Note: Descriptions are shown in the official language in which they were submitted.
~ W096~93 Y~l/rl ./Y
2 1 9723~
COMPOSITION PERMETTANT
UNE LIBÉRATION SÉLECTIVE D'UN PRINCIPE ACTIF
La présente invention a pour objet une composition
permettant une libération sélective d~un principe actif au
niveau d~un site donné, et en particulier une composition
pharmaceutique permettant une libération sélective du
principe actif au niveau d'un organe cible. La présente
invention a plus particulièrement pour objet une composition
pharmaceutique destinée à une administration par voie orale,
permettant une libération sélective du principe actif au
niveau du côlon.
De nombreuses compositions pharmaceutiques ont été
~ mises au point dans le but de permettre une libération
contrôlée du principe actif. Ces compositions sont pour la
plupart basées sur le principe d~une libération retardée ou
d~une libération prolongée du médicament au niveau ou non
d~un organe cible.
EP-A-0 250 374 décrit des mini-unités de dosage
revêtues. Le noyau de ces dernières est obtenu par
compression d~un mélange du principe actif avec des
polymères gonflant dans l~eau par un mécanisme d~osmose, le
revêtement polymère entourant le noyau contrôlant la
libération du médicament.
EP-A-0 077 956 décrit des microcapsules entériques
contenant un principe actif formant le noyau qui est entouré
d~un revêtement constitué essentiellement d~éthylcellulose
et d~un matériau polymère entérique, un matériau polymère
gonflant dans l'eau étant ~ventuellement incorporé dans le
noyau.
Gs-A-2 202 143 décrit une composition pharmaceutique
comprenant un médicament insoluble~ dans l~eau, dispersé
dans une matrice constituée de cellulose microcristalline et
d~au moins un dérivé de cellulose. une teLle composition
w096/04893 2 1 9 7 2 3 ~ rl S IU/Y ~
conduit à la libération prolongée du principe acti~ sur une
période d~au moins 8 heures. ces compositions peuvent atre
revêtues d~un revêtement entérique de façon à permettre la
libération du m~iC - -t dans le tractus intestinal plutôt
que dans l~estomac.
Les unités de dosage décrites dans ces documents ne
permettent pas d~obtenir une libération immédiate du
principe actif au niveau d~un organe précis; sa vitesse de
libération est limitée par sa vitesse de diffusion à travers
la matrice l'entourant ou le piégeant ce ~ui peut avoir pour
conséquence que la totalité de la dose administrée n~est pas
libérée au niveau de l'organe cible. En effet, dans le cas
du tractus gastro-intestinal, le temps de transit varie d~un
individu à l~autre dans de larges mesures, et les conditions
régnant dans le système gastro-intestina~, notamment le pH,
varient aussi dans de larges mesures.
Les recherches effectuées dans le but d~obtenir une
composition permettant une libération sélective des
principes actifs au niveau d~un organe cible ~ex. le côlon)
étaient axées jusqu~à présent soit sur l'utilisation de
comprimés, administrables par voie orale, enrobés de
polymères qui sont décomposés par les bactéries présentes au
niveau du côlon, soit sur l'utilisation de comprimés,
administrables par voie or~le, enrobés d~un revatement
entérigue.
Dans le premier type d'approche. on utilise le concept
de prodrogue ou ~pro-drug", à savoir précurseur du principe
actif, selon lequel l'ingrêdient actif est lié à un polymère
glucosidique par exemple (Frlend, Phillips et ~orzen, F.
Controlled Rel , 15, 47-54,(1991)). Le problème principal
dans cette approche est que le médicament lui-même est
modifié chimiquement et que la libération su principe actif
est conditionnée par une hydrolyse bactérienne in situ.
