Note: Descriptions are shown in the official language in which they were submitted.
CA 02216308 1997-09-23
FILE, ~ Pi ~
- TG~T ~RA~L~iivN Code: 295-57857
Ref. AHP-97020 PCT
PLASMID VACCINE AGAINST PSEUDORABIES VIRUS
The present invention concerns a plasmid vaccine against the
pseudorabies virus, also known under the Aujeszky's disease virus
(ADV), the porcine herpes 1 virus (PHV-1) or the Suid herpes-1
virus (SHV-1).
Aujeszky's disease is a disease of viral origin to which
most mammals are susceptible. However, humans do not seem to be
susceptible to this disease.
The causative agent of this disease is a coated virus with
two-stranded DNA of the family of the Herpesviridae, subfamily of
the alpha-herpesvirinae [sic; herpesviridae], that is the
pseudorabies virus (PRV).
The genome of the PRV virus is formed by an approximately
150-kb two-strand DNA molecule and it consists of a long unique
region (UL) and a short unique region (Us) which is flanked by
two reverse repeated sequences, a terminal sequence (TR) and an
internal sequence (IR) (T. BEN-PORAT, T. et al., 1979, Virology,
Vol. 95, pp. 285-294).
The genome of the virus of Aujeszky's disease contains at
least 70 genes.
The principal characteristics and biological properties of
the genes of PRV which have been best characterized are listed in
Table I below.
CA 02216308 1997-09-23
Table I. Properties of certain genes of PRV, their
characteristics and their biological properties (except from T.
. Mettenleiter, Acta Veterinaria Hungarica, 42 (2-3), pp. 153-177
(1994))
.
Segment Désignation DésignationProtéine Taille (kDa) Essentielle Fonction / Activité Virulence
duGénome Gène(HSV) Gène,~RV) ' ~ (réplication in
3,) (~) (~) vitro) ~
UL gB gll 913 aa 110-68-55 + pénétration, fusion +
ce!lulaire (~)
gC glll 479 aa 92 - adsorption,~ +
relargage
TK TK 320 aa 35 - thymidinekinase~ +
gH gH 686 aa 84 + pénétration, fusion n.a.
cellulaire ~)
Us prot. kinase PK 336 aa 41 protéine kina~) +
G gX 498 aa 99 - inconnu (j~ -
gD gp50 402 aa 60 + pénétration ~) n.a.
gl gp63 350 aa 63 inconnu (~ +
gE gl 577 aa 110 relargage, (~) +
transmission de
cellule à cellu~
TEG I I K 106 aa I I - protéine du (~) inco~
tegument
Key: 1 Segment of the genome
2 Gene designation (HSV)
3 Gene designation (PRV)
4 Protein
Size (kDa)
6 Essential (replication in vitro)
CA 02216308 1997-09-23
7 Function~activity
8 Penetration, cellular fusion
9 Adsorption, release
Thymidine kinase
11 Protein kinase
12 Unknown
13 Penetration
14 Release, transmission from cell to cell
Integument protein
Usually viruses are transmitted by the oral, or respiratory
route, or by direct contact between an infected animal and a
healthy animal.
The disease has become a concern in pig farms where it
manifests itself in several forms:
1. The adults develop few clinical signs, but they become
permanent sources of infection. Howeve-r, the PRV virus can cause
abortion in pregnant sows.
2. The piglets suffer a severe attack on the central nervous
system. The piglets present a high sensitivity at the birth,
followed by a decrease in sensitivity. Until the age of 10 days,
piglets which have been affected, as they do not benefit from any
passive ;mm~ln;ty from the mother, die within a few hours. Older
piglets are subjected to muscle tremors, contractions, but the
outcome is rather benign.
In other species, the disease can be fatal, and the duration
of incubation is variable, between 15 h and 12 days.
At this time there are several methods of vaccination
against Aujeszky's disease.
One of the standard methods consists in injecting live
viruses, which must be attenuated to prevent the disease from
declaring itself.
CA 02216308 1997-09-23
Thus, for the vaccination of the pigs, PRV viruses from the
Bartha strain are used, which comprise mutations in the genome
coding for the glycoproteins gI, gp63 and gIII and for a protein
of the viral capsid whose gene is in the BamHI-4 fragment-
(T. Mettenleiter, Acta Veterinaria Hungarica, Vol. 42 ~2-3),
pp. 153-177 (1994)).
The vaccines sold under the names of OMNIVAC~-PRV (FERMENTA
ANIMAL HEALTH Co., Kansas City, MO, U.S.) and OMNIMARK~-PRV
(FERMENTA ANIMAL HEALTH Co., Kansas City, MO, U.S.) are also
used, which consist of PRV viruses from the Bucharest strain,
which have been genetically manipulated. These strains comprise
deletions in the genes coding respectively for thymidine kinase
or thymidine kinase and gIII. Naturally, other vaccines against
Aujeszky's disease exist, which work on the same principle.
This vaccination method imparts a good protection to the
vaccinated subject, which is due to the intracellular action of
the live virus. Indeed, the live viruses penetrate the cells
where the viral antigens are synthesized. Then, the peptides
derived from these viral antigens are presented at the surface of
the infected cells in association with the major
histocompatibility complex of class I (MHC I). A cytotoxic
response can thus be triggered, in addition to the humoral
response, resulting in a better protection against the virus in
the vaccinated subject.
Although the viruses are attenuated by mutations, there is a
risk of the viruses recovering their pathogenicity as a result of
spontaneous mutations or recombinations with wild viruses.
The vaccinations with live viral agents can also cause,
under certain conditions, a proliferation of live viruses. This
proliferation of live viruses, even though they are attenuated,
CA 02216308 1997-09-23
constitutes a risk for more susceptible subjects, such as
newborns and pregnant subjects.
Another more recently developed technique consists in using
a nonpathogenic live vector which carries selected genes from the
PRV virus.
M. ELOIT et al. (J.T. van Oirschot (ed.), Vaccination and
control of Aujeszky's Disease, pp. 61-66, ECSC, EEC, EAEC,
Brussels and Luxembourg, 1989) have developed a vaccine based on
recombinant adenovirus type 5 (AD5) which gives rise to the
expression of the gp50 gene of the PRV virus.
W.L. MENGELING et al. (Arch. of Virology, Vol. 134, No. 3-4,
pp. 259-269, 1994) published results of tests with a vaccinia
virus (NYVAC) containing the genes of PRV coding for the
glycoproteins gp50, gII and gIII. However, the efficacy of this
type of virus is limited.
In contrast, M.L. VAN DER LEEK et al. (The Veterinary Record
(1994), Vol. 134, pp. 13-18) have reached encouraging results
with a vaccination of pigs against the PRV virus by scarification
or intramuscular injection of the recombinant of the virus of
porcine variola and of the PRV virus (rSVP-AD). This recombinant
was obtained by insertion of the PRV genes coding for the gp50
and gp63 attached to the P 7.5 promoter of the vaccinia virus in
the gene of the thymidine kinase of the SPV virus.
Naturally, other vaccines exist against Aujeszky's disease,
which work according to the same principle.
A new approach to induce an immunological response was
recently described by ULMER et al., 1993, Sci., Vol. 259,
p. 1745). Mice were immunized by intramuscular injection of a
plasmid comprising the gene of the nucleoprotein of the influenza
virus under the control of a m~mm~lian promoter. This
CA 02216308 1997-09-23
immunization by injection of the plasmid apparently has led to a
transfection of the muscle cells, followed by an in situ
expression of the protein, leading to a specific im~unological
response against the influenza virus, which response is of the
cellular and humoral type and, consequently, it is protected
against an attack by this virus.
It appears, according to the published experiments, that
parts of viral proteins produced inside the transfected cell are
taken on by the major histocompatibility complex of class I and
presented at the surface of the cell.
ROBINSON et al., describe, in 1993 in Vaccine, Vol. 11,
pp. 957-960, an ;mm~ln;zation of chicken against the influenza
virus by intravenous, intraperitoneal and subcutaneous injections
of plasmid. 28-100% of the animals so ;mmlln;zed resist a
challenge with a lethal dose of the virus. It should be noted
that the efficacy strongly depends on the route and of the system
used for the injection and a possible pretreatment of the
injection site (DANKO et al., Vaccine, Vol. 12, pp. 1499-1502
(1994)).
GRAHAM J.M. COX et al. have described a method for the
vaccination of cattle and mice against the BHV-1 virus by
injection of plasmidic DNA, in J. Virol., 1994, pp. 5685-5689. It
has been possible to show that the intramuscular injection of
cattle and mice (in the quadriceps) of plasmidic DNA containing
the gene of the glycoproteins gI, gIII and gIV of BHV-1 triggered
an immune response in the vaccinated animal. The immune response
strongly depends on the quantity of DNA injected, and on the
glycoprotein. Thus, the gene of the protein gIV triggered a
response which was superior to that of the proteins gI and gIII.
CA 02216308 1997-09-23
None of the plasmidic vaccines described to this day is
effective against viral diseases which infect pigs and other
species, such as PRV-caused Aujeszky's disease.
The purpose of the present invention is to propose a
plasmidic vaccine against the PRV virus, which is responsible for
Aujeszky's disease.
This purpose is achieved by a vaccine comprising at least
one plasmid coding for the glycoprotein gIII of the PRV virus or
for a protein presenting the same antigenicity as the
glycoprotein gIII of the PRV virus and a pharmaceutically
acceptable excipient for it.
One of the advantages of this method resides in the fact
that this method is inexpensive.
Indeed, the plasmid can be produced, using techniques which
have been well mastered, in bacteria such as E. coli.
The extraction of plasmid is well known, and a high yield
can be obtained.
