Note: Descriptions are shown in the official language in which they were submitted.
CA 02207252 1997-06-06
11713961214156
PCT/US96/01559
INHIBITORS OF PRENYL TRANSFERASES
BACKGROUND OF THE INVENTION
1. Field of the Invention
=
The present invention relates to novel
peptidomimetics and other compounds which are
useful as inhibitors of protein isoprenyl
transferases (particularly protein
farnesyltransferase and geranylgeranyltransferase)
and as anti-cancer drugs, to compositions
containing such compounds and to methods of use.
2. Background Information
Ras proteins are plasma membrane- associated
GTPases that function as relay switches that
transduce biological information from
extracellular signals to the nucleus (29-31). In
normal cells Ras proteins cycle between the GDP-
(inactive) and GTP-(active) bound forms to
regulate proliferation and differentiation. The
mechanism by which extracellular signals, such as
epidermal and platelet derived growth factor (EGF
and PDGF), transduce their biological information
to the nucleus via Ras proteins has recently been
unraveled (29-31). Binding of the growth factors
to tyrosine kinase receptors results in
autophosphorylation of various tyrosines which
then bind src-homology 2 (SH2) domains of several
signaling proteins. One of these, a cytosolic
complex of GRB-2 and a ras exchanger (m-SOS-1), is
recruited by the tyrosine phosphorylated receptor
where mS0S-1 catalyzes the exchange of GDP for GTP
on Ras, hence activating it. GTP-bound Ras
recruits Raf, a serine/threonine kinase, to the
= plasma membrane where it is activated. Raf
triggers a kinase cascade by phosphorylating
mitogen-activated protein (MAP)
- 1 -
CA 02207252 1997-06-06
WO 96/21456
PCT/US96/01559
kinase/extracellular-regulated protein kinase
(ERR) kinase (MEK) which in turn phosphorylates
MAP Kinase on threonine and tyrosine residues.
Activated MAP Kinase translocates to the nucleus
where it phosphorylates transcription factors
(31). Termination of this growth signal is
accomplished by hydrolysis of Ras-GTP to Ras-GDP.
Ras oncogenes are the most frequently
identified activated oncogenes in human tumors (1-
3). In a large number of human cancers, Ras is
GTP-locked because of mutations in amino acids 12,
13, or 61 and the above Ras pathway no longer
requires an upstream growth signal and is
uninterrupted. As a consequence, enzymes in this
pathway such as Raf, MEK and MAP Kinase are
constitutively activated.
In addition to its inability to hydrolyze
GTP, oncogenic Ras must be plasma membrane-bound
to cause malignant transformation (13). Ras is
posttranslationally modified by a lipid group,
farnesyl, which mediates its association with the
plasma membrane (10-14).
Post-translational events leading to membrane
association of p21ras have previously been
disclosed (10-14). The p21ras proteins are first
made as pro-p21ras in the cytosolyhere they are
modified on cysteine 186 of their carboxyl
terminal sequence CAAX (C = cysteine, ALI and AL2 =
isoleucine, leucine or valine and X = methionine
or serine) by the cholesterol biosynthesis
intermediate farnesyl pyrophosphate (FPP). This
farnesylation reaction is then followed by
peptidase removal of the AAX tripeptide and
carboxymethylation of the remaining cysteine. The
processed p2lras proteins associate with the inner
- 2 -
CA 02207252 1997-06-06
W096/21456
PCT/US96/01559
surface of the plasma membrane (10-14).
p21Ras farnesyltransferase, the enzyme
responsible for catalyzing the transfer of
farnesyl, a 15-carbon isoprenoid, from FPP to the
=
cysteine of the CA.1.A2X carboxyl terminus of p2lras,
has been purified to homogeneity from rat brain
=
(15,16). The enzyme is a heterodimer composed of
a and g subunits of molecular weights 49 and 46
kDa, respectively (17). The g subunit has been
shown to bind p2lras (17). Because p2lras
farnesylation and subsequent membrane association
are required for p2lras transforming activity
(13), it has been proposed that p2lras
farnesyltransferase would be a useful anticancer
therapy target. Accordingly, an intensive search
for inhibitors of the enzyme is underway (18-24,
33-44). Potential inhibitor candidates are CA1A2X
tetrapeptides which have been shown to be
farnesylated by p2lras farnesyltransferase and
appear to be potent inhibitors of this enzyme in
vitro (15,18,21-24). Competition studies have
demonstrated that CAAX peptides with the greatest
inhibitory activity are those where Al and A2 are
hydrophobic peptides with charged or hydrophilic
residues in the central positions demonstrating
very little inhibitory activity (18,21,23). A
major drawback with the use of peptides as
; therapeutic agents is their low cellular uptake
and their rapid inactivation by proteases.
The research efforts directed towards
farnesyltransferase and the inhibition of its
activity are further illustrated by the following
patents or published patent applications:
- 3 -
'
CA 02207252 1997-06-06
W096/21456 PCT/US96/01559
U.S. 5,141,851
WO 91/16340
WO 92/18465
EPA 0456180 Al
4
EPA 0461869 A2
EPA 0512865 A2
EPA 0520823 A2
EPA 0523873 Al
Of the above disclosures, EPA 0520823 A2 discloses
compounds which are useful in the inhibition of
farnesyl-protein transferase and the farnesylation
of the oncogene protein ras. The compounds of EPA
0520823 A2 are illustrated by the formula:
Cys-Xaal-dXaa2-Xaa3
or pharmaceutically acceptable salts thereof,
wherein Cys is a cysteine amino acid;
Xaal is an amino acid in natural L-isomer form;
dXaa2 is an amino acid in unnatural D-isomer form;
and
Kaa3 is an amino acid in natural L-isomer form.
The preferred compounds are said to be
CV(D1)S and CV(Df)M, the amino acids being
identified by conventional 3 letter and single
letter abbreviations as follows:
Cysteine Cys
Glycine Gly
ISoleucine Ile
Leucine Leu
Lysine Lys
Methionine Met
Phenylalanine Phe
Serine Ser
Threonine Thr
Valine Val V
- 4 -
CA 02207252 1997-06-06
WO 96/21456
PCT/US96101559
EPA 0523873 Al discloses a modification of
the compounds of EPA 0520823 A2 wherein Xaa3 is
phenylalanine or p-fluorophenylalanine.
EPA 0461869 describes compounds which inhibit
A
farnesylation of Ras protein of the formula:
Cys-Aaal-Aaa2-Xaa
where Aaal and Aaa2 are aliphatic amino acids and
Xaa is an amino acid. The aliphatic amino acids
which are disclosed are Ala, Val, Leu and Ile.
Preferred compounds are those where Aaal is Val,
Aaa2 is Leu, Ile or Val and Xaa is Ser or Met.
Preferred specific compounds are:
Cys-Val-Leu-Ser
Cys-Val-Ile-Met
Cys-Val-Val-Met
U.S. patent 5,141,851 and WO 91/16340
disclose the purified farnesyl protein transferase
and certain peptide inhibitors therefor,
including, for example, CVIM, TKCVIM and
KKSKTKCVIM.
WO 92/18465 discloses. certain farnesyl
compounds which inhibit the enzymatic methylation
of proteins including ras proteins.
EPA 0456180 Al is directed to a
farnesylprotein transferase assay which can be
used to identify substances that block
farnesylation of ras oncogene gene products while
EPA 0512865 A2 discloses certain cyclic compounds
that are useful for lowering cholesterol and
inhibiting farnesylprotein transferase.
As will be evident from the foregoing, there
is a great deal of research effort directed
towards the development of inhibitors of
= farnesyltransferase. However, there still remains
- 5 -
CA 02207252 1997-06-06
WO 96/21456
PCT/US96/01559
a need for improvements in this critically
important area.
An enzyme closely related to
farnesyltransferase, geranylgeranyltransferase I
(GGTase I), attaches the lipid geranylgeranyl to
the cysteine of the CAAX box of proteins where X
is leucine (49,69). FTase and GGTase I are cy/g
heterodimers that share the a subunit (61,62).
Cross-linking experiments suggested that both
substrates (FPP and Ras CAAX) interact with the g
submit of FTase (17,63). Although GGTase I
prefers leucine at the X position, its substrate
specificity was shown to overlap with that of
FTase in vitro (64). Furthermore, GGTase I is
also able to transfer farnesyl to a leucine
terminating peptide (65).
Although CAAX peptides are potent competitive
inhibitors of FTase, rapid degradation and low
cellular uptake limit their use as therapeutic
agents. The stragegy of the present invention to
develop superior compounds for inhibiting FTase
and GGTase has been to replace several amino acids
in the CAAX motif by peptidemimics. The rationale
behind this strategy is based on the existance of
a hydrophobic pocket at the enzyme active site
that interacts with the hydrophobic "AA" dipeptide
of the carboxyl termini CAAX of Ras molecules. In
this regard, two very potent inhibitors of FTase
(i.e. Cys-3AMBA-Met and Cys-4ABA-Met) were
disclosed by us in an earlier U.S. patent
application. The peptidomimetic Cys-4ABA-Met
incorporates a hydrophobic/aromatic spacer (i.e.
4-aminobenzoic acid) between Cys and Met. The
present application discloses several derivatives
of Cys-4ABA-Met where positions 2 and 3 of 4-amino
I
- 6 -
CA 02207252 1997-06-06
170096/21456
PCT/US96101559
benzoic acid were modified by several alkyl,
and/or aromatic groups, compounds that show great
promise of ability to selectively antagonize RAS-
dependent signaling and to selectively inhibit the
growth of human tumors with aberrant Ras function.
Of the four types of Ras proteins (H-, N-,
K4A-, and K49-Ras) PWprPRPPri hy mammalian cells,
K4B-Ras (also called K-Ras4B) is the most
frequently mutated form of Ras in human cancers
(1,3). Although several laboratories have
demonstrated potent inhibition of oncogenic H-Ras
processing and signaling (43,44), this disruption
has not been shown with K-Ras4B. Previous
studies have targeted H-Ras and not K-Ras4B as a
target for the development of inhibitors. One
recent report indicates that K-Ras4B can be
geranylgeranylated in vitro, but with relatively
low efficiency; its Km for GGTase I is 7 times
higher than its Km for FTase (67). GGTase I CAAX-
based inhibitors that can block
geranylgeranylation processing have not been
reported.
Recently, we have shown that a potent
inhibitor of FTase disrupts K-Ras4B processing but
only at very high concentrations that also
inhibited the processing of geranylgeranylated
proteins (66). This suggested that K-Ras4B may be
geranylgeranylated, and that therefore inhibitors
targeted at GGTase I would be effective in
disrupting oncogenic K-Ras4B processing and
signalling, and in treatment of cancers which were
related to this form of Ras.
- 7 -
CA 02207252 1997-06-06
WO 96/21456 PCT/US96/01559
Summary of the Invention
In accordance with the present invention
there are compounds. of the formula (A-L) :
/
I Ri
A1--, Ai 1 0
H¨ /...----1 II I
R3 \...% R3 R2 \. j
Rla (A) R2 (B) N (C)
/ i /
R1 R1 RI
-,---"--V
N i NI
I I
r=-lk..,.?
/ = / 1 0 1
Ric 1-14-- R2 /1-1--TE 1¨R2 I/ "1
-- R3 1 0 t3
(D), (E) N (F)
=
. /
Ri /
/ Ri
I Ri
,R2
D 1 1 ii .1 / Li---)S: 7\R2
R3 C )
I.--1- [I im. "2 R( C N
, ,3 ---,,,,-
. (G) 0 (H) H (I)
/ / /
R1 Ri R1
R2
iv Li--.Z =),-/- /7L1-....r r--A,V.R2
/71_1-....i.R
C) N 2
R3 R3 R3 N NH
, S'N
(J) (K) Or (L)
r
a
- 8 -
CA 02207252 1997-06-06
WO 96/21456
PCT/US96/01559
wherein R1' is
i) hydrogen;
ii) lower alkyl;
iii) alkenyl;
iv) alkoxy;
v) thioalkoxv;
vi) halo;
vii) haloalkyl;
viii) aryl-L2-, wherein L2 is absent, -CH2-, -
_
CH2CH2-, -CH(CH3)-, -0-, -S(0)g wherein q is 0, 1,
or 2, -NCR')- wherein R' is hydrogen or lower
alkyl, or -C(0)- and aryl is selected from the
group consisting of phenyl, naphthyl,
tetrahydronaphthyl, indanyl and indenyl and the
aryl group is unsubstituted or substituted; or
ix) heterocyclic-L3- wherein L3 is absent, -CH2-,
-CH2CH2-, -CH(CH3)-, -0-, -S(0),1 wherein q is 0, 1
or 2, -N(R')- wherein R' is hydrogen or
loweralkyl, or -C(0)- and heterocyclic is a
monocyclic heterocyclic wherein the heterocyclic
is unsubstituted or substituted with one, two, or
three substituents independently selected from the
group consisting of loweralkyl, hydroxy,
hydroxyalkyl, halo, nitro, oxo (.0), amino, N-
protected amino, alkoxy, thioalkoxy and haloalkyl;
Ria is hydrogen or lower alkyl;
R2' is
i) R12a
-j--R
12b
wherein R12a is hydrogen, loweralkyl or -C(0)0-R13,
wherein Ru is hydrogen or a carboxy-protecting
a
- 9 -
CA 02207252 1997-06-06
WO 96/21456
PCT/US96/01559
group and R3.2b is hydrogen or loweralkyl, with the
proviso that R3.2, and 123.2b are not both hydrogen,
ii) -C(0) NH-CH (R3.4) -C (0) OR3.5 wherein R14 is
a) loweralkyl,
b) cycloalkyl,
c) cycloalkylalkyl,
d) alkoxyalkyl,
e) thioalkoxyalkyl,
f) hydroxyalkyl,
g) aminoalkyl,
h) carboxyalkyl,
i) alkoxycarbonylalkyl,
j) arylalkyl or
k) alkylsulfonylalkyl and
R15 is hydrogen or a carboxy-protecting group or
0
iii) - C(0) - HN¨NO
(CH2)1.3
R3 11s
A
R0-CH2-CH-C-
NH2
where
A represents 0 or 2H, and
Ro represents SH, NH2, or CHy-S02-NH-, wherein
CHy is a straight chain saturated or unsaturated
hydrocarbon, with x being between 1 and 20 and y
between 3 and 41, inclusive; and
- 10 -
CA 02207252 1997-06-06
WO 96/21456
PCT/US96101559
L1 is -NH-;
or pharmaceutically acceptable salts or prodrugs
thereof.
An important embodiment of the present
invention is based on the finding that a novel
group of peptidomimetics as represented by Formula
(I) have a high inhibitory potency against human
tumor p21ras farnesyltransferase and inhibit tumor
growth of human carcinomas:
cpx (I)
where
C stands for the cysteine radical, or for the
reduced form of the cysteine radical (R-2-amino-3-
mercaptopropyl amine); p is the radical of a non-
peptide aminoalkyl- or amino-substituted phenyl
carboxylic acid; and X is the radical of an amino
acid, preferably Met. Any other natural or
synthetic amino acid can also be used at this
position. The invention also includes
pharmaceutically acceptable salts and prodrugs of
Formula (I).
A particularly preferred compound in this
regard is:
HS
-1-H3N-PM
0 0
0-
- 0
SCH3
- 11 -
CA 02207252 1997-06-06
W096/21406
PCT/US96/01559
In this compound the cysteine radical is in the
reduced form and the spacer group is 2-pheny1-4-
aminobenzoic acid.
Another preferred compound of the invention
is:
HS) 0
Y
NH 0
4H3N
0 NH
J0
0-
0
SCH3
The compounds of Formula (I) are different
from the prior art farnesyltransferase inhibitors
in that they do not include separate peptide amino
acids Al, A2 as in prior art inhibitors represented
by the formula CAIA2X. The present compounds are
consequently free from peptidic amide bonds.
It is also to be noted that the present
compounds are not farnesylated by the enzyme.
They are, therefore, true inhibitors, not just
alternative substrates. This may explain the high
inhibitory action of the present compounds
relative to their parent compounds which are
farnesylated.
A further important feature of the invention
is the provision of the compounds of Formula (I)
in the form of pro-drugs. Broadly speaking, this
is accomplished by functionalizing the terminal
end groups (amino, cysteine sulfur and carboxy
groups) of the compounds with hydrophobic, enzyme-
sensitive moieties which serve to increase the
plasma membrane permeability and cellular uptake
of the compounds and consequently their efficiency
in inhibiting tumor cell growth. In addition,
- 12 -
CA 02207252 1997-06-06
WID96/21456
PCT/US96/01559
prodrugs for amino and cysteine sulfur groups can
include loweralkycarbonyl, arylcarbony,
arylalkylcarbony, alkoxycarbonyl, aryloxycarbonyl,
cycloalkylcarbonyk, cycloalkoxycarbonyl, and other
groups well known to those skilled in the art.
In this regard, a particularly preferred
compound of the invention is the methylester form
=
of FTI-276, which is illustrated in Figure 1A.
The above-mentioned pro-drug aspect of the
invention is applicable not only to the compounds
of the invention but also to prior peptide
inhibitors CAAX as well as any other peptide with
potential for biological uses for the purpose of
improving the overall effectiveness of such
compounds, as hereinafter described.
A further modification involves the provision
of CA1A2X tetrapeptides or CflX peptidomimetics
which have been modified by functionalizing the
sulfhydryl group of the cysteine C with an alkyl
phosphonate substituent, as hereinafter described.
Another important embodiment of the invention-
contemplates replacing the A1A2X portion of the
CAAX tetrapeptide inhibitors with a non-amino
acid component while retaining the desired
farnesyltransferase inhibiting activity. These
compounds may be illustrated by Formula (II):
CA (II)
where C is cysteine or reduced cysteine and A
represents an aryl or heterocyclic substituent
such as 3-aminomethyl-biphenyl-3'-carboxylic acid,
which does not include a peptide amino acid but
corresponds essentially in size with AAX, as
hereinafter described. The invention also
includes pharmaceutically acceptable salts and
prodrugs of Formula (II).
- 13 -
CA 02207252 1997-06-06
W096/21456
PCT/US96/01559
The invention also includes compounds in
which further substitutions have been made at the
cysteine position. These compounds comprise free
cysteine thiol and/or terminal amino groups at one
end and include a carboxylic acid or carboxylate
group at the other end, the carboxylic acid or
carboxylate group being separated from the
cysteine thiol and/or terminal amino group by a
hydrophobic spacer moiety which is free from any
linking amido group as in prior CAAX mimetics. As
with other compounds of the invention, these
compounds are not subject to'proteolytic
degradation inside cells while retaining the
structural features required for FTase inhibition.
The compounds selectively inhibit FTase both in
vitro and in vivo and offer a number of other
advantages over prior CAAX peptide mimetics.
Compounds of this embodiment may be
illustrated by the formula:
C B (III)
where C is
A
11
R0-CH2-CH-C-
1
NH2
A represents 0 or 2H, and
Ro represents SH, NH2, or CHy-S02-NH-, wherein
Cxliy is a straight chain saturated or unsaturated
hydrocarbon, with x being between 1 and 20 and y
between 3 and 41, inclusive; and
B stands for -NHR, where R is an aryl group. The
invention also includes pharmaceutically
acceptable salts and prodrugs of Formula (III).
In one preferred embodiment of the invention, R is
a biphenyl substituted with one or more -COOH
- 14 -
CA 02207252 1997-06-06
WO 96/21456
PCT/US96/01559
groups and/or lower alkyl, e.g., methyl, as
represented by the formula:
H2N, AR4
410. R3
SH H
RI R2
where R/ and R3 represent H or COOH; R2 represents
H, COOH, CH3, or COOCH3; R4 represents H or OCH3;
and A represents 2H or 0. This formula represents
a series of 4-amino-3'-carboxybiphenyl derivatives
which mimic the Val-Ile-Met tripeptide but have
restricted conformational flexibility. Reduction
of the cysteine amide bond (where A is H,H)
provides a completely non-peptidic Ras CAAX
mimetic.
Preferably, R is a biphenyl group with a -
COOH substitution in the 3'- or 4'-position, most
preferably the 3'-position, with respect to the
NH-aryl group. The -COOH substituent may appear
as such or in pharmaceutically acceptable salt or
ester form, e.g., as the alkali metal salt or
methyl ester.
The features of the invention are illustrated
herein by reference to the CAAX tetrapeptide known
as CVIM (see EP 0461869 and U.S. Patent 5,141,851)
and C-4ABA-M. These compounds are, respectively,
Cys-Val-Ile-Met and Cys-4 aminobenzoic acid-Met
where Cys is the cysteine radical and Met is the
methionine radical.
A preferred non-peptide CAAX mimetic of the
invention is reduced cys-4-amino-3'-
.
biphenylcarboxylate identified as 4 in Figure 12,
- 15 -
CA 02207252 1997-06-06
W096/21456
PCT/US96/01559
which is also designated FTI-265. This derivative
contains no amide bonds and thus is a true non-
peptide mimic of the CAAX tetrapeptide.
The compounds of the invention may be used in
the carboxylic acid form or as pharmaceutically
acceptable salts or esters thereof. Lower alkyl
esters are preferred although other ester forms,
e.g., phenyl esters, may also be used.
It is also an object of the present invention
to provide a CAAX peptidomimetic that inhibits
GGTase I.
Accordingly, it is an object of the present
invention to provide a substance and means of
disrupting oncogenic K-Ras4B processing and
signaling that affects geranylgeranylation and/or
farnesylation processing.
It is a further object of the invention to
provide a pharmaceutical composition for treating
cancer which is responsive to geranylgeranyl
transferase inhibitors, such as, but not limited
to, pancreatic and colon cancer.
The latter objects are accomplished by
replacing the central "AA" of CAAX tetrapeptides
by a hydrophobic spacer and incorporating a
leucine or isoleucine residue in the C-terminal
position to optimize recognition by GGTase I.
Additionally, the cysteine moiety may be replaced
by reduced cysteine, or by other functional groups
as hereinafter disclosed.,
An important embodiment of the present
invention is based on the finding that a novel
group of peptidomimetics as represented by Formula
(IV) have a high inhibitory potency against
geranylgeranyl transferase and disrupt oncogenic
K-Ras4B processing and signalling:
(Iv)
- 16 -
CA 02207252 1997-06-06
W096/21456
PCT/US96/01559
where
C stands for the cysteine radical, or for the
reduced form of the cysteine radical (R-2-amino-3-
mercaptopropyl amine); p is the radical of a non-
. .
peptide aminoalkyl- or amino-substituted phenyl
carboxylic acid; and L is the radical of leucine
or isoleucine. The invention also includes
pharmaceutically acceptable salts and prodrugs of
the compounds of Formula (IV).
Preferred compounds of this embodiment are
derivatives of Cys-4ABA-Leu which are substituted
at the 2 and/or 3 positions of the phenyl ring of
4-aminobenzoic acid (4ABA). The substitutions at
these positions include, but are not limited to
alkyl, alkoxy and aryl (particularly to straight
chain or branched groups of 1-10 carbons of the
aforementioned) and naphthyl, heterocyclic rings
and heteroaromatic rings.
A particularly preferred compound of this
aspect of the invention, GGTI-287, is illustrated
in Figure 17. In this compound the cysteine
radical is in the reduced form and the spacer
group is 2-phenyl-4-aminobenzoic acid. Another
preferred compound, also shown in Figure 17, is
GGTI-297, which contains the spacer group 2-
naphthy1-4-aminobenzoic acid. Other spacer
groups which will be readily evident as useful are
described herein in connection with farnesyl-
transferase inhibitors. ,
A further important feature of the invention
is the provision of the compounds of the invention
in the form of pro-drugs. By "pro-drug" is meant
a compound to which in vivo modification occurs to
produce the active compound. Such compounds may,
for example, be more readily delivered to their
sites of action as pro-drugs. Broadly speaking,
the pro-drugs of the instant invention are
- 17 -
CA 02207252 1997-06-06
WO 96/21456
PCT/US96/01559
produced by functionalizing the terminal end
groups (amino, cysteine sulfur and carboxy groups)
of the compounds with hydrophobic, enzyme-
sensitive moieties which serve to increase the
plasma membrane permeability and cellular uptake
of the compounds and consequently their efficiency
in inhibiting tumor cell growth.
In this regard, a particularly preferred
compound of the invention is the methylester form
of GGTI-287, GGTI-286, also illustrated in Figure
17.
The compounds of the invention may be used in
the same manner as prior CAAX tetrapeptide
inhibitors to inhibit p2lras farnesyltransferase
or geranylgeranyl transferase in any host
containing these enzymes. This includes both in
vitro and in vivo use. Compounds which inhibit
farnesyltransferase, notably human tumor p2lras
farnesyltransferase, and consequently inhibit the
farnesylation of the oncogene protein Ras, may be
used in the treatment of cancer or cancer cells.
It is noted that many human cancers have activated
ras and, as typical of such cancers, there may be
mentioned colorectal carcinoma, myeloid leukemias,
exdcrine pancreatic carcinoma and the like.
Likewise, compounds which inhibit geranylgeranyl
transferase may be used in the treatment of cancer
which is related to K-Ras4B.
The compounds of the invention may be used in
pharmaceutical compositions of conventional form
suitable for oral, subcutaneous, intravenous,
intraperitoneal or intramuscular administration to
a mammal or host. This includes, for example,
tablets or capsules, sterile solutions or
suspensions comprising one or more compounds of
the invention with a pharmaceutically acceptable
- 18 -
CA 02207252 2009-11-19
W096/21456
PCT/US96/01559
carrier and with or without other additives.
Typical carriers for tablet or capsule use
include, for example, lactose or corn starch. For
oral compositions, aqueous suspensions may be used
with conventional suspending agents, flavoring
agents and the like.
The amount of inhibitor administered to
obtain the desired inhibitory effect will vary but
can be readily determined. It is expected that
the compounds of the present invention will be
administered to humans or other mammals as
pharmaceutical or chemotherapeutic agents in
dosages of .1 to 1000 mg/kg body weight,
preferably 1 to 500 mg/kg body weight and most
preferably 10-50 mg/kg body weight. The required
dose for a given individual or disease will vary,
but can be determined by ordinary skilled
practitioners using routine methods. The
compounds may be administered via methods well
known in the pharmaceutical and medical arts,
which include, but are not limited to oral,
parenteral, topical, and respiratory (inhalation)
routes. Pharmaceutical preparations may contain
suitable carriers or diluents. Means of
determining suitable carriers and diluents are
well known in the pharmaceutical arts.
The term "carboxy protecting group", as used
herein, refers to a carboxylic acid protecting
ester group employed to block or protect the
carboxylic acid functionally while the reactions
involving other functional sites of the compound
are carried out. Carboxy protecting groups are
disclosed in Greene, "Protective Groups in Organic
Synthesis", pp. 152-186 (1981).
In addition, a
carboxy protecting group can be used as a prodrug
whereby the carboxy protecting group can be
- 19 -
=
CA 02207252 2009-11-19
WO 96/21456PCT/US96/01559
=
readily cleaved in vivo, for example by enzymatic
hydrolysis, to release the biologically active
parent. A comprehensive discussion of the prodrug
concept is provided by T. Higuchi and V. Stella in
"Prodrugs as Novel Delivery Systems", vol. 14 of
the ACS Symposium Series, American Chemical
Society (1975).
Such carboxy protecting groups are
will known to those skilled in the art, having
been extensively used in the protection of
carboxyl groups in the penicillin and
cephalosporin fields, as described in U.S. Pat.
No. 3,840,556 and 3,719,667.
Examples of esters useful as prodrugs for
compounds containing carboxyl groups can be found
on pages 14-21 of "Bioreversible Carriers in Drug
Design: Theory and Application", edited by E.B.
Roche, Permagon Press, New York, (1987).
Representative
carboxy protecting groups are Cl to C8 loweralkyl
(e.g. methyl, ethyl or tertiary butyl and the
like); arylalkyl, for example, phenethyl or benzyl
and substituted drivatives thereof, for example 5-
indanyl and the like; dialkylaminoalkyl (e.g.
dimethylaminoethyl and the like); alkanoyloxyalkyl
groups such as acetoxymethol, butyryloxymethyl,
valeryloxymethyl, isobutyryloxymethyl,
isovaleryloxymethyl, 1-(propionyloxy)-1-ethyl, 1-
(pivaloyloxyl)-1-ethyl, 1-methyl-1-(propionyloxy)-
1-ethyl, pivaloyloxymethyl, propionyloxymethyl and
the like; cycloalkanoyloxyalkyl groups such as
cyclopropylcarbonyloxymethyl,
cyclobutylcarbonyloxymethyl,
cyclopentylcarbonyloxymethyl,
cyclohexylcarbonyloxymethyl and the like;
aroyloxyalkyl, such as benzoyloxymethyl,
- 20 -
CA 02207252 1997-06-06
114396/21456
Per1US96101559
benzoyloxyethyl and the like;
arylalkylcarbonyloxyalkyl, such as
benzylcarbonyloxymethyl, 2-benzylcarbonyloxyethyl
and the like; alkoxycarbonylalkyl or
cycloalkyloxycarbonylalkyl, such as
methoxycarbonylmethyl,
cyclohexyloxycarbonylmethyl, 1-methoxycarbony1-1-
ethyl, and the like; alkoxycarbonyloxyalkyl or
cycloalkyloxycarbonylalkyl, such as
methoxycarbonyloxymethyl, t-
butyloxycarbonyloxymethyl, 1-ethoxycarbonyloxy-1-
ethyl, 1-cyclohexyloxycarbonYloxy-l-ethyl and the
like; aryloxycarbonyloxyalkyl, such as 2-
(phenoxycarbonyloxy)ethyl, 2-(5-
indanyloxycarbonyloxy)ethyl and the like;
alkoxyalkylcarbonyloxyalkyl, such as 2-(1-methoxy-
2-methylpropan-2-oyloxy)ethyl and the like;
arylalkyloxycarbonyloxyalkyl, such as 2-
(benzyloxycarbonyloxy)ethyl and the like;
arylalkenyloxycarbonyloxyalicyl, such as 2-(3-
phenylpropen-2-yloxycarbonyloxy)ethyl and the
like; alkoxycarbonylaminoalkyl, such as t-
butyloxycarbonylaminomethyl and the like;
alkylaminocarbonylaminoalkyl, such as
methylaminocarbonylaminomethyl and the like;
alkanoylaminoalkyl, such as acetylaminomethyl and
the like; heterocycliccarbonyloxyalkyl, such as 4-
methylpiperazinylcarbonyloxymethyl and the like;
dialkylaminocarbonylalkyl, such as
dimethylaminocarbonylmethyl,
diethylaminocarbonylmethyl and the like; (5-
(loweralkyl)-2-oxo-1,3-dioxolen-4-yl)alkyl, such
as (5-t-butyl-2-oxo-1,3-dioxolen-4-yl)methyl and
the like; and (5-pheny1-2-oxo-1,3-dioxolen-4-
yl)alkyl, such as (5-pheny1-2-oxo-1,3-dioxolen-4-
yl)methyl and the like.
- 21 -
CA. 02207252 2009-11-19
W096/21456
PCT/US96/01559
Preferred carboxy-protected compounds of the
invention are compounds wherein the protected
carboxy group is a loweralkyl, cycloalkyl or
arylalkyl ester, for example, methyl ester, ethyl
ester, propyl ester, isopropyl ester, butyl ester,
sec-butyl ester, isobutyl ester, amyl ester,
isoamyl ester, octyl ester, cyclohexyl ester,
phenylethyl ester and the like or an
alkanoyloxyalkyl, cycloalkanoyloxyalkyl,
aroyloxyalkyl or an arylalkylcarbonyloxyalkyl
ester.
The term "N-protecting group" or "N-
protected" as used herein refers to those groups
intended to protect the N-terminus of an amino
acid or peptide or to protect an amino group
against undesirable reactions during synthetic
procedures. Commonly used N-protecting groups are
disclosed in Greene, "Protective Groups in Organic
Synthesis," (John Wiley & Sons, New York (1981)),
The term "alkanoyl" as used herein refers to
R29C(0)-0- wherein R29 is a loweralkyl group.
The term "alkanoylaminoalkyl" as used herein
refers to a loweralkyl radical to which is
appended R71-NH- wherein Rn is an alkanoyl group.
The term "alkanoyloxy" as used herein refers
to R.29C(0)-0- wherein R29 is a loweralkyl group.
The term "alkanoyloxyalkyl" as used herein
refers to a loweralkyl radical to which is
appended an alkanoyloxy group.
The term "alkenyl" as used herein refers to a
straight or branched chain hydrocarbon containing
from 2 to 10 carbon atoms and also containing at
least one carbon-carbon double bond. Examples of
alkenyl include -CH=CH2, -CH2CH=CH2, -C(CH3)=CH2,
-CH2CH=CHCH3, and the like.
- 22 -
CA 02207252 1997-06-06
W096/21456
PCT/US96/01559
The term "alkenylene" as used herein refers
to a divalent group derived from a straight or
branched chain hydrocarbon containing from 2 to 10
carbon atoms and also containing at least one
carbon-carbon double bond. Examples of alkenylene
include
-CH=CH-, -CH2CH=CH-, -C(CH3) =CH-, -CH2CH=CHCH2-, and
the like.
The term "alkoxy" as used herein refers to
R300- wherein Rn is loweralkyl as defined above.
Representative examples of alkoxy groups include
methoxy, ethoxy, t-butoxy and the like.
The term "alkoxyalkoxy" as used herein refers
to R310-R320- wherein Rm is loweralkyl as defined
above and R32 is an alkylene radical.
Representative examples of alkoxyalkoxy groups
include methoxymethoxy, ethoxymethoxy, t-
butoxymethoxy and the like.
The term "alkoxyalkyl" as used herein refers
to an alkoxy group as previously defined appended
to an alkyl group as previously defined. _ Examples
of alkoxyalkyl include, but are not limited to,
methoxymethyl, methoxyethyl, isopropoxymethyl and
the like.
The term "alkoxyalkylcarbonyloxyalkyl" as
used herein refers to a loweralkyl radical to
which is appended R66-C(0)-0- wherein R" is an
alkoxyalkyl group.
The term "alkoxycarbonyl" as used herein
refers to an alkoxy group as previously defined
appended to the parent molecular moiety through a
carbonyl group. Examples of alkoxycarbonyl
include methoxycarbonyl, ethoxycarbonyl,
isopropoxycarbonyl and the like.
The term "alkoxycarbonylakly1" as used herein
refers to an alkoxylcarbonyl group as previously
defined appended to a loweralkyl radical.
- 23 -
CA 02207252 1997-06-06
WO 96/21456
PCT/US96/01559
Examples of alkoxycarbonylakly1 include
methoxycarbonylmethyl, 2-ethoxycarbonylethyl and
the like.
