Note: Descriptions are shown in the official language in which they were submitted.
CA 0221~870 1997-09-19
TITLE OF THE INVENTION
METHOD FOR THE IDENTIFICATION OF ESSENTIAL
GENES AND THERAPEUTIC TARGETS
5 FIELD OF THE INVENTION
The present invention relates to the identification of
essential genes in a given genome. More specifically, the invention
relates to the identification of essential genes in a diploid organism in
which homozygocity conversion is erricie nl or in a haploid organism. The
10 present invention also relates to the identification of therapeutic targets
and more specifically to therapeutic targets in bacteria.
BACKGROUND OF THE INVENTION
The human genome project as well as genome projects
15 of model organisms have opened the area of genomics. Although
thousands of genetic sequences are available in data bases, only a small
minority thereof have a recognized function. It has become apparent that
biological functions cannot be solely deduced by computer approaches
and that even in integrated format, databases present significant
20 limitations.
Large amounts of data, from the partial or complete DNA
sequences of microbial genomes are also rapidly accumulating in
databases. There is heightened expectations that the increasingly
powerful computer analyses will be able to yield biological function from
25 these DNA sequence. However, it is becoming clear that even for
microbial genomes, the sole information in databases will not be sufficient
11229-79.DRF 19Sep. 1997-16h12
CA 0221~870 1997-09-19
to deduce the biological function. Thus, it becomes apparent that whole
genome or genome-based analysis of biological function could provide
significant results. Indeed, such analysis could be the next phase in
microbial genomics, particularly as it pertains to finding novel therapeutic
5 targets in bacteria.
It has become apparent that expression of a subset of
genes is essential for survival of the eukaryotic and prokaryotic cells;
mutations in these genes give rise to a lethal phenotype. Recently, the
number of lethal loci has been estimated in a number of life forms
10 serving as model organisms for genome projects: Drosophila (3,600
essential genes), Caenorhabditis (3,000), Arabidopsis (500),
Saccharomyces (900). Bacterial genomes comprise gene numbers which
vary from approximately 500 to more than 8000. The number of essential
genes in such genomes is unknown but can be esli",aled as being
between 100 to 150 in smaller genomes, such as that of Haemophilus
influenzae (1.83 Mb), to more than 500 in larger bacterial genomes, such
as that of Pseudomonas aeruginosa (5.9 Mb). The potential and
ramifications of using these essential genes and their products as novel
therapeutic targets is enormous for the pharmaceutical industry and could
20 open a new era in antimicrobial research. In addition, the identification
of essential genes in higher life forms could provide important
fundamental and practical information relating to cellular homeostasis,
cancer and the like.
Powerful genetic techniques such as allelic replacement
25 and gene knockouts have been developed. These technologies are
effective but can only be applied to selected and candidate genes of
11 229-79.DRF 1 9 Sep. 1 997 - 1 6h 1 2
CA 0221~870 1997-09-19
interest. Applying these genetic techniques to whole genomes, even in
the context of bacterial genomics, represents a highly inefficient and
costly task and novel whole-genome based techniques and gene-
screening assays must therefore be developed.
Comprehensive, rapid and, simple screening of bacterial
genomes for essential genes has not been possible because of the
inability to identify mutants having an attenuated or no significant growth
within pools of mutagenized bacteria. It is also impractical to separately
assess the significance of essential versus non-essential genes from
each of the several thousand mutants necessary to screen a bacterial
genome. Although genome-wide functional analysis appears to offer the
best approach for the identification of dispensable versus essential
genes, no simple, rapid and effficient identification method therefor has
been forthcoming. Genome-based analyses provide primarily a functional
classification rather than a detailed understanding of each gene. This is
a critical aspect in microbial genomics in which one can identify
therapeutic targets by identifying essential genes.
USP 5,612,180 teaches a genetic footprinting method
which, in essence, is a functional screen of genes under different
selective conditions. A PCR-based method which identifies genes
essential for survival of a cell, under the selective growth conditions used
is taught. Briefly, insertional mutagenesis is carried out on the genome to
be tested. The method is then based on the use of one set of primers for
the PCR-based genetic footprinting: one primer binding to the insertional
mutagen, the other being chosen arbitrarily as a unique sequence in the
targeted region. This genetic footprinting method is unfortunately
11229-79.DRF 1 9 Sep. 1 997 -16h12
CA 0221~870 1997-09-19
restricted to the identification of essential genes under a specific selection
scheme. Furthermore, it lacks in providing a positive control of
amplification originating solely from the targeted region (not from the
insertional mutagen). Moreover, it is dependant on the discrimination of
small differences in the extension products. Finally, it is based on the
comparison of amplification products originating from two different sub-
populations (selected vs non-selected).
