Note: Descriptions are shown in the official language in which they were submitted.
CA 02219437 1997-10-24
T.Tl~RA~ AND CYCLIC GLY-GLN-ALA-ARG, ARG-GI.N-ALA-ARG AND R13LATI~D
TETRAP~PTID~S AS POT~NT ANTINOCI~llv~ AGENTS
RAC~GROUND OF THE lNv~.llON
Histogranin (HN: H-Met-Asn-Tyr-Ala-Leu-Lys-Gly-Gln-
Gly-Arg-Thr-Leu-Tyr-Gly-Phe-COOH) was first coined by our
laboratory as an adrenal medullary peptide possessing N-
methyl-D-aspartate (NMDA) receptor antagonist activity as
assesRed by its ability to block NMDA-induced convulsions in
mice (1) and to produce phencyclidine(PCP)-like behaviourial
effects in rats (2). The radiolabelled peptide possesses a
specific receptor on rat brain membranes (3). Binding of HN
to its receptor was demonstrated to affect the activity of
specific modulators (Gly, dextromethorphan) of the NMDA
receptor (4,5).Herein, two tetrapeptides [Gly-Gln-Ala-Arg (SL-
100) and Arg-Gln-Ala-Arg (SL-101)] derived from the structure
of the m;n;~l active core peptide, HN-(7-10) (SL-99), and
their cyclic forms [cyclo-(-Gly-Gln-Ala-Arg-): SL-102~ and
[cyclo-(-Arg-Gln-Ala-Arg-): SL-103] and analogues are proposed
as pain relieving agents.
It has been known for a long time that neuropathic
pain, eg. pain induced by peripheral nerve injury as a result
of a chronic disease or some inflammatory processes, is
manifested by hyperalgesia (exaggerated nociceptive responses
to noxious stimulation), allodynia (nociceptive response to
innocuous stimulation) and ~pontaneous pain. Compelling
evidence indicates that activation of spinal cord NMDA
-- 1 --
72591-10
CA 02219437 1997-10-24
receptors contributes to the hyperalgesia that occurs
following peripheral nerve injury or inflammation. Thus,
administration of either competitive (AP-5) or non-competitive
(MK-801) NMDA receptor antagonists powerfully reduces thermal
hyperalgesia in animal models of neuropathy (6), carrageenan-
induced acute peripheral inflammation (7), heat-injury (8) and
formalin-induced pain (9). Likewise, activation of NMDA
receptors within the spinal cord has been shown to play a role
in the development of tolerance to the analgesic effects of
morphine (10). In this regard, various studies indicate that
agents antagonizing the NMDA receptor can prevent morphine
tolerance (11-13). However, currently used NMDA receptor
antagonists produce major side-effects, including motor
dysfunction, learning impairment, hallucinations etc...
In 1990, Sagan and colleagues (14) have devised an
experimental model for the alleviation of chronic pain in
which the hyperalgesic state caused by sciatic nerve injury in
rats was completely blocked by adrenal medullary implants into
the spinal cord. The beneficial effects of the transplantation
of adrenal chromaffin cells into the spinal subarachnoid space
were also observed in rats models of arthritis (15) and
depression (16). In addition, the analgesic effects o~ adrenal
medullary transplants did not display tolerance and morphine
cross-tolerance upon intermittent administration of nicotine
(which evoked the release of the analgesic factor(s) from the
adrenal medullary implant) (17) and they were accompanied by a
reduction of spinal nerve degeneration (18). At that time,
most analgesic and neuroprotective effects of adrenal
-- 2
72591-lO
CA 02219437 1997-10-24
medullary implants were thought to be produced by opioid
peptides and catecholamines released from transplanted adrenal
chromaffin cells (19), although such effects were not
completely blocked by naloxone (an opioid antagoni~t) and
phentolamine (an adrenergic antagonist), alone or in
combination.
Recently, Dr. Sagan and colleagues reported that the
analgesic effects of the adrenal implants in rat models of
chronic pain and inflammation are mimicked by the peptide
~Ser1]HN (20, 21, 22). In their experimental protocols, a
relatively low dose of [Ser1]HN (1 nmol, i.t.) was shown to
block chronic pain induced by peripheral neuropathy, formalin-
induced pain and direct application of NMDA. In the formalin
test, HN produced analgesia in the late (NMDA-dependent)
phase, but not early (NMDA-independent) phase of the pain
assay (21).
