Note: Descriptions are shown in the official language in which they were submitted.
CA 02218188 1997-10-14
wo 96/32504 PCT~US96/05136
SOLID PHASE SEQUENCING OF BIOPOLYMERS
~i~ht~ in the Invention
This invention was made with United States Government
" support under grant number DE-FG-02-93ER61609, awarded by the United
S States Department of Energy, and the United States Govçrnment has certain
rights in the invention.
Rack~round ofthe Invention
l . Field of the Invention
This invention relates to methods for ~letecting and sequenc~ng
nucleic acids using sequencing by hybridization technology and molecular
weight analysis. The invention also relates to probes and arrays useful in
sequencing and detection and to kits and apparatus for determining sequence
information.
2. Description of the Background
Since the recognition of nucleic acid as the carrier of the
genetic code, a great deal of interest has centered around determining the
sequence of that code in the many forms which it is found. Two landmark
studies made the process of nucleic acid sequencing, at least with DNA~ a
common and relatively rapid procedure practiced in most laboratories. The
20 first describes a process whereby terrnin~lly labeled DNA molecules are
chemically cleaved at single base repetitions (A.M. Maxam and W. Gilbert,
Proc. Natl. Acad. Sci. USA 74:560-64, 1977). Each base position in the
nucleic acid sequence is then determined from the molecular weights of
fr~nent~ produced by partial cleavages. Individual reactions were devised
25 to cleave preferentially at guanine, at adenine, at cytosine and thymine at
cytosine alone. When the products of these four reactions are resolved by
molecular weight. using, for example, polyacrylamide gel electrophoresis,
CA 02218188 1997-10-14
WO 96t32504 PCl[~/USg6/OS136
DNA sequences can be read from ~e pattern of f~gments on ~e resolved
gel.
The second study describes a procedure whereby DNA is
sequenced using a variation ofthe plus-minus method (F. Sanger et al., Proc.
S Natl. Acad. Sci. USA 74:5463-67, 1977). This procedure takes advantage
of the chain t~l~nin~ting ability of dideoxynucleoside triphosphates
(ddNTPs) and the ability of DNA polymerase to incorporate ddNTPs with
nearly equal fidelity as the natural substrate of DNA polymerase,
deoxynucleosides triphosphates (dNTPs). Briefly, a primer, usually an
10 oligonucleotide, and a template DNA are incubated together in the presence
of a useful concentration of all four dNTPs plus a limited amount of a single
ddNTP. The DNA polymerase occasionally incorporates a
dideoxynucleotide which termin~tes chain extension. Because the
dideoxynucleotide has no 3'-hydroxyl, the initiation point for the polymerase
15 enzyme is lost. Polymerization produces a mixture of fragments of varied
sizes, all having identical 3' termini. Fractionation of the mixture by, for
example, polyacrylamide gel electrophoresis, produces a pattern which
indicates the presence and position of each base in the nucleic acid.
Reactions with each of the four ddNTPs allows one of ordinary skill to read
20 an entire nucleic acid sequence from a resolved gel.
Despite their advantages, these procedures are cumbersome
and impractical when one wishes to obtain megabases of sequence
information. Further, these procedures are, for all practical purposes,
limited to sequencing DNA. Although variations have developed, it is still
25 not possible using either process to obtain sequence information directly
from any other form of nucleic acid.
CA 02218188 1997-10-14
WO 96t32504 PC~t~JS96~(15136
A relatively new method for obtaining sequence information
from a nucleic acid has recently been developed whereby the sequences of
groups of c~ nti~lQus bases are determined simultaneously. In comparison
to traditional techniques whereby one determines base specific inforrnation
5 of a sequence individually, this method, referred to as sequencing by
hybridization (SBH), represents a many-fold amplification in speed. Due,
at least in part to the increased speed, SBH presents numerous advantages
including re~ ce~l expense and greater accuracy. Two general approaches
of sequencing by hybridization have been suggested and their practicality
10 has been demonstrated in pilot studies. In one format, a complete set of 4"
nucleotides of length n is immobilized as an ordered array on a solid support
and an unknown DNA sequence is hybridized to this array (K.R. Khrapko
et al., J. DNA Sequencing and Mapping 1:375-88, 1991). The resulting
hybridization pattern provides all "n-tuple" words in the sequence. This is
15 sufficient to determine short sequences except for simple tandem repeats.
In the second format, an array of immobilized samples is
hybridized with one short oligonucleotide at a time (Z. Strezoska et al., Proc.
Natl. Acad. Sci. USA 88:10,089-93~ 1991). When repeated 4n times for each
oligonucleotide of length n, much of the sequence of all the immobilized
20 samples would be determined. In both approaches, the intrinsic power of
the method is that many sequenced regions are determined in parallel. In
actual practice the array size is about 104 to 105.
Another aspect of the method is that information obtained is
quite recllln(l~n~ and especially as the size of the nucleic acid probe grows.
25 Mathematical simulations have shown that the method is quite resistant to
experimental errors and that far fewer than all probes are necessary to
CA 02218188 1997-10-14
WO 96/32504 PCTIUS96/OS136
determine reliable sequence data (P.A. Pevmer et al., J. Biomol. Struc. &
Dyn. 9:399-410, 1991; W. Bains, Genomics 11:295-301,1991).
In spite of an overall optimistic outlook, there are still a
number of potentially severe drawbacks to actual implementation of
5 sequencing by hybridization. First and foremost among these is that 4n
rapidly becomes quite a large number if chemical synthesis of all of the
oligonucleotide probes is actually contemplated. Various schemes of
automating this synthesis and compressing the products into a small scale
array, a sequencing chip, have been proposed.
There is also a poor level of discrimin~tion between a
correctly hybridized, perfectly matched duplexes, and end mi~m~tches. In
part, these drawbacks have been addressed at least to a small degree by the
method of continuous stacking hybridization as reported by a Khrapko et al.
(FEBS Lett. 256:118-22, 1989). Continuous stacking hybridization is based
15 upon the observation that when a single-stranded oligonucleotide is
hybridized adjacent to a double-stranded oligonucleotide, the two duplexes
are mutually stabilized as if they are positioned side-to-side due to a
stacking contact between them. The stability of the interaction decreases
significantly as stacking is disrupted by nucleotide displacement, gap or
20 terminal mi.~m~tch Internal mi~m~tçhes are presumably ignorable because
their thermodynamic stability is so much less than perfect matches.
Although promising, a related problem arises which is the inability to
distinguish between weak, but correct duplex formation, and simple
background such as non-specific adsorption of probes to the underlying
25 support matrix.
CA 02218188 1997-10-14
wo 96/32s04 PCTJU~96l05136
Detection is also monochromatic wherein separate sequential
positive and negative controls must be run to discrimin~te between a correct
r hybridization match, a mis-match, and background. All too often,
ambiguities develop in reading sequences longer than a few hundred base
pairs on account of sequence recurrences. For example, if a sequence one
base shorter than the probe recurs three times in the target, the sequence
position cannot be uniquely determined. The locations of these sequence
ambiguities are called branch points.
Secondary structures often develop in the target nucleic acid
affecting accessibility of the sequences. This could lead to blocks of
sequences that are unreadable if the secondary structure is more stable than
occurs on the complementary strand.
A final drawback is the possibility that certain probes will
have anomalous behavior and for one reason or another, be recalcitrant to
hybridization under whatever standard sets of conditions llltim~tely used.
A simple example of this is the difficulty in finding matching conditions for
probes rich in G/C content. A more complex example could be sequences
with a high propensity to form triple helices. The only way to rigorously
explore these possibilities is to carry out extensive hybridization studies withall possible oligonucleotides of length "n" under the particular format and
conditions chosen. This is clearly impractical if many sets of conditions are
involved.
Among the early publication which appeared discussing
sequencing by hybridization, E.M. Southern (WO 89/10977), described
v 25 methods whereby unknown, or target, nucleic acids are labeled, hybridized
to a set of nucleotides of chosen length on a solid support~ and the nucleotide
CA 02218188 1997-10-14
WO 96/32S04 PCT/US96/05136
sequence of the target ~letetTnined, at least partially, from knowledge of the
sequence of the bound fragments and the pattern of hybridization observed.
Although promi~in~, as a practical matter, this method has numerous
drawbacks. Probes are entirely single-stranded and binding stability is
dependent upon the size of the duplex. However, every additional
nucleotide of the probe necessarily increases the size of the array by four
fold creating a dichotomy which severely restricts its plausible use. Further,
there is an inability to deal with branch point ambiguities or secondary
structure of the target, and hybridization conditions will have to be tailored
or in some way accounted for each binding event. Attempts have been made
to overcome or circumvent these problems.
R. Drmanac et al. (U.S. Patent No. 5,202,231) is directed to
methods for sequencing by hybridization using sets of oligonucleotide
probes with random or variable sequences. These probes, although useful,
suffer from some of the same drawbacks as the methodology of Southern
(1989), and like Southern, fail to recognize the advantages of stacking
interactions.
K.R. Khrapko et al. (FEBS Lett. 256:118-22, 1989; and J.
DNA Sequencing and Mapping 1:357-88, 1991) attempt to address some of
these problems using a technique referred to as continuous stacking
hybridization. With continuous stacking, conceptually, the entire sequence
of a target nucleic acid can be determined. Basically, the target is
hybridized to an array of probes, again single-stranded, denatured from the
array, and the dissociation kinetics of denaturation analyzed to determine the
target sequence. Although also promising, discrimination between matches "
and mis-matches (and simple background) is low and, further, as
CA 02218188 1997-10-14
WO 96/~S2504 PCT/US~6/05136
hybridization conditions are inconstant for each duplex, discrimin~tion
becomes increasingly re~lllce-l with increasing target complexity.
Another major problem with current sequencing formats is the
inability to eff1ciently detect sequence information. In conventional
S procedures, individual sequences are separated by, for example,
electrophoresis using capillary or slab gels. This step is slow, expensive and
requires the talents of a number of highly trained individuals, and, more
importantly, is prone to error. One attempt to overcome these difficulties
has been to utilize the technology of mass spectrometry.
Mass spectrometry of organic molecules was made possible
by the development of instruments able to volatize large varieties of organic
compounds and by the discovery that the molecular ion forrned by
volatization breaks down into charged fragments whose structures can be
related to the intact molecule. Although the process itself is relatively
straight forward, actual implementation is quite complex. Briefly, the
sample molecule or analyte is volatized and the resulting vapor passed into
an ion chamber where it is bombarded with electrons accelerated to a
compatible energy level. Electron bombardment ionizes the molecules of
the sample analyte and then directs the ions formed to a mass analyzer. The
mass analyzer, with its combination of electrical and magnetic fields,
separates impacting ions according to their mass/charge (m/e) ratios. From
these ratios, the molecular weights of the impacting ions can be determined
and the structure and molecular weight of the analyte deterrnined. The
entire process requires less than about 20 microseconds.
Attempts to apply mass spectrometry to the analysis of
biomolecules such as proteins and nucleic acids have been disappointing.
CA 02218188 1997-10-14
WO 96/32504 PCT/US96105136
Mass spectrometric analysis has traditionally been limit~d to molecules with
molecular weights of a few ~ousand ~ ton~. At higher molecular weights,
samples become increasingly difficult to volatize and large polar molecules
generally cannot be vaporized without catastrophic consequences. The
5 energy requirement is so significant that the molecule is destroyed or, even
worse, fragmented. Mass spectra of fragmented molecules are often
difficult or impossible to read. Fragment linking order, particularly useful
for reconstructing a molecular structure, has been lost in the fragmentation
process. Both signal to noise ratio and resolution are significantly
10 negatively affected. In addition, and specifically with regard to
biomolecular sequencing, extreme sensitivity is necessary to detect the
single base differences between biomolecular polymers to determine
sequence identity.
A number of new methods have been developed based on the
15 idea that heat, if applied with sufficient rapidity, will vaporize the samplebiomolecule before decomposition has an opportunity to take place. This
rapid heating technique is referred to as plasma desorption and there are
many variations. For example, one method of plasma desorption involves
placing a radioactive isotope such as Californium-252 on the surface of a
20 sample analyte which forms a blob of plasma. From this plasma, a few ions
of the sample molecule will emerge intact. Field desorption ionization,
another form of desorption, utilizes strong electrostatic fields to literally
extract ions from a substrate. In secondary ionization mass spectrometry or
fast ion bombardment, an analyte surface is bombarded with electrons which
25 encourage the release of intact ions. Fast atom bombardment involves
bombarding a surface with accelerated ions which are neutralized by a
CA 02218188 1997-10-14
WO 96132504 PCTIUS96/05136
charge exchange before they hit the surface. Presumably, neutralization of
the charge lessens the probability of molecular destruction, but not the
creation of ionic forms of the sample. In laser desorption, photons comprise
the vehicle for depositing energy on the surface to volatize and ionize
molecules of the sample. Each of these techniques has had some measure
of success with different types of sample molecules. Recently, there have
also been a variety of techniques and combinations of techniques
specifically directed to the analysis of nucleic acids.
Brennan et al. used nuclide markers to identify terminal
nucleotides in a DNA sequence by mass speckometry (U.S. Patent No.
5,003,059). Stable nuclides, ~ietect~ble by mass spectrometry, were placed
in each ofthe four dideoxynucleotides used as reagents to polymerize cDNA
copies of the target DNA sequence. Polymerized copies were separated
electrophoretically by size and the terrninal nucleotide identified by the
presence of the unique label.
Fenn et al. describes a process for the production of a mass
spectrum cont~ining a multiplicity of peaks (U.S. Patent No. 5,130,538).
Peak components comprised multiply charged ions formed by dispersing a
solution containing an analyte into a bath gas of highly charged droplets.
An electrostatic field charged the surface of the solution and dispersed the
liquid into a spray referred to as an electrospray (ES) of charged droplets.
This nebulization provided a high charge/mass ratio for the droplets
increasing the upper limit of volatization. Detection was still limited to less
than about 100,000 daltons.
Jacobson et al. utilizes mass spectrometry to analyze a DNA
sequence by incorporating stable isotopes into the sequence (U.S. Patent No.
CA 02218188 1997-10-14
WO 96/32504 PCT/US96/05136
5,002,868). Incorporation required the steps of enzymatically introducing
the isotope into a strand of DNA at a terminus, electrophoretically
s~a,dling the strands to determine fragment size and analyzing the
separated strand by mass spectrometry. Although accuracy was stated to
5 have been increased, electrophoresis was necessary to isolate the labeled
strand.
Brennan also utilized stable markers to label the terminal
nucleotides in a nucleic acid sequence, but added the step of completely
degrading the components of the sample prior to analysis (U.S . Patent Nos.
10 5,003,059 and 5,174,962). Nuclide markers, enzymatically incorporated
into either dideoxynucleotides or nucleic acid primers, were
eleckophoretically separated. Bands were collected and subjected to
combustion and passed through a mass spectrometer. Combustion converts
the DNA into oxides of carbon, hydrogen, nitrogen and phosphorous, and
15 the label into sulfur dioxide. Labeled combustion products were identified
and the mass of the initial molecule reconstructed. Although fairly accurate,
the process does not lend itself to large scale sequencing of biopolymers.
A recent advancement in the mass spectrometric analysis of
high molecular weight molecules in biology has been the development of
20 time of flight mass spectrometry (TOF-MS) with matrix-assisted laser
desorption ionization (MALDI). This process involves placing the sample
into a matrix which contains molecules which assist in the desorption
process by absorbing energy at the frequency used to desorp the sample.
The theory is that volatization of the matrix molecules encourages
25 volatization of the sample without significant destruction. Time of flight
analysis utilizes the travel time or flight time of the various ionic species as
CA 02218188 1997-10-14
WQ 96132504 PCTlUSg6~()S136
an accurate indicator of molecular mass. There have been some notable
successes with these techniques.
Beavis et al. proposed to measure the molecular weights of
DNA fr~rnent~ in mixtures prepared by either Maxam-Gilbert or Sanger
5 sequencing techniques (U.S. Patent No. 5,288,644). Each of the different
DNA fr~ t~ to be generated would have a common origin and tçrrnin~te
at a particular base along an unknown sequence. The separate mixtures
would be analyzed by laser desorption time of flight mass spectroscopy to
deterrnine fr~nent molecular weights. Spectra obtained from each reaction
10 would be compared using computer algc~liLhllls to determine the location of
each of the four bases and ultimately, the sequence of the fragment.
Williams et al. utilized a combination of pulsed laser ablation7
multiphoton ionization and time of flight mass spectrometry. Effective laser
desorption was accomplished by ablating a frozen film of a solution
15 containing sample molecules. When ablated, the film produces an
expanding vapor plume which entrains the intact molecules for analysis by
mass spectrometry.
Even more recent developments in mass spectrometry have
further increased the upper limits of molecular weight detection and
20 determination. Mass spectrograph systems with reflectors in the flight tube
have effectively doubled resolution. Reflectors also compensate for errors
in mass caused by the fact that the ionized/accelerated region of the
instrument is not a point source, but an area of finite size wherein ions can
accelerate at any point. Spatial differences between particle the origination
25 points of the particles, problematic in conventional instruments because
arrival times at the detector will vary, are overcome. Particles that spend
CA 02218188 1997-10-14
WO 96/32504 PCT/US96/OS136
more time in the accelerating field will also spend more time in the Lcla.ding
field. Therefore, particles emerging from the reflector are mostly
synchronous, vastly improving resolution.
Despite these advances, it is still not possible to generate
5 coordinated spectra representing a continuous sequence. Furthermore,
throughput is sufflciently slow so as to make these methods impractical for
large scale analysis of sequence information.
Sllmm~ of tlle Jnvention
The present invention overcomes the problems and
disadvantages associated with current strategies and designs and provides
methods, kits and apparatus for determining the sequence of target nucleic
aclds.
One embodiment of the invention is directed to methods for
15 sequencing a target nucleic acid. A set of nucleic acid fragments containing
a sequence which is complementary or homologous to a sequence of the
target is hybridized to an array of nucleic acid probes wherein each probe
comprises a double-stranded portion, a single-stranded portion and a
variable sequence within said single-stranded portion, forming a target array
20 of nucleic acids. Molecular weights for a plurality of nucleic acids of the
target array are determined and the sequence of the target constructed.
Nucleic acids of the target, the target sequence, the set and the probes may
be DNA, RNA or PNA comprising purine, pyrimidine or modified bases.
The probes may be fixed to a solid support such as a hybridization chip to
25 facilitate automated determination of molecular weights and identification
of the target sequence.
CA 02218188 1997-10-14
WO 96/32504 PCTIUS96~05136
Another embodiment of the invention is directed to methods
for sequencing a target nucleic acid. A set of nucleic acid fr~rnent.c
cont~inin~ a sequence which is complem~nt~ry or homologous to a
sequence of the target is hybridized to an array of nucleic acid probes
S forming a target array cont~inin ~ a plurality of nucleic acid complexes. One
strand of those probes hybridized by a fragment is extended using the
fr~grnent as a template. Molecular weights for a plurality of nucleic acids
ofthe target array are ~let~rmined and the sequence ofthe target constructed.
Strands can be enzymatically extended using chain termin~ting and chain
elon~ting nucleotides. The resulting nested set of nucleic acids represents
the sequence of the target.
Another embodiment of the invention is directed to methods
for detecting a target nucleic acid. A set of nucleic acids complementary to
a sequence of the target, is hybridized to a fixed array of nucleic acid probes.The molecular weights of the hybridized nucleic acids are determined by
mass spectrometry and a sequence of the target can be identified. Target
nucleic acids may be obtained from biological samples such as patient
samples wherein detection of the target is indicative of a disorder in the
patient, such as a genetic defect, a neoplasm or an infection.
Another embodiment of the invention is directed to methods
for sequencing a target nucleic acid. A sequence of the target is cleaved into
nucleic acid fragrnents and the fragments hybridized to an array of nucleic
acid probes. Fr~gment~ are created by enzymatically or physically cleaving
the target and the sequence of the fragments is homologous with or
v 25 complementary to at least a portion of the target sequence. The array is
attached to a solid support and the molecular weights of the hybridized
CA 02218188 1997-10-14
WO 96/32504 PCT/US96/05136
14
fr~nentr~ det~rmined by mass spectrometry. From the molecular weights
d~t~rmined, nucleotide sequences of the hybridized fragments are
~letçrmined and a nucleotide sequence of the target can be identified.
Another embodiment of the invention is directed to methods
S for sequencing a target nucleic acid. A set of nucleic acids complementary
to a sequence of the target is hybridized to an array of single-stranded
nucleic acid probes wherein each probe comprises a constant sequence and
a variable sequence and said variable sequence is determinable. The
molecular weights of the hybridized nucleic acids are determined and the
10 sequence of said target identified. The array comprises less than or equal toabout 4R different probes and R is the length in nucleotides of the variable
sequence and may be attached to a solid support.
Another embodiment of the invention is directed to methods
for sequencing a target nucleic acid by strand-displacement, double-stranded
lS sequencing. A set of partially single-stranded and partially double-stranded
nucleic acid fragments are provided wherein each fragment contains a
sequence that corresponds to a sequence of the target. These nucleic acid
fragments are hybridized to a set of partially single-stranded and partially
double-stranded nucleic acid probes, via the single-stranded regions of each,
20 to form a set of fragment/probe complexes. Prior to hybridization, either the fragments or the probes may be treated with a phosphorylase to remove
phosphate groups from the 5'-termini of the nucleic acids. 5'-termini are
ligated with adjacent 3'-termini of the complex forming a common single
strand. The complementary unligated strand contains a nick which is
25 recognized by a nucleic acid polymerase that initiates strand-displacement
polymerization~ extending the unligated strand. Polymerization proceeds,
CA 02218188 1997-10-14
WO 96/32504 PCI'IUS961(~5136
using the ligated strand as a template, in the presence of labeled nucleotides
such as mass modified nucleotides. The sequence of the target can be
~leterrnined by mass spectrometry from the molecular weights of the
ex~n~ecl strands. This process can be used to sequence target nucleic acids
5 and also to identify a single sequence in a mixed background. Selection of
the species of nucleic acid to be sequenced occurs upon hybridization to the
probe. As only fragments complementary to the single-stranded region of
the probe will form complexes, only those fragments complexes are
sequenced.
