Note: Descriptions are shown in the official language in which they were submitted.
CA 02216997 1997-09-30
TITLE OF THE INVENTION
MOLECULAR DIAGNOSTIC OF GLAUCOMAS
ASSOCIATED WITH CHROMOSOME 1
5 FIELD OF THE INVENTION
The present invention relates to the identification of
mutations in the GLC1A gene and the detection of these mutations in
individuals. The invention also relates to individuals being genotypically
homozygote mutant in an autosomal dominant inherited disease yet
10 being phenotypically normal.
BACKGROUND OF THE INVENTION
Glaucoma encompasses a complex of ocular-disease
entities characterized by an optic neuropathy in which degeneration of
15 retinal ganglion cells leads to a characteristic excavation of the head of
the optic nerve (Shields et al., 1996, The Glaucomas, _:717-725). Such
damage causes progressive narrowing of the visual fields and, when
unconl~olled, blindness. Affected people often have ocular hypertension
defined as intraocular pressures consistently >21 mm Hg in both eyes.
20 Although ocular hypertension is no longer an obligatory diagnostic
criterion for glaucoma, it is still recognized as one of the most important
risk factors (Wilson et al., 1996, The Glaucomas, _:753-763). Until now,
a diagnosis of glaucoma is made after observation of the characteristic
atrophy of the optic nerve, which is associated with typical visual field
25 defects.
In 1992, the World Health Organization estimated that,
in the global population, 5.2 million people were blind as a result of
CA 02216997 1997-09-30
glaucoma (Thylefors et al., 1994, World Health Organ. Bull., 72:323-326),
making it the third leading cause of blindness worldwide. The most
con"non form is adult-onset primary open-angle glaucoma (MIM 137760;
McKusick, 1994, Johns Hopkins University Press, p. 272), which
5 represents -50% of all cases of glaucoma. Among Caucasians, this form
of the disorcler affects ~2% of the population >45 years old (Leske, 1983,
Am. Epidemiol., 1 18:166-191; Thylefors et al., 1994, supra; Wilson et al.,
1996, supra). In African Americans, prevalence of adult-onset open-angle
glaucoma is three to four times higher than that observed in White
10 Americans. More than 15 million North Americans may have some form
of glaucoma, but at least half of them may not be aware of it.
The glaucomas traditionally have been grouped into
three categories: open angle, closed angle (also termed "angle closure"),
and congenital. Each subtype has been further arbitrarily subdivided into
15 primar~, when the anterior chamber of the eye appears normal and no
cause for glaucoma can be identified, or secondary, when glaucomas are
caused by underlying ocular or systemic conditions (Shields et al., 1996,
supra). Whereas the division between open and closed angles refers to
the configuration of the irido-corneal angle in the anterior chamber of the
20 eye, congenital glaucoma is used to define one of the many types of
developmental gl~ucoma that usually occurs within the 1 st year of life.
The majority (60%-70%) of primary glaucomas are of the open-angle
type. Primary open-angle glaucomas have been further subdivided into
two groups according to age at onset, severity, and mode of inheritance:
25 the more prevalent is middle- to late-age-onset chronic open-angle
glaucoma (COAG), by convention diagnosed after age 35 years and
characterized by its slow, insidious course (Shields et al., 1996, supra;
CA 02216997 1997-09-30
Wilson et al., 1996, supra). The less common form, juvenile open-angle
glaucoma (JOAG), occurs between 3 years of age and early adulthood
and generally manifests highly elevated intraocular pressures with no
angle abno,l"alilies (Goldwyn et al., 1970, Arch. Ophtalmol., 84:579-582;
Francois, 1980, Am. J. Ophtalmol., 3:429-449; Jo~mso" et al., 1 996a, The
Glaucomas, 1:39-54).
Although the precise molecular defects leading to
open-angle glaucomas remain partly unknown, numerous advances in
basic and clinical sciences have begun to identify the molecular basis of
glaucomas by mapping the gene loci involved in the disease process.
Due to recent mapping successes, the difrerent forms of glaucoma will be
further identified by the names of the loci to which they have been
localized. According to the Human Genome Organization/Genome
Database nomenclature, "GLC' is the general symbol for the glaucoma
genes; "1", "2", and "3" are, respectively, the symbols for the open-angle,
angle-closure, and congenital subtypes of glaucoma; and "A", "B", and
"C" refer, respectively, to the first, second, or third gene mapped in each
subgroup.
JOAG is a rare but aggressive form of glaucoma that
usually segregates in an autosomal dominant fashion with high
penetrance (Stokes, 1940, Arch. Ophthalmol., 24:885-909; Crombie et
al., 1964, Br. J. Ophtalmol., 48:143-147; Lee et al., 1985, Ann.
Ophtalmol., 17:739-741; Johnson et al., 1993, Ophthalmology,
100:524-529). In a single large American pedigree affected by an
autosomal dominant form of JOAG, Sheffield et al. (1993, Nat. Genet.,
_:47-50) located a gene responsible for this condition, at 1 q21 -q31. This
locus, being the first open-angle glaucoma locus to be mapped, was
CA 02216997 1997-09-30
named "GLC1A." The GLC1A disease gene consistently was associated
with onset of the JOAG phenotype before the age of 70 years, highly
elevated intraocular pressures, and typical excavation of the head of the
optic nerve. Gonioscopy showed open angles with no anterior-chamber
5 abnol",alities. The GLC1A has subsequently been reported by Nguyen
et al. in US Patent 5,606,043 to encode the trabecular meshwork induced
glucocorticoid response (TIGR) gene. The gene sequence was first
submitted (13-JAN-1997) by Nguyen et al. to the GeneBank accession
# U85257. The TIGR sequence was modified on 19 April 1997 in
10 GeneBank following modifications by Nguyen submitted on
02-APR-1997. The accession number stayed the same # U85257.
