Note: Descriptions are shown in the official language in which they were submitted.
a
t
- 1 -
Specification
SOLID DISPERSION AND SOLID DISPERSION DOSAGE FORM
OF XANTHINE DERIVATIVE
Technical Field
The present invention relates to a solid dispersion and a
solid dispersion dosage form of a xanthine derivati-ve or a
pharmacologically allowable salt thereof which exhibits an
antagonistic activity relative to the adenosine A1 receptor,
and which has a diuretic activity, a kidney-protecting activity,
a bronchodilating activity, a cerebral function-improving
activity, an anti-dementia activity, or the like.
Background Art
In general, drugs which are slightly soluble in water and
have high crystallinity have low bioavailability since they
have low solubility and low disolution rate in the
gastrointestinal tract. Hitherto, in order to improve their
absorbability, several methods for finely grinding drug
crystals or for transforming them into amorphous substances
have been examined.
By fine-grinding, however, particle diameters become
irregular between lots, or inter-particle force is enhanced to
cause agglomeration.
As a method for obtaining amorphous substances, grinding
zza~o~~
- 2 -
or forming a solid dispersion are considered. Drug crystals
which can become amorphous by grinding are limited. Further,
it has been known that solubility and absorbability of a
slightly water-soluble compound are improved by dispersing it
in a polymer to form a solid dispersion.
As to xanthine derivatives or pharmacologically allowable
salts thereof, which are slightly soluble, however, solid
dispersions or solid dispersion dosage forms are not yet known.
Disclosure of Invention
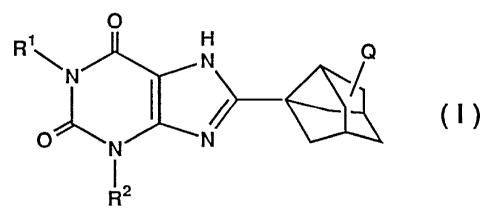
The present invention relates to a solid dispersion
comprising a xanthine derivative or a pharmacologically
allowable salt thereof and a polymer, the xanthine derivative
[hereinafter referred to as compound (I)] being represented by
the following formula (I).
O
H
N Q
m
O ~N ~N
1 2
(In the above formula, R1 and RZ are the same or different, and
represent substituted or unsubstituted lower alkyl groups, and
Q represents a hydrogen atom or a hydroxyl group.)
Furthermore, the present invention relates to a solid
dispersion dosage form comprising a xanthine derivative of a
CA 02201096 2003-02-27
- 3 -
compound (I) or a pharmacologically allowable salt thereof and
a polymer.
In the definition of the formula (I), the lower alkyl
groups are straight or branched groups having 1 to 6 carbon
atoms, and examples include a methyl group, an ethyl group, a
propyl group, an isopropyl group, a butyl group, an isobutyl
group, a sec-butyl group, a tert-butyl group, a pentyl group, a
neopentyl group, and a hexyl group. As to substituent groups
of the substituted lower alkyl groups, examples include a
hydroxyl group and an acetyl group.
The pharmacologically allowable salts of compounds (I)
include pharmacologically allowable acid addition salts, metal
salts, ammonium salts, organic amine addition salts, and
amino acid addition salts.
Examples of pharmacologically allowable acid addition
salts of compounds (I) include inorganic acid salts such as
hydrochlorides, sulfates, phosphates; and organic acid salts
such as acetates, maleates, fumarates, tartrates, and citrates.
Examples of pharmacologically allowable metal salts include
alkali metal salts such as lithium salts, sodium salts, and
potassium salts; alkaline earth metal salts such as magnesium
salts and calcium salts; aluminum salts; and zinc salts.
Examples of pharmacologically allowable ammonium salts include
salts of ammonium or tetramethyl ammonium. Examples of
pharmacologically allowable organic amine addition salts
220 ~ 09.~
- 4 -
include addition salts of morpholine or piperidine. Examples
of pharmacologically allowable amino acid addition salts
include addition salts of lysine, glycine, phenylalanine,
glutamic acid, and aspartic acid.
