Note: Descriptions are shown in the official language in which they were submitted.
CA 02205824 l997-05-22
PATENT RULES
SECTION 111(c) STATEMENT
The content of the sequence listing in computer-readable
form is the same as the content of the sequence listing
contained in the description.
Feb. 3, 1997 JDM:sbt
C:\KEEP~I10-INFO.PGS
CA 0220~824 1997-0~-22
The invention relates to a stable pharmaceutical
preparation with factor VIII procoagulant activity and
vWF binding activity.
Factor VIII functions in the intrinsic pathway of blood
coagulation as a cofactor, in the presence of Ca2+ and
phospholipids for the factor IXa-dependent conversion
of factor X to Xa. Molecular cloning of factor VIII
cDNA revealed that factor VIII consists of a series of
homologous domains which can be represented as follows:
Al-A2-B-A3-Cl-C2. In plasma, factor VIII circulates as
a metal-ion linked hetero-dimer which is noncovalently
bound to von Willebrand factor (vWF). The heavy-chain
of factor VIII with a molecular weight of 90 000 to 200
000 comprises the Al and A2-domain as well as a
variable portion of the 8-domain. The factor VIII light
chain of 80 kDa consists of the A3, Cl and C2 domain.
Activation of factor VIII occurs by limited proteolysis
by thrombin at amino acid position Arg372, Arg740 in
the heavy chain and Argl639 in the light chain.
Consequently, activated factor VIII consists of a
hetero-trimer of the separate Al and A2 domain together
with a thrombin-cleaved light chain.
The X-linked bleeding disorder haemophilia A has been
associated with (functional) absence of factor VIII.
CA 0220~824 1997-0~-22
Over the years the molecular defect in the factor VIII
gene has been localized in an impressive number of
haemophilia A patients (Tuddenham et al., NAR 22
(1994), 3511-3533; Antonarakis et al., Human Mutation 5
(1995), 1-22). Particular interest has been directed to
haemophilia A patients that contain cross reacting
material (CRM+) in their plasma. Elegant studies
employing micro-purification of factor VIII from plasma
of CRM+-haemophilia A patients have significantly
contributed to thé current knowledge of structure-
function relations of factor VIII. Mutations at the
thrombin cleavage sites at amino acid position Arg372
and Arg1689 have been shown to interfere with
activation of factor VIII, in agreement with data ob-
tained with recombinant proteins in which the cleavage
sites of thrombin have been altered by site directed
mutagenesis.
Furthermore, two mutations in the factor VIII gene were
shown to result in additional glycosilation of the
protein that interfered with factor VIII activity. The
molecular basis of the genetic defect in patients with
a reduced level of factor VIII in their plasma has been
less well explored. Higuchi and co-workers have
identified a mutation in the factor VIII gene of a
patient suffering from mild haemophilia A that resulted
in the substitution of a tyrosine for a phenylalanine
at amino acid position 1680 (PNAS 88 (1991), 8307-
CA 0220~824 1997-0~-22
8311). A recombinant factor VIII protein containing
this particular amino acid substitution was shown to be
defective in binding to vWF.
Another mutation of the factor VIII molecule in which
the arginine at amino acid position 2307 is replaced by
a glutamine has been associated with mild to moderate
haemophilia A (Gitschier et al., Science 232 (1986j,
1415-1416; Casula et al., Blood (1990), 662-670).
Functional importance of residue Arg2307 in function of
factor VIII is suggested by the ability of a synthetic
peptide containing this particular residue, to inhibit
binding of factor VIII to phospholipids. Furthermore,
evidence has been presented that this portion of the
native factor VIII molecule is capable of binding to
vWF. Initial attempts to characterise the defect that
is caused by this mutation using material purified from
plasma have been unsuccesful.
On the other side, it is known that about 20~ of
haemophilia A patients which are treated with factor
VIII concentrates develop factor VIII inhibitors (i.e.
antibodies against factor VIII), thereby inhibiting the
effects of the administered factor VIII preparations.
The treatment of factor VIII inhibitor patients is very
difficult and to date there is only a limited number of
CA 0220~824 1997-0~-22
special methods to treat those haemophilia A inhibitor
patients.
It is possible, yet very costly to apply high doses of
factor VIII, thereby neutralizing the factor VIII
antibodies in vivo. The surplus factor VIII then acts
haemostatically. In many cases desensibilisation takes
place and it is again possible to apply standard factor
VIII treatments. Such a high dose treatment, however,
requires large amounts of factor VIII, is time
consuming and may be afflicted with severe anaphylactic
side reactions.
Another cost-intensive method for removing factor VIII
inhibitors employs the extracorporal immunoadsorption
on lectins which bind to immunoglobulins (protein A,
protein G) or on immobilised factor VIII. Since the
patient has to be connected to the apherese machine
during this treatment, this method is also a big burden
in financial terms and for the patient. Despite from
that it is also not possible to achieve blood clotting
of an acute bleeding with this method.
The therapy of choice to date, however, is the
administration of activated prothrombin complex
concentrates (APCC), like FEIBA~ and AUTOPLEX~, which
is suitable to treat acute bleedings even in patients
CA 0220~824 1997-0~-22
with high inhibitor titer (see: e.g. DE-PS 31 27 318).
Other methods which are at present under intensive
investigations are the administration of immunoglobulin
preparations which contain antiidiotypic antibodies and
the administration of recombinant factor VIIa. Yet for
both methods it is not possible to make final
assesments to their clinical efficacy to date.
It is an object of the present invention to provide a
pharmaceutical preparation for the treatment of
patients with factor VIII inhibitors and related
coagulation disorders which allows a simple
administration, a high stability of the product to be
administered, an effective treatment and a longer half
life thereby lessening the burden of the patients.
This object is solved by a stable pharmaceutical
preparation containing a protein with factor VIII
procoagulant activity and vWF binding activity, which
protein has an amino acid sequence derived from the
amino acid sequence of the factor VIII protein,
preferably the human factor VIII protein, with at least
one mutation in at least one immunodominant region of
factor VIII.
An immunodominant region of the factor VIII protein is
defined as an epitope, structure or domain of the
CA 0220~824 1997-0~-22
protein which predominately causes antibody formation.
The mutation(s) may be a point mutation, which results
in the replacement of an amino acid by another,
substitutions, deletions (e.g. deletions of regions
not being essential for the action of factor VIII in
vivo) or insertions (e.g. doubling of certain regions).
Preferably, the mutation(s) are present in the C2
and/or the A2 domain of the factor VIII molecule.
According to the present invention, the protein
contained in the preparation should exhibit both,
factor VIII procoagulant activity and vWF binding
activity.
