Note: Descriptions are shown in the official language in which they were submitted.
WO 96/05810 '~~~õ9i064' PCT/GB95/01980
Drug delivery composition containing chitosan or derivative thereof having a
defined z. potential
The present invention relates to drug delivery compositions and more
particularly to compositions based on chitosan microparticles which
provide for the improved uptake of active drug material across mucosal
surfaces, such as the vagina, the small intestine, the colon, the lungs, the
rectum, the eye, the buccal cavity or the nasal cavity.
A major problem in drug delivery is the effective absorption of polar
molecules, that include high molecular weight material such as proteins
and peptides, across biological membranes. Normally such molecules are
not well taken up by the body if administered to the gastrointestinal tract,
the buccal mucosa, the rectal mucosa, the vaginal mucosa or the intranasal
mucosa. By a polar molecule, we mean a substance that has an
octanol/water partition coefficient of less than 50. Recent studies with
insulin have demonstrated that the absorption of such a compound can be
increased if it is given together with a so-called absorption enhancer.
These absorption enhancing materials have included surfactants of the non-
ionic type as well as various bile salt derivatives. An increased
permeability of membranes in the presence of these types of surfactant
materials is obtained and the literature in the field of pharmaceutical
sciences contains a wide range of such absorption promoters. (For a
review see Davis et al (editors), Delivery Systems for Peptide Drugs,
Plenum Press, New York, 1987). However, such materials are sometimes
unacceptable because of their irritant effects on membranes. These
include not only the non-ionic variety of surface active agents but also bile
salts and bile salt derivatives (eg fusidic acid).
EP-A-023 359 and EP-A-122 023 describe a powdery pharmaceutical
composition for application to the nasal mucosa and methods for
1
WO 96/05810 219 PCT/GB95l01980~
administration thereof.( The pharmaceutical composition allows
polypeptides and derivatives thereof to be effectively absorbed through the
nasal mucosa. Similarly, US-A-4 226 849 describes a method for
administering a powdery medicament to the nasal mucosa where the
preferred composition has mucoadhesive properties.
Formulations based on microspheres for mucosal delivery have been
described in WO 88/09163. The formulations contain certain enhancers
to aid effective penetration of the mucosa by the drug. W089/03207
further describes formulations which do not require an enhancer.
WO 90109780 describes a composition consisting of a drug and a
polycationic substance including chitosan that promotes the transport of the
drug across mucosal membranes. Tlle,composition can also comprise
microspheres of the polycationic substance.
Chitosan is deacetylated chitin, or poly-N-acetyl-D-glucosamine. It is
available from Protan Laboratories, Inc. Redmond, Washington 98052,
U.S.A. and, depending on the grade selected, can be soluble in water up
to pH 6Ø Chitosan has previously been used to precipitate proteinaceous
material, to make surgical sutures and as an immunostimulant. It has also
been employed previously in oral drug formulations in order to improve
the dissolution of poorly soluble drugs (Sawayanagi et al, Chem. Pharm.
Bull., 31, 1983, 2062-2068) or for the sustained release of drugs by a
process of slow erosion from a hydrated compressed matrix (Nagai et al
Proc. Jt. US Jpn. Semin. Adv. Chitin. Chitosan. Relat. Enzymes, 21-39.
Zikakis J.P. (ed), Academic Press. Orlando (1984)).
Chitosan microspheres have been produced for use for example for
enhanced chromatographic separation (Li Q. et al, Biomater. Artif. Cells
2
WO 96/05810 2197 ~ 62 PCT1GB95/01980
Immobilization Biotechnol, 21, 1993, 391-8) for topical delivery of drugs
(Machida Y, Yakugaku Zasshi, 113, 1993, 356-68) for drug targeting
after injection (Ohya Y et al, J. Microencapsul. 10, 1993, 1-9) and for
controlled release of drugs (Bodmeier R. et al, Pharm Res. 6, 1989, 413-
7, Chithambara et al J. Pharm. Pharmacol. 44, 1992, 283-286).
EP 454044 and EP486959 describe polyelectrolyte microparticles or
polysaccharide microspheres including chitosan microspheres for
controlled slow release of drugs. The drug is chemically bound or
adsorbed to the surface.
Glutaraldehyde cross linked chitosan microspheres have been described in
JP 539149 (Taisho Pharm. Co.). The use of chitosan as a biodegradable
polymer material that is then modified by an amphiphilic polymer and an
agent modifying the interface properties has been described in EP 486959
(Vectorpharma).
The standard method for the preparation of non-dissolving chitosan
microspheres or microcapsules is to use an emulsification method. For
example Chithambara et al (Cross-linked chitosan microspheres
preparation and evaluation as a matrix for controlled release of
pharmaceuticals, J. Pharm. Pharmacol. 44, 1992, 283-286) describe how
an aqueous acetic acid dispersion of chitosan in paraffin oil using
dioctylsulphosuccinate as a stabilising agent was cross-linked by
glutaraldehyde. A similar emulsion cross-linking procedure was described
by Hassan et al (Optimized formulation of magnetic chitosan microspheres
containing the anticancer agent, oxantrazole, Phann. Research 9, 1992,
390). Microspheres made from the complexation of chitosan with other
polymers but of opposite charge have been described in papers and patents
(EP 454044 to Hoechst AG).