Compounds (I) or pharmacologically allowable salts thereof
can be produced according to the method described in Japanese
Unexamined Patent Publication No. 3-173889.
Practical examples of compounds (I) are shown in Table 1.
2~~ 1096
- 5 -
TABLE 1
O
Ri H
~N N
X
O, WN ~N
1 2
R
Compound No. R~ R2 X
1 n-C3H~ n-C3H~
2 n-CsH~ n-CsH~
OH
3 n-CsH~ n-C3H~
4 CH3CHCH2 n-C H OH
3 7
OH OH
CH3CCH2 n-C3H~
p OH
CA 02201096 2006-06-28
- 6 -
In the solid dispersion or the solid dispersion dosage
form, a compound (I) or a pharmacologically allowable salt
thereof is contained at 1 to 50o by weight, and preferably, 3
to loo by weight.
The polymer to be used in the present invention is not
particularly limited so long as it can disperse compounds (I)
or pharmacologically allowable salts thereof, and accordingly,
several natural polymers and synthetic polymers can be used.
Examples of polymers include rubber compounds such as gum
arabic; gelatin; polysaccharides such as agar; cellulose
compounds or derivatives thereof such as crystalline cellulose,
micro crystalline cellulose, low-substituted hydroxypropyl
cellulose, carboxymethyl cellulose, hydroxypropylmethyl
cellulose, hydroxypropylmethyl cellulose phthalate,
hydroxypropylmethyl cellulose acetate succinate, carboxymethyl
ethyl cellulose, ethyl cellulose, and cellulose acetate
phthalate; polyvinyl derivatives such as polyvinyl alcohol,
polyvinyl pyrrolidone, and polyvinyl acetal
diethylaminoacetate; aminoalkyl methacrylate copolymers such as
aminoalkyl methacrylate copolymer E and aminoalkyl methacrylate
copolymer RS; and methacrylate copolymers such as methacrylate
copolymer L, methacrylate copolymer LD, and methacrylate
copolymer S. Among these polymers, enteric-coating polymers
are preferred. Examples of enteric-coating polymers include
TM
methacrylate copolymer L (trade name: Eudragit L100 and
~2010~fi
Eudragit L100-55; Rohm Pharma Co., Ltd.), hydroxypropylmethyl
cellulose phthalate (trade name: hydroxypropylmethyl cellulose
phthalate HP-55; Shin-Etsu Chemical Co., Ltd.),
hydroxypropylmethyl cellulose acetate succinate, and
carboxymethylethyl cellulose. Particularly, methacrylate
copolymer L (trade name: Eudragit L100) is preferably used.
Such a polymer is contained in the solid dispersion or the
solid dispersion dosage form at 3 to 50o by weight, and
preferably, 9 to 10o by weight.
In the solid dispersion or the solid dispersion dosage
form of the present invention, the blending ratio of a compound
(I) to the polymer is 3:1 to 1:5 (by weight). In particular,
when the content of a compound (I) or a pharmacologically
allowable salt thereof is 3o by weight, the blending ratio is
preferably 1:3, and when the content of a compound (I) or a
pharmacologically allowable salt thereof is 10 to 20o by weight,
the blending ratio is preferably 1:1.
The solid dispersion of the present invention can be
prepared by a co-grinding method, a solvent method, a melting
method, a heat-melt-kneading method, or the like.
In the co-grinding method, a compound (I) or a
pharmacologically allowable salt thereof is mixed with a
polymer using a mixer or the like, and the mixture is ground
using a grinder or the like. In the solvent method, a compound
(I) or a pharmacologically allowable salt thereof and a polymer
zzo1Q~~
_8-
are dissolved or dispersed in an organic solvent, and the
organic solvent is then removed according to an ordinary method
under reduced pressure or ordinary pressure.
The organic solvent to be used in the solvent method is
not particularly limited so long as it can dissolve a compound
(I) or a pharmacologically allowable salt thereof and a polymer.