It is not necessary for the gene introduced into the plasmid
to code for the entire gIII protein, rather it is sufficient for
it to code for a part or a homolog of the gIII protein of PRV,
which has the same antigenicity as the gIII protein, that is the
same effect on the immunological system as the protein gIII.
Indeed, only a part of the protein gIII's are identified by the
immunological system of the animal; the other parts of the
protein, although they certainly play a role in the life of the
virus, are not essential in the recognition of the protein by the
infected organism.
Another advantage is that it is easy to distinguish the
vaccinated animals from animals infected by the PRV virus.
Indeed, the vaccinated animals develop only antibodies against
CA 02216308 1997-09-23
the protein gIII, whereas the animals infected by the PRV virus
also develop antibodies against other proteins-of the virus.
In addition, the method is reliable; because no
proliferation of live viruses n-eed be feared, there is no
infection by viruses, and consequently, there can be no
proliferation of viruses.
Because the muscle cells have a long life and do not
circulate in the human body, the local and continuous expression
of the antigen at low concentrations can stimulate the long-term
immunological response.
The vaccination with the vaccine according to the present
invention can be used as a diagnostic tool, because it induces
the formation of monospecific antibodies. These antibodies can be
used for the detection of the antigen, for example, in ELISA
tests or other tests.
A surprising effect of the invention resides in the efficacy
of the immllnization against the PRV virus. Although the
importance of the glycoprotein gIII in the immlln;zation is well
known, the injection of the purified protein gIII does not seem
to have any pronounced ;mmlln-zation effect.
Z.H. BISEIBUTSU et al. describe, in Japanese Patent
Application No. JP 05/246888, a vaccine against the PRV virus,
based on the purified glycoprotein gIII and an oil-based
adjuvant. This approach is not used on a commercial scale.
A. MATSUDA TSUCHIDA et al. show in an article published in
J. Vet. Med. Sci., Vol. 54 (3), pp. 447-452, 1992, that a mixture
of glycoproteins gII, gIII and gIV, purified and injected
together with a conventional oil-based adjuvant, imparts a better
protectian to mice against a challenge with virulent PRV viruses
than the glycoproteins injected individually.
CA 02216308 1997-09-23
It remains to be noted that vaccines which use purified
proteins are in general very expensive, because the purification
steps are long and complicated.
According to a first advantageous embodiment, the plasmid is
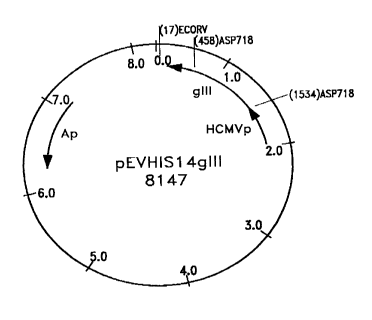
the plasmid pEVhisl4gIII.
The plasmid pEVhisl4gIII has the advantage of comprising a
gene which imparts a resistance to ampicillin, so that the
transformed bacteria, having incorporated the plasmid, can easily
be selected for by adding ampicillin to their growth medium.
Naturally, one can consider using other plasmids. It is
sufficient for the plasmid to contain a gene, which codes for a
protein having the same antigenicity as the glycoprotein gIII of
the PRV virus, inserted in the plasmid so that it is expressed in
the vaccinated organism. A marker such as a gene which imparts
resistance to an antibiotic allows the selection of bacteria
transformed by the plasmid.
To increase the efficacy of the vaccine, the plasmid can
contain, besides the gene coding for the glycoprotein gIII of the
PRV virus or for a protein presenting the same antigenicity as
the glycoprotein gIII of the PRV virus, one or more genes coding
for these cytokines.
Certain cytokines are known to exert an adjuvant activity on
the vaccines. The fact of introducing them into the plasmid so
that they can be expressed in the cells will increase the
efficacy of the vaccine.
According to another advantageous embodiment, the vaccine
also comprises a pharmaceutically acceptable excipient in which
the plasmid is incorporated. The term "pharmaceutically
acceptable excipient," mentioned in this document, refers either
to liquid media or to solid media which can be used as an
CA 02216308 1997-09-23
excipient (vehicle) to introduce the plasmid into the animal to
be vaccinated.
Let us cite, as examples of liquid media, water,
physiological serum, the phosphate-salt buffer, solutions
containing the adjuvants, detergents, stabilizers and substances
which promote the transfection, suspensions of liposomes, of
virosomes, and emulsions.
Let us cite, as examples of solid media, gold microbeads
covered with plasmid, intended to be projected by bombardment
("gene gun") into the tissue of the animals and the
microparticles containing the DNA, which can be used for
administration by the parenteral or oral route.
The vaccination with DNA can optionally be preceded by a
pretreatment of the vaccination site (for example, use of a local
anesthetic) so as to improve its efficacy.
The present invention also proposes a plasmidic vaccine
against the PRV virus which comprises a nucleic acid sequence
coding for the protein gIII of the PRV virus or for a protein
presenting the same antigenicity as the glycoprotein gIII of the
PRV virus or a DNA construct comprising an expression cassette
including:
a) a DNA sequence coding for a polypeptide containing at
least one antigenic determin~nt of the glycoprotein gIII or an
immunogenic factor of the latter, and
b) controlled sequences which are operatively connected to
said coding sequence where said coding sequence can be
transcribed and translated in a cell and where said control
sequences are homologous or heterologous with respect to said
coding sequence.
CA 02216308 1997-09-23
Advantageously, the plasmidic vaccine contains one or more
genes coding for cytokines.
According to another aspect of the present invention, the
proposal is made to use the plasmid pEVhisl4gIII in the
ma'nufacture of a vaccine.
It is also propose to use the plasmid pEVhisl4gIII in the
manufacture of a vaccine against the PRV virus.
The invention is described in detail, as an illustration, in
the following examples.
Example 1: Obtention of a vaccine
The vaccine was obtained in three steps, comprising the
construction of a plasmid (pEVhisl4gIII) containing the gene of
the glycoprotein gIII of the PRV virus, the production of this
plasmid in transformed bacteria and the formulation of the
vaccine comprising the plasmid and the pharmaceutically
acceptable excipient.
Step A: Construction of a plasmid (pEVhisl4gIII) containing the
gene of the glycoprotein gIII of the PRV virus
The DNA of the plasmid pEVhisl4gIII was obtained from the
Institute for Animal and Science and Health (ID DLO, Lelystad,
NL). It comprises the gene of the glycoprotein gIII of the PRV
virus, under the control of the HCMV promoter and the marker gene
of resistance to ampicillin; it is used as a DNA (DNA+) vaccine.
The map of the plasmid is given in Figure 1.
The plasmid was deposited according to the Budapest Treaty
on November 16, 1995, in the collection called "Belgian
CA 02216308 1997-09-23
Coordinated Collections of Microorganisms--Laboratorium voor
Moleculaire Biologie--Plasmiden collectie [Laboratory for
Molecular Biology--Plasmid collection]" (LM3P), University of
Ghent,-K.L. Ledeganckstraat 35 Ghent, Belgium B - 9000 under the
accession number LMBP3377.
Step B: Production of the plasmid in transformed bacteria
Preparation of E. coli cells treated with RbCl
Before being transformed, the E. coli cells, strain DH 5*
(Gibco), were subjected to a treatment with RbCl to increase the
effectiveness of the transformation. The procedure which was
used, with the exception that we used a strain of E. coli DH 5a,
is described in the information bulletin The NEB Transcript,
Vol. 6 (1), p. 7, May, 1994, edited by NEW ENGLAND BIOLABS, Inc.,
Beverly, MA, 01915.
Transformation of E. coli cells treated with RbCl
100 uL of cells treated with RbCl and 1 uL of DNA solution
containing 250 ng of plasmid pEVhisl4gIII were incubated for
10 min on ice (at 0~C) and then for 5 min at 37~C. The mixture
was then transferred into 2 mL of RB buffer (bactopeptone 1%
(wt/vol), 0.5% yeast extract (wt/vol), 1% NaCl (wt/vol)) and
incubated between 30 min and 2 h at 37~C with stirring. The
abbreviation % (wt/vol) represents a percentage expressed in
weight by volume.
100 and 500 uL of the cell culture were spread in a Petri
dish containing
CA 02216308 1997-09-23
RB medium + ampicillin 100 ug/mL + agar 2~ (wt/vol).
Minipreparation of pEVhisl4gIII plasmidic DNA
.
The colonies obtained above were first used to prepare
minipreparations for verifying the production and the structure
of the plasmid pEVhisl4gIII. Individual colonies were cultured
overnight in 2 mL of RB medium + ampicillin 100 ug/mL.
The cultures were then treated as described in Molecular
Cloning, a Laboratory Manual, J. Shambrook, E.F. Fritsch and
T. Maniatis, Cold Spring Harbor Laboratory Press (1989) in the
chapter, Small scale preparations of plasmid DNA, 1.21-1.28,
with the exceptions of steps 1, 4, the centrifugations were
carried out at ambient temperature, and the fact that the
composition of the solution III was modified so as to contain 3M
sodium acetate, at pH 4.8.
The pellets were dried under a vacuum and redissolved in
100-200 ~L of water (filtered using a Milli-Q apparatus,
Millipore, U.S.) without treatment with RNAse.
The analysis by digestion using various restriction enzymes,
followed by electrophoresis on agarose gel has allowed the
verification of the conformity of the plasmid (see also Molecular
Cloning, a Laboratory Manual, J. Shambrook, E.F. Fritsch and
T. Maniatis, Cold Spring Harbor Laboratory Press (1989), 6).
CA 02216308 1997-09-23
14
Maxipreparation of pEVhisl4gIII plasmidic DNA
To obtain plasmidic DNA in a sufficient quantity for the
vaccinations, the plasmidic DNA was produced in a larger quantity
according to the following two protocols.
