Note: Descriptions are shown in the official language in which they were submitted.
CA 02216~9 1997-09-2~
G2 CHECKPOINT INHIBITORS AND ASSAY
Field of Invention
This invention relates to substances which inhibit
arrest of the cell cycle during the G2 phase and which are
useful for sensitizing cancer cells to the effect of DNA
damaging agents. This invention also relates to an assay
for such substances.
Background
Normal cells respond to DNA damage in one of two ways,
depending upon their type and the degree of damage. The
cells may activate an apoptotic pathway leading to suicide
of the cell and its removal or, survival of a damaged cell
may be promoted by activating checkpoints that temporarily
halt the normal cycle of growth and division to allow time
for DNA repair. The checkpoints operate during the G1
phase of the cycle so that DNA is repaired before it is
replicated in the S phase; and, during the G2 phase so that
DNA is repaired before chromosomes are segregated in the
mitosis phase (M).
Few G2 checkpoint inhibitors are known. Two groups
have been found: the purine analogues (caffeine,
pentoxifylline, 2-aminopurine, 6-dimethylaminopurine), and
staurosporine with its derivative UCN-01
(7-hydroxystaurosporine). In addition, the protein
phosphatase inhibitors (okadaic acid and fostriecin) can
act as G2 checkpoint inhibitors but they also induce
premature mitosis in the absence of DNA damage and have
tumour-promoting activity.
CA 02216~9 1997-09-2~
Experiments employing cells deficient in the tumour
suppressor protein p53 have demonstrated the value of the
two groups of G2 checkpoint inhibitors described above, for
selectively sensitizing cancer cells. Pentoxifylline has
been shown to enhance cisplatin induced killing of p53~ MCF-
7 cells 30-fold and radiation induced killing of p53~ A549
human lung adenocarcinoma cells 5-fold. Caffeine has been
shown to enhance radiation induced killing of p53~ mouse
embryonic fibroblasts 3-fold and radiation induced killing
of p53~ A549 human lung adenocarcinoma cells 5-fold. UCN-01
has been shown to enhance cisplatin induced killing of p53~
MCF-7 cells 25-fold. UCN-01 is active in vitro as a
G2 checkpoint inhibitor in the submicromolar range.
Over 50~ of human cancers exhibit a loss of function
of the protein p53. Cells with mutated p53 are unable to
activate the G1 checkpoint in response to DNA damage.
However, the G2 checkpoint (although usually weaker than in
normal cells) still provides an opportunity to repair the
DNA damage before cell division. Inhibition of the
G2 checkpoint alone would have little effect on normal or
cancer cells. However, G2 checkpoint inhibitors used in
combination with a DNA damaging agent would increase the
killing of cancer cells which cannot activate the
G1 checkpoint.
The known G2 checkpoint inhibitors have been found
serendipitously. An assay suitable for high throughput
screening of compounds for G2 checkpoint inhibition
activity would be desirable.
CA 02216~9 1997-09-2
Summary of The Invention
In one aspect, this invention provides G2 checkpoint
inhibitor compounds having the formula of Compound I as
defined herein, including pharmaceutically acceptable salts
thereof.
In another aspect, this invention provides the use of
compounds having the formula of Compound I to inhibit the
G2 checkpoint, including; release of cells that are
arrested at the G2 checkpoint thereby permitting the cells
to proceed to mitosis; and, to prevent G2 checkpoint arrest
in cells (for example, in response to DNA damage). This
invention also provides the use of compounds of the formula
Compound I to sensitize cancer cells to the effects of DNA
damaging agents and the use of such compounds in the
formulation of agents, including medicaments, for
potentiating the effect of DNA damaging agents on cancer
cells. This invention also provides the use of compounds
having the formula of Compound I to increase the killing of
cancer cells by DNA damaging agents.
This invention provides a method of increasing the
killing of cancer cells having G1 checkpoint deficiency as
a result of DNA damage, comprising the steps of
administering a DNA damaging agent to said cancer cells
thereby damaging DNA of said cells, and administering a
G2 checkpoint inhibitor compound to said cells, wherein the
G2 inhibitor compound is a compound or a pharmaceutically
acceptable salt thereof having the formula of Compound I.
CA 02216~9 1997-09-2~
In a further aspect, this invention provides an assay
for G2 checkpoint inhibitors. This invention provides a
method for determining whether a sample contains a G2
checkpoint inhibitor, comprising the steps of:
(a) applying a DNA damaging agent to p53 deficient
cells which will arrest at G2 in response to the
DNA damaging agent;
(b) applying a sample to be tested and a microtubule
depolymerizing agent to said cells;
(c) culturing said cells for a time sufficient for
the cells to arrest at G2 in response to the DNA
damaging agent; and
(d) subsequent to step (c), lysing said cells to
provide an extract, and
(e) applying a mitotic cell specific antibody to said
extract and measuring antibody binding of said
antibody to epitopes in the extract.