Another embodiment of the invention is directed to arrays of
nucleic acid probes. In these arrays, each probe comprises a first strand and
a second strand wherein the first strand is hybridized to the second strand
forrning a double-stranded portion, a single-stranded portion and a variable
sequence within the single-stranded portion. The array may be attached to
15 a solid support such as a material that facilitates volatization of nucleic acids
for mass spectrometry. Arrays can be fixed to hybridization chips
cont~ining less than or equal to about 4R different probes wherein R is the
leng~th in nucleotides of the variable sequence. Arrays can be used in
detection methods and in kits to detect nucleic acid sequences which may
20 be indicative of a disorder and in sequencing systems such as sequencing by
mass spectrometry.
Another embodiment of the invention is directed to arrays of
single-stranded nucleic acid probes wherein each probe of the arra~
comprises a constant sequence and a variable sequence which is
25 determinable. Arrays may be attached to solid supports which comprise
matrices that facilitate volatization of nucleic acids for mass spectrometry.
CA 02218188 1997-10-14
WO 96132504 PCT/US96/05136
16
Arrays, generated by conventional processes, may be characterized using the
above methods and replicated in mass for use in nucleic acid detection and
sequencing systems.
Another embodiment of the invention is directed to kits for
S ~letecting a sequence of a target nucleic acid. Kits contain arrays of nucleic
acid probes fixed to a solid support wherein each probe comprises a double-
stranded portion, a single-stranded portion and a variable sequence within
said single-stranded portion. The solid support may be, for example, coated
with a matrix that facilitates volatization of nucleic acids for mass
10 spectrometry such as an aqueous composition.
Another embodiment of the invention is directed to mass
spectrometry systems for the rapid sequencing of nucleic acids. Systems
comprise a mass spectrometer, a computer with ay~lopliate software and
probe arrays which can be used to capture and sort nucleic acid sequences
15 for subsequent analysis by mass spectrometry.
Other embodiments and advantages of the invention are set
forth, in part, in the description which follows and, in part, will be obvious
from this description and may be learned from the practice of the invention.
20 De~cription of the Drawin~.c
Figure 1 (A) Schematic of a mass modified nucleic acid primer; and
(B) primer mass modification moieties.
Figure 2 (A) Schematic of mass modified nucleoside triphosphate
elongators and terminators; and (B) nucleoside triphosphate
mass modification moieties.
Figure 3 List of mass modification moieties.
CA 02218188 1997-10-14
WO 96132504 PCTlUS96rO5136
Figure 4 List of mass modification moieties.
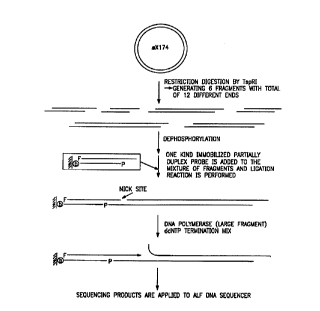
Figure 5 Cleavage site of Mwo 1 indicating bidirectional sequencing.
Figure 6 Sehem~tic of seql7encin~ strategy after target DNA digestion
by ~sp Rl.
Figure 7 Calc~ te~l Tm of matched and mi~m~tçhed complement~ry
DNA.
Figure 8 Replication of a master array.
Figure 9 Reaction scheme for the covalent attachment of DNA to a
surface.
Figure 10 Target nucleic acid capture and ligation.
Figure 11 Ligation efficiency of matches as compared to mi~m~tches.
Figure 12 (A) Ligation of target DNA with probe attached at 5'-
terrninlls; and (B) ligation of target DNA with probe attached
at the 3'-terminus.
Figure 13 Gel reader sequencing results from primer hybridization
analysis.
Figure 14 Mass spectrometry of oligonucleotide ladder.
Figure 15 Schematic of mass modification by alkylation.
Figure 16 Mass spectrum of 1 7-mer target with 0, 1 or 2 mass modified
moieties.
- Figure 17 Schematic of nicked strand displacement sequencing with
immobilized template.
Figure 18 Analysis of sequencing reaction in the presence and absence
of single-stranded DNA binding protein.
Figure 19 Schematic of nicked strand displacement sequencing with
immobilized probe.
CA 02218188 1997-10-14
WO 96/32504 PCTrUS96/05136
18
Figure 20 Results of sequencing performed using DF27- 1 as a probe.
Figure 21 Results of sequencing performed using DF27-2 as a probe.
Figure 22 Results of sequencing performed using DF27-4 as a probe.
Figure 23 Results of sequencing ~ rolmed using DF27-5-CY5 as a
probe.
Figure 24 Results of sequencing performed using DF27-6-CY5 as a
probe.
nescription of the Invention
As embodied and broadly described herein, the present
invention is directed to methods for sequencing a nucleic acid, probe arrays
useful for sequencing by mass spectrometry and kits and systems which
comprise these arrays.
Nucleic acid sequencing, on both a large and small scale, is
15 critical to many aspects of medicine and biology such as, for example, in theidentification~ analysis or diagnosis of diseases and disorders, and in
determining relationships between living org~ni.sms. Conventional
sequencing techniques rely on a base-by-base identification of the sequence
using electrophoresis in a semi-solid such as an agarose or polyacrylamide
20 gel to determine sequence identity. Although attempts have been made to
apply mass spectrometric analysis to these methods, the two processes are
not well suited because, at least in part, information is still be gathered in asingle base format. Sequencing-by-hybridization methodology has
enhanced the sequencing process and provided a more optimistic outlook for
25 more rapid sequencing techniques, however, this methodology is no more
applicable to mass spectrometry than traditional sequencing techniques.
CA 02218188 1997-10-14
WO 96t32504 PCT~US9610SI36
19
In contrast, positional sequencing by hybridization (PSBH)
with its ability to stably bind and discrimin~te different sequences with large
O or small arrays of probes is well suited to mass spectrometric analysis.
Sequence information is rapidly ~letermined in batches and with a minimllm
5 of effort. Such processes can be used for both sequencing unknown nucleic
acids and for detecting known sequences whose presence may be an
indicators of a disease or con~ tion. Additionally, these processes can
be lltili7e~1 to create coortlin~te~l patterns of probe arrays with known
sequences. Determination of the sequence of fragments hybridized to the
10 probes also reveals the sequence ofthe probe. These processes are currently
not possible with conventional techniques and, further, a coor lin~tecl batch-
type analysis provides a significant increase in sequencing speed and
accuracy which is expected to be required for effective large scale
sequenclng operatlons.
PSBH is also well suited to nucleic acid analysis wherein
sequence information is not obtained directly from hybridization. Sequence
information can be learned by coupling PSBH with techniques such as mass
spectrometry. Target nucleic acid sequences can be hybridized to probes or
array of probes as a method of sorting nucleic acids having distinct
20 sequences without having a priori knowledge of the sequences of the
various hybridization events. As each probe will be represented as multiple
copies, it is only necessary that hybridization has occurred to isolate distinctsequence packages. In addition, as distinct packages of sequences, they can
be amplified, modified or otherwise controlled for subsequent analysis.
25 Amplification increases the number of specific sequences which assists in
any analysis requiring increased quantities of nucleic acid while retainin
CA 02218188 1997-10-14
WO 96/32504 PCT/US96/OS136
sequence specificity. Modif1cation may involve chemically altering the
nucleic acid molecule to assist with later or downstream analysis.
Consequently, another important feature ofthe invention is the
ability to simply and rapidly m~s modify the sequences of interest. A mass
5 modification is an alteration in the mass, typically measured in terms of
molecular weight as daltons, of a molecule. Mass modification which
increase the discrimin~ion between at least two nucleic acids with single
base differences in size or sequence can be used to facilitate sequencing
using, for example, molecular weight determinations.
One embodiment of the invention is directed to a method for
sequencing a target nucleic acid using mass modified nucleic acids and mass
spectrometry technology. Target nucleic acids which can be sequenced
include sequences of deoxyribonucleic acid (DNA) or ribonucleic acid
(RNA). Such sequences may be obtained from biological, recombinant or
15 other man-made sources, or purified from a natural source such as a patient'stissue or obtained from environmental sources. Alternate types of molecules
which can be sequenced includes polyamide nucleic acid (PNA) (P.E.
Nielsen et al., Sci. 254:1497-1500, 1991) or any sequence of bases joined
by a chemical backbone that have the ability to base pair or hybridize with
20 a complementary chemical structure.
The bases of DNA, RNA and PNA include purines,
pyrimidines and purine and pyrimidine derivatives and modifications, which
are linearly linked to a chemical backbone. Common chemical backbone
structures are deoxyribose phosphate, ribose phosphate, and polyamide. The
25 purines of both DNA and RNA are adenine (A) and guanine (G). Others
that are known to exist include xanthine, hypoxanthine. 2- and l-
CA 02218188 1997-10-14
WO ~6132504 PCTIUS~?6~(~5136
~ minopurine, and other more modified bases. The pyrimidines are
cytosine (C), which is common to both DNA and RNA, uracil (U) found
- pre~lomin~ntly in RNA, and thymidine (T) which occurs almost exclusively
in DNA. Some of the more atypical pyrimidines include methylcytosine,
hydroxymethyl-cytosine, methyluracil, hydroxymethyluracil,
dihydroxypentyluracil, and other base modifications. These bases interact
in a complementary fashion to form base-pairs, such as, for example,
guanine with cytosine and adenine with thymidine. This invention a~so
encompasses situations in which there is non-traditional base pairing such
as Hoogsteen base pairing which has been identified in certain tRNA
molecules and postulated to exist in a kiple helix.
Sequencing involves providing a nucleic acid sequence which
is homologous or complementary to a sequence of the target. Sequences
may be chemically synthesized using, for example, phosphoramidite
chemistry or created enzymatically by incubating the target in an a~pr~liate
buffer with chain elongating nucleotides and a nucleic acid polymerase.
Initiation and termination sites can be controlled with dideoxynucleotides
or oligonucleotide primers, or by placing coded signals directly into the
nucleic acids. The sequence created may comprise any portion of the target
sequence or the entire sequence. Alternatively, sequencing may involve
elongating DNA in the presence of boron derivatives of nucleotide
kiphosphates. Resulting double-stranded samples are treated with a 3'
exonuclease such as exonuclease III. This exonuclease stops when it
encounters a boronated residue thereby creating a sequencing ladder.
Nucleic acids can also be purified, if necessary to remove
substances which could be harmful (e.g. toxins), dangerous (e.g. infectious)
CA 022l8l88 l997- lO- l4
WO g6/32504 PCTIUS96/OS136
or might interfere with the hybridization reaction or the sensitivity of that
reaction (e.g metals, salts, protein, lipids). Purification may involve
techniques such as chemical extraction with salts, chloroform or phenol,
se-liment~tion centrifugation, chromatography or other techniques known
5 to those of ordinary skill in the art.
If sufficient quantities of target nucleic acid are available and
the nucleic acids are sufficiently pure or can be purified so that any
substances which would interfere with hybridization are removed, a plurality
of target nucleic acids may be directly hybridized to the array. Sequence
10 information can be obtained without creating complementary or homologous
copies of a target sequence.
Sequences may also be amplified, if necessary or desired, to
increase the number of copies of the target sequence using, for example,
polymerase chain reactions (PCR) technology or any of the amplification
15 procedures. Amplification involves denaturation of template DNA by
heating in the presence of a large molar excess of each of two or more
oligonucleotide primers and four dNTPs (dGTP, dCTP, dATP, dTTP). The
reaction mixture is cooled to a temperature that allows the oligonucleotide
primer to anneal to target sequences, after which the annealed primers are
20 ext~nded with DNA polymerase. The cycle of denaturation, annealing, and
DNA synthesis, the principal of PCR amplification, is repeated many times
to generate large quantities of product which can be easily identified.
The major product of this exponential reaction is a segment of
double stranded DNA whose termini are defined by the 5' termini of the
25 oligonucleotide primers and whose length is defined by the distance between
the primers. Under normal reaction conditions, the amount of polymerase
CA 02218188 1997-10-14
WO 96132504 PCI'IUS9610S136
becomes limiting after 25 to 30 cycles or about one million fold
amplification. Further, amplification is achieved by diluting the sample
1000 fold and using it as the template for further rounds of amplification in
another PCR. By this method, amplification levels of 109 to 10'~ can be
S achieved during the course of 60 sequential cycles. This allows for the
detection of a single copy of the target sequence in the presence of
Co~ ";"~tin~ DNA, for example, by hybridization with a radioactive probe.
With the use of sequential PCR, the practical detection limit of PCR can be
as low as 10 copies of DNA per sample.
Although PCR is a reliable method for amplification of target
sequences, a number of other techniques can be used such as ligase chain
reaction, self sustained sequence replication, Q,B replicase amplification,
polymerase chain reaction linked ligase chain reaction, gapped ligase chain
reaction, ligase chain detection and strand displacement amplification. The
1~ principle of ligase chain reaction is based in part on the ligation of two
adjacent synthetic oligonucleotide primers which uniquely hybridize to one
strand of the target DNA or RNA. If the target is present, the two
oligonucleotides can be covalently linked by ligase. A second pair of
primers, almost entirely complementary to the first pair of primers is also
20 provided. The template and the four primers are placed into a thermocycler
with a thermostable ligase. As the temperature is raised and lowered,
oligonucleotides are renatured immediately adjacent to each other on the
template and ligated. The ligated product of one reaction serves as the
template for a subsequent round of ligation. The presence of target is
2~ manifested as a DNA fragment with a length equal to the sum of the two
adjacent oligonucleotides.
CA 02218188 1997-10-14
WO 96/32504 PCT/US96/OS136
24
Target sequences are fr~rnent~.l, if necç~s~ry, into a plurality
of fragments using physical, chemical or enzymatic means to create a set of
fragments of uniform or relatively uniform length. Preferably, the
sequences are enzymatically cleaved using nucleases such as DNases or
S RNases (mung bean nuclease, micrococcal nuclease, DNase I, RNase A,
RNase Tl), type I or II restriction endonucleases, or other site-specific or
non-specific endonucleases. Sizes of nucleic acid fragments are between
about 5 to about 1,000 nucleotides in length, preferably between about 10
to about 200 nucleotides in length, and more preferably between about 12
1 0 to about l OO nucleotides in length. Sizes in the range of about 5, 1 0,12,1 5,
18, 20, 24, 26, 30 and 35 are useful to perform small scale analysis of short
regions of a nucleic acid target. Fragment sizes in the range of 25, 50, 75,
125, 150, 175, 200 and 250 nucleotides and larger are useful for rapidly
analyzing larger target sequences.
Target sequences may also be enzymatically synthesized
using, for example, a nucleic acid polymerase and a collection of chain
elongating nucleotides (NTPs, dNTPs) and limiting amounts of chain
terrnin~ting (ddNTPs) nucleotides. This type of polymerization reaction can
be controlled by varying the concentration of chain termin~ting nucleotides
20 to create sets, for example nested sets, which span various size ranges. In
a nested set, fragments will have common one terminus and one terminus
which will be different between the members of the set such that the larger
fragments will contain the sequences of the smaller fragments.
The set of fragments created, which may be either homologous
25 or complementary to the target sequence, is hybridized to an array of nucleicacid probes forming a target array of nucleic acid probe/fragrnent
CA 02218188 1997-10-14
WO 96/32504 PCTJUS96~0S136
complexes. An array con~titllte~ an ordered or structuredplurality of nucleic
acids which may be fixed to a solid support or in liquid suspension.
. Hybridization of the fr~nen~ to the array allows for sorting of very large
eolleetions of nueleie aeid fr~ment~ into i~l~ntifi~ble groups. Sorting does
S not require a priori knowledge of the sequences of the probes, and can
greatly facilitate analysis by, for example, mass spectrophotometric
techniques.
Hybridization between complementary bases of DNA, RNA,
PNA, or combinations of DNA, RNA and PNA, occurs under a wide variety
10 of conditions such as variations in tempc-~lule, salt concentration,
electrostatic strength, and buffer composition. Exarnples ofthese conditions
and methods for applying them are described in Nucleic Acid Hybridizafion:
A Practical Approach (B.D. Hames and S.J. Higgins, editors, IRL Press,
1985). It is preferred that hybridization takes place between about 0~C and
15 about 70~C, for periods of from about one minute to about one hour,
depending on the nature of the sequence to be hybridized and its length.
However, it is recognized that hybridizations can occur in seconds or hours,
depending on the conditions of the reaction. For example, typical
hybridization conditions for a mixture of two 20-mers is to bring the mixture
20 to 68~C and let cool to room temperature (22~C) for five minutes or at very
low temperatures such as 2~C in 2 microliters. Hybridization between
nucleic acids may be facilitated using buffers such as Tris-EDTA (TE), Tris-
HCI and HEPES, salt solutions (e.g. NaCI, KCI, CaC12), other aqueous
solutions, reagents and chemicals. Examples of these reagents include
25 single-stranded binding proteins such as Rec A protein, T4 gene 32 protein,
E. coli single-stranded binding protein and major or minor nucleic acid
CA 02218188 1997-10-14
WO 96/3250~ PCT/US9~i/05136
26
groove binding proteins. Examples of other reagents and chemicals include
divalent ions, polyvalent ions and interc~l~tin~ substances such as ethidium
bromide, actinomycin D, psoralen and angelicin.
Optionally, hybridized target sequences may be ligated to a
5 single-strand of the probes thereby creating ligated target-probe complexes
or ligated target arrays. Ligation of target nucleic acid to probe increases
fidelity of hybridization and allows for incorrectly hybridized target to be
easily washed from correctly hybridized target. More importantly, the
addition of a ligation step allows for hybridizations to be performed under
10 a single set of hybridization conditions. Variation of hybridization
conditions due to base composition are no longer relevant as nucleic acids
with high A/T or G/C content ligate with equal efficiency. Consequently,
discrimination is very high between matches and mis-matches, much higher
than has been achieved using other methodologies wherein the effects of
15 G/C content were only somewhat neutralized in high concentrations of
quaternary or tertiary amines such as, for example, 3M tetramethyl
ammonium chloride. Further, hybridization conditions such as temperatures
of between about 22~C to about 37~C, salt concentrations of between about
0.05 M to about 0.5 M, and hybridization times of between about less than
20 one hour to about 14 hours (overnight), are also suitable for ligation.
Ligation reactions can be accomplished using a eukaryotic derived or a
prokaryotic derived ligase such as T4 DNA or RNA ligase. Methods for use
of these and other nucleic acid modif~ing enzymes are described in Current
Protocols in Molecular Biology (F.M. Ausubel et al., editors, John Wiley &
25 Sons, 1989).
CA 02218188 1997-10-14
WO 96/32504 PCTJUS96/OS136
Each probe of the probe array comprises a single-stranded
portion, an optional double-stranded portion and a variable sequence within
the single-stranded portion. These probes may be DNA, RNA, PNA, or any
combination thereof, and may be derived from natural sources or
5 recombinant sources, or be organically syrltl-esi7e~1 Preferably, each probe
has one or more double stranded portions which are about 4 to about 30
nucleotides in length, preferably about 5 to about 15 nucleotides and more
preferably about 7 to about 12 nucleotides, and may also be identical within
the various probes of the array, one or more single stranded portions which
10 are about 4 to 20 nucleotides in length, preferably between about 5 to about
12 nucleotides and more preferably between about 6 to about 10 nucleotides,
and a variable sequence within the single stranded portion which is about 4
to 20 nucleotides in length and preferably about 4, 5, 6, 7 or 8 nucleotides
in length. Overall probe sizes may range from as small as 8 nucleotides in
15 lengths to 100 nucleotides and above. Preferably, sizes are from about 12
to about 35 nucleotides, and more preferably, from about 12 to about 25
nucleotides in length.
Probe sequences may be partly or entirely known,
determinable or completely unknown. Known sequences can be created, for
20 example, by chemically synthesizing individual probes with a specified
sequence at each region. Probes with determinable variable regions may be
chemically synthesized with random sequences and the sequence
information determined separately. Either or both the single-stranded and
the double-stranded regions may comprise constant sequences such as, for
25 example, when an area of the probe or hybridized nucleic acid would benefit
CA 02218188 1997-10-14
WO 96132504 PCT/US96/05136
from having a constant sequence as a point of rc;fe~ ce in subsequent
analyses.
An advantage of this type of probe is in its structure.
Hybridization of the target nucleic acid is encouraged due to the favorable
5 thermodynamic conditions, including base-stacking interactions, established
by the presence of the adjacent double strandedness of the probe. Probes
may be structured with t~rmin~l single-stranded regions which consist
entirely or partly of variable sequences, internal single-skanded regions
which contain both constant and variable regions, or combinations of these
10 structures. Preferably, the probe has a single-stranded region at one
terminus and a double-stranded region at the opposite terminus.