It is advantageous that the factor VIII mutant protein
in the pharmaceutical preparation exhibits a factor
VIII procoagulant activity and/or vWF binding activity
of at least 30 ~, preferably at least 50 %, more
preferably at least 80 %, particularly at least 100 %,
of the factor VIII procoagulant activity and/or the vWF
binding activity of a factor VIII protein without the
mutation in the immunodominant region, for example, of
a commercially available factor VIII preparation based
on recombinant factor VIII:C.
CA 0220~824 1997-0~-22
The evaluation of the factor VIII procoagulant activity
and the vWF binding activity can be performed by any
suitable test for these properties, especially by those
tests which are routinely carried out when assaying
factor VIII samples, like the one stage clotting assay,
a chromogenic assay such as factor VIII IMMUNOCHROM
(IMMUNO) and adsorptin of the factor VIII on immo-
bilized vWF or vice versa, respectively (see also
Veltkamp et al., Thromb. Diath. Haemos. 19 (1968), 279-
303)-
Factor VIII coagulant activity of the mutant proteinaccording to the present invention is preferably tested
by a "one-stage ¢lotting assay" as describèd e.g. in
Mikaelsson and Oswaldsson Scand.J.Haematol.Suppl.33
(1984), 79-86.
Factor VIII activity may also be assessed by measuring
the ability of factor VIII to function as a cofactor
for factor IXa in the conversion of factor X to factor
Xa employing a chromogenic substrate for factor Xa
(Coatest Factor VIII, Chromogenix, Moelndal, Sweden).
In addition other assays that serve to determine the
amount of factor VIII activity in a sample may be
utilized to test the factor VIII activity of the mutant
proteins that are described in the present invention.
CA 0220~824 1997-0~-22
The actual assay whether any new mutant factor VIII
protein exhibits a certain percentage of factor VIII
procoagulant activity is preferably carried out in
parallel with an assay for the same factor VIII mo-
lecule without the mutation in the immunodominant
region (e.g. factor VIII wild type or a fully active B-
domain deleted factor VIII). With such a calibrated
assay of the mutated factor VIII molecule, the relative
procoagulant activity (the percentage of activity
compared to 100 ~ activity of the wild type or the B-
domain deleted factor VIII) may be assayed without the
risk of an environmental error.
The VWF-binding activity may also be assayed by any
test system, capable of determining a factor VIII:C/vWF
complex which has been formed in the course of the
assay for vWF binding. Binding of the mutant factor
VIII proteins to vWF may be performed using assays that
are described in the literature (e.g. Leyte et al.
Biochem. J., 257 (1990), pp 679-683; Donath et al.
Biochem. J., 312 (1995), pp 49-55). In these assays
purified or non-purified vWF may be used. The vWF used
is preferably purified vWF protein. These methods
include but are not limited to coating of purified vWF
onto microtiter wells (Leyte et al., Biochem. J., 257
(1990), pp 679-683). Alternatively, purified or non-
purified vWF may be immobilized with the help of
CA 0220~824 1997-0~-22
monoclonal antibodies that are directed to vWF (Leyte
et al., Biochem. J., 274 (1991), pp 257-261; Donath et
al., Biochem J., 312 (1995), pp 49-55). Subsequently
different amounts of factor VIII are added to the
immobilized vWF and the amount of bound factor VIII is
deter- mined by common methods. The assay described
above may be utilized to address the binding of factor
VIII mutant proteins to vWF. Similarly other methods
that serve to determine the affinity of a factor VIII
protein for vWF may be employed to address the vWF-
binding properties of the mutant proteins disclosed in
the present invention.
Since with in vitro-tests for factor VIII procoagulant
activity and vWF-binding activity, the results may
often be affected with errors due to their artificial
design, both properties are preferably also assayed by
in vivo or ex vivo tests to obtain more reliable
results with respect to the activity values.
As with the in vitro assays, parallel testing of the
factor VIII molecule without the mutation in the
immunodominant region is also preferred when performing
the in vivo tests. Suitable animal models for
evaluating the factor VIII:C activity in the presence
of inhibitor are described by W0 95/01570 and A 987/95.
CA 0220~824 1997-0~-22
According to a preferred embodiment, the factor VIII
mutant protein in the stable pharmaceutical preparation
of the present invention exhibits the factor VIII
procoagulant activity and/or the vWF binding activity
in the presence of a factor VIII inhibitor, which can
be isolated from inhibitor patients or an equivalent
antibody produced by hybridoma technology, especially
in the presence of anti-factor VIII antibodies which
include but are not limited to antibodies of human
origin directed against the C2-domain and A2-domain of
factor VIII, mouse monoclonal antibodies which include
but are not limited to CLB-CAg 117. It is preferred to
provide a factor VIII mutant, which is active in the
presence of a plurality of inhibitors, at least in the
presence of two antibodies, which bind to the
immunodominant region of the factor VIII molecule.
The fact that these preferred mutant proteins are not
inhibited or inhibited only to a small extent by the
factor VIII inhibitors makes them suitable for the
successful treatment of factor VIII inhibitor patients.
Preferred mutations of the factor VIII molecule in the
prepara- tion according to the present invention are in
immunodominant epitopes or regions of factor VIII,
especially in the C2 or A2 domain. Preferable regions
for site directed mutagenesis are around Arg2307 and
- 10 -
CA 0220~824 1997-0~-22
Arg593, for instance in the region of amino acid 2182
to 2332, like Thr2303-Trp2313. Arginine is then
preferably substituted to Glutamin or Cystein.
Preferable examples are the mutations R2307Q and R593C.
A further immunodominant region of the A2 domain is
located between Arg484 and Ile508 (Healey G.F. et al.,
JBC 270 (1995), 14505-14509) and the regions around
this location. Each new mutation can be assayed as
outlined above whether it exhibits the properties
necessary for the preparation according to the present
invention.
The factor VIII mutant according to the present
invention preferably exhibits a further mutation,
namely a deletion of at least part of the B-domain, a
region which is not essential for the physiological
activity of factor VIII. Deletion mutants as
exemplified in EP-0 690 126 can be used for producing
the mutation according to the present invention. The
comparative assays for factor VIII coagulant activity
and for vWF binding activity for such a B-domain de-
leted factor VIII mutant according to the invention are
then preferably carried out in parallel with a B-domain
deleted factor VIII protein without the mutation in the
immunodominant region.
Further mutants and/or fragments as well as derivatives
CA 0220~824 1997-0~-22
of the factor VIII protein can be used as long as they
have factor VIII:C activity and vWF binding activity.
It surprisingly turned out that the factor VIII mutant
according to the invention still retained vWF binding
activity even though it is not inhibited by common
inhibitors. This finding is the more surprising because
it was know in the art that the binding sites for vWF
and inhibitors overlap.
The binding to vWF in vitro or in vivo is a
prerequisite for the stability of the factor VIII
mutant in a preparation or after the administration of
the preparation to the patient. In the presence of vWF
the factor VIII:C activity is stabilized so that its
activity is substantially preserved even after a
prolonged period of storage time. Also the factor
VIII:C level in a patient is elevated even after a
prolonged period of time after administration due to
the vWF binding and a longer half-life of the thus
stabilized factor VIII mutant.
