Note: Descriptions are shown in the official language in which they were submitted.
CA 02214837 1997-09-08
WO 96/27677 PCT/US96/03369
Method of Purification of Recombinant Viral Vectors
Containing a Therapeutic Gene
Background of the Invention
The treatment of disease by gene therapy has moved from the
theoretical to the practical realm. The first human gene therapy trial was
begun in September 1990 and involved transfer of adenosine deaminasc
(ADA) gene into lymphocytes of patients with defects of this enzyme. Lack of
ADA activity results in immune deficiency. Of several methods for delivering
therapeutic genes to diseased cells, viral vectors hold out particular
promise.
to Tumor suppressor genes are being investigated for treating cancerous cells.
Viral vectors containing such tumor suppressor genes are being evaluated as
potential therapeutics in the field of cancer therapy. Recently, adenovirus
vectors have received particular attention as an advantageous vector for the
delivery of such tumor suppressor genes and other biological response
modifiers. As studies of cancer gene therapy progress to clinical trials,
larger
and larger quantities of purified viral vectors are needed. One problem in
producing suitable quantities of vectors for such trials is the purifcation of
the
particles from the cell lysates in which the viral particles are produced.
Specifically, purification of these vectors has historically been performed
2 o by using density-based ultracentrifugation methods. While this method has
proven effective for use as a research tool, it is not feasible as a method
for
industrial scale production. Meeting the demands for material in the future
would lead to prohibitive costs unless a new purification scheme could be
developed. One alternative to ultracentrifugation is chromatographic
techniques for purification of infectious viral particles.
The use of size exclusion chromatography for purification of various plant
viruses has been demonstrated either as a stand alone technique or to
augment density gradient centrifugation (Albrechtsen et al., J. ViroIQgical
CA 02214837 1997-09-08
WO 96/27677 PCTIUS96I03369
2
Methods 28:245-256 (1990); ahd Hewish et al., ,~. Virological Methods
7:223-228 (1983)). Size exclusion appears promising for bovine papilloma
virus (Hjorth and Mereno-Lopez, J. Virological Methods 5:151-158 (1982));
A
and has been shown to be a superior method for the purification of tick-borne
s encephalitis virus (Crooks stet al., J. Chrom. 502:59-68 (1990)). The use of
size exclusion chromatography has not yet become widespread, but is
currently being employed for large scale production of recombinant
retroviruses (Mento, S. J., Viagene, Inc. as reported at the 1994 Williamsburg
Bioprocessing Conference). Affinity chromatography, mostly using
to monoclonal antibodies (Mab), has been reported to be an effective method
for the purification of antigens of viral origin (Najayou stet al., J.
Virological
Methods, 32:67-77. 1991 ). Soybean mosaic virus (a virus which can survive
pH 3) can be recovered using Mab affinity chromatography (Diaco stet al., ~
den. Virol. 67:345-351. 1986). Fowler (J. Virological Methods. 11:59-74.
15 (1985)) used affinity chromatography and density gradient centrifugation to
purify Epstein Barr virus.
Adenoviruses are large (diameter of approximately 80 nm) and somewhat
fragile. A large literature base dealing with the relationship of structure to
function has accumulated (for reviews see Philipson, Viroloov 15:263-268
2 0 (1961 ) and Horwitz, Viroloav (Second Edition) Raven Press Ltd, New York
(1990)). Little has been reported in the literature about chromatographic
purification of live adenoviruses. Haruna a al. (Viroloav 13:264-267 (1961 ))
reported using DEAE ion exchange chromatography for purification of types
1, 3 and 8 adenoviruses while Klemperer and Pereir (Virolog,y 9:536-545
25 1959)) and Philipson (Viroloav 10:459-465 (1960)) reported difficulties
using
the same method with other types of adenoviruses. Poor resolution and poor ~
yield are important problems with this methodology that has prevented its use
in large-scale production. "
CA 02214837 2003-06-30
3
Thus, a need exists for a chromatographic method for purifying viral
vectors such as adenovirus vectors.
Summary of the Invention
This invention provides a method for purification of an intact viral
particle from a cell lysate, the method comprising: (a) treating said cell
lysate
which contains said intact viral particle with an enzymatic agent that
selectively
degrades both unencapsulated DNA and RNA; (b) chromatographing the treated
lysate from step a) on a first resin; and (c) chromatographing the eluant from
step
b) on a second resin; wherein one resin is an anion exchange resin and the
other
is an immobilized metal ion affinity resin.
This invention also provides a method for purification of intact viral
particles from a cell lysate, the method comprising the steps of: (a) treating
said
cell lysate which contains said intact viral particle with an enzymatic agent
that
selectively degrades both unencapsulated DNA and RNA; (b) chromatographing
the treated lysate from step a) on a first resin; and (c) chromatographing the
eluant from step b) on a second resin; wherein: one resin is an anion exchange
resin and the other is a hydrophobic interaction chromatography resin; or one
resin is a cation exchange resin and the other is either a hydrophobic
interaction
chromatography resin or an immobilized metal ion affinity resin.
This invention also provides a method of determining the number of
intact viral particles in a sample, the method comprising: (a)
chromatographing
the sample containing the intact viral particles on an anion exchange resin;
(b)
monitoring the absorbance of the eluate from the chromatography of step (a) at
a
selected wavelength; and (c) determining the total number of intact viral
particles
in the sample by comparing the absorbance value obtained in step (b) to a
standard curve which relates absorbance to number of viral particles.
CA 02214837 2003-06-30
3a
One obstacle to the successful practice of gene therapy is the
availability of purified viral vectors to deliver therapeutic genes. The
present
invention solves that problem by the unexpected and surprising discovery that
viral vectors containing therapeutic genes can be purified sufficiently for
therapeutic and/or prophylactic use using a three step process comprising:
enzymatically treating the cell lysate comprising the viral vector containing
the
therapeutic gene; chromatographing the enzymatically treated cell lysate on a
first resin; and chromatographing the eluate from the first column on a second
resin. The resulting purified viral vectors having the therapeutic gene
retained
their infectivity during and after chromatographic treatment and are able to
effect
gene transfer. For the first time it was found using the purification method
of this
invention that the chromatographically purified viral vector containing the
therapeutic gene is as pure and active as a viral vector purified using a
three day
ultracentrifugation. The purification method of this invention provides
several
advantages over existing methods including quality of purified viral vector,
consistency, decreased process time and the ability to process large amounts
of
sample. Furthermore, because this novel purification scheme is based on the
surface characteristics of the virion it is broadly applicable to other
virions using
the teaching of this invention as well as to virions containing different
internal
DNA constructs with different therapeutic genes. This break-through
purification
method is an important aspect in bulk commercialization of gene therapy.