3
CA 02197062 2005-05-31
lonotropic gelation of chitosan by tripolyphosphate has been reported by
Bodmeier et al (Pharm. Research 6, 1989, 413). The chitosan beads so
prepared disintegrated at acid pH.
Microspheres intended to be bioadhesive have been described by WO
93/21906. The microspheres were characterised by having a bioadhesive
force of at least 11 mN/cm2 as measured in a tensile test in the rat
intestine.
The attachment of positively charged ligands to microspheres to improve
adhesion due to the electrostatic attraction of the cationic groups coating
the beads, to the net negative charge of the mucus is mentioned as a
possible strategy. The preparation of chitosan microspheres is described.
The particles were prepared using a 1% chitosan concentration at pH 5Ø
They were cross-linked using tripolyphosphate (3%). The particles so
prepared were apparently of a size of approximately 2000 .m. It is noted
that chitosan microparticles were found not to be satisfactory since the
force of detachment per projected surface area (mN/cm2) for such particles
was less than 5 mN/cm2.
We have now surprisingly found that positively charged solidified or
partially cross-linked chitosan particles are able to considerably improve
the absorption of drugs across mucosal tissue. They appear to do so by
prolonging the known absorption promoting effect of chitosan resulting in a
sustained absorption. This is surprising since chitosan in solution gives rise
to a pulsed absorption profile similar to the one obtained when using
bioadhesive microparticles such as starch (WO 88/09163, WO 89/03207).
In one aspect, the invention therefore provides a drug delivery composition
for administration to mucosa comprising a pharmacologically active
4
CA 02197062 2005-05-31
compound and particles of chitosan or a chitosan derivative or salt, wherein
the particles are either solidified or partially cross-linked such that they
have a zeta potential of +0.5 to +50mV.
Preferably the particles are either microspheres or powder.
The surface charge of the particles expressed as the zeta potential is
determined at pH 7.4 and 0.1 M ionic strength.
The method by which the zeta potential is measured depends on the size of
the particles. For particles of 5 m or more, the zeta potential is measured
by microelectrophoresis using for example a Rank Mark II
microelectrophoresis apparatus (Rank Bros., Cambridge). For particles
below 5 m, the zeta potential is measured by Laser Doppler Anemometry
(LDA) using for example a Malvem Zeta Sizer IV (Malvem, UK).
According to another aspect of the invention, there is provided a drug
delivery composition for administration to mucosa comprising a
pharmacologically active compound and particles of chitosan or a chitosan
derivative or salt, wherein the particles are either solidified or partially
cross-linked such that they have a zeta potential of +0.5 to +30mV,
measured, by microelectrophoresis for microspheres of 5 m or more, and
by laser Doppler anemometry for microspheres below 5 m, at pH7.4 and
0.1M ionic strength.
5
CA 02197062 2006-09-18
Microelectrophoresis
For microelectrophoretic studies, a Rank Mark II microelectrophoresis
apparatus (Rank Bros.., Cambridge) can be used. A cylindrical glass
microelectrophoresis cell is placed in a therrnostated water bath at 25'C.
Platinum black electrodes, prepared by oxidation in platinum chloride, are
placed at either end of the cell. illumination is by a 40mW gas laser
(Scientific and Cook Electronic, UK). The movement of the particles is
viewed by means of a binocular microscope system.
The inter-electrode distance is calculated by measuring the conductivity of
standard solutions of potassium chloride of for example 0.10M and 0.0 ] M
ionic strength at 25 C.. The specific conductivity is the reciprocal of the
resistance of a 1M long and lm2 in cross-sectxon colurrxn of solution
5a
219706,2';
rx
WO 96105810 PCT/GB951019800
(ie) k
aazR
where k is the conductivity, t the inter-electrode distance and R the
resistance of the cell. The resistance is determined by measuring the
change in current for a known change in voltage.
For the determination of the particle mobility the cell is filled with the
appropriate buffer system (O.IM ionic strength, for example phosphate
buffer or McIlvaine buffer, pH = 7.4) and 1 % w/v of the particles
suspended in the buffer.
The suspension is left to equilibrate at 25 C for 5 minutes before any
measurements are taken. The electrodes are placed at each end of the cell
to form a tight seal. A voltage in the range 20-60 volt is applied across
the electrodes to give a reasonable transit time which reduces errors due
to Brownian motion and operator timing. The velocity of individual
particles is timed across a fixed distance, such as 30gm calculated from
an eyepiece graticule. Alternative measurements are taken with the
electrode polarity reversed to prevent electrode polarization. The velocity
of fifteen particles in each direction and at two voltages are measured and
the electrophoretic mobility (u) calculated from
V
u=- --_ - - - - -
E
where V is the particle velocity ( ms"'). The electrophoretic mobilities
can be converted to zeta potentials (D using the equation derived by
Smoluchowski (1903)
- - -
b_
R .'