In the assay method, the sample may be applied at
about the same time as the DNA damaging agent, or applied
after the cells are arrested at G2. The measurement of
antibody binding in step (e) may be compared to a
measurement of antibody binding with cells arrested at G2,
CA 02216~9 1997-09-2~
or the measurement of antibody binding in step (e) may be
compared to a measurement of antibody binding with cells in
a method wherein in step (b), a known G2 checkpoint
inhibitor is applied to the cells.
In the assay, the antibody may be a monoclonal
antibody specific for phospho-epitopes. The monoclonal
antibody may be TG-3 and in step (e) of the method, TG-3
not bound to epitopes in the extract may be removed and an
antibody capable of binding to TG-3 is applied with binding
of TG-3 to said epitopes being measured by detecting
production of a product of a reaction catalyzed by an
enzyme linked to the TG-3 binding antibody.
Throughout this specification, the term "G2" or
G2 phase" means the phase of the cell cycle between the end
of DNA synthesis and the beginning of mitosis. A cell in
G2 has an interphase nucleus as determined by microscopy,
and duplicated DNA (usually determined by flow cytometry).
Throughout this specification, the term "G2 checkpoint
inhibitor" means a substance which is capable of releasing
a cell from arrest in G2 phase brought about by DNA damage,
preventing a cell from arrest in G2 phase in response to
DNA damage, or both.
Throughout this specification, the term "DNA damaging
agent" means any substance or treatment which induces DNA
damage in a cell, including W irradiation, gamma
irradiation, X-rays, alkylating agents, antibiotics that
induce DNA damage by binding to DNA, inhibitors of
topoisomerase and any compound used in chemotherapy which
CA 02216~9 1997-09-2~
acts by causing DNA damage. Examples of specific compounds
are cisplatin, VM-26, and procarbazine.
Brief Description of the Drawinqs
In Fig. 1, Figs lA-E show flow cytometry analysis at
various times following DNA damage, with caffeine (Fig. lD)
and UCN-01 (Fig. lE) as G2 checkpoint inhibitors.
Figs. 2 and 3 are graphs showing the mitotic index of
irradiated cells treated with or without caffeine or UCN-01
plotted against time and drug concentration.
Fig. 4 is a graph plotting the linear relationship
between mitotic index and the optical measurements from an
ELISA assay as described herein.
Fig. 5 is a histogram showing the mitotic index of
cells subjected to gamma irradiation (~) or VM-26 treated
with nocodazole as a control (-), nocodazole plus
debromohymenialdisine (H), and nocodazole plus
caffeine (C).
Fig. 6 is a graph showing potentiation of cytotoxicity
of cisplatin by UCN-01 and debromohymenialdisine.
Detailed Description
The assay of this invention involves treating cancer
cells with a DNA damaging agent under conditions whereby
arrest of the cells at G2 will be induced. A sample to be
tested for G2 checkpoint inhibition activity and an agent
which arrests cells in mitosis are applied to the cells,
following which it is determined whether the G2 checkpoint
CA 02216~9 1997-09-2~
is overcome and the cells enter mitosis. Release of the
cells from arrest at G2 or prevention of arrest at G2, such
that the cells proceed to mitosis, is the indicator of
G2 checkpoint inhibition activity.
The assay of this invention requires the use of a
cancer cell culture in which the cells are incapable of
arrest at the G1 checkpoint in response to DNA damage. The
cells may be from any of the numerous cancer cell lines for
which it is known that the cells are incapable of arrest at
G1. Such cells include cells which are p53 deficient,
including: any cell line with a natural mutation or
deletion in the p53 gene which renders p53 inactive; any
cell line expressing a viral oncogene which disrupts p53;
any cell line expressing dominant-negative mutant p53 (some
mutant p53 proteins inhibit wild-type protein such as in
the p53~ MCF-7 cells used in the examples herein); and any
primary or established cell line derived from p53 "negative
transgenic" animals. Specific examples of suitable cancer
cells are the following: cancer cells which have
incorporated the human papillomavirus type-16 E6 gene, cell
lines which are mutant for p53 function including Burkitt's
human lymphoma cell line CA46 and the human colon carcinoma
~ell line HT-29 (CA46 and HT-29 are available from the
American Type Culture Collection). Cell lines with a
genetic deficiency which disrupts the G1 checkpoint other
than by disruption of p53, may also be employed in this
assay.
CA 02216~9 1997-09-2~
Once a cell line incapable of G1 arrest is chosen,
conditions for arresting a majority of the cells at G2 in
response to a DNA damaging agent are optimized by
determining appropriate culture conditions, incubation
time, and type and dosage of DNA damaging agent.
Preferably, at least 50~ of the cells in a culture will be
arrested at G2 in response to the DNA damaging agent.