Examples of such organic solvents include aliphatic halogenated
hydrocarbons, ketones, alcohols, ethers, aliphatic hydrocarbons,
aromatic hydrocarbons, esters, organic acids, amides, and mixed
solvents thereof. Examples of aliphatic halogenated
hydrocarbons include methylene chloride, dichloroethane, and
chloroform. Examples of ketones include acetone and methyl -
ethyl ketone. Examples of alcohols include methanol, ethanol,
propanol, and butanol. Examples of ethers include diethyl
ether, dibutyl ether, diisobutyl ether, dioxane,
tetrahydrofuran, and ethylene glycol dimethyl ether. Examples
of aliphatic hydrocarbons include n-hexane, cyclohexane, and n-
heptane. Examples of aromatic hydrocarbons include benzene,
toluene, and xylene. Examples of esters include ethyl acetate.
Examples of organic acids include acetic acid and propionic
acid. Examples of amides include dimethylformamide and
dimethylacetamide. Aliphatic halogenated hydrocarbons,
alcohols, and mixtures of aliphatic halogenated hydrocarbons
and alcohols are preferably used, and an ethanol/methylene
chloride (1/1) mixture is particularly preferably used.
a 22Q'~0.9~a
- g _
As to removal of the organic solvent, operating conditions
such as the treatment temperature and time period are
ordinarily at room temperature to 150°C and for several minutes
to more than ten hours, though they are altered depending on
the compound, the polymer, the solvent, or the like to be used.
According to the solvent method, the solid dispersion is
produced using, for example, a fluidized-bed granulator, an
agitating granulator, a spray-dry granulator, or a vacuum-dry
granulator, and specifically, is produced as follows.
A compound (I) or a pharmacologically allowable salt
thereof is dissolved in an organic solvent with a polymer to
prepare a spray solution. At this point, a surfactant may be
added to the spray solution. Examples of surfactants include
sodium lauryl sulfate, polysolbate 80, and sucrose-fatty acid
esters. Although there exist several sucrose-fatty acid esters
having different HLB values which vary depending on the carbon
chain of the fatty acid or the ester concentration, sucrose-
fatty acid esters having high HLB values such as a sucrose-
fatty acid ester having an HLB value of 22 (Ryoto-Sugar Ester
DK-SS; Mitsubishi Chemical Corporation) are preferably used.
The surfactant concentration is preferably 0.1 to S.Oo by
weight.
In such a spray solution, the solid ingredient
concentration based on the compound (I) or the
pharmacologically allowable salt thereof and the polymer is 5
Zzolo9s
- 10 -
to 23o by weight/volume, preferably 11 to 15o by weight/volume,
and more preferably 15o by weight/volume.
The solid dispersion dosage form of the present invention
can be obtained by spray-drying such a spray solution or
spraying it onto an absorbent carrier, or by injecting it into
water and isolating the resulting granules. Excipients
ordinarily used in the pharmaceutical field can be used as
absorbent carriers. Examples of excipients include saccharides
such as lactose, sucrose, glucose, reducing maltose, mannitol,
and sorbitol; starch or derivatives thereof such as corn starch,
potato starch, dextrin, and pullulane; cellulose compounds such
as cr-ystalline cellulose and micro crystalline cellulose; amino
acids such as arginine, asparagine, aspartic acid, citrulline,
cysteine, glutamic acid, glutamine, glycine, histidine,
homoserine, isoleucine, leucine, lysine, methionine, ornithine,
phenylalanine, proline, serine, threonine, tryptophan, tyrosine,
valine; aminobutyric acid; aminocapronic acid; and
glycylglycine. For example, among lactose products, those.
having a mesh size of #200 are particularly preferred.
For performing such spray-drying, a granulator may be used
which is further equipped with a vacuum-drying unit or a
microwave drying unit.
In the melting method, a compound (I) or a
pharmacologically allowable salt thereof is melted or dispersed
in a polymer, and then quenched. For melting or dispersing,
.' ~'~~'~a~6
- 11 -
for example, the mixture is heated above the melting point or
softening point of the compound (I), the pharmacologically
allowable salt thereof, or the polymer. In this case,
additives such as plasticizers or the aforementioned
surfactants can be added. Examples of plasticizers include
triethyl citrate, polyethylene'glycol, and triacetin.
