Note: Descriptions are shown in the official language in which they were submitted.
CA 02205175 1997-05-12
WO 96/15241 PCT/US95/14899
-1-
TITLE OF THE INVENTION
A POLYNUCLEOTIDE TUBERCULOSIS VACCINE
BACKGROUND OF THE INVENTION
A major obstacle to the development of vaccines against
viruses and bacteria, particularly those with multiple serotypes or a high
rate of mutation, against which elicitation of neutralizing antibodies
and/or protective cell-mediated immune responses is desirable, is the
diversity of the external proteins among different isolates or strains.
Since cytotoxic T-lymphocytes (CTLs) in both mice and humans are
capable of recognizing epitopes derived from conserved internal viral
proteins [J.W. Yewdell et al., Proc. Natl. Acad. Sci. (USA) 82, 1785
(1985); A.R.M. Townsend, et al., Cell 44, 959 (1986); A.J. McMichael
et al., J. Gen. Virol. 67, 719 (1986); J. Bastin et al., J. Exp. Med. 165,
1508 (1987); A.R.M. Townsend and H. Bodmer, Annu. Rev. Immunol.
7, 601 (1989)], and are thought to be important in the immune response
against viruses [Y.-L. Lin and B.A. Askonas, J. Exp. Med. 154, 225
(1981); I. Gardner et al., Eur. J. Immunol. 4, 68 (1974); K.L. Yap and
G.L. Ada, Nature 273, 238 (1978); A.J. McMichael et al., New Engl. J.
Med. 309, 13 (1983); P.M. Taylor and B.A. Askonas, Immunol. 58, 417
(1986)], efforts have been directed towards the development of CTL
vaccines capable of providing heterologous protection against different
viral strains.
It is known that CTLs kill virally- or bacterially-infected
cells when their T cell receptors recognize foreign peptides associated
with MHC class I and/or class II molecules. These peptides can be
derived from endogenously synthesized foreign proteins, regardless of
the protein's location or function within the pathogen. By recognition
of epitopes from conserved proteins, CTLs may provide heterologous
protection. In the case of intracellular bacteria, proteins secreted by or
released from the bacteria are processed and presented by MHC class I
and II molecules, thereby generating T-cell responses that may play a
role in reducing or eliminating infection.
CA 02205175 1997-05-12
WO 96/15241 PCT/US95/14899
-2-
Most efforts to generate CTL responses have either used =
replicating vectors to produce the protein antigen within the cell [J.R.
Bennink et al., ibid. 311, 578 (1984); J.R. Bennink and J.W. Yewdell,
Curr. Top. Microbiol. Immunol. 163, 153 (1990); C.K. Stover et al.,
Nature 351, 456 (1991); A. Aldovini and R.A. Young, Nature 351, 479
(1991); R. Schafer et al., J. Immunol. 149, 53 (1992); C.S. Hahn et al.,
Proc. Natl. Acad. Sci. (USA) 89, 2679 (1992)], or they have focused
upon the introduction of peptides into the cytosol [F.R. Carbone and
M.J. Bevan, J. Exp. Med. 169, 603 (1989); K. Deres et al., Natur=e 342,
561 (1989); H. Takahashi et al., ibid. 344, 873 (1990); D.S. Collins et
al., J. Immunol. 148, 3336 (1992); M.J. Newman et al., ibid. 148, 2357
(1992)]. Both of these approaches have limitations that may reduce
their utility as vaccines. Retroviral vectors have restrictions on the size
and structure of polypeptides that can be expressed as fusion proteins
while maintaining the ability of the recombinant virus to replicate [A.D.
Miller, Curr. Top. Microbiol. Immunol. 158, 1 (1992)], and the
effectiveness of vectors such as vaccinia for subsequent immunizations
may be compromised by immune responses against vaccinia [E.L.
Cooney et al., Lancet 337, 567 (1991)]. Also, viral vectors and
modified pathogens have inherent risks that may hinder their use in
humans [R.R. Redfield et al., New Engl. J. Med. 316, 673 (1987); L.
Mascola et al., Arch. Intern. Med. 149, 1569 (1989)]. Furthermore, the
selection of peptide epitopes to be presented is dependent upon the
structure of an individual's MHC antigens and, therefore, peptide
vaccines may have limited effectiveness due to the diversity of MHC
haplotypes in outbred populations.
Benvenisty, N., and Reshef, L. [PNAS 83, 9551-9555,
(1986)] showed that CaC12 precipitated DNA introduced into mice
intraperitoneally (i.p.), intravenously (i.v.) or intramuscularly (i.m.)
could be expressed. The intramuscular (i.m.) injection of DNA expression
vectors in mice has been demonstrated to result in the uptake
of DNA by the muscle cells and expression of the protein encoded by the DNA
[J.A. Wolff et al., Science 247, 1465 (1990); G. Ascadi et al.,
Nature 352, 815 (1991)]. The plasmids were shown to be maintained
CA 02205175 1997-05-12
WO 96/15241 PCTIUS95/14899
-3-
episomally and did not replicate. Subsequently, persistent expression
has been observed after i.m. injection in skeletal muscle of rats, fish and
primates, and cardiac muscle of rats [H. Lin et al., Circulation 82, 2217
(1990); R.N. Kitsis et al., Proc. Natl. Acad. Sci. (USA) 88, 4138
(1991); E. Hansen et al., FEBS Lett. 290, 73 (1991); S. Jiao et al., Hum.
Gene Therapy 3, 21 (1992); J.A. Wolff et al., Human Mol. Genet. 1,
363 (1992)]. The technique of using nucleic acids as therapeutic agents
was reported in W090/11092 (4 October 1990), in which naked
polynucleotides were used to vaccinate vertebrates.
Recently, the coordinate roles of B7 and the major
histocompatibility complex (MHC) presentation of epitopes on the
surface of antigen presenting cells in activating CTLs for the
elimination of tumors was reviewed [Edgington, Biotechnology 11,
1117-1119, 1993]. Once the MHC molecule on the surface of an antigen
presenting cell (APC) presents an epitope to a T-cell receptor (TCR),
B7 expressed on the surface of the same APC acts as a second signal by
binding to CTLA-4 or CD28. The result is rapid division of CD4+
helper T-cells which signal CD8+ T-cells to proliferate and kill the
APC.
It is not necessary for the success of the method that
immunization be intramuscular. Thus, Tang et al., [Nature, 356, 152-
154 (1992)] disclosed that introduction of gold microprojectiles coated
with DNA encoding bovine growth hormone (BGH) into the skin of
mice resulted in production of anti-BGH antibodies in the mice. Furth
et al., [Analytical Biochemistry, 205, 365-368, (1992)] showed that a jet
injector could be used to transfect skin, muscle, fat, and mammary
tissues of living animals. Various methods for introducing nucleic acids
was recently reviewed [Friedman, T., Science, 244, 1275-1281 (1989)].
See also Robinson et al., [Abstracts of Papers Presented at the 1992
meeting on Modern Approaches to New Vaccines, Including Prevention
of AIDS, Cold Spring Harbor, p92; Vaccine 11, 957 (1993)], where the
im, ip, and iv administration of avian influenza DNA into chickens was
alleged to have provided protection against lethal challenge.
Intravenous injection of a DNA:cationic liposome complex in mice was
CA 02205175 1997-05-12
WO 96/15241 PCT/1JS95/14899
-4-
shown by Zhu et al., [Science 261, 209-211 (9 July 1993); see also =
W093/24640, 9 Dec. 1993] to result in systemic expression of a cloned
transgene. Recently, Ulmer et al., [Science 259, 1745-1749, (1993)] reported
on the heterologous protection against influenza virus infection
by injection of DNA encoding influenza virus proteins.
Wang et al., [P.N.A.S. USA 90, 4156-4160 (May, 1993)]
reported on elicitation of immune responses in mice against HIV by
intramuscular inoculation with a cloned, genomic (unspliced) HIV
gene. However, the level of immune responses achieved was very low,
and the system utilized portions of the mouse mammary tumor virus
(MMTV) long terminal repeat (LTR) promoter and portions of the
simian virus 40 (SV40) promoter and terminator. SV40 is known to
transform cells, possibly through integration into host cellular DNA.
Thus, the system described by Wang et al., is wholly inappropriate for
administration to humans, which is one of the objects of the instant
invention.
WO 93/17706 describes a method for vaccinating an animal
against a virus, wherein carrier particles were coated with a gene
construct and the coated particles are accelerated into cells of an animal.
Studies by Wolff et al. (supra) originally demonstrated that
intramuscular injection of plasmid DNA encoding a reporter gene
results in the expression of that gene in myocytes at and near the site of
injection. Recent reports demonstrated the successful immunization of
mice against influenza by the injection of plasmids encoding influenza A
hemagglutinin (Montgomery, D.L. et al., 1993, Cell Biol., 12, pp.777-
783), or nucleoprotein (Montgomery, D.L. et al., supra; Ulmer, J.B. et
al., 1993, Science, 2_59, pp.1745-1749). The first use of DNA
immunization for a herpes virus has been reported (Cox et al., 1993,
J.Virol., 67, pp.5664-5667). Injection of a plasmid encoding bovine
herpesvirus 1 (BHV-1) glycoprotein g IV gave rise to anti-g IV antibodies in
mice and calves. Upon intranasal challenge with BHV-1,
immunized calves showed reduced symptoms and shed substantially less virus
than controls.
