Note: Descriptions are shown in the official language in which they were submitted.
~20g86q
PATENT RULES
SECTION 104(4) NOTICE
It is the applicant's wish that, until either a patent has issued on the basis of
the application or the application is refused, or is abandoned and no longer
subject to reinstatement, or is withdrawn, the Commissioner only authorize the
furnishing of a sample of any deposited biological material referred to in the
specification to an independent expert nominated by the Commissioner in
accordance with section 109 of the Patent Rules.
Feb. 3,1 997 JDM:sbf
C:~l<EEP\S10-INFO.PGS
~ WO96/12406 2 2 0 0 8 6 9 PCT~S95/13253
GENE TRERAPY INVOLVING CONCURRENT AND R~PEATED
ADMINISTRATION O~ AD~NO~lKUSES AND
IMMUNOSUPPRESSIVE AGENTS
This application is a continuation-in-part of
Application Serial No. 08/478,482, filed June 7, 1995, which
is a continuation-in-part of Application Serial No.
08/325,679, filed October 19, 1994, the disclosures of which
are incorporated by reference.
.
This invention relates to gene therapy comprising the
use of adenoviruses as the gene delivery vehicles. More
particularly, this invention relates tO gene therapy
invoi~ing the concurrent and repeated administraticn of
adenGviruses and immunosuppressive agents, wherebv the
efficiency of the gene therapy treatment is enhanced through
suppression of an immur.e response against the adenoviruses.
BACRGROUND OF T~E INVENTION
Adenovirus genomes are linear, double-stranded DNA
molecules of approximately 36 kilobase pairs. Each extremity
of the virai genome has a short sequence :~nown as the
~nverted terminal repeat (or ITR), which is necessary for
viral replication. The well-characterized molecular genetics
of adenovirus render it an advantageous vector for gene
transfer. Portions of the viral genome can be substituted
--1--
2 2 0 0 8 6 ~ PCT~S9S/13253
WO96/12406
with DNA of foreign origin. In addition, recomkinant
adenoviruses are structurally stable.
Adenoviruses can be very efficient in gene transfer into
cells in vivo, and, thus may be employed as delivery vehicles
for introducing desired genes into eukaryotic cells, whereby
the adenovirus delivers such genes to eukaryotic cells by
.
binding cellular receptors. There are, however, several
limitations to adenovirus gene transfer which are due in part
to host responses directed at either the adenovirus vector
particle, breakdown products of the vector particle, or the
transduced cells. These host responses include non-specific
responses and specifi- lmmune responses. The non-specific
responses include inflammatory and non-inflammatory changes.
An example of the latter is a change in host cell gene
expression. Specific immune responses include various
cellular responses and humorai antibody responses. Cellular
responses include those mediated by T-helper lymphocytes, T-
suppressor lymphocytes, cytotoxic T lymphocytes (CTL), and
natural killer cells.
Despite the high efficiency of adenovirus vector
mediated gene transfer, the translent nature of adenovirus
vecto- ~ediated gene transfe- ~as suggested that repeat
administrations of adenovirus ve~~ors may be necessary.
Recen. s~udies in cott^n rats however, have demonstrated
that host immune responses dire~teà towards adenoviral
vectors correlate wi~h decreased e ficiency of gene transfer
and expression after repeated administration. Yei et ~ 7 .,
Gene i~rerapy, l:l92-~Q0 (1994).
Smith, ~t al., Nature Genetics, Vol. 5, pgs. 357-402
(l9g3) discloses the administration to mice of an adenoviral
vector including a human Factor IX gene. Such administration
resulted in efficient liver transduction and plasma levels of
2 2 0 0 8 6 9
WO96/12406 PCT~S95/13253
human Factor IX that would be therapeutic for hemophilia B
patients. Human Factor IX levels, however, slowly declined
to baseline by nine wee~s after injection, and were not re-
established by a second vector injection. Smith, et al.,
also found that neutralizing antibodies to adenovirus block
successful repeat administration of the adenovirus.
Kozarsky, et al ., J. Biol. Chem., Vol. 269, No. 18, pgs.
13695-13702 (May 6, 1994) discloses the infusion of an
adenoviral vec~or including DNA encoding the LDL receptor to
rabbits. Stable expression of the LDL receptor gene was
found in the rabbits for 7 to 10 days, and diminished to
undetectable levels within 3 weeks. The development of
neutralizing antibodies to the adenovirus resulted in a
second dose being completely ineffective.
Kass-Eisler, et al ., Gene Therap~, Vol. 1, pgs. 395-402
(1994) suggest thac a T-cell response contributes to, but is
not responsible solely for, the limited duration of
expresslon in adul_s from adenovirus vectors. The authors
further show that cyclosporin A is not effective in blocking
the humoral response to the vector.
Fang, et a ., _. Cell. Biochem., Supplement 21A, C6-109,
pg. 363 ;1595i d sclose the attempted re-injection of an
adenovirus vector ~. dogs which were treated with cyclosporin
A, an immunosuppressive agent. Such attempted re-injection
was unsuccessfli.
Yang, et al . , Proc. Nat. Acad. Sci., Voi. 91, pgs. 4407-
4411 (May 1994J describe recombinant adenoviruses in which
the Ela and E'b regions have been deleted. ~uch viruses also
include a ~ransgene. When such adenoviruses are administered
to an animai hos~, cells harboring the recombinant viral
genome express the transgene as desired; however, low level
expression of viral genes also occurs. A virus-specific
--3--
220086q
W O 96/12406 PCT~US9S/13253
cellular immune response i8 stimulated that leac1s to
destruction of the genetically modified cells, thereby
limiting the duration of expression of the transgene.
The aforementioned studies clearly document the need for
a method of circumventing or blocking the host immune
response to adenovirus vectors which prevents effective
readministration o the vector, and limits the effectiveness
of expression; however, they do not describe how to
accomplish this.
It is therefore an object of the present invention to
provide for sustained efficacy of gene transfer via repeated
administration of adenoviral vectors, and for sustained
expression of the transferred gene, through the suppression
of an immune response against the adenoviral vectors.
BRIEF DESCRIPTION OF THE DRAWINGS
The invention now will be described with respect to the
drawings, wherein:
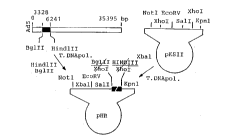
Figure 1 is a schematic of the construction of plasmid
pHR.
Figure 2 is a schematic of the construction of an
expression vehicle including an adenoviral ITR, an
encapsidation signal. a Rous Sarcoma '~Tirus promoter, an
adenoviral triparti,e leader sequence, and linking sequences;
Figure 3 is a schematic of the construction of plasmid
pAvS6;
Figure 4 is a map of plasmid pAvS6;
Figure 5 is a map of plasmid pBQ4.7;
2200869
PCT~S95/13253
WO96112406
Figure 6 is a map of plasmid pAvS6 - CFTR;
Figure 7 is a schematic of adenoviral vectors AvlLucl
and AvlCf2;
Figure 8 is a map of plasmid pGEM-luc;
Figure 9 is a map of plasmid pAVS6-luc;
Figures lOA, lOB, and lOC depict the histologic
appearance of the lung in response to AvlCf2 administration
three days after vector administration;
Figures llA and llB are graphs showing the effect of
dexamethasone administration on lung lavage cells at 3 days
and 42 days after the administration of AvlCf2;
Figure 12 is a graph of anti-adenoviral antibody titers
of lung lavage samples from rats infected with AvlCf2 and
which were ~reated or not treated with dexamethasone; and
Figure 13 is a graph of CTL responses in rats 42 days
after infection with AvlCf2;
Fi~lre 14 is a graph of luciferase enzyme activity in
ra.s infected with AvlC 2 and which were treated or no~
treated with dexamethasone, followed by infection with
AvlL~Icl;
Figure 15 is a map of plasmid pAvlHgFR;
Figure 16 is a schematic of the adenovirai vector
AvlH9F2;
2200869
WO96/12406 PCT~S95/13253
Figure 17 is a graph of plasma human Factor IX levels in
mice which were given AvlH9F2, and were or were not given one
of the immunosuppressive agents deoxyspergualin,
cyclophosphamide, or dexamethasone with or without the
administration of the vector AvlLac24 five weeks earlier;
Figure 18 is a graph of plasma Factor IX levels (ng/ml)
in mice which received from lxlO; to lxlO~ pfu of AvlLacZ4,
~ollowed by administration of AvlH9FR five weeks later;
~ igure l9 is a graph of neutralizing antibody titer in
mice that were given AvlLacZ4, and received no
immunosuppression, o- were treate~ with decxyspergualin,
cyclophosphamide, or dexamethasone;
Flgure 20 is a graph of plasma Factor VIII levels in
mice which were g ven AvlLacZ4, and received no
immunosuppression, or were given cyclophosphamide, followed
by administration of AvlH9F2 with or without
cyclophosphamide, followed by adm,nistratior. of AvlALAPH81;
and
Figure 21 is a graph of piasma Factor IX levels (ng/ml)
in mice which received AvlLacZ4 and 0 mg/kg, 5 mgJkg, lO
mg/kg, 2û mg/kg, or 3~ ~g,'kg de^~spergualir, foiiowed by
administration of AvlH5F2.
DE:TAILED D~5SCRIPTION Ol~ T~E INV~NI ION
Ir accordance with an aspect of the presen_ invention,
there is provided a method of e'fe-ting a gene therapy
treatment in a host. Th- method comprises admlnistering to
a host (i) an adenoviral vec~or ir~luding a. 'east one DNA
sequence and (ii) an immunosuppressive a~ent. T~e course of
administration of tne adenovirai vector and immunosuppressive
agen. then is discontinued. A~mi ni stration of the
.
2200869
PCT~S95/13253
WO96/12406
immunosuppressive agent and the adenoviral vector t~en is
repeated at least once. The adenoviral vector is
administered in an amount effective to produce a therapeutic
effect in the host. The immunosuppressive agent is
administered in an amount effective to prevent or suppress a
humoral and/or cellular immune response to the vector and/or
cells containing the vector.
.
The term "DNA sequence" as used herein, refers generally
to a polydeoxyribonucleotide molecule and more specifically
to a linear series of deoxyribonucleotides connected one to
the other by phosphodiester bonds between the 3' and 5
carbons of the adjacent pentoses.
Applicants have found that, when an immunosuppressive
agent is administered with the adenoviral vector, and then
adminis~ration of the vector is repeated, one achieves
enhanced efficacy of the repeat in vivo adenoviral-mediated
gene transfer through suppression of an immune response (such
as a humoral antibody response! against the adenoviral vector
and/or cells transduced with the vector, and thereby achieves
increased expression of the transferred genes.
