Note: Descriptions are shown in the official language in which they were submitted.
CA 02216809 1997-09-29
W O96/31509 PCTAEP96/01438
IMID~ZO [1,2a ~ PYRIDINE DERIVATIVES
This invention relates to imidazo~1,2-a]pyridine derivatives, to processes for
their pre,udr~lion, to pl,a""~ Jtic~l composilions containing them and to their
use in medicine.
The enzyme cyclooxygenase (COX) has recently been discovered to exist in
two isoforms, COX-1 and COX-2. COX-1 corresponds to the originally identified
constitutive enzyme while COX-2 is rapidly and readily inducible by a number of
agents including mitogens, encloloxin, hormones, cytokines and growth factors.
Prostaglandins generalted by the action of COX have both physiological and
pathological roles. It is generally believed that COX-1 is responsible for the
important physiological functions such as mainte, Idl ,ce of gastrointestinal
integrity and renal blood flow. In contrast the inducible form, COX-2, is believed
to be responsible for the pathological effects of prostaglandins where rapid
induction of the enzyme occurs in response to such agents as inflalllr"atoly
agents, hormones, growth factors and cytokines. A selective inhibitor of COX-2
would therefore have anti-inflammatory, anti-pyretic and analgesic properties,
without the potential side effects associated with inhibition of COX-1. We have
now found a novel group of compounds which are both potent and selective
inhibitors of COX-2.
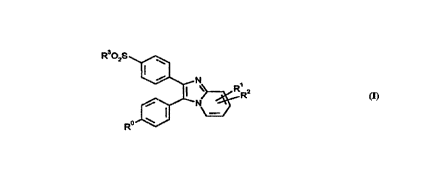
The invention thus provides the compounds of formula (I)
R 02S _~N~,
and pharmaceutically acceptable derivatives thereof in which:
R~ represents halogen;
R' and R2 are independently selected from H, halogen, C,~alkyl, C1 4alkyl
substituted by one or more fluorine atoms, C1~alkoxy, C'4hydroxyalkyl,
SC14alkyl, C(O)H or C(O)C,4alkyl; and
R3 represents C,~alkyl.
CA 02216809 1997-09-29
W O96131509 PCTAEP96/01438
By pharmaceutically acce,ulable derivative is meant any ,c l Idl "~oe~ ~tic~lly
acceptaL31e salt, solvate or ester, or salt or solvate of such ester, of the
co",,.,ounds of formula (I), or any other compound which upon administration to
the recipient is capable of providing (directly or indirectly) a cG,.,pound of
formula (I) or an active metabolite or residue thereof.
It will be appreciated that, for pl,ar",~ce~tic~l use, the salts referred to above
will be the physiologically acceptable salts, but other salts may find use, for
example in the preparalio" of ccjrnpounds of formula (I) and the physiologicallyacceplable salts thereof.
Suitable phar"~aceutically acceplable salts of the compounds of formula (I)
include acid addition salts formed with inorganic or organic acids, preferably
inorganic acids, e.g. hydrochlorides, hydrobromides and sulphates.
The term halogen is used to represent fluorine, chlorine, bromine or iodine.
The term 'alkyl' as a group or part of a group means a straight or branched
chain alkyl group, for exa",pl~ a methyl, ethyl, n-propyl, i-propyl, n-butyl, s-butyl
or t-butyl group.
The substituents R' and R2 may be at the 5-, 6-, 7- or 8- positions of the pyridine
ring of formula (I), as defined hereinbelow:
5 6
P,ererably, R' is at the 8- position; and R2 is at the 7- position or, when R' is
other than H, the 5-, 6- or 7-position. More preferably, R' is at the 8- position;
and R2 is at the 7- position or, when R' is C,4alkyl (e.g. methyl), the 5- or 7-position.
Preferably, R~ represents fluorine.
Preferably, R' represents H, chlorine, bromine, C,~alkyl (e.g. methyl), methyl
substituted by one to three fluorine atoms (e.g. CH2F or CF3), C,4hydroxyalkyl
CA 022l6809 l997-09-29
W O96/31509 PCTAEP96/01438
(e.g. CH20H or CH(OH)CH3), SC,~alkyl (e.g. SCH3), C(O)H or C(O)C.~alkyl
(e.g. C(O)CH3). Mor~ prererably, R1 represe,)ls H, chlorine, bromine, methyl,
CH2F, CF3, SCH3, C(C))H or C(O)CH3. Most ~,-erer~ly R' represents C(O)CH3.
Preferably, R2 represents H, chlorine, bromine, or C~alkyl (e.g. methyl). More
preferably, R2 represents H, bromine or methyl.
P,~rerably, R3 represents methyl.
Within the invention there is provided one group of compounds of formula (I)
(group A) wherein: R~' represents fluorine; R' represents H, chlorine, bromine,
C~alkyl (e.g. methyl), methyl substituted by one to three fluorine atoms (e.g.
CH2F or CF3), C,~hy~droxyalkyl (e.g. CH20H or CH(OH)CH3), SC,~alkyl (e.g.
SCH3), C(O)H or C(~))C,~alkyl (e.g. C(O)CH3); R2 represents H, chlorine,
bromine, or C~alkyl (e.g. methyl); and R3 represents methyl.
Within group A there is provided the sub-group of compounds wherein: R~
represents fluorine; R~ represents H, chlorine, bromine, methyl, CH2F, CF3,
SCH3, C(O)H or C(l~))CH3; R2 represents H, bromine or methyl; and R3
represents methyl.
Within the invention there is provided another group of compounds of formula (I)(group B) wherein R~ represents fluorine; R'is at the 8- position and representsH, chlorine, bromine, C,~alkyl (e.g. methyl), methyl substituted by one to threefluorine atoms (e.g. CH2F or CF3), C,~hydroxyalkyl (e.g. CH20H or
CH(OH)CH3), SC,~alkyl (e.g. SCH3), C(O)H or C(O)C,4alkyl (e.g. C(O)CH3); R2
is at the 7- position or, when R' is other than H, the 5-, 6- or 7- position, and
represents H, chlorine, bromine, or C,~alkyl (e.g. methyl); and R3 represents
methyl.
Within group B there is provided the sub-group of compounds wherein: R~
represents fluorine; R'is at the 8- position and represents H, chlorine, bromine,
methyl, CH2F, CF3, SCH3, C(O)H or C(O)CH3; R2 is at the 7- position or, when
R' is methyl, the 5- or 7- position, and represents H, bromine or methyl; and R3represents methyl.
Within the invention there is provided a further group of compounds of formula
(I) (group C) wherein R~ represents fluorine; R' is at the 8- position and
CA 02216809 1997-09-29
W O96/31509 PCT~EP96/01438
r~rese"Ls H, chlorine, Lro",i"e, C,4alkyl (e.g. methyl), methyl substituted by
one to three fluorine atoms (e.g. CH2F or CF3), C,~hydroxyalkyl (e.g. CH20H or
CH(OH)CH3), SC,4alkyl (e.g. SCH3), C(O)H or C(O)C,4alkyl (e.g. C(O)CH3); R2
represents H; and R3 represenls methyl.
Within group C there is provided the sub-group of co~pounds wherein: R~
represents fluorine; R1 is at the 8- position and represents H, chlorine, bromine,
methyl, CH2F, CF3, CH(OH)CH3, SCH3, C(O)H or C(O)CH3; R2 represents H;
and R3 represents methyl.
Within the above groups (and prerer, ed groups) of compounds, espe,_ially
preferred groups of compounds are those wherein R' represents C(O)CH3.
