Note: Descriptions are shown in the official language in which they were submitted.
CA 02211458 1997-07-24
W O 96/2S410 P~N~G~
.
DESCRIPTION
Azole Compounds, Their Production and Use
TECHNICAL FIELD
The present invention relates to a~ole compounds useful
as antifungal therapeutic agents, methods for producing the
same and use thereof.
BACKGROUND ART
A variety of azole compounds have been reported exhib-
iting antifungal activity (see, for example, EPOi22056Al,
EP03323B7Al, EP0122693Al and EP0567982A).
None of these azole compounds, however, is sari~factory
as a pharmaceutical agent in terms of its antifungai activi-
ty, antiiungal spectrum, side effect and in vivo ph~-~acokl-
netics.
There has been a demand for a safer compound ~hich
exhibits better absorption in vivo and higher antlfungal
activity as an antifungal therapeutic agent.
DISCLOSURE OF INVENTION
The present invent,ion provides
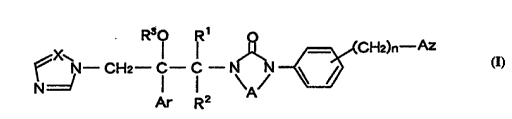
(1) a compound represented by the formula (I):
CHz--C--C--N\ / ~(CH2)n--Az
Ar R2
CA 022114~8 1997-07-24
W O 96/2S410 PCT/JF9. 003~
wherein Ar is an optionally substituted phenyl group;
Rl and R2, the same or different, are a hydrogen atom or a
lower alkyl group, or Rl and R2 may combine together to form
a lower alkylene group; R3 is a hydrogen atom or an acyl
group; X is a nitrogen atom or a methine group; A is Y=Z (Y
and Z, the same or different, are a nitrogen atom or a
methine group optionally substituted with a lower alkyl
group) or an ethylene group optionally substituted with a
lower alkyl group; n is an integer from O to 2; and Az is an
optionally substituted azolyl group,
or a salt thereof,
~ 2) a process ~or preparing a compound of the formula (I)
as defined in claim 1 or a salt thereof which comprises
(i) reacting a compound represented by the formula
(II):
~(CH2)n--Az
H2C--C--f--N\ y ~ (:~)
Ar R2
wherein the symbols have the same meanings as de~ined above;
or a salt thereof
with a compound represented by the formula (III):
gNH ( ~:
N
CA 02211458 1997-07-24
W O96/25410 PCr~JF~
wherein the symbols have the same meanings as defined
above; or a salt thereof, and, if necessary, followed by an
acylation;
(ii) reacting a compound represented by the formula
(IV):
~X~ / \ / ( ~ )
wherein the symbols have the same meanings as defined above;
or a salt thereof
with a compound repres2nted by the formula (V'):
~ ~CH2)n ~
HN~ N ~/ (~
wherein A" is -N=CH-, -CH=N- or -CH=CH-; the symbols have
the same meanings as defined above; or a salt thereof, and,
if necessary, followed by an acylation; or
(iii) reducing a compound represented by the formula
(I")
x~ f ~ ~CH2)n--A~
~N - CH2 - C - C ~
wherein the symbols have the same meanings as defined above;
or a salt thereof, and, if necessary, followed by an acyla-
CA 022114~8 1997-07-24
W O96/2S410 PCT/JP96/0032S
tion,
(3) a pharmaceutical composition to be an antifungal
preparation containing a compound represented by the above
formula (I) or a salt thereof.
Examples of the substituents for "optionally substitut-
ed phenyl group" represented by Ar in the formula (I) in-
clude halogen atoms (e.g., fluorine, chlorine, bromide and
iodine), lower (C1_4) haloalkyl, lower (C1_4) haloalkoxy,
lower (C1_4) alkylsulfonyl, lower (C1_4) haloalkyisulfonyl
and the like. Preferably, the substituent is halogen atoms
(e.g., fluorine, chlorine, bromine and iodine), and more
preferably it is fluorine. The number of the substituents
is preferably from one to three, more preferabiy from one ~o
two.
Examples of Ar include halophenyl, lower (C1_4) haloal-
kyiphenyl, lower (C1_4) haloalkoxyphenyl, lower (C1_4)
alkylsulfonylphenyl, lower (C1_4) haloalkylsulfonylphenyl
and the like.
Examples of the halophenyl groups include 2,4-difluoro-
phenyl~ 2,4-dichlorophenyl, 4-chlorophenyl, 4-fluorophenyl,
2-chlorophenyl, 2-fluorophenyl, 2-fluoro-4-chlorophenyl, 2-
chioro-4-fluorophenyl, 2,4,6-trifluoropheny, 4-bromophenyl
and the like.
Examples of the lower (C1_4) haloalkylphenyl groups
include 4-trifluoromethylphenyl group and the like.
CA 022114~8 1997-07-24
W O 96/25410 PCT/JF96~0C~
Examples of the lower (Cl_4) haloalkoxyphenyl groups
include 4-trifluoromethoxyphenyl, 4-(1,1,2,2-tetrafluoro-
ethoxy)phenyl, 4-(2,2,2-trifluoroethoxy)phenyl, 4-(2,2,3,3-
tetrafluoropropoxy)phenyl, 4-(2,2,3,3,3-pentafluoro-
propoxy)phenyl and the like.
Examples of the lower (Cl_4) alkylsulfonylphenyl groups
include 4-methanesulfonylphenyl and the like.
Examples of the lower (Cl_4) haloalkylsulfonylphenyl
groups include 4-(2,2,2-trifluoroethanesulfonyl)phenyl, 4-
(2,2,3,3-tetrafluoropropanesulfonyl)phenyl, 4-(2,2,3,3,3-
pentafluoropropanesulfonyl)phenyl and the like.
Specific examples of the phenyl groups of Ar are phenyl
~roups substituted with one to two halogen atoms such as
2,4-difluorophenyl, 2,4-dichlorophenyl, 4-chlorophenyl, 4-
fluorophenyl, 2-chlorophenyl, 2-fluorophenyl, 2-fluoro-4-
chlorophenyl, 2-chloro-4-fluorophenyl, 4-bromophenyl and the
like, among which phenyl groups substituted with one or two
fluorine atoms such as 4-fluorophenyl, 2-fluorophenyl and
2,4-difluorophenyl are more preferable and 2-fluorophenyl
and 2,4-difluorophenyl are most preferable.
Examples of the lower alkyl groups represented by Rl or
R in the formula (I) include straight or branched Cl_4
~ alkyl groups such as methyl, ethyl, propyl, isopropyl,
butyl, isobutyl, sec butyl, tert-butyl and the like, among
which methyl is more preferable. It is particularly prefer-
able that both of Rl and R2 are hydrogen atoms or methyl
CA 022114~8 1997-07-24
W O96/2S410 . PCT/JP96/00325
groups, or one of R1 and R2 is a hydrogen atom and the other
is a methyl group.
Examples of the lower alkylene groups formed by the
combination of R1 and R2 include straight lower (C2_4)
alkylene groups such as ethylene, propylene, butylene and
the like, among which ethylene is preferred.
Among them, it is pre~erable that one of R1 and R2 is a
hydrogen atom and the other is a C1_4 alkyl group such as a
methyl group and the like.
Examples of the acyl groups represented by R3 in the
formula (I) include acyl groups derived from organic carbox-
ylic acids such as alkanoyl, preferably Cl_7 alkanoyl (e.g.,
formyl, acetyl, propionyl, butyryl, isobutyryl, pentanoyl,
hexanoyl and heptanoyl), more preferably C1_3 alkanoyl;
arylcarbonyl, preferably C7_15 arylcarbonyl (e.g., benzoyl
and naphtalenecarbonyl), more preferably C7_11 arylcarbonyl
group; alkoxycarbonyl, preferably C2_7 alkoxycarbonyl (e.g.,
methoxycarbonyl, ethoxycarbonyl, propoxycarbonyl, isopro-
poxycarbonyl, butoxycarbonyl, isobutoxycarbonnyl, sec-bu-
toxycarbonyl and tert-butoxycarbonyl), more preferably C2_4
alkoxycarbonyl; aryloxycarbonyl, preferably C7_15 aryloxy-
carbonyl (e.g., phenoxycarbonyl), more preferably C7_11 ary-
loxycarbonyl; aralkylcarbonyl group, preferably C8_20 aral-
kylcarbonyl (e.g., benzylcarbonyl, phenetylcarbonyl) phenyl-
propylcarbonyl and naphthylethylcarbonyl), more preferably
CA 022114~8 1997-07-24
W O 9612S4~0 P ~/l~_.''G~-'
C8_14 aralkylcarbonyl; and the like.
Preferably, the above acyl groups are those being
capable of hydrolyzing in vivo. Examples thereof are for-
myl, acetyl, benzoyl, benzylcarbonyl and the like. Pre-
ferred R3 is a hydrogen atom.
X in the general formula (I) is preferably a nitrogen
atom.
Examples of the lower alkyl groups for "a methine group
optionally substituted by a lower alkyl group" represented
by Y or Z when A is Y=Z in the formula (I) include straight
or branched C1_4 alkyl groups (methyl, ethyl, n-propyl,
isopropyl, n-butyl, isobutyl, sec-butyl and tert-butyl),
among which methyl is preferred.
Examples for the methine group optionally substitute~
with a lower alkyl group represented by Y or Z include me-
thine, ethylidyne (-C(CH3)=), propylidyne (-C(CH2CH3)=),
butylidyne (-C(CH2CH2CH3)=) and the like, among which me-
thine, ethylidyne and the like are preferable, and methine
and the like are more preferable.
It is preferable that one of Y and Z is a nitrogen atom
and the other is methine; that both are methine; that both
are nitrogen atoms; and one is a nitrogen atom and the other
is ethylidyne. It is particularly preferable that one of Y
and Z is a nitrogen atom and the other is methine or both of
Y and Z are methine.
When A is "an ethylene group optionally substituted
CA 022114~8 1997-07-24
W O96/25410 PCTN~
with a lower alkyl group" in the formula (I), examples of
the lower alkyl groups include straight or branched Cl_4
alkyl groups (methyl, ethyl, n-propyl, isopropyl, n-butyl,
isobutyl, sec-butyl and tert-butyl), among which methyl,
ethyl and the like arepreferable, and methyl and the like
are more preferable.
Examples of the ethylene groups optionally substituted
with a lower alkyl group represented by A include ethylene,
l-methylethylene, l,l-dimethylethylene, 1,2-dimethyl-
ethylene, l-ethylethylene, 1,2-diethylethylene and the like,
among which ethylene and the likeare preferred.
Specific examples of A are -N=CH-, -CH=N-, -CH=CH-,
-N=N-, -N=C(CH3)-, -C(CH3)=N-, -CH2-CH2- and the like, among
which -N=CH-, -CH=N-, -CH=CH-, -CH2-CH2- and the like are
preferred.
In the formula (I), the groups represented by
\A
are preferably
N . N
N . N ~ N~l
CH3 CH3
= = = = ~----
CA 022114~8 1997-07-24
W O 9612S410 P~~l ~lr J C~
and the like, more preferably
and the like, still more preferably
and the like.
The integer from O to 2 represented by n is preferably
O or 1, more preferably 0.
Examples of the azolyl groups for "an optionally sub-
stituted azolyl group" represented by Az in the formula (I)
include five-membered aromatic heterocyclic groups contain-
ing one to four nitrogen atoms as ring-constituent atoms,
which may further contain a hetero atom selected from sulfur
or oxygen as a ring-constituent atom, such as pyrrolyl,
pyrazolyl, imidazolyl, 1,2,3-triazolyl, 1,2,4-triazolyl,
tetrazolyl, thiazolyl, isothiazolyl, oxazolyl, iso~azolyl,
furazanyl, 1,3,4-thiadiazolyl, 1,2,3-thiadiazolyl, 1,2,5-
thiadiazolyl, 1,2,4-thiadiazolyl and the like.
In particular, the azolyl groups are preferably pyrazo-
lyl, imidazolyl, 1,2,3-triazolyl, 1,2,4-triazolyl, tetrazo-
lyl and the like, more preferably lH-pyrazol-l-yl, lH-
imidazol-l-yl, lH-1,2,3-triazol-1-yl, 2H-1,2,3-triazol-2-yl,
lH-1,2,4-triazol-1-yl, 4H-1,2,4-triazol-4-yl, lH-tetrazol-
l-yl, 2H-tetrazol-2-yl and the like, and further more pref-
CA 022114~8 1997-07-24
W O96/2S410 ' PCT/J~9~00~
erably lH-pyrazol-l-yl, lH-1,2,3-triazol-1-yl, 2H-1,2,3-
triazol-2-yl, lH-1,2,4-triazol-1-yl, lH-tetrazol-l-yl, 2H-
tetrazol-2-yl and the like.
Examples of the substituents for "an optionally substi-
tuted azolyl group" represented by the above Az include
hydroxyl group, optionally esterified carboxyl group (e.g.,
carboxyl, Cl_6 alkoxycarbonyl such as methoxycarbonyl,
ethoxycarbonyl and butoxycarbonyl), nitro group, amino
group, acylamino group (e.g., Cl_10 alkanoylamino such as
acetylamino, propionylamino and butyrylamino), mono-Cl_10 or
di-Cl_lo alkylamino group (e.g., methylamino, dimethylamino,
diethylamino and dibutylamino), Cl_6 alkyl group (e.g.,
methyl, ethyl, propyl, isopropyl, butyl, tert-butyl, sec-
butyl, pentyl and hexyl), Cl_6 alkoxy group (e.g., methoxy,
ethoxy and butoxy), halogen atom (e.g., fluorine, chlorine
and bromine), Cl_6 haloalkyl group (e.g., trifluoromethyl,
dichloromethyl, 2,2,2-trifluoroethyl and 2,2,3,3-tetrafluo-
ropropyl), Cl_6 haloalkoxy group (e.g., trifluoromethoxy,
1,1,2,2-tetrafluoroethoxy, 2,2,2-trifluoroethoxy, 2,2,3,3-
tetrafluoropropoxy, 2,2,3,3,3-pentafluoropropoxy,
2,2,3,3,4,4,5,5-octafluoropentoxy and 2-fluoroethoxy), oxo
group, thioxo group, mercapto group, Cl_6 alkylthio group
(e.g., methylthio, ethylthio and butylthio), Cl_6 alkylsul-
fonyl group (e.g., methanesulfonyl, ethanesulfonyl and
butanesulfonyl), Cl_10 alkanoyl group (e.g., acetyl, formyl,
CA 02211458 1997-07-24
W O 96/2S410 . PCT/J1g~
propionyl and butylyl), phenyl group, Cl_6 alkylphenyl group
(e.g., p-tolyl, mesityl and p-cumenyl), Cl_6 alkoxyphenyl
group (e.g., 4-methoxyphenyl and 4-isopropoxyphenyl), halo-
phenyl group (e.g., 4-chlorophenyl and 4-fluorophenyl,
2,4,-difluorophenyl?, Cl_6 haloalkylphenyl group (e.g., 4-
trifluoromethylphenyl), Cl_6 haloalkoxyphenyl group [e.g.,
4-trifluoromethoxyphenyl, 4-(2,2,3,3-tetrafluoropropoxy)-
phenyl and 4-(1,1,2,2-tetrafluoroethoxy)phenyl] and the
like. These substituents may be substituted on the ring-
constituent carbon and/or nitrogen atom(s) of the azolyl
~roup and the number of the substituents is preferably one
or two.
Specifically, Az are preferably diazolyl, triazolyl and
tetrazolyl such as
-~3
~ N3 ' N~ '
and the like, more preferably
N~
N
and the like. 11
CA 022114~8 1997-07-24
W O96/2S410 PCT/JPg6/00325
A preferred example of the compound (I) is a compound
represented by the formula (I')
HO CH3 O
N CH2 1 IH_ N N~Az (I')
N I (R) I (R)
Al '
(wherein Ar' is a monofluorophenyl (e.g., 2-
fluorophenyl) or difluorophenyl (e.g., 2,4-difluorophenyl)
group; A' is -N=CH-, -CH=CH- or CH2-CH2-; and Az' is an
azolyl group selected from the group consisting of a diazo-
lyl, triazolyl, tetrazolyl, thiazolyl or oxazolyl which are
optionally substituted with one or two substituents selected
from the group consisting of an oxo group, a C1_6 alkyl
group (e.g., methyl, ethyl, n-propyl, iso-propyl), a C1_6
haloalkyl group (e.g., trifluoromethyl, 2,2,2-trifluoroeth-
yl, 2,2,3,3-tetrafluoropropyl) and a C1_6 haloalkyloxyphenyl
group (e.g., 4-trifluoromethoxyphenyl, 4-(2,2,3,3-tetra-
fluoropropoxyphenyl, 4-(1,1,2,2-tetrafluoroethoxyphenyl)) or
a salt thereof. In the formula (I'), A' is preferably
-CH2-CH2-, and Az' is preferably a triazolyl group and a
tetrazolyl group.
The compound represented by the formula (I), (I') may
be used as a salt thereof. Examples of such salts are
pharmacologically acceptable salts such as inorganic acid
salts (e.g., hydrochloride, hydrobromide, sulfate, nitrate
CA 022114~8 1997-07-24
WO 9612S4~0 PCT~J~C.'OOi~
and phosphorate), organic acid salts ~e.g., acetate, tarta-
rate, citrate, fumarate, maleate, toluenesulfonate and
methanesulfonate). When carboxyl group is included in the
formula (I) as a substituent, it may be an alkali methal
(sodium, pottasium and the like) salt.
The compounds represented by the formula (I) or a salt
thereof (hereinafter abbreviated as the compound of the
present inventionj have two or more stereoisomers thereof
because of having one or more asymmetric carbon atom in
their molecule. It should be understood that any of such
stereoisomers as well as a mixture thereof is within a scope
of the present invention. Among those, when R1 is hydrogen
and R2 is methyl, both the carbon atom to which the option-
ally substituted phenyl group represented by Ar is bonded
and the carbon atom to which R2 is bonded are preferred to
be in (R)-configuration.
The compound of the present invention can be produced
by, for example, reacting a compound represented b; the
formula (II):
/o\ IR1 R ~ CH2)n--AZ
H2C--f--c--N\ /N ~d ~)
Ar R2
(wherein the symbols have the same meanings as defined
above) or a salt thereof
CA 022114~8 1997-07-24
W O96/2S410 PCT/Jl~GIQ~
with a compound represented by the formula (III):
~X~
~ NH (~)
(wherein the symbols have the same meanings as defined
above) or a salt thereof. This reaction provides a compound
of the present invention in which R3 is a hydrogen atom.
The reaction can be carried out in a solvent which does
not inhibit the reaction. Examples of the solvents are
water, ketones (e.g., acetone), sulfoxides (e.g., dimethyl
sulfoxide), ethers (e.g., diethyl ether, tetrahydrofuran and
dioxane), nitriles (e.g., acetonitrile), aromatic hydrocar-
bons (e.g., benzene, toluene and xylene), halogenated hydro-
carbons (e.g., dichloromethane, chloroform and 1,2-dichloro-
ethane), esters (e.g., ethyl acetate), amides (e.g., dimeth-
ylformamide, acetamide, dimethylacetamide and l-methyl-2-
pyrrolidinone), ureylenes (e.g., 1,3-dimetyl-2-imidazolidi-
none) and the like. These solvents may be used either
singly or as a mixture of two or more solvents in a suitable
mixing ratio.
Further, the reaction is preferably carried out in the
presence of a base such as alkali metal hydroxides (e.g.,
lithium hydroxide, potassium hydroxide and sodium
hydroxide), alkali metal hydrides (e.g., potassium hydride
and sodium hydride), alkali metal carbonates (e.g., lithium
carbonate, sodium bicarbonate, cesium carbonate, potassium
CA 022114~8 1997-07-24
W O96/2S410 PC~JJP~6~0032S
carbonate and sodium carbonate), organic acid salts (e.g.,
sodium acetate), alkali metal alcoholates (e.g., sodium
methylate, potassium tert-butylate and sodium tert-
butylate), tetrabutylammonium fluoride, bis(tri-n-butylstan-
nyl)oxide and the like.
Alternatively, the desired compound can also be pre-
pared by the reaction in the above solvent using a metal
salt (e.g., alkali metal salt such as sodium and potassium
salt) of the compound (III) instead of the compound (III).
The amount of the base used is usually about 0.01 to at
100 equivalents, preferably about 0.1 to about 50 equiva-
lents per equivalent of the compound of formula (III) or a
salt thereof.
The amount of the compound (III) or a salt thereof is
about 1 to about 100 equivalents, preferably about 1 to
about 50 equivalents per equivalent of the compound of
formula (II) or a salt thereof.
The reaction temperature is not especially limited, but
usually within the range of about 0 to about 150 C, prefera-
bly about 10 to about 120 C.
The reaction time is usually within the range of about
several minutes to tens of hours (e.g., from five minutes to
fifty hours).
In another embodiment, the compound of the present
invention can also be prepared by, for example, reacting a
compound represented by the formula (IV):
CA 02211458 1997-07-24
W O 96/25410 PCT/JP96/00325
~ N- CH2- C/-\ / (~)
N I R2
(wherein the symbols have the same meanings as defined
above) or a salt thereof
with a compound represented by the formula (V'):
(CH2)n--Az
\A~/ ~ (V')
(wherein A" is -N=CH-, -CH=N- or -CH=CH-, the other
symbols have the same meanings as defined above) or a salt
thereof. The compound of the formula (V') may be a compound
represented by the formula (V"):
NQ /=v(CH2)n--Az
H-- N ~ (V )
(wherein the symbols have the same meanings as defined
above) or a salt thereof. This reaction provides a compound
of the present invention which A is Y-Z and R3 is hydrogen.
The reaction is usually carried out in a solvent which
does not inhibit the reaction. Examples of the solvents are
water, ketones (e.g., acetone), sulfoxides (e.g., dimethyl
sulfoxide), ethers (e.g., diethyl ether, tetrahydrofuran and
dioxane), nitriles (e.O.~ acetonitrile), aromatic hydrocar-
16
CA 022114~8 1997-07-24
W 09612S4~0
bons (e.g., benzene, toluene and xylene), halogenated hydro-
carbons (e.g., dichloromethane, chloroform and 1,2-
dichloroethane), esters (e.g., ethyl acetate), amides (e.g.,
dimethylformamide, acetamide, dimethylacetamide and 1-meth-
yl-2-pyrrolidinone), ureylenes (e.g., 1,3-dimetyl-2-
imidazolidinone) and the like. These solvents may be used
either singly or as a mixture of two or more solvents in a
suitable mixing ratio.