Fragmented target sequences, preferably, will have a
distribution of terminal sequences sufficiently broad so that the nucleotide
sequence ofthe hybridized frS~grnçntc will include the entire sequence ofthe
15 target nucleic acid. Consequently, the typical probe array will comprise a
collection of probes with sufficient sequence diversity in the variable
regions to hybridize, with complete or nearly complete discrimination, all
of the target sequence or the target-derived sequences. The resulting target
array will comprise the entire target sequence on strands of hybridized
20 probes. By way of example only, if the variable portion consisted of a four
nucleotide sequence (R=4) of adenine, guanine, thymine, and cytosine, the
total number of possible combinations (4R) would be 44 or 256 different
nucleic acid probes. If the number of nucleotides in the variable sequence
was five, the number of different probes within the set would be 45 or 1,024.
25 In addition, it is also possible to utilize probes wherein the variable
nucleotide sequence contains gapped segrnents, or positions along the
CA 02218188 1997-10-14
W~ 96/32504 PCT(U~i96105136
29
variable sequence which will base pair with any nucleotide or at least not
interfere with adjacent base pairing.
A nucleic acid strand of the target array may be extt?ncle~l or
elongated enzymatically. Either the hybridized fr~grnent or one or the other
5 ofthe probe strands can be e~tPn-1e~1 Extension reactions can utilize various
regions of the target array as a template. For example, when fr~nent
sequences are longer than the hybridizable portion of a probe having a 3'
single-stranded terminus, the probe will have a 3' overhang and a 5'
overhang after hybridization of the fragment. The now internal 3' terminus
10 of the one strand of the probe can be used as a primer to prime an extension
reaction using, for example, an a~lo~liate nucleic acid polymerase and
chain elon~ting nucleotides. The extended strand of the probe will contain
sequence information ofthe entire hybridized fr~nent Reaction mixtures
cont~ining dideoxynucleotides will create a set of extended strands of
15 varying lengths and, preferably, a nested set of strands. As the fragments
have been initially sorted by hybridization to the array, each probe of the
array will contain sets of nucleic acids that represent each segment of the
target sequence. Base sequence information can be determined from each
extended probe. Compilation of the sequence information from the array,
20 which may require computer assistance with very large arrays, will allow
one to ~et~rrnine the sequence of the target. Depending on the structure of
the probe (e.g 5' overhang, 3' overhang, internal single-stranded region),
strands of the probe or strands of hybridized nucleic acid containing target
sequence can also be enzymatically amplified by, for example, single primer
25 PCR reactions. Variations of this process may involve aspects of strand
displacement amplification, Q~ replicase amplification, self-sustained
CA 02218188 1997-10-14
WO 96/32504 PCT/IJS96/05136
sequence replication amplification and any of the various polymerase chain
reaction amplification technologies.
Fxt~n~led nucleic acid strands of the probe can be mass
modified using a variety of techniques and methodologies. The most
S straight forward may be to erLzymatically synthesize the extension lltili7ing a polymerase and nucleotide reagents, such as mass modified chain
elongating and chain termin~ing nucleotides. Mass modified nucleotides
incorporate into the growing nucleic acid chain. Mass modifications may
be introduced in most sites of the macromolecule which do not interfere
10 with the hydrogen bonds required for base pair formation during nucleic
acid hybridization. Typical modifications include modification of the
heterocyclic bases, modifications of the sugar moiety (ribose or
deoxyribose), and modifications of the phosphate group. Specifically, a
modifying functionality, which may be a chemical moiety, is placed at or
15 covalently coupled to the C2, N3, N7 or N8 positions of purines, or the N7
or N9 positions of deazapurines. Modifications may also be placed at the
C5 or C6 positions of pyrimidines (e.g Figures lA, lB, 2A and 2B).
Examples of useful modifying groups include deuterium, F, Cl, Br, I, biotin,
fluorescein, iododicarbocyanine dye, SiR, Si(CH3)3, Si(CH3)2(C2Hs),
20 Si(CH3)2(C2Hs)2, Si(CH )~C H ~ ,5 2Si(C H ) ~ (Ç~ ) CH, 2 ~CH )3NR, 2 n
CH2CONR, (CH2)nOH, CH2F, CHF2 and CF3; wherein n is an integer and R
is selected from the group consisting of-H, deuterium and alkyls, alkoxys
and aryls of 1-6 carbon atoms, polyoxymethylene, monoalkylated
polyoxymethylene, polyethylene imine, polyamide, polyester, alkylated
25 silyl, hetero-oligo/polyaminoacid and polyethylene glycol (Figures 3 and 4).
CA 02218188 1997-10-14
WO 96/32504 PCT/US96/05136
Mass modifying functionalities may also be generated from
a precursor functionality such as -N3 or -XR, wherein X is: -OH, -NH2, -
., NHR,-SH,-NCS,-OCO(CH2)nCOOH,-NHCO(CH2)nCOOH,-OSO20H,
-OCO(CH2)nI or -OP(O-alkyl)-N-(alkyl)2, and n is an integer from 1 to 20;
5 and R is: -H, deuterium and alkyls, alkoxys or aryls of 1-6 carbon atoms,
such as methyl, ethyl, propyl, isopropyl, t-butyl, hexyl, benzyl, benzhydral,
trityl, substituted trityl, aryl, substituted aryl, polyoxymethylene,
monoalkylated polyoxymethylene, polyethylene imine, polyamide,
polyester, alkylated silyl, heterooligo/polyaminoacid or polyethylene glycol.
10 These and other mass modifying functionalities which do not interfere with
hybridization can be attached to a nucleic acids either alone or in
combination. Preferably, combinations of different mass modifications are
utilized to maximize distinctions between nucleic acids having different
sequences.
1~ Mass modifications may be major changes of molecular
weight, such as occurs with coupling between a nucleic acid and a
heterooligo/polyaminoacid, or more minor such as occurs by substituting
chemical moieties into the nucleic acid having molecular masses smaller
than the natural moiety. Non-essential chemical groups may be elimin~ted
20 or modified using, for example, an alkylating agent such as iodoacetamide.
Alkylation of nucleic acids with iodo~cet~mide has an additional advantage
that a reactive oxygen of the 3'-position of the sugar is elimin~ted. This
provides one less site per base for alkali cations, such as sodium, to interact.Sodium, present in nearly all nucleic acids, increases the likelihood of
25 forming satellite adduct peaks upon ionization. Adduct peaks appear at a
slightly greater mass than the true molecule which would greatly reduce the
W096/32S04 CA 02218188 1997-10-14 PCT/US96105136
accuracy of molecular weight determinations. These problems can be
addressed, in part, with matrix selection in mass spectrometric analysis, but
this only helps with nucleic acids of less than 20 nucleotides. Ammonium
(+NH3), which can substitute for the sodium cation (+Na) during ion
5 exchange, does not increase adduct forrnation. Consequently, another useful
mass modification is to remove alkali cations from the entire nucleic acid.
This can be accomplished by ion exchange with aqueous solutions of
arnmonium such as ammonium ~cet~te, ammonium carbonate, diammonium
hydrogen citrate, ammonium tartrate and combinations of these solutions.
10 DNA dissolved in 3 M aqueous ammonium hydroxide neutralizes all the
acidic functions of the molecule. As there are no protons, there is a
significant reduction in fragmentation during procedures such as mass
spectrometry.
Another mass modification is to utilize nucleic acids with non-
15 ionic polar phosphate backbones (e.g. PNA). Such nucleotides can begenerated by oligonucleoside phosphomonothioate diesters or by enzymatic
synthesis using nucleic acid polymerases and alpha- (~-) thio nucleoside
triphosphate and subsequent alkylation with iodo~cet~mide. Synthesis of
such compounds is straight forward and can be performed and the products
20 separated and isolated by, for example, analytical HPLC.
Mass modification of arrays can be performed before or after
target hybridization as the modification do not interfere with hybridization
of or hybridized nucleic. This conditioning of the array is simply to perform
and easily adaptable in bulk. Probe arrays can therefore be synthesized with
2~ no special manipulations. Only after the arrays are fixed to solid supports,
-
CA 02218188 1997-10-14
WO 96/32504 PCItUS96105136
just in fact when it would be most convenient to perform mass modification,
would probes be conditioned.
Probe strands may also be mass modified subsequent to
synthesis by, for example, contacting by treating the extended strands with
S an alkylating agent, a thiolating agent or subjecting the nucleic acid to cation
exchange. Nucleic acid which can be modified include target sequences,
probe sequences and strands, extended strands of the probe and other
available fragment~. Probes can be mass modified on either strand prior to
hybridization. Such arrays of mass modified or conditioned nucleic acids
10 can be bound to fr~rnent~ cont~inin~ the target sequence with no
il~L~lr~lc,lce to the fidelity of hybridization. Subsequent extension of either
strand of the probe, for example using Sanger sequencing techniques, and
using the target sequences as templates will create mass modified extended
strands. ~he molecular weights of these strands can be determined with
15 excellent accuracy.
Probes may be in solution, such as in wells or on the surface
of a micro-tray, or attached to a solid support. Mass modification can occur
while the probes are fixed to the support, prior to fixation or upon cleavage
from the support which can occur concurrently with ablation when analyzed
20 by mass spectrometry. In this regard, it can be important which strand is
released from the support upon laser ablation. Preferably, in such cases, the
probe is differentially attached to the support. One strand may be permanent
and the other temporarily attached or, at least, selectively releasable.
Examples of solid supports which can be used include a
25 plastic, a ceramic, a metal, a resin, a gel and a membrane. Useful types of
solid supports include plates, beads. microbeads, whiskers, combs,
CA 02218188 1997-10-14
WO 96132504 PCT/US96/OS136
34
hybridization chips, membranes, single crystals, ceramics and self-
assembling monolayers. A pL~fe.led embodiment comprises a two-
~lim~n.~ional or three--1imen.~ional matrix, such as a gel or hybridization chipwith multiple probe binding sites (Pevzner et al., J. Biomol. Struc. & Dyn.
5 9:399-410,1991; Maskos and Southern, Nuc. Acids Res.20: 1679-84, 1992).
Hybridization chips can be used to construct very large probe arrays which
are subsequently hybridized with a target nucleic acid. Analysis of the
hybridization pattern of the chip can assist in the identification of the targetnucleotide sequence. Patterns can be manually or computer analyzed, but
10 it is clear that positional sequencing by hybridization lends itself to
computer analysis and automation. Algorithms and software have been
developed for sequence reconstruction which are applicable to the methods
described herein (R. Drrnanac et al.? J. Biomol. Struc. & Dyn. 5:1085-1102,
1991; P. A. Pevzner, J. Biomol. Struc. & Dyn. 7:63-73, 1989).
Nucleic acid probes may be attached to the solid support by
covalent binding such as by conjugation with a coupling agent or by,
covalent or non-covalent binding such as electrostatic interactions, hydrogen
bonds or antibody-antigen coupling, or by combinations thereof. Typical
coupling agents include biotin/avidin, biotin/streptavidin, Staphylococcus
20 aureus protein A/IgG antibody Fc fragrnent, and streptavidin/protein A
chimeras (T. Sano and C.R. Cantor, Bio/Technology 9:1378-81, 1991), or
derivatives or combinations of these agents. Nucleic acids may be attached
to the solid support by a photocleavable bond, an electrostatic bond, a
disul~lde bond, a peptide bond, a diester bond or a combination of these sorts
25 of bonds. The array may also be attached to the solid support by a
selectively releasable bond such as 4~4'-dimethoxytrityl or its derivative.
CA 02218188 1997-10-14
WO 96/32504 PCTI~JS96105136
Derivatives which have been found to be useful include 3 or 4 [bis-(4-
methoxyphenyl)]-methyl-benzoic acid, N-succinimidyl- 3 or 4 [bis-(4-
methoxyphenyl)]-methyl-benzoic acid, N-succinimidyl- 3 or 4 [bis-(4-
methoxyphenyl)]-hydroxymethyl-benzoic acid, N-succinimidyl- 3 or 4 [bis-
(4-methoxyphenyl)]-chloromethyl-benzoic acid, and salts of these acids.
Binding may be reversible or permanent where strong
~ associations would be critical. In addition, probes may be attached to solid
supports via spacer moieties between the probes of the array and the solid
support. Useful spacers include a coupling agent, as described above for
10 binding to other or additional coupling partners, or to render the attachment to the solid support cleavable.
Cleavable ~t~hments may be created by attaching cleavable
chemical moieties between the probes and the solid support such as an
oligopeptide, oligonucleotide, oligopolyamide, oligoacrylamide,
15 oligoethylene glycerol, alkyl chains of between about 6 to 20 carbon atoms,
and combinations thereof. These moieties may be cleaved with added
chemical agents, electromagnetic radiation or enzymes. Examples of
attachments cleavable by enzymes include peptide bonds which can be
cleaved by proteases and phosphodiester bonds which can be cleaved by
20 nucleases. Chemical agents such as ~-mercaptoethanol, dithiothreitol (DTT)
and other reducing agents cleave disulfide bonds. Other agents which may
be useful include oxidizing agents, hydrating agents and other selectively
active compounds. Electromagnetic radiation such as ultraviolet, infrared
and visible light cleave photocleavable bonds. Attachments may also be
25 reversible such as, for example, using heat or enzymatic treatment, or
CA 02218188 1997-10-14
WO 96/32504 PCT/US96/05136
36
reversible chemical or magnetic ~ chments. Release and re~ chment can
be performed using, for example, magnetic or electrical fields.
Hybridized probes can provide direct or indirect information
about the hybridized sequence. Direct information may be obtained from
5 the binding pattern of the array wherein probe sequences are known or can
be determined. Indirect information requires additional analysis of a
plurality of nucleic acids of the target array. For example, a specific nucleic
acid sequence will have a unique or relatively unique molecular weight
depending on its size and composition. That molecular weight can be
10 determined, for example, by chromatography (e.g HPLC), nuclear magnetic
resonance (NMR), high-definition gel electrophoresis, capillary
electrophoresis (e.g. HPCE), spectroscopy or mass spectrometry.
Preferably, molecular weights are determined by measuring the mass/charge
ratio with mass spectrometry technology.
lS Mass spectrometry of biopolymers such as nucleic acids can
be performed using a variety of techniques (e.g U.S. Patent Nos.4,442,354;
4,931,639; 5002,868; 5,130,538;5,135,870; 5,174,962). Difficulties
associated with volatization of high molecular weight molecules such as
DNA and RNA have been overcome, at least in part, with advances in
20 techniques, procedures and electronic design. Further, only small quantities
of sample are needed for analysis, the typical sample being a mixture of 10
or so fragments. Quantities which range from between about 0.1 femtomole
to about 1.0 nanomole, preferably between about 1.0 femtomole to about
1000 femtomoles and more preferably between about 10 femtomoles to
25 about 100 femtomoles are typically sufficient for analysis. These amounts
CA 02218188 1997-10-14
WO 96/32S04 PCTIUS96105136
can be easily placed onto the individual positions of a suitable surface or
attached to a support.
Another of the important features of this invention is that it is
llnn~ceSs~ly to volatize large lengths of nucleic acids to dettormine sequence
S information. Using the methods of the invention, segments of the nucleic
acid target, discretely isolated into separate complexes on the target array,
can be sequenced and those sequence segments collated m~kin,~ it
unnecçss~ry to have to volatize the entire skand at once. Techniques which
can be used to volatize a nucleic acid fragment include fast atom
10 bombardment, plasma desorption, matrix-assisted laser
desorption/ionization, electrospray, photochemical release, electrical release,
droplet release, resonance ionization and combinations of these techniques.
In eleckohydrodynamic ionization, thermospray, aerospray
and electrospray, the nucleic acid is dissolved in a solvent and injected with
15 the help of heat, air or electricity, directly into the ionization chamber. If the
method of ionization involves a light beam, particle beam or electric
discharge, the sample may be attached to a surface and inkoduced into the
ionization chamber. In such situations, a plurality of samples may be
attached to a single surface or multiple surfaces and introduced
20 ~imlllt~neously into the ionization chamber and still analyzed individually.
The a~pr~liate sector ofthe surface which contains the desired nucleic acid
can be moved to proximate the path an ionizing beam. After the beam is
pulsed on and the surface bound molecules are ionized, a different sector of
the surface is moved into the path of the beam and a second sample, with the
25 same or different molecule, is analyzed without reloading the machine.
Multiple samples may also be introduced at electrically isolated regions of
CA 02218188 1997-10-14
WO 96/32504 PCT/US96/05136
a surface. Different sectors of the chip are cormected to an electrical source
and ionized individually. The surface to which the sample is attached may
be shaped for m~x;~ efficiency ofthe ionization method used. For field
ionization and field desorption, a pin or sharp edge is an efficient solid
support and for particle bombardment and laser ionization, a flat surface.
The goal of ionization for mass spectroscopy is to produce a
whole molecule with a charge. Preferably, a matrix-assisted laser
desorption/ionization (MALDI) or electrospray (ES) mass spectroscopy is
used to deterrnine molecular weight and, thus, sequence information from
the target array. It will be recognized by those of ordinary skill that a
variety of methods may be used which are a~ropliate for large molecules
such as nucleic acids. Typically, a nucleic acid is dissolved in a solvent and
injected into the ionization chamber using electrohydrodynamic ionization,
thermospray, aerospray or electrospray. Nucleic acids may also be attached
to a surface and ionized with a beam of particles or light. Particles which
have successfully used include plasma (plasma desorption), ions (fast ion
bombardment) or atoms (fast atom bombardment). Ions have also been
produced with the rapid application of laser energy (laser desorption) and
electrical energy (field desorption).
In mass spectrometer analysis, the sample is ionized briefly by
a pulse of laser beams or by an electric field induced spray. The ions are
accelerated in an electric field and sent at a high velocity into the analyzer
portion of the spectrometer. The speed of the accelerated ion is directly
proportional to the charge (z) and inversely proportional to the mass (m) of
the ion. The mass of the molecule may be deduced from the flight
characteristics of its ion. For small ions, the typical detector has a magnetic
CA 02218188 1997-10-14
WO 96/~2504 PCT/US96/05136
39
field which functions to constrain the ions stream into a circular path. The
radii of the paths of equally charged particles in a uniform magnetic field is
directly proportional to mass. l~at is, a heavier particle with the same
charge as a lighter particle will have a larger flight radius in a magnetic
5 field. It is generally considered to be impractical to measure the flight
characteristics of large ions such as nucleic acids in a magnetic field because
the relatively high mass to charge (m/z) ratio requires a magnet of unusual
size or strength. To overcome this limitation the electrospray method, for
example, can consistently place multiple ions on a molecule. Multiple
10 charges on a nucleic acid will decrease the mass to charge ratio allowing a
conventional quadrupole analyzer to detect species of up to 100,000 daltons.
Nucleic acid ions generated by the matrix assisted laser
desorption/ionization only have a unit charge and because of their large
mass, generally require analysis by a time of flight analyzer. Time of flight
15 analyzers are basically long tubes with a detector at one end. In the
operation of a TOF analyzer, a sample is ionized briefly and accelerated
down the tube. After detection, the time needed for travel down the detector
tube is calculated. The mass of the ion may be calculated from the time of
flight. TOF analyzers do not require a magnetic field and can detect unit
20 charged ions with a mass of up to 100,000 daltons. For improved resolution,
the time of flight mass spectrometer may include a reflectron, a region at the
end of the flight tube which negatively accelerates ions. Moving particles
emering the reflectron region, which contains a field of opposite polarity to
the accelerating field, are retarded to zero speed and then reverse accelerated
25 out with the same speed but in the opposite direction. In the use of an
analyzer with a reflectron, the detector is placed on the same side of the
CA 02218188 1997-10-14
WO 96132504 PCT/US96/05136
flight tube as the ion source to detect the returned ions and the effective
length of the flight tube and the resolution power is effectively doubled.
The calculation of mass to charge ratio from the time of flight data takes into
ac~;~t sf ~e ti~le sp~t in ~ etr~n.
S Ions with the same charge to mass ratio will typically leave the
ion accelerators with a range of energies because the ionization regions of
a mass spectrometer is not a point source. Ions generated further away from
the flight tube, spend a longer time in the accelerator field and enter the
flight tube at a higher speed. Thus ions of a single species of molecule will
arrive at the detector at different times. In time of flight analysis, a longer
time in the flight tube in theory provide more sensitivity, but due to the
different speeds of the ions, the noise (background) will also be increased.
A reflectron, besides effectively doubling the effective length of the flight
tube, can reduce the error and increase sensitivity by reducing the spread of
detector impingement time of a single species of ions. An ion with a higher
velocity will enter the refleckon at a higher velocity and stay in the
reflectron region longer than a lower velocity ion. If the reflectron electrode
voltages are arranged appropriately, the peak width contribution from the
initial velocity distribution can be largely corrected for at the plane of the
detector. The correction provided by the reflectron leads to increased mass
resolution for all stable ions, those which do not dissociate in flight, in the
spectrum.
While a linear field reflectron functions adequately to reduce
noise and enhance sensitivity, reflectrons with more comple~; field strengths
offer superior correctional abilities and a number of complex reflectrons can
be used. The double stage reflectron has a first region with a u eaker electric
CA 02218188 1997-10-14
WO 96/~2504 PCTIUS96105136
41
field and a second region with a skonger eleckic field. The quadratic and
the curve field reflectron have a eleckic field which increases as a function
of the distance. These functions, as their name implies, may be a quadratic
or a complex exponential function. The dual stage, quadratic, and curve
5 field reflectrons, while more elaborate are also more accurate than the linear reflectron.
The detection of ions in a mass speckometer is typically
performed using electron detectors. To be detected, the high mass ions
produced by the mass spectrometer is converted into either electrons or low
10 mass ions at a conversion electrode. These eleckons or low mass ions are
then used to start the eleckon multiplication cascade in an eleckon
multiplier and further amplified with a fast linear amplifier. The signals
from multiple analysis of a single sample are combined to improve the
signal to noise ratio and the peak shapes, which also increase the accuracy
15 of the mass determination.