Dans le second type d~approche, il est fait usage de
revêtements constitués de polymères dont la solubilité
dépend du pH, et qui se dissolvent à un pH supérieur à celui
de l'estomac. Les produits ne peuvent se dissoudre que
lorsque la valeur du pH du milieu environnant dépasse une
certaine valeur
2 ~ 97~3~
L'utilisation de comprimés présentant ce type de __
revêtement ne conduit pas à des résultats satisfaisants et
ne permet pas d~obtenir une libération ciblée des principes
actifs. En effet, si on étudie le profil du pH du tractus
5 gastro-intestinal, on observe que le pH dans la partie
distale de l~intestin varie entre 6,8 et 7,2, puis chute à
une valeur de 4,5 à 6 au niveau de la partie ascendante du ~~
côlon. Par conséquent, la composition pharmaceutique est
concue de sorte que le revêtement polymère se dissolve à un
10 pH supérieur ou égal à 7 ; le polymère se dissout dans m
l~intestin et le principe actif est libéré à ce niveau. Or,
le pH varie grandement dlun individu à l~autre. Ainsi, si le
pH reste à des valeurs trop faibles, le revêtement entéri~ue
va rester intact et le principe actif ne va pas être libéré. , -
Il apparait donc clairement qu~un contrôle de la
libération du principe actif basé uniquement sur
l~utilisation d~un revêtement se dégradant sélectivement au
niveau ou avant d~atteindre l~organe cible ne permet pas une
libération ciblée du médicament au niveau de ce dernier,
<insérer page 3a>
Il existe donc un besoin pour une composition
permettant une libération sélective d~un principe actif au
niveau d'un site donné, et en particulier pour une
composition pharmaceutique permettant une libération
sélective d~un principe actif au niveau d'un organe cible.
~ a présente invention a pour objet d~éliminer les
inconvénients des compositions pharmaceutiques existantes, à
savoir une libération non sélective et prolongée, et de
fournir une co~position permettant une libération sélective
d~un principe actif au niveau d~un site donné, et notamment
une composition pharmaceutique permettant une lib~ration
sélective du principe actif au niveau d~un organe cible, en
particulier le côlon.
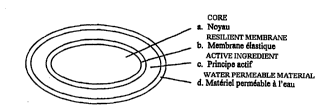
~a présente invention a pour objet une composition
comprenant, successivement:
(a) un noyau (1) constitué d~un matériau gonflant dans
l~eau;
(b) une couche (2) en un matériau élastique laissant
passer l'eau et insolu~le dans l'eau
3 FEUILLE MODIRE~
~lq~2~
WO=A-92 17165 décrit des compositions comprenant un
noyau, une couche d~excipient comprenant un matériau soluble
dans l~eau et un matériau insoluble dans l~eau, et un
revêtement entérique. Le noyau contient le principe actif
S en association avec un agent gonflant.
EP-A-0 210 5~0 décrit des compositions comprenant un
noyau inerte, une couche contenant le principe actif, une
couche en un matériau gonflant et un revêtement entérique,
ou un noyau inerte, une couche contenant le principe actif
avec un matériau gonflant et un revêtement entérique.
Ces ~e~x documents précités ne permettent pas une
libération sélective du principe actif au niveau d~un organe
cible, et de plus posent des problèmes de contamination du
principe actif par l~agent gonflant.
_
3 ~ FEUILLE MODIFIE
W096l04893 21 ~ 7 2 3 ~ /r. A /7 ~
(c) une couche (3) contenant au moins un principe
actifi et
(d) une couche (4) en un matériau devenant perméable à
l~eau dans un milieu aqueux prédéterminé, cette couche étant
5 susceptible de se rompre sous l~effet de l~expansion du
noyau (1).
Cette composition permet l~obtention d'une composition
pharmaceutique, en particulier destinée à l~administration
par voie orale, permettant une libération sélective d'un
10 principe actif au niveau d~un organe cible. -~
En présence du milieu environnant aqueux approprié, le f
matériau de la couche (4) est ou devient perméable ~ l~eau;
de l~eau pénètre dans la composition et atteint le noyau
(1). Le noyau (1) gonfle à une vitesse contrôlée par le
15 matériau élastique (2), exerçant ainsi une certaine pression
sur les couches externes. Après un certain temps, cette
pression conduit à la rupture de la couche (4) et à la mise
en contact de la couche de principe actif (3) avec le milieu
environnant, ce qui entraine la libération immédiate de la
20 totalité du principe actif. Grâce à la couche (2) qui laisse
passer l~eau mais qui est sensiblement insoluble dans l~eau,
le principe actif ne peut pas migrer dans le noyau. On évite
ainsi la formation in situ d'une forme de dosage à
libération prolongée ou retard; les inconvénients de l~art
25 antérieur sont donc évités.
selon un mode de réalisation, dans la présente - :
composition, le noyau (1) présente une expansion en volume,
de 50 à 700 ~, de préférence 100 à 500 % , plu5
préférentiellement de 150 à 300 %.