A suspension of E. coli transformed by the plasmid
pEVhisl4gIII was incubated for 1 h in 2 mL of RB medium and then
distributed in 1-4 flasks of 400 mL of RB medium containing
ampicillin at the concentration of 100 ug/mL. The cultures were
incubated for one night at 37~C with stirring at approximately
150-200 rpm.
The cultures were centrifuged in 500-mL Nalgene tubes for
7 min at 8670 G. Centrifugations were carried out in a Beckman
centrifuge, model J2-21.
The supernatant-was eliminated and the pellet resuspended in
10 mL of solution 1 (glucose 1% (wt/vol); Tris-HCL 25mM; pH =
8.0; EDTA 10mM; lysozyme 1% (wt/vol)), and transferred into a 40-
mL Nalgene tube. The tube was left for 5 min at ambient
temperature. Then 10 mL of solution 2 (NaOH 0.2M; SDS (sodium
dodecyl sulfate) 1% (wt/vol)) were added, the tubes were stirred
and left for 5 min at ambient temperature. 10 mL of solution 3
(sodium acetate 3M; pH = 4.8) were added, and the flasks were
stirred and then centrifuged for 30 min at 48,400 g at 0~C. 25 mL
of the supernatant were transferred into a 50-mL Falcon tube
covered with six layers of gauze. If necessary, the volume can be
adjusted with TE (Tris-HCL 10mM; pH = 8.0; EDTA lmM). Then, 15 mL
of isopropanol at ambient temperature were added, followed by
stirring, and transfer of the mixture into a 40-mL Nalgene tube.
After centrifugation for 15 min at 48,400 G at 0~C, the
supernatant was eliminated. After draining the liquid well, and
CA 02216308 1997-09-23
after dissolution of the pellet in 5 mL of TE, the suspensions
were incubated for 30 min at 37~C with stirring in the presence
of RNase A at a final concentration of 0.1 mg/mL. Then, 10 uL of
proteinase K in solution (10 mg/mL) were added, followed by
incubation for 30 min at minimllm 37~C, and with stirri~g.
The columns TIP 500 QIAGEN (QIAGEN, INC., CA, U S.) were
equilibrated with 10 mL of QBT buffer (NaCl 750mM; MOPS 50mM;
ethanol 15% (vol/vol); Triton X-100, 0.15% (vol/vol); pH = 7.0)
by simple gravity flow. The abbreviation MOPS represents
3-(N-morpholino)propanesulfonic acid. The samples were loaded
onto the columns, and the columns were washed 6 times with
10 mL of QC buffer (NaCl 1000mM; MOPS 50mM; ethanol 15~
(vol/vol); pH = 7.0). After elution with 20 mL of QF buffer (NaCl
1250mM; MOPS 50mM; ethanol 15~ (vol/vol); pH = 8.2), the solution
was recovered in 40-mL Nalgene tubes. 14 mL of isopropanol were
added at ambient temperature. After stirring, the mixture was
centrifuged for 15 min at 48,400 G at 0~C. The supernatant was
eliminated, and the pellet was dried under a vacuum and dissolved
in 500 uL of Milli-Q water. After three extractions of the
supernatant with 500 ,uL of phenol/chloroform/isoamyl alcohol
(25/24/1, vol/vol/vol) and an extraction with one volume of
ether, the DNA was precipitated with 50 ~L of 3M sodium acetate
and 0.7 mL of isopropanol. After centrifugation for 10 min at
18,320 G at 0~C, the pellet was washed with 1 mL of 7Q% ethanol
(vol/vol). The supernatant was eliminated after a 10-min
centrifugation at 18,320 G at 0~C, and the pellet comprising the
plasmidic DNA was dried under a vacuum and dissolved in 500 uL of
Milli-Q water. The abbreviation % (vol/vol) represents the
percentage of volume by volume.
CA 02216308 1997-09-23
Another method for producing plasmidic DNA in large quantity
using PZ523 columns (5 Prime ~ 3 Prime, INC., -Boulder, CO, U.S.)
is used below.
The 400-mL cultures were centrifuged for 10 min at 8670 g in
a 500-mL Nalgene beaker (Beckman J2-21). 10 mL of solution 1
(glucose 1~ (wt/vol); Tris-HCL 25mM, pH = 8, EDTA 10mM; lysozyme
1% (wt/vol) (SIGMA)) were added, and 10 mL of this mixture were
transferred into another 40-mL Nalgene tube. After incubation for
5 min at ambient temperature, 10 mL of a solution 2 (NaOH 0.2M;
SDS 1% (wt/vol)) which was freshly prepared were added. After
slight stirring, the mixture was incubated for 5 min on ice.
After the addition of 10 mL of cold solution 3 (sodium acetate
3M; pH = 4.8), and centrifugation of the tube for 20 min at 0~C
and 48,400 G, the supernatant, approximately 25 mL, was
transferred into a 50-mL Falcon tube, and covered with six layers
of gauze. After the addition of 15 mL of isopropanol, the 40 mL
of sample so obtained were transferred into another 40-mL Nalgene
tube and centrifuged for 20 min at 0~C and 48,400 G. The
supernatant of isopropanol was eliminated, the pellet obtained
was washed with 1 mL of 70% (vol/vol) ethanol, and the liquid was
well drained. The pellet was resuspended in 5 mL of TE to which
50 ~L of RNase A (10 mg/mL solution) were added, and incubated
for 30 min at 37~C with stirring. 10 uL of proteinase K (10 mg/mL
solution) were added, and the mixture was incubated for at least
30 min at 37~C with stirring. For the two successive extractions
with phenol/chloroform/isoamyl alcohol (25/24/1, vol/vol/vol), 5
mL of this solution of phenol were added in a Greiner tube with
threading screw cap, followed by the sample. After slight
stirring for 20-30 sec to mix, the solution was centrifuged for 5
min at 3920 G in a "swinging bucket" rotor. After having
CA 02216308 1997-09-23
transferred the solution into a Nalgene tube, 1 mL of 7.5M
ammonium acetate and 10 mL of absolute ethanol-were added. After
centrifugation for 20 min at 0~C and 48,400 G (Beckman J2-21),
the pellet was washed with 1 mL of 70% ethanol and then dried
under a vacuum and resuspended in 1.8 mL of solution 4.(Tris
1OmM; EDTA lmM; NaCl lM).
After having removed the top stopper, and then the lower
stopper of the column, the column was placed on a collection tube
and then centrifuged for 1 min at 980 G. The collection tube
which collected the equilibration buffer was discarded. The
column was placed on another collection tube and the dissolved
sample (1.8 mL) was loaded the top of the resin. The column was
centrifuged for 12 min at 980 G in a rotor of the "swinging
bucket" type. The plasmidic solution collected was divided into
two Eppendorf tubes to which 600 ~L of isopropanol were added.
After centrifugation for 15 min at 18,320 G (SIGMA 2K15
centrifuge), the pellet was washed with 300 uL of 70% ethanol
(vol/vol) and dried under a vacuum. The pellet comprising the
plasmidic DNA was redissolved in 500 uL of Milli-Q water
(Millipore, U.S.) and stored cold until the vaccination.
As far as the preparation of the DNA plasmid is concerned,
approximately 11 mg were prepared according to the method using
the PZ523 columns and approximately 16 mg using the Qiagen
columns. These two preparations were mixed and used for the
described examples.
CA 02216308 1997-09-23
18
Step C: Formulation of the vaccine comprising the plasmid and a
pharmaceutically acceptable excipient
The plasmidic DNA pellet having been resuspended in water,
the DNA concentration was determined by loading agar gel and
development with ethidium bromide (see Molecular Cloning, a
Laboratory Manual, J. Shambrook, E.F. Fritsch and T. Maniatis,
Cold Spring Harbor Laboratory Press (1989), 6 and Appendix E,
and Winnacker, E. L., "From Genes to Clones," VCH (1987),
2.1.2.2). The DNA concentration was adjusted to 0.3-1 ug of DNA
per ~L of water.
Example 2: Use of the plasmidic vaccine in mice to stimulate the
induction of an antibody response and a cytotoxic T-cell response
The experience performed on mice to prove the efficacy of
the plasmidic vaccine requires the construction of a control
plasmid derived from the plasmid pEVhisl4gIII, prior to the
immlln;zation of the animals and the analysis of the immune
response.
Step 1: Construction of a plasmid derived from the plasmid
pEVhisl4gIII by the deletion of the sequence coding fo.r the gene
gIII
A plasmid derived from the plasmid pEVhisl4gIII by deletion
of the sequence coding for the gene of the glycoprotein gIII was
used as a negative control (pEVhisl4gIII~, DNA-). This deleted
plasmid was obtained as follows: the plasmid pEVhisl4gIII was
CA 02216308 1997-09-23
digested using the restriction enzymes Asp 718 and EcoRV. The DNA
was then treated with the T4 DNA polymerase to obtain fragment
with blunt ends. The DNA so obtained was ligated with the T4 DNA
ligase (see "Molecular Cloning, a Laboratory Manual," J.
Shambrook, E. F. Fritsch, and T. Maniatis, Cold Spring.Harbor
Laboratory Press (1989), 1.53, 1.73).
With regard to the preparation of the vaccine against the
deleted plasmid, the same steps were used as those described for
the DNA+ plasmid, except that during the production of the
plasmid pEVhisl4gIII by the maxipreparation, only the PZ523
columns were used.