We have also observed that HN and related peptides
are potent analgesics in the mouse writhing test (23). In
this acetic acid-induced pain assay, central (i.c.v., 0.5-50
nmol) and peripheral (i.p., 5 ~mol/ kg) administrations of HN
and related peptides produced strong (up to 100%) analgesia
with no motor side-effect. The analgesic effects of [Ser1]HN
(50 nmol, i.c.v.) were blocked by the NMDA receptor
antagonists, CPP and MK-801, but not by the opiate antagonist,
naloxone, suggesting that the analgesic properties of HN and
related peptides involve NMDA receptor-mediated mechanisms
(23). Thus, adrenal medullary HN may be one of the factor(s)
that mediate the non-opioid antinociceptive effects of spinal
-- 3
72591-10
CA 02219437 1997-10-24
adrenal medullary implants; the adrenal peptide may also be
the mediator of some physiological phenomena such as ~tress-
induced analgesia, a physiological condition that i8 known to
involve NMDA receptor mechanisms (24).
HN and related peptides at analgesic doses in mice
do not display any noticeable behaviourial activity (in the
rotarod assay and by gross observation). However, these
peptides display marked analgesia after both central (i.c.v.)
or peripheral (i.p.) administration (23). It is presumed that
the analgesic effects of HN result from its interaction with
both central and peripheral receptors. Interestingly, HN was
shown to bind to specific receptors located in the brain (3)
and on peripheral blood lymphocytes (25). Analgesia may also
result from the blockade by HN of the formation of pain
mediators such as prostaglandins. Prel;m;n~ry results
indicate that HN blocks the synthesis of prostaglandin-E2 in
isolated rat alveolar macrophages in response to
lipopolysaccharide (unpublished observations). The mechanism
by which HN and related peptides produce analgesia is ~till
unknown, but the possible involvement of the dextromethorphan
b; n~; ng site on the NMDA receptor complex is suggested by the
close correlation that exists between the ability of HN and
related peptides to produce analgesia and potentiate the
binding of [3H]dextromethorphan, a non-competitive NMDA
antagonist, to rat brain membranes (4).
Chronic pain may result from multiple causes
including pain related to inflammation, peripheral nerve
injury, cancer, AIDS, diabetes etc... The drugs that are
-- 4
72591-10
CA 02219437 1997-10-24
being used for the treatment of chronic pain (derivatives of
aspirin and non-steroidal antiinflammatory agents) have very
limited efficacy and they produce important ~ide-effect~.
They interfere with blood coagulation, they cause and/or
exacerbate peptic ulcer etc... NMDA receptor antagonists are
effective in ~n; ~1 models of neuropathy, but these latter
compounds produce behaviourial side-effects (motor impairment,
learning impairment, locomotion, ataxia etc...) that hamper
their use as therapeutic agents. Morphine and opioid
analgesic~ show no or few beneficial effects: they produce
marked tolerance, addiction and withdrawal ~yndromes and they
are not effective against neuropathic pain. Based on the data
observed with the adrenal implants, it is expected that the
adrenal medullary peptide, HN, and its analogues will not only
be effective as analgesic agent~, but they may al~o display
neuroprotective activity and alleviate tolerance to morphine.
OBJ13CTg OF 1~S lNV~ lON
An object of the invention is to provide a peptide,
for example HN-(7-10): Gly-Gln-Gly-Arg (SL-99), a~ ;n; -1
core peptide comprising amino acids of only the L-
configuration that produces analgesia.
Another object of the invention is to provide two
analogues of HN-(7-10): [Ala9]HN-(7-10): Gly-Gln-Ala-Arg (SL-
100) and [Arg', Ala9]HN-(7-10): Arg-Gln-Ala-Arg (SL-101), with
improved analgesic potencies.