Selon un autre mode de réalisation, dans la présente
composition, les proportions relatives, en poids par rapport
au poids total de la composition, des différents éléments
constitutifs (1) à (4) sont les suivantes :
30 à 90 %, de préférence 50 à 70 %, plus
préférentiellement environ 60 ~ en poids de (1);
1 à 6 ~, de préférence 2 à 4 %, plus preférentiellement
envirOn 3 % en poids de (2);
1 à 60 %, de préférence 10 à 50 %, plus
préférentiellement environ 30 % en poids de (3);
~ W096/04893 ~ 1~ 7~ 3 4 P~llrh~ ~1079
3 à 15 %, de préférence 5 à 9 %, plus
préférentiellcment environ 7 ~ en poids de ~4).
Selon un mode de réalisation, dans la présente
composition, ledit matériau de la couche (4) étant ou
devenant perméable à l~eau dans un milieu aqueux
prédéterminé est un matériau polymère soluble dans l~eau
dans ce milieu prédéterminé, dont la solubilité dépend du
pH, ledit matériau présentant une solubilité à partir d~une
valeur de pH comprise entre pH = 6,0 et pH = 8,0.
Selon une variante de ce mode de réalisation, ledit
matériau polymère de la couche (4) comprend un matériau
polymère présentant une solubilité à partir d'une valeur de
pH de 6.8.
Selon un mode de réalisation, dans la présente
composition, le matériau de la couche (4) est un matériau
gastro-résistant et entéro-soluble.
Selon un mode de réalisation, dans la présente
composition, le principe actif est de la prednisolone ou un
de ses dérivés pharmaceutiquement acceptables.
Selon un mode de réalisation, dans la présente
composition, la couche (3) comprend 1 à 500 mg de principe
actif et de préférence de 5 à 100 mg de principe actif.
L~invention a encore pour objet ladite composition pour
son utilisation en tant que médicament.
Selon un mode de réalisation, l utilisation est une
utilisation en tant que médicament pour le traitement des
maladies du côlon.
L ' invention a encore pour objet un procédé de
préparation d~une composition selon l~invention, comprenant
les étapes suivantes :
(i) compression du noyau (1) à partir d'un mélange
pulvérulent;
(ii~ revêtement sur ledit noyau (1) de la couche (2);
(iii) revêtement sur ladite couche (2) de la couche (3)
comprenant au moins un principe actif; et
(iv) revêtement sur ladite couche (3) de la couche (4).
Tout procédé connu dans l'art approprié pour les étapes
de compression, de revêtement, et de préparation de gélule,
comprimés, etc., peut être utilisé. Les ingrédients des
2 ~
W096/04893 PCT~R9~01079 -
couches sont fournis de façon classique, par exemple sous
forme pulvérule'nte, sous forme dissoute dans un solvant
approprié, etc. -
La composition selon l~invention se présente sous une
forme classique, par exemple capsule, gélule, comprimé, etc.Cette composition est particulièrement appropriée pour une
administration p~r voie orale La dose de principe actif
devant etre administrée peut être formulée soit sous la
forme de comprimé, capsule ou gélule unique soit sous la
forme de mini-unités de dosage pouvant être administrées en
une ou plusieur's' fois, en fonction du principe actif. Ces
mini-formes sont avantageuses pour la mise en oeuvre de
l~invention.
L'invention est maintenant décrite plus en détail dans
1~ ce qui suit, et en référence aux figures l et 2.
La figure l représente une vue en coupe d~une composition
selon l~invention;
La figure 2 représente un graphe donnant le profil de
libération pour une composition selon l~invention.
En référen-ce à la figure l, le noyau et les couches
sont indiqués respectivement ~l) à (4).
Le noyau (l) comprend un matériau qui gonfle en
présence d~eau, mais qui est insoluble dans l~eau et de
préférence est un matériau polymère neutre.