Step 2: Immunization of mice
Five groups of 6-10 female mice from the consanguineous
strain Balb/c with ages from 16 to 18 weeks at the first
injection were used.
The mice must be consanguineous to measure the responses of
such toxic T cells (CTL), because the major histocompatibility
complex (MHC) between the cytotoxic T cells and the target
cells--3T3 Swiss albino cells (fibroblasts) (haplotype H-2D)--
must be guaranteed. The target cells were cultivated in a 10% DME
medium (vol/vol) in fetal calf serum.
The plasmidic DNA of the plasmid pEVhisl4gIII, containing
the gene of the glycoprotein gIII of the PRV virus, was used as
positive DNA (DNA'), whereas the equivalent plasmid, without the
gene gIII (DNA), was used as the negative control.
100 ~g of DNA were intramuscularly injected in each mouse,
in the left and right upper hind quarters in two portions
CA 02216308 1997-09-23
containing 50-150 uL of aqueous solution, according to the
following immunization protocol.
Group I comprising 10 mice labeled by a color code was
vaccinated four times, in week 0 (DNA1+), in week 3 (DNA2), in
week 5 (DNA3+), and in week 10 (DNA4+) with DNA+. Serum ~DNA3+ was
removed 2 days before the last injection; serum sDNA4+ was
removed 6 days after the first collection, that is, 5 days after
the last DNA4' injection.
In week 11, spleen cells were collected and restimulated in
vitro. The CTL tests were carried out 4 days later.
Group 2, also consisting of 10 mice labeled with a color
code, was vaccinated three times, in week 0 (DNA1+), in week 3
(DNA2+), and in week 5 (DNA3+), with DNA+. Serum sDNA2+ was
collected 2 days before the last injection and sDNA3+ serum was
collected in week 9, that is, 4 weeks after the-last DNA3+
injection.
During week 9, the spleen cells were restimulated in vitro.
The CTL test were carried out 6 days later.
Group 3, consisting of 8 mice labeled by a color code, was
vaccinated only two times--in week 0 (DNAl); and week 3 (DNA2).
Serum sDNA2+ was removed 2 weeks after the last injection and in
week 9.
During week 9, the spleen cells were restimulated in vitro.
The CTL tests were carried out 6 days later.
Group 4, or the control group, consisting of 10 mice labeled
with a color code, was vaccinated three times with DNA~--in week
0 with 200 ug of DNA (DNA1-), in week 2 with 100 ug (DNA2-), and
in week 7 with 100 ~g of DNA- (DNA3-). Serum sDNA2~ was collected
2 days before the last injection and serum sDNA3~ was collected
in week 8, that is, 1 week after the last DNA3- injection.
CA 02216308 1997-09-23
During week 8, the spleen cells were collected and
restimulated in vitro. The CTL tests were carried out 4 days
later.
Group 5 (positive control group), consisting of 6 mice, was
vaccinated three times with live virus, strain NIA3 M207
(obtained from the Institute for Animal Science and Health,
ID-DLO, NL), at a dose of 107 PE'U (Plaque Forming Units) per
mouse and per injection in the instep, in week 0, in
week 16, and in week 17.
Serum was collected 2 days after the last injection. A
mixture of serum originating from 5 animals was used for the
analyses.
During week 18, the spleen cells were removed and
restimulated in vitro. The CTL tests were carried out 4 days
later.
Step 3: Analysis of the humoral and cellular immune response (CTL
test)
Depending on the case, the animals were euthanized; the
spleen was removed under aseptic conditions between 7 days and up
to six weeks after the last DNA injection.
Part A - In vitro culturing and restimulation of the effectors
The vaccinated mice and the control mice were euthanized by
cervical dislocation and rinsed with alcohol (70% vol/vol). The
spleens of these animals were deposited in a Petri dish
containing PBS (Gibco) and they were crushed in these dishes by
means of a piece of nylon gauze and a curved plastic tube. The
CA 02216308 1997-09-23
elimination of the aggregates and of the conjunctive tissue
surrounding the spleen was done by filtration ~through the nylon
gauze) of the crushed material obtained. After a centrifugation
at 220 G-for 4 min, the pellet was recovered and the erythrocytes
were lysed by the addition of 4 mL/spleen of sterile ACK solution
(0.15M NH4Cl, lmM KHCO3, 0.1mM NaEDTA, pH = 7.2-7.4).
Two washings were then performed with sterile effector
medium (compound of DME medium (Dulbecco's modified Eagle,
Gibco), completed with 10% (vol/vol) of fetal calf serum (Gibco),
1% (vol/vol) of 200mM L-glutamine (Gibco), and 1% (vol/vol) of
the penicillin-streptomycin antibiotic solution (10,000 U/mL of
penicillin and 10,000 ,ug/mL of streptomycin (Gibco), along with
lOmM of HEPES buffer (pH = 7.4) (Sigma), 2 x 10-~ of 2-
mercaptoethanol (Gibco), and 2mM of sodium pyruvate (Merck)). The
cells were resuspended at a concentration 5 x 106 cells per mL in
this sterile medium. The spleen cells were distributed for
culturing in vitro in 25-cm2 culture flasks (Falcon) at the rate
of 25 x 106 cells/flask. A part of these cells was restimulated
in vitro by the addition of the viral strains cited above with an
infection multiplicity (MOI) equal to 2; the others were used as
nonrestimulated controls. These flasks were vertically deposited
in an incubator for 4-7 days (at 37~C, 3% (vol/vol) of CO2, and a
humidity of more than 90% of saturation level), as described
above.
Part B - The CTL test
The cells of the histocompatible line (fibroblasts) 3T3-
Swiss albino (haplotype H-2D) were used as targets.
CA 02216308 1997-09-23
.
23
The CTL test comprises several steps:
a) The target cells were either infected or not infected by
a strain of the Aujeszky virus (NIA3 M207) at an MOI equal to 10,
with the infected cells being denoted as CV+ and the noninfected
cells denoted as CV~. Ninety minutes later, the labeling of the
target cells in suspension starts (see below--Labeling of the
suspended target cells (3T3) with "Eu").
b) With regard to the effectors cultured in vitro for 4-7
days (as described above) earlier, they were dissolved in culture
flasks, washed 2 times with the effector medium and counted to be
resuspended at a concentration of 5 x 106 cells/mL. Six hours
after the start of the infection of the target cells, they were
brought in contact with the effectors (which include Tc
lymphocytes). In a plate with 96 wells with round bottoms, 5000
target cells in 50 uL of effector medium (labeled with Eu, and
either infected or not infected) were deposited on, respectively,
500,000, 250,000, 125,000, 62,500, 31,250, and 15,625 effectors
in 100 uL (that is, at effector/target ratios from 100/1 to 3/1).
Repetitions (3 or 4 times) were carried out for each condition.
The plate was centrifuged at +50 G for 4 min and kept at 37~C for
4 h. The evaluation of the quantity of target cells lysed by the
Tc lymphocytes was made by collecting the supernatant after a new
centrifugation for 4 min at +50 G. The supernatant was placed
into a plate with 96 wells with flat bottoms in 200 uL of a
fluorescence amplifying solution (DELFIA~ Enhancement solution,
Pharmacia, Sweden) in the case of labeling with Eu. The
fluorimeter count (delayed-time fluorimeter, 1234 DELFIA
Recherche, Wallac) was carried out +12 h, later taking care to
CA 02216308 1997-09-23
24
place the plates containing the mixture in darkness and at
ambient temperature.
The quantification of the specific lysis (in %) was
estimated by means of the following formula: -
(Experimental release - background noise)/(Maximum release -
background noise) x 100 = specific lysis (~)
Part C - Labeling of the suspended target cells with Eu
This type of labeling is applied both for cells that develop
in suspension and for adhering cells.
Culture flasks with maximum confluence were used for the
different labelings of the 3T3 target cells.
The growth medium-was removed from the culture flask
containing the adhering 3T3 target cells; the flask was washed
one time with PBS (Gibco). 5 mL of trypsin-EDTA (Gibco) at 37~C
were deposited in the flask on the cell lawn. After 1 min, the
cells were detached by small abrupt knocks against the flask.
Once the detachment was completed, 7 mL of sterile effector
solution (described above) were added. The cells were washed one
time with a solution consisting of HEPES buffer (50mM HEPES ((2-
hydroxyethyl)-4-piperazinyl-1,2-ethanesulfonic) acid (pH = 7.4),
(Sigma); 93mM NaCl (Merck); 5mM KCl (Merck); 2mM MgCl2 6H20
(Merck)) and resuspended at a concentration of 6 x 106 cells/min
in said solution. The live cell count was performed with Trypan
Blue (0.4% (wt/vol) solution, Sigma).
1 mL of this solution was completed with:
- 750 uL of the HEPES buffer solution (pH = 7.4),
CA 02216308 1997-09-23
- 200 ~L of the solution of Eu-DTPA (1.52 mL of the standard
solution of Eu (1000 ug/mL in 1% (vol/vol) of nitric acid)
(Aldrich); 8 mL of the solution of the HEPES buffer (pH 7.4); and
0;5 mL of DTPA diethylenetriaminepentaacetic acid (Merck) at 3.93
g in 100 mL of the HEPES buffer solution); after 2 min.it was
completed with 100 uL of dextran sulfate (50 mg of dextran
sulfate, M.W. = 500 kd, Pharmacia in 10 mL of the HEPES buffer
solution).