This invention is also directed to the detection of multiple
primary ions directly through the use of ion cyclotron resonance and Fourier
analysis. This is useful for the analysis of a complete sequencing ladder
immobilized on a surface. In this method, a plurality of samples are ionized
20 at once and the ions are captured in a cell with a high magnetic field. An RF field excites the population of ions into cyclotron orbits. Because the
frequencies of the orbits are a function of mass, an output signal
representing the spectrum of the ion masses is obtained. This output is
analyzed by a computer using Fourier analysis which reduces the combined
25 signal to its component frequencies and thus provides a measurement of the
ion masses present in the ion sample. Ion cyclotron resonance and Fourier
CA 02218188 1997-10-14
WO 96/3250~1 PCT/US96/OS136
42
analysis can cl~t~rmine the masses of all nucleic acids in a sample. The
application of this method is especially useful on a sequencing ladder.
The data from mass spectrometry, either pe,rolllled singly or
in parallel (multiplexed), can detcnnine the molecular mass of a nucleic acid
S sample. The molecular mass, combined with the known sequence of the
sample, can be analyzed to determine the length of the sample. Because
different bases have different molecular weight, the output of a high
resolution mass spectrometer, combined with the known sequence and
reaction history of the sample, will determine the sequence and length of the
10 nucleic acid analyzed. In the mass spectroscopy of a sequencing ladder,
generally the base sequence of the primers are known. From a known
sequence of a certain length, the added base of a sequence one base longer
can be clelluced by a comparison of the mass of the two molecules. This
process is continlle~l until the complete sequence of a sequencing ladder is
1 5 deterrnined.
Another embodiment of the invention is directed to a method
for detecting a target nucleic acid. As before, a set of nucleic acids
complementary or homologous to a sequence of the target is hybridized to
an array of nucleic acid probes. The molecular weights of the hybridized
20 nucleic acids determined by, for example, mass spectrometry and the nucleic
acid target detected by the presence of its sequence in the sample. As the
object is not to obtain extensive sequence information, probe arrays may be
fairly small with the critical sequences, the sequences to be detected,
repeated in as many variations as possible. Variations may have greater than
25 95% homology to the sequence of interest, greater than 80%, greater than
70% or greater than about 60%. Variations may also have additional
CA 02218188 1997-10-14
wo 96/32s04 PC rlus96JO5136
43
sequences not required or present in the target sequence to increase or
decrease the degree of hybridization. Sensitivity of the array to the target
sequence is increased while reducing and hopefully elimin~ting the number
of false positives.
Target nucleic acids to be detected may be obtained from a
biological sample, an archival sarnple, an environment~l sample or another
source expected to contain the target sequence. For exarnple, samples may
be obtained from biopsies of a patient and the presence of the target
sequence is indicative of the disease or disorder such as, for example, a
neoplasm or an infection. Samples may also be obtained from
environmental sources such as bodies of water, soil or waste sites to detect
the presence and possibly identify or~nism~ and microor~ni.~m which may
be present in the sample. The presence of particular microorg~nism.c in the
sample may be indicative of a dangerous pathogen or that the normal flora
is present.
Another embodiment of the invention is directed to the arrays
of nucleic acid probes useful in the above-described methods and
procedures. These probes comprise a first strand and a second strand
wherein the first strand is hybridized to ~he second strand forming a double-
stranded portion, a single-stranded portion and a variable sequence within
the single-stranded portion. The array may be attached to a solid support
such as a material that facilitates volatization of nucleic acids for mass
spectrometry. Typically, arrays comprise large numbers vfprobes such as
less than or equal to about 4R different probes and R is the length in
nucleotides of the variable sequence. When utilizing arrays for large scale
sequencing, larger arrays can be used whereas, arrays which are used for
CA 02218188 1997-10-14
WO 96/32504 PCTIUS96/05136
44
detection of specific sequences may be fairly small as many of the potential
sequence combinations will not be necessary.
Arrays may also comprise nucleic acid probes which are e
entirely single-stranded and nucleic acids which are single-stranded, but
possess hairpin loops which create double-stranded regions. Such structures
can function in a manner similar if not identical to the partially single-
stranded probes, which comprise two strands of nucleic acid, and have the
additional advantage of thermodynamic energy available in the secondary
structure.
Arrays may be in solution or fixed on a solid support through
streptavidin-biotin interactions or other suitable coupling agents. Arrays
may also be reversibly fixed to the solid support using, for example,
chemical moieties which can be cleaved with electromagnetic radiation,
chemical agents and the like. The solid support may comprise materials
such as matrix chemicals which assist in the volatization process for mass
spectrometric analysis. Such chemicals include nicotinic acid, 3'-
hydroxypicolnic acid, 2,5-dihydroxybenzoic acid, sinapinic acid, succinic
acid, glycerol, urea and Tris-HCl, pH about 7.3.
Another embodiment of the invention is directed to
sequencing double-stranded nucleic acids using strand-displacement
polymerization. With this method it is unnecessary to denature the double-
strands to obtain sequence inforrnation. Strand-displacement polymerization
creates a new strand while simultaneously displacing the existing strand.
Techniques for incorporating label into the growing strand are well-know
and the newly polymerized strand is easily detected by, for example, mass
spectrometry.
CA 02218188 1997-10-14
Wo 96/32504 PcTruss6~o5l36
Target nucleic acid or nucleic acids cont~inin~ sequences that
correspond to the sequence of the target are digested, for exarnple, with
restriction enzymes, in one or more steps to create a set of fragments which
are partially single-stranded and partially double-stranded. Another set of
5 nucleic acids, the probes, are also partially single-stranded and partially
double-stranded. These probes preferably contain a variable or constant
regions within the single-stranded portion of the terminus of each fragment
(5'- or 3'-ovPrh~n,~.c). Probes or fragments are treated with a phosphatase LO
remove phosphate groups from the 5'-termini of the nucleic acids.
10 Phosphatase treatment prevents nucleic acid ligation by ligase which
requires a t~nnin~l 5'-phosphate to covalently link to a 3'-hydroxyl. Single-
stranded regions ofthe fr~gm~nt~ are hybridized to single-stranded regions
of the probes forming an array of hybridized target/probe complexes.
Adjacent or abutting nucleic acid strands of the complex are ligated,
15 covalently joining a strand of the fragrnent to a strand of the probe.
Phosphatase treatment prevents both self-ligation of phosphatase-treated
nucleic acids and ligation between the 5'-termini of phosphatased nucleic
acids and the 3'-termini of untreated nucleic acids. These complexes are
treated with a nucleic acid polymerase that recognizes and bind to the nick
20 in the unligated strand to initiate polymerization. The polymerase
synthesizes a new strand using the ligated stand as a template, while
displacing the complementary strand. The reaction may be supplemented
with labeled or mass modified nucleotides (e.g mass modifications at
positions C2, N3, N7 or C8 of purine, or at N7 or N9 of deazapurine) or
25 other detectable markers that will allow for the detection of new synthesis.
Either the probes or the fragments may be fixed to a solid support such as
CA 02218188 1997-10-14
WO 96t32504 PCT/US96/05136
46
a plastic or glass surface, membrane or structure (magnetic bead) which
elimin~tes the need for repetitive extractions or other purification of nucleic
acids between steps.
Preferably, double-stranded nucleic acids cont~ining target
5 sequences are obtained by polymerase chain reaction or enzymatic digestion
(e.g restriction enzymes) of the target sequence. Target sequences may be
DNA, RNA, RNA/DNA hybrids, cDNA, PNA or modifications or
combinations thereof and are preferably from about 10 to about 1,000
nucleotides in length, more preferably, from about 20 to about 500
10 nucleotides in length, and even more preferably, from about 35 to about 250
nucleotides in length. 5'-termini ofthe nucleic acid fr~gment~ orprobes may
be dephosphorylated with a phosphatase, such as ~lk~line or calf intestinal
phosphatase, which elimin~tes the action of a nucleic acid ligase. Upon
hybridization of fragment to probe, only one of the two internal 5'-3'
15 junctions contains a 5'-phosphate and is capable of ligation. The second
junction appears as a nick in a strand of the complex. Nucleic acid
polymerases, such as Klenow, recognize the nick and synthesize a new
strand while displacing the complementary, ligated strand. Chain elongation
can proceed in the presence of, for example, nucleotide triphosphates and
20 chain termin~ting nucleotides. Nucleic acid synthesis tern~in~tes when a
dideoxynucleotide is incorporated into the elongating strand. The resulting
fragments represent a nested set of the sequence of the target. Precursor
nucleotides may be labeled with, for example, mass modifications. The
mass modified fragments can be easily analyzed by mass spectrometry to
25 determine the sequence of the target. Complexes may further comprise
single-stranded binding protein (SSB; E. coli) which increases stability of
CA 02218188 1997-10-14
wo 96/32504 PCT~S96S~5136
47
the complex and facilitate polymerase action. Bands otherwise obscured are
more easily detecte-l SSB can be used to sequence fragments of greater
than 100 nucleotides, preferably greater than 150 nucleotides and more
preferably greater than 200 nucleotides.
S This method is generally useful for m~nllAl or automated
nucleic acid sequencing, and especially useful for identifying and
sequencing a single or group of nucleic acid species in a mixed background
cont~inin~ a plurality of species of different sequences. In this method,
selection is performed upon hybridization and ligation of fragments to
probes. Probes may be designed to contain a cornrnon or variable sequence
within the single-stranded region that is complementary to a sequence of the
fragment to be identified and, if desired, sequenced. Stringency of
fragment/probe hybridization can be adjusted by methods well-known to
those of ordinary skill to match desired conditions of selection. For
l S example, the single-stranded region of the probe can be designed to contain
a specific sequence only found on the single-stranded region of the nucleic
acid frA~nent of interest. ~IternAtively, multiple probes containing multiple
variable regions may be used to select for those fragment sequences which
may be longer than the length of the single-stranded region of any one
probe. Hybridization and ligation selects the specific fragment from a
complex mixture of different fragments and only that specific fragment is
subsequently sequenced.
Probes are typically ~om about lS to about 200 nucleotides
in length, but can be larger or small depending on the particular application.
Single-stranded regions ofthe probes may be about 3, 4, 5, 6, 7, 8, 9, 10, 12,
15, 20, 22, 25 or 30 nucleotides in length or larger. For probes containing
CA 02218188 1997-10-14
WO 96/32504 PCT/US96/05136
48
a variable region within the single-stranded region, the length of this
variable region may be the same or smaller than the length of the entire
single-stranded portion. Variable regions may be distinct between probes
or common within sets of probes. The double-stranded region of the probe
5 is typically larger than the single-stranded region and may be about 4, 5, 6,
7, 8, 9, 10, 12, 14, 16, 18, 20, 22, 24, 26, 28, 30, 35 40 or 50 nucleotides in
length or larger. Probes may also be modified to facilitate attachment to a
solid support or other surfaces, or modified to be individual detectable for
identification or other purposes. Sets of nucleic acids, either fragments or
10 probes, preferably contain greater than 1o2, 103, 104, 105, 1o6, 107, 1o8, 109
or 10'~ different members.
Another embodiment of the invention is directed to kits for
detecting a sequence of a target nucleic acid. An array of nucleic acid
probes is fixed to a solid support which may be coated with a matrix
15 chemical that facilitates volatization of nucleic acids for mass spectrometry.
Kits can be used to detect diseases and disorders in biological samples by
detecting specific nucleic acid sequences which are indicative of the
disorder. Probes may be labeled with detectable labels which only become
detectable upon hybridization with a correctly matched target sequence.
20 Detectable labels include radioisotopes, metals, luminescent or
bioluminescent chemicals, fluorescent chemicals, enzymes and
combinations thereof.
Another embodiment of the invention is directed to nucleic
acid sequencing systems which comprise a mass spectrometer, a computer
25 loaded with a~ op- iate software for analysis of nucleic acids and an array
CA 02218188 1997-10-14
Wl~ ~6/;}2504 PCTJ~JS96105136
49
of probes which can be used to capture a target nucleic acid sequence.
Systems may be m~nll~l or automated as desired.
The following experiments are offered to illustrate
embodiments of the invention, and should not be viewed as limiting the
S scope of the invention.
Fx~n~les
Fx~mrle 1 Preparation of Tar~et Nucleic Acid.
Target nucleic acid is prepared by restriction endonuclease
cleavage of cosmid DNA. The properties of type II and other restriction
10 nucleases ~at cleave outside of their recognition sequences were exploited.
A restriction digestion of a 10 to 50 kb DNA sample with such an enzyme
produced a mixture of DNA fragments most of which have unique ends.
Recognition and cleavage sites of useful enzymes are shown in Table 1.
Table 1
Restriction Enzymes and Recognition Sites for PSBH
Mwo I GCNNNNN-NNGC
CGNN-NNNNNCG
~si YI CCNNNNN-NNGG
GGNN-NNNNNCC
t
Apa BI GCANNNNN-TGC
CGT-NNNNNACG
t
Mnl I CCTCN7
GGAGN6
t
CA 02218188 1997-10-14
WO 96/32504 PCI~/US96/OS136
Tsp RI NNCAGTGNN
NNGTCACNN
S Cje I CCANNNN~N-GFNNNN
GG~NN~N-CANNNN
Cje PI CCANM~N-NNTCNN
1 0 GG~NNNN-NNAGNN
One restriction enzyme, ~paB 15, with a 6 base pair
recognition site may also be used. DNA sequencing is best served by
15 enzymes that produce average fragment lengths comparable to the lengths
of DNA sequencing ladders analyzable by mass speckometry. At present
these lengths are about 100 bases or less.
BsiYI and Mwo I restriction endonucleases are used together
to digest DNA in preparation of PSBH. Target DNA from is cleaved to
20 completion and complexed with PSBH probes either before or after melting.
The fraction of fragments with unique ends or degenerate ends depends on
the complexity of the target sequence. For example, a 10 kilobase clone
would yield on average 16 fr~f~nent~ or a total of 32 ends since each double-
stranded DNA target produces two ligatable 3' ends. With 1024 possible
25 ends, Poisson statistics (Table 2) predict that there would be 3%
degeneracies. In contrast, a 40 kilobase cosmid insert would yield 64
fragments or 128 ends, of which, 12% of these would be degenerate and a
50 kilobase sample would yield 80 fragments or 160 ends. Some of these
would surely be degenerate. Up to at least 100 kilobase, the larger the target
30 the more sequence are available from each multiple~ DNA sample
CA 02218188 1997-10-14
WO 96/32504 PCTIUS96105136
preparation. With a 100 kilobase t~rget, 27% of the targets would be
degenerate.
Table 2
Poisson Distribution of Restriction Enzyme Sites
Targetsize Mwo I TspR I
(kb) Sequencing Assembly Sequencing Assembly
0.97 0.60 0.94 0.94
0.88 0.14 0.80 0.80
100 0.73 0.01 0.57 0.57
With BsiYI and Mwo I, any restriction site that yields a unique
5 base end may be captured twice and the resulting sequence data obtained
will read away from the site in both directions (Figure 5). With the
knowledge of three bases of overlapping sequence at the site, this sorts all
15 sequences into 64 different categories. With 10 kilobase targets, 60% will
contain fragments and, thus sequence assembly is automatic.
Two array capture methods can be used with Mwo I and BsiY
I. In the first method, conventional five base capture is used. Because the
two target bases adjacent to the capture site are known, they from the
restriction enzyme recognition sequence, an alternative capture strategy
would build the complement of these two bases into the capture sequence.
Seven base capture is thermodynamically more stable, but less
discrimins~ting against mi~m~tches.
TspR I is another commercially available restriction enzyme
with properties that are very attractive for use in PSBH-mediated Sanger
sequencing. The method for using TspR I is shown in Figure 6. TspR I has
a five base recognition site and cuts two bases outside this site on each
strand to yield nine base 3' single-stranded overhangs. These can be
captured with partially duplex probes with complementary nine base
CA 02218188 1997-10-14
WO 96/32504 PCI~/US96/05136
overh~n~c. Because only four bases are not specified by enzyme
recognition, TspR I digest results in only 256 types of cleavage sites. With
human DNA the average fr~gment length that results is 1370 bases. This
enzyme is ideal to generate long Sequence ladders and are useful to input to
5 long thin gel sequencing where reads up to a kilobase are common. A
typical human cosmid yields about 30 ~spR I fragments or 60 ends. Given
the length distribution expected, many of these could not be sequenced fully
from one end. With 256 possible overhangs, Poisson statistics (Table 2)
indicate that 80% adjacent fragments can be assembled with no additional
10 labor. Thus, very long blocks of continuous DNA sequence are produced.
Three additional restriction enzymes are also useful. These
are Mnl I, Cje I and CjeP I (Table 1). The first has a four base site with one
A+T should give smaller human DNA fragments on average than Mwo I or
BsiY I. The latter two have unusual interrupted five base recognition sites
15 and might supplement TspR I.
Target DNA may also be prepared by tagged PCR. It is
possible to add a preselected five base 3' terminal sequence to a target DNA
using a PCR primer five bases longer than the known target sequence
priming site. Samples made in this way can be captured and sequenced
20 using the PSBH approach based on the five base tag. A biotin was used to
allow purification of the complementary strand prior to use as an
immobilized sequencing template. A biotin may also be placed on the tag.
After capture of the duplex PCR product by streptavidin-coated magnetic
microbeads, the desired strand (needed to serve as a sequencing template)
25 could be flçn~tl~red from the duplex and used to contact the entire probe
array. For multiplex sample preparation, a series of different five base
CA 02218188 1997-10-14
WO 96/~2504 PCT~S96105136
tagged primers would be employed, ideally in a single multiplex PCR
reaction This approach also requires knowing enough target sequence for
unique PCR amplification and is more useful for shotgun sequencing or
co~ e sequencing than for de novo sequencing.
5 Fx~mrle 2 R~ic Aspects of Positional Sequenc;r~ by Hybridization.
An ex~min~tion of the potential advantages of stacking
hybridization has been carried out by both calculations and pilot
experim~nt~ Some c~lc~ te(l Tm's for perfect and mi~m~tçhed duplexes are
shown in Figure 7. These are based on average base compositions. The
10 calculations revealed that the binding of a second oligomer next to a pre-
formed duplex provides an extra stability equal to about two base pairs and
that mis-pairing seems to have a larger consequence on stacking
hybridization than it does on ordinary hybridization. Other types of mis-
pairing are less destabilizing, but these can be elimin~te~l by requiring a
l5 ligation step. In standard SBH, a terminal mi~m~tch is the least
destabilizing event, and leads to the greatest source of ambiguity or
background. For an octanucleotide complex, an average terminal mi~m~tch
leads to a 6~C lowering in Tm. For stacking hybridization, a terminal
mi~m~tch on the side away from the pre-existing duplex, is the least
20 destabilizing event. For a pentamer, this leads to a drop in Tm of 10~C.
These considerations indicate that the discrimin~tion power of stacking
hybridization in favor of perfect duplexes are greater than ordinary SBH.
Example 3 Preparation of Model Arrays.
In a single synthesis, all 1024 possible single-stranded probes
25 with a constant 18 base stalk followed by a variable 5 base extension can be
created. The 18 base extension is designed to contain two restriction
CA 02218188 1997-10-14
WO 96/32~i04 PCT/US96/05136
enzyme cutting sites. Hga I generates a S base, 5' overhan~, consisting of the
variable bases N5. Not I generates a 4 base, 5' overhang at the constant end
of the oligonucleotide. The synthetic 23-mer mixture hybridized with a
complementary 1 8-mer forms a duplex which can be enzymatically
5 extended to form all 1024, 23-mer duplexes. These are cloned by, for
example, blunt end ligation, into a plasmid which lacks Not I sites. Colonies
containing the cloned 23-base insert are selected and each clone contains
one unique sequence. DNA mi~ reL)s can be cut at the constant end of the
stalk, filled in with biotinylated pyrimidines and cut at the variable end of
10 the stalk to generate the 5 base 5' overhang. The resulting nucleic acid is
fractionated by Qiagen columns (nucleic acid purification columns) to
discard the high molecular weight material. The nucleic acid probe will then
be attached to a ~ avidin-coated surface. This procedure could easily be
automated in a Beckman Biomec or equivalent chemical robot to produce
l S many identical arrays of probes.
The initial array contains about a thousand probes. The
particular sequence at any location in the array will not be known.
However, the array can be used for statistical evaluation of the signal to
noise ratio and the sequence discrimination for different target molecules
20 under different hybridization conditions. Hybridization with known nucleic
acid sequences allows for the identification of particular elements of the
array. A sufficient set of hybridizations would train the array for any
subsequent sequencing task. Arrays are partially characterized until they
have the desired properties. For example, the length of the oligonucleotide
25 duplex, the mode of its ~ chment to a surface and the hybridization
conditions used can all be varied using the initial set of cloned DNA probes.
CA 02218188 1997-10-14
WO 96/32504 PCTIUS96~05136
Once the sort of array that works best is determined, a complete and fully
characterized array can be constructed by ordinary chemical synthesis.
Example 4 P~ al~lion of Specific Probe Arrays.
With positional SBH, one potential trick to compensate for
5 some variations in stability among species due to GC content variation is to
provide GC rich st~king duplex adjacent AT rich overhangs and AT rich
stacking duplex adjacent GC rich overh~n~s. Moderately dense arrays can
be made using a typical x-y robot to spot the biotinylated comp~unds
individually onto a ~ t~vidin-coated surface. Using such robots, it is
10 possible to make arrays of 2 x 104 samples in 100 to 400 cm2 of nominal
surface. Commercially available ~ avidin-coated beads can be adhered,
permanently to plastics like polystyrene, by exposing the plastic first to a
brief treatment with an organic solvent like triethylamine. The resulting
plastic surfaces have enormously high biotin binding capacity because of the
15 very high surface area that results.