The term "alkoxycarbonylaminoalkyl" as used
herein refers to a loweralkyl radical to which is
appended R69-NH- wherein R69 is an alkoxycarbonyl
group.
The term "alkoxycarbonyloxyalkyl" as used
herein refers to a loweralkyl radical to which is
appended R63-0- wherein R63 is an alkoxycarbonyl
group.
The term "alkylamino" as used herein refers
to R35NH- wherein R36 is a loweralkyl group, for
example, methylamino, ethylamino, butylamino, and
the like.
The term nalkylaminoalkyl" as used herein
refers a loweralkyl radical to which is appended
an alkylamino group.
The term "alkylaminocarbonylaminoalkyl" as
used herein refers to a loweralkyl radical to
which is appended R70-0(0)-..NH- wherein Rn is an
alkylamino group.
The term "alkylene" as used herein refers to
a divalent group derived from a straight or
branched saturated hydrocarbon having from 1 to 10
carbon atoms by the removal of two hydrogen atoms,
for example methylene, 1,2-ethylene, 1,1-ethylene,
1,3-propylene, 2,2-dimethylpropylene, and the
like.
The term nalkylsulfinyl" as used herein
refers to R33S(0)- wherein Rn is a loweralkyl
group.
The term nalkylsulfonyl" as used herein
refers to R34S(0)2- wherein R34 is a loweralkyl
group.
- 24 -
CA 02207252 1997-06-06
11439&21456
PCTMS96/01559
The term "alkylsulfonylalkyl" as used herein
refers to a loweralkyl radical to which is
appended an alkylsulfonyl group.
The term "alkynyl" as used herein refers to a
straight or branched chain hydrocarbon containing
from 2 to 10 carbon atoms and also containing at
least one carbon-carbon triple bond. Examples of
alkynyl include -CaCH, -CH2CECH, -CH2CaCCH3, and
the like.
The term "alkynylene" as used herein refers
to a divalent group derived from a straight or
branched chain hydrocarbon containing from 2 to 10
carbon atoms and also containing at least one
carbon-carbon triple bond. Examples of alkynylene
include
-CC-, -CH2CEC-, -CH2CECCH2, and the like.
The term "amino" as used herein refers to -
NH2.
The term "aminoalkyl" as used herein refers
to a loweralkyl radical to which is appended an
amino group,
The term "aroyloxyalkyl" as used herein
refers to a loweralkyl radical to which is
appended an aroyloxy group (i.e., R61-C(0)0-
wherein 1261 is an aryl group).
The term "aryl" as used herein refers to a mono-
or bicyclic carbocyclic ring system having one or
two aromatic rings including, but not limited to,
phenyl, naphthyl, tetrahydronaphthyl, indanyl,
indenyl and the like. Aryl groups (including
bicyclic aryl groups) can be unsubstituted or
substituted with one, two or three substituents
independently selected from loweralkyl, haloalkyl,
alkoxy, thioalkoxy, amino, alkylamino,
dialkylamino, hydroxy, halo, mercapto, nitro,
cyano, carboxaldehyde, carboxy, alkoxycarbonyl,
haloalkyl-C(0)-NH-, haloalkenyl-C(0)-NH- and
- 25 -
CA 02207252 1997-06-06
W096/21456
PCT/US96/01559
carboxamide. In addition, substituted aryl groups
include tetrafluorophenyl and pentafluorophenyl.
The term "arylalkenyl" as used herein refers
to an alkenyl radical to which is appended an aryl
group.
The term "arylalkenyloxycarbonyloxyalkyl" as
used herein refers to a loweralkyl radical to
which is appended R66-0-C(0)-0- wherein R" is an
arylalkenyl group.
The term "arylalkyl" as used herein refers to..
a loweralkyl radical to which is appended an aryl
group. Representative arylalkyl groups include
benzyl, phenylethyl, hydroxybenzyl, fluorobenzyl,
fluorophenylethyl and the like.
The term "arylalkylcarbonyloxyalkyl" as used
herein refers to a loweralkyl radical to which is
appended an arylalkylcarbonyloxy group (i.e., R62C(0)0-
wherein Ru is an arylalkyl group).
The term "arylalkyloxycarbonyloxyalkyl" as
used herein refers to a loweralkyl radical to
which is appended R67-0-C(0)-0- wherein R67 is an
arylalkyl group.
The term "aryloxyalkyl" as used herein refers
to a loweralkyl radical to which is appended R65-0-
wherein R65 is an aryl group.
The term "aryloxthioalkoxyalkyl" as used
herein refers to a loweralkyl radical to which is
appended R75-S- wherein Rm is an aryloxyalkyl
group.
The term "aryloxycarbonyalkyl" as used herein
refers to a loweralkyl radical to which is
appended R65-0-C(0)-0- wherein R65 is an aryl group.
The term "arylsulfonyl" as used herein refers
to R.36S(0)2- wherein R36 is an aryl group.
The term "arylsulfonyloxy" as used herein
refers to R57S(0)20- wherein R3.7 is an aryl group.
- 26 -
CA 02207252 1997-06-06
WOW21456
PCT/US96/01559
The term "carboxyalkyl" as used herein refers
to a loweralkyl radical to which is appended a
carboxy (-COOH) group.
The term "carboxaldehyde" as used herein
refers to the group -C(0)H.
The term "carboxamide" as used herein refers
to the group -C(0)NH2.
The term "cyanoalkyl" as used herein refers
to a loweralkyl radical to which is appended a
cyano (-CN) group.
The term "cycloalkanoylalkyl" as used herein
refers to a loweralkyl radical to which is
appended a cycloalkanoyl group (i.e., R60-C(0)-
wherein R" is a cycloalkyl group).
The term "cycloalkanoyloxyalkyl" as used
herein refers to a loweralkyl radical to which is
aPPended a cycloalkanoyloxy group (i.e., R60-C(0)0-
wherein R" is a cycloalkyl group).
The term "cycloalkenyl" as used herein refers
to an alicyclic group comprising from 3 to 10
carbon atoms and containing a carbon-carbon double
bond including, but not limited to, cyclopentenyl,
cyclohexenyl and the like.
The term "cycloalkyl" as used herein refers
to an alicyclic group comprising from 3 to 10
carbon atoms including, but not limited to,
cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl,
norbornyl, adamantyl and the like.
The term "cycloalkylalkyl" as used herein
refers to a loweralkyl radical to which is
appended a cycloalkyl group. Representative
examples of cycloalkylalkyl include
cyclopropylmethyl, cyclohexylmethyl, 2-
(cyclopropyl)ethyl, adamantylmethyl and the like.
The term "cycloalkyloxycarbonyloxyalkyl" as
used herein refers to a loweralkyl radical to
- 27 -
CA 02207252 1997-06-06
WO 96/21456
PCT/US96/01559
which is appended R64-0-C(0) -0- wherein R64 is a
cycloalkyl group.
The term "dialkoxyalkyl" as used herein
refers to a loweralkyl radical to which is
appended two alkoxy groups.
The term "dialkylamino" as used herein refers
to R38R39N- wherein R38 and R39 are independently
selected from loweralkyl, for example,
dimethylamino, diethylamino, methyl propylamino,
and the like.
The term "dialkylaminoalkyl" as used herein
refers to a loweralkyl radical to which is
appended a dialkylamino group.
The term "dialkyaminocarbonylalkyl" as used
herein refers to a loweralkyl radical to which is
appended 1273-C(0)- wherein R.73 is a dialkylamino
group.
The term "dioxoalkyl" as used herein refers
to a loweralkyl radical which is substituted with
two oxo (.0) groups.
The term "dithioalkoxyalkyl" as used herein
refers to a loweralkyl radical to which is
appended two thioalkoxy groups.
The term "halogen" or "halo" as used herein
refers to I, Br, Cl or F.
The term "haloalkenyl" as used herein refers
to an alkenyl radical, as defined above, bearing
at least one halogen substituent.
The term "haloalkyl" as used herein refers to
a lower alkyl radical, as defined above, bearing
at least one halogen substituent, for example,
chloromethyl, fluoroethyl or trifluoromethyl and
the like.
The term "heterocyclic ring" or
"heterocyclic" or "heterocycle" as used herein
refers to a 5-, 6- or 7-membered ring containing
one, two or three heteroatoms independently
- 28 -
CA 02207252 1997-06-06
V11396/21456
PCTIUS96101559
selected from the group consisting of nitrogen,
oxygen and sulfur or a 5-membered ring containing
4 nitrogen atoms; and includes a 5-, 6- or 7-
membered ring containing one, two or three
nitrogen atoms; one oxygen atom; one sulfur atom;
one nitrogen and one sulfur atom; one nitrogen and
one oxygen atom; two oxygen atoms in non-adjacent
positions; one oxygen and one sulfur atom in non-
adjacent positions; two sulfur atoms in non-
adjacent positions; two sulfur atoms in adjacent
positions and one nitrogen atom; two adjacent
nitrogen atoms and one sulfur atom; two non-
adjacent nitrogen atoms and one sulfur atom; two
non-adjacent nitrogen atoms and one oxygen atom.
The 5-membered ring has 0-2 double bonds and the
6- and 7-membered rings have 0-3 double bonds.
The term "heterocyclic" also includes bicyclic,
tricyclic and tetracyclic groups in which any of
the above heterocyclic rings is fused to one or
two rings independently selected from the group
consisting of an aryl ring; a cyclohexane ring, a
cyclohexene ring, a cyclopentane ring, a
cyclopenene ring and another monocyclic
heterocyclic ring (for example, indolyl, quinolyl,
isoquinolyl, tetrahydroquinolyl, benzofuryl or
benzothienyl and the like). Heterocyclics
include: pyrrolyl, pyrrolinyl, pyrrolidinyl,
pyrazolyl, pyrazolinyl, pyrazolidinyl, imidazolyl,
imidazolinyl, imidazolidinyl, pyridyl,
piperidinyl, homopiperidinyl, pyrazinyl,
piperazinyl, pyrimidinyl, pyridazinyl, oxazolyl,
oxazolidinyl, isoxazolyl, isoxazolidinyl,
morpholinyl, thiomorpholinyl, thiazolyl,
thiazolidinyl, isothiazolyl, isothiazolidinyl,
indolyl, quinolinyl, isoquinolinyl,
benzimidazolyl, benzothiazolyl, benzoxazolyl,
furyl, thienyl, thiazolidinyl, isothiazolyl,
- 29 -
CA 02207252 1997-06-06
WO 96/21456
PCT/US96/01559
triazolyl, tetrazolyl, oxadiazolyl, thiadiazolyl,
pyrimidyl, tetrahydrofuranyl, dihydrofuranyl,
tetrahydrothienyl, dihydrothienyl, dihydroindolyl,
tetrahydroquinolyl, tetrahydroisoquinolyl,
pyranyl, dihydropyranyl, dithiazolyl, benzofuranyl
and benzothienyl. Heterocyclics also include
bridged bicyclic groups wherein a monocyclic
heterocyclic group is bridged by an alkylene
group, for example,
CD 4
,and the
like.
Heterocyclics also include compounds of the
formula
Y*
L.%."-(21/
wherein X* is -CH2-, -CH20- or -0- and Y* is -C(0)-
or -(C(R")2),- wherein R" is hydrogen or C1-C4-alkyl
and v is 1, 2 or 3 such as 1,3-benzodioxolyl, 1,4-
benzodioxanyl and the like.
Heterocyclics can be unsubstituted or
substituted with one, two or three substituents
independently selected from the group consisting
of
a) hydroxy,
b) -SH,
c) halo,
d) oxo (=0),
e) thioxo (=S),
f) amino,
g) -NHOH,
h) alkylamino,
i) dialkylamino,
- 30 -
CA 02207252 1997-06-06
WO 96/21456
PCT/US96/01559
j) alkoxy,
k) alkoxyalkoxy.
1) haloalkyl.
hydroxyalkyl,
n) alkoxyalkyl,
o) cycloalkyl,
p) cycioaikeny1,
q) alkenyl,
r) alkynyl,
s) aryl,
t) arylalkyl,
u) -COOH,
v) -S02H,
w) loweralkyl,
x) alkoxycarbonyl,
y) -C (0) NH2
z) -C(S)NH2,
aa) -C(=N-OH)NH2,
bb) loweralkyl-C(0)-,
cc) loweralkyl-C(S)-,
=
dd) formyl,
ee) cyano, and
if) nitro.
The term "(heterocyclic)alkyl" as used herein
refers to a heterocyclic group as defined above
appended to a loweralkyl radical as defined above.
Examples of heterocyclic alkyl include 2-
pyridylmethyl, 4-pyridylmethyl, 4-quinolinylmethyl
and the like.
The term "heterocycliccarbonyloxyalkyl" as
used herein refers to a loweralkyl radical to
which is appended R72-C(0)-0- wherein R.72 is a
heterocyclic group.
The term "hydroxyalkyl" as used herein refers
to a loweralkyl radical to which is appended an
hydroxy group.
- 31 -
CA 02207252 1997-06-06
W096/21456
PCT/US96/01559
The term "hydroxythioalkoxy" as used herein
refers to R51S- wherein R51 is a hydroxyalkyl group.
The term "loweralkyl" as used herein refers
to branched or straight chain alkyl groups
comprising one to ten carbon atoms, including
methyl, ethyl, propyl, isopropyl, n-butyl,
neopentyl and the like.
The term "N-protected alkylaminoalkyl" as
used herein refers to an alkylaminoalkyl group
wherein the nitrogen is N-protected.
The term "oxoalkyloxy" as used herein refers
to an alkoxy radical wherein the loweralkyl moiety
is substituted with an oxo (.0) group.
The term "spiroalkyl" as used herein refers
to an alkylene diradical, both ends of which are
bonded to the same carbon atom of the parent group
to form a spirocyclic group.
The term "thioalkoxy" as used herein refers
to R52S- wherein R52 is loweralkyl. Examples of
thioalkoxy include, but are not limited to,
methylthio, ethylthio and the like.
The term "thioalkoxyalkyl" as used herein
refers to a thioalkoxy group as previously defined
appended to a loweralkyl group as previously
defined. Examples of thioalkoxyalkyl include
thiomethoxymethyl, 2-thiomethoxyethyl and the
like.
The present invention also relates to
processes for preparing the compounds of formula
(1)-(X11) and to the synthetic intermediates
useful in such processes.
In a further aspect of the present invention
are disclosed pharmaceutical compositions which
comprise a compound of the present invention in
combination with a pharmaceutically acceptable
carrier.
- 32 -
CA 02207252 1997-06-06
WO 96/21456
PCT/US96/01559
In yet another aspect of the present
invention are disclosed pharmaceutical
compositions which comprise a compound of the
present invention in combination with another
chemotherapeutic agent and a pharmaceutically
acceptable carrier.
In yet another aspect of the present
invention is disclosed a method for inhibiting
protein isoprenyl transferases (i.e., protein
farnesyltransferase and/or
geranylgeranyltransferase) in a human or lower
mammal, comprising administering to the patient a
therapeutically effective amount of a compound of
the invention.
In yet another aspect of the present
invention is disclosed a method for inhibiting
post-translational modification of the oncngenir
Ras protein by protein farnesyltransferase,
protein geranylgeranyltransferase or both.
In yet another aspect of the present
invention is disclosed a method for treatment of
conditions mediated by farnesylated or
geranylgeranylated proteins, for example,
treatment of Ras associated tumors in humans and
other mammals.
In yet another aspect of the present
invention is disclosed a method for inhibiting or
treating cancer in a human or lower mammal,
comprising administering to the patient a
therapeutically effective amount of a compound of
the invention alone or in combination with another
chemotherapeutic agent.
In yet another aspect of the present
invention is disclosed a method for treating or
preventing restenosis in a human or lower mammal,
comprising administering to the patient a
- 33 -
CA 02207252 1997-06-06
WO 96/21456
PCT/US96/01559
therapeutically effective amount of a compound of
the invention.
The compounds of the present invention can be
used in the form of pharmaceutically acceptable
salts derived from inorganic or organic acids.
These salts include but are not limited to the
following: acetate, adipate, alginate, citrate,
aspartate, benzoate, benzenesulfonate, bisulfate,
butyrate, camphorate, camphorsulfonate,
digluconate, cyclopentanepropionate,
dodeoylsulfate, ethanesulfonate, glucoheptanoate,
glycerophosphate, hemisulfate, heptanoate,
hexanoate, fumarate, hydrochloride, hydrobromide,
hydroiodide, 2-hydroxy-ethanesulfonate, lactate,
maleate, methanesulfonate, nicotinate, 2-
naphthalenesulfonate, oxalate, pamoate, pectinate,
persulfate, 3-phenylpropionate, picrate, pivalate,
propionate, succinate, tartrate, thiocyanate, p-
toluenesulfonate and undecanoate. Also, the basic
nitrogen-containing groups can be quaternized with
such agents as loweralky halides (such as methyl,
ethyl, propyl, and butyl chloride, bromides, and
iodides), dialkyl sulfates like dimethyl, diethyl,
dibutyl, and diamyl sulfates, long chain halides
such as decyl, lauryl, myristyl and stearyl
chlorides, bromides and iodides, aralkyl halides
like benzyl and phenethyl bromides, and others.
Water or oil-soluble or disperisble products are
thereby obtained.
Examples of acids which may be employed to
form pharmaceutically acceptable acid addition
sales include such inorganic acids as hydrochloric
acid, sulphuric acid and phosphoric acid and such
organic acids as oxalic acid, maleic acid,
succinic acid and citric acid.
Basic addition salts can be prepared in situ
during the final isolation and purification of the
- 34 -
CA 02207252 1997-06-06
PCT/US96/01559
W096/21456
compounds of formulas A-L, or separately by
reacting the carboxylic acid function with a
suitable base such as the hydroxide, carbonate or
bicarbonate of a pharmaceutically acceptable metal
cation or with ammonia, or an organic primary,
secondary or tertiary amine. Such
pharmaceutically acceptable salts include, but are
not limited to, cations based on the alkali and
alkaline earth metals, such as sodium, lithium,
potassium, calcium, magnesium, aluminum salts and
the likes, as well as nontoxic ammonium,
quaternary ammonium, and amine cations, including,
but not limited to ammonium, tetramethylammonium,
tetraethylammonium, methylamine, dimethylamine,
trimethylamine, triethylamine, ethylamine, and the
like. Other representative organic amines useful
for the formation of base addition salts include
diethylamine, ethylenediamine, ethanolamine,
diethanolamine, piperazine and the like.
Other features of the invention will also be
hereinafter apparent.
Brief Description of the Drawings
Figure 1: Ras CAAX peptidomimetics and
FTase/GGTase I activities
A. Structures of CVIM, FTI-249, FTI-276 and FTI-
277. FTI-276 and FTI-277 were synthesized as
described in Examples 10 and 11. B. FTase and
GGTase I inhibition assays were carried out as
described in Example 12 by determining the ability
of FTI-276 to inhibit the transfer of farnesyl and
geranylgeranyl to recombinant H-Ras-CVLS and H-
Ras-CVLL, respectively. The data are
representative of at least three different
experiments.
- 35 -
CA 02207252 1997-06-06
W096/21456
PCT/US96/01559
Figure 2: Inhibition of Ras and RaplA Processing
A. H-RasF cells were treated with various
concentrations of FTI-277, lysed and the lysates
immunoblotted with anti-Ras or anti-RaplA
antibodies as described in Example 13. B.
pZIPneo, H-RasF, H-RasGG, Raf and S186 cells were
treated with vehicle or FTI-277 (5 AM), lysed and
lysates immunoblotted by anti-Ras antibody. Data
is representative of 5 different experiments. The
cells were obtained from Dr. Channing Der,
University of North Carolina, Chapel Hill, North
Carolina.
Figure 3: Effects of FTI-277 on Ras/Raf
Association. pZIPneo, H-RasF, H-RasGG and S186
cells were treated with vehicle or FTI-277 (5 AM),
homogenized and the membrane (IQ and cytosolic (B)
fractions were separated and immunoprecipitated by
an anti-Raf antibody. The immunoprecipitates were
then separated by SDS-PAGE and immunoblotted with
anti-RAS antibody as described in Example 14.
Data is representative of three different
experiments.
Figure 4: Effects of FTI-277 on Ras Nucleotide
Binding and Raf Kinase Activity
A: H-RasF cells were treated with vehicle or FTI-
277, lysed and the lysates immunoprecipitated with
anti-Ras antibody. The GTP and GDP were then
released from Ras and separated by TLC as
described in Example 15. B: pZIPneo and H-RasF
cells were treated with vehicle or FTI-277, lysed
and cells lysates immunoprecipitated with an anti-
Raf antibody. Raf kinase was assayed by using a
19-mer autophosphorylation peptide as substrate as
described in Example 16. Data are representative
of three different experiments.
- 36 -
CA 02207252 1997-06-06
W4396)214156
PCTMS96101559
Figure 5: Effect of FTI-277 on Oncogenic
Activation of MAPK
A: H-RasF cells were treated with various
concentrations of FTI-277, cells lysed and lysates
run on SDS-PAGE and immunoblotted with anti-MAPK
antibody. B: pZIPneo, H-RasF, H-RasGG, Raf, and
S186 cells were treated with vehicle of FTI-277 (5
AM), lysed and cells lysates processed as for A.
Data are representative of two different
experiments.
Figure 6. FTI-276 inhibits selectively Ras
processing and oncogenic Ras activation of MAP
Kinase. NIH 3T3 cells transfected with empty
vector (pZIPneo), oncogenic (GTP-locked)
farnesylated Ras (RasF), geranylgeranylated Ras
(RasGG) or a transforming mutant of human Raf-1
were obtained from Channing Der and Adrienne Cox
(University of North Carolina, Chapel Hill, NC,
USA) (26,27). The cells were plated in DMEM/10%
CS (Dubelco's Modified Eagles Medium, 10% calf
serum) on day one and treated with vehicle or FTI-
270 (20 AM) on days 2 and 3. The cells were then
harvested on day 4 and lysed in lysis buffer (30
mM HEPES, pH 7.5, 1% TX-100, 10% glycerol, 10 mM
NaC1, 5 mM MgCl2, 25 mM NaF, 1 mM EGTA, 2 mM
Na3VO4, 10 jig/ml Trypsin inhibitor, 25 jig/ml
leupeptin, 10 jig/ml aprotinin, 2 mM PMSF). The
lysate (35 fig) was electrophoresed on 15% SDS-
PAGE, transferred to nitrocellulose membranes and
immunoblotted simultaneously with anti-Ras
antibody Y13-238 (isolated from hybridomas
purchased from ATCC, Rockville, MD) and an Anti-
MAP kinase (erk2) antibody (UBI, Lake Placid, NY)
as described previously (17, 22).
- 37 -
CA 02207252 1997-06-06
W096/21456
PCT/US96/01559
Figure 7. Antitumor efficacy of FTI-276 against
human lung carcinomas. Calu-1 (Panel A) and NCI-
H810 cells (Panel B) were purchased from ATCC and
grown in McCoy's 5A medium in 1096- FBS (Fetal
Bovine Serum) and RPMI 1640 in 10 6 FBS,
respectively. The cells were harvested,
resuspended in PBS and injected s.c. into the
right and left flank of 8 week old female nude
mice (107 cells/flank). Nude mice (Harlan Sprague
Dawley, Indianapolis, Indiana) were maintained in
accordance with the Institutional Animal Care and
Use Committee (IACUC) procedures and guidelines.
On day 32 after s.c. implantation of tumors,
animals were dosed i.p. with 0.2 ml once daily for
36 days. Control animals (filled circles)
received a saline vehicle whereas treated animals
(open triangles) were injected with FTI-276 (50
mg/kg). The tumor volumes were determined by
measuring the length (1) and the width (w) and
calculating the volume (V = (1) X (w)2/2). Data
are presented as the average volume of eight
tumors in each group for each cell line.
Statistical significance between control and
treated groups were evaluated by using student t
test (*P<0.05).
Figure 8. Antitumor Efficacy of FTI-276 and FTI-
277 in Human Lung Carcinoma (Calu-1) Cells.
Experimental procedure was the same as described
in Figure 7.
Figure 9. Inhibition of Tumor Growth in Ras
transformed cells by FTI-276 and FTI-277. Ras-
transformed NIH 3T3 cells were implanted
subcutaneously into nude mice, and daily
intraperitoneal injections with FTI-276 and FTI-
- 38 -
CA 02207252 1997-06-06
WO 96/21456
PCT/US96/01559
277 (50 mg/kg) were started when the tumors
reached 50 mm3.
Figure 10. Inhibition of Tumor Growth in Raf
transformed cells by FTI-276 and FTI-277. Raf-
transformed NIH 3T3 cells were implanted
subcutaneously into nude mice, and daily
intraperitoneal injections with FTI-276 and FTI-
277 (50 mg/kg) were started when the tumors
reached 50 mm3.
Figure 11. Dose response: Antitumor efficacy and
Ras processing correlations. A. Antitumor
efficacy was carried out as described in Fig. 3
except that animals were randomly assigned to four
groups each of 4 mice each (2 tumors per mouse).
Saline treated groups (circles); FTI-276 treated
groups: 10 mg/kg (squares), 50 mg/kg (upward
triangles), 100 mg/kg (downward triangles). B.
Ras processing was carried out 5 hours after the
last treatement on day 17. Tumors were extracted
from the animals, tissumized, and lysed in lysis
buffer as described in Fig. 1. Lysates (25 lug)
were electrophoresed on a 12.5 6 SDS-PAGE and
immunoblotted with anti-Ras antibody Y13-238 as
described previously. The blots were then
reprobed with anti-Rap1A antibody (Santa Cruze
Biotechnologies, Santa Cruz, California).
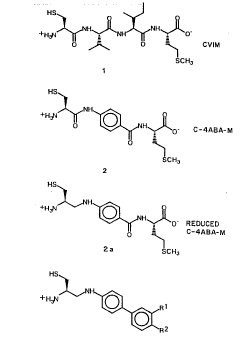
Figure 12. Structures of CVIM, C-4ABA-M, reduced
C-4ABA-M, FTI-265 (4), FTI-271 (5), and FTI-261
(8).
Figure 13. Energy-minimized structural
conformations for CVIM and farnesyltransferase
inhibitor FTI-265.
- 39 -
CA 02207252 1997-06-06
VOD96/21456
PCT/US96/01559
Figures 14A and B. Comparison of FTase and GGTase
I inhibition by FTI-265 and FTI-271.
Figure 15. Silica gel TLC relating to Ras CAAX
peptide and peptidomimetic farnesylation.
Figure 16. Ras and RaplA processing in cells
using a compound according to the invention.
Figure 17. CAAX peptidomimetic structures.
Structures of FTI-276/277, GGTI-287/286, and GGTI-
297.
Figure 18. Disruption of H-Ras and RaplA
processing. NIH 3T3 cells that overexpress
oncogenic H-Ras were treated with various
concentrations of FTI-277 (0-50 AM) or GGTI-286
(0-30 AM). The cells were lysed and the lysates
were electrophoresed on SDS-PAGE and immunoblotted
with either anti-Ras or anti-RaplA antibodies as
described in Example 3. U and P designate
unprocessed and processed forms of the proteins.
Data are representative of three independent
experiments.
Figure 19. Disruption of K-Ras4B processing.
NIH 3T3 cells that overexpress oncogenic K-Ras4B
were treated with FTI-277 or GGTI-286 (0-30 AM).
The cells were lysed and the lysates were
electrophoresed on SDS-PAGE and immunoblotted with
anti-Ras antibodies as described in Example 3. U
and P designate unprocessed and processed forms of
Ras. The data are representative of three
independent experiments.
Figure 20. Inhibition of oncogenic activation
of MAP Kinase. NIH 3T3 cells that overexpress
- 40 -
CA 02207252 1997-06-06
WC096/21456
PCT/US96/01559
either oncogenic H-Ras or K-Ras4B were treated
with either FTI-277 or GGTI-286 (0-30 AM). The
cells were lysed and the lysates were
electrophoresed on SDS-PAGE and immunoblotted with
an anti-MAP kinase antibody. P-MARK designates
hyperphosphorylated MAP kinase. The data are
representative of three independent experiments.
Figure 21. Inhibition of FTase and GGTase I
Activity by GGTI-297.
Figure 22. Antitumor Efficacy of GGTI-286 in K-
Ras4B.
Description of Preferred Embodiments
For ease of reference, the following
abbreviations may be used in the present
specification:
FTase: farnesyltransferase;
GGTase: geranylgeranyltransferase;
SDS-PAGE: sodium dodecyl sulfate
polyacrilamide gel
electrophoresis
PBS: phosphate-buffered saline;
CAAX: tetrapeptide where C is cysteine, A is
an aliphatic amino acid and X is an
amino acid
DTT: dithiothreitol;
DOC: deoxycholate
BSA: bovine serum albumin
GGTase I: geranylgeranyl transferase I;
PAGE: polyacrylamide gel electrophoresis;
MAPK: mitogen activated protein kinase;
FTI: farnesyltransferase inhibitor;
GGTI: geranylgeranyltransferase inhibitor;
- 41 -
,
CA 02207252 2012-03-19
PMSF: * phenylmethylsulfonyl fluoride.
I. Farnesyltransferase Inhibitors of the type C X
The peptidomimetics of Formula (I), one of the
preferred embodiments of the invention, may be made
using procedures which are conventional in the art.
Preferably, p is 2-phenyl-4-aminobenzoic acid although
constrained derivatives such as tetrahydroisoguinoline-7-
carbcowlic acid, 2- aminomethylpyridine-6-carbaxylic acid
or other heterocyclic derivatives, may also be used.
Compounds in which p is an aminomethylbenzoic acid
(particularly 3-aminomethylbenzoic acid) are disclosed
in U.S. Patent application No.5,602,098. The acid
component of p is conveniently reacted with cysteine
so that the amino group of p and the cysteine carboxyl
group react to fonn an amido group, other reactive
substituents in the reactants being suitably protected
against undesired reaction. In the case of the reduced-
cysteine series of cornpounds, the amino group of p is
reacted with a suitably protected cysteinal. The
amino acid represented by X, preferably Met, is then
reacted through its amino group with the deprotected and
activated carboxyl group of spacer compound p.
Following deprotect ion by removal of other protecting
groups, the compound of Formula (I) is obtained.
As an alternative, 0 may first be reacted
with the X amino acid followed by reaction with the
cysteine or cysteinal component using conventional
reaction conditions.
The invention also includes the
pharmaceutically acceptable salts of the compounds
of Formula (I). These may be obtained by reacting the
free base or acid with the appropriate amount
-42-
CA 02207252 1997-06-06
WO 96/21456
PCT/US96/01559
of inorganic or organic acid or base, e.g. an
alkali metal hydroxide or carbonate, such as
sodium hydroxide, an organic amine, e.g.
trimethylamine or the like. Acid salts include
the reaction products obtained with, for example,
toluene sulfonic acid, acetic acid, propionic acid
or the like as conventionally used in the art.
The compounds of the invention may be used to
inhibit p2lras farnesyltransferase in any host
containing the same. This includes both in vitro
and in vivo use. Because the compounds inhibit
farnesyltransferase, notably'human tumor p2lras
farnesyltransferase, and consequently inhibit the
farnesylation of the oncogene protein ras, they
may be used in the treatment of cancer or cancer
cells. It is noted that many human cancers have
activated ras and, as typical of such cancers,
there may be mentioned colorectal carcinoma,
myeloid leukemias, exocrine pancreatic carcinoma
and the like.
The compounds of the invention may be used in
pharmaceutical compositions of conventional form
suitable for oral, subcutaneous, intravenous,
intraperitoneal or intramuscular administration to
a mammal or host. This includes, for example,
tablets or capsules, sterile solutions or
suspensions comprising one or more compounds of
the invention with a pharmaceutically acceptable
carrier and with or without other additives.
Typical carriers for tablet or capsule use
include, for example, lactose or corn starch. For
oral compositions, aqueous suspensions may be used
with conventional suspending agents, flavoring
agents and the like.
The amount of inhibitor administered to
obtain the desired inhibitory effect will vary but
can be readily determined. For human use, daily
- 43 -
CA 02207252 1997-06-06
WO 96/21456
PCT/US96/01559
dosages are dependent on the circumstances, e.g.
age and weight. However, daily dosages of from
0.1 to 20 mg per kg body weight may be mentioned
for purposes of illustration.
The various aspects of the invention are
further described by reference to the following
examples. These examples illustrate, among other
things, the preparation of the present
peptidomimetics and compounds compared therewith.
In the invention, the g component is, in
general, any non-peptide amino acid combination or
other hydrophobic spacer element that produces a
compound which mimics the structure and
conformation of CVIM or like tetrapeptides CA1A2X.
A variety of hydrophobic spacers have been used as
the g component according to this aspect of the
invention. This includes, for example, 3-
aminobenzoic acid, 4-aminobenzoic acid and 5-
aminopentanoic acid as well as heterocyclic
carboxylic acids such as tetrahydroiso-quinoline-
7-carboxylic acid, 2-aminomethyl pyridine-6-
carboxylic acid or the like as mentioned earlier,
as replacements for the g component of the Formula
(I) compounds. Thus, in a broad sense, the
peptidomimetics of the invention include variants
for Formula (I) where g stands for the radical of
a non-peptide aminoalkyl or amino-substituted
aliphatic or aromatic carboxylic acid or a
heterocyclic monocarboxylia acid, for example, 3-
aminobenzoic acid (3-ABA), 4-aminobenzoic acid (4-
ABA) or 5-aminopentanoic acid (5-APA).
Other suitable g substituents which may be
mentioned include those obtained by using
aminomethyl- or aminocarboxylic acid derivatives
of other cyclic hydrophobic compounds such as
furan, quinoline, pyrrole, oxazole, imidazole,
- 44 -
CA 02207252 1997-06-06
WO 96/21456
PCT/US96/01559
pyridine and thiazole. Generally speaking,
therefore, the A substituent may be derived from
any hydrophobic, non-peptidic aminoalkyl- or
amino-substituted aliphatic, aromatic or
heterocyclic monocarboxylic acid.
According to still another feature of this
embodiment of the invention, other effective
inhibitors for farnesyltransferase may be provided
by incorporating a negatively charged residue onto
the compounds of Formula (I). This feature of the
invention is based on a consideration of the
transition state of the farnesylation reaction and
the recognition that the functional enzyme complex
must involve a farnesyl pyrophosphate binding site
close to the peptide binding region. Compounds
representative of this embodiment include peptides
prepared with a phosphonate residue linked at
different distances to the cysteine sulfur. These
derivatives have been prepared by reaction of N-
Cbz-cysteine with ethyl 2-chloroethylphosphonate
followed by condensation with the C-terminal
methionine adduct of 4-aminobenzoic acid (or N-
deprotected VIM methyl ester). Deprotection of
the phosphonate, carboxylate and amino protecting
groups gives analogs (5) and (6), respectively,
which contain elements of the tetrapeptide and
farnesyl pyrophosphate residues and hence are able
to interact with binding groups in both
recognition sites in p2lras farnesyltransferase:
-0
,0
H 0
H3N 410 1\11j- (5)
= CC
0 0
- 45 -
CA 02207252 1997-06-06
WO 96/21456
PCT/US96/01559
-0 0
=
S HO HO
p
( 6 )
+ 0 0
The above described phosphonates as
contemplated herein can be structurally
represented as follows:
Ai-c-g-x
where C, X, 0 and A are as previously described
and Ai is a phosphonate group joined to cysteine
through the cysteine sulphur atom.