Thirty minutes were required for the labeling at ambient
temperature. During this labeling, the tubes were slightly
stirred every 10 min. Afterwards, 7 mL of repair buffer (0.588 g
of CaCl2 2H20 (Merck), with 1.8 g of glucose (Merck) in 1 L of
HEPES buffer solution, pH 7.4), 3 mL of effector medium (see
above), and 12 uL of DNAase at 17,000 units/mL (Boehringer
Mannheim) were added. A pause of 8 min was observed. A washing
with the effector medium was carried out, followed by a Ficoll-
Paque gauze (Ficoll and sodium diatrizoate, Pharmacia LKB). For
this purpose, the cells were resuspended in 5 mL of effector
medium in a 50-mL tube (Falcon) and 5 mL of Ficoll-Paque were
deposited at the bottom.
After 15 min of centrifugation at 800 G and 20~C, the top
part of the solution in the tubej up to and including the
interface, was recovered.
Any traces of Ficoll-Paque were removed from the cell by
washing with the effector medium. After counting, the cells were
resuspended at a concentration of 105 cells/mL.
For the evaluation of the labeling, 5000 target cells were
deposited in a plate with 96 wells with round bottoms in the
presence of 100 uL of effector medium to determine the quantity
of the background noise of the Eu.
CA 022l6308 l997-09-23
26
The.same quantity of cells was deposited in 100 ,uL of Triton
X-100 (1% v/v, Merck), for the maximum release of Eu. After a
centrifugation of 4 min at 50 G, 20 ,uL of supernatant were
collected and deposited in 200 ,uL of a fluorescence amplifying
solution (see above); the plate with 96 wells with flat bottoms
was passed through the fluorimeter (1234 DELFIA Recherche,
Wallac) after a 1-h incubation in darkness to obtain a stable
reading.
CA 02216308 1997-09-23
Part D --Results of the CTL tests
Table II. Comparison of the specific lysis of the splènocytes
before and after passage through a Ficoll-Paque gradient
Groupes/in vitro/cibles f', Lyse spécifique (en %)
~éviation standard (en %)
(3) Rapport E/C ~
100/1 ¦ 50/1 ~1 ~ 12/1 l 6/1 l 3/1 l n
Groupe 5
SB++/MOI 2/CV+ 37,6 22,9 16,1 8,4 3,8 -0,5 4AvantFicoll i 10,0 ~ 5,8 i 4,7 i 2,2 i 1,2 i 0,2
SB++/MOI2/CV+ 42,2 35,5 20,3 17,0 9,1 11,4 4
Après Ficoll i 16,4 i 12,2 i 6,4 i6,9 i 3,3 ~4,5
Groupe I
ADN4+/V-/CV- 7,5 6,1 2,8 1,2 -0,7 -0,9 4
Avant Ficoll i 1,6 i 1,1 i 0,4 i 0,3 i 0,1 i 0,2
ADN4+/V-/CV 9,2 9,1 5,5 2,7 1,9 1,2 4
AprèsFicoll i 1,6 i 1,5 iO,6 i 0,5 iO,2 iO,2
ADN4+/V-/CV+ 1 1,9 9,3 1,8 -2,1 -2,7 -3,6 4
AvantFicoll i4,0 i3,5 +0,6 +0,6 iO,8 i 1,0
ADN4+/V/CV+ 31,8 49,4 45,6 20,6 19,8 10,7 4
AprèsFicoll i 14,2 i 20,3 l 23,8 i 9,0 i 9,0 i 4,7
ADN4+/M0I 2/CV- -0,3 -0,7 -1,0 -0,6 -1,3 -1,2 4
AvantFicoll iO,l iO,l iO,2 iO,l iO,2 iO,2 .
ADN4+/M0I 2/CV- -2,0 -1,6 -1,2 -2,7 -1,1 -1,3 4
Après Ficoll i 0,3 i 0,2 io,2 i 0,4 i 0,1 ~ 0,2
ADN4+/M0I 2/CV+ -5,3 -6,2 -5,4 -5,I 4 1 -3,3 4
Avant Ficoll i 1~6 i 1~6 i 1,4 i 1,5 i 1,1 io,g
ADN4+/M0I 2/CV+ 15,4 12,8 19,1 15,3 7,6 22,9 4
AprèsFicoll i 6,6 i 5,8 ~ 8,6 i 7,2 i2,9 i 13,3
Groupe 4
ADN3 -/MOI 2/CV+ -5, 5 -5 ,2 -3 ,9 -3 ,3 -3 ,9 -3 ,7 4
Avant Ficoll i 1,7 i 1,5 i 1,2 i 1,1 i 1,2 i 1,3
ADN3-/M01 2/CV+ -10,3 -5,8 -7,8 -2,4 -3,5 6,5 4
Après Ficoll i 4,2 i 2,1 i 3,4 i 1,1 i 1~3 i 2~8
CA 02216308 1997-09-23
28
Key: 1 Groups/in vitro/targets
2 Specific lysis (in %) + standard deviation (in %)
3 E/C ratio
4 Group 5
SB++/MOI 2/CV+
Before Ficoll
6 SB++/MOI 2/CV+
After Ficoll
7 Group 1
8 DNA4+/V-/CV-
Before Ficoll
9 DNA4+/V-/CV-
After Ficoll
DNA4+/V-/CV+
Before Ficoll
11 DN~+/V-/CV+
After Ficoll
12 DNA4+/MOI 2/CV-
Before Ficoll
13 DNA4+/MOI 2/CV-
After Ficoll
14 DNA4+/MOI 2/CV+
Before Ficoll
DNA4+/MOI 2/CV+
After Ficoll
16 Group 4
17 DNA3-/MOI 2/CV+
Before Ficoll
18 DNA3-/MOI 2/CV+
After Ficoll
CA 02216308 1997-09-23
29
Explanatory notes concerning the in vitro restimulation
treatments and the preparation of the targets:
V~: spleen cells cultured in vitro without virus
MOI 2: spleen cells cultured in vitro and restimulated by
the addition of live virus (NIA3 M207) with an
infection multiplicity (MOI) equal to 2.
CV~: noninfected target cells, labeled with Europium
CV+: target cells infected with the virus NIA3 M207 at
a MOI equal to 10, and labeled with Europium
SB++: group 5, positive control vaccinated with live
virus
DNA4+: group 1, animals injected 4 times with DNA+
DNA3-: group 4, animals injected 3 times with DNA-
n: number of repetitions
The lysis of the noninfected target cells occurred between 0
and 9%, and it was not affected by the passage through the
Ficoll-Paque gradient. In contrast, the positive control (group
5), both before and after the Ficoll treatment, presented a
significantly positive cell-lysis rate. The lysis percentage of
infected cells was increased by the passage through Ficoll-Paque
for the animals immunized with DNA.
The CTL test of group 1, injected 4 times with DNA+ (DNA4+),
showed a lytic activity.
The CTL test of group 2, injected 3 times with DNA+ (DNA3+)
did not show any lytic activity.
The CTL test of group 3, injected 2 times with DNA (DNA2)
did not show any lytic activity.
CA 02216308 1997-09-23
The CTL test of group 4, injected 3 times with DNA- (DNA3-)
did not show any lytic activity.
Naturally, other frequencies and other intervals can be
considered, as well as other dosages and routes of immunization.
.
Part E - Analysis of the humoral immunological response
The serum was collected two times during the experiment,
with the last collection occurring just before the sacrifice of
the animals to obtain the spleen cells for the CTL test, and the
antibody responses were measured by:
- a test of seroneutralization of the PRV virus (SN)
- an immunoenzymatic test (ELISA) for measuring, in the
mouse serum, antibodies directed against an extract of all the
PRV glycoproteins.
The seroneutralization test of the PRV virus was carried out
according to the protocol indicated below.
PDs cells (SOLVAY-DUPHAR, NL) and viruses of the Bartha K61
strain (SOLVAY-DUPHAR, NL) were used. The experiment was carried
out in plates with 96 microwells with flat bottoms from Greiner
(France).
The medium in which the SN test was carried out had the
following composition: 340 mL of essential Eagle minim~l medium
(flow), 100 mL of hydrolysate of lactalbumin 2.5% (wt/vol),
5-10 mL of NaHCO3 at 5.6% (wt/vol), and 50 mL of fetal calf serum
(Gibco).
The serum to be tested was subjected to in a 2-by-2 series
dilution in the medium, with the dilutions ranging from 1:2 to
1:4096 (50 ~uL of serum + 50 uL of medium, each time), in the
CA 02216308 1997-09-23
plate with 96 wells with flat bottom (Greiner). Each sample was
tested in duplicate.
The virus was diluted to 100 TCIDso (tissue culture
infectious dose at 50%) in 0.05 mL of medium, with 50 ~L of this
diluted virus solution being added to each well. The serum/virus
mixture was incubated for 24 h at 37~C. 50 uL of the PD5 cell
suspension, at a concentration of 4 x 105 cell/mL, were added to
each serum/virus sample. The plates were then incubated at 37~C
for 5 days.
The results were observed under microscope, then the titers
were calculated by taking the inverse of the dilution that
corresponds to 50% of the limit dilution.
The virus was controlled by incubating a diluted virus
sample for 24 h at 4~C and another virus sample for 24 h at 37~C.
The two virus suspensions were diluted (vol/vol) with the SN test
medium (see above) at 1:10j 1:100, and 1:1000. Subsequently, 0.05
mL of each dilution of the virus suspensions was added per well
using 8 wells for each dilution, then 0.05 mL of medium and 0.05
mL of cell suspension were added. The interpretation of the
results under the microscope took place after an incubation of 5
days at 37~C.
The humoral responses measured using the SN test--after two,
three, and four injections of DNA--showed values of zero or
weakly positive values.
It should be noted that after immunization with live virus
at high doses, measurable but relatively slow titers were
observed in the seroneutralization test. These observations were
confirmed by the literature.