Genetic maps of the human genome can be exploited
to rapidly locate human monogenic disorders. The final version of the
Généthon linkage map, which spans close to 100 % of the human
genome, was published in March 1996 (Dib et al., 1996, Nature,
380:152-154). This map consisls of 5,264 short tandem (AC/TG)n repeat
polymorphisms with a mean heterozygosity of 70%.
The nomenclature system for the markers is well known
in the field. The nomenclature used is decided by the Human Genome
20 Organization (HUGO) nomenclature committee. It is as follows: for
anonymous DNA sequences, the convention is to use D which is
equivalent to DNA followed by 1-22, X or Y to denote the chromosomal
number and location, then S stands for a unique segment and finally a
serial number. For example, marker D2S2161 is a DNA marker located
25 on chromosome 2 representing a unique segment. Its serial number is
2161 .
CA 02216997 1997-09-30
The nomenclature for the glaucoma genes is the
following:
"GLC' is the general symbol for the gl~ucoma genes; "1", "2", and "3"
are, respectively, the symbols for the open-angle, angle-closure, and
5 congenital subtypes of glaucoma; and, "A", "B" and "C" refer,
respecti~ely, to the first, second, or third gene mapped in each subgroup.
For example, the GLC1A locus was the first open-angle glaucoma locus
to be mapped, in this case to chromosome 1 q23-q25 in 1993. It was later
identified as the trabecular meshwork inducible glucocorticoid response
gene product (TIGR) (Stone et al, 1997, Science, 275: 668-670).
These markers are ~ccessible to all individuals. The
central data resource for the human gene mapping effort is the Genome
Data Base (GDB). It was established at Johns Hopkins University, School
of Medicine. GDB is updated regularly. It collects, organizes, stores and
distributes human genome mapping information. GDB is ~ccessible
electronically at WWW-URL: http://gdbwww.gdb.org/.
Alternatively, all the markers disclosed herein, except
D6S967, are short (CA)n repeat markers that have been developed in the
Généthon laboratory near Paris, France. These markers are also
accessible electronically at WWW-URL: http://www.genethon.fr/.
Therefore markers are accessible either at GDB or at
Généthon.
The first mutations identified in the TIGR gene that have
been shown to give rise to glaucoma were first reported by Stone et. al
(Science, 1997, 275:668~70). There are three mutations reported. No
other mutations relating to the TIGR gene have been reported. The
methodology used to identify these mutations was by amplifying
CA 02216997 1997-09-30
overlapping regions by polymerase chain reaction (PCR) performing
single-strand conformational polymorphism (SSCP) on the amplification
products and sequencing those DNA products that produced aber,a"l
band pattern on the SSCP. No quick method for mutational analyses for
the TIGR has been proposed.
The prior art as a whole teaches that a homozygote
mutant for an autosomal dominant disease should display a higher
penelrance than a heterozygote mutant. Heterozygote for an autosomal
dominant disease often exhibits variable penetrance.
The present description refers to a number of
documents the content of which is herein incorporated by reference.
SUMMARY OF THE INVENTION
The invention concerns the mutational analyses in the
GLC1A gene locus encoding the nGR gene (GeneBank accession
no. U85257).
The present invention provides means to identify at
least two nucleolide changes in the DNA sequence coding for TIGR that
result in an amino acid change in the TIGR gene.
The invention further demonstrates that these amino
acid changes result in mutations producing a disease state in individuals
the disease being glaucoma.
The early detection of individuals at risk for developing
glaucoma is an i",po,lanl aspect of this invention. Early detection allows
for intervention prior to the genesis of the disease process and disease
progression and may obviate the sy"~pto,ns and the onset of the disease.
A method for mutation analyses called amplification
CA 02216997 1997-09-30
rer,actory mutation system (ARMS), that is simple and quick is disclosed
herein. The proposed invention relates to the inclusion of primers and
probes for the amplification and detection of all mutations in the TIGR
gene. The invention teaches the use of the method of ARMS as related
5 to glaucoma but the invention is not limited to this method for mutation
analyses. Other methods known in the field for mutation analyses such
as allele specific oligonucleotide (ASO), denaturing gradient gel
electrophoresis (DGGE) and artificially created restriction site (ACRS)
can also be used.
These mutation detection and analyses can be
performed on either genomic DNA or DNA that has been transcribed to
cDNA by any method known to a person skilled in the art.
In addition the applicant has demonstrated for the first
time a new type of dominance in mammals in which heterozygotes have
15 a much higher penetrance rate for a disease gene mutation than their
homozygotic counterparts.
Further it is provided for the first time in an autosomal
dominant disease that a homozygote mutant is phenotypically normal.
Even though such an individual may give rise to an affected heterozygote
20 offspring.
The invention provides applications and uses for such
a discovery. These include but are not limited to:
a) treatment of heterozygote mutant affected individuals
with overexpressed mutant protein to induce protein complementation
25 such that normal protein function can be restored, this application will
apply to any autosomal dominant disease exhibiting the same mode of
action as described herein.
CA 02216997 1997-09-30
b) similarly an individual being a heterozygote for an
autosomal dominant disorder exhibiting the same mode of action as
described herein can be treated by gene therapy, such that a mutant
allele is inserted into a vector and delivered to an individual thereby
5 negating the effect of the heterozygote mutation by either allelic or
protein comple",entation.
c) with this new knowledge a transgenic animal
designed to carry a deleterious autosomal dominant mutation can be
used to assess the requirement to produce a phenotypically normal
10 animal, by either allelic complementation or protein complementation.
d) a diagnostic means to identify phenotypically normal
genotypically mutant individuals that can transmit the mutant allele to
their offsprings.
e) this knowledge can be used for showing dimerisation
15 of TIGR peptides.
