE 4
2-1 (2,6-Dipropyl-4-hydroxymethyl)phenoxyl-2-(4-phenylphenyl)acetic
acid
IH NMR(400 MHz,CD30D,ppm):~7.62-7.60 (m, 4H),7.51(br,
20 2H),7.44(t,2H,J=7.5 Hz),7.34(t,111,J=7.4 Hzj,6.99(s,2Hj,4.83
(s,llI),2.40(br,4H),1.53(br,2H),1.42(br,211),0.82(t,6H,
J=6.2 Hz).
FAB-MS m/e = 419 (M+l)
2s FXAMPI,E 5
2-[(2,6-Dipropyl-4-hydroxymethyl)phenoxy]-2-(3-carboxyphenyl)-
acetic acid
30 IH NMR(400 MHz,CD30D, ppm): ~ 8.18(s,1H)8.04(d,111,
J=7.5 Hz),7.70 (d, 111, J=7.4 Hz),7.51(d,1H~J=7.6)Hz,6.99(s,
2H),5.]5(s,1H),4.48(s,21-1),2.37(m,4H),1.52(m,2H),1.44(m,
2H),0.80(t,6H,J=7.3 Hz).
FAB-MS m/e = 387(M+I)
- 2~ ~5753
~0 96104gOS P~
- 100-
EXAIVIPI ~ 6
2-[(2,6-l:)ipropyl-4-hydroxymet}lyl)phenoxy]-2-~3 74-ethylenedioxy-
5 phenyl)acetic acid
IH NMR ~200 MHz, CD3OD7 ppm~: ~ 6.g5 (m7 3H)7 6.85 (dd7 IH7
J=8.37 2.0 Hz~7 6.72 (d7 IH7 J=8.3 Hz~7 4.76 (s7 lH)7 4.46 (S7 2H)7
4.201s7 4H)7 2.37 (t7 4H7 J=7.9 llz)7 1.44 (m7 411), 0.83 (t. 6H. J=7.3
o Hz).
FAB-MS m/e = 401 (M+l)
BXAMPLE 7
2-[(276-Dipropyl-4-hydroxymethyljphenoxy1-2-(374~5-trimethoxy-
phenyl)acetio acid
111 NMR (4()~3 MHz7 CD30D, ppm): ~ 6.97 (s, 2H), 6.80 (s, 2H),
4.88 (s. IH), 4.48 (s, 2H). 3.81 (s, 6H), 3.7~; (s~ 3H), 2.39 (t, 3H,
J=8.1 Hz)7 1.~5~ (m7 2H)7 1.41 ~m7 211)7 0.82 (t7 6H, J-7.3 I{z).
FAB-MS mle = 433 (M+l)
EXA~IPI,E 8
2-[2,6-Dipropyl-4-hydroxymethyl)phenoxy1-2-(3,4-methylenedioxy-
phenyl)acetic acid
lH NM~R (20Q lvlHz7 CD30D? ppm): o 7.03 (S7 11~1)7 6.97 (S7 211),
6.83 (d7 IH7 J=7.7 llz)7 6.73 (d, 2H7 J= 7.7 Hz), 5.94 (s, ~H)7 4.~4 (S7
3~ IH), 4.48 (s. ~H), 2.38 ~t7 4H7 J=~.0 Hz), 1.46 lm7 4H)7 0.85 ~t7 6H7
J=7.4 Hz).
PAB-MS m/e = 387 (M+l~
-- W096/0490S ~ . 9 7 5~ F~.IIL_. !i. ~5~7
- 101 -
EXAMPLE 9
2-[ (2,6-Dipropyl -4-hydroxymethyl)phenoxy] -2-(3,4-dimethoxy-
phellyl)acetic~ acid
lH NMR (400 MHz, CD30D, ppm): o 7.15 (d, IEI), 6.95 (d~ 2H),
6.~5 (dd, 2H), 4.8 (br s, IH), 4.46 (br s, 2H), 3.84 (s, 3H)~ 3.8 (s.
3H), 2.35 (t. 4H), 1.62-1.47 (m, 211), 1.45-1.3 (m, 2H), 0.85 (t, 6H).
EXAMPLE 10
2-[(2,6-Dipropyl-4-hydroxymethyl)phenoxy]-2-(3,5-dimetlloxy-
phenyl)acetic acid
I H NMR (400 Ml lz, CD30D, ppm): o 6.954 (s, 2H), 6.66 (d, 2H),
6.39 (t, lH~, 4.747 (s, IH), 4.47 (s, 2H), 3.74 (s, 6H)~ 2.39 (t, 4H),
1.60-1.51 (m, 2H), 1.437-1.35 (m, 2H), 0.82 (t, 6H).
EXAMPLE 11
2-~(2,6-Dipropyl-4-tetrazol-5-yl)phenoxy)-2-(3-bromophenyl)acetic
acid
I H NMR (400 MHz, CD30D, ppm): ~ 7.73 (s, 2H), 7.67 (t, lH, J=l .X
Hz), 7.57 (m, IH), 7.46 (m, IH), 7.33 (t, lH, J=7.9 Hz), 5.X9 (ddd, lH,
J=1.6, 10.1, 17.1 Hz), 5.41 (s, IH), 5.08 (dd, 11-1, J=10.1, 1.6 Hz), 5.01
(dd. IH, J= 1.7, 17.1 Hz), 4.93 ~s, 2H), 3.72 (s, 311), 3.36-3.30 (m, 2H).
FAB-MS mle = 470 (M+ l )
EXAMPLE 12
2-[(2,6-Dipropyl-4-hydroxymethyl)phenoxyJ-2-(3-bromophellyl)acetic
acid
WO 96/0490S ~ , 2 1 9 5 --TT 5 8
- 102-
I H NMR ~400 MHz. C'D~OD/CI:)C13,2/1, ppm): o 7.667 (s, I H~),
7.496 (d, IH), 7.3925 (d, 111)7 7.252 (t, 11-I), 6.9~ s~ 2H), 4.995 (s,
111), 4.~85 (s, 2H), 2.342 (t, 4E-1), 1.65-1.35 (m, 2H),0.803 (t,6H~.
EXAMPLE 13
2-[(2,6-Dipropyl-4-hydroxymethyl)phenoxyl-2-(2-naphthyl)acetic acid
H NMR (400 MHz, CD30D, ppm): o S.56-~.52 (m, lH), 7.~¢6-7.79
o (m, 2H), 7.47-7.4~ ~m, 2H), 7.35-7.32 (m, I H), 6.91 (s, 2Tl), 5.36
(s, 111),4.44 (~, IH~, 1.46-1.41 (m, 2 1~). 1.2-1.16 (m, 2E~), 0.5~ (t, J
= 7.37, 6 H).
EXAMPLE 14
2-[(2,6-Dipropy]-4-(2-hydroxyethyl)phenoxy]-2-(2-naphthyl)acetic acid
STEP A: t-ButvldimeLhvlsilvloxv-~.6-Diprop~TI- I-furm~lberlzerle
To a solution of 5.03 g (1~.4 mmol~of t-butyl-
dimethylsilylo~y-2,6-dipropyl-4-hydroxylnethyl benzene in 30 mL
of met:hylene chluride was aT~ded 8.7 g of pyridiniT~ dichrumale
(PDC). The reaT~tion mixture w~s stirred for 3 houls and thell
dilute.d with 30~3 ml, of ethyl ether. The sclluticm was then filtered
25 through a pad Qf a ifl mixture of florisil and celite. Concentration
of the filtrate ga~e 4.~5 g of the title compound.
] H NMR ~400 MHz. CDC13, ppm): o 9.~ (s, IH),7.51 (s, 2H), ~.59-
2.55 (m, 2Hi 1.59-1.55 ~m, 211), 0.99 (s, 9H T, 0.91 ~t, J = 7.2~ T.~Z~,
30 3H), ().20 (s,6H).
STEP B: t-l~utyldimethyl~silyloxy-2~6-flillropyl-4-vinyl benzene
Tcl a .solution of 1.0 g (2.~0 mmol) of methyl
triphenylphosphollium bromide in 5.0 mL of ether at 0 C was added
1.12 mL (2.51~,1, 2.~0 mmol) of butyllithium. The reaction mixture
-- WO 9GlQ4gO5 ~ 2 1 9 5 7 5 8
- 103 -
~ was stirred for 30 minute.s at 0 C and then 756 mg (2.33 mmol) oi
the title compound from Example 14 (Step A) was added. After
stirring for lh at room temperature. the reaction mixture was
poured into ethyl acetate and washed with water and then saturated
aqueous sodium chloride. The organic layer was dried over
anhydrous sodium sulfate, filtered and concentrated. Purification by
flash cll-ulllc~to~ld~lly (silica gel, hexane / ethyl acetate 97:3) gave
411 mg of the title compound.
IH NMR (400 MHz, CDC13, ppm): o 7.02 (s, 2H), 6.6 (dd, J = 17.6,
J = 10.7 H~, IH~, 5.55 (d, J=17.6 E~z, IH), 5.0~ (d, J = 10.7 llz, 11:1),
2.53-2.50 (m, 4 H), 1.59-1.53 (m, 4H), 0.996 (s, 9 H), 0.90 (t, J =
7.33E-Iz,6H),0.16(s,6H).
STEP C: t-E3utyldimethylsilyloxy-2,6-dipropyl-4-(2-hydroxyethyl)-
benzene
To a solution of 475 mg (1.4~ mmol) of the product of
Step B in 3 mL of THF at 0 C was added 1.6 mL (1.62 rnmol) of a
I N borane / THF solution. After I houl Tl.C indicated th.lt the
2c starting material had been consullled. The reaction mixture was
quenched Wit}l 3 drops of methanol and then 0.70 rmL (6.22 mmol)
of 30 ~'~O sodium peroxide and 6.2 mL (6.2 n~nol) of I N sodium
hydroxide were added. After two hours tlle reaction mixture was
diluted with ethyl acetate and washed with water and brine. The
25 organic layer was then dried over sodium sulfate, filtered and
concentrated. Flash chromatography (silica gel, hexane / ethyl
acetate 4: 1) gave 265 mg of the title compound as a clear oil.
I EE Nh/IR (4()0 MHz, CDC13, ppm): ~ 6.79 (s, 2 H), 3.79 (m, 2 H),
30 2.75 - 2.72 (m, 2 EE), 2.51 - 2.4~ (m, 4 H), 1.5~ - 1.51 (m. 6 E-l),
0.99(s,9H),0.90(t,J=7.33,6H),0.16(s,6H).
STEP E>: 2~6-Dipropyl-4-(2-hydroxyethyl)phenol
To a solution of 1.0 g (2.9~ mmol) of the product of
Step C in 3.0 mL of THE~' was added 3.57 mL (3.57 mmol) of 1.0 N
WO 961049U5 ! ~ 2 t 9 ~ 7 5 8 I ~ I ~ ,115 .
- I ()4 -
solution of tetrabutylammonium fluoride in ~HF. After 1~ minutes
TLC indicated that the reaction was complete. The reaction mi~ture
was ,.ol1cen~ d and then purified by flash ~ vl~ o~;ld~hy ~silica
gel, hexane/eth~l acetate 3:1) to give 1.13 g of the title compound.
I H NMR (400 MHz~ C'DC13, ppm): o 6.8 (s. 2 H~, 3.78 (t . J = 6.5
Hz, 2 E-1), 2.73 ~t, J = 6.5 Hz., H~, 2.54 - 2.50 (m, 4H), 1.66 - 1.56
(m,4H),0.96(t,J=7.3Hz,6H).
STEP E: Methyl 2-[(2,6-d;propyl-4-(2-hydroxyethyl)phenoxy]-2-(2-
n~phthvl )acetate
The title compound was prepared from 2,6-dipropyl-4-
(2-hydroxyetbyl)phenol (S~ep D) by alkylating with methyl 2-
bromo-2-(2-naphthyl)acetate using cesium carbonate or pota.ssium
carbonate in DMF. The reaction mixture was filtered through Celite
and the filter cake was washed with methylene chloride. The filtrate
was concentrated and the resultant material was purified by ~lash
columll chromatograplly to yield the titled es~er.
I H NMR (400 MH7., C'DC13, ppm~: o 7.90 - 7.82 (m, 4 H~, 7.69 -
7.67 tm. I H), 7.49 - 7.47 (m. 2H), 6.8 (s, 2 H), 2.74 (t~ J = 6.2 Hz,
2 H~. 2.36-2.3~ (m, 4 E-1~, 1.49 - 1.41 ~m, 4 H), 0.72 (t, J = 7.3, 6 El).
S'lEP E~: 2-[(2,6-Dipropyl-4-(2-hydroxyethyl,~phenoxy~-2-(2-
naphthyllacetic acid
'I'lle title eoll1pound was prepared from the producl c)f Step E
by saponification with IN aque.ous KOM in med1anol as outlined in
Step C of E~ample 1.
30 l E'l 'NMR (400 MHz, CD30D, ppm) o 7.84 - 7.7 (Irl, 4 H), 7.73 ~d,
J = 6.8 Hz, I H~, 7.45-7.43 (rm, 2 H), 6.79 (s, 2 H), 5.01 (s, 2 H~,
3.66(t,J=7.2Elz,2H),2.68(t,J=7.2Hz,2H~,2.33-, 29(m~4
H), 1.55 - 1.4~i (m, 2H~, 1.40- 1.28 (M, 2 El), 0.69 (t,3 = 7.3, 6 El).
F.4.B- MS: mle = 445 (M + K), 429 (~1 + Na), 407 (M f 1).
~ W0 96104905 ~ 2 1 q 5 7 5 P~ P~ 7
- 105-
The following phenoxyphenylacetic acid derivatives
were prepared using the general procedures outlined in Example 14.
EXAMPLE 15
2-1 (2,6-Dipropyl -4-(2-hydroxyethyl)phenoxy 1-2-(3 ,4-methylenedioxy-
phenyl)acetic acid
NMR (200 MHz, CD30D, ppm): o 7.03 (s~ IH), 6.133 (m, 3H),
6.72 (d, lH, J=7.8 I-lz), 5.93 (s, 211)7 4.80 (s, IHJ, 3.68 (t, 2H, J=7.1
Hz), 2.69 (t, 2H, J=7.1 Hz), 2.35 (t, 4H, J=7.9 l~z), 1.42 (m, 4H),
0.~3 (t, 6H, J=7.3 Hz).
5 FAB-MS m/e = 401 (IM+I)
EXAMPLE 16
2-L(2,6-Dipropyl-4-~2-hydroxyethyl)pllelloxy]-2-(3-methoxyphenyl)-
20 acetic acid
IH NMR ~400 MHz, CD30D, ppm): o 7.28 (t, IH, J=7.9 Hz), 7.07
(m, IH), 7.15 (m~ IH), 6.g4 (m, IH), 6.8i4 (s, 2H), 4.99 (s, IH), 3.79
(s, 3H), 3.G8 (t, 2H, J=7.] Hz), 2.70 (t, 2H, J=7. I Hz), 2.34 (t, 4H,
25 J=8.0 Hz), 1.54-1.40 (m, 4H), 0.~0 (t, 31-1, J=7.3 H7).
FAB-MS m/e = 387 (M+l)
EXAMPLE ]7
30 2-[(2,6-Dipr(lpyl-4(1,2-dillydroxyethyl)phelloxy~]-2-(2-llapllthyl)acetic
acid
STEP A: 2~6-dipropyl-4-vinylphenol
WOg6~W9DS ~ 21 ~57~8 r~~
- io6-
'Ille title compoulld was prepared from t-butyldimetllyl-
silyloxy-2,6-dipropyl-4-vinylbenzene (Step B, Exarnple 14) by
treatment with tetrabutyl~mmnnillm fluoride in THF for a few
hours. It was poured into ether/ethyl acetate mixture and washed
with brine. After removal of the solvent the crude product was
purified by flash column chromatography using ethyl acetate/he.xalle
as eluent.
IH NMR (400 MHz, CDCL3, ppm): o 7.01 (s, 2 H), 6.55 (dd, J =
o 17.6, J = l l.O Hz, lH). 555 (d, J = 17.6 Hz. l H), ~.06 (d, J = l l.O
Hz, I H), 2.64 ~t, J = 7.7 I{-z, 4 H~, 1.65 - 1.6() ~'m, 4 H), 0.96 (t, J =
7.2 Hz 6 H).
STEP ~B: Methyl 2-[~2,6-dipropyl-4-vinyl)phenoxyl-2-I2-naphthyl)-
acetate
The title compound was prepared by alkykatiorl of 2,6-
dipropyl-4-vinyl phenol (Step A~ with methyl 2-bromo-2-(2-
naphthyl)acetate using the procedure for alkylation described in Step
B of F,xample 1.
~H NMR (400 MHz, CDC13~ ppm): o 7.9 - 7.~ (m, 411), 7.6R ~d, J =
6.7 llz, IH), 7.50 ~ 7.4~ ~m, 2 H), 7.02 (s, 2 H), 6.57 (dd J = 18.4,
10.8Hz, I H).5.61 (d.J=18.4, I E~).5.~6~s, 1 11),~.14~;d,J=
10.8, I H)~ 3.73 ~s, I H), 2.3~ - 2.34 (m, 4 H), 154 - 1.43 Im, 4 H~,
0-74 (t, J = 7.33 H~, 6 Hj.
ST~P (~: Methyl 2-[(2,6-dipropyl-4-(1 ,2-dihydroxyethyl)phenoxyl-
2-(2-naphthyl)acer~rt~
To a solution of 6 mg (0.024 mmol) of OsO4 and 31 mg
(0.263 mmol) of N-methylmorpholine-N-oxide ('NMO) in 3 ml, oi
acetone and 2 drops of water was added 96 mg (0.239 mmol) of the
prc~duct c)f Step 13. After 90 minutes the reaction mixture was poured
intn a mixture of ether and water. The layers were separated aucl tbe
aqueous layer was extracted twice with ether. The combined aqueous
klyers were wa.stled ~ith saturated sodium chlnride, dried over
~ wO 96/049û5 ~ 19 5 7 5 ~ r~ , 7
- 107 -
~ anhydrous magnesium sulfate, filtered and concentrated. Purification
by flash chromatography (silica gel, hexane / ethyl acetate 1: 1) gave
63 mg of the title compound.
s 1H N~IR (400 MHz, CDCl3, ppm): o 7.89 - 7.80 (nl~ 4 H), 7.67 (d, J
=8.4Hz,lH),7.50-7.48(m,2H),6.9(s,2H),5.25(s,1H),
4.75-4.68 (m. I H), 3.72 (s, 3 H), 3.75 - 3.61 (m, 2 H), 2.38 - 2.34
(m, 4 H), 1.53 - 1.46 (m, 4 H), 0.75 - 0.71 (m, 6 H).
10 STEP D: 2-[(2,6-Dipropyl-4-(1,2-dihydroxyethyl)phenoxyl-2-(2-
naphthyl)acetic acid
The title compound was prepared from the product of
Step C by saponificatiol1 with IN aqueous KOH solutiol1 as described
above.
I l l NMR (400 MHz, CD30D, ppm): o 7.84 - 7.71 (m, 5 H), 7.46 -
7.42(m,2H),6.96(s,2H),5.03(s, I H),4.55(t,J=7.2Hz, I H),
3.54(d,J=7.2Hz,2H),2.34(t,J=7.9Hz,4H), 1.51-1.30(m,4
H~,0.70(t,J=7.3Hz,6H).
~XAMPLE~ 1~
2-1 (2,6-Dipropyl-4-( 1 -hydroxypentyl)phenoxy] -2-(2-naphthyl~acetic
acid
STEP A: Methyl 2-l (2,6-dipropyl-4-lormyl)phenoxy]-2-(2-
naphthylklcetateTo a solution of 262 mg (0.645 mmol) of methyl 2-
[(2,6-dipropyl4-hydroxymethyl)pllenoxyl-2-(2-naptlthyl)acetate in 2
mL of methylene chloride wa.s added 4()4 mg (0.968 mmol) of PDC.
After 4 hours the reaction mixture was diluted with 20 mL of ether
and filtered through a pad of florisil / celite and concentrated to give
235 mg of the title comllound.
wo96~90s ~ ~ ' . 2 ~ 9 5 7 5 8 1~I~ LL.. ~ '57
- 108 -
I H NMR (400 Ml lz. CDC13. ppm): ~ 9.S6 (s, Il), 7.89-7.S() (m. 4
H), 7.65 (m, 1 H), 7.5~ - 7.49 (m, 4 H), 5.35 (s. 1 H), 3.73 (s. 3 H),
2.47-~.43 (m, 4 ~), 1.54-1.43 (m, 4 H), 0.77 (t, J = 7.3 Hz, 6 H).
STEP B: Methyl 2-1(2,6-dipropyl-4-~1-hydrox~pentyl)pilelloxy]-2-
(2-naphtllyl)acetate
Tu a solution of 56 mg (0.143 mmol) of the product of
Step A in 1 mL of THF at -78 C was added 0.075 mL (2.0 M in
THF. 0.]50 mmol) of n-butyl magnesium chloride. TLC analysis
showed that the starting material remained uncollsullled so ().020 nlL
of n-butyl magnesium chloride was added. After 1 h the reaction
mi~ture wa,s diluted with saturated aqueous ~IllllllUIIiUIII chloride
solution and thcn extracted twice with ethyl acetate. The organic
layers were dried over anhydrous sodiurn sulfate, filtered and
concentrated. 'Purification by Jla.sh colunm cluomalogrdpll~ (silica
gel, hexane / ettlyl acetate 6: 1) gave 32 mg of the title cc~mpc und.
I H NMR (400 MHz, CDC13, ppm): ~ 7.~9-7.X2 (Itl, 4 H), 7.67 (m, I
I l), 7.49-7.47 ~;m, I 1), 6.9 (.s, 2 H), 5.26 (s, I H), ~.36 (t, J = 8.0
I-lz, 4 T-l). 1.75-1.2 (m, 8 1~), 0.86 (t, J = 7.2 Hz, 3 H). 0.72 (t, J =
7.3 Hz. 6 H).
STEP C: 2-L(2~6-Dipropyl-4-~1-hydroxypentyl)phenoxy)]-2-
naphth~llacetic acid
2s The title compound was prepared from the product of
Step B by saponification with aqueous lN KOH in methanol as
described above.
11-1 NMR (400 MHz, C~D3OD, ppm): o 7.S4-7.72 (m, 5 I l), 7.45 -
~.43 (m, 2 H), 6.91 {s, 2 H), 5.03 (s. 1 1-1)~ 4.45 (t. I 1-1), 2.36 - 2.32
(m, 4 H), 1.75 -1.45 (m, 4 H), 1.35 -1.29 (m, 4 H~, 0.S7 (t, J = 7.
Hz,3H),0.70(t,J=7.21-lz,6H).
FAB-MS: m/e = 487 (M+ K), 469 (M + Na).
~ W096/04905 . ~1 95758 r~"~ os c7
. ~ . i
,
- 109-
EXAMPLE 19
2-~(4-Carboxy-2.6-dipropyllphenoxyl-2-pbenylacetic acid
5 STEP A: t-Butyl 2-[(4-carbomethoxy-2,6-dipropyl~phenoxyl-2-
phenylacetate
Methyl 2,6-dipropyl-4-hydroxybenzoate (1.5 g, 6.383
mmol~ was refluxed with K2C03 (1.5 equiv) and t-butyl a-
bromophenylacetate (2.4 g. 8.856 mmol) in acetone for 16 h. The
o reaction mixture was filtered through Celite, the filter cake was
wa.shed with acetone and the combined filtrate and washillgs were
concentrated. The resulting crude oil was chromatographed (flash
column) using silica gel and lO~'~o ethyl acetate in hexane to give the
titled compound (2.7 g).
IH NMR (400 MHz, CD30D, pprn): o 7.665 (s, 2H), 7.443 (dd, 2Hj,
7.345 (dd, 3H), 5.019 ~s, IH), 3.851 (s~ 3H), 2.49-2.335 (m, 4H),
1.63- 1.4 (m, 4H), 1.364 (s, 9H), 0.803 (t, 6H).
20 STEP B: t-Butyl 2-[(4-carboxy-2,6-dipropyl)phenoxyl-2-phenyl
acetate
Saponification of the above t-butyl 2-[(4-carbomethoxy-
2,6-dipropyl)phenoxy]-2-phenylacetate (200 mg, 0.47 mmol) with
IN aqueou.s solution of LiOH in methanol gave the titled compound
2s (125 mg).
I H NMR ~400 MHz, CD30D, ppm~: ~ 7.66 (s. 2H), 7.5-7.4 (dd, 2H).
7.43-7.36 (dd, 3H) 4.88 (s, IH), 2.5-2.35 (m, 4H)~ 1.63-1.33 (m,
4H), 1.38 (t, 911). 0.83 (t, 61-1).
STEP C: 2-1(4-Carbomethoxy- ,6-dipropyl)phenoxyl-2-phenyl~lcetic
acid
t-Butyl 2-[(4-carbomethoxy-2,6-dipropyl)phenoxy]-2-
phenylacetate (Step A) (125 mg, 0.293 mmol) was treated with 3 mL
WO 96/0490!i ;~ , P~
2 ' 95/758
- 110-
of trifluoroacetic acid ~TFA) in methylene chloride for 2 h. 'llle
volatiles were removed to give the titled compound ~90 mg~.
I H NMR ~400 ~IHz, CD30D, ppm): ~ 7.67 (s. 2H), 7.463-7.44 (m,
2H), 7.387-7.362 (m, 311), 5.177 ~s, lH), 3.856 ~s, 3H), 2.377 (t,
4H), l.tj -1.366 (m, 4H), 0.773 ~t, 6H).
STEP D: 2-r~-Carboxy-2.6-dipropyl)phenoxy1-2-phenyiacetic acid
The product of Step C ~70 mg, O.lg mmol) WdS treated
o with lN aqueous solution of LiOH in methanol. The reaction was
monitored by TL,C. When the starting material was completely
consumed, the mixture was acidified at 0~C to pH 5 by addition of
IN HCI. 'rhe aqueous phase was extracted with ethyl acetate, dried
over anhydrous magnesium sulfaie and filtered. The filtrdte was
15 ~oncerltrated to yield the titled compound (25 mg).
I H NMR (400 MHz, CD30D, ppm): ~ 7.68 (~, 2H), 7.52-7.45 (m,
2H), 7.43-7.365 (m, 3H), 5.175 (s, IH), 2.43 (t, 4H), 1.o4-1.4 (m,
4H), 0.83 (t, 6H).
EXAMPLE 20
2-l(4-Carboxy-2~6-dipropyl)phenoxyl-2-~314-dichlorophellyl'~acetic
acid
The titled compound was prepared using procedures
similar to that described in Example 19.
IH NMR (20(1 MH7., CD30D, ppm): o 7.72 ~d, IH, J= 2.() H~) 7.6~:~
(s, 2H), 7.56 (d, IH, J=8.3 Hz), 7.43 (dd, lH, J=8.3, 1.9 Hz~, 5.18 (s,
IH), 2.45 (111, 4H), I.S8-1.43 (m, 4H), 0.~4 (t, 6H, J=7.3 Hz).
FAB-MS m/e = 426 ~M+l )
~ WO 96/04905 ~ , 9 5 7 5 8
111
EXAMPLE 21
2-[(4-Carboxy-2,6-dipropyl)phenoxy]-2-(3-bromophenyl)acetic acid
The titled compound was prepared using procedures
similar to that described in Example 19.
111 NMR (200 Mltlz, CD30D, ppm): o 7.73 (d, IH, J=l .8 Hzj, 7.69
(s, 2H), 7.56 (dd, lH, J=7.8, 1.9 Hz), 7.46 (d, Ill, J=7.9 Hz), 7.32 (t,
o IH, J=7.8 Hz), 5.19 (s, IH), 2.44 (t, 4H. J=7.6 Hz), 1.70-1.34 (m,
4H), 0.~4 (t, 6H, J=7.3 Hz).
FAB-MS m/e = 436 (M+l )
EXAMPLE 22
2-[(4-Carboxy-2,6-dipropyl)phenoxyl-2-[3,4-methylelledioxyphenyl'
acetic acid
The titled compound was prepared using procedures
20 similar to that described in Example 19.
IH NMR (200 MHz, CD30D, ppm): ~ 7.68 (s, 2H), 7.02 (d, IH,
J=1.6 Hz), 6.84 (m, 2H), 5.98 (s, 2H), 5.08 (s, IH), 2.44 (t, 4H,
J=7.9 Hz), 1.52 (m, 4H), 0.86 (t, 6H. J=7.3 Hz).
FAE~-MS m/e = 401 (M+l)
EXAMPLE 23
2-[(4-Carboxy-2,6-dipropyl)phenoxyl-2-(3-methoxyphenyl)acetic acid
The titled cornpc~und was prepared using procedures
similar to that described in Example 19.
IH NMR (200 MHz, CD30D, pprm) o 7.68 (s, 2H), 7.29 (t, I H, J=7.9
I-lz), 7.08 (d, 1 H, J= 2.3 l lz), 7.03 (d, lH, J=7.7 Hz), 6.95 (dd, I H,
W0 96/~4~05 ~ 2 t 9 5 7 5 ~ b l --
- 112 -
J=0.9, 8.3 Hz), 5.14 (s, lH), 3.79 (s,3H), 2.43 1t, 4H, J=7.9 Hz).
1.58-1.42 (m, 4H), 0.82 ~t, 6H, J=7.3 Hz).
FAB-MS mle - 387 ~M+I)
LXAMPLE 24
(N-Benzenesulfonyl)-2-[(4-(N-ben~enesulfonyl)carboxarnido-2,6-
dipropylpheno~y]-2-(3 -bromophenyl)acetamide .
Th~ titled compound was prepared using the procedure
descrihed for the synthesis of N-sulfr m,~lcarboxamides in US Palellt
5,177,095. The diacid 2-1(4-carboxy-2,6-dipropyl)pheno.cy~-2-~3-
bromophenyl)acetic acid (200 mg~ 0.46 rnmol; from Example 2i)
was refluxed with carbonyldiimidazole (1.5 equiv) in THF for 3-4 h.
At room 1~ ",~ llre, a mixture of 1.5 equiv of benzelle.sulfollalllide
and 1.5 e~luiv DBU in THF' was added to the abvve reaction mixture,
and the mixture was stirred overnight. The reaction mixture W.IS
diluted with ethyl acetate and washed with 5'-;~, a4. solution of citric
acid. The solvent wa.s removed and the crude product wa.s purified
by flash column chromatography to provide 238 mg of the titled
compc und.
IH NMR 140() M}lz, CD30D, ppm): ~ 8.07 (dd, 2H. J=1.4.7.2 Hz),
7.91-7.88 (m, 2H), 7.67-7.49 ~m,8H), 7.46 (s, 2H). 7.~8-7.2~ (m.
25 2H), ~.01 (s, IH~, 2.28-2.23 (m, 4H7, 1.49-1.2'9 (m, 4H7; 0.71 (t. 6H~
J=7.4 l lz').
FAB-MS mle = 713 ~M+1).
~0
l!~XAl~rlPLE 2~
N-(4-t-butylbenze:nesulfonyl)-2-(4-meihoxycarbonyl-2-propyl-
phenoxy)-2-(3,4-methylenedioxyphenyl ~acetamide
~WO 96/04905 ~ 9 5 7 5 ~
; ;.