In certain experiments, the need for attaching oligonucleotides
to surfaces may be circumvented altogether, and oligonucleotides attached
to streptavidin-coated magnetic microbeads used as already done in pilot
experiments. The beads can be manipulated in microtiter plates. A
20 magnetic separator suitable for such plates can be used including the newly
available compressed plates. For example, the 18 by 24 well plates
(Genetix, Ltd.; USA Scientific Plastics) would allow cont~inment of the
entire arrav in 3 plates. This format is well handled by existing chemical
robots. It is preferable to use the more compressed 36 by 48 well format so
25 the entire array would fit on a single plate. The advantages of this approachfor all the experiments are that any potential complexities from surface
CA 02218188 1997-10-14
WO 96/32504 PCT/US96/05136
56
effects can be avoided and already-existing liquid h~n~lling, therm~l control
and imaging methods can be used for all the experiments.
Lastly, a rapid and highly efficient method to print arrays has
been developed. Master arrays are made which direct the preparation of
S replicas or a~)pr~rlate complementary arrays. A master array is made
manually (or by a very accurate robot) by sampling a set of custom DNA
sequences in the desired pattern and then transferring these sequences to the
replica. The master array is just a set of all 1024-4096 compounds printed
by multiple he~e~l pipettes and compressed by offsetting. A potentially
10 more elegant approach is shown in Figure 8. A master array is made and
used to transfer components of the replicas in a sequence-specific way. The
sequences to be transferred are designed to contain the desired 5 or 6 base
5' variable overhang adjacent to a unique 15 base DNA sequence.
The master array consists of a set of streptavidin bead-
15 impregnated plastic coated metal pins. Immobilized biotinylated DNAstrands that consist of the variable 5 or 6 base segment plus the constant 15
base segment are at each tip. Any unoccupied sites on this surface are filled
with excess free biotin. To produce a replica chip, the master array is
incubated with the complement of the 15 base constant sequence, 5'-labeled
20 with biotin. Next, DNA polymerase is used to synthesize the complement
of the 5 or 6 base variable sequence. Then the wet pin array is touched to
the streptavidin-coated surface ofthe replica and held at a temperature above
the Tm of the complexes on the master array. If there is insufficient liquid
carryover from the pin array for efficient sample transfer, the replica array
25 could first be coated with spaced droplets of solvent, either held in concavecavities or delivered by a multi-head pipettor. After the transfer, the replica
CA 02218188 1997-10-14
WO 96/32504 PC~IUS96J053.36
chip is incllb~tecl with the complement of 15 base con~t~nt sequence to
reform the double-stranded portions of the array. The basic advantage of
this scheme is that the master array and transfer compounds are made only
once aIld the m~nllf~chlre of replica arrays can proceed almost endlessly.
5 Example 5 AttachmeIlt ofNucleic Acids Probes to Solid Supports.
Nucleic acids may be attached to silicon wafers or to beads.
A silicone solid support was d~.iv~ ed to provide iodoacetyl fimctionalities
on its surface. Derivatized solid support were bound to disulfide containing
oligodeoxynucleotides. ~ltern~tively~ the solid support may be coated with
10 ~llc~idin or avidin arld bound to biotinylated DNA.
Covalent ~ chment of oligonucleotide to derivatized chips:
Silicon wafers are chips with an approximate wei~ht of 50 mg To m~int~in
uniform reaction condition, it was necessary to determine the exact weight
of each chip and select chips of similar weights for each experiment. The
15 reaction scheme for this procedure is shown in Figure 9.
To derivatize the chip to contain the iodoacetyl functionality
an anhydrous solution of 25% (by volume) 3-aminopropyltrieshoxysilane
in toluene was prepared under argon and aliquotted (700 111) into tubes. A
50 mg chip requires approximately 700 ~11 of silane solution. Each chip was
20 flamed to remove any surface cont~rnin~nts during its manufacture and
dropped into the silane solution. The tube containing the chip was placed
under an argon environment and shaken for approximately three hours.
~fter this time, the silane solution was removed and the chips were washed
three times with toluene and three times with dimethyl sulfoxide (DMSO).
25 A 10 mM solution of N-succinimidyl(4-iodoacetyl)aminobenzoate (SIAB)
(Pierce Chemical Co.; Rockford, IL) was prepared in anhydrous DMSO and
CA 02218188 1997-10-14
WO 96/32504 PCT/US96105136
added to the tube cont~inin~ a chip. Tubes were shaken under an argon
environment for 20 minutes. The SIAB solution was removed and after
three washes with DMSO, the chip was ready for attachment to
oligonucleotides.
Some oligonucleotides were labeled so the efficiency of
~ çhment could monitored. Both 5' disulfide cont~ining
oligodeoxynucleotides and unmodified oligodeoxynucleotides were
radiolabeled using terminal deoxynucleotidyl transferase enzyme and
standard techniques. In a typical reaction, 0.5 mM of disulfide-containing
oligodeoxynucleotide mix was added to a trace amount of the same species
that had been radiolabeled as described above. This mixture was incubated
with dithiothreitol (DTT) (6.2 ,umol, 100 mM) and
ethylenediaminetetraacetic acid (EDTA) pH 8.0 (3 ,umol, 50 mM). EDTA
served to chelate any cobalt that remained from the radiolabeling reaction
that would complicate the cleavage reaction. The reaction was allowed to
proceed for 5 hours at 37~C. With the cleavage reaction essentially
complete, the free thiol-containing oligodeoxynucleotide was isolated using
a Chromaspin- 10 column.
Similarly, Tris-(2-carboxyethyl)phosphine (TCEP) (Pierce
Chemical Co.; Rockford, IL) has been used to cleave the disulfide.
Conditions utilize TCEP at a concentration of approximately 100 mM in pH
4.5 buffer. It is not necessary to isolate the product following the reaction
since TCEP does not competitively react with the iodoacetyl functionality.
To each chip which had been derivatized to contain the
iodoacetyl functionality was added to a 10 ,uM solution of the
oligodeoxynucleotide at pH 8. The reaction was allo-~ed to proceed
CA 02218188 1997-10-14
WO 96/32504 PCTIUS96JOS1~6
59
overnight at room temperature. In ~is manner, two different
oligodeoxynucleotides have been t;x~llilled for their ability to bind to the
iodoacetyl silicon wafer. T~e first was the free thiol cont~inin~
oligodeoxynucleotide already described. In parallel with the free thiol
5 cont~ining oligodeoxynucleotide reaction, a negative control reaction has
been performed that employs a S' unmodified oligodeoxynucleotide. This
species has similarly been 3' radiolabeled, but due to the unmodified 5'
terrninus, the non-covalent, non-specific interactions may be determined.
Following the reaction, the radiolabeled oligodeoxynucleotides were
10 removed and the chips were washed 3 times with water and quantitation
proceeded.
To ~letçrrnine the efficiency of ~tt~c~ment~ chips of the wafer
were exposed to a phosphorimager screen (Molecular Dynamics). This
exposure usually proceeded overnight, but occasionally for longer periods
15 of time depending on the amount of radioactivity incorporated. For each
different oligodeoxynucleotide l.tili7~-1 reference spots were made on
polystyrene in which the molar amount of oligodeoxynucleotide was known.
These reference spots were also exposed to the phosphorimager screen.
Upon sc~nning the screen, the quantity (in moles) of oligodeoxynucleotide
20 bound to each chip was determined by c~ g the counts to the specific
activities of the references. Using the weight of each chip, it is possible to
calculate the area of the chip:
(g of chip) ( 1 130 mm2/g) = x mm2
By incorporating this value, the amount of oligodeoxynucleotide bound to
25 each chip mav be reported in fmol/mm2. It is necessary to divide this value
by two since a radioactive signal of 32p iS strong enough to be read through
CA 02218188 1997-10-14
WO 96/32504 PCT/US96/05136
the silicon wafer. Thus the instrument is essentially recording the
radioactivity from both sides of the chip.
Following the initial qll~ntit~tion each chip was washed in 5
x SSC buffer (75 rnM sodium citrate, 750 rnM sodium chloride, pH 7) with
5 50% formamide at 65~C for 5 hours. Each chip was washed three times
with warm water, the S x SSC wash was repeated, and the chips
requantitated. Disulfide linked oligonucleotides were removed from the
chip by incubation with 100 rnM DTT at 37~C for 5 hours.
Example 6 Attachment of Nucleic Acids to Streptavidin Coated Solid
10 Support.
Immobilized single-stranded DNA targets for solid-phase
DNA sequencing were prepared by PCR amplification. PCR was performed
on a Perkin Elmer Cetus DNA Thermal Cycler using VentR (exo~) DNA
polymerase (New England Biolabs; Beverly, MA), and dNTP solutions
15 (Promega; Madison, WI). EcoR I digested plasmid NB34 (a PCRTM II
plasmid with a one kb target anonymous human DNA insert) was used as
the DNA template for amplification. PCR was performed with an 1~-
nucleotide upstream primer and a downstream 5'-end biotinylated 18-
nucleotide primer. PCR amplification was carried out in a 100 ~11 or 400 ~11
20 volume containing 10 mM KCI, 20 mM Tris-HCI (pH 8.8 at 25~C), 10 mM
(NH4)2SO4, 2 mM MgSO4, 0.1% Triton X-100, 250 ~lM dNTPs, 2.5 ,uM
biotinylated primer, 5 ~lM non-biotinylated primer, less than 100 ng of
plasmid DNA, and 6 units of Vent (exo~) DNA polymerase per 100 ~1 of
reaction volume. Thirty temperature cycles were performed which included
25 a heat denaturation step at 9~~C for 1 minute, followed by annealing of
primers to the template DNA for 1 minute at 60~C, and DNA chain
CA 02218188 1997-10-14
wo 96/32504 PC~IUS96105136
61
extension with Vent (exo~) polymerase for 1 minute at 72~C. For
amplification with the tagged primer, 45~C was selected for primer
~nne~lin~ The PCR product was purified through a Ultrafree-MC 30,000
NMVVL filter unit (Millipore, Bedford, MA) or by electrophoresis and
S extraction from a low melting agarose gel. About 10 pmol of purified PCR
fragment was mixed with 1 mg of prewashed magnetic beads coated with
fidin (Dynabeads M280, Dynal, Norway) in 100 ~1 of 1 M NaCl and
TE incubating at 37~C or 45~C for 30 minutes.
The magnetic beads were used directly for double stranded
10 sequencing. For single stranded sequencing, the immobilized biotinylated
double-stranded DNA fragment was converted to single-stranded form by
treating with freshly prepared 0.1 M NaOH at room temye~aLllFe fsr 5
minl-tes. The magnetic beads, with immobilized single-stranded DNA, were
washed with 0.1 M NaOH and TE before use.
15 Example 7 Hybri(1i7~tion Specificity.
Hybridization was performed using probes with five and six
base pair overhangs, including a five base pair match, a five base pair
mi~m~tch, a six base pair match, and a six base pair mismatch. These
sequences are depicted in Table 3.
W096/32504 CA 02218188 1997-10-14PCT/US96/05136
62
Table 3
Hybridized Test Sequences
Test Sc~ s.
5 bp overlap, perfect match:
3'-TCG AGA ACC TTG GCT*-S' (SEQ ID NO I )
3'-CTA CTA GGC TGC GTA GTC(SEQ ID NO 2)
S'-biotin-GAT GAT CCG ACG CAT CAG AGC TC-3' (SEQ ID NO 3)
5 bp overlap, ,..;~.,.;1l. 1) at 3' end:
3'-TCG AGA ACC TTG GCT*-5' (SEQ ID NO 1)
3'-CTA CTA GGC TGC GTA GTC(SEQ ID NO 2)
5'-biotin-GAT GAT CCG ACG CAT CAG AGC 1~-3' (SEQ ID NO 4)
6 bp overlap, perfect match:
3'-TCG AGA ACC TTG GCT*-5' (SEQ ID NO I )
3'-CTA CTA GGC TGC GTA GTC(SEQ ID NO 2)
15 5'-biotin-GAT GAT CCG ACG CAT CAG AGC TCT-3' (SEQ ID NO 5)
6 bp overlap, ~..;~ four bases ~om 3' end:
3'-TCG AGA ACC TTG GCT*-5' (SEQ ID NO 1)
3'-CTA CTA GGC TGC GTA GTC(SEQ ID NO 2)
5'-biotin-GAT GAT CCG ACG CAT CAG AGT TCT-3' (SEQ ID NO 6)
The biotinylated double-stranded probe was prepared in TE
buffer by annealing the complimentary single strands together at 68~C for
five minutes followed by slow cooling to room tempcla~u,e. A five-fold
25 excess of monodisperse, polystyrene-coated magnetic beads (Dynal) coated
with streptavidin was added to the double-stranded probe, which as then
incubated with agitation at room temperature for 30 minlltes. After ligation,
the samples were subjected to two cold (4~C) washes followed by one hot
(90~C) wash in TE buffer (Figure 10). The ratio of 32p in the hot
30 supern~t~nt to the total amount of 32p was determined (Figure 11). At high
NaCl concentrations~ mi.cms~tched target sequences were either not annealed
or were removed in the cold washes. Under the same conditions, the
matched target sequences were annealed and ligated to the probe. The final
hot wash removed the non-biotinylated probe oli~onucleotide. This
CA 02218188 1997-10-14
WO 96t32504 PCTIUS961~)5136
63
oligonucleotide contained the labeled target if the target had been ligated to
~e probe.
Fx~mple 8 Con~pens~tin~ for Variation~ in Base Con~l?osition.
The Dependence on TM on base composition, and on base
5 sequence may be overcome with ~e use of salts like te~amethyl ammonium
halides or betaines. ~It~rn~tively, base analogs like 2,6-diamino purine and
5-bromo U can be used instead of A and T, respectively, to increase the
stability of A-T base pairs, and derivatives like 7-deazaC~ can be us;,d to
decrease the stability of G-C base pairs. The initial Experiments shown in
10 Table 2 indicate that the use of enzymes will elimin~te many of the
complications due to base sequences. This gives the approach a very
significant advantage over non-enzymatic methods which require different
conditions for each nucleic acid and are highly matched to GC content.
Another approach to compensate for differences in stability is
15 to vary the base next to the stacking site. Experiments were performed to
test the relative effects of all four bases in this position on overall
hybridization discrimin~tion and also on relative ligation discrimination
other base analogs such as dU (deoxyuridine) and 7-deazaG may also be
useful to suppress effects of secondary structure.
20 Example 9 nNA T ig~tion to Oligonuçleotide Arrays.
E. coli and T4 DNA ligases can be used to covalently attach
hybridized target nucleic acid to the correct immobilized oligonucleotide
probe. This is a highly accurate and efficient process. Because ligase
absolutel~ requires a correctly base paired 3' terminus, ligase will read only
25 the 3'-terrninal sequence of the target nucleic acid. After ligation, the
resulting duplex will be 23 base pairs long and it will be possible to remove
CA 02218188 1997-10-14
WO 96/32504 PCT/US96/05136
64
unhybridized, unligated target nucleic acid using fairly stringent washing
conditions. A~r~ol)l;ately chosen positive and negative controls
n~1rate the specificity of ~is method, such as arrays which are lacking
a 5'-tçrrnin~l phosphate adjacent to the 3' overhang since these probes will
5 not ligate to the target nucleic acid.
There are a number of advantages to a ligation step. Physical
specificity~ is supplanted by enzymatic specificity. Focusing on the 3' end
ofthe target nucleic also minimi7e problems arising from stable secondary
structures in the target DNA. DNA ligases are also used to covalently attach
10 hybridized target DNA to the correct immobilized oligonucleotide probe.
Several tests of the feasibility of the ligation method shown in Figure 12.
Biotinylated probes were attached at 5' ends (Figure 12A) or 3' ends (Figure
12B) to ~,L~c~lavidin-coated magnetic microbeads, and annealed with a
shorter, complementary, constant sequence to produce duplexes with 5 or
15 6 base single-stranded overhangs. 32P-end labeled targets were allowed to
hybridize to the probes. Free targets were removed by capturing the beads
with a magnetic separator. DNA ligase was added and ligation was allo~ ed
to proceed at various salt concentrations. The samples were washed at room
temperature, again manipulating the immobilized compounds with a
20 magnetic separator to remove non-ligated material. Finally, samples w ere
incubated at a temperature above the Tm of the duplexes, and eluted single
strand was retained after the remainder of the samples were removed b~
mag~netic separation. The eluate at this point consisted of the li~~ated
material. The fraction of ligation was estimated as the amount of 3'P
25 recovered in the high temperature wash versus the amount recovered in both
the hi ~h and low temperature washes. Results indicated that salt conditions
-
CA 02218188 1997-10-14
WO 96/;~2504 PCTIUS96105136
caIl be found where the ligation proceeds efficiently with perfectly matched
5 or 6 base ov~rh~n~, but not wi~ G-T mi~m~tçl~es. The results of a more
extensive set of similar experiments are shown in Tables 4-6.
Table 4 looks at the effect of the position of the mi~m~t~h and
5 Table S examines the ef~ect of base composition on the relative
discrimination of perfect matches verses weakly destabilizing mi.~m~tçlles.
These data demonstrate that effective discrimin~tion between perfect
matches and single mi~m~t~l~es occurs with all five base overhangs tested
and that there is little if any effect of base composition on the amount of
10 ligation seen or the effectiveness of match/mi~m~tch discrimin~tion. Thus,
the serious problems of ~ lin~ with base composition effects on stability
seen in ordinary SBH do not appear to be a problem for positional SBH.
Furthermore, as the worst micm~teh position was the one distal ~om the
phosphodiester bond formed in the ligation reaction, any mi~m~tçhes that
15 survived in this position would be elimin~ted by a polymerase extension
reaction. A polymerase such as Sequenase version 2, that has no 3'-
endonuclease activity or terminal transferase activi~y would be usefill in this
regard. Gel electrophoresis analysis confirrned that the putative ligation
products seen in these tests were indeed the actual products synthesized.
Table 4
Ligation Efficiency of Matched and Mismatched Duplexes
in 0.2 M NaCI at 37~C
(SEQ ID NO 1) 3'-TCC AGA ACC TTG GCT-S'
Lip:~tion Efficicncy
- CTA CTA GGC TGC GTA GTC-S'(SEQ ID NO 2)
S'-B- GAT GAT CCG ACG CAT CAG AGC TC 0.170 (SEQ ID NO 3)
S'-B- GAT GAT CCG ACG CAT CAG AGC TT 0.006 (SEQ ID NO 4)
S'-B- GAT GAT CCG ACG CAT CAG AGC TA 0.006 (SEQ ID NO 7)
30 S'-B- GAT GAT CCG ACG CAT CAG AGC CC 0.002 (SEQ ID NO 8)
S'-B- GAT GAT CCG ACG CAT CAG AGT TC 0.004 (SEQ ID NO 9)
CA 02218188 1997-10-14
WO 96/32~i04 PCT/US96/05136
66
S'-B-GAT GAT CCG ACG CAT CAG AAC TC 0.001 (SEQ ID NO 10)
Table S
Ligation Efficiency of Matched and Mismatched Duplexes in
0.2 M NaCI at 37~C and its Dependance on AT Content of the
Overhang
Overh~ng Sequences ~T Content Li~tionFfficiency
Match GGCCC 0/5 0.30
Mi.cm~tch GGCCT 0.03
Match AGCCC 1/5 0.36
15 Mi.~m~tch AGCTC 0-.02
Match AGCTC 2/5 0.17
Mi~m~tch AGCTT 0.01
Match AGATC 3/5 0.24
Mi~m~tch AGATT 0.01
Match ATATC 4/5 0.17
Mi~m~tch ATATT 0.01
Match ATATT 5/5 0.31
Micm~tch ATATC Q.02
CA 02218188 1997-10-14
wo 96/32504 PC rnJSs6~05136
67
Table 6
a Increasing Di~.in~tion by Sequencing Extension at 37~C
T.~p~tion F.fficjency niP~fion l~xtension (c~m)
S (percent) (+) (-)
(SEQID NO 1) 3'-TCG AGA ACC TTG GCT-5'*
CTA CTA GGC TGC GTA GTC-5'(SEQID NO 2)
5'-B- GAT GAT CCG ACG CAT CAG AGA TC 0.24 4,934 29,500
0 (SEQIDNO 11)
5'-B- GAT GAT CCG ACG CAT CAG AGC TT 0.01 116 ~Q
(SEQID NO 4)
Dis.;.;.. ;.. ~;cn= x24 x42 x118
(SEQID NO 1) 3'-TCG AGA ACC TTG GCT-5'*
CTA CTA GGC TGC GTA GTC-5'(SEQID NO 2)
5'-B- GAT GAT CCG ACG CAT CAG ATA TC 0.17 12,250 25,200
(SEQID NO 12)
5'-B- GAT GAT CCG ACG CAT CAG ATA TT 0.01 ~Q ~Q
(SEQID NO 13)
Di.s~ n = x17 x51 x65
"B"--Biotin
The discrimination for the correct sequence is not as great with
an external mi.~m~tch (which would be the most difficult case to
discrimin~te) as with an internal mismatch (Table 6). A mismatch right at
the ligation point would presumably offer the highest possible
30 discrimination. In any event, the results shown are very promising. Already
there is a level of discrimin~tion with only 5 or 6 bases of overlap that is
better than the discrimin~tion seen in conventional SBH with 8 base
overlaps.