Maximizing the proportion of cells in the population which
are arrested at G2 will reduce the background signal.
Once the conditions for G2 arrest in the cell culture
are established, the assay may be carried out in one of two
ways. First, the cells may be arrested at G2 in response
to the DNA damaging agent and then treated with the sample
to determine whether there is release from G2 arrest or,
the cells may be treated with the sample prior to the time
when the majority of the cells would be expected to be
arrested at G2 and determine whether the cells are
prevented from G2 arrest.
Release from G2 arrest or prevention of G2 arrest is
detected by a quantitative determination of the cells which
proceed to mitosis. The assay culture is treated with a
agent which will arrest such cells in mitosis. Such agents
include microtubule depolymerizing agents that arrest cells
in metaphase, such as nocodazole. This will increase the
number of mitotic cells in the sample which will be
detected.
In this assay, determination of the cells which
proceed to mitosis is carried out using any of the known
CA 02216~9 1997-09-2~
immunological methods by employing antibodies which have
specificity for mitotic cells. Monoclonal antibodies
demonstrating such specificity are known and include MPM-2
which was raised against mitotic HeLa cells and recognizes
phospho-epitopes that are highly conserved in mitotic
proteins of all eukaryotic species. Other examples are the
monoclonal antibodies recognizing phospho-epitopes in the
paired helical filament proteins (PHF) found in brain
tissue of patients suffering from Alzheimer's disease as
described in: PCT International Application published
July 4, 1996 under No. WO 96/20218; and, Vincent, I. et al.
(1996) "Mitotic Mechanisms in Alzheimer's Disease?" The
Journal of Cell Biology, 132:413-425. The examples in this
specification make use of the antibody TG-3 described in
the latter two references, which may be obtained from
Albert Einstein College of Medicine of Yeshiva University,
Bronx, New York.
TG-3 has a high specificity for mitotic cells. When
TG-3 is used in the ELISA assay described herein, a very
good signal/noise ratio is achieved permitting the ELISA
assay to be easily monitored by optical absorbance.
Dying cells exhibit some characteristics which are
similar to mitotic cells. TG-3 doe5 not rea~t with dying
cells which prevents apoptotic cells from being counted in
the assay of this invention. The efficiency of the assay
is not affected by the presence of dying cells.
Immunological methods useful for determination of
mitotic cells in this assay include any method for
CA 02216~9 1997-09-2~
- 10
determining antibody-antigen binding, including:
immunocytochemistry (eg. immunofluorescence), flow
cytometry, immunoblotting, and ELISA. Several
immunological methods are described in detail in the
examples herein as well as in Vincent I. et al. [supra] .
Other immunological procedures not described herein are
well-known in the art and may be readily adapted for use in
this assay. However, high throughput testing of samples
may best be achieved by use of ELISA.
The assay of this invention has been used to screen
crude extracts from approximately 1,000 marine organisms.
As described in more detail in the examples herein,
extracts from a marine sponge tested positively in the
assay and analysis of the extracts resulted in the
identification of a family of compounds having G2
checkpoint inhibition activity. Two such inhibitor
compounds obtained from the sponge: hymenialdisine and
debromohymenialdisine, have been previously isolated from
sponge tissue and are known to have some cytotoxic activity
and activity as alpha-adrenoceptor blockers. These
compounds are now shown to be potent inhibitors of the G2
checkpoint and may be used to selectively sensitize cancer
cells to the effect of DNA damaging agents thereby
potentiating cytotoxity of DNA damaging agents. The
compounds of this invention may be administered in
conjunction with DNA damaging agents to increase killing of
cancer cells which are deficient in the G1 checkpoint.
CA 02216559 1997-09-25
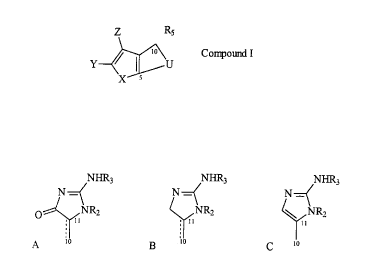
The G2 checkpoint inhibitor compounds of this
invention have the formula:
z Rs
Y~U Compound I
X
wherein R5 is selected ~om the grou~ co~
NHR3 NHR3 NHR3
N~( N~ N~=(
o~NR2 ~NR2 ~NR2
A ~ B '~ C '~
in which U is selected ~om the group CO
W'
D 5 1 I N--(CH2)n ~~
R
W
E 5 1I N--CH=CH--~~
Rl
F 5 CH=N--CH= ~~
W
G S 1I N=CH--CH= lo
CA 022l6~9 l997-09-2
- 12 -
and wherein:
X = NR4 (R4 = H, or alkyl including straight chain,
branched and cyclic aliphatic, optionally substituted
with alkyl, alkene, aklyne, hydroxyl, carbonyl, aryl,
amine, halogen, nitro, cyano, thio or sulfate) O, or
S;
Y = H, F, Cl, Br, or I;
Z = H, F, Cl, Br, or I;
W = O, or H2;
R1 and R2 independently = H, or alkyl (as defined for
R4);
R3 = H, alkyl (as defined for R4), or acyl (including
aroyl); and
n = 2 O.