Another object of the invention is to provide a
series of analogues of SL-100 and SL-101 with the following
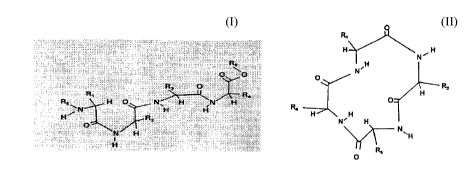
general Formula I:
-- 5
72591-10
CA 02219437 1997-10-24
Fo ~ula I .~ . ~ . .~ . . . .
and the pharmaceutically-acceptable saltR, esters and amide~,
wherein:
Rl is hydrogen, an alkyl or a basic radical (the term alkyl
as used herein mean~ a hydrocarbon radical having from
one to ten carbon atoms, which can be a straight or
branched chain, and including from zero to four carbon-
carbon double or triple bonds. Representative of such
radicals are methyl, ethyl, n-propyl, isopropyl, n-butyl,
2-ethyl-hexyl and the like. The term basic as used
herein means (CH2)n-NH2 or (CH2)n-NH-C(=NH)NH2, "n" each
independently an integer from 0 to 10);
~0 R2 is an amide radical such as (CH2)n-CONH2 "n" an integer
from 0 to 10;
R3 is hydrogen or an alkyl radical as defined above;
R~ is a basic radical as defined above;
R5 is hydrogen or an acetyl or an alkyl radical as defined
above;
R6 is hydrogen, alkyl, alkyl carbonyl, alkoxy carbonyl,
amino carbonyl, alkylaminocarbonyl, dialkylamino,
carbonyl, (CH2)n-benzyl, (CH2)n-phenyl ("n" an integer
-- 6
72591-10
CA 02219437 1997-10-24
from 1 to 10).
O-R6 is replaced by R7 (not shown), R7being amino, hydroxy,
alkoxy, alkylamino, dialkylamino, or alkoxyaryl.
O-R6 is replaced by R7, R7 being independently positions
11 to 15 in HN and represented by Thrll-Leu-Tyr-Gly-
Phel5, Thrll-Leu-Tyr-Glyl4, Thrll-Leu-Tyrl3, Thrll-Leul2
and Thrll, or homologous peptides or amino acids, for
example, Thrll may be exchanged for Ser, Leul2 may
be exchanged for Ala, Val or Ile, Tyrl3 may be
exchanged by Phe or diiodotyrosine, Glyl4 may be
exchanged for Ala, Val, Leu or Ile, and Phel5 may be
exchanged for Tyr or diidotyrosine.
Another object of the invention is to provide the
structure of the cyclic tetrapeptides, cyclo(-Gly-Gln-Ala-
Arg-) (SL-102) and cyclo(-Arg-Gln-Ala-Arg-) (SL-103), as
potent and long-lasting analgesic agents.
Another object of the invention is to provide a series
of analogues of SL-102 and SL-103, with the following general
Formula II:
Formula II R, ~
H~ N,H
N' ~_
R~ ~ H
N~
O
72591-10
CA 02219437 1997-10-24
wherein Rl, R2 R3 and R4 are defined as above.
In the peptides of general formulae I and II, the
chiral carbons of the peptide backbone may each independently
be of either the D- or L-configuration. It i8 preferred that
they are of the L-configuration.
Another object of the invention is to provide the
structure of an analogue of the pentadecapeptide tSerl, Ala9]HN
(SL-104), a~ a potent analgesic agent.
The invention further extends to fragment~ of
histogranin of greater than 4 residues, as well a~ homologues
of histogranin and homologues of the fragments. By homologue
is intended a peptide in which the sequence differs from that
of the parent by replacement of 1 to 4 amino acid residues
with other amino acids.
The cyclic peptides of formula II are cyclized in a
head-to-tail fashion.
Peptides of the general formula I and II are prepared
using techniques of peptide chemistry. They may be prepared
in solution or by solid-phase methods. Examples of preferred
synthetic methods are as follows:
Peptides of Formula I may be synthesized for example,
as described by Prasad et al. (Can. J. Physiol. Pharmacol. 73:
209-124, 1995) by the use of preformed symmetrical anhydrides
(Lemaire et al., J. Med. Chem. 21: 1232-1235, 1978; with the
exception of Boc-Arg, Boc-Asn and Boc-Gln) of Boc-amino acids
(Bachem California) with a solid-phaae method (Merrifield, J.