2~ Tout matériau polymère connu bio-compatible et/ou
biodégradable cIassiquement utilisé peut être incorporé dans
la composition selon la présente invention. On peut citer à
titre d~exemple : méthylcelluloses de différents poids
moléculaires, alcools polyvinyliques , polymères acryliques,
hydroxypropylméthylcelluloses, et de facon générale tout
polymère naturel ou de synthèse ainsi que les copolymères en
dérivant et les mélanges en dérivant capables de gonfler au
contact de l'eau ou d~un fluide aqueux .
Le noyau peut éventuellement comprendre tout excipient
classiquement utilisé dans l'art.
La couche ~2) déposée sur le noyau constitue une
barrière qui empêche la migration du principe actif dans le
noyau et qui contrôle le taux d~expansion du noyau. Cette
couche est constituée d~un matériau, de préférence polymère,
21 9~3~ .
~ W096l04893 Y~llr~ /Y
qui laisse pagser l~eau présente dans le milieu jusqu'au
noyau, qui est élastique et sensiblement insoluble dans
l~eau. Ce matériau peut être par exemple poreux. Tous les
matériaux, polymères, copolymères et les mélanges en
dérivant, présentant les caractéristiques susmentionnées
peuvent être utilisés. Le polymère (2) présente une
élasticité définit comme suit : capacité de doubler au moins
de volume, au contact de l~eau à 37~C, sans rupture du film
périphérique Des exemples de composés sont : éthylcellulose
plus hydroxypropylméthylcellulose, méthylcellulose plus
hydroxypropylméthylcellulose, Eudragit~ NE30D, etc..
Il a aussi été mis en évidence qu'une composition comprenant
au moins un matériau polymère hydrophile et un agent
plastifiant conduisait a une couche présentant une
élasticité importante.
La couche (3) comprend au moins un principe actif, seul
ou en mélange avec des exciPients classiquement utilisés
dans le domaine pharmaceutique ou tout autre domaine en
fonction de l'utilisation finale du principe actif. Le
principe actif peut être un biocide, un acaricide, un
insecticide, un bactéricide, un fongicide, un médicament,
etc., et de préférence un composé présentant une activité
pharmaceutique. Tout composé présentant une activité,
soluble ou insoluble dans l~eau, peut être utilisé.
La couche (3) peut aussi être constituée de sous-couches,
par exemple contenant des principes actifs différents,
éventuellement séparées par des sous-couches en des
matériaux appropriés.
Dans ce qui suit, et dans un souci de commodité et de
simplicité, il sera fait référence à une composition
pharmaceutique obtenue à partir d~un principe
pharmaceùtiquement actif.
La couche ~4) est constituée en un matériau devenant
perméable à l'eau, et susceptible de se rompre sous l'effet
de l'expansion du noyau. Le terme ~matériau devenant
perméable à l~eau dans un milieu aqueux prédéterminé~
signifie que le matériau laisse passer l'eau ou est
susceptible de laisser passer l~eau après exposition à ce
milieu donné. Le terme ~milieu aqueux~ est utilisé ici selon
W096/04893 21 ~ PCT~R95/01079 -
l~acceptation classique du terme. Par exemple, ce matériau
peut devenir poreux sous l~effet de sa solubilité dans ledit
milieu ou de sa dégradation dans les conditions régnant dans
ledit milieu. Ce matériau est susceptible de se rompre sous
l~effet de l~expansion du noyau (1), une fois que l~eau,
après avoir traversé les couches ~4), ~3) et (2), ait
provoqué le gonflement du noyau ~
Cette couche (4) peut être constituée d~un matériau, de
préférence polymère, susceptible d~être~dégradé ou dissous
par un élément présent spécifi~uement immédiatement en amont
de ou au niveau du site do~né où la lib~ration doit avoir
lieu. Par exemple, ce matériau peut correspondre à un
polymère entér~que se dégradant ou se solubilisant sous
l~effet d~un pH donné, ou peut correspondre à un polymère
qui est spécifiquement dégradé par une enzyme donnée, par
exemple une enzyme pancréati~ue ou intestinale telle qu'une
esterase.