According to the melting method, the solid dispersion is
produced using, for example, an agitating granulator with a
heating unit. The compound (I) or the pharmacologically
allowable salt thereof and the polymer are physically mixed
beforehand. To this mixture, the aforementioned plasticizers,
surfactants, or the like may be added as occasion demands.
Operating conditions such as the treatment temperature and time
period are ordinarily at room temperature to 150°C and for
several minutes to mare than ten hours, though these are
altered depending on the compound, the polymer, concentrations
or types of additives, or the like to be used. Subsequently,
quenching is carried out to obtain the solid dispersion. The
quenching temperature is -80°C to room temperature.
In the heat=melt-kneading method, a solid dispersion is
obtained by a mixing process under heat and compression using
an extruder having a heating unit, for example, a biaxial
extruder. Operating conditions such as the treatment
temperature, pressure, and time period are ordinarily at room
temperature to 150°C, under 10 to 200 kg/cm2, and for several
.' 201~~G
- 12 -
minutes to more than ten hours, though they are altered
depending on the compound, the polymer, concentrations or types
of additives, or the like to be used.
According to the heat-melt-kneading method, a solid
dispersion is produced using, for example, a biaxial extruder,
and is specifically produced as follows.
A compound (I) or a pharmacologically allowable salt
thereof and a polymer, and optional additives as described
above are physically mixed beforehand. This mixture is then
fed at a powder feeding rate of 10 to 200 g/min. As to
treatment conditions, the screw rotating speed is 50 to 300
r.p.m., the temperature is 20 to 200°C, and the pressure is 40
to 120 kg/cm2. Thus, a solid dispersion having a plastic
characteristic can be obtained. Solid dispersion powder can be
obtained by grinding the solid dispersion using a grinder.
Solid dispersions of compounds (I) or pharmacologically
allowable salts thereof obtained according to the co-grinding
method, the solvent method, the melting method, the heat-melt-
kneading method, or the like can be used as they are in the
form of a powder or granular dosage form. Alternatively, they
may be further transformed into solid dispersion dosage forms
through a formulating process such as a mixing process, a
granulating process, a tabletting process, an encapsulating
process, and a coating process.
The mixing process is, for example, a process for mixing
.
2201096
- 13 -
such a solid dispersion and other compound using a mixer or the
like. The granulating process is, for example, a process for
granulating such a solid dispersion using a granulating mixer
or the like. The tabletting process is, for example, a process
for making such a solid dispersion into tablets using a
tabletting machine or the like. The encapsulating process is,
for example, a process for packing such a solid dispersion in
capsules using an encapsulating machine or the like. The
coating process is, for example, a process for coating such a
solid dispersion with a coating agent using a coating machine
or the like.
Dosage forms obtained according to the above-described
manner include parvule dosage forms, granular dosage forms,
tablets, capsule dosage forms, or the like. Further, an
additive such as a coloring agent, a corrigent, an excipient, a
disintegrator, a lubricant, and a surfactant can be added as
occasion demands. Such an additive can be added in a manner
suitable for each process. Examples of coloring agents include
yellow iron sesquioxide, iron sesquioxide, several food dyes,
and copper sodium chlorophyllin. Examples of corrigents
include sucrose, saccharin, aspartame, mannitol, dextran, a
lemon flavor, menthol, and citric acid. Examples of excipierits
include the aforementioned excipients. Examples of
disintegrators include hydroxypropyl cellulose, sodium
croscarmellose, crospovidone, and sodium carboxymethyl starch.
22Q 10'9 fi
- 14 -
Examples of lubricants include magnesium stearate. Examples of
surfactants include the aforementioned surfactants.
When an automatic encapsulating or tabletting machine is
used, 0.5 to 2o by weight of magnesium stearate as a lubricant
should preferably be mixed before the encapsulating or
tabletting process in order to prevent powder adhesion onto the
piston or punch.
Advantages of the solid dispersion or the solid dispersion
dosage form according to the present invention will be
illustrated with experimental examples.