There therefore remains a need to provide a simple and
effficient method of identifying essential genes in a genome under non-
selective conditions. There also remains a need to provide a simple and
efficient method of identifying genes which are essential under specific
conditions, the method providing an amplified signal originating solely
from the non-mutagenised targeted region and in which amplification
products from a single sub-population of cells are analysed. The present
invention seeks to meet these and other needs.
The description refers to a number of documents, the
content of which is herein incorporated by reference.
SUMMARY OF THE INVENTION
Accordingly, the present invention seeks to provide an
essential gene test (EGT), an effficient and economical approach to define
the function of thousands of sequences containing a complete open
reading frame (ORF) or parts thereof, or known and/or unknown genes
encoding hypothetical proteins or products. The EGT test is particularly
effective at defining which sequences in databases contain an essential
or a non-essential (dispensable) gene. In one embodiment the EGT
1 1 229-79. DRF 1 9 Sep. 1 997 -1 6h 1 2
CA 0221~870 1997-09-19
assay is based on the premise that a mutation inactivating an essential
gene should give rise in vivo, to a lethal phenotype irrespective of the
growth conditions.
The present invention also seeks to provide an EGT test
5 which enables the categorization of gene sequences as encoding
essential and dispensable genes under selective conditions, the
categorization being based on the analysis of a single sub-population of
cells ("one tube population").
Furthermore, the present invention seeks to provide an
10 EGT test based on the detection of two basic types of extension products
originating from two primer pairs.
By enabling an idei,liricalion of essential genes in
organism, the EGT assays permits the identification of therapeutic targets
in this organism. The present invention more preferably seeks to provide
15 therapeutic targets in haploid organisms, particularly bacteria.
Nucleotide sequences are presented herein by single
strand, in the 5' to 3' direction, from left to right, using the one letter
nucleotide symbols as commonly used in the art and in accordance with
the recommendations of the IUPAC-IUB Biochemical Nomenclature
20 Commission.
The present description refers to a number of routinely
used recomb,nant DNA (rDNA) technology terms. Nevertheless,
definitions of selected examples of such rDNA terms are provided for
clarity and consistency.
11229-79.DRF 19Sep. 1997-16h12
CA 0221~870 1997-09-19
As used herein, "isolated nucleic acid molecule", refers
to a polymer of nucleotides. Non-limiting examples thereof include DNA
and RNA molecules purified from their natural environment.
The term "recombinant DNA" as known in the art refers
5 to a DNA molecule resulting from the joining of DNA segments. This is
often referred to as genetic engineering.
The term "DNA segment", is used herein, to refer to a
DNA molecule comprising a linear stretch or sequence of nucleotides.
This sequence when read in accordance with the genetic code, can
10 encode a linear stretch or sequence of amino acids which can be referred
to as a polypeptide, protein, protein fragment and the like.
The terminology "amplification pair" or "primer pair"
refers herein to a pair of oligonucleotides (oligos) of the present invention,
which are selected to be used together in amplifying a selected nucleic
15 acid sequence by one of a number of types of amplification processes,
preferably a polymerase chain reaction. Other types of amplification
processes include ligase chain reaction, strand displacement
amplification, or nucleic acid sequence-based amplification, as explained
in greater detail below. As commonly known in the art, the oligos are
20 designed to bind to a complementary sequence under selected
conditions.
The nucleic acid (i.e. DNA or RNA) for practicing the
present invention may be obtained according to well known methods.
Oligonucleotide probes or primers of the present
25 invention may be of any suitable length, depending on the particular
assay format and the particular needs and targeted genomes employed.
1 1229-79.DRF 1 9 Sep. 1 997 -1 6h1 2
CA 0221~870 1997-09-19
In general, the oligonucleotide probes or primers are at least 12
nucleotides in length, preferably between 15 and 24 nucleotides, and they
may be adapted to be especially suited to a chosen nucleic acid
amplification system. As commonly known in the art, the oligonucleotide
probes and primers can be designed by taking into consideration the
melting point of hydrizidation thereof with its targeted sequence (see
below, and in Sambrook et al., 1989, Molecular Cloning - A Laboratory
Manual, 2nd Edition, CSH Laboratories; Ausubel et al., 1989, in Current
Protocols in Molecular Biology, John Wiley & Sons Inc., N.Y.).