The adenoviral vector which is employed may, in one
embodimen_, be a. adenoviral vecto- which includes
esser-ially the complete adenoviral genome. Shenk et al .,
Curr. Tsp. Mlc~obiol. Immunol., 1'1 (3):l-39 (l984).
Alternat:veiy, the adenoviral vecto- may be a modified
adenovira; vec~cr in which at least a portion of the
adenoviral genome nas been deleted.
In another embodiment, the adenoviral vector comprises
an adenoviral 5' ITR; an adenoviral 3' ITR; an adenoviral
encapsidation signal; at least one DNA sequence encoding a
therapeutic agent; and a promoter controlling the at least
--7--
2200869
WO96/12406 PCT~S95/132S3
one DNA sequence encoding the therapeutic agent. The vector
is free of at least the majority of adenoviral El and E3 DNA
sequences, but is not free of all of the E2 and E4 DNA
sequences, and DNA sequences encoding adenoviral proteins
promoted by the adenoviral major late promoter.
In one embodiment, the vector also is free of at least
a portion of at least one DNA sequence selected from the
group consisting of the E2 and E4 DNA sequences.
In another embodiment, the vector is free of at least
the majority of the adenoviral El and E3 DNA sequences, and
is free of a portion of the other of the E2 and E4 DNA
sequences.
In still another embodiment, the gene in the E2a region
that encodes the 72 kilodalton binding protein is mutated to
produce a temperature sensitive protein that is active at
32C, the temperature at which the viral particles are
produced. This temperature sensitive mutant is described in
Ensinger et al., ~. Virology, 10: 328-339 (1972); Van der
Vliet, et al., J. Virology, 15:348-354 (1975); and Friefeld
et al., Virology, 12~:380-389 (1983~; Englehardt, et al.,
Proc.Nat. Acad. Sci., Vol. 91, pgs. 6196-6200 (June 1994);
Yang, et a'., Nature Genetics, Vol. ~, Dgs 362-369 (July
1994).
Such a vector, in a preferred embodiment, is constructed
first by constructing, according ~o standard techniques, a
shut~le plasmid which contains, beginning at the 5' end, ~he
"critical left ena elemerts," which inciude an adenoviral 5'
ITR, an adenoviral encapsidation slgna', and an Ela enhancer
sequence; a promoter (which may be an adenoviral promoter cr
a foreign promoter); a multiple cloning site (which may be as
hereinabo~e described); a poly A signal; and a DNA segment
--8--
2200869
WO96/12406 PCT~S95/132S3
which corresponds to a segment of the adenoviral genome. The
vector also may contain a tripartite leader sequence. The
DNA segment corresponding to the adenoviral genome serves as
a substrate for homologous recombination with a modified or
mutated adenovirus, and such sequence may encompass, for
example, a segment of the adenovirus 5 genome no longer than
from base 3329 to base 6246 of the genome. The plasmid may
also include a .selectable marker and an origin of
replication. The origin of replication may be a bacterial
origin of replication. Representative examples of such
shuttle plasmids include pAvS6, shown in Figure 4. A desired
DNA sequence encoding a clotting factor may then be inserted
into the multiple cloning site to produce a plasmid vector.
This construct is then used to produce an adenoviral
vector. Homologous recombination is effected with a modified
or mutated adeno~ir~s in whlch at least the ma,ority cf the
El and E3 adenoviral DNA sequences have been deleted. Such
homologous recombination may be effected through co-
transfection of the plasmid vector and the modified
adenovirus into a helper cell line, such as 293 cells, by
CaPOL precipitation. Upon such homologous recombination, a
recombinant adenoviral vector is formed that includes DNA
sequences derived from the shuttle plasmid be~ween the Not I
site and the homologous recombination fragmen,, and DNA
derived from the El and E3 deleted adenovirus between the
homologous recombination fragment and the 3~ ITR.
In one embodiment, the homologous recombinatior.fragment
overlaps with nucleotides 3329 to 6246 of tne adenovirus,5
(ATCC '~R-5) genome.
Through such homologous recombination, a ve,ctor is
~ormed which includes ar. adenoviral 5' ITR, an ader.Gviral
encapsidation signal; an Ela enhancer sequence; a promoter;
_g_
220086q
WO96/12406 PCT~S95/132~3
at least one DNA sequence encoding a therapeutic agcnt; a
poly A signal; adenoviral DNA free of at least the majority
of the El and E3 adenoviral DNA sequences; and an adenoviral
3' ITR. The vector also may include a tripartite leader
sequence. This vector may then be transfected into a helper
cell line, such as the 293 helper cell line (ATCC No.
CRL1573), which will include the Ela and Elb DNA sequences,
which are necessary for viral replication, and to generate
infectious adenoviral particles. Transfection may take place
by electroporation, calcium phosphate precipitation,
microinjection, or through proteoliposomes.
The vectors hereinabove described may include a
multiple cloning site to facilitate the insertion of the at
least one DNA sequence encoding a therapeutic agent into the
cloning vector. In general, the multiple cloning site
includes "rare" restriction enzyme sites; i.e., sites which
are found in eukaryotic genes at a frequency of from about
one in every lO,000 to about one in every lO0,000 base pairs.
An appropriate vector in accordance with th~ present
invention is thus formed by cutting the cloning vector by
standard techniques at appropriate restriction sites in the
multiple cloning site, and then ligating the DNA sequence
enccding a therapeutic agent irto the cloning vector.
The adenoviral vector, as stated hereinabove, includes
at least one DNA sequence enccding at least one therapeutic
agent. The term ~herapeutic~ i 9 used in a generic sense and
includes ~reating agents, prophylactic agents, and
replaceme-.t agents.
~ NA seouences encoding therapeutic agents which may be
placed into the adenoviral ve_tor include, but are not
limited to, DNA sequences encoding tumor necrosis factor
(TNF) genes, such as TNF-~; genes encoding interferons such
-10-
2200869
WO96/12406 PCT~S95/13253
as Interferon-~, Interferon-~, and Interferon-~ ; genes
encoding interleukins such as IL-l, IL-l~, and Interleukins
2 through 14; genes encoding GM-CSF; genes encoding adenosine
deaminase, or ADA; genes encoding antioxidants such as Mn-
SOD, catalase, CuZnSOD, extracellular superoxide dismutase,
and glutathione reductase; genes which encode cellular growth
factors, such as lymphokines, which are growth factors for
lymphocytes; genes encoding gro~th factors such as epithelial
growth ~actor (EGF) and keratinocyte growth factor (KGF);
genes encoding soluble CD4; Factor VIII; Factor IX; von
Willebrand's factor; T-cell receptors; the LDL receptor,
ApoE, ApoC, ApoAI and other genes involved in cholesterol
transpor, and metabolism; the alpha-l antitrypsin (~lAT)
gene, the ornithine transcarbamylase (OTC) gene, the CFTR
gene, lung surfactant protein genes, the B-glucuronidase
gene, the insulin gene, negative selective markers or
"suicide" genes, such as viral thymidine kinase genes, such
as the Herpes Si~plex Virus thymidine kinase gene, the
cytomegalovirus virus thymidine kinase gene, and the
varice 'a-zoster virus thymidine kinase gene; Fc receptors
for antigen-binding domains of antibodies, and antisense
seouences which inhibit viral replication, such as antisense
sequences which inhibit replication of hepatitis B or
hepat~tis non-A non-B virus; tissue piasminogen activator
~tpA?; urinary piasminogen acti~ator (urokinase)i hirudin;
nitr~c oxide syn.hase; vasoactive pep.ides; and angiogenic
peptides.
The DNA sequence encoding a therapeutic agent is under
the ccntrol of a suitable promoter. Suitable promoters which
may be employed include, but are not limlted to, adenoviral
promoters, such as the adenoviral major late promoter; or
heterologous promoters, such as the cytomegalovirus ( CMV?
promoter; the Rous Sarcoma Virus (RSV) promoter; inducible
promoters, such as the MMTV promoter, the metallothionein
220086q
PCT~S95/132~3
WO96/12406
promoter; heat shock promoters; the albumin promoter; and the
ApoAI promoter. Alternatively, the DNA sequence encoding a
therapeutic agent may be under the control of its native
promoter. It is to be understood, however, that the scope of
the present invention is not to be limited to specific
foreign genes or promoters.
Immunosuppressive agents which may be employed include
those which prevent: (i) a humoral (antibody) response
against the adenoviral vector; (ii) a cellular immune
response, such as, for example, a T-cell response to cells
containing the adenoviral vector; or (iii) a non-specific
inflammatory responses against the vector and against cells
containing the vector. By preventing a humoral and/or T-cell
and/or non-specific inflammatory response against the vector,
and/or cells containing the vector administration of the
immunosuppressive agent permits effective re-administration
of the vector in order to produce a therapeutic effect in the
host. Preferably, where repeat administration of the
adenoviral vectcr i9 desired, the immunosuppressive agent i5
an immunosuppressive agent which prevents a humoral antibody
response against the adenoviral vector. Preferably, where
longer duration or nigher levels of expression is desired,
the immunosuppressive agent is one that prevents or
suppresses a cellular sr non-specifi_ inf'ammatory response.
Host immune responses to in ~ivo adenovinls vector
administration vary ir. relation to 'i) tne dose of vector;
(ii) the route of administration; ~i i! the level of
replication (if any occurs); (iv) the nat~re of the transgene
contained in the recombinant vector; (v! the genetic and
physiological characteristics of thC host; and (vi) t;~e
existence and ievei cf pre-existing immune responses to
previously adminis~ered adenovirus vectors.
In general, host responses are dependent on the dose of
vector administered. Importantly, the magnitude of specific
-12-
22û0869
WO96/12406 PCT~S95/13253
host responses is dependent on the route of ve-tor
administration. For example, intravenous administration will
yield a higher host antibody response than that of an
equivalent amount of vector given via the respiratory route.
The diverse host responses to adenovirus vectors occur
due to separate inflammatory and immune effector mechanisms,
although most, if not all, of these distinct molecular
mecha,nisms are connected and significantly interdepen~ent.
Thus, for example, humoral antibody formation is very
dependent on certain T-helper lymphocyte support. Also, some
cell-mediated cellular toxicity is dependent on antibody
formation, e.g., opsonized macrophage cell killing.
The two principal host responses affecting the duration
o~ transgene expression are the inflammatory response and the
cellular immune response.