A further aspect of the present invention relates to a
biologically active factor VIII complex comprising a
factor VIII mutant as described above and vWF or a vWF
fragment. It is preferred to use recombinant vWF, such
as described in EP-O 197 592.
CA 0220~824 1997-0~-22
Yet another aspect of the present invention is a method
of producing a pharmaceutical preparation according to
the present invention comprising the expression of a
factor VIII mutant protein as described above in
mammalian cells, purification of this mutant protein
from these cells and formulating of the purified mutant
protein to a pharmaceutical preparation.
The expression and purification step may be performed
according to any method known for recombinant
production of factor VIII, factor VIII mutants or
related proteins. The formulating to a pharmaceutical
preparation may comprise the addition of
pharmaceutically acceptable additives, stabilizers,
further active components, as well as the providing of
a suitable dosage form and a ready-for-use package.
Expression is preferably performed at temperatures
ranging from 20 to 45~C. For production of mutant
proteins disclosed in this invention expression at a
temperature from 25 to 37~C may prove useful in
obtaining significant amounts of factor VIII in the
conditioned medium; more preferably a temperature of
28~C may be employed to facilitate expression of mutant
proteins.
- 13 -
CA 0220~824 1997-0~-22
It is also possible to co-express the factor VIII
mutant protein together with vWF or fragments thereof
using standard techniques for coexpression.
Alternatively, it is also possible to purify the mutant
factor VIII protein from plasma of a patient carrying
this special mutation in an immunodominant region of
factor VIII and use the purified protein for preparing
a stable pharmaceutical preparation according to the
invention.
The addition of vWF and/or vWF fragments is preferably
performed during the purification or formulating steps.
The invention also relates to the use of a factor VIII
mutant protein as described above for manufacturing a
pharmaceutical preparation to treat patients with
factor VIII inhibitors or patients with a risk for
factor VIII inhibitors.
According to the invention such patients are treated
with an effective dose of the present stable
pharmaceutical preparation. The effective dosage may be
easily determined for each individual patient according
to the severity of the disorders he or she suffers from
and according to inhibitor titers of the patient; the
dosages of standard factor VIII therapies may be an im-
CA 0220~824 1997-0~-22
.
portant standard to rely upon as a first dosage of the
pharmaceutical preparation according to the present
invention. It is likely (although not essentially
necessary) that during the further treatment with the
present preparation the dosage will be decreased due to
the longer half-life of the mutant protein.
According to a further aspect, an antibody preparation
comprising antibodies raised against a factor VIII
mutant protein as described above is provided by the
present invention. These antibody preparation may be
used e.g. for the purification or detection and/or
determination of a factor VIII mutant protein according
to the present invention or for distinguishing between
the factor VIII mutant and native factor VIII,
particularly in the blood or plasma of the patient.
The invention will be explained in more detail by way
of the following examples and the associated drawing
figures to which, however, it shall not be limited.
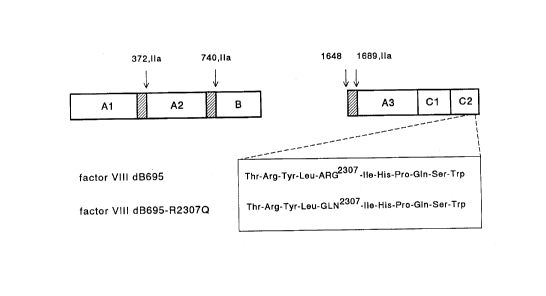
There are illustrated in: Fig.1: a schematic
representation of factor VIII dB695 and factor VIII
dB695-R2307Q; Fig.2: the pulse-chase analysis of cells
transfected with factor VIII dB695-cDNA; Fig.3: the
pulse-chase analysis of cells transfected with factor
VIII dB695-R2307Q-cDNA; Fig.4: Endoglycosidase H
CA 0220~824 1997-0~-22
digestions of factor VIII dB695 and factor VIII dB695-
R2307Q; Fig.5: the expression of factor VIII dB695 in
SKHEP cells; Fig.6 and Fig.7: the expression of factor
VIII dB695-R2307Q in SKHEP cells; Fig.8 and Fig.9:
Inhibition of factor VIII by CLB-CAgll7; Fig.10: factor
VIII-vWF binding.
E x a m p l e s :
EXAMPLE 1: Plasmid constructions
The plasmid pCLB-BPVdB695, encoding a factor VIII
mutant in which Ala867 is fused to Asp1583 has been
described by Mertens et al. (Brit.J.Haematol.85 (1993),
133-145). Plasmid pCLB-BPVdB695 has been modified by
introducing a synthetic linker (5'
TCGACCTCCAGTTGAACATTTGTAGCAAGCCACCATGGAAATAGAGCT 3'),
containing part of the 5' untranslated region of the
factor VIII cDNA linked to a consensus sequence for
initiation of translation, in front of the start-codon
(underlined) at the 5' end of the factor VIII cDNA
(Kozak, J.Biol.Chem.266 (1991), 19867-19870). At the
3'-end, the PstI site at nucleotide-position 7066 of
the factor VIII cDNA (nucleotide 1 corresponding to the
first nucleotide of the start codon), a linker was
inserted that contains a NotI site that was utilized to
clone the factor VIII dB695 cDNA into plasmid pBPV
- 16 -
CA 0220~824 1997-0~-22
yielding plasmid pCLB-dB695. The plasmid pCLB-dB695-
R2307Q was constructed by site-directed mutagenesis
using overlap extension (Ho et al., Gene 77 (1989), 51-
59). Oligonucleotide primer RQ2307-1 (5'
CGCTACCTTC_AATTCACCCC 3'; nucleotide position 6967-6987
of factor VIII; sense) and oligonucleotide primer
RQ2307-2 (5'CCATAGGTTGGAATCTAA 3'; nucleotide 1221-1239
of pBPV; anti-sense) were used to amplify a 302 bp
fragment that contained part of the factor VIII cDNA
and the plasmid pBPV. Oligonucleotide primer RQ2307-3
(5' TTAGGATCCCACTAAAGATGAGTTT 3'; nucleotide position
5530-5547; sense) was used together with
oligonucleotide primer RQ2307-4 (5'
GGGGTGAATTTGAAGGTAGCG 3'; nucleotide position 6967-6987
of factor VIII; anti-sense) to amplify a 1464 bp
fragment of factor VIII. Reaction conditions were: 2
min 90~C, 20 min 50~C, 3 min 72~C; 37 times 45 sec
90~C, 90 sec 50~C, 3 min 72~C; 5 min 65~C in the
presence of 1 mM dNTPs, Pfu-polymerase reaction buffer,
50 pMol of each oligonucleotide primer and 2.5 U of
Pfu-Polymerase (Lundberg et al., Gene 108 (1991), 1-6).