Further, the reaction is preferably carried out in the
presence of a base such as alkali metal hydroxides (e.g.,
lithium hydroxide, potassium hydroxide, sodium hydroxide),
alkali metal hydrides (e.g., potassium hydride and sodium
hydride), alkali metal carbonates (e.g., lithium carbonate,
sodium bicarbonate, cesium carbonate, potassium carbonate
and sodium carbonate), organic acid salts (e.g., sodium
acetate), alkali metal alcoholates (e.g., sodium methylate,
potassium tert-butylate and sodium tert-butylate), tetrabu-
tylammonium fluoride, bis(tri-n-butylstannyl)oxide and the
like.
Alternatively the desired compound can also be prepared
by the reaction in the above solvent using a metal salt
(e.g., alkali metal salt such as sodium and potassium salt)
of the compound (V') or (V") instead of the compound (V') or
(V " )
The amount of the base used is usually about 0.01 to
CA 022114~8 1997-07-24
W O96/2S410 PCT/J~,G~
about 100 equivalents, preferably about 0.1 to about 50
equivalents per equivalent of the compound of formula (V')
or a salt thereof or (V") or a salt thereof.
The amount of compound (V') or (V") or a salt thereof
is about 1 to about 100 equivalents, preferably about 1 to
about 50 equivalents per equivalent of the compound of
formula (IV) or a salt thereof.
The reaction temperature is not especially limited, but
usually within the range of about 0 to about 150 C, prefera-
bly about 10 to about 120 C.
The reaction time is usually within the range of about
several minutes to tens of hours (e.g., from five minutes to
fifty hours).
According to another embodiment, the compound of the
present invention can be prepared by, for e~ample, reacting
a compound represented by the formula (VI):
L--CH2 1 1 \ / ~(CH23n--Az
Ar ~2
{wherein L is a leaving group [e.g., halogen atom
(e.g., chlorine, bromine and iodine) OI' R4So3 (wherein R4 is
lower (C1_4) alkyl group or optionally substituted phenyl
group)] and the other symbols have the same meaning as
defined above} or a salt thereof
with a compound represented by the formula (III):
18
CA 022114~8 1997-07-24
WO g6/2S410 PCT~J~, ,'5 ~37
~X'NH ( )
(wherein the symbols have the same meaning as defined
above) or a salt thereof. This reaction provides a compound
of the formula (I) in which R3 is hydrogen.
Examples of the C1_4 lower alkyl group represented by
R4 are methyl, ethyl, propyl, butyl and tert-butyl.
Examples of the optionally substituted phenyl group are
the same as those of the optionally substituted phenyl group
represented by Ar.
The reaction is usually carried out in a solvent which
does not inhibit the reaction. Examples of the solvents are
water, ketones (e.g., acetone), sulfoxides (e.g., dimethyl
sulfoxide), ethers (e.g., diethyl ether, tetrahydrofuran and
dioxane), nitriles (e.g., acetonitrile), aromatic hydrocar-
bons (e.g., benzene, toluene and xylene), halogenated hydro-
carbons (e.g., dichloromethane, chloroform and 1,2-dichloro-
ethane), esters (e.g., ethyl acetate), amides (e.g., dimeth-
ylformamide, acetamide, dimethylacetamide and 1-methyl-2-
pyrrolidinone), ureylenes (e.g., 1,3-dimetyl-2-imidazolidi-
none) and the like. These solvents may be used either
singly or as a mixture of two or more solvents in a suitable
mixing ratio.
Further, the reaction is preferably carried out in the
presence of a base such as alkali metal hydroxides (e.g.,
lithium hydroxide, potassium hydroxide and sodium
19
CA 022114~8 1997-07-24
W O 96/25410 PCTIJF9G;~3~
hydroxide), alkali metal hydrides (e.g., potassium hydride
and sodium hYdride), alkali metal carbonates (e.g., lithium
carbonate, sodium bicarbonate, cesium carbonate, potassium
carbonate and sodium carbonate), organic acid salts (e.g.,
sodium acetate), alkali metal alcoholates (e.g., sodium
methylate, potassium tert-butylate and sodium tert-
butylate), tetrabutylammonium fluoride, bis(tri-n-butylstan-
nyl)oxide and the like.
Alternatively the desired compound can be prepared by
the reaction in the above solvent using a metal salt (e.g.,
alkali metal salt such as sodium and potassium salt) of the
compound (III) instead of the compound (III).
The amount of the base used is usually within the range
of about 2 to about 100 equivalents, preferably about 2 to
about 50 equivalents per equivalent of the compound of
formula (III) or a salt thereof.
The amount oftthe compound (III) or a salt thereof is
usually within the range of about 1 to about 100 equiva-
lents, preferably about 1 to about 50 equivalents per equiv-
alent of the compound of formula (VI) or a salt thereof.
The reaction temperature is not especially limited, but
usually about 0 to about 150 C, preferably about 10 to about
120~C
The reaction time is about tens of minutes to tens of
hours (e.g., from thirty minutes to fifty hours).
.
CA 022114~8 1997-07-24
~V~ 961~541a PCT/3r~
The compound of the present invention wherein A is an
ethylene group optionally substituted with a lower alkyl or
salt thereof can be prepared by, for example, subjecting to
a catalytic reduction a compound of the formula (I"):
X~ /~ CH2)n--A~
~N - CH2 - C - C
(wherein the symbols have the same meaning as defined
above) or a salt thereof, or the compound of the formula (I)
wherein Y and Z are methine groups optionally substituted
with lower alkyl (i.e., a compound (I''')):
R30 Rl
X~ I I ~ /=\~CH2)n--Az
N - CH2 - C - C - ~ N ~ (I~)
(wherein A''' is a vinylene group optionally substitut-
ed with lower alkyl group and the other symbols have the
same meanings as defined above) or a salt thereof.
The above-mentioned reaction is usually carried out in
the presence of a single or mixed solvent(s) such as water
and organic solvents which do not inhibit the reaction such
as ketones (e.g., acetone and methylethyl ketone), alcohols
(e.g., methanol, ethanol, propanol, isopropyl alcohol and
butanol), esters (e.g., ethyl acetate), hydrocarbons (e.g.,
CA 022114~8 1997-07-24
WO96/2S410 PCT/~6/00325
benzene, toluene, hexane and xylene), organic carboxylic
acids (e.g., acetic acid and propionic acid) and the like.
The reaction is usually carried out in the presence of
catalyst. A suitable metal catalyst such as palladium-
carbon is used as the catalyst. The reduction reaction is
carried out at a pressure from atmospheric pressure up to
about l50kg/cm2 at a temperature from room temperature up to
about lO0 C.
Examples of the salts of the above starting compounds
(II), (IV), (VI), (I") and (I''') are the same as those of
the compounds (I).
When a compound or a salt thereof of the present inven-
tion wherein R3 is a hydrogen atom is obtained in the above
reactions, the obtained compound or a salt thereof can be
converted into by the conventional method to provide a
compound of the formula (I) wherein R3 is an acyl group, by
treating it with an appropriate acylating agent of R3Ll
[wherein R3 is an aliphatic or aromatic carboxylic acid
residue (e.g., acetyl, propionyl, butylyl, ethoxycarbonyl,
benzoyl, substituted benzoyl) and Ll is group to be removed
(e.g., a halogen atom such as chlorine, bromine and the
like, an active ester)] in accordance with the conventional
method.
The above-mentioned reaction is usually carried out in
the presence or absence of a solvent which does not inhibit
the reaction. Examples of such solvents are water, ketones
CA 022ll4~8 l997-07-24
W 096/2S410 PCT~JF9.'~
such as acetone, sulfoxides such as dimethyl sulfoxide,
ethers such as diethyl ether, tetrahydrofuran and dioxane,
nitriles such as acetonitrile, aromatic hydrocarbons such as
benzen, toluene and xylene, halogenated hydrocarbons such as
dichloromethane, chloroform and 1,2-dichloroethane, esters
such as ethyl acetate, amides such as dimethylformamide,
acetamide, dimethylacetamide, ureylenes such as 1,3-dimeth-
yl-2-imidazolidinone, and the like. Also a base (e.g.,
dimethylaminopyridine, pyridine, pyrroline and triethyla-
mine) may be added to the reaction system for accelaration
of the reaction. The amount of the base used is usually
about 1 to about 100 equivalents per equivalent of the
compound of formula (I) or a salt thereof.
The compound of the present invention obtained as above
can be isolated and purified from the reaction mixture by a
known procedure per se such as extraction, concentration,
neutralization, filtration, recrystallization, column
chromatography, thin layer chromatography and the like.
The compound of the present invention may have at least
two stereoisomers as mentioned above. Such a stereoisomer
can be separately prepared if desired. For example, a
single isomer can be obtained by the above reaction using
each single isomer cf the starting compounds (II), (IV),
(VI), (I") and (I''') or salts thereof. Alternatively, when
the obtained product is a mixture of two or more isomers,
CA 022114~8 1997-07-24
W 096/25410 PCT/J~'G~~~
they can be separated into each isomer by the conventional
separating method such as a method for producing a salt with
an optically-active acid (e.g., camphorsulfonic acid and
tartaric acid), various types of chromatographies, fraction-
al recrystallization and the like.
The salt of the compound of the present invention can
be prepared by a known method per se such as adding the
aforesaid inorganic or organic acid to the compound (I).
The starting compound (II) or a salt thereof in the
present invention wherein Rl is hydrogen, R2 is methyl, the
carbon atom to which Ar is bonded is an (S)-configuration
and the carbon atom to which R2 is bonded is an (R)-configu-
ration [i.e., a compound (VII) or a salt thereof] can be
prepared, for example, by a method represented by the fol-
lowing reaction scheme:
CH3 1 )Ph3P,PhCOOH
A~OH EtO2CN=NCO2Et
~5T 2)NaOMe/MeOH
Ar
~OH iso Pr2NEt ~OSO2CF3
Ar ( IX) CH2CI2 Ar ( x )
NàH CH~)n--Az N N ~CHz)n--AZ
DMF Ar (~nr)
24
CA 02211458 1997-07-24
W O 961254tO PCT/J~
(wherein Me is methyl, Et is ethyl, Pr is propyl, Ph is
phenyl, (R) and (S) denote the respectively symbolized
configurations of the carbon atoms, DMF is dimethylforma-
mide, and the other symbols have the same meanings as de-
fined above).
The starting compound (VIII) in the reaction can be
prepared, for example, by a method represented by the fol-
lowing reaction scheme:
O~THP
CH3 CO ~ ~ O ~
(Xl) Ar (Xlll)
(CH ) S + ~ ~ ~N-TsOH
DMSO Ar (XIV) EtOH
NO2
~0~ /~< CH3 NO2
CH3 1)Cl-C~ ~ ~~~
(XV) 2)recrystallization ~ (XVI) N02
ÇH3
- OH ~ ~OH
Ar (Vlll)
CA 02211458 1997-07-24
W O96/2S410 PcT/Jl~cto~
[wherein THP is tetrahydropyranyl group, Ts is p-
toluenesulfonyl group, L2 is a halogen atom (e.g., chlorine,
bromine, iodine), DMSO is dimethylsulfoxide, and the other
symbols have the same meaings as defined above].
The intermediate compound (IX) can be synthesized, for
example, by a method represented by the following reaction
scheme:
02N>~
1 ) ~\ /~
CH3 ~, CH3 o /N02
~OH EtO2CN=NC02Et ~ ~OC~)
Ar 2)recrystallization ~
(XV) (XVII) 'N02
CH3
NaOMe/MeOH
Ar (IX~
(wherein the symbols have the same meanings as defined
above).
The starting compound (IV) in the present invention
wherein Rl is a hydrogen atom, R2 is a methyl group, the
carbon atom to which Ar is bonded is in an (R)-configuration
and the carbon atom to which R2 is bonded is in an (S)-
configuration [i.e., a compound (XVIII)]:
CH3
26
CA 022114~8 1997-07-24
WO96t2S410 P~llJr~S/a~?~
(wherein the symbols have the same meanings as defined
above) can be synthesized, for example, by methods described
in EP0421210A, EP0548553A or EP0567982A or by a method based
thereon.
The starting compound (VI) or a salt thereof in the
present invention wherein Rl is a hydrogen atom, R2 is a
methyl group, the carbon atom to which Ar is bonded in (S)-
configuration, the carbon atom to which R2 is bonded in
(R)-configuration, and L is a leaving group represented by
R4S03 (wherein R4 has the same meaning as mentioned above)
[i.e., a compound (XIX) or a salt thereof], and the starting
compound (II) or a salt thereof in the present invention
wherein Rl is a hydrogen atom, R2 is a methyl group, the
carbon atom to which Ar is bonded in (S)--configuration and
the carbon atom to which R2 is bonded in (R)--conf igurat ion
[i.e., a compound (VII) or a salt thereof] can be prepared,
for example, by a method represented by the following reac-
tion scheme:
CA 02211458 1997-07-24
W O96/2S410 PCT/JP96/00325
lÇH~~N~ ~CH2)n--Az
Q~s~ 0--S02--CF~ L~ \A/
~V)
ÇH~ o
~ J~ ~(CH2)n--Az
fH~ A
(CH~)2CH--O--Si--CH2 Md ~ \HzC= CH--MgL5
CH~ / \
\~
CH~ H0 ÇH~ ~ CH ) A H0 CH~ 0
(CH~)2CH 0 ~i C ~5)1 (R) \A/ ~ H2C cH(5~l (CR)H N\ / ~CH2)n--Az
()Wll) Ar
H202
H0 H~ ~
lÇ ~ /=\ ~(CH2)n--Az
H0--CH2--~ H--N N
Ar oc~n
R4So2L'
R~S02--0--CH;~ (CR)H--N\ /~( base ~ C/H2--\C--CH ~CH2)n--Az
OUX)
~1)
28
CA 02211458 1997-07-24
WO 96/2S41~ PCTJJ~,G,'01~'7s
[wherein each of L3, L4, L5 and L6 is a halogen atom
(e.g., chlorine, bromine, iodine) and the other symbols have
the same meanings as defined above].
The starting compound (XX) or a salt thereof and the
starting compound (XXI) or a salt thereof in the above
reaction wherein L3 is a chlorine atom ~i.e., a compound
(XXVI) or a salt thereof] can be each prepared, for example,
by a method represented by the following reactions scheme:
CH~ HN/ \0 CH3 ¢~ ~ 3
oR5 ~ ~ (XXVIII) ~ ~ (XXIX)
CH3 e~N ~ TsOH CH3
Ar MgL r (~OTHP ~ O~OH
Ar (XXX) Ar (X>CXI)
CH3
(CF3SO2)0 o~ O--SO2--CF3
isoPr2NEt
Ar (XX~
~ wherein R~ is a lower alkyl group, L7 is a halogen
atom (e.g., chlorine, bromine, iodine) and the other symbols
have the same meanings as defined above].
A synthesizing method for a compound (XXXI) or a salt
thereof in which Ar is 2, 4-difluorophenyl as mentioned in
the above reaction scheme is described in Japanese Patent
29
CA 02211458 1997-07-24
W O96/2S410 ~ PCTIJ~C/0
Laid-Open No. HEI 5(1993)-230038.
CHs CHs
o~ l PhCH2Br 0~ l (ChSO2)0
s)~OH K CO ~ s)~oH iSOr ~ t
OH (~OOCII) OCH2Ph (~
N (CH2)n--Az
CHs H~ ~ CH3
~ O I \A/ (V) n 1 ~ /~(CH2)n--Az
O--SO2--Ch NaH ' ~R)--N N ~)
OCHzPh (XXXIV) OCH2Ph A (XXXV)
CH3
H2/Pd-C 0~ N~(CH2)n--Az
OH A
(XXXVI)
CH3
(COCI)2 0 ~ ~ N ~ (CH2)n-Az
(XXVI)
(wherein H2/Pd-C denotes a catalytic reduction using
palladium-carbon catalyst and the other symbols have the
same meanings as defined above).
A compound (XXI) or a salt thereof wherein L3 is a
halogen atom except chlorine can be prepared using the
corresponding halogenating agent [e.g., (COBr)2, PBr3]
instead of (COCl)2 in a similar way to the above reaction.
The starting compounds (V) or a salt thereof in the
present invention wherein A is -N=CH-, -CH=CH- or -CH2-CH2-
[i.e., compounds (XXXVII), (XXXVIII) and (XXXIX) or salts
thereof respectively] can be prepared, for example, by the
CA 02211458 1997-07-24
W ~ 96125410 . PCT1JPg6/00325
method represented by the following reaction scheme:
H2N~CH2)~ ~l ~CH2)n--Az
(XXXX
*~/
~"~ (EtO)2CHCH2NH2
~R ~S~(CH2)n--Az ~ ~
H2NNHCNH~ > .~l ~ (CH2)n--Az
\=/ (XXXXII) (EtO)2CHCH2NHCNH~
~ (XXXXIII)
HN=CH-NH2 HCI/H20
(C1~12)n--Az (CH2)n--Az
Hl~ ~ Hl~
(XXXVII) (XXXVIII)
H2/Pd-C
~CH2)n--Az
(XXXIX)
(wherein the symbols have the same meanings as defined
above).
Further, the starting compound (V) or a salt thereof
wherein A is -CH=N- [i.e., a compound (XXXXIV) or a salt
thereof ] can be prepared, for example, by a method repre-
sented by the reaction scheme:
CA 022114~8 1997-07-24
W 09612S410 . PCT/JP96100325
1 ) NaNO2
H2N~ 3) NaOH H2NNH~(CH2)n-AZ 1 ) OHCCOOH H~CH2)n~z
2) (PhO)2PON3 ~
()CXXX) / (XXXXV ()O~XXIV)
Q~GG~/
~O HN=CH-NH2
~ (CH2)n--Az 1) PhCHO CONH2 (CH2)n~Az
H~ HzN-N--?~
,~rNH ~ 2) 1 kNNI I ~NO2 ~ ~XXXVII)
(wherein the symbols have the same meanings as defined
above).
The starting compounds or synthesized intermediate
compounds above-obtained can be isolated and purified from
the reaction mixtures by a known procedure per se such as
extraction, concentration, neutralization, filtration,
recrystallization, column chromatography, thin layer chroma-
tography and the like. Alternatively the reactant mixture
itself can be used as a material in the next step without
isolation.
The compound of the present invention has low toxicity
and exhibits potent antifungal activity with broad antifun-
gal spectrum against, for example, microorganisms cf genus
32
CA 022114~8 1997-07-24
WO 9612S410 PCI~ 2'~
Candida (e.g., Candida albicans, Candida utilis, Candida
grabrata, etc.), those of genus Histoplasma (e.g., Histo-
plasma capsulatum, etc.), those of genus Aspergillus (e.g.,
Aspergillus niger, Aspergillus fumigatus, etc.), those of
genus Cryptococcus (e.g., Cryptococcus neoformans, etc.),
those of genus Tricophyton (e.g., Trichophyton rubrum,
Trichophyton mentagrophytes, etc.), those of genus Microspo-
rum (e.g., Microsporum gypseum, etc.), those of genus Malas-
sezia (e.g., Malassezia furfur, etc.) and the like. Accord-
ingly, it can be used for prevention and treatment of the
systemic fungal infection and dermatomycosis (e.g., candid-
iasis, histoplasmosis, aspergillosis, cryptococcosis, tri-
chophytosis and microsporumosis) of mammals (e.g., human
being, domestic animals and fowls) and further atopic derma-
titis. Further, the compound of the invention can be used
as an antifungal agent for agricultural use.
When the compound of the present invention is adminis-
tered to a human being, it can be safely administered either
orally or parenterally in the form of pharmaceutical compo-
sitions such as oral administration preparations (e.g.,
powders, granules, tablets, capsules), parenteral prepara-
tions [e.g., injections and external preparations (e.g.,
nasal and dermatological ones), suppositories (e.g., rectal
and vaginal ones)] and the like in per se or in mixture with
suitable pharmacologically-acceptable carriers, fillers or
CA 022114~8 1997-07-24
W O96/2S410 PCT/JF~G~
diluents. The content of the compound of the present inven-
tion in a whole pharmaceutical composition is usually 5 to
100 wt%, preferably 20 to 100 wt% in an oral drug and 5 to
30 wt% in a parenteral drug.
Those preparations can be manufactured by methods which
are known per se and commonly used in the manufacture of
pharmaceutical preparations.
For example, the compound of the present invention can
be made into an injection such as aqueous injections togeth-
er with dispersing agents [e.g., Tween 80 (Atlas Powder,
U.S.A.), HCO60 (Nikko Chemicals, Japan), carboxymethylcellu-
lose or sodium alginate], preservatives (e.g., methylpara-
ben, propylparaben, benzyl alcohol and chlorobutanol), iso-
tonic agents (e.g., sodium chloride, glycerol, sorbitol and
glucose) and the like, or as oily injections by dissolving,
suspending or emulsifying in a plant oil (e.g., olive oil,
sesame oil, peanut oil, cotton seed oil and corn oil),
propylene glycol and the like.
In the manufacture of preparations for oral administra-
tion, the compound of the present invention is compression-
molded together, for example, with fillers (e.g., lactose,
sugar and starch), disintegrating agents (e.g., starch and
calcium carbonate), binders (e.g., starch, arabic gum,
carboxymethylcellulose, polyvinylpyrrolidone and hydroxypro-
pylcellulose), lubricants (e.g., talc, magnesium stearate
and polyethylene glycol 6000) and the like, followed, if
34
CA 022114~8 1997-07-24
W O 96/2S410 rCTJJl,.'l~ 5
necessary, by coating in accordance with a known method per
se with an object of taste-masking or of providing the
preparation with enteric or sustained release property.
Examples of the coating agents are hydroxypropylmethylcellu-
lose, ethylcellulose, hydroxymethycellulose, hydroxypropyl-
cellulose, polyoxyethylene glycol, Tween 80, Pluronic F68,
cellulose acetate phthalate, hydroxypropylmethylcellulose
phthalate, hydroxymethylcellulose acetate succinate, Eudra-
git (Rohm, West Germany; a copolymer of methacrylic acid
with acrylic acid) and pigments such as titanium oxide and
red iron oxide.