As indicated earlier, an important further
feature of the invention is the modification of
the compounds of the invention, as well as the
tetrapeptide p2lras farnesyl transferase
inhibitors of the formula CA1A2X, to provide pro-
drugs. This involves forming lipophilic enzyme-
sensitive derivatives from the compounds by
appropriately functionalizing the terminal groups.
For example, the terminal amino groups and the
cysteine sulfur can be reacted with benzyl
chloroformate to provide carbobenzyloxy ester end
groups while the terminal carboxy group at the
other end of the compound is converted to an alkyl
or aryl ester, e.g. the methyl ester. Other
- 46 -
CA 02207252 1997-06-06
PCTMS96/01559
WO 96/21456
examples include alkyl esters from 1 to 10 carbons
in length, activated esters such as cyanomethyl or
trifluoromethyl, cholesterol, cholate or
carbohydrate derivatives. The term "lipophilic",
when used in this context, is meant to include,
inter a/ia, methoxycarbonyl and other long chain
or carbamate groups. Examples of such groups are
well known to the ordinarily skilled practitioner.
Derivatization of the prior peptides CAAX
and the peptidomimetics described herein with
lipophilic or hydrophobic, enzyme-sensitive
moieties increases the plasma membrane
permeability and cellular uptake of the compounds
and consequently their efficiency in inhibiting
tumor cell growth.
While the carbobenzyloxy derivatives have
been referred to as one way of functionalizing the
peptides and peptidomimetics to improve
efficiency, it will be appreciated that a variety
of other groups may also be used for the purposes
noted. Typical alternatives include
cholesterolyl, aryl or aralkyl such as benzyl,
phenylethyl, phenylpropyl or naphthyl, or alkyl,
typically methyl or other alkyl of, for example,
up to 8 carbon atoms or more. It is contemplated
that such functional groups would be attached to
the cysteine sulfur and the terminal amino and
carboxy groups.
- 47 -
CA 02207252 1997-06-06
PCT/US96/01559
W096/21456
Using C-ABA-M as representative of the
present compounds, the functionalized pro-drug
embodiment of the invention may be structurally
illustrated as follows:
BEM- CABAM
0
N-11r"
14111 HO
= 0
H 0
0 R
In the above described BBM-compounds, the
"BBM" used in the formulas represents a shorthand
reference to the bis-(carboxybenzyloxy)methyl
esters of CflM and CVIM.
The functionalized derivatives of the
phosphonates described earlier herein are also
useful cell growth inhibitors. Correspondingly,
the "BMMM" designation used with compounds refers
to the carboxy benzyloxy substitution and the
three methyl groups in the methylated phosphoric
and carboxylic acid end groups.
As noted, the purpose of the functional
groups added to the parent compounds is to improve
entry of the compounds into tumor cells. Once in
the cells, the functional groups are removed to
liberate the active compound to function in its
inhibitory capacity.
As will be recognized by those in the art,
the functionalized pro-drugs of the invention can
be prepared using conventional and well-known
procedures for esterifying amino, SH and
- 48 -
CA 02207252 1997-06-06
W096/21456
PCT/US96/01559
carboxylic acid groups. Hence, details of such
procedures are not essential for the preparation
of the present pro-drugs.
EXAMPLE 1
5. SYNTHESIS OF FTI-232
A. N-B0C-4-aminobenzoic acid
4-amino-benzoic acid (10g, 72.9 mmol) was
placed into a mixture of dioxane (145.8 ml) and
0.5M NaOH (145.8 ml). The solution was cooled to
0 C and di-t-butyl dicarbonate (23.87 g, 109.5
mmol) was added. The reaction mixture was allowed
to warm to room temperature and stirred overnight.
The next day, the dioxane was removed, the residue
was made acidic and extracted into ethyl acetate.
The ethyl acetate fractions were combined and
washed with 1N HC1 to remove any unreacted
starting material. The solution was dried over
Na2SO4 and the solvent was removed in vacuo. The
crude material was recrystallized from ethyl
acetate/hexanes to yield 12.2 g (70.69s) of pure
product. mp 189-190 C; 1H NMR (CD30D) 1.52 (9H,
s), 7.49 (2H, d, J=8.6 Hz), 7.91 (2H, d, J=8.6
Hz), 9.28 (1H, s); 13C NMR (CD30D) 28.59, 81.29,
118.54, 125.30, 131.81, 145.70, 155.00, 169.80;
anal. calc. for C3.2113.5N04, C: 60.76, 1.1; 6.37, N:
5.90; found, C: 60.52, H: 6.43, N: 5.83; HRMS
calc. for C121115N04, 237.0961, found, 237.1001.
B. N-B0C-4-aminobenzoyl methionine methyl ester
Into a dried, nitrogen filled flask was
placed N-B0C-4-aminobenzoic acid (8.77 g, 36.97
mmol) in dry CH2C12 (148 ml) along with methionine
methyl ester hydrochloride (8.12 g, 40.66 mmol).
This solution was cooled in an ice bath and
triethylamine (6.7 ml), EDCI (7.80 g, 40.66 mmol)
and hydroxybenzotriazole (HOBT, 5.50 g, 40.66
- 49 -
CA 02207252 1997-06-06
WO 96/21456
PCT/US96/01559
mmol ) were added. The mixture was stirred
overnight, diluted with more CH2C12 and was
extracted 3 times each with 1M HC1, 1M NaHCO3 and
water. The CH2C12 was dried over MgSO4 and the
solvent was removed in vacuo. The solid was
recrystallized from ethyl acetate/ hexanes to
yield 9.72 g (71.3%) of pure product. mp 184-
185 C; NMR (CDC13) 1.53 (9H, s), 2.06-2.18 (4H,
m), 2.23-2.33 (1H, m), 2.59 (2H, t, J=7.6 Hz),
3.80 (3H, s), 4.92 (1H, m), 7.45 (2H, d, J=8.7
Hz), 7.77 (2H, d, J=8.7 Hz); 13C NMR (CDC10 15.59,
28.34, 30.15, 31.64, 52.10, 52.73, 81.20, 117.73,
127.8, 128.33, 141.88, 152.33, 166.50, 172.75;
anal. cald. for Ca8H26N205S, C: 56.53, H: 6.85, N:
7.29; found, C: 56.47, H: 6.86, N: 7.29; m/ z (El)
382 (M).
C. HC1-4-aminobenzoyl methionine methyl ester
N-B0C-4-aminobenzoyl methionine methyl ester
(3.53 g, 9.59 mmol) was placed into CH2C12 (30-35
ml) and to it was added 3M HC1/ Et20 (38.4 ml).
After standing a white precipitate formed. After
2 hours the solution was decanted, and the
crystals were collected by centrifugation. The
crystals were then washed several times with fresh
ether and dried overnight on the vacuum pump.
Meanwhile, the filtrate was left to stand
overnight to allow additional product to
precipitate. The second fraction was washed with
ether and dried overnight on the vacuum pump. The
total yield of pure fully deprotected material was
2.87 g (93.9%) yield. mp 158-164 C; NMR (CDC10
2.10 (3H, s), 2.12-2.29 (1H, m), 2.52-2.71 (1H,
m), 2.59 (2H, t, J=7.6 Hz), 3.75 (3H, s), 4.79
(1H, m), 7.02 (2H, d, J=8.6 Hz), 7.55 (2H, d,
J=8.6 Hz); 13C NMR (CDC10 15.23, 31.43, 31.53,
52.91, 52.43, 124.35, 130.56, 135.31, 135.76,
- 50 -
CA 02207252 1997-06-06
174/096)21456
PCT/US96/01559
168.95, 173.87; HRMS calc. for C131118N203S, 282.1038,
found 282.1009.
D. N-B0C-S-trityl-cysteine-4-aminobenzoy1
methionine methyl ester
N-BOC-S-trityl-Cys (2.86 g, 6.54 mmol) and
triethylamine (1.2 ml) were placed into a dried, N2
filled flask containing dry THF (104 ml). This
was .cooled to -10 C using an ice/ salt bath and
isobutyl chloroformate (0.9 ml), IBCF, was added.
The solution was stirred at -10 C for 40 minutes
and HC1-4-aminobenzoyl methiOnine methyl ester
(2.08 g, 6.54 mmol) in dry CH2C12 (34.1 ml) with
triethylamine (1.2 ml, 1.3 eq) was added. The
solution warmed to room temperature and was
stirred overnight under N2. The solvent was then
removed in vacuo and the residue was taken up In
CH2C12 and extracted several times each with 1M
HC1, H20 and brine (saturated NaC1). The organic
layer was dried over Na2SO4 and the solvent was
removed in vacuo. The pale. yellow foam was then
chromatographed on silica gel using a 2:1 hexanes,
ethyl acetate elution mixture to yield 2.62 g
(54.9) of pure product. mp 110-111 C; [(]2513=-
8.0 (c=1, CH3OH); NMR (CDC10 1.44 (9H,$),
2.11-2.18 (4H, m), 2.22-2.34 (1H,m), 2.59 (2H, t,
J=7.4 Hz), 2.66-2.83 ( 2H, m), 3.80 (3H, s), 3.98
(1H, m), 4.84 (1H, m), 4.92 (1H, m), 6.96 (1H, d,
J=7..7 Hz), 7.23-7.33 (9H, m), 7.43-7.46 (6H, m),
7.51 (2H, d, J=8.5 Hz), 7.74 (2H, d, J=8.5 Hz),
8.51 (1H, s); 13C NMR (CDC10 15.53, 28.34, 30.72,
30.89, 33.60, 52.23, 52.88, 54.95, 60.50, 67.13,
80.64, 118.81, 119.31, 126.94, 128.07, 128.30,
129.53, 141.06, 144.38, 156.31, 167.02, 170.13,
174.49; anal calc for C401144N306S2-H20, C: 64.50, H:
6.22, N: 5.64; found C: 64.14 H: 6.19, N: 5.56.
- 51 -
CA 02207252 1997-06-06
WO 96/21456
PCTMS96/01559
E. HC1-cysteine-4-aminobenzoyl methionine methyl
ester
N-E0C-S-trityl-cysteine-4-aminobenzoyl
methionine methyl ester (1 g, 1.37 mmol) was
placed into a flask and taken up in CH3OH (13.7
m1). To this solution was added a solution of
mercuric chloride (0.75 g, 2.74 mmol) in CH3OH
(13.7 m1). Upon addition of the mercuric
chloride, a white precipitate began to form. The
mixture was heated on a steam bath at 65 C for 35
minutes and then it was cooled and the precipitate
was filtered and washed sparingly with cold CH3OH.
After drying for several minutes on the filter,
the solid was placed into a 50 ml 3-neck flask
fitted with a gas inlet and outlet. Approximately
20-30 ml of CH3OH was added and H2S gas was bubbled
through the heterogeneous solution for 30 minutes.
Upon addition of the gas, the white solution
turned orange and then black. The solution was
centrifuged and the clear, colorless liquid was
dried to give a white foam. This solid was placed
on the vacuum pump for a short period and then was
taken up in CH2C12 (10 ml) and the product was
precipitated with a 3-4M HC1/ Et20 solution. The
precipitate was collected by centrifugation and
was washed with ether until pH was neutral. After
drying under vacuum overnight, 0.38 g (66.5%) of
product was obtained that was >9596 pure by HPLC.
mp foamed 141-143 C, decomp 195 C; [c]25D=+3 (c=1,
H20); 1H NMR (CD30D) 2.09 (3H, s), 2.14-2.26
(1H,m), 2.51-2.67 (3H, m), 3.05 (IH, dd, J=14.8
Hz, 7.3 Hz), 3.17 (1H, dd, J=14.8 Hz, 4.8 Hz),
3.74 (3H,$), 4.17 (1H, J=7.3 Hz, 4.8 Hz ), 4.75-
4.81 (1H, m), 7.74 (2H, d, J=8.6 Hz), 7.87 (2H, d,
J=8.6 Hz), 8.67 (1H, d, J=8.4 Hz); 13C NMR (CD30D)
15.23, 26.38, 31.43, 31.56, 52.88, 53.30, 56.92,
120.46, 129.58, 130.75, 142.33, 166.91, 169.66,
- 52 -
CA 02207252 1997-06-06
WO 96/21456
PCT/US96/01559
174.06; anal calc for Ci6H24C1N304S2, C: 45.55 H:
5.73, N: 9.96; found C: 45.31, H: 5.84, N: 9.79.
F. HC1-cysteine-4-aminobenzoyl methionine FT1-232
HC1-cysteine-4-aminobenzoyl methionine methyl
ester (0.51 g, 0.7 mmol) was taken up in THF (4.1
ml) and to this solution was added 0.5 M LiOH (2.9
ml) at 0 C. The heterogeneous solution was
stirred at 0 C for 35-40 minutes and then the THF
was removed in vacuo. The residue was taken up in
CH2C12 and was washed three times with 1M HC1
followed by brine. The organic solution was dried
over Na2SO4 and the solvent was removed in vacuo.
The pale yellow solid was taken up in 3 ml of
CH2C12 and the product was precipitated with 3-4 M
HC1/ Et20. The solid was collected by
centrifugation, washed several times with ether
until the ether washings were neutral and the
process repeated until the HPLC appeared pure. A
final yield of 78.6 mg (27.5 ) of pure product was
obtained. mp sub 157 C, decomp 211 C; [cd25,3= 100 _
(c=0.8, H20); 114 NMR (CD30D) 2.09 (3H, s), 2.17-
2.32 (1H,m), 2.53-2.66 (3H, m), 3.06 (1H, dd,
J=14.6 Hz, 7.2 Hz), 3.19 (1H, dd, J=14.6 Hz, 4.6
Hz), 4.21 (1H, dd, J=7.23 Hz, 4.63 Hz), 4.73-4.78
(1H, m), 7.75 (2H, d, J=8.1 Hz), 7.87 (2H, d,
J.8.1 Hz); 13C NMR (CD30D) 15.23, 26.33, 31.58,
31.86, 53.24, 56.98, 120.48, 129.59, 131.10,
142.26, 166.89, 169.66, 175.29; anal calc for
C3.5H22C1N304S2, C: 44.16, H: 5.44, N: 10.30; found C:
45.45, H: 5.62, N: 10.03; m/ z (FAB) for free
amine, 371 (141- 1).
- 53 -
CA 02207252 1997-06-06
WO 96/21456
PCT/US96/01559
EXAMPLE 2
SYNTHESIS OF FTI-260
A. N-B0C-4-amino-3-methylbenzoic acid
4-amino-3-methylbenzoic acid (5 g, 33.1 mmol)
was reacted according to the same procedure as N-
BOC-4-aminobenzoic acid. The orange-brown solid
was recrystallized from ethyl acetate and hexanes
to yield 4.99 g (60%) of tan prismatic crystals.
mp 180-182 C; 314 NMR (CD30D) 1.51 (9H, s), 2.27
(3H, s), 7.66 (1H, d, J=8.1 Hz), 7.79-7.82 (2H,
m), 8.32 (1H, s); 13C NMR (CD30D) 17.98, 28.62,
81.47, 123.12, 127.05, 129.14, 130.65, 132.99,
142.45, 155.33, 168.70; anal calc for C3.31117N04, C:
62.15, H: 6.82, N: 5.58; found C: 62.07, H: 6.86,
N: 5.46; m/ z (El) 251; HRMS calc. for C131137N04,
251.1158; found, 251.1153.
B. N-B0C-4-amino-3-methylbenzoyl methionine
methyl ester
N-B0C-4-amino-3-methylbenzoic acid (2.00 g,
7.96 mmol) was reacted with methionine methyl
ester hydrochloride (1.75 g, 8.76 mmol), EDCI
(1.68 g, 8.76 mmol), HOBT (1.18 g, 8.76 mmol) and
Et3N (1.4 ml) in dry CH2C12 (31.8 ml) according to
the procedure described for N-B0C-4-aminobenzoyl
methionine methyl ester in Example 1. The crude
material was recrystallized from ethyl acetate and
hexanes to yield 2.61 g (85.7%) of pure product.
mp 163-165 C; ]-H NMR (CDC10 1.54 (9H,$), 2.06-2.18
(4H, m), 2.23-2.34 (4H, m), 2.59 (2H, t, J=6.8
Hz), 3.80 (3H, s), 4.92 (1H, m), 6.45 (1H, s),
6.88 (1H, d, J=7.5 Hz), 7.63 (1H, d, J=8.6 Hz),
7.66 (1H, s), 8.05 (1H, d, J=8.6); 13C NMR (CDC10
15.47, 17.61, 28.22, 30.03, 31.55, 51.93, 52.57,
81.04, 118.73, 125.62, 127.66, 129.54, 139.89,
152.34, 166.58, 172.66.
- 54 -
CA 02207252 1997-06-06
W096/21456
PCT/US96/01559
C. HC1-4-amino-3-methylbenzoyl methionine methyl
ester
N-B0C-4-amino-3-methylbenzoyl methionine
methyl ester (0.99 g, 2.59 mmol) was dissolved in
CH2C12 (15-20 ml) and precipitated with 3M HC1/
Et20 (20.7 ml). 0.83 g (96.6%) of pale orange
precipitate was obtained after drying overnight on
the vacuum pump. mp 157-159 C; NMR (CD30D) 2.04
(3H,$), 2.11-2.25 (1H, m), 2.47 (3H, s), 2.52-2.68
(3H. m), 3.74 (3H, s), 4.75-4.80 (1H, m), 7.48
(1H, d, J=8.2 Hz), 7.81 (2H, d, J=8.2 Hz), 7.87
(1H, s); 1-3C NMR (CD30D) 15.23, 17.28, 31.43,
31.51, 52.91, 53.37, 124.41, 127.85, 131.99,
133.63, 134.14, 135.65, 169.05, 173.84; anal.
calc. for Cl4H21N203S, C: 50.52, H: 6.36, N: 8.42;
found C: 50.71, H: 6.40, N: 8.34.
D. N-B0C-S-trityl-cysteine-4-amino-3-
methylbenzoyl methionine methyl ester
N-BOC-S-trityl-cysteine (0.55 g, 1.25 mmol)
in dry THF (25 ml) was reacted with Et3N (0.19 ml),
IBCF (0.16 ml, 1.25 mmol) at -10 C as described
above. HC1-4-amino-3-methylbenzoyl methionine
methyl ester (0.42 g, 1.25 mmol) in dry CH2C12
(6.5 ml) with Et3N (0.26 ml) was added at -10 C and
the reaction mixture was allowed to stir overnight
under nitrogen. Workup was carried out as
described above and the crude material was
chromatographed on silica gel using a 2:1 mixture
of hexanes and ethyl acetate as an elution mixture
to give 0.12 g (13.96) of pure product. mp 83-
85 C; [c]25p=-14.0 (c=1, CH3OH); NMR (CDC13) 1.44
(9H,$), 2.10-2.17 (4H, m), 2.22-2.32 (4H, m), 2.61
(2H, t, J=6.57 Hz), 2.68-2.70 (1H, m), 2.85-2.90
(1H. m), 3.79 (3H,$), 3.93-4.08 (1H, s), 4.84-4.88
( 1H, m), 4.90-4.95 (1H, m), 6.95 (1H, d, J=7.00
Hz), 7.20-7.33 (9H,m), 7.39 (1H, d, J=6.96 Hz),
- 55 -
CA 02207252 1997-06-06
W096/21456
PCT/US96/01559
7.44-7.47 (6H,m), 7.59 (1H, d, J=8.46 Hz), 7.65 (
1H, s), 8.12 ( 1H,d, J=8.22 Hz), 8.31 (1H,$); 'C
NMR (CDC10 15.39 17.55, 27.70, 28.17, 30.00,
31.43, 31.41, 51.90, 52.51, 59.95, 67.30. 80.74,
84.54, 120.74, 125.33, 126.70, 126.83, 127.89,
128.00, 129.40, 138.92, 144.22, 166.50, 166.89,
168.87, 172.56.
E. TFA-cysteine-4-amino-3-methylbenzoyl
methionine FTI-260
N-DOC-S-trityl-cysteine-4-amino-3-
methylbenzoyl methionine methyl ester (0.27 g,
0.37 mmol) in THF (2.1 ml) was deprotected with
0.5M LiOH (2.9 ml) over 1.5 h at room temperature.
The solvent was removed in vacuo and the residue
was taken up in CH2C12 and extracted 3 times with
= 1N HC1 followed by extraction with brine. The
organic solution was dried over Na2SO4 and the
solvent was removed in vacuo to give 0.19 g (73.5
%) of the free acid. The free acid was then taken
up in CH2C12 (1.4 ml) and Et3SiH (0.04 ml) was added
followed by trifluoroacetic acid, TFA (1.4 ml).
The reaction mixture was stirred at room
temperature for 1 hour. The TFA was removed and
the residue was dissolved in H20 and extracted with
Et20 until all of the trityl derivative was
removed. The water was lyophilized and a crude
HPLC showed that the material was impure and
contained diastereomers. The product was purified
on the preparative HPLC using 0.3A TFA in water
and acetonitrile elution mixture to give 2
diastereomers and only the major component
(determined according to the major compound in the
HPLC trace) was characterized. mp sub 112 C,
foamed 158-163 C, decomp 196-197 C; [a]25p=4.12.7
(c=0.6 H20), [a]25õ=+ 21.0 (c=1 H20); 1H NMR (CD30D)
2.09-2.17 (4H, m), 2.19-2.30 (1H, m), 2.36 (3H,
- 56 -
CA 02207252 1997-06-06
V1/396/21456
PCT/US96/01559
s), 2.57-2.65 (2H, m), 3.08 (1H, dd, J=14.6 Hz,
6.9 Hz), 3.19 (1H, dd, J=14.6 Hz, 5.2 Hz), 4.25 (
1H, dd, J=6.9, 5.2 Hz), 4.70-4.75 (1H, m), 7.64
(1H, d, J=8.4 Hz), 7.69-7.73 (1H, m), 7.77 (1H,
s); I3C NMR (CD30D) 15.23, 18.28, 26.54, 31.58,
32.06, 53.53, 56.66, 125.54, 125.77, 126.74,
131.04, 133.24, 139.26, 167.53, 169.70, 175.59.
EXAMPLE 3
SYNTHESIS OF FTI-261
A. N-B0C-4-amino-3-methoxybenzoic acid
4-amino-3-methoxybenzoic acid (1 g, 5.98
mmol) was reacted with di-t-butyl dicarbonate
(1.96 g, 6.58 mmol) in dioxane (12 ml) and 0.5 M
NaOH (12 ml) according to the same procedure as N-
BOC-4-aminobenzoic acid. 1.50 g (93.7) of tan
= crystals were obtained after recrystallization
from ethyl acetate and hexanes. mp 176-178 C; IH
NMR (CD30D) 1.52 (9H, s), 3.92 (3H, s), 7.56 (1H,
s), 7.62 (1H, d, J=8.4 Hz), 7.96 (1H, s), 8.03
(1H, d, J=8.4 Hz); I3C NMR (CD30D) 28.53, 56.35,
81.78, 112.01, 118.58, 124.20, 125.76, 133.84,
149.04, 154.20, 169.60; HRMS calc. for C13111.7N05,
267.1107; found, 267.1103.
B. N-B0C-4-amino-3-methoxybenzoyl methionine
methyl ester
N-B0C-4-amino-3-methoxybenzoic acid (0.35 g,
1.31 mmol) was reacted with methionine methyl
ester hydrochloride (0.9 g, 1.43 mmol) using EDCI
as in N-B0C-4-aminobenzoyl methionine methyl
ester. After recrystallization from ethyl acetate
and hexanes, 0.36 g (57.2 -IT) of pure product was
obtained. mp 163-165 C; NMR (CDC10 1.53 (9H,
s), 2.09-2.18 CH, m), 2.23-2.35 (1H, m), 2.60
(2H, t, J=6.9 Hz), 3.80 (3H, s), 3.93 (3H, s),
4.92 (1H, br s), 6.93 (1H, d, J.7.6 Hz), 7.25 (1H,
- 57 -
CA 02207252 1997-06-06
W096/21456
PCT/US96/01559
m), 7.31 (1H, d, J=10.2 Hz), 7.44(1H, s), 8.15(1H,
d, J=8.5 Hz); 1.3C NMR (CDC10 15.47, 28.23, 30.09,
31.48, 52.06, 52.54, 55.81, 80.82, 98.06, 109.38,
116.66, 119.31, 131.52, 147.23, 152.31, 166.57,
172.58; m/ z (FAB) 413 (M+1).
C. HC1-4-amino-3-methoxybenzoyl methionine methyl
ester
N-BOC-4-amino-3-methoxybenzoyl methionine
methyl ester (0.71 g, 1.79 mmol) was taken up in
CH2C12 (4 ml) and precipitated with 3-4M HC1/ Et20
(12 m1). The precipitate was washed as usual with
Et20 and dried overnight under vacuum to result in
0.55 g (88.3) of reddish material. mp 176-177 C;
NMR (CD30D) 2.08 (3H, s), 2.21 (2H, m), 2.61
(2H, m), 3.74 (3H, s), 4.02 (3H, s), 4.79 (1H, m),
7.50 (1H, d, J=8.2 Hz), 7.57 (1H, d, J=4.1 Hz)
7.67 (1H, s); 13C NMR (CD30D) 15.26, 31.34, 31.42,
52.95, 53.38, 57.12, 112.29, 121.43, 124.57,
124.77, 136.15, 153.67: 168.79, 173.81.
D. N-B0C-S-trityl-cysteine-4-amino-3-
methoxybenzoyl methionine methyl ester
N-B0C-S-trityl-cysteine (0.76 g, 1.74 mmol)
in dry THF (27.5 ml) was reacted with Et3N (0.24
ml), IBCF (0.23 ml, 1.74 mmol) at -10 C as
described above. HC1-4-amino-3-methoxybenzoyl
methionine methyl ester (0.55 g, 1.58 mmol) in
dry CH2C12 ( 8.7 ml) with Et3N (0.30 ml) was added
to the mixture and was allowed to stir overnight
under nitrogen. It was worked up as described for
N-BOC-S-trityl-cysteine-4-aminobenzoyl methionine
methyl ester in Example 1, and the crude material
was chromatographed on silica gel using a 2:1
mixture of hexanes and ethyl acetate to give 0.18
g (15.2 5) of pure product. 1H NMR (CDC10 1.45
(9H, s), 2.05-2.33 (5H, m), 2.57-2.65 (2H, m),
- 58 -
CA 02207252 1997-06-06
WO 96/21456
PCT/US96/01559
2.68-2.72 (1H, m), 2.75-2.96 (1H, m), 3.78 (3H,
s), 3.84 (3H, s), 4.90-5.00 (1H, m), 5.03-5.18
(1H, m), 7.17-7.48 (17H, m), 8.30-8.38 (1H, m),
8.65 (1H, br s).
E. TFA-Cysteine.4-amino-3-methoxybenzoyl
methionine FTI-261
N-B0C-S-trityl-cysteine-4-amino-3-
methoxybenzoyl methionine methyl ester (0.18 g,
0.24 mmol) was_deprotected with LiOH at room
temperature as described above to give the free
acid. The free acid was then further deprotected
in CH2C12 (1.2 ml) with Et3SiH (0.04 ml, 0.24 mmol)
and TFA (1.2 ml). The product was worked up as
described for HC1-cysteine-4-aminobenzoyl
methionine in Example 1, and HPLC revealed that
the product was impure. The crude material was
then purified on the HPLC using 0.196 TFA in water
and acetonitrile as eluting solvents to result in
two pure samples that were expected to be
diastereomers. The major component (determined
according to the major compound in the HPLC trace)
was characterized as follows. mp sub 109 C,
decomp 191-193 C; [a]253)=-30.00 (c=1, H20) ,
[a]250=+19.0 (c=1, H20); 11-1 NMR (CD30D) 2.10 (3H,
s), 2.12-2.18 (1H, m), 2.20-2.32 (1H, m), 2.53-
2.71 (2H, m), 3.00 (1H, dd, J=14.6, 7.5), 3.15
(1H, dd, J=14.58, 4.8), 4.77 (1H, dd, J=7.5, 4.8),
7.50 (1H, d, J=8.4 Hz), 7.56 (1H, s), 8.23 (1H, d,
J=8.4 Hz); 13C NMR (CD30D) 15.20, 26.65, 31.60,
31.76, 53.27, 56.58, 56.76, 111.04, 121.08,
122.14, 130.85, 131.85, 150.88, 167.21, 169.61,
175.36; m/ z (FAB) for free amine, 402 (M+1).
- 59 -
'
CA 02207252 1997-06-06
WO 96/21456
PCUUS96/01559
EXAMPLE 4
SYNTHESIS OF FTI-272
A. 4-nitro-2-phenyltoluene
2-bromo-4-nitrotoluene (2.16 g, 10.00 mmol)
and phenyl boric acid (1.46 g, 12.00 mmol) were
dissolved into anhydrous DMF (25 ml) under
nitrogen. To this mixture was added Pd(Ph3P)4
(0.58 g, 5P6). The mixture was heated at 100 C
overnight. The solution was poured onto 1N HC1
and extracted with Et20. The crude material was
chromatographed on silica gel using hexanes as an
eluent. After recrystallization from ethanol,
1.23 g (57.69s) of pale orange needles were
obtained. mp 69 - 71 C; NMR (CDC13) 2.36 (3H,
s), 7.29-7.40 (2H, m), 7.41-7.49 (5H,m), 8.07-8.10
(2H, m); 13C NMR (CDC10 20.68, 121.96, 124.51,
127.78, 128.41, 128.83, 131.06, 139.44, 142.97,
143.48, 146.05; anal calc. for Ci3HuNO2, C:73.26,
H:5.20, N:6.57; found, C:73.10, H:5.12, N:6.50; m/
z (El) 213; HRMS calc. for Ci3H11NO2, 213.0790;
found, 213.0793.
B. 4-nitro-2-phenylbenzoic acid
4-nitro-2-phenyltoluene (0.50 g, 2.34 mmol)
was dissolved in water (4.6 ml) and pyridine (2.3
ml). The mixture was heated to ref lux and KMn04
(1.85 g, 11.70 mmol) was added. The reaction
mixture was heated overnight and the solution was
filtered and washed several times with boiling
water. The aqueous solution was made acidic and
the product was extracted into ethyl acetate. The
ethyl acetate was dried over Na2SO4 and the solvent
removed in vacuo to result in 0.37 g (67.9 1) of
pure yellow product. mp 174-176 C; 111 NMR (CD30D)
7.38-7.48 (5H, m), 7.96 (1H, d, J=8.5 Hz), 8.21
(1H, d, J=2.3 Hz), 8.28 (1H, dd, J=8.48, 2.37); 13C
NMR (CD30D) 122.95, 126.09, 129.27, 129.42, 129.49,
- 60 -
CA 02207252 1997-06-06
WC0915/21456
PCT/US96/01559
131.56, 139.26, 140.42, 144.41, 150.17, 170.52; m/
z (El) 243 (M).
C. 4-nitro-2-phenylbenzoyl methionine methyl
ester
4-nitro-2-phenylbenzoic acid (0.30 g, 1.23
mmol), methionine methyl ester hydrochloride salt
(0.27 g, 1.35 mmol), EDCI (0.26 g, 1.35 mmol),
HOBT ( 0.18 g, 1.35 mmol) and Et3N (0.19 ml) in dry
CH2C12 (4.9 ml) were reacted according to the above
procedure and worked up as described for N-BOC-4-
aminobenzoyl methionine methyl ester in Example 1.
After recrystallization from ethyl acetate and
hexanes, 0.41 g (85.5%) of pure product was
isolated. mp 98-101 C; 1H NMR (CDC10 1.62-1.73
(14, m), 1.79-1.88 (1H, m), 1.91 (3H, s), 1.99
(2H, t, 3=7.2 Hz), 3.59 (3H, s), 4.53 (1H, m),
6.45 (1H, d, 3=7.8 Hz), 7.33-7.40 (5H, m), 7.67
(1H, d, 3=8.3 Hz), 8.07-8.12 (2H, m); 13C NMR
(CDC10 14.92, 29.11, 30.67, 51.51, 52.29, 121.86,
124.74, 128.27, 128.60, 128.69, 129.52, 137.50,
140.56, 141.02, 148.09, 167.23, 171.23; m/ z
(FAB), 389 (M+1).
D. 4-amino-2-phenylbenzoyl methionine methyl
ester
4-nitro-2-phenylbenzoyl methionine methyl
ester (0.35 g, 0.90 mmol) was taken up in ethyl
acetate (9.0 ml). To this mixture was added
SnC12-2H20 (1.02 g, 4.50 mmol) and the reaction was
heated under nitrogen at ref lux for lh. The
mixture was poured onto ice, the solution was made
basic using NaHCO3 and the product was extracted
into ethyl acetate several times (7-8). The ethyl
acetate fractions were combined washed with brine
and dried over Na2SO4 and the solvent was removed
in vacuo to give 0.24 g (73.4%) of yellow solid.
- 61 -
CA 02207252 1997-06-06
WO 96/21456
PCT/US96/01559
NMR (CDC13) 1.58-1.70 (1H, m), 1.80-1.92 (1H,
m), 1.98 (3H, s), 2.06 (2H, t, J=7.7 Hz), 3.62
(3H, s), 4.00 (2H, br s), 4.56-4.63 (1H, m), 5.84
(1H, d, J=7.7 Hz), 6.50 (1H, s), 6.61 (1H, d,
J=8.4 Hz), 7.29-7.42 (5H, m), 7.58 (1H, d, J=8.3
Hz); 13C NMR (CDC10 15.02, 29.25, 31.25, 51.57,
52.15, 113.27, 115.88, 123.52, 127.56, 128.37,
128.44, 130.92, 140.66, 141.44, 148.53, 168.58,
171.91.
