Note: Descriptions are shown in the official language in which they were submitted.
CA 02212226 1997-08-01
~,
DESCRIPTION
BICYCLIC AMINO GROUP-SUBSTITUTED PYRIDONE-
CARBOXYLIC ACID DERIVATIVES, ESTERS THEREOF
AND SALTS THEREOF, AND BICYCLIC AMINES USEFUL
AS INTERMEDIATES THEREOF
Technical Field
This invention relates to novel compounds
useful as antibacterial agents, and bicyclic amines
useful as intermediates for the synthesis thereof.
Back~round Art
A variety of antibacterial pyridonecarboxylic
acid derivatives are known. For example, Japanese
Patent Application Laid-Open No. 56673/'89 describes
pyridonecarboxylic aGids of the general formuia
O .,
~ ~ C O O H
wherein R repre.sents a lower alkyl group, a halogenated
lower alkyl group, a lower alkenyl group, a cycloalkyl
group" or a phemyl group which may have one or more
substituents; X represents a nitrogen atom or C-A in
which A represents a hydrogen atom or a halogen atom; Y
repre:;ents a hydrogen atom or a halogen atom; and Z
repre:;ents a group of the formula
R1,
R2-N
(R,3CH)n
R4- N-
~ /
R5
CA 022l2226 l997-08-Ol
in which R~ represents a hydrogen atom, a lower alkyl-
oxycarbonyl group, or an acyl group that may be substi-
tuted by one or more halogen atoms; two of R 2~ R 3, R 4
and R5 are connected directly or through a lower alky3
chain to form a ring, and the others represent hydrogen
atoms; and n is 0 or 1, provided that R 2 and R 3 are
directly connected. However, no specific example o~ a
compound of the above ~ormula (A) in which R 4 and R5 are
connected through an ethylene chain to form a ring is
10 disclosed therein.
Moreover, European Patent Application Laid-
~pen No. 0343524 discloses pyridonecarboxylic acid
derivatives of the general formula
/ (~ H2)P
(C H2 ~ N-A (B)
R1/ (CH2)m-R2
wherein R1 is hydrogen, hydroxy, C,-C4 alkyl, C1-C4
alkoxy,l oxo, halogen, or amino which may optionally be
substi-tul;ed by C1-C4 alkyl and/or Ct-G4 alkanoyl; R2 is
azido, hydroxy, I,l-C4 alkoxy, C1-C4 alkoxycarbonyl.
25 C1 -C4 alkanoyl, or amino which may optionally be substi-
tuted by C1 -C4 aIkyl and/or C1-C4 alkanoyl; A is
R~ O R5 O
RG~3,COOR3 R6~3,COOR3
R4 0~_,X
R3 is hydrogen or a carboxyl-protecting group; R4 is
35 Ct-G4 alkyl, G2-C,5 alkenyl, C3-C5 CyC loalkyl, mono- or
difluorophenyl, or a five-membered or six-membered
CA 02212226 1997-08-01
heterocyclic gr OUp which may optionally be substituted
by halogen and/or Cl-C4 alkyl; R5 is hydrogen, amino,
hydroxy or C1-C4 alkoxy; R6 is halogen; X is CH-(C1-G4
alkyl:), C=CH2, W-H or N-(Cl-C4 alkyl); Z is CQ or N; Q
is hydrogen, C1-C4 alkoxy, halogen, ~1 -C4 alkyl or
cyano; m is an integer of O or 1; and n and p are each
an in-teger of 1 to 3. However, they do not include any
compound in which n is 0.
Furthermore, Chemical Abstract, 66, 37500b
(1967,~ and Japanese Patent Application Laid-Open No.
11729,/'81 disclose compounds of the formula
¦ ~ ,NH
where;n R represents a hy,drogen atom or a carboxyl
group.
Conventional pyridonecarboxylic acid deriva-
20 tives substituted by a bicyclic amino group, such as
those represented by the above general formulae (A) and
(B), are useful as antibacterial agents. However, their
antibacterial ac:tivities and, in particular, in vivo
antibacterial ac:tivities are not always satisfactory.
The present invention has been completed as a
result: of extensive investigations conductedL with a view
to developing a pyridonecarboxylic acid derivative
showing a furthe:r enhancement in antibacterial activity
and, in particular, in vivo antibacterial activity.
D;sclosure of thle Invention
Accordling to the present invention, there are
provided novel blicyclic amino group-substituted pyr;-
doneca!rboxylic acid derivatives of the general formula
(I)
=
CA 02212226 1997-08-01
A-Pri (I).
esters thereof ,and salts thereof, wherein:
Pri i~s a pyridonecarboxylic acid residue, and
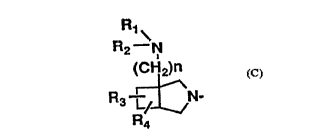
A is a bicyclic amino group represented by the
following formula (C) and joined to the 7-position o~
the pyridonecarboxylic acid or a position equivalent to
the 7--position ithereof.
F~1\
R2--N
(ClJ2)n
R3 L/ N ( C )
IR4
wher~in R1 and R2 may be,the same or different and each
represents a hyd.rogen atom, a lower alkyl group or an
amino-protecting group; R3~and R4 may be the same or
20 different and each represents a hydrogen atom, a halogen
atom, a cyano group, a hydroxyl group, an oxo group, a
lower alkoxy group or a lower alkyl group; and n is an
integer of 0 or 1.
According to the.present invention, there are
25 also provided bicy.clic amine compounds of the following
general formula (Ill) and salts thereof which are useful
as int,ermediates for the synthesis of pyridonecarboxylic
acid derivatives of the general formula (I).
R.~ -
R2 ---N
(I~ t 2)n
R3 L/ - ~NH ( m
R4
CA 022l2226 l997-08-Ol
wherein Rl, R2, R3, R4 and n have the same meanings as
described previously~
The pyridonecarboxylic acid residue repre-
sented herein by "Pri" is a group having a skeletal
struct:ure of the following formula (D) in the mol~cule.
CO~
wherein x, y ancl z may be the same or different and each
repres;ents a carbon atom or a nitrogen atom, and w
represients a carbon atom.
"The 7-position of the pyridonecarboxylic acid
or a p,osition equivaIent to the 7-position thereof"
means the position of w in the above formula (D). For
. example, this me:ans the 7-position in pyridonecarboxyIic
20 acids having the: quinoline or 1,8-naphthyridine struc-
ture, the 2-position in pyridonecarboxylic acids having
the pyrido~2,3-cl]pyrimidine structure, and the 10-posi-
tion in pyridone~carboxylic acids having the ofloxacin
structure.
Accordingly, the present invention preferably
provides bicycIic amino group-substituted pyridonecar-
boxylic acid derivatives of the following general for-
mula (I-A), esters thereof and saIts thereof.