This invention includes the E and Z configurations at
the R5 linkage when that linkage is a double bond. The
Z configuration is preferred.
Preferably, the alkyl and acyl substituents of the
compounds of this invention will be from one to ten carbon
2 0 atoms and n = 0, 1, or 2 .
More preferably, n = 1, the alkyl substituents will be
methyl, ethyl or benzyl with benzyl being optionally
substituted with OH, halogen, nitro, carboxylic acid,
carboxaldehyde, methyl, ethyl, methoxy, sulfonyl or cyano;
25 the acyl substituents will be from two to four carbon atoms
or benzoyl; and Y and Z will be H, Cl or Br.
When X = NH, Y = H or Br, Z = H, R5 = A
(Z configuration), W = O, R1 = H, R2 = H, R3 = H, and n = 1
CA 02216~9 1997-09-2~
in Compound I, the compound is hymenialdisine (Y = Br) or,
debromohymenialdisine (Y = H).
While the examples in this specification describe
purification of compounds from a natural source, the
compounds of this invention can be synthesized by routine
modification of the known methodology for total synthesis
of hymenialdisine and debromohymenialdisine such as that
described in: H. Annoura and T. Tatsuoka (1995)
Tetrahedron Letters, 36:413-416 (scheme 1 at page 414),
which used pyrrole-2-carboxylic acid as starting material,
with an amide intermediate produced by reacting the
starting material with an H2N(CH2)nCOOMe amino ester. The
X, Y, and Z variations of the compounds of this invention
arise from using substituted pyrroles (X = NR), furans (X
= O) or thiophenes (X = S) as the starting material.
Variations in "n" arise from using different H2N(CH2)nCOOMe
amino esters in the reaction with the starting material to
produce the amide intermediate. The latter agent may have
to be modified by using a protected version of the ester
for some values of "n" in order to prevent polymerization
of the amino ester. W variations arise from reduction of
the amide intermediate using LAH followed by reoxidation
and esterification of the side chain. R1, R2, and R3
variations arise from alkylation and acylation reactions on
the starting material (for R2, R3) or, on the amide
intermediate (for R1).
Compounds of this invention, including
pharmaceutically acceptable salts thereof (eg. HCl salts)
CA 022l6~9 l997-09-2
-- 14
are sufficiently water-soluble that the compounds may be
administered in vivo in aqueous form. A preferred mode of
administration to an animal (including human) would be
intervenous with the goal being to achieve a circulating
concentration of the compound in the patient of about 10-5 -
10-4 molar.
EXAMPLE I
Cell Culture: Human m~mm~ry carcinoma cells (MCF-7)
expressing a dominant-negative p53 mutant (p53~ MCF-7) as
described in Fan. s., et al. (1995) "Disruption of p53
Function Sensitizes Breast Cancer MCF-7 Cells to Cisplatin
and Pentoxifylline", Cancer Research 55:1649-1654, were
grown in monolayer cultures at 37~ in humidified 5~ C02 in
Dulbecco's Modified Eagle Medium (DMEM) supplemented with
10~ fetal calf serum, 100 U/ml penicillin and streptomycin,
2 mM L-glutamine, 1 mM sodium pyruvate, 1~ Modified Eagle
Medium (MEM), non-essential amino acids, and 1 ng/ml human
epidermal growth factor (all from Gibco, and 1 ~/ml bovine
insulin, 1 ~g/ml hydrocortisone, and 1 ng/ml ~-estradiol
(from Sigma). To arrest cells in mitotic metaphase,
cultures were treated with 50 ng/ml nocodazole (Sigma) from
a 1000-fold stock in dimethylsulfoxide stored at -20~C.
Immunofluorescence Microscopy: Cells were grown on
coverslips coated with poly-L-lysine (Sigma), centrifuged
at 1000 g for 5 minutes, fixed with 3~ formaldehyde in
CA 022l6~9 l997-09-2~
Tris-buffered saline (TBS: 10 mM Tris-HCl pH 7.4, 0.15
M NaCl) for 40 minutes at 4~C, followed by ice-cold 100~
methanol for 5 minutes and then rinsed twice with TBS. The
coverslips were blocked in 3~ dried milk (Carnation brand)
in TBS for 1-2 hours at 4~C and then incubated with
TG-3 hybridoma culture supernatant ( 15 ~g/ml IgM) diluted
1/10 in 3~ milk in TBS for 1 hour at room temperature.