The compound of the present invention can be also used
solid, semisolid or liquid preparations for external use.
For example, in the case of solid external preparation, the
compound of the present invention is made into the form of
powdered compositions as it is or in a mixture with filler
(e.g., glucose, mannitol, starch and microcrystalline cellu-
lose), thickeners (e.g., natural gum, cellulose derivatives
and acrylic acid polymers) and the like. In the case of
semisolid external preparation, aqueous or oily gel prepara-
tion or ointment is preferred. In the case of liquid exter-
nal preparation, the procedures are nearly the same as those
in the case of injections to give oily or aqueous suspen-
sions. pH Adjusting agents (e.g., carbonic acid, phosphoric
acid, citric acid, hydrochloric acid and sodium hydroxide),
CA 022114~8 1997-07-24
W 096/2S410 . PCT/J~C/QO.~
antiseptics (e.g., p-hydroxybenzoates, chlorobutanol and
benzalkonium chloride) or the like can be added to the
above-mentioned solid, semisolid or liquid preparations.
More specifically, it can be used for sterilization of
disinfection of skin or mucous membrane as an ointment
containing, for example, about 0.1 to lOOmg of the compound
of the present invention per gram using Vaseline (petroleum
jelly) or lanolin as a base material.
The compound of the present invention can be made into
oily or aqueous solid, semisolid or liquid suppositories.
Examples of the oily base materials used therefor are higher
fatty acid glycerides [e.g., cacao butter and Witepsols
(Dynamite-Nobel)], medium fatty acids (e.g., Migriols
(Dynamite-Nobel)] or plant oil (e.g., sesame oil, soybean
oil and cotton seed oil) and the like. Examples of the
aqueous base materials are polyethylene glycols, propylene
glycols, and those of the aqueous gel base materials are
natural gums, cellulose derivatives, vinyl polymers, acrylic
acid polymers.
The dose of the compound of the present invention may
vary depending upon the state of infection, the route of
administration or the like. In the case of orally adminis-
trating it to an adult patient (weight: 50 kg) for the
therapy of candidiasis, for example, it is about 0.01 to
lOOmg/kg/day and, preferably about 0.1 to 50mg/kg/day, and
more preferably about 1 to 20mg/kg/day.
36
CA 022114~8 1997-07-24
WO 9612S410 PCr~J~9~ 0~
When the compound of the present invention is used as
an agricultural antifungal agent, it may be dissolved or
dispersed in a suitable liquid carrier (e.g., solvents), or
mixed or absorbed with a suitable solid carrier (e.g.,
diluents and fillers), followed, if necessary, by addition
of an emulsifier, suspending agent, spreader, penetrating
agent, moisturizing agent, thickener, stabilizer, etc. to
give the preparation a form such as emulsion, hydrating
agent, powder, granules and the like. Such preparations can'
be prepared by known methods per se. The amount of the
compound of the present invention is, for example, about 25
to 150g, preferably about 40 to 80g per acre of irrigated
rice field for prevention of rice blast diseases.
Examples of the above liquid carrier are water, alco-
hols (e.g., methyl alcohol, ethyl alcohol, n-propyl alcohol,
isopropyl alcohol and ethylene glycol), ethers (e.g., diox-
ane, tetrahydrofran), aliphatic hydrocarbons (e.g., kero-
sene, lamp oil and fuel oil), aromatic hydrocarbons (e.g.,
benzene and toluene), haloganated hydrocarbons (e.g., meth-
ylene chloride and chloroform), acid amides (e.g., dimethyl-
formamide and dimethylacetamido), esters ( e.g., ethyl
acetate and butyl acetate), nitrils (e.g., acetonitrile and
propionitrile) and the like. They may be used either singly
or as a mixture thereof in a suitable mixture ratio.
Examples of the above solid carriers are plant powder
CA 022114~8 1997-07-24
WO96/2S410 PCT1~6/0032S
(e.g., soybean powder, tobacco powder and wheat flour),
mineral powder (e.g., kaolin and bentonite), alumina, sulfur
powder, activated charcoal and the like. They may be used
either individually or as a mixture thereof in a suitable
mixing ratio.
BEST MODE FOR CARRYING OUT THE INVENTION
The present invention is further described by way of
the foliowing Reference Examples and Working Examples.
lH-NMR spectra were measured by a spectrometer of
Varian Gemini 200 type (200MHz) using tetramethylsilane as
an internal standard. All 8 values are given by ppm. In
the mixing solvents, the figures given in ( ) are the mixing
ratio of each of the solvents by volume. Unless otherwise
specified, the symbol % means by weight. In the silica gel
chromatography, the ratio of the solvents is a ratio of the
mixed solvents by volume.
The symbols used in the examples have the following
meanings.
s: singlet; d: doublet; t: triplet; q: quartet; dd:
double doublet; tt: triple triplet; m: multiplet; br: broad;
J: coupling constant.
Reference Exam~le l
2-(2,4-Difluorophenyl)-2-[(lR)-l-(3,4,5,6-tetrahydro-
2H-pyran-2-yl)oxyethyl]oxirane (82 g) (synthesized by the
method disclosed in Japanese Unexamined Patent Publication
38
CA 022114~8 1997-07-24
W O 96/25410 PCTJJ~ 3.
No. Hei 4(1992)-74168) and pyridinium p-toluenesulfonate
(6.3 g) were dissolved in ethanol (600 ml), and the result-
ant was stlrred at 55 C for 1 hour and concentrated under
reduced pressure. The residue was dissolved in ethyl ace-
tate (1 lit.) and washed with water (200 ml x 2). The aque-
ous layer was extracted with ethyl acetate (100 ml x 2).
The combined organic layers were washed with a saturated
aqueous solution of sodium chloride, dried over magnesium
sulfate and distilled under reduced pressure to remove the
solvent. The residue was purified by silica gel chromatog-
raphy (eluent: hexane/ethyl acetate = 10/1 to 8/1 to 3/1) to
give (lR)-1-[2-(2,4-difluorophenyl)-2-oxiranyl]ethanol (31.5
g) as a pale yellow oily substance.
lH-NMR (CDC13)~: 1.14-1.23 (3H,m), 1.77, 2.22 (lH), 2.80,
2.92 (lH), 3.27-3.32 (lH), 4.00-4.20 (lH,m), 6.75-6.94
(2H,m), 7.36-7.48 (lH,m)
Reference Exam~le 2
(lR)-1-[2-(2,4-Difluorophenyl)-2-oxiranyl]ethanol (31.5
g) and 3,5-dinitrobenzoyl chloride (40 g) were dissolved in
methylene chloride (500 ml), to which trimethylamine (24.1
ml) was added dropwise at ice-bath temperature. After the
mixture was stirred at room temperature for 3.5 hours, it
was washed with water (150 ml) and 5% sodium bicarbonate
aqueous solution successively, dried over magnesium sulfate
and concentrated under reduced pressure. The precipitated
crystals were filtrated and washed with methylene chloride.
39
CA 022114~8 1997-07-24
W O96/2S410 PCT1JPg6/00325
The mother liquor and the washings were combined and dis-
tilled off under reduced pressure. Ethyl acetate (25 ml)
and methanol (300 ml) were added to the residue, and the
mixture was cooled in an ice bath. The precipitated crys-
tals were collected by filtration and recrystallized from a
mixture of ethyl acetate (25 ml) and methanol (250 ml) to
give [(lR)-1-[(2R)-2-(2,4-difluorophenyl)-2-oxiranyl]ethyl]
3,5-dinitrobenzoate (28.7 g) as colorless needles.
mp: 104-107~C (recrystallized from ethylacetate-hexane)
H-NMR (CDCl3)~: 1.46 (3H,dd,J=6.6Hz,J=1.2Hz), 3.01
(lH,d,J=4.6Hz), 3.23 (lH,d,J=4.6Hz), 5.33 (lH,q,J=6.6Hz),
6.85-7.07 (2H,m), 7.54 (lH,m), 9.13 (2H,d,J=2.2Hz), 9.25
(lH,t,J=2.2Hz)
Reference Exam~le 3
[(lR)-1-[(2R)-2-(2,4-Difluorophenyl)-2-oxiranyl]ethyl]
3,5-dinitrobenzoate (50 g) was dissolved in methanol (2
lit.), to which lN-sodium hydroxide (255 ml) was added
dropwise at room temperature. The mixture was stirred at
room temperature for 1 hour and neutralized by an addition
of 1 N-hydrochloric acid (127 ml) thereto. The resultant
was concentrated under reduced pressure, to which ethyl
acetate (1 lit.) and water (200 ml) were added. The mix-
ture was extracted with ethyl acetate. The organic layer
was washed with a saturated aqueous solution of sodium
chloride (200 ml), dried over magnesium sulfate and dis-
CA 022ll4~8 l997-07-24
W O96/2S410 PCT~3F~C~OC~
tilled under reduced pressure to remove the solvent. The
residue was purified by silica gel chromatography (eluent:
ethyl acetate/hexane = 1/3) to give (lR)-1-[(2R)-2-(2,4-
difluorophenyl)-2-oxiranyl]ethanol (25 g) as a pale yellow
oily substance.
H-NMR (CDC13)o: 1.17 (3H,dd,J=6.6Hz,1.2Hz), 1.83
(lH,d,J=8Hz), 2.80 (lH,d,J=5.2Hz), 3.30 (lH,d,J=5.2Hz),
4.01-4.17 (lH,m), 6.75-6.93 (2H,m) 7.36-7.48 (lH,m)
Reference ExamPle 4
To an ice-cooled solution of (lR)-1-[(ZR)-2-(2,4-di-
fluorophenyl)-Z-oxiranyl]ethanol (16.1 g) in tetrahydrofuran
(320 ml) were added triphenylphosphine (63.3 g), benzoic
acid (29.5 g) and diethyl azodicarboxylate (42.0 g). The
mixture was stirred at room temperature for 6 hours under an
argon atmosphere. After ethyl acetate (800 ml) and water
(500 ml) were added thereto, the separated aqueous layer was
extracted with ethyl acetate (200 ml). The combined organic
layers were washed with water and a saturated aqueous solu-
tion of sodium chloride successively, dried over magnesium
sulfate and concentrated. The residue was purified by
silica gel chromatography (eluent: hexane/ethyl acetate =
15/1 to 7/1) to give [(lS)-1-[(2R)-2-(2,4-difluorophenyl)-
2-oxiranyl]ethyl] benzoate (19.2 g) as a colorless oily
substance.
1H-NMR (CDC13)~: 1.37 (3H,d,J=6.6Hz), 2.90 (lH,d,J=~.2Hz),
3.Z8 (lH,d,J=5.2Hz), 5.36 (lH,q,J=6.6Hz), 6.74-6.94 (2H,m),
CA 022114~8 1997-07-24
W O 96/2S410 PCT/~G~
7.38-7.60 (4H,m), 7.94-8.01 (2H,m)
IR ~maxneatcm~1: 1725, 1615, 1600, 1505, 1450, 1425
[(lS)-1-[(2R)-2-(2,4-Difluorophenyl)-2-oxiranyl]-ethyl]
benzoate (15.9 g) was dissolved in methanol (800 ml), to
which 28% sodium methylate-methanol solution (12.9 ml) was
added at ice-bath temperature and stirred at room tempera-
ture for 6 hours. After lN-hydrochloric acid (63.2 ml) was
added thereto, the solvent was distilled off under reduced
pressure. The residue was purified by silica gel chromatog-
raphy (eluent: hexane/ethyl acetate = 6/1 to 2/1) to give
(lS)-1-[(2R)-2-(2,4-difluorophenyl)-2-oxiranyl]ethanol (9.7
g) as a colorless oily substance.
H-NMR (CDCl3)~: 1.20 (3H,dd,J-6.4Hz,lHz), 2.24
(lH,d,J=2Hz), 2.92 (lH,d,J=5Hz), 3.28 (lH,d,J=5Hz), 4.12
(lH,dq,J=6.4Hz,2Hz), 6.77-6.95 (2H,m), 7.32-7.44 (lH,m)
IR ~maxneat cm 1 3420, 2980, 1615, 1600, 1500, 1425
Reference ExamPle 5
2-(Z-Fluorophenyl)-2-[(lR)-1-(3,4,5,6-tetrahydro-2H-
pyran-2-yl)oxyethyl]oxirane (synthesized by the method dis-
closed in EP0548553A) was converted into [(lR)-1-[(2R)-
2-(2-fluorophenyl)-2-oxiranyl]ethyl] 3,5-dinitrobenzoate by
the method described in Reference Examples 1 and 2.
Colorless prisms (recrystallized from ethyl acetate)
mp: 183-184~C
H-NMR (CDCl3)~: 1.47 (3H,dd,J=6.6Hz,1.6Hz), 3.03
42
CA 02211458 1997-07-24
W O 9612S410 PCTJJP96~DD~25
(lH,d,J=4.7Hz), 3,23 (lH,d,J=4.7Hz), 5.35 ~lH,q,J=6.6Hz),
7.09-7.59 (4H,m), 9.13 (2H,d,J=2.2Hz), 9.23 (lH,t,J=2.2Hz)
[a]23D -24.7~C (c=l.O, in CHCl3)
Elemental analysis for C17H13FN207
Calcd (%): C,54.26; H,3.48; N,7.44
Found (%): C,54.23; H,3.25; N,7.41
Reference ExamPle 6
[(lR)-1-[(2R)-2-(2-Fluorophenyl)-2-oxiranyl]ethyl]
3,5-dinitrobenzoate was converted into (lR)-1-[(2R)-2-(2-
fluorophenyl)-2-oxiranyl]ethanol by the method described in
Reference Example 3.
Colorless oily substance
H-NMR (CDC13)~: 1.17 (3H,dd,J=6.6Hz,l.OHz), 1.78
(lH,d,J=8.2Hz), 2.81 (lH,d,J=5.3Hz), 3.32 (lH,d,J=5.3Hz),
4.09-4.23 (lH,m), 6.99-?.47 (4H,m)
Reference Exam~le 7
(lR)-1-[(2R)-2-(2-Fluorophenyl)-2-oxiranyl]ethanol was
converted into (lS)-1-[(2R)-2-(2-fluorophenyl)-2-oxiranyl]-
ethanol by the method described in Reference Example 4.
Colorless oily substance
H-NMR (CDC13)~: 1.21 (3H,d,J=7Hz), 2.27 (lH,d,J=2Hz), 2.96
(lH,d,J=5Hz), 3.30 (lH,d,J=5Hz), 4.16 (lH,dq,J=7Hz,2Hz),
~ 7.03-7.44 (4H,m)
Reference Exam~le 8
2-(2-Fluorophenyl)-2-[(lR)-1-(3,4,5,6-tetrahydro-2H-
pyran-2-yl)oxyethyl] oxirane (synthesized by the method de-
43
CA 022114~8 1997-07-24
W O96/2S410 ' . PCT/JP96/00325
scribed in EP0548553A) was converted into (lR)-1-[2-(2-
fluorophenyl)-2-oxiranyl]ethanol by the method described in
Reference Example 1. To an ice-cooled solution of this
compound (34.77 g) in tetrahydrofuran (600 ml) were added
triphenylphosphine (127.21 g), 3,5-dinitrobenzoic acid
(102.88 g) and diethyl azodicarboxylate (84.47 g). The
mixture was stirred at room~temperature for 7 hours under an
argon atmosphere, and then ethyl acetate (600 ml), diisopro-
pyl ether (100 ml) and water (800 ml) were added. The
separated aqueous layer was extracted with ethyl acetate
(600 ml, 400 ml). The organic layers were combined, washed
with water and a saturated aqueous solution of sodium chlo-
ride successively, dried over anhydrous magnesium sulfate
and concentrated. The residue was purified by silica gel
chromatography (eluent: hexane/ethyl acetate = 5/1) and
recrystallized from ethyl acetate to give [(lS)-1-[(2R)-
2-(2-fluorophenyl)-2-oxiranyl]ethyl] 3,5-dinitrobenzoate
(23.15 g) as colorless needles.
H-NMR (CDC13)~: 1.47 (3H,d,J=7Hz), 2.97 (lH,d,J=5Hz), 3.29
(lH,d,J=5Hz), 5.43 (lH,q,J=7Hz), 7.02-7.56 (4H,m), 9.06
(2H,d,J=2Hz), 9.21 (lH,t,J=2Hz)
This compound (22.91 g) was dissolved in methanol (700
ml), to which an aqueous solution of lN-sodium hydroxide
(146.5 ml) was added at ice-bath temperature. The mixture
was stirred at room temperature for 1 hour. After lN-hydro-
CA 022114~8 1997-07-24
WO 96r2S410 PCr/J~C~OO~?~
chloric acid (85.5 ml) was added thereto, the solvent was
distilled off under reduced pressure. To the residue were
added ethyl acetate (500 ml) and water (500 ml). The sepa-
rated organic layer was washed with water and a saturated
aqueous solution of sodium chloride successively, dried over
anhydrous magnesium sulfate and concentrated. The residue
was purified by silica gel chromatography (eluent:
hexane/ethyl acetate = 3/1~ to give (lS)-1-[(2R)-2-(2-fluo-
rophenyl)-2-oxiranyl]ethanol (10.76 g) as a colorless oily
substance. The product was identical with the compound
obtained in Reference Example 7.
Reference Exam~le 9
A mixture of 4-fluoronitrobenzene (3.1 g), 4-[4-
(2,2,3,3-tetrafluoropropoxy)phenyl]-3(2H,4H)-1,2,4-
triazolone (5.8 g), potassium carbonate (13.8 g) and N,N-
dimethylformamide (60 ml) was stirred at 80 C for 2 hours.
The resultant was cooled and poured into water (500 ml).
The mixture was neutralized with hydrochloric acid and the
precipitated crystals were collected by filtration. The
crystals thus obtained were dissolved in ethyl acetate (300
ml) and dried over anhydrous magnesium sulfate. After
filtration, the filtrate was concentrated under reduced
~ pressure. The residue was crystallized from a mixture of
ethyl acetate and diisopropyl ether to give 2-(4-nitrophe-
nyl)-4-[4-(2,2,3,3-tetrafluoropropoxy)phenyl]-3(2H,lH)-
1,2,4-triazolone (5.5 g, 67%) as yellow crystalline powders.
CA 02211458 1997-07-24
W O96/2S410 PCTIJl~C
mp: 161-162~C
Reference Exam~les 10 to 14
The compounds shown in Table 1 as below were obtained
in the same manner as in Reference Example 9.
Table 1
02N ~ Az
Ex~mp;e No. ~2 yield (~) mp. (9~)
--~ I ~ OCH2CF2CF2H 75 1 17-118
11 --~ ~ OCF2CF2H 70 143-145
12 --1~--CH2Ch 45 143-145
N
13 --~ q 83 198-199
14 --~1--CH2CF2CF2H 41 141-143
N
Reference ExamPle 15
4-Fluoronitrobenzene (21 g) was reacted with lH-1,2,3-
triazole (12.4 g) in the same manner as in Reference Example
9. The resultant was cooled and poured into water. The
precipitated crystals were collected by filtration and
46
CA 022114~8 1997-07-24
W O 96/2S410 . P~Nl ,.','~
purified by silica gel chromatography (eluent: dichlorometh-
ane to dichloromethane/acetone = 8/1). The first eluted
fraction was recrystallized from dichloromethane-diisopropyl
ether to give 2-(4-nitrophenyl)-2H-1,2,3-triazole (18.8 g)
as pale yellow prisms.
mp: 183-184~C
lH-NMR (CDC13)~: 7.90 (2H,s), 8.28 (2H,dt,J=9.4Hz,J=2.4Hz),
8.38 (2H,dt,J=9.4Hz,J=2.4Hz)
Further, the second eluated fraction was recrystallized
from dichloromethane-diisopropyl ether to give 1-(4-nitro-
phenyl)-lH-1,2,3-triazole (6.02 g) as pale yellow prisms.
mp: 205-206~C
H-NMR (CDCl3)8: 7.92 (lH,d,J=1.4Hz), 8.00
(2H,dt,J=9Hz,J=2.4Hz), 8.13 (lH,d,J=1.4Hz), 8.44
(2H,dt,J=9Hz,J=2.4Hz)
Reference ExamPle 16
2-(4-Nitrophenyl)-4-[4-(2,2,3,3-tetrafluoropropoxy)-
phenyl]-3(2H,4H)-1,2;4-triazolone (5.5 g) and 10%
palladium-carbon (50% wet, 0.5 g) were added to methanol
(200 ml). The mixture was subjected to catalytic hydroge-
nation at ordinary temperature under ordinary pressure.
When hydrogen absoption stopped, dichloromethane (200 ml)
was added thereto and the catalyst was removed by filtra-
tion. The catalyst was washed with dichloroethane (50 ml).
The washings and the filtrate were combined and distilled
47
CA 02211458 1997-07-24
W O9612S410 PCT/J~3cN~
under reduced pressure to give 2-(4-aminophenyl)-4-[4-
(2,2,3,3-tetrafluoropropoxy)phenyl]-3(2H,4H)-1,2,4-triazo-
lone (4.6 g, 90%) as a white solid. This compound was used
for the next process.without purification.
Reference Exam~les 17 to 21
The compounds shown in Table 2 as below were obtained
in the same manner as in Reference Example 16.
Table 2
H2N ~ Az
Roference
E~mple No. Az yield (96)
.