- 113 -
Step A: Preparation of 2-(~-carboll1ethoxy-2-propylpllenoxy)-3 4-
methylenedioxyphel1ylacetic acid
~ To ethyl 2-(4-carbomethoxy-2-propylphenoxy)-3 4-
methylenedioxyphenylacetate (Step A of Example 56) (2.04 g 5.10
mmol) in MeOH (40 mL) wa.s added 5 N NaOI-I (8 mL). The rapid
reaction was fo]lowed immf~ t~ly by TLC to monitor mono
deesterification. The reaction was quenched with 9 N HCI (4.5 mE)
after loss of the ethyl ester and before methyl ester saponification. A
saturated solution of NaHCO~ was added to the reaction until it was
basic and the ~leOH was removed in vacuo. The residue wa.s
partitioned between Et20 and uater collecting the product in the
aqueous phase and removin~ impurities with the organic phase. The
aqueous phase was then acidified with 9 N HCI (pH = I ) and the product
extracted into EtOAc. The solution was dried over ~IgSO4 filtered and
the solvent removed. yield = 1.78 g (4.7~ mmol 94~o) rf = 0.16
(~O:10:1/CHCl~:MeOH:NH40H).
Step B: Preparation of the precursor sulfonamide
To a dichloromethane solution of the sulfonyl chloride
(~ leq) cooled to 0~C was added t-butylamine (3 eq). After 3-5 hrs the
CH2CI2 was removed and replaced with EtOAc. The reaction solution
was washed with I N HCI water~ 1 N NaOH and brine. The resulting
.solution was dried over MgSO4 and filtered. The .solvent was removed.
To the resulting solid was added a couple of drops of anisole and then
TI~A to remove the t-butyl group. After all of the sulfonamide had
been deprotected. the TFA was removed in vacuo and the residue taken
up in EtOAc/Et20. The solution ~vas ~vashed with saturated l~al-{CO3
solution to remove any residual TFA then with brine dried over
- MgS04 filtered and the solvent removed.
The sulfonamide precursors used in the preparation of the
compounds of Examples 28 29 31 32~ 37 38 and 39 were prepared
from the corresponding sulf'onyl chlorides utilizing the procedure
described above. The sulfonamide precursors used in the prepardtion
W096104905 p~"~ ,57
2~ 9~75~
- 114-
of the compound.s of Examples 26 and 33-36 are commercially
available.
'I'he sult'onamide precursors used in Examples 27, 30 and
32, whose sulfonyl chlorides are not cornmercially availahle, were
prepared using standard chemistry:
Preparation of precursor sulfo~mide for Example ~7
The t-buh~lsulfonamide of 4-bromobenzenesulfonyl
chloride was prepared usin6 the procedure- described above. The t-
butylsulfonalnide was then coupled to phenylboronic acid in a p~ll"~ rn
catalyzed cross-coupling reaction with NaOH, EtO}I, toluene, and
Pd(PPh3~4 at l()(~' C' to afiord the biphenylsulfonamide. Deprotection
of the t-butylsulfonamide with TFA and anisole yielded the f'ree
sulfonamide.
Preparation of precursor.sulfonamide for Fuuln~le 30
The t-butylsulfonamide of 2-thiophenesulfollyl chloride ~vas
prepclred using the procedure described above. Treatmenl of the t-
butyl.sulf'onamide with Bul,i then i.sobutyl iodide afforded the .1-
20 isohutyl-2-thiophene-t-butylsulfonamide which was then deprutected
Witil TFA and anisole to yield the free sulfonclmide.
Preparation of prec:ursor sulfonamide for Ex ~nu;~le 32
The ~-butylsulfonamide of p-nitrober~enesult'onyl chloride
was prepared using the procedure described above. Reduction of the
nitro group to the amine W'dS accomplished with hydrogen in MeC)H
over Pd-C. Treatment of the free arnine with LiBr and (MeOi~PO and
then NaOH afforded the dimethylamine and the t-butylsulfonainide was
deprotected with TFA and ani.sole to yield the free sulfonalllide.
31)
~ : ~ '' 2~q~7~8
W0 96/04905 Y~ 7
- 115 -
Step C: Preparation of N-(4-t-butylbenzenesulfonyl~-2-(4-carbo-
methoxy-2-propylphenoxy)-3,4-methylenedio~yphenyl-
acetamide
To the product of Step A (57.2 mg. 0.154 mmol) in dry
THF (0.75 mL~ was added CDI (76.0 mg, 0.469 mmol) and the reaction
heated to 50~C for 2.5 hr. To this solution was added a solution of p-t-
butylphenyl-sulfonamide (131.7 mg, 0.618 mmol) and DBU (92.1 ~lL,
0.594 mmol) in dry TIHF (0.75 mL). The reaction continued to be
stirred at 50~C monitoring by thin layer chromatography until all of the
mono-acid was con.sumed (approx. 3 hr). The reaction ~,vas
concentrated in vacuo and the residue was taken up in
50:50/F,t20:EtOAc. The organic phase was washed with lO~o citric acid
(2~), water and brine then dried over MgSO~, filtered and the solvent
removed. Purifïcation was accomplished by radial chromatography
15 eluting with 3:2/He~:EtOAc. yield = 74.3 mg (0.131 mmol, 85~) rf =
0.32 (R0:10:1/CHCll:MeOH:NH4011) FAB mass spectrum, m/e 590.0
(M+Na calculated for C30H33NSO# 5903. See Drummond, J.T.;
Johnsoll, (G. Tetrahedron Lett., 1988, 29, 1653.
2~
5 7 5 8
W0 9G/04gO5 r~
- 116-
EXAMPLES 26-39
Example.s 26 through 39 were prepared following the
procedure.s de~cribed above in Example 25.
CO2Me
~
o~z
0~3
E ~ h,1as~ pe~ -um
CON} S02Ph (M+ ;) 5 ~.. 0
-CONH 02-~p-phen~ )Ph (M+l~ a) l ~.0
CONrrlS02-(p-CI) ~ (~1+) 5--6.0
.,CO~-lSO2-(p-Me' ~1
coN-~c --(5-isu~ hio,~rlene (~I+~IJ ~6.4
C~ !~ û,-(p-:~/eO)Eh
,_CON~ 02-(p-N YIe ) 'h Y + ) : . .
'.,0.~- 02-(o- \~ +- ) - -
2s -CC'N - -(o-CO2~ e,Ph ~ ~+~ . 7 .u
::C~~- 02-~o-C ):'h v +~: 4 .
02-lm-C ~h ~ +') 4
Cf~ ~ -I 02CH2 'h ( ~' + ~ ~h,
C~ ~ ~-dansy ( v + )
COI~-. 02-~-quinoline 1':.~ + )
- ~. . ' 2 ~ 9 5 7 5 8
WO 96/0490~ 7
- 117-
The proton NMR data for Example 29 is given below:
EXAMPLE 29
5 N-(4-methylben~enesulfonyl)-2-(4-methoxycarbonyl-2-propylphenoxy)-
2-(3.4-methylenedioxyphenyl)acetamide
~H NMR (400 MHz~ CD30D, ppm): o 0.89 (t, 3H), 1.59 (m, 2H), 2.34
(s, 3H), 2.63 (m, 2H), 3.85 (s, 3H), 5.49 (s, 11-~), 5.97 (s, 2H), 6.55 (d,
0 111), 6.79 (d, 11-1), 6.91 (d, 11-1), 6.96 (dd, 111), 7.26 (d, 2H), 7.58 (dd, IH), 7.70 (d, 2H), 7.74 (d, lH).
EXAMPLE 40
N-(4-t-butylbenzenesulfonyl)-2-(4-carboxy-2-propylphenoxy)-2-(3,4-
methylenedioxyphenyl)acetamide
To the product of Example 25 (51.1 mg, 0.090 mmol) in
MeOltl (2 mL) was added 5 N NaOH (0.5 mL). The reaction was
2c monitored by TLC. When the reaction was complete the MeOH was
removed .md the residue partitioned between water .md Et20:EtOAc.
The water layer was acidified with HCI solution and the product
extracted into the organic phase. The organic phase was washed with
brine then dried over MgSO4, filtered and the ~olvent removed.
25 Trituration wittl Et20/Hex provided a wllite solid. yiekl = 25.8 mg
(0.047 mmol, 52~,o) I~AB mass spectrum, m/e 554.2 (M+l calculated
f'or C2~ NSOi 554).
75~
~ ' 21q5
WO9C/OJ90~ r~l,.u,
- 118 -
EXAMPLES 41-~4
E~amples 41 through ~4 were prepared following the~ A
procedures described above in E~ample 40.
s
CO2H
~~
o~z
0~
h'x. Z Mass Spectrum
C~IN~ SOzPh ~'v1+1~ 4
CONH ~ 'p-phenyl)Ph ~ M+~
~C~ - S _7-(p-C~l)P} I 1~+) ~ ~.C
CO\ ~SC~2-(p-M~)P I (' vi+l~ 3, 3
~'C()l~ iBu)-hor~ene ~'~+1~ ) 5 2.0
~n C ~ ~.. O~-~p-~v ~ ~ h , ~/+ ~''Y.(l
ON~S~2-(P-Nve.)'h ~,v+ M ~ .
C~ l O~-(o-h~ J~/ + ~,.
~C~ S~ O-c~2~ 1 ( V + . ' ,.
')~f-~ 3. ~2-(~-~ V+~
(_QI' O~-(m-Cl)r~ v + '
", ~ iH o2CH2Ph (.v+ ' _.
T- -dansyl ( v+
~ CO1~1~07-8-quinoline (v+ )~
2 ~ 9 ~ 7 5 ~
WO 96/04905 : I _IlVv .l ,_ _ I
119-
The proton NMR data for several of the Examples is given
below:
EXAMPLE 47
N-(4-dimethylamillobenzenesulfonyl~-2-(4-carboxy-2-propylphenoxy)-
2-(3,4-methylenedioxyphenyl)acef:lmi~l~
H NMR(300 MHz,CD3OD,ppm):o0.89 (t. 3H),1.59(m,2H),2.62
(m,2H),3.03(s,6Hj,5.48(s,1H),5.96(s,2EI),6.43 (d, lH),6.64 (dd,
2H)~6.79 (d. IH),6.91 (m, 2H),7.55 (dd, lH),7.64 (dd. 2H),7.74 (d,
IH).
EXAMPLE 49
lS
N-~2-carboxybenzenesulfonyl)-2-(4-carboxy-2-propylphenoxy)-2-(3,4-
methylenedioxypheny l)acetamide
IH NMR(400 MHz,CD30D, ppm): o0.92 ~t, 3H),1.62(m,2EI),2.67
(m,2H),5.66(s,1H),5.g5(s,2H),6.74 (d, 111), 6.77 (d, 11-1), 6.93 (d,
IH),6.98 (dd. IH), 7.60(m,2H),7.69(m,1E~),7.75(m,2H),8.09(d,
IE-I).
EXAMPLE53
2s
N-(dansylsulfonyl)-2-(4-carboxy-2-propylphelloxy)-2-(3,4-
methylenedioxyphenyl)acetamide
IEINMR(400 MHz, CD~OD, ppm): ~ 0.83(t,3H),1.51(m,2H),2.57
(m,2H),2.88(s,6H),5.40(.s,1H),5.95 (dd, 2H),6.26 (d, IH),6.65 (d,
IH),6.79 (d, IH),6.85 (dd, lH),7.19 (dd, IH),7.25 (d, IH), 7.48(t,
lH),7.54(t,IH),7.66 (d, IH),8.13 (d, IH), 8.30 (dd, IH), 8.53 (d,
IH~.
q ~ 7
W0961U490~i 58 1~l" ~,
- 120-
EXAMPLE 54
N -(8-quinolinesulfonyl i-2-(4-carboxy-2-propylphelloxy)-2-(3,4-
methylenedic~xyphenyl)~ e.l~lmi~
IH NMR (300 M11z, CD~OD, ppm): o 0.S5 ~t, 31{), 1.54 (m, 2H), 2.59
Im~ 2H), 5.57 ~s. IE-1), 5.94 (s~ 21H), 6.47 (d, IE-I), 6.66 (d, Il{), 6.71 ~d~
IH), 6.87 (dd, lH), 7.34 (dd, IH), 7.58 (dd, ]H~, 7.6~ (m, 2H)1 ~.20
(dcl, IH), 8.39 ~dd~ ]H), 8.47 (dd, lH), 8.~3 ~dd~ IH).
EXAMPLE 55
N-(8-quinoline~uli'onyl)-2-(4-carbo~amido-2~ Jyl~ o~y)-2-(3,4-
methylenedioxyphenyl)~e~rni~f~
To N-~8-quinolinesulfonyl)-2-l4-carboxy-2-propyl-
pheno~y l-2-(3 ,4-1nethylenedioxyphenyl)~ ei:lm i~, Example 54,125.X
mg, 0.047 mmol~ dry DMF (0.~ mL) was added 1-~3-dimethyk~ o-
propyl)-3-ethylcarbodiimide hydrochloride, also referred to as EDC,
20 (18.4 mg, 0.0~ mmol), NH4CI ~6.7 m$, 0.125 mrnol), ;~d TEA (17.
mL, 0.125 mmol). Reaction was followed by thin layer
chromatography ~100:15:1.5/CH2Cl,:MeOH:HOAc). When iiIe l~actioll
was completed the DMF was remc ved in vacuo and the residue taken uy
in Et~O/EtOAc. The solution was washed with 109~, citric acid, water
2s and brine then dried over MgSO4, filtered and the solvent removed.
The prodllct was purihed by chromatograplly eluting with
200:5:1.~1CH2Cl2:MeOH:HOAc. rf = 0.35
(100:5:1.5/CI12CI2:MeOr-l:HOAc) FAB mass spectrum. m/e :548.01~1+1
calculate(i fc~r C2~H25N3SO7 ~48).
~ WO 96104905 ~ 2 ~ 9 5 7 5 ~ p~,l/LI~_lb, j
- 121 -
EXAMPI F 56
- a-(4-carbomethoxy-2-n-propylphenoxy)-3.4-methylenedioxy- phenyl)acetic acid
Step A: Preparation of ethyl (x-(4-carbomethoxy-2-n-propyl-
phenoxv)-3.4-methvlenedioxyphenylacetate
To a 2 L three necked 24/40 round bottom flask equipped
with a mechanical stirrer, a nitrogen inlet and a dropping funnel wa.s
first added a solulion of 36.0 g (0.1 ~5 ItlOI) of methyl 4-hydroxy-3-n-
propylben~oate dissolved in 700 mL of anhydr()us DMF followed by
66.4 g (0.204 mol) of cesium carbonate. The flask was purged wittl
nitrogen and the reaction mixture was stirred at room temperature tor 2
hours. A solution of 58.5 g (0.204 mol) of ethyl o~-bromo-3,4-
5 methylenedioxyphenylacetate dissohr7ed in 100 mL of DMF was theadded via ;m addition funnel over a 15 minute period. The reaction
mixture was stirred an additional I hour at room temperature then
quenched by addition to 5 I of a 5'Yc, aqueous citric acid .solution. The
organic product was extracted into diethylether (2 x 4 L), the organic
20 layers were separated, wa.shed with saturated aqueous NaCI, dried
(1\1gS04), filtered, and evaporated. The residue was applied to a .silica
gel (2 kg; 70-230 mesh) column equillibrated in 1()~! C112Cl2-hexane.
The column was then eluted succes.sively with 12 L of 10~ Cl-12CI2-
hexane, 12 L of 5'~c, EtOAc-hexane, 4 L of 7.5C~c ~tOAc-hexane, 12 L
25 of 10'~c EtOAc-hexane, and finally 8 L of 2()~o F,tOAc-hexane.
Combination of the purified fractions and evaporation in vacuo afforded
76.3 g (74.2 theoretical) of the title compound as ~l p~lle yellow oil
which was used without further purification in the nexl step.
30 Step B: Preparation of a-~4-carbomethoxy-2-n-propylphenoxy)-
3,4-methylenedioxyphenvlacetic acid
A I L 3 necked 24/40 round bottom flask equippe.d with a
mechanical stirrer, a dropping funnel, and a nitrogen inlet was charged
with a solution of 76.3 g 0. l 85 mol) of the semi-purified product of
~V0 96/0491}5 2 1 9 5 7 5 8 ~ L . ~
- 122-
Step A dissolved in 500 mL of methanol. The flask was purged witl
nitrogen, the stirrer was started, and 37 mL of a 5.0 N aqueou~ .~iolutio:n
of sodium hydroxide was added over a 30 minute period via an addition
~fu:lmel. The reaction mixture was stirred at room temperature for an
add:itional 30 mimltes at which point TLC analysis (CH2C12-l\leOH-
:NH4OH 90:10: 1) iodicated that the starting material had been con.sumed.
The reaction mixture was adjusted to pH=4 with 6 N HCI. and the bulk
of the organic solvent was removed in vacuo. The precipitated or,gallic
product and t:he ~queous layer were next partitioned between Cl-1~2CI1 ( I
L) and water (I L) which produced a copious emulsion. '1'he, react:ion
mixture was then aged ovemight in ~I refridgerator which resulted ir
crystallization of the organic product. The crystalline solid was
se~larated from the two phase mixture by filtration and washed with
CH2C12. The solid was slurried again in diethylether7 filtered, wa.shed
with hexane, and then dried in a ~acuum to at'ford 65 g (94~o) of the
title compound as a white crystalline .solid.
I E-l-NMR (400 MHz, CD30D, ppm): ~ ().93 (t, J=7.2() H~., 3H), 1.62-
1.75 ~m, 21-1), 2.63-2.70 (m. lH), 2.77-2.81 (m, lH), 3.~4 (s~ 3H)~ 5.54
(s~ 1), 5.94 (~, 21-1), 6.~1 (d, J=7.60 Hz, IH)~ 6.89 (d, J=g.20 Hz, I ~iJ,
7 0~ ~d, J=1.60 1-1z, Il{), 7.11 ~br s, lH), 7.78-7.RI (m, 2H).
E~X~4MPLE 57
N-(4-iso-propylbenzenesulfonyl)-a-~4-carbomethoxy-2-n-propyl-
phelloxy)-3~4-methylenedioxyphenyl~ret:~m~
Step A: Preparation of IV-(4-iso-propylben~enes~ onyl)-o~-(4-
carbomelhoxy-2-rl-propylphenoxy i-3,4-methylenedioxy-
phenylacetamide
An oven dried three-necked 24/40 1 L round-bottom flask
V~,'.lS eqllipped ~ith a mechanica:l stirrer, a nitrogen inlet, and a septum.
The flask was flushed with nit:rogen, then char~ed witll 20.06 g (53.9
mmol) of the product of Example ~6, 400 mL of anhydrous THF. and
9.76 mL ~70.0 nurlol) of triethylamine. The flask and its contents were
~ , 2~9~7~8
W0 96/04905 ~ - r~
- 123 -
stirred and cooled to -78~C with an extemal dry ice-acetone bath and
then 7.30 mL (59.3 mmol) c~f trimethylacetyl chloride was added slowly
via a syrin~,e. After the addition was complete, the dry ice-acetone bath
was replaced with an ice-water bath and the reaction was stirred at 0'C
for 1 hour. A separate oven dried 3 necked 24/40 2 L round-bottom
ilask was equipped with a mechanical stirrer, a septum and a nitrogen
inlet. The flask was flushed with nitrogen then charged with 16.102 g
(80.8 mmol) of 4-iso-propylbenzellesulfonamide and 300 mL of
anhydrous methyl sulfoxide. The stirrer WLIS started and a 162 mL of a
1Q 1.0 M solution of lithium bis(trimethylsilylamide) in TIIF was .slowly
~mildly exothermic) added via a syringe through the .septum. After the
addition was complete, the reaction nli~ture was stirred at room
temperature for ;m additional 30 minutes. The contents of the first
reaction mixture including a fine white precipitate that was suspended in
the reaction mixture were then slowly transfered to the stirred solution
of the deprotonated sulfonamide in the second flask via a wide diameter
cannula. The combined reaction mixture was then stirred for an
additional l4 hours under a nitrogen atmosphere. The reaction wlls the
quenched ~,ith 1.0 N HCI llnd the majority of the volatile solvents \i,~ere
20 removed in vacuo. The residue wa.s partitioned bet~A~een E~tOAc and 1.0
N HCI. then organic layer wa.s separated. washed ~vith saturated a~lueous
NaCI, dried (MgSO4), filtered and evaporated in vacuo. The residue
was purified on a silica gel (3 kg; 70-230 mesh) chromatography
column (15 cm ~ 150 cm) eluted with (90:10:1 CH2C12-MeOII-
25 NH40H). Combhlation of the purified fractions and evaporation hvacuo afforded 18.367 g (629'o) of the title cumpound.
IH-NMR (400 Mll~, CD30D, ppm): ~ 0.8X (t, J=7.60 Hz,3H),1.24 (d,
J=7.00 Hz,3H)~ 1.25 (t. J=7.00 Hz,3H),1.55-1.60 (m,2H),2.59-2.66
- (m,2H),2.97 (.sept, J=7.00 Hz,111),3.83 (s,311),5.52 (s, IH),5.97 (s~
3~ 211),6.50 (d, .1=8.80 Hz~ I 1-1), 6.80 (d, J=8.00 Hz, IH),6.8~ (d, J=1.60
Hz, IH),6.~4 (dd, J=2.00, 8.00 Hz7 IH), 7.14 ~d, J=8.80 Hz,2H),7.59
(dd, J=2.20, 8.80 Hz, I }-1), 7.75 (d, J=2.20.1 H),7.79 (d, J=8.80 Hz,
2H).
W0 961~4905 ~C ~ / r~ u~
- 124-
EXA~IPLE :58
N-~4-iso-propylh~n7.f n~.slllfonyl)-a-(4-carboxy-2-n-prc pyll)helloxy')-
3,4-methylenedioxyphenylacetamide dipotassium salt
~; F~reparation of N-(4-iso-prc pylbenz,enesull'onyl)-a-(4-
carboxy-2-/1-propylphenoxy)-3,4-methylenedioxyphenyl-
~,t.~mi~lP dipota.ssium salt
To a solution of 18.367 g (33.2 mmol') of the product of
o Example 57 dissolved in 100 mL of methanol was added a solution of
6.56 g (116.9 mmol~ of potassium hydroxide in ?5 mL of water alld the
reaction mixture was stirred at 60~C under a nitrogen atmosphere.
After 6 hours TLC analysis (80:15:1 CHC13-MeOH-NH4011) indicated
that ester hydrolysis was complete. The reaction mi~ture ~vas cooled to
15 room temperatllre. diluted with 100 mL water7 filtered tllrou~h a 0.45
micron filter and then divided into two equal volume pc~rtions. '1'he
fractions were indi~r~idually desalted and purified on a Waters Millipore
Delta Prep 30QO liquid chromatograph equipped with an MIOQO Prep-
Pak module containing a 47 x 300 mm Delta-Pa}c C18 1511m IOOA
20 column cartridge. Two solvent resevoirs were employed: solvent
system A ~95-5 water-acetonitrile), and solvent .sy.stem B ~.'i-95 water-
acetollitrile), and the column effluent was monitored sirnultaneously at
210 and 280 nm Wit'tl a Waters model 490 UV-visible detector. Each
fraction was pump-injected onto the colurnn and desalted by elutioll 150
2s mL/min) with ~several column ~olumes of solvent system A. A gradient
elution was then begun which had as initial cr~n~itir)n~ lOO~o solvent
system A-O~o solvent system B and reached after 30 minutes 50~o
solvent system A-50~~, solvent system B, and the ~fractions were
collected u!ith an ISC~O Foxy 200 fractioll collector. The purified
30 fraction.s were combined in round bottom flasks, frozen in a -78~C dry
ice-acetone bath. and Iyophilized. Combination of the purified product
afforded 18.719 g (92%) of the title compound a.s a white Iyophili~ed
powder.
2 7 ~5~5~
-- W096N~491}5 r~ J,,.;h,, 7
- i25-
IH-NMR (400 MHz, ~D30D, ppm): o 0.88 (t, J=7.20 Hz, 3H), 1.21 (d,
J=7.00 Hz, 3H), 1.22 (d, J=7.00 Hz, 3H~, 1.56-1.63 (m, 2H), 2.52-259
~ (m, lHj, 2.67-2.74 (m, lH), 2.91 (sept, J=7.00 Hz, lH), 5.33 (s, IH),
5.92 (d, J=1.20 Hz, I H), 5.93 (d, J=1.20 Hz, IH), 6.72 (d7 J=8.50 Hz,
IH), 6.76 (d, J=8.50 llz, IH~, 7.04 (d, .1=7.50 I-lz, IH), 7.05 (s, IH),
7.21 (d, J=8.50 Hz, 2}{), 7.64 (dd, J=2.00, 8.50 Hz, 111), 7.67 (d, J=~.50
Elz, 2H), 7.73 (d, J=2.00 Hz, IH).
Microanalysis for C28H27Nso8K2-H2o~
0 Calc'd: C = 53.06; H = 4.61; N = 2.21; K = 12.34.
Found: C = 52.81; H = 4.56; N = 2.17; K = 12.02.
EXAMPLE 59
a-(2-iso-butyl-4-carbomethoxyphenoxy)-3.4-1nethylelledioxy-
phenylacetic acid
Step A: Yreparation of ethyl a-(2-i~v-butyl-4-carbometlloxy-
phenoxv)-3.4-methYlenedio.~ypheny1acetate
To a solution of 1.008 g (4.84 mmol) c~f methyl 3-isz~-
butyl-4-hydloxyberlzoate and 1.737 g (6.05 mmol) of ethyl oc-bromc~-
3,4-methylenedioxyphenylacetate in 10 mI of acetone W.ls added 1.33
g (10 mmol) of finely powdered potassium carbonate. The reaction
mixture was m~gn~lically stirred and refluxed fclr 4 hours, then cooled
2 to room temperature, filtered and evaporated. The residue was purified
on a silica gel fla.sh chromatography column eluted with 10% EtOAc-
hexane; combination of the purified fractions and drying in vacuo
afforded 1.518 g (76~/o) of the title cormpound as an amorphous powder.
1 H-NMR (400 l~lE-lz, CDC13, ppm): o 0.90 (d. J=6.60 Hz, 3H), 0.94 (d,
- J=6.60 Hz, 3H), 1.17 (t, .J=7.20 llz. 3H), 2.02-2.08 (m, IH), 2.55 (dd,
J=7.20, 13.20 Elz, IH)~ 2.64 (dd, J=7.20, 13.20 llz., 11-~), 3.~ (s, 3H),
4.11-4.19 (m, 2H), 5.56 ~s. 111), 5.96 (s, 2H), 6.70 (d,J=9.20 Hz, lH),
6.6~ (d, J=7.60 Hz, lE~), 7.02 (dd, J=1.60, 8.00 Hz, lH), 7.05 (d, .1=2.00
Hz, IH). 7.78-7.81 (m, 2H~.
2 ' q 5 7 ~ ~
WO ~C/~4905 r~
- 126-
Step B: Preparation of u-(2-iso-butyl-4-carborlletl1oxyphenoxy)
3~4-methylenedioxyphenylacetic acid
To a solution of 1.518 g ( 3.66 mmol) of the product vf
Step A dissolved in 8.0 mL of methanol was added 1.0 mL of a 5.0 M
5 solution of aqueous sodium hydroxide. The reaction was stirred at
room temperature and monitored by TLC (80:15:1 C~HC13-MeOll-
NH4OH). After 1.5 hours the reaction was ;judged to be complete and
the reaction mixture was adjusted to pH=5 with 1.0 N HCI. 'l~he
reaction mixture ~h~as then partitioned between EtOAc and water,
separated~ dried (MgSO4)~ filtered, and evaporated. The residue wa.s
purified on a silica gel flash chromatography cc~lumn eluted with
CHC13-MeOEI-NH4OH (80:15:1); evaporation of the purified f}actions
and drying in vacuo afforded the title compound as an amorphous foam.
I H-NMR (400 MH~.. CD30D, ppm): o 0.86 (d, J=6.R0 H~, 3EI), 0.89
15 (d,./=6.80 llz, 3E-1), 1.96-2.04 (m, IH). 2.4'~ (dd,J=7.20, 12.~0 llz, 11-1
2.69 (dd,.l=7.20, 12.80 E-lz, 1~1), 3.84 (s, 3H), 5.49 (s, lH), 5.92 (d,
.l=1.2() H~, lH), S.g3 (d,.l=1.20 ~Iz, IH~, 6.79 (d,J=~.OO l-lz, IH'~, 6.89
id, J=8.~0 Elz, 11-1), 7.08 (dd, J=1.60, 8.00 Hz, IHj, 7.11 (d, .l=1.60 Hz.
I El j, 7.74 (d, .r=2.40 Hz, IH). 7.7X (dd, J=2.40, X.80 Hz. 11-1).
20 Cl-MS mle = 386.2 (M+)-
EX~MP!.~. 60
N-(4-iso-propylhPn7,P,nPsulfonyl)-a-(2-iso-butyl-4-ca}bomethoxy-
25 plletlox,r~)-3,4-rlleth!,~lenedioxyp~lenylacetaLlIide
Step A: Preparation of 1~,'-(4-iso-propylbeiIzenesulfonyl)-cx-(2-iso-
butyl-4-carhomethoxyphenoxy)-3,4-mettlylenedio,Yypllellyl-
a~etaltlide,
To a solution of U.727 g ~ 8 mmol) of the product of
Step B in Example 58 dissol~ed in 4 mL of ~mbyrdmus THF was added
0.458 g (2.82 mmol) of 1,1 ~ -carbonyldiimidazole and the mixtun~ \~'ilS
ma~netically s~irred and refluxed for 2 hours. Tlle reaction mixture
was then cooled to room temperature, and 0.56~ g (2.82 mmol) of 4
.
~ wo 96/W905 ~ ; 2 1 9 ~ 7 5 8 ~",, .~ ,
- 127-
iso-propylbenzenesulfonamide and 0.42 mL (2.~2 mmol) of l,S-
diazabicyclo[5.4.0]undec-7-ene were added. The reaction mixture was
stirred an additional 3 hours at room temperdture, then was evaporated
in vacuo. The residue was partitioned betweell EtC)Ac and 1.0 N HCI
and extracted. The organic layer was separated, dried (MgSO4),
filtered, and evaporated and the residue was purified on a silica gel flash
chromatography column eluted with CHC13-MeOH-NH4011 (S0:15:1).
Evaporation of the purified fractions and drying in vacuo afforded
0.666 g (63~o) of the title compound.
1 I-NMR (400 MHz, CD30D, ppm): ~ 0.R I (d, J=6.80 HZ! 3H), 0.S4
(d, J=6.S0 Hz, 3H). 1.23 (d, J=6.80 llz, 311), 1.24 (d, J=6.S0 HZ! 3H),
I.S8-1.94 (m, 111~, 2.45 (dd, J=7.00, 13.00 Hz, 111), 2.~S (dd, ~=7.00,
13.00 H~, lH), 2.95 (sept, J=6.80 Hz, IH), 3.S4 (s, 31-1), 5.46 (s, IH),
5.95 (d, J=l .20 H~, I H). 5.96 (d, J= 1.20 II~, IH), 6.59 (d, J=8.60 Hz,
IH), 6.79 (d, .1=S.00 1 Iz, 11-1), 6.98 (br s, I H), 6.99 (dd, J=I .60, S.00
Hz, 1 H), 7.30 (d, J=8.40 Hz, 211), 7.60 (dd, J=2.00, S.60 I Lz, 11-1)~ 7.70
(d, .1=2.00 Hz, IH), 7.72 (d, J=8.40 H~., 2H).