Therefore, the invention provides a method of purifying viral vectors
containing therapeutic genes for use in gene therapy. In one embodiment, the
invention comprises a method of purification from a cell lysate of a
CA 02214837 1997-09-08
WO 96/27677 PCT/US96103369
4
recombinant viral vector containing a therapeutic gene, which comprises: a)
treating said lysate with an enzymatic agent that selectively degrades both
unencapsulated DNA and RNA; b) chromatographing the treated lysate from ,
step a) on a first resin; and c) chromatographing the eluant from step b) on a
second resin; wherein one resin is an anion exchange resin and the other is
an immobilized metal ion chromatography (IMAC) resin.
In an alternative embodiment a hydrophobic interaction chromatography
resin may be substituted for the immobilized metal ion chromatography resin
and/or a cation exchange resin may be substituted for the anion exchange
1 o resin.
In an alternative embodiment the enzymatically treated cell lysate
undergoes a filtration step adding a fourth step to the three step method.
In a more preferred embodiment the viral vector being purified is an
adenoviral vector containing a therapeutic gene. In a still more preferred
15 embodiment the adenoviral vector is a recombinant type 5 adenovirus with an
internal DNA construct including a tumor suppressor gene.
Brief Description of the Figures
Figure 1: Recovery of ACN53 from the supernatant of centrifuged crude
infected cell lysate which was centrifuged for 5 minutes at different speeds
in
2 o an Eppendorf centrifuge model 5415c at 4 °. Analysis for ACN53 was
by
analytical anion exchange.
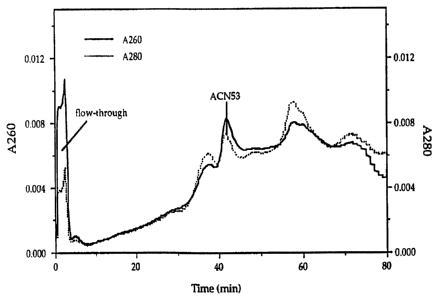
Figure 2: Identification of ACN53 infected cell lysate components and
separated by DEAE chromatography by dual wavelength UV absorbance
using their 260/280 nm absorbance ratio.
CA 02214837 1997-09-08
WO 96/27677 PC'T/US96/03369
Figure 3: Comparison of CsCI-ACN53 and host-cell contaminant retention
times during DEAE purification. Elution of CsCI-ACN53 is compared to
elution of uninfected H293 host-cell lysate blank chromatographed over a
DEAE column.
s Figure 4: Butyl-Hydrophobic Interaction Chromatography of a
DEAE-ACN53 fraction pool. A semi-pure DEAE purified ACN53 fraction pool
was mixed with an equal volume of 50 mM tris/pH 8.0/3 M ammonium sulfate
and injected onto a TosoHaas Butyl-650M column and eluted with a 1.5 - 0 M
decreasing ammonium sulfate gradient.
to Figure 5: Immobilized metal affinity chromatography of a DEAE-ACN53
fraction pool- A semi-pure DEAE purified ACN53 fraction pool was injected
onto a 6.6 x 50 mm TosoHaas AF chelate 650M column charged with ZnCl2
and eluted with a linear 0-500 mM glycine gradient.
Figure 6: SDS-PAGE comparison of ACN53 derived from column
chromatography and CsCI-ultracentrifugation. Samples were
electrophoresed on an 8-16% gradient polyacrylamide gel and silver stained
for analysis. Lanes 2 and 3 are DEAE and IZAC eluate pools respectively.
Lanes 4-6 represent 3 different lots (CsCI-1, CsCI-2, and CsCI-3) of
CsCI-ACN53 run side by side in order to examine lot to lot consistency. Lane
2 0 7, CsCI-ion exchange treated lot, represents the ACN53 peak recovered
when a sample of CsCI-ACN53 derived ion-exchange treated lot is purified
over a Resource Q anion exchange HPLC column (Resource Q, Pharmacia).
Lanes 1 and 8 are standard molecular weight markers.
Figure 7: Western Blot Comparison of ACN53 Derived from Column
Chromatography and CsCI-Ultracentrifugation. Samples, identical to those
previously described, were electrophoresed on an 8-16% gradient gel and
transferred to a PVDF membrane. The blot was incubated first with 5 ~g/ml
Cytimmune rabbit IgG anti adenovirus type 5 antibody, then with Amersham's
CA 02214837 1997-09-08
WO 96/27677 PCT/CTS96/03369
6
horseradish peroxidase conjugated anti-rabbit Ig (NA934) and developed
using electrochemical detection.
Figure 8: Expression of p53 gene product in Saos-2 cells. Two different
lots of chromatographically produced ACN53 were assayed by western blot
s for their ability to affect gene transfer to p53-null Saos-2 cells. The semi-
pure
DEAE fractions are shown in lanes 7 & 8, the final product in 5 & 6. P53-
expressing SW480 cells were used as a positive control; uninfected Saos-2
cells were used as a negative control.
Detailed Descria~tion of the Invention
to This invention is directed to a method for purification of a recombinant
viral
vector containing a therapeutic gene from a cell lysate, which comprises:
a) treating the cell lysate with an enzymatic agent that selectively
degrades both unencapsulated DNA and RNA; b) chromatographing the
treated lysate from step a) on a first resin; and c) chromatographing the
15 eluant from step b) on a second resin; wherein one such resin is an anion
exchange resin and the other is an immobilized metal ion affinity resin.
The term "viral vector" means a recombinant virus that has had some or all
of the genes in the native genome removed such that the virus is replication-
incompetent. Furthermore, the viral vector shall mean viruses wherein the
2 o recombinant viral genome contains a DNA encoding a therapeutic gene such
that the viral vector is used to transfer the therapeutic gene to a desired
human cell for gene therapy. Representative vectors include those that will
infect mammalian, and especially human, cells, and are derived from viruses
chosen from the group consisting of retroviruses, adenoviruses, herpes
CA 02214837 1997-09-08
WO 96/27677 PCT/ITS96I03369
7
viruses and avipox viruses. Retroviral and adenoviral vectors are preferred.
Adenoviral vectors, especially type 2 and type 5 adenoviral vectors, are
especially preferred. The recombinant type 5 adenoviral vectors are the
most preferred.
The term "therapeutic gene" means genes or functional fractions thereof
encoding molecules which have a desired therapeutic effect. For example, a
gene which either by its absence or mutation causes an increase in
pathological cell growth or proliferation of cells. A therapeutic gene as used
to herein would replace such an absent or mutated gene. Therapeutic genes
may give rise to their therapeutic effect either by remaining extrachromosomal
such that the gene will be expressed by the cell from the extrachromosomal
location or the gene may be incorporated into the genome of the cell such
that it recombines with the endogenous gene. Such genes include tumor-
suppressor genes, including those chosen from the group consisting of Rb,
Rb mutants, p53, p53 mutants, DCC, NF-1, Wlm's tumor, NM 23, BRCA-1,
BRCA-2, BRUSH-1, p56, H-NUC, thyroid hormone receptor gene, retenoic
acid receptor gene, genes encoding p130, p107, and p85. Other gene
replacement or supplementation strategies include adenosine deaminase
(ADA), thymidine kinase (TK), genes encoding various cytokines such as Y-
interferon, a-interferon, IL-2 and other hormones.