CA 02205175 1997-05-12
WO 96/15241 PCT/US95/14899
-5-
Tuberculosis (TB) is a chronic infectious disease of the
lung caused by the pathogen Mycobacterium tuberculosis. TB is one of
= the most clinically significant infections worldwide, with an incidence of
3 million deaths and 10 million new cases each year. It has been
estimated that as much as one third of the world's population may be
infected and, in developing countries, 55 million cases of active TB have
been reported. Until the turn of the century, TB was the leading cause
of death in the United States. But, with improved sanitary conditions
and the advent of antimicrobial drugs, the incidence of mortality
steadily declined to the point where it was predicted that the disease
would be eradicated by the year 2000. However, in most developed
countries, the number of cases of active TB has risen each year since the
mid-1980's. Part of this resurgence has been attributed to immigration
and the growing number of immunocompromised, HIV-infected
individuals. If left unabated, it is predicted that TB will claim more
than 30 million human lives in the next ten years. As alarming as these
figures may seem, it is of even greater concern that multidrug-resistant
(MDR) strains of M. tuberculosis have arisen. These MDR strains are
not tractable by traditional drug therapy and have been responsible for
several recent outbreaks of TB, particularly in urban centers.
Therefore, one of the key components in the management of TB in the
long-term will be an effective vaccine [for review see Bloom and
Murray, 1993, Science 257, 1055].
M. tuberculosis is an intracellular pathogen that infects
macrophages and is able to survive within the harsh environment of the
phagolysosome in this type of cell. Most inhaled bacilli are destroyed
by activated alveolar macrophages. However, the surviving bacilli can
multiply in macrophages and be released upon cell death, which signals
the infiltration of lymphocytes, monocytes and macrophages to the site.
Lysis of the bacilli-laden macrophages is mediated by delayed-type
hypersensitivity (DTH) and results in the development of a solid caseous
tubercle surrounding the area of infected cells. Continued DTH causes
the tubercle to liquefy, thereby releasing entrapped bacilli. The large
dose of extracellular bacilli triggers further DTH, causing damage to
CA 02205175 1997-05-12
WO 96/15241 PCT/US95/14899
-6-
the bronchi and dissemination by lymphatic, hematogenous and
bronchial routes, and eventually allowing infectious bacilli to be spread
by respiration. =
Immunity to TB involves several types of effector cells.
Activation of macrophages by cytokines, such as interferon-y, is an
effective means of minimizing intracellular mycobacterial
multiplication. However, complete eradication of the bacilli by this
means is often not achieved. Acquisition of protection against TB
requires T lymphocytes. Among these, both CD8+ and CD4+ T cells
seem to be important [Orme et al, 1993, J. Infect. Dis. 167, 1481].
These cell types secrete interferon-y in response to mycobacteria,
indicative of a Thl immune response, and possess cytotoxic activity to
mycobacteria-pulsed target cells. In recent studies using (3-2
microglobulin- and CD8-deficient mice, CTL responses have been
shown to be critical in providing protection against M. tuberculosis
[Flynn et al, 1992, Proc. Natl. Acad. Sci. USA 89, 12013; Flynn et al,
1993, J. Exp. Med. 178, 2249; Cooper et al, 1993, J. Exp. Med. 178,
2243]. In contrast, B lymphocytes do not seem to be involved, and
passive transfer of anti-mycobacterial antibodies does not provide
protection. Therefore, effective vaccines against TB must generate cell-
mediated immune responses.
Antigenic stimulation of T cells requires presentation by
MHC molecules. In order for mycobacterial antigens to gain access to
the antigen presentation pathway they must be released from the
bacteria. In infected macrophages, this could be accomplished by
secretion or bacterial lysis. Mycobacteria possess many potential T-cell
antigens and several have now been identified [Andersen 1994, Dan.
Med. Bull. 41, 205]. Some of these antigens are secreted by the
bacteria. It is generally believed that immunity against TB is mediated
by CD8+ and CD4+ T cells directed toward these secreted antigens. In =
mouse and guinea pig models of TB, protection from bacterial
challenge, as measured by reduced weight loss, has been achieved using a
mixture of secreted mycobacterial antigens [Pal and Horowitz, 1992
CA 02205175 1997-05-12
WO 96/15241 PCTIUS95/14899
-7-
Infect. Immunity 60, 4781; Andersen 1994, Infect. Immunity 62, 2536;
Collins, 1994, Veterin. Microbiol. 40, 95].
Several potentially protective T cell antigens have been
identified in M. tuberculosis and some of these are being investigated as
vaccine targets. Recent work has indicated that the predominant T-cell
antigens are those proteins that are secreted by mycobacteria during
their residence in macrophages, such as: i) the antigen 85 complex of
proteins (85A, 85B, 85C) [Wiker and Harboe, 1992, Microbiol. Rev.
56, 648], ii) a 6 kDa protein termed ESAT-6 [Andersen 1994, Infect.
Immunity 62, 2536], iii) a 38 kDa lipoprotein with homology to PhoS
[Young and Garbe, 1991, Res. Microbiol. 142, 55; Andersen, 1992, J.
Infect. Dis. 166, 874], iv) the 65 kDa GroEL heat-shock protein [Siva
and Lowrie, 1994, Immunol. 82, 244], v) a 55 kDa protein rich in
proline and threonine [Romain et al, 1993, Proc. Natl. Acad. Sci. USA
90, 5322], and vi) a 19 kDa lipoprotein [Faith et al, 1991, Immunol. 74,
1].
The genes for each of the three antigen 85 proteins (A, B,
and C) have been cloned and sequenced [Borremans et al, 1989, Infect.
Immunity 57, 3123; Content et al, Infect. Immunity 59, 3205; DeWit et
al 1994, DNA Seq. 4, 267]. In addition, these structurally-related
proteins are targets for strong T-cell responses after both infection and
vaccination [Huygen et al, 1988, Scand. J. Immunol. 27, 187; Launois et
al, 1991, Clin. Exp. Immunol. 86, 286; Huygen et al, 1992, Infect.
Immunity 60, 2880; Munk et al, 1994, Infect. Immunity 62, 726;
Launois et al, 1994, Infect. Immunity 62, 3679]. Therefore, the antigen
85 proteins are considered to be good vaccine targets.
SUMMARY OF THE INVENTION
To test the efficacy of DNA immunization in the prevention
of M.tb disease, M.tb protein-coding DNA sequences were cloned into
eukaryotic expression vectors. These DNA constructions elicit an
immune response when injected into animals. Immunized animals are
infected with mycobacteria to evaluate whether or not direct DNA
immunization with the gene (or other M.tb genes) could protect them
CA 02205175 1997-05-12
WO 96/15241 PCT/US95/14899
-8-
from disease. Nucleic acids, including DNA constructs and RNA transcripts,
capable of inducing in vivo expression of M.tb proteins
upon direct introduction into animal tissues via injection or otherwise are
therefore disclosed. Injection of these nucleic acids may elicit
immune responses which result in the production of cytotoxic T
lymphocytes (CTLs) specific for M.tb antigens, as well as the generation
of M.tb-specific helper T lymphocyte responses, which are protective
upon subsequent challenge. These nucleic acids are useful as vaccines
for inducing immunity to M.tb, which can prevent infection and/or
ameliorate M.tb-related disease.
BRIEF DESCRIPTION OF THE DRAWINGS
Fig. 1. General principle for cloning M.tb genes into
expression vectors is shown.
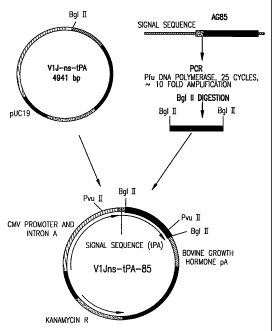
Fig. 2. Vector map of V1Jns.tPA85A.C1 is shown.
Fig. 3. Vector map of VlJns.85A.C2 is shown.
Fig. 4. Vector map of VlJns.85A.C3 is shown.
Fig. 5. Vector map of VlJns.tPA85B.C1 is shown.
Fig. 6. Vector map of V 1Jns.tPA85C.C1 is shown.
Fig. 7 N-Terminal sequence verification of constructs is
shown.
Fig. 8 Expression of M.tb proteins in tissue culture is shown. Fig. 9
Production of antigen 85A-specific antibodies in DNA-
vaccinated mice is shown.
CA 02205175 1997-05-12
WO 96/15241 PCT/US95/14899
-9-
Fig. 10 IL-2 production in BALB/c mice by a Th DNA vaccine is
shown.
Fig. 11 IL-2 production in C57BL/6 mice by a Th DNA vaccine is
shown.
Fig. 12 IFN-,y production in BALB/c mice by a Tb DNA vaccine is
shown.
Fig. 13 IFN-y production in C57BL/6 mice by a Th DNA
vaccine is shown.
Fig. 14 Lack of IL-4 production in BALB/c mice by a Tb
DNA vaccine is shown.
Fig. 15 Lack of IL-6 production in mice by a Th DNA vaccine is
shown.
Fig. 16 Lack of IL-10 production in mice by a Tb DNA vaccine is
shown.
Fig. 17 Reduction of BCG multiplication in lungs of C57BL/6 mice
vaccinated with a Th DNA vaccine is shown.
Fig. 18 Reduction of BCG multiplication in lungs of BALB/c mice
vaccinated with a Tb DNA vaccine is shown.
Fig. 19 Reduction of BCG multiplication in spleens of BALB/c
mice vaccinated with a Th DNA vaccine is shown.