~ WO 96/05810 2197062 PCT/6B95/01980
U= -
. 4~
where E is the permittivity and 17 the viscosity of the dispersion medium.
Assumptions are made as to the values of E and n in the electrical double
layer. As a guideline the zeta potential can be obtained from the
electrophoretic mobility by multiplying by approximately 15.
Laser poppler Anemometry
Laser Doppler Anemometry involves the detection of scattered laser light
from particles which are moving in an applied electric field. The
equipment used can for example be a Malvern Zetasiser II(Malvem
Instmments). The sample is illuminated with a laser light source (15mW
Helium-Neon laser) which is split into two beams of equal intensity. The
split beams are then forced to cross to give an ellipsoid measuring volume;
consisting of dark and light bands. The interference fringe pattern will be
dependent on the beam crossing angle and the laser frequency. Particles
moving under the application of an electric field will scatter light from the
incident beam. The frequency shift- the Doppler frequency, Fd- is a
ftrnction of the particle velocity as described by
Fd = 2 sin (AB12)u
where B is the detection angle, X the laser wavelength and u the particle
velocity, respectively. The Malvern instrument detects the scattered light
intensity and converts this to a Fd and allows for the calculation of u.
Particle velocities are usually expressed as electrophoretic mobilities. The
electrophoretic mobility (EM) of a particle is defined as follows:-
7
W096/05810 219I 06v;,'~
PCT/GB95/01980~
EM = nifield strength
The zeta potential (ZP) can be calculated by application of the
Smoluchowsld equation since the measurements are to be carried out at
low ionic strengths and with relatively large particles,
ZP=4,q ule
where n is the viscosity of the medium, e is the dielectric constant and u
is the particle velocity, respectively.
Electrophoretic mobility measurements are carried out by dispersing
microspheres in lOml buffer solution such as phosphate or Mcllvaine
buffer from 100lig - lmg, with a constant ionic strength 0.1M. Four
readings are taken using a PC4 wide capillary cell at a voltage of 100 volt,
electrode spacing of 50mm and a dielectric constant of 78.54.
By "solidified particles" we mean particles made from a water soluble salt
of chitosan that has been made insoluble in non-acid containing water by
exposure to an alkaline agent. Exposure to the alkaline agent brings the
chitosan out of solution so that it is no longer in its soluble salt form.
Suitable alkaline agents for use include sodium hydroxide, calcium
hydroxide, aluminium hydroxide, potassium hydroxide, sodium phosphate,
sodium carbonate and ammonia. Particles, either powder or
microspheres, treated in this way still retain a positive charge. For
example, solidified chitosan microspheres can be prepared by emulsifying
a chitosan or chitosan derivative salt into soya oil or a similar organic
medium and adding an alkaline agent such as sodium hydroxide that
solidifies (precipitates) the chitosan during mixing. The resultant solid
microspheres will be positively charged in an aqueous suspension since no
8
. .. .. ~+
= WO 96/05810 121{970 U"' PCT/GB95/01980
crosslinking has taken place.
If the particles are cross-linked, the degree of cross-linking must be such
that the particles retain a positive charge in the range of +0.5 mV to
+50 mV. If the particles are completely cross-linked and all available -
NH2 groups are used, they will become neutral or negatively charged. By
partially cross-linking the chitosan, it is possible to leave the particles
positively charged and partly soluble.; The degree of cross-linlcing
required is determined by measuring the zeta potential.
Partially crosslinked microspheres can be prepared using for example
either emulsification or spray drying techniques. In the emulsification
technique the chitosan solution is emulsified in an organic medium such
as toluene or soya oil with an emulsifier such as Span 80. A crosslinldng
agent in the form of for example glutaraldehyde or formaldehyde is either
added to the organic phase before mixing with the chitosan solution or can
be added after emulsification has taken place. The partly crosslinked
microspheres can be harvested by filtration and washing.
The chitosan microspheres can be prepared by spray drying a solution of
chitosan (0.05% w/v - 0.5% at pH 3 - 7) containing an appropriate
amount of glutaraldehyde or formaldehyde or similar crosslinidng agent.
For obtaining positively charged chitosan microspheres the ratio of the
crosslinking agent to chitosan should be 0.01 to 1.00 more preferably 0.05
to 0.75 and most preferably 0.1 to 0.6.
The zeta potential of the particles is preferably +1.OmV to 45mV and
more preferably + 1.5mV to +4OmV, more preferably 2.OmV to 35mV
and especially +3.OmV to +30mV.
9
WO 96105810 -P("PIGB95101981
The chitosan or chitosan derivative or salt used preferably has a molecular
weight of 4 000 or more, preferably in the range 25 000 - 2 000 000 and
most preferably about 50 000 - 300 000. Chitosan or salts of chitosan
may be used.