Therapeutic gene is also understood to include DNA encoding a ribozyme.
Such DNA constructs encode an RNA enzyme which binds and destroys the
RNA of selected positively-acting growth regulatory genes such as (1)
oncogenes or proto-oncogenes selected from the group consisting of, but not
limited to, the following: c-myc, c-fos, c jun, c-myb, c-ras, Kc and JE; (2)
growth factor receptor genes selected from the group consisting of, but not
limited to, epidermal growth factor, platelet derived growth factor,
transferrin
and insulin.
CA 02214837 1997-09-08
WO 96!27677 PCT/LTS96/03369
8
Preferred therapeutic genes are the tumor suppressor genes, with most
preferred tumor suppressor genes being Rb, Rb mutants, p53, p53 mutants,
BRUSH-1, p56, BRCA-1, BRCA-2, p16 and p21.
The term "cell lysate" means a collection of cells, including host cells
s which contain a vector, preferably a viral vector, which cells have been
removed from their growth environment and have had their cell membranes
disrupted by physiological or chemical means.
The term "enzymatic agent" means a compound or mixture that selectively
degrades unencapsulated DNA and RNA, while not disrupting the
1 o recombinant viral vectors to the extent they are not infectious and are
unable
deliver an intact copy of the therapeutic gene to the desired target cell.
Such
an enzymatic agent is generally a combination of one or more enzymes
referred to as endonucleases or DNAses or RNAses. BenzonaseTM is a
preferred enzymatic reagent.
15 The term "anion exchange resin" shall mean a positively-charged organic
moiety covalently cross-linked to an inert polymeric support. Representative
organic moieties are drawn from primary, secondary, tertiary and quaternary
amino groups; such as trimethylaminoethyl (TMAE), diethylaminoethyl
(DEAE), dimethylaminoethyl (DMAE), and other groups such as the
2 o polyethyleneimine (PEI) that already have, or will have, a formal positive
charge within the pH range of approximately 5 to approximately 9.
Similarly, representative negatively changed organic groups are chosen
from the group consisting of carboxymethyl and sulfomethyl and other groups
that have, or will have, a formal negative charge within the pH range of about
2 s 5 to about 9
CA 02214837 1997-09-08
WO 96/27677 PC"T/US96/03369
9
The support material should be one that is easily derivatizable and
possess good mechanical strength. The material can be a natural polymeric
substance, a synthetic polymer or co-polymer, or a mixture of natural and
synthetics polymers. The support can take the shape of porous or non-
porous particles, beads, membranes, disks or sheets. Such supports include
silica, hydrophilic polymer (MonoBeads~, Pharmacia, Piscataway, New
Jersey), cross-linked cellulose (e.g. Sephacel~), cross-linked dextran (e.g.
Sephadex~) cross-linked agarose (e.g. Sepharose~), polystyrene, or a co-
polymer such as polystyrene-divinylbenzene or one composed of
to oligoethyleneglycol, glycidylmethacrylate and pentaerythroldimethacrylate,
to
which are grafted polymerized chains of acrylamide derivatives (the latter co-
polymer is known as a "tentacle" support).
The resins can be used in a traditional (gravity) column chromatography or
high pressure liquid chromatography apparatus using radial or axial flow,
fluidized bed columns, or in a slurry (batch) method. In the latter method,
the
resin is separated from the sample by decanting or centrifugation or
filtration
or a combination of methods. The viral vectors can be purified then eluted
from these resins by an increasing salt gradient, preferably a gradient of
sodium chloride.
2 o Examples of suitable anion exchange resins include Fractogel~ (E. Merck,
Gibbstown, New Jersey) resins derivatized with either DEAE or DMAE;
Fractogel~ EMD Tentacle resins derivatized with DEAE, DMAE, or TMAE;
Toyopearl~ (TasoHaas, Montgomeryville, Pennsylvania) resins derivatized
with DEAE or QAE; Acti-Disk~ (Whatman, Clifton, New Jersey) supports
derivatized with Quat, DEAE or PEI; Sepharose~ (Pharmacia, Piscataway,
New Jersey) resins derivatized with DEAE; Sephacel~ (Pharmacia,
Piscataway, New Jersey) resins derivatized with DEAE; and Sephadex~
resins derivatized with DEAE and QAE. Preferred anion exchange resins are
derivatized with the DEAE group, and further preferred columns are the
CA 02214837 1997-09-08
WO 96/27677 PCT/LTS96/03369
Fractogel, Toyopearl and Streamliner"" (Pharmacia, Piscataway, New Jersey)
DEAF resins.
Similarly, the term "cation exchange resin" means a negatively-charged
organic moiety covalently cross-linked to an inert polymeric support.
s Representative negatively-charged moieties include the carboxymethyl (CM)
and the sulfomethyl (SP) groups. Other organic moieties that have, or will
have, a formal negative charge in the ph range of about 5.0 to about 9.0 is
also included within the definition. The above discussion for the support and
the method of use for the anion exchange resin applies equally to the cation
to exchange resin. Examples of suitable commercially- available cation
exchange resins include Sepharose, Sephacel and Sephadex covalently
linked with either a CM or an SP group.
The term "Immobilized metal ion affinity chromatography" ("IMAC") resin
refers to an inert natural or synthetic polymeric support covalently cross-
es linked with a metal chelating group. The metal chelating groups are those
known in the art to bind to zinc, nickel, copper, cobalt, calcium or magnesium
ions in the formal (+2) oxidation state. Such groups include the iminodiacetic
(IDA) group, the tris(carboxymethyl)ethylenediamine (TED) group, the N-
(hydroxyethyl)ethylenediaminetriacetic group, and derivatives such as the N-
(methyl), and the N-(hydroxymethyl) IDA groups. These groups can be cross-
linked to the natural or synthetic polymeric support by standard aliphatic
ether
linkages and reagents, such as bisoxirane, epichlorhydrin, and 1,4-bis-(2,3-
epoxypropoxy)butane. For the method of the invention, the chelating groups
can be bound to any of the above-listed metals, with zinc the preferred metal.
2 s The description of the chemical composition, physical form and uses of the
polymeric supports described above for the term "anion exchange resin"
applies to the IMAC resin as well. Cross-linked agarose and the "tentacle"
supports are preferred. The viral vectors can be eluted from the IMAC
column by adding increasing concentrations of competing chelating agents
CA 02214837 2003-06-30
11
such as imidazole, histamine, glycine, or ammonium chloride, or, alternatively
the
pH of the eluant can be raised or lowered, as long as the extremes of the
range
of the pH gradient used remain from about 5 to about 9. Examples of
commercially-available products that can be used in the instant method
include the Acti-Disk IDA cartridge (Whatman), Fractogel AF chelate, and
Toyopearl AF chelate IMAC resins.