17 ~ --CH2CF~ 97
18 --~ 94
19 _~ 96
-~ 100
21 --~1--CH2CF2CF2H 95
N
Reference Exam~le 22
Ferric chloride (0.2 g) and activated carbon (2.0 g)
48
CA 022114~8 1997-07-24
W O 96/2S410 PC~Jr~r'D~ S
were added to a solution of 1-(4-nitrophenyl)-3-[4-(2,2,3,3-
tetrafluoropropoxy)phenyl]-2(1H,3H)-imidazolone (20.5 g) in
methanol-tetrahydrofran (75 ml:75 ml), to which hydrazine
hydrate (8.0 ml) was added dropwise over the period of 10
minutes. After the mixture was refluxed with stirring for
14 hours, ferric chloride (0.2 g), activated carbon (2.0 g)
and hydrazine hydrate (8.0,ml) were added thereto and the
reaction mixture was refluxed with stirring for further 6
hours. The activated carbon was filtered off and washed
with methanol (100 ml). The filtrate and the washing were
combined and distilled off under reduced pressure. The
residue thus obtained was dissolved in ethyl acetate (700
ml). The ethyl acetate layer was washed with water (200 ml
X 4), dried over anhydrous magnesium sulfate and distilled
off under reduced pressure to give 1-(4-aminophenyl)-3-[4-
(2,2,3,3-tetrafluoropropoxy)phenyl]-2(lH,3H)-imidazolone
(18.1 g, 95%) as a pale yellow powder.
mp: 178-179~C
Elemental analysis for C18H15F4N3O2
Calcd (%): C,56.70; H,3.96; N,11.02
Found (%): C,56.58; H,3.93; N,11.21
Reference Example 23
In the same manner as in Reference Example 22, starting
from 1-(4-nitrophenyl)-3-[4-(1,1,2,2-tetrafluoroethoxy)
phenyl]-2(lH,3H)-imidazolone, 1-(4-aminophenyl)-3-[4-
49
CA 022114~8 1997-07-24
W O 96/2S410 PCT/JP96/0032S
(1,1,2,2-tetrafluoroethoxy)phenyl]-2(1H,3H)-imidazolone was
obtained.
mp: 150-151~C
Elemental analysis for C17H13F4N302
Calcd (%): C,55.59; H,3.57; N,11.44
Found (%): C,55.74; H,3.40; N,11.49
Reference Example 24
2-(4-Aminophenyl)-4-[4-(2,2,3,3-tetrafluoropropoxy)
phenyl]-3(2H,4H)-1,2,4-triazolone (4.6 g) and pyridine (1.43
g) were dissolved in ethyl acetate (200 ml). To the result-
ant was added dropwise at room temperature a solution of
phenyl chlorocarbonate (2.83 g) in ethyl acetate (20 ml).
After the addition was completed, the reaction solution was
stirred at room temperature for 2 hours. Water (200 ml),
ethyl acetate (600 ml) and tetrahydrofuran (300 ml) were
added thereto. The separated organic layer was washed with
5% phosphoric acid (200 ml X 2) and water (200 ml) succes-
sively, dried over anhydrous magnesium sulfate and filtrat-
ed. The filtrate was concentrated to about 50 ml and the
precipitated crystals were collected by filtration. The
crystals thus obtained were washed with diethyl ether and
dried to give phenyl 4-[5-oxo-4-[4-(2,2,3,3-tetrafluoropro-
poxy)phenyl]-lH,4H-1,2,4-triazol-1-yl]phenylcarbamate (5.6
g, 93%) as colorless scaly crystals.
mp: 204-206~C
Elemental analysis for C24H18F4N404
CA 02211458 1997-07-24
WO 961254~0 PCT~ 0
Calcd (%): C,57.37; H,3.61; N,11.15
Found (%): C,57.50; H,3.67; N,11.13
Reference Exam~les 25 to 31
The compounds shown in Table 3 as below were obtained
in the same manner as in Reference Example 24.
Table 3
~ OCNH ~Az
Reference A2 yield(%) mp. (~)
--~I~OCH2CF2CF2H 89 243-244
26 _~OCF2CF2H 95 208-211
27 --~--CH2CFs 87 183--184
N~
28 --~ 90 157-160
29 --~J 96 195-200
r~
--~N~ 89 143-144
31 --1~--CH2CF2CF2H 84 173-175
CA 022114~8 1997-07-24
W O96/2S410 PCT/JF9GI'~G325
Reference Exam~le 32
Phenyl 4-[5-oxo-4-[4-(2,2,3,3-tetrafluoropropoxy)-
phenyl]-lH,4H-1,2,4-triazol-1-yl~phenylcarbamate (5.6 g) was
added to a mixture of ethanol (100 ml) and tetrahydrofuran
(100 ml). To the resulting mixture was added hydrazine
hydrate (3 g) with stirring. The resultant was stirred at
80~C for 2 hours and concentrated under reduce pressure to
about 20 ml. After water (100 ml) was added, the precipi-
tated crystals were collected by filtration, washed with
ethanol and dried under reduced pressure t~ give 4-[4-[5-
oxo-4-[4-(2,2,3,3-tetrafluoropropoxy)phenyl]-lH,4H-1,2,4-
triazol-1-yl]phenyl]semicarbazide (4.8 g, 98%) as colorless
prisms.
mp: >350~C
Elemental analysis for C18H16F4N6O3
Calcd (%): C,49.10; H,3.66; N,19.08
Found (%): C,48.95; H,3.72; N,19.20
Reference ExamPles 33 to 39
The compounds shown in Table 4 as below were obtained
in the same manner as in Reference Example 32.
CA 02211458 1997-07-24
W 096/2S410 PCT~Jr3~DO~
Table 4
H2NNHCNH~Az
Roforo~co A2 yield(~) mp. (~)
33 --I~I~OCH2CF2CF2H 95 >280
34 ~ OCF2CF2H 92 >250
--~1--CH2CF!~ 90 275-280
36 _N,Nq 95 228-232
37 --1~ 98 225-234
38 --~ 96 275-277
39 --~1--CH2CF2CF2H 95 265-274
Reference Exam~le 40
4-[4-t5-Oxo-4-t4-(2~2~3~3-tetrafluoropropoxy)phenyl]-
lH,4H-l,Z,4-triazol-1-yl]phenyl]semicarbazide (4.75 g) was
added to N,N-dimethylformamide (60 ml). To the mixture were
-
CA 022114~8 1997-07-24
W O96/25410 PCTIJ~C~
added acetic acid (4 g) and formamidine acetate (6 g), and
the resulting mixture was stirred at room temperature for 3
hours and then at 80~C for 1.5 hours. After cooling, the
reaction solution was diluted with water (30 ml). The
precipitated crystals were collected by filtration and
washed with water (100 ml). The crystals were dried and
dissolved in a mixture of tetrahydrofuran (300 ml) and ethyl
acetate ~600 ml) with warming. The solution thus obtained
was dried over anhydrous magnesium sulfate, filtrated and
concentrated under reduced pressure. Ethyl acetate (50 ml)
was added to the residue and the precipitated crystals were
collected by filtration and recrystallized from tetrahydrof-
uran to give 4-[4-[5-oxo-4-[4-(2,2,3,3-tetrafluoropropoxy)-
phenyl]-lH,4H-1,2,4-triazol-1-yl]phenyl]-3(2H,4H)-1,2,4-
triazolone (2.4 g, 49%) as a white crystalline powder.
mp: 297-298~C
Elemental analysis for C1gH14F4N603
Calcd (%): C,50.67; H,3~.13; N,18.66
Found (%): C,50.49; H,3.20; N,18.50
Reference Exam~les 41 to 47
The compounds shown in Table 5 as below were obtained
in the same manner as in Reference Example 40.
54
CA 02211458 1997-07-24
W O 96/2S410 PCT/J~ 3
Table 5
H ~ ~ Az
a-~ r;nc A~ y~eld (%) mp.
41 - ~ ~ OCHzCFzCFzH 52 >260
42 - ~ ~ OCF2CF2H 49 >260
43 - ~ - CH~CF 71 >300
44 - ~ 40 >300
- ~ 58 >300
46 - ~ ~ 54 .281-283
47 - ~ - CHzCF2CF2H 52 Z46-248
~eference Exzm~le 48
A mixture of phenyl 4-(lH-1,2,4-triazol-1-yl)phenylcar-
bamate ~13 g), 2,2-diethoxyethyl amine (7.4 g) and pyridine
(3.67 g) was heated at 50 C for 3 hours. The resultant was
CA 02211458 1997-07-24
WO 96/2S410 PCrlJ~ OD~
cooled and the precipitated crystals were washed with a
mixture of diisopropyl ether and petroleum ether (1:1, 100
ml x 2) to give 1-(2,2-diethoxyethyl)-3-[4-(lH-1,2,4-tria-
zol-l-yl)phenyl]urea (14.5 g) as a colorless crystalline
powder.
mp: 139-140~C
lH-NMR (CDC13)~: 1.25 (6H,t,J=7.2Hz), 3.43 (2H,t,J=5Hz),
3.52-3.85 (4H,m), 4.57 (lH,t,J=5Hz), 5.08-5.18 (lH,m), 7.16
(lH,br), 7.49 (2H,d,J=9.4Hz), 7.57 (2H,d,J=9.4Hz), 8.08
(lH,s), 8.48 (lH,s)
Reference Exam~les 49 to 50
The compounds shown in Table 6 as below were obtained
in the same manner as in Reference Example 48.
Table 6
(CHsCH20)2CHCH2NHCNH~ Az
R~eronce A2 yield (%) mP- (~)
49 - ~ 95 194-196
- ~ 84 175-176
56
CA 02211458 1997-07-24
W O 96/2S410 . P ~r~
Reference Exam~le 51
1-(2,2-Diethoxyethyl)-3-[4-(lH-1,2,4-triazol-1-
yl)phenyl]urea (14.5 g) was dissolved in a mixture of metha-
nol (214 ml) and water (85 ml), to which diluted hydrochlor-
ic acid (0.48 M, 104 ml) was added dropwise. After the
reaction solution was stirred at room temperature for 14
hours, the precipitated crystals were collected by filtra-
tion to give 1-[4-(lH 1,2,4-triazol-1-yl)phenyl]-2-
(lH,3H)-imidazolone (8.0 g) as a colorless crystalline
powder. The filtrate was concentrated under reduced pres-
sure to about 200 ml and the precipitated crystals were
collected by filtration to give an additional amount (1.08
g! of the product.
mp: 294-296~C
Reference Exam~les 52 to 53
The compound shown in Table 7 as below were obtained in
the same manner as in Reference Example 51.
Table 7
Ref erence
E~ple Noi Azyield (g63 mP- (~C)
52 - ~ 86255-257
(~f ~C _ -sition)
53 - ~ 85 ~00
CA 022114~8 1997-07-24
W 096/2S410 PCT/Jl~G~
Reference Exam~le 54
To a solution of (lS)-1-[(2R)-2-(2,4-difluorophenyl)-2-
oxiranyl]ethanol (1.0 g) in dichloromethane (14 ml) was
added diisopropylethylamine (0.96 ml) at -78 C in a nitrogen
atmosphere, to which trifluoromethanesulfonic anhydride
(0.93 ml) was added dropwise over the period of 5 minutes.
After the reaction solution was stirred at -78 C for 20
minutes and tnen at -25 C for 25 minutes, the reaction
solution was concentrated at -10 C to about 10 ml. The
concentrated solution was subjected to flash column chroma-
tography using silica gel and eluted with dichloromethane-
hexane (1:1). The desired fraction was concentrated to
about 10 ml, and the residue was added at -14 C to a solu-
tion prepared from 4-[4-[5-oxo-4-[4-(2,2,3,3-tetrafluoropro-
poxy)phenyl]-lH,4H-1,2,4-triazol-1-yl]phenyl]-3(2H,4H)-
1,2,4-triazolone (2.1 g), dimethylformamide (40 ml), dimeth-
yl sulfoxide (50 ml) and sodium hydride (60% in oil: 180
mg). The resulting mixture was stirred at -14 C for 20
minutes and then at -5 C for 20 minutes. The reaction
solution was diluted with water (500 ml) and extracted with
dichloromethane (300 ml X 2). The dichloromethane layer was
washed with water (200 ml X 2) and a saturated aqueous
solution of sodium chloride successively, dried over anhy-
drous magnesium sulfate and distilled off under reduced
pressure to give a colorless powder. The product was puri-
fied by silica gel chromatography (eluent: hexane/ethyl
58
CA 02211458 1997-07-24
W O96125410 . PCT1JPg6~0032~
acetate = 1/1 to 1/2) and crystallized from ethyl acetate-
hexane to give 2-[(lR,2S)-2-(2,4-difluorophenyl)-2,3-
epoxy-l-methylpropyl]-4-[4-[5-oxo-4-[4-(2,2,3,3-tetrafluoro-
propoxy)phenyl]-lH-1,2,4-triazol-1-yl]phenyl]-3(2H,4H)-1,2,4-
triazolone (0.29 g) as colorless crystalline powders.
mp: 181-183~C
Elemental analysis for C29H22F6N604
Calcd (%): C,55.07; H,3.51; N,13.29
Found (%): C,55.12; H,3.34; N,13.24
Reference Exam~les 55 to 63
The compounds shown in Table 8 as below were obtained
in the same manner as in Reference Example 40.
59
CA 02211458 1997-07-24
WO 96/2S410 PCr/J1 ,"~
Table 8
H~(CH2)n--Az
ofor;nc n Az mp- (~C)
1 - ~ 249-250
56 0 - ~ 269-270
57 1 - ~ 201-205
58 0 - ~ 2~4-286
59 0 - ~ 245-248
1 - ~ 250-252
61 0 ~H3 282-284
62 0 ~H3 280-283
63 0 - ~ 243-248
CA 02211458 1997-07-24
W~ 961"54~0 PCr~JFg~ 0
Reference ExamPles 64 to 72
The compounds shown in Table 9 as below were obtained
in the same manner as in Reference Example 51.
Table 9
Hl~ ~CH2)n--AZ
E~mp;- No. n Az mp. (~C)
64 0 - ~ 239-240
1 - ~ 170-171
66 1 - ~ 190-191
67 o - ~ 210-211
68 o - ~ 235-240
69 1 - ~ 213-215
o ~ 206-208
71 0 ~ 216-218
72 o - ~ 251-255
61
CA 022114~8 1997-07-24
WO96/25410 PCT/~610
Reference Exam~le 73
1-[4-(lH-1-Tetrazolyl)phenyl]-2(1H,3H)-imidazolone (5.0
g) was dissolved in acetic acid (500 ml) and 10% palladium- -
carbon (50% wet, 5.0 g) was added. The resulting mixture
was stirred at 40 C for 4 hours under a hydrogen atmosphere.
The catalyst was filtered and washed with acetic acid. The
filtrate and the washings were combined and distilled off
under reduced pressure. The residue was crystallized from
ethanol to give 1-[4-(lH-1-tetrazolyl)phenyl]-2-imidazolidi-
none (4.1 g) as colorless crystals.
mp: 237-240 C (dec.)
lH-NMR (d6-DMS0)~: 3.45 (2H,t,J=7Hz), 3.93 (2H,t,J=7Hz),
7.20 (lH,s), 7.82 (4H,s), 10.02 (lH,s)
Elemental analYSis for C1OHlON6O
Calcd (%): C,52.17; H,4.38; N,36.50
Found (%): C,51.99; H,4.33; N,36.41
Reference ExamPle 74
Diisopropylethylamine (1.15 ml) was added to a solution
of (lS)-1-[(2R)-2-(2,4-difluorophenyl)-2-oxiranyl]ethanol
(1.20 g) in dichloromethane (26 ml) at -78 C under a nitro-
gen atmosphere, to which trifluoromethanesulfonic anhydride
(1.10 ml) was added dropwise over the period of 5 minutes.
The mixture was stirred at -78 C for 20 minutes and then at
-30 C for 15 minutes. After addition of hexane (26 ml), the
mixture was subjected to flash column chromatography using
silica gel and eluted with dichloromethane-hexane (1:1).
62
CA 022114~8 1997-07-24
W O96/25410 - PCTJ~9GJ~ s
The desired fraction was concentrated to about 20 ml, and
the residue was added to a solution prepared from 1-[4-
(lH-1-tetrazolyl)phenyl]-2(lH,3H)-imidazolone (940 mg),
dimethylformamide (20 ml), dimethyl sulfoxide (10 ml),
tetrahydrofuran (10 ml) and sodium hydride (72% in oil: 126
mg) at -30~C. The resulting mixture was stirred for 20
minutes at -30~C and then fOr 40 minutes at ice-bath temper-
ature. Water (100 ml) was added and the mixture was ex-
tracted with ethyl acetate (150 ml). The ethyl acetate
layer was washed with water t100 ml X 2) and a saturated
aqueous solution of sodium chloride successively, dried over
anhydrous magnesium sulfate and distilled off under reduced
pressure to give a colorless powder. The product was puri-
fied by silica gel chromatography (eluent: hexane/ethyl
acetate = 1/3) to give 1-[(lR,2S)-2-(2,4-difluorophenyl)-
2,3-epoxy-1-methylpropyl]-3-[4-(lH-1-tetrazolyl)phenyl]-
2(lH,3H)-imidazolone (0.13 g) and (2R)-2-(2,4-
difluorophenyl)-2-[(lR)-1-[1-[4-(lH-1-tetrazolyl)phenyl]-2-
imidazolyl]oxy]ethyl]oxirane (0.05 g).
1-[(lR,2S)-2-(2,4-Difluorophenyl)-2,3-epoxy-1-meth-
ylpropyl]-3-[4-(lH-1-tetrazolyl)phenyl]-2(lH,3H)-imidazo-
lone: colorless crystalline powder.
mp: 205-207~C
H-NMR (CDC13)8: 1.39(3H,d,J=7Hz), 2.73 (lH,d,J=5Hz), 2.83
(lH,d,J=5Hz), 5.09 (lH,q,J=7Hz), 6.52 (lH,d,J=3Hz), 6.66
63
CA 022114~8 1997-07-24
W O96/2S410 PCT/J~9-'00
(lH,d,J=3Hz), 6.81-6.96 (2H,m), 7.36-7.48 (lH,m), 7.78
(2H,d,J=9Hz), 7.94 (2H,d,J=9Hz), 9.02 (lH,s)
SIMS (MH+): 411
Reference ExamPle 75
Diisopropylethylamine (1.27 ml) was added to a solution
of (lS)-1-[(2R)-2-(2-fluorophenyl)-2-oxiranyl]ethanol (1.21
g) in dichloromethane (25 mli at -78~C under a nitrogen
atmosphere, to which trifluoromethanesulfonic anhydride
(1.22 ml) was added dropwise over a period of 5 minutes.
The reaction solution was stirred at -78 C for 15 minutes
and then at -30~C for 15 minutes. The resultant was diluted
with hexane (25 ml), subjected to flash column chromatogra-
phy using silica gel and eluted with dichloromethane-hexane
(1:1). The desired fraction was concentrated to about 20
ml, and the residue was added to a solution prepared from
1-[4-(lH-1-tetrazolyl)phenyl]-2(lH,3H)-imidazolone (1.14 g),
l-methyl-2-pyrrolidone (30 ml) and 72% sodium hydride in oil
(150 mg) at -30 C. The reaction solution was stirred at
-30 C for 15 minutes and then at -10 C for 15 minutes.
Water (100 ml) was added and the mixture was extracted with
ethyl acetate (150 ml). The ethyl acetate layer was washed
with water (100 ml) and a saturated aqueous solution of
sodium chloride successively, dried over anhydrous magnesium
sulfate and distilled off under reduced pressure to give a
colorless powder. The product was purified by silica gel
chromatography (eluent: hexane/ethyl acetate = 1/3) to give
64
CA 022114~8 1997-07-24
WO 9612S410 PCI'JJ~
l-[(lR,2S)-2-(2-fluorophenyl)-2,3-epoxy-1-methylpropyl]-3-
[4-(lH-l-tetrazolyl)phenyl]-2(1H,3H)-imidazolone (0.39 g)
and (2R)-2-(2-fluorophenyl)-2-[(lR)-1-[1-[4-(lH-l-tetrazo-
~ lyl)phenyl]-2-imidazolyloxy]ethyl]oxirane (0.18 g).
l-[(lR,2S)-2-(2-Fluorophenyl)-2,3-epoxy-1-methylpro-
pyl]-3-[4-(lH-l-tetrazolyl)phenyl]-2(1H,3H)-imidazolone:
colorless crystalline powder.
H-NMR (CDC13)ô: 1.39(3H,d,J=7Hz), 2.76 (lH,d,J=5Hz), 2.84
(lH,d,J=5Hz), 5.15 (lH,q,J=7Hz), 6.55 (lH,d,J=3Hz), 6.67
(lH,d,J=3Hz), 7.06-7.49 (4H,m), 7.79 (2H,d,J=9Hz), 7.96
(2H,d,J=9Hz), 9.04 (lH,s)
Reference Exam~le 76
A mixture of (S)-ethyl lactate (75 g) and morpholine
(164 g) was heated at 80~C for 64 hours. The reaction
solution was concentrated and the residue was subjected to
silica gel chromatography (eluent: hexane/ethyl acetate =
4/1 to ethyl acetate) to give 4-[(S)-2-hydroxypropionyl]-
morpholine (69.4 g) as a pale yellow oily substance. p-
Toluenesulfonic acid monohydrate (0.82 g) was added to a
solution of 4-[(S)-2-hydroxypropionyl]morpholine (69.4 g) in
dichloromethane (300 ml), to which 3,4-dihydro-2H-pyran
(40.3 g) was added dropwise at ice-bath temperature. The
reaction solution was stirred at 0 C for 30 minutes and
washed with a 5% aqueous solution of sodium bicarbonate.
After the organic layer was dried over anhydrous magnesium
CA 022114~8 1997-07-24
W O9612S410 PCT/JPg6/00325
sulfate and concentrated, the residue was subjected to
silica gel chromatography (eluent: hexane/ethyl acetate =
8/1 to ethyl acetate) to give 4-[(2S)-2-(3,4,5,6-tetrahy-
dro-2H-pyran-2-yloxy)propionyl]morpholine (89.1 g) as a pale
yellow oily substance.
l-Bromo-2-fluorobenzene (15 g) and 4-[(2S)-2-(3,4,5,6-
tetrahydro-2H-pyran-2-yloxy)propionyl]morpholine (40 g) were
dissolved in tetrahydrofuran (200 ml), to which magnesium
(turnings: 4.4 g) was added. The mixture was stirred vigor-
ously. The reaction flask was cooled when the temperature
of the reaction solution reached to 35 C, and l-bromo-2-
fluorobenzene (16.7 g) was added thereto over the period of
10 minutes while the temperature of the reaction solution
was kept at 35 to 37 C. After the reaction solution was
stirred at 30 to 35~C for 2 hours, it was cooled in an ice-
bath. A saturated aqueous solution of ammonium chloride
(100 ml) was added thereto and the mixture was extracted
with ethyl acetate (200 ml x 2, 100 ml). The extract was
washed with water and a saturated aqueous solution of sodium
chloride successively, dried over anhydrous magnesium sul-
fate, and distilled under reduced pressure to remove the
solvent. The residue was subjected to silica gel chromatog-
raphy (eluent: hexane/ethyl acetate = 10/1 to 5/1) to give
(2S)-2'-fluoro-2-(3,4,5,6-tetrahydro-2H-pyran-2-yloxy)-
propiophenone (22.4 g) as a pale yellow oily substance.