EXPERIMENTAL EXAMPLE 1
One point six seven gram of each of the solid dispersions
obtained in Examples 1 and 3, and 0.50 g of the solid
dispersion obtained in Example 2 were each put in 900 ml of a
solution containing 0.3o by weight of sodium lauryl sulfate in
order to perform an dissolution test by a puddle method at 37°C
and 100 r.p.m. The results are shown in Fig. 1. For comparison,
similar dissolution tests were performed using 50 mg of
Compound No..l.crystals and tablets obtained in Comparative
Example 1 instead of the solid dispersion. The results are
shown in Fig. 1. As is revealed in Fig. 1, the solubility of
the solid dispersions according to the present invention was
significantly higher than those of the Compound No. 1 crystal
and the dosage form obtained in Comparative Example 1.
EXPERIMENTAL EXAMPLE 2
zzo X096
- 15 -
The solid dispersion dosage forms obtained in Examples 6,
7, and 8 were each orally administrated to a male beagle with
20 ml of water. For comparison, dosage forms each comprising 5
mg of Compound No. 1 crystals packed in a gelatin hard capsule
and tablets of Comparative Example 1 were administrated to
beagles in a similar manner. The dosage of each dosage form
(the weight of Compound No. 1 per 1 kg body weight), and the
number of dogs per group are shown in Table 2.
zoo joss
i
- 16 -
TABLE 2
Dosage (mg/kg) Number of Dogs (per
group)
Example 6 3 5
Example 7 3 4
Example 8 3 5
Crystal of ~ 6 2
Compound No. 1
Comparative 4 5
Example 1
Pharmacokinetic parameters in the results of this
~'vWparati~T~-absOrbab111ty test are shown in Table 3.
TABLE 3
The Area under the Plasma Concentration Curve
(0 - 24, ng ~ h/ml)
Administrated Formulation Intact Drug -
(in terms of 3 mg/kg)
Example 6 404.3~ 80.1
Example 7 156.2 t 141.5
Example 8 110.3 ~ 53.6
Crystal of Compound No. 1 69.9~ 2.6
Comparative Example 1 77.3 ~ 61.8
As is revealed from the results of thiscomparative
absorbability test, the solid dispersions of the present
invention have greater absorbability than the crystal of
Compound No. 1 and the dosage form of Comparative Example 1.
Brief Description of the Drawings
220 109 6
- 17 -
Fig. 1 shows the results of the dissolution tests for the
solid dispersions of Examples 1 to 3, the crystal of Compound
No. 1, and the dosage form of Comparative Example 1. The
results for the dosage form of Example 1 are represented by
black dots, those for the solid dispersion of Example 2 are
represented by black squares, those for the solid dispersion of
Example 3 are represented by black triangles, those for the
crystal of Compound No. 1 are represented by white squares, and
those for the dosage form of Comparative Example 1 are
represented by white dots.
Fig. 2 shows the results of measurement by a differential
scanning calorimeter (DSC) for Compound No. 1.
Fig. 3 shows the results of DSC measurement for a -
physically mixed composition according to the same recipe as
the solid dispersion of Example 3.
Fig. 4 shows the results of DSC measurement for the solid
dispersion of Example 3.
Fig. 5 shows the results of DSC measurement for the solid -
dispersion of Example 4.
Fig. 6 shows the results of DSC measurement for the solid
dispersion of Example 5.
Best Mode for Carrying Out the Invention
EXAMPLE 1
Thirty grams of Compound No. 1 and 90 g of methacrylate
zZO~o~s
- 18 -
copolymer L (Eudragit L100; Rohm Pharma Co., Ltd.) were
dissolved in 700 g of an ethanol/methylene chloride (1/1 by
weight) mixed solution, and the resultant was then sprayed on
880 g of lactose (#200 mesh) by a fluidized-bed granulating
method to obtain a solid dispersion. Concerning the conditions
for the fluidized-bed granulating at this time, the intake air
temperature was 60°C, the solid ingredient concentration in the
spray solution was 15o by weight/volume, and the spraying
pressure was 2.0 kg/cm2. The powder characteristics of the
solid dispersion containing 3o by weight of Compound No. 1 are
shown in Table 4.