We use the term chitosan derivatives to include ester, ether or other
derivatives formed by interaction of acyl or alkyl groups with the OH
groups and not the NHZ groups. Examples are 0-alkyl ethers of chitosan,
0-acyl esters of chitosan. Suitable derivatives are given in G.A.E.
Roberts, Chitin Chemistry, MacMillan Press Ltd, London, 1992. Suitable
salts of chitosan include nitrates, phosphates, sulphates, xanthates,
hydrochlorides, glutamates, lactates, acetates.
The composition is preferably administered as a freeze dried formulation
of the particles together with the active compound. The composition can
also be prepared as a physical/mechanical mixture of the dried
microspheres with the drug.
The microspheres may be prepared by spray drying, emulsification,
solvent evaporation, precipitation or other methods known to a person
sldIled in the art. The active drug can be incorporated into the
microspheres during their production or sorbed onto the microspheres after
their production. The microspheres or powder can be partially cross-
linked by glutaraldehyde, formaldehyde, benzydianone, benzoquinone,
tripolyphosphate or other cross-linking agents known to the person skilled
in the art. The conditions for carrying out the cross-linking, such as the
amount of cross-linking agent required, are determined by monitoring the
zeta potential and adjusting the conditions until the required zeta potential
is obtained.
-
219706~
= WO 96/05810 PCT/GB95/01980
The size of the cross-linked or solidified microspheres are 1- 200 m,
more preferably I - 100 m.
If desired, other materials may be included in the composition, for
example absorption enhancers. Suitable absorption enhancers include
phospholipids such as lysophosphatidylcholine, lysophosphatidylglycerol
and generally those mentioned in WO 88/09163.
The term '~)harmacologically active compound" includes drugs, genes
(DNA) or gene constructs, vaccines and components thereof (for example
isolated antigens or parts thereof) and monoclonal antibodies.
The compositions may be used with drugs selected from the following
non-exclusive list: insulin, PTH (parathyroid hormone), PTH analogues,
calcitonins (for example porcine, human, salmon, chicken or eel) and
synthetic modifications thereof, enkephalins, LHRH (lutenising hormone
releasing hormone) and analogues (nafarelin, buserelin, leuprolide,
goserelin), glucagon, TRH (Thyrotrophine releasing hormone),
Vasopressin, Desmopressin, growth hormone, heparins, GHRH (growth
hormone releasing hormone), nifedipine, THF (thymic humoral factor),
CGRP (calcitonin gene related peptide), atrial natriuretic peptide,
metoclopramide, ergotamine, Pizotizin, vaccines (particularly AIDS
vaccines, measles, rhinovirus Type 13 and respiratory syncytial virus,
influenza vaccines, pertussis vaccines, meningococcal vaccines, tetanus
vaccines, diphtheria vaccines, cholera vaccines, DNA vaccines),
pentamidine and CCK (cholecystokinin).
Further drugs include: antibiotics and antimicrobial agents such as
tetracycline hydrochloride, leucomycin, penicillin, penicillin derivatives,
erythromycin, sulphathiazole and nitrofurazone; antimigraine compounds
11
R'O 96/05810 21976 " PCT/GB95J01980
such as sumatriptan or other 5-HTi agonists; vasoconstrictors such as
phenylephrine hydrochloride, tetrahydrozoline hydrochloride, naphazoline
nitrate, oxymetazoline hydrochloride and tramazoline hydrochloride;
cardiotonics such as, digitalis and digoxin; vasodilators such as nitro-
glycerine and papaverine hydrochloride; bone metabolism controlling
agents such as vitamin D and active vitamin D,; sex_hormones;
]ypotensives; sedatives anti-tumor agents; steroidal anti-inflammatory
aQents such as hydro-cortisone, prednisone, fluticasone, prednisolone,
triamcinolone, triamcinolone acetonide, dexamethasone, betamethasone,
beclomethasone and beclomethasone dipropionate; non-steroidal anti-
inflammatory agents such as acetaminophen, aspirin, aminopyrine,
phenylbutazone, mefanic acid, ibuprofen diclofenac sodium, indomethacin,
colchicine and probenecid; enzymatic anti-inflammatory agents such _ as
chymotrypsin and bromelain seratiopeptidase; anti-histaminic agents such
as diphenhydramine hydrochloride, chloropheniramine maleate and
clemastine; antitussive-expectorants such as codeine phosphate and
isoproterenol hydrochloride; anaigesics such as morphine and its polar
metabolites such as morphine-6-glucuronides and morphine-3-sulphate;
antiemetics such as metoclopramide, ondansetron, chlorpromazine; drugs
for treatment of epilepsy such as Clonazepam; drugs for treatment of
sleeping disorders such as melatonin; drugs for treatment of asthma such
as salbutamol.