EXAMPLE 61
N-(4-i.s(l-propylbenzenesulfollyl)-a-(2-iso-butyl-4-carboxyphenoxy)-
3 ,4-methylenedioxypllenylacetdmide
Step A: Preparatioll of N-(4-iso-prc pylbell~enesulfonyl)-a-(2-iso-
butyl-4-carboxyphenoxy)-3,4-rmethylenedioxyphenyl-
acetamide
To a solutiolI of 0.294 g (0.52 mmol) of the product of
Example 60 dissolved in 3.0 mL of methanol was added 1.0 mL of a 5.0
- N a~lueous .solution of .sodium hydroxide. The reaction mixture was
maglletically stirred at 60~C. After 3 hours TI.C analysis (CHC13-
MeOH-NH40H S0:15:1) indicated complete hydrolysis of the ester. The
reaction was cooled to room t~ peld1ul~, adjusted to pl 1=5 with
dropwise addition of 1.0 N HCI, then partitioned behveen EtOAc and
water. The organic layer was separated, washed witb .saturated a~ueous
2 1 Y ~ ~5 ,~ --
W0 ~6/04905 ~ r~
- 128-
NaCI, dried ~MgSO4~ filltered and evapor.lted. The residue was driecl
in vacucl to afford 0.238 g (X3~70) Of the title compound a.s an
amorphous po~der.
I H-NMR (400 MHz. CD30D, ppm): o 0.82 ~d, J=6.80 l lz, 3H), 0.85
(d, J=6.80 Hz, 31 i), 1.24 (d, J=7.20 Hz, 3H), i.75 (d, ~=7.~0 Hz., 3H),
1.91 (.sept, J=~ 0 Hz, I 1-1), 2.48 ~dd, J=7.20, 13.2û Hz~ IH), 2.~6 ~dd,
J=7.20~ 13.20 Hz, IHj, 2.97 (sept,J=7.20 Hz, IH), ~.51 (s, IH~, 5.97 (s,
111),6.50(d,J=8.4011z, IH),6.81 (d,J=8.00Hz, IH),6.91 (d,J=1.6(1
Hz, IH), 6.9:5 ~dd, J=1.60, 8.00 Hz, IH~, 7.36 (d, J=S.40 TIz, 2H~, 7.62
(dd. J~2.20, 8.40 Hz, I H), 7.72 (d, J=2.20 l lz, IH), 7.79 (d, J=8.40 Itlz~
2E-1).
FAB-MS nlle = 554 (M + 1).
EXAMPLE 62
/~-(4~ o-propy3henzenesulfonyl)-o~-(2-n-propyl-4-methoxycarbonyl-
~henoxy~-o~-methyl-3 ,4-met~lylenedioxyphenyl~et,lmi-l~
Step A: Preparation of N-~4-iso-propylbell~enesulfonvl~l-(x ~7-n-
propyl-~-methoxycarbonylphenoxy~ methyl-3,4
m~thylenedioxvphenvlacetamide
To a sohltioll of 0.516 ~ ( 0.93 rnrnol) of the producl of
Example 57 dissolved in 1.0 mL of anhydrous THF was added ~ 80 mL
(2.80 mmol) ol a 1.0 M solution of lithium bis~trimeth~isilylami(ie) in
25 THF at -78~C under ~a nitrogen atmosphere. The reaction mixture was
magnetically stirred at -78~C for I hour, then 174 !lL (2.80 mmol~ of
ioclometllane was added ~/ia syringe. The reaction was allowed to wa
to room temperah~re and was stirred an additional 14 hours. The
reaction was next quenched with excess 10~c aqueous NaHSO4 and
30 partitiolled between EtOAc and water. The organic Ltyer was washed
with saturated aqueou.s NaCI, dried (MgSO4), filtered ~md evapotaled.
The residue was purified on a silica gel flasiI chlomatography column
eluted with Cl-lC13-MeOH-NH40H (90: 10~ . Evaporation of the
95758
W096~04905 ~ r~ . .. 7
- 129 -
purified fractions and drying in vacuo afforded 0.293 g (55~o) of the
title compound as an amorphous solid.
I I l-NMR (400 MH~, CD30I:), ppm): o 0.g9 (t, J=7.20 Hz,3H~, 1.31 (d,
J=6.80 Hz, 6H), 1.64-1.72 (m, 2H), 1.66 (s, 3H), 2.64-2.73 (m, IH),
2.81-2.88 (m, lH), 3.02 ~sept, J=6.~0 llz, ll-l), 3.85 (s, 3H), 5.96 (s,
2H)~ 6.36 (d, J=8.40. 11-1), 6.77 (d, J=8.40 Hz, 2H), 6.99 (m, I H), 7.05
(br s, IH), 7.37 (d, J=7.60 Hz, IH), 7.43 (dd, J=2.40, 8.60 Hz, IH),
7.76 (d, J=8.40 Hz, 2H), 7.81 (br s, I H).
FAB-MS mlc = 568 (M + 1).
EXAMPLE 63
N-(4-i.s()-propylbenzetlesulfonyl)-c~-(2-n-propyl-4-carboxyptlenoxy)-oc-
methyl-3,4-methylenedioxyphenyl~cets mi~ dipotassium salt
Step A: Preparation of N-(4-iso-propylbenzenesulfollyl)-oc-(2-n-
propyl-4-clrboxyphenoxy)-(x-methyl-3,4-methy lenedioxy-
phenylacetamide dipotassium salt
To a solution of 0.293 g (0.52 mmol) of the product of
20 F.xample 62 dissolved in 2.0 mL of methallol was added a solution of
0.143 g (2.54 mmol) of potassiurll hydroxide dissolved in 1.0 mL of
water. The reaction mixture was ms~n~tic~lly stirred at 60~C for 4
hours until TLC anlysi~ (CHC13-MeOH-NH4OH 80:15:1) indicated
complete hydrolysili of the .starting material. The reaction mixture was
2s then cooled to room temperature, dilwted with 5.0 mL of water and
filtered through a 0.45 micron filter. The filtrate was then purified
a Waters l~lillipore Delta Prep 3000 liquid chromatograph equipped
with two DuPont Zorbax(~ 21.2 mm x 25 cm ODS reversed phase
HPLC columns connected in series. Tivo solvent resevoirs were
3c employed: solvellt system A (95-5 wclter-acetonitrile), and solvent
systetm B (5-95 water-acetollitrilej, and the column effluent was
mc~llitored sirnlllt:~neously at 210 and 2~0 nm with a Waters model 490
UV-visible detector. The reaction mixture was injected onto the colurnn
and desalted by elution (50 ml /min) with approximately IL of .solvent
W0 96/049U5 2 1 5~ 5 ~ ~7 8 T ~
- 130-
system A. A gradient elution was tl~e~n begun whiuh had as initial
conditions 100~ solYent system A-O~ro solvent sy.stem B and reached
after 30 Inillutes 50~Y~7 solvent system A-50% s01veIlt system B, and the
fractions were collected with an ISCO l:~oxy 200 fractioll collector. The
purified fractions were combined in round bottom flaslis, fro7en in a
-7~'C dry ice ac~tone bath, ~md lyophilized. Combination of the
purified product afforded 0.273 g (~4~o) of the title compound as a
white lyophili_ed powder.
lH-NMR (400 Mllz, CD30D, ppm): ~ 0.96 (t, J=~.20 l lz, 3Hj, 1. - 5 (d.
J=7.20 }-Iz, 3H~ 6 (d, J=7.20 Hz, 3H), 1.64- 1.71 (m, 2H j, 1.67 (s,
3H~ 2.58-2.65 (m, 11-1~, ~.74-2.82 (m, lH), 2.96 (sept, J=7.~0 Hz, lHh
5.91 (d, .1=1.20 11_, 1II), 5.92 (d, J=1.20 Hz, I H), 6.52 (d, J=8.40 llz,
lH), 6.72 (d, .J=~.00 Hz, IH), 7.12 (dd, J=l .80, 8.00 Hz, 11~), 7.17 ~d,
J=2.00 H_. IH), 7.2~ (cl, J=~.80 Hz, 211), 7.50 (dd, J=2.2(), 8.40 H.z.,
L 5 l H), 7.72 (d, J=8.80 H~, 2H~, 7.74 (d, J=2.00 Hz, 111).
E~AB-MS m/f' = ~91.6 (M + K+~.
EXAl~PLE 64
N-(4-lso-prop~lbenzelleslllfonyl)-a-(2-n-propyl-4-carboxami(l{)-
phenoxy)-3 ,4-methylenedioxyphenylacetamide
Step A: Preparation of N-(4-iso-propylbenzenesulfonyl)-o-(2-rl-
pr~pyl-4-c.Lrboxamidophenoxy3-3,4-methylenedioxy-
s phenylacetamide
Tu a sOIUt:iOtl of 0.162 g (0.30 mmol) of N-(4-iscJ-
propylben7.en~sll1fc)llyl)-a-(4-carboxy-2-n-propylphenoxy~-3,4-
methylenedio~yphenylacetamide (free acidic form of the product Or
Exampie 5~) di.ssolved in 1.5 rnL of anhydruus THF was added 0.0~3 ~
(045 mmol~ of l,l'-citrbonyl~iimid~i7O1e and the resulting mixtule was
magnetically stirred and refluxed for 50 rm~r:iutes. The reaction mixture
was cooled to room lc,l,L,elalul~, and then addecl at O~C to e~cess TH}-~
that had been preYiously saturated with anhydrous ga.seous ~Ininloni~
The reaction mixture was se.aled and then stirred at room ~ -,alu
~ W096104905 /~ - ' '. 219575~ .~"~ cg~ ~
- 131 -
for 14 hours. The reaction mixture was then poured into water (70
mL) and extracted with EtOAc (150 mL). The organic layer was
separated, washed with saturated aqueous NaCI. dried (MgSO4),
filtered, and evaporated in vacuo to afford the title compound as an
5 amorphous solid.
I I-I-NMR (400 MHz, CD30D, ppm): o 0.X8 (t, J=7.60 lHz, 311), 1.21 (d,
J=6.80 Hz, 6H), 1.55-1.66 (m, 2H)~ 2.54-2.62 (m, IH), 2.70-2.77 (m,
lH), 2.89 (sept, J=6.80 l lz, III), 5.36 (s, 111), 5.93 (d, ~=1.20 Hz. IH),
5.94 (d, J=1.20 ~Iz, III), 6.75 (d, .1=~.40 ~-Iz, 11 ~), 6.78 (d, J=8.80 Hz,
IH), 7.02-7.04 (nl, 2H), 7.06 (br s, 2H), 7.20 (d, J=8.40 Hz, 2H), 7.55
(dd, J=2.20, 8.60 Hz, I H), 7.62-7.66 (m, 2H), 7.71 (s, I H).
FAB-MS mle = 539 (M + 1).
EXAMPLE 65
N-(4-iso-propylbenzenesulfonyl)-o!-(2-n-propyl-4-hydroxymethyl-
phenoxy~-3,4-metlIylenedioxyphenylacetamide
20 ~tep A: Preparation of methyl a-(4-hydroxymethyl-2-/t-propyl-
phenoxv)-3,4-methvlenedioxyphenylacetate
To a solution of 3.~4 g (23.13 mmol) of 4-hydroxy-3-t1-
propylbenzyl alcohol dissolved in 70 mL of anhydrou.s DMF was added
9.04 g (27.7 mmol) of cesium carbonate and the reaction mixture was
25 magnetically stirred at room temperature i'or 15 minutes. Methyl a-
bromo-3,4-methylenedioxyphenylacetate (7.58 g, 27.7 mmol) was added
and the reaction mixture wa.s then stirred for an additional 14 hours at
room temperature under a nitrogen atmosphere. The reaction W.IS t}lell
- partitioned between 5% a~lueolls citric acid (700 mL) and EtOAc (100
30 mL) and extracted. The organic Iayer was separated, washed with
saturated aquec us NaCI, dried (MgSO4), filtered and evaporated. The
residue WLIS PUr;f;ed On ;I silica gel ilash chrolIlcltography column eluted
with 40% EtOAc-hexane. The purified fractions were combined,
WO 96/04905 ~ 2 ? ~ 5 ~ 5 8 r ~"~
- 132-
evaporated~ and dried in vacuo to afford 6.74 g (81 ~G) of the title
compound as a yellow oil.
I H-NMR (200 I\IHz~ CDC13, ppm): o 0.97 (t7 J=7.60 1 L 3~). 2.55-
2.75 ~m, 2H), 2.71 ~t, J=7.20 Hz, 2H), 3.71 (s, 3H), 4.59 (s, 2H), 5.55
(s, 111). 5.97 (,s, '2ll), 6.69 (d, J=8.20 Hz. lH), 6.82 (d, l=7.80 Hz. IH),
7.02-7.28 ~m~ 4~1).
FAB-MS mle = 359 (M + 1).
Step ~: Prepaiation of methyl a-(4-tcrt-butyldimethylsilyloxy-
methyl-2-n-propylphenoxy)-3 ,4-methylenedioxyphenyl-
acetate
To a solution of 2.50 g (6.g8 mmol) of the product of Step
A dissolved in 20 mL of dichloromethane was added 1.95 mL (14.0
mmol) of triethylamine, 1.26 g (8.38 mmol) of ~ert-
l S butyldimeth~lchlorosilane, 85 mg (0.1 eq) of 4-dimethylaminopyridine
and the reaction mixture was stirred at room te.l.pelalu,e for 30
minute.s under a nitrogen abnosphere. The reactic~ll was thell dilute(l
with I00 nlI. E~tOAc, washed ~ith water~ 1.0 N HCI, saturated aqueous
NaHCO3, .satulated NaCI, dried (M~,SO4). filtered and evaporated in
vacuo to affi3rd 3.2() g (97~o) of the title compolmd.
El-MS m~e = 472 (M+).
Step (:~: Prep;3ration af o~-(4-tert-butyldimethylsilyloxymetl~yl-2
propylphenoxy)-3~4-methylerlediQxyphenylacetic acid
To a solution of 3.20 g (6.7~ n~nol) of the product of Step
B dissolved in IV mL of methanol and 3 mL of dichloromethane ~,vas
added 1.42 mL ~7.17 mmol) of a 5.0 N aquec~ll.s solution of sodium
hydroxide and the reaction mixture wa.s magnetically stirred at room
temperature. After 4 hours TLC analysis (CHC13-MeOH-NH4O~I
3~ 80:15:1 j indicated complete hydrolysis and the reaction mixttlre was
adjusted to pH=4 with 1.0 N ~Cl. The reaction mixture was then
compl(,tely evapDIated and dried in vacuo to afford the Grude product
which was used directly in the next step.
FAB-MS nllc = 48I (M + Na+).
~ WO96/04905 ~ . . 2 f q575~ L .,c7
- 133 -
Step D: Preparation of N-(4-iso-propylben_ene.sulfonyl)-c;~-(4-tert-
butyldimethylsilyloxymethyl-2-~i-propylphenoxy)-3 ,4-
methylenedioxyphenvlacetamide
To a solution of 3.30 g (7.21 mmol~ of the crude produc~
from Step C dissolved in 40 mL of anhydrous THF was added 1.75 g
(10.8 mmol) of l,1'-carbonyldiimidazole and the reaction mixture was
magnetically stirred and heated at reflux for 10 minutes. The reaction
was then cooled to room le~ ure, 2.15 g (10.R mmol) of 4-iso-
propylben_enesulfonamide and 1.61 mL (10.8 mrnol) of 1,~-
diazabicyclol5.4.01undec-7-ene were added and the reaction was stirred
for an additional 30 minutes at room temperature. The mixture was
then diluted with EtOAc (80 mL), washed with 10% aqueous citric acid,
saturated aqueous NaCI, dried (MgSO4), filtered and evaporated. The
residue was partially purified on a silica gel flash chromatography
column eluted with (:IHC13-MeOH-NH4OH (92:8:0.5). The semi-
purified material was combined and repurified on a second silica gel
flash chromatograplly collmm eluted initially with 35% EtOAc-hexane,
later witll 50% EtOAc-hex~me, and finally with 70~70 EtO,t~C-heXalle.
2Q Combination of the purified fractions and evaporatic)n afforded 3.20 g
(69C~o) of the title comE~ound as a yellou, oil.
FAB-MS mi~ = 67Rs (M + K+).
Step E: Preparation of N-(4-rso-propylllen7enf slllfollyl) o~ (4
2s hydroxymethyl-2-n-propylphenoxy)-3,4-methylenedioxy-
phenylacetamide
To a solution of 3.20 g (5.01 mmol) of the product of Step
D disscllved in 5.0 mL of anhydrous THF \~v~lS added 5 06 mL (5.06
- mmol) of a 1.0 ~1 solution of tetrabutylammonium fluclride in THF and
the reaction mixture was stirred at room ternperature under a nitroge
atmosphere. After 2.5 hours 1.0 mL additional tetrabutylammonium
fluoride in THF was added and the reaction mixture was stirred for a
additional 14 hours. The reaction mixture was then concentrated in
vacuo and applied to a silica gel flash chromatograplly column and
W096/WgO5 ~ 5 75~ r~ D;. /--
- 134-
eluted with 60~~to EtOAc-he~ane. Comhin~tinn of the purified fractions
and drying in vacuo afforded 0.691 g (26%) of the title compound dS an
amorphous powder.
I hl-Nl~R (400 MHz, CD30D. ppm): ~ 0.87 (t, J=7.60 Hz, 3H~, 1.26 (d.
J=6.80 Hz, 3H). 1.27 (d, J=6.80 Hz, 3H), 1.51-1 63 (m, 2H), 2.54-2.68
m, 2H~ 2.98 ~sept. J=6.~0 Hz, IH), 4.46 (s, 2H), 5.37 Q~ 1), 5.95 ~s.
1 H), 6.51 ~d, J=8.40 Hz, IH), 6.77 (d, J=8.00 Hz, lH), 6.8X-6.95 (m,
3H), 7.10 (d, J=2.00 Hz, l H), 7.36 (d, J=8.4() Hz. 2H), 7.77 (d, J=8.40
Hz, 2H).
FAB-MS ~??le = 548 (M + Na+).
F.XAMPLE 66
N-(4-iso-propylbenzenesulfonyl)-a-(4-formyl-2-n-propylphenoxy)-3,4-
methylenedioxyphenyl:~ret~mi~
Step A: Preparation of N-(4-i.so-propylbenzenesulfonyl)-a-(4-
formyl -2-n-propylphenoxy)-3,4-metllylenedioxyphenyl-
acetamide
2c To a solution of 0.573 g (].09 mmvl~ of tl~e product vf
Ex;{mple 65 dis~vlved in 5.0 mL of dichloromethane W~IS added 2.8(~ r
(32.9 mmol) of m~ng~n~.~e dioxide and 1.15 ~ of finely powdered 3A
mc)lecular sieves alld the reaction mixture was magnetically stirred at
room temperature for 14 hours. The reaction mixture wa.s thell filtered
25 through a bed of celite and MgSO4 and the filtratç was evaporated in
vacuo. The residue was dissolved in dichloromethane ~md applied to a
silica gel flash chromatography column and then eluted with 3~ MeC)H-
CH2C12. Ev.lpvrdtion of the purified fractions and drying in vacu
afforded 0.149 ~ (26~o) of the title compound.
3~ IH-NM:R ~400 ~Hz, CD30D, ppm): ~ 0.89 (t, ~=7.60 H7, 3H), 1.24 (d.
J=7.2U Hz, 3H). 1.25 (d,.J=7.20 Hz, 3H), 1.57-1.68 (nl, 2H), 2.63-2.74
(m, 2H~, 2.96 lsept, J=7.20 Hz, IH), 556 (s, 1~L), 5.97 (s, 2H)~ 6.70 (d,
J=8.40 H7, lH~, 6.80 (d, J=8.00 11~, IH), 6.91 (d,J=1.60 Hz, lH), 6.96
(dd, J=1.60. 8.Q() Hz, lH), 7.34 (d, .J=8.40 Hz, 2H~, 7.53 (dd, ~=2.()0,
, ~ ~ t ~, 2 ~ ~5753
~ WO 96/04905 : ~ ~ ' P~ '115
- 135-
8.40 Hz, lH), 7.66 (d, J=2.00 Elz, lH), 7.76 (d, .1=8.40 Hz, 2H), 9.77 (s,
111).
FAB-MS mle = 546 (M + Na+).
EXAMPLE 67
a-(4-acetyl-2-n-propylphenogy)-3,4-methyienedioxyphenylacetic acid
Step A: Preparation of 4-hydroxy-2-n-propylacetophellone
0 A Parr hydrogenation apparatus flask was charged with a
soluticln of 2.00 g (11.36 mmol) of 3-allyl-4-hydroxyacetophenone
dissolved in 10 mL of ethanol and 200 mg of a 10~~'c palladium on
carbon catalyst. The nask was mounted in the Parr apparatus and
shaken under a 46 psig hydrogen atmosphere for 15 minutes. At the
5 end of this period TLC analysis (15~o EtOAc-hexane) indicated that the
starting material had been completely consumed, and the reaction
mixture was filtered ~und evaporated. The residue was purified on a
silica gel flash chromatography column eluted with 25% EtOAc-llexane.
Evaporation of the purified fractions and drying in vacuo afforded 1.83
20 g (91%) of the title compound.
II-I-NMR (200 MHz. CDC13, ppm): oO.98 (t,J=7.40 Hz, 3H). 1.56-
1.78 (m. 2H), 2.57 (s, 3H), 2.63 (t, J=7.20 Hz, 2H), 6.08 (br s, IH),
6.84 (d, J=8.20 Hz, IH), 7.74 (dd, ./=2.20, X.20 Hz, IH), 7.79 (d, J=2.20
Hz, IH).
25 FAB-MS m~e = 178 (M+)-
Step B: Preparation of methyl o~-(4-acetyl-2-n-propylphenoxy)-3,4-
methylelledio~yphenylacetate
- To a solution of 0.250 g (1.40 mmol) of the product of
3~ Step A dissol~ed in 3.0 mL of DMF was added 0.504 g (1.54 mmol) of
cesium carbonate and the reaction mixture was magnetically stirred at
room temperature under a nitrogen atmosphere for 15 minutes. Methyl
o~-bromo-3,4-methylenedioxypherlylacetate (0.422 g, 1.54 mmol) was
then added and the resulting mixture was stirred at room temperature
wog6,049~ i ~ i 2 ~ q5 7~8 . ~ 7--
- 136-
for an additional 24 hours. The reaction mixture WdS then partilioned
between 105'o aqueous citric acid and EtOAG. The organic layer was
washed with saturated aqueous NaHCO3, saturated aqueous NaCI, dried
(MgSO4). filtered and evaporated in vacuo to afford the title compound.
IH-NMR (300 MHz~ DC13, ppm~: o 0.96 (t, .1=7.50 Hz. 3H), 1.ti2-
1.74 (m, 2H), 2.52 (s, 3H), 2~68-2.75 (m, 2H), 3.71 (s. 3H)~ 5.61 ~s,
I H), 5.98 (s. 2H~, 6.71 ~d, ./=8.60 Hz, lH'), 6.81 ~d, J= 8.20 Hz. lH~
7.02 (dd, J=l .80, 8.20 Hz, IH), 7.04 (d, J=l .80 Hz, ] H), 7.73 (dcl,
J=2.20, 8.60 Hz, lI-i), 7.79 (d, J=2.20 Hz7 lH).
FAB-MS mie = 371 (M + ]).
Step C: Preparatiotl of cx-(4-acet~ 2-n-propylpl1eno~y)-3,4-
me:thylenedio~yphenylacetic acid
To a solutioll of 0.556 g ~1.50 rnmol) of the product of
Step R dissol~ed in 4.0 mL of methanol WdS added 0.45 mL (2.15
mrllol) of a 5.0 N ayueous solution of sodium hydro~ide. The reactio
mi~ture was stirred at room lell-pe~ and monitored by TLC
(CHCl3-MeOH-NH4OH 80:15:l). After4 hour.s the reaetion was
judged to be complete and the reaction mixture was adjusted to pH=7
Wit}l 6.0 N I ICI. The mixture was then evapo~ated in vacuo and the
residue was purified on a silica gel flash chromatography eolulrll1 eluted
with ClIC13-MeOH-NH4OH (80:15:1). Evaporation of the puril~ied
fraction6 and drying in ~acuo afforded 0.416 g ~78~o'J of the title
compoulld.
I I I-NMR (400 ~ lz~ CD30D, ppm): o 0.94 ~t, J=7.60 Hz, 3H), 1.62-
1.70 (m, 2H), 2.53 (s, 3H), 2.61-2.69 (m, IH), 2.80-2.88 9m, IH), ~i.39
(s, 11~1), 5.93 (d, J=1.20 Hz, IH), 5.94 (d, J=1.20 Hz, lH), 6.79 ~d.
./=8.00 Hz, lH), 6.91 (d,.l=8.80 Hz, lH), 7.10 ~dd,J=1.60, 8.00 lIz.
111~, 7.15 (d~ J=l .60 Hz. I H~), 7.78 Id, J=2.40 Hz, 1 H), 7.81 ~dd, J=2.4(),
3~ 8~01-Iz IH)
~ WO 96104905 ~ 2 1 9 5 75 ~ I ~",J~
- ]37 -
- EXAMPLE 68
N-(4-iso-propylbenzenesulfonyl)-a-(4-acetyl-2-n-propylphenox~ )-3 ,4-
methylenedioxyphenylacet~mide
Step A: Preparation of N-(4-iso-propylbenzenesulfonyl)-a-(4-
acetyl -2-n-propylphenoxy)-3,4-metllylenedioxyphenyl-
acetamide
To a solution of 0.181 g (0.51 mmol) of the product of
0 Example 67 dissolved in 2.5 mL of anhydrous DMF was added 0.248 g
(1.53 mmol) of l,l'-c.lrbonyldiimidazole and the reaction mixture was
magnetically stirred and heated at X0'C under a nitrcgen atmosphere in
an oil bath. After 20 minutes the reaction mixture was cooled to room
temperature and 0.152 g (0.77 mmol) of 4-~.s(J-propylbenzene-
15 sulfonamide and 381 ,uL (2.55 mmol) was added. The reaction mixturewa.s heated at ~0~C for an additional 10 minutes then cooled again to
room temperature and partitioned between EtOAc and 10% aqueous
citric acid. The organic layer was separated, washed with saturated
aqueous NaHCO3, saturated aqueous NaCI, dried (MgSO4)~ filtered and
20 evaporated. The residue was purifïed on a silica gel flash
chronnatography column eluted with CHC13-MeOH-NE14OII (80:15:1);
evaporation of the purified fractions and drying in v~cuo afforded
0.128 g (47C~!) of the title compound as an amorphous solid.
I H-NMR (400 MHz, CD30D, ppm): o 0.88 (t, J=7.60 l lz, 3H), 1.21 (d,
25 .l=6.~0 Hz, 3H), 1.22 (d,./=6.$0 Hz, 3H), 1.55-1.65 (m, 2H), 2.51 (s,
3H), 2.54-2.64 (m, IH), 2.67-2.75 (m, IH), 2.92 (sept,J=6.80 Hz, lH),
5.43 (s, I H), 5.94 (s, 2H), 6.75 (d, J=8.80 Hz" I H), 6.77 (d, J=8.40 Hz,
IH), 7.01-7.03 (m, 2H), 7.23 (d, J=8.40 1 Iz, 2Hj, 7.66 (dd, J=2.40, 8.80
- Hz, I H), 7.67 (d, ~=8.40 Hz, 2H), 7.73 (d7 J=2.40 Hz, 1 H).
30 FAB-MS m/e = 538 ~M + 1).
EXAMPI~E 69
a-(2-n-propylpheno~y)-3,4-methylenedioxyphenylacetic acid
, " . : ! ' ~
W096tOJ905 ~ 2~9575~ r~
- 138 -
EAB-MS for C1~H1 8~5: mle = 337 (:M + Na+).
EXAMPLE 70
s
N-(4-i.s~o-propyl'bellzenesulfonyl)-o -~2-n-propylphelloxy)-3,4-
methylenedioxyphenylacetamide
FAB-MS for C27H29NS06: mle = 534 (M + K+~.
EXAMPLE 71
(x-(3-methoxyphenoxy)-3,4-methylenedioxyphenylacetic acid
El-MS for C16:H1406: n~le = 30~ (M+).
EXAMPLE 72
0!-(2-(2-h~ oxyr,lllylJphenoxy)-3,4-methylenedioxyphenyl~lcetic acid
FAB-MS for C17H1606: ~?tle = 317 (M + 1).
EXAMPLE 73
25 c~ 2-(2-carbomethoxyethyl)phenoxy)-3,4-methylenedioxyphenytacetic
acid
Cl-MS l'or C'19H18~7: lille = 3~9 (M + I).
EXAMPLE 74
~-(4-hydroxymethyl-2-/1-propylphenoxy)-3 ,4-methylenedioxyptlenyl-
acetic acid
Cl-MS for C19112006: mle = 326 (M+ - H20~.
~ W0 96/049~5 ~ .' 2 ! 9 5 7 5 8 r~l~u~. . . c~
- 139-
EXAMPLE 75
a-(4-(2-hydroxyethyl)-2-n-propylpllenoxy)-3,4-methylelledioxyphenyl-
acetic acid
Cl-MS for C20H22o6: mle = 359 (M + 1~.
EXAMPLE 76
N-(4-iso-propylhe,n7.t~nt~cl~1fonyl)-a-~2-(2-carbomethoxyetllyl)phenoxy)-
3 ,4-methylenedioxyphenylacetamide
ESI-MS for C2gH29NSOg: m/e = 540 (M + 1).
EXAMPLE 77
N-(4-i.so-propylbenzenesulfonyl)-ol-(2-(2-carboxyethyl)phenoxy)-3,4-
methylenedioxyphenylacetamide
Cl-MS for C27H27NSOX: mle = 526 (M + 1).
:EXAMPLE 78
~ 2-(2-carboxyethyl)pllerloxy)-3,4-1llethylenedioxyphenylacetic acid
Cl-MS for ClXH16O7: mle = 345 (M + 1).
EXAMPLE 79
N-(4-~so-propylbenzenesulfonyl)-2-(4-carbomethoxy-2-n-propyl-
phenoxy)-2-(5-methoxy-3 ,4-methylenedioxyphenyl)acetamide
W0961D4905 ~ ~ 2195758 ~ .7--
- 140-
Step A: Ethyl 2-(4-carbomethoxy-2-/1 -propylphenoxy)-2-( 5 -
methoxy-~ ~4-methylenedioxvphenyl)acetate
To a mixutre of methyl 4-hydroxy-3-n-propylbenzoate (3.0
g. 15.46 mmol3 and C.s2C03 (5.1 g, 16 mmol) in dry
dhnethylfcl~namide (50 mL) was added ethyl 2-bromo-2-(5-methoxy-
3,4-methylenedioxy)phenylacetate (4.3 g, 15.56 mrnol~, and the
resulting mixture was stirred at room temperature for 6 h. At the end
Or this period~ the reaction mixture was diluted with ice water (300 mL)
;md extracted with ethyl acetate (3 x 60 mL). The combined organic
phase was washed with water and brine, and then dried over anhydrous
MgS04, filtered and solvent removed to give the crude~ product.
Purification of the crude product by silica-gel flash column
chromatograplly using ethyl acetate-llexane (1:9) aiforded the titled
pr)duct as an oil (5.1 g).
lll NMR (200 MHz, CDC13. ppm) ~ 7.82 ~m, 2H). 6.75 (m, 3H); 6.61
(d, 111, J = 1.5 Hz); 5.93 (~s, 2Hl; S.~S4 ~s, IH); ~ (m, 21-1); 3.84 (s.
3H); 3.~3 ~.s, 3H); 2.68 (m, 2~1~; 1.69 (ln. 2H~ 0 ~t, 311, J = 7.4 llz?;
0.90 (t, 3H, J = 7.4 1 Iz).