~\ X ~
(C~2)11 D ~ C O O H (I - A)
R ' ~Nl'T N,G
R4 R5
CA 02212226 1997-08-01
-;
wherein R5 represents a lower alkyl group, a lower
alkeny~l group, al lower cycloalkyl group, a phenyl group
or a heterocyclic group ~all of which may further be
substituted); G represents ~-E or a nitrogen atom in
which E represents a hydrogen atom or combines with Rs
to form a bridge~ represented by the formula -S-CH(CH 3) -;
T represents C-Z: or a nitrogen atom in which Z repre-
sents a hydrogen a~om, a halogen atom, a cyano group, a
lower alkoxy group, a halogenated lower alkoxy group, a
lower alkyl growp or a halogenated lower alkyl group, or
combines with R5 to form a bridge represented by the
formula -0-CH2-C'H(CH3)-; X represents a hydrogen atom, a
halogen atom, a hydroxyl group, a lower alkyl group or
an amino group which may be protected; D represents C-Y
15 or a nitrogen atom in which Y represents a hydrogen atom
or a halogen atom; and Rl, R 2~ R3, R 4 and n have the
same ~leanings as; describe~ previously.
The compounds (I) of the present invention are
structurally characterized by the fact that a speeific
20 b;cyclic amino g:roup is chosen as a substituent joined
to the! 7-position of the pyridonecarboxylic acid or a
position equivalent to the 7-position thereof.
The terms as used herein in connection with
substituents andl the like are described below.
Although no particular limitation is placed on
the scope of the term "halogen atom", fluorine, chlorine
and bromine are preferred. The term "lower" means
groups containing 1 to 7 carbon atoms, unless otherwise
specified. The term "lower alkyl" comprehends, for
30 example, straight-chain and branched alkyl groups such
as methyl, ethyl, propyl, isopropyl, butyl, t-butyl and
pentyl, of which methyl is preferred. The term "lower
alkenyl" comprehends, for example, vinyl, allyl, 1-pro-
penyl and isopropenyl, of which vinyl is preferred. The
35 term "lower cycloalkyl" comprehends, for example, cyclo-
propyl, cyclobutyl. cyclopentyl and cyclohexyl, of which
CA 02212226 1997-08-01
cyclopropyl is preferred. The term "lower alkoxy"
comprehends, for exan-ple, methoxy and ethoxy.
In the ~definition of R 5, examples of the
substituent(s) used in the "lower alkyl group which may
further be substituted", the S'lower alkenyl group which
may further be substituted", or the "lower cycloalkyl
group which may further be substituted" include halogen
atoms such as fluorine and chlorine. Examples of the
substituent~s) used in the "phenyl group which may
10 further be substituted" include halogen, lower alkyl,
lower alkoxy, hydroxy, nitro and amino. The term
"heterocyclic group" as used in the definition of R5
comprehends, for example, five-membered and six-membered
heterocyclic groups having N, 0 or S as a heteroatom,
15 such as pyrrole, furan, thiophene. thiazole, isothia-
zole, oxazole, isoxazole, pyrazole, imidazole, pyridine,
pyridazine, pyrimidine an,d pyrazine. The heterocyclic
groups may further be substituted. for example, by
halogen, lower alkyl, IDwer alkoxy, hydroxy, nitro
20 and/or amino.
As the "protecting group" or "amino-protecting
group" used in the "amino group which may be protected",
there may be employed any of various groups which can
readily be eliminated by a common deprotection reaction
25 such as hydrolysis or hydrogenolysis, without exerting
no substantial influence on the other structural part.
Examples of easily hydrolyzable amino-protect-
ing groups which can readily be eliminated by hydrolysis
inclu,de oxycarbonyl groups such as ethoxycarbonyl,
30 t-butoxycarbonyl (abbreviated as Boc), benzyloxycar-
bonyl, p-methoxybenzyloxycarbonyl, vinyloxycarbonyl and
~-(p-toluenesulfonyl)ethoxycarbonyl; acyl groups such as
formyl, acetyl ;and trifluoroacetyl; and o-nitrophenyl-
sulfenyl, trimethylsilyl, tetrahydropyranyl and di-
35 phenylphosphinyl.
Examples of easily hydrogenolyzable amino-
CA 02212226 1997-08-01
protec:ting groups which can readily be eliminated by
hydrogenolysis include arytsulfonyl groups such as
p-toluenesulfon~l; phenyl- or benzyloxy-substitut~d
methyl groups such as benzyl, trityl and benzyloxy-
methyl; arylmethoxycarbonyl groups such as benzyloxy-
carbonyl and o-methoxybenzyloxycarbonyl; and halogeno-
ethoxycarbonyl groups such as ~ -trichloroethoxy-
carbonyl and ~-iodoethoxycarbonyl.
"Esters" should preferably be those which can
1~ be converted in~:o the corresponding free carboxylic
acids (I) of th~: present invention by chemical or
enzymological means. The esters which can be converted
into the corresponding free carboxylic acids by c~emical
means such as hydrolysis include, for example, lower
aikyl esters such as methyl esters and ethyl esters.
The esters which can be converted into the corresponding
free carboxylic acids not,only by chemical means but
also by enzymological means include, for example, lower
alkanoyloxy-lower alkyl esters such as acetoxymethyl
20 esters, 1-aceto,cyethyl esters and pivaloyloxymethyl
esters; lower alkoxycarbonyloxy-lower alkyl esters such
as 1-ethoxycarbonyloxyethyl esters; aminoethyl esters
such as 2-dimethylaminoethyl esters and 2-(1-piperi-
dinyl'~ethyl esters; and other esters such as 3-butyro-
2~ lactonyl esters, choline esters, phthalidyl esters and(5-methyl-2-oxo--1,3-dioxol-4-yl)methyl esters.
As the salts of the compounds (I) of the
present invention, pharmaceutically acceptable salts
thereof are especially preferred. Examples thereof
include salts formed with organic acids such as tri-
fluoroacetic acid, aGetic acid, lactic acid, succinic
acid, methanesulfonic acid, maleic acid, malonic acid,
gluconic acid and amino acids (e.g., aspartic acid and
glutamic acid); salts formed with inorganic acids such
as hydrochloric acid and phosphoric acid; metallic salts
such ;as sodium, potassium, zinc and s'iIver salts; ammo-
CA 02212226 1997-08-01
.
nium salts; and salts formed with organic bases such as
trimethylamine, triethylamine and N-methylmorpholine.
Salts of the compounds (111) of the present
invent:ion which are useful as intermediates include
salts formed wit:h inorganic acids such as hydrochloric
acid alnd s~lfuric acid; and salts formed with organic
acids such as formic acid, acetic acid, trifluoroacetic
acid, methanesulfonic acid and p-toluenesulfonic acid.
The compounds (I) and ~111) of the present
invention may sometimes exist in the form of hydrates
and solvates. Moreover, they may exist in the form of
optical isomers and stereoisomers ~cis- and trans-
forms). These compounds are also within the scope of
the present invention~
1~ Preferred examples of the compounds ~I) of the
present invention are the compounds represented by the
following general formula.(l-B).
X; O
zo H2N V~COOH
25 wherein ~5, G, T, X, Y and n have the same meanings as
described previously.
More preferred examples of the compounds (I)
of the present invention are the compounds represented
by the following general formula (I-C).
X' O
~ ~COOH ( I - C)
R5
CA 02212226 1997-08-01
wherein R5' is a, cyclopropyl group which may be substi-
tuted by f I uorine, a 2,4-di f I uorophenyl group or a
t-butyl group, X' is a hydrogen atom, a halogen atom or
an amino group, and T' is C~, CF, CCI, C-OCH3, C-~C~F2
or a nitrogen atom.