"Nucleic acid hybridization" refers generally to the
hybridization of two single-stranded nucleic acid molecules having
complementary base sequences, which under appropriate conditions will
form a thermodynamically favored double-stranded structure. Examples
of hybridization conditions can be found in the two laboratory manuals
referred above (Sambrook et al., 1989, supra and Ausubel et al., 1989
supra) and are commonly known in the art. In the case of a hybridization
to a nitrocellulose filter, as for example in the well known Southern
blotting procedure, a nitrocellulose filter can be incubated overnight at
65~C with a labeled probe in a solution containing 50% formamide, high
salt ( 5 x SSC or 5 x SSPE), 5 x Denhardt's solution, 1% SDS, and 100
,ug/ml denatured carried DNA ( i.e. salmon sperm DNA). The non-
specifically binding probe can then be washed off the filter by several
washes in 0.2 x SSC/0.1% SDS at a temperature which is selected in
view of the desired stringency: room temperature (low stringency), 42~C
(moderate stringency) or 65~C (high stringency). The selected
temperature is based on the melting temperature (Tm) of the DNA hybrid.
11229-79.DRF 19 Sep. 1997 - 16h12
CA 0221~870 1997-09-19
Of course, RNA-DNA hybrids can also be formed and detected. In such
cases, the conditions of hybridization and washing can be adapted
according to well known methods by the person of ordinary skill. High
stringency conditions will be preferably used (Sambrook et al.,1989,
5 supra).
Probes of the invention can be utilized with naturally
occurring sugar-phosphate backbones as well as modified backbones
including phosphorothioates, dithionates, alkyl phosphonates and
a-nucleotides and the like. Modified sugar-phosphate backbones are
generally taught by Miller, 1988, Ann. Reports Med. Chem. 23:295 and
Moran et al., 1987, Nucleic acid molecule. Acids Res., 14:5019. Probes
of the invention can be constructed of either ribonucleic acid (RNA) or
deoxyribonucleic acid (DNA), and preferably of DNA.
The types of detection methods in which probes can be
used include Southern blots (DNA detection), dot or slot blots (DNA,
RNA), and Northern blots (RNA detection). Although less prepared,
labelled proteins could also be used to detect a particular nucleic acid
sequence to which it binds. Other detection methods include kits
containing probes on a dipstick setup and the like.
Although the present invention is not specifically
dependent on the use of a label for the detection of a particular nucleic
acid sequence, such a label might be beneficial, by increasing the
sensitivity of the detection. Furthermore, it enables automation. Probes
can be labelled according to numerous well known methods (Sambrook
et al.,1989, supra). Non-limiting examples of labels include 3H,14C,32p,
and 35S. Non-limiting examples of detectable markers include ligands,
11229-79.DRF 19Sep. 1997-16h12
CA 0221~870 1997-09-19
fluorophores, chemiluminescent agents, enzymes, and antibodies. Other
detectable markers for use with probes, which can enable an increase in
sensitivity of the method of the invention, include biotin and
radionucleotides. It will become evident to the person of ordinary skill that
5 the choice of a particular label dictates the manner in which it is bound to
the probe.
As commonly known, radioactive nucleotides can be
incorporated into probes of the invention by several methods. Non-limiting
examples thereof include kinasing the 5' ends of the probes using gamma
10 32p ATP and polynucleotide kinase, using the Klenow fragment of Pol I of
E. coli in the presence of radioactive dNTP (i.e. uniformly labelled DNA
probe using random oligonucleotide primers in low-melt gels), using the
SP6/T7 system to transcribe a DNA segment in the presence of one or
more radioactive NTP, and the like.
As used herein, "oligonucleotides" or "oligos" define a
molecule having two or more nucleotides (ribo or deoxyribonucleotides).
The size of the oligo will be dictated by the particular situation and
ultimately by the particular use thereof, and adapted accordingly by the
person of ordinary skill. An oligonucleotide can be synthetised chemically
or derived by cloning according to well known methods.
As used herein, a "primer" defines an oligonucleotide
which is capable of annealing to a target sequence, thereby creating a
double stranded region which can serve as an initiation point for DNA
synthesis under suitable conditions.
Amplification of a selected, or target, nucleic acid
sequence may be carried out by a number of suitable methods. See
11229-79.DRF 19Sep. 1997-16h12
CA 0221~870 1997-09-19
generally Kwoh et al., 1990, (Am. Biotechnol. Lab. 8:14-25). Numerous
amplification techniques have been described and can be readily adapted
to suit the particular needs of a person of ordinary skill. Non-limiting
examples of amplification techniques include polymerase chain reaction
5 (PCR), ligase chain reaction (LCR), strand displacement amplification
(SDA), transcription-based amplification, the Q~ replicase system and
NASBA (Kwoh et al., 1989, Proc. Natl. Acad. Sci. USA 86, 1173-1177;
Lizardi et al., 1988, BioTechnology 6:1197-1202; Malek et al., 1994,
Methods Mol. Biol., 28:253-260; and Sambrook et al., 1989, supra).