Step B: 2-(4-carbomethoxy-~-n-propylphenoxy)-2-(5-methoxy-3.4-
m~tll~ylenediQxyphel1y])acetic acid
To a sclluti(ll1 of the product of Step A (4.3 g, 12 rmnol) in
methanol (25 mL) was added aqueous 2N NaOH (10 rmL) and the
reaction mixture was stirred at room ~ e~ u~. The rapid progress
2s of mono-deesterification was monitored by TL-C analy.sis Usillg CHC13-
MeOH-NH40H:(80~ 1). After 1~ min, the reaction nlixture was
cooled to 0~C and neutralized with aqueous 2N i ICI. l~,Iethanol was
rellloved in vacuo and the resulting mixture was acidified with aqueous
2N HCI. The oily product which precipitated was extracted hltc~ -
30 methylene chloride (l3 x 40 mL~ and the combined organic phase was
wa.shed with water, brine and then dried over MgS04. Removal of' the
solvent in vacuo afforded the crude product which was then purified by
fla.sh-chromato~raphy on silica gel using CHC13-~,leOH-
Nl 1401-1:(80:10:1 ) to give desired product as the arnmonium salt. The
2~9~7~8
~ W096/0490~ r~,l/Liv....................................... .,~7
- 141 -
- salt was treated with aqueous I N HCI (20 mL) to provide the titled
compound as a white solid (3.4 g).
I H NMR (200 MHz, CD30D, ppm) o 7.78 (m, 21-1~, 6.77 (m, 3H), 6.61
(d, lH, J = 1.5 Hz), 5.93 (s, 2H), 5.54 (s, IH), 3.84 (s, 3H), 3.83 (s,
3H), 2.68 (m, 2H), 1.69 (m, 2H)~ 0.90 (t, 3H, J = 7.4 Hz).
Step C: N-(4-isc/-Propylbenzenesulfonyl)-2-(4-carbomethoxy-2-n-
propylphenoxy)-2-(5-metho~y-3 ,4-1nethylelledioxyphenyl )-
acetamide
To the product of Step B (0.12 g, 0.30 mmol) in d~ THF
(1.5 mL) was added l,l'-carbonyl-liimid i7nle (0.1 g, 0.61 mmol) and
the reaction stirred at 50QC fc r 3 hr. To this solution was added a
solution of 4-is~o-plulJyl~r-lr7enesl~1fonamide (0.17 g, 0.9 mmol) and
DBU (0.14 mL, 0.94 mmol) in dry THF (1.5 mL), and the reaGtion
5 continued at 50~C for 4 hr. The reaction was diluted with ice water and
acidified with aqueous IN HCI. The precipitated material was taken up
in EtOAc and the organic phase was washed with water, brine, and then
dried over MgSO4, filtered and the sol~{ent removed. The product was
purified by flash-chromatography on silica-gel USillg
20 CHCI3:MeOH:NH40H (80:10:1) as the eluting solvent to ~/ielcl the titled
product as the ammonium salt. Acidificatio n of the anmlonium salt
afforded the titled product as a white solid (0.14 g).
11-1 NMR (400 Ml~z, CD30D, ppm): o 7.78 (d, 2H, J = 8.4 Hz),7.76 (d,
ll-I,J=2.3Hz),7.62(dd, IH,J=8.6,2.21-lz),7.37(d,2H,J=8.4Hz),
6.70 (d, lH, J = 1.4 llz), 6.61 (d, IH, J = 1.5 Hz), 5.97 (s, 211),5.49 (.s,
IH), 3.84 (s, 3H), 3.853 (s, 3H), 2.98 (sept. 111. J = 6.9 Hz), 2.65 (m,
2H), 1.59 (m, 2H), 1.25 (dd, 6H, J = 7.0, 2.5 Hz), 0.90 (t, 3H, J = 7.4
~)
C3"H3~NO~)N: Calc: C 59.50 ll 5.33 N 2.31. Found: C 59.60 H 5.34 N
30 2.59
;; 2t957
W0 96/0490S r~
- 142-
EXAMPLE ~0
N -(4-i~o-propylben7,enesll1fonyl)-2-(4-carboxy-2-propylphelloxy)-2-(5-
methoxy-3,4-methylenedioxyphenyl)~et~m i~L~
To tlle product of Example 79 (0.6 g, 1.02 mlmol~ in MeOH
~15 mL) was added aqueous 2N NaOH (5 mL) and the reaction was
stirred at 60~C ~or 3 h. When the reaction was complete the MeOH was
removed in vacuo and the aqueous phase was acidified with 2N HCI.
Ti1e product precipitated was e~;tracted il1tO methylene chloride (3 X 50
mL) ~md the comhined org~mic phase vvas wa.shed with brine tllen dried
o~er MgSO4, filtered and the solvent removed. The residue upo n
trituration with ether provided the titled product as a white solid (0.45
O)
IH NMR (400 MHz" DM~SO-d~, ppm): o 12.67 (br, IH), 12.63 (br~ IH~,
7.70 Id, 2H, J = 8.4 Hz), 7.66 (d, IH, J = 2.1 Hz)~ 7.5B (dd, IH, J = 8.5,
2.2 Hz), 7.40 (d, 2EI, J = ~s.4 Hz), 6.78 (d, IH, J = 1.2 Hz}, 6.66 (d, IH,
J = 1.2 Hz), 6.51 l~d, 1 EI, J = B.5 Hz), 6.02 (d, 2EI. J = ~.1 Hz), 5.69 (s,
111)~ 3.79 (s, 31-13, 2.93 (.sept, IH, J = 6.9 Hz)~ 2.56 Im. 2H1, 1.53 (m,
2'~ 21-1), 1.17(d,6H,J=6.9Hz),0.B3(t,3H,J=7.4Hz).
FAB mass .spectrurrl: m/e 570 (M+l).
C~qH3lNOgS: Calc: C 61.15 11 5.49 N 2.46 Found: C 60.X6 H 5.64 N
2.71.
2s
EXAMPLEI 81
N-~4-;so-propyrl~nzenesult'onyl)-2-(4-(N-(4-iso-propylbenzene-
sulfonyl)carboxalllido)-2-propylphenoxy)-2-(5-n1ethox~ -3 ~4-methylene-
dioxyphenyl~,~ref~ltlid~
The titled compound was prepared from N-(4-iso-
prc~pyll~en7.~nt-slllfonyl)-2-(4-carboxy-2-prop,vlpheno~y)-5~ ethoxy-
W0 96/04905 ,. .~ = 2 t 9 ~7 58 r~ .._7
- 143-
- 3,4-methylenedioxyphenyl)acetaniide (Example 80) using a procedure
similar to that de.scribed in Step C of Example 79.
FAB mass spectrum: m/e 751 (M+l).
C3gH42N2O~oS2 ~0.5 H2O: Calc.: C 60.06; 1-15.70, N 3.69.
Found. C 60.15; H 5.73; N 3.58.
EXAMPLE 82
N-(4-lso-propylbenzellesulfonyl)-2-(4-carboxamido-2-prc)pylphenoxy)-
2-(5 -methoxy-3.4-methylenedioxyphenyl)acetamide
To N-(4-i.so-propylbenzenesulfonyl)-2-(4-carboxy-2-
propylphenoxy~-2-(5-methoxy-3,4-methylenedioxyphen~Yl)~l~et~m i(l~
(Exaniple 80) (0.12 g, 0.21 mmol) in dry THF (1.5 rnl.~ was added
l,l'-carbonyldiimidazole (0.1 g, 0.61 mmol) and the mixture was
stirred at 50~C for 21h The reaction wa.s cooled to room le,~ e~,ltu~e
and was .saturated with dry NH3 The reaction mixture was stirred at
room temperature for Ih and then acidified. The crude product isolated
was purified by silica-gel flash column chromatography usin~ CHCI3-
MeOH-NH40H: (40:10:1 i to give the product as the ammonium salt.
Acidification provided tlle desired titled product as a white solid (0.06
g)-
IH NMR (300 MHz, [:~MSO-d6, ppm): o 7.76 (br, IH), 7.68 (d, 2H, J =
8 4 llz), 7.64 (d, lH, J = 2.1 Hz), 7.55 (dd, lH, J = 8.6, 2.3 Hz), 7.40
(d, 2H, J = 8.4 Hz), 7.16 (br, IH), 6.76 (s, IH), 6.65 (d, IH, J = 1.2
Hz), 6.53 (d, 111, J = 8.7 Hz), 6.01 (d, 2H, J = 2.9 Hz), 5.67 (s. 111),
3.78 (s, 3H), 2.93 (sept, I iH, J = 6.8 Hz~, 2.55 (m, 2H), 1.54 (m, 2H~,
1.17 (d, 6H, J = 6.9 Hz), 0.84 (t, 31-I, J = 7.3 llz).
FAB mas.s spectrum: m/e 569 (M+l).
W0 96t04905 ' ~ 2 ~ 9 5 7 5 8 . ~
- 144-
EXAMPLE 83
N-(4-is~J-propylhP,Il7,~nt~s-~lfollyl)-2-(4-~N-metllyl)carbQxamido-2-
propylphenoxy3-2-(5-met~hoxy-3,4-methylenedioxypherlyl~ 3~er ~m;~
s
The titled compound was prepared using procedures
similar to those described in Example 82.
IH NMR (3~0 hIHz, DMSO-d~, ppm): o 8.21 ~q. 111, J = 4.7 Hz)~ 7~68
(d,2H,J=8.4Hz),7.59(d, lH,J= 1.9Hz),7.49(dd, IH,J=8.6,2.1
o Hz), 7.40 (d, 2H. J = 8.4 Hz3, 6.77 (s, IH), 6.65 (s, lH), 6.53 (d. IE-I, :I =
8.7 Hz), 6.01 (d. 2H, J = 2.9 Hz), 5.67 ~s, 11-1~, 3.78 (s, 3H), 2.93 (sept.
lH, J = 6.8 Elz), 2.73 (d, 3H, J = 4.4 Hz3, 2.56 (m, 2EI), 1.54 (m, 21-I~,
1.16 (d, 6H, J = 6.9 Hz), 0.84 (t, 3H, 7.3 Hz).
C3(,H34N20~S: Calc: C 61.84; H 5.88; N 4.81.
Found: C 61.84; H 6.03; N 4.5g.
EXAMPLE 84
20 N-(4-i~ J-prOpylbenZeneSUIf(~nyl)-2-(4-(N-2-tl~drOXyet:h~lcarbo~ ido)
2-propylpherlo~y)-~-(5-methc xy-3,4-methylenedio~yphellyl)acetamide
The titled compound was prepared USillg procectures
25 .simiLIr to those described in Example 82.
I H NMR (4()0 MHz, CD30D, ppm): o 7.77 (d, 2H, J = 8.4 Hz), 7.64 (d,
IH, J = 2.3 E-lz), 7.51 (dcd, lH, J = 8.5, 2.4 Hz), 7.36 (d, 2H, J = X.5 Hz),
6.68 (d, IH, J = 1.4 Hz), 6.60 ~m, 2H), 5.96 ts, 211), 5.41~ (s, IH~, 3.82
(s, 3H), 3.68 (t, 2H, J = 5.9 Hz). 3.46 (t. 2H. J = 5.9 Hz), 2.97 Isept~ IE-I,
30 J = 6.9 Hz), 2.66 (m, 2H), 1.62 (m, 2H), 1.25 ~cld, 6H, J = 6.9, 1.2 1 Iz),
0.90 (t, 3H, J = 7.4 Hz).
C3~ 6N20~S: Calc: C 60.77; H 5.92; N 4.57.
Found: C 60.49; H 6.04; N 4.45
~ W0 96/04905 ' 2 1 9 ~ 7 5 ~
- 145 -
- EXAI~,IPLE 85
N-(4-i.sO-propylbenzenesulfonyl~-2-(4-(N-morpholinylcarboxamido)-2-
propylphenoxy)-2-(5-methoxy-3.4-methylenedioxyphenyl)acetamide
The titled compound was prepared using procedures
similar to those described in Example 82.
I H NMR (400 MHz, CD3OD. ppm): o 7.77 (d, 2H, J = 8.5 Hz), 7.37 (d,
2H, J = 8.4 Hz), 7.22 (d, lH. J = 2.1 ~Lzj, 7.09 (dd, IH, J = ~.4, 2.2 Hz),
o 6.66(d, IH,J= 1.5Hz),6.62(d, 1~-I,J=8.5Hz),6.57(d, lH,J= 1.5
Hz), 5.95 (s, 2H). 5.46 (s, IH), 3.81 (s~ 3H), 3.65 (m, 81~), 2.98 (m,
IH), 2.66 (m, 2H), 1.60 (m, 2H), 1.26 (d, 6H, J = 7.1 llz), 0.90 (t, 311, J
= 7.4 Hz).
C331-13~N20c~S: Calc: C 62.05; H 6.00; N 4.39.
Found: C 61.96; H 5.98; N 4.55.
EXAMPLF, 86
N-(4-i.~o-prclpylbenzeneslllf'onyl)-2-(4-(N-3-methylbutylcarboxamido)-
2-propylphenoxy)-2-(5-methoxy-3,4-methylelledioxyphenyl)acetarllide
The titled compound was prepared using procedures
similar to those described in Example 82.
I H NMR (400 MHz, CD30D7 ppm): ~ 7.77 (d, 2H, J = 8.5 Hz,), 7.60 (d,
IH, J = 2.3 Hz), 7.47 (dd, 111, .1 = 2.3, X.S llz), 7.36 (d, 2H, J = 8.5 Hz),
6.68 (d, IH, J = 1.5 Hz), 6.59 (d, IH, J = 1.4 Hz), 6.58 (d, IH, J = 8.6
~ Hz), 5.96 (s. 2H), 5.48 (s, IH), 3.82 (s, 3H), 3.36 (t, 2H, J = 7.5 Hz)~
2.97 (m, IH), 2.66 (m, 2H), 1.62 (m, 3H), 1.49 (q, 2H, J = 7.2 Hz),
1.25(cld,2H.J= 1.2,6.9Hz),0.95(d,6H,J=6.6Hz),0.9(J(t,311,J=
7.4 Hz).
C3~H~2N2O~S: Calc: C 63.93; H 6.63; N 4.39.
Found: C 63.81; H 6.73; N 4.44.
2~ 95~5~
W096104905 ~ 3 ~ 5~3~7 ~
- 146 -
LXAMPLE 87
5 N-(4-i.~ u,uyl~-r~ culfonyl)-2-(4-(N-carboxymethylcarbo,~ i
2-propylplle,rloxy)-2-(5-metlloxy-3,4-mettlylenedioxvphellyl)~ mi
~tep A: N-[4-is~i-propylbel1zenesulfonyl)-2-~4-(N-t-butoxy-
carbonylmethylcarboxarllido)-2-propylphenoxy)-2-(5-
m~thoxy-3.4-methylelledioxyphen~l lacetalnide
The titled compound wa.s prepared u.sing procedare.s
.similar to those described in Example 827 where ~Iycine-t-butyl ester
was the amine .starting material.
111 NMR (300 M13z, CDC13 ppm): o 7.70 (d~ 2E-I, J = X.2 llz), 7.66 (d,
Il~,J= 1.3H~,7.56(m, IH),7.41 (d,2H,J=~.2Hz3,6.79(s. IH),
6.67 (.s, IH), 6.59 (d~ IH, J = 8.5 Hz), 6.03 (s, 2H), 5.71 (s, lH). 3.X8
(d, 21-1, J = 5.5 Hz), 3.R0 (s. 3H), 2.95 (sept, IH, J = 6.9 Hz), 2.78 (m~
2H), 1.56 (m, 2H), 1.32 (s, 91-1'~, 1.17 (d, 6H, J = 6.~ Hz'), 0.86 (t, 3}-1, J
=7.3Hz).
Step B: N-(4-iso-propylbellzenesulfonyl)-2-(4-~N-c~arboxymethyl-
carboxamido)-2-propylphenoxy)-2-(5-methoxy-3,4-
m~lh~ylerledioxyphenvl)acetamide
2s A solution of the product of Step A (0.069 g, 0.1 mmol~ in
anhydrou.s trifluoroacetic acid (1.5 mL) was stirred at room
temperature for 4h. 'I'he excess reagent was evaporated in vacuo ialld
the resulting residue was triturated with dry ether to give the titled
product as white solid (0.6 g~.
IH NMR (300 MHz, DMSO-d~, ppm): o 7.70 (d, 2H~ J = 8.2 Hz~, 7.66
(d, 11-1, J = 1.3 llz), 7.56 (m, IH), 7.41 (d, 2H, J = 8.2 Hz), 6.79 (s, 11-1~,
6.67 (5, lH), 6.59 (d, lH, J = 8.5 Hz), 6.03 (~, 2H), 5.71 (~t IH), 3.88
(d, 2H, J = 5.5 Hz), 3.80 (~, 311), 2.95 (sept, IH, J = 6.9 Hz), 58 (m,
2H), 1.56 (m, 21-1), 1.17 (d, 6H, J = 6.8 Hz), 0.86 (t, 3H, J = 7.3 llz).
f i 21 957~g
W0 96104~05 ~ r~ )s
- 147 -
- C3lH34N2OloS Ø4 H2O: Calc.: C 58.74; H 5.53, N 4.42.
Found: C 58.79; lH 5.83; N 4.37.
EXAMPLE 88
N-(4-i.so-propylbenzenesulfonyl)-2-t4-(N-(L-Ala-OEt)carboxamido)-2-
propylphenoxy)-2-(5-methoxy-3,4-methylenedioxyphenyl)~eet~mide
The titled compound was prepared using procedures
o similar to those described in Example 82, where l.-aklnine ethyl ester
was the amine starting material.
I H NMR (400 MHz, DMSO-d6, ppm): ~ 8.55 (d, I H, J = 6.1 Hz), 7.69
(m,3H),7.57(q, IH,J=9.2Hz),7.40(m,21[),6.78(d, IE~,J=3.8
Hz), 6.63 (s, IH), 6.55 (m, lH), 6.02 (s, 2H), 5.70 (s, IH), 4.39 (m,
5 lH), 4.08 (q, 2H, J = 6.8 Hz), 3.79 (d, 3H, J = 2.9 Hz), 2.93 (sept, IH, J
= 6.9 Hz), 2.57 (m, 2H), 1.55 (nl, 2H), 1.37 (d, 3H, J = 5.5 Hz), 1.16
(m, 9H), 0.~¢5 (t, 3H, J = 7.5 Hz).
EXAMPLE 89
N-(4-is~l-propylbenzenes~ onyl)-2-(4-(N-2-ethoxycarbonylethyl-
carboxamido)-2-propylphenoxy)-2-(5 -methoxy-3,4-methylenedioxy-
phenyl)acetamide
'I'he tiLled compound was prepared using procedures
similar to those described in Example 82, where ~-aLmine ethyl ester
was the amine starting material.
I H NMR (400 MHz, DMSO-d(j, ppm): o 8.34 (t, I H, J = 5.4 Hz), 7.68
(d,2H,J=8.3Hz),7.58(d, lH,J=2.2Hz),7.49(dd, IH,J=8.6,2.3
3~ Hz), 7.39 (d, 2H, J = 8.4 Hz), 6.77 (d, IE~, J = 1.4 Elz), 6.65 (d, 111, J =
1.3 Hz), 6.53 (d, IH, J = 8.8 llz), 6.01 (s, 2EI), 5.67 (s, IE-I), 4-05 ('1,
2H, J = 7.1 Hz), 3.78 (s, 3H), 3.44 (m, 2H), 2.92 (sept, IH, J = 6.9 Elz),
2.53 (m, 2H), 1.54 (m, 2H), 1.16 (111, 9H), 0.84 (t, 3H, J = 7.4 Hz).
W0 96/04905 ~ 2 ~ 9 ~ 7 5 8 ~ L /~t l ~
- 148-
EXAMPLE gO
N-~4-i.so-prop), 11~el~ellesultonyl)-2-(4-(N-(L-Ala)carboxamido)-2-
propylphenoxy)-2-~5 -methoxy-3,4-methyle.nedioxyphenyl~acetalllide
The product from Example 8R was saponifiled to give the
tit]ed product.
IH NMR (400 MHz, DMSO-d6, ppm): ~ 12.64 (br. IH), 12.51 (br, IHi,
8.44 ~dd, IH, J = 7.1, 2.7 Hz), 7.69 (m, 3H~, 736 ~m, lH), 7.40 (ro,
211), 6.77 (d, IH, J = 1.6 Hz), 6.66 (d, IH, J = 1.7 Hz), 6.55 (m, 11~1),
6.01 ~d, 2H, J = 2.6 lIz), 5.69 (s, lH), 4.37 (pn, lE~, J = 7.4 Hz~, 3.79
(d, 3H, J = 1.9 Hz), 2.93 ~sept, IH, J = 6.9 Hz), 2.57 ~m, 211), 1.54 (m,
2H), 1.36 ~dd, 3H, J = 7.3, 2.7 Hz), 1.16 (d, 6H, J = 6.X Hz), ().85 ~t,
3H,J=7.2Hz).
EXAMPLE 91
N-(4-iso-propylbenzenesulfonyl)-2-(4-(N-2-carbclxyei:hylcarboxalnido3-
2-propvlphenoxy)-2-~5-metlloxy-3 ,4-methylenedioxypllel3yl'~acetalllide
Tile product fiom Example ~9 was .saponified to give the
titled product.
IH NMR (400 MHz, D~I~O-d~, ppm): ~ 12.64 ~br, Il{), 12. 21 (br,
IH). 8.32 ~t, IH, J = 5.5 lHz), 7.68 (d, 2H, J = 8.4 Hz), 7.~9 (d, I H, J =
1.9 Hz), 7.49 (dd, IH, J = 8.5, 2.1 Hz), 7.40 (d, 2H, J = 8.4 Hz), 6.77 (s,
lli), 6.65 (d, 111, J = 1.2 Hz), 6.01 (d, 2H, J = 2.9 Hz), 5.68 (s, 11:1),
3.79 Is, 3H), 3.:?tg ~Ill, 2H), 2.93 (sept, IH, J = 6.B Hz), 2.55 (m, 2H).
1.5~1 (m, 211), 1.16 (d, 6H, J = 6.9 Hz), 0.84 ~t, 3H, J = 7.3 llz).
30 C32H36N2OI(~S: ('alc: C 59.g9; H 5.66; N 4.37.
Found: C 59.72; H 5.77; N 4.49.
W096/04905 ~ 2 ~ 95758 r~"~ 3 1
- 149-
EXAI\/IPLE 92
N-(4-iso-propylbenzenesulfonyl)-2-(4-(N-3-hydroxypropyl-
carboxamido)-2-propylphenoxy)-2-(5 -methoxy-3,4-methylenedioxy-
5 phenyljacetamide
The titled cc~mpound was prepared using procedures
similar to those described in Example 82, where 3-aminopropanol was
the amine starting material.
IH NMR (400 MHz, C'D30D, ppm): o 8.33 (m, 11-1), 7.77 (d, 2H, J =
8.5 Hz), 7.60 (d, IH, J = 2.3 Hz), 7.48 (dd, IH, J = 8.5, 2.3 Hz), 7.36
(d, 2H, J = 8.4 Hz), 6.68 (d, lH, J = 1.5 Hz), 6.60 (d, 111, J = 1.4 Hz),
6.59 (d, llI, J = 8.6 Hz), 5.96 (s, 2H), 5.48 (s, IH), 3.82 (.c, 3H), 3.63 (t,
2H, J = 6.3 1 Iz), 3.43 (t, 2H, J = 5.8 Hz), 2.97 (sept, I H, J = 7.0 Hz),
2.66 ~m, 2H), 1.80 (pn, 2H, J = 6.7 Hz~, 1.61 (m~ 21~), 1.25 (dd, 6H, J =
6.9, 1.3 l lz), 0.90 (t, 3H, J = 7.4 Hz).
C32H3gN2O~)S: Calc: C 61.33; H 6.11, N 4.47.
Found: C 61.07, H 6.09: N 4.48.
EXAMPLE 93
N-(4-i.so-propylben7,t~.nPslllfollyl)-2-(4-(N-tetrazol-S-ylcarboxamido)-2-
propylphenoxy)-2-(5 -methoxy-3 ,4-methylene.dioxyphenyl)acetamide
The titled compound was prepared using procedures
simiklr to those described in Example 82, where 5-aminotetrazole was
the amine starting material.
30 FAE~-MS m/e = 640 (M+l)
WO96104905 2 1 ~5758 1~1~ 7 --
- 150-
EXAMPLE 94
N-(4-i.s o-propylbenzenesulfonyl)-2-(4-(N -3 -(~morpholin-4-yl)propyl-
carboxamido)-2-propylphenoxy)-2-(5-methoxy-3 ,4-meihylenedioxy-
phenyl)~-~el,lmifli~
The titled compound was prepared using procedures
similar to those described in Example 82, where 3-(N-morpholinyl)-
aminopropane was the amine starting material.
o IH NMR (400 MHz~ CD30D, ppltl) ~ 7.65 ~d, 2~, J = 8.3 Hz), 7.65 (s,
lH~, 7.58 (dd, IH. J = 2.4, 8.6 Hz), 7.24 (d, 2H, J = R.4 Hz), 6.~1 (d,
lH~ J = 8.6 Hz~, 6.78 Sd, lH, J = 1.4 Elz). 6.69 (d, 111, J = 1.4 Hz~, .Ci.9 l
(s~ 2EI), 5.40 ~s, lHj, 3.132 (s, 7H), 3.54 (m, 2H), 3.12 (m, 6 H), 2.9'~
(sept, lH, J = 6.9 Hz), 2.66 (m, 2H), 1.62 (m, 2H), 1.22 (d, 6E{, J = 6.9
1 IZ) 0.90 (t, 3H, J = 7.4 Hz).
C3sH43N3OgS Ø75 H2O: Calc.: C 60.46: H 6.45; N 6.C)4.
l~ound: C 60.39; E-l 6.43. N cj 93
EXAMPLE 95
N-(4-is~-propylbellzellesulfollyl)-2-(~ N-(D-Ala-OMe)c~rboxamido i-
2-propylphenoxy)-2-(:5-methoxy-3,4-methylenedioxyphenyl)acel "m i~
The titled compound was prepared using procedures
2s similar to those described in Example 82, where D-alanine methyl ester
W.lS tlie amine .s~tarting material.
lH NMR (400 MH~, CD3OD, ppm): o 854 (d, lH, J = 6.8 Hz); 7.77 (d,
2}1, J = 8.3 Hz), 7.66 (.s, IH), 7.53 (m~ lH)~ 7.36 (d, 21{, J = ~.3 Ikl,
30 6.68 (s~ IH~, 6.60 (m, 2EI), 5.96 (s, 2H), 5.49 (s. IH), 4.57 (Ill, IH),
3.82 (s~ 3H), 3.i3 (s, 3H), 2.97 (sept, IEI, J = 6.8 Hz), 2.67 (m, 2H),
1.62 (m, 2H),. 1.47 ~d, 3H, J = 7.4 Hz), 1.24 (d, 6EI, J = 7.0 llz), 0.91
(t, 3~-1, J = 7.4 Hz).
2~9~7~8
W0 96104905 ' ~ , f7
- 151 -
EXAMPLE 96
N-(4-iso-propylbenzenesulfonyl)-2-(4-(N-(D-Ala)carboxamido)-2-
5 propylphenoxy)-2-(5-methoxy-3,4-methylenedioxyphenyl)acetamide
The product from Example 95 was saponified to give the
titled product.
0 IH NMR (40û MHz, DMSO-d~" ppm) o 12.64 (br7 IH), 12.48 (br7 IH)7
~.44 (dd7 lH7 J = 7.3, 2.6 Hz), 7.68 (m, 3H), 7.56 (m, lH)7 7.40 (dd,
2H7 J = 4.0, X.4 llz), 6.77 (d, IEI7 J = 2.3 Hz)7 6.66 (m7 lH)7 6.55 (dd,
IH,J=21.0~8.~Hz),6.01 (d,2H,J=3.6Hz),5.70(d,111,J=3.8Hz),
4.37 (pn, IH, J = 7.3 Hz), 3.78 (d, 3H, J = 1.8 Hz), 2.93 (sept, IH, J =
7.0 Hz), 2.57 (m, 2H), 1.55 (m, 2H), 1.36 (dd, 3H, J = 7.3, 2.7 Hz),
1.16(d,6H,J=6.9Hz),0.85(t73H7J=7.3Hz).
EXAMPLE 97
N-(4-is~J-propylbenzenesulfonyl)-2-(4-(N-(3-carbc xymethylpropyl)-
carboxamido)-2-propylphenoxy)-2-(5 -methoxy -3 ,4-
methylenedioxypllenyl)~( et lmi~lP
The titled compound was prepared using procedures
2s similar to those described in Example 82, where methyl ~-
aminobutyrate was the amine starting material.
IH NMR (400 MHz, CD30D7 ppm): ~ 7.77 (d, 2H7 J = 8.5 Hz), 7.61 (d7
IH, J = 2.2 Hz), 7.48 (dd7 lH7 J = 8.5, 2.3 Hz)7 7.36 (d7 2H7 J = 8.5 Hz)7
6.68 (s, IH), 6.59 (s, IE-1), 6.59 (d. IH, J = 8.3 Hz). 5.95 (s. 2H)7 5.48
(S7 lH), 4.09 (q, 2H, J = 7.1 Hz), 3.X2 (.s, 3H), 3.37 (m, 2H), 2.97 (sept,
I H, J = 6.9 Hz,), 2.66 (m, 2H), 2.38 (t, 2H, J = 7.4 Hz.), 1.89 (pn, 2H, J
= 7.1 Hz~, 1.61 (m, 2H), 1.23 (d, 6H, J = 6.9 Hz), 0.90 (t, 3H, J = 7.3
l~z).
WO 96104905 2 1 9 5 7 5 8 r~ . Js 7 ~
- 152-
E:XAMPLF 9~
N-(4-is~)-prc~pylbellzelIesulfonyl)-2-(4-(N-(3 -carboxypropyl~-
5 carboxamido~)-2-n-propylphenoxy)-2-(5-methoxy-3.4-methylenedioxy-
phenyl)acetamide
The product f rclm Example 97 was saponified to give the
tilled product.
lH NMR (400 MHz, DM-SO-d6, ppm): o 12.fi3 (br, lH;, 12.06 (br, IH)~
8.27(t, IH,J=5.5Hz),7.68(d,2H,J=8.4Hz).7.60(d, IH,J=2.2
Hz), 7.50 (dd, I H, J = 8.5, 2.2 Hz), 7.40 (d, 2H. J = 8.4 Hz), 6.77 (d,
IH,J=1.211z),6.66(d,1H,J=1.3Hz),6.52(d,1H,J=g.8Hz),6.01
(d, 2H, J = 2.9 Hz), 5.68 (s, IH), 3.7g (s, 3H), 3.22 (q, 2H, J = 6.5 H~.),
2.92 (sept, 6.B llz~, 2.54 (m, 2H), 2.2.S (t, 2H, J = 7.4 Hz), 1.71 (pn, 7.1
~izj, 1.~4 (m, 211), 1.16 (d, 6H, J = 6.8 H~l, 0.84 (t, 3H, J = 7.3 11z).
C33H~xN~OIl~S: Calc: C 60.54; H ~ ; N 4.28.
Found: C 60.26; H 6.17; N 4.0~.