More specific examples thereof are the Gom-
pounds described in the Examples which will be given
later.
The following compounds and physiologically
10 acceptable salts thereof are also preferred examples of
the compounds (I) of the present invention.
7-(1-Amino-3-azabicyclo~3.2.0]hept-3-yl)-8-cyano-1-
cyclopropyl-B-fluoro-1,4-dihydro-4-oxoquinoline-3-car-
boxylic acid.
7-~1-Amino-3-azabicyclo[3.2.0~hept-3-yl)-8-bromo-1-
cyclopropyl-6-fluoro-1,4-dihydro-4-oxoquinoline-3-car-
boxylic acid. t
7-(1-Amino-3-azabicyclo[3.2~0]hept-3-yl)-1-cyclo-
propyl-6-fluoro-1,4-dihydro-8-methyl-4-oxoquinoline-3-
20 carboxylic acid.
7-~1-Amino-3-azabicyclo[3.2.0~hept-3-yl)-1-cyclo-
propyl-6-fluoro-1,4-dihydro-4-oxo-8-trifluoromethyl-
quinoline-~-carboxylic acid.
The compounds (I) of the present invention may
25 be prelpared, for example, by the following (a) amination
reacti,on, (b) hy,drolysis reaction and (c) ring closure
reacti,on.
(a) Amination reaction
The compounds (I) of the present invention,
30 esters thereof and salts thereof may readily be prepared
by reacting a compound of the general formula (Il)
L-Pri ~II)
35 wherein Pri has ithe same meaning as described previ-
ously, L is a leaving group joined to Pri at the 7-posi-
CA 02212226 1997-08-01
11
tion c~f Pri Qr al position equivalent to the 7-position
thereof, and the carboxyl and oxo groups present in the
pyridonecarboxylic acid residue represented by Pri may
form a boron chelate bond therebetween, an ester thereof
or a s,alt thereof with a bicyclic amine compound of the
generall formula (Ill)
R1
F~ N
(C~ 2)n
( III )
R3 L/ NH
R4
15 wherein Rl, R2, R3, R4 and n have the same meanings as
described previously; and if a boron chelate part is
present in the product, hydrolyzing it.
Examples of the leaving group L in the gereral
formula (Il) inc:lude halogein atoms, lower alkoxy groups,
lower alkylthio groups, lower alkylsulfinyl groups,
lower alkylsulfonyl groups, lower alkylsulfonyloxy
groups and arylsulfonyloxy groups. Among others, halo-
gen atoms such clS fluorine and chlorine are preferred.
This reaction may be carried out by stirring a
25 mixture of the compound (Il) and the compound (Ill) in
an inert solvenl: at a temperature of 10 to 130~C and
preferably 20 to 130~~, for a period of time ranging
from 10 minutes to 24 hours and preferably from 30
minute~s to 3 hours. Useful solvents include water,
30 methanol, ethanol, acetonitrile, chloroform, pyridine,
dimethylformamide, dimethyl sulfoxide, 1-methyl-2--
pyrrolidone and the like. These solvents may be used
alone or in admixture.
This reaction is generally carried out in the
35 presence of an acid acceptor by using the compound (Ill)
in an amount equivalent to or slightly in excess of that
CA 02212226 1997-08-01
12
of the! compound (Il). The compound (lli) may be used in
excess so as to function as an acid acceptor too~
Preferred examples of the acid acceptor include organic
bases such as triethylamine, 1,8-diazabicyclo~5~4.0]-7-
undecene (DBU), pyridine, quinoline and picoline; and
inorganic bases such as sodium hydroxide, potassium
hydroxide, sodium carbonate, potassium carbonate, sodium
hydrogen carbonate and potassium hydrogen carbonate.
Compounds (Il) are well known or may be pre-
10 pared according to well-known processes. Bicyclic amine
compounds (Ill) are all novel and the processes for the
preparation thereof will be described later.
(b) Hydrolysis reaction
0~ the compounds (I) of the present inventi on,
15 those in carboxylic acid form may also be prepared by
hydrol~yzing a compound of the general formula (IV)
r
~ X O
R2---N ~ (IV~
R3 L ~ N 1 T N
25 wherein U represents a group which can be converted into
a carboxyl group by hydrolysis, and R1, R 2~ R 3, R 4~ R 5,
n, G, 1, X and D have the same meanings as described
previ OLIS I y.
In this, case, examples of the group U convert-
ible into a carboxyl group include an ester group, acyano group, an amido group, an amidino group, and a
group of the formula -C(=NH)-O-(lower alkyl).
The above hydrolysis reaction may be carried
out by bringing the aforesaid compound (IV) into contact
35 with water in a suitable solvent. In order to acceler-
ate this reaction, it is usuallY carried out in the
CA 02212226 1997-08-01
13
presence of a catalyst such as an acid or a base.
Usable acid catalysts include inorganic acids such as
hydrochloric acid, hydrobromic acid, sulfuric acid and
phosphoric acid; and organic acids such as acetic ac f d,
trifluoroacetic acid, formic acid and p-toluenesulfonic
acid. Usable base catalysts include metal hydroxides
such as sodium hydroxide and barium hydroxide; carbon-
ates such as soclium carbonate and potassium carbonate;
and sodium acetalte.
Usually, water is used as the solvent. How-
ever, according to the properties of the compound ~IV),
a water-miscible organic solvent such as ethanol, ethyl-
ene glycol dimethyl ether, benzene or dioxane may be
used in combination with water. The reaction tempera-
15 ture may usually range from 0 to 150~C and pre~erably
from 30 to 100~C.
This reaction may also be carried out by ,
heating the compound (IV) directly in the presence of an
acid as described above. an~ then adding water thereto.
(c) Ring closure reaction
Furthermore, the compounds (I) of the present
invention may also be prepared by subjecting a compound
of the general formula (V)
R1
~, X O
(Cl- 2)n Ds~COoR
R3 ~ ~Nl T" L' 'NHR5"
wherein L' repre'sents a leaving group, R5" represents a
lower alkyl group, a lower alkenyl group, a lower cyclo-
alkyl group, a phenyl group or a heterocyclic group (all
of whi ch may fur-ther be substituted), G' represents CH
or a n;trogen atom, T" represents C-Z or a nitrogen atom
CA 02212226 1997-08-01
..
14
in which ~ has the same meaning as described previously,
R6 represents a lower alkyl group, an alIyl group or a
benzyl group, amd R1, R2, R3, R4, n, X and D have the
same meanings as described previously, to a ring closure
reaction.
In this case, examples of the leaving group L'
inclulde the same groups as described previously for ~he
leavimg group L Among others, halogen atoms such as
fluorine and chlorine are preferred.
This ring closure reaction may be carried out
by stirring a mixture of the compound (V) and a solvent
at a temperature of 30 to 150~C and preferably 30 to
100~C for a period of time ranging from 1 to 6 hours in
the presence of a base (e.g., potassium carbonate,
sodium carbonate, sodium hydride, potassium t-butoxide
or potassium fluoride) which is used in an amount of 1
to 3 moles per mole of the compound (V). Preferred
examples of -the solvent include ethanol, dioxane,
tetrahydrofuran, dimethylformamide and dimethyl sul-
20 foxide.
