Note: Descriptions are shown in the official language in which they were submitted.
CA 02218808 1997-11-07
W096/35798 PCT~n~6100195
Title: Improved retroviral vectors, especially suitable for
gene therapy.
The invention relates to improved retroviral vectors
which are especially useful for methods of gene therapy. The
invention further relates to such vectors in combination with
suitable packaging cell lines, as well as virus-like particles
which can be produced using said combination and methods of
providing cells with genetic information of interest using
said virus-like particles.
Retrovirus-based vectors are highly favoured tools to
achieve stable integrated gene transfer of foreign genes in
10 m~m~ 1 ian cells. Especially in the area of gene therapy their
use has attracted considerable attention. Retrovirus-based
vectors have been used in both ex vivo and in vivo gene
transfer procedures and were shown to be capable of yielding
long-term expression of foreign genes in culture and in vivo
in ~ n i m~ 1 studies as well as in man.
Retroviral vector svstems for (safe) gene therapv
purposes comprise two building blocks:
the recnmhi~nt retrovir~l vector that carries the genetic
information which is to be transduced plus all of the elements
required in cis for the packaging and integration of the viral
genome, and; retroviral packzqing cells that provide the viral
proteins encoded by the genes gag, pol and env. These
polypeptides are required in trans for the production of
viable virus particles but by themselves, the packaging cells
are incapable of releasing infectious virus. Packaging cells
transduced with the recombinant vector will therefore generate
recombinant retroviruses carrying the genetic information
contained in the aforementioned vector. These viruses can
subsequently be used to transduce cells in which the
recombinant material will be integrated following the natural
retrovirus life cycle. The current invention in one aspect
relates to recombinant vectors with improved characteristics
as compared to the previously described vectors.
SUBSTITUTE SHEET (RULE 26)
CA 022l8808 l997-ll-07
W096/35798 PCT~96/00195
A favoured design of the recombinant virus with
properties that permit long term expression in ~nimAl5 and
which is based on a non-pathogenic retrovirus, has previously
been described (Valerio et al ., 1989; International patent
application WO9307281)-. In these constructs the gene of
interest is under the transcriptional control of a viral long
t~minAl repeat tLTR). In the favoured LTR the retroviral
enhancer is replaced by an enhancer active in undifferentiated
cells such as embryo carcinoma (EC) cells. An example is the
enhancer of a polyoma virus mutant (PyF101) that was selected
to grow on undifferentiated EC cells. This results in
mutations in its enhancer that permit activitv of heterologous
promoters in primitive cells. An LTR based on Moloney murine
leukemia virus (Mo-MLV) in which the enhancer is replaced for
by the PyF101 enhancer is called ~Mo+PyF101. Since replication
competent viruses harbouring the ~Mo~PyF101-LTR can cause
viremia in newborn mice without causing disease, the
properties of ~Mo+Py~101-carrying vectors are superior to the
commonly used vectors based on viral backbones of disease-
inducing viruses such as Moloney murine leukemia virus,myeloproliferative sarcoma virus or Harvey murine sarcoma
virus (Davis et al ., 1985).
Our work with ~Mo+PyF101-based retroviral ~ectors has led
to the conclusion that thev are capable of introducing aenes
~S into primary haemopoietic stem cells of mice, non-human
primates and man. Following transplantation of such modified
stem cells sustained expression of the transduced gene was
observed in all haemopoietic lineages analvzed in the absence
of any gene transfer-related toxicity (Einerhand et al .,
1991; Van Beusechem et al., 1990, 1992, 1993, 1994 and 1995).
Based on these results the world first clinical gene therapy
study in man aimed to correct the haemopoietic stem cells of
three patients with an inherited deficiencv of adenosine
~e~min~se was performed using the aforementioned retroviral
vector carrying a correct -~ersion or the human adenosine
~mi n~e gene (Hoogerbrugge et al ., 1995)-
Despite the many favourable characteristics of thepreviously described vector, several limitations of the
SUBSTITUTE 5t~EET (RULE Z6)
CA 02218808 1997-11-07
W096/35798 PCT~n96/00195
original design will still limit its general usefulness as a
gene therapy product.
Identification of these short~mings as well as remedies
for these shortcomings are one aspect of the present
invention. These shortcomings of the recombinant vectors are:
1) The presence of superfluous retroviral sequences which do
not contribute to the efficient packaging of the recombinant
virus, which limit the space available for genes to be
transduced and which increase the risk of recombination events
during the generation of producer cells possibly leading to
the production of helper virus.
2) The presence of a gag open reading frame which may result
in the translation of truncated viral proteins that would
cause unwanted immune responses and will probably increase the
potential of helper-virus formation in packaging cells
carrying homologous gag sequences.
3) The presence of superfluous non-coding sequences of the
gene(s) of interest, limiting space and possibly negatively
~0 affecting gene expression and/ or messenger stabillty.
4) Expression in vitro and in vivo is still influenced by
the integration site (e~g~ ~inerhand et al ., 1993) and
therefore variable. The inclusion of elements that could
control site independent expression such as specif-c boundary
'5 elements (Chung et al ., 1993) and Locus Control Reaions
(Grosveld et al ., 1987; Greaves et al., 1989/WO 9101329) have
been suggested, but were shown to be the cause of
rearrangements (e.g. Novak et al ., 1990).
5) Sequences contained within the viral construct can
function as unwanted cryptic splice sites, resultlr.g in
packaging and expression of aberrant RNA molecules. (e.g.
McIvor et al ., 1987; Sorrentino et al ., 1993).
6) Given that in the preferred configuration of the vector
the gene of interest is under transcriptional control of the
S viral LTR, the expression of more than one gene can only be
achieved through bicistronic messengers or in the form of
fusion proteins. Currently used intercistronic sequences are
derived from viruses (Pelletier and Sonenberg, 1988; Jang et
al., 1988), they are in general large and contain extensive
SUBSTITUTE S~EET (RULE 26)
CA 022l8808 l997-ll-07
PCT~n96/0019S
W096/35798
secondary structures which render them less favourable than
the synthetic intercistrons according to the present
invention.
The problems as mentioned under nos. 1 and 2 have been
S solved in the prior art, in vectors designated as the LN
series (Miller and Rosman, 1989), but despite the large body
of work on retroviral vectors that has been published since
the availability of the LN-based vectors, to our knowledge,
they were never combined with constructs in combination with
~Mo+PyF101 or other enhancer-replaced LTRs.
The current invention discloses a basic vector and
derivatives thereof that will result in improvements as
related to the shortcomings stated above.
Thus, the current invention provides, ln one aspect, a
vector derived from a retrovirus, comprising a se~uence
responsible for transcriptional control, including an
enhancer, which vector further comprises a site for insertion
of at least one gene of interest, a packaging signal, said
vector having no superfluous retroviral sequences and no open
~0 reading frame encoding at least parts of viral proteins,
characterized in that the enhancer is an enhancer that is
active in undifferentiated cells.
The vectors according to the invention have all the
benefits of the so-called LN vectors as earlier mentioned, but
~5 they have the additional advantage that they can be used to
functionally tr~nsfer material into undifferentiated cells and
retaining said function throughout the differentiation and other
processes. Preferably the modified LIR comprising the invented
enhancer will not be able to give rise to signiricantly
pathogenic viruses. The ~Mo+PyF101 enhancer is a good example of
such an enhancer and is a preferred embodiment of the present
invention. Based on the disclosure of the present invention
however, the skilled worker will be able to design other
Simi l~rly suitable enhancers. In designing vectors according to
the invention, the skilled worker will be able to modify the
concept of the invention in order to suit his needs. Therefore
it will be clear that the definitions of the present application
should be interpreted in a broad sense. For instance a gene will
read upon any DNA-like material to be transduced into a cell.
SUBSTITUTE S~EET (RULE 26)
~ ,
CA 02218808 1997-11-07
W096/35798 PCT~L96/00195
Thus non-coding DNA or cDNA are within that definition. Similar
defintions shoud be given a similar scope. The skilled worker
will now also be able to work around the problems or
shortcomings as identified above, because once identified
according to the invention, in most cases the solution presents
itself. For instance, now that a problem has been identified in
that a viral sequence may contain a cryptic splice site, the
skilled worker can ~mi ne the viral ~equence for such a site
and delete or mutate it. The same is of course true for
untranslated parts of the gene of interest. The skilled worker
may now identify such untranslated superfluous sequences and
remove them from the gene of interest.
The advantages to be gained by providing a short stretch of
intercistronic linker-basepairs, which has a number of bases
dividable by three is clear to the skilled worker.
The other characteristics of the vectors according to the
invention as defined in the claims and the specification by
themselves or in combination lead to clear advantages over the
prior art. Insofar as they have not been clearly defined herein,
they are clear to the skilled workers in the field.
Of course the ultimate goal of the vectors according to the
invention lies in providing a safe and viable system of
providing certain subsets of cells with additional genetic
material, especially in the context of gene therapy.
~5 For efficient transfer of the vectors according to the
invention it is usually necessary that they be presented in a
virus-like infectious particle. For providing such a particle it
is of course necessary that a packaging signal is present on the
vectors. Such a packaging signal may be any functional one, i.e.
one that works with the packaging material. The packaging
material will usually be provided by a cell into which the
vector is transferred. Said packaging material will of course
have to be functional in packaging the vectors according to the
invention. The most logical and most preferred combination is
that of the retroviral packaging signal together with a cell
that constitutively produces the retroviral proteins necessary
for packaging, a so-called packaging cell line. The many
possible combinations of the two (kit of parts) are of course
part of the present invention.
SUBS 111 UTE SHEET (RULE 26)
=
CA 022l8808 1997-ll-07
W096/35798 PCT~n96/00195
Both the positive mode and the negative mode of gene
therapv can be realized using the vectors according to the
invention. For the present application the positive mode of gene
therapy is int~n~e~ to read upon any deficiency in a group of
cells that can be treated by providing at least a number of said
cells with a gene capable of removing said deficiency, such as
for instance providing hematopoietic cells with a gene encoding
factor vIII for correcting haemophilia. The negative mode of
gene therapy for this application includes the functional
removal of any subset of cells within an individual by
introducing genetic material into at least a number of cells
from the subset. Examples are suicide genes for tumour cells.
Descr~tion of s~ecific embodiments
In one aspect the invention relates to several retroviral
~ectors that share a number of basic characteristics and that
can be used to efficiently generate infectious recombinant virus
particles when transfected into packaging cells. Hereunder a
description of the basic embodiments present in the vectors
according to this invention is given, as well as the specific
characteristics of some of the preferred retroviral vectors that
are included in this invention together with examples of their
applications.
The described retroviral vectors conlain a 5' LTR,
preferabl~ from Moloney murine sarcoma virus (MoMsv base l to
541; numbering according to van Beveren et al., 1985), including
part of the packaging signal. The remainder of the packaging
signal is preferably derived from Moloney murine leukemia virus
(MoMLv base 566 to 1038) with the start codon of the gag coding
domain preferably mutated to a stop codon (Miller and Rosman,
1989) The LTR and the ext~n~e~ packaging signal with the point
mutations together ensure efficient packaging of the recombinant
~rirus whithout anv production of virus-derived proteins in the
targe~ cells. Furthermore, the reduced sequence homology between
the MoMSV LTR and the 5' part of the packaging cons~ructs (MoMLV
LTR) generally used in packaging cells like PA317 and PG13 will
reduces the chance of recombination between the constructs and
thus reduces the chance of helper virus formation. A splice
donor site located downstream of the 5' LTR can in combination
SUBSTITUTE S~EET (RULE 26)
CA 022l8808 ls97-ll-07
Wos6/3s7s8 PCT~96/00195
with the splice acceptor site just upstream of the insertion
site of the gene of interest lead to enhanced translation of the
inserted gene in case splicing occurs. To further reduce chances
of recombination of retroviral vectors with packaging
constructs, the vectors contain no overlap with the 3' end of
the env constructs used in the above mentioned packaging cell
lines. Such a construct is again a preferred embodiment.
The resulting vector (shown in Figure 1) having all
preferred features, contains a 5' and 3' LTR, a packaging signal
ext~n~ing into the gag coding region and a poly cloning site for
the insertion of (a) gene(s) of interest and thus meets the
basic requirements a retroviral vector according to the
invention should meet.
In another preferred embodiment, the above basic retroviral
vector is modified by deleting (parts of) the gag coding
se~uences without losing packaging function. Such a vector
further reduces the probability of recombination events in the
packaging cells that may lead to replication competent
retroviruses. Thus this improves the general safety features of
the retroviral vectors.