Furthermore, the preferred conditions for the immobilized metal
affinity ion resin for the purification of the viral vectors such as type 5-
adenovirus
derived vector on DEAF had the resin charged with a divalent metal cation
chosen from the group consisting of cobalt, nickel, copper, zinc, calcium and
magnesium and further wherein the resin is an IDA or TED cross-linked agarose
resin, especially an IDA agarose resin that is charged with zinc ion.
A hydrophobic interaction chromatography ("HIC") can be
substituted for the IMAC resin in the instant method. Such a resin has lower
alkyl
or aryl groups covalently bound through a non-polar group, such as an
aliphatic
ether, to an inert polymeric support. Lower alkyl groups such as methyl,
propyl, n-
butyl, neo-pentyl, and octyl and the phenyl group are examples of the
interactive
group on the instant resin. Butyl is the preferred interactive group. The
chemical
composition, physical form and method of use for the supports described in the
definition of the term "anion-exchange resin" above also apply to the HIC
resins.
Cross-linked agarose, hydroxylated polyether, hydrophilic media and silica are
the preferred supports with cross-linked agarose the most preferred. The viral
vectors can be purified then eluted from the HIC resin by a decreasing salt
gradient, with ammonium sulfate the preferred salt. Examples of commercially
available HIC columns useful for the current invention include the cross-
linked
agarose columns such as Phenyl-, Butyl and Octyl Sepharose, and Toyopearl
(Phenyl, Propyl, and Butyl) and Fractogel (Propyl or Phenyl).
CA 02214837 1997-09-08
WO 96/27677 PCT/LTS96/03369
12
As mentioned above, in the instant invention the IMAC or HIC resin can be
used before or after the anion exchange resin. If used before, the salt
concentration of the eluant from the HIC or IMAC resin should diluted to about
450 millimolar or less in order to prevent premature stripping of viral
particles
s from the exchange resin.
The term "buffer" or "buffered solution" refers to a mixture of acid and
base which when present in a solution reduces or modulates changes in pH
that would otherwise occur in the solution when acid or based is added. In
one embodiment, the cell lysate is maintained in a buffered solution. Suitable
to buffers are those that can maintain the pH of the resultant solution
between
about 5.0 and about 9. 0. Such buffers are commercially available and
include phosphate, MES, HEPES, MOPS, Borate, TRIS, BES, ADA, ACES,
PIPES, MOPSO, BIS-TRIS PROPANE, BES, TES, DIPSO, TAPSO, TRIZMA,
HEPPSO, POPSO, TEA, EPPS, TRICINE, GLYCYLGLYCINE, BICINE,
15 TAPS, and the like. Preferred buffers include the phosphate, MES, HEPES,
MOPS, borate, and TRIS, with HEPES being the most preferred.
The use of chromatography for the purification of viral vectors, such as an
adenoviral vector referred to as Type 5, for use in gene therapy has been
shown to be an effective alternative to cesium chloride density gradient
2 o ultracentrifugations. There are several advantages related to this
methodology, including quality, consistency, decreased process time, system
automation, and the ability to process large amounts of crude lysate. The
purification scheme described in this invention selects for product based on
the surface characteristics of the virion. These characteristics do not change
25 with different internal DNA constructs, e.g. having different therapeutic
genes
in the construct, therefore leading to similar chromatographic behaviors. In
other words, the chromatography is unaffected by the nature of the
therapeutic gene inserted inside the vector.
CA 02214837 2003-06-30
13
The anion exchange resins immobilized metal ion affinity
chromatography resins cation exchange resins and hydrophobic interaction
chromatography resins are cleaned using methods known to the ordinarily
skilled
artisan. By way of example, DEAE is treated first with NaOH then HCI and
finally
with NaCI. The IZAC is first treated with EDTA then NAOH, HCL, NaCI flushing
with H20 in between each step. The columns are equilibrated in appropriate
buffers using appropriate binding conditions. Columns are loaded with sample
in
a buffer such that the product will bind to the resin by controlling pH and
salt
concentration for DEAE and by controlling pH, salt concentration and
divalent metal ion concentration for the immobilized metal ion affinity
column.
The columns are washed to remove contaminants and may be reused.
Other embodiments of the instant invention include the additional
step of filtering the cell lysate after it is treated with the enzymatic
agent. In an
alternative embodiment the cell lysate is buffered before treatment with
Benzonase at a pH between about 5.0 and about 9.0 before applying it to the
first
resin. These embodiments are preferred for purifying a recombinant viral
vector
derived from either a retrovirus or an adenovirus, and especially so when the
vector is derived from an adenovirus, and such a vector contains a tumor
suppressor gene.
Further embodiments for purifying an adenoviral-derived vector
containing a tumor suppressor gene are those when the treated, buffered cell
lysate is first chromatographed over an anion exchange resin followed by
chromatography over an immobilized metal affinity resin. These conditions are
preferred especially when the recombinant viral vector is derived from either
a
type 2 or type 5 adenovirus, and especially a type 5 adenovirus.
The enzymatic agent used to treat the cell lysate is one or more
enzymes, especially those chosen from the group consisting of RNAse and
DNAse or a mixture of endonucleases as would be known to the ordinarily
skilled
artisan.
CA 02214837 1997-09-08
WO 96/27677 PCT/LTS96/03369
14
The preferred enzymatic agent for use in this embodiment is BenzonaseT"", a
recombinant non-specific nuclease which cleaves both RNA and DNA.
Finally, an especially preferred method of the one described in the
preceding paragraphs is where the type 5 adenoviral-derived recombinant
s viral vector has a genome containing the wild-type p53 gene.
~xlaerimental Section
Procedure 1
Preaaration of AGN53 Standard MateriaI~GsCI-ACN53~
Standard recombinant Adenovirus Type 5 designated ACN53 is a vector
to derived from a Type 5 Adenovirus which has the F1 coding sequence with a
1.4-kb full length p53 cDNA driven by the human cytomegalovirus promoter
~Ils etet al., Numan ene Therapy 5:1079-1088 (1994)). Virus was prepared
by a 3-step centrifugation procedure as described (Layer et al., Viroloav
45:598-614 (1971 )) with the following modifications. Infected cells were
lysed
is by 3 cycles of freeze-thaw and centrifuged at 15,000 rpm for 10 min,
4°C in a
Sorvall RC-5B centrifuge. The pellet was discarded, and the supernatant was
treated with BenzonaseT"" (American International Chemical, Natick, Mass.)
at 133 U/mL for 30 min at room temperature. The treated material was then
layered onto a 1.25 gm/mL and 1.40 gm/mL CsCI discontinuous step gradient
2 o in 10 mM Tris pH8.1 and centrifuged at 30,000 rpm for 75 min, 10°C
in a
Sorvall TST 41-14 rotor. The virus band from each tube was collected,
pooled, mixed with 1.35 gm/mL CsCI (in 1 OmM Tris pH8.1 ) and centrifuged
overnight at 45,000 rpm in a Beckman VTi 50 rotor at 10°C. The virus
band
from each tube was collected and recentrifuged at 45,000 rpm as before for
25 an additional 4 hrs. The final virus pool from this step was dialyzed
extensively against phosphate buffered saline (PBS) supplemented with 2%
sucrose and 2 mM MgCl2 at a temperature of 4°C. The purified virus was
used to infect human embryonic kidney 293 cells (available from American
Type Culture Collection), as described below in Procedure 2.