Both the 302 bp and 1464 bp fragment were purified and
used as template in a second PCR using primer RQ2307-1
and RQ2307-4 in order to amplify a 1738 bp fragment
that contains the carboxy-terminus of the factor VIII
cDNA in which Arg2307 has been replaced by a Gln. The
amplified fragment was digested with SalI and ApaI and
CA 0220~824 1997-0~-22
the resulting 879 bp fragment containing a mutation at
amino acid position Arg2307 was used to replace the
corresponding ApaI-SalI fragment of pCLB-dB695. The
resulting construct was termed pCLB-dB695-R2307Q. The
nucleotide sequence of the SalI-ApaI fragment used for
construction of pCLB-dB695-R2307Q was verified.
EXAMPLE 2: Tissue culture and transfection
C127 cells were maintained in Iscove's medium
supplemented with 10 % fetal calf serum, 100 U/ml
penicillin and 100 pg/ml streptomycin. Subconfluent
monolayers of C127 cells were transfected essentially
as described in Donath et al. (Biochem.J.312 (1995),
49-55). The presence of factor VIII protein in the
culture medium was monitored by measuring both factor
VIII activity as well as factor VIII antigen (Mertens
et al. (1993)). Factor VIII cofactor activity was
assessed by the ability of factor VIII to function as a
cofactor for the factor IXa-dependent formation of
factor Xa, employing a chromogenic substrate for factor
Xa (Coatest Factor VIII, Chromogenix, Molndal, Sweden).
Factor VIII antigen was determined using monoclonal
antibodies that have been characterized previously
(see: Lenting et al., J.Biol.Chem. 264 (1994), 7150-
7155). Monoclonal antibody CLB-CAg 12, directed against
the factor VIII light chain was used as a solid phase,
- 18 -
CA 0220~824 1997-0~-22
while peroxidase-labeled monoclonal antibody CLB-CAg
117, also directed against the light chain was used to
quantify the amount of factor VIII bound. The amount of
factor VIII heavy chain was determined as follows:
monoclonal antibody ESH-5 (2.5 ~g/ml) was immobilized
to microtiter wells overnight at 4~C in 50 mM NaHC0 3
(pH 9.5). Wells were washed with 50 mM Tris-HCl (pH
7.4). 100 mM NaCl, 0.1% (v/v) Tween-20 and blocked for
1 hour in the same buffer containing 1 % (w/v) HSA.
Factor VIII samples were diluted into blocking buffer
and incubated for 2 hours at 37~C with the immobilized
antibody. Subsequently the wells were washed and the
amount of factor VIII heavy chain bound was determined
employing peroxidase-labeled monoclonal antibody CLB-
CAg 9 at a concentration of 0.7 pg/well. The detection-
limit of this particular ELISA is 10 mU/ml. Normal
plasma from a pool of 40 donors was used as a standard.
The results of these expression experiments are
depicted in table 1 (values expressed as mU/ml:
-- 19 --
CA 0220~824 1997-0~-22
Table 1:
factor VIII factor VIII
dB695 dB695-R2307Q
cofactor activity 181.4 + 2.4 1.73 + 0.06
antigen (light chain) 199 + 4.4 not detectable
antigen (heavy chain) 225 + 39 not detectable
In contrast to factor VIII dB695, only limited amounts
of cofactor acitivity could be detected in the medium
of cells transfected with factor VIII dB695-R2307Q
cDNA. No immuno-reactive material could be detected
with the ELISA specific for factor VIII light chain.
Since the presence of the Arg2307 to Gln mutation may
affect binding of the monoclonal antibodies used,
factor VIII antigen was examined employing monoclonal
antibody CLB-CAg 9, directed against an acidic region
at the carboxy-terminus of the A2-domain of factor
VIII, in conjuction with monoclonal antibody ESH-5
directed against the heavy chain of factor VIII
(Griffin et al., Thromb.Haemostasis 55 (1986), 40-46).
No factor VIII antigen could be detected in the medium
of cells that have been stably transfected with factor
VIII dB695-R2307Q cDNA, while factor VIII dB695 could
- 20 -
CA 0220~824 1997-0~-22
be detected using this particular combination of
antibodies.
EXAMPLE 3: Metabolic labeling and immunoprecipitation
Transfected cells, maintained in Iscove's medium
supplemented with 10% fetal calf serum and 100 U/ml
penicillin and lOOpg/ml streptomycin were metabolically
labeled upon 80% confluency. Cells were washed twice
with PBS and incubated for 30 min in RPMI (Voorberg et
al., J. Cell. Biol. 113 (1991), 195-205), lacking
methionine, which was supplemented with 10% fetal calf
serum dialyzed against 25 mM HEPES (pH 7.0).
Subsequently, the cells were labeled for 30 min in the
presence of [35S]-methionine (50 ~Ci/ml, spec. act.
>800 Ci/mmol). Medium of metabolically labeled cells
was collected in an equal volume of 2 times
concentrated immunoprecipitation buffer (IPB)
consisting of 1% NP-40, 50 mM Tris-HCl (pH 7,5), 150 mM
NaCl, 0.5% SDS, 10 ~g/ml soybean trypsin inhibitor, 10
mM benzamidine and 5 mM N-ethylmaleimide. Cells were
washed twice with phosphate buffered saline (PBS) and
the cells were lysed in IPB. Lysates and conditioned
media were either stored at -20~C or used immediately
for immunoprecipitation. Cell extracts and conditioned
media were precleared by incubation for one hour at
room temperature with gelatin Sepharose and two
successive incubations with protein A Sepharose.
CA 0220~824 1997-0~-22
Specific adsorption was performed overnight at 4~C, by
preformed complexes of protein A Sepharose with
polyclonal rabbit anti-factor VIII antiserum raised
against factor VIII purified from plasma.
Immunoprecipitates were extensively washed with IPB and
finally with 20 mM Tris-HCl (pH 7.4) and analyzed under
reducing conditions on a 7.5 % (w/v) SDS-polyacrylamide
gel. Following electrophoresis gels were fixed in 30%
methanol, 10% acetic acid and treated with 20%
diphenyloxazol in acetic acid for 30 minutes.
Subsequently, gels were incubated in H 2~~ dried and
exposed for variable times. Endoglycosidase H
digestions of immunoprecipitated material were
performed according to Voorberg et al. (J.Cell Biol.113
(1991), 195-205).