This invention also discloses further modifications to this
basic retroviral vector to allow more efficient transcription
and translation. First, a consensus Kozak sequence is introduced
around the ATG of the gene of interest to improve transiation of
that gene. Second, the viral enhancer in the 3' L~R is replaced
by a mutant form of the Polyoma virus enhancer that is
specifically selected for activity in F9 embryonal carcinoma
ceLls (Linney et al., 1984) and that is known to be less
sensitive for promoter inactivation in haemopoietic stem cells
and early haemopoietic progenitor cells cnmp~ red to the wild
type MoMLV enhancer (Valerio et al., 1989; Van Beusechem et al .,
1990). Another advantage arising from the replacement of the
wild type MoMLV enhancer for the polyoma enhancer is that the
resulting ~Mo+PyF101 LTR renders murine leukemia viruses into
non-pathogenic viruses (Davis et al ., 1985). Following one round
of replication this alteration contained within the U3 region of
the 3' LTR is also tranferred to the S' LTR and is thus present
in both LTR's of the proviral sequence.
SUBSTITUTE 5t~EET tRULE 26)
=
CA 02218808 1997-11-07
W096/3S798 PCT~n96/00195
As a consequence, after infection of an amphotropic
packaging cell line, the ~Mo+PyFl0l LTRs contain less sequence
homology with the packaging constructs as compared to LTRs with
wtMoLV enhancers.
Additional modifications to improve expression in target
cells include:
- incorporation of a Locus Control Region (LCR) e.g. the CD2 LCR
for high and controled expression in T-cells (see example 5).
- Incorporation of a selectable marker preferably as a second
gene in a dicistronic transcription unit. Selection genes
include the neomyciner gene, the hyyLulllyciner gene, a gene
encoding a fluorescent protein or a gene coding for a
biologically inactive tr~n~m~mhrane molecule that all allow for
efficient selection in vitro. AlternatiVely, selection markers
may be included that also allow for selection of transgene
expression in vivo like for example, without limitation, the
human genes for Multi Drug Resistance (MDR-l see examples 2 and
3), a UDP-Glucuronosyl Transferase, Thvmidylate Synthetase,
canalicular Multispecific Organic Anion Transporter, y-GLutamyl
Cysteine Synthetase, as well as biologically active mutants of
these genes.
- additional (regulatory) sequences that can influence the
expression of the integrated provirus in the target cells e.g.
boundary elements and/or (tissue-specific) promoters or
enhancers.
Preferred or additional properties of the retroviral vectors
described in this invention include:
- a gene of interest with no or a mi nimllm of 5' and 3' non-
translated sequences necessary for ~imllm RNA stability and
translation.
- a dicistronic transcription unit whereby the two coding
regions are separated by a short non-coding linker aIlowing
efficient reinitiation of the ribosomal complex on the start
codon of the second gene. This non-coding linker can have a
variable length but is devoid of any ATG sequences or sequences
that form strong secondairy structures in the RNA. For ~;mtlm
efficiency of translation, the length of a favourable
intercistron is a multiplicit~ of 3, the stop codon of the first
SUBSTITUTE SHEET (RULE 26)
CA 022l8808 l997-ll-07
W096/35798 PCT~96/00195
gene thus placed in frame with the start codon of the second
gene.
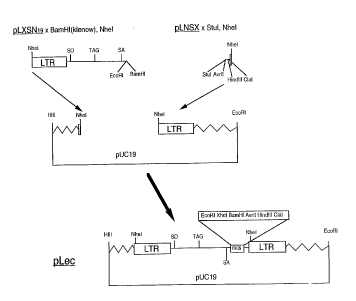
Co~struction of the retroviral vector pnec
The retroviral vector pLXSN (Miller and Rosman, 1989) was
digested with NheI and the insert con~ining the viral sequences
was ligated into the vector backbone pUCl9 obtained from pSFG-
tpa ( R. Mulligan and I. Riviere, Whitehead Institute for
Biomedical Research, Cambridge, MA) after digestion with NheI.
IO The resulting construct, named pLXSN1g, was digested with BamHI
and the ends were filled in using the Klenow enzyme. After
removal of the enzymes, the DNA was digested with NheI after
which the 1452 bp NheI/BamHI fragment containing the 5' LTR and
the extended packaging signal was isolated. A 98 bp fragment
cont~;ning the 3' LTR was isolated from the vector pLNSX (Miller
and Rosman, 1989) following digestion with NheI and StuI.
Ligation or these fragments into the NheI fragment from pSFG-tpa
cont~;n~ng the pUCl9 backbone resulted in the viral construct
pLec (Figure l).
Mo~ification of the vir~1 ~nh~ncer in the 3' rlTR of pTec
pLec was digested with NheI and the fragment conr~'ning the
viral sequences -~as ligated into the NheI site of
pSK~Zip~Mo+PyFlOl resulting in pLec~Mo (Figure 2). This vector
carries the modifications in its 3' LTR which will result in
conversion of these alterations into the 5' LTR after one round
of replication (Valerio et al ., 1989). pSK/Zip~Mo-PyFlOl has
been generated by subcloning of the ClaI-EcoRI fragment from
pZip~Mo+P~IFlOl(N~) into the pBluescript vector (M. Einerhand,
TNO, Radiobiological Institute). pZip~Mo+PyFlOl(N~) is a low
copy vector (pBR 322 based) cont~ining a 3' LTR that has been
made by combining the ClaI/KpnI fragment from pML~-
C/R/B~Mo+PyFlOl (Linney et al., 1984) with a KpnI/ EcoRI
fragment from pZipSV(X)l (Cepko et al., 1984). The first
contains part of the R region and a U3 region in ~hich the
enhancer sequences have been replaced by a mutant form (F101) of
the Polyoma virus enhancer, the second contains the remaining R
and U5 sequences of the 3' LTR. After destroying the NheI site
in the tetracycliner gene in the vector sequences, the unique
SUBSTITUTE SltEET (RULE ~6)
CA 02218808 1997-11-07
W096/35798 PCT~n96/00195
NheI site can be used to swap recombinant vec~ors with wild t~pe
enhancers into one that contains the mutant form (Valerio et
al., 1989).
5 ~r~ ,7e 1: Monocistro~ic and bicistro~ic ,~LLo~iral vectors
for suicide gene therapy
A promising method for the treatment of solid tumours is
the introduction of suicide genes into the tumour cells in vivo.
Suicide genes, like the Herpes simplex virus type 1 thymidine
kinase (HSV-tk) gene or the cytosine ~mi n~e gene, encode
proteins that are capable of transferring a non-toxic prodrug
into a toxic drug. For example the prodrug ganciclovir is not
toxic for eukaryotic cells but after (mono)phosphorvlation by
the HSV-tk gene it will be converted into a nucleotide analog by
cellular enzymes. Incorporation of this analog into the DNA of
replicating cells results in chain termination and cell death.
The attractivity of this system as an anti tumour therapy has
become apparent from the notice that transduced cells that are
dying due to the prodrug treatment can trigger death of
untransduced cells in their close vicinity: 'the byst~n~er
effect' (Moolten, 1986). Several groups have used tumour models
in rats to show that after transfer of a suicide gene into
tumour cells in vivo, only a minority of the tumour cells need
to express the suicide gene in order to establish an erfective
anti-tumour respons (reviewed in Moolten, 1994). Efficient
transduction of tumour cells can be achieved i~ vivo bv direct
injection of retrovirus producer cells into the tumour (Culver
et al., 1992).
As an example for the application of the retroviral vectors
outlined in this invention we describe the vector pIGTk and its
use in suicide gene therapy for malignant brain tumours in a rat
model.
Co~struction of pIGTk
The retroviral vector pLec was digested with XhoI, blunted
with Rlenow and subsequently digested with BamHI. After
dephosphorylation using calf intestine phosphatase (CIAP), this
fragment was ligated to a fragment containing the coding region
of the Herpes simple~ virus type 1 thvmidine kinase gene (HSV-l
SUBSTITUTE 5~tEET (RULE 26)
CA 022l8808 l997-ll-07
W096/35798 PCT ~ 96/00195
tX) obtained from pAdTK (Bram Bout, IntroGene; European Patent
Application 94202322.7) by digestion with HindIII, blunting with
Klenow and digestion with BamHI. The resulting viral vector was
named pLTk.
pLTk was linearised with BsiWI, partially digested with
EcoRI and dephosphorylated with CIAP. The 5' part of the tk gene
was then reintroduced as a EcoRI/BsiWI pcr fragment obtained
after amplification of the 5' part of the tk gene in the ~ector
TNFUS69 (Schwartz et al., 1991) using the primers TKkozUp: 5'-
CGGAATTCGCCGCCACCATGGCTTCGTACCCCGGCCATCAG-3' and TkDo-l: 5'-
CGGCTCGGGTACGTAGACGATATCG-3' followed by digestion with EcoRI
and BsiWI. The resulting retroviral construct was named pLTKkoz
(Figure 3). Using this strategie for cloning, a retroviral
vector was created that only contains the coding sequences of
the HSV-l tk gene with an optimized Kozak sequence around the
start codon. The NcoI site 5' and the BamHI, AvrII, HindIII and
ClaI sites 3' of the inserted gene are useful cloning sites to
swap inserts in this vector (Figure ~a).
The NheI fragment from pLTKkoz was introduced into the
unique NheI site from pSK/Zip~Mo+(PyF101) to generate construct
pIGTk (Figure 4b).
The retroviral vector pIGTX was cotransfected into the
amphotropic packaging cell line PA317 (ATTC No. CRL 9078)
together with an expression construct containing the neomyciner
gene (~Mo+PyFlOlLTR-Neo, M. Einerhand unpublished) and G418
resistant clones were isolated. One of these produced around
lx105 infectious virus particles/ml that were capable of
transferring the HSV-l tk gene to thymidine kinase deficient
Rat-2 cells. This virus producer (term~ IG-RV-TK) has been
succesfully used in a preclinical study aimed at curing
experimental brain tumours in rats (vincent et al ., 1996 ) . In
this study 344 Fischer rats were inoculated with 4x104 9L rat
gliosarcoma brain tumour cells (Weizsaeker et al., 1981) in the
left forebrain using a stereotaxic apparatus. After 3 days the
growing tumours were inoculated once with 5X106 IG-RV-TK
producer cells, 5X106 control cells (PA317 non-producer cells,
IL-2 retrovirus producer cells or LacZ retrovirus producer
cells)~ supernatant from IG-RV-TK cells or PBS. Treatment with
ganciclovir 15 mg/kg twice daily intraperitoneally for ten days
SUB5TITUTE SH EET (RULE 26)
CA 02218808 1997-11-07
W096/35798 PCT~n96100195
was initiated 8 days after inoculation of the 9L tumour cells.
Figure 5 clearly shows the prolonged survival of rats treated
with IG-RV-TK producer cells in combination with ganciclovir
(Vincent et al ., 1996 ) .
S The tk/ganciclovir system can also be of great value in the
treatment of leukemia. Currently, patients treated for leukemia
often receive an allogeneic bone marrow transplantation (BMT),
using a BM graft from an (un)related, but closely MHC-matched
donor. The presence of T-cel s in such a graft has turned out to
be a ma~or factor determining the succes of the treatment. In
addition, it has been demonstrated that in patients receiving an
allogeneic BMT the likelihood of a leukemia relapse is reduced
due to a so called graft-versus-leukemia (GVL) reaction (Antin~
1993). However, these patients often suffer from a severe life-
threatening graft-versus-host disease (GVHD). Unfortunately, it
seems not possible to separate the GVL and GVHD reactions in
human patients. Thus, although the patients clearlv benefit from
the presence of allogeneic T-cells in the graft, this treatment
is seriously hampered by the occurrence of GVHD. A solution to
this problem could be to isolate (allogeneic) peripheral blood
lymphocytes from the donor prior to the B~T, transduce them in
vitro with a suicide gene and use these cells together with the
T-cell depleted BM graft (Tiberghien et al ., 1994). In case GVHD
develops treatment of the patients with ganciclovir will result
in selective killing of the activated (transduced) T-cells
prospectively leading to abrogation of GVHD. Using this method
patients may still benefit from a GVL reaction resulting in a
decreased rate of leukemia relapse.