Tout revêtement, de préférence entérique, classiquement
utilisé dans le domaine pharmaceutique peut être utilisé
dans le cadre-de l~invention Des exemples de composés
sont : polymères acryligues tels que Eudragit~ en
combinaison avec un agent plastifiant, tel que
dibutylphtalate ou triethyl citrate, etc..
Le noyau (1~ et les co-uches (2) à (4) peuvent contenir
éventuellement ~des additifs appropriés connus dans l~art,
tels que par exemple stabilisant, antioxydant, colorant,
plastifiant, lubrifiant, conservateur, arôme, agents de
compression, etc. Par ailleurs, des couches supplémentaires
peuvent être prévues, comme par exemple une couche externe
conférant un arôme et/ou une couleur et/ou améliorant
l~acceptabilité du m~dicament etJou permettant un marquage.
Comme il a été indiqué ci-dessus, le mécanisme est le
suivant :en présence du milieu environnant aqueux approprié,
le matériau de.-la couche (4) laisse passer l~eau. Le noyau
(1) en présence~d~eau gonfle à une vitesse contrôlée par le
revêtement polymère (2), exercant une certaine pression sur
les couches externes. Après u~ certain temps, cette pression
conduit à la rupture de la couche (4) et à la mise en
contact de la couche de principe actif r3) avec le milieu
7 ~ q7~3 7~
environnant, ce qui entraîne la libération immédiate de L~
totalité du principe actif. Grâce à la couche (2~ qui laisse
passer l~eau~ mais qui est sensiblement insoluble dans
l~eau, le principe actif ne peut pas migrer dans le noyau.
1a présente composition permet, par un double contrôle,
à savoir un contrôle de l~p~n~i~n du noyau ~1) réalisé par
la couche (2) élastique et un contrôle~ du lieu de
dégradation de la couche (4), d~obtenir une libération
sélective et immédiate du principe actif (3) au niveau d~un
site donné, tel que par exemple et de préférence un organe
cible lorsque la composition est de préférence une
composition pharmaceutique.
La composition des couches (1), (2) et (4) va être
modifiée en fonction de la vitesse d~expansion du noyau (1)
qui est requise. On peut remarquer notamment que plus les
matériaux, de préférence polymères, (2) et (4) sont
hydrophiles, plus la vitesse de pénétration de l~eau est
importante et plus la vitesse de gonflement du noyau est
grande. Par ailleurs, la vitesse de libération du principe
actif peut être contrôlée par la nature du constituant du
noyau qui offre une expansion plus ou moins forte.
Si l~on souhaite par exemple que la composition
pharmaceutique libère le principe actif au niveau du côlon,
alors, la couche (4) peut être constituée d~un revêtement
entérique (à savoir gastro-résistant et entéro-solublel dont
la solubilité dépend du pH. Dans un tel cas, lorsque le
système pharmaceutique va être en présence d~un milieu
environnant dont le p~ est supérieur ou égal à la valeur à
partir de laquelle le matériau (polymère) (4) se dissout, la
couche (4) va 'commencer à se dissoudre, ce qui conduit à la
formation de pores permettant la pénétration du fluide
gastro-intestinal dans la composition. Le fluide gastro-
intestinal va provoquer le gonflement du noyau, la vitesse
d~expansion du noyau doit alors être contrôlée de sorte que
la rupture du revêtement (4) n~apparaisse qu~au niveau du
côlon.
Les exemples suivants illustrent l~invention sans
toutefois en limiter la portée.