After 2 rinses in TBS, the coverslips were incubated with
CY3-conjugated goat anti-mouse secondary antibody (Jackson
Immunoresearch Laboratories, West Grove, Philadelphia:
Cat. #115-165-006) diluted 1/500 in 3~ milk in TBS for
1 hour at room temperature. They were then rinsed in TBS,
stained with the DNA dye Hoechst 33258TM (Sigma), mounted on
slides in 10~ TBS in glycerol containing 0. 2 M n-propyl
gallate, and photographed on Kodak TMax 400TM film with a
Zeiss AxiophotTM microscope.
Preparation for Cell Sorting: Cultured cells were detached
using trypsin, and fixed in 3~ formaldehyde in TBS for
40 minutes at 4~C, followed by 70~ ethanol for 5 minutes at
4~C. The cells were blocked in 1~ bovine serum albumin
(Sigma) in TBS (B-TBS) for 1 hour at 4~C and then incubated
overnight at 4~C with TG-~ hybridoma culture ~upernatant
diluted 1/10 in B-TBS. After two rinses in TBS, they were
incubated for 1 hour at 4~C with fluorescein-isothiocyanate
(FITC)-conjugated goat anti-mouse secondary antibody
(Pierce, Rockford, Illinois; Cat. #31560) diluted 1/100 in
B-TBS, rinsed twice in TBS and then resuspended in 5 ~g/ml
CA 022l6~9 l997-09-2
- 16 -
PI in TBS. The cells were subjected to flow cytometry for
sorting.
Preparation for Cell Cycle Analysis: To determine the
percentage of cells in each of the G1, S, G2, and M phases
of the cell cycle, cultured cells were incubated for
20 minutes with 30 ~M debromodeoxyuridine (BrdUrd). The
cells were detached using trypsin, fixed in 70~ ethanol for
30 minutes at 4~C, treated with 2 N HC1 in 0.5~ Triton
0 X-lOOTM at room temperature for 30 minutes, followed by 0.1
M Na2B4O7, pH 8.5 for 5 minutes. After 1 hour at 4~C in 0.5~
TweenTM 20 and 1~ bovine serum albumin in TBS (TB-TBS), they
were incubated at room temperature for 30 minutes with
monoclonal anti-BrdUrd antibody (Becton Dickinson, San
Jose, California; Cat #347580) diluted 1/3 in TB-TBS, to
which was then added TG-3 hybridoma culture supernatant
diluted 1/10 in TB-TBS and the cells were left overnight at
4~C. After two rinses in TBS, the cells were incubated in
FITC-conjugated goat anti-mouse secondary antibody diluted
1/25 in TB-TBS for 2 hours at 4~C. Cells were then rinsed
twice in TBS, resuspended in 5 ~g/ml PI in TBS and
subjected to flow cytometry.
Flow Cytometry: A Coulter Epics Elite ESPTM flow cytometer
(Coulter Corp., Miami, Florida) equipped with an
EnterpriseTM ion laser (Coherent, Santa Clara, California)
with an output of 400 mW at 488 nm was used. Forward and
side scatter were measured simultaneously and used to gate
CA 02216~9 1997-09-2~
out debris. Time of flight was used to gate for single
cells. A minimum of 20,000 gated cells was collected.
FITC fluorescence was collected using a 525 nm bandpass
filter, and propidium iodide fluorescence was collected
using a 610 nm longpass filter. All data was saved in
listmode for further gating and analysis. TG-3 positive
cells were sorted according to fluorescence levels
determined empirically. For sorting, a minimum of
50,000 cells was collected and 5,000 were reanalysed by
flow cytometry to determine the purity of the sort.
Mitotic Index: Cycling cells stained both with the
monoclonal antibody TG-3 using indirect immunofluorescence
and with the DNA dye Hoechst 33258TM were examined to
determine their mitotic stage. In interphase cells,
TG-3 staining is extremely weak and is restricted to small
speckles within the nucleus. Cells in mitosis show a
dramatic increase in staining. Prophase cells, in which
the DNA has condensed into chromosomes but the nuclear
lamina has not yet broken down, show intense staining
throughout the nucleus. The intense staining is maintained
in metaphase cells but is also spread throughout the
cytoplasm. Staining is particularly strong around the
chromosomes. Cytoplasmic staining is reduced slightly in
anaphase cells and further reduced in telophase cells. In
telophase cells, discrete speckles of fluorescence reappear
both within the nucleus and extranuclearly, but the latter
disappear after cytokinesis. The relative intensity of
CA 022l6~9 l997-09-2
- 18 -
TG-3 immunofluorescence in interphase and mitotic cells can
be seen by comparing these cells (for example, in the same
photograph) and is at its most extreme in metaphase.