Fig. 20 Reduction of BCG multiplication in spleens of C57BL/6
mice vaccinated with a Th DNA vaccine is shown.
DETAILED DESCRIPTION OF THE INVENTION
CA 02205175 1997-05-12
WO 96/15241 PCT/US95/14899
-10-
This invention provides polynucleotides which, when =
directly introduced into a vertebrate in vivo, including mammals such as
humans, induces the expression of encoded proteins within the animal. =
As used herein, a polynucleotide is a nucleic acid which contains
essential regulatory elements such that upon introduction into a living
vertebrate cell, and is able to direct the cellular machinery to produce
translation products encoded by the genes comprising the
polynucleotide. In one embodiment of the invention, the polynucleotide
is a polydeoxyribonucleic acid comprising Mycobacterium tuberculosis
(M.tb ) genes operatively linked to a transcriptional promoter. In
another embodiment of the invention the polynucleotide vaccine
comprises polyribonucleic acid encoding M.tb genes which are
amenable to translation by the eukaryotic cellular machinery
(ribosomes, tRNAs, and other translation factors). Where the protein
encoded by the polynucleotide is one which does not normally occur in
that animal except in pathological conditions, (i.e. an heterologous
protein) such as proteins associated with M.tb, the animals' immune
system is activated to launch a protective immune response. Because
these exogenous proteins are produced'by the animals' own tissues, the
expressed proteins are processed by the major histocompatibility system
(MHC) in a fashion analogous to when an actual M.tb infection occurs.
The result, as shown in this disclosure, is induction of immune
responses against M.tb. Polynucleotides for the purpose of generating
immune responses to an encoded protein are referred to herein as
polynucleotide vaccines or PNV.
There are many embodiments of the instant invention
which those skilled in the art can appreciate from the specification.
Thus, different transcriptional promoters, terminators, carrier vectors
or specific gene sequences may be used successfully.
The instant invention provides a method for using a polynucleotide which, upon
introduction into mammalian tissue, induces
the expression, in vivo, of the polynucleotide thereby producing the
encoded protein. It is readily apparent to those skilled in the art that
variations or derivatives of the nucleotide sequence encoding a protein
CA 02205175 1997-05-12
WO 96/15241 PCTIUS95/14899
-11-
can be produced which alter the amino acid sequence of the encoded
protein. The altered expressed protein may have an altered amino acid
sequence, yet still elicits immune responses which react with the
mycobacterial protein, and are considered functional equivalents. In
addition, fragments of the full length genes which encode portions of
the full length protein may also be constructed. These fragments may
encode a protein or peptide which elicits antibodies which react with the
mycobacterial protein, and are considered functional equivalents.
In one embodiment of this invention, a gene encoding an
M.tb gene product is incorporated in an expression vector. The vector
contains a transcriptional promoter recognized by eukaryotic RNA
polymerase, and a transcriptional terminator at the end of the M.tb gene
coding sequence. In a preferred embodiment, the promoter is the
cytomegalovirus promoter with the intron A sequence (CMV-intA),
although those skilled in the art will recognize that any of a number of
other known promoters such as the strong immunoglobulin, or other
eukaryotic gene promoters may be used. A preferred transcriptional
terminator is the bovine growth hormone terminator. The combination
of CMVintA-BGH terminator is preferred. In addition, to assist in
preparation of the polynucleotides in prokaryotic cells, an antibiotic
resistance marker is also optionally included in the expression vector
under transcriptional control of a suitable prokaryotic promoter.
Ampicillin resistance genes, neomycin resistance genes or any other
suitable antibiotic resistance marker may be used. In a preferred
embodiment of this invention, the antibiotic resistance gene encodes a
gene product for neomycin/kanamycin resistance. Further, to aid in the
high level production of the polynucleotide by growth in prokaryotic
organisms, it is advantageous for the vector to contain a prokaryotic
origin of replication and be of high copy number. Any of a number of
commercially available prokaryotic cloning vectors provide these
elements. In a preferred embodiment of this invention, these
functionalities are provided by the commercially available vectors
known as the pUC series. It may be desirable, however, to remove non-
essential DNA sequences. Thus, the lacZ and lacI coding sequences of
CA 02205175 1997-05-12
WO 96/15241 PCTIUS95/14899
-12-
pUC may be removed. It is also desirable that the vectors are not able =
to replicate in eukaryotic cells. This minimizes the risk of integration
of polynucleotide vaccine sequences into the recipients' genome. =
In another embodiment, the expression vector pnRSV is
used, wherein the Rous sarcoma virus (RSV) long terminal repeat
(LTR) is used as the promoter. In yet another embodiment, V1, a
mutated pBR322 vector into which the CMV promoter and the BGH
transcriptional terminator were cloned is used. In a preferred
embodiment of this invention, the elements of Vl and pUC19 have been
been combined to produce an expression vector named V1J.
Into V1J, V1JtPA or another desirable expression vector is
cloned an M.tb gene, such as one of the antigen 85 complex genes, or
any other M.tb gene which can induce anti-M.tb immune responses
(CTLs, helper T lymphocytes and antibodies). In another embodiment,
the ampicillin resistance gene is removed from V 1 J and replaced with a
neomycin resistance gene, to generate V 1J-neo, into which any of a
number of different M.tb genes may be cloned for use according to this
invention. In yet another embodiment, the vector is VlJns, which is the
same as V 1Jneo except that a unique Sfi 1 restriction site has been
engineered into the single Kpnl site at position 2114 of V 1J-neo. The
incidence of Sfil sites in human genomic DNA is very low
(approximately 1 site per 100,000 bases). Thus, this vector allows
careful monitoring for expression vector integration into host DNA,
simply by Sfil digestion of extracted genomic DNA. In a further
embodiment, the vector is V 1 R. In this vector, as much non-essential
DNA as possible is "trimmed" to produce a highly compact vector.
This vector allows larger inserts to be used, with less concern that
undesirable sequences are encoded and optimizes uptake by cells when
the construct encoding specific virus genes is introduced into
surrounding tissue. The methods used in producing the foregoing vector
modifications and development procedures may be accomplished
according to methods known by those skilled in theyart.
From this work those skilled in the art will recognize that
one of the utilities of the instant invention is to provide a system for in
CA 02205175 1997-05-12
WO 96/15241 PCT/US95/14899
-13-
vivo as well as in vitro testing and analysis so that a correlation of M.tb
sequence diversity with CTL and T-cell proliferative responses, as well
as other parameters can be made. The isolation and cloning of these
various genes may be accomplished according to methods known to
those skilled in the art. This invention further provides a method for
systematic identification of M.tb strains and sequences for vaccine
production. Incorporation of genes from primary isolates of M.tb
strains provides an immunogen which induces immune responses against
clinical isolates of the organism and thus meets a need as yet unmet in
the field. Furthermore, if the virulent isolates change, the immunogen
may be modified to reflect new sequences as necessary.
In one embodiment of this invention, a gene encoding an
M.tb protein is directly linked to a transcriptional promoter. The use of
tissue-specific promoters or enhancers, for example the muscle creatine
kinase (MCK) enhancer element may be desirable to limit expression of
the polynucleotide to a particular tissue type. For example, myocytes
are terminally differentiated cells which do not divide. Integration of
foreign DNA into chromosomes appears to require both cell division
and protein synthesis. Thus, limiting protein expression to non-dividing
cells such as myocytes may be preferable. However, use of the CMV
promoter is adequate for achieving expression in many tissues into
which the PNV is introduced.
M.tb and other genes are preferably ligated into an
expression vector which has been specifically optimized for
polynucleotide vaccinations. Elements include a transcriptional
promoter, immunogenic epitopes, and additional cistrons encoding
immunoenhancing or immunomodulatory genes, with their own
promoters, transcriptional terminator, bacterial origin of replication
and antibiotic resistance gene, as described herein. Optionally, the
vector may contain internal ribosome entry sites (IRES) for the
expression of polycistronic mRNA. Those skilled in the art will
appreciate that RNA which has been transcribed in vitro to produce
multi-cistronic mRNAs encoded by the DNA counterparts is within the
scope of this invention. For this purpose, it is desirable to use as the
CA 02205175 1997-05-12
WO 96115241 PCTIUS95114899
-14-
transcriptional promoter such powerful RNA polymerase promoters as the T7 or
SP6 promoters, and performing in vitro run-on transcription
with a linearized DNA template. These methods are well known in the
art.
The protective efficacy of polynucleotide M.tb immunogens
against subsequent challenge is demonstrated by immunization with the
DNA of this invention. This is advantageous since no infectious agent is
involved, no assembly/replication of bacteria is required, and
determinant selection is permitted. Furthermore, because the sequence
of mycobacterial gene products may be conserved among various strains
of M.th, protection against subsequent challenge by another strain of
M.tb is obtained.
The injection of a DNA expression vector encoding antigen
85A, B or C may result in the generation of significant protective
i.mmunity against subsequent challenge. In particular, specific CTLs and
helper T lymphocyte responses may be produced.
Because each of the M.tb gene products exhibit a high
degree of conservation among the various strains of M.tb and because
immune responses may be generated in response to intracellular
expression and MHC processing, it is expected that many different M.tb
PNV constructs may give rise to cross reactive immune responses.
The invention offers a means to induce heterologous
protective immunity without the need for self-replicating agents or
adjuvants. The generation of high titer antibodies against expressed
proteins after injection of viral protein and human growth hormone
DNA, [Tang et al., Nature 356, 152, 1992], indicates this is a facile and
highly effective means of making antibody-based vaccines, either
separately or in combination with cytotoxic T-lymphocyte and helper T
lymphocyte vaccines targeted towards conserved antigens.