10 Preferably, amplification will be carried out using PCR.
Polymerase chain reaction (PCR) is carried out in
accordance with known techniques. See, e.g., U.S. Pat. Nos. 4,683,195;
4,683,202; 4,800,159; and 4,965,188 (the disclosures of all three U.S.
Patent are incorporated herein by reference). In general, PCR involves,
15 a treatment of a nucleic acid sample (e.g., in the presence of a heat
stable DNA polymerase) under hybridizing conditions, with one
oligonucleotide primer for each strand of the specific sequence to be
detected. An extension product of each primer which is synthesized is
complementary to each of the two nucleic acid strands, with the primers
20 sufficiently complementary to each strand of the specific sequence to
hybridize therewith. The extension product synthesized from each primer
can also serve as a template for further synthesis of extension products
using the same primers. Following a sufficient number of rounds of
synthesis of extension products, the sample is analysed to assess
25 whether the sequence or sequences to be detected are present.
Detection of the amplified sequence may be carried out by visualization
1 1229-79.DRF 1 9 Sep. 1 997 -1 6h1 2
CA 0221~870 1997-09-19
following EtBr staining of the DNA ~ol'ow;"g gel electrophoresis, or using
a detectable label in accordance with known techniques, and the like. For
a review on PCR techniques (see PCR Protocols, A Guide to Methods
and Amplifications, Michael et al., Eds, Acad. Press, 1990).
Ligase chain reaction (LCR) is carried out in accordance
with known techniques (Weiss,1991, Science 254:1292). Adaptation of
the protocol to meet the desired needs can be carried out by a person of
ordinary skill. Strand displacement amplification (SDA) is also carried out
in accordance with known techniques or adaptations thereof to meet the
particular needs (Walker et al., 1992, Proc. Natl. Acad. Sci. USA
89:392-396; and ibid., 1992, NucleicAcids Res. 20:1691-1696.
As used herein, the term "gene" is well known in the art
and relates to a nucleic acid sequence defining a single protein or
polypeptide. A "structural gene" defines a DNA sequence which is
transcribed into RNA and translated into a protein having a specific amino
acid sequence thereby giving rise the a specific polypeptide or protein. It
will be readily recognized by the person of ordinary skill, that the nucleic
acid sequences of the present invention can be incorporated into anyone
of numerous established kit formats which are well known in the art.
The term "vector" is commonly known in the art and
defines a plasmid DNA, phage DNA, viral DNA and the like, which can
serve as a DNA vehicle into which DNA of the present invention can be
cloned. Numerous types of vectors exist and are well known in the art.
The term "expression" defines the process by which a
structural gene is transcribed into mRNA (transcription), the mRNA is then
being translated (translation) into one polypeptide (or protein) or more.
11229-79.DRF 19Sep. 1997-16h12
CA 0221~870 1997-09-19
The terminology "expression vector" defines a vector or
vehicle, as described above, but designed to enable the expression of an
inserted sequence following transformation into a host. The cloned gene
(inserted sequence) is usually placed under the control of control element
5 sequences such as promoter sequences. The placing of a cloned gene
under such control sequences is often referred to as being "operably
linked" to control elements or sequences.
Expression control sequences will vary depending on
whether the vector is designed to express the operably linked gene in a
10 prokaryotic or eukaryotic host or both (shuttle vectors) and can
additionally contain transcriptional elements such as enhancer elements,
termination sequences, tissue-specificity elements, and/or translational
initiation and termination sites.
As used herein, the designation "functional derivative"
15 denotes, in the context of a functional derivative of a sequence, whether
nucleic acid or amino acid sequence, a molecule that retains a biological
activity (either functional or structural) that is substantially similar to thatof the original sequence. This functional derivative or equivalent may be
a natural derivative or may be prepared synthetically. Such derivatives
20 include amino acid sequences having substitutions, deletions, or
additions of one or more amino acids, provided that the biological activity
of the protein is conserved. The same applies to derivatives of nucleic
acid sequences which can have substitutions, deletions, or additions of
one or more nucleotides, provided that the biological activity of the
25 sequence is generally maintained. When relating to a protein sequence,
the substituting amino acid has chemico-physical properties which are
11229-79.DRF 19 Sep. 1997 - 16h12
CA 0221~870 1997-09-19
similar to that of the substituted amino acid. The similar chemico-physical
properties include, similarities in charge, bulkiness, hydrophobicity,
hydrophylicity and the like. The term "functional derivatives" is intended
to include "fragments", "segments", "variants", "analogs" or"chemical
5 derivatives" of the subject matter of the present invention.
Thus, the term "variant" refers herein to a protein or
nucleic acid molecule which is substantially similar in structure and
biological activity to the protein or nucleic acid of the present invention.