It is to be understood that the present invention encompasses all isomers of thecompounds of formula (I) and their pharmaceutically acceptable derivatives,
including all geometric, tautomeric and optical forms, and mixtures thereof (e.g.
racemic mixtures).
Prefer, ed compounds of the invention are:
3-(4-fluoro-phenyl)-2-(4-methanesulfonyl-phenyl)-imidazo[1 ,2-a]pyridine;
3-(4-fluoro-phenyl)-2-(4-methanesulfonyl-phenyl)-8-methyl-imidazo[1 ,2-
a]pyridine;
3-(4-fluoro-phenyl)-2-(4-methanesulfonyl-phenyl)-8-trifluoromethyl-imidazo[1 ,2-a]pyridine;
3-(4-fluoro-phenyl)-2~4-methanesulfonyl)-7-methyl-imidazo[1 ,2-a]pyridine;
8-chloro-3-(4-fluoro-phenyl)-2-(4-melha"esulfonyl-phenyl)-imidazo[1 ,2-
a]pyridine;
3-(4-fluoro-phenyl)-2-(4-methanesulfonyl-phenyl)-8-methanesulfanyl-
imidazo[1,2-a]pyridine;
8-bromo-3-(4-fluoro-phenyl)-2-(4-methanesulfonyl-phenyl)-imidazo[1 ,2-
a]pyridine;
8-fluoromethyl-3-(4-fluoro-phenyl)-2-(4-methanesulphonyl-phenyl)-imidazo[1 ,2-
a]pyridine;
3-(4-fluoro-phenyl)-2-(4-methanesulfonyl-phenyl)-7,8-dimethyl-imidazo[1,2-
a]pyridine;
3-(4-fluoro-phenyl)-2-(4-methanesulfonyl-phenyl)-imidazo[1 ,2-a]pyridine-8-
carbaldehyde;
CA 02216809 1997-09-29
W O 96/31509 PCTAEP96/01438
5-bromo-3-(4-fluoro-phenyl)-2-(4-methanesulfonyl-phenyl)~-methyl-
i~ la~o[1,2-a]pyridine;
6-bromo-3-(4-fluoro-phenyl)-2-(4-methanesulfonyl-phenyl)~-methyl-
il l IIJd~0[1 ,2-a]pyridine;
[3-(4-fluoro-phenyl)-2-(4-l l l~:Li ,~nesulfonyl-phenyl)-imidazo[1 ,2-a]pyridin-8-yl]-
methanol;
(:t) 1-[3-(4-fluoro-phen~1)-2-(4-",elh dl ~esulphonyl-phenyl)-imidazo[1,2-a]pyridin-
8-yl]-ethan-1-ol;
and phal " ,aceutically acceptable derivatives thereof.
A particularly preferred co",pound of the invention is:
8-acetyl-3-(4-fluoro-phenyl)-2-(4-methanesulfonyl-phenyl)-imidazo[1 ,2-
a]pyridine; and pha",)aceutically accep~able derivatives thereof.
Compounds of the invention are potent and selective inhibitors of COX-2. This
activity is illu~ ated by their ability to selectively inhibit COX-2 over COX-1.
In view of their selective COX-2 inhibitory activity, the compounds of the present
invention are of interest for use in human and veterinary medicine, particularlyin the treatment of the pain, fever and inflan",ldlion of a variety of conditions
and diseases. Such conditions and diseases are well known in the art and
include rheumatic fe~er; sy,llptoms ~ssoci~ted with influenza or other viral
inre.;lions, such as the com,llon cold; lower back and neck pain; headache;
toothache; sprains and sl,ai"s; myositis; neuralgia; synovitis; alllllilis, including
rheumatoid arthritis; degenerative joint diseases, including osteoarthritis; gout
and ankylosing spondylitis; tendinitis; bursitis; skin related conditions, such as
psoriasis, eczema, burns and dermatitis; injuries, such as sports injuries and
those arising from surgical and dental procedures.
The compounds of the invention may also be useful for the treatment of other
conditions mediated by selective inhibition of COX-2.
For example, the compounds of the invention may inhibit cellular and neoplastic
ll cl ,src,r" ,ation and metastatic tumour growth and hence be useful in the
treatment of certain cancerous diseases, such as colonic cancer.
Compounds of the invention may also prevent neuronal injury by inhibiting the
generation of neuronal free radicals (and hence oxidative stress) and therefore
CA 02216809 1997-09-29
W O96131509 PCT~EP96/01438
may be of use in the treatment of stroke; epilepsy; and epileptic seizures
(including grand mal, petit mal, myoclonic epilepsy and partial seizures).
Compounds of the invention also inhibit proslanoid-induced smooth muscle
conl,aclion and hence may be of use in the treatment of dysmenorrhoea and
premature labour.
Compounds of the invention inhibit inflar"r"alo,y processes and therefore may
be of use in the treatment of asthma, allergic rhinitis and respiratory distresssyndrome; gastroinleslinal conditions such as inflammatory bowel disease,
Chron's disease, gastritis, irritable bowel syndrome and ulcerative colitis; andthe inflammation in such diseases as vascular disease, migraine, periarteritis
nodosa, thyroiditis, aplastic anemia, Hodgkin's disease, sclerodoma, type I
diabetes, myasthenia gravis, multiple sclerosis, sorcoidosis, nephrotic
syndrome, Bechet's syndrome, polymyositis, gingivitis, conjunctivitis and
myocardial ischemia.
Compounds of the invention may also be useful in the treatment of ophthalmic
diseases such as retinitis, retinopathies, uveitis and of acute injury to the eye
tissue.
Compounds of the invention may also be useful for the treatment of cognitive
disorders such as dementia, particularly degenerative dementia (including
senile dementia, Alzheimer's disease, Pick's disease, Huntington's chorea,
Parkinson's disease and Creutzfeldt-Jakob disease), and vascular dementia
(including multi-infarct dementia), as well as dementia associated with
intracranial space occupying lesions, trauma, infections and related conditions
(including HIV i"rec~io"), metabolism, toxins, anoxia and vitamin deficiency; and
mild cognitive impairment associated with ageing, particularly Age Associated
Memory Impairment.
According to a further aspect of the invention, we provide a compound of
formula (I) or a pharmaceutically acceptable derivative thereof for use in humanor veterinary medicine.
According to another aspect of the invention, we provide a compound of formula
(I) or a pharmaceutically acceptable derivative thereof for use in the treatment of
a condition which is mediated by selective inhibition of COX-2.
CA 02216809 1997-09-29
W O96/31509 PCT~EP96/01438
Accc,rdi. ,9 to a further aspect of the invention, we provide a method of 1, ealing a
human or animal subject suffering from a condition which is medi~ted by
selective jIIhj~ ;GII of COX-2 which comprises administering to said subject an
effective amount of a compound of formula (I) or a pl,d,-,!aceutically acceptable
derivative.
Acc~r~ir~g to another aspect of the invention, we provide the use of a compound
of formula (I) or a ,~l ,dr",aceutically accep~able derivative thereof for the
manufacture of a therapeutic agent for the treatment of inflammatory disorders.
Accor-ling to a further aspect of the invention, we provide a method of treating a
human or animal subject suffering from an inflammatory disorder, which method
comprises administering to said subject an effective amount of a compound of
formula (I) or a pharmaceutically acceptable derivative thereof.
It is to be understood that reference to treatment includes both treatment of
established sy",pLo",s and prophylactic treatment, unless explicitly stated
1 5 otherwise.