A prerequisite for the success of this approach is that
virtually all T-cells that are infused into the patients stably
express the suicide gene to be able to eradicate them in vivo
with ganciclovir. The efficiency by which human peripheral blood
lympnocytes can be transduced in vitro with retroviral vectors
is unlikely to ever become 100%. As a consequence it is
necessary to incorporate a selection marker in the retroviral
vector. In former e~periments with retroviral constructs
harboring an internal promoter driving the expression of a
second gene ~often the selection gene), it was noticed that
interference between the 2 promoters in the retrovirus often
SUBST1TUTE SHEET (RULE 26)
CA 022l8808 l997-ll-07
W096/3S798 PCT~L96/00195
I _ .~,~
resulted in (unwanted) shut-off of one of the promoters (Fm~rm~n
and Temin, 1984, 1986). Therefore, in the constructs described
~in this invention both genes are located in a dicistron driven
by one (viral) promoter. Dicistronic mRNAs allow for efficient
S translation of the second gene if the genes are separated by
either a short intercistronic linker depending on ribosome
sc~nning, or by specialised sequences triggering internal
binding of ribosomes. According to the ribosome sc~nn'ng model
(Kozak, 1987a, 1989) the small ribosomal subunit binds to the 5'
end of the capped mRNA and scans for the presence of ATG
sequences. Initiation of translation occurs at the first ATG in
a favourable context (Kozak, 1987b) and the translation complex
dissociates when a stop codon is encountered. Translation of a
second coding region is possible presumably because the small
subunit (or another factor of the elongation complex) continues
to scan along the mRNA and, when an ATG is recognised, is able
to reinitiate translation. Experiments using different
intercistronic linker sequences have shown that the sequence
itself is of more importance than the length (Kozak, 1987a;
Levine et 21 ., 1991 ) . The use of internal ribosomal entry sites
(IRES) from picornaviruses to express two genes in a dicistron
following retroviral infection has been described bv W. Anderson
in patent application WO 9303143. These viral sequences are
however relatively large and have strong secondary structures
which could affect the packaging capacity and stability of the
construct and its RNA product.
As an example of a favourable dicistronic mRNA we describe
here the construct pLTk+Neo~Mo in which the tk gene and the neor
gene are separated by a 36 nucleotide linker. The tk or the neor
gene can both be replaced by other genes. In general, a
favourable intercistronic linker: (1) Has the start codon of the
second gene inserted in frame with the stop codon of the first
-gene: the length thus being a multiplicitv of 3, (2) should not
contain any ATG sequences, (3) can vary in length between 9 and
iS 200 bp and (4) should not contain sequences that form strong
secondary structures in the RNA. The two genes with their
intercistrons can subsequently be introduced into the retroviral
construct.
SUBS 111 UTE SHEET (RULE 26)
.
CA 022l8808 1997-11-07
W096/35798 PCT~96/00195
14
Construction of ~r~Tk+Ne~A~o
The HSV-tk gene was excised from pLTKkoz using EcoRI and
BamHI and subcloned into pUCll9. An EcoRI/HincII fragment
contA i n i ng the promoter of the human phosphoglycerate kinase
S gene (Singer-Sam et al., 1984; Michelson et al., 1983) was
provided with an EcoRI linker and introduced into the EcoRI site
generating pPGK-Tk. To optimise translation of the neor gene and
introduce an NcoI site at the start of the neor gene, the
sequence around the ATG codon was changed into the consensus
Kozak se~uence (Kozak, 1987b) by a pcr reaction on pMClneopA
(Thomas and Capecchi, 1987) using the primers 5'-
CCCTGCAGCGCCACCATGGGATCGGCCATTGAACAAGATGG-3' (forward) and 5'-
GCCAGTCCCTTCCCGCTTC-3~ (reverse). The 280 ~p pcr fragment was
digested with ~stI and subcloned into the pBluescript KS~ ~ector
lS (stratagene). A PstI/AsuII fragment from pLNCX (Miller and
Rosman, 1989) cont~i ni ng the 3' part of the neor gene was then
introduced into this vector after digestion with ClaI followed
by dephosphorylation and partial PstI digestion. The modified
neor gene was isolated as a BamHI/SalI fragment and cloned into
~0 the corresponding sites in pPGK-TK, resulting in pPTk+Neo/IF.
Lastly, a blunted EcoRI/BamHI fragment from pAMG-1 (Valerio et
al., 1985) con~ining an poly Adenglation signal from hepatitisB
virus (HBV) was introduced into the blunted HindIII site,
generating pPTk+NeopA/IF. In this construct the stop codon of
'5 the tk gene is positioned in frame with the start codon of the
neor gene. Using a different forward primer in the pcr reaction,
lacking the C just 3' from the PstI site, a second Tk-Neo
dicistron has been generated in which the two coding regions are
separated by a 35 nucleotide linker in stead of 36 nucleotides
(pPTK-NeopA/OF; Figure 6).
The Tk+Neo dicistron from pPTk.NeopA/IF was e~cised by
EcoRI and SalI digestion and ligated into the pLec vector after
digestion with EcoRI (partial) and XhoI. pLTk+Neo~Mo was
completed after subcloning of the NheI fragment into the
corresponding site in pZip~Mo+PyF101(N~)
SUBSTITUTE 5~EET (RULE 26)
CA 02218808 1997-11-07
W096/35798 PCT~n96/00195
~fficient tra~slatio~ of the seco~ aene in a bicistron
Two monocistronic (control) constructs were generated
carrying either a single T~ coding region (pPTkpA) or the neor
coding region (pPNeopA; Figure 6). The first originated after
- 5 digestion of p~Tk+NeopA/IF with BamHI and SalI, blunting with
Klenow enzyme and religation, the second after partial digestion
with NcoI and religation of the vector fragment lacking the tk
gene. To test the efficiency of translation of both coding
regions in the bicistronic constructs, plasmid DNA (16 ~g) was
cotransfected with 4 ~g pCMVLuc (L. Fortunati and M. Scarpa,
unpublished) using the CaPO4 coprecipitation method described by
Chen and Okayama (1987). Addition of equal amounts of the
luciferase expression construct to each DNA/CaPO4 mixture
enables correction for differences in transfection efficiency.
Two days after transfection cells were trypsinised and half was
used to measure luciferase activity. The other half of the cells
was partly plated in different dilutions (1/100 and 1/S00) in 6
cm dishes and selected for either tk (HAT-supplemented medium)
or neor (medium plus lmg/ml G418 sulphate) activity and the
r~m~;n;ng cells were pooled and selected for neor activity
(except for the transfection with pPTkpA wich was selected in
HAT supplemented medium). Table 1 shows the relative number of
colonies obtained after selection in HAT or G418 cont~; n; ng
medium and correction for the luciferase activity. From these
results it is clear that the tk activitv in the bicistronic
- constructs is comparable whereas the translation of the second
gene (neor) is less efficient when the first and the second gene
are not in frame with each other.
SUBSTITUTE St~EET (RULE 26)
CA 02218808 1997-11-07
WO 96/35798 PCT/N196/0019S
16
# o~ c~l ~n; es corrected ~or luciferase activity
HAT selection G418 selection
pPTk+NeopA~IF12 16
12
pPTk-NeopAIOF ll 2
8 3
pPNeopA - 61
96
15 pPTkpA S
Table 1: Relative number of colonies obtained after
transfection of the different constructs
into Rat-2 cells grown in indicated selective medium. Each
transfection has been performed ~~~~~
in duplo. See text for details.
~5
The G~18 selected pools con~ining the constructs
pPTk+NeopA/IF, pPTk-NeopA/OF or pPNeopA and the HAT selected
pool pPTkpA can be used to test the sensitivity of the cells to
the prodrug ganciclovir. To test this, cells were plated at low
densitv (lx104 cells) in 75 cm2 flasks in the presence of 5 ~M
Ganciclovir (GCV). Growing colonies were scored and represent
tk- cells present in the pools of transfected cells. Cells were
plated at low density to avoid negative influences of the
byst~n~r effect i.e. the death of untransduced cells due to the
transmittance of the toxic activity from transduced cells.
Plating efficiency was determined by mixing increasing numbers
of tk- cells (100- 5000) with or without lx10~ tkT cells and
culture in medium cont~;ning 5 ~M GCV. From these experiments
~0 it became clear that the pool of cells transrected with
pPTk NeoPA/IF contained -12% t~- cells whereas the pool of cells
SUBSTITUTE S~EET (RULE 26)
CA 022l8808 l997-ll-07
W096/35798 PCT~ng6/00195
transfected with pPTk-NeopA/OF contained -45% tk- cells. The
appearance of t~- cells can be partly explained by the fact that
circular plasmid DNA was used for the transfection resulting in
integration events that inactivate the t~ gene. The difference
between the two bicistronic constructs however, can only be
explained by differences in translation of the neor gene due to
the different intercistronic linker. The outcome of the above
described experiments lead to the conclusion that the situation
in which the two coding regions in a bicistron are placed in
frame to each other is favourable compared to an out -of- frame
situation.
To be able to directly compare the performance of the
bicistronic constructs with an intercistronic linker sequence
with bicistronic constructs containing an IRES sequence, two
additional bicistronic expression constructs were made. The
first, pPTkpolioNeo, contains an IRES sequence derived from
poliovirus and the second, pPTkemcvNeo containes an IRES
sequence from EMCV (encephalomyocarditis virus). pPTkpolioNeo
was generated by ligation of a 750 bp Klenow treated
HindIII-EcoRV fragment from pP2-5' (Pelletier et al., 1988)
into pPTk+NeopA/IF digested with XbaI followed by Klenow
treatment. The resulting construct was then digested with
Bam~I and partially digested with NcoI, Klenow treated and
religated to ~ .O~ most of the linker sequences present in
pPTk+NeopA. pPTkemcvNeo was generated by inserting a 582 bp
blunt EcoRI, MscI fragment from pBS-ECAT (Jang et al., 1989)
into a pBr322 based pPTk-NeopA clone. The resulting vector was
modified by exchanging the sequences between the HindIII site
ln the IRES sequence and the NcoI site at the 5' end of the
neo coding sequences for a fragment from the 3' end of the
EMCV IRES with the sequences around the translation start site
modified to an NcoI site thus placing the ATG in the neo gene
onto the starting ATG from the EMCV IRES. The EMCV IRES with a
NcoI site on the ATG has been made by exchanging the 3' end of
an EMCV IRES clone in pUC119 for a pcr fragment generated with
the following primers: 5'-CCCAGTGCCACGTTGTGAGTTGG-3' and
5'-GCGGATCCGGCCATGGTATCATCGl~Lllll~-3'.
SUBSTITUTE 5HEET (RULE 26)
CA 022l8808 l997-ll-07
W096/35798 PCT~n96/00195
18
To test the efficiency of translation or both coding
regions in the different bicistronic constructs Rat-2
fibroblasts were cotransfected as described above with pRSVLuc
and one of the following bicistronic constructs:
S pPTk+NeopA/IF, pPTk-NeopA/OF, pPTkpolioNeo, pPTkemcvNeo,
pPTkpA or pPNeopA (Figure 6). Forty eight hrs after
transfection half of the cells was used to measure luciferase
activity and the other half was partly plated in different
dilutions in medium containing HAT or G418, and partly pooled
and subjected to G418 selection 1.4 mg/ml (bicistronic
constructs and pPNeopA) or HAT selection (pPTkpA). In three
independent cotransfection experiments the number of colonies
formed with G418 selection was calculated from the different
dilutions tha~ were plated and correcled for differences in
transfection efficiency in two ways: 1) bv making use of the
luciferase activity measured in each of the transfections
(Figure 6A) and 2) by making use of the number of colonies
formed under HAT selection (Figure 6B). In the latter case the
assumption is made that the differences in intercistronic
sequences do not influence the translation of the tk gene. As
is evident from the figures 6A and B in both cases there are
no significant differences in the efficiency by which neor
colonies are formed after transfection of the bicistronic
constructs. The monocistronic COnStrUCI pPNeopA is, however,
~5 about 4x as efficient compared to the bicistronic constructs.
The transfected Rat-2 cells that were pooled and selected
with G418 can be used to analyse co-expression of the tk gene
by monitoring cell kill after growth in medium supplemented
with ganciclovir (GC~). Hereto cells from the different pools
were plated in quadruple in 96 well plates at 800 cells/well
and grown for 4 days in the presence of 0, 5, 10 or 25 ~M GC~.
The number of GCV resistant, viable cells was then determined
colorimetrically by means of MTS st~ining (Promega). The
results are presented in table 2 below. The absorbance at 490
nm in the wells containing no GC~ is set to 100 %.
SVBST1TUTE SHEET (RULE 26)
CA 022l8808 l997-ll-07
W096/35798 PCT~96/0019S
19
Canciclo~ir concentration (mM)
S Cell ~ool 0 ~M 5 ~M 10 ~M 25 ~M
pPTk+NeopA/IF100 + 11.1 3.4 + 1.0 2.6 + 2.0 2.8 + 2.1
pPTk-NeopA/OF 100 + 15.7 4.2 + 2.1 4.5 + 2.5 5.1 + 2.9
pPTkpolioNeo 100 + 13.8 2.3 + 2.0 1.8 + 1.4 2.3 + 1.6
pPTkemcvNeo 100 + 15.20.2 + 0.4 0.2 + 0.2 0
pPTkpA 100 + 19.60.2 + 0.5 1.0 + 2.0 0
pPNeopA 100 + 12.396 + 3.0 102 + 25 87 f 17
Table 2: ~ of surviving cells grown in medium with
different concentrations GCV relative to cells grown
in normal medium
Cell kill is almost complete at 5 ~M GCV, a concentration
that can be reached in the blood of a patient without any
effect on normal cells. A Simi 1 ~r e~perimental set up was used
to test lower concentrations of GCV ranging from 0.125 to
1.0 ~M. As is shown in table 3 below, pools of cells
containing an IRES sequence to express the neor gene in a
bicistronic transcription unit have a slightlv lower LCso
(GCV concentration at which 50% of the cells are killed) as
~5 compared to the cell pools generated with the bicistronic
constructs containing an intercistronic linker. In addition,
complete cell kill is reached at slightly lower concentrations
of GCV in case of the IRES containing constructs.