The functional derivatives of the present invention can
10 be synthesized chemically or produced through recombinant DNA
technology. All these methods are well known in the art.
As used herein, "chemical derivatives" is meant to cover
additional chemical moieties not normally part of the subject matter of the
invention. Such moieties could affect the physico-chemical characteristic
15 of the derivative (i.e. solubility, absorption, half life and the like, decrease
of toxicity). Such moieties are exemplified in Remington's Pharmaceutical
Sciences (1980). Methods of coupling these chemical-physical moieties
to a polypeptide are well known in the art.
The term "allele" defines an alternative form of a gene
20 which occupies a given locus on a chromosome.
As commonly known, a "mutation" is a detectable
change in the genetic material which can be transmitted to a daughter
cell. As well known, a mutation can be, for example, a detectable change
in one or more deoxyribonucleotide. For example, nucleotides can be
25 added, deleted, substituted for, inverted, or transposed to a new position.
Spontaneous mutations and experimentally induced mutations exist. The
11229-79.DRF 19 Sep. 1997 - 16h12
CA 0221~870 1997-09-19
14
result of a mutations of nucleic acid molecule is a mutant nucleic acid
molecule. A mutant polypeptide can be encoded from this mutant nucleic
acid molecule.
As used herein, the term "purified" refers to a molecule
5 having been separated from a cellular component. Thus, for example, a
"purified protein" has been purified to a level not found in nature. A
"substantially pure" molecule is a molecule that is lacking in all other
cellular components.
The mutagenesis of the DNA or of the cells is carried out
10 in accordance with well-known methods (Sambrook et al., 1989, supra),
such that the total DNA population or cell population has statistically at
least an insertion mutation in each and every gene of the genome.
Essentially, the one tube collection of mutants obtained by mutagenesis
covers the complete genome. A typical mutagenesis experiment can yield
mutants at frequencies varying from 10,000 clones to more than
1,000,000 clones. Such mutants can be recovered in a single tube.This
mutagenesis scheme is based on the premise that the genome size is
known, that mutagenesis is a random event and that a typical gene has
an average size of 1 kilobase. For example and on a statistical basis, the
20 5.9 Mb Pseudomonas aeruginosa genome would require a minimum of
5,900 mutants to cover the genome at least once. This is herein defined
as a 1 X genome coverage. Thus, a collection of 17,500 mutants (3 X),
29,500 mutants (5 X) or 59,000 mutants (10X) could be utilized for
screening in a typical EGT assay for this particular microorganism. Of
25 course, the person of ordinary skill could also screen more than 10X.
11229-79.DRF 19Sep. 1997-16h12
CA 0221~870 1997-09-19
As used herein, the designation "therapeutic target"
refers to any gene or product thereof that when blocked by known or
novel molecules will affect the growth of the organism coding for the
target.
As used herein, the designation "Non-selective
conditions" refers to in vitro and/or in vivo growth conditions wherein all
the paramaters and factors which are required for optimal growth are
present. Non-limiting examples of such parameters/factors include
growth media nutrients, temperature, pH, cell line, and the like. Under
such conditions, one would expect the organism to be maintained prior
to the mutagenesis step.
As used herein, the designation "Selective conditions"
refers to conditions which are defined by the nature of the experiment
done in vitro and/or in vivo and in which one specific parameter or factor
or set of conditions are modified (in comparison to non-selective
conditions) to determine if essentials genes or gene products can be
identified in that particular condition. A non-limiting example of a
selective condition includes growth at a restrictive temperature.
It will be clear to the person of ordinary skill, that
insertional mutagenesis of an essential gene, within the context of a cell,
will result in the death of that cell. Consequently, the genome of this
particular cell will not be available as a substrate for the amplification
process in accordance with the EGT method of the present invention.
The DNA molecule analysed may be a gene, a fragment
thereof cloned into a vector or preferably a genome.
11229-79.DRF 19 Sep. 1997 - 1 6h12
CA 0221~870 1997-09-19
16
As used herein, the terminology "target region" defines
a DNA region for which preliminary sequence data is sufficiently available
to enable the design of a first primer pair which will, under appropriate
conditions, give rise to a recognizable extension product. The target
region is determined and defined by the available sequence data
available for the particular genome analysed, and by the limits in the
amplification method used. For PCR, for example, the conditions permit
extension products to reach about 2000 nucleotides. The target region
should thus be between about 50 to about 2000 nucleotides. Preferably
between about 200 and about 1000. Since sequence information can be
clustered, some genes might have several target regions. In any event,
the mutagenesis conditions should be adapted so as to enable an
insertional mutagenesis of all targeted regions. In essence, a person of
ordinary skill will adapt the mutagenesis scheme so as to permit
saturation mutagenesis of the DNA to be analysed.