(2S)-2'-Fluoro-2-(3,4,5,6-tetrahydro-2H-pyran-2-
CA 022114~8 1997-07-24
W O 961254~0 PCTJJF9CJOD~
yloxy)propiophenone t25 g) was dissolved in ethanol (200
ml), to which pyridinium p-toluenesulfonate (1.28 g) was
added. The reaction solution was stirred at 55 C for 2.5
hours and then concentrated. The residue was subjected to
silica gel chromatography (eluent: hexane/ethyl acetate =
9/1 to 5/1) to give (2S)-2'-fluoro-2-hydroxypropiophenone
(16.4 g) as a colorless oily substance.
IR(neat): 1690 (C=0) cm~1
H-NMR (CDCl3)8: 1.41(3H,dd,J=7Hz,J=1.4Hz), 3.78
(lH,d,J=6Hz), 4.98-5.15 (lH,m), 7.12-7.36 (2H,m), 7.54-7.68
(lH,m), 7.90-8.00 (lH,m)
Reference Exam~le 77
(2S)-2',4'-Difluoro-2-hydroxypropiophenone (synthesized
by the method disclosed in Japanese Unexamined Patent Publi-
cation No. Hei 5(1993)-230038: 26.01 g) was dissolved in di-
chloromethane (300 ml), to which diisopropylethylamine
(19.90 g) was added at -60~C under a nitrogen atmosphere,
and then trifluoromethanesulfonic anhydride (25.90 ml) was
added thereto dropwise over the period of 20 minutes. After
the reaction temperature was gradually raised to -30 C, the
reaction solution was further stirred for 30 minutes. The
reaction solution was purified by silica gel chromatography
(silica gel 400 g, eluent: dichloromethane/hexane = 1/1) to
give (2S)-2',4'-difluoro-2-trifluoromethanesulfonyloxypro-
piophenone (39.21 g) as a pale yellow oily substance.
CA 022114~8 1997-07-24
W O96/2S410 PCT/JPg6/00325
H-NMR (CDC13)~: 1.73(3H,dd,J=7.0Hz,1.6Hz), 5.93
(lH,q,J=7.0Hz), 6.90-7.12 (2H,m), 8.03
(lH,dt,J=6.4Hz,8.6Hz)
[a]D23 + 29.2 (c=1.12, in MeOH)
Reference ExamPle 78
(2S)-2'-Fluoro-2-hydroxypropiophenone (synthesized by
the method disclosed in Reference Example 76: 3.36 g) was
dissolved in dichloromethane (30 ml). To the resultant was
added diisopropylethylamine (4.18 ml) at -60 C under a
nitrogen atmosphere, and then trifluoromethanesulfonic anhy-
dride (4.03 ml) was added dropwise to the mixture over the
period of 2 minutes. After the reaction temperature was
gradually raised to -25 C, the reaction solution was stirred
for 30 minutes. The reaction solution was purified by
silica gel chromatography (silica gel 60 g, eluent: dichlo-
romethane/hexane = 1/1) to give (2S)-2'-fluoro-2-
trifluoromethanesulfonyloxypropiophenone.(5.30 g) as a pale
yellow oily substance.
H-NMR (CDC13)~: 1.73(3H,dd,J=7Hz,J=1.6Hz), 6.49
(lH,q,J=7Hz), 7.15-7.38 (2H,m), 7.58-7.72 (lH,m), 7.97
(lH,dt,J=1.8Hz,J=7.6Hz)
Reference Exam~le 79
1-[4-(lH-1,2,4-Triazol-l-yl)phenyl]-2(1H,3H)-
imidazolone (3.39 g) was dissolved in 1-methyl-2-pyrrolidone
(220 ml), to which 72% sodium hydride in oil (528 mg) was
added. The mixture was stirred at room temperature for 1
68
CA 022114~8 1997-07-24
W O9612S410 PCTJJ~ Q~3~s
hour. The reaction solution was cooled in an ice-bath and
added dropwise over the period of 15 minutes to a solution
of (2S)-2'-fluoro-2-trifluoromethanesulfonyloxypropiophenone
(4.7 g) in tetrahydrofuran (100 ml) which had been cooled to
-20 C. After the addition was complete, the reaction tem-
perature was raised to 10~C over 30 minutes and the reaction
solution was further stirred for 12 hours. The reaction
solution was diluted with acetic acid (10 ml) and ethyl
acetate (500 ml), washed with water (250 ml x 2), 0.5 hydro-
chloric acid (250 ml x 2) and a saturated aqueous solution
of sodium chloride (250 ml) successively, dried over anhy-
drous magnesium sulfate and distilled under reduced pressure
to remove the solvent. The residue was purified by silica
gel chromatography (silica gel, eluent: hexane/ethyl ace-
tate/acetic acid = 1/4/0.06) and recrystallized from diiso-
propyl ether (25 ml) to give 1-[(lR)-2-fluorophenyl)-2-oxo-
l-methylethyl]-3-[4-(lH-1,2,4-triazol-1-yl)phenyl]-2(1H,3H)-
imidazolone as a colorless crystalline powder.
lH-NMR (CDC13)~: 1.65(3H,d,J=7.2Hz), 5.82 (lH,q,J=7.2Hz),
6.64 (lH,d,J=3.2Hz), 6.70 (lH,d,J=3.2Hz), 7.14-7.31 (2H,m),
7.53-7.94 (6H,m), 8.11 (lH,s), 8.56 (lH,s)
Reference Exam~le 80
1-[4-(lH-l-Tetrazolyl)phenyl]-2(1H,3H)-imidazolone
(0.94 g) was dissolved in 1-methyl-2-pyrrolidone (25 ml), to
which 72% sodium hydride in oil (0.126 g) was added. The
69
CA 022ll4~8 l997-07-24
W O 96/2S410 PCTIJF9C~
reaction solution was stirred at room temperature for 30
minutes. The resultant was ice-cooled and added dropwise
over the period of 10 minutes to a solution of (2S)-2'-
fluoro-2-trifluoromethanesulfonyloxypropiophenone (1.57 g)
in tetrahydrofuran (25 ml) which had been cooled to -10 C.
After the addition was complete, the reaction temperature
was raised to 0~C over 15 minutes and the reaction solution
was stirred for 30 minutes. The reaction solution was
diluted with acetic acid (3 ml) and ethyl acetate (100 ml),
washed with water (50 ml x 2), 0.5 N-hydrochloric acid (50
ml x 2) and a saturated aqueous solution of sodium chloride
(50 ml) successively, dried over anhydrous magnesium sulfate
and distilled under reduced pressure to remove the solvent.
The residue was purified by silica gel chromatography
(eluent: hexane/ethyl acetate/acetic acid = 1/3/0.05) and
recrystallized from diisopropyl ether (20 ml) to give 1-
[(lR)-2-fluorophenyl)-2-oxo-1-methylethyl]-3-[4-(lH-l-tetra-
zolyl)phenyl]-2(lH,3H)-imidazolone (0.22 g) as a colorless
crystalline powder.
mp: 162-164~C
lH-NMR (CDCl3)~: 1.66 (3H,d,J=7.2Hz), 5.83 (lH,q,J=7.2Hz),
6.67 (lH,d,J=3.2Hz), 6.74 (lH,d,J=3.2Hz), 7.16-7.33 (2H,m),
7.54-7.98 (2H,m), 7.77 (2H,d,J=9Hz), 7.91 (2H,d,J=9Hz), 9.03
(lH,s)
Reference ExamPle 81
Chloromethylisopropoxydimethylsilane (2.14 g) and
CA 02211458 1997-07-24
W O 96t~5410 P~T/J~9CJOO.
magnesium (for Grignard reaction, 313 mg) were added to
tetrahydrofuran (15 ml), and the mixture was heated to 60~C.
To the mixture was added magnesium in the form of turnings
which had been activated by methyl iodide, and then the
mixture was stirred in a bath at 60~C for 3 hours.
The solution of the Grignard reagent thus obtained was
added dropwise to a solution of l-[(lR)-2-(2-fluorophenyl)-
2-oxo-1-methylethyl]-3-[4-(lH-l-tetrazolyl)phenyl-2(lH,3H)-
imidazolone (1 g) in tetrahydrofuran (150 ml) over the
period of 10 minutes at ice-bath temperature, and the mix-
ture was stirred for 30 minutes. A cooled saturated aqueous
solution of ammonium chloride (30 ml) and cooled water (100
ml) were added thereto at ice-bath temperature and the
mixture was extracted with ethyl acetate (200 ml). The
extract was washed with a saturated aqueous solution of
~odium chloride, dried over magnesium sulfate and concen-
trated under reduced pressure. The residue was recrystal-
lized from a mixture of diisopropyl ether and ethyl acetate
to give l-[(lR,2S)-2-(2-fluorophenyl)-2-hydroxy-3-
(isopropoxydimethylsilyl)-l-methylpropyl]-3-[4-(lH-l-
tetrazolyl)phenyl]-2(1H,3H)-imidazolone (637 mg) as a color-
less crytalline powder.
H-NMR (d6-DMSO)~: -0.30(3H,s), -0.28 (3H,s), 0.99-1.64
(llH,m), 3.83 (lH,quintet,J=6Hz), 4.81 (lH,q,J=7Hz), 5.21
(lH,br), 6.93-7.77 (6H,m), 8.05 (2H,d,J=9Hz), 8.17
71
CA 022114~8 1997-07-24
W O96/25410 PCT/JF9C/0~2
(2H,d,J=9Hz), 10.17 (lH,s)
Reference ExamPle 82
l-[(lR,2S)-2-(2-Fluorophenyl)-2-hydroxy-3-(isopropoxy
dimethylsilyl)-l-methylpropyl]-3-[4-(lH-l-tetrazolyl)phe-
nyl]-2(lH,3H)-imidazolone (1 g) was dissolved in a mixture
of methanol and tetrahydrofuran (1:1, 20 ml), to which an
30% aqueous solution of hydrogen peroxide (2 ml) and sodium
bicarbonate (157 mg) were added. The mixture was heated at
50 C for 4 hours, then cooled and extracted with ethyl
acetate (100 ml). The extract was washed with water (30
ml), an aqeous solution of Na2S203 (30 ml x 2) and a satu-
rated aqueous solution of sodium chloride (30 ml) succes-
sively, dried over magnesium sulfate and distilled under
reduced pressure to remove the solvent. The residue was
purified by silica gel chromatography (eluent: hexane/ethyl
acetate = 1/4) and recrystallized from diethyl ether (20 ml)
to give l-[(lR,2S)-2-(2-fluorophenyl)-2,3-dihydroxy-1-methyl
propyl]-3-[4-(lH-l-tetrazolyl)phenyl]-2(1H,3H)-imidazolone
(440 mg) as a colorless crystalline powder.
lH-NMR (CDC13)~: 1.17(3H,d,J=7Hz), 3.52-3.62 (lH,m), 4.05-
4.18 (2H,m), 5.01 (lH,q,J=7Hz), 6.72 (lH,d,J=3.2Hz), 6.82
(lH,d,J=3.2Hz), 7.01-7.33 (3H,m), 7.70-7.78 (lH,m), 7.90
(2H,d,J=9Hz), 7.99 (2H,d,J=9Hz), 9.55 (lH,s)
Reference ExamPle 83
l-[(lR,2S)-2-(2-Fluorophenyl)-2,3-dihydroxy-1-methyl
propyl]-3-[4-(lH-l-tetrazolyl)phenyl]-2(1H,3H)-imidazolone
CA 022114~8 1997-07-24
W 0961~54~0 PCT/~P~6/~032S
(440 mg) was dissolved in a mixture of ethyl acetate and
tetrahydrofuran (1:2, 30 ml), to which methanesulfonyl
chloride (0.18 g) and triethylamine (0.16 g) were added
dropwise at ice-bath temperature. The reaction solution was
stirred at 0 C for 30 minutes and washed with water (15 ml x
2) and a saturated aqueous solution of sodium chloride (15
ml) successively. The organic layer was dried over magnesi-
um sulfate and distilled under reduced pressure to remove
the solvent. The residue was purified by silica gel chroma-
tography (eluent: hexane/ethyl acetate = 1/4) to give 1-
[(lR, 2S) - 2 - (2 - f luorophenyl)-2-hydroxy-3-methanesulfonyloxy-
l-methylpropyl]-3-[4-(lH-l-tetrazolyl)phenyl]-2(1H,3H)-
imidazolone (330 mg) as a colorless crystalline powder.
H-NMR (CDC13)~: 1.27(3H,d,J=7Hz), 2.87 (3H,s), 4.54
(lH,d,J=12Hz), 4.73-4.88 (2H,m), 6.63 (lH,d,J=3.2Hz), 6.72
(lH,d,J=3.2Hz), 7.09-7.39 (3H,m), 7.75-7.94 (lH,m), 7.81
(2H,d,J=9Hz), 7.93 (2H,d,J=9Hz), 9.04 (lH,s)
Reference ExamPle 84
l-[(lR,2S)-2-(2-Fluorophenyl)-2-hydroxy-3-methanesulfo-
nyloxy-l-methylpropyl]-3-[4-(lH-l-tetrazolyl)phenyl]-
2(1H,3H)-imidazolone (100 mg) was dissolved in dimethylfor-
mamide (4 ml), to which potassium carbonate (42 mg) was
added, and the mixture was heated at 40 C for 1 hour. The
resultant was diluted with ethyl acetate (20 ml) and washed
with water (10 ml) and a saturated aqueous solution of
CA 022114~8 1997-07-24
W O 96/2S410 PCT/JF9G/00~
sodium chloride (10 ml) successively. The organic layer was
dried over magnesium sulfate and distilled under reduced
pressure to remove the solvent. The residue was purified by
silica gel chromatography (eluent: hexane/ethyl acetate =
1/4). The desired fraction was concentrated and the residue
was recrystallized from diisopropyl ether to give 1-
[(lR,2S)-2-(2-fluorophenyl)-2,3-epoxy-1-methylpropyl]-3-[4-
(lH-1-tetrazolyl)phenyl]-2(lH,3H)-imidazolone (58 mg) as a
colorless crystalline powder.
H-NMR (CDCl3)~: 1.40 (3H,d,J=7Hz), 2.76 (lH,d,J=5Hz), 2.84
(lH,d,J=5Hz), 5.15 (lH,q,J=7Hz), 6.55 (lH,d,J=3.2Hz), 6.67
(lH,d,J=3.2Hz), 7.07-7.48 (4H,m), 7.79 (2H,d,J=9Hz), 7.95
(2H,d,J=9Hz), 9.05 (lH,s)
Workin~ Exam~le 1
60% sodium hydride in oil (108 mg) was dispersed in
dimethylformamide (4 ml), to which 1,2,4-triazole (207 mg)
was added at ice-bath temperature, and the mixture was
stirred at room temperature for 10 minutes. To the result-
ant was added a solution of 2-[(lR, 2S)-2-(2,4-difluorophe-
nyl)-2,3-epoxy-1-methylpropyl]-4-[4-[5-oxo-4-[4-(2,Z,3,3-
tetrafluoropropoxy)phenyl]-lH-1,2,4-triazol-1-yl]phenyl]-
3(2H,4H)-1,2,4-triazolone (560 mg) in dimethylformamide (2
ml), and the mixture was heated at 60 C for 11 hours. After
cooling, water (40 ml) and ethyl acetate (40 ml) were added
to the mixture. The separated aqueous layer was extracted
74
CA 022ll4~8 l997-07-24
W O 96/25410 P~-lJJ~
with ethyl acetate twice. The combined ethyl acetate layers
were washed with water and a saturated a~ueous solution of
sodium chloride successively, dried over anhydrous magnesium
sulfate and distilled under reduced pressure to remove the
solvent. The residue was purified by silica gel chromatog-
raphy (eluent: ethyl acetate/hexane = 19/1 to ethyl acetate)
and then by reverse phase chromatography (eluent:
ethanol/water = 4/1) to give 2-[(lR,2R)-2-(2,4-difluorophe-
nyl)-2-hydroxy-1-methyl-3-(lH-1,2,4-triazol-1-yl)propyl]-4-
[4-[5-oxo-4-[4-(2,2,3,3-tetrafluoropropoxy)phenyl]-lH-
1,2,4-triazol-1-yl]phenyl]-3(2H,4H)-1,2,4-triazolone (Com-
pound l; 0.21g) as a colorless powder.
[a]20D -16.9~ (c=1.0% in MeOH)
Elemental analysis for C31H25F6NgO4-0.5H2O
Calcd (%): C,52.40; H,3.69; N.17.74
Found (%): C,52.59; H,3.67; N,17.69
Workin~ Exam~le 2
(2R,3S)-2-(2,4-Difluorophenyl)-3-methyl-2-(lH-1,2,4-
triazol-l-yl)methyloxirane (1.2g), 4-[4-[2-oxo-3-[4-
(2,2,3,3-tetrafluoropropoxy)phenyl]-lH,3H-imidazol-l-
yl]phenyl]-3(2H,4H)-1,2,4-triazolone (2.2 g) and potassium
carbonate (powder: 3.5 g) were added to N,N-dimethylforma-
mide (50 ml), and the mixture was heated with stirring at
90 C for 42 hours. After cooling, the resultant was diluted
with ethyl acetate (150 ml) and tetrahydrofuran (50 ml).
Ice water (150 ml) was added thereto to separate the ethyl
CA 022114~8 1997-07-24
W O96/25410 . PCT1JP96/0032S
acetate layer. The aqueous layer was extracted with ethyl
acetate (100 ml). The ethyl acetate layers were combined
and washed with 0.5N-sodium hydroxide (100 ml), lN-
hydrochloric acid (100 ml) and a saturated aqueous solution
of sodium chloride (100 ml) successively. The ethyl acetate
layer was dried over anhydrous magnesium sulfate and dis-
tilled under reduced pressure to remove the solvent. The
residue was purified by silica gel chromatography
(elute:ethyl acetate/acetone = 10/1) and crystallized from
tetrahydrofuran-diisopropyl ether to give 2-[(lR,2R)-2-(2,4-
difluorophenyl)-2-hydroxy-1-methyl-3-(lH-1,2,4-triazol-1-
yl)propyl]-4-[4-[2-oxo-3-[4-(2,2,3,3-tetrafluoropropoxy)-
phenyl]-lH,3H-imidazol-l-yl]phenyl]-3(2H,4H)-1,2,4-triazo-
lone (Compound 2; 0.26g) as a colorless crystalline powder.
mp: 181-183~C
Elemental analysis for C32H26F6N8O4
Calcd (%): C,54.86; H,3.74; N.15.99
Found (%): C,54.58; H,3.75; N,15.71
[a]20D -18.9 (c=1.0% in MeOH)
Workin~ Exam~le 3
2-[(lR,2R)-2-(2,4-Difluorophenyl)-2-hydroxy-1-methyl-
3-(lH-1,2,4-triazol-1-yl)propyl]-4-[4-[2-oxo-3-[4-
(1,1,2,2-tetrafluoroethoxy)phenyl]-lH,3H-imidazol-1-yl]
phenyl]-3(2H,4H)-1,2,4-triazolone (Compound 3) was obtained
in the same manner as in Working Example 2,
76
CA 022114~8 1997-07-24
W O 96/2S410 PCTJJ~ D~
Colorless crystalline powder
mp: 214-215~C
Elemental analysis for C31H24F6N8O4
Calcd (%): C,54.23; H,3.52; N,16.32
Found (%): C,54.05; H,3.37; N,16.32
[a]20D -19.0 (c=1.0% in MeOH)
Workin~ Ex~m~le 4
A mixture of 60% sodium hydride in oil (0.24 g) and di-
methyl sulfoxide (60 ml) was stirred at 80~C for 30 minutes.
To the mixture was added 4-[4-[5-oxo-4-(2,2,2-trifluoroeth-
yl)-lH,4H-1,2,4-triazol-1-yl]phenyl]-3(2H,4H)-1,2,4-triazo-
lone (1.94 g), and the mixture was stirred for 5 minutes.
To the resultant was added (2R,3S)-2-(2,4-difluorophenyl)-3-
methyl-2-(lH-1,2,4-triazol-1-yl)methyloxirane (1.0 g), and
the mixture was stirred at 80 C for 24 hours under an argon
atmosphere. The reaction solution was cooled, diluted with
ethyl acetate (300 ml) and washed with water (50 ml X 2) and
a saturated aqueous solution of sodium chloride (50 ml)
successively. The organic layer was dried over magnesium
sulfate and concentrated under reduced pressure. The resi-
due was purified by silica gel chromatography (eluent:
hexane/ethyl acetate = 1/2 to ethyl acetate) to give 2-
[(lR,2R)-2-(2,4-difluorophenyl)-2-hydroxy-1-methyl-3-(lH-
1,2,4-triazol-1-yl)propyl]-4-[4-[5-oxo-4-[4-(2,2,2-trifluo-
roethyl)-lH,4H-1,2,4-triazol-1-yl]phenyl~-3-(2H,4H)-1,2,4-
triazolone (Compound 4; 0.46 g) as a pale yellow powder.
CA 022114~8 1997-07-24
W O96/2S410 PCT/J~3G;~
H-NMR (CDCl3)~: 1.31(3H,d,J=7Hz), 4.36 (2H,q,J=8.4Hz), 4.37
~lH,d,J=14Hz), 5.03 (lH,d,J=14Hz), 5.10 (lH,q,J=7Hz), 5.44
(lH,s), 6.75-6.88 (2H,m), 7.48-7.65 (lH,m), 7.67
(2H,d,J=9Hz), 7.68 (lH,s), 7.69 (lH,s), 7.83 (lH,s), 7.94
(lH,s), 8.16 (2H,d,J=9Hz)
Workin~ Exam~le 5
(2R,3S)-2-(2,4-Difluorophenyl)-3-methyl-2-(lH-1,2,4-
triazol-l-yl)methyloxirane (1.0 g) was reacted with 4-[4-
(lH-1,2,4-triazol-1-yl)phenyl]-3(2H,4H)-1,2,4-triazolone
(0.91 g) in the same manner as in Working Example 4 to give
2-[(lR,2R)-2-(2,4-difluorophenyl)-2-hydroxy-1-methyl-3-(lH-
1,2,4-triazol-1-yl)propyl]-4-[4-(lH-1,2,4-triazol-1-
yl)phenyl]-3(2H,4H)-1,2,4-triazolone (Compound 5; 0.54 g).