g F~UlLLE MODIFIE
Wog6/o~s3 ~ 1 9 7 ~3 ~ . ~llrl Q ~1079 -
~x le 1 :
Par des procédés classlques, on pr~pare un comprimé
présentant les caractéristiques suivantes :
- composition du noyau (1) :
5 hydroxypropyl méthylcellulose 26,00 mg
cellulose microcristalline 52,00 mg
hydroxypropylcellulose 35,00 mg
(faible degré de substitution, L-HPC~)
silice colloidale 1,50 mg
10 st~arate de magnésium 0,50 mg
- composition de la couche (2) :
éthyl cellulose 3,00 mg
hydroxypropyl méthylc=ellulose 2,50 mg
dibutyl phtalate 1,50 mg
lS - composition de la couche (3) :
métasul~obenzoate de prednisolone 31,43 mg
hydroxypropyl méthylcellulose ~20,00 mg
- composition de la couche (4) :
Eudragit~ S100 14,00 mg
20 dibutyl phtalate 3,50 mg
F.x le 2 : . ~~
Par des procédés classiques, on prépare un comprimé
présentant les caractéristiques suivantes :
- composition du noyau (1) :
25 hydroxypropyl méthylcellulose :26,00 mg
cellulose microcristalline 52,00 mg
hydroxypropylcellulose 35,00 mg
(faible degré de substitution, L-~PC~)
silice colloidale 1,50 mg
30 stéarate de magnésium 0,50 mg
- composition de la couche (2) :
éthyl cellulose 3,00 mg
hydroxypropyl méthylcellulose 2,50 mg
triéthyl citrate 1,50 mg
35 - composition de la couche (3~ :
métasul~obenzoate de prednisolone 31,43 mg
hydroxypropyl méthylcellulose 20,00 mg
~0
~1 9723~
W096/04893 r_llr-_.l/Y
- composition de La couche (4) :
Eudragit~ slOO lo, Oo mg
hydroxypropyl methylcellulose 4,00 mg
triéthyl citrate 3,50 mg5 E ~e 3 :
Par des procédés classiques, on prépare un comprimé
présentant les caractéristiques suivantes :
- composition du noyau tl) :
hydroxypropyl méthylcellulose 26,00 mg
10 cellulose microcristalline 52,00 mg
hydroxypropylcellulose 35,00 mg
(faible degré de substitution, L-HPC~)
silice colloidale 1,50 mg
stéarate de magnésium 0,50 mg
15 - composition de la couche t2) :
Eudragit~ ME30D 5,00 mg
- composition de la couche t3) :
métasulfobenzoate de prednisolone 31,43 mg
hydroxypropyl méthylcellulose 20,00 mg
20 - composition de la couche t4) :
Eudragit~ S100 10,00 mg
hydroxypropyl méthylcellulose 4,00 mg
triéthyl citrate 3,50 mg
~x~ le 4 :
Par des procédés classiques, on prépare un comprime
présentant les caractéristiques suivantes :
- composition du noyau (1) :
hydroxypropyl méthylcellulose 26,00 mg
cellulose microcristalline 52,00 mg
30 hydroxypropylcellulose 35,00 mg
(faible degré de substitution, L-HPC~)
silice colloldale 1,50 mg
stéarate de magnésium 0,50 mg
- composition de la couche (2) :
Eudragit~ NE30D 5,00 mg
- composition de la couche (3) :
métasulfobenzoate de prednisolone 31,43 mg
hydroxypropyl méthylcellulose 20,00 mg
21 97~
W096/04893 r~llrl__[l~lY -
- composition de la couche (4) :
hydroxypropyl méthylcellulose 15,0Q mg
dibutyl phtalate 3,00 mg
E~~-~,le 5 :
On procède à une étude clinigue in vivo sur six
volontaires sains (3 hommes, 3 femmes). On prépare des
comprimés placebo contenant 2 mg d~oxyde de samarium enrichi
en isotope Sm 153, a partir de la composition de
l~exemple 1, dans laquelle le principe ac~if a été remplacé
par un dérivé cellulosique. De tels comprimés sont
administrés aux patients. A l~aide d'une technique classique
de scintigraphie, on détermine la position anatomique et le
temps de désintegration du comprimé.
Après un léger petit déjeuner, chaque sujet reçoit un
comprimé avec 2~0 ml d~eau. Les résultats sont enregistrés
périodiquement.~ Ensuite chaque sujet reçoit un léger
déjeuner et l~enregistrement est poursuivi. Les résultats
sont donnés à -la figure 2. Cette fiaure, qui indique les
temps d~arrive~e dans le côlon et le moment de la
désintégration initiale du comprimé, montre clairement que
le placebo radiomarqué se désint~gre au niyeau du côlon, ce
qui signifie _ qu~un principe actif serait libéré
sélectivement e~ immédiatement aussi au niveau du côlon.
12