In accordance with the present invention there is
therefore provided the means to easily identify novel mutations in the
TIGR gene, wherein these mutations give rise to glaucoma. These
mutations can also be identified by any other means known to a person
20 skilled in the art. As well these means disclosed in this application can
easily be part of a kit comprising probes, primers, oligonucleotides and
any reagents required in the methodologies for detecting mutations that
may cause glaucoma. These mutation analyses are useful for screening
individuals at risk for glaucoma. Such individuals may have a family
25 history of glaucoma, and, identifying individuals carrying a mutation in
glaucoma gene would permit early treatment that may obviate or minimise
the progression of the disease.
CA 02216997 1997-09-30
The invention and the al-plic~lions thereof will be made
obvious with the foregoing disclosure.
DEFINITIONS AND TECHNOLOGICAL BACKGROUND
Nucleotide sequences are presented herein by single
strand, in the 5' to 3' direction, from left to right, using the one letter
nucleotide symbols as commonly used in the art and in accordance with
the recommendations of the IUPAC-IUB Biochemical Nomenclature
Commission.
The present description refers to a number of routinely
used recombinant DNA (rDNA) technology terms. Nevertheless,
definitions of selected examples of such rDNA terms are provided for
clarity and consistency.
As used herein, "isol-~ed nucleic acid molecule", refers
to a polymer of nucleotides. Non-limiting examples thereof include DNA
and RNA molecules purified from their natural environment.
The term "recombinant DNA" as known in the art refers
to a DNA molecule resulting from the joining of DNA segments. This is
often referred to as genetic engineering.
The term "DNA segment", is used herein, to refer to a
DNA molecule comprising a linear stretch or sequence of nucleotides.
This sequence when read in accordance with the genetic code, can
encode a linear stretch or sequence of amino acids which can be referred
to as a polypeptide, protein, protein fragment and the like.
The terminology Uamplification pair" or Uprimer pair"
refers herein to a pair of oligonucleotides (oligos) of the present
invention, which are selected to be used together in amplifying a selected
CA 02216997 1997-09-30
nucleic acid sequence by one of a number of types of amplification
processes, preferably a polymerase chain reaction. Other types of
amplification processes include ligase chain reaction, strand
~ispl~cement amplification, or nucleic acid sequence-based amplification,
5 as explained in greater detail below. As commonly known in the art, the
oligos are designed to bind to a complei"enta~y sequence under selected
conditions.
The nucleic acid (i.e. DNA or RNA) for practising the
present invention may be obtained according to well known methods.
Oligonucleotide probes or primers of the present
invention may be of any suitable length, depending on the particular
assay format and the particular needs and targeted genomes employed.
In general, the oligonucleotide probes or primers are at least 10
nucleotides in length, preferably between 15 and 24 nucleotides, and
15 they may be adapted to be especially suited to a chosen nucleic acid
ampliricalion system. As commonly known in the art, the oligonucleotide
probes and primers can be designed by taking into consideration the
melting point of hydrizidation thereof with its targeted sequence (in
Sambrook et al., 1989, Molecular Cloning - A Laboratory Manual, 2nd
20 Edition, CSH Laboratories; Ausubel et al., 1989, in Current Protocols in
Molecular Biology, John Wiley & Sons Inc., N.Y.).
"Nucleic acid hybridization" refers generally to the
hybridization of two single-stranded nucleic acid molecules having
co",ple,ne"laly base sequences, which under appropriate conditions will
25 form a thermodynamically favored double-stranded structure. Examples
of hybridization conditions can be found in the two laboratory manuals
referred above (Sambrook et al., 1989, supra and Ausubel et al., 1989
CA 02216997 1997-09-30
supra) and are cor"",only known in the art. In the case of a hybridization
to a nitrocellulose filter, as for example in the well known Southern
blotting procedure, a nitrocellulose filter can be incubated overnight at
65~C with a labeled probe in a solution containing 50% formamide, high
salt ( 5 x SSC or 5 x SSPE), 5 x Denhardt's solution, 1% SDS, and 100
,ug/ml denatured carried DNA ( i.e. salmon sperm DNA). The
non-specifically binding probe can then be washed off the filter by several
washes in 0.2 x SSC/0.1% SDS at a temperature which is selected in
view of the desired stringency: room temperature (low stringency), 42~C
(moderate stringency) or 65~C (high stringency). The selected
temperature is based on the melting ter",~eralure (Tm) of the DNA hybrid.
~ Of course, RNA-DNA hybrids can also be formed and detected. In such
cases, the conditions of hybridization and washing can be adapted
according to well known methods by the person of ordinary skill. High
stringency conditions will be preferably used (Sambrook et al.,1989,
supra).
Probes of the invention can be utilized with naturally
occurring sugar-phosphate backbones as well as modified backbones
including phosphorothioates, dithionates, alkyl phosphonates and
a-nucleotides and the like. Modified sugar-phosphate backbones are
generally taught by Miller, 1988, Ann. Reports Med. Chem. 23:295 and
Moran et al., 1987, Nucleic acid molecule. Acids Res., 14:5019. Probes
of the invention can be constructed of either ribonucleic acid (RNA) or
deoxyribonucleic acid (DNA), and preferably of DNA.
The types of detection methods in which probes can be
used include Southern blots (DNA detection), dot or slot blots (DNA,
RNA), and Northern blots (RNA detection). Although less prepared,
CA 022l6997 l997-09-30
12
labelled proteins could also be used to detect a particular nucleic acid
sequence to which it binds. Other detection methods include kits
containing probes on a dipstick setup and the like.