It will be appreciated that the compounds of the invention may advantageously
be used in conjunction with one or more other therapeutic agents. Exampies of
suitable agents for adjunctive therapy include pain relievers such as a glycine
antagonist, a sodium channel inhibitor (e.g. Iamotrigine), a substance P
antagonist (e.g. an NK~ antagonist), aceta")il,ophen or phenac~tin; a matrix
metalloproteinase inhibitor; a nitric oxide synthase (NOS) inhibitor (e.g. an iNOS
or an nNOS inhibitor); an inhibilor of the release, or action, of tumour necrosis
factor a; an antibody therapy (e.g. a monoclonal antibody therapy); a stimulant,including caffeine; an H2-anl&go"ist, such as ranitidine; an antacid, such as
2~ aluminium or magnesium hydroxide; an antiflatulent, such as simethicone; a
decongestant, such as phenylephrine, phenylpropanolamine, pseudoephedrine,
oxymetazoline, epinephrine, naphazoline, xylometazoline, propylhexedrine, or
levo-desoxyephedrine; an antitussive, such as codeine, hydrocodone,
carmiphen, carbetapentane, or dextramethorphan; a diuretic; or a sedating or
non-sedating antihistamine. It is to be understood that the present invention
covers the use of a compound of formula (I) or a pharmaceutically acceptable
derivative thereof in combination with one or more other therapeutic agents.
CA 02216809 1997-09-29
W O96/31509 PCTAEP96/01438
The compounds of formula (I) and their ,ul,d,."~Ge~tic~lly acceplable derivatives
are conveniently administered in the form of pl ,a" "~ce~ ~tic~l compositions.
Thus, in another aspect of the invention, we provide a ,uha~ m~cel lti~l
co~ JosiliGn comprising a compound of formula (I) or a ~I ,d""aceutically
acceplable derivative thereof adapted for use in human or veteri"dry medicine.
Such coi ",oosilions may convenientiy be ,urt:se, ~led for use in conventional
manner in admixture with one or more physiologically acceplable carriers or
excipients.
The compounds of formula (I) and their pharmaceutically acceplable derivatives
may be formulated for admini~l,dlion in any suitable ~"d""er. They may, for
example, be formulated for topical administration or adminislralio" by inhalation
or, more prererably, for oral, transdermal or parenteral administration. The
pharmaceutical composition may be in a form such that it can effect controlled
release of the compounds of formula (I) and their pharmaceutically acceplable
1 5 derivatives.
For oral administration, the pharmaceutical composition may take the form of,
for example, tablets (including sub-lingual tablets), capsules, powders,
solutions, syrups or suspensions prepared by conventional means with
acceptable excipients.
For transdermal administration, the pharmaceutical composilion may be given in
the form of a transdermal patch, such as a l, ~"sde" "al ion~opl ,o, elic patch.
For parenteral administration, the pharmaceutical composition may be given as
an injection or a continuous infusion (e.g. intravenously, intravascularly or
subcutaneously). The compositions may take such forms as suspensions,
solutions or emulsions in oily or aq~ ~eous vehicles and may cGnlai,) formulatory
agents such as suspending, stabilising and/or dispersing agents. For
administration by injection these may take the form of a unit dose presentation
or as a multidose presentation preferably with an added preservative.
Alternatively for parenteral administration the active ingredient may be in
powder form for reconstitution with a suitable vehicle.
The compounds of the invention may also be formulated as a depot preparation.
Such long acting formulations may be administered by implantation (for example
CA 02216809 1997-09-29
W O96/31509 PCTAEP96/01438
sl~hclIt~neously or intraml'sc~ rly) or by intram~sc~ 3r injection. Thus for
~3xa..~ple the cc,-,pounds of the invention may be formulated with suitable
polymeric or hyd~o,ch~L~ic ~. IdLel ials (for example as an emulsion in an
acc~pla~le oil) or ion ~exo~,dnge resins or as sparingly soluble derivatives forexample as a spa, ingly soluble salt.
As stated above th0 compounds of the invention may also be used in
combination with other therapeutic agents. The invention thus provides in a
further ~cpect a combination comprising a compound of formula (I) or a
l l,ar",aceutically acceptable derivative thereof together with a further
therapeutic agent.
The combinations referred to above may conveniently be presented for use in
the form of a ,cl,a""aceutical formulation and thus pharmaceutical formulations
comprising a combination as defined above together with a pharmaceutically
acceptable carrier or excipient ccsi",crise a further aspect of the invention The
individual components of such co")binations may be administered either
sequentially or simulLaneously in separate or combined pharmaceutical
formulations.
When a compound of formula (I) or a pharmaceutically acceptable derivative
thereof is used in combination with a second therapeutic agent active against
the same clise~-se state the dose of each co"",ound may differ from that when
the compound is used alone. App(c,priate doses will be readily appreciated by
those skilled in the art.
A proposed daily dosage of a compound of formula (I) for the treatment of man
is 0.01 mg/kg to 500 mg/kg such as 0.05mg/kg to 100mg/kg e.g. 0.1mg/kg to
50mg/kg which may be conveniently administered in 1 to 4 doses. The precise
dose employed will depend on the age and condition of the patient and on the
route of administration. Thus for example a daily dose of 0.25mg/kg to
1 Omg/kg may be suitable for systemic administration.
Compounds of formula (I) and pharmaceutically acceptable derivatives thereof
~ 30 may be prepared by any method known in the art for the preparation of compounds of analogous structure.
CA 02216809 1997-09-29
W O96/31509 PCT~EP96/01438
Suitable methods for the preparalion of compounds of formula (I) and
pha",~aceutically acce,utable derivatives thereof are desc,iL,ed below. In the
formulae that follow R~ to R3 are as d~ined in formula (I) above unless otherwise
stated and Lg represents a leaving group such as a sulphonate (e.g.
",~ll,d"esulphonate) or a halogen (e.g. br~r"ine).
Thus accor(:li. ,9 to a first prc,cess (A) compounds of formula (l~ may be prepared
by reacting a compound of formula (Il)
R O2
'~0
~--Lg
R~ (ll~
or a protected derivative thereof with a cor"pound of formula (Ill)
R~ R2
H2N~
N~ (~
or a protected derivative thereof. The reaction is conveniently carried out in asolvent such as a polar solvent (e.g. acetonitrile or isoprc,,uar,ol); at elevated
temperature e.g. reflux; and optionally in the presence of a base such as an
alkali metal bicarbonate or carbonate (e.g. potassium carbonate).
Suitable leaving atoms or groups in respect of Lg in formula (Il) are described in
many standard texts on organic chemistry for example in table 10.10 on page
357 of Advanced Organic Chemistry by Jerry March fourth edition (Wiley
1992). It will be appreciated by a person skilled in the art that the choice of a
particular leaving group in the above reaction may depend upon the meanings
of R~ to R3 (and hence the compound of formula tl) desired~ and the reaction
conditions employed.
CA 02216809 1997-09-29
W O96/31509 PCTAEP96/01438
11
According to a another process (B), cc",pounds of formula (I) may be prepared
by reacting a con"~ound of formula (IV)
P~3S~
R~~ RR2
or a protected derivative thereof with an oxidising agent. Conveniently the
oxidation is effected using a mol,o,~,er~ulfate compound, such as potassium
peroxymonosulfate (known as OxoneTM) and the reaction is carried out in a
solvent, such as an aqueous alcohol, (e.g. aqueous methanol), and at between
-78~C and ambient temperature.
Acc~rding to another process (C) compounds of formula (I) may be prepared by
interconversion, utilising other compounds of formula (I) as precursors.
Thus, for example, ~ompounds of formula (I) wherein R1 or R2 represent
chlorine, bromine or iodine may be prepared from the corresponding compound
of formula (I) wherein R1 or R2 represent H, by treatment with an appropriate
halogenating agent (i.e. chloro-, bromo- or iodinating agent). Suitable agents
include the corresponding N-halosuccinimdes. Conveniently the reaction is
effected in the presence of a solvent, such as a halogenated hydrocarbon (e.g.
chlol L~ror~ and at ambient temperature.