E. N-BOC-S-trityl-oysteine-4-amino-2-
phenylbenzoyl methionine methyl ester
N-BOC-S-trityl-cysteine (0.31 g, 0.66 mmol)
in dry THF (11 ml) was reacted with Et2N (0.10 ml),
IBCF (0.09 ml, 0.73 mmol) at -10 C as described
for N-B0C-S-trityl-cysteine-4-aminobenzoyl
methionine methyl ester in Example 1. 4-amino-2-
phenylbenzoyl methionine methyl ester (0.24g, 0.66
mmol) in dry CH2C12 (3.5 ml) was added and the
mixture was allowed to stir overnight under
nitrogen. It was worked up as described as
further described for N-B0C-S-trityl-cysteine-4-
aminobenzoyl methionine methyl ester in Example 1,
and after drying the crude material was
chromatographed on silica gel using a 2:1 mixture
of hexanes and ethyl acetate to give 84.70 mg
(16.096) of pure product. mp 100-103 C; 11.1 NMR
(CDC10 1.41 (9H,$), 1.61-1.78 (1H, m), 1.84-1.95
(1H, m), 2.00 (3H, s), 2.05 (2H, t, J=7.6 Hz),
2.63 (1H, dd, J=12.7 Hz, 6.9 Hz), 2.72 (1H, dd,
J=12.7 Hz, 5.51 Hz), 3.64 (3H, s), 4.02 (1H, br
s), 4.58-4.63 (1H, m), 4.90 (1H, d, J=7.4 Hz),
6.10 (1H, d, J=6.6 Hz), 7.18-7.30 (10H, m), 7.37-
7.44 (11H, m), 7.50 (11-1, s), 7.58 (1H, d, J=8.2
Hz), 8.69 (1H, s); 13C NMR (CDC10 15.21, 28.20,
29.38, 31.24, 33.00, 51.77, 52.35, 54.15, 67.30,
80.85, 118.18, 120.86, 126.88, 127.90, 128.03,
- 62 -
CA 02207252 1997-06-06
W096/21456
PCT/US96/01559
128.56, 128.66, 129.44, 129.79, 130.14, 156.00,
168.52, 169.11, 171.85.
F. TFA-Cysteine-4-amino-2-phenylbenzoyl
methionine FTI-272
N-B0C-S-trityl-cysteine-4-amino-2-
phenylbenzoyl methionine methyl ester (84.70 mg,
0.11 mmol) of was taken up in THF (0.62 ml) and to
this was added 0.5 M LiOH (0.32 ml) at 0 C. The
mixture was stirred at 0 C for 35 minutes. The
solvent was removed in vacuo using a cold water
bath on the rotovap. The residue was worked up as
described for HC1-cysteine-4-aminobenzoyl
methionine in Example 1, and 60 mg of the free
acid was obtained. This was then dissolved into
CH2C12 (0.8 ml) and EtSiH (0.01 ml) was added
followed by TFA (0.8 ml). The reaction mixture
was stirred at room temperature for 1 h and worked
up as described for TFA.cysteine-4-amino-3-
methylbenzoyl methionine in Example 2. After
lyophilization, 0.0387 g (84.096) was_obtained.
HPLC revealed that no epimerization had occurred,
however the material was purified on the HPLC to
eliminate baseline impurities. mp 150-154 C;
[a] 25D=+21.5 (c=0.7, H20/ CH3OH) ; 111 NMR (CD30D)
1.62-1.79 (1H, m), 2.00-2.10 (5H, m), 2.16-2.18
(1H, m), 3.03 (1H, dd, J=14.7 Hz, 7.3 Hz), 3.15
(1H, dd, J=14.7 Hz, 4.8 Hz), 4.46 (1H, br s),
7.37-7.41 (5H, m), 7.52- 7.55 (1H, m), 7.65-7.67
(2H, m); 13C NMR (CD30D) 1.03, 26.35, 31.78,
32.79, 57.01, 119.40, 122.35, 128.95, 129.62,
129.71, 130.15, 133.49, 140.50, 141.36, 142.53,
167.05, 167.76, 172.51; anal. calc. for
C231126F3N306S2, C: 49.20, H: 4.67, N: 7.48; found, C:
49.14 H: 4.71, N: 7.42.
- 63 -
CA 02207252 1997-06-06
W096/21456
PCI7US96/01559
EXAMPLE 5
HC1-cysteine-4-amino-2-phenylbenzoyl methionine
methyl ester FTI274
N-B0C-S-trityl-cysteine-4-amino-2-
phenylbenzoyl methionine methyl ester (0.06 g,
0.075 mmol) was dissolved into methanol (2 ml) and
to it was added HgC12 (0.04 g) in methanol (1 ml).
The reaction was carried out as described above to
yield 15.7 mg of slightly impure compound by HPLC.
mp 130-132 C; NMR (CD30D) 1.72-1.84 (1H, m),
1.90-2.24 (6H, m), 3.05 (1H, dd, J=14.6 Hz, 8.5
Hz), 3.19 (1H, dd, J=14.6 Hz, 3.6 Hz), 3.69 (3H,
s), 4.22 (1H, dd, J=.5 Hz, 3.6 Hz), 4.48-4.53 (1H,
m), 7.33-7.43 (5H, m), 7.51 (1H, d, J=8.9 Hz),
7.70-7.72 (2H, m); 13C NMR (CD30D) 15.04, 26.36,
30.88, 31.36, 52.85, 53.05, 56.93, 119.42, 122.38,
128.88, 129.55, 129.73, 130.05, 133.17, 140.55,
141.32, 142.52, 166.92, 172.61, 173.58; anal.
calc. for C24H29C1N306S2-2H20, C: 51.20, H: 5.86, N:
8.14; found, C: 51.23 H: 5.60, N: 8.22.
EXAMPLE 6
SYNTHESIS OF FTI-275
A. 2-bromo-4-nitrobenzoic acid
2-bromo-4-nitrotoluene (5.00 g, 23.14 mmol)
was dissolved into pyridine ( 23 ml) and water (46
ml). The heterogeneous mixture was heated to 60
wC and KMn04 (18.29 g, 115.7 mmol) was added
carefully. The mixture was then heated under
ref lux overnight. The reaction mixture was
filtered and washed with boiling water. The
solution was then made acidic and extracted into
ethyl acetate, dried over Na2SO4, and the solvent
was removed in vacuo. A crude NMR revealed
remaining starting material so the solid was taken
up in NaOH and washed with hexanes. The aqueous
phase was made acidic and the product was
-64-
CA 02207252 1997-06-06
WO 96121456
PCT/US96/01559
extracted into ethyl acetate. The ethyl acetate
fractions were combined and dried over Na2SO4 and
the solvent was removed in vacuo to yield 3.72 g
(65.49.). mp 158-160 C; 1H NMR (CD30D) 7.81 (1H, d,
J=8.5 Hz), 8.08 (1H, d, J=8.5 Hz), 8.30 (1H, s);
13C NMR (CD30D) 121.96, 122.75, 129.36, 132.24,
139.52, 149.54, 167.75; anal. calc. for C7H4BrN04 +
0.1 ethyl acetate, C: 34.88, H: 1.90, N: 5.50;
found, C: 34.68, H: 1.86, N: 5.82.
B. 3,5-dimethylphenyl boronic acid
Mg turnings (1.44 g, 59:43 mmol) were covered
with dry THF (18.8 ml) in a dried, N2 filled flask
fitted with an addition funnel and ref lux
condenser. To this was added 5-bromo-m-xylene (10
g, 54.03 mmol) in THF (15 ml) after initiation of
the Grignard reaction. The addition was carried
out over several minutes and the reaction mixture
was heated at reflux for 1-2 h until most of the
Mg had reacted. The reaction mixture was then
cooled and transferred to an addition funnel
fitted to a N2 filled flask containing triisopropyl
borate (24.9 ml) at -70 C. The dropwise addition
was carried out over several minutes and the
mixture warmed to room temperature and stirred
overnight. The grey solution was poured onto 2 M
HC1 and immediately turned yellow. The solution
was extracted into Et20 and the Et20 fractions were
combined, dried over MgSO4 and the solvent was
removed in vacuo to yield 2.41 g (29.79d. mp 249-
251 C; 114 NMR (CDC10 2.44 (6H, s), 7.23 (1H, s),
7.84 (2H,'s); 13C NMR (CD30D) 21.36, 133.28,
134.39, 137.48.
C. 4-nitro-2-(3,5-dimethylphenyl)benzoic acid
2-bromo-4-nitrobenzoic acid (0.50 g, 2.03
mmol) and 3,5-dimethylphenyl boronic acid (0.34 g,
- 65 -
CA 02207252 1997-06-06
W096/21456
PCT/US96/01559
2.23 mmol) were dissolved into anhydrous DMF
(dimethylformamide) (25 ml) under nitrogen. To
this mixture was added Cs2CO3 (1.66 g, 5.08 mmol)
followed by Pd(Ph3P)4 (0.12 g, 59.). The mixture
was heated at 100 C overnight. The solution was
poured onto 1N HC1 and extracted into Et20. It was
dried over MgSO4 and the solvent was removed in
vacuo. The crude material was chromatographed on
silica gel using a 9:1 mixture of hexanes and
ethyl acetate to yield 0.34 g (61.7) of pure_
product. 1H NMR (CDC10 2.36 (6H, s), 6.99 (2H,
s), 7.07 (1H, s), 8.03 (1H, d, J=9.0 Hz), 8.23-
8.25 (2H, m); 13C NMR (CDC10 21.28, 121.68,
123.68, 125.74, 126.07, 130.22, 131.19, 131.31,
135.04, 138.21, 144.74, 170.75.
= D. 4-nitro-2-(3,5-dimethylphenyl)benzoyl
methionine methyl ester
4-nitro-2-(3,5-dimethylphenyl)benzoic acid
(0.15 g, 0.55 mmol), methionine methyl ester
_20 hydrochloride salt (0.11 g, 0.55 mmol), EDCI
(0.11 g, 0.55 mmol), HOBT (0.07 g, 0.55 mmol) and
Et3N (0.08 ml) in dry CH2C12 (2.2 ml) were reacted
and worked up according to the procedure for N-
BOC-4-aminobenzoyl methionine methyl ester in
25. Example 1. After recrystallization from ethyl
acetate and hexanes, 0.13 g (58.4Ps) of pure
product was isolated. mp 122-124 C; 114 NMR (CDC10
1.2-1.84 (1H, m), 1.85-1.97 (1H, m), 2.01 (3H, s),
2.05 (3H, t, J.7.7 Hz), 2.38 (6H, s), 3.70 (3H,
30 s), 4.67-4.74 (1H, m), 6.03 (1H, d, J=7.9 Hz),
7.05 (2H, s), 7.09 (1H, s), 7.84-7.87 (1H, m),
7.84-7.87 (1H, m), 8.23-8.26 (2H, m); 13C NMR
(CDC13), 15.20, 21.26, 29.22, 31.15, 51.79, 52.57,
122.07, 25.11, 126.27, 130.03, 130.53, 137.77,
35 138.82, 140.29, 141.56, 148.41, 167.14, 171.53.
- 66 -
CA 02207252 1997-06-06
V1/0915/21456
PCT/US96/01559
E. 4-amino-2-(3,5-dimethylphenyl)benzoyl
methionine methyl ester
4-nitro-2-(3,5-dimethylphenyl)benzoyl
methionine methyl ester (0.11 g, 0.26 mmol) was
taken up in ethyl acetate (3.0 ml). To this
mixture was added SnC12=2H20 (0.30 g, 1.30 mmol)
and the reaction was heated under nitrogen at
ref lux for 6h. The mixture was worked up as
described for 4-amino-2-phenylbenzoyl methionine
methyl ester in Example 2, to give 0.15 g of a
yellow film that was wet with solvent. The
material was otherwise pure by NMR and was used
without further purification. 1H NMR (CDC13) 1.60-
1.70 (1H, m), 1.80-1.90 (1H, m), 1.99 (3H, s),
2.05 (2H, t, J=7.6 Hz), 2.33 (6H, s), 3.64 (3H,
s), 3.93 (2H, br s), 4.61-4.64 (1H, m), 5.82 (1H,
d, J=7.7 Hz), 6.49 (1H, d, J=2.3 Hz), 6.62 (1H,
dd, J=8.4 Hz, 2.4 Hz), 6.98 (2H, s), 7.00 (1H, s),
7.65 (1H, d, J=8.3 Hz); 13C NMR (CDC12) 15.08,
21.17, 29.28, 31.49, 51.70, 52.18, 113.30, 115.94,
123.55, 126.36, 129.32, 131.23, 138.15, 140.72,
141.92, 148.40, 168.45, 172.01.
F. N-B0C-S-trityl-cysteine-4-amino-2-(3,5-
dimethylphenyl)benzoyl methionine methyl ester
4-amino-2-(3,5-dimethylphenyl)benzoyl
methionine methyl ester (0.10g, 0.26 mmol) was
dissolved into dry CH2C12 (1.4 ml) and it was
allowed to stand. In another flask, N-BOC-S-
trityl-Cys (0.12 g, 0.26 mmol) was dissolved into
THF (.4.4 ml) and was reacted with IBCF (0.03 ml)
and Et3N (0.04 ml) as described above. The product
was worked up as described for N-B0C-S-trityl-
cysteine-4-aminobenzoyl methionine methyl ester in
Example 1 and chromatographed on silica gel using
a 1:1 hexanes and ethyl acetate elution mixture to
give 0.12 g (56.0%) of pure material. 1H NMR
- 67 -
CA 02207252 1997-06-06
W096/21456
PCTMS96/01559
(CDC10 1.33 (9H, s), 1.61-1.68 (1H, m), 1.73-1.91
(4H, m), 1.96 (2H, t, J=7.6 Hz), 2.24 (6H, s),
2.57- 2.64 (2H, m), 3.57 (3H, s), 4.00 (1H, br s),
4.54-4.58 (1H, m), 5.84 (1H, d, J=7.8 Hz), 5.97
(1H, br d), 6.90 (1H, s), 6.92 (2H, s), 7.18-7.22
(9H, m), 7.27-7.40 (7H, m), 7.55 (1H, m), 7.61
(1H, m), 8.58 (1H, br s); 13C NMR (CDC10 15.11,
21.20, 27.79, 29.25, 31.28, 51.70, 52.28, 54.08,
60.32, 71.45, 80.75, 118.01, 120.80, 126.38,
126.82, 127.98, 129.41, 129.87, 130.22, 138.11,
139.18, 139.79, 141.06, 144.17, 168.38, 169.04,
171.82.
G. TFA-Cysteine-4-amino-2-(3,5-
dimethylphenyl)benzoyl methionine FTI275
N-B0C-S-trityl-cysteine-4-amino-2-(3,5-
dimethylphenyl)benzoyl methionine methyl ester
(0.12 g, 0.15 mmol) was placed into THF (0.9 ml)
and was reacted with 0.5 M of LiOH (0.6 ml) at 0 C
as described above, followed by deprotection with
=
TFA (1.5 ml) and Et3SiH (0.24 ml). Addition of
excess scavenger does not appear to affect the
result. The product was purified by preparative
HPLC to give 23.8 mg (27.3%). mp 135-138 C; 111 NMR
(CDC10 1.76-1.84 (1H, m), 2.00-2.17 (6H, m), 2.33
(6H, s), 3.05 (111, dd, J=14.6 Hz, 7.3 Hz), 3.17
(1H, dd, J=14.6 Hz, J=4.9 Hz), 4.15 (1H, dd,
J=7.3, 4.9 Hz), 4.45-4.48 (1H, m), 7.02 (3H, s),
7.53 (1H, d, J=8.0 Hz), 7.66 (2H, m); 13C (CD30D)
14.96, 21.51, 26.28, 30.91, 31.70, 53.03, 56.98,
119.27, 122.30, 127.52, 130.07, 130.57, 133.37,
139.28, 140.39, 141.29, 142.86, 166.89, 172.60,
174.81.
- 68
CA 02207252 1997-06-06
WO 96/21456
PCTIUS96/01559
EXAMPLE 7
SYNTHESIS OF FTI-266
A. 4-amino-1-naphthoic acid
4-amino-1-naphthalenecarbonitrile (1.50 g,
8.91 mmol) was dissolved into a 50% KOH solution
(18 ml). The heterogeneous solution was heated at
reflux for 9-1 ri=yc nnr",= the solution became
homogenous and TLC showed no more starting
material, the deep red solution was cooled and
poured over 200 ml of water. The solution was
then filtered and the acid was precipitated with
concentrated HC1. The red crystals were filtered
and the filtrate was ref iltered to give pink
crystals. The first fraction was treated with
activated carbon to remove some of the red color.
1.51 g (90.6%) of product was obtained. mp 169-
171 C; 11i NMR (CD30D) 6.69 (1H, d, J=8.2 Hz), 7.38-
7.43 (1H, m), 7.48-7.54 (1H, m), 8.03 (1H, d,
J=8.5 Hz), 8.13 (1H, d, J=8.2 Hz), 9.09 (1H, d,
J=8.5 Hz); 1.3C NMR (CD30D) 107.39, 114.61, 122.99,
123.92, 125.21, 127.40, 128.48, 135.04, 151.35,
171.44; HRMS calc. for C11H7NO2, 187.0633; found,
187.0642.
B. N-B0C-4-amino-1-naphthoic acid
4-amino-1-naphthoic acid (0.86 g, 4.61 mmol)
was dissolved into dioxane (9.2 ml) and 0.5 M NaOH
(9.2 ml). Di-t-butyl dicarbonate (1.11 g, 5.07
mmol) was added and the mixture was stirred
overnight. The reaction mixture was worked up as
described for N-B0C-4-aminobenzoic acid in Example
1 to give 0.76 g (56.7%) of reddish pink solid.
mp 194-195 C; NMR (CD30D) 1.56 (911, s), 7.53-
7.62 (2H, m), 7.79 (1H, d, J=8.1 Hz), 8.12 (1H, d,
J=8.0 Hz), 8.22 (1H, d, J=8.18 Hz), 9.02 (1H, d,
J=8.9 Hz); 13C NMR (CD30D), 26.68, 81.62, 119.06,
123.40, 124.57, 127.03, 127.37, 128.49, 128.77,
- 69 -
CA 02207252 1997-06-06
W096/21456
PCT/US96/01559
131.89, 133.76, 139.86, 155.95, 170.73; anal.
calc. for C371137N04, C: 66.90, H: 5.96, N: 4.88;
found C: 66.49, H: 6.08, N: 4.79; m/ z (El), 289;
HRMS calc. for C361137N04, 287.1158; found, 287.1151.
C. N-B0C-4-amino-1-naphthoyl methionine methyl
ester N-B0C-4-amino-1-naphthoic acid (0.46 g,
1.60 mmol), methionine methyl ester hydrochloride
(0.35 g, 1.76 mmol), EDCI (0.43 g, 1.76 mmol),
HOBT (0.24 g, 1.76 mmol) and Et3N ( 0.27 ml) in
CH2C12 (6.4 ml) were reacted as described for N-
BOC-4-aminobenzoyl methionine methyl ester in
Example 1. After workup and recrystallization
from ethyl acetate and hexanes, 0.44 g (63.6%) of
pale pink crystals were obtained. mp 131-132 C; 1H
NMR (CDC10 1.57 (9H, s), 2.11-2.21 (4H, m), 2.29-
2.41 (1H, m), 2.65 (2H, t, J=7.1 Hz), 3.83 (3H,
s), 4.99-5.06 (1H, m), 6.68 (1H, d, J=8.0), 7.02
(1H, s), 7.56-7.59 (2H, m), 7.69 (1H, d, J=7.9
Hz), 7.87-7.90 (1H, m), 8.02 (1H, d, J=7.9 Hz),
8.44-8.48 (1H, m); 2.3C NMR CDC13) 15.56, 28.31,
30.19, 31.65, 52.06, 52.64, 81.17, 115.82, 120.18,
125.79, 126.37, 126.53, 127.18, 131.02, 135.65,
152.93, 169.04, 172.40; HRMS calc. for C22H28N205S,
432.1719; found 432.1702; m/ z (FAB) 433 (M+1).
D. HC1-4-amino-1-naphthoyl methionine methyl
ester
N-B0C-4-amino-l-naphthoyl methionine methyl
ester (0.57 g, 1.31 mmol) was deprotected with
HC1/ ether to yield 0.31 g (64.1%) of white solid.
mp 178-181 C; 111 NMR (CD30D) 2.08-2.16 (41-1, m),
2.20-2.30 (1H, m), 2.57-2.75 (2H, m), 3.82 (3H,
s), 4.87-4.91 (1H, m), 7.59 (1H, d, J=7.5 Hz),
7.67 (1H, d, J=7.5 Hz), 7.71-7.80 (2H, m), 8.03
(1H, dd, J=7.1 Hz, 2.0 Hz), 8.35 (1H, dd, J=6.8
Hz, 1.8 Hz); 13C NMR (CD30D) 15.23, 31.40, 53.01,
- 70 -
CA 02207252 1997-06-06
V0)9101456
PCT/US96101559
53.33, 119.90, 122.20, 126.15, 127.41, 127.77,
129.09, 129.31, 131.50, 132.33, 135.64, 171.77,
173.83; m/ z (FAB), 369 (M+1).
E. N-B0C-S-trityl-cysteine-4-amino-1-naphthoyl
methionine methyl ester
N-B0C-S-trityl-Cys (0.31 g, 0.67 mmol) in dry
THF (11.2 ml) was reacted with Et3N (0.10 ml) and
IBCF (0.10 ml, 0.74 mmol) at -10 C as described
above. HC1.4-amino-1-naphthoyl methionine methyl
ester (0.25 g, 0.67 mmol) in dry CH2C12 (3.5 ml)
was added and the mixture was stirred overnight
under nitrogen. The mixture was worked up as
described for N-BOC-S-trityl-cysteine-4-
aminobenzoyl methionine methyl ester in Example 1,
and the crude material was chromatographed on
silica gel using a 2:1 mixture of hexanes and
ethyl acetate to give 0.20g (37.5 9s) of pure
product. III NMR (CDC13) 1.48 (9H, s), 2.10-2.20
(4H, m), 2.30-2.37 (1H, m), 2.63 (2H, t, J=7.4),
2.74 (1H, J=12.9 Hz, J=5.3 Hz), 2.90 (1H, J=12.9
Hz, 6.2 Hz), 3.81 (3H, s), 4.96-5.03 (2H, m), 6.77
(1H, d, J=8.0 Hz), 7.18-7.33 (11H, m), 7.44-7.56
(7H, m), 7.60 (1H, d, J=7.7 Hz), 7.88 (1H, d,
J=8.0 Hz), 8.00 (1H, d, J=7.1 Hz), 8.37 (1H, d,
J=8.4 Hz), 8.94 (1H, br s); 13C NMR (CDC10 15.23,
26.52, 31.41, 31.50, 52.98, 53.31, 56.79, 68.15,
122.52, 123.54, 126.16, 126.99, 128.03, 128.39,
129.52, 132.30, 134.04, 135.24, 168.08, 172.38,
173.94.
F. TFA-cysteine-4-amino-l-naphthoyl methionine,
FTI-270
N-B0C-S-trityl-cysteine-4-amino-l-naphthoyl
methionine methyl ester (83.3 mg, 0.11 mmol) was
taken up in THF (0.7 ml) and to this mixture was
added 0.5 M LiOH (0.43 ml) at 0 C. The mixture
- 71 -
CA 02207252 1997-06-06
W096/21456
PCT/US96/01559
was stirred at 0 C for 35 minutes. The solvent
was removed in vacuo using a cold water bath. The
residue was worked up as described for
TKA.cysteine-4-amino-3-methylbenzoyl methionine in
Example 2, and 74.1 mg of the free acid was
obtained. This was then dissolved into CH2C12 (1
ml) and Et3SiH (0.015 ml) was added followed by TFA
(1 m1). The reaction mixture was stirred at room
temperature for 1 h and worked up as further
described for TFA-cysteine-4-amino-3-methylbenzoyl
methionine in Example 2. After lyophilization,
42.4 mg of crude material waa obtained which was
then purified on the HPLC using 0.19s TFA in water
and acetonitrile. mp 121-125 C; [cd251,=+2.4
(c=0.8, H20); 11-1 NMR (CD30D) 2.03-2.13 (4H, m),
2.22-2.36 (1H, m), 2.59-2.74 (2H, m), 3.16-3.33
(2H, m), 4.42 (1H, m), 4.84-4.89 (1H, m), 7.57-
7.61 (2H, m), 7.64 (1H, d, J=7.7 Hz), 7.70 (1H, d,
J=7.7 Hz), 8.08-8.11 (1H, m), 8.29-8.32 (1H, m),
8.98 (1H, d, J=7.7 Hz); 13C NMR (CD30D) 15.19,
26.45, 31.50, 31.63, 53.20, 56.72, 122.52, 123.43,
126.43, 126.12, 127.02, 127.96, 128.32, 129.49,
132.27, 134.15, 135.12, 168.11, 172.41, 175.17;
anal. calc. for C23.1123F3N306S2, C: 47.19, H: 4.34, N:
7.86; found, C: 46.53, H: 4.56, N: 7.59; Note:
difference for C is 0.65.
G. HC1-cysteine-4-amino-1-naphthoyl methionine
methyl ester FTI-270-11C1
TFA-cysteine-4-amino-1-naphthoyl methionine
(0.12 g, 0.15 mmol) was dissolved in CH3OH (4.3
ml). To this solution was added a solution of
HgC12 (0.23 g, 0.86 mmol) in CH3OH (4.3 ml). The
procedure was continued as described above and
after HC1/ Et20 precipitation and several
reprecipitations 31.0 mg (18.3 9s) of pure white
material was obtained. mp sub 137 C, decomp 214-
- 72 -
CA 02207252 1997-06-06
V1/096/21456
PCT/US96/01559
215 C; [a)25D=-32.00 (c=1 CH3OH); NMR (CD30D) 2.12
(3H, s), 2.21-2.28 (1H, m), 2.57-2.73 (3H, m),
3.20-3.34 (2H, m), 3.82 (3H, s), 4.39-4.43 (1H,
m), 7.61-7.68 (3H, m), 7.78 (1H, d, J=7.7 Hz),
8.13-8.16 (1H, m), 8.28-8.32 (1H, m); 13C (CD30D)
15.23, 26.52, 31.41, 31.50, 52.98, 53.31, 56.79,
122.52, 123.54, 126.16, 126.99, 128.03, 128.39,
129.52, 132.30, 134.04, 135.24, 168.08, 172.38,
173.94.
EXAMPLE 8
SYNTHESIS OF FTI-254
A. N-Boc-S-trityl cysteinal
Triethylamine (2.22 mL, 16 mmoL) and N,0-
dimethylhydroxylamine hydrochloride (1.57 g, 16.1
mmol) were added to a solution of N-Boc-S-trityl
cysteine (7.44 g, 16 mmol) in 85 mL of methylene
chloride. This mixture was cooled in an ice bath
and 1-(3-dimethylaminopropy1)-3-ethylcarbodiimide
hydrochloride (EDCI, 3.08 g, 16.0 mmol) and HOBT
(2.17 g, 16 mmol) was added. The mixture was
stirred at 0 C for 1 hr and at room temperature
for a further 10 hr. The mixture was extracted
with methylene chloride and 0.5 N HC1. The
organic layer was washed consecutively with 0.5 N
HC1, concentrated NaHCO3 and brine. The organic
layer was dried and evaporated. The residue was
purified by flash column chromatography (1.5 : 1 =
hexane : ethylacetate) to give a white foam (7.40
g, 919). m.p. 59-60 C (decomp). 11.1 NMR (CDC10 6
7.41 (m, 6H), 7.20-7.31 (m, 9H), 5.13 (d, 8.9 Hz,
1H), 4.76 (br s, 1H), 3.64 (s, 3H), 3.15 (s, 3H),
2.56 (dd, 4.7 and 12.1 Hz, 1H), 2.39 (dd, 7.8 and
12.1 Hz, 1H), 1.43 (s, 9H). 13C NMR (CDC10 6
170.7, 154.9, 144.2, 129.3, 127.6, 126.4, 79.3,
66.4, 61.2, 49.5, 33.8, 31.8, 28.1. This
carboxyamide (2.02 g, 4.0 mmol) was dissolved in
- 73 -
CA 02207252 1997-06-06
W096/21456
PCT/US96/01559
30 mL of ether and cooled to -10 C. Lithium
aluminum hydride (167 mg, 4.40 mmol) was added and
the mixture was stirred for 15 min under the
nitrogen. Then 40 mL of 0.5 N HC1 was added and
the solution was extracted with ether. The ether
layer was washed with 0.5 N HC1 and dried. The
evaporation of solvents gave a white foam (1.80 g)
which was used for further reaction without
purification. The '11 NMR spectrum of this compound
was complex. The percentage of the aldehyde was
about 65-70%, which was calculated according to
the integration of the sharp singlet (6 9.17) and
the trityl peak (6 7.40, m, 6H; 7.28, m, 9H).
Lowering the temperature to -45 C did not improve
the aldehyde percentage.
B. 4-N- [2 (R) -tert-Butoxycarbonylainino-3-
triphenylmethyithiopropyl] aminobenzoyl methionine
methyl ester.
One equivalent of N-Boc-S-trityl cysteinal in
10 mL of methanol was added to a solution of 4-
aminobenzoyl methionine methyl ester hydrochloride
(1.7836 g, 5.6 mmol) in 60 mL of methanol and 4 mL
of glacial acetic acid. Sodium cyanoboronhydride
(0.528 g, 8.40 mmol) was added to this deep
colored solution at 0 C. The mixture was stirred
at room temperature for 15 hr. After the
evaporation of solvents, the residue was extracted
with ethyl acetate and concentrated sodium
bicarbonate. The organic phase was dried and the
solvents were evaporated. The residue was purified
through flash column chromatography (ethyl acetate
/ hexane = 1:1) to give a pure desired product
(2.52 g, 65%). 1I1 NMR (CDC13) 6 7.63 (d, 8.6 Hz,
2H), 7.43 (m, 6H), 7.21-7.32 (m, 9H), 6.73 (d, 7.6
Hz, 1H, Met amide), 6.50 (d, 8.6 Hz, 2H), 4.91
(ddd, 5.1 Hz, 5.3 Hz and 7.6 Hz, 1H, Met a H),
- 74 -
CA 02207252 1997-06-06
111396/21456
PCT1US96101559
4.59 (d, 8.9 Hz, 1H, Boc amide), 4.25-4.40 (br,
1H, NHPh), 3.80 (m, 1H, Cys H), 3.78 (s, 3H,
OCH3), 3.09 (d, 6.3 Hz, 2H, CH2NH), 2.55-2.60 (m,
2H, CH2SCPh3), 2.46 (d, 5.0 Hz, 2H, CH2SCH3), 2.23-
*
2.28 (m, 1H, Met CH2), 2.07-2.12 (m, 1H, Met CH2),
2.09 (s, 3H, SCH3), 1.43 (s, 9H, Boc).
C. 4-N-(2(R)-Amino-3-mercaptopropyl]aminobenzoyl
methionine methyl ester.
The fully protected 4-N-[2(R)-tert-
Butoxycarbonylamino-3 triphenylmethylthiopropyl]
amino-benzoyl methionine methyl ester (1.31 g,
1.83 mmol) was dissolved into 20 mL of methanol.
To this solution wad added mercuric chloride (1.09
g, 4.04 mmol) in 5 mL of methanol. The mixture
was ref luxed for 20 min and then cooled down. The
clear solution was removed and the solid
precipitate was washed with 5 mL of methanol. The
solid was dried and then suspended in 15 mL of
methanol. The suspension was stirred and reacted
with hydrogen sulfide gas for 1 hr. The black
precipitate was removed by centrifugation. The
clear solution was evaporated to dryness. The
residue was dissolved in 6 mL of methylene
chloride followed by the addition of 20 mL of 3N
HCl in ether. The white precipitate was filtered
and dried to give a hydrochloride salt of the
desired product (0.60 g, 73%). NMR (CD30D)
7.73 (d, 8.8 Hz, 2H), 6.75 (d, 8.8 Hz, 2H), 4.74
(dd, 4.9 Hz and 4.3 Hz, 1H, Met a H), 3.72 (s, 3H,
OCH3), 3.43-3.59 (m, 3H, CH2NH and Cys a H), 2.93
(dd, 3.9 Hz and 14.4 Hz, 1H, CH2SH), 2.81 (dd, 5.2
Hz and 14.5 Hz, 1H, CH2SH), 2.49-2.66 (m, 2H,
CH2SCH3), 2.07-2.20 (m, 2H, Met CH2), 2.10 S, 3H,
sCH3).
- 75 -
CA 02207252 1997-06-06
W096/21456
PCT/US96/01559
D. 4-N-(2(R)-Amino-3-mercaptopropyl]aminobenzoyl
methionine
The fully protected peptide 4-N-[2(R)-tert-
Butoxycarbonylamino-3-triphenylmethylthiopropy1)-
aminobenzoyl methionine methyl ester (567 mg, 0.79
mmol) was dissolved into 3.0 mL of 0.5 N lithium
hydroxide and 3.0 mL of tetrahydrofuran. The
mixture was stirred at 0 C for 1 hr. After the
evaporation of solvents, the residue was dissolved
in water and extracted with methylene chloride and
1N hydrochloric acid. The organic phase was dried
and the solvents were evaporated. The residue was
dissolved in a mixture of lmL of methylene
chloride and 2 mL of trifluoroacetic acid.
Triethylsilane was added dropwise until the deep
brown color disappeared. The mixture was kept at
rt for 1 hr. The solvents were evaporated and the
residue was dried. This residue was dissolved in 1
mL of 1.7N HC1 in acetic acid followed by the
addition of 20 mL of 3N HCl in ether. The white
precipitate was filtered and dried to give a
hydrochloride salt of the desired product (159 mg,
4690. Analytical HPLC showed purity over 98%. 1H
NMR (CD30D) (5 7.74 (d, 8.7 Hz, 2H), 6.75 (d, 8.7
Hz, 2H), 4.73 (dd, 4.5 Hz and 4.7 Hz, 1H, Met a
H), 3.45-3.58 (m, 3H, CH2NH and Cys a H), 2.93 (dd,
4.5 Hz and 14.6 Hz, 1 H, CH2SH), 2.80 (dd, 5.3 Hz
and 14.6 Hz; 1H, CH2SH), 2.53-2.64 (m, 2H,
CH2SCH3), 2.15-2.23 (m, 1H, Met CHO, 2.07-2.13 (m,
1H, Met CHO, 2.10 (s, 3H, SCH3).