Colorless crystalline powder
mp: 182-184~C
H-NMR (CDC13)~: 1.32(3H,d,J=7Hz), 4.40 (lH,d,J=14.4Hz),
5.03 (lH,d,J=14.4Hz), 5.11 (lH,q,J=7Hz), 5.41 (lH,s), 6.75-
6.90 (2H,m), 7.50-7.65 (lH,m), 7.69 (lH,s), 7.79
(2H,d,J=9Hz), 7.88 (2H,d,J=9Hz), 7.92 (lH,s), 7.96 (lH,s),
8.14(1H,s), 8.65 (lH,s)
Elemental analysis for C22HlgF2N902
Calcd (%): C,55.11; H,3.99; N,26.29
Found (%): C,55.05; H,4.01; N,26.14
IR(KBr): 1714, 1618, 1556, 1527, 1394cm~
CA 022114~8 1997-07-24
W 096t254~0
Workin~ Exam~le 6
(2R,3S)-2-(2,4-Difluorophenyl)-3-methyl-2-(lH-1,2,4-
triazol-l-yl)methyloxirane (1.0 g) was reacted with 4-[4-
(lH-1,2,3-triazol-1-yl)phenyl3-3(2H,4H)-1,2,4-triazolone
(1.09 g) in the same ~anner as in Working Example 4 to give
2-[(lR,2R)-2-(Z,4-difluorophenyl)-2-hydroxy-1-methyl-3-(lH-
1,2,4-triazol-1-yl)propyl]-4-[4-(lH-1,2,3-triazol-1-yl)
phenyl]-3(2H,4H)-1,2,4-triazolone (Compound 6; 0.27 g).
Colorless crystalline powder
mp: 219-220~C
lH-NMR (CDC13)~: 1.32(3H,d,J=7Hz), 4.40 (lH,d,J=14.2Hz),
5.03 (lH,d,J=14.2Hz), ~.10 (lH,q,J=7Hz), 5.38 (lH,s), 6.7~-
6.90 (2H,m), 7.50-7.65 (lH,m), 7.70 (lH,s), 7.82
(2H,d,J=9Hz), 7.88 (lH,s), 7.90 (lH,s), 7.94 (2H,d,J=9Hz),
7.94 (lH,s), 8.05 (lH,s)
Elemental analysis for C22HlgF2N902
Calcd (%): C,55.11; H,3.99; N,26.29
Found (%): C,54.91; H,3.97; N,26.26
IR(KBr): 1700, 1675, 1618, 1556, 1527, 1502cm 1
Workin~ Exam~le 7
(2R,3S)-2-(2,4-Difluorophenyl)-3-methyl-2-(lH-1,2,4-
triazol-l-yl)methyloxirane (1.0 g) was reacted with 4-[4-
(2H-1,2,3-triazol-2-yl)phenyl]-3(2H,4H)-1,2,4-triazolone
(1.09 g) in the same manner as in Working Example 4 to give
2-[(lR,2R)-2-(2,4-difluorophenyl)-2-hydroxy-1-methyl-3-(lH-
1,2,4-triazol-1-yl)propyl~-4-[4-(2H-1,2,3-triazol-2-
79
CA 022114~8 1997-07-24
W O96/2S410 PCTIJ~3C/Q~
yl)phenyl]-3(2H,4H)-1,2,4-triazolone (Compound 7; 0.65 g).
Pale yellow plates
mp: 213-215~C
H-NMR (CDC13)~: 1.32(3H,d,J=7Hz), 4.38 (lH,d,J=14.2Hz),
5.04 (lH,d,J=14.2Hz), 5.11 (lH,q,J=7Hz), 5.42 (lH,s), 6.?5-
6.90 (2H,m), 7.50-7.64 (lH,m), 7.69 (lH,s), 7.74
(2H,d,J=9Hz), 7.85 (2H,s), 7.86 (lH,s), 7.95 (lH,s),
8.25(2H,d,J=9Hz)
Elemental analysis for C22HlgF2NgO2
Calcd (%): C,55.11; H,3.99; N,26.29
Found (%): C,54.97; H,3.96; N,26.29
IR(KBr): 1697, 1623, 1602, 1564, 1519, 1510cm~
Workin~ ExamPle 8
2-[(lR,2R)-2-(2,4-Difluorophenyl)-2-hydroxy-1-methyl-3-
(lH-1,2,4-triazol-1-yl)propyl]-4-[4-[5-oxo-4-[4-(2,2,3,3-
tetrafluoropropyl)-lH,4H-1,2,4-triazol-1-yl]phenyl]-3-
(2H,4H)-1,2,4-triazolone (Compound 8) was obtained in the
same manner as in Working Example 4.
Pale yellow powder
H-NMR (CDCl3)8: 1.31(3H,d,J=7Hz), 4.34 (2H,t,J=14Hz), 4.37
(lH,d,J=14Hz), 5.04 (lH,d,J=14Hz), 5.10 (lH,q,J=7Hz), 5.46
(lH,s), 5.98 (lH,tt,J=53Hz,J=2.4Hz), 6.75-6.90 (2H,m),
7.50-7.65 (lH,m), 7.67 (2H,d,J=9Hz), 7.68 (lH,s), 7.72
(lH,s), 7.86 (lH,s), 7.96 (lH,s), 8.16 (2H,d,J=9Hz)
CA 022114~8 1997-07-24
W 096/2S4tO PC~rh.,~J5 ~2
Workin~ Exam~le 9
(2R,3S)-2-(2,4-Difluorophenyl)-3-methyl-2-(lH-1,2,4-
triazol-l-yl)methyloxirane (2.5 g), 1-[4-(lH-1,2,4-tria-
zol-l-yl)phenyl]-2(1H,3H)-imidazolone (2.72 g) and cesium
carbonate (powder: 9.7 g) were added to N,N-dimethylforma-
mide (150 ml), and the mixture was heated at 80 C with
stirring for 9.5 hours. After cooling, the reaction mixture
was diluted with ethyl acetate (400 ml). Ice water (150 ml)
was added thereto to separate the ethyl acetate layer. The
aqueous layer was extracted with ethyl acetate (100 ml).
The ethyl acetate layers were combined and washed with
0.5N-sodium hydroxide (100 ml), lN-hydrochloric acid (100 ml
x 2) and a saturated aqueous solution of sodium chloride (50
ml) successively. The ethyl acetate layer was dried over
anhydrous magnesium sulfate and distilled under reduced
pressure to remove the solvent. The residue was purified by
silica gel chromatography (eluent: ethyl acetate/acetone =
2/1) to give 1-[(lR,2R)-2-(2,4-difluorophenyl)-2-hydroxy-1-
methyl-3-(lH-1,2,4-triazol-1-yl)propyl]-3-[4-(lH-1,2,4-
triazol-l-yl)phenyl]-2(1H,3H)-imidazolone (Compound 9; 1.03
g) as a pale yellow powder.
lH-NMR (CDC13)8: 1.21 (3H,d,J=7Hz), 4.22 (lH,d,J=14.2Hz),
5.01 (lH,q,J=7Hz), 5.12 (lH,d,J=14.2Hz), 5.50 (lH,br), 6.72
(lH,d,J=3.2Hz), 6.73-6.90 (2H,m), 6.83 (lH,d,J=3.2Hz),
7.40-7.55 (lH,m), 7.75 (lH,s), 7.78 (2H,d,J=9.4Hz), 7.86
(lH,s), 7.86 (2H,d,J=9.4Hz), 8.13 (lH,s), 8.59 (lH,s)
81
CA 022114~8 1997-07-24
W O 96/2S410 PCTlJ~JG/O
IR(KBr): 3400, 3118, 1683, 1616, 1527, 1500cm~
Workin~ ExamDle 10
l-t(lR,2R)-2-(2,4-Difluorophenyl)-2-hydroxy-1-methyl-3-
(lH-1,2,4-triazol-1-yl)propyl]-3-[4-(lH-1,2,4-triazol-1-
yl)phenyl]-2(lH,3H)-imidazolone (0.50 g) obtained in Working
Example 9 was dissolved in acetic acid (25 ml), to which 10%
palladium-carbon (200 mg) was added. The resultant was
stirred under a hydrogen atmosphere at room temperature for
3 hours and then at 50 C for 3 hours. After the catalyst
was filtered off, the filtrate was concentrated. The resi-
due was purified by silica gel chromatography (eluent: ethyl
acetate/acetone = 5/1 to 2/1) and recrystallized from ethyl
acetate-diisopropyl ether to give l-[(lR,2R)-2-(2,4-
difluorophenyl)-2-hydroxy-1-methyl-3-(lH-1,2,4-triazol-1-
yl)propyl]-3-[4-(lH-1,2,4-triazol-1-yl)phenyl]-2-
imidazolidinone (Compound 10; 0.37 g) as a colorless crys-
talline powder.
H-NMR (CDCl3)~: 1.08(3H,d,J=7.2Hz), 3.68-4.18 (4H,m), 4.52
(lH,d,J=14Hz), 4.58-4.80 (lH,m), 5.12 (lH,d,J=14Hz), 5.38
(lH,br), 6.70-6.86 (2H,m), 7.35-7.50 (lH,m), 7.66
(2H,dt,J=9.4Hz,J=2.4Hz), 7.75 (2H,dt,J=9.4Hz,J=2.4Hz), 7.77
(lH,s), 7.87 (lH,s), 8.11 (lH,s), 8.53 (lH,s)
Elemental analysis for C23H22F2NgO2
Calcd (%): C,57.50; H,4.62; N,23.32
Found (%): C,57.46; H,4.47; N,23.19
CA 022114~8 1997-07-24
wos6ns4l0 PCTJJ~C,'Or'~
IR(KBr): 3390, 3106, 1677, 1614, 1523, 1484cm~
Workin~ Exam~le 11
To a mixture of 1-[4-(lH-1,2,3-triazol-1-yl)phenyl]-
2(1H,3H)-imidazolone (2.72 g) and 1-methyl-2-pyrrolidone
(100 ml) was added sodium hydride (70% in oil, 0.40 g), and
the mixture was stirred at room temperature for 1 hour.
(2R,3S)-2-(2,4-Difluorophenyl)-3-methyl-2-(lH-1,2,4-
triazol-l-yl)methyloxirane (2.51 g) was added thereto, and
the mixture was stirred at 100~C for 8 hours under an argon
atmosphere. After cooling, the reaction solution was dilut-
ed with ethyl acetate (400 ml), and washed with water (100
ml), lN-hydrochloric acid (100 ml X 2) and a saturated
aqueous solution of sodium chloride (50 ml) successively.
The ethyl acetate layer was dried over anhydrous magnesium
sulfate and concentrated under reduced pressure. The resi-
due was purified by silica gel chromatography (eluent: ethyl
acetate to ethyl acetate/acetone = 5/1) and recrystallized
from ethyl acetate-diisopropyl ether to give l-[(lR,2R)-2-
(2 7 4-difluorophenyl)-2-hydroxy-1-methyl-3-(lH-1,2,4-triazol-
l-yl)propyi]-3-[4-(lH-1,2,3-triazol-1-yl)phenyl]-2(1H,3H)-
imidazolone (Compound ~1; 1.82 g) as a pale yellow crystal-
line powder.
mp: 178-181~C
~ lH-NMR (CDC13)~: 1.22(3H,d,J=7Hz), 4.22 (lH,d,J=14.4Hz),
5.01 (lH,q,J=7Hz), 5.12 (lH,d,J=14.4Hz), 5.38 (lH,br),
6.70-6.88 (4H,m), 7.40-7.55 (lH,m), 7.76 (lH,s), 7.80-7.93
CA 022114~8 1997-07-24
W O9612S410 PCT/J1 3G/Q0
(6H,m), 8.03 (lH,s)
Elemental analysis for C23H20F2N8O2
Calcd (%): C,57.74; H,4.21; N,23.42
Found (%): C,57.46; H,4.25; N,23.30
IR(KBr): 1691, 1656, 1619, 1527, 1502, 1430cm~
Workin~ Exam~le 12
l-[(lR,2R)-2-(2,4-Difluorophenyl)-2-hydroxy-1-methyl-3-
(lH-1,2,4-triazol-1-yl)propyl]-3-[4-(lH-1,2,3-triazol-
l-yl)phenyl]-2(lH,3H)-imidazolone (0.80 g) obtained in
Working Example 11 was subjected to catalytic hydrogenation
in the same manner as in Working Example 10 to give 1-
[(lR,2R)-2-(2,4-difluorophenyl)-2-hydroxy-1-methyl-3-(lH-
1,2,4-triazol-1-yl)propyl]-3-[4-(lH-1,2,3-triazol-1-
yl)phenyl]-2-imidazolidinone (Compound 12; 0.69 g).
Colorless crystalline powder
H-NMR (CDC13)~: 1.08(3H,d,J=7Hz), 3.70-4.14 (4H,m), 4.52
(lH,d,J=14.2Hz), 4.60-4.78 (lH,m), 5.12 (lH,d,J=14.2Hz),
5.38 (lH,br), 6.70-6.86 (2H,m), 7.35-7.50 (lH,m), 7.68-7.82
(4H,m), 7.77 (lH,s), 7.86 (2H,s), 7.97 (lH,s)
Elemental analysis for C23H22F2N8O2
Calcd (%): C,57.50; H,4.62; N,23.32
Found (%): C,57.38; H,4.59; N,23.41
IR(KBr): 1697, 1664, 1618, 1527, 1502, 1427cm~
84
CA 022114~8 1997-07-24
W O9612S410 PCT/J~C~. ~~ 5
Workin~ Exam~le 13
(2R,3S)-2-(2,4-Difluorophenyl)-3-methyl-2-(lH-1,2,4-
triazol-l-yl)methyloxirane (2.51 g) was reacted with 1-[4-
(2H-1,2,3-triazol-2-yl)phenyl]-2(1H,3H)-imidazolone (2.72 g)
in the same manner as in Working Example 11 to give 1-
[(lR,2R)-2-(2,4-difluorophenyl)-2-hydroxy-1-methyl-3-(lH-
1,2,4-triazol-1-yl)propyl]-3-[4-(2H-1,2,3-triazol-2-
yl)phenyl]-2(lH,3H)-imidazolone (Compound 13; 1.41 g).
Pale yellow needles
mp: 182-185~C
lH-NMR (CDCl3)8: 1.22(3H,d,J=7Hz), 4.22 (lH,d,J=14.4Hz),
4.99 (lH,q,J=7Hz), 5.0~ (lH,d,J=14.4Hz), 5.13 (lH,br),
6.70-6.88 (4H,m), 7.40-7.56 (lH,m), 7.75 (lH,s), 7.81
(2H,d,J=9.2Hz), 7.84 (2H,s), 7.86 (lH,s), 8.18
(2H,d,J=9.2Hz)
Elemental analysis for C23H20F2N802
Calcd (%): C,57.74; H,4.21; N,23.42
Found (%): C,57.67; H,4.20; N,23.59
IR(KBr): 3328, 1664, 1614, 1519, 1430, 1384cm 1
Workin~ ExamPle 14
l-[(lR,2R)-2-(2,4-Difluorophenyl)-2-hydroxy-1-methyl-3-
(lH-1,2,4-triazol-1-yl)propyl]-3-[4-(2H-1,2,3-triazol-2-
yl)phenyl3-2(1H,3H)-imidazolone (0.80 g) obtained in Working
~ Example 13 was subjected to catalytic hydrogenation in the
same manner as in Working Example 10 to give 1-[(lR,2R)-2-
(2,4-difluorophenyl)-2-hydroxy-1-methyl-3-(lH-1,2,4-triazol-
8~
CA 022114~8 1997-07-24
W O 96/2S410 PCT/JPg6/00325
l-yl)propyl]-3-[4-(2H-1,2,3-triazol-2-yl)phenyl]-2-imidazol-
idinone (Compound 14; 0.70 g).
Colorless crystalline powder
mp: 196-197~C
H-NMR (CDC13)~: 1.08(3H,d,J=7.4Hz), 3.68-4.12 (4H,m), 4.53
(lH,d,J=14Hz), 4.58-4.76 (lH,m), 5.13 (lH,d,J=14Hz), 5.42
(lH,br), 6.70-6.85 (2H,m), 7.36-7.50 (lH,m), 7.71
(2H,d,J=9Hz), 7.76 (lH,s), 7.81 (2H,s), 7.87.(lH,s), 8.07
(2H,d,J=9Hz)
IR(KBr): 3426, 1687, 1658, 1616, 1517, 1484cm 1
Workin~ ExamPle 15
(2R,3S)-2-(2,4-Difluorophenyl)-3-methyl-2-(lH-1,2,4-
triazol-l-yl)methyloxirane was reacted with l-[4-(lH-l-
pyrazolyl)phenyl]-2(1H,3H)-imidazolone in the same manner as
in Working Example 11 to give 1-[(lR,2R)-2-(2,4-difluoro-
phenyl)-2-hydroxy-1-methyl-3-(lH-1,2,4-triazol-1-yl)propyl]-
3-[4-(lH-l-pyrazolyl)phenyl]-2(lH,3H)-imidazolone (Compound
lH-NMR (CDC13)~: 1.21(3H,d,J=7Hz), 4.22 (lH,d,J=14.4Hz),
4.98 (lH,q,J=7Hz), 5.12 (lH,d,J=14.4Hz), 5.56 (lH,br),
6.47-6.54 (lH,m), 6.68-6.88 (4H,m), 7.40-7.56 (lH,m?, 7.70-
7.85 (6H,m), 7.85 (lH,s), 7.94 (lH,d,J=2.4Hz)
Elemental analysis for C24H2lF2N7o2
Calcd (%): C,60.37; H,4.43; N,20.53
Found (%): C,60.29; H,4.42; N,20.50
86
CA 022114~8 1997-07-24
W~ 96~2S4~ PCT~JP9~0032S
Workin~ ExamPle 16
l-[(lR,2R)-2-(2,4-Difluorophenyl)-2-hydroxy-1-methyl-3-
(lH-1,2,4-triazol-1-yl)propyl]-3-~4-(lH-l-pyrazolyl)phenyl]-
2(1H,3H)-imidazolone (Compound 15) obtained in Working
Example 15 was subjected to catalytic hydrogenation in the
same manner as in Working Example 10 to give 1-[(lR,2R)-2-
(2,4-difluorophenyl)-2-hydroxy-1-methyl-3-(lH-1,2,4-triazol-
1- yl)propyl]-3-[4-(lI~-l-pyrazolyl)phenyl]-2-imidazolidinone
(Compound 16).
H-NMR (CDCl3)8: 1.08 (3H,d,J=7Hz), 3.65-4.10 (4H,m), 4.53
(lH,d,J=14.2Hz), 4.55-4.75(1H,m), 5.12 (lH,d,J=14.2Hz), 5.45
(lH,br), 6.46-6.48 (lH,m), 6.70-6.85 (2H,m), 7.35-7.50
(lH,m), 7.60-7.75 (5H,m), 7.76 (lH,s), 7.88 (lH,s), 7.90
(lH,d,J=2.6Hz)
Elemental analysis for C24H23F2N7O2
Calcd (%): C,60.12; H,4.83; N,20.45
Found (%): C,60.02; H,4.95; N,20.34
Workin~ ExamPle 17
(2R,3S)-2-(2,4-Difluorophenyl)-3-methyl-2-(lH-1,2,4-
triazol-l-yl)methyloxirane was reacted with l-[4-(lH-1,2,4-
triazol-l-ylmethyl)phenyl]-2(1H,3H)-imidazolone in the same
manner as in Working Example 11 to give 1-[(lR,2R)-2-(2,4-
difluorophenyl)-2-hydroxy-1-methyl-3-(lH-1,2,4-triazol-1-
yl)propyl]-3-[4-(lH-1,2,4-triazol-1-ylmethyl)phenyl]-
2(lH,3H)-imidazolone (Compound 17).
lH-NMR (CDC13)~: 1.20 (3H,d,J=7Hz), 4.19 (lH,d,J=14.4Hz),
CA 022114~8 1997-07-24
W O96/2S410 PCT/Jl ,CI'~f ~
4.97 (lH,q,J=7Hz), 5.10 (lH,d,J=14.4Hz), 5.37 (2H,s), 5.55
(lH,br), 6.65 tlH,d,J=3.2HZ), 6.70-6.90 (3H,m), 7.37
(2H,d,J=8.6Hz), 7.35-7.55 (lH,m), 7.69 (2H,d,J=8.6Hz), 7.74
(lH,s), 7.85 (lH,s), 7.99 (lH,s), 8.10 (lH,s)
Workin~ ExamPle 18
- (2R,3S)-2-(2,4-Difluorophenyl)-3-methyl-2-(lH-1,2,4-
triazol-l-yl)methyloxirane was reacted with 4-[4-(lH-1,2,4-
triazol-l-ylmethyl)phenyl]-3(2H,4H)-1,2,4-triazolone in the
same manner as in Working Example 11 to give 2-[(lR,2R)-2-
(2,4-difluorophenyl)-2-hydroxy-1-methy1-3-(lH-1,2,4-
triazol-l-yl)propyl]-4-[4-(lH-1,2,4-triazol-1-ylmethyl)-
phenyl]-3(2H,4H)-1,2,4-triazolone (Compound 18).
lH-N~ (CDC13)~: 1.30 (3H,d,J=7.2Hz), 4.36 (lH,d,J=14.2Hz),
5.02 (lH,d,J=14.2Hz), 5.09 (lH,q,J=7.2Hz), 5.42 (2H,s), 5.43
(lH,s), 6.75-6.90 (2H,m), 7.43 (2H,d,J=8.6Hz), 7.50-7.67
(lH,m), 7.63 (2H,d,J=8.6Hz), 7.69 (lH,s), 7.82 (lH,s), 7.94
(lH,s), 8.01 (lH,s), 8.15 (lH,s)
Workin~ ExamPle 19
(2R,3S)-2-(2,4-Difluorophenyl)-3-methyl-2-(lH-1,2,4-
triazol-l-yl)methyloxirane was reacted with 4-[4-(lH-l-
pyrazolyl)phenyl]-3(2H,4H)-1,2,4-triazolone in the same
manner as in Working Example 11 to give 2-[(lR,2R)-2-(2,4-
difluorophenyl)-2-hydroxy-1-methyl-3-(lH-1,2,4-triazol-1-
yl)propyl]-4-[4-(lH-l-pyrazolyl)phenyl]-3(2H,4H)-1,2,4-
triazolone (Compound 19).