The compositions can be administered via the nasal route as a powder
using a nasal powder device, via the vaginal route as a powder using a
powder device, formulated into a vaginal suppository or pessary or vaginal
tablet or vaginal gel and via the pulmonary route using a powder inhaler
or metered dose inhaler, via the rectal route formulated into suppositories
and via the small intestine or colonic route formulated in tablets or
capsules. The compositions may gel on the mucosa at least to some extent
12.
CA 02197062 2005-05-31
and this may facilitate retention of the composition on the mucosa.
A further aspect of the invention provides a method of treating a human or
other mammal by administering a composition as described above to a
mucosal surface of that human or other mammal, for example the vagina,
rectum, lungs, eye, colon or nasal cavity.
According to another aspect of the invention, particles of chitosan or a
chitosan derivative or salt are used for delivering a pharmacologically
active compound across a mucosal surface, wherein the particles are either
solidified or partially cross-linked such that they have a zeta potential of
+0.5 to +30mV, measured, by microelectrophoresis for microspheres of 5
gm or more, and by laser Doppler anemometry for microspheres below 5
m, at pH7.4 and 0.1 M ionic strength.
Preferred embodiments of the invention will now be described by way of
example and with reference to the figures in which:
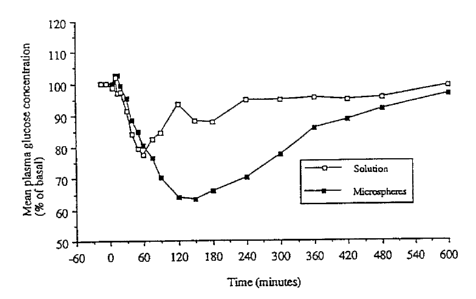
Figure 1 shows the mean plasma glucose/time curves after
administration to sheep of 2 IU/kg insulin in chitosan solution and with
chitosan microspheres (prepared by emulsion heat method followed by
formaldehyde treatment);
Figure 2 shows the mean plasma glucose/time curves after
administration to sheep of 2 IU/kg insulin in chitosan solution and with
chitosan microspheres (prepared by adding glutaraldehyde to chitosan
emulsion);
Figure 3 shows the mean plasma glucose/time curves after
administration to sheep of 2 IU/kg insulin in chitosan solution and with
cross-linked chitosan powder;
13
CA 02197062 2005-05-31
Figure 4 shows the mean plasma glucose/time curves after
administration to sheep of 2 IU/kg insulin in chitosan solution and with
chitosan microspheres (prepared by precipitation with sodium hydroxide);
Figure 5 shows the mean plasma calcium concentrations after nasal
administration to sheep of 20 IU/kg salmon calcitonin in chitosan solution
and with cross-linked chitosan powder;
Figure 6 shows the plasma morphine concentrations following nasal
administration of morphine HC 1 in chitosan solution or with cross-linked
chitosan;
13a
CA 02197062 2005-05-31
Figure 7 shows the plasma calcium concentrations in sheep
following the vaginal administration of salmon calcitonin (1600 IU) in a
pessary containing cross-liked chitosan or as a solution;
Figure 8 shows the mean plasma glucose/time curves after
administration to sheep of 2 IU/kg insulin in chitosan solution and as a
freeze-dried chitosan/lactose powder;
Figure 9 shows the plasma concentration of LHRH agonist in sheep
after intranasal and subcutaneous administration;
Figure 10 shows the plasma concentration of LHRH agonist in sheep
after vaginal administration of formulations containing chitosan
microspheres;
Figure 11 shows the plasma concentration of LMWH following
administration of subcutaneous solution and nasal microsphere
formulation; and
Figure 12 shows the plasma concentration of PTH analogue
following nasal administration of two chitosan powder formulations to
sheep.
Example 1
Partially cross-linked microspheres were prepared as follows:
1 g of Span 80 was mixed into 200 ml of soya oil. The oil/Span mixture
was divided into two; one half was heated to 120 C. Into the remaining
100 ml of oil/Span was emulsified (Silverson homogeniser, 7000 rpm/3
min) 5 ml of a 10% w/v aqueous solution of low viscosity chitosan
hydrochloride (Sea CureTM CL1 13, Pronova, Drammen, Norway.) The
emulsion was poured into the hot oil/Span mixture and stirred to 1500 rpm
using an overhead stirrer. After 10 minutes, the oil/Span/chitosan mixture
was transferred to an ice bath and allowed to cool <40 C while stirring
14
CA 02197062 2005-05-31
continued. 100 ml of acetone was added and the mixture centrifuged (2500
rpm/ 5 min). The microspheres were washed with acetone, collected by
filtration and allowed to dry. The entire process was repeated until 800 mg
of microspheres had been prepared. The microspheres were stirred into a
mixture of 40 ml of acetone and 10 ml of 38% w/v formaldehyde solution.
After 24 hours the microspheres were recovered by filtration and
resuspended in 100 ml of acetone. After a further 24 hours, the
microspheres were recovered by filtration and dried at room temperature.
The size of the microspheres was in the range 1-50 m.