Inflammation is one of the first host responses that
occurs following vector administration. Cytokine release ls
very likely involved in the subsequent in lux of in~lammatory
cells. Such cytokines likely include IL-1, IL-6, IL-8, and
TNF .
The amount cf inflammation seen ~ollowing in vivo
adenov~ral vector administration increases ~ith incresing
doses cf vector given. Higher doses lead to a more rapid
decline in transgene expression than do smaller doses.
CTL responses directed towards the transduced cells are
believed to be important in reducins the duration of
transgene expression. It is believed ~ha~ the CTL are
direc~ed against low level adenovirus ger.e expression by the
cells, which induces the CTL.
The principal host response affecting the ability to
administer adenovirus vectors repeatedly to a host is the
humoral antibody response. It develops tc adenovirus
-13-
220086q
WO96/12406 PCT~S95/132~3
administered by a variety of routes, including oral,
intravenous, intraperltoneal, and intrapulmonary. In
general, the level of antibody response achieved i5 very
dependent on the dose of vector administered. The antibody
response is also dependent on the route of vector
a~i n; stration. Intravenous vector administration results in
higher antibody levels than pulmonary administration for a
given dose of vector. In contrast, wild type adenovirus
elicits high antibody levels irrespective of the amount of
virus given due to virus replication in vivo. The ability to
repeat successfully adenovirus vector administration is
inversely correlated with the level of circulating anti-
adenovirus vector antibody present.
Pharmacologic modulation of host immune responses to
adenovirus vectors involves the use o' anti-inflammatory
agents, celllular immune modifiers, and humoral antibody
immune modifiers.
An.i-inflammatory agents include steroids,
cyclophosphamide, and azothiophrine.
Steroids have potent anti-inflammatory properties.
Applicants have showr. that steroids, such as dexamethasone,
given parenterally prolong th- durar i5n of transgene
expression following in vivo adm nistratlon or vector~ia the
lung route. Steroids also bloc~ the function of lymphocytes.
Thus, dexamethasone reduces the CTL responses observed after
pulmonary vector administratior. Dexamethasone also blocks,
at ieast in part, the host antibody response to adenovirus
vec-or administration.
Antibodies directed at -ellular components of the immune
system reduce cellular immune response. For example, anti T-
cell receptor antibody (such as, for example anti-CD4 and
anti-CD3 antibodies) administration prolongs transgene
expression. CTLA4 immunoglobulin is another example. Anti-
-14-
220086~
WO96112406 PCT~S95/13253
CD4 antibody is directed against the T-helper lymphocytes and
reduces their function. Other agents directed primarily at
the cellular immune response include cyclosporins such as
cyclosporin A; rapamycin binding protein ligands such as
FK506; and steroids such as dexamethasone.
Agents which affect humoral antibody responses are
generally directed at antibody producing B lymphocytes (B-
cells) or at the T-cells which are responsible for in~ucing
B-cell antibody production to high levels.
Examples of immunosuppressive agents which prevent a
humoral antibody response against the adenoviral vector
include, but are not limited to, deoxyspergualin, or DSG,
which has the following structure:
H.NCNH(CH~)lCH7CH.CONHCHCONH(CH2)lNH(CHJ)3NH,
NH OH
The terms "deoxyspergualin and "DSG" as used herein,
mean deoxyspergualin or DSG and derivatives or analogues
therecf, such as salts of deoxyspergualin, including but not
limited tO , trihydrochlorides thereof, and any other
analogues which have immunosuppressive activity. Such
compounds are described further in U.S. Patent Nos.
4,525,259; 4,817,299; 5,162,581; and 5,196,453.
In o..- embodiment, the immunosuppressive agent which
prevents a r,umoral antibody response is a steroid. Steroids
which may be employed include, but are nol limited to,
dexamethasone, and any adrenocortical hormones, such as,for
example corticosteroidsi hydrocortisone; prednisolone; and
methylpre~n solone.
In another embodimen~, the immunosuppressive agent which
prevents a humoral antibody response is a cyclosporin, such
as, for example, cyclosporin A. Other immunosuppressive
agents which prevent a humoral antibody response and which
may be employed include, but are not limited to,
220086~
WO96/12406 PCT~S9S/132~3
azathioprine; cyclophosphamide; brequinar; leflunGmide;
mycophenolate mofetil; anti-CD40 antibodyi anti-CD40 ligand
antibody; cyclophosphamine; rapamycin; anti-CD4 antibody;
CTLA-4 immunoglobulin; Interleukin -12; Interferon -~;
rapamycin binding protein (FEBP) ligands, such as, for
example, FK506, as described in Bierer, et al., Proc. Nat.
Acad. Sci., Vol. 87, pgs. 9231-9235 (1990); Dumont, et al.,
J. Immunol., Vol. 144, pgs. 1418-1424 (1990); and Bierer, et
al., Science, Vol. 250, pgs. 556-559 ~1990); anti-lymphocyte
function antigen-l (LFA-l) antibody; and anti-T-cell receptor
antibody.
Applicants have found that, when compounds which
prevent, suppress, or eliminate humoral immune responses to
foreign antigens (such as, for example, deoxyspergualin,
cyclophosphamide, brequinar, leflunomide, mycophenolate,
mofetil, anti-CD40 antibody, or anti-CD40 ligand antibody)
are administered at a short time prior to, and/or during,
and/or for a short time after adenoviral vector
administration, to a host, such compounds prevent the
production of anti-adenoviral neutralizing antibodies in the
host. The prevention of the production of such neutralizing
antibodies enables the efficient re-administration of the
adenoviral vector to the host.
It is to be understood that, withln the scope of the
present invention, that an immunosuppressive agent may
prevent more than one of the immune responses hereinabove
described. It also is to be understood, however, that the
scope of the present invention is not intended to be limited
to any specific immunosuppressive agents.
.
It is also contemplated that within the scope of the
present invention, a combination of immunosuppressive agents
may be employed.
2 2 ~ q ~ l
WO96/12406 PCT~S95/13253
The adenoviral vector and immunosuppressive agent, in
general, are administered concurrently in an amount effective
to produce a therapeutic effect in the host while preventing
an immune response against the vector or against cells
transduced with the vector. The term "concurrently," as
used herein, means that the administration of the adenoviral
vector and administration of the immunosuppressive agent are
begun at approximately the same time, i.e., within a brief
time frame of each other, and the administration of the
adenoviral vector and the administration of the
immunosuppressive agent are parts of a unitary course of
treatment. Thus, for example, the immunosuppressive agent is
administered at approximately the same time the adenoviral
vector is administered, i.e., the administration of the
immunosuppressive agent is begun at a short time (for
example, about 24 hours) before, or during, or at a short
time (e.g., 24 hours) after the administration of the
adenoviral vector. In general, the ;mmllnosuppressive agent
is administered according to standard dosage schedules
established for that agent, and for a period of time which in
general does not exceed 14 days, and preferably does not
exceed ll days, and more preferably does not exceed 8 days.
Thus, long-term administration of the immunosuppressive agent
is not required for enabling repeated administration of the
adenovirus.
At the conclusion of the course of administration of the
immunosuppressive agent, the course of administration of the
adenoviral vector and immunosuppressive agent is discontinued
for a period of time. The period of time between courses of
administration of the adenoviral vector and the
immunosuppressive agent, and the number of courses of
administration of the adenoviral vector and immunosuppressive
agent is dependent upon a variety of factors, including the
age, weight, and sex of the patient, the disease or disorder
-17-
SUBSTITUTE SHEET (RULE 26)
220086q
WO96/12406 PCT~S95/13253
being treated, and the severity of the disease or disorder
being treated.
It is to be understood that the above course of
administration of the immunosuppressive agent is repeated
with each administration of the adenoviral vector.
In one embodiment, the adenoviral vector may be
administered, at each administration, in an amount of from l
plaque forming unit ~o about lO;' plaque forming units,
preferably from about lO~ plaque forming units to about lo;t
plaque forming units, more preferably from about lO~ to about
lOI~ plaque forming units per kg. The host may be a human or
non-human animal host.
The adenoviral vector may be administered systemically
or topically. Examples of systemic administration include,
but are not limited to, intravenous administration (such as
for example, portal vein injection or peripheral vein
injec~ion), intramuscuiar administration, intraperitoneai
administration, intranasal administration, or encapsulated
oral administration.
The immunosuppressive agent is ad~._r.is.ered in an amo~n~
effective to produce a desired immunosuppressive effec~ in
tne host. The immunosuppressive agent may be administere~,
at eacn administration, in an amount cf from about l ~gjkg -o
about 15 mg/kg, when dexamethason- is employed, or at ~ne
dose equivalents for other steroids. When deoxyspergualln is
employed, the deoxyspergualin may be administered in an
amoun_ of from about l mg/kg to about 33 mg/kg, preferabiy
from about 3 mg/kg to about 7 mg/ka. When cyclophcsphamide
is employed, the cyclophopham1de may be administered in an
amount of from about 5 mg/kg to about 300 mg/kg, preferably
from about 50 mg/kg to about lO0 mg/kg.
-18-
2200869
WO96/12406 PCT~S95/132S3
The adenoviral vector particles and the
immunosuppressive agent each may be administered in
combination with a pharmaceutically acceptable carrier
suitable for administration to a patient. The carrier may be
a liquid carrier such as, for example, a saline solution.
The adenoviral vector particles also may be administered in
combination with a solid carrier, such as, for example,
microcarrier beads, or a sustained drug delivery material,
such as, for example, a polyol.
Cells which may be transduced by the adenoviral
particles include, but are not limited to, lung, airway, or
alveolar epithelial cells; primary cells, such as primary
nucleated blood cells, such as leukocytes, granulocytes,
monocytes, macrophages, lymphocytes (including T-lymphocytes
and B-lymphocytes), totipotent stem celis, and tumor
infiltrating lymphocytes (TIL cells); bone marrow cells;
endothelial cells; activated endothelial cells; epithelial
cells; keratinocytesi stem cells; hepatocytes, including
hepatocyte precursor cells; fibroblasts; mesenchymal cells;
mesothelial cells; parenchymal cells; vascular smooth muscle
cells; brain cells and other neural cells; gut enterocytes;
gut stem cells; and myoblasts.