The ease of producing and purifying DNA constructs
compares favorably with traditional protein purification, facilitating the
generation of combination vaccines. Thus, multiple constructs, for
example encoding antigen 85 complex genes and any other M.tb gene
also including non-M.tb genes may be prepared, mixed and co-
CA 02205175 1997-05-12
WO 96/15241 PCTIUS95/14899
-15-
administered. Additionally, protein expression is maintained following
DNA injection [H. Lin et al., Circulation 82, 2217 (1990); R.N. Kitsis et
al., Proc. Natl. Acad. Sci. (USA) 88, 4138 (1991); E. Hansen et al.,
FEBS Lett. 290, 73 (1991); S. Jiao et al., Hum. Gene Therapy 3, 21
(1992); J.A. Wolff et al., Human Mol. Genet. 1, 363 (1992)], the
persistence of B- and T-cell memory may be enhanced [D. Gray and P.
Matzinger, J. Exp. Med. 174, 969 (1991); S. Oehen et al., ibid. 176,
1273 (1992)], thereby engendering long-lived humoral and cell-
mediated immunity.
The amount of expressible DNA or transcribed RNA to be
introduced into a vaccine recipient will have a very broad dosage range
and may depend on the strength of the transcriptional and translational
promoters used. In addition, the magnitude of the immune response
may depend on the level of protein expression and on the
immunogenicity of the expressed gene product. In general, an effective
dose ranges of about 1 ng to 5 mg, 100ng to 2.5 mg, 1 gg to 750 .g, and
preferably about 10 g to 300 g of DNA is administered directly into
muscle tissue. Subcutaneous injection, intraderrnal introduction,
impression through the skin, and other modes of administration such as
intraperitoneal, intravenous, or inhalation delivery are also suitable. It
is also contemplated that booster vaccinations may be provided.
Following vaccination with M.tb polynucleotide immunogen, boosting
with M.tb protein inimunogens such as the antigen 85 complex gene
products is also contemplated. Parenteral administration, such as
intravenous, intramuscular, subcutaneous or other means of
administration of interleukin-12 protein (or other cytokines, e.g. GM-
CSF), concurrently with or subsequent to parenteral introduction of the
PNV of this invention may be advantageous.
The polynucleotide may be naked, that is, unassociated with
any proteins, adjuvants or other agents which affect the recipients'
immune system. In this case, it is desirable for the polycucleotide to be
in a physiologically acceptable solution, such as, but not limited to,
sterile saline or sterile buffered saline. Alternatively, the DNA may be
associated with liposomes, such as lecithin liposomes or other liposomes
CA 02205175 1997-05-12
WO 96/15241 PCT/US95/14899
-16-
known in the art, as a DNA-liposome mixture, or the DNA may be =
associated with an adjuvant known in the art to boost immune responses,
such as a protein or other carrier. Agents which assist in the cellular
uptake of DNA, such as, but not limited to, calcium ions, may also be
used. These agents are generally referred to herein as transfection
facilitating reagents and pharmaceutically acceptable carriers.
Techniques for coating microprojectiles coated with polynucleotide are
known in the art and are also useful in connection with this invention.
For DNA intended for human use it may be useful to have the final
DNA product in a pharmaceutically acceptable carrier or buffer
solution. Pharmaceutically acceptable carriers or buffer solutions are
known in the art and include those described in a variety of texts such as
Remington's Pharmaceutical Sciences.
In another embodiment, the invention is a polynucleotide
which comprises contiguous nucleic acid sequences capable of being
expressed to produce a gene product upon introduction of said
polynucleotide into eukaryotic tissues in vivo. The encoded gene
product preferably either acts as an immunostimulant or as an antigen
capable of generating an immune response. Thus, the nucleic acid
sequences in this embodiment encode an M.tb immunogenic epitope, and
optionally a cytokine or a T-cell costimulatory element, such as a
member of the B7 family of proteins.
There are several advantages of immunization with a gene
rather than its gene product. The first is the relative simplicity with
which native or nearly native antigen can be presented to the immune
system. Mammalian proteins expressed recombinantly in bacteria,
yeast, or even mammalian cells often require extensive treatment to
insure appropriate antigenicity. A second advantage of DNA
immunization is the potential for the immunogen to enter the MHC class
I pathway and evoke a cytotoxic T cell response. Immunization of mice
with DNA encoding the influenza A nucleoprotein (NP) elicited a CD8+
response to NP that protected mice against challenge with heterologous
strains of flu. (Montgomery, D.L. et al., supra; Ulmer, J. et al., supra)
CA 02205175 1997-05-12
WO 96/15241 PCT/1JS95/14899
- 17-
There is strong evidence that cell-mediated immunity is
important in controlling M.tb infection [Orme et al, 1993, J. Infect. Dis.
167, 1481; Cooper et al 1993, J. Exp. Med. 178, 2243; Flynn et al,
1993, J. Exp. Med. 178, 2249; Orme et al, 1993, J. Immunol. 151, 518].
Since DNA immunization can evoke both humoral and cell-mediated
immune responses, its greatest advantage may be that it provides a
relatively simple method to survey a large number of M.tb genes for
their vaccine potential.
Immunization by DNA injection also allows, as discussed
above, the ready assembly of multicomponent subunit vaccines.
Simultaneous immunization with multiple influenza genes has recently
been reported. (Donnelly, J. et al., 1994, Vaccines, pp 55-59). The
inclusion in an M.tb vaccine of genes whose products activate different
arms of the immune system may also provide thorough protection from
subsequent challenge.
The vaccines of the present invention are useful for
administration to domesticated or agricultural animals, as well as
humans. Vaccines of the present invention may be used to prevent
and/or combat infection of any agricultural animals, including but not
limited to, dairy cattle, which are susceptible to Mycobacterial infection.
The techniques for administering these vaccines to animals and humans
are known to those skilled in the veterinary and human health fields,
respectively.
The following examples are provided to illustrate the
present invention without, however, limiting the same thereto.
EXAMPLE 1
Vectors_ for Vaccine Production
A) V 1 Expression Vector
The expression vector V 1 was constructed from pCMVIE-
AKI-DHFR [Y. Whang et al., J. Virol. 61, 1796 (1987)]. The AKI and
DHFR genes were removed by cutting the vector with EcoR I and self-
ligating. This vector does not contain intron A in the CMV promoter,
CA 02205175 1997-05-12
WO 96/15241 PCTIUS95/14899
- 18-
so it was added as a PCR fragment that had a deleted intemal Sac I site
[at 1855 as numbered in B.S. Chapman et al., Nuc. Acids Res. 19, 3979
(1991)]. The template used for the PCR reactions was pCMVintA-Lux,
made by ligating the Hind III and Nhe I fragment from pCMV6a120
[see B.S. Chapman et al., ibid.,] which includes hCMV-IEl
enhancer/promoter and intron A, into the Hind III and Xba I sites of
pBL3 to generate pCMVIntBL. The 1881 base pair luciferase gene
fragment (Hind III-Sma I Klenow filled-in) from RSV-Lux [J.R. de Wet
et al., Mol. Cell Biol. 7, 725, 1987] was cloned into the Sal I site of
pCMVIntBL, which was Klenow filled-in and phosphatase treated.
The primers that spanned intron A are:
5' primer, SEQ. ID:1:
5'-CTATATAAGCAGAG CTCGTTTAG-3'; The 3' primer, SEQ ID:2:
5'-GTAGCAAAGATCTAAGGACGGTGA CTGCAG-3'.
The primers used to remove the Sac I site are:
sense primer, SEQ ID:3:
5-GTATGTGTCTGAAAATGAGCGTGGAGATTGGGCTCGCAC-3'
and the antisense primer, SEQ ID:4:
5'-GTGCGAGCCCAATCTCCACGCTCA=CAGACACA TAC-3'.
The PCR fragment was cut with Sac I and Bgl II and
inserted into the vector which had been cut with the same enzymes.
B) V1J Expression Vector
The purpose in creating V 1J was to remove the promoter
and transcription termination elements from vector V 1 in order to place
them within a more defined context, create a more compact vector, and
to improve plasmid purification yields.
V 1 J is derived from vectors V 1 and pUC 18, a
commercially available plasmid. V 1 was digested with Sspl and EcoRI
restriction enzymes producing two fragments of DNA. The smaller of
these fragments, containing the CMVintA promoter and Bovine Growth
CA 02205175 1997-05-12
WO 96/15241 PCT/US95/14899
-19-
Hormone (BGH) transcription termination elements which control the
expression of heterologous genes, was purified from an agarose
electrophoresis gel. The ends of this DNA fragment were then
"blunted" using the T4 DNA polymerase enzyme in order to facilitate
its ligation to another "blunt-ended" DNA fragment.
pUC 18 was chosen to provide the "backbone" of the
expression vector. It is known to produce high yields of plasmid, is
well-characterized by sequence and function, and is of small size. The
entire lac operon was removed from this vector by partial digestion
with the HaeII restriction enzyme. The remaining plasmid was purified
from an agarose electrophoresis gel, blunt-ended with the T4 DNA
polymerase treated with calf intestinal alkaline phosphatase, and ligated
to the CMVintA/BGH element described above. Plasmids exhibiting
either of two possible orientations of the promoter elements within the
pUC backbone were obtained. One of these plasmids gave much higher
yields of DNA in E. coli and was designated V1J. This vector's
structure was verified by sequence analysis of the junction regions and
was subsequently demonstrated to give comparable or higher expression
of heterologous genes compared with V 1.