It could be shown by the present experiments that in
the cell factor VIII dB695 appears as a single-chain
molecule that disappears from the cell gradually in
time (Fig.2A: analysis of cell extracts of transfected
cells at different periods of chase: lane 1: 0 hour
chase, lane 2: 1 hour chase, lane 3: 3 hour chase, lane
4: 4 hour chase, lane 5: 6 hour chase, lane 6: control;
Fig.2B: analysis of conditioned medium at different
periods of chase: lane 1: 0 hour chase, lane 2: 1 hour
chase, lane 3: 3 hour chase, lane 4: 4 hour chase, lane
5: 6 hour chase, lane 6: control; single chain (sc),
CA 0220~824 1997-0~-22
light chain (lc) and heavy chain (hc) of factor VIII
are indicated in the figure; molecular weight markers
are indicated at the right of the figure).
Concomitant with the decrease in signal observed in the
cell, an increase in factor VIII-reactive material is
observed in the medium. In agreement with previous
data, factor VIII dB695 is secreted in part as a
single-chain protein, while two forms can be detected
that have previously been characterized as light chain
and heavy chain containing a portion of the B-domain of
factor VIII dB695 (Mertens et al.(1993)). Biosynthesis
of factor VIII dB695-R2307Q inside the cell is similar
to that observed for factor VIII dB695. However, no
immuno-reactive material could be detected in the me-
dium of the stably transfected cells, indicating that
factor VIII dB695-R2307Q is not secreted from the cell
(Fig.3A: analysis of cell extracts of transfected cells
at different times of chase: lane 1: 0 hour, lane 2: 1
hour, lane 3: 3 hour, lane 4: 4 hour, lane 5: 6 hour,
lane 6: control; Fig.3B: analysis of conditioned medium
at different periods of chase: lane 1: 0 hour, lane 2:
1 hour, lane 3: 3 hour, lane 4: 4 hour, lane 5: 6 hour,
lane 6: control; molecular weight markers are indicated
at the right and the single chain factor VIII dB695-
R2307Q is indicated at the left of the figure). This
observation suggests that the vast majority of the
CA 0220~824 1997-0~-22
initially synthesized factor VIII dB695-R2307Q is
processed within the cell via a mechanism different
from factor VIII dB695.
EXAMPLE 4: Biochemical analysis of factor VIII inside
the cell
In order to assess the intracellular localization of
both factor VIII dB695 and factor VIII dB695-R2307Q,
pulse-chase analysis was performed. Sensitivity of
immuno-purified proteins towards endo H provides a
marker for the intracellular localization of both
proteins. As shown in Fig.4 both factor VIII dB695 as
well as factor VIII dB695-R2307Q remain sensitive
towards endo H at the different time-points indicated
(Fig.4A: cells transfected with factor VIII dB695 cDNA:
lane 1: 0 hour chase + endo H, lane 2: 0 hour chase -
endo H, lane 3: 2 hour chase + endo H, lane 4: 4 hours
chase + endo H, lane 5: 6 hour chase + endo H; Fig.4B:
cells transfected with factor VIII dB695-R2307Q cDNA:
lane 1: 0 hour chase - endo H, lane 2: 0 hour chase +
endo H, lane 3: 2 hour chase + endo H, lane 4: 4 hours
chase + endo H, lane 5: 6 hour chase + endo H; molecu-
lar weight markers are indicated at the right of the
gels.
These results indicate that both factor VIII dB695 and
- 24 -
CA 0220~824 1997-0~-22
factor VIII dB695-R2307Q are predominantly present in a
compartment prior to the medial-Golgi.
EXAMPLE 5: Expression of factor VIII dB695 and factor
VIII dB695-R2307Q at 28~C and 37~C
The results given in the previous paragraphs indicate
that factor VIII dB695-R2307Q is poorly secreted from
mouse fibroblasts C127 cells. To assess whether
intracellular retention of factor VIII dB695-R2307Q is
a phenomenon specific for C127 cells factor VIII dB695-
R2307Q and factor VIII dB695 cDNA was expressed in
SKHEP cells. Both factor VIII dB695 and factor VIII
dB695-R2307Q cDNA were used to replace the factor VIII
dB928 cDNA in plasmid pCMV-dB928 (EP-0 711 835-A).
The plasmid pCLB-dB695 (see example 1) was modified as
follows a synthetic double stranded linker 5'
GGCCGCCCGGGC 3' was inserted into the NotI-site of
pCLB-dB695 and pCLB-dB695-R2307Q. Subsequently, a KpnI-
XmaI fragment derived of pCLB-dB695 corresponding to
the carboxy-terminus of the factor VIII CDNA was used
to replace the corresponding KpnI-XmaI fragment of
pCMV-dB928. The resulting plasmid was termed pCLB-CMV-
dB695. Similarly, a BglII-XmaI fragment that
corresponds to the carboxy terminus of the factor VIII-
dB695-R2307Q cDNA was used to replace the corresponding
- 25 -
CA 0220~824 1997-0~-22
fragment of pCMV-dB928 which resulted in the plasmid
pCLB-CMV-dB695-R2307Q. SKHEP cells were maintained in
Iscove's medium supplemented with 10~ fetal calf serum,
100 U/ml penicillin and 100 ~g/ml streptomycin.
Subconfluent monolayers of SKHEP cells were transfected
essentially as described in Donath et al.
(Biochem.J.312 (1995), 49-55). Transfected cells were
selected at hygromycin concentrations of 100-1050 mg/ml
and individual clones were isolated and propagated in
selective medium. The presence of factor VIII protein
in the conditioned medium of the transfected cells was
monitored by measuring factor VIII activity as well as
factor VIII antigen. Factor VIII cofactor activity was
measured by probing the abilty of factor VIII to
function as a cofactor for the factor IXa-dependent
formation of factor Xa, employing a chromogenic
substrate for factor Xa (Coatest Factor VIII,
Chromogenix, Molndal, Sweden). Factor VIII antigen was
determined using monoclonal antibodies that have been
characterized previously (see: Leyte et al., Biochem
J.263 (1989) 187-194). Monoclonal antibody CLB-CAg 12
directed against the light chain of factor VIII was
used as a solid phase, while peroxidase conjugated
monoclonal antibody CLB-CAg 69 or CLB-CAg 117, also
directed against the light chain of factor VIII was
used to quantify the amount of factor VIII bound.
CA 0220~824 1997-0~-22
Transfected SKHEP cells expressing factor VIII dB695
were grown at 28~C and 37~C and the amount of factor
VIII protein secreted into the medium was monitored by
determining factor VIII cofactor activity as outlined
above. At 37~C factor VIII-dB695 is secreted from
transfected SKHEP cells at a level of 2 U/ml. On day 4
the expression of factor VIII dB695 is somewhat higher
and reaches a value of 5 U/ml (Figure 5). At 28~C the
amount of factor VIII activity encountered in the
conditioned medium ranges from 2-10 U/ml (Figure 5).