The compound ~V)'used as the starting materialis also novel, and this may be prepared, for example,
according to the following reaction formula (1).
CA 02212226 1997-08-01
Reaction formula (1)
X X O
5L~ L LJ~COOR6
Rl~ I IK R, ~I o
R2--N D~.l~ COOR~' R2--N ~,,COOR6
~CI- 2)n ll ~C~ 2~ D
10~ --~~Nl' r~L~ R3--~NlT"~L~
R4 R4
152 N ~ COOR6' R2--N ~ COOR6
R --~N ~' T" ~ L' G~ N ~ R7 R --~N 1' T" ~. G' NHR
4 ' R4
t
20 wherein R6' is a hydrogen atom or has the same meaning
as described previously for R6, R7 and R8 may be the
same or different and each represents a lower alkyl
group, and R1, R2, R3, R4, R5", R6, n, G', T", D, X, L
and L' have the same meanings as described previously.
When the compound (l~ of the present invention
prepare~d according to any of the above-described pro-
cesses (a), (b) and (c) has an amino-protecting group,
it may be subjected to a hydrolysis reaction or a hydro-
genolys;is reaction as desired. Thus, there can be
30 obtaine!d a compound (I) of the present invention in
which t:he amino-protecting group has been converted into
a hydrogen atom.
The realction for eliminating the amino-pro-
tecting group by hydrolysis may be carried out in the
same manner as de~scribed in the above process (b).
Alternatively, the reaction for eliminating
CA 02212226 1997-08-01
16
the amino-protecting group by hydrogenolysis may advan-
tageously carried out by treating a compound (I) of the
present invention having an easily hydrogenolyzable
amino--protectingr group with hydrogen gas in a solvent in
the presence of a catalyst. Thus, there can be obtained
a compound of the present invention in which the amino-
protec:ting group has been converted into a hydrogen
atom. The catalysts which can be used in this re~ction
inclucle, for example, platinum, palladium and Raney
10 nickél catalyst. Usable solvents include, for example,
ethylene glycol, dioxane, dimethyl~ormamide, ethanol,
acetic acid and water~ This reaction may be carried out
at a t:emperature: of 60~C or below and is usually carried
out at: room temperature.
When the easily hydrogenolyzabl e amino-pro-
tecting group is; benzyl, trityl, benzyloxycarbonyl or
p-toluenesulfonyl and the,like, the protecting group may
be eliminated by metallic sodium treatment in liquid
ammonia at a temperature of;-50 to -20~C.
The ccmpound (Ill) used as the starting mate-
rial in the above-described process (a) may be prepared
by tre!ating a ccmpound of the general formula (Vl)
R
R2 - N
(C~ 2)n
~N R (VI)
L/ 11
F~10
wherein R9 and R1o have the same meanings as described
previously for R3 and R4, or represent groups convert-
ible into R3 and R4, Rl1 represents an amino-protecting
group, and R1, R2 and n have the same meanings as de-
scribed previously, so as to eliminate the amino-pro-
tecting group R1, and convert it into a hydrogen atom;
CA 02212226 1997-08-01
17
and w~en Rg and,~or R10 are groups convertible into R3
and/or R4, convlsrting Rg and/or Rlo into R3 and/or R4~
In this case, examples of the amino-protecting
group Rt1 inclu~e the above-described easily hydrogeno-
Iyzable amino-protecting groups and easily hydrolyzable
amino--protecting groups. When Rl or R 2 i n the compound
(Vl) ;s an amino-protecting group, it is desirable for
subsequent reactions to employ, for R1l, an amino-pro-
tecting group differing in character from the amino-
10 protecting group represented by R1 or R 2 For exampie,when l:he amino-protecting group represented by Rl or ~2
is an easily hydrolyzable amino-protecting group such as
t-butoxycarbonyl, an easily hydrogenolyzable amino-
protecting group such as benzyl or trityl is pre~erably
15 chosen for Rll
This elimination reaction may be carried out
by subjecting the compound (Vl) to a hydrogenolysis or
hydrolysis react:ion which has previously been explained.
With regard to Rg and R1o, examples of the
"group,s convertible into R3 and R 4 i nclude methane-
sulfonyloxy, p-toluenesulfonyloxy, benzyloxy, carboxyl,
carbamloyl, hydroxyiminomethyl, benzylidene, cyclic
acetal and dithioacetal.
Metha~esulfonyloxy and p-toluenesulfonyloxy
25 may be converted, by a nucleophilic substitution reac-
tion, into a halogen atom, a cyano group or a lower
alkoxy group as used for R3 or R4.
Benzyloxy may be converted into a hydroxyl
group as used for R3 or R4, by a hydrogenolysis reaction
30 or a hydrolysis reaction. Carboxyl may be converted
into a halogen atom as used for R3 or R4, by deriving an
acid halide therefrom and then treating it with the
Wilkinson catalyst {RhCl[P(C6H 5) 3]3}.
Furthermore, carbamoyl and hydroxyiminomethyl
35 may be converted into a cyano group as used for R3 or
R4, by treatment with thionyl chloride or chlorosulfonyl
CA 02212226 1997-08-01
18
isocyanate. Benzylidene, cyclic acetal and d;thioacetal
may be convert~d into an oxo group as used for R 3 or R 4,
by an oxidation reaction, a hydrolysis reaction using an
acid catalyst cand a hydrolysi 5 reaction in the presence
of me~rcuric chloride, respectively.
Compc~unds (Vl) are also novel and may be
prepalred, for e:xample, according to the processes de-
scribed in Examples I to lll which will be given later
or processes equivalent thereto.
The compounds (I) of the present invention and
the intermediate compounds ~III) thereof, which have
been prepared in the above-described manner, may be
isolalted an~ pulrified according to any conventional
proce~dure. The!se compounds are obtained in the form of
salts;, free acids or amines, or hydrates, depending on
the c:onditions of isolation and purification. According
to the intendeal purposes,r these forms may be converted
into each other to obtain the compounds of the present
invemtion in desired forms.
The stereoisomers of the compounds (I) and
(Ill) of the present invention may be separated from
each other by any conventional method such as fractional
crystallization or chromatography. Their optical iso-
mers may be i so lated by an optical resolution method
25 whichl is known per se.
The compounds (I) of the present invention and
salts thereof, which have been obtained in the above-
described manner, are all novel compounds and are valu-
able as antibacterial agents because of their high
30 antibacterial activities. The compounds (I) of the
present invention and salts thereof can be used not only
as drugs for human beings and animals, but also as fish
drugs, agricultural chemicals and food preservatives.
Esters of the compounds (I) of the present
invention are valuable as raw materials for the synthe-
sis of the compounds (I) of the present invention in
CA 02212226 1997-08- 01
19
carboxylic acid form. However, if these esters them-
selves may readily be converted into the correspondin~
carboxylic acids in the body, they exhibit the same
action and effect as the carboxylic acids and are hence
useful as antibacterial agents.