C) V 1 Jneo Expression Vector
It was necessary to remove the ampr gene used for
antibiotic selection of bacteria harboring V 1J because ampicillin may
not be desirable in large-scale fermenters. The ampr gene from the
pUC backbone of ViJ was removed by digestion with Sspl and
Eam 11051 restriction enzymes. The remaining plasmid was purified by
agarose gel electrophoresis, blunt-ended with T4 DNA polymerase, and
then treated with calf intestinal alkaline phosphatase. The commercially
available kanr gene, derived from transposon 903 and contained within
the pUC4K plasmid, was excised using the Pstl restriction enzyme,
purified by agarose gel electrophoresis, and blunt-ended with T4 DNA
polymerase. This fragment was ligated with the V1J backbone and
plasmids with the kanr gene in either orientation were derived which
were designated as VlJneo #'s 1 and 3. Each of these plasmids was
CA 02205175 1997-05-12
WO 96/15241 PCT/US95114899
-20-
confirmed by restriction enzyme digestion analysis, DNA sequencing of
the junction regions, and was shown to produce similar quantities of
plasmid as V 1J. Expression of heterologous gene products was also
comparable to V 1J for these VlJneo vectors. VlJneo#3, referred to as
V 1 Jneo hereafter, was selected which contains the kanr gene in the same
orientation as the ampr gene in V 1J as the expression construct.
D) VIJns Expression Vector
An Sfi I site was added to VlJneo to facilitate integration
studies. A commercially available 13 base pair Sfi I linker (New
England BioLabs) was added at the Kpn I site within the BGH sequence
of the vector. VlJneo was linearized with Kpn I, gel purified, blunted
by T4 DNA polymerase, and ligated to the blunt Sfi I linker. Clonal
isolates were chosen by restriction mapping and verified by sequencing
through the linker. The new vector was designated VlJns. Expression
of heterologous genes in V 1Jns (with Sfi I) was comparable to
expression of the same genes in VlJneo (with Kpn I).
E) VlJns-tPA
In order to provide an heterologous leader peptide
sequence to secreted and/or membrane proteins, V 1Jns was modified to
include the human tissue-specific plasminogen activator (tPA) leader.
Two synthetic complementary oligomers were annealed and then ligated
into V 1Jn which had been BglII digested. The sense and antisense
oligomers were 5'-GATC ACC ATG GAT GCA ATG AAG AGA GGG
CTC TGC TGT GTG CTG CTG CTG TGT GGA GCA GTC TTC GTT
TCG CCC AGC GA-3', SEQ. ID:5:, and 5'-GAT CTC GCT GGG CGA
AAC GAA GAC TGC TCC ACA CAG CAG CAG CAC ACA GCA
GAG CCC TCT CTT CAT TGC ATC CAT GGT-3', SEQ. ID:6. The
Kozak sequence is underlined in the sense oligomer. These oligomers
have overhanging bases compatible for ligation to Bglll-cleaved
sequences. After ligation the upstream BgIII site is destroyed while the
downstream BglII is retained for subsequent ligations. Both the
junction sites as well as the entire tPA leader sequence were verified by
CA 02205175 2006-04-03
WO 96/15241 PCT/US95/14899
-21-
DNA sequencing. Additionally, in order to conform with the consensus
optimized vector VlJns (=VlJneo with an SfiI site), an SfiI restriction
site was placed at the Kpnl site within the BGH terminator region of
V1Jn-tPA by blunting the KpnI site with T4 DNA polymerase followed
by ligation with an SfiI linker (catalogue #1138, New England Biolabs).
This modification was verified by restriction digestion and agarose gel
electrophoresis.
F) pGEM-3-X-IRES-B7
(where X = any antigenic gene) As an example of a
dicistronic vaccine construct which provides coordinate expression of a
gene encoding an immunogen and a gene encoding an inununo-
stimulatory protein, the murine B7 gene was PCR amplified from the B
lymphoma cell line CH 1(obtained from the ATCC). B7 is a member of
a family of proteins which provide essential costimulation T cell
activation by antigen in the context of major histocompatibility
complexes I and II. CH 1 cells provide a good source of B7 mRNA
because they have the phenotype of being constitutively activated and B7
is expressed primarily by activated antigen presenting cells such as B
cells and macrophages. These cells were further stimulated in vitro
using cAMP or IL-4 and mRNA prepared using standard guanidinium
thiocyanate procedures. cDNA synthesis was performed using this
mRNA using the GeneAmp RNA PCR kit (Perkin -Elmer Cetus) and a
priming oligomer (5'-GTA CCT CAT GAG CCA CAT AAT ACC
ATG-3', SEQ. ID:7:) specific for B7 located downstream of the B7
translational open reading frame. B7 was amplified by PCR using the
following sense and antisense PCR oligomers: 5'-GGT ACA AGA TCT
ACC ATG GCT TGC AAT TGT CAG TTG ATG C-3', SEQ. ID:8:,
and 5'-CCA CAT AGA TCT CCA TGG GAA CTA AAG GAA GAC
GGT CTG TTC-3', SEQ. ID:9:, respectively. These oligomers provide
Bg1II restriction enzyme sites at the ends of the insert as well as a Kozak
translation initiation sequence containing an Ncol restriction site and an
additional Ncol site located immediately prior to the 3'-terminal BglII
site. Ncol digestion yielded a fragment suitable for cloning into pGEM-
* Trademark
CA 02205175 1997-05-12
WO 96/15241 PCTIUS95/14899
-22-
3-IRES which had been digested with NcoI. The resulting vector,
pGEM-3-IRES-B7, contains an IRES-B7 cassette which can easily be
transferred to V 1 Jns-X, where X represents an antigen-encoding gene.
G) pGEM-3-X-IRES-GM-CSF
(where X = any antigenic gene) This vector contains a
cassette analogous to that described in item C above except that the gene
for the immunostimulatory cytokine, GM-CSF, is used rather than B7.
GM-CSF is a macrophage differentiation and stimulation cytokine which
has been shown to elicit potent anti-tumor T cell activities in vivo (G.
Dranoff et al., Proc. Natl. Acad. Sci. USA, 90, 3539 (1993).
H) pGEM-3-X-IRES-IL-12
(where X = any antigenic gene) This vector contains a
cassette analogous to that described in item C above except that the gene
for the immunostimulatory cytokine, IL-12, is used rather than B7. IL-
12 has been demonstrated to have an influential role in shifting immune
responses towards cellular, T cell-dominated pathways as opposed to
humoral responses jL. Alfonso et al., Science, 263, 235, 1994].
EXAMPLE 2
Vector V 1 R Preparation
In an effort to continue to optimize the basic vaccination
vector, a derivative of V l Jns, designated V 1 R, was prepared. The
purpose for this vector construction was to obtain a minimum-sized
vaccine vector without unneeded DNA sequences, which still retained
the overall optimized heterologous gene expression characteristics and
high plasmid yields that V 1J and VlJns afford. It was determined from
the literature as well as by experiment that (1) regions within the pUC
backbone comprising the E. coli origin of replication could be removed
without affecting plasmid yield from bacteria; (2) the 3'-region of the
kanr gene following the kanamycin open reading frame could be
CA 02205175 1997-05-12
WO 96115241 PCT/US95/14899
- 23 -
removed if a bacterial terminator was inserted in its place; and, (3)
~300 bp from the 3'- half of the BGH terminator could be removed
without affecting its regulatory function (following the original KpnI
restriction enzyme site within the BGH element).
V1R was constructed by using PCR to synthesize three
segments of DNA from VlJns representing the CMVintA
promoter/BGH terminator, origin of replication, and kanamycin
resistance elements, respectively. Restriction enzymes unique for each
segment were added to each segment end using the PCR oligomers:
Sspl and Xhol for CMVintA/BGH; EcoRV and BamHI for the kan r
gene; and, Bcll and SaII for the ori r. These enzyme sites were chosen
because they allow directional ligation of each of the PCR-derived DNA
segments with subsequent loss of each site: EcoRV and Sspl leave blunt-
ended DNAs which are compatible for ligation while BamH1 and Bc1I
leave complementary overhangs as do SaII and Xhol. After obtaining
these segments by PCR each segment was digested with the appropriate
restriction enzymes indicated above and then ligated together in a single
reaction mixture containing all three DNA segments. The 5'-end of the
ori r was designed to include the T2 rho independent terminator
sequence that is normally found in this region so that it could provide
termination information for the kanamycin resistance gene. The ligated
product was confirmed by restriction enzyme digestion (>8 enzymes) as
well as by DNA sequencing of the ligation junctions. DNA plasmid
yields and heterologous expression using viral genes within V 1 R appear
similar to V 1Jns. The net reduction in vector size achieved was 1346 bp
(VlJns = 4.86 kb; V1R = 3.52 kb).