Am. Chem. Soc., 85: 2149-2154, 1963) on chloromethylated
polystyrene-divinylbenzene resin (benzhydrylamine or oxime
-- 8
72591-10
CA 02219437 1997-10-24
resins can also be used to generate the various C-terminal
substituted groups of Formula I according to Bodanszky and du
Vigneaud (J. Am. Chem. Soc. 81: 5688, 1958). The various
steps of the automatic coupling cycles are described by St.
Pierre, Gaudreau, Drouin, Regoli and Lemaire (Can. J. Biochem.
57: 1084-1089, 1979). Boc-Arg, Boc-Asn and Boc-Gln are
coupled to the deprotected N-terminal group of the growing
peptide-resin by the method of Coste et al. (Tetrahedron Lett.
31: 205-208, 1990). Side-chain protecting groups are a~
follows: Arg, Tos: Lys, 2-Cl-Z; Thr and Ser, Bz: Tyr, 2,6-
dichloro-Bz; His, Boc; Asp and Glu, Obzl. The completed
peptides are cleaved from the resin and deprotected with
liquid hydrogen fluoride (HF) and purified by successive
chromatographies on Sephadex G-10 and high performance liquid
chromatography (HPLC) on Bio-Sil C18 column (Waters, Milford,
MA). The purity and identity of synthetic peptides is
verified by analytical HPLC on ~-Bondapak C18 column (waters,
Milford, MA), amino acid analysis of acid (HCI) hydrolysates
and fast atom bombardment mass spectrophotometry.
The synthesis of the cyclic peptides included in
Formula II may be achieved, for example by a solid-phase
procedure using Kaiser' 8 oxime-resin and following procedures
of Osapay et al., (Tetrahedron Letters, 31, 6121-6124, 1990)
and Nishino et al., (J. Chem. Soc. Kin Trans. 1, 939-946,
1986).
Another object of the invention is to provide
pseudopeptides based on the structures of peptides of general
Formulae I and II, wherein (CO-NH) bonds between amino acids
g
72591-10
CA 02219437 1997-10-24
are replaced each independently by (CS-NH) or (CH2-NH) bonds
known as pseudopeptide bonds, said pseudopeptides possessing
one or two pseudopeptide bonds of the same or different type~
for Formula I, and one, two or three pseudopeptide bonds of
the same or different types for Formula II. Pseudopeptides
may be obtained by a solid-phase procedure (Le, Michelot,
Dumont, Shukla, Mayer, and Lemaire (Can. J. Physiol.
Pharmacol., 75: 9-14, 1997), for example, according to the
method of Michelot et al., (In "Innovation and perspectives in
solid-phase synthesis, biological and biomedical applications
Edited by R. Epton. Mayflower Worldwide Inc., Birmingham, in
press.)-
Another object of the invention is to provide retro-
verso forms of tetrapeptides of general Formulae I and II,
such peptides comprising, for example, Arg-Gly-Gln-Gly, Arg-
Ala-Gln-Gly, Arg-Ala-Gln-Arg, cyclo(-Arg-Ala-Gln-Gly-) and
cyclo(-Arg-Ala-Gln-Arg-) for the retro-verso forms of SL-99,
SL-100, S-101, SL-102 and SL-103, respectively. The synthesis
of these peptides can be as described above for peptides of
Formulae I and II.
Another object of the invention is to provide a
mechanism for a tetrapeptide to produce analgesia, said
mechanism consists in bloaking the activity of the central
excitatory amino acid NMDA receptor.
Another object of the invention is to provide a method
which consists in administering, centrally or peripherally, a
peptide, said peptide HN fragments or analogues of HN
- 10 -
72591-10
CA 02219437 1997-10-24
fragments represented in Formula I and Formula II, to treat
pain.
A further aspect of the invention is a phArr-ceutical
compo-qition comprising a peptide of general formula I or II,
or a phArm~ceutically acceptable salt thereof, in admixture
with a pharmaceutically acceptable diluent or carrier. The
preparation and A~; n; qtration of pharmaceutical compositions
may be by known methods, such as those described in U.S.
Patent No. 5,169,833, which is herein incorporated by
reference.
In yet another aspect the invention provides a
commercial package, contA;n;ng a peptide of general formula I
or II, with instructions for its use in the treatment of pain.
BRIBF DFSCRIPTION OF THF DRAWINGS
The invention is illustrated by the following non-
limiting examples, which can be better understood with the aid
of the figures.