Ganc,clovir conce, IL, dLion (~lM)
Cell pool o 0.125 025 0.5 0.75 1.0
pPTl<+NeapAJlF 100+ 7.190.8+ 6.948.8+ 1.717.8+ 2.313.1 + 2.4 9.7+ 5.3
pPTk-NeOpA/OF 100+ 6.380.5+10.945.6+ 8.818.7+ 2.111.4_ 3.7 9.9+1.4
pPr~ " N~:o100+ 8262.9+ 3.527.4 1.38.5+ 0.4 3.1 + 0.7 1.4_1.3
40pPTl~e~100 + 11.5672 + 7.034.1 + 2.2 9.7 _ 1.02.6 + 1.9 0.3 + 0.3
pPlkpA 100 + 10.844.9 + 10.2 21.4 t 1.8 8.1 + 2.7 8.7 ~ 3.6 6.6 + 2.3
SUBSTITUTE SHEET (RULE 26)
CA 02218808 1997-11-07
W096/35798 PCT~96100195
Table 3: ~ of surviving cells grown in medium with
different concentrations GCV relative to cells grown
in normal medium
Mixing of HSV-t~+ cells with HSV-tk- cells showed that,
at the cell densities used in these experiments, there is no
influence of the so called 'bystander effect', proving that
cell kill is due to endogenous expression of HSV-tk and not
due to transfer of the toxic substance to t~- cells (results
not shown).
Above experiments demonstrate that intercistronic linkers
can be used in a bicistronic transcription unit in stead of
IRES sequences without losing functional expression of either
gene in the bicistron. Synthetic intercistronic linkers are
preferred over virus-derived IRES sequences because they are
shorter in size and do not form strong secondary structures in
the RNA.
Example 2: Retroviral vectors useful for transfer of the human
multidrug resistance-l gene
Multidrug resistance may result from svnthesis of a
multidrug transporter (P-glycoprotein) encoded bv the
'multidrug resistance (~DRl ) ' gene. It is possible to confer a
multidrug-resistant phenotype to drug-sensitive cells by
~S transfection and subsequent expression of the MDRl aene. An
attractive approach therefore would be to introduce the MDRl
gene into haemopoietic stem cells (HSC), with the objective to
protect patients from drug-induced mvelotoxicity.
Alternatively, the MDRl vector could be used to introduce yet
,0 another gene of interest (e.g. Glucocerebrosidase gene or HIV
inhibiting genes/sequences) allowing selection in vivo, using
cytotoxic drugs efficientLy pumped out of the cell by P-
glycoprotein. Successful circumvention of myelosuppression bv
transduction of the MDRl gene in bone marrow cells is
dependent on an efficient gene transfer system. Currentl~,
retrovirus-mediated gene transfer is the only technique that
allows efficient and stable gene transfer into HSC.
An optimal retroviral construct for introduction of the
human MDRl gene in haemopoietic cells has besides the
SUBSTITUTE 5~EET ~RULE 26)
CA 02218808 1997-11-07
w096/35798 PCT~n96/00195
~1
properties of the basic pLec~Mo construct the following
additional properties:
- only the full length coding DNA sequence of the MDRl gene is
inserted to avoid negative influence of non-coding flanking
nucleotide sequences on gene expression and to allow mA~i~
space for additional gene(s) of interest.
- The wild-type MDRl is utilised instead of mutant forms with
altered substrate specificity (Choi et al., 1988) as mutant
MDRl P-glycoproteins may result in vivo in an immunogenic
response to transduced cells.
Co~struction of DIGm~rl-G
To facilitate cloning of the ~DRl gene, the high-copy
number plasmid backbone in pLTKkoz was replaced by a low-copy
number plasmid by subcloning of the NheI fragment of pLTRkoz
in pZIP~Mo+PyFlOl(N-). The resulting construct was named LTK-
~Mo. The wild-type human MDRl cDNA (van der Bliek et al.,
1988) was inserted in LTK-~Mo by ligation of 3 fragments:
Fragment 1: NcoI-BamHI fragment or LTK-~Mo.
Fragment 2: NcoI-EcoRI MDRl fragment.
This 1178 bp fragment was generated by PCR using Pfu DNA
polymerase. Two primers were used: 'mdr5'(thio)' (5'-
CCTCTAGACCATGGATCTTGAAGGGGACCGCAA
TGGAGGA-3') spanning the start-codon of MDRl, in which a
~5 cytosine was placed before the ATG star~-codon, thereby
creating a NcoI site at this position. The second primer
(4728A: 5'-CCAACCAGGGCCACCGTCTGCCC.~-3') is positioned 3' of
the EcoRI site in MDRl. This fragment was first digested with
~coRI and subsequently partially digested with NcoI. The 1.2
kbp fragment was isolated from an agarose gel.
Fragment 3: EcoRI-BamHI 3' MDRl fragment.
This fragment was isolated ~rom subclone Kl. Kl resulted
from a previous 4-part ligation to generate another retroviral
vector expressing the MDRl gene wnich was performed as
~ 3S follows. The wild-type MDRl gene, inserted in pJ3 ~ DRl.l is
described in S~hinkel et al., 1991. A 3.8 kbp DraI-HhaI
fragment was isolated from this construct. This fragment was
ligated to two double strand linkers: a 5'-linker (5'-
CTCTGAGCTCCcATGGATcTTGAAGGGGAccGcAATGc~cc~crAAAG
SUBSTITUTE SHEET (RULE ~6)
CA 02218808 1997-11-07
W096/3s798 PCT~96/00195
TAAATCTC-3') which was cut with NcoI and DraI and isolated
from a agarose gel and a 3'-linker (5'-
CAGGCTGGAACAAAGCGCCAGTGAGGATCCTCTCT-3') which was cut with
HhaI and Bam~I and also isolated from a agarose gel (bold
sequence indicates fragment to be inserted). After ligation,
the product was recut with BamHI and ligated to a NcoI-BamHI
retroviral fragment. After transformation in competent DH5a
cells, a clone was isolated, K1, which had a correctly
inserted 3' HhaI-BamHI MDRl fragment as confirmed by sequence
analysis. However, the 5' oligonucleotide was not inserted.
Therefore, K1 has a Bam~I-site directly positioned after the
TGA stop-codon which could be used to isolate the EcoRI-BamHI
MDRl fragment.
Ligation of the three fragments 1,2 and 3 resulted in
pIGmdrl-G and was used to generate ~Rl producer cell lines.
Generation of the l~.m~rl-5 retroviral producer cell line:
IGvpO10
IGmdrl-G was transfected in the PA317 packaging cell line
O (obtained from the American Type Culture Collection: ATCC No.
CRL 9078). PA317 cells were selected in ~AT medium to select
for cells that retain the packaging function. The cells were
grown in HT medium for 4 days to dilute residual amethopterin.
6x105 cells were transfected with 6~g IGmdrl-G DNA using
~5 Lipofect~MTN~ as reagent. The following day cells were
trypsinized and l.Ox106 cells were seeded per 75 cm2 dish. The
next day 70 nM of vincristine was added. Medium with
vincristine was refreshed every 3 days. Two weeks after
trypsinization vincristine resistant colonies were
trypsinized, pooled, and further cultured. The resulting cell
line was called IGvpO10.
Tr~ncduction and expression studies with IGm~rl-G
a) Test for the presence of helpervirus
IGvpO10 was proven to be free of replication competent
retroviruses (RCR). This was det~rmined by co-cultivation of
5X106 cells for 5 passages with Mus dunni cells which permits
amplification of RCR by the feline (PG-4) S+L- focus assay.
SVBSTIT~JTE 5HEET (RULE 26)
CA 022l8808 l997-ll-07
W096l35798 PCT~n96/00195
~3
b) Expression of IGmdrl-G in a human drug-sensitive A2780
cell line.
To simulate bulk transduction conditions that are
empLoyed for haemopoeitic target cells we tested supernatant
harvested from the IGvpO10 producer cell line on a P-
~glycoprotein negative human ovarian tumour cell line (A2780).After a two-hour transduction and a 48 hour culture period, it
was possible to accurately determine the proportion of
transduced cells by their ability to exclude the fluorescent
dye Rh-123 which is an efficient substrate of the P-
glycoprotein. Under these conditions the IGvpO10 supernatant
yielded 29.37 ~ Rh-123dUll cells (corrected for background
activity, Figure 7).
c) Transduction of mobilised human peripheral blood
progenitor cells (PBPC).
CD34T selected PBPC were transduced over 96 hours with
IGvpO10 supernatant at a ceLl concentration of 1 x 106/ml in
the presence of human inte_leukin-3. IGvpO10 supernatant was
refreshed every 24 hours. 2rotamine sulphate (4 ~g/ml) was
added with every supernatan~ change. MDRl -transduced and mock-
transduced PBPC were plate~ in duplicate at 5 x 103/ml in 1 ml
methylcellulose medium in _he presence of IL-3 and GM-CSF.
Screening for MDRl overexpressing progenitor cells was
performed with vincristine which is an efficient substrate for
~5 the P-glycoprotein. Freshl-.- thawed vincristine was added to
the dishes in increasing amounts. At all concentrations, cells
were plated in duplicate. Colony forming units (CFU-C) were
scored after 14 days. M~Rl-transduced and mock-transduced were
compared by dividing the number of colonies in the dishes with
vincristine by the number of colonies in the dishes without
vincristine. Figure 8 shows the result (error bars give the
minimum and m~imllm value _btained). At a dose of 20 nM, 47%
of the IGmdrl-G colonies survived compared to 3% of the mock
infected cells. A dose of 30 nM killed all colonies in the
control group, while still 9% of the MDRl- infected colonies
survived This experiment not only clearlv demonstrates that
IGmdrl-G efficiently infects haemopoietic cells, but also has
a high expression level of the inserted gene in haemopoietic
precursor cells
SUBSTITUTE 5HEET (RULE ;26)
CA 022l8808 l997-ll-07
W096/35798 PCT~n96/00195
24
d) Transduction of normal human bone marrow cells.
CD34+ selected normal human bone marrow cells were
transduced as described for PBPC. After transduction, cells
were seeded for CFU-C formation in the presence of increasing
amounts of vincristine. Individual CFU-Cs were picked and DNA
isolated from the colonies was subjected to a provirus-
specific PCR. Seven independent experiments demonstrated that
8+9 percent of the CFU-C was transduced with the IGmdrl-G
retrovirus. From one e~periment, also vincristine resistant
colonies were analyzed. This experiment showed that the
percentage PCR+ CFU-C increased from 30% without vincristine
to 44% (20 nM), 71% (30 nM) and 100% at 40 nM drug. This
experiment clearly demonstrates that in vitro selection of
transduced hemopoietic progenitor cells at increasing doses or
cytostatic drug actually occurs.
In accordance with results presented bv Soren~ino et al .
(1993), we detected aberrant splicing of the RNA derived from
the MDRl cDNA inserted in our vectors. An (obvious) impL~vc...ent
~0 ~ould be the modification of cryptic splice sites without
altering amino acid coding sequences.
E~ample 3: Gene therapy ~or AIDS/~ ViYo selection of transduced
HSC
~5 Infection of CD4- T-cells bv the human immunodericiency
virus (HIV) is the first and causative event in the development
of AIDS. As a member of the large family of retroviridae HIV has
an RNA genome and a life-cycle like other retroviruses. Spread
of the virus is depending on infection, reverse transcription,
integration, transcription and packaging of the viral genome.
Gene therapy strategies have been developed that interrere with
the life c~cle of the retrovirus using so called genetic
antivirals like e.g. intracellular antibodies, ribozymes,
antisense molecules or decoys (reviewed in Gilboa and Smith,
1994). These molecules have to be delivered to the cells
primarily susceptible to infection by HIV i.e. CD4+ T-cells and
monocytes/macrophages in the blood. Protection of these cells to
HIV infection may limit or even prevent the spread of the virus
and limit the pathogenic effect of the virus onto the immune
SIJBSTITUTE 5~EET (RULE 26)
CA 022l8808 l997-ll-07
W096/35798 PCT~ng6/0019S
~5 ~.
system. Ideally, haemopoietic stem cells are the targets for
such protective therapy as they will provide the patient with a
continuous source of protected T-cells. However, the stable
infection of HSC may not be very efficient and following
transplantation of the transduced cells a multitude of non-
transduced endogenous stem cells will continue to generate
mature cells resulting in many unprotected cells in the
peripheral blood and thus facilitating replication of HIV.