PCR oligomer sequences used to synthesize V IR (restriction enzyme
sites are underlined and identified in brackets following sequence):
(1) 5'-GGT ACA AAT ATT GG CTA TTG GCC ATT GCA TAC G-
3'
[SspI], SEQ.ID:10:,
(2) 5'-CCA CAT CTC GAG GAA CCG GGT CAA TTC TTC AGC
ACC-3' [Xhol], SEQ.ID:11:
CA 02205175 1997-05-12
WO 96/15241 PCT/US95/14899
-24-
(for CMVintA/BGH segment)
(3) 5'-GGT ACA GAT ATC GGA AAG CCA CGT TGT GTC TCA
AAA TC-3'[EcoRV], SEQ.ID:12:
(4) 5'-CCA CAT GGA TCC G TAA TGC TCT GCC AGT GTT ACA
ACC-3' [BamHI], SEQ.ID:13:
(for kanamycin resistance gene segment)
(5) 5'-GGT ACA TGA TCA CGT AGA AAA GAT CAA AGG ATC
TTC TTG-3'[Bcll], SEQ.ID:14:,
(6) 5'-CCA CAT GTC GAC CC GTA AAA AGG CCG CGT TGC
TGG-3' [SaII], SEQ.ID:15:
(for E. coli origin of replication)
EXAMPLE 3
Cell Culture and Transfection
For preparation of stably transfected cell lines expressing M.tb
antigens RD cells (human rhabdomyosarcoma ATCC CCL 136) were
grown at 370C, 5% C02 in Dulbecco's modified Eagle's medium
(DMEM) supplemented with 10% heat inactivated fetal bovine serum,
20mM HEPES, 4mM L-glutamine, and 100 g/mL each of penicillin
and streptomycin. Cells were seeded at 1.5x 106 cells/100 mm2 plate
and grown for 18 hours. Cell were transfected with 10 g/plate of the
TB construct and 10 g of co-transfected Cat construct using the
CellPhect kit (Pharmacia), and glycerol shocked (15% glycerol in PBS,
pH 7.2 for 2.5 min) 5 hours after DNA was added to the cells. Cultures
were harvested 72 hours after transfection by washing the plates 2x- 10
mL of cold PBS, pH 7.2, adding 5 mL of cold TEN buffer (40 mM
TRIS-Cl, pH 7.5, 1 mM EDTA, 150 mM NaCI) and scraping. For
analysis of protein expression, cell pellets were lysed in 50 gL of Single
Detergent Lysis Buffer (50 mM Tris-Cl, pH 8.0, 150 mM NaCI, 0.02%
NaN3, 1 %Nonidet P-40, 100 mM PMSF, 2 g/mL aprotinin, 2 g/mL
leupeptin, and 1 g/mL Pepstatin A) and sonicated on ice (2-15 second
CA 02205175 1997-05-12
WO 96/15241 PCTIUS95/14899
-25-
bursts). Lysates were centrifuged at 13,000xg, 40C, for 10 minutes.
Protein concentration was determined by the Bradford method and 20
g of cell extract protein per lane was applied to a 10% TRIS-glycine
polyacrylamide gel (Novex), then transferred to Immobilon P
(Millipore) membrane. Immunoblots were reacted overnight with a
1:20 dilution of the mouse monoclonal antibody TD 17-4 [Huygen et al,
1994, Infect. Immunity 62, 363], followed by a 1.5 hours reaction with
a 1:1000 dilution of goat anti-mouse IgGFc peroxidase (Jackson). The
blots were developed using the ECL kit (Amersham).
EXAMPLE 4
Cloning and DNA preparation
1. Construction of VlJns-tPA-85A (contains mature Ag85A with
tPA signal sequence) was done using the following primers:
sense 85A.C1 primer [SEQ.ID.NO.:16]
GG AAG ATC TTT TCC CGG CCG GGC TTG CCG
Bgl II
antisense 85A primer [SEQ.ID.NO.:17]
GGAAGATCTTGTCTGTTCGGAGCTAGGC.
The Ag85A from M. tuberculosis was amplified from
plasmid p85A.tub, which was prepared by ligating an 800 bp HindIII
fragment to a 1600 bp Hindlll-Sphl fragment from Figure 2 of
Borremans et al, 1989 [Infect. Immunity 57, 3123]. The resulting 2400
bp insert was subcloned in the HindIII and Sphl sites of the BlueScribe
M13+. The entire coding sequence and flanking regions in BlueScribe
M13+ (VCS/Stratagene) were amplified by PCR with the indicated
primers in the following conditions. Each 100 l reaction contains 2.5
Units Cloned Pfu DNA Polymerase (Stratagene), 200 mM dNTP, 0.5 g
of each primer and 250 ng of template DNA in the reaction buffer
CA 02205175 1997-05-12
WO 96/15241 PCT/US95/14899
-26-
supplied with the enzyme (Stratagene). The Hybaid Thermal Reactor
was programmed as follows: 5 minutes denaturation at 94 C followed
by 25 cycles (1 minute at 94 C, 2 minutes at 55 C and 3 minutes at
72 C) ending with 10 minutes extension at 72 C.
Amplified DNA was digested with 50 g/ml Proteinase K
(Boehringer Mannheim) for 30 minutes at 37 C, heated 10 minutes at
95 C followed by 2 phenol (Chloroform-Isoamyl alcohol) extractions
and precipitated with 1 volume of isopropanol, washed twice with 70%
ethanol, dried and dissolved in 20 gl H20. 3 g of amplified DNA was
digested with 40 Units of Bgl II(Boehringer Mannheim) and the 907
bp fragment (in the case of 85A-C 1) was isolated on a 1% agarose gel
and extracted on "Prep a Gene" (BioRad) following the manufacturer's
instructions.
Fifty ng of this fragment was ligated to 20 ng of the Bgl II
digested and dephosphorylated VlJns.tPA vector in a 10 l reaction
containing 2.5 Units T4 DNA ligase (Amersham) in ligation buffer for
16 hours at 14 C, transformed into competent DH5 E. coli (BRL) and
plated on Kanamycin (50 gg/ml) containing LB Agar medium.
Transformants were picked up and their plasmidic DNA was restricted
with Bgl II(to confirm the presence of insert) and with Pvu II to define
its orientation.
2. Construction of VlJns-85A [C2] (contains mature Ag85A with no
signal sequence) was done using the following primers:
Sense 85A C2 [SEQ.ID.NO.: 18]
GGAAGATCTACC ATG GGC TTT TCC CGG CCG GGC TTG C
Antisense 85A [SEQ.ID.NO.:17]
GGAAGATCTTGCTGTTCGGAGCTAGGC.
The same procedure as 1 above was followed, except that cloning was in
V 1Jns.
CA 02205175 1997-05-12
WO 96/15241 PCTIUS95/14899
-27-
3. Construction of VlJns-85A [C3] (contains Ag85A with its own
signal sequence) was done using the primers:
Sense 85A C3 [SEQ.ID.NO.:19]
GGAAGATCTACC ATG GCA CAG CTT GTT GAC AGG GTT
Antisense 85A [SEQ.ID.NO.:17]
GGAAGATCTTGCTGTTCGGAGCTAGGC.
The same procedure as 1 above was followed, except that cloning was in
VlJns.
4. Construction of VlJns-tPA-85B [C1] (contains Ag85B with tPA
signal sequence) was done using the following primers:
Sense 85B [C l ] [SEQ.ID.NO.:20]
GGAAG ATC TCC TTC TCC CGG CCG GGG CTG CCG GTC GAG
Antisense 85B [SEQ.ID.NO.:21 ]
GGAAGATCTAACCTFCGGTTGATCCCGTCAGCC.
The same procedure as 1 above was followed, except that the template
for PCR was p85B.tub.
5. Construction of VlJns-tPA-85C [Cl] (contains Ag85C with tPA
signal sequence) was done using the following primers:
Sense 85C [C 1 ] [SEQ.ID.NO.:22]
GGAAG ATC TCC TTC TCT AGG CCC GGT CTT CCA
Antisense 85C [SEQ.ID.NO.:23]
GGAAGATCTTGCCGATGCTGGCTTGCTGGCTCAGGC.
CA 02205175 1997-05-12
WO 96/15241 PCTlUS95/14899
-28-
The same procedure as 1 above was followed, except that the template
for PCR was p85C.tub.
6. Construction of VlJns-85B [C2] (contains Ag85B with no signal
sequence) is done using the following primers:
Sense 85B [C2] [SEQ.ID.NO.:24]
GGA AGA TCT ACC ATG GGC TTC TCC CGG CCG GGG CTG C
Antisense 85B [SEQ.[D.NO.:21 ]
GGAAGATCTAACCTCGGTTGATCCCGTCAGCC.
The same procedure as 1 above is followed, except that template for
PCR is p85B.tub and that cloning is in VlJns.
7. Construction of VlJns-85C [C2] (contains Ag$5C with no signal
sequence) is done using the following primers:
Sense 85C [C2] [SEQ.ID.NO.:25]
GGA AGA TCT ACC ATG GGC TTC TCT AGG CCC GGT CTT C
Antisense 85C [SEQ.ID.NO.:23]
GGAAGATCTTGCCGATGCTGGCTTGCTGGCTCAGGC.
The same procedure as 1 above is followed, except that template for
PCR is p85C.tub and that cloning is in V 1Jns.
After restriction analysis all of the constructions are
partially sequenced across the vector junctions. Large scale DNA
preparation was essentially as described (Montgomery, D.L. et al.,
supra).
The plasmid constructions were characterized by restriction
mapping and sequence analysis of the vector-insert junctions (see
Figures 1-6). Results were consistent with published M.th sequence
CA 02205175 2006-04-03
WO 96115241 PGT/US95/14899
-29-
data and showed that the initiation codon was intact for each construct
(Figure 7). Also shown are the various additional amino acid residues
unrelated to M.tb Ag85 that were inserted as a result of cloning.