CA 02214837 1997-09-08
WO 96/27677 PCT/US96/03369
Procedure 2
Production of Infected ATCC 293 ells Harvest and Lysis
ATCC 293 (ATCC CRL 1573) cells were grown in a Cell FactoryT"" (Nunc,
Ruskilde, Denmark) in a C02 incubator in 1.5L of medium consisting of DME
s high glucose medium containing 10% Hyclone bovine serum defined
supplemented, 2 mM glutamine (Irvine Scientific, Santa Ana, California), 1
mM sodium pyruvate (Irvine Scientific). No antibiotics were added to the
medium.
Two to two and a half days after seeding the Cell FactoryTM, when cell
1 o monolayers reached about 50-60% confluency, the cells were infected with a
multiplicity of infection (MOI) of 5 to 10 infectious units per cell in 500 mL
of
fresh medium. The virus was added to the medium, mixed thoroughly, and
introduced to the cells in the unit.
When cell monolayers from Preparation 1 showed signs of detachment
15 from the surface of the Cell FactoryTM (usually at 3 to 4 days post-
infection),
the cells were harvested by gentle tapping and were centrifuged in a
Beckman TJ-6 at 1500 rpm for 5 min. They were washed once with serum
free media, pelleted again, and resuspended in 25 mL of 50mM Tris buffer pH
8.0/150 mM NaCI, 2 mM MgCl2, 2% sucrose for use in the preparation of
2 o ultracentrifuge-derived standard virus. Samples destined for use in the
procedures of Examples 1 through 3 were resuspended in 25 mL of 50mM
HEPES buffer pH 7.5/150 mM NaCI, 2 mM MgCl2, 2% sucrose. The cells
were lysed at this point by 3 cycles of freeze-thaw. Following the third
cycle,
cellular debris was removed by centrifugation in a Beckman TJ-6 at 1500 rpm
for 5 min. At a temperature of 4°C.
CA 02214837 2003-06-30
16
Procedure 3
Western Blot Analysis of Samales Containing Recombinant Adenovirus
Particles
An SDS-PAGE gel was run as described in Example 3, Procedure
F with approximately the same loading as that of a silver stained gel. The
bands
were then transferred to a PVDF membrane pre-wetted in 100% methanol and
equilibrated in Tris-buffered saline (TBS). The gel was also equilibrated in
TBS.
The proteins were transferred to the membrane using a Bio-Rad semi-dry
transfer apparatus at 25 V for 30 minutes. The membrane was then blocked in
1 % casein/0.01 % sodium azide overnight at 4° or at room temperature
for I hr,
and washed 3 times with TBS. The membrane was incubated with the primary
antibody (Cytimmune rabbit IgG a-adenovirus type 5, Lee Biotechnology
Research: San Diego, CA) at 5 Ng/mL (in TBS) for 1 hr at room temperature.
Following primary incubation, the membrane was washed 3 times with TBS and
incubated with the secondary antibody (Amersham Life Sciences, Arlington
Heights, IL) Horseradish peroxidase conjugated anti-rabbit Ig) diluted to 1 pL
stock antibody/1 mL TBS for 1 hr at room temperature. A final three time wash
was performed with TBS and the membrane incubated with Amersham ECL
detection reagents for 1 minute, exposed in the dark to Hyperfilm-ECLT""
(Amersham) for various times (several seconds to minutes to give a selection
of
various contrasts) and developed in an X-ray film developer.
Using this western blot analysis, differences between banding
patterns of ACN53 in various states of purification can be seen in Figure 7.
CA 02214837 1997-09-08
WO 96!27677 PCTlUS96/03369
17
Procedure 4
Assay for Recombinant Adenoviral Particle Number b_y Absorbance at 260nm
in Presence of SDS.
For this measurement, concentrated virus was diluted 1 in 10 in 0.1 % SDS
s in phosphate buffered saline (PBS). The sample was vortexed for 1 minute
and then centrifuged at 14,000 rpm in an Ependorf microfuge to remove any
precipitate. A matched pair of cuvettes were blanked with 0.1 % SDS in PBS
buffer by running a baseline scan on Shimadzu UV160U spectrophotometer
(Shimadzu Scientific Instrument, Columbia, MD.). The SDS-treated virus
1 o sample was then placed in the sample cuvette and scanned from 220 to
340 nm. If there was absorbance between 310 and 320 nm, the sample was
too concentrated and was diluted further and remeasured. The A~/A2~ ratio
was also determined from this scan, and must be between 1.2-1.3 in order to
ensure that the product is pure enough to calculate particle number. If this
15 condition is met, the absorbance value at 260 nm only is used to calculate
the
number of virions/mL. A conversion factor of 1.1 x 10~z particles per
absorbance unit at 260 nm (Maize! et al., it I 36:115-125 (1968)) was
used to calculate particle number with approximately 10% error. Typical
values for samples subjected only to Anion Exchange Chromotography (i.e.,
2 o Examples 1 and 2 using a DEAE resin) were between 1.14 to 1.19. When
such samples were subjected to an IZAC as described in Example 3, the
ratios were in the 1.22 to 1.25 range.
Example 1
Lysis of Unencapulated Nucleic Acids
2s Nuclease Treatment
Infected cell lysate is comprised of contaminants both host-cell and viral in
origin. Some of these contaminants were removed prior to chromatography.
CA 02214837 1997-09-08
WO 96/27677 PCT/L1S96103369
18
Specifically, host cell, non encapsulated or incomplete ACN53 nucleic acids
were enzymatically degraded at this stage of the process with a nuclease
such as BenzonaseTM. This may be done early in the process for two
reasons. Early BenzonaseT"" treatment resulted in better yields in anion
s exchange chromatography. BenzonaseTM was removed by subsequent
process steps. The presence of Benzonase during the process can be .
assayed by a commercially available ELISA kit (American International
Chemical, Natick, MD.)
Clarification of the treated lysate was accomplished by filtration rather than
Zo centrifugation. A slow speed spin was used to remove cellular debris (Fig.
1).
Filtration was then used to prepare the product for loading onto the first
column. The type of filter used (i.e. composition and pore size) and its
effective product recovery was assayed by the quantitative anion exchange
assay described in Example 3, Procedure A.