In one embodimen~, the adenovira pa~ti_les may be
targeted tO blood ceils, whereby such adenoviral vector
particles infect the blood cells with a gene which directly
or ir.directly enhances the therapeutic effects of the blood
cells. The gene carried by the blood cells can be any gene
which allows the blood cells to exert a therapeutic efrect
that it would not ordinarily have, such as a gene encoding a
clotting factor useful ir the treatment of hemophilia. The
gene can encode one or more products having therapeutic
effects. Examples of suitable genes include those that
encode the CFT~ gene; cytokines such as TNF, interleukins
-19-
2 2 0 0 8 6 q PCT~S95/13253
WO96/12406
(interleukins 1-14), interferons (~, B, ~-interferons), T-
cell receptor proteins and Fc receptors for antigen-binding
domains of antibodies, such as immunoglobulins. Other
examples of suitable genes include genes encoding soluble CD4
which is used in the treatment of AIDS and genes encoding ~-
antitrypsin, which is useful in the treatment of emphysema
caused by ~-antitrypsin deficiency.
,
The transduced cells are useful in the treatment of a
variety of diseases including but not limited to, cystic
fibrosis, adenosine de~m;n~.ce deficiency, sickle cell
anemia, thalassemia, hemophilia, diabetes, ~-antitrypsin
deficiency, brain disorders such as Alzheimer's disease,
phenylketonuria and other illnesses such as growth disorders
and heart diseases, for example, those caused by alterations
in the way cholesterol is metabolized and defects of the
immune system.
In another emDodiment, the adenoviral vector particles
may transduce liver cells, and Suc:rL adenoviral vector
particles may include gene(s) encoding polypeptides or
proteins which are useful in preventicn and therapy of an
acquired or ar inherited defect in hepatocyte (liver)
function. For example, they can be used to correct an
inhe_iteà de~iciency of the low densi~-v l poprotein (T DL)
receptor, and/or to correct an inherited dericiency of
ornithine transcarbamylase (OTC), which results in eongenital
hyperam~.onemia.
Ir. another embodimen., the adenovira_ parti_les may
transduce liver cells, whereby tne adenoviral particles
include a gene encoding a therapeutic agent emp'oyed to treat
acquired infectiQus diseases, such as diseases resulting from
viral infection. For example, the infectious adenoviral
particles may be employed to treat viral hepatitis,
-20-
220~6~
WO96/12406 PCT~S95/132~3
particularly hepatitis B or non-A non-B hepatitis. For
example, an infectious adenoviral particle containing a gene
encoding an antisense gene could be employed to infect liver
cells to inhibit viral replication. In this case, the
infectious adenoviral particle, which includes a vector
including a structural hepatitis gene in the reverse or
opposite orientation, would be introduced into liver cells,
resulting in production in the infected liver cells of an
anti-sense gene capable of inactivating the hepatitis virus
or its RNA transcripts. Alternatively, the liver cells may
be infected with an infectious adenoviral particle which
includes a gene which encodes a protein, such as, for
example, ~-interferon, which may confer resistance to the
hepatitis virus.
The vector particles also may be employed in treating
Hodgkin's lymphoma. An infectious adenoviral vector particle
may be targeted to neoplastic cells of Hodgkin's lymphoma.
The adenoviral vector particle also includes a negative
selective marker or "su cide gene, such as the Herpes Simplex
thy~idine kinase gene. The adenovirus may be administered ln
vivo tG a patient, whereby the virus infects neoplastic cells
of Hodgkin's lymphoma. After the adenovirus is administered
to the patient, the patient is given an interaction agen.
such as gancyclovir or acyclovir, whereby the neoplastic
Hodgkin's lymphoma cells infected with the adenovir~s are
killed.
In addition, a vector may be constructed which includes
the CFLR gene. The vector then may b^ administered to the
respiratory epithelium in an effective therapeutic amount for
the correction of the pulmonary defici~ in patients with
cysti^ fi~rosis. In another example, vectors containing
functional proteins may be delivered to the respiratory
epithelium in order to correct deficiencies in such proteins.
-21-
WO96/12406 2 2 0 0 8 6 9 PCT~S9S/13253 ~
Such functional proteins include antioxidants,
antitrypsin, CFTR, lung surfactant proteins, cytokines, and
growth factors such as BGF and KGF, and may also include
adenosine deaminase for treatment of severe combined immune
de~iciency, von Willebrand's ~actor for treatment of
Christmas disease, and ~-glucuronidase for treatment of
Gaucher's disease. Also, vectors including genes encoding
anti-cancer agents or anti-inflammatory agents may be
administered to lung cells of a patient for the treatment of
lung cancer or inflammatory lung disease.
EXAMPLES
The invention now will be described with respect to the
following examples; however, the scope of the present
invention is not intended to be iimited thereby.
Exam~le l
Construction of AvlCf2 and AvlLucl
A. C~nstruction of PAvS5
The adenoviral construction shuttle plasmid pA~S5 was
constructed in several steps using standard cloning
techniques including polymerase chain reaction based cloning
techniques. First, the 2913 bp BglII, HindIII fragment was
removed from Ad-dl327 and inserted as a blurt fragment into
the XhoI site of pBluescript II KS-(Stra~gene, La Jolla, C~)
(Figure l).
Ad-dl327 is identical to adenovirus 5 except .hat an
XbaI ~ragment including bases 28591 to 33474 (or map units
78.5 to 84.7) of the Adenovirus 5 ge.Lome, and which is
located in the E3 region, has been deieted. The E3 deletion
in Ad-dl327 is similar to the E3 deletion in Ad-dl324, wh ch
is described in Thimmappaya et al., Celi, 3i:543 (1983). The
complete Adeno~irus 5 genome is registered as Genba.~k
accession #M73260, incorporated herein by rererence, and the
.
-22-
2200869
WO96/12406 PCT~S95/13253
virus is available from the American Type Culture Collec'ion,
Rockville, Maryland, U.S.A. under accession number VR-5.
Ad-dl327 was constructed by routine methods from
Adenovirus 5 (Ad5). The method is outlined briefly as
follows and previously described by Jones and Shenk, Cell
13:181-188, (1978). AdS DNA is isolated by proteolytic
digestion of the virion and partially cleaved with Xba I
restriction endonuclease. The Xba I fragments are then
reassembled by ligation as a mixture of fragments. This
results in some ligated genomes with a sequence similar to
Ad5, except excluding sequences 28591 bp to 30474 bp. This
DNA is then transfected into suitable cells (e.g. KB cells,
HeLa celis, 293 cells) and overlaid with soft agar to allow
plaque formation. Individual plaques are then isolated,
amplified, and screened for the absence of the 1878 bp E3
region Xba fragment.
The orientation of this fragment was such that the BglII
site was nearest the T7 RNA polymerase si~e of pBluescript II
KS- and the HindIII site was nearest the T3 RNA polymerase
site cf pBluescript II KS-. This plasmid was designated PHR.
~Figure 1).
Second, the ITR, e~capsidation signa', Rous Sarcoma
Virus promoter, the adenoviral ~r~partite leader (TPL)
sequence and linking sequences were asse~bled as a block
using PCR amplification (Figure 2). The ITR and
encapsidation signal (sec~ences 1-392 of Ad-dl327 [identical
to sequences from AdS, GenbanX accession #M73260]
incorporated herein by reference ! were amplified
(amplification 1~ together from Ad-dl327 using primers
containing NotI or AscI ~estriction sites. The Rous Sarcoma
Virus LTR promoter was amplified (amplification 2) from the
plasmid pRC/RSV (sequences 209 to 605; Invitrogen, San Diego,
-23-
~ 2200869
WO96/12406 PCT~S95/13253
CA) using primers containing an AscI site and an SfiI site.
DNA products from amplifications 1 and 2 were joined using
the "overlap" PCR method (amplification 3) (Horton et al .,
BioTechni~ues, 8:528-535 (1990)) with only the NotI primer
and the SfiI primer. Complementarity between the A~cI-
containing end of each initial DNA amplification product from
reactions 1 and 2 allowed joining of these two pieces during
amplification. Next the TPL was amplified (amplification 4)
(sequences 6049 to 9730 of Ad-dl327 [identical to similar
sequences from AdS, Genbank accession #M73260]) from cDNA
made from mRNA isolated from 293 cells (ATCC Accession No.
CRL 1573) infected for 16 hrs. with Ad-dl327 using primers
containing SfiI and XbaI sites respectively. DNA fragments
from amplification reactions 3 and 4 were then joined using
PCR (amplification 5) with the NotI and XbaI primers, thus
creating the complete gene biock.
Third, the ITR-encapsidation signal-TPL fragment was
then purified, cleaved with NotI and XbaI and inserted into
the NotL, XbaI cleaved PHR plasmid. This plasmid was
designated pAvS6A- and the orientation was such that the NotI
site of the fragment was next to the T7 RNA polymerase site
(Figure 3).
Fourtn, the SV40 early polyA signal was removed from
SV40 DNA as an HpaI-BamHI fragment, treated with T4 DNA
pol~merase and inserted into the Sall site of the plasmià
pAvS6a-;Figure 3) to create pAvS6 (Figures 3 and 4).
B. Construction of AvlCf2 and AvlLucl
Av'Cf2 tFigure 7) (Yei et al., Gene Therapy, 1:192-20C
(1994)), an E1-deleted (i.18 map units ~o 9.2 map units), E3-
deleted (78.5 map units to 84.7 map units) adenoviral vector
constructed first by inserting the normal human CFTR cDNA
coding sequence fragment into the EcoRV si~e of pAvS6 so that
-24-
2 2 0 0 8 6 9 PCT/USg5/13253
WO96/12406
the 5' end of the CFTR coding sequence was closest .o the
Adenovirus 5 tripartite leader- The CFTR cDNA was removed as
a PstI fragment (nucleotides 75 to 4,725; for numbering see
GenBank Accession No- M28688) from the plasmid pBQ4.7 (Figure
5) (provided by L.-C- Tsui, The Hospital for Sick Children,
Toronto, Canada), and inserted as a blunt fragment. The
resulting plasmid, pAvS6-CFTR (Figure 6) was linearized with
KpnI and recombined with the large (35 kb) ClaI fraglnent of
Ad-dl327 in 293 cells as described in Trapnell, Advanced Drug
Delivery Reviews, 12:185-199 (1993) to form AvlCf2 (Figure
7) .
After double-plaque purification, the identify of the
clonal isolates was confirmed by Southern analysis,
immunoprecipitation of CFTR, as prevlously described.
(Tolstoshev, et al., Proceedings of the Ninth Nagoya
International Symposium on Cancer Treatment, September 17-18,
1993, Nagoya, Japan (in press).
AvlLucl (Figure 7) (Yei e~ al., Gene ~herapy, Vol. 1,
pgs. 192-200 (1994) ) is an adenoviral reporter vector
identical in genomic organization and sequence to AvlCf2,
except that it expresses the firefly luciferase gene (Genbank
Accession No. M15077).