Transfected SKHEP cells expressing factor VIII dB695-
R2307Q were analyzed in a similar manner. At 37~C only
limited amounts of factor VIII activity are secreted
from the transfected cell (+ 10 mU/ml). Surprisingly at
28~C significant amounts of factor VIII activity are
encountered in the conditioned medium of cells
transfected with factor VIII dB695-R2307Q cDNA (Figure
6). Next the amount of factor VIII antigen in the
conditioned medium of SKHEP cells stably transfected
with factor VIII dB695-R2307Q cDNA was determined using
the monoclonal antibodies that have been described
above. The amount of factor VIII antigen as determined
with the monoclonal antibodies CLB-CAg 12 and CLB-CAg
69 was found to be similar to the amount of cofactor
activity present in the conditioned medium (Figure 7).
CA 0220~824 1997-0~-22
This observation reveals that factor VIII dB695-R2307Q
can be secreted from transfected SKHEP cells at 28~C as
functional fully active factor VIII protein. Next the
amount of factor VIII antigen using monoclonal
antibodies CLB-CAg 12 and CLB-CAg 117 was assessed.
Surprisingly, no factor VIII antigen could be detected
in the conditioned medium of cells transfected with
factor VIII dB695-R2307Q cDNA using this particular
combination of monoclonal antibodies (Figure 7). These
data indicate that monoclonal antibody CLB-CAg 117 does
not react with factor VIII dB695-R2307Q. Analysis of
transfected SKHEP cells employing immunofluorescence
confirmed that CLB-CAg 117 did not react with factor
VIII dB695-R2307Q. The observations on the lack of
reactivity of CLB-CAg 117 with factor VIII-dB695-R2307Q
revealed that substitution of amino acid Arg2307~Gln in
factor VIII interferes with binding of monoclonal
antibody CLB-CAg 117.
EXAMPLE 6: Characterization of monoclonal antibody CLB-
CAg 117.
In the previous example it was shown that monoclonal
antibody CLB-CAg 117 is not reactive with factor VIII
dB695-R2307Q. In addition it could be shown that factor
VIII dB695-R2307Q is a functionally fully active factor
VIII protein that can be expressed in SKHEP cells at
- 28 -
CA 0220~824 1997-0~-22
28~C. The properties of monoclonal antibody CLB-CAg 117
were further characterized. The epitope of monoclonal
antibody CLB-CAg 117 has been localized to the factor
VIII light chain. The observation that monoclonal
antibody CLB-CAg 117 does not react with factor VIII
dB695-R2307Q strongly suggests that the epitope of this
antibody is localized in the C2-domain of factor VIII.
Plasmids pCLB-GP67-80K and pCLB-GP67-C2 were
constructed. Plasmid pCLB-GP67-80K encoding the light
chain of factor VIII was constructed by amplifying a
689 bp fragment using oligonucleotide primers 80K-1 (5'
GCCCCATGGGGGAAATAACTCGTACTACTC 3'; nucleotide position
5000-5020 of factor VIII, sense) and 80K-2 (5'
CTGTACTGTCACTTGTCTCCC 3'; nucleotide position 5659-5679
of factor VIII; antisense). The 689 bp product was
purified and digested with NcoI and NdeI. The plasmid
pCLB-dB695 was digested with NdeI (nucleotide position
5521 of factor VIII) and NotI resulting in a fragment
that corresponds to the carboxy terminal part of the
factor VIII light chain. The NcoI-NdeI fragment and the
NdeI-NotI fragment were cloned together into plasmid
pAcGP67B (Pharmingen, San Diego, CA, USA) and this
yielded plasmid pCLB-GP67-80K. Plasmid pCLB-GP67-C2 was
constructed using oligonucleotide primer C2-1 (5'
GTGCCATGGGTAGTTGCAGCATGCCATTG 3'; nucleotide position
6574-6591 of factor VIII; sense) and primer C2-2 (5'
CCATAGGTTGGAATGTAA 3'; nucleotide position 1222-1239 of
- 29 -
CA 0220~824 1997-0~-22
pBPV; anti-sense) which were used to amplify a fragment
which corresponds to the C2-domain of factor VIII.
Following amplification the fragment was digested with
NcoI and NotI and cloned into plasmid PAcGP67B. The
nucleotide sequence of the cloned sequence was verified
by oligonucleotide sequencing. Recombinant baculo-
viruses expressing the factor VIII light chain and the
C2-domain of factor VIII were obtained by transfection
in Sf-9 cells in conjunction with BaculogoldTM
Baculovirus Autographa californica DNA (Pharmingen, San
Diego, CA, USA). Recombinant viruses expressing the
factor VIII light chain and the C2-domain of factor
VIII were used to infect High FiveTM cells at a multi-
plicity of infection of 7. The cells were maintained in
culture medium which consisted of 25% (v/v) Grace's
insect medium and 75 % (v/v) of EX-CELL 401 medium
supplemented with 2.5 % fetal calf serum, 100 U/ml
penicillin and 100 mg/ml streptomycin. At 24 hours
post-infection the cells were pulse-labeled with
[35S]methionine (50 ~Ci/ml, spec. act. > 800 Ci/mmol)
for 24 hours in a similar culture medium lacking
methionine. Medium of metabolically labeled cells was
collected in an equal volume of 2 times concentrated
immunoprecipitation buffer (IPBB). IPBB consists of 50
mM Tris-HCl (pH 7.6); 1 M NaCl; 1.2% (v/v) Triton-X-
100; 0.1~ (w/v) Tween-20; 1.0 % (v/v) BSA; 35 mM EDTA;
10 ~g/ml soybean trypsin inhibitor; 10 mM benzamidine
- 30 -
CA 0220~824 1997-0~-22
and 5 mM N-ethylmaleimide. Immunoprecipitation with
monoclonal antibody CLB-CAg 117 was performed as
follows. Conditioned media were precleared by
incubation for two hours at room temperature with
Gelatin Sepharose 4B and two successive incubations
with Protein G Sepharose 4FF. Specific adsorption was
performed overnight at 4~C by adding 1 ~g/ml of
monoclonal antibody CLB-CAg 117 to Protein G Sepharose.