15 Centrifugation was followed by filtration through a Gelman Sciences
AcrodiscT"" 0.8/0.2 lcm 2-stage syringe filter. Recovery of ACN53 depended
on the pore size and type of membrane used for filtration. Filters such as
polysulfone, PVDF membranes and cellulose acetate based membranes
were used. Polysulfone and PVDF were preferred.
2 o The supernatent from this step was made 2 mM in MgClz, 2% (wt/vol) in
sucrose and 2.5% (wt/vol) t3-cyclodextrin. BenzonaseTM (American
International Chemical, Inc.) was added to a final concentration of 100
units/mL and allowed to incubate for 1 hr at room temperature. The treated
material was clarified by centrifugation in a Beckman TJ-6 at 3000 rpm for 10
25 min and filtration through a Gelman Sciences AcrodiscTM 0.8/0.2 ,um filter.
The resultant supernatant was taken on to Example 2. '
CA 02214837 1997-09-08
WO 96/27677 PCT/US96/03369
19
exam Ip a 2
Anion Exchange Ghromatogra~hv
Overall, the characteristics of DEAE chromatography were found to be
consistent, and loading studies with high titer lysate (3 x10'2 ACN53
s particles/mL) showed a linear response between volume injected and AGN53
peak area recovered. Elution of a DEAE column by introduction of a linear
salt gradient gave three major peaks. The first of these was a protein peak
with a A~/AZ~ratio of ca. 0.5. Next was the ACN53 peak (A~Az~= 1.23)
followed by a DNA (A~A2~= 2) peak at the end of the gradient. The same
to three peaks were obtained whether run in a HEPES, Tris buffer or phosphate
buffer system. The pH can be varied, however, if run at pH 7.5 using HEPES
pH 7.5/NaCI/sucrose/MgCl2 less contaminating material bound to the
columns. DEAE chromatography yielded a high degree of initial purification.
The immobilized metal ion purification chromatography step removed these
15 contaminants.
Column resins were tested for their separation characteristics in 6.6 x
50 mm (1.7 mL) borosilicate OmnifitT"" columns (Omnifit Ltd., Cambridge,
England). The columns were mounted on a PerSeptive Bioystems BiocadT""
(Cambridge, MA) chromatography workstation. The chromatography was
2 o monitored on-line for pH, conductivity, and dual wavelength optical
density
detection at 280nm and 260nm.
Anion exchange resins were equilibrated in 50 mM HEPES, pH 7.5, 300
mM NaCI, 2 mM MgCl2, 2% sucrose at 1 mUmin. A 50 mM Tris buffer pH 8.0
(with 300 mM NaCI, 2 mM MgCl2, 2% sucrose) was also used. After the cell
2 s lysate was loaded and washed to baseline as monitored by absorbance,
elution was performed with a 20 column volume 300-600 mM linear NaCI
gradient and collected in 0.5 mL fractions. In preparation for future use, the
CA 02214837 1997-09-08
WO 96/27677 PCT/US96/03369
column was then cleaned with 2 column volumes of 0.5 M NaOH, a 1.5 M
NaCI wash and re-equilibrated.
In order to obtain an approximate control retention time, CsCI ACN53 was
injected onto a DEAE column equilibrated in 50mM Tris pH 8 at 2 mUmin
s (350 cm/hr) and eluted with a 10 min (11.7 column volume) 0-1.5 M linear
NaCI gradient. A single peak was detected with an A~/A2~ of 1.23. The
protein bands present in this fraction reacted with Ad 5 polyclonal antibody
upon slot-blot analysis.
Several peaks were resolved when an infected cell lysate sample was
to applied to the DEAE column (Fig. 2). The composition of the peaks were
deduced from the A~A2~ absorbance ratio. For example, the first peak has
an A~/A2~ ratio of 0.5, and is mainly protein. The third peak has an A~/AZ~
ratio of 2, suggesting that this material was nucleic acid. The ACN53 virus
peak eluted second with a ratio of 1.23. The identities of these peaks were
15 confirmed by spiking experiments and by running SDS gels of each peak. In
typical experiments using the above conditions an A~/A2~ratio of 1.23 t.08
was found to be characteristic of virus peaks.
In order to assess the purification capabilities of DEAF chromatography,
experiments were performed in which both non-infected ATCC-293 cell lysate
2 o and CsCI-ACN53 were applied to the column (Fig. 3). Most of the host cell
material either passed through the column during the load or eluted at an
earlier retention time than that of ACN53; however, a small peak eluted with
the same retention time as ACN53. From these data it appeared that
non-viral contamination of the ACN53 peak might be expected from host cell
2s material. Examination of the contaminant peak, the peak eluting last in the
cell lysate sample shown in Fig. 3, revealed an A~/AZeo nm ratio of
approximately 2. This indicated that the peak had a high nucleic acid content
and could possibly be reduced or eliminated by treatment with nuclease.
CA 02214837 1997-09-08
WO 96/27677 PCT/L1S96/03369
21
DEAE runs with and without BenzonaseT"" pretreatment demonstrated that
the enzyme reduced the amount of contamination of host cell material in the
ACN53 fraction pool.
Examl to a 3
Immobilized Metal Affinity Chromatograa~hv
s
Immobilized metal affinity chromatography (IMAC) using zinc as the
divalent cation (IZAC) gave higher product recovery and did not require any
sample manipulation of the DEAE fraction pool prior to loading. Impurities
removed by this method eluted in the flow through peak and were well
to resolved from product, leading to simpler pooling criteria. IZAC was
reproducible, and when used with DEAE provided a two column purification
protocol capable of delivering pure ACN53 as specified by SDS-PAGE gels
and westerns, A~A2~ ratios and infectious to non-infectious particle ratios. A
summarized overall protocol is as follows:
Harvest cells
1
Nuclease treatment
[3-Clyclodextrin treatment
1
2 o Clarification
1
DEAE chromatograhy
1
IZAC chromatography
Buffer exchange
Stepwise recoveries in terms of total particles and infectious units are
summarized in Tables 1 and 2 in the experimental section.
The Immobilized zinc affinity chromatography (IZAC) system was
3 o prepared for metal charging by washing the column sequentially with 1
volume of 100 mM EDTA and 1 volume of 0.2 M NaOH, flushing with water
CA 02214837 2003-06-30
22
after each step. The matrix was subsequently charged with zinc by injecting 1
column volume of 100 mM ZnCl2 in H20 acidified with 0.5 NL/mL glacial acetic
acid. The matrix was then thoroughly washed in water prior to equilibration in
50
mM HEPES pH 7.5/450 mM NaCI/2% sucrose/2 mM MgCl2. Sample loading did
not require any manipulation; DEAF pool fractions or CsCI derived material
could
be injected directly onto the column. After loading, the column was washed
with a
column volume decreasing NaCI linear gradient from 50 mM HEPES pH
7.5/450 mM NaCI/2% sucrose/2 mM MgCl2to 50 mM HEPES pH 7.5/150 mM
10 NaCI/2% sucrose/2 mM MgCl2. Elution was performed with a linear 0-500 mM
glycine gradient (in 150 mM NaCI) applied over 10 column volumes.