The firefly luciferase gene was obtained from pGEM-luc
(Figure 8 - Promega! pGEM-luc was digested with StuI and
HindIII in order to splice out the firefly luciferase gene.
The firefly luciferase gene was inserted into the
EcoRV si~e of pAvS6 so that the 5' end of the firefly
luciferase coding sequence was closest to the Adenovirus 5
tripartite leader- The resulting plasmid, pAvS6-Lucl (Figure
9) was linearized with KpnI and recombined with the large (35
kb) ClaI fragment of Ad-dl327 as hereinabove described.
-25-
.~. 2200869
WO96/12406 PCT~S95/13253
Clonal isolates then were identified as herei~above
described.
Both viral vectors were propagated, purified by double-
banding in CsCl gradients~ and titered in 293 cells as
described in Rosenfeld et al ., Cell, 68:143-155 (1992).
ExamPle 2
Adenoviral-mediated qene transfer with
concurrent intermittent steroid administration
Cotton rats (weight approximately 150g) were divided
into four groups with 9 rats in each group. AvlCf2 was
administered by intranasal inhalation (Yei et al., Human Gene
Therapy, Vol. 5, pgs. 731-744 (1994)) to the lungs of cotton
rats at a low dose (10~ pfu) or at a high dose (101 pfu),
either with or without coadministration of dexamethasone by
intraperitoneal injection in an amount of 2 mg/kg daily,
beginning 1 day prior to and continuing for 10 days after
administration of the vector. A control group of rats was
given PB~ ir.stead of AvlCf2, either w~th or without
coadministration of dexamethasone as hereinabove described.
At 3 days and at 42 days after vector administration, 3
rats from each group were evaluated fo~ hosr responses to the
AvlCC2 vector. Evaluations sf hos. responses included
pulmonary histopathology appearance, ~o-al lung lavage
cellularity, lung lavage anti-adenovirus antibody production,
and cytotoxic .-lymphocyte (CTL' response.
Evaluation of pulmonary histopathoio~y was performed as
described -n Yei et al., Human Gene ~herapy, Vol. 5, pgs 731-
744 (1994j. The lungs were removed, fixed in 2.5~ (wt./vol.`~
paraformaldehyde, 0.25~ (wt./vo' . ~ giutaraldehyde al 4C
overnight, embedded in paraffin, and 6~m sections were
stained with hematoxylin and eosin.
-26-
220086~ ^
WO96/12406 PCT~S95/13253
The histologic appearance of the lungs of the ~otton
rats in response to AvlCf2 injection, with and without daily
administration of immunosuppression therapy, at three days
after adenoviral infection, is shown in Figures lOA, lOB, and
lOC. Figure lOa is a section of the lung of a control rat
that did not receive adenovirus and instead received PBS.
Figure lOB is a section of the lung of a rat which received
AvlCf2 without immunosuppression therapy. Figure 10- is a
section of the lung of a rat which received AvlCf2 with daily
administration of dexamethasone. All sections have been
magnified 100 times. As shown in Figures lOA, lOB, and lOC,
there was less pulmonary parenchymal inflammation in the rat
which received adenovirus and immunosuppressive therapy as
compared with the control rat and the rat infected with
adenovirus, but did not receive immunosuppression therapy.
Lung lavage fluid was collected by lavaging the lung
with 4.0 ml of PBS, and the total number of cells determined
by counting in a hemocytometer, or the cells were evaluated
in cytocentrifuge preparations for the percentage of
neutrophils by light microscopy.
Figure llA is a graph cf the lung lavage cell count from
rats infected with AvlCf2 three days after infection, as
compared with con~rcl ra.s which rece~ved PBS. The control
rats either received dexamethasone or did not receive
dexamethasone. Figure llB is a grapn of the lung lavage cell
coun~ from rats infected with AvlCf2 at 42 days after
infection, as compared w~h control rats which received PBS.
The rats either receivea dexamethasone ~r did not receive
immunosuppressant therapy.
As shown in Figure llA, dexamethasone significantly
reduced the non-specific host cellular inflammatory responses
(represented by total lung lavage cellularity) at three days
-27-
- 220086~
WO96/12406 PCT~S95/13253
after vector administration, which is the peak of
inflammation.
Lung lavage anti-adenovirus production was measured by
an ELISA assay carried out as follows.
10~1 of AvlLacZ4 (Yei, et al., Human Gene Therapy, Vol.
5, pgs. 731-744 (1994)) at lxlOIl pfu/ml was added to 9~ ~l of
double distilled H~0 in a 0.5 ml Eppendorf tube. The tube was
irradiated with ultraviolet light for 30 minutes in order to
kill the adenovirus, and protein concentration was measured
with a Bio-Rad kit. 8 ml of O.lM Na.C03 (pH 9.6) was added to
the tube to provide a protein yield of 10 ~g/4 ml or 125 ng
of adenoviral antigen per 50 ~l per well.
50~1 of antigen then was added to each well of a 96-well
microtiter plate (Immulon 2); and the plate was incubated at
37C for 1 hour, or room temperature for 2 hours, or at 4C
overnight. The plate then was washed twice with PBS or
doub_e disti;ied HØ
300~1 of blocking buffer (1~ BSA in PBS) was added to
each well, and the plate was incubated for ' hour at room
temperature. The plate then was rinsed wi~h ~ouble distilled
H 0.
Blocking agent then was added to the background wells.
5C~l of antibody samples (i.e., lung lavage samples prepared
as hereinabove described) then were added to ccated wells at
seria' two-fold dilutions, beginning at 1/1 and ending at
1/8192. 50 !~1 of negative control sampies of serum from an
uninfected cotton rat were added to another set of coated
welis at the same serial dilutions. The plate the was
inc~bated for 2 hours at room temperature, and 300 ~l of
0.05~ Tween 20/PBS then was added. The plate was incubated
22 00869 - ~
WO96/12406 PCT~S95113253
for 5 minutes at room temperature, emptied, and 300~1 of
O.05~ Tween 20/PBS again was added. The plate again was
incubated at room temperature for 5 minutes, and wa~ emptied.
The plate then was washed twice with double distilled H~0 or
PBS.
Peroxidase-labeled goat anti-hamster IgG (l0~g/l0~l) was
diluted with l0 ml BSA and PBS to make a working solution of
l mg/ml concentration. (l:l,000 dilution). l00 ~l of this
solution then was added to each well, and the plate was
incubated at room temperature for 2 hours. The plate then
was washed five times with 300 ~l of 0.0l~ Tween 20/PBS. The
plate then was emptied and dried.
100 ~1 of tetramethyl benzidine (TM3) substrate was
added to each well at room temperate, and the color was
developed immediately. The blue color was monitored by
reading at ODo~. with an ELISA reader. The reaction was
stopped when the OD~,o was 0.5 to 0.6 by adding l00 ~l of TMB
sto~ solution to the wells. The ODI~, then was read betwee..
5 minutes and l hour after stopping the reaction. The
antibody titer is the reciprocal of the dilution that gave an
OD value of 0.l larger than background OD. Alternatively,
antibody titer also may be determined as 3 standard
devia ions above the OD of non-specifi- ackground. Th~
average results, expressed in antibody titer, for the rats in
each group at 3 days and 42 days after vector administration
are shown in Figure 12. As shown in Figure 12, the rats
which we~e given dexamethasone showed a decreased antibody
titer ~2 days after vector administration as compared with
rats ~hat we~e not given dexamethasone.
C~' assays were carried out at 42 days after vector
administration.
-29-
2200869
WO96/12406 PCT~S95/13253
Sensitizer cells were prepared by infecting cotton rat
lung fibroblasts with Ad-dl327 at a multiplicity of infection
of lO0. The cells were incubated for 3 days, and checked for
hexon expression by FACS. The cells then were washed with
PBS/EDTA, contacted with trypsin, washed, spun, and
resuspended in l ml RPMI medium. The cells then were
irradiated with 13 Cs at 5,000 rads in order to inactivate the
DNA.
Spleens then were isolated from uninfected (control)
rats and adenovirus-infected rats 42 days after infection.
The spleens were kept in sterile HBSS and ice. lO ml of HBSS
then was injected into each spleen with a 25/27 gauge needle.
The spleen was mashed, and filtered with a cell strainer into
a 50 ml tube. The ~olume then was brought to 40 ml in RPMI
plus lO~ FCS. The tube was spun at l,500 rpm for lO minutes.
Red blood cells then were lysed by adding 2.5 ml of ACK lysis
buffer, and the liquid was swirled for less than l minute.
The volume was brought up to 53 ml with RPMI-lO. The tube
then was spun again at l,500 rpm for 0 m nutes. The cell
pellet then was resuspended, and celis were counted at a l:lO
dilution. The splenocytes then were plated with the
sensitizer cells at a ratio of splenocytes to sensitizer
cells of 4:l in ~PMI. The splenocytes and sensitizer cells
were incubated at 37C in th~ presence Or 20-50 un tsi~; of
Interleukin-2. In~erleukin-2 was adde~ daily for 5 ~o
days.
Target cells were prepared by in~ecring 3XlO~ cotton rat
lung fibroblasts with Ad-dl327 at a multiplicily of infectior
of lO0 for l hour. ~ulture medium is added to tne cells, and
`iCr in an amount greater than 50 ~Ci is adde ~or 18 hours.
Target cells are harvested by washing the cotton ra~
lung fibroblasts with EDTA/PBS, followed by trypsinization.
-30-
220086q
WO96/12406 PCT~S95/13253
The cells then were washed, spun, resuspended in 5 ml culture
medium, and counted. The cells were resuspended to 105
cells/ml and 10~ cells/0.1 ml well were used for the CTL
assay.
Effector cells (i.e., the combination of splenocytes and
sensitizing cells (also sometimes referred to as Es cells)
were spun at 1,500 rpm for 10 minutes at 4C. The ce ls were
resuspended in 2 ml of HBSS-10, loaded onto 7 ml Ficoll
Hypaque, and spun at 1,500 rpm for 10 minutes. The top
portion (4 ml) was harvested, and 5 ml of culture medium was
added. This material was spun, the cell pellet was saved,
and resuspended in 1 ml of culture medium. The effector
cells were counted by mixing 50 ~l of effector cells with 50
~1 Trypan blue.
The effector cells then were added to wells containing
10' target cells, at effector:target (E:T) ratios of 3.125,
6.25, 12.5, 25, 50, and 100. The cells then were spun at 500
rpm for 5 mlnutes. The ceiis t~en were incubated at 37C for
4 hours. The cells then were spun, and 100 ~l of supernatant
was analyzed for ~icr release with a WALLAC gamma counter.