Example 10 Capture and Sequencin~ of a Tar~et Nucleic Acid.
A mixture oftarget DNA was prepared by mixing equal molar
ratio of eight different oligos. For each sequencing reaction, one specific
- partially duple~; probe and eight different targets were used. The sequence
of the probe and the targets are shown in Tables 7 and 8.
CA 02218188 1997-10-14
WO 96/32504 PCT/US96/05136
68
Table 7
Duplex Probes Used
(DF25) 5'-F-GATGATCCGACGCATCAGCTGTG (SEQID NO 14)
53'-CTACTAGGCTGCGTAGTC (SEQID NO 2)
(DF37) 5'-F-GATGATCCGACGCATCACTCAAC(SEQID NO 15)
3'-CTACTAGGCTGCGTAGTG (SEQID NO 2)
0(DF22) 5'-F-GATGATCCGACGCATCAGAATGT(SEQIDNO 16)
3'-CTACTAGGCTGCGTAGTC (SEQID NO 2)
(DF28) 5'-F-GATGATCCGACGCATCAGCCTAG(SEQID NO 17)
53'-CTACTAGGCTGCGTAGTC (SEQID NO 2)
(DF36) S'-F-GATGATCCGACGCATCAGTCGAC(SEQID NO 18)
3'-CTACTAGGCTGCGTAGTC (SEQID NO 2)
(DFlla)5'-F-GATGATCCGACGCATCACAGCTC(SEQID NO 19)
203'-CTACTAGGCTGCGTAGTG (SEQID NO 2)
(DF8a) 5'-F-GATGATCCGACGCATCAAGGCCC(SEQID NO 20)
3'-CTACTAGGCTGCGTAGTT (SEQID NO 2)
25Table 8
Mixture of Targets
(NB4) 3'-TTACACCGGATCGAGCCGGGTCGATCTAG (DF22)
(SEQID NO 1)
(NB4-5) 3'-GGATCGACCGGGTCGATCTAG (DF28) (SEQID NO '')
(DF5) 3'-AGCTGCCGGATCGAGCCGGGTCGATCTAG (DF36)
(SEQID NO ~3)
(TSI0) 3'-TCGAGAACCTTGGCT (DFlla) (SEQID NO 24)
35 (NB3.10) 3'CCGGGTCGATCTAG (DF8a) (SEQID NO ~5)
Micm~trh
(NB3.4) 3'-CCGGATCAAGCCGGGTCGATCTAG(DF8a) (SEQID NO ~6)
(NB3.7) 3'-TCAAGCCGGGTCGATCTAG (DFlla) (SEQID NO ~7)
40 (NB3-9) 3'-AGCCGGGTCGATCTAG (DF36)(SEQID NO '8)
Two pmol of each of the two duplex-probe-forming
oligonucleotides and 1.5 pmol of each of the eight different targets were
45 mixed in a 10 ~11 volume containing 2 ,ul of Sequenase buffer stock (200 mM
CA 02218188 1997-10-14
WO 96/32504 PCTIUS96105136
69
Tris-HCl, pH 7.5, 100 mM MgC12, and 250 mM NaCl) from the Sequenase
kit. The ~nn~lin~ mixture was heated to 65 ~C and allowed to cool slowly
to room tt~ ~d~ . While the reaction mixture was kept on ice, 1 ~11 0.1
M dithiothreitol solution, 1 ~Ll Mn buf~er (0.15 M sodium isocitrate and 0.1
5 M MnCl2), and 2 ~1 of diluted Sequenase (1.5 units) were mixed, and the 2
~11 of reaction mixture was added to each of the four teImination mixes at
room temperature (each consisting of 3 ~1 of the a~l~liate termination
mix: 16 ~lM dATP, 16 IlM dCTP, 16 ,uM dGTP, 16 ~LM dTTP and 3.2 ~M
of one of the four ddNTPs, in 50 mM NaCl). The reaction mixtures were
10 fur~er incubated at room temperature for 5 minl~tes, and termin~t~rl with the addition of 4 ~11 of Ph~rm~cia stop mix (deionized formamide cont~ining
dextran blue 6 mg/ml). Samples were denatured at 90-95 ~C for 3 minl~tes
and stored on ice prior to loading. Sequencing samples were analyzed on
an ALF DNA sequencer (Pharmacia Biotech; Piscataway, NJ) using a 10%
15 polyacrylamide gel containing 7 M urea and 0.6 x TBE. Sequencing results
from the gel reader are shown in Figure 13 and summarized in Table 9.
Matched targets hybridized correctly and are sequenced, whereas
mism~tched targets do not hybridize and are not sequenced.
W096/32504 CA 02218188 1997-10-14 PCT/US96/05136
Table 9
Sllmm~ry of Hybridization Data
Reaction ~Iybridization Sequence Comment
S 1 Probe: DF25 Target: mixture No mi~ tch
2 Probe: DF37 Target: mixture No micm~tch
3 Probe: DF22 Target: mixture Yes match
4 Probe: DF28 Target: mixture Yes match
Probe: DF36 Target: mixture Yes match
6 Probe: DFl la Target: mixture Yes match
7 Probe: DF8a Target: mixture Yes match
8 Probe: DF8a Target: NB3.4 No mismatch
9 Probe: DF8a Target: TS12 No mi~m~tch
Probe: DF37 Target: DF5 No mismatch
Example 11 Elon~tion of Nucleic Acids Bound to Solid Su~orts.
Elongation was carried out either by using Sequenase version
2.0 kit or an AutoRead sequencing kit (Ph~ cia Biotech; Piscataway, NJ)
employing T7 DNA polymerase. Elongation of the immobilized single-
20 stranded DNA target was performed with reagents from the sequencing kitsfor Sequenase Version 2.0 or T7 DNA polymerase. A duplex DNA probe
containing a 5-base 3' overhang was used as a primer. The duplex has a 5'-
fluorescein labeled 23-mer, containing an 1 8-base 5' constant region and a
5-base 3' variable region (which has the same sequence as the 5'-end of the
25 corresponding nonbiotinylated primer for PCR amplification of target DNA,
and an 1 8-mer complementary to the constant region of the 23-mer. The
duplex was formed by annealing 20 pmol of each of the two
oligonucleotides in a 10 ,ul volume containing 2 ~11 of Sequenase buffer
stock (200 mM Tris-HCI, pH 7.5, 100 mM MgCI2, and 250 mM NaCI) from
30 the Sequenase kit or in a 13 ,ul volume containing 2 1ll of the annealing
buffer ( 1 M Tris-HCI, pH 7.6, 100 mM MgCI2) from the AutoRead
CA 02218188 1997-10-14
WO g6/32504 PCT/USS~610SI36
71
sequencing kit. The ~nne~ling mixture was heated to 65 ~C and allowed to
cool slowly to 37~C over a 20-30 minllte time period. The duplex primer
was ~nnto~led with the immobilized single-stranded DNA target by adding
the ~nne~lin~ mixture to the DNA-cont~ining magnetic beads and the
5 resulting mixture was further incubated at 37~C for 5 minlltes, room
temperature for 10 minlltes, and finally 0~C for at least 5 minlltes. For
Sequenase reactions, 1 ,ul 0.1 M dithiothreitol solution, 1 ~11 Mn buffer (~.15
M sodium isocitrate and 0.1 M MnCl2) for the relative short target, and 2 ~11
of diluted Sequenase (1.5 units) were added, and the reaction mixture was
10 divided into four ice cold termination mixes (each consists of 3 ~11 of the
al.~r~liate termin~tion mix: 80 ~lM dATP, 80 ~lM dCTP, 80 ,uM dGTP, 80
~lM dTTP and 8 ~lM of one of the four ddNTPs, in 50 mM NaCl). For T7
DNA polymerase reactions, 1 ~1 of extension buffer (40 mM McCk, pH 7.5,
304 mM citric acid and 324 mM DTT) and 1 ,ul of T7 DNA polymerase (8
15 units) were mixed, and the reaction volume was split into four ice cold
termination mixes (each consisting of 1 ,ul DMSO and 3 ~11 of the
appropriate termination mix: 1 mM dATP, 1 mM dCTP, 1 mM dGTP, 1
mM dTTP and 5 IlM of one of the four ddNTPs, in 50 mM NaCI and 40 mM
Tris-HCl, pH 7.4). The reaction mixtures for both enzymes were further
20 incubated at 0~C for 5 minutes, room temperature for 5 minutes and 37~C
for 5 minutes. After the completion of extension, the supernatant was
removed. and the magnetic beads were re-suspended in 10 ,ul of Pharmacia
stop mi.x. Samples were denatured at 90-95~C for S minutes (under this
harsh condition, both DNA template and the dideoxy fragments are released
25 from the beads) and stored on ice prior to loading. A control experiment
was performed in parallel using a 1 8-mer complementarv to the 3 ' end of
CA 02218188 1997-10-14
WO 96/32504 PCT/US96/OS136
72
target DNA as the sequencing primer instead of the duplex probe and the
annealing of 18-mer to its target was carried out in a similar way as the
annealing of the duplex probe.
Example 12 Chain Flon~tion of Tar~et Sequences.
Sequencing of immobilized target DNA can be performed
with Sequenase Version 2Ø A total of S elongation reactions, one with
each of 4 dideoxy nucleotides and one with all four simultaneously, are
performed. A sequencing solution, cont~ining (40 mM Tris-HCI, pH 7.5,
20 mM MgCI2, and 50 mM NaCl, 10 mM dithiothreitol solution, 15 mM
sodium isocitrate and 10 mM MnCk, and 100 u/ml of Sequenase (1.5 units)
is added to the hybridized target DNA. dATP, dCTP, dGTP and dTTP are
added to 20 ~LM to initiate the elongation reaction. In the separate reactions,
one of four ddNTP is added to reach a concentration of 8 ~lM. In the
combined reaction all four ddNTP are added to the reaction to 8 ~M each.
The reaction mixtures were incubated at 0~C for S minutes room
temperature for 5 minutes and 37~C for 5 minlltes. After the completion of
extension, the supernatant was removed and the elongated DNA washed
with 2 mM EDTA to terminate elongation reactions. Reaction products are
analyzed by mass spectrometry.
CA 02218188 1997-10-14
Wo 96/32S04 PCTIUS9~105136
73
Fx~mI~le 13 Capillary Electrophoretic An~ysi~ of Tar~et Nucleic Acid.
Molecular weights oftarget sequences may also be determined
by capillary electrophoresis. A single laser capillary eleckophoresis
instrument can be used to monitor the performance of sample preparations
5 in high pclrolmance capillary electrophoresis sequencing. This instrument
is designed so that it is easily converted to multiple channel (wavelengths)
detection.
An individual element of the sample array may be engineered
directly to serve as the sample input to a capillary. Typical capillaries are
10 250 rnicrons o.d. and 75 microns i.d. The sample is heated or denatured to
release the DNA ladder into a liquid droplet. the silicon array surfaces is
ideal for this purpose. The capillary can be brought into contact with the
droplet to load the sample.
To facilitate loading of large numb~rs of samples
15 simultaneously or seqllenti~lly, there are two basic methods. With 250
micron o.d. capillaries it is feasible to match the dimensions of the target
array and the capillary array. Then the two could be brought into contact
manually or even by a robot arm using a jig to assure accurate alignment.
An electrode may be engineered directly into each sector of the silicon
20 surface so that sample loading would only require contact between the
surface and the capillary array.
The second method is based on an inexpensive collection
system to capture ~actions eluted from high performance capillary
electrophoresis. Dilution is avoided by using designs which allow sample
25 collection without a perpendicular sheath flow. The same apparatus
designed as a sample collector can also serve inversely as a sample loader.
CA 02218188 1997-10-14
WO 96/32504 PCT/US96/05136
74
In this case, each row of the sample array, equipped with electrodes, is used
directly to load samples automatically on a row of capillaries. Using either
method, sequence information is ~letermined and the target sequence
constructed.
5 Example 14 Mass Spectrolnetry of NucleicAcids.
Nucleic acids to be analyzed by mass spectrometry were
redissolved in ultrapure water (MilliQ, Millipore) using amounts to obtain
a concentration of 10 pmoles/~ll as stock solution. An aliquot ( 1 ,ul) of this
concentration or a dilution in ultrapure water was mixed with 1 ~1 of the
10 makix solution on a flat metal surface serving as the probe tip and dried
with a fan using cold air. In some experiments, cation-ion exchange beads
in the acid form were added to the mixture of matrix and sample solution to
stabilize ions formed during analysis.
MALDI-TOF spectra were obtained on different commercial
15 instruments such as Vision 2000 (Finnigan-MAT), VG TofSpec (Fisons
Instruments), LaserTec Research (Vestec). The conditions were linear
negative ion mode with an acceleration voltage of 25 kV. Mass calibration
was done externally and generally achieved by using defined peptides of
appropriate mass range such as insulin, gramicidin S, trypsinogen, bovine
20 serum albumen and cytochrome C. All spectra were generated by
employing a nitrogen laser with 5 nanosecond pulses at a wavelength of 33 7
nrn. Laser energy varied between 1 o6 and 107 W/cm2. To improve signal-
to-noise ratio generally, the intensities of 10 to 30 laser shots were
accumulated. The output of a typical mass spectrometry showing
25 discrimination between nucleic acids which differ by one base is shown in
Figure 14.
CA 02218188 1997-10-14
WO 96/32504 PC~/US96/OS1:}6
CA 02218188 1997-10-14
WO 9G/32504 PCT/US96/OS136
76
Example 15 Sequel-ce Determin~tion from M~ Speckornet~
Elongation of a target nucleic acid, in the presence of dideoxy
chain termin~ting nucleotides, generated four families of chain-te~nin~ted
fr~ nt~. The mass difference per nucleotide addition is 289.19 for dpC,
5 313.21 for dpA, 329.21 for dpG and 304.20 for dpT, respectively.
Comparison of the mass differences measured between fragments with the
known masses of each nucleotide the nucleic acid sequence can be
determined. Nucleic acid may also be sequenced by performing polymerase
chain elongation in four separate reactions each with one dideoxy chain
10 tPrmin~tingnucleotide. To examine mass differences, 13 oligonucleotides
from 7 to 50 bases in length were analyzed by MALDI-TOF mass
spectrometry. The correlation of calculated molecular weights of the ddT
fragments of a Sanger sequencing reaction and their experimentally verified
weights are shown in Table 10. When the mass spectrometry data from all
15 four chain termination reactions are combined, the molecular weight
difference between two adjacent peaks can be use to determine the
sequence.
CA 02218188 1997-10-14
WO 96t32504 PCT~US96105136
Table 10
Sl~mm~ry of Molecular Weights Expected v. Measured
Fr~nent ~-mer) Calculated Mass Fx~?erirnental Mass Difference
7-mer 2104.45 2119.9 +15.4
10-mer 3011.04 3026.1 +15.1
11-mer 3315.24 3330.1 +14.9
19-mer 5771.82 5788.0 +16.2
20-mer 6076.02 6093.8 +17.8
24-mer 7311.82 7374.9 +63.1
26-mer 7945.22 7960.9 +15.7
33-mer 10112.63 10125.3 +12.7
37-mer 11348.43 11361.4 +13.0
38-mer 11652.62 11670.2 +17.6
42-mer 12872.42 12888.3 +15.9
46-mer 14108.22 14125.0 +16.8
50-mer 15344.02 15362.6 +18.6
Example 16 Reduced Pass Sequencin~-
To maximize the use of PSBH arrays to produce Sanger
ladders, the sequence of a target should be covered as completely as possible
with the lowest amount of initial sequencing redundancy. This will
maximize the performance of individual elements of the arrays and
m~Ximi7e the amount of useful sequence data obtained each time an array
is used. With an unknown DNA, a full array of 1024 elements (Mwo I or
BsiYI cleavage) or 256 elements (TspR I cleavage) is used. A 50 kb target
DNA is cut into about 64 fragments by Mwo I or BsiYI or 30 fragments by
~spR I, respectively. Each fragment has two ends both of which can be
captured independently. The coverage of each array after capture and
ignoring degeneracies is 128/1024 sites in the first case and 60/256 sites in
- the second case. Direct use of such an array to blindly deliver samples
CA 02218188 1997-10-14
WO 96/32504 PCT/US96/05136
78
element by element for mass spectrometry sequencing would be inefficient
since most array elements will have no sarnples.
In one method, phosph~t~ecl double-stranded targets are used
at high concentrations to saturate each array element that detects a sample.
5 The target is ligated to make the caplule irreversible. Next a different
sample mixture is exposed to the array and subsequently ligated in place.
This process is repeated four or five times until most of the elements of the
array contain a unique sample. Any tandem target-target complexes will be
removed by a subsequent ligating step because all of the targets are
1 0 phosphatased.
Alternatively, the array may be monitored by confocal
microscopy after the elongation reactions. This reveals which elements
contain elongated nucleic acids and this information is communicated to an
automated robotic system that is l-ltim~tely used to load the samples onto a
15 mass speckometry analyzer.
Example 17 Synthesis of Mass Modified Nucleic Acid Primers.
Mass modification at the 5' sugar: Oligonucleotides were
synthesized by standard automated DNA synthesis using ~-
cyanoethylphosphoamidites and a 5'-amino group introduced at the end of
20 solid phase DNA synthesis. The total amount of an oligonucleotide
synthesis, starting with 0.25 micromoles CPG-bound nucleoside, is
deprotected with concentrated aqueous ammonia, purified via OligoPAKTM
Cartridges (Millipore; Bedford, MA) and Iyophilized. This material with a
5'-terminal amino group is dissolved in 100 ~11 absolute N, N-
25 dimethylformamide (DMF) and condensed with 10 ,umole N-Fmoc-glycine
pentafluorophenyl ester for 60 minutes at 25 ~C. After ethanol precipitation
CA 02218188 1997-10-14
WO 96132504 PCTJUS96,'0S136
79
and centrifugation, the Fmoc group is cleaved off by a 10 minute treatment
with 100 ~Ll of a solution of 20% piperidine in N,N-dimethylforrnamide.
, Excess piperidine, DMF and the cleavage product from the Fmoc group are
removed by ethanol precipitation and the precipitate lyophilized from 10
5 mM TEAA buffer pH 7.2. This material is now either used as primer for ~e
Sanger DNA sequencing reactions or one or more glycine residues (or other
suitable protected amino acid active esters) are added to create a series of
mass-modified primer oligonucleotides suitable for Sanger DNA or RNA
sequencing.
10Mass modification at the heterocyclic base with glycine:
Starting material was 5-(3-aminopropynyl-1)-3'5'-di-p-tolyldeoxyuridine
prepared and 3' 5'-de-O-acylated (Haralambidis et al., Nuc. Acids Res.
15 :4857-76, 1987). 0.281 g (1.0 mmol) 5-(3-aminopropynyl-1)-2'-
deoxyuridine were reacted with 0.927 g (2.0 mmol) N-Fmoc-glycine
15 pentafluorophenylester in 5 ml absolute N,N-dimethylformamide in the
presence of 0.129g (1 mmol; 174 1ll) N,N-diisopropylethylamine for 60
minll1es at room temperature. Solvents were removed by rotary evaporation
and the product was purified by silica gel chromatography (Kieselgel 60,
Merck; column: 2.5 x 50 cm, elution with chloroform/methanol mixtures).
20 Yield was 0.44 g (0.78 mmol; 78%). To add another glycine residue, the
Fmoc group is removed with a 20 minutes treatment with 20% solution of
piperidine in DMF, evaporated in vacuo and the remaining solid material
extracted three times with 20 ml ethylacetate. After having removed the
remaining ethyl~cet~te, N-Fmoc-glycine pentafluorophenylester is coupled
25 as described above.5-(3(N-Fmoc-glycyl)-amidopropynyl-1)-2'-deoxyuridine
is transformed into the 5'-O-dimethoxytritylated nucleoside-3'-O-~-
CA 02218188 1997-10-14
WO 96/32504 PCT/US~6/OS136
cyanoethyl-N,N-diisopropylphosphoamidite and incorporated into
automated oligonucleotide syn~esis. This glycine modified thymidine
analogue building block for chemical DNA synthesis can be used to
substitute one or more of the thymidine/uridine nucleotides in the nucleic
S acid primer sequence. The Fmoc group is removed at the end of the solid
phase synthesis with a 20 minute treatment with a 20% solution of
piperidine in DMF at room temperature. DMF is removed by a washing
step with acetonitrile and the oligonucleotide deprotected and purified.
Mass modification at the heterocyclic base with ~ ol~nine
10 0.281 g (1.0 -mmol) 5-(3-Aminopropynyl-1)-2'-deoxyuridine was reacted
with N-Fmoc-~-alanine pentafluorophenylester (0.955 g; 2.0 rnmol) in 5 ml
N,N-dimethylformamide (DMF) in the presence of 0.129 g (174 ~11; 1.0
mmol) N,N-disopropylethylamine for 60 minutes at room temperature.
Solvents were removed and the product purified by silica gel
15 chromatography. Yield was 0.425 g (0.74 mmol; 74%). Another ~-alanine
moiety can be added in exactly the same way after removal of the Fmoc
group. The ~el~a~ion of the S'-O-dimethoxytritylated nucleoside-3'-O-~-
cyanoethyl-N,N-diisopropylphosphoamidite from 5-(3-(N-Fmoc-~-alanyl)-
amidopropynyl-1)-2'-deoxyuridine and incorporation into automated
20 oligonucleotide synthesis is performed under standard conditions. This
building block can substitute for any of the thymidine/uridine residues in the
nucleic acid primer sequence.