Although the present invention is not specifically
dependent on the use of a label for the detection of a particular nucleic
acid sequence, such a label might be beneficial, by increasing the
sensitivity of the detection. Furthermore, it enables automation. Probes
can be labelled according to numerous well known methods (Sambrook
et al., 1989, supra). Non-limiting examples of labels include 3H, 14C, 32p,
and 35S. Non-limiting examples of detectable markers include ligands,
fluorophores, chemiluminescent agents, enzymes, and antibodies. Other
detectab'E markers for use with probes, which can enable an increase in
sensitivity of the method of the invention, include biotin and
radionucleotides. It will become evident to the person of ordinary skill that
the choice of a particular label dictates the manner in which it is bound
to the probe.
As commonly known, radioactive nucleotides can be
incorporated into probes of the invention by several methods.
Non-limiting examples thereof include kinasing the 5' ends of the probes
using gamma 32p ATP and polynucleotide kinase, using the Klenow
fragment of Pol I of E. co/i in the presence of radioactive dNTP (i.e.
uniformly labelled DNA probe using random oligonucleotide primers in
low-melt gels), using the SP6/T7 system to transcribe a DNA segment in
the presence of one or more radioactive NTP, and the like.
As used herein, "oligonucleotides" or"oligos" define a
molecule having two or more nucleotides (ribo or deoxyribonucleotides).
The size of the oligo will be dictated by the particular situation and
CA 02216997 1997-09-30
ultimately by the particular use thereof, and adapted accordingly by the
person of ordinary skill. An oligonucleotide can be synthetised chemically
or derived by cloning according to well known methods.
As used herein, a Uprimer" defines an oligonucleotide
5 which is capable of annealing to a target sequence, thereby creating a
double stranded region which can serve as an initiation point for DNA
synthesis under suitable conditions.
Amplification of a selected, or target, nucleic acid
sequence may be carried out by a number of suitable methods. See
generally Kwoh et al., 1990, (Am. Biotechnol. Lab. 8:14-25). Numerous
amplification techniques have been described and can be readily
adapted to suit the particular needs of a person of ordinary skill.
Non-limiting examples of amplification techniques include polymerase
chain reaction (PCR), ligase chain reaction (LCR), strand displacement
amplification (SDA), transcription-based amplification, the Q,~ replicase
system and NASBA (Kwoh et al., 1989, Proc. Natl. Acad. Sci. USA 86,
1173-1177; Lizardi et al.,1988, BioTechnoloyy _:1197-1202; Malek et al.,
1994, Melhods Mol. Biol.,28:253-260; and Sar"brook et al.,1989, supra).
Preferably, amplification will be carried out using PCR.
Polymerase chain reaction (PCR) is carried out in
accordancewithknowntechniques. See, e.g., U.S. Pat. Nos. 4,683,195;
4,683,202; 4,800,159; and 4,965,188 (the disclosures of all three U.S.
Patent are incorporated herein by reference). In general, PCR involves,
a treatment of a nucleic acid sample (e.g., in the presence of a heat
stable DNA polymerase) under hybridizing conditions, with one
oligonucleotide primer for each strand of the specific sequence to be
detected. An extension product of each primer which is synthesized is
CA 02216997 1997-09-30
14
compler"e"lary to each of the two nucleic acid strands, with the primers
sufficiently complementary to each strand of the specific sequence to
hybridize therewith. The exte"sion product synthesized from each primer
can also serve as a template for further synthesis of extension products
using the same primers. Following a sufficient number of rounds of
synthesis of extension products, the sample is analysed to assess
whether the sequence or sequences to be detected are present.
Detection of the amplified sequence may be carried out by visualization
following EtBr staining of the DNA following gel electrophoresis, or using
a detectable label in accordance with known te~ln.~ ~es, and the like. For
a review on PCR techniques (see PCR Protocols, A Guide to Methods
and Amplifications, Michael et al., Eds, Acad. Press,1990).
Ligase chain reaction (LCR) is carried out in accordance
with known techniques (Weiss, 1991, Science 254:1292). Adaptation of
the protocol to meet the desired needs can be carried out by a person of
ordinary skill. Strand d;spl~cement amplification (SDA) is also carried out
in accordance with known techniques or adaptations thereof to meet the
particular needs (Walker et al., 1992, Proc. Natl. Acad. Sci. USA
89:392-396; and ibid.,1992, NucleicAcids Res. 20:1691-1696).
As used herein, the term "gene" is well known in the art
and relates to a nucleic acid sequence defining a single protein or
polypeptide. A "structural gene" defines a DNA sequence which is
transcribed into RNA and translated into a protein having a specific
amino acid sequence thereby giving rise the a specific polypeptide or
protein. It will be readily recognized by the person of ordinary skilll that
the nucleic acid sequences of the present invention can be incorporated
CA 02216997 1997-09-30
into anyone of numerous established kit formats which are well known in
the art.
The term "vector" is commonly known in the art and
defines a plasmid DNA, phage DNA, viral DNA and the like, which can
5 serve as a DNA vehicle into which DNA of the present invention can be
cloned. Numerous types of vectors exist and are well known in the art.
The term "expression" defines the process by which a
structural gene is transcribed into mRNA (transcription), the mRNA is
then being translated (translation) into one polypeptide (or protein) or
1 0 more.
The terminology "e3~ression vector" defines a vector or
vehicle, as described above, but designed to enable the expression of an
inserted sequence following transformation into a host. The cloned gene
(inserled sequence) is usually placed under the control of control element
15 sequences such as promoter sequences. The placing of a cloned gene
under such control sequences is often referred to as being "operably
linked" to control elements or sequences.
Expression control sequences will vary depending on
whether the vector is designed to express the operably linked gene in a
20 prokaryotic or eukaryotic host or both (shuttle vectors) and can
additionally contain lrar,scri,c,lional elements such as enl ,ancer elements,
termination sequences, tissue-specificity elements, and/or translational
initiation and termination sites.