Example 2
Partially cross-linked microspheres were prepared as follows:
To 200 ml of soya oil was added 1 g of Span 80. The oil was stirred using
an overhead mixer at 1000 rpm and 0.5 ml of 25% aqueous glutaraldehyde
was added. After stirring for 30 minutes, 10 ml of a 5% w/v aqueous
solution of low viscosity chitosan hydrochloride (Sea CureTM CL113,
Pronova, Drammen, Norway) was added to the oil/glutaraldehyde
emulsion. After stirring at 1000 rpm for a further 75 minutes, 200 ml of
acetone was added to the emulsion. The mixture was then centrifuged
(2500 rpm/5min). The microsphere pellets were resuspended in acetone,
recovered by filtration, rinsed with further acetone and dried at room
temperature. The majority of the microspheres were in the size range 10-
50 m.
Example 3
Partially cross-linked powder was prepared as follows:
CA 02197062 2005-05-31
Into a beaker was weighed 1 g of chitosan hydrochloride (Sea CureTM
CL113) powder. To the chitosan powder was added 80 ml of acetone and
20 ml of formaldehyde solution 38% w/v in water/methanol). The beaker
contents were stirred for 24 hours. The cross-linked chitosan powder was
recovered by filtration, and suspended in 200 ml of acetone. After 24
hours, the chitosan was recovered by filtration and dried in an oven at 50 C
for 48 hours. The mean particle size of the cross-linked powder was 20
m.
Example 4
Solidified chitosan microspheres were prepared as follows:
A 2% w/v aqueous solution of medium viscosity chitosan glutamate (Sea
CureTM +210) was prepared. 10 ml of chitosan solution was emulsified
(8000 rpm/10min) into 100 ml of soya oil. 100 ml of 10% w/v sodium
hydroxide solution was added and stirring continued at 8000 rpm for 5 min.
The mixture was then mixed with a magnetic stirrer bar for a further 30
min. The microspheres were collected by centrifugation and washed with
petroleum ether, then ethanol, and finally hot distilled water. Microspheres
of mean diameter 25 m were obtained with a surface charge of +3.7mV.
Example 5
Partially cross-linked microspheres were prepared as follows:
A 3% w/v aqueous solution of medium viscosity chitosan glutamate (Sea
CureTM +210) was prepared. 10 ml of chitosan solution was emulsified
(8000 rpm/2min) into a mixture of 100 ml of toluene and 1 g of Span 85.
16
2197062
= WO 96/05810 PCT/GB95/01980
2 ml of 8% w/v glutaraldehyde solution was added and the emulsion left
to gently mix, using a magnetic stirrer bar, for 12 hours. The
microspheres were collected by filtration, washed with toluene, and then
ethanol, and left to dry.
Example 6
Partially cross-linked microspheres were prepared as follows:
250 ml of a solution of 0.2% w/v chitosan in 1% acetic acid was
prepared. 2 ml of 4% glutaraldehyde solution was added and the solution
was spray-dried (Lab-Plant SD 04 spray-drier) using a drying temperature
of 160 C and a flow rate of 5-10 ml/min. Chitosan microspheres of 5 gm
diameter and a zeta potential of +5.7 mV were obtained.
Example 7
600 mg of microspheres prepared using the method described.in Example
1 were weighed into a 100 ml volumetric flask. To the microspheres were
added 30 ml of water and 10 ml of sodium insulin solution (60 IU/ml).
The flask contents were swirled intermittently for 20 minutes and then
frozen by immersing the flask into liquid nitrogen. The frozen contents
were transferred to a freeze-drier and lyophilised. The lyophilised
formulation was administered nasally to each of four sheep at an insulin
dose of 2 IU/kg (equivalent to 2 mg/kg of chitosan microspheres). As a
control, a solution of 200 IU/ml insulin in 5 mg/ml medium viscosity
chitosan solution was administered intranasally at 2 IU/kg. Blood samples
were taken and plasma glucose concentrations measured. The mean
changes in plasma glucose concentration with time for the two
formulations is shown in Figure 1. It can be seen that the fall in plasma
17
2197062
WO 96/05810 PCTlGB95l019819
glucose was of greater magnitude and more prolonged with the
microsphere formulation of the invention than with the chitosan solution.
Example 8
A lyophilised formulation containing insulin was prepared from 600 mg
of the microspheres described in Example 2 using the method described
in Example 7. The lyophilised formulation was administered nasally to
each of four sheep at an insulin dose of 2 IU/kg (equivalent to 2 mg/kg
of chitosan microspheres). The chitosan solution formulation described
in Example 7 was also administered. Blood samples were taken and
plasma glucose concentrations measured. The mean changes in plasma
glucose with time for the two formulations are shown in Figure 2.
Although the minimum glucose concentration achieved was similar for
both formulations, the duration of action achieved with the microsphere
formulation was significantly prolonged compared with the chitosan
solution.