Although in a preferred embodiment, the present
invention is adapted for use with a whole genome, a DNA molecule
inserted into a vector can also be used in accordance with the present
invention. In such an embodiment, the vector should permit an expression
of the DNA molecule in order to permit an ~ssessment of the essentiality
of the gene product. In such a scheme, it will be understood that only
dominant insertional mutation can provoke the lethality since,
presumably, a copy of a wild type or homologous copy of the gene which
is present on the vector, is present in the host cell. Consequently, it will
be clear to the person of ordinary skill that although the present invention
is not limited to haploid genomes, the method of the present invention is
11229-79.DRF 19Sep. 1997-16h12
CA 0221~870 1997-09-19
favorably used in a context of a haploid organism, and more preferably
a haploid microorganism. Organisms in which conversion to
homozygocity is efficient and/or complete are also covered by the scope
of the present invention. In a preferred embodiment therefore, prokaryotic
genomes and lower eukaryotic genomes such as the haploid genomes
of parasites and protista are used. Non-limiting examples of such lower
embryotic genomes include that of tachyzoite form of Toxoplasma gondii,
of Plasmodia, Schistosoma and Leishmania species, as well as those of
fungi such as that of Candida, Aspergillus, Neospora and other disease
causing (in plants, in animals and in humans) relevant fungi are especially
preferred genomes. In addition, all disease causing agents such as
Influenzae, Hl\/, Herpes and other viruses may also be used in the
context of the present invention.
It shall be understood that although the saturation
insertional mutagenesis of the present invention is carried out by a
shotgun approach (without specifically directing the insertion to specific
sequences), a rational design of insertion mutation could also be carried
out, especially with DNA molecules inserted into vectors.
Since the design of the first pair of primers depends on
known sequence data from the genome to be analysed, it follows that
minimum stretches of sequence data must be available in order to enable
the EGT method of the present invention. Preferably, contiguous nucleic
acid sequence data of approximately twelve nucleotides, to approximately
twenty-four nucleotides in the targeted region must be available.
Although in a preferred embodiment, the method of the
present invention relates particularly to genomes of organisms which do
11 229-79.DRF 19 Sep. 1997 - 1 6h12
CA 0221~870 1997-09-19
not contain or contain few introns, the present invention could be adapted
by a person of ordinary skill for intron-containing genomes. Briefly, the
level of mutagenesis would have to be increased in order to enable
saturation to occur. Saccharomyces cerevisiae is one non-limiting
5 example of an organism which contains introns.
Numerous insertional mutagenesis method are known
in the art. It will be clear to the person of ordinary skill that the method
should be adapted to enable the insertion of the sequence which is
complementary to that of a primer binding thereto (generally described
10 herein as primer 3).
The term "saturation mutagenesis" as used herein with
reference to a genome, refers to an insertion mutagenesis in substantially
every gene thereof and/or every target region thereof. Based upon
statistical analysis and well known methods, at least 90%, preferably,
95% and more preferably 100% of the genes and/or target regions will
have been mutagenised. Briefly, to esli,nate the conditions to permit the
aiming of a complete population of mutagenised genes, the statistical
analysis utilised is based on a number of criterions: 1) a completely
random insertion of the insertion element (i.e. a mobile element); 2) an
average size of 1 Kb for a typical gene in a prokaryote genome; 3)
knowledge a priori of the genome size (Megabases). For example, a
complete 1 X coverage of the P. aeruginosa 5.9 Mb genome would
require a minimum of 6000 clones after the mutagenesis experiment.
Preferably, a minimum of 5 X coverage of the genome should be used by
using 60,000 clones. When relating to DNA molecules present on a
11229-79.DRF 19 Sep. 1997 - 16h12
CA 0221~870 1997-09-19
19
vector, saturation mutagenesis refers preferably to the insertion element
being present at every nucleotide position thereof.
Mutational methods include, without being limited
thereto, insertional mutations in which a DNA molecule is inserted without
5 loss of native sequences, or substitutional mutations in which the DNA
molecule inserted replaces native DNA molecule of the targeted region.
It shall be understood that the choice of a particular
insertional element can be adapted to particular needs, provided that it is
absent from the genome which is to be analysed, that it is sufficiently long
10 to permit the generation of a primer which binds thereto (hence the need
for known sequence data of about 12 contiguous nucleotides for the
primer target on the genome, and disrupts the gene or target region it is
inserted into. In a preferred embodiment, the insertional mutagenesis is
provided by a insertional element such as transposons (i.e. Tn5, Tn10,
15 Tn916, Ty). In such cases, the insertional mutagenesis will be carried out
with the insertional elements in accordance with known methods.