Immunoprecipitates were washed extensively with IPBB
and finally with 20 mM Tris-HCl (pH 7.6). Bound protein
was eluted by boiling for 5 minutes in SDS PAGE-sample
buffer and analyzed under reducing conditions on a 10 %
(w/v) SDS-polyacrylamide gel. Following electrophoresis
gels were fixed in 30% methanol, 10% acetic acid and
treated with 10% diphenyloxazol in acetic acid for 30
minutes. Finally, gels were incubated in H20, dried and
exposed for variable times. Immunoprecipitation with
monoclonal antibody CLB-CAg 117 revealed that this
particular antibody reacted both with the radiolabeled
factor VIII light chain and the C2-domain. The epitope
of CLB-CAg 117 is located in the C2-domain of factor
VIII. As such CLB-CAg 117 resembles anti-factor VIII
antibodies that develop in haemophilia A patients
following treatment with factor VIII. The epitope of a
significant portion of these anti-factor VIII
antibodies is located in the C2-domain. Clearly, CLB-
CAg 117 provides an useful model to mimic the action of
CA 0220~824 1997-0~-22
the anti-factor VIII antibodies that develop upon
treatment with factor VIII in patients with haemophilia
A. The capacity of CLB-CAg 117 to inhibit factor VIII
activity was monitored. Different amounts of CLB-CAg
117 were incubated with normal plasma for 2 hours at
37~C. Subsequently the factor VIII activity of normal
plasma was determined using a one-stage clotting assay
(Mertens et al., Brit. J. Haematol. 85. (1993), 133-
145). The results of the experiments are depicted in
Figure 8 and show that monoclonal antibody CLB-CAg 117
is a strong inhibitor of factor VIII activity. As
little as 10 ~g/ml of CLB-CAg 117 is capable of
reducing factor VIII activity to 10 % of its initial
value. In summary, these results reveal that CLB-CAg
117 has its epitope on the C2-domain of the factor VIII
light chain and acts as a strong inhibitor of factor
VIII activity. The above experiments show that the
properties of CLB-CAg 117 closely resemble that of
human allo or auto-antibodies directed against the C2-
domain of factor VIII, that develop either
spontaneously or as a consequence of factor VIII
replacement therapy of patients with haemophilia A.
EXAMPLE 7: Inhibition of factor VIII dB695-R2307Q by
CLB-CAg 117
In the previous examples monoclonal antibody CLB-CAg
- 32 -
CA 0220~824 1997-0~-22
117 an inhibitory antibody directed against the C2-
domain of factor VIII have been characterized. In
example 5 the expression of factor VIII dB695-R2307Q in
SKHEP cells has been described. Surprisingly, secretion
of factor VIII dB695-R2307Q was found to be temperature
dependent and significant amounts of factor VIII dB695-
R2307Q were secreted into the conditioned medium at
28~C. Secreted factor VIII dB695-R2307Q was found to be
functionally active. Inspection of the reactivity of
CLB-CAg 117 employing both a factor VIII antigen assay
and immunofluorescence revealed that factor VIII dB695-
R2307Q did not react with monoclonal antibody CLB-CAg
117. This observation suggests that factor VIII dB695-
R2307Q was also insensitive towards the factor VIII
inhibiting activity of CLB-CAg 117. To test this
hypothesis the ability of monoclonal antibody CLB-CAg
117 to inhibit factor VIII dB695-R2307Q was determined.
As a control inhibition of factor VIII dB695 by
monoclonal antibody CLB-CAg 117 was tested. Different
amounts of CLB-CAg 117 were incubated for 2 hours at
room temperature with factor VIII dB695 or factor VIII
dB695-R2307Q. Subsequently, the residual factor VIII
activity was determined using a one-stage clotting
assay (Mertens et al. Brit. J. Haematol. 85 (1993),
133-145). Inspection of the pattern obtained for the
inhibition of factor VIII dB695 by monoclonal antibody
CLB-CAg 117 revealed that as little as 0.2 ~g/ml of
- 33 -
.
CA 0220~824 1997-0~-22
CLB-CAg 117 resulted in a residual activity of 20~.
Higher amounts of CLB-CAg 117 did not yield an extra
inhibition of factor VIII activity. The pattern
obtained following incubation of CLB-CAg 117 with
factor VIII dB695-R2307Q was entirely different. The
activity of factor VIII dB695-R2307Q could not be
inhibited by CLB-CAg 117. Even high amount of antibody
(20 ~g/ml) were not able to block the factor VIII
activity of factor VIII dB695-R2307Q (Figure 9). These
results show that point mutations at immuno-dominant
regions of factor VIII may be used to selectively
interfere with the binding of inhibiting anti- bodies
while retaining factor VIII activity. Similar to
substitution of Arg2307 by Gln, other amino acid
substitutions may be utilized to interfere with binding
of factor VIII inhibiting antibodies.
EXAMPLE 8: Determination of vWF bin~in~ activity of
factor VIII dB695-R2307Q
In the previous example it was shown that CLB-CAg 117
is not able to inhibit the biological activity of
factor VIII dB695-R2307Q. The vWF-binding properties of
factor VIII dB695-R2307Q were determined using a
binding assay that has been described previously (Leyte
et al. Biochem. J. (1990) vol. 257, 679-683; Donath et
- 34 -
CA 0220~824 1997-0~-22
al. Biochem. J. (1995) vol. 312, 49-55)). Purified von
Willebrand factor (5 ~g/ml) was immobilized onto
microtiter wells overnight at 4~C. Subsequently serum
free medium containing different amount of factor VIII
dB695-R2307Q and factor VIII dB695 were incubated for
one hour with the immobilized von Willebrand factor.
The amount of factor VIII that binds to the immobilized
von Willebrand factor was quantified. The results are
displayed in Figure 10 and from these data it appears
that factor VIII dB695 is capable of binding to immo-
bilized von Willebrand factor in accordance with
previous data (Mertens et al. Brit. J. Haematol., 85
(1993), 133-142). Surprisingly, also factor VIII dB695-
R2307Q was bound in a quantitative manner to the
immobilized von Willebrand factor. These results
indicate that similar to factor VIII dB695, factor VIII
dB695-R2307Q is capable of binding with high affinity
to von Willebrand factor. The present data show that an
Arg2307~Gln substitution in factor VIII does not effect
the von Willebrand factor binding properties of the
molecule. Taken together, these results show that
factor VIII dB695-R2307Q possess normal factor VIII
activity and is capable of binding to von Willebrand
factor. In addition it was shown that factor VIII
dB695-R2307Q is resistant to the inhibitory effect of
an anti-factor VIII antibody. This may render factor
VIII dB695-R2307Q useful in the treatment of patients
- 35 -
CA 0220~824 1997-0~-22
anti-factor VIII antibodies which are capable of
inhibiting the coagulant activity of factor VIII.
Consequently, factor VIII dB695-R2307Q may be used as
part of a pharmaceutical preparation that is used in
treatment of patients with acquired haemophilia A or in
treatment of haemophilia A patients that have developed
allo-antibodies following factor VIII replacement
therapy.