Example 9
A lyophilised formulation containing insulin was prepared from 600 mg
of the cross-linked powder described in Example 3 using the method
described in Example 7. The lyophilised formulation was administered
nasally to each of four sheep at an insulin dose of 2 IU/kg (equivalent to
2 mg/kg of chitosan powder). The chitosan solution formulation described
in Example 7 was also administered. Blood samples were taken and
plasma glucose concentrations measured. The mean changes in plasma
glucose with time for the two formulations are shown in Figure 3. The
fall in plasma glucose was of greater magnitude and more prolonged with
the microsphere formulation of the invention compared with the chitosan
18
~ WO 96/05810 PCT/GB95101980
solution. 2 ~ ~ ( ~ 6 ~
Example 10
A lyophilised formulation containing insulin was prepared from 600 mg
of the microspheres described in Example 4 using the method described
in Example 7. The lyophilised formulation was administered nasally to
each of four sheep at an insulin dose of 2 IU/kg (equivalent to 2 mg/kg
of chitosan microspheres). The chitosan solution formulation described
in Example 7 was also administered. Blood samples were taken and
plasma glucose concentrations measured. The mean changes in plasma
glucose with time for the two formulations are shown in Figure 4.
Example 11
Into a 100 ml conical flask was weighed 880 mg of the cross-linked
powder described in Example 3. To the powder was added 53.7 ml of
water and 5 ml of salmon calcitonin solution (1760 IU/ml). The flask
contents were swirled intermittently for 20 minutes and then frozen by
immersing the flask into liquid nitrogen. The frozen contents were
transferred to a freeze-drier and lyophilised. The lyophilised formulation
was administered nasally to four sheep at a salmon calcitonin dose of 20
IU/kg (equivalent to 2 mg/kg of chitosan powder). As a control, a
solution of 2000 IU/mi salmon calcitonin in 5 mg/ml medium viscosity
chitosan solution was administered intranasally at 20 IU/kg. Blood
samples were taken and plasma calcium concentrations measured. The
changes in plasma calcium concentration with time for the two
formulations are shown in Figure 5. For the chitosan powder formulation,
the fall in plasma calcium, indicative of calcitonin absorption, was
markedly greater and more prolonged than for the chitosan liquid
19
CA 02197062 2005-05-31
formulation.
Example 12
Into a 250 ml conical flask was weighed 800 mg of the cross-linked powder
described in Example 3. To the powder was added 48.3 ml of water and 5
ml of 24 mg/ml morphine hydrochloride solution. The flask contents were
swirled intermittently for 20 minutes and then frozen by immersing the flask
into liquid nitrogen. A solution was prepared containing 30 mg/ml morphine
hydrochloride in medium viscosity chitosan glutamate solution, adjusted to
pH 4. The cross-linked chitosan powder and the chitosan liquid formulation
were each dosed intranasally to a group of four sheep at a morphine
hydrochloride dose of 0.3 mg/kg. Plasma samples were collected and
analysed for morphine content using a radioimmunoassay. The
concentration time curves for the two formulations are shown in Figure 6.
The peak plasma concentration achieved was approximately 50% higher for
the chitosan powder formulation than for the chitosan liquid formulation.
Example 13
Into a 500 ml conical flask was weighed 1400 mg of the cross-linked powder
described in Example 3. To the powder was added 90 ml of water and 3.1
ml of salmon calcitonin solution (9000 IU/ml). The flask contents were
swirled intermittently for 20 minutes and then frozen by immersing the flask
into liquid nitrogen. The frozen contents were transferred to a freeze-drier
and lyophilised. 18.54 g of SuppocireTM BS2X (Gattefosse) was weighed
into a beaker and melted at 35 C. 0.36 g of the freeze-dried
calcitonin/chitosan mixture was mixed into the melted SuppocireTM. The
mixture was poured into each of four 5 g pessary moulds.
2197062
~ WO 96/05810 PCT7GB95/01980
The pessaries were allowed to set, removed from the mould, and trimmed
to a weight of 4.2 g. The pessaries were administered intravaginally to
each of four sheep. An aqueous solution containing 1600 IU/ml of salmon
calcitonin was also administered intravaginally to four sheep. The changes
in plasma calcium concentration with time for the pessary formulation and
aqueous solution are shown in Figure 7. The fall in plasma calcium,
indicative of calcitonin absorption, was markedly greater in magnitude and
duration for the pessary formulation than for the solution.
Example 14
The effect of a chitosan solution formulation on the nasal absorption of
insulin in sheep was compared with the effect of a chitosan powder
formulation in which the chitosan had not been treated in any way. A
solution of 200 IU/ml insulin in 5 mg/ml medium viscosity chitosan
solution was administered intranasally at 2 IU/kg to sheep. The powder
formulation was prepared by mixing 640 IU insulin with 80 mg chitosan
HCl 211 and 720 mg lactose and administered to the sheep at 2 IU/kg.
Blood samples were taken and plasma glucose concentrations measured.