As used herein, the designation "functional derivative"
25 denotes, in the conlext of a functional derivative of a sequence, whether
nucleic acid or amino acid sequence, a molecule that retains a biological
activity (either functional or structural) that is subslanlially similar to that
CA 02216997 1997-09-30
16
of the original sequence. This functional derivative or equivalent may be
a natural derivative or may be prepared synthetically. Such derivatives
include amino acid sequences having substitutions, deletions, or
additions of one or more amino acids, provided that the biological activity
5 of the protein is cGnserved. The same applies to derivatives of nuclei
acid sequences which can have substitutions, deletions, or additions of
one or more nucleotides, provided that the biological activity of the
sequence is generally maintained. When relating to a protein sequence,
the substituting amino acid has chemico-physical properties which are
10 similar to that of the substituted amino acid. The similar chemico-physical
properties include, similarities in charge, bulkiness, hydrophobicity,
hydrophylicity and the like. The term Ufunctional derivatives" is intended
to include "fragmentsn, "segments", "variants", "analogs" or"chemical
derivatives" of the subject matter of the present invention.
Thus, the term "variant" refers herein to a protein or
nucleic acid molecule which is substantially similar in structure and
biological activity to the protein or nucleic acid of the present invention.
The functional derivatives of the present invention can
be synthesized chemically or produced through recombinant DNA
20 technology. All these methods are well known in the art.
As used herein, "chemical derivatives" is meant to cover
additional chemical moieties not normally part of the subject matter of the
invention. Such moieties could affect the physico-chemical characteristic
of the derivative (i.e. solubility, absorption, half life and the like, decrease25 of toxicity). Such moieties are exemplified in Remington's Pharmaceutical
Sciences (1980). Methods of coupling these chemical-physical moieties
to a polypeptide are well known in the art.
CA 02216997 1997-09-30
The term "allele" defines an alternative form of a gene
which occupies a given locus on a chromosome.
As commonly known, a "mutation" is a detectable
change in the genetic material which can be transmitted to a daughter
5 cell. As well known, a mutation can be, for example, a detectable change
in one or more deoxyribonucleotide. For example, nucleotides can be
added, deleted, substituted for, inverted, or transposed to a new position.
Sponlaneous mutations and experi"len~ally induced mutations exist. The
result of a mutations of nucleic acid molecule is a mutant nucleic acid
10 molecule. A mutant polypeptide can be encoded from this mutant nucleic
acid molecule.
As used herein, the term "purified" refers to a molecule
having been separated from a cellular component. Thus, for example, a
"purified protein" has been purified to a level not found in nature. A
15 "substantially pure" molecule is a molecule that is lacking in all other
cellular components.
The term "autosome" defines any chromosome other
that the sex chromosomes, X and Y.
The term "dominant" refers to an allele that determines
20 the phenotype displayed in a heterozygote with another (recessive)
allele.
The terminology "transgenic animal" defines an animal
that has had its germ line genetically modified to give rise to a progeny
animal that is different from the parental type and carrying the
25 modification in its germ line.
"Single Strand Conformational Polymorphism (SSCP)"
refers to a Ille~hod for detecting the presence of a base pair change in an
CA 02216997 1997-09-30
amplified DNA fragment. The method involves denaturing the double
stranded amplified DNA and comparing the band pdller" in a known
non-mutant rray",enl to that of an unknown fragment. A shift in the band
pattern is indicative of a base pair change.
The designation ~gene therapy" defines an attempt to
treat disease by genetic modification of the cells of a patient.
UAllele Specific Oligonucleotide (ASO)" are designed to
detect known and identified base pair change by designing
oligonucleotides that are specific to the DNA fragment with and without
the base change. These oligonucleotides are used as probes in
hybridisation protocols under stringent conditions. Differences in the
hybridization patterns is indicative of the presence or absence of the
base change.
"Artificially Created Restriction Site (ACRS)" refers to a
method for detection a known base change in a DNA sequence. It
involves the designing of a primer that may either create or obviate a
restriction site in the vicinity of known base change, such that the
resl, i~;tion endonuclease used can have a different digestion pattern for
the changed and unchanged base.
BRIEF DESCRIPTION OF THE DRAWINGS
Having thus generally described the invention, reference
will now be made to the accompanying drawings, showing by way of
illustration a prefel,ed embodiment thereof, and in which:
Figure 1 shows the sequence that encodes the wild-type
GLC1A/T1GR cDNA sequence.
CA 02216997 1997-09-30
19
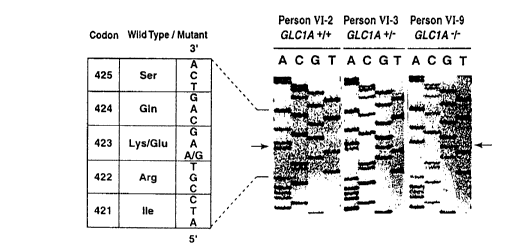
Figure 2 shows the characteri alion of a carrier
homozygous for the Lys423Glu TIGR mutation. a, Structure of the TIGR
encoded protein. The leucine zipper domain (amino acids 117-166) is
shown within the N-terminal half of the protein. The Lys423Glu mutation
5 is depicted by an open circle in the olfactomedin homology domain
represented by a striped boxe within the C-terminal half of the protein.
Amino acids comparison between human TIGR protein (amino acids
415437), human neuronal olfactomedin, rat and bullfrog neuronal
olraclon,edin-related proteins (GeneBank accession U79299, U03417,
L13595, respectively) and, C. elegans F11c3.2 protein (GeneBank
accession Z81499) is represented. Identical amino acids are shaded in
black, conserved amino acids are further boxed by white squares. The
codon numbers correspond to those of the TIGR protein, b, Identification
of an homozygous carrier of the Lys423Glu TIGR mutation. Direct
15 sequencing of genomic DNA revealed that persons Vl-3 and Vl-9 were,
respectively, heterozygotic and homozygotic carriers of the Lys423Glu
TIGR mutation. The arrows indicate the A to G transition. Person Vl-2
carried two wild-type TIGR alleles.