The interaction of ACN53 with a metal affinity column was shown to
be metal specific (with zinc preferred); injection of CsCI-ACN53 onto an
uncharged column (a column not pre-loaded with zinc) resulted in a shift of
the
product peak to the flow through. An ACN53-DEAF from Example 2 fraction pool
purified over IZAC is shown in Fig.S. Analysis of an IZAC fraction pool
produced
a yield of 49-65% and an A26o/AZ8o ratios of 1.22-1.25. A gel and western blot
comparison of CsCI-ACN53, DEAF purified and DEAE/IZAC purified material can
be seen in Figs 6 and 7. The CsCI-ACN53 and DEAE/IZAC materials were very
similar, and the DEAE-only purified material was less pure by these criteria.
The effect of different IZAC buffer and elution systems was
evaluated by splitting a DEAE-ACN53 pool in half and purifying both halves
over
IZAC in HEPES pH 7.5 and Tris pH 8 buffer systems. IZAC was also run in the
presence of sucrose and MgCl2 without affecting the separation. The use of
copper as the metal ion and imidazole as the elution agent were also tested
(for a
general review of metal affinity chromatography, see Belew et al. (1987) Anal.
Biochem. 164:457-465; and, Kato et al. (1986) J. Chrom. 354:511-517). Various
systems: zinc/glycine, zinc/imidazole, copper/glycine and copper/imidazole,
all
worked on the metal affinity
CA 02214837 1997-09-08
WO 96/27677 PCT/US96I03369
23
purification columns. ACN53 can be eluted using a pH 7 to pH9 gradient.A:
Assay for Recombinant Adenoviral Particle N tuber by Anion Exchange
HPLC.
A 1 mL Resource Q- (Pharmacia, Piscataway, NJ) anion exchange
s column was used to quantitate the number of viral particles in various
samples. The column was equilibrated in 300 mM NaCI, 50 mM HEPES, pH
7.5. at a flow rate of 1 mL lmin on a Waters 625 chromatography system
equipped with 717p1us autosampler and a 991 photodiode array detector
(PDA). The chromatography was monitored on the PDA detector (Milford,
to MA) scanning from 210 to 300 nm. A standard curve was constructed by
injecting CsCI purified ACN53 virions characterized for total particles at a
selected absorbance, A~ in 0.1 %SDS.
The assay was independent of injected sample volume. After the sample
loaded, the column was washed with two column volumes of equilibration
15 buffer followed by a linear gradient from 300 to 600 mM NaCI in 50 mM
HEPES, pH 7.5, over 10 column volumes. The gradient was followed with a 2
column volume wash with 600 mM NaCI in 50mM HEPES, pH 7.5. After each
chromatographic run, the column was cleaned with 2.6 column volumes of 1.5
M NaCI in 50 mM HEPES, pH 7.5, and then re-equilibrated for the next
2 o injection. The column was cleaned more vigorously after injection of crude
samples by injecting 0.25 to 1 column volume of 0.5 N NaOH followed by a
wash with 1.5 M salt. Injecting NaOH and then running the gradient was a
convenient way to accomplish cleaning. The use of such an assay in
measuring the total number of adenovirus particles present are set forth in
the
25 following Table 1.Table 1. Yield and Purity Data Based on Total ACN53
Particles
CA 02214837 1997-09-08
WO 96/27677 PCT/US96/03369
24
Step Viral Vol. (ml) Total Step Total
Particle Particles Yieldb Yieldb
#/111 L
Lysate 6x10" 5.0 3x10'2 100%
(Procedure
2)
DEAE load 6x10" 5.0 3x10'2
(Example
1 )
DEAE 4x10" 5.0 2x10'2 67%
eluate
(Example
2)
IZAC load 4x10" 3.8 1.52x10'2
(Example
3)
IZAC eluate~2.38x10" 3.0 7.14x10" 47% 31
(Example
3)
aAs determined by Analyzical Ion Exchange
bln terms of viral particle number
°After dialysis into final formulation
~: Buffer Cnnrtitinne
A variety of buffer conditions have been used in the purification of ACN53
by column chromatography in this study in the pH range of 7.0-8Ø (e.g. Seth
Viroloav 68:1204-1206 (1994)). Checking for degradation at various pH limits
2 o is accomplished by assaying for degradation by TCID50 (Procedure C below)
and analytical anion exchange analysis (Procedure A above). The effect of
buffer salt concentration on the viability of ACN53 showed that abrupt
changes of ionic strength can lead to loss of virus. Salt concentration should
be carefully controlled and monitored.
CA 02214837 2003-06-30
C: Measurement of Infectious Recombinant Adenovirus Particles by TCIDSo
Assay
The quantitation of infectious Adenovirus Type 5 particles, before,
5 during and after the purification methods taught in these Examples is
accomplished by an end point titer assay (tissue culture infective dose-50%;
abbreviated TCIDSO). (See Philipson, Viroloay 15:263-268 (1961)). Reagents, a
materials list and instructions are available from Chemicon International,
Inc.
(cat.# 3130: "Adenovirus Direct Immunofluorescence Assay", Temecula, CA).
ATCC 293 cells were plated into a 96-well plate: 100 NL of 5 x 105
cells/mL for each well in complete MEM (10% bovine calf serum; 1 % glutamine)
media. In a separate plate, a 250 pL aliquot of virus sample diluted 1:106 was
added to the first column and is serially diluted two-fold across the plate.
One row
was reserved for a positive control (CsCI-purified ACN53); one for a negative
control. A 100 NL aliquot of each well was transferred to its identical
position in
the ATCC-293 seeded plate and allowed to incubate a 37° in a humidified
incubator for 2 days. The media was then decanted by inversion and the cells
were fixed with the addition of 50% acetone/50% methanol. After washing with
PBS, the fixed cells were incubated for 45 min with a FITC labeled anti-ads
antibody (Chemicon International #5016) prepared according to the kit
instructions. After washing with PBS, the plate was examined under a
fluorescent
microscope (490 nm excitation, 520 nm emission) and scored for the presence of
label. The titer was quantitated using the Titerprint Analysis program (Lynn,
BioTechnigues, 12:880-881 (1994)). Results from assays performed in
accordance with this procedure are set forth in the following Table 2, which
Table
includes a ratio calculated using the values from Table I above. Table 2.