EXAMPLE 99
N-(4-i.s(l-propylbenzenesulfonyl)-2-(4-(N-iso-propylcar~a:moyl)a~ o-
25 2-n-propylphenoxy)-2-~3,4-1Ilethylelledic xyphenyl)acetamide
Step A: 4-Nitro-2-(propen-3-yl)phenol
A mixture of 4-nitrophenoxyallyl ether ~4.0 g, 22.35
mmol) and 1,2-dichl-,lube~ e (15 mL) was heated to reflux for 6h.
3~ The reaction mixture was cooled and purified by silica-gel tlash column
cllromatography using hexanes and EtOAc-hexanes ( 1 6) as eluents,
respectively. 1'he pure product was obtained as an yellow oil ~2.6 g).
I H NMR (200 l\~Hz~ CDC13, ppm): o 8.05 (d, 2H~. 6.92 (d, I H)7 6.0 i
(m. lH), 5.18 (111, 2H), 3.42 (d, 2H, J = 7.3 Hz).
2 1 95758
~ W096/r4905 ~ ' r~"............................... ~., c7
- 153-
Step B: Methyl 2-(4-nitro-2-(propen-3-yl)phenoxy)-2-(3,4-
methylenedioxyphenyl)acetate
The titled compound was prepared using the procedures
5 similar to that described in Step A of Example 79. Methyl 2-bromo-2-
(3,4-methylenedioxyphenyl)acetate was used as the alkylating agent.
Purification of the crude product was accomplished by silica-gel flash
column chromatography USillg ethyl acetate-hexane (l:S).
IH NMR (200 MHz, CDC13, ppm): o 8.05 (m, 2H), 7.02 (m. 2H), 6.78
(m, 2H), 5.96 (s, 2H), 5.6 (s, IH). 5.15 (m, 2H), 4.15 ~m, 2H), 3.75 (s,
3H), 3.47 (m, 2H).
Step C: 2-(4-(N-iso-propylcarbamoyl)amino-2-n-propylph~noxy)-
2-(3~4-methylenedio?~yphenvl)acetic acid
To a solution of the product c f Step B (0.5 g) in methanol
(6 mL) was added Pd-C(10%)(0.05g), and the reaction mixture was
stirred at room temperature for 6h under an atmosphere of hydrogen
gas. The catalyst was filtered off and the filtrate was concentrated in
vacuo to give the desired methyl a-(4-amilIo-2-ll-propylphenoxy)-2-
(3.4-methylenedioxyphenyl)acetate (0.5 g) as a solid. This material
without further purification was dissolved in dry THF (S mL) and
reacted with N-is~-propylisocyanate (0. I mL) at room temperature f'clr
12h. Purification of the crude product by flash chromatclgraphy using
EtOAc-hexanes (I :2) gave the titled compound as white solid (0.36 g).
I H NMR (300 MHz, CDC13, ppm): o 7.1-6.77 (m. 611), 6.61 (d, lH, J =
1.5 I-lz), 5.93 (s, 2H), 5.54 (s, IH), 3.98 (m, IH), 3.78 (s, 3H), 3.63 (m,
IH) 2.6~ lm. 2H), 1.69 (m, 211), 1.]5 ~dd~ 611, J = 7.0, 2.5 Hz~, 0.90 (t,
3H, J = 7.4 Hz).
WO 96/04905 2 1 9 5 7 5 8 P~
- 154-
SteF D: N-~4-iso-propylbenzenesulfonyl)-2-(4-(N-i~o-propyl-
carbanloyl~amino-2-n-propylpheno~y)-2-~3 ,4-methylene-
dioh~lphenyl~acetamide
The titled product ~-.IS prepared frotn the prodllct obtail1eci
5 in Step C usin~ procedures similar to those described in Steps B and C'
of Example 79.
I H N~IR (300 MHz, CD30D, ppm): ~ 7.78 (d, 2H. J = 8.4 H~), 7.76
(m. IH), ~.52 ~rll. lH), 7.32 ~d, 2H, J = 8.4 Hz~, 7.07 (d, IHJ = 1.4
Hz), 6.75-6.90 (m, 2H), 6.75 ~d, lH, J = 8.2 Hz), 6.42 (d, IH, J = 8.2
Hz~, 5.97 ~s. 21-1), 5.21 ~.s. IH), 3.8~ (m, IH1, 2.82 (m, IH), 2.54 (m,
2H), 1.69 (m, ~H), 1.26 (dd, 6H, J = 7.0, 2.5 liz), 1.15 Idd, 61-1. J = 7.0,
2.5 11z), 0.90 ~t. 3H, J = 7.4 Hz).
FAB-MS- rn/e ~96 (M+1).
EXAMPLE 100
c~-(, -n -propyl -4-methylaminosulfollylphelloxy)-~ ,4-methylenedioxy-
phenylacetic ac~id
~0 Step A: Preparation of 3-allyl-4-hydroxybenzenesult'onamide
TD a solut;on of 5.00 g (28.9 rnmol) of 4-
hydroxybenzellesulfonamide dissolved in 30 mL of anhydrous DMF was
added 10.36 g (31.8 mmol) of cesium carbonate and the reactic~l1
mixture was magnetically stirred at room lellll~,.<ltUlC'- under a nitrogen
2s at1llospl1e~l-e for 10 minutes. Allyl brornide ~2.75 mL. 31.8 mmol) was
added and the reaction mixture was then slirred For an additional 14
hours. The reaction mixture wa.s then partitioned between EtC)Ac (6(1
mL) and 10% aqueous citric acid (200 mL) and extracted. The organic
layer was separated, washed with saturated aqueous N?aHCO3, saturated
3~ aLIueolls NaCI, dried ~MgSO4~, filtered, evaporated and dried in ~acuo
to afiord 5.40 g (8~%) of a yellow solid. The crude O-allyl ether
(5.36, 25.~ mmol) WLIS then dissolved in 10 ml. of 1,2-dichloroben7elle
in a 50 mL round bottom fla.ck and magnetically stirred at reflux under
a nitrogen ~llno~llhPre for 15 hours. The reaction mixture WaS then
cooled to room temperature and diluted with methanol. ~ e 1,2-
1 9 ~ 7 5 8
~ , .. ~ ,
WO 96/0490S - ' ~ r .l,lJ..,...~., ~7
- 155-
- dichlorobenzene was removed by extraction of the rmethanol layer with
hexane, the methanol layer was separated, thell evaporated. The residue
was then purifed on a silica gel flash chromatography colurnn eluted
with 5% MeOH-CE~2C12. Combination of the purified fractions,
5 evaporation and drying in vacuo afforded 3.04 g (57~o) of the title
compound.
IH-NMR (400 MHz, CD30D. ppm~: o 3.38 (d, J=6.40 Hz.2H), 5.02-
5.10 (m, 2H), 5.94-6.04 (m, lH). 6.84 (d, J=8.40 Hz, IEI), 7.58 (dd~
.J=2.40,8.40 Hz, IH),7.61 (d, J=2.40 I-lz, lH).
o cl-Ms l~lle = 213 (M+)
Step B: Preparation of 4-hydroxy-3-~?-propvlbenzellesulfollamide
A Parr hydrogenation flask was charged with a solution of
3.04 g (14.30 mmol) of the product of Step A di.ssolved in 25 mL of
5 ethanol and 0.300 g of a 10~ palladium on carbon catalyst was added.
The flask was mounted in the hydrogelIatioll apparatus, freed of air,
pressurized with hydrogen (40 psig) and shaken for 15 minutes. At the
end of this period TLC analysis (3C~o MeOH-CH2C12, 2 elutions)
indicated that the reaction was complete and the reaction mixture was
20 filtered and evaporated. The product was dried in vacuo to afford 3.06
g (99~c,) of the title compolmd.
IH-NMR (400 MHz, CD30D, ppm): ~ 0.94 (t, J=7.20 Hz, 3Hh 1.58-
1.68 (m, 2H), 2.01 -2.62 (rn, 2EI). 6.82 (d, J=8.40 E~z, IH), 7.55 (dd,
J=2.40, ~.40 E Iz, 111), 7.60 (d, J=2.40 Hz, IH).
25 FAB-MS mle = 216 (M + 1).
Step C: Preparation of methyl o~-(2-n-propyl-4-amillosulfolIyl-
pheno~v)-3,4-methvlenedi(IxvpllellY lacetate
To a solution of 3.06 g (14.23 rnmol) of the product of
3 ~ Step B dissolved in 25 mL of anhydrous DMF was added 4.~7 g (1 :; .0
mmol) of ce.sium carbonate and the reaction mixture was magnetically
stirred under a nitrogen atmosphere at room temperature for 15
minutes. Methyl a-bromo-3,4-1Ilethylelledioxyphenylacetate (4.08g,
15.0 mmol) was then added and the reaction mixture was stirred t'or an
additional 3 hours. The reaction mixture was then partitioned between
~ ~1 q57~ --
W0 96/04905 -- ~ ~ S ~
- 156-
EtOAc (80 mL~ and 1(11~6 aqueous citric acid (300 mL). The organic
layer was ~separated. washed with saturated aqueous NaHCO3, satual-ated
aqueous NaCI. dried (MgSO4), filtered and evaporated. Th~ residue
wa.s driecl in VclCllO to afford 5.90 g (5.79 theoretical) of the t;tle
5 compound which was used in the next step without further purifilcation.
H-NMR (400 MHz, CD30D, ppm): ~ 0.97 (t, J=7.20 Elz, 3H), 1.64-
1.76 (m, 2H), 2.74 (t, J=7.20 Hz, 2H), 3.70 ~s, 311), 5.87 (s, lH), 5.97
(.s, 2H), 6.8~ ~d, J=8.1)0 Hz, IH), 6.93 (d, .l=8.40 Hz. 11-1;, 7.03 ~d,
J=1.60 Hz, IH~ 7.06 (dd, J=1.60, R.00 E-lz, 1~1). 7.65 (dd7 J=2.40, ~.40
HZ. IH), 7.69 ~d, J=2.40 Hz, IH).
FAB-MS nll~ = 408 (M + 1).
Step ~: Preparation of rmethyl o-(2-n-propyl-4-methylamino-
sul~onylphenoxy)-3.4-methylenediQItypher~lacetate
To a so!ution of 2.19 g (5.38 mmol~ of ~e product of Step
15 C dissolved in ~0 mL of anhydrous THF was added 2.4l mL (16.1
mrnol) of 1,8-diazilbicyclol5.4.0-1undec-7-ene and the reaction mixture
was magnetically stirred under a nitrogen atmosphere for 2~ minutes at
room temperature. Iodomethane (1.00 mL; 16.1 mlnol) was added ial)d
the reaction mixture was stirred an aclditional 15 hours at room
20 temperature. The reaction mixture wa.s diluted with EtOAc alld a
precipitate formed which was redissolved by additioll of' metharlc)l. The
mixture was furthel diluted with warm EtOAc (150 mL total),
rel'ridgerated overnight and a solid separated whiGh was removed by
filtratioiu The fi!trate was evaporated in vacuo and the residue v~as
25 purified on a silica gel flash chromatography collumn eluted with 5~6
EtOAc-CHCl3. Combinatioll of the purified fractions and evaporation
in vacuo afforded 0.164 g of the title compound and a number of
iml)ure fractions whictl were reserved l~or repurification.
IH-NMR (400 ~IHz, Cl)3OD, ppm): ~ 0.97 ~t, J=7.20 Hz, 3E~), 1.65-
30 1.77 (m, 2H)~ 2.48 (s. 3H~, 2.74 (t, J=7.20 Hz, 2H), 3.71 (s, 3H), 5.87
(s, IH), 5.98 (s, 2H), 6.85 ~d, ./=X.00 Hz, lH~ 6.96 (d, J=8.80 Hz, I E~
7.04 (d, J=1.60 Hz, 1~, 7.07 (dd, J=1.60, 8.00 Hz, 111), 7.58-7.61 (m,
2H).
ESI-MS mle = 421 (M+).
2 1 ~ 5 7 ~ 8
~. WO 96/04905
- 157-
~ ~ Preparation of o -(2-n-propyl-4-1llethylaminosulfonyl-
phenoxy)-3~4-methylenedioxyphenylacetic acid
To a solution of 0.372 g (0.884 mrnol) of the product of
Step D dissolved in 3.0 mL of methanol was added 212111, (1.06 mmol)
5 of a 5.0 N aqueous sodium hydroxide solution which resulted in a
cloudy suspension. The reaction wa.s warmed to assist solution,
methanol (1 mL) was added followed by dichloromethane (0.5 mL),
however a clear solution was not obtained. Additional 5 N sodium
hydroxide solution was added (212 IlL), and finally 0.5 mL of THF was
added which resulted in a clear solution. After stirring an additional 15
hours at room temperature, Tl.C analysis (CHC13-MeOH-N1140H
80: 15: 1) indicated complete hydrolysis of the starting material and the
reaction ivas adjusted to pH=7 with 6 N HCI. The reaction mixture was
then concentrated in vacuo and the residue was purified on a silica gel
5 flash chromatography column eluted with CHC13-MeOII-HOAc
(92:7:1). Combination of the purified fractions ~md drying in vacuo
afforded 0.335 g (93~) of the title compound as an amorphous solid.
IH-N~R ~400 MHz, Cr~3OD, ppm): ~ 0.96 (t, J=7.20 Hz. 3}-1), 1.66-
1.78 (m, 2H), 2.48 ~s. 3H), 2.73-2.77 (m, 2H), 5.74 ~s, 11-1), 5.97 (s,
20 2H), 6.85 (d, J=7.60 H~, 111), 6.98 (d, J=9.20 Hz, IH), 7.07-7.10 ~m,
2H), 7.59-7.62 (m, 2H).
ESI-MS m~e = 407 (M+).
EXAMPLE 101
N-(4-isr~-propylbenzeneslllfonyl)-f~-(2-17-propyl-4-methylarnino-
.sulfonylphenoxy)-3,4-methylenedioxyphenylacetamide potassium salt
To a solutiotl of 0.298 g (0.73 mmol) of the product of
Example 100 dissolved in 4.() mL of anhydrous THF was added 0.237 g
30 (1.46 mrnol) of l,l'-carbonylfliimifl~7r)1e and the reaction mixture was
magnetically stirred and refluxed l'or 2 hours under a nitrogen
atmosphere. The reactioll mixture was then cooled to room
temperature, 0.219 g (1.10 mmol) 4-iso-propylhf n7f neslllfonamide and
16411L (1.10 mrllol) 1,8-diazahicyclo[5.4.0]undec-7-ene were added auld
2 ~ 9 5 7 ~ 8
W0 96/04905 r~
- 158-
the reaction w~s stirred and heated at reflux for an additional l 0
minutes. The reaction mixture was then ccloled to room temperature.
partitioned between 105'o aqueous citric acid and EtOAc and extracted.
The organic layer u,hich separated was washed with saturated aqueous
NaCI, dried ~MgSO4l, filtered and e~ aporated. The residue was
redis.solved in 1.0 mL of methanol and treated with 2.2C~ mL (3 e~l) of a
1.1 hl aqueous solution of potassium hydroxide. The mixture was then
diluted with ~ mL of water and filtered through a 0.45 micron filter.
The filtrate w-as desalted and purified on a VVater.s Millipore Delta Prep
3000 liquid chrt)lllatograph equipped witll an M1000 Prep-Pak module
containing a 47 x 300 mm Delta-Pak C18 ISIlm 100A colunm cartridge.
Two solvent resevoirs were employed: solvent system A (95-5 water-
acetonitrile), and .solvent system B (5-95 water-acetonitrile), .uld the
column et'fluent was monitored ~irnlllf:ln?ously at 210 and 280 mn with
a Waters model 490 UV-visible detector. The column was
preequillibrated with solvent system A and the filtrate wa.c injected.
The product wa.s desalted by elution with 0.5 L of solvent system A (50
mLfmin) then a gradient elution was be~un wllich had .I.S illitial
conditions 100~ sol~ent system A-0% solvent systelII B arlcl reached
after 1~ minute~ 60~, solvent ~ystem A-40~i sol~ent sy.stern B, ~md the
f'rdctions were collected with an ISCO ~oxy 2()0 fr~ction collectl~r. Thoe
purified frac~tions were combined in round bottom flasks, fro7en in a
-7~C dly ice-acetone bath. and Iyophilized. Combination of the
purified product afforded 0.284 g (62%) of the title compound as a
white lyophilized powder.
I H-NMR (400 MHz, CD30D, ppm): o 0.89 (t, J=7.60 llz, 3H), 1.21 (d.
.l=6.80 Hz. 3H), 1.22 (d, .J=6.80 Hz. 3H), 1.57-1.64 (m, 2H), 2.45 ~s,
3H), 2.56-2.63 (m, IH), 2.70-2.76 (m, IH), 5.37 (s, IH), 5.93 (d,
J=l .?0 H z, III), 5.94 ~d, .J=1.20 lIz, IEE), 6.76 ~d, J=8.40 H7., II-IJ. 6.85
(d, J=8.80 Hz, I H), 7.02-7.04 (m, 2H), 7.21 (d, .J=S.40 llz, 2H), 7.47
(dd, J=2.40, 8.80 Hz, lH), 7.52 (d. J=2.40 Hz, IH), 7.65 (d, J=8.40 Hz,
211).
ESI-MS mle = 627 (I\I + 1).
21 9~7~Q
W096/049~ /C~
- 159-
- EXAMPLE 102
N-(4-iso-propylbenzenesuli'onyl)-c~-L4-(cyanomethyl)-2-n-
propylphenoxy)]-3 ,4-methylenedioxyphenylacetamide
5 Step A: Preparation of ~T-(4-i.so-propylben~enesulfonyl)-o~-(4-
bromomethyl-2-~?-propylphenoxy)-3 ,4-methylenedioxy-
phenvlacetamide
To a solution of 0.200 g ~.381 mmole) of the product of
Example 65 dissolved in l.:'j mL of diethyl ether was added 0.837 mL
(.837 mmole) of 1.0 M phosphorus tribromide in methylene chloride
solution under nitrogen at 0~C. The reaction mixture was stirred at
0~C for 2 hours when TLC analysis (~():15:1 CHC13-~feOH-NI-14OI-I)
indicated that the reaction was nearly complete. The reaction was
quenched at 0~C with water and then partiLioned with EtOAc. The
5 aqueous portion was separated and the EtOAc portion was washed with
brine (2 X 10 mL). The EtOAc portion was then dried over MgSO4,
filtered, evaporated to a residue, and then used in the next step of the
reaction scheme.
Step B: Preparation of N-(4-i.so-propylbt n 7~n~s- 11fonyl)-a-(4-
cyanomethyl-2-n-propylphelloxy)-3 ,4-methylenedioxy-
phenylacetamide
To a solution of the crude product of Step A dissolved in
1.5 mL of methyl sulfoxide was added 0.050 g (.762 mmole) of
potassium cyanide at room temperature under nitrogen. The reaction
mixture was stirred at room temperature for 1 hour when TLC analysis
(80:15:1 CHC13-MeOI l-NI-1401-1) indicated that the reaction had gone to
completion. The reaction mixture was diluted with EtOAc and 10
NaHSO4 aqueous solution. The aqueous phase was separated and the
EtOAc portion was washed with brine (2 X 10 mL). The EtOAc
portion was then dried ov er MgSO4, filtered, evaporated to a residue.
and purifled. Purification was done by reversed phase HPLC (Waters
Millipore Delta Prep 4000 with Delta-Pal~ C18 15 llm 100 A column
cartridge) with a solvent system of 30:70 water-acetonitrile and 0.1 ~!
TFA buffer. The purified fractions collected were combined in a round
~ ~ 2 1 ~57
WO961D4905 - 58 .~ lu~ ,,'7
- 160-
bottom flask, freeze in a -78~C dry ice-acetone bath, and Iyopbilized.
The resulting Iyophilized powder afforded 0.071 g (35 ~c~ 2-step yield~.
I l-l-NMR (400 MHz, CD3OD. ppm): S 0.8R (t, J=7.37 Hz, 3H), I .25 (d,
J=6.92 Hz, 3HI, 1.27 (d, .1=6.97 Hz, 3H), 1.~5 Im.2H)~ 2.60 ~m, 2H),
2.99 (m, IH), 3.75 (s, 2H), 5.40 (s, IH), 5.96 (s, 2H), 6.~3 (d, J=8.39
Hz. lH), 6.78 (d? J=8.03 Hz, lH), 6.86-6.96 (m, 3EI), 7.10 (s, lH), 7.37
(d, J=6.h4 Hz, 2H), 7.7R (d, J=8.48 Hz, 2H).
MS ~ESI): C29H30N206S 534.63 Foulld: [535.1, M+l].
EXAMPLE 103
N-(4-i.s~o-propylhPn7e~ sulfollyl)-(x-l4-(tetraz(71-~-ylmethyl~-2-/1-
propylpheno~y)l-3,4-methylenedioxyphenylsl- et:lmi~
A solution of 0.120 g (.224 mmole~ of the produet of
Example 102 and 0.139 g (.673 mmole) of trimethyltin azide dissolved
in 1.5 mL of toluene was heated in sealed pressure reaction tube and
.stirrecl for 5 hours at 120~C. Analytical HP~C analysis (30:70 water-
acetollitrile with 0.1 ~~ TFA) ;ndicated that the reaction had gone to
20 ec)mpletioD. The reaction mixture was cooled to room temperature
when 2 M 11C l solution was added. The reaction mixture wa.s
piartitioned with EtOAc and the aqueous ponion was separated. ~he
EtC)Ac portion was washed with brine (2 X 10 ml,). dried over r~lgSO4
. filtered~ e~raporated to a residue, and purified. Purification was done
25 by reverse(l phase HP1,C (Waters Millipore Delta Prep 400Q rwitll Delta-
Pak ClR 15 ,~m 100 A column cartridgre) with a solvent system of 30:70
watel Acetl)nitrile and 0.1 % TFA buffer. The purified fractions
collected were combined in a round bottom flask, freeze in a -7~~C' dry
ice-acetone bath. and Iyophilized. The resulting Iyophilized powder
30 affordeo 0.0306 g (24 ~,10 yield).
IH-NMR (40Q MHz, CD30D, ppm): ~ 0.B6 ~t, J=7.47 lIz, 311), 1.25
(d7 J=6.82 Hz, 6H), 1.~4 (m, 2H), 258 (m, 2H), 2.96 (m, lHI, 4.19 ~s,
2H), 5.38 (.s, IH~, ~.96 (s, 2H), 6.51 (d, J=R.30 Hz, 111), 6.76 (d, J=8.~3
Hz, III), 6.84-6.89 ~m, 3H), 7.04 (.s, lH~, 7.3~ (d, J=8.30 Hz, 2H), 7.76
(d, J=8.3~ Hz, 2H).
~ WO9G10490S ~t 95758 r~l,~J M~l ............................ -,
- 161 -
~ MS (ESI~: C29H31N506S 577.66 Found: j578.2~ M+l].
EXAMPLE 104
N-(4-iso-propylbenzenesulfonyl)-a-[N-(4-carbomethoxyphen~lamino)l-
3 ,4-methylenedioxypilenylacetamide
~: Preparation of ethyl a-[N-(4-carbomethoxyphenylamino)l-
3 ~4-methylenedioxyplletlvlacetate
o To a .solution of 5.034 g (33.4 mmoles) of methyl 4-
aminobel~oate dissolved in 50.0 mL of DMF was added 10.526 g (36.7
mmoles) of ethyl a-bromo-3,4-methylenedioxyphenylacetate. The
reaction mixture was heated to 85~C and stirred in a sealed pressure
reaction tube for 16 hours. TLC analysis (25~o EtOAc:Hexanes)
5 indicated that the reaction had gone to completion. The reaction
mixture was transferred to a sep. funnel and partitioned between EtOAc
and water. The aqueous portion was separated and the organic portion
was washed with brine (2X25 mL), dried over MgS01, filtered, and
evaporated to a residue. Purification was done by flash
20 chromafography eluting with 20~rO EtOAc:EEexanes. The purified
fractions collected were combined and evaporated to afford 7.90 g of
the titled product.
MS (ESI): C19H19N06 357.36 E~ound: [357.9. M+l].
Step B: Preparation of a-[N-(4-carbomethoxyphenylamino)]-3~4-
methvlenedioxyphenykacetic acid
To a solution of 2.12 g (5.93 mmoles) of the product of
Step A dissolved in 10 mL of methanol was added 6.52 mL (6.52
30 mmoles) of 1.0 N KOH solution in methanol. The reaction mixture vvas
stirred at room t~.l.pe.atul~ for I hour. TLC analysi.s (25~7o
EtOAc:Hexanes) at that time indicated that there was no more starting
material present in the reaction mixture. The reaction mixture was
diluted with EtOAc and quenched witlI 10~~ NaHS04 aqueous solution.
t ~ 5 7 5 8
W096/04905 ~ ~ P~~ 13.::7--
- 162-
The aqueous phase was separated and the organic portion wa.s washed
with brine (2 X t5 mL), and evaporated to a residue. Purificatioll ~as
done by reversed phase HPLC (Waters Millipore Delta Prep 400() witll
Delta-Pak C18 1~ llm lû0 A column cartridge) with a solvent system of
50:50 water-acetonitrile and 0.1 % TFA bufter. The purified fractic)ns
collected were combilIed in a round bottom flask, freeze in a -7~~C dry
ice-acetone bath. and Iyc)philized. The resulting Iyophili~ed powder
afforded 1.11 ~ (57O,lo yield) of the titled product.
MS (C~l): C17Hl~NO6 329.13 Found: [330-5, M+11.
Step C: Preparation of N-(4-i.so-propylbenzelIesulfonyl)-a-[N-(4-
c~arbomethox),phenylarllirlo)~-3,4-1nethylenedioxyllhenyl-
acetarnide
l'o a solutiolI of 0.180 g ~.~47 mmole) of the product of
5 Step B di.ssolved in 1.5 ml, of methylene chloride wa.s added 0.080 g
(.656 mmole) of 4-dimethylaminopyridine, 0.147 g (.766 mmole'~ clf 1-
ethyl-3-(3-dimethylamilIo-propyl3carbodiimide hydrochloride, and
().121:) g (,60? mmole) of the sulfonalnide respectively. The reaction
mixture was .stirred at room te"-pe,dLu,~ under nitrogen i-or ~4 hours.
20 TLC analysis ~0:15:1 CHC13-CI-1301-1-NH40H) indicated that the
reaction had ~one to completion after 24 hours of stirring. The
reaction mixture was diluted with E~tOAc and transferred to a sep.
funnel. The organic pclrtion was washed with 2 N HC~I (2X 10 mL) and
brine (IX10 mL). dried over MgSO4, filtered. and evaporated to a
2s residue. Purification was done by reversed phase HPLC (Waters
Millipore Delta l'rep 4000 with Delta-Pak CIR 15 ~lm 100 A colulIm
cartridge3 with a solvent .system of 40:60 water-acetonitrile and 0.1 ~,fo
1'FA buffer. ~he purified fractions collected were combined in a round
bclttom flask, t'reeze in a -78~C dry ice-acetone bath~ and Iyophilized.
30 The resulting Iyophilized powder afforded 0.060 g (21C~c yieldj of titled
product.
I H-NMR (40Q MHz, CD30D, pprm3: o 1.25 (d. J=6.96 llz, 3H). 1.2t~
id, J=6.~7 Hz~ 3H~, 2.97 (sept., J=7.06 Hz. 1~31 3.~0 (s, 311), 1.86 (.s~
2? 9~758
~ WO 96/04905 ~ i7
- 163 -
~ lH), 5.g4(s, 2H), 6.45 (d, J=8.81 Hz, 211), 6.87-6.75 (m, 3H~, 7.36 (d,
J=8.39 Hz, 2H), 7.65 (d, J=8.85 Hz, 2H), 7.78 (d, J=8.48 Hz, 2H).
MS (ESI): C261T26N207S 510.57 Found: [511.0, M+ll.
EXAMPLE 105
N-(4-iso-~ pylbel~e-lesulfolly1)-a-[N-(4-carboxyphenylamino)~-
3 ,4-methylenedioxyphenylacetamide
Following the hydrolysis procedure described in Example
58 the titled compound is prepared from N-(4-iso-propylbenzene-
sulfonyl)-a-[N-(4-carbomethoxyphenylamino)] -3 .4-methylenedioxy-
phenyl~re~milt~
~ r ~ 2 1 ~ 5 jr 5 8
WO 9610490!; ~ I ~,11-!~. '. ............................ _ I _
- 164-
EXAI~PLES 106-121
Ex~mple~ 106 throu~h 121 were prepared follovving the
5 procedures de~cribed i:n E~ample 40.
CO2H
~
O~Z
0~
~x. # Z Ma~s
Spectrum
1~+1)
2~ 106 C.ONHS~,-3-pyridyl fM+I~ 513
107 CONHSO2-(2-Me)-3-4uininolinyl (M+l) 563
1~)8 CoN~so2-3~ui~ yl (M+l) 54~,
109 CONHS02-~4-OH)-3-pyridyl ~M+1) 529
1 10 CONHS02-(4-OEt)Ph (M+NH4+) 559
111 CONHS02-(4-CONH2)Ph (M+l) 542
1 12 CONHS02-[4-CO(NiMej2)]Ph (M+:l ) 569
113 coNHso2-(4- SEt~- 3-pyridyl 1 M+ I ) 5 5
114 CONllS02-(4-OEt)-3-pyndyl (M+l i 543
3~ 115 CONHS02-(4-~rnine, 2,5-di-OMe~Ph (M+:l~ 573
116 CONHS02-(2~5-di-OMe)Ph (M+l) 558
1]7 CONHSO2-i3,4-di-OMelPh (M+l) 558
118 CONHS02-[5-(4- , ~ ~ yl~l-2-~Li~J~J~ +l) 639
I 1~} CONI'lSO2-(4-O~ e!-2-bl-n~ f+l ) 585
120 coNHso2-~4-(fcH2)2NHcsz))]ph ~M+ l 1 675
2 ~ 9 5 7 5 8
~ WO 96/0490S P~, I /LI.~ 'T,'O,.l 7
- 165 -
121 CoNHso2-(2~s-di-oMe~4-NHcoNTHipr!ph (M+l) 658
122 CoNHso2-(2~4-di-oMe)ph
123 CoNHso2-(2~4~6-tri-oMe)ph
EXAMPLES 124- 129
~
o)~ R
Ex # R 12 R3a Z Ma~
Spectrum
(M+l )
124 CO2H H CoNHso2-x-quininolinyl 579
125 CO2H H CoNHso2-3-quininolinyl 579
126 CON112 OMe CONHSO2-8-quininolinyl 57
127 CONH2 OMe CONHSO2-(4-t-butyl)Ph 553
12~ CON112 OMe CoNHso2-(4-amine~2~s-di-oMe)ph 572
129 CO2H H CONHSO2NH-(4-iPr)Ph 5~5
9 ~ 7 ~ 8
W0 96104905 - - r~ . , 7'--
- 166 -
EXAMPLE 130
N-IN'-(4-iso-propylbenzene)aminosulfonyl~cx-[(4-c,lrboxy-2-n-
propyl)phenoxy]-3,4-methylenedioxyphenyl~oetsimi~lP
Step A: Preparation of N-~4-isopropylbenzene)-N'-tert-hsutyl-
sulfamide
To a solution of p-isopropylaniline (1.69 g, 11.77 mmol) in
CH2C12 (0.5 ml~ was added N,N-diisopropylethylamine (2 ml) followed
by the dropwise addition of a solution of N-t-butylsulfamoyl chloride
(1.01 g. 5.88 mmol) [prepared according to the procedure described b~
W.L. Matier and W.T. Comer, J. I~ied. Chem., 15:5~ 538 (1972)~ in
CH2C12 (0.4 ml~ via a ~yringe at 0~C. The reaction mixture was then
maglletical}y stirred at room temperature for i8 hrs. The reactioll was
diluted with C1~2C12 smd quenched with aqueouslN HCI. The organic
phase was separated~ washed with water, saturated aqueous NaCI. dried
(MgS04)~ filtered~ evaporated and dried in vacuo to yield an impure
solid. The residue was puri~led bv trituratin~ with Hex:EtOAc (4:1) fo
yield 930 mg (58~~c,) of the titled product as a white solid.