Therefore, the constructs that we describe here and that are
designed to express genetic antivirals in HSC and descendents
thereof, are all based on the pIGmdrl-G retroviral construct
(see example 2). In this construct the human MD~-1 gene allows
for selection of transduced stem cells ln vivo.
Construct~on of DIGmdrl-G/HIVasTAR and pIGmdr1-G/HIVasTARaaa and
S i m ilar vectors
As an example of the construction of recombinant retroviral
vectors specially designed to deliver anti-HIV-l molecules to
the HSC and their desc~n~Ants, we describe recombinan~
retroviral constructs generating antisense RNAs direc~ed to the
5' end of HIV-l. The use of the polymerase III-dependent
adenoviral VAl promoter (Fowlkes and Shenk, 1980) ensures high
levels of expression of short inserted sequences.
Firstly, a subclone from pIGmdrl-G -~as generated by
?5 digestion with BamHI and religation or vector sequences. his
clone, pIGmdrARAm~T was used to introduce the Adenoviral VAl
gene and promoter sequences that were obtained by amplification
of Ad5 sequences with the primers: 5'-CCTGCTAGCTCIAGACCGTGCAAAA-
3' and 5'-AAAGCTAGCAAAAAAGCGGCCGCGGGGCTCGAACCCCGGTCGTCC-3'.
Digesllon of the pcr product with NheI allowed for cloning into
either the unique AvrII or NheI site of pIGmdrAR~m~T. Clones
were selected that contained the VAl promoter in either
orientation. The unique NotI site that was introduced into the
VAl gene during the pcr amplification, then served as an
35 insertion site for HIV-l sequences. ~hese were obtained by
amplification of HIV-l sequences in the pBRU2 vector (XbaI/ClaI
fragment from pLAI/pBru from B.Klaver, AMC A' dam) using the pcr
primers: 5' TAR 5'-AATCGCGGCCGC~l~L~l~lGGTTAGAC-3' with 3' TAR
5'-AATCGCGGCCGCGGTTCCCTAGCTAGCC-3' to amplify the TAR loop from
SUBSTITUTE S~EET (RULE 26)
CA 02218808 1997-11-07
W096/3~798 PCT~L96/00195
_6
+l to+57 (pIGmdrl-G/HIV-TAR) and with 3' gag 5'-
AATCGCGGCCG~l~l~GCACCCAT-3' to amplify the 5' end up to the gag
start codon from +l to +348 (pIGmdrl-G/HIV-gag). The pcr
fragments were digested with NotI and cloned into the
S pIGmdr~3amHI/VA constructs. Depending on the orientation of
inserted HIV sequences with respect to the internal WAl
promoter, the constructs pIGmdrl-G/HIVasTAR, /HIVsTAR, pIGmdrl-
G/HIVasTARgag or /HIVsTARgag were generated (where s=sense and
as=antisense).
In addition, all constructs were derived in two
orientations: one in which the VAl promoter is drivlng
transcription in the same direction as the viral LTR and one in
the reversed direction.
Esample 4: Retroviral vectors for gene therapy of G~llrh~r
disease and 7 n ~ivo marking studies
Retroviral constructs for the treatment of Gaucher
disease should be based on vectors working favourablv in the
haematopoietic system particular following stem cell gene
~0 tranfer. The construction of IGGC therefor carries the
glucocerebrosidase (GC) sequence in the retroviral back bone
as disclosed in this invention.
Moreover, the construction of IG-GC retroviral vectors
that differ in the length of the inserted Glucocerebrosidase
'5 (GC) cDNA (described below) was undertaken in order to perform
in vivo gene marking studies. The difference in length of the
inserted GC cDNA's allows for the discrimination between
multiple retroviruses after ex vivo infection and reinfusion
of the infected graft into the same Ani~l or human. To
iO optimize gene delivery to CD34- primitive progenitor cells
these vectors can be used to study transduction efficiency
differences between viruses produced bv different packaging
cell lines. In addition, different transduction protocols i.e.
different growth factors and the role of virus titers can be
i5 studied. C~mpAred to single vector gene marking studies, gene
marking with multiple, disting~ h~hle vectors enables one to
rapidlv assess the role of crucial parameters in determining
transduction efficiency of CD34+ primitive progenitor cells.
Furthermore, an additional advantage of the use of therapeutic
SUBSTITUTE SHEET (RULE ~6)
CA 022l8808 l997-ll-07
W096/35798 PCTQ~L96/00195
27
cDNA's, such as hGC, in gene marking studies instead of cDNA's
encoding foreign proteins, for instance Neor (Brenner et al.,
1993), is the absence of unwanted host immune responses
against the expressed foreign protein.
Construction of ~etroviral Glucocerebrosidase vectors, IG-GC
The complete cDNA sequence (1888 bp) of the human
placental glucocerebrosidase was digested to completion by
XhoI and separated from the 7549 bp pGB12S backbone (Genzyme
cooperation) by agarose gel electrophoresis. The DNA fragment
was electroeluted from the agarose and purified by
phenoL/chloroform/ isoamylalcohol extraction.
The pLec plasmid (5~73 bp) was linearized by XhoI
digestion. The XhoI digested DNA was treated with calf
intestinal phosphatase (CIAP) and subjected to agarose gel
electrophoresis. The linearized DNA fragment was excised and
purified. The isolated XhoI hGC cDNA fragment of 1888 bp was
ligated to the dephosphorylated 5773 kb XhoI DNA fragment of
pLec using T4-DNA ligase. The resulting 7661 bp retroviral
vector is designated IG-GC-1 (Figure 9). To construct
retroviral vector IG-GC-2, the IG-GC-l vector was digested
~~ith NheI. The resulting 3431 bp NheI DNA fragment was
ligated to the linearized and dephosphorylated 4375 kb NheI
DNA fragment of pSK/Zip~Mo+PyF101 using T4-DNA ligase. The
resulting 7806 bp retroviral DNA construct is designated IG-
GC-2 (Figure 10).
In addition to IG-GC-l and IG-GC-2, two variants were
constructed designated IG-GC-3 and IG-GC-4. These variants are
identical to IG-GC-1 and IG-GC-2 respectively except for the
3'-untranslated region of the hGC coding sequence. From this
region a 160 bp fragment (from nt. 1728 to nt. 1888) was
deleted using PCR. Construction or IG-GC-3 was done as shown
in figure 11. Briefl~, two oligonucleotides were synthesized,
GCo3 with sequence 5'-CGGGATCCTAGAGGGGAAAGTGAG-3' and GCo4
with sequence 5'-CAGCCCAl~ll~lACCAC-3'. GCo3 contains a BamHI
restriction site. These two oligonucleotides were used to
amplify a 420 bp DNA fragment using IG-GC-2 plasmid DNA as
template. The 420 bp PC~ fragment was digested with BamHI and
the 220 bp PCR fragment was isolated and ligated to IG-GC-1
SUBSTITUTE SHEET (RULE 26)
CA 02218808 1997-11-07
W096/35798 PCT~n96/00195
~8
DNA that was linearized with Bam~I (Figure 11). The resulting
vector, designated IG-GC-3, now contains a human GC cDNA
which lacks 160 bp in the 3' noncoding region (Figure 12).
To construct IG-GC-4, IG-GC-3 was digested with NheI and a
3231 bp DNA fragment was isolated. This fragment, which
includes the hGC cDNA se~uence was cloned into the NheI
linearized and dephosphorylated psK/zip~Mo+pyFlol 4375 kb DNA
fragment (Figure 13). The resulting 7606 bp retroviral vector
is designated IG-GC-4.
G~nerat;o~ of IG-GC-2 a~d IG-GC-4 rec~mhinant retrovirus
~roduce_ c~lls
IG-GC-2 and IG-GC-4 plasmid DNAs were introduced into
ecotropic retroviral packaging cell lines GP I E86 (Markowitz
et al ., 1988) and ~-CRE (Danos et al ., 1988) respectively.
Selection of transfected cells was achieved by cotransfection
of expression plasmids pSV2neo (Southern and Berg, 1982) with
ICG-GC-2 and pPGKneo (R. Vogels, see e~ample 1) with IG-GC4
at a ratio of 1 : 10. G418 resistant cell pools GP2b and ~4c
were generated. Ecotropic IG-GC-2 and IG-GC-4 virus was
produced by growing confluent layers of ecotropic producer
cells in fresh medium at 32~C for 24 hours. Virus containing
supernatants were collected, passed through a 0.45 ~m filter,
aliquoted and immediately frozen in liquid nitrogen followed
bv storage at -80 ~C.
To test whether the G418 resistan~ eco~ropic cell pools
also express the human GC protein, cell lysates were made and
the GC enzvme activity level was measured using an artificial
GC substrate (fluorescence: 4-Mu-~-glucoside or colorimetric:
PNP-~-glucoside). Cell lysates were made from untransfected GP
30 - E86/~-CRE cells, transfected GP - 86/~-CRE cells, and 3T3
mouse fibroblast cells infected with the GP2b/~4c virus
supernatant. The results of these measurements showed that the
transfected ecotropic GP - E86 /~-CRE cells and the infected
3T3 cells had 1.8-2.0 times elevated GC activity levels
compared to the non-transfected packaging cells (Figure 14A).
A Western blot of cell pools A, B and C (3 independent IG-GC-2
transfections) using the human GC specific monoclonal antibod~
8E4 (Aerts et al ., 1985) showed that the 59 kDa hGC protein is
expressed in the GP - E86 packaging ceLl line (Figure 14B).
SUE~5TITUTE SHEET (RULE Z6)
CA 022l8808 l997-ll-07
W096/35798 PCT~96/00195
~9
To show that the increased GC enzyme activity levels
after infection of cells with ecotropic virus is caused by
- expression of the human GC protein, ecotropic virus obtained
from the GP2b pool was used to multiply infect amphotropic
S PA317 cells. Cell lysates of the infected PA317 cells were
made and the GC activity level was measured. It was shown
that the infected cell pool had approximately 1.8-2.0 times
elevated GC activity levels compared to the non-infected
packaging cells (data not shown)~ A Western blot of the cell
lysates of these infected PA317 cell pools (Bl and B2), using
the human GC specific antibody 8E4 (Aerts et al ., 1985),
clearly shows that ecotropic virus, carrying the IG-GC-2
vector, transfers the human GC protein at significant levels
(Figure 15).
lS Using the same ecotropic IG-GC2/IG-GC4 virus supernatant,
Gibbon ape leukaemia virus (GALV) packaging cells (PG13) were
infected. In the generated PG pools hGC activity was measured
as described above and again proved to be 2 to 3 times
elevated c~mr~red to parental PG13 cells (data not shown).
Next, producer clones were isolated from these pools. In
order to achieve this two rounds of limiting dilution
(<1 cell/well) were performed in 96-well microtiter plates.
Virus production from these clones was initially tested by
measuring hGC activity in NIH/3T3 cells (PA317 derived) or
~5 Rat-2 fibroblasts (PG13 derived) after incubation with the
cell culture supernatant. Clones giving the highest increase
in hGC activity were selected and designated
PA2 (PA3 17/IG-GC2), PA4 (PA3 17/IG-GC4), PG2 (PG13/IG-GC2),
and PG4 (PG13/IG-GC4). These cell lines are deposited at the
ECACC under Nos: PG4 - 96050256, PG2 - 9605025/, PA4 -
96050258 and PA2 - 96050259 according to the Budapest treaty.
~fficac~ testina of the isolated retrovirus ~roducer cell
clo~es
To characterise endogenous hGC expression lysates were
prepared from the producer cells. In addition, lysates were
prepared from Gaucher type II fibroblasts infected with PA2,
PA4, PG2, and PG4 virus supernatants to c~mr~e virus titer.
A Western analysis or these protein lysates indicates a
SUBSTITUTE 5HEET (RULE 26)
=
CA 022l8808 l997-ll-07
W096/35798 PCT~n96/00195
~0
correlation between endogenous hGC expression in the producer
clones and virus titer i.e. the producers with high endogenous
hGC levels yield high titers (Fig. 16).
The virus titer of the PA2 producer was det~mined by
incubating 105 NIH/3T3 cells (6-well plates) with 1 ml of PA2
virus supernatant over a 48 hour period. Subsequently, the
infected NIH/3T3 cells were sub~ected to one round of limiting
dilution (>l cell/well) and 50 individual clones were expanded
in order to measure hGC activity and to isolate genomic DNA
for copy number determ~n~tion. All of these 50 NIH/3T3 clones
expressed hGC. Southern analysis revealed that in each
transduced individual NIH/3T3 clone at least three separate
integrations took place. From these results it was concluded
that the titer of the PA2 producer is at least 3 x 105
15 functional virus particles/ml. (Fig. 17).