Yield
and Purity Data Based on Infective ACN53 Particles
CA 02214837 1997-09-08
WO 96!27677 PCT/ITS96/03369
26
Step Step YieldA2so~A2eo Viral Particle
(TCIDso~ Ratio Purity by
HPLC
at AZ8o -
Lysate - _ 3%
DEAE load 49% 1.17 92%
IZAC Eluate44% 1.23 98%
s The ratio of total virus particles to infectious viral particles can vary
widely
from preparation to preparation. Values for CsCI derived viruses in the range
of 60 to 80. The calculation of this ratio in crude lysates or semi-pure
fractions has been made possible by the anion exchange particle assay (see
Example 3[A]). Using analytical anion exchange changes in the ratio of ideal
to virus particles to infectious virus particles can be monitored. Using the
purification method of this invention the total virus particle to infectious
virus
particle ratio of the crude lysate and subsequently purified material is
comparable to that obtained using ultracentrafugation. Within the error of the
assay these values are equivalent. By this criteria, chromatographic
is purification is well tolerated by the virus. By other criteria, namely SDS-
PAGE
and western blot analysis (Figs. 6 and 7), chromatographic purification is
superior to ultracentrifugation based methods.
D: Assav for Infectious Recombinant Adenovirus Particles by Expression of
P53 Protein.
2 o The activity of virus preparations was also tested by assaying for the
expression of the p53 gene product in Saos-2 cells, a p53-negative
osteosarcoma cell line. Saos-2 cells were seeded into a 6-well tissue culture
plate at a concentration of 5 x 105 cellslwell in 3 mL of media: Kaighn's
nutrient mixture F12: DME High Glucose (1:1 mixture), supplemented with 2
2s mM L-glutamine and 10% fetal bovine serum. The cells were incubated in
humidified air, 7t1 % COZ at 37° for 16-24 hrs. The spent media were
CA 02214837 1997-09-08
WO 96/27b77 PCT/ITS95/03369
27
removed and replaced with 1 mL of fresh media, and the cells were infected
at 20,40, or 60 MOI of purified virus. After incubation for 1 hr, an
additional 2
. mL of media is added and allowed to incubate for 8 hrs. The cells were then
washed once with Dulbecco's-PBS and lysed by adding of 250 ~cL of: 50 mM
- s tris/0.5% Noridet P-4.0/250 mM NaCI/5 mM EDTA/5 mM NaF/5 ~g/mL
Leupeptin and 5 ~g/mL Aprotinin/2 mM PMSF. The plate was incubated' on
ice for 5 min after which the lysates were transferred to individual 1.7 mL
microcentrifuge tubes. They were spun down for 45 sec at 14000 rpm in a
microfuge and the supernatants were assayed for the presence of p53 protein
io by western blot analysis with the primary anti-p53 monoclonal antibody 1801
(Vector Laboratories, Burlingame, California) and a 1:1 mixture of sheep
anti-mouse IgG-HRP and streptavidin-HRP (Amersham). The p53 protein
band was detected using Amersham's ECL detection kit according to the
manufacturer's instructions. The results of the use of such assay are shown
15 in Figure 8. p53 expression can be seen in ACN53 purified only by DEAE
chromatography as well as by both DEAE and IZAC chromatography. (The
lower levels of expression in lanes 5 and 6 are due to the fact that these
samples were run at a lower MOI than the DEAE samples.)
~: Host Cell Protein Contamination
2 o Host cell contamination was assessed by western blot analysis using
polyclonal antibodies developed against ATCC-293 cell components.
Polyclonal antibodies were commercially obtained and were raised against
various 293 cellular antigens (HTI, Ramona, CA). The results indicated that
the final products of either CsCI or DEAE-IZAC purification contained no
2s detectable host cell contaminants. In the case of semi-pure DEAE-ACN53,
there was a small amount of immunogenic contamination seen in the product
pool: 3 major bands and several minor ones. The majority of host cell
contamination is recovered in the flow-through portion of the DEAE step.
CA 02214837 2003-06-30
28
Contaminants which copurified with ACN53 in the DEAF step were removed by
zinc affinity chromatography, and were recovered in IZAC flow-through
fractions
prior to the introduction of the glycine gradient.
F: SDS-PAGE Analysis Samples Containing Recombinant Adenovirus
Particles
For Coomassie blue staining, 100-200 pL of an adenovirus
containing sample (at approximately 1 x 10" particles/mL) was collected,
desalted by trichloroacetic acid precipitation or by dialysis followed by
concentration in a speed-vac. The sample was then resuspended in SDS-PAGE
reducing buffer
(12S mM Tris-HCL, pH 6.8, 20% glycerol, 4% (w/v) SDS, 0.005% bromophenol
blue, 0.5% ~i-mercaptoethanol) to approximately 30 pL, boiled for 5 min and
loaded onto a 1 mm x 10 well NovexT"~ 8-16% gradient Tris-Glycine minigel.
Samples were electrophoresed for 1.5 hrs at 140 V. The gel is then fixed in
40%
methanol/10% acetic acid for 30 min, and then Coomassie stained with the Pro-
Blue staining system (Integrated Separation Systems, Natick MA.) according to
the vendor's procedure.
Gels to be silver stained were loaded with 5-15 NL of sample. The
sample was boiled with an equal volume of reducing buffer and electrophoresed
as described for Coomassie detection. Fixing was performed by treating the gel
in 10% trichloroacetic acid for 1 hr followed by a 3x wash in water purified
to
18 megohms. The gel was stained with the Daiichi silver staining kit according
to
the instructions provided (Integrated Separation Systems).
CA 02214837 1997-09-08
WO 96/27677 PCT/US96/03369
29
Example IV
Vector InfectiviW Transfer of Therapeutic Gene
Characterization of DEAE-IZAC-ACN53 has shown that virions retained
their infectivity during chromatographic treatment as measured by TCID50
s analysis (Table 1,2) and were able to effect gene transfer as assayed by'
P53
protein expression in Saos-2 cells (Fig 8). Comparison of this
DEAE-IZAC-ACN53 to CsCI-ACN53 in SDS-PAGE analysis has shown that
there were more lower molecular weight protein bands present in the
CsCI-ACN53 (Figs. 6 and 7). Side-by-side comparison of different
1 o CsCI-ACN53 lots has shown some batch-to-batch variability whereas
chromatographically produced material has been very consistent. The total to
infectious particle ratio and absorbance spectrum of both types of material
are
directly comparable. Characterization of chromatographically produced
ACN53 in terms of purity and activity have shown that a 1 day 2-column
15 procedure can replace a 3 day ultracentrifugation protocol.
A preferred purification scheme for ACN53 as shown above is to treat
infected cell lysate with BenzonaseT"" prior to the chromatographic steps.
Clarification is then accomplished by step filtration through 0.8 ~cm followed
by
0.2 ,um membranes. If necessary, a larger pore (e.g. 5 ~cm) prefiltration step
2 o can be added for more viscous suspensions. Adjustment to pH 7.5/300 mM
NaCI is then performed in preparation for loading onto a DEAE column. The
product peak, as detected by the A~A28o nm ratio or the characteristic
photo-diode array spectrum, is pooled and directly injected onto a
zinc-charged, iso-osmotically equilibrated metal affinity column. The ionic
2s strength of the buffer is then gradually lowered to approximate
phosphate-buffered saline (ca,150 mM NaCI) prior to elution of product with a
glycine gradient. This material is then dialyzed into the final formulation.