Insertional mutagenesis of DNA can also be carried out
by using the integrases protein of retroviruses to mediate the insertion of
a selected primer into a target region. Following amplification, the
20 amplified product or extension product can be detected. In a preferred
embodiment they can be sized-fractionated by gel electrophoresis as well
known in the art. In another embodiment the extension products can be
detected after separation on columns and the like. Hybridization capture
and the triplex DNA technology are non-limiting examples of technologies
25 which could be used to detect the amplified products (Lanbiewicz et al.,
11229-79.DRF 19 Sep. 1997 - 16h12
CA 0221~870 1997-09-19
1997, Nucl. Acids Res.25; 2037-38; and Ito et al.,1992, Proc. Natl, Acad.
Sci 89: 495-8).
A kit for identifying essential genes in a genome
contains at least three oligonucleotide primers, constituting at least two
5 primer pairs, a mutated genome, and solutions for enabling hybridization
between the mutated genome sequences and the oligonucleotide primers
and for enabling amplification of the extension product. Oligonucleotide
primers can be suspended in solution or provided separately in
Iyophilized form. The components of the kit can be packaged together in
10 a common container, the kit typically including an instruction sheet for
carrying out a specific embodiment of the method of the present
invention. Additional optional components of the kit include detection
probes, and means for carrying out a detection step (for example, a probe
or primer is labelled with a detectable marker).
Other objects, advantages and features of the present
invention will become more apparent upon reading of the following non
restrictive description of preferred embodiments thereof, given by way of
example only with reference to the accompanying drawings.
20 BRIEF DESCRIPTION OF THE DRAWINGS
In the appended drawings:
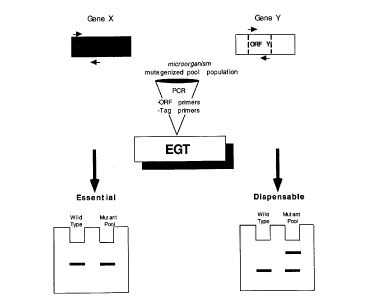
Figure 1 shows a sumll,a, i~ed schematic representation
of the essential gene test (EGT) according to the present invention; and
Figure 2 shows a more detailed view of the EGT shown
25 in Figure 1.
11229-79.DRF 19 Sep. 1997 - 16h12
CA 0221~870 1997-09-19
Other objects, advantages and features of the present
invention will become more apparent upon reading of the following
non-restrictive description of preferred embodiments with reference to the
accompanying drawing which is exemplary and should not be interpreted
5 as limiting the scope of the present invention.
DETAILED DESCRIPTION
Insertional Mutagenesis of the Targeted Genome
First, insertional mutagenesis must be performed so as
10 to cover most if not all genes of a particular genome in a population of
cells. Under these conditions, one would expect the one tube
mutagenized population to cover the spectrum of each and every gene
coded by a particular organism.
Insertional Mutagen
In one embodiment in which a bacterial genome is
targeted, a bacterial population is mutagenized using for example a
mobile element having a high frequency of transposition (Tn5, Tn10,
Tn916, IS elements or any other known mobile genetic element) creating
insertional mutations at diverse sites. Depending on the conditions and
20 mobile element utilized, one may produce a single tube population
containing cells having an insertion in essentially all the genes. Any
particular type of mutagenesis scheme including insertion elements, PCR
mutagenesis, random insertion of DNA by synthetic or biological methods
would be amenable to genetic analysis by the EGT test or assay.
1 1229-79.DRF 19 Sep. 1 997 -16h12
CA 0221~870 1997-09-19
The assay can also be applied to any simple organisms
such as viruses. The EGT has excellent potential in disease causing
viruses from plants, from animals and from humans. Non-limiting
examples include the potato blight virus in plants, the equine encephalitis
5 virus in animals and the cytomegalovirus in humans. Additional examples
include single eukaryotic cells of fungi and of yeasts causing diseases
such as mycoses and include Candida, Cryptococcus, Histoplasma,
Blastomyces, Coccioides, Aspergillus, Fusarium, and Trychophyton, and
the like. Thus, the EGT assay could be applied to all disease causing
10 organisms (See the listing of the Manual of Clinical Microbiology, 1995,
ASM Press). The person of ordinary skill will adapt the EGT accordingly.
For the targeting of the yeast genome the insertional element Ty is a
representative example of an insertional mutagen which can be used in
accordance with the present invention. In addition, the EGT assay can be
15 utilized to dissect metabolic and genetic pathways by assessing
mutagenized populations in different in vitro and in vivo conditions.