To investigate whether the biochemical phenotype in
Gaucher type I and type II primar~ fibroblasts could be
reversed, these cells and two normal human primary fibroblast
cells were incubated with virus supernatant of the PA2, PA4,
~0 PG2, and PG4 producer cell clones (Fig 18). The data show that
a single infection with either PA2 or PG2 virus supernatant is
sufficient to augment hGC activity levels in the Gaucher type
I and II infected cell pools to those comparable to uninfected
normal human fibroblasts. ~hus, correction of the biochemical
~5 Gaucher phenot~pe is accomplished by infection with these
recombinant viruses carrying the IG-GC constructs. The data
also show that a single incubation with either PA4 or PG4
supernatant on Gaucher t~pe I and II fibroblasts increases hGC
activity levels to 50-70% compared to normal.
~0
Tra~sduction of hl-m~n C334- cells
CD34+ cells were isolated from total bone marrow
harvested from a Gaucher tvpe I patient. These CD34+ cells
were seeded at a concentration of 105 cells in 24-well plates
35 in 400 ml virus supernatant supplemented with 4 mg/ml
protamine sulphate, IL-3, and pen/strep. Daily, for four
executive days, the virus supernatant was rerreshed. After
this transduction period 2.5 x 104 cells were seeded in a
liquid culture assay (medium containing IL-3, IL-6, SCF,
SUBSTITUTE 5HEE~ (RULE 26)
CA 02218808 1997-11-07
W096/3S798 PCT~ng6/00195
31
GM-CSF, and G-CSF). After a 10 day incubation at 37~C/10% C02
the cells were harvested and counted. The number of cells
obtained after this 10 day period normally was between
5 x 105-1 x 106 showing a proliferative capacity factor of 20
to 40 times. Nine-tenth of the cells was used to measure
elevation of hGC activity. One-tenth of the cells was pelleted
and lysed for PCR. Figure 19 shows the elevation of hGC
activity in the differentiated Gaucher type I cells derived
from the liquid culture after transduction with either
IG-GC2/IG-GC4 or MDR virus. PCR and subsequent Southern
analysis of the cells derived from the liquid culture shows
that the provirus is present in the IG-GC2/IG-GC4 transduced
cells (fig. 20).
Besides the liquid culture, CD34+ cells were also seeded
lS for a colony forming unit (CFU) assay at a concentration of
5xllO3/ml in semi-solid methylcellulose. After a 14 day period
in an incubator set at 37~C/10% C02 approx. 100 individual
colonies were picked to investigate the transduction
efficiency of the virus supernatants on these primitive human
?0 cells. By means of PCR it could be shown that with either PA2
or PG2 recombinant --irus approx. 40-50~ of all CF~s contained
the provirus. Infection of this cell type with PA4 proved to
be less efficient with approximately 10~ infected CFUs (data
not shown).
From these data it can be concluded that the IG-derived
viruses are potent gene delivery vehicles capable of
correcting the biochemical Gaucher phenotype in primary
fibroblasts and CD34~ hemopoietic Gaucher cells.
~onstruction of ret-ovirAl Glucocerehrosidase-dihvdrofolAte
reductAse ~lIA 1 vectors
Low transduction efficiency of the human hemopoietic stem
cells poses a serious limitation with respect to successful
gene therapy for diseases such as Gaucher disease. A potential
strategy to circumvent this problem is to use retroviral
vectors carrying a therapeutic cDNA and a ~;n~nt selectable
marker. Such retroviral vectors make it possible to select
transduced cells in vivo. For this reason we constructed
bicistronic retroviral constructs carrying the hGC cDNA and
SUBSTITUTE SHEET (RULE 26)
CA 02218808 1997-11-07
W096/3S798 PCT~96/00195
the methotrexate resistant cDNA of human dihydrofolate
reductase (hDHFR).
The human wild type DHFR was amplified with Pwo enzyme
from single stranded cDNA synthesized from mRNA of human
liver. The 5'oligonucleotides DHFRl
(5~cccaagcttcccgggctgcagcgccaccatggttggttcgctaaactg-3l) and
the 3' oligonucleotide DHFR2
(5~ccatcgatctcgagtcattcttctcatatacttcaaa-3~) yielded the
expected DNA fragment of 550 bp. To obtain a methotrexate
resistant version of hDHFR a point mutation, Phe32Ser, was
introduced in the wild type hDHFR cDNA. Therefore, a 120 bp
5' part of hDHFR was amplified with oligonucleotides DHFRl and
DHFR4 (5'-gaaatatctagattcattcctg-3') ~hich carries the desired
mutation, full length hDHFR PCR product as template, and Pwo
enzyme. In an additional PCR reaction a 130 bp 3' part of
hDHFR was amplified with DHFR2 and DHFR3
(5'-caggaatgaatctagatatttc-3') which is the re~rerse complement
of DHFR4. Both 5'-part and 3'-part were denatured and annealed
to each other and subsequently amplified with DHFR1 and 2 to
~0 reassemble the 550 bp full length hDHFR (Fig.21). The presence
of the desired mutation was analyzed by digestion with EcoRI
which cuts in the wild type hDHFR but not in the Phe32Ser
mutant.
To delete the entire 3'-noncoding domain or hGC, ~G-GC2
plasmid was utilized for the amplification of a 300 bp hGC DNA
fragment with oligonucleotides GColl
(5~-gatcgagggatgcagtac-3l) and oligonucleotide GCol4
(5'-tstggcgtcgccagtgaggatcctctagaagcttggg-3'). Oligonucleotide
GCol4 contains the stopcodon of hGC. Downstream of
oligonucleotide GColl a uni~ue SalI site is present and
oligonucleotide GCol4 contains a HindIII site. For cloning
purposes, the wildtype and Phe32Ser mutant hDHFR PCR fragments
were digested with HindIII (present in DHFRl) and ClaI
(present in DHFR2). The 300 bp 3'-hGC PCR fragment was
~5 diges~ed with SalI and HindIII. These t~o DNA fragments were
ligated in a three point ligation reaction into a SalI, ClaI
digested IG-GCl construct (Fig. 22). The resulting bicistronic
vectors were coded IG-GCS (wildtype hDHFR) and IG-GC6
(phe32ser hDHFR). In these ~rectors the hGC coding region is
SUBSTITUTE SHEET (RULE 26)
-
CA 022l8808 l997-ll-07
W096/35798 PCT~96/00195
i3
separated from the hDHFR coding region by a 36 bp
intercistronic linker enabling translation of both proteins
~ from one single mRNA. To obtain bicistronic retroviral vectors
containing the mutant polyoma enhancer PyF101 in the 3'-LTR,
the NheI fragments were isolated and cloned into
pSK/ZipDMo+PyF101 (IG-GC7 and IG-GC8 respectivily). These
bicistronic retroviral vectors can be transfected into
retroviral packaging cell lines to generate viruses that upon
infection of target cells render these cells resistant to
methotrexate, a potent cytotoxic drug which inhibits DNA
synthesis by depleting the pool of pyrimidines.
~Y~ _le 5: I~Llodhction of ~ocus Control Region se~uences of
the human CD2 gene in the rec~h;n~nt retroviral vector
pLgAL(~Mo~PyF101).
For this example the re~roviral vector construct
pLgAL(AMo+PyF101) (Van Beusechem et al ., 1990) was used,
wherein A represents the human cDNA gene encoding adenosine
in~e (hADA), which is further referred to as "the
~0 vector". Additionall~, the Locus Control Region (LCR) sequence
from the 3'regionof the human CD2 gene (Lang et al ., 1988) was
used, which is further referred to as "CD2-LCR". In the CD2-
LCR a 2076 nt HindIII fragment (nt 2-2077) has been identified
which in transgenic mice e~erts all the characteristic
~5 features of the CD2-LCR on the CD2 promotor as well as on
heterologeous promoters (Lang et al., 1988, Lang et al.,
1991). Within this fragmen~ lies a 880 nt AflIII fragment (nt
433-1314), of which it has been shown in human T-cell lines in
vit~o that it act as a CD2-LCR (Lake et al ., 1990). In the
.0 vector the HindIII CD2-LCR fragment, further referred to as
"L2", or the AflIII CD2-LCR fragment, further referred to as
"L0.8", was cloned. Thereto the L2 and L0.8 fraaments were
isolated from the construct GSE1502 (D. Kiousis, MRC) and
provided with a blunt end with Klenow-polymerase. The vector
~5 was digested with ClaI (nt 767S of Mo-MuLV, in en~) or NheI
(nt 7846 of Mo-MuLV, in the 3'LTR) and also provided with a
blunt end. The fragments L2 or L0.8 were cloned into the ClaI
site (resulting constructs are further referred to as "CL2" or
"CL0.8"), or into the NheI site (resulting constructs are
SUBSTITUTE S~ EET (RULE 26)
-
CA 02218808 1997-11-07
PCT~96/00195
Wo96/35798
further referred to as "NL2" or "NL0.8"). They were cloned in
the normal 5'-->3' orientation of the CD2-LCR (forward,
further referred to as "F"~ as well as in the 3'-->5'
orientation (reverser further referred to as "R"). In this way
8 different novel retroviral constructs were made, referred to
as "CL2F", "CL2R", "CL0.8F", "CL0.8R", "NL2F", "NL2R",
"NL0.8F", and "NL0.8R" (Figure 2).
The 8 new constructs were packaged into recombinant
retroviruses. Thereto 20 ~g DNA of the constructs was
transfected into the ecotropic packaging cell line GP+E-86
(Markowitz et al., 1988), using the method as described by
Chen and Okayama (1977). Prior to the transfection the GPf~-86
cells were cultured in a medium conta; n ing 15 ~g/ml
hypoxanthine, 250 ~g/ml x~nhine, and 25 ~g/ml mycophenolic
acid, in order to select for retaining the DNA sequences which
are responsible for the production or viral proteins.
Transfectants producing a functional hADA enzym were isolated
through a selective culture in medium containing ~ ~M
xy~ofuranosyl-adenine (Xyl-A) and l0 nM deoxycoformycin (dCF)
(Kaufman et al ., l986). Culture supernatant of Xyl-A/dCF-
resistant transfectants was, after filtration through a filter
with a pore size of 0.45 ~m, used to transduce the amphotropic
packaging cell line GP+envAm12 (Markowitz et al, 1988) with
the ecotropic recombinant retroviruses present in that culture
'5 supernatant. The amphotropic packaging cells were selected for
ret~in;ng the DNA sequences encoding viral proteins prior to
use (2S described for GP'E-86 cells, with the addition of
200~g/ml hygromycine B) and preinc~lbated with 4 ~g/ml
polybrene to promote retrovirus transduction. GPfenv Aml2
cells producing a functional hADA enzym were isolated through
Xyl-A/dCF-selection as described above. Individual hADA-
positive GP+envAml2 clones were isolated and e~panded,
Before the hADA-positive GP+envAml2 clones were characterized
for integrity of the integrated recombinant retroviruses and
the production of amphotropic recombinant retroviruses it was
first verified that all clones were derived from individual
transductions with recombinant retroviruses. Hereto
chromosomal DNA from the clones was digested with ~glIII
(whicn cuts once in the construct, -~ithin the hADA gene) and
SUE3STtTUTE 5HFEl- (RULE 26)
-
CA 022l8808 l997-ll-07
W096/35798 PCT~L96tO019S
hybridised with an hADA probe (551 nt BglIII-SstI fragment of
pAMGl (Valerio et al ., 1985)) in a Southern anal~rsis. Herewith
3'junction fragments with a length which depends on the
insertion site in the genome are identified. All clones were
5 shown to have one single insertion of one of the recombinant
retroviruses. Clones having junction fragments of the same
length were excluded from further analysis. Thereafter all
r~m~i n i ng clones were tested for the correct structure of the
integrated recombinant retroviral construct through Southern
analysis. Hereto the chromosomal DNA of the clones was
digested with KpnI (cuts once in both retroviral LTRs,
resulting in fragments which hybridise with de hADA probe of
3.5 kb for the vector and 5.5 kb and 4.3 kb for constructs
having L2 and LO.8 insertions, respectively). The result of
this analysis is shown in Table 2. When the L2 fragment was
cloned in the ClaI site the fragment length in all analysed
clones was correc~, independent of the orientation of the
in$ertisn. Cloning in the NheI site, however, resulted in
instability of the resulting constructs. This result was most
~0 serious after insertion in the forward orientation. Insertion
of the LO.8 fragment resulted in stable recombinant retrovirus
constructs in most cases, except after the cloning into the
ClaI site in the reverse orientation. All clones packaged
recombinant retroviruses; even the clones which harboured
25 damaged retrovirus insertions. The different insertions had no
significant effect on the titers with which viruses were
produced (Table 4). A number of clones harbouring truncated
retrovirus insertions was subjected to a more thorough
analysis. Three independent NL2R clones were all shown to have
3~ a 2 kb deletion in a fragment covering the area from the 3'end
of the hADA gene until the 3'end of the L2R insertion. In 7
analysed NL2F clones the retrovirus insertions were s-hown to
have deletions varying from 200 bp to 2 kb in size. In all 7
clones the deletion comprised (a part of) the L F fragment, in
2 of these clones the deletion extended into the vector (3~ of
hADA and 5' of L2F in the 3'LTR). The 5 damaged CLO.8 clones
had deletions of different lengths in 4 cases and an insertion
in one case. These results show that the L2 fra~ment comprises
sequences which influence the stability of the vector
SUBSTITUTE SHEET (RULE 26)
CA 022l8808 l997-ll-07
W096/35798 PCTA~L96/00195
36
negatively when this fragment is incorporated the LTR of the
vector. Therefore, L2 fragments must be placed between the
LTRs, whereby it is preferred to use the ClaI site for that
purpose. As an alternative for insertion into the LTR the
smaller L0.8 fragment can be used.