When cycling cells stained with TG-3 antibody and
propidium iodide are subjected to flow cytometry, a
histogram of the fluorescence emitted from propidium iodide
shows the standard distribution of cells consisting of a
peak of G1 cells with diploid DNA, a diffuse plateau of
S phase cells with increasing fluorescence, and a peak of
cells in G2+M in which the mean fluorescence intensity is
double that of the G1 peak. A histogram of FITC
fluorescence associated with TG-3 immunoreactivity shows a
major peak of cells with low fluorescence and a minor peak
of cells with about 50-fold higher fluorescence. Dual
parameter analysis of FITC and propidium iodide shows that
the subpopulation with intense FITC fluorescence is within
the G2+M phase. Such TG-3 positive cells represented
2.2 + 0.3~ of the total cell population, comparable to the
value of 3.16 + 0.9~ mitotic cells obtained independently
from the same samples by fluorescence microscopy using
chromosome condensation as the marker for mitosis. The
TG-3 positive subpopulation can be seen most clearly in
nocodazole-treated samples. Cells previously treated with
nocodazole for 4 hours before antibody staining when
subjected to flow cytometry gave 17.3~ _ 0.9~ TG-3 positive
cells which agrees well with the value of 19.7 + 2.9~
mitotic cells obtained by microscopy. Similar results are
obtained with cells arrested in mitosis with other
CA 02216~9 1997-09-2~
antitubulin agents, including demecolcine, hemiasterlin,
and hemiasterlin A.
Cycling and nocodazole-treated preparations were
subjected to cell sorting based on their TG-3
immunofluorescence. The sorted fractions where subjected
to flow cytometry, giving 94.6~ and 96.8~ TG-3 positive
cells. Samples of the sorted TG-3 positive fractions were
prepared for microscopy and the percentage of mitotic cells
determined to be 93.5% and 95.0~ respectively. The TG-3
positive fraction sorted from cycling cells comprised 13.8~
prophase, 73.1~ metaphase, 6.9~ anaphase, 0~ telophase, and
6.2~ interphase cells, while the fraction sorted from
nocodazole-treated cells consisted almost exclusively of
cells in metaphase, with 2~ prophase, 94~ metaphase and 4~
interphase. Microscopic ex~m~n~tion of 150 mitotic cells
from a cycling population revealed 16~ prophase, 81
metaphase, and 2~ telophase.
Although the TG-3 antigen is present throughout
interphase at a very low level as nuclear speckles, high
levels of antigen are found only in mitotic cells. This
dramatic difference can be used to distinguish mitotic
cells from nonmitotic cells. 1~ mitotic cells may be
detected with accuracy comparable to that of conventional
microscopy.
The TG-3 antibody and cell cycle analysis: As the ability
to determine the proportion of cells in each of the G1, S,
G2 and M phases of the cell cycle would be useful, the
CA 02216~9 1997-09-2
- 20 -
above-described flow cytometry protocol was used in which
standard methods using antibodies to incorporated BrdUrd to
identify S phase cells and propidium iodide labelling of
total DNA to identify Gl and G2+M phase cells was combined
5 with TG-3 antibody binding to distinguish G2 and M phase
cells. Cycling cells that had incorporated BrdUrd for
2 0 minutes were fixed and their DNA was denatured. The
cells were then incubated with either mouse monoclonal TG-3
antibody, or mouse monoclonal antibody to BrdUrd, or both,
followed by FITC-con~ugated goat anti-mouse secondary
antibodies. Propidium iodide was used to label total DNA.
Dual parameter analysis of the fluorescence emitted by FITC
versus propidium iodide was carried out. The antigen
recognised by TG-3 was able to withstand the acid
15 denaturation required for detection of S phase cells.
Cells stained with TG-3 alone show the same pattern as
described above for cycling cells not exposed to BrdUrd.
The mitotic subpopulation of the G2+M phase showing intense
FITC staining, indicating that 1~ of the cells are mitotic.
20 Antibodies to BrdUrd alone show a typical horseshoe-shaped
pattern of FITC-staining cells spanning the DNA content
range of diploid to tetraploid with 35~ in Gl, 50~ in S,
and 15~ in G2+M. When both TG-3 antibody and antibody to
BrdUrd are used, two discrete FITC-positive populations are
25 observed, one like that obtained with antibodies to BrdUrd
alone and another that is more intense, like that obtained
with TG-3 antibodies alone. The percentage of cells in the
different phases of the cells cycle agrees with those
CA 02216~9 1997-09-2~
observed with TG-3 antibody or antibody to BrdUrd alone:
29~ in G1, 54~ in S, 16~ in G2, and 1~ in M.
EXAMPLE II
Preparation of Sponge Extracts: Specimens (87 g, wet) of
Stylissa flabelliformis (PNG94-51: S. flabelliformis
Hentschel, 1912; Class Demospongiae, Order Halichondrida,
Family Axinellidae; registered as ZMA POR, 11416) were
thawed and extracted exhaustively with MeOH (250 ml x 5,
each about one day). The MeOH extract was filtered and
concentrated in vacuo to give a dark brown solid (~5 g).