Il-i-NMR (300 MHz, CD30C), ppm~: ~ 1.1-1.3 (m, 15H), 2.7-2.9 ~m~
IH). 6.9-7.2 (IU, 4H).
ESI-MS mle = 271 (M + 1~.
Step B: Preparatic n of N -4-isopropvlht~n7~n~culfamide
2s A solution of 0.9 g (3.32 mmol) of the product of Step A in
tritluoroacetic acid (15 ml) was mslgn~ticiqlly stirred at ruom
temper.lture ullt;l TLC indicated the reaction was collIplete. The solvent
was removed in vacuo, washed with cold Et20, smd filtered to y;eld 553
mg (78 ~~i) of the titled product as a white solid.
IH-NMR (300 MHz, CD30D, ppm): ~ 1.1-1.3 (d. 6EI), 1.7-1.9 (m,
IH). 7.1 (s~ 4H).
Cl-MS, mle = 232 (M ~ NH4+).
9 J 7 5 ~
~ WO 96/~4905 ' ' ~ J ~ 5/~
- 167 -
Step C: Preparation of N-[N'-(4-iso-propylbenzene)aminosulfonyll-
a-l (4-carbomethoxy-2-n-propyl)pllenoxy]-3 ,4-methylene-
dioxyphenylacetamide
The compound from Step B was reacted with the
carboxylic acid, obtained in Step B of F.xample 56, according to the
procedure de.scribed in Step C of Example 25 to provide the titlted
compound.
IH-NMR (300 MHz~ CD30D, ppm): o O.X-0.9 (t, 3H), 1.1-1.3 (d. 6H),
1.4-1.7 (m, 2H), 2.5-2.9 (m, 2H), 2.7-2.9 (m, lH), 3.~ (s. 3H), 5.3 (s,
IH), 5.9 (s, 2H). 6.5-6.7 (dd, 2H), 6.8-7.1 (m, 6H), 7.5-7.6 (dd, IH),
7.7 (s, 111).
ESI-MS, m/~ = 659 (M+l).
Step D: Preparation of N-[N'-(4-iso-propylbenzene~aminosulfonyl]-
a-l(4-CarbOXy-2-tl-prOpyl)phenOXyl-3.4-lllethylenedioxy-
phenylacetamide
The titled compound was prepared following the procedure
described in Example 40.
IH-NMR (300 MHz, CD30D, ppm): ~ O.X-0.9 (t, 3H), 1.1-1.3 (d. 6H),
1.4-1.7 (m, 2H), 2.5-2.9 (m, 2H), 2.7-2.9 lm, IH), 5.3 (s, IH), 5.9 (s,
2H), 6.5-6.7 (dd, 2H), 6.8-7.1 (Ul, 6H), 7.5-7.6 (dd, IH), 7.7 o;, 11-1).
EXAMPLE 131
N-(4-i..~o-propylbenzenesulfollyll-o -[4-methanesulfonylamino-2-n-
propylphenoxyl-3,4-methylenedioxyphenyl~ et lmide
o-(4-Amino-2-n-propylphenoxy)-3,4-rnethylenedioxy-
phenylacetate (Product of Step C Example 99) was reacted with
methanesulfonyl chloride in ~ mixture of pyridine and methylene
chloride to pro~ide o~-[4-(N-methanesulfonyl)amino-2-n-propyl-
phenoxy]-3,4-methylenedioxy- phenylaeetate. which upon further
2~ 957C;~ ~'
WO ~610~905 J u I ~
- 168 -
elaboration following the procedures described in Example 99 provided
the titled compoulld.
I H-NMR ~300 I~lHz, CDC13, ppm): ~ 0.8-O.g (t, 3H), 1.1-1.3 (d, 611)~
1.4-1.7 (m, 2H), 2.4-2.69 (m, 2H), 2.72 (s, 3H), 2.75-Z.9 (m, lH), 5.3
(s, IE-I), 5.85 (s. 2H), 6.6-6.7 (dd, ,H), 6.8-7.1 (m, 6H), 7.6-7.75 (dd,
IH~, 7.9 (s, 111).
EXAMPLE~ 132
N-(4-i.~o-propylben~enesulfonyl)-cc-l(4-(N,N-dilnetilylcarh3Jnclyl)-
amino-2-n-propylphenoxy l-3 ,4-methylenedioxy-phenylacetamide
The title compound '~Y.LS prepared by reacting the amine
from Example 99 (Step C) with N,N-dimethylcarbamoyl chloride.
1 H-NMR (300 MHz, CDC13, ppm): o U.95 (t, 3H), 1.1-1.3 (d, 6H).
].45-1.8 (Itl, 2H), 2.5-2.7 ~m, 2H), 2.75-2.9 (m, 11-1)~ 3.0 l~s. 61-1), 5.3 (s,IH), 5.85 (s, 2H), 6.6-6.7 (dd, 2H~, 6.8-7.1 (m, 6H), 7.6-7.75 (dd. 11-1),
7.9 (d, 2H).
EXAMPLE 133
N-(4-i~o-propylhenzenesulfonyl)-~x-[4-methoxycarbonylaminu-2-1l-
propylphenoxy]-3.4-methylenedic~xy-pllenyl i~e~:lmiflP
2s 1 he title compound was prepared by reacting the amine
from Example 99 (Step C) with ethylchlorc)iorlll~te.
H-NMR (300 MHz, CDC13, ppm)- o 0.9 (t, 3H), I .I -1.3 lm, 9H),
1.45-1.7 (m, 2H). 2.45-2.7 (m, 2E-1), 2.8-3.0 ~m, IH), 4.1-4.25 (Ll, 2}1),
5.3 ~s, IH), 5.9 (s, 2H), 6.5-6.75 (m, 2H), 6.8-7.0 (m, 6H~, 7.6-7.75
30 (dd, IH), 7.8 (d, 211)-
2 1 q ~i 7 5 PJ
~ W096/04905 ,~"~
- 169-
EXAMPLE 134
Methyl 3-allyl-4-hydroxybenzoate
5 Step A: Preparation of methyl 4-allyloxybenzoate
To a nitrogen flushed 5 L three neck round bottom flask
fitted wilh a mech~llir 1l stirrer, condenser, and a nitrogen inlet was
charged 60~ g ~4 mol~ of methyl 4-hydoxybenzoate, 520 ml ( 727 g,
6.00 mol, 1.5 eq) of allyl bromide, 663 g (9.6 mol of anhydrous
0 potassium carbonate, and 2.3 L of acetone. The mixture was refluxed
with vigorous stirring for ~0 min. Additional potassium carb~mate, (50
g) was added, and 25 g added again after an additional 50 min. At'ter
~0 min (total reaction time of 160 min), the suspension was allowed to
cool to ambient t~-llL)el~lule and stirred overnight. The mixture was
5 filtered and the cake washed with 3 L of acetone. The solution was
concentrated to obtain 7~8.6 g (theoretical yield 76~.9 g) of a pale
yellow, almost colorless oil which was used without purification in the
next step. The product was a single spot on TLC (silica-l:l
EtOAc/Hex) and the MNR was consistent with methyl 4-
20 allyloxybenzoate.
Step B: Preparation of methyl 3-allyl-4-hydroxybenzclllte
To a nitrogen flushed magnetically stirred 3 L single neck
round bottom flask fitted with a condenser, and a nitrogen inlet was
25 charged the methyl 4-allyloxybenzoate, 4()0 mL of 1.2-dichlolol,~ elle,
and 10 g of BEIT. The solution was heated and distillate collected untill
the head temperature reached tl~at c~f 1,2-dichlorobenzene (1~0~C). The
solution was then refluxed for 6.5 hr, then cooled to 140~C and aged
ovemight. The hot solution was then poured into 2.5 L of hexanes and
30 the re.sulting suspension aged overnight with stirring. The suspension
was filtered, ~md the cake waslle(l with hex~mes. The solid wa.s air dried
affording 747.7g (~7.3~ yield) as a white solid haYing a faint odor of'
o-dichlorobenzene.
7 1 0 ~ 7 ~ 0
;, ~ L I ~ J I J U j~
W096/04905 ' I ~ Jv rr; _ ~ _
- 170-
I :H-I~MR (3(1() ~IHz7 CDC13. ppm): ~ 3.42 (dt .J=6.4,1.4 Hz, 2L~), 3.~7
(~s, 311), ~.11-~.18 (m, 2H~, S.X7 (hs, IH), 5.93-6.06 ~m,lll), 6.R3 (d,
J=7.9 Hz~ lH), 7.79-7.85 ~m, 21-1).
EXAMPLE 135
Methyl 4-hydoxy-3-n-propylbenzoate
o Step A: Preparation of methyl 4-hvdoxy-3-n-propvlbenzoate
A solutioll of 3~3 g of methyl 3-allyl-4-hydoxybenzoate in 1.5 L of
methallol was hydrogellated for I hr in a ParrR type shaker at 40 psi
and ambient temperature using 1.5 g c~f 10~ p~lladium on carbon as the
catalyst. The reaction was filterd through Sulka-FlocR and the caL e
washed with I L of methanol. The combined filtrate was concentrated
and the oil tlushed with ether. Hexanes (1.5 L~ were added and the
resulting suspension cooled to 0~C. The product was collected by
filtrati(m, washecl with hexanes ancl dried affolding 176.6 g of methyl
4-hydoxy-3-1l-propylbenzoate. A secc~lld crop of 166.4 g was obt;lilled
hy c(JIlcentratin~; tile filtrate, cliluting with hexan~s ;md filtelillg,
bringillg the total to 343 g (94.3% yield).
I H-NMR (3()0 MHz, C'DC13, ppm): ~ 0.94 (t .1=7.4 Hz; 3H), 1.63 ~m,
2H)t 2.59 (t .1=7.7 Hz, 2H), 3.86 (s, 3H), ~.87 (s, IH), 6.~4 ~d J=8.4 Hz,
111~. 7.76 (dd 1=8.4. 2.2 Hz, IH~, 7.81 (d ./=2.2 Hz, ] H).
~XAMPLE 136
Ethyl 3,4-methylenedioxy-d,l-mandelate
Step A: a-1'rimethylsilvloxy-3.4 methylenedioxyphenvl~lcetorlitrile
To a nitrogen fTushed magnetically stirred 3 L single neck
rou1ld bc~ttom fla.sk fitted with a nitrogen inlet was charged 2RSg (1.9
mol) of piperonal, 200g (2.0 mol) of trrmethylsilylyc.yanide, 0.2 g of
potassium cyanide, 0.2 g of 1~-crown-6 and 500 mL of methylene
chloride. The mixture was stirred at amhient temperature for 75 min,
2 ~ ~ 5 7 5 ~
W0 96/1~4905 r~ ,3;7
171
_
during which time the reaction exothermed to 35~C. A second charge
of 5 g of piperonal was added and the reaction stirred an additional 75
min. The reaction mixture was diluted with ether and 250 mL of
saturated sodiuim biGarbonate solution u!as added. The mixture wa.s
stirred for 20 min before partitioning. The organic Llyer was washed
with another 250 ml, portion of saturated sodiuim bicarbonate, twice
with brine (300 mL)~ dried with sodiurm .sulfate, filtered and
concentrated leaving 48g.6 g (481.4 g theoretical yield) of the title
compound as a pale yellow oil. This was used as is without purification
in the next step.
Step B: Preparation of ethyl 3~4-meth~/lenediox~-dd-mandelate
To a nitrogen flushed magnetically stirred 3 L single necl;
round bottom flask fitted with a gas inlet was charged the product
obtained from the previous step and I L of absolute ethanol. The
solution was cooled to 0~C and HCI gas gentiy bubbled through the
solution for 1 hr. After a few minutes the reaction solidified to a white
mass which was aged at room temperature ovemight. I L of methylene
chloride, and I L of water were added. The mixture wa.s shaken for ca
5 min dis.solving some of the white solid. l'he mixture wa.s deccmted
and the procedure repeated several more time.s until all of the .solid had
been dissolved. The layers were separated and the aqueous Iayer back
extracted once with methylene chloride. The combined organic layer
was washed with brine, dried with magnesium sulfate and filtered
through a pad of .silica . The solution wa.s concentrated, flu.shed witl
ether and diluted with hexanes. The white slurry was cooled to 0~C
then filtered. The cake was wa.shed with 1:2 ether/tlexalles followed by
hexanes. The product was dried affording 347.2 g of the title
compound as a white solid. A second crop of 24 g was obtained by
3 concentrating the mother liquors, bringing the total to 371.4 g (85.8~,
yield).
IH-NMK (300 MHz, Cl:)C13, ppm): ~ 1.22 (t, J=7.2 Hz, 3H), 3.41.(d,
J=5.6 1 Iz, IH), 4.10-4.31 (nl, 2H), 5.03 (d, J=5.6 Hz, IH), 5.94 (s, 2H),
6.77 (d, J=8.5 Hz, IH), 6.8~S-6.gO (m, 2H).
W0961049U~ 2 t 9 5758 . ~
- 172 -
F.XAMPLE 137
Ethyl o~-bromo-3,4-methylenedioxyphenylacetate
Step A Ple~ lati~ of ethyl o-bronlo-3,4-rllethylenedioxypilellyl-
acetate
To a nitrogen flushed 5 L three neck round botlc~nl fla.sk
fitted with a mechanical stirrer, a dropping funnel and a nitrogen inlet
was charged 433.8g (1.93 mol) of ethyl 3,4-methylenedioxy-d,l-
mandelate and 3.51, o f ether. The suspellsion ~lla.s caoled to 0-5~C and
a solution of 179~ ~0.66 mol) of PBr3 in 500 mL of ether was added
over a period of 30 min. The reaction was aged for ~.5 hr at 0-5~C
during which time, an additional 24.2 g (0.09 mol) of PBr3 wa~s added.
15 The solid initially plesent slowly dissolved leaving a clear yellow
solutioll. The reaction was quencl~ed by careful addition of 800 mL of
saturated sodiuim bicarbonate solution and 200 nlL o~ water. The
layers were separated and the aqueous layer e~itracted once witll ether.
The combined organic phase was washed once with satumted sodiuim
20 bicarbonate solution, lO~o sodium bisulfite solution. brinel dried with
magnesium sul~'ate, and filtered throu~h a pad of silica. The solution
was concentrdted to 507.6 g (91.4~~to ) of a pale yellow oil. Es.sentially a
single spot on TLC (silica-l:l Et2O/Hex)~ NMR indic~tei a small
annount of ether is present. This was used as is without purification in
5 the next step.
lH-NMR (300 l\lHz, CDC13, ppm)- ~ 1.27 (t, .I=7.2 Hz, 3H~. 4.1( -4.35
(m, 2H), 5.26 (s, lH), 5.96 (s, 2H~, 6.72 (d, J=8. Hz, IE-13, 6.94 (~dd,
J=8.0, 1.8 I-lz, IH). 7.11 (d,J=1.8 Hz, IH~.
3~ EXAMPLE 138
cc-(4-Carbometlloxy-2-~1-propylpheno1i;y)-3 ,4-methylenedioxy-
phenyl)acetic acid sodium salt
~ WO 96tO4!~05 ~ ~2 1 9 5 7 5 8 r~ e ~7
- 173 -
Step A: Preparation of ethyl oc-(4-carbomethoxy-2-n-propyl-
phenoxy)-3.4-methylenedioxyphenylacetate
To a 2 L three necked 24/40 round bottom flask e4uipped
with a mechanical stirrer, a nitrogen inlet and a dropping furmel was
first added a solution of 36.0 g (0.1~5 mol) of methyl 4-hydroxy-3~
propylben~oate dissolved in 700 ml. of anhydrous DMF follo~ed by
66.4 g (0.204 mol) of cesium carbonate. The flask was purged with
nitrogen and the reaction mixture was stirred at room te~ eld~ul~ ~or 2
hours. A solution of .~i8.5 g (0.204 mol) of ethyl (x-bromo-3.4-
methylenedioxyphenylacetate clissolved in 100 mL of DMF wa.s then
added via an addition i'unnel over a 15 minute period. The reaction
rmixture W.ls stirred an additional I hour at room telulu~.dtulc then
quenched by addition to 5 L of a 5% aqueous citric acid solution. The
organic product wa.s extracted into diethylether (2 x 4 L). the organic
layers were separated, wa.shed with saturated aqueous NaCI, dried
(MgSO4), filtered, .und evaporated. The residue was applied to a silica
gel (2 kg; 70-230 mesh) column equillibrated in 10% CH2C12-hexane.
The column was then eluted successively with 12 1. of lO~o CH2C12-
hexane, 12 L of 5~ EtOAc-hexane, 4 L of 7.5~o EtOAc-hexane, 12 1,
Of 1O~! FtOAc-hexane, and finally X L of 20~i' F.tOAc-hexane.
Combination of the purified fractions and evaporation in vacu(i afforded
76.3 g (74.2 theoretical) of the title compound as a pale yellow oil
which wa~s used without further purification in the next step.
Step B: Preparation of o~-(4-carbomethoxy-2-n-propylphenoxy)-
3~4-metnylenedioxvphenvlacetic acid .sodium salt
A I L 3 necked 24/40 round bottom flask e~uipped with a
mechanical stirrer, a dropping funnel, and a nitrogen inlet was charged
with a solution of 76.3 g 0.1X5 mol) of the semi-puriried product of
Step A dissolved in 500 mL of methanol. The flask was purged with
nitrogen, the stirrer was started. and 37 mL of a 5.0 N aqueous .solution
of sodium hydroxide was added over a 30 minute period via an addition
funnel. The reaction mixture was stirred at room temperature for an
additional 30 minutes at W}liCh point TLC analysis (CH2C12-h,leOH-
WO 9610490~ .' . 2 1 ~ 5 7 5 8 , ~
- 174-
NH40H 90:10 1 ) indicated that the starting material had been consumed.
The reaction mixture was adjusted to pH=4 with 6 N HCI, and the bull~
of the organic solvent was removed in vacuo. The precipitated c7rgallic
product and the aqueous layer were next partitioned betweell CH~CI ( I
L) and water (I L) which producecl a copious emulsion. Tbe reaclion
mixture was th~n aged overnight in a refridgerator which resulted in
crystallization of the organic product The eïystalline solid was
separated from the two phase mixture by filtration and washed with
CH2C12. The solid was slurried again in diethylether. filtered, waslled
~~ vvith hexane, and then dried in a vacuum to afford 65 g (85 3~to) of Ihe
title compound as a white crystallhle solid
I l Z-NMR (40Q l~,lHz, CD30D, ppm): ~ 0.~3 (t, J=7.2 Hz, 3H3, 1.62-
1.75 (m~ 2H), 2h3-2.70 (m, IH), 2.77-2.81 ~m, IH), 3 ~s4 (s, 3H3, :5.54
(s. I H~, 5.94 (s, 2H)~ 6.81 (d, J=7 6 Hz, lH), 6.89 ~d, J=9.2 Hz. I H),
7 O~i ~d, J=l .6 1 Iz, lH)~ 7.11 (hr s, IH3, 7.78-7 81 (m. 2H).
Microanalysis for C20H20o7Nao~7~-L25 H20.
Calc'd. C = 58 29; H = 5.~0: Na = 4.1
Found: C = 58.19; I-l = 5.17; Na = 3.93
I~XAMPLE 139
N-(4-is(~-propylbellzellesulfonyl)-a-(4-carbomethoxy-2-n-propyl-
phenoxy)-3 t4-methylenedioxyphenyl :~cet~ln~
25 Step A: Preparation of ethyl a-(4-~ u~ Ll.oxy-2-il-propyl-
phenoxy~-3.4-methylenedic xyphenylacetate
To a nitrogen flushed 5 L three neck round bottom flasli
fittecl witll a mecllanical stirrer, condenser, and a nitrogen inlet was
Ghargecl 326g (1.68 mol) of methyl 4-hydoxy-3-n-propvlbenzoate,
30 ~(}7.6 g (1.73 mol3 of ethyl o~-bromo-3,4-methyleneclioxyphenylacetate
from abo~ie. 23~g ( 1.70 mol) of anhydrous potassium carbonate, and
1.7 L of acetone. The mixture was refluxed with vigorous stirring f'c~r
9 hr The suspension was allowed to cool to ambient temperdture aod
stirred overnight. The mixture diluted with ~ L of ether, cooled to 0~C
W0 96/OJ905 2i } 9 ~i 7 5 8 T ~ 7
- 17~ -
and filtered through Super-CelR. The cake washed with ether and the
combined filtrate concentrated. l'he residue was redissolved in ether
and the organic layer washed with ollce with I N HCI~ saturated
sodiuim bicarbonate solution. lO~o sodium bisulfite solution, brine,
dried with magnesium sulfate, treated with charcoal ~md filtered
through a plug of silica. The pale yellov.~ solution was concentrated to
697.3 g (theoretical 678 g) of a thick yellow oil which was used without
purification in the next step. NMR was ccnsistent with the title
compound.
I H-NMR (300 I\IHz, CDC13, ppm): ~ 0.95 (t, J=7.3 Hz, 311), 1.17 (t,
J=7.1 Hz, 3H), 1.61-1.81 (m, 2H)j 2.63-2.80 (m, 2H), 3.85 (s, 3H~,
4.07-4.23 (rm, 2H), 5.58 ~s. lH), ~.96 (s, 2H), 6.71 (d, J=8.5 Hz, IH),
6.76 (d, J=8.0 1 Iz, IH), 7.02 (d,d, J=8.0, 1.7 Hz. I H), 7.05 (d, J=l .7 Hz,
I H), 7.79 (d,d, J=8.5. 2.2 1 Iz, IH), 7.~4 ~d, J=2.2 Hz, IH).
Step B: Preparation of a-(4-carbolIlethoxy-2-n-propyipheno7sy)-
3.4-methylenedioxyphenylacetic acid
To a nitrogen flushed 5 L 3 neck round bottom fklsk
e~luipped with a mPch,lni~sll stirrer, a dropping fulmel, and a nitrogen
inlet was charged with 697.3 g ( 1.68 mol) of the crude product of Step
A and 2 L of methanol. 500 mL of 5.0 N (1.5 eq) aqueous sodium
hydroxide was added over a 20 minute period via an addition funnel.
The reaction mixture was stirred at room temperature for an additional
I hr at which point TLC ~malysis (CH2C12-MeOH-NH4OH 90:1():1 )
indicated that the starting material had been consumed. The reaction
mixture neutralized with 420 mL of 6 N HCI, and the bulk of the
organic solvent was removed in vacuo. The residue was dissolved in
ether and extracted with a ~u~ ation of aqueous NaOH and NaHCO3
The aqueous layer was extracted with ether and the combined organic
layer was washed with aqueous Nal-lCO3 ~e aqueous klyer was
acidified with HCI and extracted with ether. The ether solution was
dried with magnesium sulfate, filtered, and concentrated tc afford
708.9g ~theoretical 62~ gi of the title compound as a viscous orange oil.
WO96/04905 . ~ 2 1 9 5 7 ~8
- 176-
NMR rndicated that it was ca 85'~o product by weight (I~'~rc ether) thus
pro~iding a corrected yield of 602.6 g (96.4~ yield)
I H-NMR (300 MHz, CD30D, ppm): ~ 0.93 ~t, J=7.4 1{~, 3H), 1.56-
1.77 (m, 2~ .6~ (t, 2H), 3.84 (s, 311), ~.57 (s, IH~, 5.95 (s. 2H)~ 6.4
(bs, lH), 6.71 ~d, J=8.5 Hz, lH), 6.79 (d, J=7.9 Hz, III), 6.99-7.05 (m,
2H~ 7.78 (d,d, J= 8.5, 2.2 Hz, IH), 7.82 (d, J=2.2, lH).
Step C: Plep,aratioll of N-(4-is~-propylbenzenesulfonyl~-a-(4-
carbomethoxy-2-~-propylphenoxy3-3 ,4-methylenedioxy-
phenylacetamide pota~siulll salt.
To~ a nitrogen ilu.shed S L 3 neck round bc1ttOm flas~
equiped with a mechanical stirrer, a dropping funnel, a condenser and a
nitrogen inlet was charged I L of THF and 3~û g (2.16 mol, 1.~2 eql c f
carbonyl diimidazole (CDI~. The mixture was heated to reflux and a
solution of 663.6 g ( 1.52 mol) c f acid trom Step B an~d I L of THF was
added dropwise over a period of 30 min. The reaction was monitored
for coversic)ll of the acid to the acyl imidazolidc bv NrMR. An
additional S5 g of COI was added over 45 min. 11le solutioll W.IS cooled
and 291 g (1.4~ mol~ 4-isl)-propylbenzenesulfonamide was ~Idcled as a
solid in one portion and the solutioll aged 20 min. DBli 2~0 mL (?34g,
1.54 moll W;iS added drop~;vise over 10 min resulting in an exotherm to
45~C. The reaction ~a.s aged at room temperatyurc for 3 hr then
concentraEed in vacuc . The residue was partitioned between ~.751, oi'
2.5 N I~CI and 3 L of ether. The aqueous layer was extracted with I L
25 of ether, and the combined organic layer washed with 2 N HCI ;md
saturated potassillm bicarbonate solution. The etherial layer was
l-d~ e,lt;d to a ~ L 3 neck round bottom flask equipped with a
mechanical stirrer. I L of aqueous pOtaSSiUIll bicarbonate solution was
added and the mixture stirred overnight at room te~ GIdlul~. The
30 resulting thick suspension wa,r; filtered and the cake ~h~ashed with 500 mL
of water followed by 1 L of ether. l'he product was then slurried in the
fulmel with additional ether and sucked dry yielding 741g of a tan .solicl
The scllid was recharged to a ~ L 3 neck round hottorn flask equipped
with rl mechanical st;rrer to which WEIS added I L of ethyl acetate alld
~¦ W0 96104905 ~ ' 2 ? 9 5 7 5 8 ~ . _., '7
- 177-
500 mL of saturated potassium bicarbonate .solution. The slurry was
,stirred at room lC~ JCld~Ul~ for I hr, diluted with 3 L of ether, and the
,~ slurried stirred at room temperature overnight. The yroduct was
filtered, washed with 500 mL of water and I L of ether and dried in
5 vacuo. The yield was 592 g of the title compound as a white crystalline
solid. A second crop of 47.6 g was obtained frc m the mother liqours
bringing the total to 639.6 g (74% of theory)
IH-NMR (300 ~IHz, CD30D, ppm): o 0.88 (t, J=7.4 El-, 3H),1.21 (d,
J=6.~ Hz, 6H), 1.52-1.66 (m, 2H), 2.50-2.76 lrll, 21-1), 2.90 (sept, J=6.9
llz, IH~, 3.X4 (s~ 3H),5.35 (s. IE~ .94 (s, 2H), 6.69 (d, J=8.6 Hz, 111)?
6.76 (d, J=85 Hz. l H), 7.()4 (m, 2H), 7.20 (d, J=, 8.4 Hz, 2H), 7.61 (dd,
J=8.5, 2.20, Hz, 111), 7.67 (d, J=~.4, 2H), 7.71 (d, J=2.1 Hz, 111).
EXAMPLE 140
N-(4-i.sc~-propylbenzeneslllfollyl)-a-(4-carboxy-2-n-propylphelloxy)-
3,4-methylelledioxyphenylacetamide dipotassium salL
Method A:
20 ~tep A: Preparation of N-(4-i~c~-propylbellzenesulfonyl)-o~
carboxy-2-/7-propylphenoxy)-3,4-methylelledioxyphenyl-
acetamide dipotassium salt
A mixture of 204 g (0.345 mol) of the product of Example
139, 420 mL of 1.0 N KOH in methanol and 500 mL of water was
25 .stirred at 60~C under a nitrogen atmo.sphere. After 3 hours TLC
analysis (90:10: I CH2C12-MeOH-NH4OH) indic,lted that ester
hydroly.si.s wa.s complete. The reaction mixture was cool ~slightly, then
concentrated on a rotary evapc~rator to a weight of 500 g. 2.5 L of
isopropanol was added alld the solution re-oncentrated to an oil. The
3() residue was flushed with an additional 2-3 L of isoprc~panol until
crystallizatioll began. The slurry wa.s concentrated to ca 1.5 L and
cooled to 30~C, filtered and wasllecl with 300 mL of IPA and 500 mL, of
ether. The product was dried affording 185 g of semi-pure title
compound a.s a white crystalline solid. A second crop of 17 g was
WO g61049U5 ~ ' ' 2 ~ 9 5 7 ~ 8 P~
- 178-
obt~ined from the filtrate after cooling. The material was recrystallized
as follows: 16~ g was dissolved in 3 L of absolute etharlol at retlux,
filtered hot, and the flask and funnel rinse~ with an additiollal 50() mL
of ethanol. 70 rnL of ~vater was added and the solution cooled to 0~C
5 over 2 hr then aged at 0~C ior 6 hr. The product was collected by
filtration, washed with ethanol, then air. The yield was 1~0.8 g of the
title compound as a white crystalline solid.
I H-NMR ~400 MHz, CD30D, ppm): o 0.88 (t, J=7.2 Hz. 3H~, 1. 2 I (d,
J=7.0 Hz, 3H), 1.22 ~d~ J=7.0 Hz, 3H). 1.56-1.63 (m, 2H), 2.52-2.59 ~m,
IH), 2.67-2.74 (m, lH), 2.91 ~sept, J=7.0 Hz, IH), 5.33 (s, I H), 5.92 (d,
J=l .2 H~, I H!t 5.93 (d, J=1.2 Hz, I H), 6.~2 (d, J=R.5 Hz. 11-1~, 6.7h (d,
J=X.5 l-lz, lH), 7.04 (d, J=7.5 Hz, lH), 7.05 (s, I H). 7.21 (d, J=R.5 1 Iz,
211), 7.64 (dd. J=2.0, 8.5 Hz~ IH), 7.67 (d, .1=8.5 Hz, 21-1), 7.73 (d, J=2.0
Hz, lH).
5 Microanalysis for C2~H27NSO~K2-3.4 H20.
KF = 9.00 (calc for 3.4 H20 = 9.04)
Calc'd: C = ~J,9.67; H = 5.03; N = 2.07; K = i 1.55; S = 4.74.
Found: C = 49.30, H = 4.95; N = 2.06; K = I I.g5; S = 4.~2
20 ~lethod B:
Step A Preparat;on of N-(4-i.s~v-propyl~-~n7en~ iollyl)-o~-{4-
carboxy-2-n-propylp'ileno~y)-3 ,4-methylenedioxyptlenyl-
ace~.amide
A mi~ture of 20~ g (0.345 mol i of the product clf E~ample
25 139, 425 mL of 1.0 N KOH in methanol and 500 mL Cli' water wasstirred at 60~C under a nitrc~gell atmosphere. After 1.75 hours l'LC~
analy.sis (90:1~:1 CH2C12-MeOH-NH4O}I) indicated that ester
hydrolysis was cormplete. The reaction mixture wa.q cooled slightly,
then cc~ncentrated on a rotary evaporator. The concentrate was
30 acidified with 400 mL of 2 ~ HCI and extracted first with 6 L oi ether-
EtOAc-CI-12C12 4:1:1, then with 3 L of 1 :2 EtOAc-CH2CI The
organic layers were washed with 250 mL of 2N HCI, then with 3 X 500
mL of water, dried with magnesium sulfate, filtered, and concentrated,
during whicll~ the product began to crystallize. The solution ~IS
9 5 7 5 8
~ WO 96104905 ' P~
- 179 -
concentrated to a white sluury of ca 750 mL, diluted with I L of
hexanes, cooled to 0CC, aged I hr then filtered. The product wa.s air
dried affording 170.0 g (919~ yield) of the title compound as a white
crystalline solid.