CA 022114~8 1997-07-24
WO96/2S410 PCT1~6~003~
lH-NMR (CDCl3)~: 1.32 (3H,d,J=7Hz), 4.38 (lH,d,J=14.4Hz),
5.05 (lH,d,J=14.4Hz), 5.11 (lH,q,J=7Hz), 5.45 (lH,s), 6.52-
6.54 (lH,m), 6.76-6.90 (2H,m), 7.50-7.65 (lH,m), 7.65-7.93
(7H,m), 7.96 (lH,s), 7.98 (lH,d,J=2.6Hz)
Workin~ Exam~le 20
(2R,3S)-2-(2,4-Difluorophenyl)-3-methyl-2-(lH-1,2,4-
triazol-l-yl)methyloxirane was reacted with l-[4-(2H-1,2,3-
triazol-2-ylmethyl)phenyl]-2(1H,3H)-imidazolone in the same
manner as in Working Example 11 to give 1-t(lR,2R)-2-(2,4-
difluorophenyl)-2-hydroxy-1-methyl-3-(lH-1,2,4-triazol-1-
yl)propyl]-3-[4-(2H-1,2,3-triazol-2-ylmethyl)phenyl]-
2(1H,3H)-imidazolone (Compound 20).
lH-NMR (CDC13)~: 1.19 (3H,d,J=7Hz), 4.17 (lH,d,J=14.4Hz),
4.95 (lH,q,J=7Hz), 5.09 (lH,d,J=14.4Hz), 5.55 (lH,br), 5.63
(2H,s), 6.63 (lH,d,J=3.2Hz), 6.70-6.86 (3H,m), 7.40-7.55
(lH,m), 7.42 (2H,d,J=8.6Hz), 7.64 (ZH,s), 7.64
(2H,d,J=8.6Hz), 7.73 (lH,s), 7.85 (lH,s)
Workin~ ExamPle 21
(2R,3S)-2-(2,4-Difluorophenyl)-3-methyl-2-(lH-1,2,4-
triazol-l-yl)methyloxirane was reacted with 4-[4-(2H-1,2,3-
triazol-2-ylmethyl)phenyl]-3(2H,4H)-1,2,4-triazolone in the
same manner as in Working Example 11 to give 2-[(lR,2R)-2-
(2,4-difluorophenyl)-2-hydroxy-1-methyl-3-(lH-1,2,4-
triazol-l-yl)propyl]-4-[4-(2H-1,2,3-triazol-2-ylmethyl)-
phenyl]-3(2H,4H)-1,2,4-triazolone (Compound 21).
H-NMR (CDCl3)~: 1.29 (3H,d,J=7Hz), 4.34 (lH,d,J=14.4Hz),
89
CA 022114~8 1997-07-24
W O96/2S410 PCT/JP96/00325
5.02 (lH,d,J=14.4Hz), 5.08 (lH,q,J=7Hz), 5.44 (lH,s), 5.66
(2H,s), 6.73-6.87 (2H,m), 7.46 (2H,d,J=8.6Hz), 7.50-7.62
(lH,m), 7.58 (2H,d,J=8.6Hz), 7.66 (2H,s), 7.68 (lH,s), 7.78
(lH,s), 7.94 (lH,s)
Elemental analysis for C23H21F2N902
Calcd (%): C,55.98; H,4.29; N,25.55
Found (%): C,55.87; H,4.18; N,25.42
Workin~ ExamPle 22
(2R,3S)-2-(2,4-Difluorophenyl)-3-methyl-2-(lH-1,2,4-
triazol-l-yl)methyloxirane was reacted with 4-[4-(lH-l-
imidazolyl)phenyl~-3(2H,4H)-1,2,4-triazolone in the same
manner as in Working Example 11 to give 2-[(lR,2R)-2-(2,4-
difluorophenyl)-2-hydroxy-1-methyl-3-(lH-1,2,4-triazol-1-
yl)propyl]-4-[4-(lH-l-imidazolyl)phenyl]-3(2H,4H)-1,2,4-
triazolone (Compound 22).
H-NMR (CDC13)~: 1.32 (3H,d,J=7Hz), 4.40 (lH,d,J-14Hz), 5.03
(lH,d,J=14Hz), 5.11 (lH,q,J=7Hz), 5.42 (lH,s), 6.73-6.88
(2H,m), 7.26 (lH,s), 7.33 (lH,s), 7.51-7.65 (lH,m), 7.57
(2H,d,J=9Hz), 7.71 (lH,s), 7.76 (2H,d,J=9Hz), 7.86 (lH,s),
7.91 (lH,s), 7.95 (lH,s)
Workin~ ExamPle 23
(2R,3S)-2-(2,4-Difluorophenyl)-3-methyl-2-(lH-1,2,4-
triazol-l-yl)methyloxirane was reacted with l-~4-(lH-l-
imidazolyl)phenyl]-2(1H,3H)-imidazolone in the same manner
as in Working Example 11 to give 1-[(lR,2R)-2-(2,4-
CA 022114~8 1997-07-24
W O 96/2S410 PCr/JP9~/00325
difluorophenyl)-2-hydroxy-1-methyl-3-(lH-1,2,4-triazol-1-
yl)propyl]-3-[4-(lH-l-imidazolyl)phenyl]-2(1H,3H)-imidazo-
- lone (Compound 23).
H-NMR (CDC13)ô: 1.21 (3H,d,J=7Hz), 4.22 (lH,d,J=14Hz), 5.00
(lH,q,J=7Hz), 5.12 (lH,d,J=14Hz), 5.56 (lH,br), 6.70
(lH,d,J=3Hz), 6.76-6.~6 (3H,m), 7.23 (lH,s), 7.30 (lH,s),
7.42-7.54 (lH,m), 7.49 t2H,d,J=8HZ), 7.75 (lH,s), 7.78
(lH,s), 7.84 (2H,d,J=~Hz), 7.86 (lH,s)
Workin~ ExamPle 24
l-{(lR,2R)-2-(2,~-Difluorophenyl)-2-hydroxy-1-methyl-3-
(lH-1,2,4-triazol-1-yl)propyl]-3-[4-(lH-imidazolyl)phenyl]-
2(1H,3H)-imidazolone (Compound 23) obtained in Working
Example 23 was subjected to catalytic hydrogenation in the
same manner as in Working Example 10 to give 1-[(lR,2R)-2-
(2,4-difluorophenyl)-2-hydroxy-1-methyl-3-(lH-1,2,4-triazol-
l-yl)propyl]-3-[4-(lH-l-imidazolyl)phenyl]-2-imidazolidinone
(Compound 24).
H-NMR (CDC13)8: 1.08 (3H,d,J=7Hz), 3.70-4.08 (4H,m), 4.52
(lH,d,J=14Hz), 4.55-4.76 (lH,m), 5.11 (lH,d,J=14Hz), 5.40
(lH,br), 6.73-6.84 (2H,m), 7.20 (lH,s), 7.26 (lH,s), 7.36-
7.50 (lH,m), 7.39 (2H,d,J=9Hz), 7.69 (2H,d,J=9Hz), 7.76
(lH,s), 7.82 (lH,s), 7.87 (lH,s)
Workin~ ExamPle 25
(2R,3S)-2-(2,4-Difluorophenyl)-3-methyl-2-(lH-1,2,4-
triazol-l-yl)methyloxirane was reacted with 4-[4-(2H-2-
tetrazolyl)phenyl]-3(2H,4H)-1,2,4-triazolone in the same
91
CA 022114~8 1997-07-24
W O96/2S410 PCT1J~9C/00~
manner as in Working Example 11 to give 2-[(lR,2R)-2-(2,4-
difluorophenyl)-2-hydroxy-1-methyl-3-(lH-1,2,4-triazol-1-
yl)propyl]-4-[4-(2H-2-tetrazolyl)phenyl]-3(2H,4H)-1,2,4-
triazolone (Compound 25).
H-NMR (CDCl3)~: 1.33 (3H,d,J=7Hz), 4.40 (lH,d,J=14Hz), 5.04
(lH,d,J=14Hz), 5.11 (lH,q,J=7Hz), 5.37 (lH,s), 6.77-6.88
(2H,m), 7.52-7.64 (lH,m), 7.71 (lH,s), 7.87 (2H,d,J=9Hz),
7.92 (lH,s), 7.95 (lH,s), 8.34 (2H,d,J=9Hz), 8.71 (lH,s)
Elemental analysis for C21H18F2N10O2
Calcd (%): C,52.50; H,3.78; N,29.15
Found (%): C,52.36; H,3.85; N,29.02
Workin~ ExamPle 26
(2R,3S)-2-(2,4-difluorophenyl)-3-methyl-2-(lH-1,2,4-
triazol-l-yl)methyloxirane was reacted with l-[4-(2H-2-
tetrazolyl)phenyl]-2(1H,3H)-imidazolone in the same manner
as in Working Example 11 to give 1-[(lR,2R)-2-(2,4-
difluorophenyl)-2-hydroxy-1-methyl-3-(lH-1,2,4-triazol-1-
yl)propyl]-3-[4-(2H-2-tetrazolyl)phenyl]-2(lH,3H)-imidazo-
lone (Compound 26).
H-NMR (CDC13)~: 1.21 (3H,d,J=7Hz), 4.22 (lH,d,J=14Hz), 5.02
(lH,q,J=7Hz), 5.12 (lH,d,J=14Hz), 5.49 (lH,br), 6.75
(lH,d,J=3Hz), 6.75-6.85 (2H,m), 6.85 (lH,d,J=3Hz), 7.42-7.54
(lH,m), 7.76 (lH,s), 7.85 (lH,s), 7.93 (2H,d,J=9Hz), 8.25
(2H,d,J=9Hz), 8.68 (lH,s)
92
CA 022114~8 1997-07-24
WO g6/2S410 PCTJJ~G~ 4
Workin~ Exam~le 27
l-[(lR,2R)-2-(2,4-Difluorophenyl)-2-hydroxy-1-methyl-3-
(lH-1,2,4-triazol-1-yl)propyl]-3-[4-(2H-2-tetrazolyl)
phenyl]-2(1H,3H)-imidazolone (Compound 26) obtained in
Working Example 26 was subjected to catalytic hydrogenation
in the same manner as in Working Example 10 to give 1-
[(lR,2R)-2-(2,4-difluorophenyl)-2-hydroxy-1-methyl-3-(lH-
1,2,4-tri-azol-1-yl)propyl]-3-[4-(2H-2-tetrazolyl)phenyl]-
2-imidazolidinone (Compound 27).
lH-NMR (CDC13)~: 1.08 (3H,d,J=7Hz), 3.69-3.81 (lH,m), 3.94-
4.10 (3H,m), 4.52 (lH,d,J=14Hz), 4.62-4.80 (lH,m), 5.13
(lH,d,J=14Hz), 5.25-5.50 (lH,br), 6.72-6.84 (2H,m), 7.36-
7.49 (lH,m), ? 77 (lH,s), 7.80 (2H,d,J=9Hz), 7.86 (lH,s),
8.13 (2H,d,J=9Hz), 8.64 (lH,s)
Workin~ Exam~le 28
(2R,3S)-2-(2,4-Difluorophenyl)-3-methyl-2-(lH-1,2,4-
triazol-l-yl)methyloxirane was reacted with l-[4-(lH-1,2,3-
triazol-l-ylmethyl)phenyl]-2(1H,3H)-imidazolone in the same
manner as in Working E~ample 11 to give 1-[(lR,2R)-2-(2,4-
difluorophenyl)-2-hydroxy-1-methyl-3-(lH-1,2,4-triazol-l-
yl)propyl]-3-[4-(lH-1,2,3-triazol-1-ylmethyl)phenyl]-
2(lH,3H)-imidazolone (Compound 28).
H-NMR (CDC13)~: 1.20 (3H,d,J=7Hz), 4.19 (lH,d,J=14Hz), 4.97
(lH,q,J=7Hz), 5.09 (lH,d,J=14Hz), 5.55 (lH,br), 5.59 (2H,s),
6.65 (lH,d,J=3.2Hz), 6.75-6.90 (3H,m), 7.35-7.55 (4H,m),
7.66-7.75 (4H,m), 7.84 (lH,s)
CA 022114~8 1997-07-24
W O96/2S410 PCT/JPg6/00325
Workin~ ExamPle 29
(2R,3S)-2-(2,4-Difluorophenyl)-3-methyl-2-(lH-1,2,4-
triazol-l-yl)methyloxirane was reacted with 4-[4-(lH-1,2,3-
triazol-l-ylmethyl)phenyl]-3(2H,4H)-1,2,4-triazolone in the
same manner as in Working Example 2 to give 2-[(lR,2R)-2-
(2,4-difluorophenyl)-2-hydroxy-l-methyl-3-(lH-1,2,4-
triazol-l-yl)propyl]-4-[4-(lH-1,2,3-triazol-1-ylmethyl)-
phenyl]-3(2H,4H)-1,2,4-triazolone (Compound 29).
H-NMR (CDC13)8: 1.30 (3H,d,J=7Hz), 4.36 (lH,d,J=14Hz), 5.00
(lH,d,J=14Hz), 5.08 (lH,q,J=7Hz), 5.41 (lH,s), 5.63 (2H,s),
6.75-6.90 (2H,m), 7.40-7.64 (6H,m), 7.69 (lH,s), 7.76
(lH,s), 7.80 (lH,s), 7.94 (lH,s)
Workin~ ExamPle 30
(2R,3S)-2-(2,4-Difluorophenyl)-3-methyl-2-(lH-1,2,4-
triazol-l-yl)methyloxirane was reacted with l-[4-(2-methyl-
4-thiazolyl)phenyl]-2(lH,3H)-imidazolone in the same manner
as in Working Example 11 to give 1-[(lR,2R)-2-(2,4-
difluorophenyl)-2-hydroxy-1-methyl-3-(lH-1,2,4-triazol-1-
yl)propyl]-3-[4-(2-methyl-4-thiazolyl)phenyl]-2(1H,3H)-
imidazolone (Compound 30).
H-NMR (CDCl3)~: 1.21 (3H,d,J=7Hz), 2.78 (3H,s), 4.22
(lH,d,J=14Hz), 4.98 (lH,q,J=7Hz), 5.12 (lH,d,J=14Hz), 5.60
(lH,br), 6.70-6.85 (4H,m), 7.33 (lH,s), 7.40-7.55 (lH,m),
7.69-8.00 (6H,m)
94
CA 022114~8 1997-07-24
W O 96125410 PCTJ~ 5
Workin~ Exam~le 31
(2R,3S)-2-(2,4-Difluorophenyl)-3-methyl-2-(lH-1,2,4-
triazol-l-yl)methyloxirane was reacted with 4-[4-(2-
methyl-4-thiazolyl)phenyl]-3(ZH,4H)-1,2,4-triazolone
in the same manner as in Working Example 2 to give
2-[(lR,2R)-2-(2,4-difluorophenyl)-2-hydroxy-1-methyl-3-(lH-
1,2,4-triazol-1-yl)propyl]-4-[4-(2-methyl-4-thiazolyl)phe-
nyl]-3(2H,4H)-1,2,4-triazolone (Compound 31).
H-NMR (CDCl3)~: 1.31 (3H,d,J=7Hz), 2.79 (3H,s), 4.37
(lH,d,J=14Hz), 5.04 (lH,d,J=14Hz), 5.11 (lH,q,J=7Hz), 5.47
(lH,s), 6.77-6.90 (2H,m), 7.39 (lH,s), 7.50-7.70 (4H,m),
7.85-8.05 (4H,m)
Workina Exam~le 32
(2R,3S)-2-(2-Fluorophenyl)-3-methyl-2-(lH-1,2,4-
triazol-l-yl)methyloxirane was reacted with 1-[4-(lH-1,2,3-
triazol-l-yl)phenyl]-2(1H,3H)-imidazolone in the same manner
as in Working Example 11 to give 1-[(lR,2R)-2-(2-fluorophe-
nyl)-2-hydroxy-1-methyl-3-(lH-1,2,4-triazol-1-yl)propyl]-3-
[4-(lH-1,2,3-triazol-1-yl)phenyl]-2(1H,3H)-imidazolone
(Compound 32).
l-NMR (CDCl3)~: 1.21 (3H,d,J=7Hz), 4.23 (lH,d,J=14.2Hz),
5.08 (lH,q,J=7Hz), 5.17 (lH,d,J=14.2Hz), 5.36 (lH,br), 6.73
(lH,d,J=3.2Hz), 6.86 (lH,d,J=3.2Hz), 6.99-7.09 (2H,m),
7.19-7.52 (2H,m), 7.51-7.92 (6H,m), 7.80 (lH,s), 8.03 (lH,s)
Elemental analysis for C23H21FN8O2
Calcd (%): C,59.99; H,4.60; N,24.23
CA 022114~8 1997-07-24
W O9612S410 PCT1JP96/00325
Found (%): C,59.62; H,4.61; N,24.13
Workin~ ExamPle 33
(2R,3S)-2-(2-Fluorophenyl)-3-methyl-2-(lH-1,2,4-
triazol-l-yl)methyloxirane was reacted with 1-[4-(2H-1,2,3-
triazol-2-yl)propyl]-2(lH,3H)-imidazolone to give
l-[(lR,2R)-2-(2-fluorophenyl)-2-hydroxy-1-methyl-3-(lH-
1,2,4-triazol-1-yl)propoxy]-3-[4-(2H-1,2,3-triazol-2-
yl)phenyl]-2(lH,3H)-imidazolone (Compound 33).
H-NMR (CDC13)~: 1.21 (3H,d,J=7Hz), 4.23 (lH,d,J=14Hz), 5.07
(lH,q,J=7Hz), 5.18 (lH,d,J=14Hz), 5.37 (lH,br), 6.70
(lH,d,J=3.2Hz), 6.83 (lH,d,J=3.2Hz), 6.98-7.08 (2H,m),
7.19-7.51 (2H,m), 7.73 (lH,s), 7.75 (2H,d,J=9.2Hz), 7.83
(2H,s), 7.85 (lH,s), 8.17 (2H,d,J=9.2Hz)
Elemental analysis for C23H21FNgO2
Calcd (%): C,59.99; H,4.60; N,24.33
Found (%): C,59.80; H,4.58; N,23.87
Workin~ Exam~le 34
l-[(lR,2R)-2-(2-Fluorophenyl)-2-hydroxy-1-methyl-3-(lH-
1,2,4-triazol-1-yl)propyl]-3-[4-(lH-1,2,3-triazol-1-
yl)phenyl]-2(1H,3H)-imidazolone (compound 32) obtained in
Working Example 32 was subjected to catalytic hydrogenation
in the same manner as in Working Example 10 to give 1-
[(lR,2R)-2-(2-fluorophenyl)-2-hydroxy-1-methyl-3-(lH-1,2,4-
triazol-l-yl)propyl]-3-[4-(lH-1,2,3-triazol-1-yl)phenyl]-2-
imidazolidinone (Compound 34).
_ 96
CA 022114~8 1997-07-24
W O96/2S410 PC~/JP~/00~2S
H-NMR (CDC13)o: 1.08 (3H,d,J=7Hz), 3.72-4.14 (4H,m), 4.54
(lH,d,J=14.2Hz), 4.70-4.84 (lH,m), 5.17 (lH,d,J=14.2Hz),
.30 (lH,br), 6.96-7.08 (2H,m), 7.18-7.50 (2H,m), 7.68-7.78
(4H,m), 7.75 (lH,s), 7.84 (2H,s), 7.98 (lH,s)
Elemental analysis for C23H23FN8O2
Calcd (%): C,59.73; H,~.01; N,24.23
Found (%): C,59.32; H,4.99; N,24.00
Workin~ ExamPle 35
l-[(lR,2R)-2-(2-Fluorophenyl)-2-hydroxy-1-methyl-3-(lH-
1,2,3-triazol-1-yl)propyl]-3-[4-(2H-l,Z,3-triazol-2-yl)phe-
nyl]-2(1H,3H)-imidazolone (Compound 33) obtained in Working
Example 33 was subjected to catalytic hydrogenation in the
same manner as in Working Example lQ to ~ive l-[(lR,2R)-2-
(2-fluorophenyl)-2-hydroxy-1-methyl-3-(lH-1,2,4-triazol-1-
yl)propyl]-3-[4-(2H-1,2,3-triazol-2-yl)phenyl]-2-imidazoli-
dinone (Compound 35).
mp.: 178-179~C
H-NMR (CDCl3)o: 1.08 (3H,d,J=7Hz), 3.71-4.07 (4H,m), 4.54
(lH,d,J=14Hz), 4.74-4.77 (lH,m), 5.18 (lH,d,J=14Hz), 5.38
(lH,br), 6.96-7.06 (2H,m), 7.16-7.51 (2H,m), 7.71
(2H,d,J=9Hz), 7.74 (lH,s), 7.80 (2H,s), 7.83 (lH,s), 8.07
(2H,d,J=9Hz)
Elemental analysis for C23H23FNgO2
Calcd (%): C,59.73; H,5.01; N,24.23
Found (%): C,59.49; H,5.23; N,24.01
97
CA 022114~8 1997-07-24
W O96/2S410 PCTIJ~3C
Workin~ Exam~le 36
(2R,3S)-2-(2,4-Difluorophenyl)-3-methyl-2-(lH-1,2,4-
triazol-l-yl)methyloxirane (0.50 g), 4-[4-(2-methyl-4-
oxazolyl)phenyl]-3(2H,4H)-1,2,4-triazolone (0.56 g) and
potassium carbonate (powder: 1.38 g) were added to a mixture
of l-methyl-2-pyrrolidone (5 ml) and N,N-dimethylformamide(4
ml), and the mixture was heated with stirring at 90 C for 20
hours. After cooling, the reaction solution was diluted
with ethyl acetate (40 ml). Ice water (40 ml) was added
thereto to separate the ethyl acetate layer. The ethyl
acetate layer was washed with O.SN-sodium hydroxide (40 ml),
lN-hydrochloric acid (40 ml) and a saturated aqueous solu-
tion of sodium chloride (40 ml) successively. The ethyl
acetate layer was dried over anhydrous magnesium sulfate and
distilled under reduced pressure to remove the solvent. The
residue was purified by silica gel chromatography (eluent:
ethyl acetate to ethyl acetate/methanol = 9/1) and crystal-
lized from ethyl acetate-diisopropyl ether to give 2-
[(lR,2R)-2-(2,4-difluorophenyl)-2-hydroxy-1-methyl-3-(lH-
1,2,4-triazol-1-yl)propyl]-4-[4-(2-methyl-4-oxazolyl)phe-
nyl]-3(2H,4H)-1,2,4-triazolone (Compound 36, 0.33 g).