Moreover, the compounds (Ill) of the present
invention are useful as direct intermediates for the
synthesis of the compounds (I) of the present invention~
10 ~est l~ode for C,arrYin~ Out t~e Invention
Now, the in vitro and in vivo antibacterial
activities of the compounds (I) of the present invention
are described with reference to the following experimen-
tal data. The results are summarized in Table 1. The
15 figures given in th;s table indicate minimum inhibitory
concentrations (MIC; ~g/ml) as measured according to the
procel~ure described in ~hemotherapy, 2g(1), 76 (19&1),
and tl~e figures given in brackets indicate effects
(ED50; mg/kg~ on systemic infection in mice.
The eFfects (ED 5 o; mg~kg) on systemic infec-
tion in mice were determined as follows: Male Std-ddy
strain mice (weighing about 20 g) were infected with
each of the pathogenic bacteria shown in Table 1 by
administering 5 x 10 3 Vi able microorganisms intraper~-
25 toneally to each mouse. Then, a suspension of each test
compo~nd in o, 4a~ carboxymethylcellulose was orally
adminiistered twiice, i.e., immediately after infection
and 6 hours after infection. Seven days after infec-
tion, the ED 5 0 value was calculated from the survival
rate of each mouse group by probit analysis.
As re1:erence compounds, there were used pipe-
midic acid that is an excellent antibacterial agent
currently on the market, and 7-(1-amino-3-azabicyclo-
[3.1.0]hex-3-yl)-1-cyclopropyl-6,8-difluoro-1,4-dihydro-
35 4-oxoquinoline-3-carboxylic acid that has the following
structure and is disclosed in Example 6 of the aforemen-
CA 02212226 1997-08-01
tioned Japanese Patent Application Laid-Open No.
~673/'89.
H2N ~_COOH
CA 02212226 1997-08-01
21
Tab I e 1 Ant i bacter i a I act i v i t i es
X O Bacterial s train
F ~ StaphY- Pseudo-
T ~ lococcus monas
Rs
aureus aeru
nosa
R s X T Salt 50774 No. 12
1 H C F - 0.025 0.39
~ [0.928] [~.oo
2 H C-Cl CF3C02H 0.013 0.39
~ [0.682] [~.28
3 H C-O~le CF3C02H 0.013 0.78
A [1.41 ] [8.62
4 H C H CF3C02H 0.1 0.78
~ [~.42 ] [6.92
NH2 C F CF3C02H 0.025 0.39
~ [1.16 ] [5.08
6 F C F - 0.05 1.56
~ [~.81 ]
7 Ph-F2 H C H - 0.05 1.56
[2.25 ]
8 Ph-F2 H N CF3C02H 0.05 0.39
[0.799] [3.25
g H N - 0.05 0.78
~ [1.18 ] [~.88
12 ~ H C-O~le H C 1 0.013 0.78
F [2.32 ]
Compound of Exa~r~le 6 of 0. 025 0.1
Japanese Patent Application[ 6.82 ] [12 5
Re:ference ];aid OPe~ No. 56673/'89
compound
Pipemidic acid [215 ] [70 8
Ph-F2: 2,~-di~luorophenyl; Me: methyl
CA 02212226 1997-08-01
22
As shown in Table 1 above, the compounds (1)-
of the present invention exhibit an excellent antibacte-
rial activity not only in vitro but also in animal
experiments. E.specially in the in vivo experimer-ts, the
compounds of the present invention exhibit a more excel-
lent antibacterial activity than the reference com-
poundls.
The compounds (I) of the present invention
have low toxicity and can hence be used as antibacterial
10 ~gents for the prophylaxis and treatment of bacterial
diseases in mam,mals including man.
When the compounds (I) of the present inven-
tion àre used as antibacterial agents in human beings,
~heir dosage may vary according to the age and body
1~ weight of the patient, the severity o~ symptoms, the
route of administration, and the like. However, it is
recommended to administer them in a daily dose of 5 mg
to 5 g which may be given once or in several divided
' doses. The route of administration may be either oral
20 or parenteral.
The compounds (I) of the present invention,
may be administered alone to human beings and other
mammals. However, they are usually combined with one or
more Ipharmaceutically acceptable additives and adminis-
25 tered in the form of pharmaceutical preparations. S~lchpharmaceutical preparations include tablets, solutions,
capsules, granules, powders, syrups, injections, oint-
ments artd the like. These pharmaceutical preparations
may be made in the usual manner by using common addi-
30 tives. For example, as additives for oral preparations,there may be used various carriers or diluents which are
commonly used irt the field of pharmaceutics and do not
react with the compounds (I) of the present invention,
such as starch, mannitol, crystalline cellulose, car-
35 boxymethylcellulose calcium, water and ethanol. More-
over, as additives for injections, there may be used
CA 02212226 1997-08-01
23
various additives which are commonly used in the field
of injections, such as water, physiological saline,
glucose solutions and trans~usions.
The aforesaid solutions and ointments may also
be used for purposes of therapy and treatment in the
fields of otorhinolaryngology and ophthalmology.
The present invention is further illustrated
by thle following examples. Examples I to lll relate to
the preparation of intermediates ~III), Examples 1 to 1
relate to the p;reparation of desired compounds (I~, and
Example A relates to a pharmaceutical preparation.
ExamPle I
1-~t-Butoxvc;3rbonvlamino)-3-azabicYclo r3. 2.01hePtane
(A~ :35.1 g of 1-cyclobutene-1-carboxylic acid
1~ (J. ~hem. Soc., p. 3002, 1953) was dissolved in 250 ml
of melhylene chloride, and a solution of diphenyldiazo-
methar~e in methylene chlorride was added dropwise thereto
at room temperature until a red color did not disappear
any longer. Afl:er this mixture was stirred at room
20 temperature for one hour, the dichloromethane was dis-
tillecl off. To the resulting crude product were added
750 ml of tetrahydrofuran, 93.4 g of N-benzyl-N-
(methoxymethyl)t:rimethylsilylmethylamine (Chem. Pharm.
B~ll., Vol. 33, p. 2762, 1985), 10.9 g of cesium fluo-
2~ ride and 15.9 g of trimethylsilyl triflate, followed byheating at 60~C for 18 hours. The reaction mixture was
cooled to ~~C, and 350 ml of a 15% aqueous solution of
sodium hydroxide was added dropwise thereto, followed by
stirring at that temperature for 30 minutes. After the
30 organic layer was separated and dried (over anhydrous
magnesium sulfate), the solvent was distilled off under
reduced pressure. The resulting residue was purified by
silica gel column chromatography (using a 30:1 mixture
o~ n-hexane and ethyl acetate as the eluent) and
3~ recrystallization (from diisopropyl ether) to obtain
52.1 g of 3-benzyl-1-diphenylmethoxycarbonyl-3-
CA 02212226 1997-08-01
24
azabicyclo[3.2.0]heptane.