The protocol of the ELISA test is described by M. ELOIT et
al. in ARCH. virol. (1992), Vol. 123, pp. 135-143.
CA 02216308 1997-09-23
In addition to the serum originating from the treated animal
groups as described under "Immunization of the mice," 2
additional sera were included in the analysis--a positive serum
(serum +) and a negative serum (serum -). The serum - originates
from noninjected mice (OF1 mice, 3 weeks old ). A mixture of
serum from 10 mice was prepared. The serum + is a mixture of
serum originating from 10 OF1 mice having received, after 3
weeks, 109 TCID (tissue culture infectious dose) of recombinant
adenovirus expressing the gene gD by intramuscular injection,
with collection/sampling 3 weeks later.
Tables III and IV, reproduced below, show the optical
density (OD) as a function of the dilutions of the serum. The
samples were first tested at a dilution of 1/10 (Table III--test
1) to identify the positive examples, that is, the samples for
which the OD was greater than the optical density of the serum of
the negative control (OD x 100 = 509). The positive samples so
identified were tested a second time at variable dilutions
(Table IV).
In the serum of the animals that had received an injection
of plasmidic DNA without the gene gIII (DNA - pEVhisl4gIII~), no
antibodies directed against the glycoprotein gIII were found. It
has been shown that after 4 injections of DNA+ plasmids, at
intervals of 2 weeks or longer, the mice showed a good humoral
response against the virus. Seven animals out of 9 tested showed
anti-gIII antibodies after four injections.
Even after three injections of DNA', the sera of 9 animals
out of 10 showed measurable titers of anti-gIII antibodies.
Similarly, after 2 injections of DNA+, the sera of 7 animals out
of 8 showed positive responses in the ELISA test.
CA 02216308 1997-09-23
Table III. ELISA test: Optical density (OD) as a function of the
dilutions of the serum--identification of the positive examples
~ OD(xl00)
n~desouns ~ dilution1/10
( ~ ) Témoinpositif(NIA3 M207)(~) mélange 2000
SAI~N2- 1+2+3 552
4+5+6 545
7+8+9+10 395
r~
)sAI~N3- 1+2+3 587
4+5+6 679
7+8+9+10 697
SA~DN3+ 1 2000
2 2000
- 3 .2000
4 511
1263
6 2000
7 2000
CA 02216308 1997-09-23
34
~ 2000
9 . 2000
0 2000
SA~DN4~ 2 2000
3 2000
4 874
759
6 2000
7 2000
8 2000
9 2000
_ . .
2000
~\
(~ sérum- (~ melange 509
~'
sémm + (~) mélange 2000
CA 02216308 1997-09-23
Explanatory notes:
. .
sDNA2: serum originating from animals injected~ 2 times
with DNA-
sDNA3~: serum originating from animals injected 3 times
with DNA
sDNA3+: serum originating from animals injected 3 times
with DNA+
sDNA4+: serum originating from animals injected 4 times
with DNA+
Positive control (NIA3 M207): serum originating from
animals injected with
live virus NIA3 M207
serum -: serum originating from noninjected AnimAl5
serum +: serum originating from animals injected with
adenovirus expressing the gene gD
Key: 1 Mouse No.
2 OD (x 100) 1/10 dilution
3 Positive control
4 Mixture
sDNA2
6 SDNA3
7 SDAN3+
8 sDNA~~
9 serum.~ _
mixture
11 serum +
CA 02216308 1997-09-23
Table IV.. ELISA test--optical density (OD) as a function of the
dllutions of the serum test 2
.. ' ~,). .
.(~) groupe sourls N~dll. 1/100 1/3001/900 1/2700
témoin positif (~)moyenne 2000 20002000 2000
(~) (M207)
(~ sADN2+ 3 1087 599 287 171
4 1378 622 303 192
1506 866 405 261
6 673 261 152 136
7 1676 913 353 205
8 1731 837 355 229
9 1710 778 274 167
Négatif àdil 1/10
~)2 et 10 ~ non testé
(~j sADN3+ 1 2000 IC29 329 142
2 2000 2000 879 296
3 2000 2000 2000 2000
CA 02216308 1997-09-23
S 197 123 89 .83
6 2000 1 704 528 244
7 2000 2000 1084 435
8 2000 809 188 122
9 2000 1 50 1 285 1 69
0 2000 2000 722 263
4( ~) Négatif àdil 1/10
sAI~N4+2 2000 2000 684 262
3 2000 2000 2000 2000
4 263 138 98 103
143 110 96 104
6 2000 1641 566 256.
7 2000 2000 1 122 419
8 2000 1097 350 143
9 2000 1739 462 211
0 2000 2000 549 249
(~) non testé
sé~m - ~élange 155 96 83 92
sé~m +~élange 827 305 146 106
CA 02216308 1997-09-23
Explanatory notes:
sDNA2: serum originating from animals injected 2 times
with DNA-
sDNA3: serum originating from animals injected 3 times
with DNA
sDNA3+: serum originating from animals injected 3 times
with DNA+
sDNA4: serum originating from animals injected 4 times
with DNA+
Positive control (NIA3 M207): serum originating from
animals injected with
live virus NIA3 M207
serum -: serum originating from noninjected animals
serum +: serum originating from animals injected with the
adenovirus expressing the gene gD
Key: 1 Group
2 Mouse No.
3 Positive control (M207)
4 Mean
sDNA2
6 Negative at 1/10 dilution
7 2 and 10
8 Not tested
9 sDNA3+
Negative at 1/10 dilution
11 sDNA4+
13 Mixture
14 serum -
serum +
CA 022l6308 l997-09-23
39
Example 3: Induction of a protection in mice against a challenge
inoculation with virulent viruses
Part A - Immunization protocol
Five groups of ten female mice (Charles River, Germany) aged
16 weeks and of the consanguineous strain Balb c were used.
For the group G0, the negative control, only a PBS buffer
(Gibco) was used.
The group G1 (the positive control) was vaccinated using an
attenuated virus (strain NIA3 M207) by the intraperitoneal route.
10' PFU (plaque forming units) were used for each mouse.
This is a very high dose compared to the doses used for the
vaccination of pigs with the attenuated vaccination strains
traditionally used at the dose of 105'5 PFU.
The three other groups received the plasmidic DNA obtained
according to the method described in Example 1.
The vaccination with plasmidic DNA was carried out by the
intramuscular injection of 100 ug in 2 x 100 luL of water/mouse in
the left and right hindquarters for several consecutive days.
The group C was vaccinated two times, on Friday and Monday,
respectively.
The group B received four consecutive injections,
distributed from Wednesday to Monday.
The group A received six doses distributed from Monday to
the next Monday.
The animals were housed in cages, separated by groups, and
the individuals were labeled with a blue felt tip pen.
The serum collected from each animal was coded so as to be
able to track each animal individually, as far as the antibody
CA 02216308 1997-09-23
dosage and the protection conferred by the vaccinations are
concerned.
Part B - Analysis of the immune responses
The development of the humoral responses was controlled
according to the ELISA protocol presented in Example 2.
The test of serum virus neutralization was carried out
according to the method described in Example 2.
Serum samples were collected at the beginning of the
immunization period, that is 3 days after the last injection of
vaccine, at the end of the im~llnization period, that is one month
after the last injection of the vaccine, and just before the test
inoculation with virulent virus that took place 9 days later.
Finally, one month after the challenge, the serum was again
sampled.
Part C - Challenge inoculation
Approximately 37 days after the last vaccination, all the
mice were exposed to infection with a live virulent virus of the
NIA3 strain, in the amount of 7000 PFU in 200 mL per ~nim~l,
injected peritoneally. The dosage is very high because the 50%
lethal dose for the animals is approximately 100 times lower
(LDso = 70 PFU).
The animals were observed, and the deaths were recorded
during the 15 days after the challenge, as indicated in Table V.
The results show the variations of the survival rate
expressed in percentages as a function of the days after the
challenge.
CA 022l6308 l997-09-23
41
All the animals of the negative control group were dead
after the challenge.
Table V. Monitoring of the survival rate of mice after a
challenge inoculation with virulent viruses
Groupes de r~
souris (Dix ~a~x de surv~(en %) en fonction des jours ~rès l'épr~,ve
par groupe) /3) (~ ) (7) C~J
4 jo~ 5 jours 6 jours 7 jours 10 lours 15 jours
G0 0 0 0 0 0 0
Gl 100 80 80 80 80 80
(~ ADN+ 50 20 0 0 0 0
2x lOOmg
ADN+ 80 40 30 30 30 30
4x lOOmg
ADN+ 90 60 60 60 60 60
6x lOOmg
Key: 1 Groups of mice (ten per group)
2 Survival rate (in %) as a function of the days after
the challenge
3 Days
4 5 days
6 days
6 7 days
7 10 days
8 15 days
9 DNA+
2 x 100 mg
DNA+
4 x 100 mg
11 DNA+
6 x 100 mg
CA 02216308 1997-09-23
The results show that a protection is given, even at the
lowest vaccination dose, that is, 2 x 100 ug. Indeed, the death
of these animals was delayed in comparison to the negàtive
control group (G0). -
At a dosage of 4 x 100 ~g, 30% of the animals surYived,whereas at a dosage of 6 x 100 ~g, 60% of the animals survived.
The results show that the protection induced by this new
plasmidic vaccination method is effective, especially if one
compares it to the survival rate of 80% observed in the G1 group
(positive control), that is, the group of animals vaccinated with
the attenuated virus at a very high dose (Io7 PFU).