Mass modification at the heterocyclic base with ethylene
monomethyl ether: 5-(3-aminopropynyl-1)-2'-deoxyuridine was used as a
25 nucleosidic component in this example. 7.61 g (100.0 mmol) freshly
distilled ethylene glycol monomethyl ether dissolved in 50 ml absolute
CA 02218188 1997-10-14
wo 96132~04 PcTruss6Josl36
81
pyridine was reacted with 10.01 g (100.0 mmol) recryst~lli7e~1 succinic
anhydride in the presence of 1.22 g (10.0 mmol) 4-N,N--
dimethylaminopyridine overnight at room temperature. The reaction was
terrnin~1 by the addition of water (5.0 ml), the reaction mixture evaporated
5 in vacuo, co-evaporated twice with dry toluene (20 ml each) and the residue
redissolved in 100 ml dichloromethane. The solution was twice extracted
sllccessively with 10% aqueous citric acid (2 x 20 ml) and once with water
(20 ml) and the organic phase dried over anhydrous sodium sulfate. The
organic phase was evaporated in vacuo. Residue was redissolved in 50 ml
10 dichloromethane and preci~i~led into 500 ml pentane and the precipitate
dried in vacuo. Yield was 13.12 g (74.0 mmol; 74%). 8.86 g (50.0 mmol)
of succinylated ethylene glycol monomethyl ether was dissolved in 100 ml
dioxane cont~ining 5% dry pyridine (5 ml) and 6.96 g (50.0 mmol) 4-
nitrophenol and 10.32 g (50.0 mmol) dicyclohexylcarbodiimide was added
15 and the reaction run at room temperature for 4 hours. Dicyclohexylurea was
removed by filtration, the filtrate evaporated in vacuo and the residue
redissolved in 50 ml anhydrous DMF. 12.5 ml (about 12.5 mmol 4-
nitrophenylester) ofthis solution was used to dissolve 2.81 g (10.0 mmol)
5-(3-aminopropynyl-1)-2'-deoxyuridine. The reaction was performed in the
20 presence of 1.01 g (10.0 mmol; 1.4 ml) triethylamine overnight at room
tempcl~Lure. The reaction mixture was evaporated in vacuo, co-evaporated
with toluene, redissolved in dichloromethane and chromatographed on
silicagel (Si60, Merck; column 4 x 50 cm) with dichloromethane/methanol
mixtures. Fractions containing the desired compound were collected,
25 evaporated, redissolved in 25 ml dichloromethane and precipitated into 250
ml pentane. The dried precipitate of 5-(3-N-(O-succinyl ethylene glycol
CA 02218188 1997-10-14
WO 96/32504 PCT/US96/05136
monomethyl ether)-amidopropynyl-1)-2'-deoxyuridine (yield 65%) is 5'-O-
dimethoxytritylated and transformed into the nucleoside-3'-O-~-cyanoethyl-
N, N-diisopropylphosphoamidite and incorporated as a building block in the
automated oligonucleotide synthesis according to standard procedures. The
5 mass-modified nucleotide can substitute for one or more of the
thymidine/uridine residues in the nucleic acid primer sequence.
Deprotection and purification of the primer oligonucleotide also follows
standard procedures.
Mass modification at the heterocyclic base with diethylene
10 glycol monomethyl ether: Nucleosidic starting material was as in previous
examples, 5-(3-aminopropynyl-1)-2'-deoxyuridine. 12.02 g (100.0 mmol)
freshly distilled diethylene glycol monomethyl ether dissolved in 50 ml
absolute pyridine was reacted with 10.01 g (100.0 mmol) recrystallized
succinic anhydride in the presence of I .22 g ( 10.0 mmol) 4-N, N-
15 dimethylaminopyridine (DMAP) overnight at room temperature. Yield was18.35 g (82.3 mmol; 82.3%). 11.06 g (50.0 mmol) of succinylated
diethylene glycol monomethyl ether was transformed into the 4-
nitrophenylester and, subsequently, 12.5 mmol was reacted with 2.81 g ( 10.0
mmol) of 5-(3-aminopropynyl -1)-2'-deoxyuridine. Yield after silica gel
20 column chromatography and precipitation into pentane was 3.34 g (6.9
mmol; 69%). After dimethoxytritylation and transformation into the
nucleoside-~-cyanoethylphosphoamidite, the mass-modified building block
is incorporated into automated chemical DNA synthesis. Within the
sequence of the nucleic acid primer, one or more of the thymidine/uridine
25 residues can be substituted by this mass-modified nucleotide.
CA 02218188 1997-10-14
wo 96/32504 PcTruss6l0sl36
83
Mass Modification at the heterocyclic base with glycine:
Starting material was N6-benzoyl-8-bromo-5'-0-(4,4'-dimethoxytrityl)-2'-
deox~yadenosine (Singh et al., Nuc. Acids Res.18:3339-45,1990). 632.5 mg
(1.0 mmol) ofthis 8-bromo-deoxyadenosine derivative was suspended in 5
5 ml absolute ethanol and reacted with 251.2 mg (2.0 mmol) glycine methyl
ester (hydrochloride) in ~e presence of 241.4 mg (2.1 mmol; 366 ,ul) N,N-
diisopropylethylamine and refluxed until the starting nucleosidic material
had disappeared (4-6 hours) as checked by thin layer chromatogranhy
(TLC). The solvent was evaporated and the residue purified by silica gel
10 chromatography (column 2.5 x 50 cm) using solvent mixtures of
chloroform/methanol cont~inin~ 0.1% pyridine. Product fractions were
combined, the solvent evaporated, the fractions dissolved in 5 ml
dichloromethane and precipitated into 100 ml pentane. Yield was 487 mg
(0.76 mmol; 76%). Transformation into the corresponding nucleoside-~-
15 cyanoethylphospho amidite and integration into automated chemical DNAsynthesis is performed under standard conditions. During final deprotection
with aqueous concentrated ammonia, the methyl group is removed from the
glycine moiety. The mass-modified building block can substitute one or
more deoxyadenosine/adenosine residues in the nucleic acid primer
20 sequence.
Mass modification at the heterocyclic base with
glycylglycine: 632.5 mg (1.0 mmol) N6-Benzoyl-8-bromo-5'-O-
(4,4'dimeethoxytrityl)2'-deoxyadenosine was suspended in 5 ml absolute
ethanol and reacted with 324.3 mg (2.0 mmol) glycyl-glycine methyl ester
25 in the presence of 241.4 mg (2.1 mmol; 366,~1) N, N-diisopropylethylamine.
The mixture was refluxed and completeness of the reaction checked by
CA 02218188 1997-10-14
WO 96132504 PCI~/US96/OS136
84
TLC. Yield after silica gel column chromatography and precipitation into
pentane was 464 mg (0.65 mmol; 65%). Transformation into the
nucleoside-~-cyanoethylphosphoamidite and into synthetic oligonucleotides
is done according to standard procedures.
S Mass Modi~lcation at the heterocyclic base with glycol
monomethyl ether: Starting material was 5'-0-(4,4-dimethoxytrityl)-2'-
amino-2'-deoxythymidine synthesized (Verheyden et al., J. Org. Chem.
36:250-54, 1971; Sasaki et al, J. Org. Chem. 41:3138-43, 1976; Imazawa et
al., J. Org. Chem. 44:2039-41, 1979; Hobbs et al., J. Org. Chem. 42:714-19,
1976; Ikehara et al., Chem. Pharm. Bull. Japan 26:240-44, 1978). 5'-0-(4,4-
Dimethoxytrityl)-2'-amino-2'-deoxythymidine (559.62 mg; 1.0 mmol) was
reacted with 2.0 mmol of the 4-nitrophenyl ester of succinylated ethylene
glycol monomethyl ether in 10 ml dry DMF in the presence of 1.0 mmol
(140 ,ul) triethylamine for 18 hours at room tempeldlule. The reaction
mixture was evaporated in vacuo, co-evaporated with toluene, redissolved
in dichloromethane and purified by silica gel chromatography (Si60, Merck;
column: 2.5 x 50 cm; eluent: chloroform/methanol mixtures containing 0.1 %
triethylamine). The product containing fractions were combined, evaporated
and precipitated into pentane. Yield was 524 mg (0.73 mmol; 73%).
Transformation into the nucleoside-~-cyanoethyl-N,N--
diisopropylphosphoamidite and incorporation into the automated chemical
DNA synthesis protocol is performed by standard procedures. The mass-
modified deoxythymidine derivative can substitute for one or more of the
thymidine residues in the nucleic acid primer.
In an analogous way, by employing the 4-nitrophenyl ester of
succinylated diethylene glycol monomethyl ether and triethylene glycol
CA 02218188 1997-10-14
WO 96/32504 PCI-IUS9610SI36
monomethyl ether, the corresponding mass-modified oligonucleotides are
l)r~aled. In the case of only one incorporated mass-modified nucleoside
within the sequence, the mass difference between the ethylene, diethylene
and triethylene glycol derivatives is 44.05, 88.1 and 132.15 daltons,
5 respectively.
Mass modification at the heterocyclic base by allylation:
Phosphorothioate-c(mtAining oligonucleotides were prepared (Gait et al.,
Nuc. Acids Res. 19:1183, 1991). One, several or all internucleotide linkages
can be modified in this way. The (-)M13 nucleic acid primer sequence (17-
10 mer) 5'-dGTAAAACGACGGCCAGT (SEQ ID NO 29) is synthesized in
0.25 ,umole scale on a DNA synthesizer and one phosphorothioate group
introduced after the final synthesis cycle (G to T coupling). Sulfurization,
deprotection and purification followed standard protocols. Yield was 3 l .4
nmole ( 12.6% overall yield), corresponding to 31.4 nmole phosphorothioate
15 groups. Alkylation was performed by dissolving the residue in 31.4 ,ul TE
buffer (0.01 M Tris pH 8.0, 0.001 M EDTA) and by adding 16 ,ul of a
solution of 20 mM solution of 2-iodoethanol (320 nmole; 10-fold excess
with respect to phosphorothioate diesters) in N,N-dimethylformamide
(DMF). The alkylated oligonucleotide was purified by standard reversed
20 phase HPLC (~P-18 Ultraphere, BeckmAn; column: 4.5 x 250 mm; 100 mM
triethyl ammonium A~etAte, pH 7.0 and a gradient of 5 to 40% acetonitrile).
In a variation of this procedure, the nucleic acid primer
contAining one or more phosphorothioate phosphodiester bond is used in the
Sanger sequencing reactions. The primer-extension products of the four
25 sequencing reactions are purified, cleaved offthe solid support, Iyophilized
and dissolved in 4 ,ul each of TE buffer pH 8.0 and alkylated by addition of
CA 022l8l88 l997- lO- l4
WO 96t32504 PCTtUS96/05136
86
2 ,~1 of a 20 mM solution of 2-iodoethanol in DMF. It is then analyzed by
ES and/or MALDI mass spectrometry.
In an analogous way, employing instead of 2-iodoethanol, e.g.,
3iodopropanol, 4-iodobutanol mass-modified nucleic acid primer are
5 obtained with a mass difference of 14.03, 28.06 and 42.03 daltons
respectively compared to the unmodified phosphorothioate phosphodiester-
containing oligonucleotide.
Example 18 Mass Modification of Nucleotide Triphosphates.
CA 02218188 1997-10-14
WO 96/32S04 PCT/~JS96/OS136
87
Mass m~slifi~ of nucleoffde triphosphates at the 2' and
3'aminofunction: Startingmaterialwas2'-azido-2'-deoxyuridine~ d
according to literature (Verheyden et al., J. Org. Chem. 36:250, 1971),
which was 4,4- dimethoxytritylated at 5'-OH with 4,4-dimethoxytrityl
S chloride in pyridine and acetylated at 3'-OH with acetic anhydride in a one-
pot reaction using standard reaction conditions. With 191 mg (0.71 mmol)
2'-azido-2'-deoxyuridine as starting material, 396 mg (0.65 mmol; 90.~%)
5'-0-(4,4-dimethoxytrityl)-3'-O-acetyl-2'-azido-2'-deoxyuridine was
obtained after purification via silica gel chromatography. Reduction of the
10 azido group was performed (Barta et al., Tetrahedron 46:587-94, 1990).
Yield of 5'-0-(4,4-dimethoxytrityl)-3'-O-acetyl-2'-amino-2'-deoxyuridine
after silica gel chromatography was 288 mg (0.49 mmol; 76%). This
protected 2'-amino-2'-deoxyuridine derivative (588 mg, 1.0 mmol) was
reacted with 2 equivalents (927 mg; 2.0 mmol) N-Fmoc-glycine
15 pentafluorophenyl ester in 10 ml dry DMF overnight at room temperature
in the presence of 1.0 mmol (174,ul) N,N-diisopropylethylamine. Solvents
were removed by evaporation in vacuo and the residue purified by silica gel
chromatography. Yield was 711 mg (0.71 mmol; 82%). Detritylation was
achieved by a one hour treatment with 80% aqueous acetic acid at room
20 temperature. The residue was evaporated to dryness, co-evaporated twice
with toluene, suspended in 1 ml dry acetonitrile and 5'-phosphorylated with
POCI3 and directly transformed in a one-pot reaction to the 5'-triphosphate
using 3 ml of a 0.5 M solution (1.5 mmol) tetra (tri-n-butylammonium)
pyrophosphate in DMF according to literature. The Fmoc and the 3'-O-
25 acetyl groups were removed by a one-hour treatment with concentrated
aqueous ammonia at room temperature and the reaction mixture evaporated
CA 02218188 1997-10-14
WO ~6132S04 PCT/US96105136
g8
and lyophili7e~1 Purification also followed standard procedures by using
anion-exch~n~e chromatography on DEAE Sephadex with a linear gradient
of ~iethylammoniurn bicarbonate (0.1 M - 1.0 M). Triphosph~te colltS~ g
fractions, checked by thin layer chromatography on polyethyleneimine
S cellulose plates, were collected, evaporated and Iyophili7ed Yield by W-
absorbance of the uracil moiety was 68% or 0.48 mmol.
A glycyl-glycine modified 2'-amino-2'-deoxyuridine-5'-
triphosphate was obtained by removing the Fmoc group from 5'-0-(4,4-
dimethoxytrityl)-3'-O-acetyl-2'-N(N-9-fluorenylmethyloxycarbonyl-glycyl)-
2'-amino-2'-deoxyuridine by a one-hour treatment with a 20% solution of
piperidine in DMF at room t~ e.dlure, evaporation of solvents, two-fold
co-evaporation with toluene and subsequent condensation with N-Fmoc-
glycine pentafluorophenyl ester. Starting with 1.0 mmol of the 2'-N-glycyl-
2'-amino-2'-deoxyuridine derivative and following the procedure described
above, 0.72 mmol (72%) of the corresponding 2'-(N-glycyl-glycyl)-2'-
amino-2'-deoxyuridine-5'triphosphate was obtained.
Startingwith 5'-0-(4,4-dimethoxytrityl)-3'-O-acetyl-2'-amino-
2'deoxyuridine and coupling with N-Fmoc-~-alanine pentafluorophenyl
ester, the corresponding 2'-(N-~-alanyl)-2'-amino-2'-deoxyuridine-5'-
triphosphate are synthesized. These modified nucleoside triphosphates are
incorporated during the Sanger DNA sequencing process in the primer-
extension products. The mass difference between the glycine, ~-alanine and
glycyl-glycine mass-modified nucleosides is, per nucleotide incorporated,
58.06, 72.09 and 115.1 daltons, respectively.
When startingwith 5'-0-(4,4-dimethoxytrityl)-3'-amino-2',3' 1 -
dideoxythymidine, the corresponding 3'-(N-glycyl)-3'-amino-,3'-(-N-glycyl-
CA 02218188 1997-10-14
WO 96/32504 PCIJUS96S~S136
89
glycyl)-3'-amino-, and 3'~ -ala~yl)-3'-amino-2',3'-dideoxythymidine-5'-
triphosphates can be obtained. These mass-modified nucleoside
triphosphates serve as a terrnin~ting nucleotide unit in the Sanger DNA
sequencing reactions providing a mass difference per tçrrnin~ted fragment
5 of 58.06, 72.09 and 115.1 daltons respectively when used in the
multiplexing sequencing mode. The mass-differentiated fragments are
analyzed by ES and/or MALDI mass spectrometry.
Mass mo~lifi~ n of nucleotide triphosphates at C-5 of the
heterocyclic base: 0.281 g (1.0 mmol) 5-(3-Aminopropynyl-1)-2'-
10 deoxyuridine was reacted with either 0.927 g (2.0 mmol) N-Fmoc-glycine
pentafluorophenylester or 0.955g (2.0 mmol) N-Fmoc-~-alanine
pentafluorophenyl ester in 5 ml dry DMF in the presence of 0.129 g N, N-
diisopropylethylamine (174 ~11, 1.0 mmol) overnight at room temperature.
Solvents were removed by evaporation in vacuo and the condensation
15 products purified by flash chromatography on silica gel (Still et al., J. Org.,
Chem. 43: 2923-25, 1978). Yields were 476 mg (0.85 mrnol; 850%) for the
glycine and 436 mg (0.76 mmol; 76%) for the ~-alanine derivatives. For the
synthesis of the glycyl-glycine derivative, the Fmoc group of 1.0 mmol
Fmoc-glycine-deoxyuridine derivative was removed by one-hour treatment
20 with 20% piperidine in DMF at room temperature. Solvents were removed
by evaporation in vacuo, the residue was coevaporated twice with toluene
and condensed with 0.927 g (2.0 mmol) N-Fmoc-glycine pentafluorophenyl
ester and purified as described above. Yield was 445 mg (0.72 mmol; 72%).
The glycyl-, glycyl-glycyl- and ~-alanyl-2'-deoxyuridine derivatives, N-
25 protected with the Fmoc group were transformed to the 3'-O-acetyl
derivatives by tritylation with 4,4-dimethoxytrityl chloride in pyridine and
CA 02218188 1997-10-14
WO 96/32504 PCT/US96/05136
acetylation with acetic anhydride in pyridine in a one-pot reaction and
subsequently detritylated by one hour treatment with 80% aqueous acetic
acid according to standard procedures. Solvents were removed, the residues
dissolved in 100 ml chloroform and extracted twice with 50 ml 10% sodium
5 bicarbonate and once with 50 ml water, dried with sodium sulfate, the
solvent evaporated and the residues purified by flash chromatography on
silica gel. Yields were 361 mg (0.60 mmol; 71%) for the glycyl-, 351 mg
(0.57 rnrnol; 75%) for the ~-alanyl- and 323 mg (0.49 mmol; 68%) for the
glycyl-glycyl-3-0'-acetyl-2'-deoxyuridine derivatives, respectively.
10 Phosphorylation at the 5'-OH with POCI3, transformation into the 5'-
triphosphate by in situ reaction with tetra(tri-n-butylammonium)
pyrophosphate in DMF, 3'-de-0-acetylation, cleavage of the Fmoc group,
and final purification by anion-exchange chromatography on DEAE-
Sephadex was perforrned and yields according to W-absorbance of the
15 uracil moiety were 0.41 mmol 5-(3-(N-glycyl)-amidopropynyl-1)-2'-
deoxyuridine-5'-triphosphate (84%), 0.43 mmol 5-(3-(N-~-alanyl)-
amidopropynyl-1)-2'-deoxyuridine-5'-triphosphate (75%) and 0.38 mmol 5-
(3-(N-glycyl-glycyl)-amidopropynyl-1)-2'-deoxyuridine-5'-triphosphate
(78%). These mass-modified nucleoside triphosphates were incorporated
20 during the Sanger DNA sequencing primer-extension reactions.
When using 5-(3-aminopropynyl)-2',3'-dideoxyuridine as
starting material and following an analogous reaction sequence the
corresponding glycyl-, glycyl-glycyl-and ~-alanyl-2',3'-dideoxyuridine-5'-
triphosphates were obtained in yields of 69%, 63% and 71 %, respectively.
25 These mass-modifled nucleoside triphosphates serve as chain-termin~ting
nucleotides during the Sanger DNA sequencing reactions. The mass-
CA 02218188 1997-10-14
WO 96J325~4 PCTIUS96/05136
91
modified sequencing ladders are analyzed by either ES or MALDI mass
spectrometry.
, Mass modification of nucleotide triphosphates: 727 mg
(1.0 mmol) of N6-(4-tert-butylphenoxyacetyl)-8-glycyl-5'-(4,4--
5 dime~oxytrityl)-2'- deoxyadenosine or 800 mg (1.0 mmol) N6-(4-tert-
butylphenoxyacetyl)-8-glycyl-glycyl-5'-(4,4-dimethoxytrityl)-2'-
deoxyadenosine ~l~cd according to literature (Koster et al., Tetrahedron
37:362, 1981) were acetylated with acetic anhydride in pyridine at the 3'-
OH, detritylated at the 5'-position with 80% acetic acid in a one-pot reaction
10 and transformed into the 5'-triphosphates via phosphorylation with POCI3
and reaction in situ with tetra(tri-n-butylamrnonium) pyrophosphate.
Deprotection ofthe N6 tert-butylphenoxyacetyl, the 3'-O-acetyl and the O-
methyl group at the glycine residues was achieved with concentrated
aqueous ammonia for ninety minutes at room temperature. Ammonia was
15 removed by Iyophilization and the residue washed with dichloromethane,
solvent removed by evaporation in vacuo and the remaining solid material
purified by anion-exchange chromatography on DEAE-Sephadex using a
linear gradient of triethylammonium bicarbonate from 0.1 to 1.0 M. The
nucleoside triphosphate cont~ining fractions (checked by l~C on
20 polyethyleneimine cellulose plates) were combined and Iyophili7e~1 Yield
of the ~-glycyl-2'-deoxyadenosine-5'-triphosphate (determined by W-
absorbance of the adenine moiety) was 57% (0.57 mmol). The yield for the
8-glycyl-glycyl-2'-deoxyadenosine-5'-triphosphate was 51% (0.51 mmol).