Melting point: 96-99~C
1H-NMIR (CDCI 3), (j: 1. 69-2.32 (m, 4H), 2.40-3.50 (m,
5H), 3.71 (s, 2H), 6.87 (s, 1H), 7.17-7.44 (m, 15H~
IR ~KI3r), cm~~: 1720
MS (m,~z): 398 (MH )
(B) 45.8 g of the compound obtained in the
preceding step (A) and 92 ml of a 20% aqueous solution
o~ sodium hydro,cide were added to 700 ml of methanol,
10 fol lowed by hea~ting under reflux for 5 hours. After
cooling, this miixture was neutralized by the addition o~
20% hydrochloric: acid, and concentrated under reduced
pressure. After the addition of water and diisopropyl
ether, the resulting mixture was vigorously stirred and
1~ the aqueous phase was separated. After this aqueous
layer was continuously extracted with chloroform, the
extract was dried (over a~7hydrous magnesium sulfate) and
the chloroform was distilled off. The resulting crude
product was dissolved in 450 ml of t-butanol, and 88.2 g
20 o~ diphenylphosphoryl azide (DPPA) and 32.4 g of tri-
ethylamine were added thereto. followed by heating under
reflux ~or 15 hours. The reaction mixture was concen-
trated under reduced pressure, mixed with ethyl acetate,
and washed twice with a 10% aqueous solution of sodium
25 hydroxide. The organic layer was dried (over anhydrous
magnesium sulfate) and then concentrated under reduced
pressure. The resulting residue was purified by silica
gel column chromiatography (using a 9:1 mixture of
n-hexane and ethyl acetate as the eluent) and recrystal-
lization (from n-hexane) to obtain 17.6 g of 3-benzyl-1-
(t-butoxycarbonylamino)-3-azabicyclo[3.2.0~heptane.
Melting point: Ij9-70~C
1H-NMR (~DCI 3), ~j: 1. 42 (s, 9H), 1.50-1.77 (m, lH),
2.03-2~45 (m, 5H~, 2~60-2~80 (m, 1H), 2~73 (d, 1H, J=
3~ 9Hz), 3.01 (d. 1H, J=9Hz), 3.68 (s, 2H), 4.73 (br s,
1H), 7~19-7~41 (m, 5H)
CA 02212226 1997-08-01
..
~ 25
IR (K:Br), cm 1 3380, 1~85
MS (m/z): 303 (MH )
(C) 5 g o-F the compound obtained in the
preceding step (B) was dissolved in 100 ml of ethanol,
and 1 g of 10% palladium-carbon was added thereto.
Then, a stoichiometric amount of hydrogen was added
there!to. After the catalyst was separated by fiItra-
tion, the solve~nt was distilled off. The resulting
crude! crystals were recrystallized from n-hexane diiso-
10 propyl ether to obtain 3 g of 1-(t-butoxycarbonylamino)-
3-azabicyclo~3.2.0]heptane.
Melting point~ 116~C
1H-NMIR (CDGI 3), ~: 1. 25-1.41 (m, 1H), 1.45 (s, 9H),
1.96-2.3~ (m, 4H~, 2.65-2.87 (m, 3H), 2.95-3.15 (m, 2H),
1~ 4.82 (br, 1H)
IR (KBr), cm-1: 3294, 3185, 2982, 1692
MS (m/z): 213 (MH+)
~xample ll
1-Methvlamino-3-azabicYclor3.2.01he~tane
(A) 6.8 g of 3-benzyl-1-(t-butoxycarbonyl-
amino)-3-azabicyclo[3.2.0]heptane was dissolved in 20 ml
of methylene chloride, and ~0 ml of trifluoroacetic acid
was added thereto. followed by stirring for 3 hours.
The reaction mixture was concentrated, mixed with an
Z5 aqueous solution of sodium hydroxide under cooling with
ice, and extracted with chloroform. After the extract
was dried over anhydrous magnesium sulfate. the chloro-
form was distilled off. The resulting crude product was
dissolved in 47 ml of formic acid, and 17 g of acetic
30 anhydride was added dropwise thereto, under cooling with
ice, over a period of 90 minutes. After the addition of
ice water, the resulting mixture was neutralized by the
addition of an aqueous solution of sodium hydroxide
under cooling with ice, and extracted with ethyl ace-
3~ tate. The organic layer was dried over anhydrous magne-
sium sulfate and then concentrated under reduced pres-
CA 022l2226 l997-08-Ol
26
sure. The resulting residue was purified by silica gel
column chromatography (using a 30:1 mixture of chloro-
form and methanol as the eluent) to obtain 3.1 g of 3-
benzyl-1-formylamino 3-azabicyclo[3.2.0]heptane.
1H-NMIR (CDCI 3), ~: 1. 61-1.82 (m, 1H), 2.08-2.50 ~m,
5H), .2.61-3.14 ~m, 3H), 3.69 (s, 2H), 5.90 (br, lH),
7.18-7.42 (m, 5iH), 8.~5 ~d, 1H, J=2Hz)
IR ~neat), cm 1 3270, 3028, 2940, 2791, 1659, 1530
MS (m,/z): 231 ~MH )
(B) :3.1 g o~ the compound obtained in the
preceding step (A) was dissolved in 30 ml of toluene,
and 20 ml of a 10% solution of sodium bis(2-methoxy-
ethoxy)aluminum hydride in toluene was added thereto,
followêd by heal:ing under reflux for 2 hours. After
15 cooling, the reaction mixture was slowly added to 20%
sulfuric acid under cooling with ice, and the insoluble
matter was separated by f,iItration. The fiItrate was
adjust;ed to pH 11 by the addition of a 20% aqueous
' solution of sodium hydroxide, and then extracted with
20 chloroform. After the extract was dried over anhydrous
magnesium sulfate, the solvent was distilled off under
reduced pressure. The resulting residue was purified by
silica gel column chromatography (using a 100:1 mixture
of n-hexane and ethyl aceta~e as the eluent) to obtain
1.97 g of 3-benzyl-1-methylamino-3-azabicyclo[3.2.0~hep-
tane.
1H-NM~ (CDCI 3), ~: 1. 40-1.72 (m, 2H), 1.89-2.55 (m,
6H~, 2.33 (s, 3H), 2.79 (d, 1H, J=20Hz), 2.83 (d, 1H,
J=20Hz), 3.66 (s, 2H), 7.18-7.43 (m, 5H)
IR (neiat), cm I 3270, 2937, 2788
MS ~m/z): 217 (MH'), 187
(C) 1.95 g of the compound obtained in the
preceding step ~13) was dissolved in 30 ml of ethanol,
and 0.4 g of 10% palladium-carbon and 2 ml of concen-
3~ trated hydrochloric acid were added thereto. Then, astoichiiometric arnount o~ hydrogen was added thereto at
CA 02212226 1997-08-01
50~C. The catalyst was separated by fiItration and
washed with methanol. After the solvent was distilled
off, the resulting residue was adjusted to pH 11 by the
additlon of an aqueous solution of sodium hydroxide, and
then extracted with chloroform. After the extract was
dried over anhydrous magnesium sulfate, the solvent was
distilled of~ under reduced pressure to obtain 0.96 g of
1-methylamino-3--azabicyclo~3~2.0~heptane.