Table 4: Analysis of GP+envAml2 clones harbouring a single
copv of a recombinant CD2-LCR comprising retrovirus construct,
obtained through infection with ecotropic recombinant
retrovirus supernatant.
Recombinant Stability Recombinant
Retrovirus Retrovirus Retrovirus
ConstruCl Structure*Titer rmean (range)]**
CL2F 9/9 9E2 (lE2-2E3)
CL2R 4/4 2E3 (lE3-4E3)
CL0.8F 7/7 9E3 (lE3-2E4)
20 CL0.8R 7/12 N.A.
NL2F 0/7 2E4 (lE3-lE5)
NL2R 8/10 3E3 (5El-lE4)
NL0.8F 6/6 2E3 (lE2-6E3)
NL0.8R 4/4 5E3 (2E3-8E3)
~5
* Number of clones having the correct recombinant retrovirus
integration / number of independent clones analysed.
*~ ~ean titer of GP+envAml2 cells harbouring the vector
without the CD2-LCR insertion was 1.5E3. N.A., not
30analyzed.
SVBSTITUTE SHEET (RULE 26)
- - - - - - - - - - - - -
CA 02218808 1997-11-07
PCT~ng6/00195
W096/35798
37
Refer~nces:
Aerts, J. M. F.G., Donker-Koopman, W.E., van Vliet, M.,
Jonsson, L.M.V., Ginns, ~.I., Murray, G.J., Barranger, J.A.,
~ager, J.M. and Schram, A.W. (1985). Relationship between the
~ two im.~unologically distinguishable forms of
glucocerebrosidase in tissue extracts. ~ur. J. Biochem. 150,
565-574.
Antin, J.H. (1993). Graft-versus-leukemia: No ionger an
epiphenomenon. Blood 82, No.8, 2273-2277.
Brenner, M.K., Rill, D.R.R., Holladay, M.S., Heslop, H.E.,
Moen, R.C., Buschle, M., Krance, R.A., Santana,V.M., French
Anderson, W. and Ihle, J.N. (1993). Gene marking to determine
whether autologous marrow infusion restores long-term
haemopoiesis in cancer patients. Lancet 342, 1134-1137.
Chen, C. and Okayama, H. (1987). High-efficiency
transformation of mAmmAlian cells by plasmid DNA. MCB 7, No.
8, 2745-2752.
Chen, C., Chin, J.E., Ueda, K., Clark, D.P., Pastan, I.,
Gottesman, M.M. and Roninson, I.B. (1986). Internal
~5 duplication and homology with bacterial transport proteins in
the MDR1 (P-glycoprotein) gene from multidrug-resistant human
cells. Cell 47, 381-389.
Cepko, C.L., Roberts, B.E. and Mulligan, R.C. (1984).
Construction and applications of a highlv trAncm;~sible murine
retrovirus shuttle vector. Cell 37, 1053-1062.
Choi, K., Chen, C., Kriegler, M., Roninson, I.B. (1988). An
~ altered pattern of cross-resistance in multidrug-resistant
human cells results from spontaneous mutations in the MDR1 (P-
glycoprotein) gene. Cell 53, 519-529.
Chung, J.H., Whiteley, M. and Felsenfeld, G. (1993). A 5'
element of the chicken b-globin ~m~; n serves as an insulator
SUBSTITUTE SHEET (RULE 26)
CA 022l8808 l997-ll-07
W096/35798 PCT~L96/00195
38
in human erythroid cells and protects against position effect
in Drosophila. Cell 74, 505-514.
Culver, K.W., Ram, Z., Wallbridge, S., Ishii, H., Oldfield,
E.H. and Blaese, R.M. (1992). In vivo gene transfer with
retroviral vector-producer cells for treatment of experimental
brain tumours. Science 256, 1550-1552.
Danos, O. and Mulligan, R.C. (1988). Safe and efficient
generation of recombinant retroviruses with amphotropic and
ecotropic host ranges. Proc. Natl. Acad. Sci. USA. 85, 6460-
6464.
Davis, B., LinneY, E. and Fan, H. (1985). Suppression of
leukaemia virus pathogenicitv by polyoma virus enhancers.
Nature 314, 550-553.
Einerhand, M.P.W., Bakx, T.A. and Valerio, D. (l991). IL-6
production by retrovirus packaging cells and cultured bone
marrow cells. Hum. Gene Ther. 2, 301-306.
Einerhand, M.P.W., Bakx, T.A., Kukler, A. and Valerio, D.
(1993). Factors affecting the transduction of pluripotent
haemopoietic stem cells: Long term expression of a human
'5 adenosine ~e~min~e gene in mice. Blood 81, 254-263.
Emerman, M. and Temin, H.M. (1984). Genes with promoters in
retrovirus vectors can be independentl~ suppressed by an
epigenetic me~h A ni ~m . Cell 39, 449-467.
~m~rm~n, M. and Temin, H.M. (1986). Comparison of promoter
suppression in avian and murine retrovirus vectors. Nucleic
Acids Res. 14, 9381-9396.
Fowlkes, D.M. and Shenk, T. (1980). ~ranscriptional control
regions of the Adenovirus VAl RNA gene. Cell 22, 405-413.
Gilboa, E. and Smith, C. (1994). Gene therapv for infectious
diseases: the AIDS model. TIG 10, No. 4, 139-144.
SUB STITUTE S~ EET (RULE 26)
CA 02218808 1997-11-07
W096/3S798 PCT~96/00195
39
Greaves, D.R., Wilson, F.D., Lang, G. and Kioussis, D. (1989).
Human CD2 3'-flanking sequences confer high-level, T-cell-
specific, position-independent gene e~pression in transgenic
mice. Cell 56, 979-986.
Grosveld, F., Blom van Assendelft, G., Greaves, D.R. and
Kollias, G. (1987). Position-independent high-level expression
of the ~-globin gene in transgenic mice. Cell 51, 975-985.
Hoogerbrugge, P.M., Van Beusechem V.W., Kaptein L.C.M.,
Einerhand M.P.W. and Valerio D. (1995). Gene therapy for
adenosine de~min~se deficiency. Britisch Medical Bulletin Vol.
51, No.1, pp. 72-81.
Jang, S.K., Krausslich, H.-G., Nicklin, M.J.H., Duek, G.M.,
pAlm~n~erg, A.C. and Wimmer, E. (1988). A segment of the 5'
nontranslated region of encephalomyocarditis virus RNA directs
internal entry of ribosomes during in vitro translation. J.
Virology 62, 2636-2643.
Jang, S.K., Davies, M.V., Kaufman, R.J. and Wimmer, E. (1989).
Initiation of protein synthesis by internal entry of ribosomes
into the 5' nontranslated region of encephalomyocarditis virus
RNA in vivo. J. Virology 63, 1651-1660.
Kaufman, R.J., Murtha, P., Ingolia, D.E., Yeung, C. and
Kellems, R.E. (1986). Selection and amplification of
heterologous genes encoding adenosine de~min~ce in m~mm~lian
cells. Proc. Natl. acad. Sci. USA 83, 3136-3140.
Kozak, M. (1987a). An analysis of 5'-noncoding seque~ces from
~ 699 vertebrate messenger RNAs. Nucl. Acid Res. 15, No. 20,
8125-8148.
~5
Kozak, M. (1987b). Effects of intercistronic length on the
efficiency of reinitiation by eukaryotic ribosomes. MCB 7, No.
10, 3438-3445.
SUB5TITUTE SHEET (RULE 26)
CA 02218808 1997-11-07
W096/35798 PCT~96/00195
~0
Kozak, M. (1989). The sc~nning model for translation: an
update. J. of Cell Biol. 108, 229-241.
Lang, G., Wotton, D., Owen, M.J., Sewell, W.A., Brown, M.H.,
Mason, D.Y., Crumpten, M.J. and Kioussis, D. (1988). The
structure of the human CD2 gene and its expression in
transgenic mice. EMBO J. 7, No. 6, 1675-1682.
Lang, G., Mamalaki, C., Greenberg, D., Yannoutsos, N. and
Kioussis, D. (1991). Deletion analysis of the human CD2 gene
locus control region in transgenic mice. Nucleic Acids Res.
19, No. 21, 5851-5856.
Lake, R.A., Wotten, D. and Owen, M.J. (1990). A 3'
transcriptional enhancer regulates tissue-specific expression
of the human CD2 gene. EMBO J. 9, No. 10, 3129-3136.
Levine, F., Yee, J.K. and Frie~m~nn, T. (1991). Eff cient gene
expression in mAmmA 1 i ~n cells from a dicistronic transcription
unit in an improved retroviral vector. Gene 108, 167-174.
Linne7, E., Davis, B., Overhauser, J., Chao, E. and Fan, H.
(1984). Non-function of a Moloney murine leukemia -.-irus
regulatory sequence in F9 embrvonal cercinoma cells. Nature
308, 470-472.
Markowitz, D.G., Goff, S.P. and Bank, A. (1988). A safe
packaging line for gene transfer: separating viral genes of
two different plasmids. J. of Virology 62, 1120-1124.
McIvor, R.S., Johnson, M.J., Miller, A.D., Pitts, S.,
Williams, S.R., Valerio, D., Martin, D.W. and Verma, I.M.
(1987). Human purine nucleoside phosphorvlase and adenosine
~e~in~-~e: Gene transfer into cultured cells and murine
hematopoietic stem cells by using recombinant ampho~ropic
retroviruses. MCB 7. No. 2, 838-846.
Michelson, A.M., Markham, A.F. and Orkin, S.H. (1983).
Isolation and DNA sequence of a full-length cDNA clone for
SVBSTITUTE SHEET (RULE 26)
CA 022l8808 1997-11-07
PCT~96/00195
Wo96l35798 41
human X chromosome-encoded phosphoglycerate kinase. Proc.
Natl. Acad. Sci. USA 80, No. 2, 472-476.
~ Miller, A.D. and RosmanG.J. (1989). Improved retroviral
vectors for gene transfer and expression. BioTechniques 7, No.
9, 980-990
Moolten, ~.L. (1986). Tumour chemosensitivity conferred by
inserted herpes thymidine kinase genes: paradigm for a
prospective cancer control strategy. Cancer Research 46, 5276-
5281.
Moolten, F.L. (1994). Drug sensitivitv ("suicide") genes for
selective cancer chemotherapv. Cancer Gene Therapy 1, no. 4,
279-287
Novak, U., Harris, E.A.S., Forrester, W., Groudine, M. and
Gelinas, R. (1990). High-level b-globin expression after
retroviral transfer of locus activation region-conr~;ning
human b-globin gene derivatives into murine erythroleukemia
cells. Proc. Natl. Acad. Sci. USA 87, 3386-3390.
Pelletier, J. and Sonenberg, N. (1988). Internal binding of
ribosomes to the 5' noncoding region of a eukaryotic mRNA:
Translation of poliovirus. Nature 334, 320-325.
Pelletier, J., Kaplan, G., Racaniello, V.R., Sonenberg, N.
(1988). Cap-independent translation of poliovirus mRNA is
conferred by sequence elements within the S' noncoding region.
MCB 8, No. 3, 1103-1112.
Schinkel, A.H., Roelofs, M.E.M. and Borst, P. (1991).
Characterization of the human MDR3 P-glycoprotein and its
recognition by P-glycoprotein-specific monoclonal antibodies.
Cancer Research 51: 2628-2635.
Schwart_, F., Maeda, N., Smithies, O., Hickev, R., ~elm~nn~
W., Skoultchi, A. and Kucherlapati, R. (1991). A dom; n~t
SUBSmUTE SHEEl (RULE 26)
CA 022l8808 l997-ll-07
pCT~n96/00195
W096/35798
42
positive and negative selectable gene for use in m~mm~ 1 ian
cells. Proc. Natl. Acad. Sci. USA 88, 10416-10420.