These mass-modified nucleoside triphosphates were incorporated during
25 primer-extension in the Sanger DNA sequencing reactions.
CA 02218188 1997-10-14
WO 96/32504 PCTIUS96/05136
92
When using the corresponding N6-(4-tert-
butylphenoxyacetyl)-8-glycyl- or-glycyl-glycyl-5'-0-(4,4-dimethoxytrityl)-
2',3'-dideoxyadenosine derivatives as startirlg materials (for the introduction
ofthe 2',3'-fimction: Seela et al., Helvetica Chimica Acta 74: 1048-58, 1991).
S Using an analogous reaction sequence, the chain-tçnnin~tin~ mass-modified
nucleoside triphosphates 8-glycyl- and 8-glycyl-glycyl-2'.3'-
dideoxyadenosine-5'-triphosphates were obtained in 53 and 47% yields,
respectively. The mass-modified sequencing fragment ladders are analyzed
by either ES or MALDI mass spectrometry.
10 Example 19 ~ Modification of Nucleotides by Alkylation A~er Sanger
Sequencin~
2',3'-Dideoxythymidine-5'-(alpha-S)-triphosphate was
prepared according to published procedures (for the alpha-S-triphosphate
moiety: Eckstein et al., Biochemistry 15: 1685, 1976) and Accounts Chem.
15 Res. 12:204, 1978) and for the 2',3'-dideoxy moiety: Seela et al., Helvetica
Chimica Acta 7~:1048-58, 1991). Sanger DNA sequencing reactions
employing 2'-deoxythymidine-5'-(alpha-S)-triphosphate are performed
according to standard protocols. When using 2',3'-dideoxythymidine-5'-
(alpha-S)-triphosphates, this is used instead of the unmodified 2',3'-
20 dideoxythymidine-5'-triphosphate in standard Sanger DNA sequencing. The
template (2 picomole) and the nucleic acid M13 sequencing primer (4
picomole) are annealed by heating to 65~C in 100 1ll of 10 mM Tris-HCl~
pH 7.5, 10 mM MgCl2, 50 mM NaCl, 7 mM dithiothreitol (DTT for 5
minutes and slowly brought to 37~C during a one hour period. The
25 sequencing reaction mixtures contain, as exemplified for the T-specific
termination reaction, in a final volume of 150 1ll, 200 ~lM (final
CA 02218188 1997-10-14
WO 96/3251)4 PcTJus96Jo5136
93
conc~ lion) each of dATP, dCTP, ~ , 300 ~lM c7-deaza-dGTP, S IlM
2',3'dideoxythymidine-5'-(alpha-S)-triphosphate and 40 units Sequenase.
Polymerization is pe~ro~ ed for 10 minutes at 37~C, the reaction mixture
heated to 70 ~C to inactivate the Sequenase, ethanol precipitated and coupled
5 to thiolated Sequelon membrane disks (8 mm diameter). Alkylation is
performed by treating the disks with 10 ~Ll of 10 mM solution of either 2-
iodoethanol or 3-iodopropanol in NMM (N-methylmorpholine/water/2-
propanol, 2/49/49, v/v/v) (three times), washing with 10 ~11 NMM (three
times) and cleaving the alkylated T-termin~ted primer-extension products
10 off the support by tre~ttnent with DTT. Analysis of the mass-modified
fragment families is performed with either ES or MALDI mass
spectrometry.
Example 20 Mass Modification of an Oligonucleotide.
This method, in addition to mass modification, also modifies
15 the phosphate backbone of the nucleic acids to a non-ionic polar form.
Oligonucleotides can be obtained by chemical synthesis or by enzymatic
synthesis using DNA polymerases and a-thio nucleoside triphosphates.
This reaction was performed using DMT-TpT as a starting
material but the use of an oligonucleotide with an alpha thio group is also
20 al~p~ liate. For thiolation, 45 mg (0.05 mM) of compound 1 (Figure 15),
is dissolved in 0.5 ml acetonitrile and thiolated in a 1.5 ml tube with 1.1-
diozo-l-H-benzo[1,2]dithio-3-on (Beaucage reagent). The reaction was
allow to proceed for 10 minlltes and the produce is concentrated by thin
layer chromatography with the solvent system dichloromethane/96%
25 ethanol/pyridine (87%/13%/1%; v/v/v). The thiolated compound 2 (Figure
15) is deprotected by treatment with a mixture of concentrated aqueous
CA 02218188 1997-10-14
WO 96/32504 PCT/US96105136
94
ammonia/acetonitrile (1/1; v/v) at room temperature. This reaction is
monitored by thin layer chromatography and the qu~ e removal of the
beta-cyanoethyl group was accomplished in one hour. This reaction mixture
was evaporated in vacuo.
To synthesize the S-(2-amino-2-oxyethyl)thiophosphate
triester of DMT-TpT (compound 4), the foam obtained after evaporation of
the reaction mixture (compound 3) was dissolved in 0.3 ml
acetonitrile/pyridine (5/1; v/v) and a 1.5 molar excess of iodoacetamide
added. The reaction was complete in 10 minutes and the precipitated salts
10 were removed by centrifugation. The supernatant is lyophilized, dissolved
in 0.3 ml acetonitrile and purified by preparative thin layer chromatography
with a solution of dichloromethane/96% ethanol (85%/15%; v/v). Two
fractions are obtained which contain one of the two diastereoisomers. The
two forms were separated by HPLC.
15 Example 21 MALDI-MS Analysis of a Mass-Modified Oligonucleotide.
~ 1 7-mer was mass modified at C-5 of one or two
deoxyuridine moieties. 5-[1 3-(2-Methoxyethoxyl)-tridecyne- I -yl]-5 '-O-
(4,4 ' -dimethoxytrityl)-2 ' -deoxyuridine-3 '-~-cyanoethyl-N,N-
diisopropylphosphoamidite was used to synthesize the modified 1 7-mers.
CA 02218188 1997-10-14
Wo 96/32504 pcTruss6Josl36
The modified 17-mers were:
5d~AAAACGACGGCCAGUG) (molecul~m~s:5454) (SEQIDNO30)
X X
d(UAAAACGCGGCCAGUG) (molecul~ m~s 5634) (SEQ IDNO 31)
where X = -C=C-(CH2)"-OH
(nnmorlifi~d 17-mer: molecularmass: 5273)
The samples were ~ ed and 500 fmol of each modified 17-
15 mer was analyzed using MALDI-MS. Conditions used were reflectron
positive ion mode with an acceleration of S kV and post-acceleration of 20
kV. The MALDI-TOF spectra which were generated were superimposed
and are shown in Figure 16. Thus, mass modification provides a distinction
detectable by mass spectrometry which can be used to identify base
20 sequence information.
Example 22 Capture and Sequencing of a Double-Stranded Target Nucleic
Acid.
In another experiment, a nucleic acid was captured and
25 sequenced by strand-displacement polymerization. This reaction is shown
schem~tically in Figure 17. Double-stranded DNA target was prepared by
PCR and attached to magnetic beads as described in Example 6. EcoR I
digested plasmid NB34 was used as the DNA template for amplification.
NB34 comprises a PCRTM II plasmid (Invitrogen) with a one kb target
30 human DNA insert. PCR was performed with an 16-nucleotide upstream
~ primer (primer I, 5'-AACAGCTATFACCATG-3'; SEQ ID NO. 32), and a
downstream 5 -end biotinylated 1 8-nucleotide primer (primer II, 5'-biotin-
CA 02218188 1997-10-14
WO 96/32504 PCT/US96/OS136
96
CTGAATTAGTCAGGTTGG-3', SEQ ID NO. 33). Five hundred basepair
PCR products, cont~inin~ a single BstX I site, were immobilized by
çhment to magnetic beads which were resuspended in a total of 300 ~11 ?
reaction buffer cont~ining 200 units of BstX I restriction endonuclease
S (Boehringer Mannheim; Tntli~n~polis, IN), 50 mM Tris-HCl pH 7.5, 10 mM
MgCl2, 100 mM NaCl and 1 mM dithiothreitol. The mixture was incubated
at 45~C for three hours or until digestion was complete which was
monitored by agarose gel electrophoresis. After digestion, magnetic beads
were washed twice with 300 ~11 of TE to remove digested and non-
immobilized fragments, excess nucleotides and restriction endonuclease.
This immobilized DNA was dephosphorylated by
resuspending the beads in 100 ,ul buffer (500 mM Tris-HCl, pH 9.0, 1 mM
MgCl2, 0.1 mM ZnCl2, and 1 mM spermidine) containing five units of calf
intes~in~l alkaline phosphatase (Promega; Madison, WI). The reaction was
incubation at 37~C for 15 minutes and at 56~C for 15 minutes. Five
additional units of calf intestinal alkaline phosphatase was added and a
second incubation was performed at 37~C for 15 minutes and at 56~C for
15 minl~tes. Beads were washed twice with TE and resuspended in 300 ~11
of fresh TE containing 1 M NaCI.
Loading of the beads was checked by incubating 10 1ll of the
beads with 10 ~1 of formamide at 95~C for 5 minutes (or by boiling in TE).
The mixture was analyzed by 1% agarose gel electrophoresis with ethidium
bromide staining. A 10 ~1 bead aliquot generally contains about 80 ng of
immobilized double stranded DNA.
A partial duplex DNA probe containing a four base 3 '
overhang was used as a sequencing primer and was ligated with BstX I
CA 02218188 1997-10-14
WO 96t32504 1~CTJUS96J~5136
97
digested DNA fr~ ont~ which were immobilized on magnetic beads. The
partial duplex had a 5'-fluorescein labeled 23 mer (DF25-SF) cont~ininp: a
5' base paring region and a 4-base 3' single stranded region (which is
complementary to the sequence of the 5'-protruding end of the
S corresponding BstXI digested target DNA as l)r~al ed above and a 19 mer
(G-CMl) complementary to the base pairing region ofthe 23 mer. The 19
mer was 5' phosphorylated by the T4 DNA Polymerase and annealed to the
corresponding 23 mer in TE at the same molar ratio. Beads, prepared from
~lk~line phosphatase treatment which have about 10 pmol immobilized
10 DNA template, were ligated to 25 pmol of partially duplex probe in an 100
~11 volume cont~ining 200 units of T4 DNA ligase (New Fngl~n(l Biolabs;
Beverly, MA), 50 mM Tris-HCI, pH 7.8, 10 mM MgCI2, 10 mM
dithiothreitol, 1 mM ATP, 25 ,ug/ml bovine serum albumin. Ligation
reactions were pelrollned at room temperature for two hours or 4~C
15 overnight. Beads were washed twice with TE and resuspended in 300 ~LI of
the same buffer.
Sequencing reactions: Thirty ,ul of beads containing the
ligation product were used for each sequencing reaction. Beads were
resuspended in a 13 ~11 volume containing 1.5 ~11 of 10 x Klenow buffer (100
20 mM Tris-HC1, pH 7.5, 50 mM MgCI2, and 75 mM dithiothreitol) and with
or without one ~1 of single stranded DNA binding protein (SSB, S llg/,ul;
USB; Cleveland, Ohio). Mixtures were incubated on ice for 5 minutes
followed with the addition of 5 units of Klenow Fragment (New England
Biolabs). The reaction volume was split into four termination mixes, each
25 consisting of I ~I DMSO and 3 ,ul ofthe apl)lol,l;ate termination mixture.
CA 02218188 1997-10-14
WO 96/32504 PCT/US96/OS136
98
Termination mixtures were made in Klenow buffer and comprise the
nucleotide concentrations shown below in Table 11.
Table 11
Termin~tion dATP dGTP dCTP dTTP ddNTPs
Mix inmM in mM inmM inmM
ddATP mix 10 100 100 100100 mM ddATP
ddGTP mix 100 5 100 100120 mM ddGTP
ddCTP mix 100 100 10 100100 mM ddCTP
ddTTP mix 100 100 100 5500 mM ddTTP
Termination mixtures were incubated for 20 minutes at
ambient tempe~ e. Two ~11 of chase solution (0.5 mM of each of four
dNTPs in Klenow buffer) were added to each reaction tube and mixtures
were incubated for another 15 minutes, again at ambient temperature.
15 Magnetic beads were precipitated with a magnetic particle concentrator (or
centrifugation) and the supernatant discarded. Beads were resuspended in
a solution containing 10 ~1 of deionized forrnamide, 5 mg/ml dextran blue
and 0.1% SDS, and heated to 95~C for 5 minutçs, and stored on ice for less
than 10 minlltçs. Samples were analyzed on a DNA sequencing gel and on
20 an ALF DNA sequencer (Pharmacia; Piscataway, NJ) using a 6%
polyacrylamide gel with 7 M urea and 0.6 x TBE. Surprisingly, sequencing
reactions performed in the presence of single-stranded DNA binding protein
showed considerable improvement in resolution. Only 50 bases were
resolved from reactions performed without single-stranded DNA binding
25 protein (Figure 18, bottom panel) whereas 200 bases could be resolved from
CA 02218188 1997-10-14
WO 96/32!j04 PCI/US96/05136
re~r,ti~n~ ~)tl rO- ..,e-1 iIl the presence of single-stranded DNA binding prol~(Figure 18, top panel).
Exarnple 23 Specificity of Double-Strand Se~llencin~ by Strand
Displacement.
S Another experiment was performed to delf . .. ,i~,e the
specificity and applicability of the nick translation strand displacement
method of sequencing double-stranded nucleic acids. A schem~tic of the
experimPnt~l design is shown in Figure 19. Briefly, a double-stranded target
DNA was ~l~pared by digesting double-stranded q?X174 phage DNA with
0 TspR I restriction en~lonllclease. TspR I has a recognition site of
NNCAGTGNN and cleaves q~X174 into 12 fr~ment~ each with distinctive
3' protruding ends. Possible ends are shown in Table 12.
Table 12
5'-AACACTGAC-3' 7 5'-GTCAGTGTT-3'
2 5'-AACAGTGGA-3' 8 5'-GTCAGTGGT-3'
3 5'-ACCACTGAC-3' 9 5'-GTCACTGAT-3'
4 5'-AACACTGGT-3' 10 5'-TCCACTGTT-3'
5'-ATCAGTGAC-3' 11 5'-TGCAGTGGA-3'
6 5'-ACCAGTGTT-3' 12 5'-TCCACTGCA-3'
q~X174 DNA (5 pmol) was dephosphorylated using calf
inteshn~l aLkaline phosphatase. Briefly, q~X174 DNA was resuspended in
100 ,ul buffer (500 rnM Tris-HCl, pH 9.0, 1 mM MgCl2, 0.1 mM ZnCl2, and
1 mM sperrni~line) cu~ 5 units of calf intestinal aLkaline phosphatase
25 (Promega; Madison, WI). The reaction was incubation at 37~C for 15
es and at 56~C for 15 minutes. Five additional units of calf intestinal
SUB~ ITE SHEET ~RULE 26)
CA 02218188 1997-10-14
WO 96/32504 PCI/US96/OS136
100
~lk~line ph~ ~ph~t~e was added and a second incubation was performed at
37~C for lS minlltes and at 56~C for 15 minl-tçs DNA in the samples was
extracted once with phenol, once with phenol/chloroform, and once with
chloroform, after which nucleic acid was precipitated in 0.3 M sodium
5 acetate/2.5 volumes ethanol. Precipitated ~X174 DNA was washed twice
with TE and resuspended in 300 ,ul of IE cont~inin~ 1 M NaCl.
Double-stranded probes, comprising biotin (B), fluorescein
(F), and infra dye (CY5) labels, were synthesized and anchored to magnetic
beads as shown in Table 13.
CA 02218188 1997-10-14
WO 96/325~4 PCTIU~9C1~5136
101
Table 13
-
DF27-1 5~-GATGATCCGACGCATCACATCAGTGAC-3' (SEQ ID NO. 34)
3~-CTACTAGGCTGCGTAGTG-p-5' (SEQID NO.35)
DF27-2 5'F-GATGATCCGACGCATCACTCCACTGTT-3' (SEQ ID NO.36)
3~-CTACTAGGCTGCGTAGTG-p-5' (SEQ ID NO. 37)
DF27-3 5~-GATGATCCGACGCATCACGTCAGTGTT-3' (SEQ ID NO. 38)
3~-CTACTAGGCTGCGTAGTG-p-5' (SEQ ID NO. 39)
DF27-4 5~-GATGATCCGACGCATCACTGCAGTGGA-3' (S~Q ID NO. 40)
3~-CTACTAGGCTGCGTAGTG-p-5' (SEQ ID NO. 41)
DF27-5-CY5 5'CY5-GATGATCCGACGCATCACGTCACTGAT-3' (SEQ ID NO. 42)
3~-CTACTAGGCTGCGTAGTG-p-5' (SEQ ID NO. 43)
DF27-6-CY5 S'CY5-GATGATCCGACGCATCACAACAGTGGA-3' (SEQ ID NO. 44)
3'B-CTACTAGGCTGCGTAGTG-p-5' (SEQ ID NO. 45)
DF27-7 5'-F-GATGATCCGACGCATCACGTCAGTG~T-3' (SEQ ~ NO. 46)
3~-CTACTAGGCTGCGTAGTC-p-5' (SE~Q ID NO. 47)
DF27-8 5'-F-GATGATCCGACGCATCACAACACTGGT-3' (SEQ ID NO. 48)
3'B-CTACTAGGCTGCGTAGTG-p-S' (SEQ ID NO. 49)
0 DF27-9 5'-F-GATCATCCCAGGGATCACAAGAGTGAC-3' (SEQ ID NO. 50)
3'B-CTACTAGGGTCCCTAGTG-p-5' (SEQ ID NO.51)
DF27-10 5'-F-GATGATCCGACGCATCACACCACTGAC-3' (SEQID NO.52)
3~-CTACTAGGCTGCGTAGTG-p-S' (SEQID NO.53)
Beads with about 25 pmol of immobili7~d primer were ligated
to 3 pmol of digested TspR I ~Xl 74 DNA in 50 ~11 co~ g 400 units of
15 T4 DNA ligase (New Fngl~n~l Biolabs; Beverly, MA), 50 mM Tris-HCl, pH
7.8, 10 mM MgC12, 10 mM dithio~eilol, 1 mM ATP and 25 ~Lg/ml bovine
serum albumin. Ligation reactions were performed at 37~C for 30 mimltes,
at 50~C to 55~C for one hour (thermal ligase), at room temperature for 2
SlJB~ E SHEET (F~ULE 26)
CA 02218188 1997-10-14
WO 96132504 PCI~/US96/OS136
102
hours or at 4~C for overni~ht After ligation, beads were washed twice with
TE and resuspended in 300 ,ul ofthe same buffer.
SeqllPncin~ reactions: For each sequencing reaction, 30 ,ul of
beads cv. ,~ the ligation product was used. Beads were resuspended in
5 a 13 ,ul volume co~ 1.5 ,ul of 10 x Klenow buffer (100 mM Tris-HCl,
pH 7.5, 50 mM MgCl2 and 75 mM dithiothreitol), and with or without 1 ~11
of single-stranded DNA binding protein (SSB, 5 ,ug/~ll; USB; Cleveland,
Ohio). Reaction ~ cs were incubated on ice for S mimltes, followed by
the addition of 5 units of Klenow Fr~ ment (New Fngl~nd Biolabs). The
reaction volume was split into four termin~tion mixes, each consisting of 1
~11 DMSO plus 3 ~Ll ofthe al)pr~liate t~rmin~tion mix. Termin~tion mixes
were made in Klenow buffer and comprise the nucleotides concentrations
shown in Table 11.
Termin~tion mixtures were incubated for 20 minutes at
ambient temperature. Two ~11 of a chase solution cont~ining 0.5 mM of each
of the four dNTPs in Klenow buffer, was added to each reaction tube and
mixtures were incubated for another 15 minntes at ambient temperature.
Beads were precipitated by magnetic particle concentrator or centrifugation
and the supern~t~nt discarded. Precipitated beads were resuspended in TE
or in a solution cO,~t~ 10 ,ul deionized form~mi-1e, 5 mg/ml dextran blue
and 0.1% SDS, and heated to 95~C for 5 minlltes Mixtures were stored on
ice for less than 10 min~tes and analyzed by a DNA sequencing gel and on
an ALF DNA sequencer (Pharmacia; Piscataway, NJ) using a 6%
polyacrylamide gel with 7 M urea and 0.6 x TBE.
One double stranded primer was used for each reaction and the
results achieved using primers DF27- 1, DF27-2, DF27-4, DF27-5-CY5 and
.~ CA 02218188 1997-10-14
WO Sf~2''01 1 ~iu~ 5
lQ3
DF27-6-CY5, are shown in Figures 20, 21, 22, 23 and 24, respectively.
Each prirner was capable of ~n~ratin~ sequencing inf~ ion of up to 200
b~eep~;rs without significant interference from the 11 f~agments with non-
compk;.--F............ ........~ , ende.
Other embo.iim~ontc and uses ofthe invention will be a~a.c.lt
to those skilled in the art from consideration ofthe specification and practice
of the invention disclosed herein. All U.S. Patents and other references
noted herein are specifically incorporated by reference. The specification
and examples should be coneitlered exemplary only wi~ the true scope and
spirit of the invention indicated by the following claims.