IH-NMR (400 MHz, CD30D, ppm): o 0.8~ (t, J=7.2 Hz, 3H), 1.21 (d,
J=7.00H7" 3H), 1.22 (d, J=7.0 Hz, 3H), 1.56-] .63 (m, 211), 2.52-2.59
(m, IH), 2.67-2.74 (m, IH), 2.91 (sept, J=7.0 Hz, lH), ~.33 (s, IH),
5.92 (d, J=1.2 Hz, I H), 5.93 (d, J=1.2 Hz, lH), 6.72 (d, J=8.5 Hz5 IH~),
6.76 (d, J=R.5 Hz, I H), 7.04 (d, J=7.5 ~z, I H), 7.05 (s, I H), 7.21 (d,
0 J=8.5 ~tz, 2H~, 7.64 (dd, J=2.0~ ~.5 Hz, I H), 7.67 (d, J=~.5 Hz, 2H),
7.73 (d, J=2.0 Hz, lH).
Microanalysis i'or C28H29N08S
Calc'd: C = 62.33; H = 5.42; N = 2.60; S = 5.94.
Found: C = 62.15; H = 5.4~; N = 2.54; S = 5.99
Step B: Preparation of N-(4-iso-propylbenzenesuifonyl)-a-(4-
carbox y-2-n-propylphenoxy)-3,4-methylenedioxyphenyl-
acetamide dipota.ssium salt
15g.7 g (0.296 mol) of acid fro~n Step A was suspended in
20 3 L of absolute ethanol. To this was added 590 mL of 1.0 N KOH in
metha[lol over 20 min while simultaneously warming the mixture to
50~C. The clear and colorless solution was cooled to 0~C during wllicl
it wa.s seeded with 20 mg of the title compound. The suspension was
stirred for 2 hr at 0~C, I L of ether was added and the suspension
25 filtered. 1~e .solid was dried affording 16~.4g of the title compound as
a white crystalline solid. A second crop Or 22.3 g of comparable quality
material was obtained by concentrating the ML to ca. I L, diluting with
I L of ether, filtering, and recrystallizing the solid (27 g) so obtained
from 200 mL of 98C~'C! ethanol. Thus affording after drying a total of
30 190.7 g (96.8~ yield corr!d for water content) of the title compound.
Microanalysis for C2XH27K2No8s-2.75 H O.
KF = 7.45 (calc for 2.75 H2O = 7-44)
Calc'd: C = 50.55; H = 4.92; N = 2.11; K = 11.75;
Found: C = 50.69; H = 4.56; N = 2.05; K = 11.20; S = 4.71
7 5 8 ; ~
WO 96/04905 , ~ , . P~
EXAMPLE 1~i3
1~-14-iso-propylb~ e~ulfonyl)-a-(4-carboxy-2-n-prc)pylphenoxy)-
3.4-methylenedioxyphenyl.~et~mi~f~ dipotassium salt
Step A: Preparatio:n of N-(~-iscj-propylbenzenesulfonyl~-a-(4-
carbc~xy-2-n-propylphenoxy)-3,4--nettlyleIlediox~ phenyl -
acetamide di-S-(-)-a-methvlbe.nzylamine~ salt
32.4 g of the acid from Example 139 wa.s dis.solved in 50()
nlL c~f isop:ropallol, and 15.5 mL of S -(-)-cx-meti-~ Ibenzyl amine was
added. The solution was allowed to stand at room ~emperature
overnight. The mixture was filtered and the cake washed with a small
amount of isopropanol. The solid was recryastallized 4 more times
from i.sopropanol affording 45 of the title compound.
Step 1~: Preparation of hl-(4-iso-propylbe:nzenesulfollyl j-a-~4-
carboxy-2-n-propylphenoxy)-3,4-methylenedioxyphenyl-
acetamide d;potassium salt
'I:'he c~-metl1ylbellzylamirle salt :from step A wa.s partitiol1ed
between ethyl acetate and NaHSO~i, dried with h~gSO~, filtered .ind
concf~ntr~t~-l The residue was dissolved in methanol-water at roc)m
)ela~ul~, and basicified with ca. 12 mL of 1 N NaOII iu methanol,
diluted with water and filtered through a 0.45 micron. The solution
was desalted and purified on a Waters Millipore Delta Prep 3000 liquid
chromatograph equipped with an MlOVO Prep-Pak module cont:ai~ g a
47 x 300 nm1 Delta-Pak Cl~ 1511m IOOA colurnu cartridge. Two
solvent resevoirs were en-ployed: solvent system A (95-5 water-
acetonitrile), and sol~ent system B (5-95 water-acetol1itrile), arld the
column effluent was monitored simultaneously at 210 and 2RO nm with
a Waters model 490 W-visible detector. Each fraction was pump-
injected onto the c.olumn and desalted by elution (50 mL/mill) witll
several colun1n volumes of solvent system A. A gradient elution wa.s
then begun which llad as initial conditions lOO~o solvel1t sy~stem A~
W096/04905 "~ 21~758 r~", ~ ~
- 181 -
solvent sy.stem B and reached after 30 minutes 50% solvent system A-
50% solvent system B, and the fractions were collected with an ISCO
Foxy 200 fraction collector. The purified fractions were combined in
round bottom flasks, frozen in a -78~C dry ice-acetone bath, and
5 Iyophilized. Combination of the purified product afforded _ of the
title compound as a white Iyophilized powder.
IH-NMR (400 MHz, CD30D, ppm): o 0.88 ~t,J=7.2 Elz, 3H), 1.21 (d,
J=7.0 Hz, 3H), 1.72 (d, J=7.0 Hz, 3H), 1.56-1.63 (m~ 2H), 2.52-2.59 (m,
IH j, 2.67-2.74 (m, IE-I), 2.91 (sept, J=7. Hz, I H), 5.33 (s, I H), 5.92 (d,
o J= 1.2 Hz, I H), 5.93 (d, J= 1.2 Hz, IH), 6.72 (d, J=8.50}-lz, 111), 6.76 (d,
.1=8.5 Hz, IH), 7.04 (d, J=7.5 Hz, IEI), 7.05 (s, IH), 7.21 (d, J=8.5 Hz,
2H), 7.64 (dd, J=2.0, 8.5 Hz, IH), 7.67 (d,./=8.5 Hz, 2H), 7.73 (d,.l=2.0
Hz, IH).
Microanalysis for C28H27Nso8K2-H2o~
Calc'd: C = 53.06; H = 4.61; N = 2.21; K = 12.34.
Found: C = 52.81; El = 4.56; N = 2.17; K = 12.02.
EXAMPLE 142
20 N-(4-i~so-plopylbenzelles~llton~ 2-[[4-[N-[2-(carbethoxy)et
carbamoyl l]-2-propylphenoxy]-2-(3 ,4-metllylenedioxyphenyl)zl~ e
The titled compound was prepared using procedures
similar to those de.scribed in Example 82 except that ,B-alanine ethyl
25 ester (liberated from the corresponding hydrochloride in situ) was the
amine .starting material. The crude product was flash chromatographed
o~,~er silica gel ~gradient elution, 1-5~, Meolllcl-l2cl2) Eo give the
desired product as a white foam in 78~o yield; homogeneous by TLC
(10190 MeOH/CH2C12); mp 167- 168~C; MS (ESI) 639 (M+H)+.
30 Analysis (C33}13~N2OgS-0.75H2O):
Calcd: C, 60.78; H, 6.10; N, 4.30
Found: C, 60.69 H,5.88 N, 4.30
I H NMR (400 MHz, CD30D, ppm): ~ 8.36 (t, 111~ J = 5 Hz), 7.72 (d,
2H, J = 8.4 Elz), 7.58 (d, lEI, J = 2.3 Hz)~ 7.46 (dd, IH, .1 = 8.5, 2.3 Hz),
WO96/04905 .~ t ~ 575~ ,s~l ~
~, ~ ,. .,;,
- 182-
7.30 (cd, 211 J = R.4 Hz), 6.93-6.98 (m, 211), 6.77 (d. 11~, J = 7.9 }Iz), F
6.65 (d, IH, J = 8.7 Hz), 5.95 (s, 2H), 5.47 (s, IH), 4.13 (q, 2H, J = 7.1
Hz), 3.57-3.63 ~m, 2H), 2.g3-2.98 (m, IH), 2.68 ~m, 2H), 2.62 (t, 2H, J
= 6.9 Hz), 1.60 ~m, 2H), 1.21-1.25 (m, 6H), 0.88 (t, 3H. J = 7.4 llz).
EXAMPLE 143
N-(4-iso-propylh.on~n~ lfonyl)-2-[14-[N-(2-carboxyethyl)carbamoyl] 1-
2-propylphenoxyl-2-(3,4-methylenedioxyphenyl)acetamide
The product from Example 142 was saponifïe{l (e~cess
NaO1 i in MeOl-1, 60~C, 4 h) to give the titled product as a white solid in
quantitative yield; mp Ig9-201~C, MS (ES1) 611 (I\l+M)~.
Analysis (C311134N2O95 0.4M20):
Calcd: C, 60.25; M, 5.67, N, 4.55
Found: C, 60.49 H, 5.48 N, 4.18
I H NMR (400 MHz, CD3OD, pprm): ~ 7.76 ~dd, 2M, J = 1.8, 8.5 1-17.),
7.5R (d, lH, J = 2.3 Hz), 7.46 (dd. IH, .1 = 2.4, 8.6 HY.), 7.3~ (d, ~H J =
8 4 Hz), 6.~2 I'dd, IH, J = 1.7. 8.0 Hz), 6.R7 (d, IH, J = 1.6 llz), 6.7R
(d~ IH, J = 8.0 H~). 6.56 (d, 11-1, J = ~.7 Hz)? 5.g6 (s, 2H), 5.51 ~s, IH),
3.~R (m~ 211), 2.95 (m, IH), ~.68 (m, 2H), 2.61 ~t, ?H, J = 7.'' Hz
(m, 21-1~), 1.21-1.25 (-m, 6M), 0.88 (t, 3H, J = 7.4 llz).
2 s EXAMPLE 144
N -(4-iso-propylbenzenesulf'onyl)-2-[ [4-~N-(2-carbamoy]ethyl)-
cal bamoyl] ] -2-propylphenoxy~ -2-~3,4-methylenedioxyphenyl)acetamide
The titled compound wa.s prepared using procedllres
similar tc those described in Example 82. The crude product ~hras fla.sh
chromatographecl over silica gel (gradient elution, 2-10~o
~leO~llCH2C12) to yield the desired product as a white foam in 42
~ W0 96~04905 ~ 219 5 7 5 8 r~
- 183 -
~ yield;homogeneousbyTLC(10190MeOII/CH2C12~,mp 110-112~C;
MS (ESI) 610 ~M+H)+.
Analysi~ (C31 H35N3O8S 0.75H20 i:
Calcd: C, 59.73; H, 5.90; N, 6.76
Found: C, 59.74 H, 5.61 N, 6.62
I H NMR (400 MI-lz, CD30D, ppm): o 7.77 (d, 2H~ J = 8.3 Hz),7.59 ~d,
IEI, J = 2.3 Elz),7.46 (dd, IH, J = 2.4, 8.6 Hz), 7.36 (d, 2H J = 8.7 Hz),
6.92 (dd, IEI, J = 1.5, 8.0 Hz), 6.87 (d, IH, J = 1.6 Elz), 6.78 (d, IH, J =
o 8.1 Hz), 6.58 (d, IH, J = $.6 }Iz),5.97 (s, 2H),5.50 (s, IH), 3.. S9 (t,21-I~
J = 7.0 Hz), 2.98 (m, IH), 2.68 (m, 2H), 2.51 (t, 2H, J = 6.9 Hz), 1.60
(m, 2H), 1.21-1.28 (m, 6H), 0.89 (t, 3H, J = 7.4 H~).
EXAMPLE 145
N-(4-iso-propylbenzenesulfonyl)-2-~4-1N-~2,2,2-trifluoroethyl)-
carbamoyll-2-propylphenoxy]-2-(3,4-methylenedioxyphenyl)acet:alllide
The titled compound wa.s prepared using procedures
similar to those descrihed in Example 82 except that trifluoroettlylamine
was used as the amine starting material. The crude product was flash
chromatographed over silica gel (gradient elution, 1-4~i~o
MeOH/CI-12Cl2) to give the desired product as a white foam in 79~k,
yield; homogeneous by Tl,C (5195 MeOH/CH2C12), mp I lU-112~C; MS
(ESI) 621 (M+H)+.
Analysis (C30H31 F3N2o7s 0~5H20):
Calcd: C, 57.22; H, 5.12; N, 4.46
Foulld: C, 57.33 H,4.87 N, 4.52
3c I H NMR (400 MHz, CD30D, ppm): o 7.71 (d, 2}I, J = 8.3 l lz),7.63 (d,
IH~J=2.4Elz),7.52(dd, lH.J=2.2,8.4Hz),7.29(d,211J=8.3Hz).
6.97 (m, 2H), 6.77 (d, I H, J = 7.9 Hz), 6.68 (d, lH, J = 8.7 Hz), 5.96 (s,
2H), 5.47 (s, IH), 4.05 (dq, 2H, J = 3.0, 9.2 Hz), 2.92 (m, III), 2.6R (m,
2EI), 1.6] (m, 2H), 1.19-1.30 ~m, 6H~, 0.89 (t, 3H, J = 7.3 Mz).
~o s6A74sns . ~ , 2 1 ~ 5 7 5 8 P~lll
- 184-
E~XAMPI r 146
N-(4-iso-propylbenzenesulfonyl~-a-(4-N-t-butyklxycarbonyl-
aminosulfonyl-2-n-propylphenoxy)-3.4-methylenedioxvphenylacetamide
Step A: Preparation of methyl o -(4-i'~ -butyloxycarbonylamino-
sulfonyl-2-n-propylphenoxy)-3 ~4-rmethylenedioxyphenyl-
acetate
To a stirred solution of 76 mg tO.lP,7 mmol) of methyl a-
(2-n-propy 1-4-~alrlillc)slllfonylpheno~y)-3 ,4-methylenedioxyphenylacetate
(the product of Lxample 100 Step C), 29 uL (0.206 mmol) of
triethylamine and 2.3 mg (0.0187 rnmol) of DMAP llydrocllloride inl
ml, of methylene chloride was added 47 mg (0.215 mmol) of di-te.rt-
butyl-dicarbor~te. After 1.5 hours, TLC analysis ~5~! methanol /
methylene chloride) indic ated that the couplillg vvas complete and the
reaction mi~ture wa~ diluted with eth~ l acetale, p~rtitiollecl with water,
washed another time with water, and wa.i}lcd Wil}l brine. The organic
layer uas then driecl o~er magnesiulll ~ult'ate, fiitered, .uld the filtrate
2G concentrated in l~acuo and dried on vacuunl to afford 95 m~ (lOO''~o) of
the titie compu~md as an amorphous solid.
1 H-NMR (400 MHz, CDC13,ppm) o 0.97 (tJ=7.40 1~z,3H~, 1.35 (~,9H),
1.62-1.75 (m,2H), 2.66-2.79 ~m,2H~, 3.71 (s,3H~, 5.60 (s,ll-l), 5.98
(s,2H), 6.75 ~d,J=9.2 Hz,lH), 6.81 (dJ=8.0 Hz,l l-l), 6.99-7.04 ~m,2H),
7.74-7.76 (m,2H).
Step B: Preparatioll of meth~l a-(4-/V-~-butyloxycarbollylamin(7-
sulfonyl-2-n-propylpheno~y)-3,4-methylelledic)xy-
p~henylacetic acid
3~7 To a stirred solution of 95 mg {0.187 mrnol) of the prclduct
of Step A in 0.75 mL of methylene chloride and 0.75 mL of methanol
wa.~ aclded 45 aL (0.225 mmol) of 5N sodiurn hydroxide. Afler S
hours, TL(~ analysis (5~ methanol / methylene chlc7ride) indicated slo~
ester hydrolysis and an additional 45 uL (0.225 mrnol) of 5N sodium
W0 96~0490!i ~ 2 ~ 9 5 7 5 8 I ~r~ J
- 185-
hydroxide was added. The reaction mixture stirred 2 days, at whic}
time TLC analysis (5% methanol / methylene chloride) indicated the
ester hydrolysis was complete. The re.tction was extracted with ethy!
following acidification to pH4-5 with 10~~, citric acid and dilution with
water. The extract was washed with brine, dried over m~gn~silln~
sulfate, filtered and the filtrate was evaporated in vacuo and dried to
afford 75 mg (82%) of the title compound as an amorphous solid.
lH-NMR (400 MHz, CD30D,ppm) o 0.96 (t,~=7.40 Hz,3H), 1.34
(s,911), 1.62-1.75 (m,2H), 2.75 (t~/=7.40 H_,2H), 5.78 (s,lH), 5.97
o (s,2H), 6.~4 (dJ=8.00 H_,IH), 6.99 (d,J=9.20 Hz,lH), 7.06-7.10
(m,2H), 7.72-7.74 (m,2H).
APCI-MS 1721e = 51 1 (M+NH4).
Step C: Preparation of N-(4-i.io-propylbenzenesulfonyl)-lx-(4-N-f-
butyloxycarbonyl-aminosulfonyl-2-n-propylphenoxy)-3,4-
methylenedioxyphenylacetamide
A solution of 68 mg (0.138 mmol) of the product of Step B
and 34 mg (0.207 mmol) of carbonylfliirnid"7- 1e in 0.~ mL of dry
tetrahydrofuran was refluxed using a heated oil bath. After 2 hours,
'I'LC analysis (20% methanol / methylene cl-loride) indicated the desired
intermediate had formed. The reaction mixture was then cooled to
room temperature and 41 mg (0.207 mmol) of dry 4-iso-
propylbenzene.sulfonamide and 31 uL (0.207 mmol) of DBU were
added. The reaction mixture was refluxed again for 25 minutes,
allowed to cool, after which TLC analy.sis (20~,~o methanol I methylene
chloride) indicated the desired product had been fonned. The reaction
mixture was poured into 10% citric acid and extracted with ethyl
acetate. The extract was washed with brine, dried over magnesium
sulfate, and evaporated in vacuo. The residue was purified by si]ica-gel
flash chromatography eluting first with 3~r~c, methanol / methylene
chloride .md then with 5'J~o methanol / methylene chloride. The mixed
fractions cont,~ ing both the title compound and the des-~-
butyloxycarbonyl version of the title compound were combined,
evaporated and used in Example 147. The purified t'ractions were
WO 9610490~? ? 9 5 7 5 ~ Jr 7 S
- 186 -
combined and evaporated to afford 11 mg ( l 29;'o) Of the title compound
as an amorphous solid.
IH-MvlR (400 MHz, CD3OD,ppm) ~ 0.~8 (t,J=7.40 Hz,3H), 1.24 ,,
(d,3=7.20 Hz,311)~ 1.25 (d?J=7.20 Hz,3H), 1.3~ (s,9H), 1.56-1.68
S (m,2H), 2.60-2.69 ~m,l I-I), 2.70-2.77 (m,lH), 2.95 (sept., J=7.20
Hz,lH), 5.46 ~s,lH). 5.95 (s,2H~, 6.77 (d,J=8.4Q Hz,lH), 6.81 (dJ=8.40
I-lz,l 1{), 7.00-7.03 (m,2H), 7.27 (drl=8.00 Hz,2H), 7.60 (dd,J=2.4(1, ~¢.4()
Hz,lH), 7.64 (d?J=8.00 Hz,2H~, 7.6X (dJ-2.4 Hz.lll).
F,SI-MS m/c~ = 697 ~M+Na).
EXAMPLE 147
N-~4-iso-propylbenzenesulfonyl)-o~-~4-N-f-batylc~xycarbonylamino-
sul fonyl-2-n-propylphenoxy)-3 ,4-rnethylenedioxyphenylacetamide
A solution of 1~ m~, iO.û267 rnmoli of mixed fractions
from Step C of Example 146 in 0.8 mL of dimeh1ylsultoxide Wah he~lted
to reflux for ~ minutes ~mcl cooled to room temperature. After the
20 lleatin~. TLC analysis (205~o methanol I methylene chloride) indicated
them1al deprotection was complete. The reactic~n was diluted with 6
mL of water and filtered through a 0.45 mrn filter disc. The filtrate
was purified U.sil1g a Waters 600E HPLC system with a 9.4 ~ 250 n~n 5
mm Zorbax-RX C8 at 40~C eluting at 5.0 mL/min first usin~ 100'~
25 (95 5 water-acetonitrile) with 0.1 ~Yo TFA for 12 minutes and then
switching 65~io A (95-5 acetonitrile-water) 35~o B (95-5 water-
acetonitrile) each with 0.1% TFA where the column effluent was
monitored simultaneously at ~10 and 277 nM with a Waters model 490
UV-visible detector. The purified fractions were cormbined in a round
30 bottom flasl~, frozen in a -78~C dry ice-acetone bath and lyophylized to
affold 10.7 mg (7]'Yo) of the title comyound as a white lyophilized
powder.
I l I-NMR (400 M~Hz, CD30D,ppm) ~ 0.90 (t,J=7.20 Hz,3H)? I .2G
(d,J=6.80 Hz.3H), 1.27 (dJ=6.80 Hz,3H), 1.54-1.6~ (m~2TI), 2.60-2.72
~ WO96/04905 ~ . F~IJ~ ,; 7
2~9~75~
- IX7-
(m,21~ 3.00 (sept., J=6.80 Hz,IH), 5.54 (s,1Hj,5.97 (s,~H), 6.65
(dl=~.40 Hz,1H). 6.78 (dJ=8.00 Hz,lH),6.84 (d,J=1.60 Hz,lH),6.91
(dd,J=1.60, R.00 Hz,lH), 7.37 (d,J=8.40 Hz,2H), 7.53 (dd,J=2.40, 8.40
Hz,lI-l),7.66 (dJ=2.4 Hz,l~),7.76 (dJ=8.40 Hz,2H).
5 ESI-MS m/e = 575 (M~H).
EXAMPLE 148
N-(~-iso-propylbenzene.sulfonyl)-a-(4-(N-methylacetamido
aminosulfonyl)-2-n-propylpheno~y)-3,4-methylenedioxyphenyl-
acetarnide
~: Preparation of methyl a-(4-(N -methylacet~rnido-
arninosulfonyl)-2-n-propylprlelloxy)-3,4-methylene-
dioxvphenyla~;etate
To a stirred solution Or 500 mg (1.23 mmol) of methyl o-
(~4-aminosulfonyl-2-n-pr( pylphenoxy)-3,4-methylenedioxyphenylacetate
20 (the product of Example 100, Step C) in dry dimethylfclrmllmide (2
mL) wa.s added ~()ul. (1.35 mmol) of methyl isocyanate l'ollowed by 6
mg (0.06 mmc l) of cuprous chloride. The reaction mixture W;IS stirred
overnight, after which TLC analysis (5 ~, methanol I methylene
chloride) indicated the reaction had not proceeded to completion.
25 Subsequently, an additional 80 uL (1.35 mmol) of methyl isocyanate, 6
mg (0.06 mmol) of cuprous chloride as well as 342 uL (2.46 mmol) of
triethylamine was added and the reaction mixture wa.s again stirred
overnight. Tl.C analysis (5 ~Jc methanol / methylene chloride) indicated
the reaction had proceede(i to completion. The reaction nlixture was
30 poured intc~lN IICI and extracted with ethyl acetate. The extract was
washed with brine, dried over magnesium sulfate, filtered and the
filtrate concentrated in vaullo and dried to afford 57() mg (100%) of the
title compound as an amorphous solid.
W096/(~ 2 1 95 758 1~ u~ 7--
- 188 -
NMR (400 MHz, CD30I:),ppm) o 0.9fi ~tJ=7.20 Hz,3H). 1.64-1.77
~m.2H), 2.65 (s,3H~ 2.74 (tJ=7.00 11z,2H3. 3.71 (s.3H3, 5.88 (5,11H).
5.98 ~s,21-1), 6.~5 (d~/=8.00 Hz,lH~, 6.95 (d,J=8.40 Hz,11~1), 7.04
(dd,J=1.60, 8.40 Hz,lH~. 7.07 (d~l=1.60 Hz,lH3, 7.72-7.76 (m,21-1~.
ESI-~IS m~e = 464 (M+13.
Step E~: Preparation of oc-(4-(h -methylacetamidoaminosulfonyl)-2-
n-propylphenoxy)-3.4-methylenedioxvphenyhlcetic acid
To a stirred solution of 570 mg (1.2~ mmol-l of the product
of Step A in G mL of methanol was added 54Q uL (2.71 mmol) of 5N
sodium hydroxide. The reaction mi.xture was allowed to stir overrligllt
after WlliCIl TLC analysis (90:10:1 c:hlc)rof'oml I medla1lol / acetic acid)
indicated the saponification had proceeded to completioll. Ttle reaction
mixtule was acidified to pH=2 using 6 N IICI, poured into water, and
extracted with ethyl acetate. The e~tract was dried over rma~llesium
sulfate, filtered Imd evaporated in l~acuo to give the crude product.
Purification of the cmde product by silica-~el f~ash chromatograpll.y
U.sillg (92:7:1 chloroform / methanol I acetic acid) affcrdecl 3X4 mg
(70%~ of the t;tle compc und as an amorphvus solid.
I E-1-NMR (400 MHz, CD3OD,ppm) ~ 0.96 (t,J=7.40 H~.3H), 1.62-1.77
(m,2H), ~.65 (~3H~, 2.74 (tJ=7.6V Hz,2H), ~.76 (s,lH~, 5.98 ~s,2H),
6.85 (d,J=8.()0 H~,11-1j, 6.95 (d,~=X.40 Hz,lH)~ 7.06 (dJ=1.60 Hz,ll-l),
7 .0~ (dd,J= 1.60, ~ .00 I-lz, ] H~, 7.72-7.75 (m,2H) .
Cl-MS m/~ = 538 (M+l ).
Step C: Preparation of N-i4-iso-propylbenze1lesulfollyl3-c)c-(4~
methylacetarnido-aminosulfonyl~-2-n-propylphenc xy)-3,4-
methvlenedioxvpllellvlacetamide
A solution of 384 mg (0.853 mmol3 of the product of Step
B and 20~ mg (1.28 mmol) of carbonyldiim~ 7nle in 2 mL of dry
tetrahydrofuran s~a.s refluxed 2 minutes by placing the reaction nlixture
into a preheated oil bath. After brief refluxing and gas evollltion, TLC
analysis (90:1():1 chloroforrn / methanol I acetic acid) indicated that the
desired intermediate had formecl. The reaction mixture was then cooled
~ W0 96/04905 ~ 2 ~ q ~ 7 5 ~ r~
- 189-
to room le-l~pe~aLu/t; and 255 mg (1.28 mmol) of dry 4-i.so-
propylbenzenesulfonamide, 10 mg (0.085 mmol) of DMAP followed by
191 uL (1.2R mmol) of DBU was added. The reaction mixture was
refluxed for 3 minutes, allowed to cool and stir at room te,,,pc,dLu,~ I
hour after which TL,C analys;s ~96:3:1 chloroform / methanol / acetic
acid) indicated the desired product had been formed. The reaction
mixture was poured into 10% citric acid and extracted with ethyl
acetate. The extract wa.s washed with brine, dried over m~nPiillm
sulfate, and evaporated in vaclf o. The residue was partially purified by
o silica-gel fla.sh chromatography eluting fir.st with ethyl acetate and then
with (92:7:1 chlorofonrl I methanol / acetic acid). The purified
fractions were combined to afford 48 mg (9~o) of the title compound.
Ilpon standing, material precipitated from the ethyl acetate fractions to
afford an addition 55 mg (IO~?o) of the title compound. The remclinder
of semi-purified material was combined and puril'ied using a Waters
Delta Prep 3000 HPI.C by applying the residue in 6 ml total volume
(4.5 mL methanol and 1.5 mL water) to an M1000 Prep-Pak module
containing a 47 x 300 mm 15 uM DeltaPak Cl~ column and eluting~
isocratically at 50 mL/;nin using 60~o A (95-5 acetonitile-water) and
40~, B (95-5 waler-acetonitrile) each with 0.15~c! TFA. The colullm
effluent ~.s monitored simultaneously at 210 and 277 nM with a Waters
model 490 VV-visible detector and the purified fractiolls were
combined in a round bottom flasl~, frozen in ;1 -78~C dry ice-acetone
hath and Iyophylized. 'I'he HPLC purified Iyophylizatel55 mg (29%)~
the silica gel purilïed amorphous solid 48 mg (99~) and the precipitated
amorphous solid 55 mg (105~o) were combined to afford 258 mg (48%'~
of the title ~ompound.
IH-NMR (400 MHz, CD3OD,ppm) o 0.89 (t,J=7.40 Hz,3H), 1.26
(d,J=6.80 Hz, 3H), 1.27 (d,J=6.80 Hz, 3H), 1.55-1.65 (m~2H), 2.63-2.70
(m,5H), 3.00 (sept.J=6.80 Hz, IH), 5.56 (s,lH), 5.97 (s,2h), 6.67
(d,J=~.40 Hz. IH)7 6.79 (d~l=7.60 Hz, 11-1), 6.84 (dJ=1.60 Hz, IH), 6.91
(dd~l=1.60. 7.60 Hz, 111), 7.38 (dJ=8.40 Hz~ 211), 7.62 (dd,J=2.40. 8.40
Hz, lH), 7.71 (d~l=2.40 Hz, IH), 7.77 (d~l=8.40 Hz. 211).
Cl-MS mle = 632 (M+1).
WO 96/0490~ ' 2 1 9 5 7 5 8
.
- IgO-
Step D: Preparation of /\~-~4-iso-propylbenzene.sulfonyl)-a-(4-~N-
m~thylacetamido-aminosult'onyl ~-2-n-propylphenoxy)-3,4-
methylenedio7;vphenylacetamide dipotassium salt
To a solution Or 250 mg (0.3~6 rnmol) of the product of
Step C in 3 mL of methanol was added 1.~8 mL (1.58 mmol) of a IN
potassium hydloxide rn methanol solution. l~e reaction mixture was
.stirred 15 mim~tes at RT, diluted with 7 mL of water and filtered
through a 0.45 uM filter disc. The filtrate WtdS purif1ed using a Waters
Delta ~Prep 3000 HPLC by applying the compound in a 15 ml total
~olume (R mL methanol and 7 mL water) to an MI000 Prep-Pak
module containing a 47 x 300 mm 15 u~l DeltaPak C l ~ colun~n and
elutin~ at 50 rnL/min first using lOO~o B (95-5 water-acetc~rlitrile) for
10 minutes and then a 30 minute linear gradient to 60~. A (95-5
acetonitile-water) and 40~ B (95-5 water-acetonitrile). The column
effluent was monitored iimlllt:7ln~?ollsly at 210 and 277 nM with a Waters
model 490 UY-vi.sible detector and the purified fr~ctions were
combined in a round hottom flask, fro7el1 in a -7~~C' dry ice-acetoIle~
2~3 bath and Iyophylized to afford 221 Illg (79aro) of the title compound as a
white Iyophillzed powder.