Compounds of formula (I) wherein R' or R2 represent C1~alkyl substituted by
one or more fluorine atoms may be prepared from the compound of formula (I)
wherein R1 or R2 represents the corresponding C,~hydroxyalkyl by treatment
with a suitable source of fluorine. Suitable sources of fluorine include, for
exampie, diethylaminosulphur trifluoride. Conveniently the reaction is effected
in the presence of a solvent, such as a halogenated hydrocarbon (e.g.
dichloromethane), anci at reciuce~ temperature, such as -78~C.
Compounds of formula (I) wherein R' or R2 represent C(O)H may be prepared
from the corresponding compound of formula (I) wherein R1 or R2 represent
CH2OH by oxidation. Suitable oxidising agents include, for example,
CA 02216809 1997-09-29
W O96/31509 PCTAEP96101438
12
manganese (IV) oxide. Conveniently the oxidation is effected in the presence of
a solvent, such as a halogenaled hyd~ocalL,G,I (e.g. chicsrorc".,), and at elevated
temperature (e.g. reflux).
Compounds of formula (I) wherein R' or R2 represent C,4hydroxyalkyl, and
wherein the hydroxy group is attached to the carbon linked to the pyridine ring,may be p,t:~ared by reduction of the compound of formula (I) wherein R' or R2
represent the cor,esponding aldehyde or ketone. Suitable reducing agents
include hydride reducing agents, such as diisobutylaluminium hydride.
Conveniently the reduction is effected in the presence of a solvent, such as a
halogenated h~/dlocalL,oll (e.g. dichloromethane), and at reduced temperature,
such as -78~C.
As will be appreciated by those skilled in the art it may be necessary or
desirable at any stage in the above described processes to protect one or more
sensitive groups in the molecule to prevent undesirable side reactions.
Another process (D) for preparing compounds of formula (I) thus comprises
deprotecting protecled derivatives of compounds of formula (I).
The protecting groups used in the preparalion of compounds of formula (I) may
be used in conventional manner. See, for example, those described in
'P, otecli"e Groups in Organic Synthesis' by Theodora W. Green, second
edition, (John Wiley and Sons, 1991), which also describes methods for the
removal of such groups.
Compounds of formula (Il) may be prepared from compounds of formula (V)
R302S ~
W~~
R~
by conventional means.
Thus, compounds of formula (Il) wherein Lg represents a halogen may be
CA 02216809 1997-09-29
W O96/31509 PCTAEP96/01438
13
udl~d from cc,r"pounds of formula (V) by treatment with a halo~~e"~li"~
agent at red~ ~c~l ten ,per~ re and in a solvent such as a ol ,lori"dled
h~llu~lLJull. For ex~"",le, where Lg represents bromine, the reac~ion is
conveniently ~rracled with a brominating agent, such as bromine in the
presence of a strong acid (e.g. hyd~ol~rcr,.ic acid in acetic acid).
Compounds of formula (Il) wherein Lg represents a sulphonate may be
~,re~.ared from compounds of formula (V) firstly by oxidation to the
corresponding a-hydroxy ketone, followed by treatment with a sulphonating
agent. Suitable oxidising agents include for example Pb(OAc)4
dimethyldioxirane and those desc,il.ed in F A Davis, J. Org. Chem., 1984,
49~17) 3284. Suitable sulphonating agents include sulphonylhalides such as
sulphonylchlorides (e.g. " ,eLi ,anesulphonylchloride). The sulphonylation i
conveniently effected in the presence of a base, such as an amine (e.g.
triethylamine); and in a solvent, such as a halGyelldled hydrocarbon.
Compounds of formula (V) may be l.re~.ared from compounds of formula (Vl)
F~
~0
,~
il
R~~ (Vl)
by treatment with an alkali sulphinate such as a sodium sulphinate.
Conveniently, the reaction is carried out in a polar solvent such as dimethyl
sulphoxide, and at elevated temperature.
Compounds o~ formula (Ill) are either known compounds or may be prepared by
literature methods such as those described in for example J A Turner, J. Org.
Chem., 1983, ~, 3401; M Malinowski, Bull. Soc. Chim. Belg., 1988, ~Z, 51; W
0 Siegl, J. Hef. Chem., 1981, ~, 1613-18; or F.Trecourt et al, J. Chem. Soc.,
Perkin Trans.1 1990, ~, 2409-2415.
Compounds of formula (Vl) are either known compounds or may be prepared by
literature methods such as those described in for example N Seko ~L Chem.
Pharm. Bull., 1991, ;~, (3) 651-7 or I Lalazori et al, J.Med.Chem. 1971, 14,
CA 02216809 1997-09-29
W O96/31509 PCTAEP96/01438 14
1 138-40.
Compounds of formula (IV) or protected derivatives thereof may be prepared
using conventional chemistry, for example chemistry analogous to that herein
described for the preparalion of co",~Jounds of formula (I)
Certain intermediates des~,iL,ed above are novel compounds, and it is to be
understood that all novel i"ler" ledidles herein form further aspects of the
present invention. Compounds of formula (Il) and (IV) are key intermediates
and represent particular aspects of the present invention.
Conveniently, compounds of the invention are isolated following work-up in the
form of the free base. Pharmaceutically accepta~le acid addition salts of the
compounds of the invention may be prepared using conventional means.
Solvates (e.g. hydrates) of a co",pound of the invention may be formed during
the work-up procedure of one of the aforementioned process steps.
The following Examples illustrate the invention but do not limit the invention in
any way. All temperatures are in ~C. Flash chror"atography was carried out
using Merck 9385 silica. Thin layer chromatography (Tlc) was carried out on
silica plates. NMR was carried out on a Brucker 300Mhz spectrometer, using
CDCI3 as solvent. Chemical shifts are given in ~ ppm with respect to
tetramethylsilane as intemal chemical shift reference. The following
abbreviations are used: Et = ethyl, s = singlet, d = doublet, t = triplet and m =
multiplet.
Interrnediate 1
2-(4-Fluoro-phenyl)-1 -(4-methanesulfonvl-PhenYl)-ethanone
A mixture of the 2-(4-fluoro-phenyl)-1-(4-fluoro-phenyl)-ethanone' (3g) and
sodium methane sulphinate (1.589) in dry dimethyl sulphoxide (1 Oml) was
heated to 105-110~ under nitrogen for 18h. The cooled reaction mixture was
poured into water (500ml) and the mixture extracted with ethyl acetate. The
combined organic extracts were adsorbed onto silica and purified by flash
cl ,romalography eluting with ethyl acetate:hexane (1 :1) to give the title
compound as a white solid (2.19).
m.p.115-116~
' Ref: I Lalazori et al, J.Med.Chem. 1971, 14, 113840.