The average results for CTB response in splenocytes taken
~rom in~ected rats (with and without dexamethasone
_reatment), and frcm tws un_r.fected controi rats, are shown
in Figure l3.
As shown in Figure 13, a lower CTL response was obtained
from splenocytes obtained from infected rats that were
treated with dexamethasone.
A~ 42 days after vector aaministration, the remaining
three rats in each gro ? received an intrar.asal pulmona~y
administration of AvlLu_1 at a dose of 2xlO pfu. Remaining
control rats, which initially received PBS, also received
-31-
220086q
WO96/12406 PCT~S95/132~3
AvlLucl at a dose of 2xlO9 pfu. Lung lavage anti-adenovirus
antibody production was evaluated three days after
administration of AvlLucl, according to the procedure
hereinabove described, and the results are shown in Figure
12. As shown in Figure 12, the rats which initially were
given 10l~ pfu of AvlCf2, and were treated with dexamethasone,
showed a decreased antibody titer as compared with rats that
were not given dexamethasone.
The efficacy of transfer and expression of the firefly
luciferase gene was evaluated three days after administration
by quantifying directly intrapulmonary luciferase enzyme
activity in light units (lu) by routine luminometry as
described in Yei et al ., Gene Therapy, Vol . 1 , pgs 192-200
(1994). The results are shown in Figure 14. As shown in
Figure 14, each dot represents the luciferase enzyme activity
for one rat, and the cross-bar represents the mean for each
group. The efficiency of repeat adenovirus-mediated gene
transfer was significantly higher in the rats which received
AvlCf2 ar.d dexamethasone than those which did not receive
dexamethasone at the time of the first adenoviral
administration (11,786 + 3523 lu vs. 622 + 192 lu,
respectively). The efficiency of gene transfer from AvlLucl
also was higher in the control group which initially received
PBS in conjunction with dexamethasone.
ExamPle 3
Suppression of humoral immune response with
DS~ or hiqh dose cyclophos~hamide, Permittinq
effec-ive re~eat administration of an adenoviral vector
This example describes the intravenous administration of
the adenoviral vectors AvlLacZ4 and AvlH9F2 to C57BL/5 ma'e
mice (Harian Sprague Dawley, Indianapolis, Indiana) at 5 to
6 weeks of age at the start of the experimen.. AvlLacZ4 is
an adenoviral vector which includes a nuclear targeted B-
-32-
~ WO96/12406 2 2 0 0 ~ 6 ~ PCT~S95/132~3 _
galactosidase gene, (lacZ) and is described in PCT
application No. W095/09654, published April 13, 1995.
AvlH9F2 is constructed from a derivative of the adenoviral
shuttle plasmid vector pAvlH9FR (Figure 15), which includes
human Factor IX DNA, and is described in PCT application No.
W094/29471, published December 22, 1994.
. .
To construct AvlH9F2, the shuttle plasmid pAvlH9FR was
digested with the restriction enzyme Sfil, the-DNA ends were
made blunt using T4 DNA Polymerase, and the DNA molecule was
recircularized by ligation. Competent DH5 cells were
transformed and ampicillin-resistant clones were amplified
and screened by restriction enzyme digestion of miniprep DNA.
A positive clone was identified and the resulting shuttle
plasmid was referred to as pAvS17H9F.
Subsequently, 293 cells were cotransfected with
pAvS17H9F and the large DNA fragment of ClaI digested Ad-
dl327. Recombinant adenoviral vector plaques were picked,
expanded, and screened for expression of Factor IX by ELISA.
A positive clone was identified and amplified, thus
generating the vector AvlH9F2. A schematic of the left end
of the vector is shown in Figure 16. AvlH9F2 has a base pair
deletion at the beginning of the tripartite leader, or TPL,
which effectively changes the ATG into a CTG. The structure
of the vector was verified by restriction enzyme diagnostics
and by DNA sequence analysis of the region between the RSV
promoter and the 3' untranslated region of the Factor IX
cDNA.
Fifteen mice were ;mmllnosuppressed with 33 mg/kg of
deoxyspergualin DSG (Nippon Kayaku Co. LTD, Tokyo, Japan),
delivered intraperitoneally (ip), once daily, beginning the
day before vector administration and continuing for a`total
of eight days. A vial containing 100 mg of lyophilized DSG
SUBSTITUTE SHEET (RULE 26)
220086q
PCT~S95/13253
WO96/12406
was reconstituted with 3.8 ml of injection grade water to
yield a 25 mg/ml solution, which was aliquoted and frozen at
-20C. Each day, immediately before imml~nosuppression, an
aliquot was thawed at room temperature and 0.7 ml wa~ mixed
with 6.3 ml of Hank's Balanced Salt Solution (B SS) to yield
a 2.5 mg/ml solution. The mice were weighed once,
immediately prior to the first dose of DSG. Six mice
received l x lO~ pfu of AvllacZ4 via tail vein injection on
the second day of the immunosuppression regimen and l x 108
pfu of AvlH9F2 five weeks later. Another six mice received
only A~lH9F2, five weeks after immunosuppression. Three mice
were immunosuppressed, but received no adenoviral vector.
Six mice were immunosuppressed with a low dose (lO0
mg/kg) cyclophosphamide (Sigma) and fifteen mice were treated
with a high dose (300 mg/kg). The animals received a single
ip injection of cyclophosphamide one day before
administration of adenoviral vector. All six mice which were
treated with a low dose of cyclophosphamide also received l
x lC~ pfu of AvlIacZ4 the day after cyclophosphamide and l x
lOd pfu of AvlH9F2 five weeks later. Six of the mice
immunosuppressed with a high dose of cyclophosphamide
received a l x lO~ pfu of AvllacZ4 the next day and l x lO~
pfu of AvlH9F2 five weeks later. Another six did not receive
AvllacZ4 but did receive AvlH9F2. Finally, three mice were
immunosuppressed, but received no adenoviral vector.
~ elve mice were immunosuppressed with 5 mg/kg
dexamethasone (American Reagent Laboratories, Inc., Shirley,
New York), delivered ip, once daily, Deginnlng the day ~efore
vector adminisrration and continuing for a total of eight
days. S x of these mice receive l x lO~ pfu of AvllacZ4 on
the second d~y of dexamethasone treatment and l x lQ~ Ffu of
AvlH9F2 five weeks later. Five immunosuppressed mice
received only AvlH9F2 and one mouse received no vector.
2200869
WO96/12406 PCT~S95/13253 -~v
Five weeks after administration of AvllacZ4, but prior
to ~m; n; stration of AvlH9F2, plasma was prepared from some
mice and analyzed for antiadenovirus neutralizing antibodies.
Neutralizing antibodies were detected in the plasma of mice
which receive AvlLacZ4 without ;mmllnosuppression, however,
mice which received vector and either DSG or high dose
cyclophosphamide had no detectable neutralizing antibodies.
In contrast, mice ;mml~nosuppressed with low dose
cyclophosphamide or dexamethasone developed neutralizing
antibodies.
Neutralizing antibody titers in 20 mice are given in
Table I below. As indicated in Table I, DSG is
deoxyspergualin, Cy is cyclophosphamide, and Dex is
methasone.
Table I
mouse vector ;mmllnosuppression neutralizing
Ab
l none none 0
2 none none 0
3 lxlO8 pfu none 64
AvllacZ4
4 lxlO8 pfu none 32
AvllacZ4
lxlO8 pfu none 32
AvllacZ4
6 lxlO8 pfu none 32
AvllacZ4
7 none 33 mg/kg DSG o
8 none 33 mg/kg DSG o
9 lxlO8 pfu 33 mg/kg DSG 0
AvllacZ4
lO lxlO8 pfu 33 mg/kg DSG 0
AvllacZ4
ll none 300 mg/kg Cy o
12 none 300 mg/kg Cy 0
-35-
SUBSTITUTE SHEET (RULE 26)
220086~
WO96/12406 . PCT~S95/132S3
13lx108 pfu 100 mg/kg Cy 64
AvllacZ4
14lx10~ pfu 100 mg/kg ~y 8
AvllacZ4
15lx10~ pfu 300 mg/kg ~y 0
AvllacZ4
16lx10~ pfu 300 mg/kg ~y 0
AvllacZ4
17 none 5 mg/kg Dex 0
18 none .5 mg/kg Dex 0
19lx108 pfu 5 mg/kg Dex 8
AvllacZ4
20lx108 pfu 5 mg/kg Dex 8
AvllacZ4
One week after administration of AvlH9F2, plasma was
prepared and analyzed by ELISA to determine ~he levels of
hl-m~n Factor IX. The results are shown in Figure 17. Mice
which received AvllacZ4 without immunosuppression, then
received AvlH9F2 five weeks later, expressed no human Factor
IX. Mice which received neither immunosuppression nor
AvllacZ4, but were treated with AvlH9F~, expressed an average
of 9.2 ~g/ml. Mice which were immunosuppressed with DSG at
the time of AvllacZ4 deiive~y, and the- rece'ved AvlH9F2,
expressed an average of 6.6 ~g/ml. ~ c- which were
immunosuppressed with DSG, but received ~.o AvllacZ4, and then
treated with. AvlH9F2, had an average level of 5.2 ~g/ml.
Finally, mice which were treated wi~h DSG, but received
neither vector, expressed no human Facto~
Mice immunosuppressed with a ~ow dose of
cyclophosphamide at the time of AvllacZ~ admln stration, did
not express human Factor IX after delive~y of AilH9F2. Mice
treated with a high dose of cyclophosphamide, but not
administered AvllacZ4, expressed an average of 8 ~g/ml one
-36-
2200~69
WO96/12406 PCT~S95/13253
week after delivery of AvlH9F2. Mice immunosuppressed with
a high dose of cyclophosphamide at the time of AvllacZ4
treatment, expres-~ed an average of 15.4 ~g/ml of human Factor
IX after treatment with AvlH9F2. Mice treated with
cyclophosphamide, but not treated with either adenoviral
vector, did not express human Factor IX.
Mice immunosuppressed with dexamethasone, bu. not
treated with AvllacZ4, expressed an average of 5.5 ~g/ml of
hl~m~n Factor IX one week after administration of AvlH9F2.
However, mice which received AvllacZ4 at the time of
immunosuppression did not express Factor IX after delivery of
AvlH9F2. Mice which were immunosuppressed, but not treated
with either vector, did not express human Factor IX.
ExamPle 4
Su~Dresslon of humoral immune resDonse to adenoviral
vectors to enable the re~eat administrations thereof
This example is an ela~oration and expansion of the data
contained in Example 3. In this example, the following
materials and methods were employed.