'H-NMR (CDCI 3), ~: 1. 21-1.45 (m, 1H), 1.77-3.00 ~m,
10 10H), 2.38 (s, 3H)
IR ~neat), cm 1 3270, 2942. 1692
Exam~le 111
1-~t-Butoxycarbonvlaminomethvl)-3-azabicvclor3.2.0l-
he~tane
(A) 13 g of 3-benzyl-1-diphenylmethoxycarbon-
y3-3-azabicyclo[3.2.0]heptane was dissolved in 100 ml of
tetrahydro~uran, and 2.8 g of lithium chloride was added
thereto. After 2.5 g of sodium borohydride was added
' little by little, 30 ml of methanol was slowly added
zO thereto and the resulting mixture was stirred at room
temperature for 15 hours. The reaction mixture was
concentrated under reduced pressure, mixed with ice
water, and extracted with ethyl acetate. After the
extract was mixed with 10% hydrochloric acid and stirred
25 vigorously, the aqueous layer was separated. This
aqueouC; layer was adjusted to pH 8 by the addition of a
10% aqueous solu1:ion of sodium hydroxide, and then
extracted with el:hyl acetate. After the extract was
dried over anhydrous magnesium sulfate, the solvent was
30 distilled off uncler reduced pressure. The resulting
residue! was puri~ied by silica gel column chromatography
(using a 4:1 mixture o-F n-hexane and acetone as the
eluent) to obtain 5.4 g o~ 3-benzyi-1-hydroxymethyl-3-
azabicyclo~3.2.0]heptane.
1H-NMR (CDCI 3), ~: 1. 64-2.19 (m. 6H), 2.28-2.80 (m,
4H), 3.52-3.77 (m, 4H), 7.18-7.43 (m, 5H)
CA 022l2226 l997-08-Ol
28
IR ~neat), cm 1: 3346, 2934, 2785
MS (m/z): 218 ~MH )
(B) 5.3 g of the compound obtained in the
preceding step (A) was dissolved in 200 ml of tetra-
hydrofuran, and 7.1 g of triphenyIphosphine, 5.~ g ofdiethyl azodicarboxylate and 7.4 g of diphenylphosphoryl
azide were successively added thereto, followed by
stirring at room temperature for 48 hours. After the
reaction mixture was concentrated under reduced pres-
10 sure, ethyl acetate and 10% hydrochloric acid were addedthereto. The resulting mixture was vigorously stirred
and the aqueous phase was separated~ This aqueous layer
was adjusted to pH 1~ by the addition of a 20% aqueous
solution of sodium hydroxide, and then extracted with
15 chloroform. After the extract was dried over anhydrous
magnesium sulfate, the solvent was distilled off under
reduced pressure. The resulting residue was purified by
silica gel column chromatography (using a 30:1 mixture
of n-hexane and ethyl acetate as the eluent) to obtain
20 5.4 g of 1-azidomethyl-3-benzyl-3-azabicyclo~3.2~0]-
heptane.
1H-NMR (CDCI 3), ~: 1. 65-2.26 (m, 6H), 2.39-2~52 (m,
1H), 2.80 (d, 1H, J=9Hz), 2.81 (d, lH, J=9Hz), 3.37 (d,
1H, J=20Hz), 3.43 (d, 1H, J=20Hz), 3.64 (d, 1H, J=
16.5Hz), 3.71 (d, 1H, J=16.5Hz), 7.18-7.44 (m, 5H)
IR (neat~, cm 1 2937. 2787, 2Q96
MS (m~z~: 243 ~MH )
(C) 3.4 g of the compound obtained in the
preceding step (B) was dissolved in 70 rnl of tetrahydro-
30 furan, and 5.9 g of triphenylphosphine was added there-
to, followed by stirring at 50~C for 2 hours. Then, 55
ml of a 28% aqueous solution of ammonia was added there-
to, f,ollowed by stirring at 50~C for 3 hours. The
organic layer was separated and the aqueous layer was
35 extracted with ~diethyl ether. After 1N hydrochloric
acid was added to the combined organic layers, the
CA 02212226 1997-08-01
29
resulting mixture was vigorously stirred and the aqueous
phase was separated~ This aqueous layer was adjusted to
pH 11 by the addition of a 10% aqueous solution of
sodiulm hydroxide, and then extracted with methylene
chloride. After the extract was dried over anhydrous
magnesium sulfate, the solvent was distilled off under
reducled pressurle. The resulting residue was dissolved
in 100 ml of tetrahydrofuran~ Then, 7.7 g of di-t-butyl
dicarlbonate was added thereto, followed by stirring at
room temperature for 15 hours. After the reaction
mixture was conoentrated under reduced pressure, the
resulting residue was purified by silica gel column
chromatography (using a 9:1 mixture of n-hexane and
ethyl acetate as the eluent~ and recrystallization (from
n-hexane) to ob-tain 3.92 g of 3-benzyl-1-(t-butoxycar-
bonylaminomethyl)-3-azabicyclo~3.2.0~heptane~
Melting point: 78-79~C
7H-NMR ~CDCI 3), ~: 1. 44 (s, 9H), 1~64-2.47 (m, 7H~,
2.72 (d, 1H, J=t3Hz), 2.74 (d, 1H, J=9Hz), 3.20 (dd, 1H,
20 J=23Hz, 6Hz), 3.28 (dd, 1H, J=23Hz, 6Hz)
IR ~KE3r), cm~': 3372, 2972, 2932, 2797, 1688, 1530
MS (m,'z): 317 (Mh ), 259
(D) 2.9 g of the compound obtained in the
preceding step l~C) was dissolved in 60 ml of ethanol,
25 and 0.6 g of 1O9D palladium-carbon was added thereto.
Then, a stoichiometric amount of hydrogen was added
thereto at 50~C. After the catalyst was separated by
filtration, the solvent was distilled off. The result-
ing crude crystals were recrystallized from n-hexane-
30 diisopropyl ether to obtain 1.58 g of 1-(t-butoxycar-
bonylaminomethyl)-3-azabicyclo[3.2.0~heptane.
Melting point: 83-86~C
IR (KEir), cm 7: 3311, 3192, 2960, 1720, 1556
CA 02212226 1997-08-01
Examplle 1
7-(1-Amino-3'-azabicvclor3.2~0]he~t-3-vl)-1-cvclo~ro-
Pvl-6.8-difluoro-1,4-dihvdro-4-oxoauinoline-3 car-
ba,xvlic acicl
~ A) 1.9 g of t-(t-butoxycarbonylamino)-3-
azabicycl'o~3.2.0]heptane and 0.83 g of 1-cyclopropyl-
6,7,8-trifluorc,-1,4-dihydro-4-oxoquinoline-3-carboxylic
acid were addecl to 1~ ml of pyridine, followed by heat-
ing under reflulx for 4 hours. After the solvent was
10 distilled off ulnder reduced pressure, the resulting
residue was purified by silica gel column chromatography
(using a 100:1 mixture of chloroform and methanol as the
eluent) and recrystallization (from ethyl acetate-diiso-
propyl ether) to obtain 0.99 g of 7-[1-(t-butoxycarbon-
1~ ylamino)-8-azabicyclo[3.2.0]hept-3-yl]-1-cyclopropyl-
6,8-difluoro-1,4-dihydro-4-oxoquinoline-3-carboxylic
acid (m.p. 211-215~C~. ,
(B) 0.97 g of the compound obtained in the
preceding step ~A) was dissolved in 20 ml of a 35%
20 solution of hydrogen chloride in ethanol, and this
solution was heated at 80~C for 10 minutes. After the
reaction solution was concentrated under reduced pres-
sure, acetonitrile and diethyl ether were added thereto,
a'n'd the formed crystals were collected by fiItration.