Figure 3 shows the amplification refractory mutation
20 system (ARMS) as a method to type specific alleles at a polymorphic
locus. In the present invention, this method, ARMS, was used for
detecting a specific pathogenic mutation. The allele-specific
oligonucleotide pri",ers were designed to discriminate between two target
DNA sequences (wild-type (normal) versus pathogenic) that differed by
25 a single nucleotide in the region of interest (either one of the two
mutations). Designed primers that differed at the extreme 3' terminus
were synthetised. This was done because the DNA synthesis step in the
CA 02216997 1997-09-30
PCR reaction is crucially dependent on correct base pairing at the 3' end.
The primers that were designed are differing in their 3' ends and can
ll ,er~rore specifically amplify the DNA fragment of interest, either normal
or mutated. This figure is a pictorial representation of ARMS for the
5 adenine to guanine transition at nucleotide 1267. The amplification
strategy is demonstrated for the wild-type or non-mutant allele and the
mutant allele.
Figure 4 shows the phenotypic status and segregation
analyses of the GLC1A disease haplotype and Lys423Glu in family
10 GV-510. All living individuals were investigated for glaucoma, genotyped
with microsatcllitc markers spanning the GLC1A locus and tested for the
presence of the Lys423Glu TIGR mutation using ARMS. Selected AFM
markers with their corresponding GDB number, number of alleles
observed for each marker in pedigree GV-001 and sizes of the allele
15 associated with the GLC1A disease haplotype are represented on top.
The position of the TIGR gene is indicated relative to genetic markers.
Sex-averaged reco"l~.nalio,l distances, depicted between marker loci in
ce"lil\1organs, were not drawn to scale. Glaucoma patients are depicted
by solid black symbols, unaffected individuals by open symbols, and
20 deceased subjects reported as blind by at least two independent family
members by a black quadrant in the upper left corner of their respective
symbols. OHT persons are represented by open symbols containing a
central solid dot. Present ages of normal and OHT patients as well as
ages of affected carriers at time of diagnosis are depicted above their
25 respective symbols. A solid black box indicates the common GLC1A
~ise~se haplotype. The right side of each phased haplotype indicates the
haplotype inherited from the father; the left side indicates the haplotype
CA 02216997 1997-09-30
inherited from the mother. An asterisk in the genotype of person Vl1-5
represents a microsatellite mutation at locus D1 S2790. Person Vl1-5 also
inherited a patemal recombination between loci D1 S2815 and D1 S2790.
Results of the ARMS tests are depicted below each subject's genotype;
5W, ARMS test performed using the wild-type primers; M, ARMS test
pe, rur" ,ed using the Lys423Glu mutant pri" ,er~. The internal control PCR
product is shown. Persons Vl-2, Vl-5, Vl~, Vl-10 and Vl-12 carried the
wild-type allele on both chromosomes 1. Persons Vl-1, Vl-7, Vl-9 and
Vl-11 are wild-type negative and mutant positive, therefore, homozygous
10for the Lys423Glu mutation. All other individuals are both wild-type
positive and mutant positive, thererore, heterozygotes for the mutation.
Figure 5 shows the characterization of carriers for the
HlS366Gln and Gln368Stop TIGR mutations. a, Structure of the TIGR
encoded protein. The leucine zipper domain (amino acids 117-166) is
15shown within the N-terminal half of the protein. The His366Gln mutation
is depicted by a black circle in the olfactomedin homology domain
represented by a striped boxe within the C-terminal half of the protein.
The Gln368Stop mutation is depicted by a stop codon in the olfactomedin
homology domain. The codon numbers correspond to those of the TIGR
20protein. b, Identification of carriers for the His366Gln and Gln368Stop
TIGR mutations. Direct sequencing of genomic DNA revealed that
persons CT 003 and LA402 were, respectively, heterozygotic carriers of
the Gln368Stop and His366Gln TIGR mutations. The arrows indicate the
C to T transition or C to G tranversion.
25Other objects, advantages and features of the present
invention will become more apparent upon reading of the following
non-restrictive desc; iplion of prefe~ l~d embodiments with reference to the
CA 02216997 1997-09-30
accor"panying drawing which is exe,npla~ and should not be inter"reted
as limiting the scope of the present invention.
DESCRIPTION OF THE PREFERRED EMBODIMENT
At its broadesl, the invention con ,~rises novel mutations
in the TIGR gene, a quick method for an easy detection of identified
mutations and the teachings for the first time of mutant homozygote being
phenotypically normal in an autosomally dominant inherited disease.
The present invention is illustrated in further detail by
the following non-limiting examples.
EXAMPLE 1
Pedi~rees and o~hthalmolo~ic assessments
1.1 Pedigree reconstitution
The pedigree genealogy was reconstituted using the registers compiled
from the Catholic parish records, which systematically list births,
marriages, and deaths of 98% of the Quebec population. Validation of the
family tree and new data on recent births were obtained through
interviews with key family members. The Archives Nationales du Quebec,
the Quebec Civil register, and the Institut de recherche sur l'étude des
populations (IREP) data base (Bouchard et al., 1991, Histoire d'un
génome. Population et génétique dans l'est du Quebec, Presses de
I'Université Laval, Sillery, Quebec, pp 607) were also consulted.