Figure 1 shows a dose-response curve of the analgesic
effects of HN, closed square, SL-100, open square, SL-101 open
circle and SL-102 closed circle.
Figure 2 shows time response curves for the analgesic
effects of [Ser1]HN, closed square, 50 nmol/mouse, SL-100,
closed triangle, 10 nmol/~ use and cyclo-(SL-100 or SL-102)
open triangle 50 nmol/~ use.
Figure 3 shows the effect of naloxone (N), NK-801 (MR)
and CPP on the analgesic effects of [Ser1]HN (SHN, 50
nmol/mouse, i.c.v.).
Figure 4 shows the analgesic effect of peripheral
A~m;n;stration of morphine, H4-(86-100), [Ser1]HN, HN-(7-15),
SL-101 and cyclic SL-100.
- 11 -
72591-10
CA 02219437 1997-10-24
EXAMPLE 1
C~NTRAL AND PERIPU~AT- NON-OPIOID ANTINOCI~K~llv~ ~k~l~ OF
GLY-GLN-ALA-ARG, ARG-GLN-ALA-ARG, CYCLO(-GLY-GLN-ALA-ARG-) AND
R~LAT~D ~h~ll~ IN TH~ MOUS~ WRl~l~lN~ PAIN ASSAY: COMPARISONS
WITH HISTOGRANIN AND MORPHIN~.
MATERIALS AND METHODS
~n;~-7S
Mice (male 20-25 g, SWi8S Webster) were obtained from
Charles River (Canadian Breeding Farm, St. Constant, Quebec).
They were housed five per cage in a room with controlled
temperature (22 + 2~C), humidity and artificial light (06.30-
l9h). The ~n;m~l 8 had free access to food and water and were
used after a minimum of 4 days of acclimation to housing
conditions. Experiments were carried out between 10:00 a.m.
and 4:00 p.m. in an air-regulated and soundproof laboratory
(23 ~ 1~C, 40 % humidity), in which mice were habituated at
least 30 min before each experiment. The experiment~ were
authorized by the ~nim~l care committee of the University of
Ottawa in accordance with the guidelines of the Canadian
Council on ~n; ~ ~1 Care.
Drugs and peptides
(+)3-(2-carboxypiperazine-4-yl)-propyl-1-propionic acid
(CPP) and (~)-5-methyl-10,11-dihydro-5H-dibenzo[a,d] cyclo-
hepten-5,10-imine maleate (MK-801) were obtained from Tocris
Neuramin, Essex, England. HN and related peptide analogues and
fragments were synthesized in our laboratory by the ~olid-
phase procedure (26) as described previously (27). The purity
of the synthetic peptides was assessed by analytical HPLC on
- 12 -
72591-10
CA 02219437 1997-10-24
~ -Bondapak C18 (Waters) and by thin-layer chromatography on
- silica gel plates (60 F 254: BDH Chemicals, Darmstadt,
Germany) in the following solvent system (v/v) : l-butanol
/acetic acid / water / pyridine (15/3/10/12). Their
composition and molecular weight were determined by amino acid
analysis of acid (HCl) hydrolysates and fast atom bombardment
mass spectrophotometry (FAB MS), respectively.