CA 02216809 1997-09-29
W O 96131S09 PCT~EP96/01438
Intermediate 2
2-Bromo-2-(4-fluoro-PhenYI)-1 -(4-methanesulfonvl-PhenYI)~L! Idl 1~1 ,e
A solution of the 2-~4-fluoro-phenyl)-1-(4-~eli Idl ~esulfonyl-phenyl)-ethanone
(1.69) in dichloromethane (30ml) and glacial acetic acid (.15ml) was cooled to 0~
and llt:dled with 48% h~ lJrc,mic acid (3 drops). A solution of bromine
(875mg) in acetic acid (2ml) was added and stirring continued for 4 hours. The
mixture was diluted with dichlGro",t:ll,ane (35ml) and the solution was washed
with water dried and co"ce"l,dlec~ to give the title compound as a yellow solid
(2.09).
m.p. 124-126~
Intermediate 3
3-Trifluoromethvl-pvridin-2-vlamine
A mixture of 2-chloro-3-trifluorùinelhyl-pyridine (59) copper(1) iodide (59) andliquid ar"",o,1ia (50ml) was heated in an autoclave at 80~ (internal temperature)
for 28 hours. The cooled reaction mixture was slurried with
methanol/chlo,urul,l, (1/1-250 ml) and filtered. The filtrate was absorbed onto
silica and purified by flash chromatography eluting with ethyl acetate/hexane
(1/1) to yield the title comPound as a white solid (1.49).
m.p. 71-72~
MH~ = 163
Tlc sio2, Rf 0.50 (ethyl acetate/hexane (1/1)) detection UV/KMnO4
Intermediate 4
2-(4-Fluoro-phenyl)-1 -~4-methanesulfanYI-PhenYI)~ll Idl lone
A mixture of 2-(4-fluoro-phenyl)-1 -(4-fluoro-phenyl)-ethanone (10.0g) sodium
methanethiolate (3.0g~ and di,netl,),rl sulphoxide (10ml) was heated at -100~ for
8h under nitrogen. The cooled mixture was added to water (250ml) stirred for
10 min and filtered. The resulting solid was crystallised from isopropanol
(100ml) twice to give the title compound as a white solid (5.0g).
m.p.143-144~
Intermediate 5
2-Bromo-2-(4-fluoro-phenyl)-1 -(4-methanesulfanvl-phenYI)-ethanone
A solution of 2-(4-fluoro-phenyl)-1-(4-methanesulfanyl-phenyl)-ethanone (4.89)
in dichloromethane (75ml) and acetic acid (30ml) at 0~ containing 48% HBr (15
drops) was treated dropwise with bromine (2.69) in acetic acid (6ml). The
CA 02216809 1997-09-29
W O96/31509 PCTAEP96/01438
16
solution was stirred at 0~ for 10 min and at room temperature for 3h, diluted with
dichloromethane (100ml) and washed with water (2 x 100ml). The dried
(MgSO4) orga~ic phase was evapo,~led and the residue was triturated with
diethyl ether (30ml) to give the title cor,ll)ound as a white $olid (4.59).
m.p.117-119~.
Tlc SiO2 (Et20:hexane 1:1) Rf 0.6 det u.v. KMnO4, isopropanol.
Intermediate 6
3-(4-Fluoro-Phenvl)-2-(4-l l lelhal1esulfanvl-Phenvl)-imidazor1,2-a1Pvridine
A solution of 2-bromo-2-(4-fluoro-phenyl)-1-(4-methanesulfanyl-phenyl)-
ethanone (4.09) and 2-aminopyridine (1.2g) in acetonitrile (25ml) was relluxed
under nitrogen for 2h and left at room temperature for 16h. The soluticn was
evaporated and the residue purified by flash column chromatography eluting
with diethyl ether (applied in CH2CI2) to give the title compound as a white solid
(1.69).
m.p. 115-118~
Tlc SiO2 (Et2O) Rf 0.5 det u.v., isopropanol.
Intermediate 7
8-Chloro-3-(4-fluoro-PhenYI)-2-(4-methanesulfanvl-PhenYl)-imidazo~1,2-
alpyridine
n-Butyllithium in hexane (1.6m; 0.35ml) was added dropwise to a solution of 3-
(4-fluoro-phenyl)-2-(4-methanesulfanyl-phenyl)-imidazo[1,2-a]pyridine (167mg)
in tetrahydrofuran (2ml) at -78~ under nitrogen. The solution was stirred at -78~
for 1 h and was treated with l ,exachloroethane (142mg) in tetrahydrofuran
(0.25ml). The solution was allowed to warm to room temperature during 30 min
and water (2ml) was added. The mixture was extracted with ethyl acetate (2 x
5ml) and the dried (MgSO4) extract was evaporated to give the title compound
as a cream solid (180mg).
m.p.148-149~
MH ' = 369
Intermediate 8
3-Methvlsulfanyl-pvridin-2-ylamine
A solution of 2,2-dimethyl-N-pyridin-2-yl-propionamide2 (890mg) in dry
tetrahydrofuran (30ml) was cooled to -78~ and treated with a solution of n-butyllithium (6.25 ml,1.6M). This solution was stirred at 0~ for 4 hours and a solution
CA 02216809 1997-09-29
W O 96/31509 PCTAEP96101438
17
of dimethyldisulphide (235mg) in tetrahydrofuran (5ml) was added and stirring
continued at ~",~ienl ter"perdlure for 30 minutes. 2N Hydrochloric acid (1ml)
was added and the solution col lce~ led in vacuo. A mixture of the residue and
2N hy~ ric acid (15ml) was heated at reflux for 4 hours. The cooled
reaction mixture was made basic by addition of solid potassium carbonate. The
basic mixture was e~L~a~ed with ethyl ~cet~e (15ml),the organic extract was
dried (Na2SO4) and absorbed onto silica. The title compound was obtained by
flash c~,rc""dlog,~ully eluting with ethyl acetate/cyclohexane (1/1) as a colourless
oil (490mg).
MHt = 141
Tlc, SiO2, Rf 0.48,(ethyl acetate/cyclohexane(1/1)) detec~ion u.v., KMnO4
2 Ref: J A Turner J. Or~3. Chem.1983,48.3401
I. ,l~r " ,ediate 9
3-(4-Fluoro-phenyl)-2-~4-methanesulfonyl-Phenyl)-imidazor1,2-alpvridine-8-
ca, ~oxYlic acid methvl ester
A solution of 2-bromo-2-(4-fluoro-phenyl)-1-(4-methanesulfonyl-phenyl)-
ethanone (700mg), and 2-amino-nicotinic acid methyl ester (287mg) in dry
aceto, lill ile was heated at reflux overnight. The reaction mixture was
concentrated onto silica and the title comPound obtained by flash
chromatography eluting with ethyl acetate:cyclohexane 5:1 as a yellow solid
(176mg).
MH+ = 425
Tlc, SiO2 Rf 0.21 (ethyl acetate:cyclohexane 5:1) detection uv
Intermediate 10
8-Acetyl-3-(4-fluoro-phenyl)-2-(4-methanesulfanvl-Phenyl)-imidazo~1,2-
alpvridine
n-Butyllithium in hexane (1.6m; 0.5ml) was added dropwise to a solution of 3-(4-fluoro-phenyl)-2-(4-methanesulfanyl-phenyl)-imidazo[1,2-a]pyridine (250mg) in
tetrahydrofuran (3ml) at -78~ under nitrogen. The solution was stirred at -78~ for
1 h and was treated with N-methyl,N-methoxyacetamide (9Omg) in
tetrahydrofuran (0.25ml). The solution was allowed to warm to room
temperature during 60min and water (10ml) was added. The mixture was
extracted with ethyl acetate (2 x 5ml) and the dried (MgSO4) extract was
CA 02216809 1997-09-29
W O~6/31509 PCTAEP96/01438
18
evaporated. The residue was purified on a column of silica, eluting with
hexane:diethylether (3:1) to give the title co""~ound as a cream solid (130mg).
MH+ = 377
Tlc SiO2 (Et20) Rf 0.9 det u.v.
ExamPle 1
3-14-Fluoro-PhenYI)-2-(4-methanesulfonYl-phenv~ midazor1,2-alPvridine
A solution of the 2-bromo-2-(4-fluoro-phenyl)-1-(4-methanesulphonyl-phenyl)-
ethanone (233mg) and 2-aminopyridine (59mg) in dry acetonitrile was heated at
reflux overnight. The reaction mixture was adsorbed onto silica and purified by
flash cl ,rumalography eluting with ethyl ~cet~te to afford the title compound as a
cream solid (100mg).