The mean changes in plasma glucose concentration with time for the two
formulations is shown in Figure 8. The fall in plasma glucose was less
pronounced for the powder formulation than for the chitosan solution.
Example 15
Into a 100 ml conical flask were weighed 640 mg of the microspheres
described in Example 2. To the microspheres were added 26.7 ml of
water and 16 ml of a solution containing I mg/ml of a LHRH agonist.
The suspension was frozen and lyophilised. The lyophilised formulation
30' was administered nasally to each of four sheep at a dose of 2.05 mg/kg
21
2197062 ...
Wd 96/05810 PCT/GB95/01980~
(= 0.05 mg/kg of LHRH agonist~: As controls, four sheep received,
nasally, 0.05 mg/kg of LHRH agonist as an aqueous solution and four
sheep received 0.01 mg/kg as a subcutaneous injection. Plasma samples
were collected and the LHRH agonist content was measured using a
radioimmunoassay. In Figure 9, the concentration vs. time profiles are
shown for the microsphere formulation, the nasal solution and the
subcutaneous injection. The microsphere formulation resulted in a marked
enhancement in nasal absorption of the LHRH agonist. Compared to the
subcutaneous injection, the mean bioavailabilities of the microsphere
formulation and control solution were1.5% and 36.6% respectively.
Example 16
Into a 100 ml conical flask was weighed 640 mg of the microspheres
described in Example 2. To the powder was added 26.7 ml of water and
16 ml of a solution containing 1 mg/ml of a LHRH agonist. 28 g of
Suppocire BS2X (Gattefosse) was weighed into a beaker and melted at
35 C. 0.56 g of the freeze-dried LHRFi/microsphere mixture was mixed
into the melted Suppocire. The mixture was poured into each of five 5 g
pessary moulds. The pessaries were allowed to set, removed from the
mould, and trimmed to a weight of 4.2 g (= 2 mg of LHRH
agonist/pessary). The pessaries were administered intravaginally to each
of four sheep.
A gel formulation was prepared by suspending 0.6 g of the microspheres
described in Example 2 in 10 ml of a solution containing 1.5 mg/ml
LHRH agonist. 1.42 g of the gel formulation (= 2 mg of LHRH agonist)
was administered to each of four sheep from two syringes (two I ml
syringes, each containing 0.71 g of gel).
22
2197062
a., _.
0 WO 96/05810 - PCT/GB95/01980
Four sheep were administered, intravaginally, 0.4 ml of a 5 mg/ml
solution of LHRH agonist.
Plasma samples were collected and assayed for LHRH agonist content.
Plasma concentration vs. time profiles for the control solution and two
vaginal formulations are shown in Figure 10.
The two formulations containing chitosan microspheres substantially
enhanced the vaginal absorption of LHRH agonist. Compared to the
subcutaneous control (Example 15), the bioavailabilities of the control
solution, gel formulation and pessary formulation were 4.7 %, 46.0 % and
32.94b respectively.
Example 17
Into a 100 ml flask was weighed 640 mg of the microspheres described in
Example 2. To the flask was added 27 ml of water and 16 ml of a 50
mg/ml aqueous solution of low molecular weight heparin (LMWH). The
suspension was frozen and lyophilised. The lyophilised formulation was
administered intranasally to four sheep at 4.5 mg/kg (= 2.5 mg/kg
LMWH). As a control, four sheep received 1.25 mg/kg LMWH as a
subcutaneous injection. Plasma samples were collected and the anti-factor
Xa activity measured using a proprietary assay kit. By measuring the anti-
factor Xa activity in standards containing known quantities of LMWH, the
LMWH content of the sheep plasma samples was calculated. The plasma
LMWH concentration vs. time profiles for the nasal and subcutaneous
formulations are shown in Figure 11. Relative to the subcutaneous dose,
the mean bioavailability of the nasal chitosan formulation was 19%.
23
W O 96105810 2197062 PCTlGB95/01980
Example 18
Into a 100 ml flask was weighed 560 mg of the cross-linked chitosan
powder described in Example 3. To the flask was added 37 ml of an
aqueous solution containing 28 mg of a parathyroid hormone (PTH)
analogue. The suspension was frozen and lyophilised. A physical mixture
of cross-linked chitosan powder and PTH analogue was prepared by
blending together 560 mg of cross-linked chitosan powder (Example 3)
and 28 mg of PTH analogue. Blending was performed using a pestle and
mortar. The two powder formulations were administered intranasally to
four sheep at 2.1 mglkg (= 0.1 mg/kg PTH analogue). As a control,
four sheep received 0.01 mg/kg PTH analogue as an intravenous injection.
Plasma samples were collected and the PTH analogue content measured
using a radioimmunoassay technique. The plasma PTH analogue
concentration vs. time profiles for the intravenous and two nasal and doses
are shown in Figure 12. Relative to the intravenous dose, the mean
bioavailabilities of the lyophilised and physical mixture formuIations were
20.7% and 18.0% respectively.
24