An ageous solution of approximately 3 g of the solid was
chromatographed on a SephadexTM LH-20 column using MeOH as
the eluent, yielding nine fractions which were subjected to
the G2 checkpoint inhibitor assay described below. Samples
(10 ~g/ml) of three of the fractions gave positive results
and those factions were further purified as described
below. Each purification step was monitored using the
G2 checkpoint inhibitor assay.
G2 Checkpoint Inhibition Assay: p53~ MCF-7 cells as
described in Example I are cultured as monolayers in DMEM
supplemented with 10~ fetal bovine serum, 2 Mm L-glutamine,
50 units/ml penicillin, 50 ~g/ml streptomycin, 1 Mm sodium
pyruvate, MEM non-essential amino acids, 1 ~g/ml bovine
insulin, 1 ~g/ml hydrocortisone, 1 n/ml human epidermal
growth factor, and 1 ng/ml ~-estradiol at 37~C in
CA 02216~9 1997-09-2~
humidified 5~ C02. The cells are seeded at 10,000 cells per
well of 96-well polystyrene tissue culture plates (Falcon)
in a volume of 200 ~l cell culture medium. The cells are
allowed to grow for 24 hours and irradiated with 6.5 Gy
using a 60Co source (Gammacell 200TM, Atomic Commission of
Canada) delivering ~-rays at a dose rate of 1.4 Gy/min.
Immediately after irradiation, 100 ng/ml nocodazole is
added (Sigma, from a 1000-fold stock in DMSO) together with
samples to be tested at about 1-10 ~g/ml (from 1000-fold
stocks in DMSO). The cells are incubated for 20 hours (G2
arrest preventer assay). Alternatively, nocodazole and
extracts may be added 16 hours after irradiation and the
cells incubated for 8 hours (G2 arrest releaser assay).
Caffeine at 2 mM (Sigma, from a 100 mM solution in PBS) may
be used as a positive control. After treatment, the cell
culture medium is removed completely and the 96-well tissue
culture plates frozen at -70~C for 1-14 hours). The frozen
cells are thawed by addition of 100 ~l of ice-cold lysis
buffer (1 mM EGTA, Ph 7.4, 0.5 mM PMSF) and lysed by
pipeting up-and-down 10 times. Cell lysates are
transferred to 96-well PolySorpTM Elisa plates (Nunc) and
dried completely by blowing warm air at about 37~C with a
hair drier positioned about 3 feet above the plates.
Protein binding sites are blocked by adding 200 ~l per well
of TBSM (10 mM Tris HCl pH 7.4, 150 mM NaCl, 0.1 mM PMSF,
3~ (w/v) dried non-fat milk (Carnation)) for 1 hour at room
temperature. The blocking medium is removed and replaced
with 100 ~l TSM containing 0.1-0.15 ug/ml TG-3 monoclonal
CA 02216~9 1997-09-2
- 23 -
antibody and horseradish peroxidase-labelled goat
anti-mouse IgM (1021-05, Southern Biotechnology Associates)
at a dilution 1/500. After overnight incubation at 4~C,
the antibody solution is removed and the wells rinsed
3 times with 200 ~1 rinse buffer (10 mM Tris Hcl Ph 7.4,
0.02~ TweenTM 20). 100 ~1 ABTS buffer (120 mM Na2HPO4, 100
mM citric acid, pH4.0) containing 0.5 mg/ml 2,2'-azino-bis
(3-ethylbenzthiazoline-6-sulfonic acid) and 0.01~ hydrogen
peroxide added for 1 hour at room temperature. The plates
are read at 405 nm using a BioTekTM plate reader. Caffeine
positive controls give absorbance readings of about 1.0,
corresponding to about 60~ mitotic cells.
Purification and Characterization of Active Compounds: The
sponge extract fractions exhibiting G2 checkpoint inhibitor
activity (typical absorbance reading in assay = 0.5 - 1.0)
were individually preabsorbed onto NP silica gel columns
and subjected to silica-gel flash chromatography using
stepwise gradient elution (100~ CH2Cl2 to 50~ CH2Cl2:MeOH
saturated with NH3 (g)). Further purification was achieved
by repeated fractionation on reversed-phase HPLC using
80:20:0.05 H2O/MeOH/CF~COOH (TFA) as eluent. One pure
product was debromoaxinohydantoin which was inactive as a
G2 checkpoint inhibitor. Two pure products, recovered as
TFA salts were converted to the hydrochloride salts by
acidification with 3N HCL and concentrated i~ vacuo. The
latter two products were active G2 checkpoint inhibitors
(typical absorbance reading in assay = 1.0 - 1.5). The
CA 02216~9 1997-09-2
- 24 -
products were identified as debromohymenialdisine and
hymenialdisine and were present in the SephadexTM fractions
as taurine salts. An inactive cleavage product,
debromopyrrololactam was obtain in pure form from one of
the active SephadexTM fractions by subjecting the fraction
to a 5% K2CO3 solution, followed by concentration in vacuo
and then reversed-phase HPLC using 80: 20 H2O/MeOH as eluent.