Amplification
A sample of the mutagenized population is then
submitted to nucleic acid amplification. In a preferred embodiment, the
20 amplification is carried out by PCR using either cells directly or by
preparing an aliquot of DNA. A collection of two primers specific to the
sequence under investigation (from a genomic database and assumed to
encode an essential or dispensable gene where only part of the ORF is
known) and defining a first primer pair, gives rise to an amplification
25 product of a defined size. A third primer specific to the insertional
mutagen is also used. This three primer assay will give specific
11229-79.DRF 19Sep. 1997-16h12
CA 0221~870 1997-09-19
amplification products defining a sequence as essential or dispensable.
The EGT assay is performed as summarized in Figure 1 using a wild-type
and a mutagenized population. The role of a particular sequence as
essential or dispensable is visu~ ed as the presence (non-essential) or
5 depletion of defined satellite amplification products (essential) (Fig. 1).
A more detailed representation is shown in Fig. 2.
Intcr~,ret~lion of the results of EGT assay
The primer pairs selected from the sequence of interest
defines an amplification product that will be present both in essential
10 genes and in dispensable genes irrespective of the growth conditions
since in the context of a population of cells, individual cells having no
insertions in the targeted sequence of interest will always be present.
Thus, the first primer pair serves as an internal control for the assay
conditions. If the insertion occurs in a dispensable gene, the second
15 primer pair, constituted by a primer specific to the targeted sequence and
one specific to the insertional mutagen, gives rise to a specific extension
product and a series of additional band products. Thus, in addition to the
expected product originating from the first primer pair, additional
amplification products will be visible. The difference in the size of the
20 additional product will reflect the distance between the target region of thethird primer (the insertion "point") and that of the first primer (or second
primer). In contrast, insertion of an element in an essential gene will not
yield an amplification product (lethal phenotype) and the only visualized
amplification product will be generated by the amplification of
25 mutagenized cells containing no insertions in the essential sequence of
interest (originating from the first primer pair).
11229-79.DRF 19Sep. 1997-16h12
CA 0221~870 1997-09-19
24
As alluded to above, the EGT assay enables
automation. For example, by using fluorescent primers (labelled with
distinct fluorochromes) the EGT assay could be used in conjunction with
the ABI GENESCAN.
The following examples are offered by way of illustration
and not by way of limitation.
E~CAMPLE 1
EGT assay on two Pseudomonas aeruginosa genes
The EGT assay was applied to the Pseudomonas
aeruginosa strain PAO1 5.9 Mb genome in the following way. First, a
library of insertion mutants was constructed with the miniTn5 Km insertion
element using standard methods. A collection of 60,000 clones (10 X
genome coverage) obtained were pooled into a single tube.
A first primer pair of 21-mers specific and internal to the
ftsZ gene sequence (ftsZ1 :5'-ATC ACC ATC CCG MC GAG MG-3') and
(ftsZ2:5'-TAT CCA GGT MT CCA GGT CAT-3') give a 669 bps amplified
PCR product.The PCR conditions for DNA amplification were carried out
in accordance with the manufacturer's recommendations (Perkin Elmer
Cetus and Applied Biosystems). In a typical EGT assay, one would
expect the 669 bps to be present irrespective of the mutagenesis or
growth conditions.
The EGT assay was performed for ffsZ by using the
following primers :(KanaputR1: 5'-GCG GCC TCG AGC MG ACG
TTT-3') and (KanaputF4: 5'-TTG GTT GTA ACA CTG GCA GAG-3') in
combination with one and\or the two above-mentioned primers (ftsZ1 and
1 1229-79.DRF 1 9 Sep. 1 997 -1 6h12
CA 0221~870 1997-09-19
ftsZ2). The result of the EGT assay showed a product of 669 bps and no
satellite bands, irrespective of the mutagenesis scheme. Thus, only the
first primer pair gave rise to an extension product. Thus, ftsZ is therefore
defined as an essential gene by the EGT method.
The EGT assay was tested with the ampC gene using
primers (ampcF1: 5'- CAT CGC TTC CAC ACT GCT-3') and (ampcR1:
5'-TGC CGG GM CAC TTG CTG CTC-3') constituting a first primer pair
giving rise to a PCR product of 592 bps irrespective of the mutagenesis.
When used in conjunction with the KanaputR1 and KanaputF1 primers,
a PCR product of 592 bps (positive control) and additional DNA bands
(due to insertions in the ampC gene) could be visualized in the agarose
ethidium bromide stained gel. Thus, the EGT assay would define the
ampC gene as non-essential.
Although the foregoing invention has been described in
some detail by way of illustration and example for purposes of clarity of
understanding, it will be readily apparent to those of ordinary skill in the
art in light of the teachings of this invention that certain changes and
modifications may be made thereto without departing from the spirit or
scope of the appended claims.
11229-79.DRF 19Sep. 1997-16h12