EXAMPLE 9
Synthesis of FTI-284
A. 4-Nitro-2-phenylbenzoy1-[1'(5)-
methoxycarbony1-3'-methylsulfonyl]propyl amide
4-nitro-2-phenylbenzoyl methionine methyl
ester (525 mg, 1.28 mmol), N-methylmorpholine
- 76 -
CA 02207252 1997-06-06
114396/21456
PCT1US96101559
oxide (453 mg, 3.87 mmol) and 0.5 mL of osmium
tetroxide (2.5 wt.% solution in tert-butanol) were
added to a mixture of 40 mL of acetone and 10 mL
of water. The mixture was stirred at rt
overnight. After the addition of excess sodium
sulfite, the reaction mixture was extracted with
ethyl acetate and washed with concentrated sodium
bicarbonate. The organic phase was dried and the
solvents were evaporated to give a solid (570 mg,
100%). 1H NMR (CDC10 5 8.29 (d, 7.7 Hz, 1H), 8.25
(s, 1H), 7.83 (d, 7.7 Hz, IH), 7.43-7.55 (m, 5H),
6.15 (d, 7.3 Hz, 1H, Met amide), 4.68 (ddd, 5.0
Hz, 5.1 Hz and 7.3 Hz, 1H, Met a H), 3.70 (s, 3H,
OCH3) , 2.85 (s, 3H, SCHO, 2.69-2.81 (m, IH,
CH2S02), 2.58-2.66 (m, 1H, CH2S02) , 2.21-2.33 (m,
1H, Met CHO, 1.96-2.08 (m, 1H, Met CHO.
B. 4-N-[2(R)-tert-Butoxycarbonylamino-3-
triphenylmethylthiopropyl]amino-2-phenylbenzoyl-
(1'(8)-methoxycarbony1-3'-methylsulfonyl]propyl
amide The_4-Nitro-2-phenylbenzoy1-[1'(S)-
methoxycarbony1-31-methylsulfonyl]propyl amide
(430 mg, 1.02 mmol) was dissolved in 20 mL of
methanol. A catalytic amount of 5% palladium on
carbon was added and the mixture was hydrogenated
at 40 PSI for 1.5 hr. The mixture was filtered and
the filtrate was evaporated to dryness to give 4-
amino product (400 mg, 100%). 1.14 NMR (CD30D) (5 7.70
(d, 8.0 Hz, 1H), 7.38-7.47 (m, 7H), 4.53 (dd, 4.6
Hz and 4.8 Hz, 1H, Met a H), 3.72 (s, 3H, OCHO ,
2.89.(s, 3H, SO2CH3), 2.79-2.85 (m, 1H, CH2S02),
2.58-2.68 (m, 1H, CH2S02) , 2.19-2.29 (m, 1H, Met
CH2), 1.93-2.04 (m, 1H, Met CHO . This amine was
dissolved in 15 mL of methanol and 0.8 mL of
acetic acid. One equivalent of N-Boc-S-trityl
cysteinal was added followed by the addition of
sodium cyanoboronhydride (97 mg, 1.5 eq). The
- 77 -
CA 02207252 1997-06-06
W096/21456 PCT/US96/01559
mixture was stirred at rt overnight. After the
evaporation of solvents, the residue was extracted
with ethyl acetate and concentrated sodium
bicarbonate. The organic phase was dried and
=
solvents were evaporated. The residue was
purified through flash column chromatography
(ethyl acetate / hexane / methanol = 15:15:2) to
give a pure product (500 mg, 60%). 'H NMR (CDC10 6
7.64 (d, 8.5 Hz, 1H), 7.37-7.46 (m, 11H), 7.18-
7.33(m, 9H), 6.53 (d, 8.5 Hz, 1H), 6.34 (s, 1H),
5.74 (d, 7.5 Hz, 1H, Met amide), 4.64 (ddd, 4.9
Hz, 5.1 Hz and 7.5 Hz, 1H, Met a H), 4.55 (d, 7.5
Hz, 111, Boc amide), 4.26 (br, 1H, NHPh), 3.79 (In,
1H, Cys a H), 3.68 (s, 3H, OCH3), 3.10 (t, 5.7 Hz,
2H, CH2NHPh), 2.84 (s, 3H, SO2CH3) , 2.62-2.82 (m,
2H, CH2S02) , 2.45 (d, 2H, CH2SCPh3) , 2.19-2.27 (m,
1H, Met CH2), 1.84-1.95 (m, 1H, Met CH2), 1.41 (s,
9H).
C. 4-N-[2(R)-Amino-3-mercaptopropy1]amino-2-
=
phenylbenzoy1-(1'(8)-methoxycarbonyl-3'-
methylsulfonul]propyl amide, FTI-284
The fully protected peptide 4-N-[2(R)-tert-
Butoxycarbonylamino-3-triphenylmethyl-
thiopropyllamino-2-phenylbenzoy1-[1'(S)-
methoxycarbony1-3'-methylsulfonyl]propyl amide (277
mg, 0.33 mmol) was dissolved into 5 mL of
methanol. To this solution was added mercuric
chloride (229 mg, 2.50 eq) in 2 mL of methanol.
The mixture was ref luxed for 20 min. The
precipitate was dried and then suspended in 10 mL
of methanol. This mixture was reacted with
hydrogen sulfide gas. The reaction mixture was
centrifuged and the clear solution was evaporated.
The residue was dissolved in 2 mL of methylene
chloride followed by the addition of 20 mL of 3N
HC1 in ether. The white precipitate was collected
- 78
CA 02207252 1997-06-06
W096/21456
PCTMS96/01559
and dried to give a hydrochloride salt of the
desired product (165 mg, 89%). 114 NMR (CD30D) 6
7.44 (d, 8.4 Hz, 1H), 7.32-7.40 (m, 5H), 6.77 (d,
8.4 Hz, 1H), 6.68 (s, 1H), 4.45 (dd, 4.5 Hz and
4.7 Hz, 1H, Met a H), 3.69 (s, 3H, OCH3), 3.40-3.57
(m, 3H, CH2NHPh and Cys a H), 2.78-2.96 (m, 3H,
CH2SH and CH2S02), 2.89 (S, 3H, SO2CH3), 2.60-2.69
(m, 1H, CH2S02), 2.15-2.24 (m, 1H, Met CH2), 1.91-
2.02 (m, 1H, Met CH2) .
EXAMPLE 10
Synthesis of FTI-277
A. 4-N-[2(R)-tert-Butoxycarbony1-3-
tripheny].methylthiopropyl]amino-2-phenylbenzoyl
methionine methyl ester =
. The coupling of 4-amino-2-phenylbenzoyl
methionine methyl ester (3.88 g, 10 mmol) with one
equivalent of N-Boc-S-trityl cysteinal in the
presence of 1.5 equivalent of sodium
cyanoboronhydride gave a crude mixture which was
purified through flash column chromatography
(ethyl acetate / hexane = 1:1) to give a pure
desired product (5.83 g, 74%). 1H NMR (CDC10 6
7.65 (d, 8.5 Hz, 1H), 7.32-7.45 (m, 11H), 7.18-
7.30 (m, 9H), 6.50 (d, 8.5 Hz, 111), 6.33 (s, 1H),
5.65 (d, 7.6 Hz, 1H, Met amide), 4.62 (ddd, 5.0
Hz, 5.2 Hz and 7.6 Hz, 1H, Met a H), 4.54 (d, 8.1
Hz,. Boc amide), 4.18 (br, 1H, NHPh), 3.78 (m, 1H,
Cys a H), 3.64 (s, 3H, CHO, 3.10 (t, 6.1 Hz, 2H,
CH2NHPh), 2.45 (d, 5.0 Hz, 2H, CH2SCPh3), 2.04-2.10
(m, 2H, CH2SCH3), 2.00 (s, 3H, SCHO, 1.81-1.92 (m,
1H, Met CH2), 1.61-1.70 (m, 1H, Met CH2), 1.40 (s,
9H). 13C NMR (CDC10 6 172.0, 168.3, 155.7, 149.4,
144.3, 141.6, 141.1, 131.3, 129.5, 128.7, 128.5,
127.9, 127.7, 126.8, 122.6, 113.6, 111.3, 79.8,
67.1, 52.2, 51.7, 49.5, 47.2, 34.3, 31.6, 29.4,
28.3, 15.2.
- 79 -
CA 02207252 1997-06-06
W096/21456
PCT/US96/01559
B. 4-N-[2(R)-Amino-3-mercaptopropyl]amino-2-
phenylbenzoyl methionine methyl ester, FTI-277.
The fully protected peptide 4-N-[2(R)-tert-
Butoxycarbony1-3-triphenylmethyl-thiopropyl]amino-
2-phenylbenzoyl methionine methyl ester(1.57 g,
2.0 mmol) was first reacted with mercuric chloride
(1.36 g, 5.0 mmol) and then reacted with hydrogen
sulfide gas in methanol to give a hydrochloride
salt of the desired product (0.808 g, 84%).
Analytical HPLC showed_purity over 98%. [a]251) = -
12.10 (c=0.008, CH3OH) . 214 NMR (CD30D) 6 7.42 (d,
8.3 Hz, 1H), 7.30-7.38 (m, 5H), 6.78 (d, 8.3 Hz,
1H), 6.71 (s, 1H), 4.47 (dd, 4.2 Hz and 5.1 Hz,
1H, Met a H), 3.68 (s, 3H, CHO, 3.44-3,54 (m, 3H,
CH2NHPh and Cys a H), 2.94 (dd, 4.1 Hz and 14.6 Hz,
1H, CH2SH) , 2.81 (dd, 5.0 Hz and 14.6 Hz, 1H,
CH2SH), 2.12-2.22 (m, 1H, CH2SCH3), 2.03-2.10 (m,
1H, CH2SCH3), 2.00 (s, 3H, SCHO, 1.90-1.97 (m, 1H,
Met CH2), 1.73-1.82 (m, 1H, Met CHO. 13C NMR
(CD30D) 6 173.7, 173.4, 150.7, 143.5, 142.3, 131.2,
129.8, 129.5, 128.6, 125.6, 115.6, 112.2, 53.7,
53.2, 52.8, 45.0, 31.3, 30.8, 25.3, 15Ø
EXAMPLE 11
Synthesis of FTI-276
4-N-[2(R)-Amino-3-mercaptopropyl] amino-2-
phenylbenzoyl methionine
The fully protected peptide 4-N-[2(R)-tert-
Butoxycarbony1-3-triphenylmethylthiopropy1]-amino-
2-phenylbenzoyl methionine methyl ester (2.36 g, 3
mmol) was first reacted with lithium hydroxide and
then with trifluoroacetic acid to give a crude
product (1.30 g, 77% yield, 85% purity shown by
HPLC) which was further purified through
preparative HPLC to give a pure product (0.98 g,
75%). kW% = -13.6 (c=0.005, CH3OH) . NMR
(CD30D) 6 7.44 (d, 8.4 Hz, 1H), 7.30-7.41 (m, 5H),
- 80 -
CA 02207252 1997-06-06
14,096)21406
PCTIUS96/01559
6.75 (d, 8.4 Hz, 1H), 6.68 (s, 1H), 4.43 (dd, 4.2
Hz and 5.1 Hz, 1H, Met a H), 3.44-3.58 (m, 3H,
CH2NHPh and Cys a H), 2.95 (dd, 4.4 Hz and 14.5 Hz,
1H, CH2SH), 2.83 (dd, 5.0 Hz and 14.5 Hz, 1H,
CH2SH), 2.14-2.23 (m, 1H, CH2SCH3), 2.05-2.11 (m,
1H, CH2SCH3), 2.00 (s, 3H, SCHO, 1.91-1.99 (m, 1H,
Met CHO, 1.72-1.81 (m, 1H, Met CH2). 13C NMR
(CD30D) 6 176.4, 173.5, 150.4, 143.0, 141.5, 131.0,
129.7, 129.4, 128.9, 124.6, 115.0, 112.2, 53.3,
44.4, 30,8, 30.1, 24.9, 14.8.
Other compounds of the invention (in
particular those of claims [[14-18) are
synthesizable by modifications of the procedure
described for the 2-phenyl-4-aminobenzoic acid
derivative of claim 3. In particular,
modifications of the Suzuki couping method will
allow the incorporation of an alkoxy-, chloro,
bromo or methyl substituted phenyl group onto the
4-aminobenzoic acid spacer. As with the
unsubstituted derivative, 4-nitro-2-bromotoluene
will be coupled with the corresponding substituted
phenyl boronic acid derivative (alkoxyphenyl or
chloro-, bromo- or methylphenylboronic acid) under
paladium catalyzed conditions. The appropriately
substituted 2-(substituted) phenyl-4-nitro toluene
derivative will be incorporated into the
peptidomimetic synthesis as described for the 2-
phenyl case.
In a similar way the precursor to the 2-
naphthyl-, 2-thiophene-, 2-pyrrole-, and 2-
pyridy1-4-aminobenzoic acid spacers can be
prepared by reaction of 4-nitro-2-bromotoluene
with naphthalene-2-boronic acid, thiophene-2-
boronic acid, pyrrole-2-boronic acid, pyridine-
2,3- or 4-boronic acid.
- 81 -
CA 02207252 1997-06-06
W096/21456
PCT/US96/01559
EXAMPLE 12
FTase and GGTase I Activity Assay
FTase and GGTase I activities from 60,000 X g
supernatants of human Burkitt lymphoma (Daudi)
cells (ATCC, Rockville, MD, USA) were assayed as
described previously for FTase (41). Briefly, 100
Ag of the supernatant was incubated in 50 mM Tris,
pH 7.5, 50 AM ZnC12, 20 mM KC1 and 1 mM
dithiothreitol (DTT). The reaction was incubated
at 30 C for 30 min with recombinant Ha-Ras-CVLS (11
AM) and PHIFPP (625 nM; 16.3 Ci/mmol) for FTase,
and recombinant Ha-Ras-CVLL (5 AM) and
[3H]geranylgeranylpyrophosphate (525 nM; 19.0
Ci/mmol) for GGTase I. The peptidomimetics were
mixed with FTase and GGTase before adding to the
reaction mixture.
EXAMPLE 13
Ras and RaplA Processing Assay
H-RasF cells (45) were seeded on day 0 in 100
mm Dishes (costar) in Dulbecco's modified Eagles
medium (GIBCO) and allowed to grow to 40-609s
confluency. On days 1 and 2, cells were fed with
4 ml of medium per plate plus various concentra-
tions of FTI-277 or vehicle. On day 3, cells were
washed one time with ice cold PBS and were
collected and lysed by incubation for 30-60 min on
ice in lysis buffer (41). Lysates were cleared
(14,000 rpm, 4 C, 15 min) and supernatants
collected. Equal amounts of lysate were separated
on a 12.59s SDS-PAGE, transfered to nitrocellulose,
and a western blot performed using a anti-Ras
antibody (Y13-238, ATCC) or anti-RaplA antibody
(Santa Cruz Biotechnology, Santa Cruz, CA).
Antibody reactions were visualized using
peroxidase-conjugated goat anti-rat IgG for Y13-
238 and peroxidase-conjugated goat anti-rabbit IgG
- 82 -
=
ak 02207252 2009-11-19
WO 96/21456 ,
PCT/US96/01559
for Rap1A and an enhanced chemiluminescence
detection system (ECL; Amersham Corp.)
EXAMPLE 14
Co-immunaprecipitation of Raf and Ras
Cells were seeded on day 0 in 100 mm dishes
in 10 ml Dulbecco's Modified Eagles Medium
(GIBCO) supplemented with 10% calf serum (Hyclone)
and 1%- pen/strep (GIBCO). On days 1 and 2 cells
were treated with FTI-277 (5 AM) or vehicle
(confluency of cells 40-60%). On day 3, cells
were collected by centrifugation in ice cold PBS.
Cell pellets were then resuspended in ice cold
hypotonic buffer (10 mM Tris, pH 7.5, 5 mM MgC12, 1
mM DTT, 1 mM PMSF) and cells were sonicated to
break up cell pellet to promote separation of
cytosol and membrane. The cell suspension was
then centrifuged at 2,000 rpm for 10 min to clear
debris after which the supernatant was loaded in
ultracentrifuge tubes and spun for 30 min at
100,000 X g to SW Ti55 Rotor to separate membrane
and cytosol fractions. The cytosol and membrane
fractions were lysed on ice for 60 min in buffer
containing 30 mM HEPES, pH 7.5, 1% TX-100, 10%
glycerol, 10 mM NaC1, 5 mM MgC12, 2 mM Na3VO4, 25
mM NaF, 1 mM EGTA, 10 AM soybean trypsin
inhibitor, 25 Ag/ml leupeptin, 10 Ag/ml aprotinin,
2 mM PMSF). The lysates were clarified by
centrifugation. Equal amounts of cytosol and
membrane fractions were immunoprecipitated using
50 Al of a 25% Protein-A Sepharose C1-4B
suspension (Sigma) with 1 Af/ml anti-c-Raf-1
(SC1-33, Santa Cruz Biotechnology, Santa Cruz, CA).
The samples were tumbled at 4 C for 60 min and then
washed 5 times in 50 mM HEPES, pH 7.5, 100 mM
NaC1, 5 mM MgC12, 0.1% TX-100, 10% glycerol, 20 mM
NaF. The final pellets were run on 12.5% SDS-
* Trademark
- 83 -
CA 02207252 2009-11-19
=
WO 96/21456 , PCT/US96/01559
PAGE, transferred to nitrocellulose, and
immunoblotted for the presence of Ras using anti-
Ras antibody (Y13-238) and immunoblotted for the
presence of Raf (c-Raf-1, SC133, Santa Cruz
= 5 Biotechnology, Santa Cruz, CA). Detection was the
same as above for Ras and Rap1A processing.
EXAMPLE 15
Detection of GTP and GDP bound to Ras (FTI-277)
H-RasF cells were seeded and treated as above
for Ras/Raf interaction and Ras and RaplA
processing. On day 2, however, cells were labeled
overnight with (32P3 orthophosphate at 100 gCi/mo
(Amersham PBS13) in 10 ml DMEM-phosphate
supplemented with 10% calf serum, 1 mg/ml BSA and
20 mM HEPES, pH 7.5. On day 3, the medium was
removed and cells were washed one time in ice-cold
PBS, scraped from the plate with a cell scraper,
collected and centrifuged. The cell pellet was
resuspended in ice-cold hypotonic buffer listed
above and the cytosol and membrane fractions were
separated according to the above description for
Ra/Raf association. The cytosol and membrane
fractions were lysed on ice for 60 min in 50 mM
Tris, pH 7.5, 5 mM MgC12, 1% Triton X-100 (TX-100),
0.5% DOC, 0.05% SDS, 500 mM NaC1, 1 mM EGTA, 10
gg/ml aprotinin, 10 gg/ml soybean trypsin
inhibitor, 25 gg/m1 leupeptin, 1 mM DTT, 1 mg/ml
BSA. Lysates were cleared and equal amounts of
protein were immunoprecipitated using anti-Ras
antibody (Y13-259) along with 30 Al Protein A-
Agarose goat anti-rat IgG complex (Oncogene
Science) for 60 min at 4 C. Immunoprecipitates
were washed 6 times in 50 mM HEPES, pH 7.5, 0.5 M
NaC1, 0.1% TX-100, 0.0005 SDS, 5 mM MgC12, drained
using a syringe and bound nucleotide eluted in 12
Al of 5 mM DTT, 5 mM EDTA, 0.2% SDS, 0.5 mM GDP
*Trademark
- 84 -
=
CA 02207252 2009-11-19
W096/21456
PCT/US96/01559
and 0.5 mM GTP at 68 C for 20 min. Immune
complexes were spun down quickly and 6 Al of the
supernatent was loaded onto PEI cellulose thin
layer chromatography plates (20 cm X 20 cm).
Nucleotides were separated by chromatography in 78
g/linter ammonium formate, 9.6% (v/v) concentrated
HC1. Plates were analyzed by autoradiogram.
EXAMPLE 16
Analysis of Raf-I Kinase Activity
Raf-1 kinase was assayed by determining the
ability of Raf to transfer phosphate from (1,-32P)
ATP to a 19-mer peptide containing an
autophosphorylation site. Membrane and cytosol
fraction isolation and Raf immunoprecipitates were
washed three times with cold HEPES buffer and
twice with kinase buffer (50 mM Tris, pH 7.5, 150
mM NaCl, 12 mM MnC12, 1 mM DTT, 0.21; Tweeri4-20.
Immune complex kinase assays were performed by
incubating immunoprecipitaes from membrane and
cytosol fractions in 96 Al of kinase buffer with
20 ACi of (y-32P)ATP (10 mCi/ml, Amersham) and 2
Al of the Raf-1 substrate peptide (1 mg/ml,
Promega) for 30 min at 25 C. The sequence of the
Raf-1 substrate peptide is IVQQFGFQRRASDDGKLTD.
The phosphorylation reaction was terminated by
spotting 50 Al aliquots of the assay mixture onto
Whatman*P81 for 40 min in 0.5% orthophosphoric
acid and air dried. The amount of 'P incorporated
was determined by liquid scintillation counting.
EXAMPLE 17
Inhibition of FTase by FTI-276 and other compounds
Fig. 1B shows that FTI-276 inhibited the
transfer of farnesyl from PHIFPP to recombinant H-
Ras-CVLS with an IC50 of 500 pM. FTI-249, the
parent compound of FTI-276, inhibited FTase with
*Ttademark
- 85 -
CA 02207252 1997-06-06
WO 96/21456
PCT/US96/01559
an IC50 of 200,000 pM. Thus, a phenyl ring at the
2 position of the benzoic acid spacer increased
inhibition potency of FTase by 400 fold confirming
our prediction of a significant hydrophobic pocket
within the CAAX binding site of FTase. This
extremely potent inhibitor was also highly
selective (100-fold) for FTase over the closely
related GGTase I (Fig. 1B). FTI-276 inhibited the
transfer of geranylgeranyl from PHJGG-PP to
recombinant H-Ras-CV11 with an IC50 of 50 nM (Fig.
1B). This 100-fold selectivity is superior to the
15-fold selectivity of the parent compound, FTI-
249. Data for a number of other compounds of
interest are shown in Table 1.
- 86 -
CA 02207252 1997-06-06
WO 96/21456 PCT/US96/01559
Table 1
FTase GGTase I IC"
Compound IC"
Ulm] [nm] GG/F
FTI
232 CABAM 213 1200 6
260 3-Me-CABAM 825 9000 11
261 3-0Me-CABAM 2550 50000 20
270 CANAM 143 3150 22
272 2-Ph-CABAM 5 267 53
274 2-Ph-CABAM-0Me 2050 30000 15
275 2-Xy-CABAM 405 400 1
249 red.CABAM 272 3967 15
254 red.CABAM-0Me 1000 19000 19
276 red.2-Ph-CABA14 0.5 57
114
277 red.2-Ph-CABAM-0Me SO 1600 32
EXAMPLE 18
Inhibition of Ras Processing by FTI-277
To facilitate cellular uptake, FTI-277, the
methylester of FTI-276, was used in experiments to
measure inhibition of Ras processing. H-RasF
cells (NIH 3T3 cells transformed with oncogenic
(61 leucine) H-Ras-CVLS (45) were treated with
FTI-277 (0-50 AM) and the lysates blotted with
anti-Ras or anti-RaplA antibodies. As shown in
Fig. 2A, concentrations as low as 10 nN inhibited
Ras processing but concentrations as high as 10 AM
did not inhbit processing of the
geranylgeranylated RaplA. FTI-277 inhibited Ras
processing with an ICso of 100 nM. In contrast,
the ICso of FTI-249 is 100 AM, and the most potent
CAAX peptidomimetics previously reported inhibited
- 87 -
,
CA 02207252 1997-06-06
WO 96/21456
PCT/US96/01559
Ras processing at concentrations of 10 AM or
higher (44).
The selectivity of FTI-277 for farnesylation
but not geranylgeranylation processing is further
demonstrated in Fig 2B. H-RasGG cells (NIH 3T3
cells transformed with oncogenic (61 leucine) H-
Ras-CVLL (45) were treated with FTI-277.
Processing of RasGG was not affected, whereas that
of RasF was completely blocked. The processing of
endogenous Ras is also blocked in pZIPneo cells
(NIH 3T3 cells transfected with the same plasmid
as H-RasF and H Ras FF except the vector contained
no oncogenic Ras sequences) and Raf cells (NIH 3T3
cells transformed by an activated viral Raf (48)).
Mechanism of disruption of Ras oncogenic
signalling by FTI-277
Ras relays biological information from
tyrosine kinase receptors to the nucleus by
activation of a cacade of MAPKs (reviewed in 29-
31). Upon growth factor stimulation, Ras becomes
GTP bound and is then able to recruit the ser/thr
kinase c-Raf-1 to the plasma membrane where it is
activated. c-Raf-1 then phosphorylates and
activates MEK, a dual thr/tyr kinase, which
activates MAPK. Recently, epidermal growth factor
has been shown to induce association of Raf with
Ras (46).
= In order to determine the mechanism by which
FTI-277 disrupts Ras oncogenic signaling, NIH 3T3
cells were transfected with activated (GTP-locked)
Ras and the effects of FTI-277 on the interaction
of Ras with its immediate effector, Raf, were
investigated. Various NIH 3T3 cell transfectants
(pZIPneo, H-RasF, and H-RasGG) were treated with
vehicle or FTI-277, membrane and cytosolic
fractions were isolated and immunoprecipitated
- 88 -
CA 02207252 1997-06-06
WO 96/21456
PCT/US96/01559
with anti-Raf antibody as described above. Raf
did not associate with Ras in pZIPneo cells which
did not contain GTP-locked Ras, as shown in Fig.
3. In contrast, H-RasF and H-RasGG cells contain
Ras/Raf complexes in the membrane, but not in the
cytosolic fractions, as shown in Fig. 3.
Treatment of these cells with FTI-277 resulted in
the accumulation of Ras/Raf complexes in the
cytosolic but not membrane fractions of H-RasF
cells, but not in the H-RasGG cells (Fig 3).
Thus, the disruption of Ras/Raf interaction at the
cell membrane and accumulation of these complexes
in the cytoplasm occurred only in Ras-F but not
Ras-GG cells, in agreement with the Ras processing
selectivity results of Fig. 2. .Thus, these
results demonstrate that inhibition with FTI-277
results in the accumulation of non-farnesylated
cytosolic Ras that is capable of binding to Raf.
The fact that non-processed Ras can associate with
Raf in a non-membranous cytoplasmic environment
was confirmed by transfecting NIH 3T3 cells with a
GTP-locked Ras that lacks a farnesylated site and,
therefore, remains in the cytoplasm (Ras mutant
with a 61 leucine oncogenic mutation and a 186
serine mutation) and showing that these cells
contained only cytoplasmic Ras/Raf complexes when
immunoprecipitated with Raf and blotted with
antiRas antibodies (Fig. 3). In short,
farnesylation is not required for Ras to bind to
Raf.
EXAMPLE 19
Determination of nucleotide state of Ras
The fact that Raf binds Ras-GTP with much
higher affinity than Ras-GDP was used to determine
the nucleotide state of Ras in the cytoplasmic
Ras/Raf complexes, as described above. In Ras-F
- 89 -
CA 02207252 1997-06-06
V0396/21456
PCT/US96/01559
cells, only membrane fractions contained GTP-
locked Ras, as shown in Fig 4A. Upon treatment
with FTI-277, however, the non-farnesylated
cytosolic Ras was found to be GTP bound. Thus,
binding of GTP to 61 leucine Ras does not require
Ras processing and subsequent plasma membrane
association. The ser/thr kinase activity of Raf
in Ras/Raf complexes was then determined by
immunoprecipitating Raf and assaying for its
ability to phosphorylate a 19-mer
autophosphorylated peptide. Fig. 4B shows that
oncogenic Ras-F induced activation of Raf in the
plasma membrane and that treatment with FTI-277
suppressed this activation. More importantly, the
cytoplasmic Ras/Raf complexes that were induced by
FTI-277 (Fig. 3) had basal levels of Raf kinase
activity that were comparable to those of the
parental NIH 3T3 cell line pZIPneo (Fig. 4B).
Taken together, Figures 3 and 4 demonstrate that
oncogenic transformation with GTP-locked Ras
results in the constitutive recruitment to the
plasma membrane and subsequent activation of Raf.
Furthermore, FTase inhibition by FTI-277
suppresses this activation by inducing the
accumulation of Ras/Raf complexes in the cytoplasm
where Ras is GTP-bound but Raf kinase is not
activated. The fact that Raf kinase is not
activated when bound to Ras in a non-membranous
environment is consistent with recent reports that
indicate that Raf activation requires an as yet to
be determined activating factor at the plasma
membrane (47).
Experiments were then performed to
investigate the effects of FTI-277 on oncogenic
Ras activation of MAPK, a Raf downstream
signalling event (29-31). Oncogenic activation of
MAPK can be easily detected since activated MAPK
- 90 -
CA 02207252 1997-06-06
WID96/21456
PCT/US96/01559
migrates slower in SDS-PAGE. Fig. 5A shows that
NIH 3T3 cells transfected with pZIPneo contain
only inactive MAPK but that upon transformation
with oncogenic H-Ras, MAPK is activated (Fig. 5A).
Pretreatment with FTI-277 results in a
concentration dependent inhibition of oncogenic
Ras activation of MAPK. Concentrations as low as
300 nM were effective and the block was complete
at 1 M. Taken together, Figs. 3 and 5
demonstrate that at least 50% inhibition of Ras
processing is required for complete suppression of
MAPK activation but that less than a 100%
inhibition of Ras processing is required for
complete suppression of MAPK activation by Ras. A
series of NIH 3T3 cell lines transformed with
various oncogenes was used to determine whether
the inhibition of MAPK activation is due to
selectively antagonizing Ras function. Fig. 5B
shows that FTI-277 was able to block H-RasF but
not H-RasGG activation of MAPK. This is
consistent with its ability to inhibit H-RasF but
not H-RasGG processing (Fig. 2). Selectivity of
FTI-277 towards antagonizing Ras-dependent
activation of MAPK was substantiated by using NIH
3T3 cells where MAPK is constitutively activated
by transformation with the Raf oncogene. Fig.
5B shows that oncogenic Raf activation of MAPK is
not blocked by FTI-277 even though processing of
endogenous Ras was inhibited in these cells.
Similar results were also obtained with FTI-276
(Fig. 6). Taken together these results clearly
demonstrate that FTI-276 and FTI-277 are highly
effective and selective in disrupting contitutive
Ras-specific activation of MAPK.
Thus, FTI-277 is an extremely potent and
highly selective FTase inhibitor. This compound
inhibited Ras processing with concentrations as
- 91 -
CA 02207252 1997-06-06
W096/21456
PCT/US96/01559
low as 10 nM and processing was blocked at 1 M.
The most potent inhibitor previously reported BZA-
5B, blocked Ras processing only at 150 AM (44).
EXAMPLE 20
Antitumor Efficacy and Selectivity of FTI-276 and
FTI-277 In order to demonstrate the efficacy of
these inhibitors as anticancer agents and show
that they can inhibit tumor growth of human tumors
which have multiple and complex genetic
alterations, antitumor efficacy experiments were
performed using a human tumor cell line. A
critical issue connected with the potential use of
the compounds of the invention is whether the
growth of human tumors which harbor K-Ras
mutations can be blocked. This is important for
further development of FTase inhibitors as
anticancer drugs since K-Ras mutations are most
common in human cancers and since K-Ras processing
is more difficult to inhibit than the processing
of the less prevalent H-Ras (1-3, 15).
Furthermore, the majority of human tumors have
multiple genetic alternations; notably a delation
in the tumor suppressor gene p53 is most
prevalent. It is therefore extremely important to
determine whether or not inhibition of Ras
function is sufficient to halt the growth of human
tumors which harbor K-Ras mutation as well as
deletions in p53.
To evaluate the antitumor efficacy of FTI-
276, a nude mouse xenograft model was used. In
this model, tumors from two human lung carcinoma
cell lines are implanted subcutaneously. One of
these (Calu-1) harbors a K-Ras oncogenic mutation
and has a deletion of the tumor suppressor gene
p53. The other human lung carcinoma (NCI-H180)
has no Ras mutations. Thirty two days after s.c.
- 92 -
CA 02207252 1997-06-06
WO 96/21456
PCT/US96/01559
implantation when the tumors reached sizes of 60
to 80 mm3, the mice were randomly separated into
control and treated groups (4 animals per group;
each animal had a tumor on both the right and the
left flank). Figure 7A shows that tumors from
control animals treated with saline once daily
starting on day 36 grew to an average size of 566
mm3 over a period of 64 days from tumor
implantation. In contrast, tumors treated once
daily with FTI-276 (50 mg/kg) grew very little and
the average tumor size was 113 mm3 (Fig. 7A). In
another experiment, FTI-277, the methylester of
FTI-276, inhibited the growth of Calu-I cells to
the same extent (Figure 8). Although the animals
were treated once daily with 50 mg/kg for 36 days
(total cumulative does of 1.8 g/kg), no weight
loss was observed and the animals appeared normal
with no evidence of gross toxicity. This lack of
toxicity was also observed in separate experiments
where the dose was escalated to 180 mg/kg once
daily. Thus, FTI-276 and FTI-277 essentially
blocked tumor growth of Calu-I carcinoma with no
evidence of gross toxicity.
The effect of FTI-276 on the tumor growth of
another human lung carcinoma, NCI-H810, that does
not harbor an oncogenic Ras mutation was also
determined. Figure 7B shows that tumors from
animals treated with saline or FTI-276 grew at a
similar rate. Over a period of 14 days of
treatment the average tumor size of the control
and FTI-276 treated groups were 919 mm3 and 815
mm3, respectively. These results clearly
demonstrate that in contrast to Calu-1, NCI-H810
carcinomas were not sensitive to FTI-276 treatment
suggesting that FTI-276 inhibition of tumor growth
of human lung carcinomas is Ras-dependent.
- 93 -
CA 02207252 1997-06-06
W096/21456
PCT/US96/01559
Furthermore, FTI-276 inhibited tumor growth even
though Calu-1 does not express p53.
To further establish the selectivity of FTI-
276 to inhibit selectively Ras-dependent tumors,
the anti-tumor efficacy of FTI-276 and FTI-277
against H-RasF and Raf transformed NIH 3T3 in the
same nude mouse xenograft model was examined.
Figure 9 shows that a once daily injection of FTI-
276 or FTI-277 (50 mg/kg) inhibited tumor growth
of H-RasF transformed NIH 3T3 cells. In contrast,
an identical treatment regimen with FTI-276 and
FTI-277 had no effect on the growth of Raf-
transformed NIH 3T3 cells (Fig. 10), further
confirming the conclusion from the results of
Figures 7 and 8 that FTI-276 and FTI-277 are
selective for Ras-dependent tumors.
= In addition, the question of whether FTI-276
inhibition of tumor growth correlated with
inhibition of Ras processing in vivo was
addressed. To so this, mice having subcutaneous
H-RasF cells were treated with various doses of
FTI-276 (0, 10, 50 and 100 mg/kg) and tumor size
and Ras processing in the HRasF tumors in vivo
were examined. Figure 11A shows that throughout
the 11 day treatment period, FTI-276 inhibited
tumor growth in a dose dependent fashion. The
tumor sizes at the end of 17 days were 2490 mm3 for
saline, 1793 mm3 for 10 mg/kg, 1226 mm3 for 50
mg/kg and 624 mm3 for 100 mg/kg treated animals.