EXAMPLE III
Correspondence between microscopic and ELISA determination
of mitotic index: p53~ MCF-7 cells grown in 10 cm diameter
cell culture dishes were treated with 0-200 nM nocodazole
for 16 hours. The cells were harvested by trypsinization,
washed once with PBS and a small sample processed for
microscopic determination of the percent mitotic cells as
follows: cells were swelled in hypotonic medium (75 mM
KCl), fixed with methanol:acetic acid (3:1), spotted onto
microscope slides, stained with bisbenzimide and observed
using a ZeissTM standard microscope (Guo et al. (1995). At
least 300 cells were counted for each sample. The
remaining cells were lysed in ice-cold lysis buffer (1 mM
EGTA, pH 7.4, 0.5 mM PMSF). The protein concentration of
the cell lysates was determined using a BioRadTM protein
assay and BSA as a standard, and the extract was kept at
-70~C as aliquots. Volumes of cell extracts corresponding
to 10,000 cells were processed for ELISA according to the
method described in Example II. The standard curve
CA 02216~9 1997-09-2
- 25 -
(Fig. 4) shows a linear response which correlates highly
(r~0.99) with results obtained with the microscopy
analysis.
Analy~is by flow cytometry and microscopy: p53~ MCF-7 cells
were cultured and irradiated as described in Example II.
After irradiation, ~90~ of the cells arrest in G2/M within
16 hours as determined by flow cytometry, and the cells
remain arrested for an additional 8 hours. Fig. 1 shows
analysis by flow cytometry of the cells irradiated at
0 hours (Fig. lA) and left for 16 hours (Fig. lB). At
16 hours, cell samples were left untreated (Fig. lC), or
were treated with 2 mM caffeine (Fig lD) or 100 nM UCN-01
(Fig. lE). After 8 hours, most of the treated cells had
gone from G2/M to G1 but not the untreated cells.
Flow cytometry per se cannot distinguish between cells
in G2 and M. For analysis by microscopy, cells were
irradiated at 0 hours, nocodazole (see Example II) was
added immediately and cell samples were harvested at
different time for microscopic determination of the
percentage of cells in mitosis showing condensed metaphase
chromosomes (mitotic index). At either 0 or 16 hours, 2 mM
caffeine wa~ added to ~ome sample~ and the mitotic index
determined at various times. The results shown in Fig. 2
show that no cells entered mitosis in the 24 hours
following DNA damage (open circles), but cells treated with
caffeine at the time of irradiation (checkpoint inhibitor
assay; closed circles) and at 16 hours (checkpoint releaser
CA 02216~9 1997-09-2
- 26 -
assay; closed rectangles) entered mitosis within 8 hours
following treatment. In a parallel procedure shown in
Fig. 3, cells were irradiated at 0 hours, and at 16 hours
nocodazole and different concentration of caffeine
(circles) and UCN-01 (rectangles) were added. The mitotic
index was measured 8 hours later. Within a 16-24 hour
post-damage period, the majority of cells can be made to
enter mitosis by a G2 checkpoint inhibitor while a
negligible number of cells do so in the absence of the
inhibitor.
EXAMPLE IV
G2 Checkpoint Inhibition by Debromohymenialdisine: Using
the assay procedure described in Example II, p53~ MCF-7
cells were exposed to 6.5 Gy of ~-irradiation; or, to 1 ~M
VM-26 (Bristol-Myers) for 100 minutes. After 16 hours, 100
ng/ml nocodazole was added to all samples. Controls
received no further treatment. Other samples also received
caffeine or debromohymenialdisine with the nocodazole.
Eig. 5 shows the results obtained 8 hours after treatment
with no further addition (-); 20 ~g/ml
debromohymenialdisine (H); and, 2 mM caffeine (C).
EXAMPLE V
Potentiation of the Cytotoxicity of a DNA-Damaging Agent by
Debromohymenialdisine: p53-MCF-7 cells were plated at 1000
CA 02216~9 1997-09-2
- 27 -
cells per well of a 96-well plate and exposed to different
concentrations of cisplatin for 2 hours. The cisplatin was
washed away and fresh medium containing: (with reference to
Fig. 6) 2 0 ~g/ml debromohymenialdisine (filled circles);
100 nM UCN-O1 (closed triangles); or, nothing (open
circles). The cultures were grown for 5 days after which
cell survival was measured optically. Average and S.D. of
triplicate measurements are shown in Fig. 3.
Although various aspects of the present invention have
been described in detail, it will be apparent that changes
and modification of those aspects described herein will
fall within the scope of the appended claims. All
publications referred to herein are incorporated by
reference.