TABLE 4
Apparent Specific Volume
Loose Specific Volume (ml/g) 2.32
Tapped Specific Volume (ml/g) 1.75
EXAMPLE 2
A solid dispersion containing 20~ by weight of Compound No.
1 was obtained in a manner similar to that in Example 1 except
that the quantity of Compound l~was 200 g, that of methacrylate
copolymer L was 200 g, and that of lactose was 600 g. The
results of DSC measurement on Compound No. 1 alone are shown in
Fig. 2, those on a physically mixed composition according to
the same recipe as a solid dispersion containing 20o by weight
of Compound No. 1 are shown in Fig. 3, and those on a solid
zz~ ~o9s
- 19 -
dispersion containing 20o by weight of Compound No. 1 are shown
in Fig. 4. Since the melting point peak of Compound No. 1,
which appears in Figs. 2 and 3, disappears in Fig. 4, it is
suggested that the solid dispersion according to the present
invention became amorphous.
EXAMPLE 3
A solid dispersion containing 3o by weight of Compound No. 1
was obtained in a manner similar to that in Example 1 except
that a water-soluble polymer, polyvinyl pyrrolidone K30, was
used instead of methacrylate copolymer L.
EXAMPLE 4
Using a biaxial extruder (manufactured by Kurimoto, Ltd.),
75 g of Compound No. 1 and 225 g of methacrylate copolymer L
(Eudragit L100; Rohm Pharma Co., Ltd.) were heat-melt-kneaded
to obtain a solid dispersion in which the concentration of
Compound No. 1 was 25o by weight. The results of DSC
measurement on this solid dispersion are shown in Fig. 5.
Since the melting point peak of Compound No. 1 disappears, it
is suggested that the solid dispersion became amorphous.
EXAMPLE 5
Using a biaxial extruder (manufactured by Kurimoto, Ltd.),
150 g of Compound No. 1 and 150 g of methacrylate copolymer L
(Eudragit L100; Rohm Pharma Co., Ltd.) were heat-melt-kneaded
to obtain a solid dispersion in which the concentration of
220096
- 20 -
Compound No. 1 was 50% by weight. The results of DSC
measurement on this solid dispersion are shown in Fig. 6.
Since the melting point peak of Compound No. 1 disappears, it
is suggested that the solid dispersion became amorphous.
EXAMPLE 6
The solid dispersion obtained in Example 1 was packed in
Size 0 of gelatin hard capsules at 333.3 mg to obtain solid
dispersion dosage forms each containing 10 mg of Compound No. 1.
EXAMPLE 7
The solid dispersion obtained in Example 2 was packed in
Size 2 of gelatin hard capsules at 150.0 mg to obtain solid
dispersion dosage forms each containing 30 mg of Compound No. 1.
EXAMPLE 8
The solid dispersion obtained in Example 3 was packed in
Size 0 of gelatin hard capsules at 333.3 mg to obtain solid
dispersion dosage forms each containing 10 mg of Compound No. 1.
COMPARATIVE EXAMPLE 1
Forty point zero grams of a finely ground product of
Compound No. 1, 137.6 g of lactose, 59.0 g of corn starch, and
13.0 g of polyvinyl pyrrolidone were fluidized-bed-granulated
using a binder solution comprising 7.8 g of polyvinyl alcohol
dissolved in 31.2 g of distilled water. To this granulated
composition, 2.6 g of magnesium stearate was admixed to produce
tablets. At this time, each tablet contained 20.0 mg of the
finely ground product of Compound No. 1, 68.8 mg of lactose,
,
zzo logs
- 21 - -
29.5 mg of corn starch, 6.5 mg of polyvinyl pyrrolidone, 3.9 mg
of polyvinyl alcohol, and 1.3 mg of magnesium stearate.
Industrial Applicability
According to the present invention, there is provided a
solid dispersion or a solid dispersion dosage form of a
slightly-soluble and crystalline drug, compound (I), wherein
the solid dispersion or the solid dispersion dosage form has
high solubility and absorbability, and also has powder
characteristics suitable for production at an industrial scale.