I H-NMR ~400 MHz, (:~D30D,ppm) ~ 0.89 (t~/=7.10 Hz~ 3H), 1.22
(d 1=6.80 Hz. 3H), 1.23 ~d,J=6.80 I-lz, 3H), 1.53-1.68 (m~211~, 2.54-2.6
(m,41-1), 2.67-2.74 (m,lH~, 2.91 (sept., J=6.8U Hz, lH~, 5.34 (s,ll-l),
~.92 (s,2H~, 6.73-6.79 (m,2H), 7.00-7.02 ~m~2H')~ 7.20 ~dl =8.00 llz,
2H), 7.57 (ddl=1.80, 8.60 Hz, lH3, 7.61-7.64 (m.3H).
F,SI-MS mic = 701 (M+l ).
EXAMP~E 14~
~IV-(4-i.s~/-propylbenzenesulfonyl j-oc-(4-(methylsul~onyklmino-1~'-
oxo1nethyl)-2-~7-propylphenoxy)-3,4-meti1ylenedioxyphenylacetamide
dipotassiurn salt
~ W096104905 ~- ' ' 21 ;~575~ 7
~: Preparation of N-(4-i.so-propylbenzellesultonyl )-a-(4-
(methyl~sulfonykLmino-N'- I -oxomethyl)-2-n-propyl-
phenoxv)-3 .4-meth~ienedioxvphenylacetamide
A solution of 146 mg (0.271 mmol) of N-(4-iso-
5 propylbenzelle.sulfonyl)-ot-~4-carboxy-2-n-prc pylphenoxy)-3,4-
methylenedioxyphenylacetamide (t'ree acidic form of the product of
Example 58) and 66 mg (0.406 mmol) of carbonyldiimidazole in I mL
of dry tetrahydrofuran was refluxed t'or 1.5 hour.s. The reaction
mixture was cooled to room temperature ancl 39 mg (0.406 mmol) of
methallesulfonamide and 101 uL (0.667 mmol) oi' DBU were added and
the mixture was reiluxed a~aill. The reaction progless was nlonitored
by analytical HPLC analysis u.sin~ a Water.s 60()E HPLC' .systenl with a
4.6 x 250 mm Sum Zorbax-RX Cl~ at 40C~C ~nd eluting isocratically
1.5 mL/min using 60~? A (95-5 acetonitrile-water) 40~c B (95-5 water-
5 acetonitrile) each with 0.1% TFA where tbe column effluent wasmonitored simultaneously at 210 and 277 nM with a Waters model 490
UV-visible detector. Al'ter 1.0 hour of additiollal refluxing, analvtic;il
HPLC analysi.s indicated that the coupling was cc~mplete. The reaction
mixture poured into lN HCI and extracted with ethyl acetate. washed
20 with hrine. dried o~er magne.sium sult'ate, filtere(l, and the filtrate
evaporated ill l'~lC110 to ai'ford 160 mg (96%) of the title compound as an
amorphous solid.
I l l-N~IR (400 MHz, CD3OD,ppin) o 0.90 (t,J=7.40 Hz. 3E-1). 1.25
(d,.J=6.80 Hz, 3H), 1.26 (dJ=6.~0 E~z, 3H), 1.53-1.66 (m,2H), 2.5~-2.63
25 (m,2H), 3.U0 (.sept., J=6.~0 Hz, IH), 3.33 (.s,3H), 5.54 (s,lH), 5.97
(s,211), 6.61 (d,.l=~.~0 Hz, IH), 6.79 (d~ .00 Hz, lH), 6.87 ~d~J=1.60
Hz. IH), 6.93 (dd~/=1.60, 8.80 llz, IEI), 7.36 (dJ=8.40 I-lz, 2H), 7.56
(d,.J=2.40, 8.2'30 Hz, IH), 7.70 (dl =2.40 E~z. IHI, 7.77 (dJ=~.40 Hz,
2H).
30 Cl-MS i721c' = 634 (M+NH4).
'Jl q5758
w0 96f049û~ .l .,C7 --
- 19~-
Step B Pr~paratiorl of N-(~-is(J-propylbenz,enesuli'orlyl)-~x-(4-
(methylsulfonylamino-l~ oxomethyl~- -tl-
propylphenoxy)-3,4-1llethylenedioxyphell3llacetamide~
~ otas~iium salt
Tv a solution of 160 m~ (0.260 mmoll of the product of
Step E~ in I mL of methanol wa~ added 1.04 mL ( 1.04 mmol~ of a I N
potassium hydroxide in melhal~ol solutioll. The reaction mixture. WilS
stirred ] 5 millutes at RT, diluted with 4 mL, of water and filtered
through a 0.4.S uM filter disc. The filtrate was purified using a Water~
Delta Prep 3000 1 {PL,C by applying tlle compound in a 10 ml tok
volume (6 mL methanol cmcl 4 nll, wateri to an tvf 1000 Prep-Pak
module containil~g a 47 x 300 mrn 15 m:M I)eltaPal; C1~ column and
eluting lat 50 mL/min first using 100% B (95-~ water-acetonitrile) i'or
10 minutes and then a 30 minute linear gradient to 60~, A (95-5
5 ~Icetonitile-water) and 4()~c B (95-~; water-acetonitrile). The column
effluellt wa.s monitored simultaneously at 210 a:lld 277 nl\/l Witil a Water.i
model 490 UV-visible detector and the purified fr.~ctions vvere
corllbined in a rouncd bottom fla.ik. frozen in a -7S~C drs~ ice-acetolle
bath and Iyophylized to afford 1 3S mg (77~,~ro 1 of the title colllpvund ;~
20 white Iyophilized powder.
II-I-NMK (40() IMHz, CD30D,ppm) o 0.~7 (t,J=7.40 Hz, 3~{), 1.21
(d~l=6 ~U Hz, 6H),1.52-1.67 ~m,2H), 2.~1-2.~ (m,lH), 2.67-2.74
(m,llH), 2.91 (~ept.~l=6.P~0 Hz, IH), 3.07 (s,3H~, 5.33 (:;,IH), ~.92
(s,21-1), 6~70 (drl=~3.S0 Hz, 11~. 6.74 (ddJ=2.20. 6.20 1Iz, 1 H),7.02-7~04
25 (m,2H), 7.21 (dJ=~.40 Hz"2H), 7.6~ (d,J=~.40 Hz,2H), 7.70 (ddJ
=2.20, ~.40 Hz. 111), 7.7~s ~d,J=2.()0 Hz, IH).
ESI-MS m/~ = 693 (M+l ).
EXA~IPLE 150
N-(4-isc~-propylbellzenesulfonyl~-a-(4-(aminosulfollylaminc~-N'-
oxomethyl)-2-/l-propylphenoxy~-3,4-nlethylenedioxyphellylacetamide
dipotassium s~lt
W0~6/0490s ~ 2195753 ,~ 7
- 193 -
~: Preparation of N-(4-iso-propylbenzenesuli'onyl)-~x-i4-
(aminosulfonylarnino-N~-l -oxomethyl)-2-n-
propvlphenoxv)-3 .4-methylenedioxyphenylacetarnide
A solution of 158 mg (0.293 mmol) of N-(4-iso-
S propylbenzellesulfonyl)-a-(4-carboxy-2-/1-propylphenoxy)-3,4-
methylenedioxyphenylacetamide (free acidic form of the product of
Example 5X) and 71 mg (0.440 mmol) of carbonykliimi~ 7ole in l mL
of dry tetrahydrofuran was refluxed for 3.5 hours. The reaction
mixture was cooled to room temperature and 141 mg (1.47 tmmol) of
0 sulfamide and 110 ul (0.733 mmol) of DBU were added and the
mixture was refluxed again. The reaction progress was monitored by
analytical HPLC analysis using a Waters 600L I-IPLC system with a 4.6
x 250 mm 5um Zorbax-RX C8 at 40~C and eluting isocratically 1.5
mLlmin using 60% A (95-5 acetonitrile-water) 40% B (95-5 water-
acetonitrile) each with 0.1% TFA where the column effluent was
monitored at 254 nM with a Waters model 490 UV-visible detector.
After 2.0 hour of additional refiuxing, analytical HPLC analysis
indicated that the coupling was complete. The reaction mixture was
diiuted with 2.5 mL of methanol and 2 mL of water, filtered and the
filtrate partially purified was purified using a Varian 550() HPLC by
applying the compound in a 4.5 ml total volurrle (2.5 mL methanol and
2 mL water) onto two in series 21.2 x 250 mrn 7mm Zorbax ODS
columns and eluting at 15 mL/min with 60''fo acetonitrile and 40'~o watel
both with 0,15to TFA. The column effiuent was monitored 25411M with
a Kratos Spectrotlow 783 IJV detector. Combination and evaporltion
of the purified fractions afforded 50 mg (28C~) of the title compound.
The mi~ed fractions were combined and sub,jected to a second
preparative HPLC chromatography using a linear gradient over 35
minutes from 65C~C water and 35C~o acetonitrile both with 0.1C~oTFA to
65~o acetonitrile and 35% water hoth ~ith 0.1 ~cTFA, holding all other
conditions from the previous chromato~raph~;. The purified fractions
were combined and concentrated to afford 57 mg (31 %) of the title
compound, which was combined with the previollsly purified material
WO96/04905 ~ 2l~575~ P~llu ~67--
- 194-
to provide a total of 107 mg (S9~o) of the title compound ax ar
amorphous solid.
IH-NMR ~400 MHz, CD30D,ppm) o 0.89 (t,~=7.40 H~, 3H), 1.25 "~
(dl=6.80 Hz, 311), 1.26 (dJ=6.80 Hz, 3H), 1.53-1.66 (m,2H), 2.58-2.73
(m,2H), 2.9S (.sept.. J=6.80 I-lz, lH), 5.54 (s,lH), 5.97 (s,2H), 6.62
(d,J=8.80 Hz, 111), 6.79 (dJ=8.00 Hz, Il-I), 6.~6 (d,J=1.60 Hz, lH). 6.92
(dd,J=1.60, 8.00 Hz, lH), 7.36 (d~=8.50 Hz, 21-1), 7.56 (d.J=2.40, 8.R0
Hz. IH), 7.69 ~d,J =2.40 Hz, IH), 7.77 (d~l=8.50 Hz. 2H).
ESI-MS m/~ = 61R (1\1+1).
Step B: Preparation of N-~4-i~s(l-propyll:en7~ slllfony~ c-(4
(aminosulfonylarllin(l-N'- I -oxomethyl)-2-n-propyl-
phenoxy)-3~4-methylenedioxyphenylacehlmide dipotassiu:m
salt
lS To a .solution of 107 mg (0.173 mmol) of the product of
Step B in 1.5 mL of methanol wa.s added 0.691 mL (0.691 1I~llOI) of a
I N potassium hydro~cide in methanol solution. The reactioll mi.~iure
was stirred 15 millutes at R'l', diluled with 1 rmL of water .md filtered
throllgh a 0.45 uM filter disc. The filtrate wa~s purii'ied Usillg a Varian
20 5500 HPLC by applying the compound in a 4.0 ml total ~olume (3 mL
methallol and I mL water') onto two in series 21.2 x 250 mm 7mm
Zorbax ODS culumns and eluting at 15 ml,/min first llsing 95~1i water
and ~o acetonitrile tor 10 minutes and therl a 30 minute linear gradiellt
to 60ac acetonhrile and 40% water. The column effluent was
25 nlonitored 254 nM with a Kratos Spectroflow 783 W detector alld the
purified fractions were combined in a round bottom flask, frozen in a
-78~C dry ice-acetone bath and Iyophylized tc aff'ord 84 mg (73~/o) Qf
the title compound as a white Iyophilized powder.
1H-NMR (4('K) MHz, CD30D,ppm) o 0.87 (tJ=7.40 Hz., 3H), 1.2(-)
30 ~dl=6.80 Hz, 3H), 1.21 (d,J=6.80 H7., 3H~? 1.53-1.66 (m,211), 2.51-2.59
fm,lH), 2.67-2.74 (m,lH), 2.91 (sept.J=6.R0 Hz~ IH). 3.07 (s,3H), 5.33
(s~lH)~ 5.g2 (s,~H), 6.71-6.75 (m,2H), 7.01-7.04 (m,2H), 7.19-7.22
(m,. H), 7.64-7.70 ~m,3EI), 7.77 (d,J=2.4 Hz, IH).
ESI-MS m/c = 694 (~M+I ).
~ Y~'09611~49~ 2 1 9~7 5~ P~
- 195-
EXAMPLE 151
5 N-(4-iso-propylbenzenesulfonyl)-a-(4-cyarto-2-n-propylphenoxy)-3,4-
methylenedioxyphenylacetarnide potassium salt
Step A: Preparation of 3-allyl-4-hydroxvbenzonitrile
To a stirred solutioll of 25.00 g (210.1 mmol) of 4-
o cyanophenol (Aldrich) in 100 nlL of acetone was added 30.49 g (220.6
mmol) of powdered potassium carbonate followed by 19.09 mL (220.6
mmol) of allyl bromide and the reaction mixture refluxed overnight.
TLC ~unalysis (15% ethyl acetate / hexane) indicated that the alkylation
was complete and the reaction mixture was filtered, the filtrated
5 evaporated in vacuo to afford 33.20 g (99%) of a light yellow oil. The
33.20 (209 mmol) of crude O-allyl ether was dissolved in 100 ml of
1,2-dichlorobenzene and stirred at reflux for 56 hours until TI,C
analysis (15% ethyl acetate /hexane) indicated essentially no starting O-
allyl ether remained. The reaction mixture was poured intO 300 mL of
20 hexane, cooled in a freezer overnight, and the precipitate was filtered
on and dried on vacuum to provide 29.76 g (90~) of the title compound
as a light tan amorphous solid. IH-NMR (300 MHz, C[~C13,ppm) d
3.36 (D,J=6.30 Hz,2H), 5.09-5.19 (m,2H), 5.79 (bs,lH), 5.~6-6.00
(m,lH), 6.82 (dd,J=1.80, 7.20 Hz,lJI), 7.3R-7.41 (m,2H).
25 El-MS mle = 159 (M+~
Step B: Preparation of 4-hYdroxy-3-n-propylbenzonitrile
A Parr hydrogenation shaker was charged with a solution
30 of 29.76 8 (1~7 mmol) of the product of Step A in 100 mL of ethanol
and 3.00 g of a lOS~o palladium on carbon cataly.st was added. The flask
~h~as mounted in the hydrogenation appartus, freed of air, pressurized
with hydrogen (40 psig) ~md shaken 80 minutes. At the end of this
preiod, TLC analysis (15C7O ethyl acetate / hexane) indicated that the
reaction was complete and the reaction mixture was filtered and
_
WO96104905 ~ ~ 219575~ ~ }~
5 ~
- 196-
evaporated. The product was dried in vacllo to afford 30.01 g (99'Y~,) of
the title compaund as a yellow oil.
IH-NMR (300 MHz, CDCl3,ppm~ ~ 0.92 (t,.l=7.40 H~,3EI), 1.53-1.~S5
(m,2H), 2.~4 (t,J=7.60 Hz,2EI), 6.77 (dJ=8.10 Hz"l H), 7.32-7.37
5 (m~2H)~
EI-MS m/e = 161 ~M+)
Step C: Preparation of methyl o!-(?-n-propyl-4-cyanophel1t~xy)-3,4-
meth vlel1edioxvphenylacetate
'I'o a stirred solution of 4.50 g (27.95 rrm1ol) of the product
of Step B in 30 mL of acetone was added 4.64 g (33.~4 mmol) of
powdered potassiurn carbonate and the reaction mixture ~A~as stirred fol
1() mil1utes. ~lethyl a-bromo-3,4-methylenedioxyphenylacetate (R.()l g;
29.35 lmnol) was then added and the reaction mi~ture refluxed
5 overnight. The reactiorl mixture was cooled, filtered. and the filtrate
evaporated irl vacuo, dried on vacuum to a~fford 10.30 g (9.~7g
theoretical) of the t;tle compound which waS used without purificatio
in the next .step.
IEI-NIvlR (300 MHz, C'DC13,pprm) ~ 0.92 (tTI=7.40 Elz,31~), 1.S8-1.6
(m,2H)~ 2.60-2.71 (m,2H), ~.53 (s,lH~, 5.95 (s,~H), 6.68 (dJ=8.10
ll~,lH~, 6.7~ ~d?l=7.~0 lIz,lE-I), 6.95-6.9~ (m.2H), 7.35-7.39 (rm, H).
El-MS ml~ = 353 ~M+)
Step D: Prepa~.ltion of methyl ~-~2-n-propyl-4-cyanophenoxy)-3,4-
methvlenedioxyphenylacetic acid
TQ a solution of 3.5 g (8.64 rnmol) of the product of Step C'
in 30 mL of methanol v.~as added '7.07 mL (10.37 mmol) of 5.0 N
aqueou.s sodium hydroxide solution. After .stirring overnight, TLC
analysis ( ~0: 15:1 chloroform / methanol l ammonium hy(lloxide)
indicated that the saponificatiou was complete. The reaction mixture
wa.s acidified to pEI 3 with 6 N aqueous HCI and concentrated in vac~io.
With heating and sonnication, the residue was redissolved in ethyl
acetate, washed with brine, dried over magnesium .sulfate, filtered, and
tlle filtrate evaporated in vauJo to afford 2.92 g ~99~) of the title
~ WO 96/04905 r . ' ; ~ ,, 2 I q ~ 7 5 8 1 ~ - "~
- 197-
compound as an amorphous solid which was used without further
purification in the next step.
? IH-NMR (300 Mllz, CD3OD,ppm) ~ 0.90 ~t,J=7.40 Hz,3H), 1.58-1.68
(m,2H), 2.66 (tJ=7.60 Hz,2H), 5.72 (s,lH), 5.92 (s,2H), 6.60 (d,J=8.10
Hz,l~), 6.90-6.93 (m,lH), 7.00-7.04 (m,2H), 7.45-7.47 (m,2H).
El-MS mle = 339 (M+)
Step E: Preparation of N-(4-iso-propylbenzene.sulfonyl)-a-(4-
cyanc~-2-n-propylpheno.xy)-3,4-methylened:ioxypllenyl-
acetamide
'I'o a stirred solution of 2.92 g (X.61 mmol) of the the
product of Step D in 40 mL of methylene chloride was added 2.31 g
(12.05 mmol) of 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide
5 hydrochloride, 1.26 g (10.33 rnrnol) of 4-dimethylaminopyridine, and
1.8g g ((9.47 mmol) of 4-iso-propylbenzenesulfonamide. The reaction
mixture was stirred overnight and after TLC analysis (80:15: 1
chlorofonn / methanol / ammonium hydroxide) was poured into I N
HCI and extracted with ethyl acetate. The extract was washed with
20 brine, dried over m; gn~sium sulfate, filtered. and evaporate(l irlV~fc uo.
The residue was partially purified with silica gel flash chromatography
eluting first with 40% ethyl acetate-hexane and then with 3~7O methanol-
methylene chloride to flush the column. All of the product product
fractions were tainted with a con~minAnt and combined and
25 concentrated for a subsequent chromatography. The 3.86 g of semi-
purified material was dissolved in 15 mL of acetonitrile and 15 mL of
water and filtered through a 0.45 uM filter disc. The compound was
purified using A Waters Delta Prep 3000 IIPLC by applying the filtered
solution io an M 1000 Prep-Pak module containing a 47 x 30(i mm 15
30 mM DeltaPak C18 column and eluting at 50 mL/min first using 50% A
(95-5 acetonitile-water) and 50~o B (95-5 water-acetolIitrile) with O.l~
TFA for 15 minutes and then 70C~o A (95-5 acetoniti]e-water) and 30%
B ~95-5 water-acetonitrile) both with 0.1% TFA. The column effluellt
was monitored simultaneously at 210 and 277 nM witll a Waters model
W0 961~14905 ; ~ 2 1 q 5 7 5 ~ 7--
- Ig8-
490 UV-visible deLectar and the purified fractions were combined in a
round bottom flask and concentiated in vacuv to afford 1.45 g (38%) ol
the title compound as an amorphous solid.
IH-NMR (500 MHz. CD30D,ppm) ~ 0.8~) ~t~/=7.50 Hz,3H~), I.26
(d,J=7.00 Hz,3H3, 1.27 (dJ=7.00 Hz,3H), 1.53-1.61 ~m,2E{), 2.59-2.66
(m,2H?, 3.00 (sept.l=7.00 Hz,lH), 5.55 (s,lH~, 5.97 (s,2H), 6.60
~d~/=7.50 llz,lHj, 6.7g (dJ=8.00 Hz~lH), 6.85 (d,~=2.50 Hz,lH), 6.92
(dd,J=2.00, 8.21:) Hz~lll), 7.31 (ddJ=2.50, 8.00 Hz,lH), 7.38 (d~=8.50
Hz~2H~, 7.45 ~d~J=2.00 Hz,lH), 7.78 (d,J=8.50 Hz,2H).
Step F: Preparation of N-(4-iso-propylbc~ le~ulfonyl)-a-~4-
cyano-2-n-propylphenoxy)-3.4-methylenedioxy-
pllenyLacetamide potassium salt
To a stirred solution of 393 mg (0.756 mmol) of the the
product of Step E in I ml of methanol was added 2.26 mL (2.27 mmol)
of a I N potassium hydroxide solution in methanol. The reaction
mixture was stirred 15 minutes, diluted with water and filtered through
a 0.45 mm filter disc. The filtrate was purified usin~ a Varian .~500
20 HPLC by apply;ng the compound in a 4.0 ml tot~l volume (3 mL
methanol and I nlL water) onto two in series 21.2 x 250 mn1 7mm
Zo}bax ODS columns and eluting at 15 m~/min first u.sin~ 85~i water
and 15~c acetonitrile for 10 minutes and then a 30 minute linear
gradient to 50% acetonitrile and 50% water. ~he column eflluent was
25 nnonitored 254 nM with a ~Kratos Spectroflow 783 UV detector and the
purified fractians were combined in a roulld bottom flask. frozen in a
-78~C dry ice-acetone bath and Iyophylized to afford 177 mg (42~o) of
the title compound as a wllite Iyophilized powder.
IH-NMR (500 MHz? CD3OD,ppm) o 0.88 (tJ=7.50 Hz,31-I). 1.23
30 (s,3H)~ 1.24 (s,3H), 1.55-1.62 (m,2H~ 2.53-2.5~ (m~lH~ 2.67-2.73
(m~ 3.90-2.94 (m~lH)~ 5.37 (s,lH), 5.94 (:s,21-1~, 6.76-6.78 (m~2H)~
7.02-7.04 (m~2H1~ 7.21 (dJ=8.50 Hz,2H0~ 7.27 (dd~/=2.0()~ 8.50
Hz~lH), 7.38 (dJ=2.00 Hz,lH), 7.66 (d,J=8.50 Hz,2H~.
ESI-M,S m/e = 5~9 (M+l).
wo96104905 ~ , 2 1 9575~ r~
- 199-
~XAMPLE 152
N-(4-iso-propylbenzenesulfonyl)-a-(4-tetrazo-5 -yl-2-r/-propyl-
phenoxy)-3,4-methylenedioxyphenylacetamide dipotassium salt
Step A: Preparation of N-(4-iso-propylben7~n~snlfollyl~ -(4-
tetrazo-5-yl-2-n-propylphenoxy)-3,4-methylenedioxy-
phenylacet~lmide
A stirred solution of 600 mg ( I .15 mmol) of the product of
Example 151, Step F and 284 mg (1.38 mmol) of trimethyltinazide in 2
mL of toluene was heated with an oil bath at reflux overnight. The
reaction was evaporated in v~cuo~ purified by silica gel flash
chromatography eluting with methylene chloride I methanol / acetic
acid 100:3:1, and the purified fractions concentrated in VClCUo to afford
244 mg (38C~o) of the title compound as ,m amorphous solid.
Ill-NMR (500 MHz, CD30D,ppm) o 0.90-0.94 (rm,2H), 1.16-1.19
Im,6~1), 1.60-1.68 (m,2H), 2.64-2.75 (m,2H), 2.89-2.95 (m,lll), 5.56
(s,lH), 5.96 (~s,2Hi, 6.73 (d,J=8.50 Hz,lH), 6.79 (dJ=8.00 Hz,lH), 6.89
(d,.7= 2.00 Hz,lH), 6.94 (dd~/=2.00, 8.00 Hz,lH), 7.34 (ddJ=1.50, 8.50
Hz,211), 7.64 (dd,J=2.00, 8.50 Hz,lH), 7.76 (d,J=2.00 Hz,lll),
(ddJ=2.00, 8.50 Hz,2H).
ESI-MS mie = 564 (M+l).
Step B: Preparation of N-(4-i.so-propylbenzenesulfonyl)-o~-(4-
tetrazo-5 -yl-2-n-propylphe.noxy)-3 ,4-methy lenedioxy-
phenylacetamide dipota.ssium salt
To a stirred solution of 240 mg (0.426 rmmol) of the
product of Step A in 3 ml of methanol was added 2.41 mL (2.41 mmol)
of a I N potassium hydroxide solution in methanol. The reaction
mixture was stirred 15 minutes, diluted with 4 mI, of water ~md filtered
WO 96/~490~ 2 1 9 5 7 r; 8 ~ 7 ~~
;. ~, .
. ~ .i
- 200 -
thlough a 0.45 mrn filter disc. The filtrate was purified using a Varian
5500 HPLC by applying the compound in a 8.0 ml total volume (4 ml
methanol and 4 mL water) OlltO two in series 21.2 x 250 mm 7mrn
Zorbax ODS columns and eluting at 15 mL/min first using 90~o water
5 and 10% acetonitrile for 5 minutes and then a 30 minutç linear gradiellt
to 40~Vo acetonitrile and 60% water. The column ef~luent was
monitored 254 nM with a Kratos Spectroflow 783 UV cletector and the
purifiecl fractions were combined in a round bottom flask! frozen in a
-7R~C dry ice-acetone bath and Iyophylized to afford 196 mg (72~o) of
the title compoulld as a white Iyophilized powder.
NMR (500 Ml Iz, CD30D,ppm) o 0.91 (tJ=7.20 E~z,3H~, 1.15
(d,J=7.0 Hz,3H~, 1.16 (d~l=7.0 Hz,3~), 1.62-1.68 ~rll,2H), 2.58-2.64
(m,lH), 2.74-21~0 (m,lH~, 2.86 (sept.,J=7.00 Hz,lH), 5.36 (s,lH), 5.93
(d,.J=1.20 Hz,lH), 5.94 (drl=l .20 Hz,lH), 6.76 (d~l=7.50 Hz,ll~). 6.82
5 (d~ .50 Hz,lE~), 7.04-7.07 (m~2H), 7.20 (dJ=~3.50 Hz,211). 7.65-7.69
(m,3H), 7.78(dJ=2.()0 H~,lH).
LSI-MS mle = 640 (M+l~.
EXAMPLE 153
N-(4-iso-propylbenzenesulfonyl)-a-(4-N-tnethyl-l~i~-methc~xy-
carboxamido-2-~-propylphenoxy)-3 ,4-methylenedioxyphenylacetamide
Step A: N-~4-i~o-propylbenzenesulfollyl)-~-~4-,'V-methyl-A,1-
methoxycarboxamido-2-n-propylphelloxy)-3,4-
methylenedioxvphenylacetamide
To a 0~ C suspension of 2.00 g (3.25 mmol~ of the product
of Example 58 and 951 mg (9.75 mmol) N,O- dimetbylhydroxylamille
hydrochloricle in 15 mL of methylene chloride and 1.36 mI, (9.75
30 ml:llOI) of triethylamine was added 1.49 g (9.75 mrllol~ of
l-hydroxybenzotria~ole hydrate and 1.87 g (g.7~ mmol~ of 1-(-3-
dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride. The
reaction mixture was stirred 16 hour and alklwed to wann to room
temperature after which TLC analysis (10~ methanollmethylene
~i Wo g6l0490s ~ p~
7 5 ~
- 201 -
chloride) indicated that the couplihg reaction was complete. The
reaction was diluted with ethyl acetate (30 mL) and partitioned with
water (150 mL). The organic layer was washed with I N HCI (50 mL),
brine (50 mL), dried over MgSO4, filtered, evaporated and dried in
5 vacuo affording 1.64 g ~7%) of the title compound as an amorphous
powder.
IH-NMR(400 MHz, d6-DMSO,ppm): o 0.82 (t~=7.20 Hz,3H), 1.17
(d,~=7.20 Hz,6H), 1.43-1.58 (m,2H), 2.4~S-2.60 (m,2H), 2.93 (sept,
J=7.20 Hz,lH), 3.19 (s,1}1), 3.51 (s,3H), 5.66 (s,lH), 6.03 (d,J=1.20
Hz,lH), 6.04 (drl=1~20 Hz,lH), 6.47 (d,J =8.40 lIz,lH), 6.91-6.9~S
(m,3H), 7.25 (dd,J= 2.00, S.40 H~, lH), 7.3~ (d,J= 2.00 Hz, lH), 7.41
(d,/= 8.40 ~Iz,2H), 7.69 ~d,J= 8.40 Hz,2H).
ESI-MS m/e = 5~3 (M+l).
EXAMPLE154
N-(4-i.so-propylbenzenesulfonyl)-o~-(4-( 1 -oxoethyl)-2-n-propyl-
phenoxy)-3,4-methylenedioxyphenylacetamide dipc tassium salt
Step A: Yreparation of N-(4-i.so-propylbenzenesulfonyl)-oc-(4-(1-
oxoethyl)-2-n -propylphenoxy)-3,4-methylenedioxy-
pheny lacetamide
To a 0~ C stirred suspension of 594 mg (1.02 mrnol) of the
25 product of Example 153 in 8 mL of dry tetrahydrofuran was added
1.19 mL (3.57 mmol) of methyln-~gn~sillm chloride as a 3.0 M solution
in tetrahydrofuran. Following the grignard addition, a homogellous
reaction mixture was achieved and tlhell allowed to uarm to room
t~ dlUI~. After stirring 3 hour~, TLC analysis (10~, methanol /
30 methylene chloride) indicated that the couplillg reaction wa.s complete.
The reaction mixture was poured into 5'~o 6N HCI / ethanol (20 mL)
and then partitioned between brine (6() ml,) and ethyl acetate (30 mL).
The extract was dried over m;l~n~siilm sulfate, filtered and the filtrate
W096fO490~ '? ~ 9 57 ~8
- 202 -
concentrated in va~ uo affording 373 mg (68~o) of the title colllpoulld as
an amorphou.s powder.
l l-I-NMR (400 MHz, CD30D,ppm) o 0.88 (tJ=7.20 E Iz,3H), 1.21
(d,J=6.80 Hz,611), 1.54-1.66 ~m,2H), 2.51 (s3H), 2.55-2.63~m,1H),
2.67-2.75 (m,lEI), 2.91 (.sept.,J=6.80 Hz,lM), 5.43 (s,lH~, 5.94
(d,J=1.20 1-lz,21-1), 6.74-6.77 (m,2H), 7.01-7.03 tm,2H), 7.23 ~d,J=8.40
Elz,2H~, 7.65-7.69 ~m,3H), 7.73 (d,J=2.40 Hz,lH).
Cl-MS mlc = 5~38 (M+l).
Step B: Preparation of N-(4-isO-propylbenzene6ulfotlyl)-a-(4-(l-
oxoethyl)-2-n-propylphenoxy)-3,4-methylenedio~yphellyl-
acetamide potas.sium salt
'rhe titled compound is prepared u6ing the product of Step
A according to the procedure described in E~ample 152, Step 13.