CA 02216997 1997-09-30
23
1.2 OPhthalmoloqic investiqations
All subjects, affected or not, gave inror"~ed consent before entering the
study. Clinical ~ssessments comprised complete ophthalmologic
evaluation, including best co~ c~ed visual acuity; optic disk examination;
5 slit-lamp biomicroscopy; applanation tonometry; gonioscopy; and
visual-field evaluation. Three criteria were required for primary
open-angle glaucoma (POAG) diagnosis: a) intraocular pressures above
22 mm Hg in both eyes, b) characteristic optic disk damage and/or visual
field impairment, and c) grade lll or IV (open-angle) gonioscopy. In the
10 absence of optic disk damage or visual-field alteration, subjects with
intraocular pressures above 22 mm Hg in both eyes and grade lll or IV
gonioscopy were diagnosed with ocular hypertension (OHT). Members
of the families were considered normal when they presented normal optic
disks and showed highest intraocular pressures ever recorded at 22 mm
15 Hg or less. Persons with other forms of glaucomas, including grade 0
(closed angle); grade I or ll (narrow-angle); congenital; and secondary
glaucomas, or with other nonglaucomatous ocular disorders were
considered unaffected. Blindness in deceased ancestors was confirmed
by at least two independent sources.
EXAMPLE 2
2.1 Source of DNA
Blood samplcs were obtained from direct descendants of the founder as
25 well as spouses of affected patients with children; from each, 20 ml of
blood was drawn by venipuncture in heparinized tubes. One additional
10 ml blood sample was drawn from each subject to establish
CA 02216997 1997-09-30
24
Iymphoblastoid cell lines using the method of Anderson et al. (1984, In
Vitro, 20:856-858).
2.2 Isolation of DNA
5 DNA was extracted from whole blood using the guanidine
hydrochloride-proteinase K method dcveloped by Jeanpierre (1987, Nucl.
Acids. Res.15:9611-9611).
2.3 GenotvPin~ Procedures
10 To accelerate genotyping, we used a protocol similar to the procedure of
Vignal et al. (1993, Methods in molecular genetics, Academic Press,
1 :211 -221) which was derived from the multiplex sequencing technique
of Church and Kieffer-Higgins (1988 Science 240:185-188). Briefly,
polymerase chain reactions (PCR) were performed in a total volume of
50 ~ul containing 100 ng of genomic DNA, 50 pmol of each primer, 125
mM dNTPs, 50 mM KCI, 10 mM Tris (pH 9), 1.5 mM MgCI2, 0.01%
gelatin, 0.1 % Triton X-100, and 1 U Taq polymerase
(Perkin-Elmer-Cetus). Amplifications were carried out using a "hot-start"
procedure. Taq polymerase was added after a 5-min denaturation step
20 at 96~ C. Samples were then processed through 35 cycles of denaturation
(94~C for 40 s) and annealing (55~ C for 30 s), followed by one last step
of elongation (2 min at 72~ C). Usually, three amplification products
synthesized with separate primer sets on identical DNA samples were
coprecipitated and comigrated in a single lane of 6% polyacrylamide
25 denaturing gels. Separated products were then transferred onto Hybond
N' nylon membranes (Amersham), hybridized with a (CA)20 oligomer 3'
labeled with Digoxigenin-11-ddUTP, and detected by chemiluminescence
CA 02216997 1997-09-30
using the DIG system (Boehringer-Mannheim) with Kodak XAR-5 films.
Genotypes were scored relative to rererence alleles of the mother of the
CEPH family 1347 (individual 134702). Genotyping was repeated upon
detection of i,lco",,,~alibilities or recombination events.
2.4 Selection of microsatellite markers
In Figure 4, the markers used for haplotype analyses are shown. With the
exception of two markers (AFMGLC21 and AFMGLC22), all AFM
(Généthon) markers reported above were described in Dib et al. (1996,
10 supra). For AFMGLC21, the sequences were primer a:
GATCTCTTATCAGTCAGGCA, and primer m:
mCTMGGCTGMTMTATTCG. For AFMGLC22, the sequences were
primer a: TTMCTCACCACTCCCTGCC, and primer m:
MTTATGGCCTTCGCCC. Assignment of the genetic location of these
15 markers was established according to the method of Weissenbach et al.
(1992, Nature, 359:795-801) and has been validated by construction of
a 10-cM physical map (Clépet et al., 1996, Eur. J. Hum.Genet.,
4:250-259).
20 2.5 HaPlotYPe analysis
Haplotypes were analysed to phase the marker genotypes with the
disease gene. The haplotype inherited by an affected child constituted
the "disease" haplotype and was compared with the common disease
haplotype inherited from the founder. The remaining three haplotypes
25 were considered the "normal" haplotypes.
CA 02216997 1997-09-30
26
EXAMPLE 3
Discovery of the ~henotypic nonnal-homozYqote mutant
5 3.1 PhenotyPic normal-homozY~ote mutant (Fioure 4)
Phenotypic status and segregation analyses of the GLC1A disease
haplotype and Lys423Glu TIGR mutation in family GV-510. All living
individuals were investigated for glaucoma, genotyped with microsatellite
markers spanning the GLC1A locus and tested for the presence of the
10 Lys423Glu TIGR mutation using ARMS. Selected AFM markers with their
corresponding GDB number, number of alleles observed for each marker
in pedigree GV-001 and sizes of the allele associated with the GLC1A
rlise~se haplotype are represenled on top. The position of the TIGR gene
is indicated relative to genetic markers. Sex-averaged recombination
15 d;slances, depi 1ed between marker loci in cer,lihlorgans, were not drawn
to scale. Glaucoma patients are depicted by solid black symbols,
unaffected individuals by open symbols, and deceased subjects reported
as blind by at least two independent family members by a black quadrant
in the upper left corner of their respective symbols. OHT persons are
20 represented by open symbols containing a central solid dot. Present ages
of normal and OHT patients as well as ages of affected carriers at time
of diagnosis are depicted above their respective symbols. A solid black
box indicates the common GLC1A disease haplotype. The right side of
each phased haplotype ind ~tes the haplotype inherited from the father;
25 the left side indicates the haplotype inherited from the mother. An asterisk
in the genotype of person V11-5 represents a microsatellite mutation at
locus D1S2790. Person V11-5 also inherited a paternal recombination
CA 02216997 1997-09-30
between loci D1S2815 and D1S2790. Results of the ARMS tests are
depicted below each subject's genotype; W, ARMS test performed using
the wild-type primers; M, ARMS test performed using the Lys423Glu
mutant p, imer~. The inte, "al control PCR product is shown. Persons Vl-2,
5 Vl-5, Vl~, Vl-10 and Vl-12 carried the wild-type allele on both
chromosomes 1. Persons Vl-1, Vl-7, Vl-9 and Vl-11 are wild-type
negative and mutant positive, therefore, homozygous for the Lys423Glu
mutation. All other individuals are both wild-type positive and mutant
positive, therefore, heterozygotes for the mutation.