MH+ = 367.2
m.p.198-200~
Example 2
3-(4-Fluoro-Phenvl)-2-(4-methanesulfonvl-Phenvl)-8-methvl-imidazor1,2-
alPyridine
A solution of the 2-bromo-2-(4-fluoro-phenyl)-1-(4-methanesulphonyl-phenyl)-
ethanone (233mg) and 2-amino-3-methylpyridine (68mg) in dry acetonitrile
(10ml) was heated at reflux overnight. The reaction mixture was adsorbed onto
silica and purified by flash cl ,ru" ~dlography eluting with
dichloromethane/methanol (19:1) to afford the title cGm~ound as a pale brown
solid (61mg).
MH+ = 381
m.p.180-182~
Exampie 3
3-(4-Fluoro-Phenvl)-2-(4-methanesulfonYI-PhenYl~-8-trifluoromethyl-imidazo~1,2-
alpyridine
A solution of 2-bromo-2-(4-fluoro-phenyl)-1-(4-methanesulphonyl-phenyl)-
ethanone (742mg), and 3-trifluoromethyl-pyridin-2-ylamine (324mg,) in dry
acetonitrile containing potassium carbonate (276mg) was heated at reflux
overnight. The reaction mixture was conce,llrdled onto silica and purified by
flash chromatography eluting with ethyl acetate, to give a yellow solid. Further
CA 02216809 1997-09-29
PCT~EP96/01438
WO 9613150g
19
purification by recrystallisation from propan-2-ol yielded the title comPound asyellow crystals (21 Omg).
m.p. 260-261~
Analysis: Found: C,57.7; H,3.0; N,6.2; S,7.3
C2,H,4F4N202S Requires: C,58.1; H,3.25;N,6.45;S,7.4%
MH' = 435
Tlc, sio2, Rf 0.25, (ethyl ~cet~te) detection u.v.
Example 4
3-(4-Fluoro-PhenY1)-2-(4-methanesulPhonvl)-7-methvl-imidazor1 ~2-alPvridine
A solution of the 2-bromo-2-(4-fluoro-phenyl)-1 -(4-methanesulphonyl-phenyl)-
ethanone (233mg) an~ 2-amino4-methylpyridine (68mg) in acetonitrile 10ml)
was heated at reflux for 18 hours. The cooled reaction mixture was absorbed
onto silica and the title comPound obtained by flash chromatography, eluting
with ethyl acetate /hexane (111), as a bu~f solid (105mg).
m.p.194-196~
MH+ = 381
Tlc, sio2, Rf 0.26, (ethyl acetate/hexane (2/1)) detection u.v.
Example 5
8-Chloro-3-(4-fluoro-Phenvl)-2-(4-methanesulfonvl-Phenvl~-imidazo~1 2-
alpyridine
A suspension of 8-chloro-3-(4-fluoro-phenyl)-2-(4-methanesulfanyl-phenyl)-
imidazo~1,2-a]pyridine (150mg) in methanol (8ml) and water (2ml) was treated
with Oxone TM (561mg) and stirred for 2h at room temperature. The resulting
suspension was treated with water (50ml) and extracted with ethyl acetate (2 x
50ml). The dried (MgSO4) extract was evaporated and the resulting solid was
treated with boiling isopropanol (8ml) for (10min) cooled and filtered to give the
title compound as a white solid (85mg).
m.p. 242-244~
Tlc SiO2(Et2O) RF 0.5 det u.v., KMnO4
MH+ = 401
CA 02216809 1997-09-29
W O96/31509 PCTAEP96/01438
Example 6
3-(4-Fluoro-Phenvl)-2-(4-, 1 ,eli Idl ,esulfonyl-phenyl)-8-meU Idl ,esulfanvl-
imidazo~1 2-alpvridine
A solution of 3-methylsulfanyl-pyridin-2-ylamine (140 mg) and 2-bromo-2-(4-
fluoro-phenyl)-1-(4-methanesulfonyl-phenyl)-ethanone (371 mg) in acetonitrile
(1 5ml) was heated at reflux ove, l ligl ,L. The cooled reaction mixture was
absorbed onto silica and the title compound obtained by flash chromatography
eluting with ethyl acetdle/cyclohexane (1/1) as a white solid (179 mg).
m.p. 224-226~
MH+=413
Tlc, SiO2, Rf 0.60 (ethyl acetate/cyclohexane(1/1)) detection u.v./KMnO4
Example 7
8-Bromo-3-(4-fluoro-phenYI)-2-(4-l, letl ,a, ~esulfonvl-Phenvl)-imidazo~1 2-
alpvridine
A solution of 3-bromo-pyridin-2-ylamine3 (173 mg) and 2-bromo-2-(4-fluoro-
phenyl)-1-(4-rllell,a"esulfonyl-phenyl)-ethanone (371 mg) in acetonitrile (15ml)was heated at reflux overnight. On cooling to room temperature the title
compound crystallised from solution and was isolated by filtration as a white
solid (241 mg).
m.p 266-268~
MH ' = 446
Analysis: Found: C,53.5;H,3.0;N,6.2;F,4.3;S,7. 1
C20H,4BrFN2O2S Requires: C,53.9;H,3.2;N,6.3;F,4.3;S,7.2%
Tlc, sio2, Rf 0.61 (Ethyl acetale/cyclohexane(2/1 )) detection u.v., KMnO4
3 Ref: M.Malinowski,Bull. Soc. Chim. Belg. 1988, 97, 51
Example 8
8-Fluoromethvl-3-(4-fluoro-phenvl)-2-t4-methanesulPhonvl-Phenyl)-imidazo~1 ,2-
alpyridine
A solution of 3-(4-fluoro-phenyl)-2-(4-methanesulphonyl-phenyl)-imidazo[1,2-
a]pyridine-8-methanol (200mg) in dichloromethane (1 Oml) was added to a
cooled solution of diethylaminosulphur trifluoride (0.067 ml) in dichloromethane(4 ml) at -78~ over 5 minutes. The solution was allowed to warm to room
temperature over 30 minutes. Water (15 ml) was added cautiously with stirring,
the organic phase was separated, dried (Na2SO4) and absorbed onto silica.
The title compound was obtained by flash chromatography eluting with ethyl
CA 02216809 1997-09-29
W O 96131509 PCT~EP96101438
21
ace~ale/cyclol ,e~dne (1/1) as a white solid which was further purified by
crystallisation from ,~r~a, l-2~1 (10 ml) yielding white crystals (85 mg).
m.p. 221-222~
MH ' = 399
Analysis Found: C,63.1; H,3.9; N,6.8, F,9.4; S,8.1
C2,H16F2N202S Requires: C,63.3; H,4.05; N,7.1, F;9.5: S,8.05%
Example 9
3-(4-Fluoro-PhenYI)-2 -(4-methanesulfonYI-phenyl)-7~8-dimethyl-imidazor1,2-
alPvridine
A solution of 3,4~imethyl-pyridin-2-ylamine4 (122 mg) and 2-bromo-2-(4-fluoro-
phenyl)-1 -(4-rneli ,anesulphonyl-phenyl)-ethanone (371 mg) in acetonitrile
(15ml) containing potassium carbonate (138 mg) was heated at reflux overnight.