To determine the levels of inhibition of Ras
processing, the animals were sacrificed 5 hrs
after the last injection, the tumors were excised
and processed for immunoblotting with anti-Ras
antibody as described in legend to Fig. 11.
Tumors from control animals contained only fully
processed Ras which migrates faster in SDS-PAGE
gels (Fig. 11B). As the dose of FTI-276 increases
- 94 -
CA 02207252 1997-06-06
W096/21456
PCTIUS96101559
from 10 to 100 mg/kg there was a progressive
accumulation of unprocessed Ras which was
paralleled by a decrease in the relative ratio of
fully processed Ras. Thus, the extent of tumor
growth inhibition correlated with the extent of
inhibition of Ras processing. Furthermore, the
inhibition of Ras processing in vivo was selective
in that FTI-276 did not inhibit RaplA processing
even at 100 mg/kg.
II. Farnesyltransferase Inhibitors of the type
CA
Compounds of another major embodiment of the
invention are represented by formula II. Several
examples are shown in Figure 12. These and other
compounds of this embodiment may be prepared using
procedures which are conventional in the art. For
example, compounds 4 and 5 of Figure 12 may be
prepared by reductive amination of 4-amino-3'-
tert.butoxy-carbonyl biphenyl or 4-amino-4'-
tert.butoxy carbonyl biphenyl, respectively, with
N-Boc-S-trityl cysteinal followed by deprotection
with, for example, trifluoroacetic acid and
purification.
This embodiment of the invention is
illustrated but not limited by the following
examples:
EXAMPLE 21
The compound C-4ABA-Met of formula (2) (see
Figure 12) was prepared as described in reference
(27).. The protected form of the peptidomimetic
(2a) was prepared through the reductive amination
of 4-aminobenzoyl methionine methyl ester and N-
Boc-S-trityl cysteinal in methanol solution
containing NaBH3CN and 5% acetic acid. This
reaction gave the N-Boc-S-trityl, methyl ester of
(2a) with a yield of 65%. The protected
- 95 -
CA 02207252 1997-06-06
W096/21456
PCIMS96/01559
peptidomimetic was deesterified by LiOH in THF and
then deprotected by trifluoroacetic acid in
methylene chloride with two equivalents of
triethylsilane to give crude (2a) which was
purified by reverse phase HPLC. The biphenyl-
based peptidomimetic (8) was prepared by the
reductive amination of 4-amino-3'-methyl biphenyl
with N-Boc-S-trityl cysteinal, to give the N-Boc-
.
S-trityl derivatives of (8), which was then
deprotected by trifluoroacetic acid and purified
by reverse phase HPLC. The peptidomimetics (4)
and (5) were prepared from the reductive amination
of 4-amino-3'-tert.butoxycarbonyl biphenyl and 4-
amino-4'-tert.butoxycarbonylbiphenyl,
respectively, with N-Boc-S-trityl cysteinal, to
give the N-Boc-S-trityl, tert-butyl ester of (4)
and (5). Deprotection by trifluoroacetic acid and
purification by reverse phase HPLC gave pure (4)
and (5).
Synthesis
The basic approach used for the preparation
of the compounds of the invention is illustrated
in Scheme 1 with the synthesis of compound 4.
[Compound numbers in the following discussion
refer to Schemes 1 and 2.]
- 96 -
,
CA 02207252 1997-06-06
PCT/US96/01559
WO 96/21456
Scheme 1. Representative Synthesis of FTase Inhibitorsa
HQ
02N Br + B. a411 02N 41 *
02N. 10 IP
= HO
. =
CH3
14 CH3
15 COOH
lc
Ph3CS12
___________________________________________________________ 02N *
BocNH CO0But
CO0But
18
16
I e
H2N COOH
4
Reagents: (a) Pd(OAc)2; (b) KMnO4, pyridine/H20; (c) (1) (C0C1)2, (2) tert-
butyl alcohol, n-BuLi; (d) (1) H2, Pd/C, (2) N-Boc-S-
tritylcysteinal 17, (3) NaB(CN)113; (e) TFA, EtsSiH.
Scheme 2. Synthesis of Compounds 11 and 12'
111 41, a 41 it b Ph3CS-140
H3C CODE& N3 CO0But BocNH N
CO0But
19 20 21
d
Ph3CS¨ * H5-)_40
BocNH N CO0But H2N N
COOH
IC II
c
H2N N COOH
12
- 97
CA 02207252 2009-11-19
WO 96/21456 PCT/US96/01559
1-Bromo-4-nitrobenzene was coupled to 3-
methylphenylboronic acid (34) through an modified
Suzuki coupling (30) to afford compound 14 (35).
Compound 14 was oxidized to carboxylic acid 15
which was converted to the acid chloride and
reacted with lithium tert-butoxide (36) to give
the tert-butyl ester 16. Reduction of 16 by
hydrogenation and subsecient reductive amination
(37) of the resulting amine with N-Boc-S-trityl-
cysteinal 17 (38) gave the fully protected
derivative 18, which was deprotected by
trifluoroacetic acid in the presence of
triethylsilane (39). Compound 4 =was purified by
reverse phase HPLC and isolated as its
trifluoroacetate salt by lyophilization.
The synthesis of 11 and 12 is described in
Scheme 2. Compound 19 was made from 3-methy1-3'-
carboxybiphenyl (itself formed from aryl-aryl
coupling of methyl 3-bromobenzoate with 3-
=
methylphenylboronic acid followed by a
saponification) via the same method as compound
16. Bromination of 19 followed by reaction with
sodium azide gave 20 which as catalytically
hydrogenated to give the corresponding amine.
Reaction of Boc-trityl protected cysteine with
this amine through the mixed anhydride method gave
21, while compound 22 was made by reductive
amination with Boc-trityl protected cysteinal.
Experimental
114 and 1-3C NMR spectrum were recorded on a
Bruker AM-300 spectrometer. Chemical shifts were
reported in 8 (ppm) relative to tetramethylsilane.
All coupling constants were described in Hz.
Elemental analyses were performed by Atlantic
Microlab Inc., Georgia. Optical rotations were
measured on a Perkin-Elmer 241 polarimeter.
*Trademark
- 98 -
CA 02207252 1997-06-06
NVID96/21456
PCTIUS96101559
Concentrations are expressed in g/mL. Flash
column chromatography was performed on silica gel
(40-63 gm) under a pressure about 4 psi. Solvents
were obtained from commercial suppliers and
purified as following: tetrahydrofuran and ether
were distilled from sodium benzophenone ketyl,
methylene chloride was distilled over lithium
aluminum hydride. Preparative HPLC was performed
using a Waters 600 E controller and a Waters 490 E
Multi-Wavelength UV detector with a 25x10 cm
Delta-Pak C-18 300 A cartridge column inside a
Waters 25x10 cm Radial Compression Module.
Analytical HPLC was performed using a Rainin HP
Controller and a Rainin UV-C detector with a
Rainin 250x4.6 mm 5 gm Microsorb C-18 column.
High resolution mass spectra (HRMS) and low
resolution mass spectra (LRMS)- were performed on a
Varian MAT CH-5 and VG 7070 mass spectrometer.
The purity of all the synthesized inhibitors was
more than 98% as indicated by analytical HPLC.
A. 4-Nitro-3'-methylbiphenyl (14).
To a mixture of 4-nitrobenzene (3.0 g, 14.8
mmol) and 3-methylphenylboronic acid (2.06 g, 15.1
mmol) in 35 mL of acetone and 40 mL of water was
added K2CO3-1.5H20 (5.93 g, 37.5 mmol) and Pd(OAc)2
(101 mg, 0.50 mmol). The deep black mixture was
refluxed for 6 hr and then cooled. The mixture
was extracted with ether and the organic layer was
passed through a layer of celite. The pale yellow
solution was dried over Na2SO4 and evaporated to
dryness. The residue was recrystallized from hot
methanol to give pale yellow crystals (2.68 g,
85%). m.p.59-60 C.1H NMR (CDC1.3) 6 8.26 (d, 8.7
Hz, 2H), 7.70 (d, 8.7 Hz, 2H), 7.41 (m, 3H), 7.26
(d, 7.1Hz, 1H), 2.43 (s, 1H). 13C NMR (CDC13) 6
147.6, 146.8, 138.8, 138.6, 129.6, 128.9, 128.0,
- 99 -
CA 02207252 1997-06-06
W096/21456
PCT/US96/01559
127.6, 124.4, 123.9, 21.4. LRMS (El) for
Ci3HliN02213 (IC, intensity 100); HRMS (El) calcd
213.0789, obsd 213.0778.
B. 4-Nitro-3'-carboxybiphenyl (15).
Compound 14 (2.31 g, 10 mmol) was suspended
in a mixture of 10mL of pyridine and 20 mL of
water. The mixture was heated to ref luxing and
then KMn04 (7.9 g, 50 mmol) was added in portions.
This mixture was refluxed for 1 hr and then
stirred at room temperature for 4 hr. The hot
mixture was filtered and the'black solid was
washed with hot water. The filtrate was acidified
with 6 N HC1. The precipitate was collected and
dried (2.16 g, 899,5). m.p. 265 C (decomp). NMR
(DMSO-d0 6 11.1-11.4 (br s, COOH), 8.32 (d, 8.7
Hz, 2H), 8.27 (s, 1H), 8.02 (m, 4H), 7.66 (t, 7.8
Hz, 1H). 13C NMR (DMSO-d0 6 167.1, 148.9, 145.6,
138.2, 131.8, 131.5, 129.6 (br), 127.9, 124.2
(br). LRMS (El) for Cl3H904N 243 (M+, 100), 152
(60); HRMS (El) calcd 243.0531, obsd 243.0544.
Anal. (Ci3H9N04) C, H, N.
C. 4-Nitro-3'-tert-butoxycarbonylbiphenyl (16).
To a solution of 15 (1.215 g, 5 mmol) in 30
mL of methylene chloride was added oxalyl chloride
(0.65 mL, 7.45 mmol) and one drop of DMF. The
mixture was stirred until no further bubbling was
observed. The clear solution was exaporated to
dryness to give the crude 'acid chloride. To
another flask containing 7.0 mL of tert-butanol
was added n-BuLi (1.8 M in hexane, 2.8 mL, 5.04
mmol) under a water bath. The turbid solution was
stirred for 5 min at room temperature and then the
above acid chloride in 20 mL of THF was added
through a dropping funnel. The mixture was
stirred overnight before the solvents were
- 100 -
CA 02207252 1997-06-06
1111)96/21456
PCTIUS96101559
evaporated. The residue was dissolved into
methylene chloride and washed with 0.5 N NaOH.
The organic layer was dried over MgSO4 and
evaporated. The residue was recrystallized from
methanol to give pale yellow crystals (851 mg,
57%). m.p. 110.5-111.0 C. 11-1 NMR (CDC10 6 8.32
A
(d, 7.8 Hz, 2H), 8.24 (s, 1H), 8.06 (d, 7.7 Hz,
1H), 7.77 (m, 3H), 7.56 (t, 7.7 Hz, 1H), 1.63 (s,
9H). 13C NMR (CDC10 6 165.1, 147.1, 146.5, 138.7,
132.8, 131.0, 129.6, 129.0, 128.2, 127.8, 124.0,
81.4, 28.0, LRMS (El) for C3.7113.704N 299 (Mt, 20), 243
(70), 266 (30), 152 (25; HRMS (El) calcd 299.1157,
obsd 299.1192. Anal. (C17112.7N04) C, H, N.
D. N-Boc-S-trityl cysteinal (17).
To a solution of N-Boc-S-trityl cysteine
(7.44 g, 16 mmol) in 85 mL of methylene chloride
was added triethylamine (2.22 mL, 16 mmoL) and
N,0-dimethylhydroxylamine hydrochloride (1.57 g,
16.1 mmol). This mixture was cooled in an ice
bath and 1-(3-dimethylaminopropy1)-3-
ethylcarbodiimide hydrochloride (EDCI, 3.08 g,
16.0 mmol) and HOBT (2.17 g, 16 mmol) was added.
The mixture was stirred at 0 C for 1 hr and at
room temperature for a further 10 hr. The mixture
was extracted with methylene chloride and 0.5 N
HC1. The organic layer was washed consecutively
with 0.5 N HC1, concentrated NaHCO3 and brine. The
organic layer was dried and evaporated. The
residue was purified by flash column
chromatography (1.5:1=hexane:ethylacetate) to give
a white foam (7.40 g, 91%).m.p.59-60 (decomp). 11.1
NMR (CDC10 6 7.41 (m, 6H), 7.20-7.31 (m, 9H), 5.13
(d, 8.9 Hz, 1H), 4.76 (br s, 1H), 3.64 (s, 3H),
3.15 (s, 3H), 2.56 (dd, 4.7 and 12.1 Hz, 1H), 2.39
(dd, 7.8 and 12.1 Hz, 1H), 1.43 (s, 9H). 13C NMR
(CDC13 6 170.7, 154.9, 144.2, 129.3, 127.6, 126.4,
- 101 -
CA 02207252 1997-06-06
1,1,096/21456
PCT/US96/01559
79.3, 66.4, 61.2, 49.5, 33.8, 31.8, 28.1. This
carboxyamide (2.02 g, 4.0 mmol) was dissolved in
30 mL of ether and cooled to -10 c. Lithium
aluminum hydride (167 mg, 4.40 mmol) was added and
the mixture was stirred for 15 min under the
nitrogen. Then 40 mL of 0.5 N HC1 was added and
the solution was extracted with ether. The ether
layer was washed with 0.5 N HC1 and dried. The
evaporation of solvents gave a white foam (1.80 g)
which was used for further reaction without
purification. The 1H NMR spectrum of this compound
was complex. The percentage of the aldehyde was
about 65-70%, which was calculated according to
the integration of the sharp singlet Co 9.17) and
the trityl peak (0 7.40, m, 6H;7.28,m,9H).
Lowering the temperature to -45 C did not improve
the aldehyde percentage.
E. 4-N-[2(R)-tert-butoxycarbony1amino-3-
triphenylmethylthipropyl]amino-3'-tert-
butoxycarbonylibiphenyl (18).
Compound 16 (768 mg, 2.56 mmol) was dissolved
in THF. A catalytic amount of 10% Pd on activated
carbon (78 mg) was added. The mixture was
hydrogenated (40 psi) for 30 min. The black
mixture was passed through a thin layer of celite
and the pale yellow solution was evaporated. The
residue was dissolved in 10 mL of methanol. To
this solution was added 0.5 mL of acetic acid and
a solution of the same equivalents of aldehyde 17
(according to the 1H NMR determination) in 6 mL of
methanol. Sodium cyanoborohydride (241 mg, 3.84
mmol, 1.5 eq) was added and the mixture was
stirred overnight. After the evaporation of
solvents, the residue was extracted with ethyl
acetate and concentrated sodium bicarbonate. The
organic layer was dried and evaporated. The
- 102 -
CA 02207252 1997-06-06
WO 96/21456
PCT/US96/01559
residue was purified by flash column
chromatography (3.5:1-hexane:THF) to give a white
foam (1.09 g, 61%). m.p. 75.0-76.0 C(decomp).
[a]25D=-2.13 (c=0.01, CH3C00C2H5). 1H NMR (CDC10 6
8.14 (s, 1H), 7.86 (d, 7.7 Hz, 1H), 7.66 (d, 7.8
Hz, 1H), 7.40 (m, 9H), 7.22-7.30 (m, 9H), 6.61 (d,
8.5 Hz, 2H), 4.58 (d, 7.1 Hz, 1H), 3.83 (br m, 2H,
Cys a proton and the amine), 3.12 (br m, 2H, CH2N),
2.48 (br, m, 2H, CH2S) , 1.60 (s, 9H), 1.44 (s, 9H).
1-3C NMR (CDc10 6 165.9, 155.6, 147.5, 144.4,
141.2, 132.3, 130.1, 129.5, 129.2, 128.5, 128.0,
127.9, 127.1, 126.8, 112.9, 80.9, 79.7, 67.0,
49.4, 47.1, 34.3, 28.3, 28.2 (expect 14 aromatic
C, observed 13). Anal. (C44H45N204S=1.2H20) C, H, N,
S.
= F. 4-N-[2(R)-amino-3-mercaptopropyl]amino-3'-
carboxybiphenyl (4).
Compound 18 (600 mg, 0.85 mmol) was dissolved
in 2mL of TFA and 2 mL of methylene chloride.
Triethylsilane was added dropwise to the deep
brown mixture until the brown color had
disappeared. The mixture was then kept at room
temperature for 1 hr. Then solvents were
evaporatee and the residue was dried under vacuum.
The solid was triturated with 30 mL of ether and
3mL of 3 N Hcl in ether. The white precipitate
was filtered and washed with ether to obtain a
crude product (270 mg, 84%). This crude product
was dissolved into 30 mL of dilute Hcl solution
(0.01N) and was lyophilized. Analytical HPLC
showed the purity to be 95%.m.p. 105-106 C
(decomp). [a]2513=+13.16 (c=0.01 in methanol. IJ M<R
(CD3OD 6 8.18 (s, 1H), 7.89 (d, 7.7 Hz, 1H), 7.78
A
(d, 7.3 Hz, 1H), 7.49 (m, 3H), 6.82 (d, 8.5 Hz,
2H), 3.56 (m, 2H CHN and CH2N) , 3.42 (dd, 8.9 and
15.2 Hz, 1H, CH2S) . 13C NMR (D20 and CD30D) 6
- 103 -
CA 02207252 1997-06-06
W096/21456
PCT/US96/01559
171.1, 147.1, 141.4, 131.9, 130.9, 130.1, 128.8,
128.5, 127.0, 115.2, 53.2, 45.4, 25.0 LRMS (FAB,
glycerol) for C3.61118N202S (M+1) 303. Anal.
(C161118N202S=2HC1) C, H, N, S. Further purification
by preparative HPLC (Waters C-18, 40%
acetonitrile, 60% water, 0.1% TFA, 40 min
gradient) gave product 4 (120 mg) with a purity
over 99.9%.
G. 3-Methyl-3'-tert-butoxycarbonylbiphenyl (19).
The coupling of 3-methylphenylboronic acid
with 3-bromobenzoic acid methyl ester gave a 3-
methy1-3'-methoxycarbonylbiphenyl (79% yield),
which was then hydrolyzed to yield a 3-methyl-3'-
= carboxybiphenyl (97% yield). Compound 19 (an oil)
was prepared from this acid using the same method
as for the preparation of compound 16 (65% yield).
NMR (CD13) a 8.21 (s, 1H), 7.95 (d, 7.8 Hz, 1H),
7.73 (d, 6.6 Hz), 1H), 7.46 (m, 3H), 7.35 (t, 7.5
Hz, 1H), 7.20 (d, 7.4 Hz, 1H), 2.43 (s, 3H), 1.62
(s, 9H). 13C NMR (CDC13)_(5 165.6, 141.3, 140.2,
138.3, 132.4, 140.0, 128.7, 128.5, 128.3, 128.0,
127.9, 124.2, 81.0, 28.1, 21.4. Lams (El) for
Ci8H2002 268 (M+, 35), 212(100), 195 (20); HRMS (El)
calcd 268.1463, obsd 268.1458.
H. 3-Azido-3'-tert-butoxycarbonylbiphenyl (20).
Compound 19 (2.18 g, 8.13 mmol) and N-
bromosuccinimide (1.70 g, 9.50 mmol) was suspended
in 60 mL of CC14. Dibenzoyl peroxide (20 mg) was
added and the mixture was ref luxed for 1.5 hr.
After removing the solid, the filtrate was washed
with concentrated sodium bicarbonate and dried
over sodium sulfate. 1H NMR showed the crude
material contained 70% of monobrominated and 30%
of dibrominated product. This material was
dissolved in 20 mL of DMSO and sodium azide (3.70
- 104 -
CA 02207252 1997-06-06
WO 96/21456
PCT/US96/01559
g, 57 mmol) was added. The mixture was heated to
80 C for 4 hr before being poured into a mixture
of methylene chloride and water. The organic
layer was dried and evaporated. The residue was
purified by flash column chromatography (5% of
ethyl acetate in hexane) to give 20 (2.14 g, 78%,
two steps) as colorless oil. 111 NMR (CDC13) 6 8.22
(s, 1H), 8.00 (d, 7.7 Hz, 1H), 7.76 (d, 8.2 Hz,
1H), 7.58 (m, 2H), 7.50 (m, 2H), 7.33 (d, 7.6 Hz,
1H), 4.43 (s, 2H), 1.62 (s, 9H). 13C NMR (CDC13) 6
165.2, 140.5, 140.3, 135.8, 132.3, 130.7, 129.1,
128.5, 128.2, 127.8, 127.1, 126.7, 126.6, 80.9,
54.3, 27.8.
I. N-Boc-S-trityl-cysteiny1-3-aminomethy1-3'-
tert-butoxycarbonylbiphenyl (21).
Compound 20 (0.75 g, 2.43 mmol) was dissolved
in 30 mL of methanol. A catalytic amount of 5%
palladium on barium sulfate (0.30 g) was added.
The mixture was hydrogenated at 1 atm for 5 hr.
The catalyst was removed by filtration and the
methanol was evaporated. This residue was
dissolved in 40 mL of methylene chloride. N-Boc-
S-trityl cysteine (1.12 g, 2.43 mmol) was added at
0 C followed by EDCI (1 eq) and HOBT (1 eq). The
mixture was stirred for 24 hr. After workup and
evaporation of solvents, the residue was purified
by flash column chromatography (hexane:ethyl
acetate=3.2:1) to give 21 (570 mg, 44%). m.p. 84-
86 C. 11.1 NMR (CDC13) 6 8.17 (s, 1H), 7.95 (d, 7.7
Hz, 1.11), 7.70 (d, 7.7 Hz, 1H), 7.50-7.30 (m, 9H),
7.30-7.10 (m, 11H), 6.44 (br, 1H), 4.86 (br, 1H),
4.45 (d, 4.0 Hz, 2H, CH2Ph) , 3.87 (br, 1H, Cys a
H), 2.75 (dd, 7.2 and 12.8 Hz, 1H, CH2S) , 2.55 (dd,
A
5.3 and 12.8 Hz, IH, CH2S) , 1.62 (s, 9H), 1.36 (s,
9H) . Anal. (C45H48N205S) C, H, N, S.
- 105 -
CA 02207252 1997-06-06
W096/21456 PCT/US96/01559
J. Cysteiny1-3-aminomethy1-3'-carboxybiphenyl
(11).
Compound 21 (150 mg) was deprotected using
the same method as for the preparation of compound
4. Final purification by preparative HPLC gave 11
as a white solid (42 mg, 46%). m.p. 88-89 C
#
(decomp). MNR (CD30D) 6 8.26 (s, 1H), 8.01 (d,
7.7 Hz, 1H), 7.86 (d, 7.7 Hz, 1H), 7.64 (s, 1H),
7.56 (m, 2H), 7.46 (t, 7.6 Hz, 1H), 7.35 (d, 7.6
Hz, 1H), 4.53 (s, 2H), 4.00 (t, 5.2 Hz, 1H, Cys_a
H), 3.06 (dd, 14.6 and 5.2 Hz, 1H, CH2S), 2.97 (dd,
14.6 and 6.8 Hz, 1H, CH2S) . 'LRMS (El) for
C3.71-13.8N203S 331 (M+1, 8), 281 (100), 226 (75), Anal.
(C17H/8N203S= HC1- 0 . 6H20) C, H, N.
K. 3-N-(2(R)-tert-Butoxycarbonylamino-3-
triphenylmethylthiopropyl]aminomethy1-3'-tert-buto
xy-carbonylbiphenyl (22).
The azide 20 (900 mg, 2.91 mmol) was
dissolved in 20 mL of methanol. A catalytic
amount of 5% Pd on barium sulfate (90 mg) was =
added. This mixture was hydrogenated at 1 atm
overnight. The catalyst was removed and the
methanol was evaporated. The remaining residue
was dissolved in a mixture of 0.5 N HC1 (20 mL)
and ether (20 mL). The aqueous phase was
neutralized with 1 N NaOH and extracted into
methylene chloride. After the evaporation of
solvents, a viscous oil was obtained (600 mg,
73%). NMR (CDC10 6 8.22 (s, IH), 7.97 (d, 7.8
Hz, 1H), 7.75 (d, 7.7 Hz, 1H), 7.57 (s, 1H), 7.50
(m, 2H), 7.43 (t, 7.7 Hz, 1H), 7.33 (d, 7.4 Hz,
1H), 3.96 (s, 2H), 1.62 (s, 9H), 1.46 (br s, 2H,
NHO. This amine (581 mg, 2.05 mmol) was dissolved
*
in 10 mL of methanol and 0.5 mL of acetic acid
before N-Boc-S-tritylcysteinal (leq, according to
'14 NMR determination of the aldehyde percentage)
- 106 -
CA 02207252 1997-06-06
W096/21456
PCT/US96/01559
was added. Sodium cyanoborohydride (193 mg, 1.50
eq) was added to the above solution and the
mixture was stirred at room temperature overnight.
After workup, the crude residue was purified by
flash column chromatography (1: 1 = ethyl acetate:
hexane) to give a white foam (602 mg, 41A). m.p.
66-68 C (decomp). NMR (CDC13) 6 8.21 (s, 1H),
7.96 (d, 7.7 Hz, 1H), 7.73 (d, 8.0 Hz, 1H),
7.37-7.51 (m, 10H), 7.15-7.31 (m, 10H), 4.69 (br
d, 1H), 3.75 (br s, 3H, PhCH2N and Cys a H), 2.68
(dd, 6.0 and 123 Hz, 1H, CH2S), 2.56 (dd, 5.5 and
12.3 Hz, 1H, CH2S), 2.47 (m, CH2N), 2.35 (m,
1H, CH2N), 1.62 (s, 9H), 1.42 (s, 9H), 1.12 (br s,
1H, NH).
L. .3-N-[2(R)-amino-3-mercaptopropyl]
aminomethy1-3'-carboxybiphenyl (12).
Compound 22 (480 mg, 0.672 mmol) was
dissolved in a mixture of 2 mL of methylene
chloride and 2 mL of trifluoroacetic acid.
Several drops of triethylsilane were added until
the deep brown color had disappeared. This
mixture was kept at room temperature for 1.5 hr,
and then the solvents were evaporated, and the
residue was dried under vacuum. The solid residue
was dissolved in 1 mL of acetic acid and 2 mL of
HC1 (1.7 M) in acetic acid. Finally 5 mL of HC1
(3 M) in ether and 10 mL of ether were added. The
white precipitate was washed with dry ether and
dried to give a hydrochloride salt (215 mg, 8130.
31.1 NMR (D20) 6 8.16 (s, 1H), 7.94 (d, 7.7 Hz, 1H),
7.85 (d, 7.7 Hz, 1H), 7.70 (s, 2H), 7.55 (t, 7.8
Hz, 2H), 7.46 (d, 7.5 Hz, 1H), 4.36 (s, 2H, PhCH2),
3.81 (m, 1H, Cys a H), 3.57 (dd, 5.7 and 13.7 Hz,
1H, CH2N), 3.44 (dd, 6.5 and 13.7 Hz, 1H, CH2N),
2.97 (dd, 5.3 and 15.1 Hz, 1H, CH2S), 2.86 (dd, 5.9
and 15.1 Hz, 1H, CH2S) .
- 107 -
CA 02207252 1997-06-06
WO 96/21456
PCT/US96/01559
H. 2-Methoxy-4-nitro-3' -tert-butoxycarbonyl-
biphenyl (23).
The coupling of 1-bromo-2-methoxy-4-
nitrobenzene with 3-methylphenylboronic acid
followed by the oxidation gave the 2-methoxy-4-
nitro-3'-carboxybiphenyl. The reaction of acid
chloride with lithium tert-butoxide gave 23 (3
steps, 35%). m.p. 88.0-88.5 C. NMR (CDC13) 6
8.13 (s, IH), 8.00 (d, 7.7 Hz, 1H), 7.89 (d, 8.3
Hz, 1H), 7.81 (s, 1H), 7.69 (d, 7.7 Hz, 1H), 7.48
(m, 2H), 3.90 (s, 3H), 1.60 (s, 9H). 13C NMR
(CDC13) 6 165.2, 156.7, 148.0, 136.3, 136.2, 133.2,
132.0, 130.8, 130.1, 129.0, 127.9, 115.8, 106.0,
81.1, 55.9, 27.9. LRMS (El) for C181119N05 329 (Yr,
30), 273 (100).
N. 2-Methoxy-4-N-(2(R)-N-tert-butoxycarbonyl-
amino-3-triphenylmethylthiopropyliamino-
3'-tert-butoxycarbonylbiphenyl (24).
Compound 24 was prepared using the same
method as for the preparation of compound 18
(yield 63%). m.p. 76.0-77.0 C (decomp). [a]25D=-
11.25 (c=0.01, CH3C00C2H5) . 11.1 NMR (CDC13) 6 8.09
(s, 1H), 7.86 (d, 7.0 Hz, 1H), 7.65 (d, 7.0 Hz,
1H), 7.37 (t, 7.7 Hz, 1H), 7.43 (m, 6H), 7.21-7.32
(m. 9H), 7.11 (d, 8.1 Hz, 1H), 6.21 (s, 1H), 6.18
(d, 8.1 Hz, 1H), 4.58 (d, 6.1 Hz, 1H), 3.86 (br s,
1H), 3.76 (s and m, 4H), 3.14 (br d, 4.9 Hz, 2H),
2.49 (br d, 5.1 Hz, 2H), 1.59 (s, 9H), 1.43 (s,
9H). 13C NMR (CDC13) 6 165.9, 157.3, 155.5, 148.8,
144.3, 138.9, 133.3, 131.5, 131.2, 130.0, 129.4,
127.8, 127.5, 126.7, 118.7, 104.7, 96.2, 80.5,
79.4, 66.8, 55.2, 49.3, 47.0, 34.1, 28.2, 28.1.
Anal. (C451150N205S) C, H, N, S.
0. 2-Methoxy-4-N-[2(R)-amino-3-mercaptopropyl]
amino-3'-carboxybiphenyl (10).
- 108 -
CA 02207252 1997-06-06
WO 96/21456
PCTIUS96/01559
Compound 10 was obtained from the
deprotection of compound 24. m.p. 120-121 C
(decomp). [a]2sD=+12.62 (c=0.01, in methanol). 1H
NMR (CD30D) 6 8.09 (s, 1H), 7.89 (d,7.8 Hz, 111),
7.67 (d, 7.8 Hz, 1H), 7.43 (t, 7.7 Hz, 1H), 7.20
(d, 8.1 Hz, 1H), 6.56 (s, 1H), 6.53 (d, 8.1 Hz,
1H), 3.81 (s, 3H), 3.60 (m, 2H, Cys a H and CH2N)
3.48 (m, 1H, CH2N), 2.96 (dd, 4.9 and 13.7 Hz, 1H,
CH2S), 2.86 (dd, 5.4 and 13.7 Hz, 1H, CH2S) . 13C
NMR (D20 and CD30D) 6 171.1, 158.2, 149.3, 139.7,
135.1, 132.2, 131.1, 130.4, 129.4, 128.4, 120.5,
106.2 (broad, due to deuterium exchange), 98.8,
56.3, 53.4, 45.1, 24.9. LRMS (El) for C17H20N203S 332
(M+). Anal. (C17H20N203S=1.2HC1=1120) C, H, N, S.
P. Cysteiny1-4-amino-3'-carboxybiphenyl (6).
Compound 6 was purified through preparative
HPLC. Purity was shown to be over 9996-. m.p.
120.0-121.0 C. 314 NMR (CD30D) 6 8.25 (s, 1H), 7.98
(d, 7.6 Hz, 1H), 7.84 (d, 7.7 Hz, 1H), 7.74 (d,
7.0 Hz, 2H), 7.54 (t, 7.7 Hz, 1H), 7.66 (d, 8.6
Hz, 2H), 4.16 (q, 5.0 Hz, 1H), 3.19 (dd, 5.20 and
14.8 Hz, 1H), 3.07 (dd, 7.7 and 14.7 Hz, 1H); 13C
NMR (CD30D) 6 169.8, 166.7, 141.9, 138.7, 137.8,
132.6, 132.2, 130.2, 129.5, 128.8, 128.5, 121.6,
56.9, 26.4. LRMS (El) for C16113.603N2S 316 (M+, 25),
213 (100); HRMS (El) calcd 316.0882, obsd
316.0867. Anal. (C161116N203S=CF3C00H=H20) C, H, N.
Q. 4-N-[2(R)-Amino-3-mercaptopropyl]amino-2'-
carboxybiphenyl (3).
m.p. 129-130 C (decomp). [a]25D=+12.58
(c=0.01, CH3OH). 1H NMR (CD30D) 6 7.73 (d, 7.6 Hz,
1H), 7.50 (d, 7.6 Hz, 1H), 7.35 (m, 2H), 7.21 (d,
8.5 Hz, 2H), 6.86 (d, 8.5 Hz, 2H), 3.58 (m, 2H),
3.46 (m, 1H), 2.97 (dd, 4.8 and 14.6 Hz, 1H), 2.86
(dd, 5.4 and 14.6 Hz, 1H). 13C NMR (D20 and CD30D) 6
- 109 -
CA 02207252 1997-06-06
WO 96/21456
PCT/US96/01559
174.3, 146.3, 141.9, 132.8, 132.7, 131.4, 130.6,
130.2, 127.9, 115.3, 52.9, 45.8, 25Ø LRMS (FAB,
glycerol) for C161118N202S (M+1) 303. Anal.
(C261118N202S= 1. 6HC1) C, H, N, S.
R. 4-N-[2(R)-Amino-3-mercaptopropyl]amino-4'-
carboxybiphenyl (5).
m.p. 260 C (decomp). [aF25D.+12.20 (c=0.01,
CH3OH). 174 NMR (CD30D) 6 8.03 (d, 8.5 Hz, 2H),:1.66
(d, 8.4 Hz, 2H), 7.56 (d, 8.4 Hz, 2H), 6.85 (d,
8.5 Hz, 2H), 3.57 (m, 2H), 3.45 (m, 1H), 2.98 (dd,
4.8 and 14.5 Hz, 111), 2.85 (dd, 5.7 and 14.5 Hz,
1H). 13C NMR (D20 and CD300) 6 169.8, 146.4, 146.1,
133.6, 131.4, 129.8, 129.4, 127.1, 117.0, 53.3,
47.2, 25.5. LRMS (El) for Ca6113.802N2S 302 (M+, 15),
285 (15), 226 (100), 213 (50). HRMS (El) calcd
302.1088, obsd 302.1089. Anal. (C261-118N202S=2HC1) C,
H, N.