EXAMPLE 5
Expression of M.tbproteins from V 1Jns.tPA plasmids
Rhabdomyosarcoma cells (ATCC CCL136) were planted
one day before use at a density of 1.2 X 106 cells per 9.5 cm2 well in
six-well tissue culture clusters in high glucose DMEM supplemented
with 10% heat-inactivated fetal calf serum, 2 mM L-glutamine, 25 mM
HEPES, 50 U/ml penicillin and 50 gg/mi streptomycin. (All from
BRL-Gibco) Phenol : chloroform extracted cesium chloride purified
plasmid DNA was precipitated with calcium phosphate using Pharmacia
CellPhect reagents according to the kit instructions except that 5 - 15 g
is used for each 9.5 cm2 well of RD cells. Cultures were glycerol
shocked six hours post addition of calcium phosphate-DNA precipate;
after refeeding, cultures were incubated for two days prior to harvest.
Lysates of transfected cultures were prepared in 1 X RIPA
(0.5% SDS, 1.0% TRITON X-100, 1% sodium deoxycholate, 1mM
EDTA, 150mM NaCI, 25 mM TRIS-HCl pH 7.4) supplemented with
1 M leupeptin, 1 M pepstatin, 300nM aprotinin, and 10 M TLCK, and
sonicated briefly to reduce viscosity. Lysates were resolved by
electrophoresis on 10% Tricine gels (Novex) and then transferred to
nitrocellulose membranes. Immunoblots were processed with M.tb
monoclonal antibodies 17/4 and 32/15 [Huygen et al, 1994, Infect.
Immunity 62, 363] and developed with the ECL detection kit
(Amersham).
Expression of M.tb antigen 85 complex genes was
demonstrated by transient transfection of RD cells. Lysates of
transfected or mock transfected cells were fractionated by SDS PAGE
and analyzed by immunoblotting. Figure 8 shows that V 1Jns.tPA-
85A(C 1), V 1Jns.tPA-$5A(C2), V I Jns.tPA-S5A(C3), and V 1Jns.tPA-
* Trademark
CA 02205175 1997-05-12
WO 96/15241 PCT/US95/14899
-30-
85B(C 1) transfected RD cells express an immunoreactive protein with
an apparent molecular weight of approximately 30-32kDa.
EXAMPLE 6
Immunization with PNV and Expression of Antigen 85 Proteins h1 Vivo
Five- to six-week-old female BALB/c and C57BL/6 mice
were anesthetized by intraperitoneal (i.p.) injection of a mixture of 5
mg ketamine HCl (Aveco, Fort Dodge, IA) and 0.5 mg xylazine
(Mobley Corp., Shawnee, KS.) in saline. The hind legs were washed
with 70% ethanol. Animals were injected three times with 100 l of
DNA (2 mg/ml) suspended in saline: 50 l each leg. At 17-18 days
after immunization, serum samples were collected and analyzed for the
presence of anti-Ag85 antibodies. Figure 9 shows specific immunoblot
reactivity of sera from Ag85 DNA-injected mice (C1) but not from
mice that received a control DNA not containing a gene insert (V1J).
Reactivity was detected to a serum dilution of at least 1:160 against 300
ng of purified antigen 85A (Figure 9b). This demonstrates that
injection of Ag85 DNA resulted in Ag85 expression in vivo such that it
was available for the generation of antibody responses in both BALB/c
and C57BL/6 (B6) mice.
EXAMPLE 7
Antigen 85-Specific T-Cell Responses
Spleen cells from vaccinated. mice were analyzed for
cytokine secretion in response to specific antigen restimulation as
described in Huygen et al, 1992 [Infect. Immunity 60, 2880].
Specifically, spleen cells were incubated with culture filtrate (CF)
proteins from M. bovis BCG purified antigen 85A or a 20-mer peptide
(p25) corresponding to a known T-cell epitope for C57BL/6 mice
(amino acids 241-260). Mice were immunized with VlJns.tPA85A (C 1)
(100 g) three times with three week intervals and analyzed 17 days
CA 02205175 1997-05-12
WO 96/15241 PCTIUS95/14899
-31 -
after the final injection. Cytokines were assayed using bio-assays for
IL-2, interferon-y (IFN-y) and IL-6, and by ELISA for IL-4 and IL-10.
Substantial IL-2 and IFN-y production was observed in both BALB/c
and C57BL/6 mice vaccinated with VlJns.tPA85A (Cl) (Figures 10-
13). Furthermore, C57BL/6 mice also reacted to the H-2b-restricted T-
cell epitope (Figure 13). IL-4, IL-6 and IL-10 levels were not
increased in VlJns.tPA85A-vaccinated mice (Figures 14-16). These
results indicate that a Thl type of helper T-cell response was generated
by the DNA vaccine.
EXAMPLE 8
Protection from Mycobacterial Challenge
To test the efficacy of an M.tb DNA vaccine, mice were
challenged with an intravenous injection of live M. bovis BCG (0.5 mg)
and BCG multiplication was analyzed in the spleens and lungs. As
controls, BCG multiplication was measured in challenged naive mice
(primary infection) and challenged mice that were vaccinated with BCG
at the time of DNA injection (secondary infection). The number of
colony-forming units (CFU) in lungs of V 1 Jns.tPA85A (C 1)-vaccinated
mice was substantially reduced compared to mice with primary
infection or mice vaccinated with control DNA V1J. In C57BL/6 mice,
CFU were reduced by 83% on day 8 after challenge (Figure 17) and in
BALB/c mice CFU was reduced by 65% on day 20 (Figure 18). In
spleen, CFU was reduced by approximately 40% at day 20 after
challenge in BALB/c mice (Figure 19) and day 8 in C57BL/6 mice
(Figure 20). Therefore, the inunune responses observed after injection
of an M.tb DNA vaccine provided protection in a live M. bovis
challenge model.
CA 02205175 1997-05-12
WO 96/15241 PCTIUS95/14899
-32-
SEQUENCE LISTING
(1) GENERAL INFORMATION:
(i) APPLICANT: CONTENT, JEAN
HUYGEN, KRIS
LIU, MARGARET A.
MONTGOMERY, DONNA
ULMER, JEFFREY
(ii) TITLE OF_INVENTION: A POLYNUCLEOTIDE TUBERCULOSIS VACCINE
(iii) NUMBER OF SEQUENCES: 25
(iv) CORRESPONDENCE ADDRESS:
(A) ADDRESSEE: JACK L. TRIBBLE
(B) STREET: 126 E. LINCOLN AVE., P.O. BOX 2000
(C) CITY: RAHWAY
(D) STATE: NEW JERSEY
(E) COUNTRY: USA
(F) ZIP: 07065-0907
(v) COMPUTER READABLE FORM:
(A) MEDIUM TYPE: Floppy disk
(B) COMPUTER: IBM PC compatible
(C) OPERATING SYSTEM: PC-DOS/MS-DOS
(D) SOFTWARE: PatentIn Release #1.0, Version #1.25
(vi) CURRENT APPLICATION DATA:
(A) APPLICATION NUMBER: US 08/338,992
(B) FILING DATE: 14-NOV-1994
(C) CLASSIFICATION:
(viii) ATTORNEY/AGENT INFORMATION:
(A) NAME: TRIBBLE, JACK L.