Singer-Sam, J., Keith, D.H., Tani, K., Simmer, R.L., Shively,
L., Lindsay, S., Yoshida, A. and Riggs, A.D. (1984). Sequence
of the promoter region of the gene for human X-linked 3-
phosphoglycerate kinase. Gene 32, 409-417.
Sorrentino, B.P., McDonagh, K.T. and Orlic, D. (1993). P-
glycoprotein mRNA levels in murine hematopoietic cells
transduced with retroviral vectors e~pressing the human
multidrug resistance 1 cDNA. Blood 82, No. 10 Suppl. l.
Southern, P.J. and Berg, P. (1982). Transformation or
lS mammalian cells to antibiotic resistance with a bacterial gene
under control of the SV40 earL~ region promoter. J. of Mol.
and Appl. Genetics 1, 327-341.
Thomas, K.R. and Capecchi, M.R. (1987). Site-directed
mutagenesis by gene targeting in mouse embryo-derived stem
cells. Cell 51, 503-512.
Tiberghien, P., Reynolds, C.W., Keller, J., Spence, S.,
Deschaseaux, M ., Certoux, J.-M., Contassot, E.M., Murphv
W.J., Lyons, R., Chiang, Y., Hervé, P., Longo, D.L. and
Ruscetti, F.W. (1994). Ganciclovir treatment of Herpes simplex
thymidine kinase-transduced primary T lymphocytes: An approach
for specific in vivo donor T-cell depletion after bone marrow
transplantation. Blood 84, 1333-1341
Valerio, D., Duyvesteyn, M.G.C., Dekker, B.M.M., Weeda, G.,
Berkvens, Th.M., Van der Voorn, L., Van ormondt, H. and Van
der Eb, A.J. (1985). Adenosine ~e~m;n~ce: characterization and
expression of a gene with a remarkable promoter. EMBO J. ~,
437-443.
Valerio, D., Einerhand, M.P.W., Wamsley, P.M., Bakx, T.A., Li,
C.L. and Verma, I.M. (1989). Retrovirus-mediated gene transfer
into embryonal carcinoma and haemopoietic stem cells:
SUB STITUTE 5H EET (RULE ~6)
CA 02218808 1997-11-07
PCT~96/00195
WO96/35798
~3
expression from a hybrid long terminaL repeat. Gene 84, 419-
427.
~ Van Beusechem, V.W., Kukler, A., Einerhand, M.P.W., Bakx,
T.A., van der Eb, A.J., van Bekkum, D.W. and Valerio, D.
(1990). Expression of human adenosine deAminAse in mice
transplanted with haemopoietic stem cells infected with
amphotropic rettroviruses. J. Exp. Med. 172, 329-336.
Van Beusechem, V.W., Kukler, A., Heidt, P.J. and Valerio, D.
(1992). Long-term expression of human adenosine ~e~min~se in
rhesus monkeys transplanted with retrovirus-infected bone-
marrow cells. Proc. Natl. Acad. Sci. USA 89, 7640-7644.
IS Van Beusechem, V.W., Bart-Baumeister, J.A.~., Bakx, ~.A.,
Kaptein, L.C.M., Levinsky, R.J. and Valerio, D. Retro~iral
vector-mediated gene transfer into non-human primate
CD34+CDllb- bone marrow progenitor ceLls capable of
repopulating lymphoid and m~eloid lineages. Hum.Gene Ther.5
(1994) 295-305.
Van Beusechem, V.W., Bakx, T.A., Bart-Baumeister, J.A.K.,
Braa~man, E., Kaptein, L.C.M., Kukler, A. and Valerto, D.
Retrovirus-mediated gene transfer into rhesus monkey
~5 haemopoietic stem cells: the effect of -~iral titers on
transduction efficiency. Hum.Gene Ther.4 (1993) 239-247.
Van Beusechem, V.W., Bart-Baumeister, J.A.K., Hoogerbrugge,
P.M. and Valerio, D. Influence of Interleukin-3, Interleukin-6
and Stem Cell Factor on retroviral transduction of rhesus
monkey CD34+ haematopoietic progenitor cells measured in vitro
and in vivo., Gene Ther.2 (1995) 1-1
Van Beveren, C., Coffin, J. and Hughes, S. (1985). Nucleotide
sequences complemented with functional and structural
analysis, p.766-782 and p.900-911. In R. Weiss, N. Teich,
H.Varmus and J. Coffin (Eds.), RNA Tumour Viruses , Vol.2. Cold
Spring Harbor Laboratory, Colg Spring Harbor, NY>
SUBSTITUTE SHEET (RULE 26)
CA 02218808 1997-11-07
W096/35798 PCT~L96/O019
44
Van der Bliek, A.M., Kooiman, P.M., Schneider, C., Borst, P.
(1988). Sequence of mdr3 cDNA encoding a human P-glycoprotein.
Gene 71, 401-411.
Vincent, A.J.P.E., Vogels, R., Van Someren, G., Esandi, M.C.,
Noteboom, J.L., Avezaat, C.J.J., Vecht, C., Van Bekkum, D.W.,
Valerio, D., Bout, A. and Hoogerbrugge, P.M. 1996). Herpes
simplex virus thymidine kinase gene therapy for rat malignant
brain tumors. Hum.Gene Ther. 7, 197-205.
Weizsaecker, ~., Deen, D.F., Rosenblum,M.L., Hoshino, T.,
Gutin, P.H. and Barker, M. (1981). The 9L rat brain tumour:
description and application of an ~nim~l model. J. Neurol.
224, 183-192.
SUE~STITUTE SHEET (RULE 26)
CA 02218808 1997-11-07
W096/35798 PCT~n96/00195
~5
J,eqe~ to fig--res
Figure l. Schematic representation of the cloning strategy
used to construct pLec. LTR, long terminal repeat;HIII,
HindIII;mcs, multiple cloning site; SD, splice donor site; SA,
splice acceptor site; TAG, stop codon; waved lines represent
mouse genomic flanking sequences.
Figure 2. Schematic representation of the cloning strategy
IO used to construct pLec~Mo.
x, XbaI and see legend figure l.
Figure 3. Schematic representation of the cloning strategy
used to construct pLTKkoz.
see also legend figure l.
Figure ~a and b. Schematic representation of the constructs
pIGTR and pLTT~Mo. see also legend to figure 1.
Figure 5. Kaplan-Meier survival curves of rats with brain
tumour treated with single dosis recombinant retrovirus
producer cell lines and subsequent GCV a~min;~tration.
Intracere~ral injection of tumour cells at day 0. IG-RV-TK was
injected 3 days after tumour cell injection. One group was
~5 injected with IG-RV-TK producer cells (n=lo)( ). The
controls (n=5/ group) were injected with PBS ( ), RV-lacZ
producer cells (--------) TK+RV- non producer cells (- -
)~ supernatant of IG-RV-TK producer cells (- - - ) and RV-
IL2 producer cells ( - - - -). One group was injected with
IG-RV-TK producer cells without subsequent GCV treatment (- -
- - - -) . Five days after virus injection 15 mg/kg GCV was
a~ministered twice a day for ten days i.p.. IG-R~-TK treated
~ rats Lived significantly longer than controls (p<0.0l; log
rank test). One rat died of superficial leptomeningeal tumour
(*)~ This rat is censored in the survival analysis.
Figure 6. Schematic representation of the constructs used to
test the intercistronic linkers. TGA, stop codon; E, EcoRI; S,
SalI.
SUBSTITUTE SHEET (RULE 26)
CA 02218808 1997-11-07
PCT~96/00195
WO96/35798
Figure 6A. Bar diagram representing the average number of
colonies obtained after G41~ selection of transfected Rat-2
cells corrected for (arbitrary units of) Luciferase activity.
Figure 6B. Bar diagram representing the average number of
colonies obtained after G418 selection of transfected Rat-2
cells corrected for the num~er of colonies o~tained after
selection in HAT medium.
Figure 7. Rh-123 exclusion analysis on A2780 cells after
transduction with IGmdrl-G virus supernatant. Transduced A2780
cells (A) are compared to mock-infected controls (B).
Figure 8. Vincristine survival of CD34~ peripheral blood cells
after transduction with IGmdrl-G virus supernatant. CD34
selected PBPC were transduced with (squares) or without
(circles) IGmdrl-G supernatant for 4 days in the presence of
IL-3 and 4 ~g/ml protamine sulphate. Supernatant -.vas refreshed
daily. After transduction, PBSC were seeded for in vitro
colony formation (GM-CFU) in the presence of increasing
amounts of vincristine. Colonies were scored after 14 days.
The survival was calculated by dividing the number of GM-CFU
in the dishes with drugs by the number of GM-CFU in the dishes
without drugs.
Figure 9. Physical map of retroviral construct IG-GC-l. SD,
splice donor: SA, splice acceptor. TAG, mutated slartcodon of
gag coding sequence; LTR, long t~rminAl repeat.
Figure lO. Physical map of retroviral construct IG-GC-2. SD,
splice donor; SA, splice acceptor; TAG; mutated szartcodon of
gag coding sequence; LTR, long termi n~ 1 repeat.
Figure ll. PCR strategy to delete part of the 3'- untranslated
region of the human Glucocerebrosidase cDNA (see text for
details).
SUBSTITUTE S~EEJ (RULE 26)
CA 022l8808 l997-ll-07
W096/35798 PCT~n96/00195
47
Figure 12. Physical map of retroviral construct IG-GC-3. SD,
splice donor; SA, splice acceptor; TAG, mutated startcodon of
~ gag coding sequencel; LTR, long terminal repeat.
Figure 13. Physical map of retroviral construct IG-GC-4. SD,
splice donor; SA, splice acceptor; TAG, mutated startcodon of
gag coding sequence; LTR, long tPrm;nAl repeat.
Figure 14A. Increase in glucocerebrosidase enzyme activity
after transfection of retroviral constructs IG-GC-2 and IG-GC-
4 in packaging cell lines GP + E 86 and Psi-CRE respectively
and after infection of 3T3 cells.
Figure 14B. Western blot with primate specific monoclonal
antibody 8E4 (80 mg total protein/lane). A, B, C: Cell lysates
prepared from GP + E86 cells after transfection with
retroviral construct IG-GC-2. +/-: With or without protease
inhibitors. GP: Cell lysate prepared from non-transfected GP +
E86 cells.
Figure 15. Western blot with primate specific monoclonal
antibody 8E4 (80 mg total protein/lane). Bl/82: Cell lysates
prepared from PA317 cells after repeated infection (1, 4, 10
times) with ecotropic GP2b virus (duplicate). GP: cell lysate
prepared from GP - E86 cells after transfection with
retroviral construct IG-GC-2 as positive control. PA: Cell
lysate prepared from non-infected PA317 cells. Cer: 16.5 mg
(0.7 units) recombinant glucocerebrosidase (Genzyme Corp.) as
positive control.
~igure 16. Western analysis on hGC expression in producer
(PA2, PA4, PG2 and PG4) and of the parental cell line PA317
(PA) was loaded on a 10~ acrylamide gel. As positive control
approx. 1 unit of Cerezyme (Genzyme corp.) was loaded. B) 20~g
3~ total protein of each infected Gaucher type II cell pool (PA2,
PA4, PG2 and PG4) and of the parental cell line (T-II) was
loaded.
SUBS 11 l UTE S~tEET (RULE 26)
CA 022l8808 l997-ll-07
WO96/357g8 PCT~96/00195
48
Figure 17. DNA analysis of PA2 infected individual 3T3 cell
clones. Isolated DNA was digested with EcoRI (unique
restriction site just 5' of hGC sequence) and Southern blots
were probed with the complete hGC sequence. In the genomic DNA
of each individual 3T3 clone a mean of three bands is visible
resulting in an estimated functional titer of at least
3 x 105/ml.
Figure 18. hGC-activity assay (PNP-~-Glu) on normal and
Gaucher type I and II fibroblasts as well as on type I and II
Gaucher fibroblasts infected with virus supernatant.
A) Infected Gaucher type I fibroblasts (grey bars) versus non-
infected (white bar) and normal (blac~ bar) cells (n=4).
B) Infected Gaucher type II fibroblasts (grey bars) versus
non-infected (white bar) and normal (blac~ bar) cells (n=4).
Figure 19. hGC-activity assay (PNP-~-Glu) in lysates of
infected CD34+1iquid culture cells of Gaucher bone marrow
(n=4)-
~0Figure 20. PCR and subsequent Southern analysis show the
presence of the recombinant provirus in infected CD34+ Gaucher
bone marrow cells. Southern blot was probed with a 300 bp
BamHI fragment from the 3' end of the hGC gene.
~5
Figure 21. Strategy for the introduction or a point mutation
(Phe32Ser) in the human dihydrofolate reductase cDNA.
Figure 22. Schematic drawing of the map of recombinant
retroviral vectors IG-GC5 and IG-GC6.
SUBSTITUTE S~EET ~RULE 26)