H-NMR (CDC13)~: 1.31 (3H,d,J=7Hz), 2.54 (3H,s), 4.36
(lH,d,J=14Hz), 5.04 (lH,d,J=14Hz), 5.10 (lH,q,J=7Hz), 5.47
(lH,s), 6.77-6.88 (2H,m), 7.51-7.69 (4H,m), 7.85 (lH,s),
7.86 (2H,d,J=8Hz), 7.88 (lH,s), 7.96 (lH,s)
98
CA 022114~8 1997-07-24
WO 96/2S410 PCT1JPg6JOD325
Workin~ ExamPle 37
(2R,3S)-Z-(2,4-Difluorophenyl)-3-methyl-2-(lH-1,2,4-
triazol-l-yl)methyloxirane was reacted with 1-[4-(2-methyl-
4-oxazolyl)phenyl]-2(lH,3H)-imidazolone in the same manner
as in Working Example 11 to give 1-[(lR,2R)-2-(2,4-
difluorophenyl)-2-hydroxy-1-methyl-3-(lH-1,2,4-triazol-
l-yl)propyl]-3-[4-(2-methyl-4-oxazolyl)phenyl]-2(1H,3H)-
imidazolone (Compound 37).
H-NMR (CDC13)o: 1.21 (3H,d,J=7Hz), 2.53 (3H,s), 4.21
(lH,d,J=14Hz), 4.97 (lH,q,J=7Hz), 5.12 (lH,d,J=14Hz), 5.60
(lH,br), 6.69-6.86 (4H,m), 7.42-7.55 (lH,m), 7.69
(2H,d,J-9Hz), 7.73 (lH,s), 7.80 (2H,d,J=9Hz), 7.84 (lH,s),
7.86 (lH,s)
Workin~ Exam~le 38
(2R,3S)-2-(2-fluorophenyl)-3-methyl-2-(lH-1,2,4-
triazol-l-yl)methyloxirane (2.24 g), 1-[4-(lH-1,2,4-tria-
zol-l-yl)phenyl]-2(1H,3H)-imidazolone (2.17 g) and cesium
carbonate (powder: 7.76 g) were added to dimethylsulfoxide
(100 ml), and the mixture was heated with stirring at 100 C
for 17 hours. After being cooled, the reaction solution was
diluted with ethyl acetate (200 ml). Ice water (200 ml) was
added thereto to separate the ethyl acetate layer. The
aqueous layer was extracted with ethyl acetate (100 ml).
The ethyl acetate layers were combined and washed with
O.~N-sodium hydroxide (100 ml), lN-hydrochloric acid (100 ml
x 2) and a saturated aqueous solution of sodium chloride
99
CA 022114~8 1997-07-24
W O96/2S410 PCT/JP96/0032S
(100 ml) successively. The ethyl acetate layer was dried
over anhydrous magnesium sulfate and distilled under reduced
pressure to remove the solvent. The residue was purified by
silica gel chromatography (eluent: ethyl acetate/acetone =
4/1) to give 1-[(lR,2R)-2-(2-fluorophenyl)-2-hydroxy-1-
methyl-3-(lH-1,2,4-triazol-1-yl)propyl]-3-[4-tlH-1,2,4-
triazol-l-yl)phenyl]-2(lH,3H) imidazolone (Compound 38; 0.45
g)-
H-NMR (CDC13)~: 1.21 (3H,d,J=7Hz), 4.23 (lH,d,J=14Hz), 5.02
(lH,q,J=7Hz), 5.17 (lH,d,J=14Hz), 5.37 (lH,br), 6.71
(lH,d,J=3.2Hz), 6.86 (lH,d,J=3.2Hz), 6.99-7.10 (2H,m),
7.20-7.52 (2H,m), 7.70-7.89 (6H,m), 8.13 (lH,s), 8.58 (lH,s)
Workin~ ExamPle 39
l-[(lR,2R)-2-(2-Fluorophenyl)-2-hydroxy-1-methyl-
3-(lH-1,2,4-triazol-1-yl)propyl]-3-[4-(lH-1,2,4-triazol-
l-yl)phenyl]-2(lH,3H)-imidazolone (Compound 38) obtained in
Working Example 38 was subjected to catalytic hydrogenation
in the same manner as in Working Example 10 to give 1-
[(lR,2R)-2-(2-fluorophenyl)-2-hydroxy-1-methyl-3-(lH-1,2,4-
triazol-l-yl)propyl]-3-[4-(lH-1,2,4-triazol-1-yl)phenyl]-2-
imidazolidinone (Compound 39).
H-NMR (CDC13)~: 1.08 (3H,d,J=7Hz), 3.72-4.10 (4H,m), 4.53
(lH,d,J=14.2Hz), 4.76-4.79(1H,m), 5.17 (lH,d,J=14.2Hz), 5.19
(lH,br), 6.97-7.09 (2H,m), 7.17-7.46 (2H,m), 7.63-7.77
(5H,m), 7.82 (lH,s), 8.10 (lH,s), 8.52 (lH,s)
100
CA 022114~8 1997-07-24
W O 96125410 PCT~JFg. ~. 5
Elemental analysis for C23H23FN8~2
Calcd t%): C,59.73; H,6.01; N,24.23
Found (X): C,59.33; H,5.03; N,23.98
Workin~ Exam~le 40
(2R,3S)-2-t2-Fluorophenyl)-3-methyl-2-(lH-1,2,4-tria-
zol-l-yl)methyloxirane (2.34 g), 1-[4-(2H-2-tetrazolyl)-
phenyl]-2(1H,3H)-imidazolone (2.28 g) and cesium carbonate
(powder: 6.52 g) were added to 1-methyl-2-pyrroli done (100
ml), and the mixture was heated at 80~C for 18 hours with
stirring. The reaction solution was cooled, diluted with
ethyl acetate (200 ml) and added to ice water (200 ml) to
separate the ethyl acetate layer. The aqueous layer was
extracted with ethyl acetate (100 ml). The combined ethyl
acetate layers were washed with lN-hydrochloric acid (100 ml
x 2) and a saturated aqueous solution of sodium chloride
(100 ml) successively. The ethyl acetate layer was dried
over anhydrous magnesium sulfate and distilled under reduced
pressure to remove the solvent. The residue was purified by
silica gel chromatography (eluent: hexane/acetone = 1/1) to
give l-[(lR,2R)-2-(2-fluorophenyl)-2-hydroxy-1-methyl-3-
(lH-1,2,4-triazol-1-yl)propyl]-3-[4-
(2H-2-tetrazolyl)phenyl]-2(1H,3H)-imidazolone (Compound 40;
0.494 g) as a colorless crystalline powder.
H-NMR (CDC13)~: 1.21 (3H,d,J=7Hz), 4.23 (lH,d,J=14Hz), 5.10
(lH,q,J=7Hz), 5.19 (lH,d,J=14Hz), 5.34 (lH,br), 6.75
(lH,d,J=3.2Hz), 6.88 (lH,d,J=3.2Hz), 6.99-7.09 (2H,m),
101
CA 022114~8 1997-07-24
W O96/25410 PCT1J~6/0
7.20-7.51 (2H,m), 7.75 (lH,s), 7.81.(lH,s), 7.94
(2H,d,J=9Hz), 8.25 (2H,d,J=9Hz), 8.68 (lH,s)
Elemental analysis for C22H20FNgO2
Calcd (%): C,57.26; H,4.37; N,27.32
Found (%): C,57.19; H,4.29; N,27.07
Workin~ Exam~le 41
l-[(lR,2R)-2-(2-Fluoropkenyl)-2-hydroxy-1-methyl-3-
(lH-1,2,4-triazol-1-yl)propyl]-3-[4-(2H-2-tetrazolyl)phe-
nyl]-2(lH,3H)-imidazolone (Compound 40) obtained in Working
Example 40 was subjected to catalytic hydrogenation in the
same manner as in Working Example 10 to give 1-[(lR,2R)-
2-(2-fluorophenyl)-2-hydroxy-1-methyl-3-(lH-1,2,4-tria-
zol-l-yl)propyl]-3-[4-(2H-2-tetrazolyl)phenyl]-2-
imidazolidinone (Compound 41) as a colorless crystalline
powder.
lH-NMR (CDCl3)~: 1.08 (3H,d,J=7Hz), 3.74-3.81 (lH,m), 3.94-
4.13 (3H,m), 4.53 (lH,d,J=14Hz), 4.63-4.81 (lH,m), 5.13
(lH,d,J=14Hz), 5.15-5.30 (lH,br), 6.97-7.07 (2H,m), 7.17-
7.45 (2H,m), 7.78 (lH,s), 7.79 (2H,d,J=9Hz), 7.83 (lH,s),
8.13 (2H,d,J=9Hz), 8.65 (lH,s)
Elemental analysis for C22H22FNgO2
Calcd (%): C,57.01; H,4.78; N,27.20
Found (%): C,56.96; H,4.86; N,26.84
Wor~in~ Exam~le 42
72% sodium hydride in oil (17 mg) was dispersed in
102
CA 022114~8 1997-07-24
W O96/25410 P~J1~6~0~32S
dimethylformamide (3 ml), to which 1,2,4-triazole (42 mg)
was added at ice-bath temperature, and the mixture was
stirred at room temperature for 40 minutes. To the result-
ant was added a solution of l-[(lR, 2S)-2-(2,4-difluorophe-
nyl)-2,3-epoxy-1-methylpropyl]-3-[4-[(lH-l-tetrazolyl)phe-
nyl]-2H(lH,3H)-imidazolone (0.205 g) in dimethylformamide (2
ml), and the mixture was heated at 50 C for 6 hours. After
the mixture was cooled, cold water (30 ml) and ethyl acetate
(30 ml) were added thereto. The separated aqueous layer was
extracted with ethyl acetate twice. The combined ethyl
acetate layers were washed with water and a saturated aque-
ous solution of sodium chloride successively, dried over
anhydrous magnesium sulfate~ and distilled under reduced
pressure to remove the solvent. The residue was purified by
silica gel chromatography (eluent: ethyl acetate) to give
l-[(lR,2R)-2-(2,4-difluorophenyl)-2-hydroxy-l-methyl-3-(lH-
1,2,4-triazol-1-yl)propyl]-3-[4-(lH-l-tetrazolyl)phenyl]-
2(1H,3H)-imidazolone (Compound 42; 0.15g) as a colorless
crystalline powder.
H-NMR (CDCl3)~: 1.21 (3H,d,J=7Hz), 4.22 (lH,d,J=14Hz), 5.03
(lH,q,J=7Hz), 5.13 (lH,d,J=14Hz), 5.45 (lH,br), 6.74-6.88
(4H,m), 7.42-7.55 (lH,m), 7.77 (lH,s), 7.82 (2H,d,J-9Hz),
7.86 (lH,s), 7.96 (2H,d,J=9Hz), 9.06 (lH,s)
The same product (Compound 42) was obtained when a
reaction was carried out in the same manner as in the above
except that sodium hydride was replaced by potassium carbon-
103
CA 022114~8 1997-07-24
W O96/2S410 PCT/JP9G~
ate.
Workin~ ExamPle 43
l-t(lR,2R)-2-(2,4-Difluorophenyl)-2-hydroxy-1-methyl-3-
(lH-1,2,4-triazol-1-yl)propyl]-3-[4-(lH-l-tetrazolyl)phe-
nyl]-2(1H,3H)-imidazolone (compound 42) obtained in Working
Example 42 was subjected to catalytic hydrogenation in the
same manner as in Working Example 10 to give l-[(lR,2R)-2-
(2,4- difluorophenyl)-2-hydroxy-1-methyl-3-(lH-1,2,4-tria-
zol-l-yl)propyl]-3-[4-(lH-l-tetrazolyl)phenyl]-2-imidazoli-
dinone (compound 43) as a colorless crystalline powder.
H-NMR (CDC13)~: 1.08 (3H,d,J=7Hz), 3.69-4.14 (4H,m), 4.52
(lH,d,J=14Hz), 4.65-4.80 (lH,m), 5.12 (lH,d,J=14Hz), 5.35
(lH,br), 6.74-6.84 (2H,m), 7.36-7.49 (lH,m), 7.68
(2H,d,J=9Hz), 7.77 (lH,s), 7.82 (2H,d,J=9Hz), 7.87 (lH,s),
8.98 (lH,s)
Workin~ ExamPle 44
72% sodium hydride in oil (120 mg) was dispersed in di-
methylformamide (10 ml), to which 1,2,4-triazole (290 mg)
was added at ice-bath temperature, and the mixture was
stirred at room temperature for 30 minutes. To the result-
ant was added a solution of l-[(lR, 2S)-2-(2-fluorophenyl)-
2,3-epoxy-1-methylpropyl]-3-[4-[(lH-l-tetrazolyl)phenyl]-
2H(lH,3H)-imidazolone (0.82 g) in dimethylformamide (5 ml),
and the mixture was heated at 50 C for 5 hours. After the
reaction solution was cooled, cold water (30 ml) and ethyl
104
CA 022114~8 1997-07-24
W O96J25410 PCTJJr,.'~0~
acetate (40 ml) were added. The separated aqueous layer was
extracted with ethyl acetate twice. The ethyl acetate
layers were combined, washed with water and a saturated
aqueous solution of sodium chloride successively, dried over
anhydrous magnesium sulfate and distilled under reduced
pressure to remove the solvent. The residue was purified by
silica gel chromatography (eluent: ethyl acetate) to give
1-[(lR,2R)-2-(2-fluorophenyl)-2-hydroxy-1-methyl-3-(lH-
1,2,4-triazol-1-yl)propyl]-3-[4-(lH-1-tetrazolyl)phenyl]-
2(1H,3H)-imidazolone (Compound 44; 0.30 g) as a colorless
crystalline powder.
H-NMR (CDCl3)~: 1.21 (3H,d,J=7Hz), 4.22 (lH,d,J=14H7), 5.10
(lH,q,J=7Hz), 5.18 (lH,d,J=14Hz), 5.31 (lH,br), 6.75
(lH,d,J=3Hz), 6.90 (lH,d,J=3Hz), 6.99-7.32 (3H,m). 7.43-7.51
(lH,m), 7.75 (lH,s), 7.80-7.85 (3H,m), 7.97 (2H,d,J=9Hz),
9.07 (lH,s)
Workin~ Exam~le 45
1-[(lR,2R)-2-(2-Fluorophenyl)-2-hydroxy-1-methyl-3-
(lH-1,2,4-triazol-1-yl)propyl]-3-[4-(lH-1-tetrazolyl)phe-
nyl]-2(1H,3H)-imidazolone (compound 44) obtained in Working
Example 44 was subjected to catalytic hydrogenation in the
same manner as in Working Example 10 to give 1-[(lR,2R)-2-
(2-fluorophenyl)-2-hydroxy-1-methyl-3-(lH-1,2,4-triazol-1-
yl)propyl]-3-[4-(lH-1-tetrazolyl)phenyl]-2-imidazolidinone
(Compound 45).
H-NMR (CDC13)~: 1.08 (3H,d,J=7Hz), 3.70-4.19 (4H,m), 4.53
105
CA 022114~8 1997-07-24
W O 96/2S410 PCT/J~G~
(lH,d,J=14Hz), 4.72-4.88 (lH,m), 5.10-5.26 (2H,m), 6.97-7.45
(4H,m), 7.68 (2H,d,J=9Hz), 7.76 (lH,s), 7.82 (lH,s), 7.83
(2H,d,J=9Hz), 8.97 (lH,s)
Workin~ Example 46
1-[(lR,2S)-2-(2-Fluorophenyl)-2-hydroxy-3-methane-
sulfonyloxy-1-methylpropyl]-3-[4-(lH-1-tetrazolyl)phenyl]-
2(1H,3H)-imidazolone (200 mg) was dissolved in dimethylfor-
mamide (10 ml), to which lH-1,2,4-triazole (83 mg) and
potassium carbonate (168 mg) were added, and the mixture was
heated at 50 C for 20 hours. The reaction solution was
diluted with ethyl acetate (30 mi), washed with water (15
ml), lN-hydrochloric acid (15 ml x 2) and a saturated aque-
ous solution of sodium chloride (15 ml). The organic layer
was dried over magnesium sulfate and distilled under reduced
pressure to remove the solvent. The residue was purified by
silica gel chromatography (eluent: ethyl acetate). The
desired fraction was concentrated and recrystallized from a
mixture of ethyl acetate and diisopropyl ether to give 1-
[(lR,2R)-2-(2-fluorophenyl)-2-hydroxy-1-methyl-3-(lH-1,2,4-
triazol-1-yl)propyl-3-[4-(lH-1-tetrazolyl)phenyl]-2(1H,3H)-
imidazolone (Compound 44, 65 mg) as a colorless crystalline
powder.
H-NMR (CDCl3)~: 1.21 (3H,d,J=7Hz), 4.22 (lH,d,J=14Hz), 5.11
(lH,q,J=7Hz), 5.18 (lH,d,J=14Hz), 5.32 (lH,br), 6.75
(lH,d,J=3Hz), 6.90 (lH,d,J=3Hz), 7.00-7.27 (3H,m), 7.43-7.51
106
CA 022ll458 l997-07-24
W O96~2S410 PCT/Jl,-'~0~
(lH,m), 7.75 (lH,s), 7.80-7.84 (3H,m), 7.97 (2H,d,J=9Hz),
9.06 (lH,s)
Tables 10 to 14 show a group of preferred compounds
belonging to the compound (I) of the present invention but
the present invention is not limited to those compounds.
107
CA 02211458 1997-07-24
W O96/25410 PCT/J~3G~
Table 10
HO CH~
I~N--CH2~ CR)--Q
D
Compo~nd No. D Q
2,4-F2 --h~ ~OCH2CF2CF2H
O O
2 2,4-F2 ~ 3 OCH2CF2CF2H
O ' O
3 2,4-F2 --~ ~OCF2CF2H
4 2,4--F2 --1~ ~--CH2CF3
2,4-F2 _~ ~N'~
6 2,4-F2 --
2,4-F2 --~
2,4-F2 --~ CH2CF2CF2H
2,4-F2 _~
2,4--F2 - --~ N~
108
CA 02211458 1997-07-24
WO 9612S410 PCI'~Jr~C,'00
Table 11
Compound No. D Q
11 2,4-F
12 2,4-F2
13 2,4-F2 --
1 4 2,4-F2
2,4-F2
16 2,4-F2 --1
17 2,4--F2 --~CH2--
18 2,4-F2 --~I~CH2--1
19 2,4--F2 --
~Q 2,4-F2 ~ ~H2--1
109
CA 02211458 1997-07-24
W O96/2S410 PCTlJ~_''vC3
Table 12
Compound No. D Q
2,4-Fz --~I~CH2--1
22 2,4-F2 --t~
23 2,4-F2
24 2,4-F2
2,4-F2
26 2,4-F2 --1
27 ~,4-F2 --
2,4-F2 --I~I~CH2--1
29 2,4-F2 --~I~CH2--1
2,4-F2 --~ ~CH3
110
CA 02211458 1997-07-24
WC~ 9612S410 PCr/JP96/00325
Table 13
ComPound No. D Q
3 l 2,4-F2 ~ WH3
32 2-F
33 2--F --t~
34 2-F --
2-F ~
36 2,4-F2 --~ /cH3
37 2,4-F2 --~H3
38 2-F
39 2-F --~
111
CA 02211458 1997-07-24
W O96t2S410 PCT/JF~6/00
Table 14
Compound No. D Q
2-F
41 2-F
42 2,4-F2
43 2,4-F2 --
44 2-F --
2-F
Formulation Exam~le 1
All the components for the below prescription using the
Compound 7 obtained in Working Example 7 were mixed, and
filled into gelatine capsules to prepare capsuled drugs each
containing Compound 7 in the amount of 50 mg.
112
CA 022114~8 1997-07-24
WO 96/2S410 PCT/Jr~C/~ 7~;
Compound 7 in Working Example 7 50 mg
Lactose 100 mg
Corn Starch 40 mg
Magnesium Stearate 10 mg
Total 200 mg
Formulation ExamPle 2
The compound lO obtained in Working Example 10 and
magnesium stearate were made into granules by using an
aqueous solution of soluble starch, dried and mixed with
lactose and corn starch. The mixture was molded under
compression to form a tablet for the below prescription.
Compound 10 of Working Example 1050 mg
Lactose 65 mg
Corn Starch 30 mg
Soluble Starch 35 mg
Magnesium Stearate 20 mg
Total 200 mg
Ex~eriment ExamPle 1
Test Method: Five-week-old Crj: CDF1 mice were inoculated
with the minimum lethal dose of Candida al bicans TA intrave-
nously. The test compounds were administered orally once
immediately after infection as a 30% HPCD (hydroxypropyl-~-
cyclodextrin) solution. The activity was expressed in terms
of ED50 value calculated by the Reed and Muench method from
the survival rate 7 days after infection.
Result: Tables 15 and 16 shows the protective effects of the
113
CA 02211458 1997-07-24
W O96/2S410 PCT1JP96/00325
present compounds against Candida albicans infection in
mice.
Table 15
Compound No. ED~(mg/~) PO
0.22
6 0.35
7 0.096
0.16
0 0.32
1 1 0.45
12 071
13 0.18
4 0.32
19 0.39
0.088
26 O.t6
0.18
34 0.80
0.71
PO: oral ~rini~tration
114
CA 022ll458 l997-07-24
W0 961'~S41~ PC'r~J
Table 16
Compound No. ED~(mg/kg) PO
38 s ~'
39 0.5
0.35
41 0.80
42 0.22
43 0.89
44 0.39
0.45
P0: oral ~ ni~tration
115
- .
CA 02211458 1997-07-24
W 096t2S410 PCTIJ~CI~C~
INDUSTRIAL APPLICABILITY
The present compounds or their salts have low toxicity
and excellent antifungal activity. Therefore, they are
useful in protection and treatment for fungal infections of
m~mm~lS as antifungal preparations. In addition, they can
serve as antifungal preparations for agricultural use.
116