3.2 Initial screeninq for mutations
To obtain a wild-type TIGR cDNA, RT-PCR was performed using the
Superscript RT protocol (Gibco/BRL), 500 ng of oligo-dT and 10 ,ug of
total RNA isolated from a pool of trabecular meshwork tissue dissected
15 from 10 pairs of human eyes. To obtain the mutated TIGR cDNA, the
same protocol was followed using 10 ,ug of total RNA isolated from
homozygote Vl-9 immortalized Iymphoblasts. One to 3 ,ul of first strand
cDNA synthesis was amplified with primers 41 F:
AGAGCTTTCCAGAGGAAGCC, and 1 731 R:
20 GGTCTACGCCCTCAGACTAC, before a second round of PCR with
internal primers 31 F: AGAGACAGCAGCACCCMCG, and 21 R:
TCTGCCATTGCCTGTACAGC. PCR products were directly cloned into
the pCRII vector using the TA cloning kit (InVitrogen) according to the
manufacturer's protocol. Cloned products were sequenced using the T7
25 sequencing kit (Pharmacia).
CA 02216997 1997-09-30
28
3.3 Sequencin~
To corlf" ") mutations, genomic DNA sequencing was also performed on
selected individuals by direct asymmetric PCR sequencing using
modificaliol-s of the protocol described by Gyllensten et al. (1988, Proc.
Natl. Acad. Sci., 85:7652-7656). The mutation was recognized by the
approximately equal peak intensity of the bands on the autoradiogram.
All sequencing was performed bidirectionally.
EXAMPLE 4
Two mutations includinq ARMS
4.1 ARMS test for the LYs423Glu mutation (Fi~ure 3)
To test for the presence of the Lys423Glu mutation, we developed an
amplification refractory mutation system (ARMS) exploiting procedures
described by Little (1997, Current Protocols in human genetics, Eds.
Dracopoli, N.C. et al., 9.8.1. - 9.8.12). Two co,nplementary PCR reactions
were conducted with the same substrate. The first reaction contained a
forward primer specific for the wild-type allele, SEQ. NO. 3,
GLC1A1313M: TCGMCAAACCTGGGAGACAAACATCCGM. The
second reaction contained a forward primer specific for the Lys423Glu
Tl GR allele, SEQ. NO. 2, GLC1A1 31 3GG:
TCGMCAAACCTGGGAGACAAACATCCGGG. In each reaction, a
common reverse primer, GLC1A1479R, was used; its
sequence was:
SEQ. NO. 4 CAAAGAGCTTCTTCTCCAGGGGGTTGTAGT. Both
reactions gave a 225 bp amplified fragment. To serve as internal control,
CA 02216997 1997-09-30
29
a second pair of primers that co amplified a 438 bp fragment within TIGR
exon 1 was added to the ARMS reaction. The forward TIGR exon 1
primer was: AGAGC I I I CCAGAGGMGCC, the reverse TIGR exon 1
primer was TTGGGmCCAGCTGGTC. PCR was performed using
5 standard protocols, annealing temperature was at 60~C. Amplification
products were electrophoresed in 1,5% agarose gels before ethidium
staining and scored by two independent observers.
4.2 ARMS test for the His366Gln mutation
10 To test for the presence of the His366Gln mutation, we developed an
amplification refractory mutation system (ARMS) exploiting procedures
described by Little (1997). Two complementary PCR reactions were
conducted with the same substrate. The first ,ea~io,1 contained a forward
primer specific for the wild-type allele, SEQ. NO. 6 GLC1A1098CT:
15 GAGMGGAAATCCCTGGAGCTGGCTACCTC. The second reaction
contained a forward primer specific for the His366Gln TIGR allele, SEQ.
NO. 5, GLC1A1098GT: GAGMGGAAATCCCTGGAGCTGGCTACCTG.
In each reaction, a con1mG~I reverse primer, GLC1A1479R, was used; its
sequence was:
20 SEQ. NO. 7, CAAAGAGCTTCTTCTCCAGGGGGTTGTAGT. Both
reactions gave a 393 bp amplified fragment. To serve as internal control,
a second pair of ,c,i",er~ that co-amplified a 438 bp fragment within TIGR
exon 1 was added to the ARMS reaction. The forward TIGR exon 1
primer was: AGAGCmCCAGAGGMGCC, the reverse TIGR exon 1
25 primer was TTGGG I I I CCAGCTGGTC. PCR was performed using
standard protocols, annealing temperature was at 60~C. Amplification
CA 02216997 1997-09-30
products were elect,ophoresed in 1.5% agarose gels before ethidium
staining and scored by two independent observers.
Although the present invention has been described hereinabove
by way of prefer,ed embodiments thereof, it can be modified, without
5 departing from the spirit and nature of the subject invention as defined in
the appended claims.