It remains to be noted that the vaccination method used in
this example can be further optimized. According to the results
of the cytotoxicity test against the PRV virus, the vaccination
by plasmidic injection for several consecutive days did not
induce any CTL response, whereas the injection of the same
quantity of DNA at intervals of 3 weeks or longer, as used in
Example 2, induced a pronounced CTL response. In addition, the
induced humoral response, measured by the response of the anti-
gIII antibodies in the ELISA test, was weaker due to the
plasmidic injections over several consecutive days than the
humoral response induced by injections of the same quantity of
DNA at 3-week intervals, described in Example 2.
Example 4: Induction of a protection in mice against a challenge
inoculation with virulent viruses
For these experiments, the immunization protocol of
Example 3 was used, with the only difference being that plasmidic
CA 02216308 1997-09-23
43
DNA injections were carried out every three weeks, and the age of
the mice was 19 weeks.
For the group G0 (negative control), only PBS buffer ~Gibco)
was used.
The group G1 (positive control) was vaccinated using the
attenuated virus (strain NIA3 M207) in the instep with a dose of
107 PFU per mouse. Four injections of plasmidic DNA were
administered by the intramuscular route in the two hindquarters
of the mice groups, each consisting of ten animals, labeled with
a color code.
Serum was collected either on the same day, or within three
days prior to the next immunization.
Three weeks after the last injection of plasmid and six
weeks after the immunizations of the control groups, all the mice
were exposed to the virulent virus NIA3, using a dose of 7000
PFU/animal, and peritoneal injections. The deaths were recorded
during 15 days after the challenge; they are listed in Table VI.
The results show the survival rates expressed in percentages as a
function of the days after the challenge.
Table VI. Monitoring of the survival rate of the mice after a
challenge inoculation with virulent viruses
Groupes de ,~ux de survie (en %) en fonction des jours après l'épreuve
souris (Dix _J
par groupe)
4 lours 5 l~urs 6 jours 7 lours l~ours l~ours
G0 40 0 0 0 0 0
Gl 100 90 90 80 80 80
(~)ADN+ 80 70 60 50 40 40
4 x 100 ~L~
CA 02216308 1997-09-23
44
Key: 1 Groups of mice (ten per group)
2 Survival rate (in %) as a function of the days after
the challenge
3 4 days
4 5 days
6 days
6 7 days
7 10 days
8 15 days
9 DNA+
4 X 100 ,ug
All the animals of the G0 group (negative control) were dead
after the challenge. A survival rate of 80~ was observed in the
group G1, that is, the animals vaccinated with the attenuated
virus at a very high dose.
40% of the animals survived a dosage of 4 x 100 mg of
plasmidic DNA. The immunization method used for the vaccination
of the animals with plasmidic DNA (four injections) every three
weeks was shown to be more effective in comparison to daily
injections (better survival rate of 40% instead 30%, and a
relative delay before the death of the animals).
Example 5: Induction of a humoral response in pigs
Three groups of pigs, each consisting of 3 animals aged 5
weeks, were used. The animals originated from a PRV-free farm and
they were part of the same litter. They were individually labeled
with an earring.
One group of 3 animals (Nos. 1, 2, and 3) that were not
vaccinated was used as the negative control.
CA 02216308 1997-09-23
For the 2 other groups, three intramuscular injections of
the plasmid pEVhisl4gIII were administered at the ages of 5, 7,
and 9 weeks.
Three animals (Nos. 4, 5, and 6) received a dose of 75 ~g of
plasmid, whereas 3 other pigs (Nos. 7, 8, and 9) received 560 ug
of plasmid at each injection. The dose of DNA was diluted in 4 mL
of PBS buffer (Gibco, U.S.) and administered in 4 portions of 1
mL at 4 inoculation sites: on the 2 sides of the neck and in the
center of the left and right hindquarters. The injection was
carried out using a syringe with a Terumo needle having a length
of 40mM and an opening of 0.9mM.
The serum was collected at the time of each injection and 2
weeks after the last injection. The analysis of the humoral
responses (antibody against the protein gIII) before and during
the immunization was carried out by a seroneutralization test
(test sensitive to mediation by the complement, see Bitsch and
Eskilsen, Curr. Top. Vet. Med. Animi. Sci., 12, 41, 49, 1982) and
by an IPMA test (Immuno Peroxidase Monolayer Assay).
The IPMA test was carried out according to the protocol
listed below. To each well of 96-well plates (Corning, U.S.)
covered with a confluent lawn of SK 6 cells (ATCC, U.S.), 500
TCID50Of PRV virus of strain 89V87 (Nauwynck H., Pensaert M., Am.
J. Vet. Research, 53 (4) 489 (1992)) were added in MEM medium
(Gibco, U.S.). As soon as a cythopathic effect was obs~erved, the
plates were thermofixed: after a washing with a PBS buffer, the
plates were dried at 37~C until evaporation of the liquid. They
were then incubated for 1 h at 80~C.
The serum to be tested, sub]ected to 2-by-2 series dilution
in PBS, was distributed into the wells, then the plates were
incubated for 1 h at 37~C.
CA 02216308 1997-09-23
46
The serum was removed, then the plates were subjected to 2
washings with PBS, followed by a 1-h incubation in the presence
of anti-porcine antibodies labeled with peroxidase (Nordic,
Holland) and diluted 100 times in PBS. A-fter 1 h, the plates were
incubated in the presence of 3-amino-9-ethylcarbazole substrate
(2 mg of AEC in 10 mL of sodium acetate buffer (0.05M at pH 5)
and 75 uL of H202 at 30%).
The appearance of a red coloration was observed under an
optical microscope. The reaction was interrupted after 15 min
with 3 washings of tap water. The IPMA titers were calculated
using the inverse of the strongest dilution that produces a red
coloration of foci of viral antigen on the infected cells.
The results of the seroneutralization test and of the IPMA
test are listed in Table VII.
CA 02216308 1997-09-23
47
Table VII. Titers of antibodies against the PRV virus, determined
by the seroneutralization test and by the IPMA test
Ti~res en anticorps en fonction de l'âge
âge
N~ Dose de ~ 5 semaines 7 semaines 9 semaines l l semaines
du plasmide ~ (~
porc pEVhis l 4gIII
~3 administrée
SN IPMA SN IPMA SN IPMA SN IPMA
l Omg <2 <5 <2 <5 <2 <5 <2 <5
2 Omg <2 <S <2 <5 ~2 <5 <2 <5
3 Omg C2 <5 - <2 <5 <2 <5 <2 <5
4 (75mg) <2 <5 <2 <5 4 <5 6 128
(75mg) <2 <5 <2 <5 <2 <5 <2 <5
6 (75mg) <2 <5 <2 <5 C2 <5 4 128
7 (560mg) <2 <5 <2 <5 <2 <5 <2 <5
8 (560mg) <2 <5 <2 <5 <2 <5 <2 <5
9 (560mg) <2 <5 <2 <s <2 <5 3 128
Key: 1 Antibody titers as a function of age
2 Age
3 Pig No.
4 Dose of plasmid pEVhisl4gIII administered
5 weeks
6 7 weeks
7 9 weeks
8 11 weeks
CA 02216308 1997-09-23
48
Explanatory notes:
SN: titer of seroneutralizing antibodies
IPMA: titer of antibodies determined by the IPMA method
The results show that none of the nonvaccinated animals
developed antibodies against the PRV virus. The results indicate
that even at the lowest vaccination dose, that is, 3 x 75 ug, a
humoral response was induced in 2 pigs out of 3. Only 1 animal
out of 3 reacted at a dosage of 560 ug. The responses were weak,
and the differences between the two immunized groups were not
significant.
Example 6: Induction of a protection against a challenge with a
virulent virus in pigs
The protocol of Example 5 was repeated exactly.
Three groups of 3 pigs aged 5 weeks were used. The animals
originated from a PRV-free farm and they belonged to the same
litter. They were individually labeled by an earring.
A group of 3 animals that were not vaccinated was used as
negative control.
For the 2 other groups, three intramuscular injections of
the plasmid pEVhisl4gIII were carried out at the ages of 5, 7,
and 9 weeks, according to the technique described in Example 5.
Three animals (pig Nos. 4, 5, and 6) received a dose of 75 ug of
plasmid, whereas the three other animals (pig Nos. 7, 8, and 9)
received a dose of 560 ug of plasmid.
CA 02216308 1997-09-23
The challenge with virulent virus was carried out according
to the method described in Vaccine, 1994, 12 (7), pp. 661-665,
and it is described below.
27 weeks later, that is, 18 weeks after the last injection
of plasmid, all the animals were transferred into an isolation
unit for exposure to virulent PRV virus of strain 75V19 (Andries,
K., Pensaert, M. B., Vandeputte, J., Am. J. Vet. Res., 1978, Vol.
39, pp. 1282-1285).
The temperature of the isolation unit was kept at 18~C, and
the ventilation at 0.2 m/sec.
105~ TCID50 of virus PRV (strain 75V19) suspended in 5 mL of
phosphate buffer were administered to all the animals, 2 mL of
this suspension were administered orally and 1.5 mL of this
suspension were instilled into each nostril.
All the animals were observed daily during the two weeks
following the challenge. The temperature of the animal bodies was
recorded, as well as the animal weights. The relative weight gain
(RDWG) was calcùlated according to the formula indicated below,
for the comparison of the performances of the three groups.
RDWG from the day of the challenge to day x:
weight at day x weight on the day of the challenge
RDWG = ----~~~~~~~~-~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
weight on the day of the challenge
The nasal secretions of all the animals were collected with
a swab every two days for 14 days after the challenge. The weight
of the nasal secretions collected was recorded. The collected
nasal secretions was suspended in a phosphate buffer. The decimal
[1/10] dilutions of these suspensions were inoculated into
monolayer cell cultures, where the cells originated from a
CA 02216308 1997-09-23
continuous cell line of porcine testicles (ST). The presence of a
cytolytic effect for these cell cultures was then checked for
five days. The lethal dose 50 [LD50] was calculated using the
method of Reed and Muench (Amer. J. Hyg., 1938, Vol. 27, pp. 493-
497). The viral titers were expressed in TCIDso per gra~ of nasal
secretion.