The cooled mixture \A~as absorbed onto silica and the title compound obtained
by flash chromatography eluting with ethyl acetate/cyclohexane (1/1) as a white
solid. Cryst~llis~tion lrom propan-2-ol (10 ml) yielded white crystals (136 mg).m.p. 228-230~
MH+ = 395
Analysis Found: C,66.6; H,4.7; N,6.9; F,4.8; S,8.2
C22H1gN2FO2S Requires: C,67.0; H,4.85; N,7.1; F,4.8; S8.1%
4 Ref: W O Siegl, J Het. Chem. 1981, 18,1613-18
Example 10
3-~4-Fluoro-phenyl)-2-(4-methanesulfonyl-phenyl)-imidazor1 ,2-alPyridine~-
carbaldehyde
A solution of the [3-(4-fluoro-phenyl)-2-(4-methanesulfonyl-phenyl)-imidazo[1,2-a]pyridin-8-yl]-methanol (500mg) and manganese (IV) oxide (1.329) in
chlororor", (50ml) was heated at reflux for 16 hours. The cooled reaction
mixture was filtered and the filtrate conce, Ill dled onto silica. The title compound
was obtained by flash chromatography eluting with ethyl acetate/cyclohexane
(4/1) as a bright yellow solid (315mg).
MH+ = 395
Analysis Found: C,63.71; H,3.55; N,6.89; S,8.07;
C2,H,5FN203S Requires: C,63.95; H,3.83; N,7.10; S,8.13%.
CA 02216809 1997-09-29
W O96/31509 PCTAEP96/01438
22
ExamPle 1 1
5-Bromo-3-(4-fluoro-PhenYI)-2-(4-methanesulfonvl-PhenYI)-8-methvl-
imicl~o~1.2-alPYridine
A solution of 3-(4-fluoro-phenyl)-2-(4-mell Idl ~esulfonyl-phenyl)-8-methyl-
imidazo[1,2-a]pyridine (380 mg ) in chloror~,lll (20 ml) was treated with solid
N-L,r~.r"osl~ccinimide (178 mg) in one portion at ambient temperature, and the
resulting solution stirred overnight. Water (50 ml) was added and the organic
phase collected, dried (Na2SO4) and absorbed onto silica. The title compound
was obtained by flash ~;I,ro",é,lography eluting with cyclohexane/ethyl acetate
(1/1) as a white solid (106 mg).
m.p. 277-279~ (dec)
MH~ = 461
Tlc, sio2, Rf 0.22(Ethyl acetate/cyclohexane(1/1)) detection uv, KMnO4
ExamPle 12
8-Acetvl-3-(4-fluoro-PhenYI)-2-(4-methanesulfonYl-phenyl)-imidazor1,2-
alPYridine
A suspension of 8-acetyl-3-(4-fluoro-phenyl)-2-(4-methanesulfanyl-phenyl)-
imidazo[1,2-a]pyridine (130mg) in methanol (8ml) and water (2ml) was treated
with Oxone TM (436mg) and stirred for 3h at room temperature. The resulting
suspension was treated with water (50ml) and extracted with ethyl acetate
(50ml). The dried (MgSO4) extract was evapor~led and the resulting solid was
treated with boiling isopropanol (3ml) for (10min), cooled and filtered to give the
title comPound as a white solid (85mg).
m.p. 236-237~
Tlc SiO2 (Et20) Rf 0.5 det u.v., KMnO4
MH+ = 401
Example 13
6-Bromo-3-(4-fluoro-phenyl)-2-(4-methanesulfonYI-phenYI)-8-methYI-
imidazo~1,2-alpyridine
A mixture of 1-amino4-bromo-2-methylpyridine (500mg), 2-bromo-2-(4-fluoro-
phenyl)-1 -(4-methanesulfonyl-phenyl)-ethanone (1.0g) and acetonitrile (1 Oml)
was refluxed under nitrogen for 20h and evaporated. The residue was triturated
CA 02216809 1997-09-29
W O96/31509 PCTAEP96/01438
23
with diethyl ether (20ml) for 5 min, cooled and flltered. The procedure was
repeated to give the title co""~ourld as a beige powder (580mg).
MH+ = 461
Tlc SiO2 (Et20) Rf O.6 ~lel~ctiGn u.v., KMnO4
Example 14
8-Acetvl-3-(4-fluoro-PhenYI)-2-f4-methanesulfonYl-phenvl)-imidazor1,2-
alpyridine
A solution of 3-acetyl-2-ar"i"o~yridine5 (2.50g) and 2-bromo-2-(4-fluorophenyl)-1-(4-methanesulphonylphenyl)elll~llolle (6.829) in acetonitrile (125ml)
containing sodium biG~ Grldle (2.31g) was heated at reflux overnight. The
mixture was filtered hot and the filtrate left to cool to room temperature. The title
compound crystallise~l from solution and was isolated by filtration as a yellow
solid (3.589).
Tlc SiO2 Rf 0.34 (ethyl acetate/hexane,1.3:1) detection u.v., iodine
'H NMR 3.04(3H,s~cH3sor)~ 3.16(3H,s,CH3CO-), 6.90(1H,t,J=7.0Hz,H~),
7.31 (2H,t,J=8.8Hz) & 7.45(2H,dd,J= 5.2Hz,8.8Hz) - C6H4F-, 7.85(2H,d,J=8.4Hz)
& 7.91 (2H,d,J=8.4Hz) - C6H4SO2, 7.93(1 H,dd,J=1.1,7.0Hz, H-7),
8.02(1H,dd,J=1.1,7.0~1z, H-5).
5 Ref: F.Trecourt et al, J. Chem. Soc., Perkin Trans.1 1990, 9, 2409-2415
Example 15
r3-(4-Fluoro-Phenvl)-2-(4-methanesulfonYI-PhenYI)-imidazo~1,2-alpyridin-8-yll-
methanol
A solution of 3-(4-fluoro-phenyl)-2-(4-methanesulfonyl-phenyl)-imidazo[1,2-
a]pyridine-8-carboxylic acid methyl ester (1.859) in dry tetrahydrofuran (130ml)was cooled to -78~ and l,~dled with diisobutylaluminium hydride (17.4ml;1.0M
solution in dichloromethane). Once the addition was complete the mixture was
allowed to warm up to 25~ and stirring continued for 2 h. Methanol (80ml) was
added and the mixture co"ce, lll dled onto silica. The title compound was
obtained by flash chromatography eluting with
dichloromethane/ethanol/ammonia, 100/8/1, as a white foam (1.549).
MH+ = 397
Tlc (sio2) Rf 0.28 (dichloromethane/ethanol/ammonia,100/8/1) detection U.V.
CA 022l6809 l997-09-29
W 096/31509 PCT~P96/01438
24
ExamPIe 16
r3~4-fluoro-PhenYI)-2-(4~ ll Id nesulPhonvl-PhenYI)-imidazo~1 2-alPyridin-
8-vll-ethan-1-ol
A solution of [3-(4-fluoro-phenyl)-2-(4-r,lt:ll,a"esulfonyl-phenyl)-imidazo[1 2-a]pyridin-8-yl]-l"~:ll,anol (204 mg) in dry dichlGro",etl,al,e (10 ml) was cooied to -
78~. A solution of ~iisob~tylaluminium hydride (1.0 M in dichloromethane 1ml)
was added dropwise and the mixture allowed to warm to ambient te",,~ eralure
over 30 minutes. M_;ha~-ol (10 ml) was added in one portion and stirring
continued for a further 30 minutes. The reaction mixture was absorbed onto
silica and the title co",pound obtained by flash chromatography eluting with
ethyl acetale as a white solid (120 mg)
m.p.201 -203~
T.l.c. SiO2 Rf 0.45 (ethyl ace~ate) detection U.V.