S. 4-N-(2(R)Amino-3-Mercaptopropyl]aminobiphenY1
(7) .
m.p. 216 C (decomp). [a]25D=+13.27 (c=0.01,
CH3OH) . NMR (CD30D) a 7.54 (m, 4H), 7.39 (m,
2H), 7.26 (m, 1H), 6.82 (br s, 2H), 3.56 (br m,
2H), 3.45 (m, 1H), 2.98 (m, 1H), 2.87 (m, 1H); 3.3C
NMR (CD30D) 6 144.8, 141.8, 135.7, 129.9, 129.1,
127.8, 127.4, 117.4, 53.2, 47.6, 25.5. LRMS (El)
for Ci5Hi8N2S 258 (M+, 15), 182 (100); HRMS (El)
calcd 258.1190, obsd 258.1183. Anal.
(C3.51-13.8N2S-1.6HC1) C, H, N, S.
T. 4-N-(2(R)-Amino-3-mercaptopropyl)amino-
3'-methylbiphenyl (8).
NMR (CD30D) 6 7.50 (d, 8.2 Hz, 2H), 7.35
(m, 2H), 7.27 (t, 7.6 Hz, 1H), 7.08 (d, 7.3 Hz,
1H), 6.95 (d, 8.2 Hz, 2H), 3.60 (m, 2H), 3.46 (m,
1H), 2.99 (dd, 4.9 and 14.6 Hz, 1H), 2.88 (dd, 5.5
- 110 -
CA 02207252 1997-06-06
PCT/US96/01559
W096121456
and 14.6 Hz, 1H) , 2.37 (s, 3H) . LRMS (El) for
C16H20N2S 272 (M+, 15) , 196 (100) . HRMS (El) calcd
272.1341, obsd 272.1347.
U. 4-N- [2(R) -Amino - 3 -mercaptopropyl] amino - 3 ' -
methoxycarbonylbiphenyl ( 9 ) .
m.p . 86-89 C (decomp) 1H NMR (CD30D) (5 8.18
(s, 1H) , 7.90 (d, 7.7 Hz, 1H) , 7.79 (d, 6.6 Hz,
1H) , 7.55 (d, 8.6 Hz, 2H) , 7.49 (t, 7.7 Hz, 1H) ,
7.01 (d, 8.6 Hz, 2H) , 3.92 (s, 3H) , 3.543.65 (m,
2H) , 3.44-3.52 (m, 1H) , 2.97 (dd, 4.7 and 14.7 Hz,
1H) , 2.88 (dd, 5.5 and 14.7 Hz, 1H) . LRMS (El) for
C17H2002N2S 316 (M+ 15) , 299 (20) , 240 (100) . HRMS
(El) calcd 316.1240, obsd 316.1239. Anal.
( Cr7H20N202S = 2HC1 ) C, H, N, S.
- 111 -
CA 02207252 1997-06-06
WO 96/21456
PCT/US96/01559
Table 2
Table of Microanalysis Data
. Compd Formulae C% (cal, obs) H% (cal, obs) N%
(cal, obs) S% (cal, obs)
3 C16HI8N2028.1.6HCI 53.27 (53.41) 5.44 (5.78)
7.77 (7.35) 8.87 (8.47)
4 C 16H 8N2025.2HCI 51.20 (51.60) 5.37 (5.30)
7.47 (7.07) 8.53 (8.22)
C16HI8N202S=2HCI 51.20 (51.62) 5.37 {5.56) 7.47 (7.00)
6 Ci6Hi6N203S=CF3COOH.H20 48.21 (48.24) 4.24 (4.20) 6.25
(6.32)
7 CI5HI8N2S.1.6HC1 56.89 (57.04) , 6.19 (6.46)
8.85 (8.74) 10.11 (10.03)
9 C17H2ON2025-2HC1 52.44 (52.84) 5.65 (5.92)
7.19 (7.37) 8.22 (8.53)
=
C17E1201s1203S.1.21-1C1+120 51.80 (51.91) 5.89 (5.96)
7.11 (6.81) 8.12(7.77)
11 Ci7HigN203S=HC1-0.6H20 54.07 (54.11) 5.35 (5.39) 7.42
(7.35)
CI3H9N04 64.19 (64.05) 3.70 (3.75) 5.76 (5.80)
16 CI7H17N04 68.23 (68.07) 5.68 (5.73) 4.68
(4.64) ,
18 C44H48N204S-1.2H20 73.17 (72.82) 6.98 (6.83)
3.88 (3.87) 4.43 (4.50)
21 C45H48N205S 74.14 (73.74) 6.64 (6.74) 3.84
(3.79) 4.39 (4.32)
24 C45H50N205S 73.97 (73.72) 6.85 (7.04) 3.83
(3.66) 4.38 0.32)
- 112 -
CA 02207252 1997-06-06
WO 96/21456
PCI113S96101559
Table 3
Examples of Peptidomimetics of the Invention
Compound Structure
2 FTI-232 Cys-4-aminobenzoyl-Met
2a FTI-249 red.Cys-4-aminobenzoyl-Met
3 FTI-273 red.Cys-4-amino-2'-carboxybiphenyl
4 FTI-265 red.Cys-4-amino-3'-carboxybiphenyl
5 FTI-271 red.Cys-4-amino-4'-carboxybiphenyl =
6 FTI-278 Cys-4-amino-3'-carboxybiphenyl
7 FTI-268 red.Cys-4-aminobiphenyl
8 FTI-263 red.Cys-4-amino-3'-methylbiphenyl
9 FTI-259 red.Cys-4-amino-3'-carboxymethylbiphenyl
10 FTI-281 red.Cys-4-amino-2-0Me-3'-carboxybiphenyl
11 FTI-285 red.Cys-4-amino-2-phenyl-3'-methylbiphenyl
12 FTI-238 Cys-3-aminomethy1-3'-carboxybiphenyl
FTI-283 red.Cys-3-aminomethy1-3'-carboxybiphenyl
FTI-282 (DL)4-(2,3-diaminopropy1)-amino-3' carboxybiphenyl
FTI-288 red.Cys-4-amino-2-0Pr-3'-carboxybiphenyl
FTI-289 red.Cys-4-amino-2-pheny1-3'-carboxybiphenyl
FTI-291 4-(3-aminoalany1)-amino-3'-carboxybiphenyl
FTI-292 (L)4(2,3-diaminopropy1)-amino-3'-carboxybiphenyl
FTI-295 4-(Ethylsulfony1-3-aminoalany1)-amino-3'-carboxy-
biphenyl
FTI-296 4-(Vinylsulfony1-3-aminoalany1)-amino-3'-
2 5 carboxybiphenyl
FTI-2102 red.Cys-4-amino-3'-tetrazolylbiphenyl
- 113 -
CA 02207252 1997-06-06
WO 96/21456 PCT/US96/01559
Number designations used for compounds of the
invention discussed below is shown in Table 3.
EXAMPLE 22
FTase and GGTase I Activity Assay
Human Burkitt lymphoma (Daudi) cells (ATCC, A
Rockville, MD) were grown in suspension in RPMI
1640 medium containing 109s fetal bovine serum
(FBS) and 19 Pen-Strep in a humidified 109s CO2
incubator at 37 C. The cells were harvested and
sonicated in 50 mM Tris, pH 7.5, 1 mM EDTA, 1 mM
EGTA, 25 Ag/m1 leupeptin, 1 mM
phenylmethylsulfonyl fluoride. Homogenates were
then spun at 12,000 x g and the resulting
supernatant further spun at 60,000 x g. The
supernatant was assayed for both FTase and GGTase
I. Briefly, 100 Ag of the supernatants was
incubated in 50 mM Tris, pH 7.5, 50 AM ZnC12, 20 mM
KC1, 3 mM MgC12 and 1 mM DTT. For FTase assays,
the reaction was incubated at 37 C for 30 minutes
with recombinant H-Ras-CVLS (11 AM) and [3H] FPP
(625 nM; 16.3 Ci/mmol). For GGTase assays, the
reaction was also incubated for 30 minutes at 37 C
but with recombinant H-Ras-CVLL (5 AM) and PH]
GGPP (525 nM; 19.0 Ci/mmol). The reaction was
stopped and passed through glass fiber filters to
separate free and incorporated label. For
inhibition studies, the peptidomimetics were
premixed with FTase or GGTase I prior to adding
the remainder of the reaction mixture.
Recombinant H-Ras-CVLS was prepared as described
previously (26) from bacteria (31). Recombinant
H-Ras-CVLL was prepared from bacteria (32).
- 114 -
CA 02207252 1997-06-06
WO 96/21456
PCT1US96101559
EXAMPLE 23
Peptidomimetics Farnesvlation Assay
The ability of human Burkitt lymphoma (Daudi)
FTase to farnesylate peptides and peptidomimetics
was determined as described previously (34, 35).
Briefly, 25 Al of reaction mixture containing 50
Ag of 60,000 xg supernatants and 20 AM
peptidomimetic in 50 mM Tris, pH 7.5, 50 AM ZnC12, =
20 mM KC1, 3 mM MgCl2, 1 mM DTT and 0.2% octy1S-D-
glucoside was incubated for 30 minutes at 37 C,
then spotted onto silica gel G TLC sheets (20 x 20
cm, Brinkmann Instruments), and developed with n-
propano1/5 N ammonium hydroxide/water (6:1:1).
The dried sheets were sprayed with En3Hance (DuPont
NEN) and exposed to x-ray film for detection of
PH] farnesylated products.
EXAMPLE 24
Ras and Rap1A Processing Assay
EJ3 cells were treated with peptidomimetics
or vehicle for 20-24 h. Cells were lysed in lysis '
buffer (10 mM Na2HPO4, pH 7.25, 150 mM NaCl, 0.1%
sodium dodecyl sulfate, 1% Triton X-100, 12 mM
sodium deoxycholate, 1 mM NaF, 0.2% NaN3, 2 mM
PMSF, 25 Ag/ml leupeptin) and the lysates were
cleared by spinning at 13,000 rpm for 15 minutes.
Ras protein was immunoprecipitated overnight at
4 C with 50 lig of anti-Ras antibody (Y13-259;
hybridoma from ATCC, Rockville, MD) along with 30
Al Protein A-agarose goat anti-rat IgG complex
(Oncogene Science, Uniondale, NY).
Immunoprecipitates were washed 4 times with lysis
buffer and the bound proteins were released by
heating for 5 minutes in 40 Al SDS-PAGE sample
buffer and subsequently electrophoresed on a 12.5%
SDS-PAGE. Proteins were transferred onto
nitrocellulose and subsequently blocked with 5%
- 115
CA 02207252 1997-06-06
VM396/21456
PCTMS96/01559
non-fat dry milk in PBS (containing 1% Tween 20
(PBS-T) and probed with Y13-259 (50 Ag/m1 in 3%
non-fat dry milk in PBS-T). Positive antibody
reactions were visualized using peroxidase-
conjugated goat anti-rat IgG (Oncogene Science,
Uniondale, NY) and an enhanced chemiluminescence
detection system (ECL; Amersham).
For RaplA processing assays, 50 Ag of cell
lysates were electrophoresed as described above
for Ras processing and transferred to
nitrocellulose. These membranes were then blocked
with 5% milk in Tris-buffered saline, pH 8.0,
containing 0.5% Tween-20 and probed with anti-
RaplA (1 Ag/m1 in 5% milk/TBS-T; Santa Cruz
Biotechnology, Santa Cruz, CA). Antibody
reactions were visualized using peroxidase-
conjugated goat anti-rabbit IgG (Oncogene) and ECL
chemiluminescence as described above.
Structural Modeling (Figure 13)
The calculation of the.energy minimized
conformations was carried out using the AMBER
force field within the MacroModel program, version
3.5a.
EXAMPLE 25
The potency of the peptidomimetics of Figure
12 and Table 3 for inhibiting partially purified
FTase was evaluated by determining their ability
to inhibit the transfer of farnesyl to recombinant
H-Ras as described above. The results are
summarized in Table 4, which indicates the ICsos
obtained for FTase activity and GGTase-I activity,
and the selectivity for a number of
peptidomimetics of the invention. The IC5c, values
given in Table 4 represent inhibition of FTase and
GGTase I in vitro by the listed compounds.
- 116 -
CA 02207252 1997-06-06
WO 96/21456
PCT/US96/01559
Table 4
In vitro Activity of CAAX Mimetic Inhibitors of FTase
_
_______________________________________________________________________________
FTase IC50 GGTase-I ICõ,
Inhibitor (nM) (nM) Selectivity
Substrate
4
FTI-232 150 1500 10
FTI-249 3004400 15
,
FTI-273 543(3)* 140,000 (2)a 258
ndb
FTI-265 114 (10) 100,000 (6) 877 no
-
FTI-271 4575(4) >100,000 (2) >22 no
_
FTI-278 13,500(2) 100,000 (2) 7 nd
FTI-268 1,070(3) >100,000 (3) 93 no
FTI-263 710 (3) >100,000 (3) 141 no
FTI-259 917 (3) >100,000 (3) 109 no
FTI-281 40 (6) 43,600 (5) 1090 =
nd
1
FTI-238 100,000 (2) >100,000 (3) >1 no
FTI-283 11,000 (1) 35,000 (2) 3 nd
FTI-285 2075 (4) 8500 (2) 4
FTI-282 50,000 (1) >>100,000(1) >2
FTI-288 41 (6) 2375 (4) 59
FTI-289 16 (5) 643 (4) 40
FTI-291 210,000 (1) 0
FTI-292 60,000 (1) 0
9'TI-295 200,000 (1) >1,000,000 (1) >5
FTI-296 430,000 (1) 1,000,000 (1) >2
FTI-2102 30 (1) 5,000 (1) 187
aNumbers in parentheses indicate number of determinations.
Where no number is given, at least two determinations were
made.
.. bnd indicates not determined.
_
- 117 -
CA 02207252 1997-06-06
W096/21456
PCT/US96/01559
The results obtained showed that compound 2,
i.e. Cys-4ABA-Met (1-10 AM) inhibited FTase in a
concentration-dependent manner with an IC50 of 150
nM (FTI-232, Table 4). This value is similar to
the previously reported IC50 values for CVIM and
Cys-4ABA-Met (35). Reduction of the amide bond
between cysteine and aminobenzoic acid gave the
red-Cys-4ABA-Met (2a, FTI-249) which had an IC50 of
300 nM. However, replacing the methionine and the
C-terminal amide bond in (2a) by another aromatic
ring to obtain the biphenyl-based peptidomimetic
(4) improved potency by twofold (FTI-265, Table
4). Peptidomimetic 4 had an IC50 of 114 nM towards
partially purified FTase from human Burkitt
lymphoma cells and 50 nM towards rat brain FTase
purified to homogeneity. Thus, despite major
structural differences between the compound CVIM
(1) and 4, the latter (4) retained the potent
FTase inhibitory activity of the tetrapeptide CVIM
(1) and the peptide mimetics 2 and 2a.
As noted, Figures 14A and 14B graphically
illustrate the results of FTase and GGTase I
inhibition studies. In these studies, partially
purified FTase and GGTase I were incubated with
the peptidomimetics to be tested and their ability
to transfer [3H] farnesyl to H-Ras-CVLS (FTase) and
[3H] geranylgeranyl to H-Ras CVLL (CCTase I) was
determined as described. Figure 14A shows FTase
inhibition by: 0, (4) and R, (5) while Figure 14B
plots FTase (0) and GGTase I OF inhibition by
(4). Each curve is representative of at least
four independent experiments.
Geranylgeranylation is a more common protein
prenylation than farnesylation (49). It is,
therefore, advantageous for CAAX peptidomimetics
targeting farnesylation to have high selectivity
towards inhibiting FTase compared to GGTase. In
- 118 -
CA 02207252 1997-06-06
14/096/:11456
PCT/US96/01559
the CAAX tetrapeptides, the X position determines
whether the cysteine thiol will be farnesylated by
FTase or geranylgeranylated by GGTase I. Those
= proteins or peptides with Leu or Ile at the X
position are geranylgeranylated. As shown in
Table 4, the present compounds do not
significantly inhibit GGTase I and demonstrate
much greater selectivity for FTase.
Figure 14B shows that compound 4, which is a
potent FTase inhibitor, is a very poor GGTase I
inhibitor. The ability of compound 4 to inhibit
the transfer of geranylgeranY1 to Ras-CVLL (IC50
100,000 nM) was found to be 877-fold less than
that of 4 to inhibit the transfer of farnesyl to
Ras-CVLS (IC50 = 114 nM) (Table 4). This
selectivity was much more pronounced than in the
peptidomimetics 2 and 2a which were more selective
for FTase relative to GGTase I by only 10 and 15-
fold, respectively. It is also noted that the
free carboxylate of compound 4 is not responsible
for this selectivity since replacement of this
group by a methyl in compound 8 did not increase
affinity towards GGTase I (Table 4). These
results indicate that the FTase and GGTase I
binding sites are quite different and that
differences between Leu, Ile and Met side chains
cannot be the only predictors of selectivity.
Regardless of the explanation, it is clear that
the compounds of the invention are much more
selective to inhibition of FTase.
Besides having poor cellular uptake and being
rapidly degraded, another disadvantage of natural
CAAX peptides is that they are farnesylated by
FTase. This results in metabolic inactivation
since farnesylated CAAX derivatives are no longer
inhibitors of FTase (34). Figure 15 shows that
the natural peptide CVLS (carboxyl terminal CAAX
- 119 -
CA 02207252 1997-06-06
W096/21456
PCT/US96/01559
of H-Ras) is farnesylated by FTase from Burkitt
lymphoma cells. Replacing the tripeptide VLS with
4-amino-3'-hydroxycarbonylbiphenyl, as in 4 did
not affect potency towards FTase inhibition (Table
4) but prevented farnesylation of the cysteine
thiol (Figure 15). None of the peptidomimetics of
the invention is metabolically-inactivated by
FTase (Figure 15). Thus, although AAX tripeptides
are not necessary for potent FTase inhibition,
they appear to be required for farnesylation.
With reference to Figure 15, it is to be
noted that the transfer of [1] farnesyl to
peptides and peptidomimetic by FTase was
determined by silica G TLC as described below.
FPP, F-peptide, and ORIGIN designate farnesyl
pyrophosphate, farnesylated peptide and origin,
respectively. Figure 15 shows: Lane 1, FPP only;
lane 2, FPP and CVLS but no FTase; lane 3, FPP and
FTase but not peptide. Lanes 4-9 all contained
FTase and FPP with lane 4, CVIM; lane 5, CVLS;
lane 6, compound 2a; lane 7, compound 4; lane 8,
compound 5; lane 9, compound 8. The results shown
indicate that the compounds of the invention are
not farnesylated in contrast to the CAAX
compounds. Data given are representative of two
independent experiments.
The foregoing results show that the novel
peptidomimetics described herein have two very
important features, namely, they are potent FTase
inhibitors, and they are resistant to metabolic
inactivation by FTase. Another important feature
is that the present compounds inhibit Ras
processing in whole cells. This is shown by the
following with reference to Figure 16 which
illustrates Ras and RaplA processing. To this
end, Ras transformed 3T3 cells were treated with
inhibitors, lysed and the lysate PO
- 120 -
CA 02207252 1997-06-06
14/396/21456
PCT/US96/01559
immunoprecipitated with anti-Ras antibody or B)
separated by SDS-PAGE. Immunoprecipitates from A)
were separated by. SDS-PAGE and blotted with anti-
Ras antibody whereas samples from B) were blotted
with anti-RaplA antibody as described hereafter.
Figure 16 shows: Lane 1, control; lane 2,
lovastatin; lane 3, reduced 2a (200 AM); lane 4, 4
(100 AM); lane 5, 4 (50 AM); lane 6, 4 (25 AM);
lane 7, 5; lane 8, 8. Data are representative of
3 independent experiments. Farnesylated Ras runs
faster than unprocessed Ras on SDS-PAGE (23-25,
28, 29). Figure 16A (lane 1) shows that cells
treated with vehicle contain only processed Ras
whereas cells treated with lovastatin (lane 2)
contained both processed and unprocessed Ras
indicating that lovastatin inhibited Ras
processing. Lovastatin, an HMG-CoA reductase
inhibitor which inhibits the biosynthesis of
farnesylpyrophosphate and
geranylgeranylpyrophosphate, is used routinely as
a positive control for inhibition of processing of
both geranylgeranylated and farnesylated proteins
(36, 37, 39, 40, 51). Cells treated with reduced
Cys-4ABA-Met 3 in its free carboxylate forms did
not inhibit Ras processing. However, in contrast,
the corresponding methyl ester of 2a (200 AM)
inhibited FTase (Figure 16A, lane 3). This is
consistent with previous work that showed that
neutralization of the carboxylate of CAAX peptides
enhances their ability to inhibit Ras processing
(37, 40, 51). Although compound 4 has a free
carboxylate negative charge, it was able to enter
cells and potently inhibit Ras processing (lane 4,
= 100 AM compound 4). It was found that compound 4
inhibited Ras processing with concentrations as
low as 50 AM (lane 5), whereas its corresponding
parent compound 2a did not inhibit Ras processing
- 121 -
CA 02207252 1997-06-06
WO 96/21456 PCT/US96/01559
at concentrations as high as 200 M. Compound 4
was as potent as the methylester of its parent
compound (2a) (Figure 16A, lane 3). Furthermore,
4 appears to be the first CAAX peptidomimetic that
effectively inhibits Ras processing in whole cells
directly without relying on cellular enzymes for
activation. The hydrophobic character of the
.
biphenyl group apparently compensates for the free
carboxylate negative charge thus allowing the
peptidomimetic to penetrate membranes and
promoting its cellular uptake.
The selectivity of the Present Ras
farnesylation inhibitors has also been
investigated by determining their ability to
inhibit processing of RaplA, a small G-protein
that is geranylgeranylated (49, 50). Cells were
treated with lovastatin or peptidomimetics exactly
as described for Ras processing experiments.
Lysates were then separated by SDS-PAGE and
immunoblotted with anti-RaplA antibody as
described below. Control cells contained only the
geranylgeranylated RaplA (Figure 16B, lane 1)
whereas lovastatin-treated cells contained both
processed and unprocessed forms of RaplA
indicating, as expected, that lovastatin inhibited
the processing of RaplA (Figure 16B, lane 2).
Compound 4, which inhibited Ras processing, was
not able to inhibit RaplA geranylgeranylation
(Figure 16B, lanes 4-6). Compounds 5 and 8 also
did not inhibit RaplA processing (Figure 16B,
lanes 7 and 8).
Structures for a number of compounds of the
invention are summarized in Table 3, and their
=
relative effectiveness in inhibiting FTTase and
GGTase shown in Table 4. The activity of the
inhibitors is reported in Table 4 as ICso values,
the concentration at which FTase or GGTase I
- 122 -
CA 02207252 1997-06-06
WO 96121456
PCUUS96/01559
activity was inhibited by 50%-. Some of the
inhibitors were further characterized for their
ability to serve as substrates for farneylsation
by thin layer chromatography, as shown in Table 4
(41).
The published sequence dependence studies on
FTase have shown a strong preference for
methionine in the terminal position of CAAX. In
the compounds of the present invention, no
methionine residue is present and the tripeptide
AX is completely replaced by a simple hydrophobic
moiety. The most potent inhibitor in the CAAX
series is Cys-Ile-Phe-Met (18, 22) with an IC50
value of 30 nM. Peptidomimetic inhibitor 10 is as
potent as CIFM despite the large difference
between their structures. These results confirm
the hydrophobic strategy for AAX replacement
according to the invention.
As previously reported, the incorporation of
an aromatic amino acid into the A2 position of
CA1A2X (such as CIFM) prevents the tetrapeptide
from serving as a substrate for farneyslation
(22). Table 4 shows that the designed non-peptide
CAAX mimetics (such as compound 4) are not
substrates for farnesylation. This lack of
farnesylation by FTase may be due to the inhibitor
binding to the enzyme in a conformation that does
not permit farnesyl transfer to the thiol group.
III. Geranylgeranyl tranferase Inhibitors
The carboxyl terminal CAAX tetrapeptide of
Ras is a substrate for FTase and serves as a
target for designing inhibitors of this enzyme
with potential anticancer activity (33). Our
earlier application describes a highly potent
(IC50=500 pM) inhibitor of FTase, FTI-276 (Fig. 17)
- 123 -
CA 02207252 1997-06-06
W096/21456
PCT/US96/01559
(66). Its cell-permeable methyl ester FTI-277
inhibits H-Ras processing in whole cells with an
IC50 of 100 nM (66). Furthermore, FTI-276 is
highly selective (100-fold) for FTase over GGTase
I (Table 5).
TABLE 5
In Vitro (ICõ UN) In Vivo Processing UN)
FTase GGTase I H-Ras K-Ras RaolA
FTI-276 0.5 50 FTI-277 0.1 10 50
GGTI-287 25 5 _GGTI-286 >30 2 2
GGTI-297 270 40 GGTI-298 >20 3 3
EXAMPLE 26
Synthesis of FTase and GGTase I Inhibitors
Peptidomimetics FTI-276 and FTI-277 were
prepared as described above. GGTase I inhibitors
GGTI-287 and GGTI-286 were prepared from 2-phenyl-
4-nitrobenzoic acid (66) by reaction with L-
leucine methyl ester followed by reduction with
stannous chloride. The resulting 4-amino-2-
phenylbenzoyl leucine methyl ester was reacted
with N-Boc-S-trityl-cysteinal and deprotected by
procedures similar to those described for the
FTase inhibitors (66) to give GGTI-286 and GGTI-
287 as their hydrochloride salts.
A. 4-Amino-2-phenylbenzoy1-(S)-methionine
methyl ester hydrochloride
To a mixture of 70 mL of acetone and 85 mL of
water was added 2-bromo-4-nitrotoluene 6.84 g (30
mmol), phenylboronic acid 3.84 g (31.5 mmol),
potassium carbonate 10.35 g (75 mmol) and
palladium acetate 336 mg (1.5 mmol). The mixture
was ref luxed for 10 hr and then extracted with
ether and dilute hydrochloric acid. After
evaporating solvents, the solid residue was
recrystallized from methanol to give 5.64 g of 4-
- 124 -
CA 02207252 1997-06-06
WO 96/21456
PCT/US96101559
nitro-2-phenyltoluene (88% yield). 11.1 NMR (CDC10 6
8.09-8.11 (m, 2H), 7.40-7.49 (m, 4H), 7.30-7.33
(m, 2H), 2.37 (s, 3H).
The above 4-nitro-2-phenyltoluene (4.46 g, 21
mmol) was suspended in 21 mL of pyridine and 42 mL
of water. The mixture was heated to boiling
followed by addition of potassium permanganate
(19.8, 126 mmol). The mixture was ref luxed for 2
hr and then filtered to remove the solids. The
filtrate was acidified with 6N HC1 to give 4.48 g
of 4-nitro-2-phenylbenzoic acid (89% yield). 31.1
NMR (CDC10 6 8.25-8.33 (m, 2H0, 8.08 (d, 8.9 Hz,
1H), 7.41-7.51 (m, 3H), 7.31-7.39 (m, 2H).
The above 4-nitro-2-phenylbenzoic acid (2.43
g, 10 mmol) was suspended in 50 mL of methylene
chloride. To this solution was added (L)-
methionine methyl ester hydrochloride (2.0 g,
10 mmol), triethylamine (1.38 mL, 10 mmol), 1-(3-
dimethylaminopropy1)-3-ethylcarbodiimide
hydrochloride (EDCI, 2.01 g, 10.5 mmol), I-
hydroxybenzotriazole (HOBT, 1.35 g, 10 mmol). The -
mixture was stirred for 12 hr and then extracted
with methylene chloride and 1N hydrochloric acid.
After the evaporation of solvents, the residue was
recrystallized from ethyl acetate and hexane to
give 3.22 g of 4-nitro-2-phenylbenzoly1-(L)-
methionine methyl ester (yield 83%). 11.1 NMR
(CDC10 6 8.24-8.28 (m, 2H), 7.85 (d, 8.9 Hz, 1H),
7.43-7.52 (m, 5H), 6.01 (d, 7.5 Hz, 1H), 4.69
(ddd, 1H), 3.68 (s, 31-i), 2.05 (m, 2H), 1.98 (s,
3H), 1.88-1.96 (m, 1H), 1.72-1.81 (m, 1H).
The 4-nitro-2-phenylbenzoyl-W-methionine
methyl ester (3.04 g, 7.83 mmol) was dissolved
into 100 mL of ethyl acetate followed by the
addition of stannous chloride hydrate (8.84 g, 39
mmol). The mixture was refluxed for 2 hr and then
extracted with a mixture of ethyl acetate and
- 125 -
CA 02207252 1997-06-06
W096/21456
PCT/US96/01559
concentrated sodium bicarbonate. After the
evaporation of solvents, the residue was dissolved
in methylene chloride followed by addition of 3N
hydrogen chloride in ether. The solid was
filtered and dried to give 2.96 g of 4-amino-2-
phenylbenzoy1-(L)-methionine methyl ester
hydrochloride (yield 96%). 1H NMR (CD30D) (5 7.65
(d, 8.1 Hz, 1H), 739-7.46 (m, 7H), 4.53 (dd, 4.3
and 9.5 Hz, 1H), 3.69 (s, 3H), 2.15-2.23 (m, 1H),
2.00 (s, 3H), 1.93-2.11 (m, 2H), 1.74-1.83 (m,
1H); 13C NMR (CD30D) .5 173.4, 171.7, 143.4, 140.1,
137.4, 134.0, 130.9, 129.7, 129.4, 125.5, 122.7,
53.0, 52.9, 31.3, 30.9, 15.1.
B. 4-[2(R)-tert-butoxycarbonylamino-3-
triphenylmethylthiopropyl]amino-2-
*phenylbenzoy1-(S)-methionine methyl
ester
To a mixture of 4-amino-2-phenylbenzoy1-(S)-
methionine methyl ester hydrochloride (1.27 g,
3.22 mmol) in 20 mL of methanol was added N-Boc-S-
trityl-(L)-cysteinal (1.0 eq; according to 111 NMR
determination of aldehyde percentage) and sodium
cyanoborohydride (400 mg, 2.0 eq). The mixture
was stirred for 12 hr. After the evaporation of
solvents, the residue was extracted with ethyl
acetate and concentrated sodium bicarbonate.
After removing solvents, the residue was purified
through flash column chromatography (1:1 = hexane:
ethyl acetate, silica) to give the product 1.67 g
(yield 67%). 1}-1 NMR (CDC10 .5 7.65 (d, 8.6 Hz,
1H), 7.34-7.42 (m, 11H), 7.18-7.29 (m, 9H), 6.52
(dd, 2.3 and 8.1 Hz, 1H), 6.34 (d, 2.3 Hz, 1H),
5.65 (d, 7.7 Hz, 1H), 4.64 (ddd, 1H), 4.55 (d, 8.1
Hz, 1H), 4.19 (br t, 1H), 3.78 (br m, 1H), 3.64
(s, 3H), 3.09 (t, 6.1 Hz, 2H), 2.44 (m, 2H), 2.04-
2.10 (m, 2H), 2.00 (s, 3H), 1.81-1.90 (m, 1H),
1.60-1.70 (m, 1H), 1.41 (s, 9H); 13C NMR (CDC10
- 126 -
CA 02207252 1997-06-06
WO 96/21456
PCT/US96/01559
172.0, 168.3, 155.7, 149.4, 144.3, 141.6, 141.1,
131.3, 129.5, 128.7, 128.5, 127.9, 127.7, 126.8,
122.6, 113.6, 111.3, 79.8, 67.1, 52.2, 51.7, 49.5,
47.2, 34.3, 31.6, 29.4, 28.2, 15.2.
C. 4-[2(R)-amino-3-mercaptopropyl]amino-
.
2-phenylbenzoy1-(S)-methionine methyl
ester hydrochloride
The above N-Boc-S-trityl protected peptide
methyl ester (900 mg) was dissolved in 5 mL of
methanol. To this mixture was added a solution of
mercuric chloride (774 mg, 2,50 eq) in 5 mL of
methanol. The mixture was ref luxed for 20 min.
The precipitate was collected and dried. This
solid was suspended in 10 mL of methanol and
reacted with gaseous hydrogen sulfide. After the
removal of black solid, the clear solution was
evaporated to dryness. The residue was then
dissolved in methylene chloride followed by
addition of 3N hydrogen chloride in ether. The
white solid was collected and dried to give the
pure product 476 mg (yield 8196). 111 NMR (CD30D) 6
7.42 (d, 8.4 Hz, 1H), 7.30-7.38 (m, 5H), 7.77 (d,
8.4 Hz, 1H), 6.71 (s, 1H), 4.48 (dd, 4.2 and 5.1
Hz, 1H), 3.68 (s, 3H), 3.44-3.58 (m, 3H),
2.90-2.95 (dd, 4.1 and 14.5 Hz, 1H), 2.79-2.85
(dd, 4.7 and 14.5 Hz, 1H), 2.18-2.22 (m, 1H),
2.03-2.16 (m, 1H), 2.00 (s, 3H), 1.91-1.97 (m,
1H), 1.73-1.82-(m, 1H); 13C NMR (CD30D) 6 173.7,
173.4, 150.7, 143.5, 142.3, 131.2, 129.8, 129.5,
128.6, 125.6, 115.6, 112.2, 53.7, 53.2, 52.8,
45.0, 31.4, 30.9, 25.3, 15Ø
D. 4-[2(R)-amino-3-mercaptopropyl]amino-
. 2-phenylbenzoy1-(S)-methionine
The N-Boc-S-trityl protected peptide methyl
ester (500 mg) was hydrolyzed with 2.0 eq of
lithium hydroxide at 0 C for 1 hr. The product
- 127 -
CA 02207252 1997-06-06
WO 96/21456 PCT/U596/01559
was deprotected with trifluoroacetic acid (2 mL)
in methylene chloride (1 mL). Triethylsilane was
added dropwise until the deep yellow color
disappeared. The mixture was kept at r.t. for 1.5
hr. After the evaporation of solvents, the
residue was dried and washed with dry ether. The
solid was purified through preparative HPLC to
give a pure product 270 mg (yield 78 O. NMR
(CD30D) (5 7.44 (d, 8.4 Hz, 1H), 7.30-7.39 (m, 5H),
6.75 (d, 8.4 Hz, 1H), 6.67 (s, 1H), 4.45 (dd, 4.2
and 5.1 Hz, 1H), 3.42-3.58 (m, 3H), 2.90 (dd, 4.3
and 14.5 Hz, 1H), 2.81 (dd, 5.5 and 14.5 Hz, 1H),
2.17-2.23 (m, 1H), 2.09-2.15 (m, 1H), 2.00 (s,
3H), 1.90-1.99 (m, 1H), 1.71-1.81 (m, 1H); 13C NMR
(CD30D) (5 176.4, 173.5, 150.4, 143.0, 141.5, 131.0,
129.7, 129.4, 128.9, 124.6, 115.0, 112.3, 53.3,
49.6, 44.4, 30.8, 30.1, 24.9, 14.8.