Adenoviral vectors
A~;LacZ4 and AvlH9F2 were aescribed in Example 3
nereinabove. AvlH9FR was made by cotransfecting 293 cells
with pAvlH9FR (Figure l5) with tAe large DNA fragment from
ClaI d1gested Ad dl327. Recombinan~ adenoviral vector
plaques were picked, expanded, and screened for expression of
Factor IX by ELISA. A positive clone was identified and
amplified, thus generating the vector AvlH3FR. This vector,
like AvlH9F2, contains a centrally truncated first intron and
the compleee 5' and 3' untranslated regions from the human
Factor IX gene. The centrally truncated first intron and 3'
untranslated region are essentially the same sequences
-37-
22~0869
WO96112406 PCT~S95/132~3
described by Jallat, et al., EMBO J., Vol. 9, pgs. 3295-3301
( 1990 ) .
AvlALAPH81 is an adenoviral vector which contains the B-
domain deleted human Factor YIII cDNA expressed from the
mouse albumin promoter, and is described in published PCT
Application No. WO94/29471.
All vector stocks contained less than 1 in 106 wild-type
adenovirus, as determined by quantitative PCR analysis of Ela
sequences.
Im}nunosuppressants
Deoxyspergualin (manufactured by Nippon Rayaku Co.,
Ltd., Tokyo, Japan) was a gift from Bristol-Myers-Squibb,
Princeton, N.J. A 100 mg vial of deoxyspergualin was
reconstituted with water to a final concentration of 25
mg/ml, aliquoted, and frozen at -70C. Frozen stocks were
thawed at room temperature and diluted with Hanks Balanced
Salt Solu~ion (BSS) prior to in~ectior..
Deoxyspergualin is an immunosuppressant currently being
tested clinically in organ transplantation. It has a potent,
long ter~ effect on antigen specific B cells and has been
showr to prevent effectively the production of specific
antibody when co-administered with protein antigens.
(Alegre, et al ., TransPlantation, Vol. 57, pgs. 1786-1794
(1994'; Tepper, Ann. N. Y. Acad. Sci., pgs. 123-132 (1993);
Tufveson, et al., TransPlant. Proc., Vol. 26, pgs. 3029-3039
(1994)j. The mode of action cf DS5 is not fully understood
at the molecular level. The data suggest that it may
interfere with differentiation of t3 and T cells and also with
antigen processing. Recent studies showed that DSG inhibited
K light chain expression and therefore blocked IgM expression
on the surface of pre B cells. (Tepper, 1993). In addition,
-38-
22008~9
WO96/12406 PCT~S95/13253
the data showed that DSG inhibited nuclear translocation of
NFKB (Tepper, 1993), which could be the mechanism by which
DSG inhibits differentiation of B and T cells. Finally, it
has been ~o~Rtrated that DSG binds to Hsc70, a heat shock
protein. (Tepper, 1993). Heat shock proteins are involved
in protein folding, molecular chaperoning, peptide loading of
MHC molecules, and antigen presentation. Therefore, binding
of DSG to Hsc70 may explain the effect on antigen
presentation.
Cyclophosphamide (Cy) was obtained from Sigma and
Dissolved in HBSS. Dexamethasone (Dex) solution from
American Regent Laboratories, Inc., Shirley, N. ~-. was
diluted in HBSS prior to injection. All three
immunosuppressants were delivered intraperitoneally,
according to the doses indicated hereinbelow.
~n; m~ 1 procedures
C57BL/6 mice were obtained from Harlan Sprague Dawley
(Indianapolis, IN). Adenoviral vectors were administered via
tail vein injection after diluting the appropriate amount of
vector stock to 0.5 ml with Hanks Balanced Salt Solution
(HBSS). At the time points indicated in the text, blood was
obtained from the retroorbital plexus. For preparation of
plasma samples, sodium citrate was added immediately to a
final concentration of 0.38~ (w/v). To prepare sera samples,
the biood was allowed to clot. Samples were centrifuged for
5 min. in an Eppendorf Microfuge after which the plasma or
serum was collected, aliquoted, and frozen.
Human Factor VIII ELISA
Plasma levels of human Factor VIII were determined by
ELISA, as described in Connelly, et al., Human Gene Therapy,
Vol. 6, pgs. 185-193 (1995). The limit of sensitivity with
mouse plasma samples containing B domain deleted human Factor
-39-
SUBSTITUTE SHEET (RULE 26)
220086~
WO96/12406 PCT~S95/13253
VIII was 3 to 6 ng/ml. Mouse plasma samples were diluted 1:5
prior to the assay, therefore, the actual limit of detection
was 15 to 30 ng/ml.
Human Factor IX ELISA
Plasma levels of human Factor IX were determined by
ELISA. Asserachrom IX:Ag ELISA kits were purchased from
American Bioproducts Company (Parsippany, NJ) and assa~-s were
performed according to the manufacturer's instructions. The
limit of sensitivity was 1.6 ng/ml.
Anti-adenoviral antibody assay
Mouse plasma or serum samples were heat inactivated at
55C for 30 minutes and then diluted in Improved Minimal
Essential Medium (Biofluids, Rockville, MD) plus 2~ FBS
(IMEM/2~FBS) in two-fold steps beginning at 1:2. 55 ~1 of
each sample were mixed with 10 ~1 of AvllacZ4 (containing 4
x 10' pfu), incubated for 1 hour at 37C and applied to nearly
confluent 293 cells in 96 well plates (4 x lG~ cells per
well). After 50 minutes in the tissue culture incubator, the
virus was aspirated from each well and re~laced with 150 ~1
of IMEM/lO~FBS. The following day, cells were fixed and
stained for ~-galactosidase expression, as described
previously. (Smith, e~ al., Nature Genetics, Vol. 5, pgs.
397-402 (1993?). In the absence of inactivating antibodies,
all of the cells stained blue. The titer of inactivating
antibodies for each sample was reported as the reciprocal of
the highest dilution with which less than 25~ of the cells
stained blue.
Results
Dose dependence of the humorai im~une
rçsPonse to adenovirus vectors
Previous studies which have ~ml ned repeat delivery of
adeno~irus vectors have employed relatively high doses of
-40-
2200869
WO96112406 PCT~S95113253
vector, which would be expected to maximize the strength of
the immune response. (Smith, et al ., 1993; Kozarsky, et al .,
J. Biol. Chem., Vol. 269, pgs. 13695-13702 (1994); Kay, et
al., Proc. Nat. Acad. Sci., Vol. 91, pgs. 2353-233357 (1994~;
Yei, et al., Gene TheraPy, Vol. 1, pgs. 192-200 (1994); Yang,
et al., J. Virol., Vol. 69, pgs. 2004-2015 (1995); Dai, et
ai., Proc. Nat. Acad. Sci., Voi. 92, pgs. 14ûi-14û5 [i995~;
Barr, et al., Gene TheraPY, Vol. 2, pgs. 151-155 (1995)).
To determine whether the production of neutralizing
antibodies and the block to repeat delivery is dependent on
the dose of vector inoculated, various doses of the
adenovirus vector AvllacZ4 to C57BL/6 mice via tail vein were
administered. The vector inoculum ranged from 1 x 103 pfu to
1 x lû~ pfu in single log increments. Thirty-four days after
vector delivery, the serum levels of anti-adenovirus
neutral i zing antibodies were determined (~ig. 18) for mice
which received lx10' pfu or greater of vector. A minus sign
indicates that none of the mice in the cohort had detectable
antibody. The plus sign corresponding to mice which received
lxl o3 pfu of vector indicates that three of tne five mice had
an antibody titer of 8, while two mice had r.o detectable
antibody. Thus, three of five mice which received 1 x 108pfu
had a level of anti-adenovirus antibody wnich was sufficient
to neutralize 4xlC` pfu of AvlLacZ4, whiie two mice had
undetectable levels. None of the mice which received lower
doses of vector had detectable antibodies using this
relatively stringent neutralization assa-y.
Thirty-five days after administraticn of AvllacZ4, each
mouse received 2 x 10~ pfu of AvlH9FR, an adenoviral vector
encoding human Factor IX. One week later, the plasma levels
of human Factor IX were determined by ELISA (Fig. 18). An
average of approximately 2 ~g/ml of Factor IX was detected in
mice which received either no AvllacZ4, or up to 1 x 105 pfu
-41-
WO96/12406 2 2 0 0 8 6 9 PCT~S95/13253 ~
of the first vector. Factor IX was also readily detected in
the mice which had received a first dose of l x 106 and l x
107 pfu, although the levels were reduced. Mice which
received l x 108 pfu of AvllacZ4 yielded little or no human
Factor IX after a~m; n; ~tration of AvlH9FR. Thus effective
gene transfer and expression can be achieved with a second
vector ~m; n; stration, provided the initial vector dose is
below a certain threshold level. The data indicate that for
intravenous delivery in C57BL/6 mice, this value is between
107 and 108 pfu.
Transient immunosuppression increases the efficiency of
vector re-administration. ~-
To determine whether transient immunosuppression wouldallow readministration of an adenoviral vector, C57BL/6 mice
were immunosuppressed with either deoxyspergualin (DSG),
cyclophosphamide (Cy), or ~ methasone (Dex) at the time of
administration of l x 108 pfu of AvliacZ4. As shown above,
this dose completely prevented an effective second delivery.
Mice were injected daily with 33 mg/kg of DSG, beginning one
day before vector delivery and continuing for seven more
days. Dexamethasone was delivered over the same time course,
at a dose of 5 mg/kg. Cyclophosphamide was administered
once, the day before vector delivery, at a dose of either lO0
mg/kg or 300 mg/kg. Control mice received AvllacZ4 without
immunosuppression, or were immunosuppressed without initial
vector delivery.
Five weeks after vector ~m; n; stration, the plasma
levels of anti-adenovirus neutralizing antibodies were
determined (Fig. l9). Mice immunosuppressed with DSG or 300
mg/kg of cyclophosphamide had no detectable neutralizing
antibodies, but all other mice which received AvllacZ4
generated neutralizing antibodies. Thirty-five days after
the first vector in~ection, the mice received l x 108 pfu of
-42-
SU85T~IlJTE SHEET(RULE 26)
~ WO96/12406 2 2 0 0 8 6 ~ PCT~S9~/13253
AvlH9F2. One week after AvlH9F2 injection, plasma samples
were prepared and the levels of human Factor IX were
determined by ELISA (Fig. 17). Mice immunosuppressed with
DSG or 300 mg/kg cyclophosphamide expressed human Factor IX
at levels which were approximately the same as levels in mice
which had not been treated with the first vector. No human
Factor IX was detected in the plasma of the mice which were
not immunosuppressed at the time of AvllacZ4 administration.