ExamPle 17 - Tablets
a) Compound ofthe invention 5.0mg
I ~ctose 95.0mg
Microcrystalline Cellulose 90.0mg
Cross-linked polyvinylpyrrolidone 8.0mg
Magnesium Stearate 2.0mg
Compression weight 200.0mg
The compound of the invention microcrystalline cellulose lactose and cross-
linked polyvinylpyrrolidone are sieved through a 500 micron sieve and blended
in a suitable mixer. The magnesium stearate is sieved through a 250 micron
sieve and blended with the active blend. The blend is col"pressed into tablets
using suitable punches.
b) Compound ofthe invention 5.0mg
Lactose 165.0mg
Pregelatinised Starch 20.0mg
Cross-linked polyvinylpyrrolidone 8.0mg
Magnesium Stearate 2.0m~
Compression weight 200.0mg
CA 02216809 1997-09-29
W O96/31509 PCTAEP96/01438
The compound of the invention l~tose and pre~el~tinised starch are blended
together and granulated with water. The wet mass is dried and milled. The
magnesium ~leardle and cross-linked polyvinylpyll~lido,le are screened
through a 250 micron sieve and blended with the granule. The resultant blend
is co""~ressed using suitable tablet punches.
Example 18 - CaPsules
a) Compound ofthe invention 5.0mg
Lactose 1 93.0mg
Magnesium Stearate 2.0m~
Fill weight 200.0mg
The compound of the invention and pregelatinised starch are screened through
a 500 micron mesh sieve blended together and lubricated with magnesium
sLearale (meshed through a 250 micron sieve). The blend is filled into hard
gelatine capsules of a suitable size.
b) Compound o~ the invention 5.0mg
Lactose 1 77.0mg
Polyvinylpyrrolidone 8.0mg
Cross-linked polyvinylpyrrolidone8.01Tg
Magnesium Stearate 2.0m~
Fill weight 200.0mg
The compound of the invention and lactose are blended together and
granulated with a solution of polyvinylpyrrolidone. The wet mass is dried and
milled. The magnesium stearate and cross-linked polyvinylpyrrolidone are
screened through a 250 micron sieve and blended with the granules. The
resultant blend is filled into hard gelatine capsules of a suitable size.
CA 02216809 1997-09-29
W O96/31509 PCT~EP96/01438
26
ExamPle 19 - SYruP
a) Compound ofthe invention 5.0mg
Hydroxypropyl Methylcellulose 45.0mg
Propyl Hydroxybe" oale 1.5mg
Butyl Hydroxyber, odle 0.75mg
Saccharin Sodium 5.0mg
Sorbitol Solution 1 .Oml
Suitable Buffers qs
Suitable flavours qs
Purified Waterto 10.ml
The hydroxypropyl methylcellulose is dispersed in a portion of hot purified water
together with the hydroxyben~o~es and the solution is allowed to cool to
ambient temperature. The sac~harin, sodium flavours and sorbitol solution are
added to the bulk solution. The compound of the invention is dissolved in a
portion of the remaining water and added to the bulk solution. Suitable buffers
may be added to control the pH in the region of maximum stability. The solution
is made up to volume, filtered and filled into suitable containers.
ExamPle 20 - Iniection Formulation
% w/v
Compound of the invention 1.00
Waterfor injections B.P. to 100.00
Sodium chloride may be added to adjust the tonicity of the solution and the pH
may be adjusted to that of maximum stability and/or to facilitate solution of the
compound of the invention using dilute acid or alkali or by the addition of
suitable buffer salts. Antioxidants and metal chelating salts may also be
included. The solution is clarified, made up to final volume with water and the
pH remeasured and adjusted if necessaly, to provide 10mg/ml of the compound
of formula (I).
The solution may be packaged for injection, for example by filling and sealing in
ampoules, vials or syringes. The ampoules, vials or syringes may be
aseptically filled (e.g. the solution may be sterilised by filtration and filled into
sterile ampoules under aseptic conditions) and/or terminally sterilised (e.g. by
CA 022l6809 l997-09-29
W O 96/31~09 ~CT~EP96/01438
27
heali,~g in an ~l~t~ol~ve using one of the acce~,taL,le cycles). The solution may
be packed Llnder an inert ~ll"os,~l ,er~ of nitrogen.
r~ ~rer~bly the soll ~tion is filled into ampoules, sealed by fusion of the glass and
terminaily sterilised.
Further sterile formulations are ~lepared in a similar manner containing 0.5, 2.0
and 5% w/v of the co",,,~ound of formula (I), so as to provide respectively 5, 20
and 50mg/ml of the c4",,l~0und of formula (I).
Biolo~ical Data
Inhibitory activity against human COX-1 and COX-2 was assessed in COS cells
which have been stably l,;ansrected with cDNA for human COX-1 and huma
COX-2. 24 Hours prior to experiment, COS cells were l,ansrerred from the
175cm2 flasks in whi~h they were grown, onto 24-well cell culture plates using
the following procedure. The incubation medium, being Dulbecco's modified
eagles medium (DME~M) to which had been added heat inactivated foetal calf
serum (10%v/v), penicillin (100U Iml), streptomycin (10011g/ml) and geneticin
(600~g/ml), was removed from a flask of confluent cells (1 flask at confluency
contains approximately 1x10' cells). 10ml of phosphate buffered saline (PBS)
was added to the flask to wash the cells. Having removed the PBS, cells were
rinsed in 10ml trypsin for 20 seconds, after which the trypsin was removed and
the flask placed in an inc~h~tor (37~) for 1-2 minutes until cells became loose.The flask was then removed from the inc~h~tor and cells resuspended in 10ml
of fresh incubation medium. The conlenls of the flask was transferred to a
250ml sterile conlai"er and the volume of incubation medium subsequently
made up to 100ml. 1ml cell suspension was pipetted into each well of 4x24-well
cell culture plates. The plates were then placed in an incubator (37~C, 95%
air/5% CO2) overnight. If more than 1 flask was used, the cells were combined
before being dispensed into the 24-well plates.
Following the overnight incubation, the incubation medium was completely
, 30 removed from the 24-well cell culture plates and replaced with 250111 fresh
DMEM (37~C). Test compounds were made up to 250x the required test
concentration in DM~O and were added to the wells in a volume of 1~L1. Plates
were then mixed gently by swirling and then placed in an incubator for 1 hour
CA 02216809 1997-09-29
W O96/31509 PCTAEP96/01438
28
(37~C, 95% air/5% C02). Following the incubation period, 10111 of arachidonic
acid (750~1M) was added to each well to give a final aracl .i ~' . ,ic acid
conce"l~lion of 3011M. Plates were then ina Ih:~ted for 15 minutes, after which
the inc~ Ih~tion medium was removed from each well of the plates and stored at
-20~C, prior to ~leter",i"dlion of prost~ ,din E2 (PGE2) levels using enzyme
immunoassay. The inhibitory potency of the test compounds was expressed as
an IC50 value, which is defined as the conce, lll dlion of the compound required to
inhibit the PGE2 release from the cells by 50%. The selectivity ratio of inhibition
of COX-1 versus COX-2 was calculated by con,~arir,g respective IC50 values.
The following IC50 values for inhibition of COX-2 and COX-1 were obtained for
compounds of the invention:
. . .
Example l~Jo~COX-2: lC~0~nM)COX-1,. IC~OlnM~
428 >100,000
2 54 >10,445
3 144 >100,000
4 159 >100,000
38 >100,000
6 212 >100,000
7 71 >100,000
8 38 >100,000
9 38 >100,000
476 >100,000
11 18 6,295
12 142 >100,000
13 275 >100,000
750 >100,000
16 650 >100,000
-