E. 4-Nitro-2-phenylbenzoyl-
(S)-leucine methyl ester
This compound was prepared through the
coupling of 4-nitro-2-phenylbenzoic acid with
(L)-leucine methyl ester hydrochloride as for the
preparation of the methionine derivative (see
Example 26, section A). 'H NMR (CDC13) (5 8.24-8.26
(m, 2H), 7.86 (d, 8.7 Hz, 1H), 7.41-7.46 (m, 5H),
5.71 (d, 7.4 Hz, 1H), 4.57 (ddd, 1H), 3.67 (s,
3H), 1.37-1.46 (m, 1H), 1.08-1.25 (m, 2H), 0.78
(dd, 6H).
F. 4-(2(R)-tert-butoxycarbonyl-
3-triphenylmethylthiopropyl]amino-2-
phenylbenzoy1-(S)-leucine methyl ester
This compound was prepared using the same
method as for the preparation of methionine
derivative (See Example 26, section B), using 4-
amino-2-phenylbenzoy1-(S)-leucine methyl ester and
N-Boc-S-trityl-(L)-cysteinal as starting
- 128 -
CA 02207252 1997-06-06
WO 96/21456
PCTIUS96/01559
materials. 1H NMR (CDC10 6 7.68 (d, 8.6 Hz, 1H),
7.33-7.41 (m, 11H), 7.17-7.29 (m, 9H), 6.50 (d,
8.6 Hz, 1H), 6.31 (s, 1H), 5.43 (d, 7.8 Hz, 1H),
4.60 (d, 6.1 Hz, 1H), 4.47 (ddd, 1H), 4.19 (br t,
1H), 3.77 (br m, 1H), 3.62 (s, 3H), 3.09 (t, 5.9
Hz, 2H), 2.45 (br m, 2H), 1.40 (s, 9H), 1.27-1.33
(m, 1H), 1.03-1.18 (m, 2H), 0.75 (dd, 6H); 1.3C
(CDC10 6 173.2, 168.2, 155.6, 149.4, 144.4, 141.7,
141.2, 131.4, 129.5, 128.8, 128.5, 127.9, 127.6,
126.8, 122.7, 113.6, 111.3, 79.6, 67.1, 51.9,
50.9, 49.5, 47.1, 41.2, 34.3, 28.3, 24.4, 22.7,
21.8.
G. 4-(2(R)-amino-3-mercaptopropyl]amino-
2-phenylbenzoy1-(S)-leucine methyl ester
hydrochloride
This compound was prepared with the same
method as for the preparation of methionine
derivative (see Example 26, section C), using 4-
[2(R)-tert-butoxycarbony1-3-
triphenylmethylthiopropyllamino-2-phenylbenzoy1-
(S)-leucine methyl ester and mercuric chloride. 1H
NMR (CD30D) 6 7.42 (d, 8.5 Hz, 1H), 7.31-7.38 (m,
5H), 6.76 (d, 8.5 Hz, 1H), 6.68 (s, 1H), 4.33 (t,
7.8 Hz, 1H), 3.67 (s, 3H), 3.46-3.55 (m, 3H), 2.95
(dd, 4.4 and 14.5 Hz, 1H), 2.81 (dd, 5.1 and 14.5
Hz, 1H), 1.44 (t, 7.6 Hz, 2H), 1.18-1.25 (m, 1H),
0.76-0.83 (dd, 4.1 and 6.6 Hz, 6H).
H. 4-(2(R)-amino-3-mercaptopropyl]amino-
2-phenylbenzoy1-(S)-leucine
This compound was prepared with the same
method as for the preparation of the methionine
derivative (see Example 26, section D), using 4-
.,
[2(R)-amino-3-mercaptopropyl]amino-2-
phenylbenzoy1-(S)-leucine methyl ester
hydrochloride and lithium hydroxide. 114 NMR
(CD30D) 6 7.42 (d, 8.5 Hz, 1H), 7.29-7.38 (m, 5H),
- 129 -
CA 02207252 1997-06-06
PCT/US96/01559
W096/21456
6.73 (d, 8.5 Hz, 1H), 6.66 (s, 1H), 4.32 (dd, 3.3
and 5.9 Hz, 1H),. 3.41-3.57 (m, 3H), 2.94 (dd, 4.3
and 14.5 Hz, 1H), 2.78 (5.2 and 14.5 Hz, 1H), 1.45
(t, 6.7 Hz, 2H), 1.17-1.26 (m, 1H), 0.78-0.83 (t,
8.5 Hz, 6H).
I. 4-Nitro-2-naphthylbenzoic acid
The coupling of 4-nitro-2-bromobenzoic acid
methyl ester (1.92 g, 7.4 mmol) with 1-
naphthylboronic acid (2.53 g, 14.7 mmol) in the
presence of anhydrous sodium phosphate (3.64 g,
22.2 mmol) and palladium
tetrakistriphenylphosphine (426 mg, 0.368 mmol) in
50 mL of DMF at 100 C gave the
4-nitro-2-naphthylbenzoic acid methyl ester (1.66
g, 73% yield). 1H NMR (CDC10 6 8.34 (d, 8.5 Hz,
1H), 8.28 (s, 1H), 8.14 (d, 8.5 Hz, 1H), 7.92 (d,
8.2 Hz, 2H), 7.47-7.56 (m, 2H), 7.41 (d, 3.8 Hz,
2H), 7.34 (d, 6.8 Hz, 1H), 3.40 (s, 3H). After
the hydrolysis of methyl ester, 1.44 g of product
was collected (yield 91%). NMR (CDC10 6 8.33
(d, 8.6 Hz, 1H), 8.23 (s, 1H), 8.17 (d, 8.6 Hz,
1H), 7.88 (d, 8.2 Hz, 2H), 7.46-7.52 (m, 2H),
7.38-7.42 (m, 2H), 7.33 (d, 7.0 Hz, 1H); 13C NMR
(CD3C0CD3) 6 167.2, 150.1, 143.1, 139.0, 138.2,
134.4, 132.5, 132.1, 129.2, 127.3, 126.7, 125.8,
125.9, 123.3.
J. 4-Nitro-2-naphthylbenzoy1-
(3)-methionine methyl ester
The coupling, of 4-nitro-2-naphthylbenzoic
acid with (L)-methionine methyl ester in the
presence of EDCI and HOBT provided the desired
product (yield 95%). TLC of the product showed
single spot, but 1H NMR showed the presence of
diastereomers caused by the restricted rotation
between naphthyl and phenyl rings. 111 NMR (CDC10
6 8.33-8.38 (m, 1H), 8.26 (ss, 1H), 8.14 (d, 8.5
- 130 -
CA 02207252 1997-06-06
WO 96/21456
PCT/US96/01559
Hz, 0.5H), 8.00 (d, 8.5 Hz, 0.5H), 7.94-7.98 (m,
2H), 7.42-7.65 (m, 5H), 5.98 (t, 1H), 4.42 (m,
1H), 3.56 (s, 1.5H), 3.51 (s, 1.5H), 1.83 (s,
1.5H), 1.74 (s, 1.5H), 1.56-1.64 (m, 1H),
1.33-1.45 (m, 2H), 1.09-1.14 (m, 1H).
K. 4-[2(R)-tert-butoxycarbony1-
3-triphenylmethylthiopropyl]amino-
2-naphthyl-(B)-methionine methyl ester
The reduction of
4-nitro-2-naphthylbenzoyl-W-methionine methyl
ester gave a quantitative yield of the amino
derivative which was reacted with N-Boc-S-trityl
cysteinal in the presence of sodium
cyanoborohydride. After flash column
chromatography (1:1 = ethyl acetate : hexane)
purification, a desired product was obtained
(yield 409). TLC showed single spot, but 11.1 NMR
showed diastereomers caused by the restricted
rotation between the naphthyl and phenyl rings. 111
NMR (CDC13) (5. 7.84-7.96 (m, 3H), 7.49-7.66 (m, 4H),
7.37-7.43 (m, 7H), 7.14-7.27 (m, 9H), 6.60-6.63
(d, 8.6 Hz, 1H), 6.33 (m, 1H), 5.67 (d, 7.8 Hz,
0.6H), 5.60 (d, 7.8 Hz, 0.4H), 4.56 (br d, 6.2 Hz,
1H), 4.35-4.44 (m, 1H), 4.30 (br, 1H), 3.78 (br m,
1H), 3.55 (s, 1.9H), 3.38 (s, 1.1H), 3.06 (t, 5.8
Hz, 2H), 2.44 (m, 2H), 1.90 (s, 1H), 1.79 (s, 2H),
1.57-1.68 (m, 0.5H), 1.36-1.45 (m, 10H), 1.23-1.32
(m, 2H), 0.94-0.98 (m, 0.7H).
L. 4-[2(R)-amino-3-mercaptopropy1]amino-
2-naphthylbenzoyl-W-methionine
This compound was prepared from the
N-Boc-S-trityl protected form (section K) by
saponification followed with acidic cleavage by
trifluoroacetic acid. The pure compound was
obtained through preparative HPLC. NMR showed
complicated diastereomers caused by the restricted
- 131 -
CA 02207252 1997-06-06
PCT/US96/01559
W096/21456
rotation of aryl-aryl bond. 111 NMR (CD30D) 6
7.86-7.94 (m, 2H), 7.73 (d, 8.6 Hz, 0.6H),
7.35-7.67 (m, 5.4H), 6.83-6.88 (m, 1H), 6.63-6.67
(m, 1H), 4.17-4.23 (m, 1H), 3.41-3.58 (m, 3H),
2.91 (dd, 4.2 and 14.5 Hz, 1H), 2.80 (dd, 5.3 and
14.5 Hz, 1H), 1.82 (s, 1.4H), 1.80 (s, 1.6H),
1.65-1.77 (m, 1H), 1.41-1.52 (m, 2H), 1.09-1.32
(m, 1H).
M. 4-Nitro-2-naphthylbenzoyl-
(S)-leucine methyl ester
This compound was prepared with the same
method as for the preparation of the methionine
derivative (section J) using 4-nitro-2-
naphthylbenzoic acid, (S)-leucine methyl ester,
EDCI and HOBT. NMR (CDC10 6 8.34-8.39 (m, 1H),
8.25 (s, 1H), 8.18 (d, 8.6 Hz, 0.6H), 8.02 (d, 8.6
Hz, 0.4H), 7.91-8.00 (m, 2H), 7.62 (t, 7.0 Hz,
O.611), 7.48-7.58 (m, 3H), 7.41 (t, 7.0 Hz, 1.411),
5.71 (d, 7.9 Hz, 0.6H), 5.60 (d, 7.9 Hz, 0.4H),
4.29 (m, 1H), 3.57 (s, 1.7H), 3.52 (s, 1.3H),
1.05-1.11 (m, 0.511), 0.88-0.97 (m, 0.7H),
O.69-0.78 (m, 0.511), 0.41-0.59 (m, 7.0H),
O.19-0.26 (m, 0.611).
N. 4-(2(R)-tert-butoxycarbonylamino-3-
triphenylmethylthiopropyl]amino-2-
naphthylbenzoy1-(S)-leucine methyl ester
This compound was prepared with the same
method as for the preparation of the methionine
derivative (section K). 'H NMR (CDC10 15 7.85-8.00
(m, 3H), 7.47-7.67 (m, 4H), 7.39-7.43 (m, 711),
7.14-7.37 (m, 911), 6.61 (d, 8.6 Hz, 111), 6.32 (s,
111), 5.46 (d, 7.6 Hz, 0.6H), 5.36 (d, 7.6 Hz,
O.4H), 4.55 (d, 7.2 Hz, 1H), 4.20-4.27 (m, 211),
3.76 (br, 111), 3.56 (s, 2H), 3.38 (s, 111), 3.06
(t, 5.9 Hz, 2H), 2.43 (m, 211), 1.36-1.43 (m, 9H),
- 132 -
CA 02207252 1997-06-06
WO 96/21456
PCT/US96/01559
0.81-1.03 (m, 1H), 0.55-0.67 (m, 2.8H), 0.36-0.45
(m, 4.7H), 0.00-0.09 (m, 0.6H).
0. 4-[2(R)amino-3-mercaptopropyl]amino-
,
2-naphthylbenzoy1-(S) -leucine methyl
ester
This compound was prepared from the
N-Boc-S-trityl methyl ester of the corresponding
compound, using the method of section L. 1H NMR
(CDC13) 6 7.98 (d, 8.5 Hz, 0.6H), 7.84-7.90
(m, 2.4H), 7.5 (d, 8.5 Hz, 0.4H), 7.43-7.58 (m,
3.6H), 7.34-7.39 (m, 1H), 6.72 (m, 1H), 6.45 (ss,
1H), 5.46 (d, 7:8 Hz, 0.6H), 5.40 (d, 7.7 Hz,
0.4H), 4.64 (m, 1H), 4.23 (m, 1H), 3.54 (s, 2H),
3.30 (s, 1H), 3.25 (m, 1H), 2.97-3.06 (m, 2H),
2.67 (dd, 3.7 and 13.1 Hz, 1H), 2.47 (dd, 6.5 and
13.2 Hz, 1H), 1.45-1.65 (br s, 2H), 0.81-1.03 (m,
1.2H), 0.54-0.67 (m, 3H), 0.36-0.39 (m, 4.3H),
0.00-0.10 (m, 0.7H).
EXAMPLE 27
FTase and GGTase I Activity Assay
FTase and GGTase I activities from 60,000 x g
supernatants of human Burkitt lymphoma (Daudi)
cells (ATCC, Rockville, MD) were assayed exactly
as described previously for FTase (41).
Inhibition studies were performed by determining
the ability of Ras CAAX peptidomimetics to inhibit
the transfer of PH]-farnesyl and Pin-
geranylgeranyl from PHIFPP and [3H]GGPP to H-ras-
CVLS and H-Ras-CVLL, respectively (41).
EXAMPLE 28
Ras and Rap.IA Processing Assay
H-Ras cells (45) and K-Ras4B Cells (32) were
kind gifts from Dr. Channing Der and Dr. Adrienne
Cox (University of North Carolina, Chapel Hill).
- 133 -
CA 02207252 1997-06-06
WO 96/21456
PCT/US96/01559
Means of obtaining these cell lines will be easily
recognized by the skilled practitioner.
Cells were seeded on day 0 in 100 mm dishes in
Dulbecco's modified Eagles medium supplemented
with 10% calf serum and 1% penicillin-
streptomycin. On days 1 and 2, cells were refed
with medium containing various concentrations of
FTI-277, GGTI-286 or vehicle (10 mM DTT in DMSO).
On day 3, cells were washed and lysed in lysis
buffer containing 50 mM HEPES, pH 7.5, 10 mM NaCl,
1% TX-100, 10% glycerol, 5 mM MgCl2, 1 mM EGTA, 25
gig/m1 leupeptin, 2 mM PMSF, 2 mM Na3VO4, 1 mg/ml
soybean trypsin inhibitor, 10 Ag/m1 aprotinin, 6.4
mg/ml Sigma-104e phosphatase substrate. Lysates
were cleared (14,000 rpm, 4 C, 15 min) and equal
amounts of protein were separated on a 12.5% SDS-
PAGE, transferred to nitrocellulose, and
immunoblotted using an anti-Ras antibody (Y13-259,
ATCC) or an anti-RapIA antibody (SC-65, Santa Cruz
Biotechnology, Santa Cruz, CA). Antibody
=
reactions were visualized using either peroxidase-
conjugated goat anti-rat 1gG (for Y13-259), or
peroxidase-conjugated goat anti-rabbit lgG (for
RaplA) and an enhanced chemiluminescence detection
(ECL, Amersham Corp.), as described previously
(41).
EXAMPLE 29
MAP Kanase Immunoblotting
Cells were treated with FTI-277, GGTI-286, or
vehicle and lysed as previously described for Ras
and Rap1A processing. Equal amounts of protein
were separated on a 15% SDS-PAGE, transferred to
nitrocellulose, and immunoblotted using an anti-
MAP kinase antibody (erk2, monoclonal, UB1, Lake
Placid, NY). Antibody reactions were visualized
using peroxidase-conjugated donkey anti-mouse IgG
- 134 -
'
CA 02207252 1997-06-06
WO 96/21456
PCT/US96/01559
(Jackson ImmunoResearch Laboratories Inc., West
Grove, PA) and an enhanced chemiluminescence
detection system (ECL, Amersham Corp.)
EXAMPLE 30
Inhibition of GGTase I by GGTI-286
GGTI-287 potently inhibited GGTase I in vitro
(IC50=5 nM) and was selective towards inhibiting
GGTase I over FTase (IC50=25 nM) (Table 5). Thus,
the substitution of methionine in FTI-276 by a
leucine in GGTI-287 (Fig. 17) increased the
potency towards GGTase I by approximately 10-fold
(Table 5). More importantly, it reversed the
selectivity from a FTase to a GGTase I-specific
inhibitor by a factor of 500 (Table 5). To
determine whether this selectivity is respected in
whole cells, the cell-permeable methyl ester .
derivative of GGTI-287, GGTI-286 (Fig. 17), was
synthesized and used to treat NIH 3T3 cells which
overexpress oncogenic H-Ras-CVLS (31). Cell
lysates were electrophoresed. on SDS-PAGE and
immunoblotted with an anti-Ras antibody as
described in Example 28. Figure 18 shows that
accumulation of unprocessed H-Ras did not occur at
concentrations lower than 30 AM GGTI-286.
Therefore, GGTI-286 is not a good inhibitor of H-
Ras processing in whole cells. However, GGTI-286
was a very potent inhibitor of the processing of
the geranylgeranylated RaplA protein (IC50=2 AM)
(Fig. 18). Thus, GGTI-286 is more than 15-fold
selective for inhibition of geranylgeranylation
over farnesylation processing (Table 5). This
data is in direct contrast to the FTase specific
inhibitor FTI-277 which inhibited H-Ras and RapIA
processing with IC50s of 100 nM and 50 AM,
respectively (Fig. 18). Thus, GGTI-286 is 25-fold
- 135 -
CA 02207252 1997-06-06
WO 96/21456
PCT/US96/01559
more potent than FTI-277 at inhibiting
geranylgeranylation in whole cells (Table 5).
EXAMPLE 31
Inhibition of GGTase I by GGTI-297 and GGTI-298
To determine the effect of replacing the
phenyl substituent with a naphthyl on GGTase I
inhibition, 4-[2(R)-amino-3-mercaptopropyl] amino-
2-naphthyl benzoyl-W-leucine (GGTI-297) and its
methylester (GGTI-298) were tested. GGTI-297
inhibited GGTase I in vitro with an IC50 of 40 nM
and was selective towards inhibiting GGTase I over
FTase (IC50 = 270 nM) (Fig. 21, Table 5). Thus, the
substitution of phenyl in GGTI-287 by naphthyl in
GGTI-297 decreased the potency towards GGTase I by
8 fold and towards FTase by over 10-fold.
However, more importantly, the selectivity for
GGTase I over FTase increased from 5-fold (GGTI-
287) to 7-fold (GGTI-297), as shown in Table 5.
This selectivity was also respected in vivo since
concentrations as high as 20 AM did not inhibit H-_
Ras processing whereas Rap1A and Ras4B were
completely blocked at 10 AM GGTI-298 (Table 5).
EXAMPLE 32
Inhibition of K-Ras4B Function by GGTI-286
The ability of GGTI-286 to inhibit the
processing and signaling of oncogenic K-Ras4B was
then evaluated. NIH 3T3 cells which overexpress
oncogenic K-Ras4B (32) were treated either GGTI-
286 (0-30 AM) or FTI-277 (0-30 AM) and the lysates
were immunoblotted with an anti-Ras antibody as
described under Example 28. Figure 19 shows that
GGTI-286 inhibited potently the processing of K-
Ras4B with an IC50 of 2 M. The ability of GGTI-
286 to inhibit the processing of K-Ras4B was much
closer to its ability to inhibit the processing of
- 136 -
CA 02207252 1997-06-06
W096/21456
PCT/US96/01559
geranylgeranylated Rap1A (IC50.2 AM) than that of
farnesylated H-Ras (IC50>30 M) (Fig. 18) (Table 5).
This suggested that K-Ras4B might be
geranylgeranylated. Consistent with this is the
fact that K-Ras4B processing was very resistant to
the FTase-specific inhibitor FT-277 (1050=10 AM)
(Fig. 19). Furthermore, GGTI-286 inhibited K-
Ras4B processing at concentrations (1-3 AM) (Fig.
19) that had no effect on the processing of
farnesylated H-Ras (Fig. 18).
EXAMPLE 33
Effects of GGTI-286 an Oncogenic K-Ras 4B
Constitutive Activation of MAP Kinase
To determine whether inhibition of K-Ras4B
processing by GGTI-286 results in disruption of
oncogenic signaling, the ability of GGTI-286 to
antagonize oncogenic K-Ras 4B constitutive
activation of MAP kinase was examined. Activated
MAP kinase is hyperphosphorylated and migrates
slower than hypophosphorylated (inactive) MAP
kinase on SDS-PAGE (43, 66). Figure 20 shows that
K-Ras4B transformed cells contained mainly
activated MAP kinase. Treatment of these cells
with the FTase-specific inhibitor FTI-277 (0-30
AM) did not inhibit MAP kinase activation until 30
gM (Fig. 20). In contrast, GGT1-286 inhibited MAP
kinase activation with an IC50 of 1 AM and the
block was complete at 10 M. Thus, GGT1-286
blocked oncogenic K-Ras4B MAP kinase activation at
a concentration (10 AM) where FTI-277 had no
effect. In contrast, oncogenic H-Ras activation
of MAP kinase was inhibited only slightly by GGT1-
286 whereas FTI-277 completely blocked this
activation at 3 AM (Fig. 20). Furthermore, GGTI-
286 blocked K-Ras4B activation of MAP kinase at a
- 137 -
CA 02207252 2009-11-19
WO 96/21456
PCT/US96/01559
concentration (10 AM) that had little effect on H-
Ras activation of MAP kinase (Fig. 20).
EXAMPLE 34
Antitumor Efficacy
The above examples demonstrate that GGTI-286
is a potent and highly selective inhibitor of K-
Ras4B processing and activation of oncogenic
signalling. In order to demonstrate the efficacy
of these inhibitors as anticancer agents, K-Ras4B
transformed NIH-3T3 cells were implanted
subcutaneously in nude mice. When the tumors
reached sizes of 50-100 mm3, the mice were randomly
separated into control and treated groups (5
animals per group, each animal had a tumor on both
the right and the left flank). Figure 22 shows
that tumors from control animals treated with
saline once daily grew to an average size of 2900
mm3 over a period of two weeks. In contrast,
tumors from animals treated once daily with GGTI-
286 (25 mg/kg or 50" mg/kg) grew to a size of 1600
mm3 or 900 mm3, respectively (Fig. 22). Thus,
GGTI-286 inhibited tumor growth by 5094. and 70%,
respectively.
In summary, the data clearly identifies GGTI-
286 not only as a potent antagonist of K-Ras4B =
oncogenic signaling in cultured cells, but also as
an inhibitor of tumor growth in whole animals.
References cited herein are listed below for
convenience.
- 138 -
CA 02207252 1997-06-06
WO 96/21456
PCTfUS96101559
REFERENCES
1. Barbacid, M., Annu. Rev. Biochem.,
56:779-828, (1987)
2. Grand, R.J.A. and Owen, D., Biochem. J.,
279:609-631, (1991)
3. Barbacid, M., Important Advances in
Oncolocry, eds. Devita, Hellman &
Rosenberg (Lippincott, Philadelphia,
PA), pp. 3-22, (1986)
4. Mulcahy, L.S., Smith, M.R., and Stacey,
D.W., Nature, 313:241-243, (1985)
5. Noda, M., Ko, M., Ogura, A., Liu, D.G.,
Amano, T., Takano, T. and Ikawa, Y.,
Nature, 318:73-75, (1985)
6. Bar-Sagi, D. and Feramisco, J.R., Cell,
42:841-848, (1985)
7. Kataoka, T, Powers, -S-M''1, C.,
Fasano, O., Strathern, J., Broach, J.
and Wigler, M., Cell, 37:437-445, (1984)
8. Willumsen, B.M., Christensen, A.,
Hubbert, N.C., Papageorge, A.G. and
Lowy, D.R., Nature, 310:583-586, (1984)
9. Willumsen, B.M., Norris, K., Papageorge,
A. G., Hubbert, N. C. and Lowy, D.R., EMBO
J., 3:2581-2585, (1984)
10. Hancock, J.F., Magee, A.I., Childs, J.E.
and Marshall, C.J., Cell, 57:1167-1177,
(1989)
11. Gutierrez, L., Magee, A.I., Marshall,
C.J. and Hancock, J.F., EMBO J, 8:1093-
1098, (1989)
12. Casey, P.J., Solski, P.A., Der, C.J. and
Buss, J.E., Proc. Natl. Acad. Sci. USA,
86:8323-8327, (1989)
13. Jackson, J.H., Cochrane, C.G., Bourne,
J.R., Solski, P.A., Buss, J.E. and Der,
C.J., Proc. Natl. Acad. Sci. USA,
87:3042-3046, (1990)
14. Hancock, J.F., Paterson, H. and
Marshall, J.C., Cell, 63:133-139, (1990)
- 139 -
CA 02207252 1997-06-06
W096/21456 PCT/US96/01559
15. Reiss, Y., Goldstein, J.L., Seabra,
M.C., Casey, P.J. and Brown, M.S., Cell,
62:81-88, (1990)
16. Reiss, Y., Seabra, M.C., Goldstein, J.L.
and Brown, M.S., METHODS: A Companion
to Methods in Enzymology, 1:241-245,
(1990)
17. Reiss, Y., Seabra, M.C., Armstrong,
S.A., Slaughter, C.A., Goldstein, J.L.
and Brown, M.S., J. Biol. Chem.,
266:10672-10877, (1991)
18. Reiss, Y., Stradley, S.J., Gierasch,-
L.M., Brown, M.S. and Goldstein, J.L.,
Proc. Natl. Acad. Sci. USA, 88:732-736,
(1991)
19. Manne, V., Roberts, D., Tobin, A.,
O'Rourke, E., DeVirgillio, M., Meyres,
C., Ahmed, N.,.Kurz, E., Resh, M., Kung,
H.F. and Barbacid, M., Proc. Natl. Acad.
Sc!. USA, 87:7541-7545, (1990)
20. Gibbs, J.B., Cell, 65:1-4, (1991)
21. Moores, S.L., Schaber,M.D., Mosser,
S.D., Rands, E., O'Hara, M.B., Garsky,
V.M., Marshall,M.S., Pompliano, D.L. and
.25 Gibbs, J.B., J. Biol. Chem., 266:14603-
14610, (1991)
22. Goldstein, J.L., Brown, M.S., Stradley,
S.J., Reiss, Y. and Gierasch, L.M., J.
Biol. Chem., 266:15575-15578, (1991)
23. Brown, M.S., Goldstein, J.L., Paris,
K.J., Burnier, J.P. and Marsters, J.C.,
Proc. Natol. Acad. Sci. USA, 89:8313-
8316, (1992)
24. Pompliano, D.L., Rands, E., Schaber,
M.D., Mosser, S.D., Neville, J.A. and
Gibbs, J.B., Biochemistry, 31:3800-3807,
(1992)
25. Lacal, J.D., Santos, E., Notario, V.,
Barbacid, M., Yamazaki, S., Kung, H.F.,
Seamans, O., McAndrew, S. and Crowl, R.,
Proc. Natl. Acad. Sc!. USA, 81:5305-
5309, (1984)
26. Stewart, F.H.C., Aus. J. Chem., 36:2511,
=
(1983)
- 140 -
CA 02207252 1997-06-06
WO 96/21456
PCI1US96/01559
27. Brown, M.J., Milano, P.D., Lever, D.C.,
Epstein, W.W. and Poulter, C.D., J. Am.
Chem. Soc., 113:3176, (1991)
28. Yang, C.C., Marlowe, C.K. and Kania, R.,
J. Am. Chem. Soc., 113:3177, (1991)
29. McCormick, F. (1993) Nature 363:15-
16.
30. McCormick, F. (1994) Current Opinion in
Genetics & Development 4:71-76
31. Marshall, C.J. (1994) Current Opinion in
Genetics & Developmbnt 4: 82-89
32. Kato, K., Cox, A.D., Hisaka, M.M.,
Graham, S.M., Buss, J.E., and Der, C.J.
(1992) Proc. Natl. Acad. Sci. USA
89:6403-6407.
33. Gibbs, J.B., Oliff, A., and Kohl, N.E.
(1994) Cell 77:175-178.
34. Nigam, M., Seong, C., Qian, Y.,
Hamilton, A.D., and Sebti, S. M. (1993)
J. Biol. Chem. 268:20695-20698.
35. Qian, Y., Blaskovich, M.A., Saleem, M.,
Seong, C., Wathen, S.P., Hamilton, A.D.,
and Sebti, S.M. (1994) J. Biol. Chem.
269:12410-12413.
36. Qian, Y., Blaskovich, M.A., Seong, C.M.,
Vogt, A., Hamilton, A.D., and Sebti,
S.M. (1994) Bioorq. Med. Chem. Lett
4:2579-2584.
37. Kohl, N.E., Mosser, S.D., deSolms, S.J.,
Giuliani, E.A., Pompliano, D.L., Graham,
S.L., Smith, R.L., Scolnick, E.M.,
Oliff, A., and Gibbs, J.B. (1993)
Science 260:1934-1937.
38. James, G.L., Goldstein, J.L., Brown,
M.S., Rawson, T.E., Somers, T.C.,
McDowell, R.S., Crowley, C.W., Lucas,
B.K., Levinson, A.D., and Marsters,
Jr. (1993) Science 260:193-194.
39. Graham, S.L., deSolms, S.J., Giuliani,
E.A., Kohl, N.E., Mosser, S.D., Oliff,
A. I., Pompliano, D. L., Rands, E.
Breslin, M.J., Deana, A.A., Garsky,
V.M., Scholz, T.H., Gibbs, J.B., and
- 141 -
CA 02207252 1997-06-06
V0396/21456
PCT/US96/01559
Smith, R.L. (1994) J. Med. Chem. 37:725-
732.
40. Garcia, A.M., Rowell, C. Ackerman, K.,
Kowalczyk, J.J., and Lewis, M.D. (1993)
J. Biol. Chem. 268:18415-18418.
41. Vogt, A., Qian, Y., Blaskovich, M.A.,
Fossum, R.D., Hamilton, A.D., and Sebti,
S.M. (1995) J. Biol. Chem. 270:660-664.
42. Kohl, N.E., Wilson, F.R., Mosser, S.D.,
Giuliani, E., deSolms, S.J., Conner,
M.W., Anthony, N.J., Holtz, W.J., Gomez,
R.P., Lee, T.J., and et al. (1994) Proc.
Natl. Acad. Sci. USA 91:9141-9145.
43. Cox, A.D., Garcia, A.M., Westwick, J.K.,
Kowalczyk, J. J., Lewis, M.D., Brenner,
D.A., and Der, C.J. (1994) J. Biol.
Chem. 269:19203-19206.
44. James, G.L., Brown, M.S., Cobb, M.H.,
and Goldstein, J.L. (1994) J. Biol.
Chem. 269:277705-277714.
45. Cox, A.D., Hisaka, M.M., Buss, J.E., and
Der, C.J. (1992) Mol. Cell. Biol.
12:2606-2615.
46. Hallberg, B. Rayter, S.I., and Downward,
J. (1994) J. Biol. Chem. 269:3913-3916.
47. Leevers, S.J., Paterson, H.F., and
Marshall, C.J. (1994) Nature 369:411-
414.
48. Stanton, V. P., Jr., Nichols,
D. W., Laudano, A. P., Cooper,
G. M. (1989) Mol. Cell. Biol.
9: 639-647.
49. Casey, P., J. Lipid. Res., 88:1731-1740
(1992)
50. Cox et al, Curr. Op. Cell Biol., 4:1008-
1016 (1992)
51. Goldstein et al, Science, 260:1937-1942
(1993)
52. Watanabe et al, Syn. Lett., 3:207-210
(1992)
- 142 -
CA 02207252 1997-06-06
WO 96/21456
PCT/US96101559
53. Stradley et al, Biochemistry, 32:12586-
12590 (1993)
54. Gardino, J. et al, J. Org. Chem., 49:
5237-5243 (1984)
55. Wallow, T. I. et al. J. Org. Chem.: 59,
5034-5037 (1994)
56. Crowther, G. P. et al., Org. Syn., 51:
96-100 (1971)
57. Fincham, C. I. et al., J. Med. Chem.,
35: 1472-1484 (1992)
58. Goel, 0. P. et al., Org. Syn., 67: 69-75
(1989)
59. Pearson, D. A. et al., Tetrahedron
Lett., 30: 2739-2742 (1989)
60. Casey, P.J., Solski, P.A., Der, C.J.,
and Buss, J.E. (1989) Cell 57, 1167-1177
61. Seabra, M.C., Reiss, Y.,-Casey, P.J.,
Brown, M.S., and Goldstein, J.L. (1991)
Cell 65, 429-434
62. Zhang, F.L., Diehl, R.E., Kohl, N.e.,
Gibbs, J.B., Giros, B., Casey, P.J., and
=
Omer, C.A. 91994) J. Biol. Chem. 269,
3175-3180
63. Omer, C.A., Kral, A.M., Diehl, R.E.,
Prendergast, G.C., Powers, S., Allen,
C.M., Gibbs, J.B., and Kohl, N.E. (1993)
Biochemistry 32, 5167-5176
64. Yokoyama, K., Goodwin, G.W., Ghomashchi,
F., Glomser, J.A., and Gelb, M.H. (1991)
Proc. Natl. Acad. Sci. USA 88, 5302-5306
65. Trueblood, C.E., Ohya, Y., and Rine, J.
(1993) Mbl. Cell. Biol. 13, 4260-4275
66. Lerner, E.G., Qian, Y., Blaskovich,
M.A., Fossum, R.D., Vogt A., Sun, J.,
Cox, A.D., Der, C.J., Hamilton, A.D.,
and Sebti, S.M. (1995) J. Biol. Chem.
270, 26802-26806
67. James, G.L., Goldstein, L.L., and Brown,
M.S. (1995) J. Biol. Chem. 270, 6221-
6226
- 143 -
CA 02207252 1997-06-06
W096/21456 PCT/US96/01559
68. Sun, J., Qian, Y., Hamilton, A.D., and
Sebti, S.M. (1995) Cancer Research 55,
= 4243-4247.
69. Moomaw, J.P. and Casey, P.J. (1992) J.
=
.Biol. Chem. 267, 17438-17443
It will be appreciated that various
modifications may be made in the invention as
described above without departing from the scope
and intent of the invention as defined in the
following claims wherein:
=
=
- 144 -