(B) REGISTRATION NUMBER: 32,633
(C) REFERENCE/DOCKET NUMBER: 19342
(ix) TELECOMMUNICATION INFORMATION:
(A) TELEPHONE: (908) 594-5321
(B) TELEFAX: (908) 594-4720
(2) INFORMATION FOR SEQ ID NO:"l:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 23 base pairs
(B) TYPE: nucleic acid
(C) STRANDEDNESS: single
(D) TOPOLOGY: linear
(ii) MOLECULE TYPE: DNA (genomic)
CA 02205175 1997-05-12
WO 96/15241 PCTlUS95/14899
-33-
(xi) SEQUENCE DESCRIPTION: SEQ ID NO:l:
CTATATAAGC AGAGCTCGTT TAG 23
(2) INFORMATION FOR SEQ ID NO:2:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 30 base pairs
(B) TYPE: nucleic acid
(C) STRANDEDNESS: single
(D) TOPOLOGY: linear
(ii) MOLECULE TYPE: DNA (genomic)
(xi) SEQUENCE DESCRIPTION: SEQ ID NO:2:
GTAGCAAAGA TCTAAGGACG GTGACTGCAG - 30
(2) INFORMATION FOR SEQ ID NO:3:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 39 base pairs
(B) TYPE: nucleic acid
(C) STRANDEDNESS: single
(D) TOPOLOGY: linear
(ii) MOLECULE TYPE: DNA (genomic)
(xi) SEQUENCE DESCRIPTION: SEQ ID NO:3:
GTATGTGTCT GAAAATGAGC GTGGAGATTG GGCTCGCAC 39
(2) INFORMATION FOR SEQ ID NO:4:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 39 base pairs
(B) TYPE: nucleic acid
(C) STRANDEDNESS: single
(D) TOPOLOGY: linear
(ii) MOLECULE TYPE: DNA (genomic)
(xi) SEQUENCE DESCRIPTION: SEQ ID NO:4:
GTGCGAGCCC AATCTCCACG CTCATTTTCA GACACATAC 39
(2) INFORMATION FOR SEQ ID NO:5:
(i) SEQUENCE CHARACTERISTICS:
CA 02205175 1997-05-12
WO 96/15241 PCT/US95114899
-34-
(A) LENGTH: 78 base pairs
(B) TYPE: nucleic acid
(C) STRANDEDNESS: single
(D) TOPOLOGY: linear
(ii) MOLECULE TYPE: DNA (genomic)
(xi) SEQUENCE DESCRIPTION: SEQ ID NO:5:
GATCACCATG GATGCAATGA AGAGAGGGCT CTGCTGTGTG CTGCTGCTGT GTGGAGCAGT 60
CTTCGTTTCG CCCAGCGA 78
(2) INFORMATION FOR SEQ ID NO:6:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 78 base pairs
(B) TYPE: nucleic acid
(C) STRANDEDNESS: single
(D) TOPOLOGY: linear
(ii) MOLECULE TYPE: DNA (genomic)
(xi) SEQUENCE DESCRIPTION: SEQ ID NO:6:
GATCTCGCTG GGCGAAACGA AGACTGCTCCACACAGCAGC AGCACACAGC AGAGCCCTCT 60
CTTCATTGCA TCCATGGT 78
(2) INFORMATION FOR SEQ ID NO:7:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 27 base pairs
(B) TYPE: nucleic acid
(C) STRANDEDNESS: single
(D) TOPOLOGY: linear
(ii) MOLECULE TYPE: DNA (genomic)
(xi) SEQUENCE DESCRIPTION: SEQ ID NO:7:
GTACCTCATG AGCCACATAA TACCATG . 27
(2) INFORMATION FOR SEQ ID NO:8:
(i) SEQUENCE-CHARACTERISTICS:
(A) LENGTH: 40 base pairs
(B) TYPE: nucleic acid
(C) STRANDEDNESS: single
(D) TOPOLOGY: linear
CA 02205175 1997-05-12
WO 96115241 PCT/US95/14899
-35-
(ii) MOLECULE TYPE: DNA (genomic)
(xi) SEQUENCE DESCRIPTION: SEQ ID NO:8:
GGTACAAGAT CTACCATGGC TTGCAATTGT CAGTTGATGC 40
(2) INFORMATION FOR SEQ ID NO:9:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 42 base pairs
(B) TYPE: nucleic acid
(C) STRANDEDNESS: single
(D) TOPOLOGY: linear
(ii) MOLECULE TYPE: DNA (genomic)
(xi) SEQUENCE DESCRIPTION: SEQ ID NO:9:
CCACATAGAT CTCCATGGGA ACTAAAGGAA GACGGTCTGT TC 42
(2) INFORMATION FOR SEQ ID NO:10:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 33 base pairs
(B) TYPE: nucleic acid
(C) STRANDEDNESS: single
(D) TOPOLOGY: linear
(ii) MOLECULE TYPE: DNA (genomic)
(xi) SEQUENCE DESCRIPTION: SEQ ID NO:10:
GGTACAAATA TTGGCTATTG GCCATTGCATACG 33
(2) INFORMATION FOR SEQ ID NO:11:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 36 base pairs
(B) TYPE: nucleic acid
(C) STRANDEDNESS: single
(D) TOPOLOGY: linear
(ii) MOLECULE TYPE: DNA (genomic)
(xi) SEQUENCE DESCRIPTION: SEQ ID NO:11:
CCACATCTCG AGGAACCGGG TCAATTCTTC AGCACC 36
CA 02205175 1997-05-12
WO 96/15241 PCT/US95/14899
-36-
(2) INFORMATION FOR SEQ ID NO:12:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 38 base pairs
(B) TYPE: nucleic acid
(C) STRANDEDNESS: single
(D) TOPOLOGY: linear
(ii) MOLECULE TYPE: DNA (genomic)
(xi) SEQUENCE DESCRIPTION: SEQ ID NO:12:
GGTACAGATA TCGGAAAGCC ACGTTGTGTC TCAAAATC 38
(2) INFORMATION FOR SEQ ID NO:13:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 37 base pairs
(B) TYPE: nucleic acid
(C) STRANDEDNESS: single
(D) TOPOLOGY: linear
(ii) MOLECULE TYPE: DNA (genomic)
(xi) SEQUENCE DESCRIPTION: SEQ ID NO:13:
CCACATGGAT CCGTAATGCT CTGCCAGTGT TACAACC 37
(2) INFORMATION FOR SEQ ID NO:14:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 39 base pairs
(B) TYPE: nucleic acid
(C) STRANDEDNESS: single
(D) TOPOLOGY: linear
(ii) MOLECULE TYPE: DNA (genomic)
(xi) SEQUENCE DESCRIPTION: SEQ ID NO:14:
GGTACATGAT CACGTAGAAA AGATCAAAGG ATCTTCTTG 39
(2) INFORMATION FOR SEQ ID NO:15:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 34 base pairs
(B) TYPE: nucleic acid
(C) STRANDEDNESS: single
(D) TOPOLOGY: linear
CA 02205175 1997-05-12
WO 96/15241 PCTIUS95/14899
-37-
(ii) MOLECULE TYPE: DNA (genomic)
(xi) SEQUENCE DESCRIPTION: SEQ ID NO:15:
CCACATGTCG ACCCGTAAAAA GGCCGCGTTG CTGG 35
(2) INFORMATION FOR SEQ ID NO:16:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 29 base pairs
(B) TYPE: nucleic acid
(C) STRANDEDNESS: single
(D) TOPOLOGY: linear
(ii) MOLECULE TYPE: DNA (genomic)
(xi) SEQUENCE DESCRIPTION: SEQ ID NO:16:
GGAAGATCTT TTCCCGGCCG GGCTTGCCG 29
(2) INFORMATION FOR SEQ ID NO:17:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 28 base pairs
(B) TYPE: nucleic acid
(C) STRANDEDNESS: single
(D) TOPOLOGY: linear
(ii) MOLECULE TYPE: DNA (genomic)
(xi) SEQUENCE DESCRIPTION: SEQ ID NO:17:
GGAAGATCTT GTCTGTTCGG AGCTAGGC 28
(2) INFORMATION FOR SEQ ID NO:18:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 37 base pairs
(B) TYPE: nucleic acid
(C) STRANDEDNESS: single
(D) TOPOLOGY: linear
(ii) MOLECULE TYPE: DNA (genomic)
(xi) SEQUENCE DESCRIPTION: SEQ ID NO:18:
GGAAGATCTA CCATGGGCTT TTCCCGGCCG GGCTTGC 37
CA 02205175 1997-05-12
WO 96/15241 PCT/US95/14899
-38-
(2) INFORMATION FOR SEQ ID NO:19:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 36 base pairs
(B) TYPE: nucleic acid
(C) STRANDEDNESS: single
(D) TOPOLOGY: linear
(ii) MOLECULE TYPE: DNA (genomic)
(xi) SEQUENCE DESCRIPTION: SEQ ID NO:19:
GGAAGATCTA CCATGGCACA GCTTGTTGAC AGGGTT 36
(2) INFORMATION FOR SEQ ID NO:20:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 38 base pairs
(B) TYPE: nucleic acid
(C) STRANDEDNESS: single
(D) TOPOLOGY: linear
(ii) MOLECULE TYPE: DNA (genomic)
(xi) SEQUENCE DESCRIPTION: SEQ ID NO:20:
GGAAGATCTC CTTCTCCCGG CCGGGGCTGC CGGTCGAG 38
(2) INFORMATION FOR SEQ ID NO:21:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 33 base pairs
(B) TYPE: nucleic acid
(C) STRANDEDNESS: single
(D) TOPOLOGY: linear
(ii) MOLECULE TYPE: DNA (genomic)
(xi) SEQUENCE DESCRIPTION: SEQ ID NO:21:
GGAAGATCTA ACCTTCGGTT GATCCCGTCA GCC
33
(2) INFORMATION FOR SEQ ID NO:22:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 32 base pairs
(B) TYPE: nucleic acid
(C) STRANDEDNESS: single
(D) TOPOLOGY: linear
CA 02205175 1997-05-12
WO 96/15241 PCT/US95/14899
-39-
(ii) MOLECULE TYPE: DNA (genomic)
(xi) SEQUENCE DESCRIPTION: SEQ ID NO:22:
GGAAGATCTC CTTCTCTAGG CCCGGTCTTC CA 32
(2) INFORMATION FOR SEQ ID NO:23:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 36 base pairs
(B) TYPE: nucleic acid
(C) STRANDEDNESS: single
(D) TOPOLOGY: linear
(ii) MOLECULE TYPE: DNA (genomic)
(xi) SEQUENCE DESCRIPTION: SEQ ID NO:23:
GGAAGATCTT GCCGATGCTG GCTTGCTGGC TCAGGC 36
- - - -
(2) INFORMATION FOR SEQ ID NO:24:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 37 base pairs
(B) TYPE: nucleic acid
(C) STRANDEDNESS: single
(D) TOPOLOGY: linear
(ii) MOLECULE TYPE: DNA (genomic)
(xi) SEQUENCE DESCRIPTION: SEQ ID NO:24:
GGAAGATCTA CCATGGGCTT CTCCCGGCCG GGGCTGC _ 37
(2) INFORMATION FOR SEQ ID NO:25:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 37 base pairs
(B) TYPE: nucleic acid
(C) STRANDEDNESS: single
(D) TOPOLOGY: linear
(ii) MOLECULE TYPE: DNA (genomic)
(xi) SEQUENCE DESCRIPTION: SEQ ID NO:25:
GGAAGATCTA CCATGGGCTT CTCTAGGCCC GGTCTTC 37