EXAMPLE 9: Preparation of an A2-domain mutant
In the previous examples it was have established that
factor VIII dB695-R2307Q cannot be inhibited by CLB-CAg
117, an anti-factor VIII antibody that is directed
against the C2-domain of factor VIII. Anti-factor VIII
antibodies may also be directed against other regions
of the factor VIII protein. An immunodominant epitope
of factor VIII is present on the A2-domain of factor
VIII. A panel of patients was screened for the
occurrence of anti-factor VIII antibodies that are
directed against the A2-domain of factor VIII. To
facilitate screening of patients, recombinant
baculoviruses expressing the heavy chain and A2-domain
of factor VIII were constructed. The plasmid PCLB-GP67-
A2 encoding the A2 domain of factor VIII was
constructed by amplifying a 1135 bp fragment employing
oligonucleotide primers A2-1 (5'
- 36 -
CA 0220~824 1997-0~-22
ATTCCATGGGATCAGTTGCCAAGAAGCAT 3'; nucleotide position
1174-1191 of factor VIII, sense) and A2-2 (5'
CTTGCGGCCGCGGAGAATCATCTTGGTTCAATGGC 3'; nucleotide
position 2263-2277 of factor VIII, antisense). The 1135
bp fragment was purified, digested with NcoI and NotI
and cloned into plasmid pAcGP67B. In the resulting
construct designated pCLB-GP67-A2, amino acid sequence
Ser373-Arg740 of factor VIII is fused to the leader
peptide of the acidic glycoprotein GP67. The plasmid
pCLB-GP67-9OK encoding amino acid Alal-Arg740 of the
heavy chain of factor VIII was constructed by
amplifying a 548 bp fragment using oligonucleotide
primers 90K-1 (5' TCTCCATGGGTGCCACCAGAAGATACTAC 3';
nucleotide position 58-75 of factor VIII sense) and
90K-2 (5' ACATACTAGTAGGGCTCC 3'; nucleotide position
577-594 of factor VIII; antisense). The 548 bp PCR
product was purified and digested with NcoI and NdeI.
Plasmid pCLB-BPVdB695 was digested with NdeI
(nucleotide position 461 of factor VIII) and KpnI
(nucleotide position 1811 of factor VIII) resulting in
a 1351 bp fragment. Plasmid pCLB-GP67-A2 was digested
with KpnI and NotI and the resulting frgament was
cloned into pAcGP67B together with the NcoI-NdeI
fragment and the KpnI-NotI fragments described above.
The resulting construct was termed pCLB-GP67-9OK.
Recombinant viruses expressing the factor VIII heavy
chain and the A2 domain were prepared as described in
- 37 -
CA 0220~824 1997-0~-22
Example 5. Immunoprecipitation experiments employing 20
~l of patients plasma were performed as described in
Example 5. Metabolically labeled A2-domain and factor
VIII heavy chain were used in the studies described
below. Screening of plasma of a panel of patients with
haemophilia A revealed that plasma derived of a patient
with mild haemophilia A contained anti-factor VIII
antibodies that were reactive with both metabolically
labeled A2-domain and factor VIII heavy chain.
Development of anti-factor VIII antibodies in patients
affected with mild haemophilia is relatively rare,
since significant amount of endogeneous factor VIII are
often present in plasma of these patients. This
suggests that the anti-factor VIII antibodies that
develop under these condities are directed against an
epitope that is present on exogeneous factor VIII and
that is not localized on endogeneous factor VIII. To
assess this possibility the mutation in the factor VIII
gene of the patient with the anti-factor VIII antibody
was determined by amplification of the 26 exons of
factor VIII employing the polymerase chain reaction.
The presence of point mutations in the factor VI I I gene
was investigated using single stranded conformation
polymorphism (SSCP) in combination with direct
sequencing of the amplified PCR-fragments. Following
SSCP an aberrant migration of a PCR fragment
corresponding to exon 12 of the factor VIII gene was
CA 0220~824 1997-0~-22
observed. Direct sequencing revealed a point mutation
(C~T) which predicted replacement of Arg593 by a Cys.
It is likely that the missense mutation could cause a
conformational change in the endogeneously synthesized
factor VIII protein. To address this issue the
Arg593~Cys mutation was introduced into the construct
that encodes the factor VIII A2-domain. Plasmid pCLB-
GP67B-A2-R593C encoding the A2 domain of factor VIII in
which Arg593 is replaced by a Cys was constructed by
site-directed mutagenesis using overlap extension.
Oligonucleotide primer R593C (5'
GAGAATATACAATGCTTTCTCCC 3'; nucleotide position 1822-
1844 of factor VIIIi sense) was used together with
oligonucleo-tide primer A2-2 to amplify a fragment of
476 bp. Oligonucleotide primer R593C-2 (5'
GGGAGAAAGCATTGTATATTCTC 3'; nucleotide position 1822-
1844 of factor VIII; antisense) and oligonucleotide
primer A2-1 were used to amplify a 682 bp fragment.
Both amplified fragments were purified and used as a
template in a PCR employing oligonucleotide primer A2-1
and A2-2 in order to amplify a DNA fragment which was
purified and subsequently digested with NcoI and NotI
and cloned into the plasmid pAcGP67B yielding pCLB-
GP67-A2R593C. The nucleotide sequence of the modified
A2-domain was verified by oligonucleotide sequencing.
Conditioned medium containing metabolically labeled A2-
domain with the Arg593~Cys mutation was prepared as
- 39 -
CA 0220~824 1997-0~-22
described in examples 2 and 3. Immunoprecipitation of
the metabolically labeled modified A2 domain, termed
A2R593C, with monoclonal antibody CLB-CAg 9 showed that
A2R593C was present in the conditioned medium in
amounts similar to the wild type A2 domain of factor
VIII. However, the antibody present in the plasma of
the patient did not react with the modified A2 domain
(A2R593C). This observation suggests that the anti-
factor VIII antibody that developed in the patient
during treatment with factor VIII is solely directed to
wild type factor VIII. The anti-factor VIII antibody
does not react with the recombinant A2 domain that
contains the Arg593~Cys mutation. The data presented
above show that binding of anti-factor VIII antibodies
can be modulated by selective modification of immuno-
dominant sites on the factor VIII protein. Similar to
the example given above selective modification of other
immuno-dominant regions of factor VIII may be utilized
to interfere with the binding of anti-factor VIII
antibodies.
EXAMPLE 10: Eormulating the purified mutant protein
preparation to a pharmaceutical preparation
The mutant proteins produced according to Example 2 and
9 have been purified by conventional means among them
- 40 -
CA 0220~824 1997-0~-22
chromatographic procedures and/or precipitation
techniques. The purified protein preparation was then
formulated to a physiologically acceptable preparation
using standard techniques like sterile filtration and
ultra/diafiltration. The preferred buffer used in the
pharmaceutical preparation is a physiological NaCl
buffer or a tris-buffer, having a pH in the range of 6-
8, preferably between 7 and 7.5. The concentration of
the protein in the pharmaceutical preparation is chosen
according to the dose required to treat a patient.
The dose of the factor VIII preparations according to
the invention is dependent on the nature, extend and
duration of the bleeding lesion as well as on the
severity of the hemophilia and the inhibitor titer,
respectively. The initial dose usually lies between 1
and 500 U/kg, preferably between 10 and 200 U/kg of
body weight.
- 41 -