The results of the serological examinations are combined in
Table VIII.
Table VIII. Titers of antibodies against the PRV virus determined
by the seroneutralization test (SN) and the IPMA test
N- Do~c da (~) 'ritrcs ~n anlicorps cn ~OIICtiOII dc l'age
~Ic pl~s~ lc
p(lrc pl'Vllis 14gUI
~dmil)islrcc
5 scm~incs7 scll-aines9 semaines 11 scmaincs27 scmaines 27semztlncs+ 29~1nes
(Ircv:lccil~Dtio~l)(2cv~lccillaliol1)(3evttccin~ticn) (eprcllve) 5 jours (14 jours~prcs
(~ )(5 jo~lr prcs I éprcuvc)
SN iPi~SA SN iPMA SN iPMA SN iPMA SN SN SN
Co- Sc- Co- Sc' Co' Se- Co- Se- Co' Sc' Co' Sc' Co'i Sc-
0,~1g <2 <2 <5 a <2 <5 a a <5 a a <5 a q a q ~G 238J
2 () p5 <2 <2 <5 <2 <2 <5 a a <5 a <2 <5 a a a <2 ~ CJ 238J
3 0 us c2 a <5 <2 c2 ~5 a a <5 <2 <2 <5 q a <2 a 128 238~
4 75,ug <2 c2 <2 <2 <5 a 4 <5 a 6 128 3 8 3 8 238-1 238J
75,u5 q <2 <2 <2 <S a <2 <5 a <2 <5 a a a a 128 238J
. fi 75,ug c2 <2 <2 <2 <5 a a <5 a ~ 128 6 24 J 32 238J 238J
7 560 llg <2 a <5 <2 <2 <5 a a <5 a <2 <5 a a a 2 128 238J
X 560 llg <2 <2 <5 <2 <2 <5 a a <5 a <2 ~5 J 21 ~ ~8 I'J2 238J
') 56() IIG <2 <2 <5 ~2 a <5 a a <5 a 3 128 a 8 a 2J 1~2 238J
Co* conventionncl~)
Se~ sensible
CA 02216308 1997-09-23
Key: 1 Pig No.
2 Dose of plasmid pEVhisl4gIII administered
3 Antibody titers as a function of the age
4 5 weeks (first vaccination)
- 5 7 weeks (second vaccination)
6 9 weeks (third vaccination)
7 11 weeks
8 27 weeks (challenge)
9 27 weeks + 5 days (5 days after the challenge)
29 weeks (14 days after the challenge)
11 Co* conventional
Sc* sensitive
The analysis of the humoral responses was carried out by a
sensitive seroneutralization test (test sensitive to the
mediation by the complement according to the technique described
by Bitsch and Eskilsen, Curr. Top. Vet. Anim. Sci., 1982, Vol.
12, pp. 41-49) and by a conventional seroneutralization test
(according to the technique described by Andries, K., Pensaert,
M. B., Vandeputte, J., Am. J. Vet. Res., 1978, Vol. 39,
pp. 1282-1285).
These results show that a serological response was found for
only one of the six animals vaccinated at the time of the last
vaccination. Two and eighteen weeks later, titers of
seroneutralizing antibodies between 3 and 24 were found,
respectively, for three of the animals (Pig Nos. 4, 6, and 9) and
for four of the animals (Pig Nos. 4, 6, 8, and 9).
14 days after the challenge, the titers of seroneutralizing
antibodies were observed to be between 64 and 128 for the animals
that belonged to the negative control group, between 128 and >384
for the animals that received a dose of 75 ~g of plasmid, and
CA 02216308 1997-09-23
between 128 and 192 for the animals that received a dose of 560
,ug of plasmid.
All the animals were listless and anorexic. They sneezed
from the third day after the challenge to the eighth or ninth day
after the challenge. Vomiting and nervous disorders were observed
in two animals (Pig Nos. 3 and 5).
A11 the animals, with the exception of one (pig No. 8), had
a fever (>40~C) from the third day to the seventh day after the
challenge. The mean maximum temperature was 41.3~C for the
negative control group and 41~C for the two other groups.
The results concerning the changes in the animal weight are
collected in Ta~le IX.
Table IX. Changes in the weights of the animals
(~) GroupeN~ de po~ ~ G7 ~ G14 E~DWG 7 RDWG 14
Contrôle 1 -5,4 2,8 -0,861 0,223
(~) négatif 2 -- -5,6 3,4 -0,845 0,256
3 -5 -10,7 -1,26 -1,352
"~ 75~g 4 -0,4 9,7 -0,092 1,110
vacciné 5 -5,4 -8,1 -1,467 -1,099
6 -3,3 4,3 -0,630 0,41 1
C~ 560,ug 7 0,8 7,7 0,178 0,857
vacciné 8 -1,1 7 -0,185 0;587
9 -3,5 7,6 -0,661 0,718
~)~ G7 = poids 7 jours après l'épreuve - poids le jour de l'épreuve
~ G14 = poids 14 jours après l'épreuve - poids le jour de l'épreuve
RDWG7 = RDWG 7 jours après l'épreuve
RDWG14 = RDWG 14 jours après l'épreuve
CA 02216308 1997-09-23
Key: 1 Group
2 Pig No.
3 Negative control -
4 75 ug
Vaccinated
5 560 ug
Vaccinated
6 ~ G7 = weight 7 days after the challenge - weight on
the day of the challenge
G14 = weight 14 days after the challenge - weight on
the day of the challenge
RDWG7 = RDWG 7 days after the challenge
RDWG14 = RDWG 14 days after the challenge
All the animals, except for one (pig No. 8), lost weight
during the first week after the challenge.
Fourteen days after the challenge, only two animals (pig
Nos. 3 and 5) had not regained weight to return to their pre-
challenge weight. Moreover, they continued to lose weight. The
three animals having received a dose of 560 ug of plasmid, and
one animal (pig No. 4) having received a dose of 75 ug of
plasmid, showed a compensatory weight gain between the seventh
and fourteenth day after the challenge, so that they had returned
to their initial growth curve between the eleventh and fourteenth
day after the challenge. The two groups of vaccinated animals
presented a significantly positive average weight gain, whereas
the control group, nonvaccinated animals, presented a negative
mean weight gain value (= weight loss).
The results of the virus titrations in the nasal secretions
are collected in Table X.
CA 02216308 1997-09-23
54
Table X. Secretion of virus after the challenge
pe N~ de (~) Sécrétion de virus expnmé en
~) porc (loglOTCID 50) en fonction du nombre de jou~s après
(~) I'épreuve
~jours après l'épreuve
02 4 6 8 101214
Contrôle I < 1,53,25,55,52,0 < 1,51,7 < 1,5
(~) négatif 2 S l,S < 1,55,07,53,02,32,0 < 1,5
3 < 1,52,76,77,34,54,54,3 < 1,5
75 !lg 4 < 1,52,06,57,31,7 s 1,5 < l,S c l,S
vacciné 5 < 1,54,76,33,72,0 < 1,5 < 1,5 < 1,5
6 < 1,5 S 1,56,77,5 < 1,5 < 1,5 S 1,5 < 1,5
56011g 7 < 1,52,76,38,04,5 < 1,5 < 1,5 < 1,5
(~) vacciné 8 < 1,5 < 1,57,37,0 < 1,5 < 1,5 < 1,5 < 1,5
9 < 1,55,36,36,3 < 1,5 < 1,5 < 1,5 < 1,5
Key: 1 Group
2 Pig No.
3 Secretion of virus expressed in (loglO TCID50) as a
function of the number of days after the challenge
4 Days after the challenge
Negative control
6 75 ,ug
vaccinated
7 560 ~g
vaccinated
The first viruses were isolated from nasal secretions two
days after the challenge, for two of the three animals in each
. CA 022l6308 l997-09-23
~ . .
group. The viral titers reached a maximum level between the
fourth and sixth day after the challenge, with-the viral titers
being between 105-5 and 108~ TCIDso~ The viral secretion stopped
between the sixth and eighth day after the challenge f~r the
vaccinated animals, whereas it continued until the twelfth day
after the challenge for the animals of the negative control
group.
Claims
\ 1. Vc~i~ ~n~L--L~il-ly d ~ld~lLLid ~ll~dillilly d ~n~ coding for
the g ~ oprotein gIII of the PRV virus, or for the protein
presentin~the same antigenicity as the glycoprotein gIII of the
PRV virus, a ~ a pharmaceutically acceptable excipient for said
vacclne.
2. Vaccine ac~rding to Claim 1, characterized in that the
plasmid also contains ~ "promoter obtained from human
cytomegalovirus."
3. Vaccine according t ~Claim 1, characterized in that the
plasmid used is the pEVhisl4gI ~ plasmid.
4. Vaccine according to Clai ~ 1-3, characterized in that
the plasmid also contains at least o ~ gene, or a portion of a
gene, coding for at least one cytokine ~ a fragment of a
cytokine. ~
5. Vaccine according to any one of Claim ~ -4, characterized
in that the pharmaceutically acceptable excipien~omprises
minibeads made of or coated with vaccine, which are ~ troduced
into the tissue of the animal to be vaccinated. ~
6. Use of a plasmid for a vaccine against the PRV vi ~s,
wi~n sala pla~l--ia Col~L~Llsi~ly d llU~ d ~ '' ly i~l