Under the conditions used in these studies, immunosuppression
with dexamethasone or l00 mg/kg cyclophosphamide was not
effective in permitting expression of Factor IX on repeat
injection. Thus the ability to achieve transgene expression
on a second vector injection correlated with the suppression
of neutralizing antibodies by DSG or high dose
cyclophosphamide treatment.
At the time of administration of the second vector,
AvlH9F2, half of the mice in each cohort which had been
immunosuppressed with cyclophosphamide or dexamethasone were
immunosuppressed again, using the same regimen as at the
first vector delivery. Five weeks later, plasma levels of
anti-adenovirus neutralizing antibodies were determined. The
mice immunosuppressed with 300 mg/kg cyclophosphamide had no
detectable neutralizing antibodies, while mice
im~.unosuppressed with l~. mg/kg cyclophosphamide or 5 mg/kg
dexamethasone had a measl~rable response (data not shown).
Thir~y-five days after AvlH9F~ injection, l x 109 pfu of a
Factor VIII vector, AvlAllPH81, were administered to the mice
which had been immunosuppressed with 300 mg/kg
cyclophosphamide. In addition, Factor VIII vector was
administered to control mice which had received AvllacZ4 and
AvlH9F2 without immunosuppression and also to naive mice.
One week later, plasma levels of human Factor VIII were
determined by ELISA ~Fig. 20). Control mice which received
only the AvlALAPH81 vector, and mice immunosuppressed with
22008 6q
Wo96/12406 PCT~S95/13253
300 mg/kg of cyclophosphamide at the time of the two prior
vector injections expressed human Factor VIII. Mice which
received AvllacZ4 and AvlH9F2 without imunosuppression, as
well as mice which were immunosuppressed only at the time of
AvllacZ4 delivery, did not express human Factor VIII.
DSG ~ermits effective rePeat administration at a clinically
relevant dose.
The previous experiment demonstrated that a high dose of
either DSG or cyclophosphamide permitted readministration of
an adenoviral vector, but that clinically relevant doses of
cyclophosphamide and dexamethasone were not effective. The
next objective was to determine whether lower doses of DSG
would be effective. The dose of DSG used in the initial
experiment (33 mg/kg) is near the maximum tolerated dose in
mice and is significantly higher than the 5-7 mg/kg dose used
in human clinical trials for organ transplantion. (Suzuki,
et al., Ann. N.Y. Acad. Sci., Vol. 696, pgs. 263-269 (1993);
Jindal, et al., Mt. Sinai J. Med., Voi. 61, pgs. 51-56
(1994~). To determine if lower doses would also be effective
in allowing vector readministration, mice were
immunosuppressed with 5, 10, 20, and 33 mg/kg of DSG at the
time of administration of ' x 10~ pfu of AvllacZ4.
Immunosuppression was started the day before vector deli~ery
and continued for a total of 8 days. Or. the day of vector
d~livery, DSG was given after injection of the adeno~irus
since it is most effective when administered after antigen.
(Takahara, et al., TransPlantation, Vol. 53, pgs. 514-918
(lg92) ) .
Thirty-five days later each mouse received 1 x 10~ pfu
of AvlH9F2. One week after AvlH9F2 injection, human Factor
IX plasma levels were determined by ELISA (Fig. 21j. Control
mice, which were not pre-immunized with AvlLacZ~, expressed
an average of 9 ~g/ml of human Factor IX. Other control
~200869
~ WO~6/12406 PCT~S95tl3253
`_
mice, which received AvllacZ4 but were not immunosuppressed,
expressed no hllm~n Factor IX after AvlH9F2 a~m; n; ~tration.
The one mouse which was ;mmllnosuppressed with 33 mg/kg DSG
- expressed 12 ~g/ml of human ~actor IX. Five of six mice
;mmllnosuppressed with 20 mg/kg DSG expressed an average of
3.0 ~g/ml of hllm~n Factor IX, and one mouse expressed none.
~ice immunosuppressed with a dose of 10 mg/kg expressed a
wide range of Factor IX, extending from 30 ng/ml to 8 ~g/ml.
Three mice immunosuppressed with 5 mg/kg expressed no human
Factor IX, while three others expressed levels ranging from
2 to 7 ~g/ml. Mice which were not immunosuppressed at the
time of AvllacZ4 administration expressed no hl7m~n Factor IX.
Discussion
The data ~emo~trate that multiple intravenous
administrations of adenovirus vectors with resulting
transgene expression can be accomplished in immune competent
animals treated with a short course of immunosuppression at
the time of vector delivery. This observation is significant
because several recent studies have demonstrated that a
humoral immune response directed against adenoviral vector
prevents readministration. (Smith, et al., 1993; Kay, et
al., 1994; Yei, et al., 1994; Yang, et al., 1995.) The
inability to readminister vector has presented a major
obstacle to the clinical utility of adenoviral vectors, since
effective readministration will almost certainly be required
for the clinical application of these vectors to gene therapy
of chronic diseases.
The failure to obtain expression following repeat dosing
in the absence of immunosuppression correlates with anti-
adenovirus neutralizing antibodies. Evidence that such
antibodies are sufficient to block readministration was
provided by Yang et al., 1995, who showed that passive
transfer of serum from a mouse previously treated with vector
SUBSTlTlJTE SHEET (RULE 26)
22oo86q
PCT~S95/13253
WO96/12406
into the venous circulation of a naive mouse was able to
prevent vector-mediated gene expression in the liver. The
role of the immune system in preventing repeat administration
also was demonstrated by the observation that repeat
administration of vector was effective in immunodeficient
mice. (Yang, et al ., 1995; Dai, et al ., 1995; Barr, et al .,
Gene TheraPy, Vol. 2, pgs. 151-155 (1995)). This result has
been confirmed by demonstrating effective administration of
AvlH9F2 to scid mice 5 weeks after delivery of 5 x 108 pfu of
AvlALAPH81 (data not shown).
Since previous reports describing humoral responses to
adenovirus administration used relatively high vector doses,
we evaluated the relationship between initial vector dose and
the ability to achieve an effective repeat gene transfer.
The results indicated that the magnitude of the immune
response was dependent on the initial dose of vector and that
if the dose is below a threshold level, a second
a~m;n;stration is possible. The finding that this level was
only one to two orders of magnitude below a clinically
relevant vector dose further suggested that immunosuppression
at the time of vector delivery would permit readministration.
In addition, these results emphasize the need for potent
vectors which would be efficacious at low doses.
Applicants have observed that an~i-adenovlrus
neutralizing antibody titers are maintained for at least ten
monthc in mice after a single administration of vector via
tail vein. The long-term maintenance of titer may have been
due to a low level of ongoing adeno~iral backbone gene
expression n ~,ansduced cells. Vectors designed to red~ce
or eiiminate backbone gene expression may eiicit a weaker
immune response and therefore may require less
immunosuppression for successfui readministration.
-46-
220086q
WO96/12406 PCT~S95/132~3
An important property of DSG is that it does not produce
a general suppression of the immune system, but rather
results in a selective lack of response to specific antigens
presented at the time of drug treatment. We found that
immunosuppression with DSG over a 7 day period following
vector delivery efficiently inhibited the humoral response to
the vector and permitted an effective second administration.
The initial experiment with DSG employed a high dose of 33
mg/kg, which is close to the m~ m tolerated dose in mice
and several fold higher than the doses used in human trials.
When administered over the same 8 day course, including 7
days post vector treatment, lower doses of DSG were also
effective in permitting repeat delivery of vector. A greater
degree of individual variability in levels of Factor IX
expression was seen with reduced doses, although even at the
lowest dose tested (5 mg/kg) significant Factor IX expression
was obtained in 3 of 6 animals.
~ yclophosphamide, administered at a dose of 300 mg/kg
the day before vector in~ection, was also effective in
blocking the humoral response and allowed a completely
effective second injection with a Factor IX adenovirus
vector Furthermore, a third injection with a Factor VIII
encoding vector was also completely efficacious when the
previous two vector administralions we~e each preceded by a
singie dose of cyclophosphamide. ~yclophosphamide is used
clinicaliy as an anti-cancer agent in the treatment of
Hodgkins disease and otner leukemias. It is also empioyed as
an i~unosuppressive agent in the treatmer- of hemophilia
patients who develop inhibitors to Factor VIII protein
replacement therapy. (Aledort, Am. J. Xemat., Vol. 47, pgs.
208-2,7 (1994); Nilsson, et al ., N. Enql. ~. Med., Vol. 318,
pgs. 947-950 (1988)i. W~ile the dose used t C successfully
obtain readministration in mice is substantially higher than
is generally used in humans, it remains tO be established
-47-
WO96/12406 2 2 0 0 8 6 ~ PCT~S95/13253 ~
whether lower, clinically acceptable doses, might be
effective in humans. One possibility suggested by experience
in the organ transplantation setting is that combinations of
immunosuppressants would yield more potent suppression of the
immune system with less toxicity. For example,
cyclophosphamide may be effective at lower doses when used in
combination with dexamethasone. It is also possible that
the degree of immunosuppression required will depend on the
dose of vector which is needed to effect therapy. The dose
of AvlH9F2 used in this study, l x l0~ pfu, yielded plasma
levels of human Factor IX of 5-l0 ~g/ml, which i8 20 to 50
times above a level that would be therapeutic in a
hemophiliac. Vectors, such as AvlH9F2, which express high
levels of transgene product and which can be administered at
relatively low doses, should reduce the extent of immune
stimulation and the degree of immunosuppression required.
In summary, Applicants have shown that effective
repetitive delivery of systemically administered adenovirus
vectors can be a_hieved with short term immunosuppression.
Importantly, this can be accomplished using pharmacologic
agents wnich are either approved for use in humans, or are in
clinical testing.
The disclosure of all patents, publicarions, (including
published patent applicationsi, and database accession
numbers, and deposi~ory accession numbers referenced in this
specifica.ion are speci~ically incorporated herein by
reference in their entirety to the same extent as if each
such individual patent, publication, and d~tabase accession
number, and depository accession number were specifically and
individual~y indicated .c be ncorporate~ by reference.
.
It is to be understood, ;nowever, that the scope of the
present invention is no. to be limited to the specific
-48-
22008~9
096/12406 PCT~S95/132~3
embodiments described above. The invention may be practiced
other than as particularly described and still be within the
scope of the accompanying claims.