25 These crystals were purified by CHP-20P column chroma-
tography (using a 7:3 mixture of water and acetonitrile
as the eluent) to obtain 244 mg of the desired product
[m.p. 242-247~C (dec.)].
ExamDle 2
7-(1-Amino-3-azabicvclor3.2.01hePt-3-vl~-8-chloro-1-
cvcloProPvl-6-fluoro-1.4-dihvdro-4-oxoquinoline-3-
carboxvlic acid trifluoroacetate
(A) 1.22 g of 1-(t-butoxycarbonylamino)-3-
azabicyclo[3.2.0]heptane, 0.96 g of 8-chloro-1-cyclo-
35 propyl-6,7-difluoro-1,4-dihydro-4-oxoquinoline-3-car-
boxylic acid and 0.58 g of 1,8-diazabicyclo[5.4~0]-7-
CA 02212226 1997-08-01
31
undecene were added to 20 ml of acetonitrile, followed
by heating under reflux for 3.5 hours~ After the sol-
vent was distilled off under reduced pressure, the
resulting residue was purified by silica gel column
chromatography ~using a 100:1 mixture of chloroform and
methanol as the elue~t) and recrystallization (from
ethyl acetate-diisopropyl ether) to obtain 0.96 g of 7-
rl - (t-butoxycarbonylamino)-3-azabicyclo~3.2~0]hept-3-
yl]-8-chloro-1-cyclopropyl-6-fluoro-1,4-dihydro-4-
10 oxoquinoline-3-carboxylic acid (m.p 200-202~C).
(B) ID.94 g of the compound obtained in the
preceding step (A~ was dissolved in 10 ml of methylene
chloride, and 2l0 ml of trifluoroacetic acid was added
thereto, followed by stirring at room temperature for 15
15 hours. After the reaction mixture was concentrated
under reduced pressure, acetonitrile and diethyl ether
were added ther~eto and the formed crystals were col-
lected by fiItration. These crystals were washed with
acetonitrile to obtain 650 mg of the desired product
[m.p. 236-240~C (dec.)~.
Exam~le 3
7-(1-Amino-3-azabicvclo[3.2.01hept-3-vl)-1-cYcloPro-
Pvl-6-fluorQ-1.4-dihvdro-8-methoxv-4-oxoauinoline-3-
carboxvlic acid trifluoroacetate
~A) 1.07 g of 1-~t-butoxycarbonylamino)-3-
azabhcyclo[3.2.0]heptane, 1.15 g of 1-cyclopropyl-6,7-
difluoro-1,4-dilhydro-8-methoxy-4-oxoquinoline-3-car-
boxylic acid-BF 2 chelate and O.B8 g of triethylamine
were ;added to 1,B ml of dimethyl sulfoxide, followed by
30 stirring at room temperature for 16 hours. After the
addition of wat~er, the resulting mixture was extracted
with chloroform and the solvent was distilled off. To
the r,esulting r,es;due were added 300 ml of 80% ethanol-
water and 50 ml of triethylamine, followed by heating
under reflux for 3 hours. After the reaction mixture
was concentrate,d under reduced pressure, the residue was
CA 02212226 1997-08-01
32
mixed with water and extracted with chloroform. AFter
the extract was dried over anhydrous magnesium sulfate,
the solvent was distilled off. The resulting residue
was purified by silica gel column chromatography (using
a 10~:1 mixture of chloroForm and methanol as the
eluent~ and recrystallization (from ethyl acetate-diiso-
propyl ether) to obtain 1.17 g of 7-[1-~t-butoxycarbon-
ylamino)-3-azabicyclo[3.2.0]hept-3-yl]-1-cyclopropyl-6-
Fluoro-1,4-dihydro-8-methoxy-4-oxoquinoline-3-carbox-
10 ylic acid (m.p. 211-213~C).
(B) The compound obtained in the preceding
step (A) was treated in the same manner as described in
step (B) oF Example 2 to obtain the desired product
[m.p. 230-23~~C (dec.)].
15 ExamPles 4-11
The compounds shown in Table 2 below were
obtained by carrying out .reaction and treatment in the
same manner as described in Example 2.
CA 02212226 1997-08-01
t
.
33
Table 2
Exam- R~--N ~ COOtl po nt
R , R z n R s X T Salt
A Boc H 1 234-236
4 0 ~ H CH
B H H CF3C02H255-260
(dec.)
A Boc H 229-231
0 ~ NH2 CF
B H H CF3C02H250-257
(dec.)
A Boc H ~ 230-235
6 0 ~ F CF
B H H 272-275
(dec.)
A Boc H 147-150
7 0 Ph-F2 H CH
B H H 216-218
A Boc H 131-13
8 0 Ph-F2 H N
B H H CF3C02H219-222
A Boc H 247-250
9 0 I N
B H H ~ H CF3C02H261-267
- (dec.)
A H H 0 t-Bu H N 245-253
(dec.)
A Boc H 210-215
11 0 1 NH2 CH (dec.)
B H H CF3C02H231-235
(dec.)
Boc: t-butoxycarbonYI; Ph-F2: 2,4-difluorophenyl
Me: methyl; t-Bu: t-butyl
CA 02212226 1997-08-01
34
Examcles 12-16
The compounds shown in Table 3 below were
obtained by carrying out reaction and treatment in the
same manner as described in Example 3.
Table 3
Rl
Exam- R2 \N X O Melting
ple (C~2)n ~ COOH po nt
Rs
R I R 2 n X R s T Salt
A Boc H
Non-crys-
12 0 H ~ ~C-OMe talline
B H H F HCl 218-223
(dec.)
13 A Me H O H ~ C-OAle - 205-207
A Boc H - 188-190
1~ 1 k ~ C-OMe
E, H H HCl 162-166
A Boc H . C 205-210
0 H
B, H H O ~ HCl 273-283
Me (S)(dec.)
A Boc H - 1~6-152
16 0 H ~ C-OCHF2
B H H - 237-2~2
(dec.)
Boc: t-butoxYcarbonyl; Me: methyl
CA 02212226 1997-08-01
Exam~le A (Formation of tablets)
Compound of Example 1 or 2 250 g
Corn sitarch 54 g
Carboxymethylcellulose calcium 40 g
Microc:rystalline cellulose 50 g
Magnes;ium stearate6 g
The above ingredients were blended together
with ethanol~ lhe resulting blend was granulated and
tablet:ed in the usual manner. Thus, there were obtained
1,000 tablets each weighing 400 mg~
Ex~loitabilitv in Industrv
As des,cribed above, the compounds (I) of the
present invention are useful as drugs (antibacterial
agents) for mammals including man. Moreover, the com-
pounds (111) of the present invention are useful as
direct: intermediates for ~the synthesis of the compounds
(I) .