For the synthesis of cyclo(-Gly-Gln-Ala-Arg-) (SL-102),
the Kaiser's oxime-resin was used following the procedures of
Osapay et al (35) and Nishino et al (36). The starting
compound, Boc-Ala-Oxim-Resin, was prepared from Oxim-Resin
(Novabiochem, lg, 0.47 meq/g) by using Boc-Ala-OH in the
presence of PyBOP (3 eq), HOBt(l eq), in DMF for 2h (repeated
2 times), and the excess oxim groups were capped by
acetylation. The peptide chain was then assembled according to
the following coupling steps: (i) two washes with DCM, (ii)
one wash with 25% TFA-DCM, (iii) deprotection with 25% TFA-
DCM (30 min), (iv) two washes with DCM, (v) one wash with
propanol-2, (vi) three washes with DCM, (vii) one wash with
DMF, (viii) coupling of Boc-amino-acids (consecutively Boc-
Gln, Boc-Gly and Boc-Arg(Tos)(3 eq, each)) in presence of
PyBOP (3 eq), HOBt(l eq) and DIEA (5 eq) in DMF (45 min), (ix)
three washes with DMF, (x) two washes with DCM. Solvent
volumes were 15 cm3g~l resin. Coupling efficiency was checked
by the Kaiser test (34). The free amino group cleaved the
peptide from the polymer support by intrachain aminolysis in
the presence of AcOH (2 eq) and DIEA (2 eq) in DMF at room
temperature. After 24 h reaction time, the product wa~
- 13 -
72591-10
CA 02219437 1997-10-24
obtained from the solution phase by filtration. Protecting
group (To~) of the peptide was removed with anhydrous HF at 0~
C for 30 min. This crude product was purified by RP-HPLC
(Bondapak C18 column, lOum x 125A, 25 x 100 mm, with the
gradient of 30%-40% acetonitrile-ammonium acetate 5mM over 50
min) with final yield 15%, based on starting resin. The
purity and identity of the synthetic peptide was assessed by
analytical HPLC on Bondapak C18 column, lOum x 125A, 3.9 x 300
mm, with the gradient of 30%-40% acetonitrile-ammonium acetate
over 50 min, tR: 35 min, molecular mass by FAB-MS: 412 (calc.:
412.5, Dr. J. Wang, Mass Spectrometry ~ab, Medical Sciences
Bldg., Toronto, Canada), amino acid analysis: Ala(1)0.9,
Arg(1)1.1, Gln(1)0.8, Gly(1)1 (Dr. R. Interior, The
Biotechnology Service Centre, Department of Clinical
Biochemistry, Toronto, Canada).
The i.c.v. administrations of the peptides were
performed as described by Shukla et al. (28). Peptides were
dissolved in double-distilled sterile water (vehicle) and 10
~1 of the peptide solution or vehicle were dellvered gradually
within approximately 3 sec, mice exhibiting normal behaviour
within 1 min after injection. The administration site was
confirmed by injecting Indian ink in prel;m;nAry experiments.
Antinociceptive assay
Antinociceptive activity of HN and related compounds
was evaluated using the acetic acid-induced writhing test
according to a modification (28) of the method of Hayashi and
Takemori (29). Male Swiss Webster [(SW)f BR] mice were
injected intraperitoneally (i.p.) with 1.0% acetic acid
- 14 -
72591-10
CA 02219437 1997-10-24
(lOml/kg) 5 min after i.c.v. in~ection of O (saline), 0.5, 1,
10 , 25, 50, 75 and 100 nmol of HN or related peptides. The
number of writhes displayed by each mouse was counted for a
period of 10 min after the injection of the acetic acid
solution. An ab~om;nAl stretch is characterized by the
contraction of the ab~nminAl muscles, the arching of the back
ventrally such as the abdomen touches the bedding surface and
the extension of one or both hind limbs. Mice were used once
and then killed immediately. Groups of 10 mice were u~ed for
each dose. The compound was said to be active at a given dose
if after its administration, the number of writhes elicited by
a mouse injected with acetic acid was equal to, or less than,
one-half the median number of writhes recorded for the saline-
treated control group of mice that day, as described by R.I.
Taber (37). The re~ults are expressed in terms of either the
number of mice out of ten in which a given dose of a peptide
was considered to be active or the EDso value (the dose of the
peptide that produced analgesia in 50% of the An;mAls). The
ED50 values with 95% confidence limits (95% CL) and potency
ratios with 95% CL were measured by the method of Lichfield
and Wilcoxon (30) using procedure 47 of the computer program
of Tallarida and Murray (31). In order to determine the
length of action of [Serl]HN and related peptides, the acetic
acid solution was administered at different times after the
administration of the peptide, as indicated. For verifying
the blockade of the analgesic effect of the peptides with
receptor antagonists, naloxone (lnmol), MK-801 (0.3 nmol) or
CPP (0.1 nmol) were A~m;n;stered i.c.v. in an aliquot of 10
- 15 -
72591-10
CA 02219437 1997-10-24
~1, alone or in combination with tSer1]HN (50 nmol) or related
peptides. The experiments for assessment of the peripheral
antinociceptive activity of the peptides were performed by
i.p. administration of 5 ~mol/kg of the tested compounds 10
min prior to the injection of the acetic acid solution. Data
were analyzed by the Wilcoxon~s paired non-parametric test.
The criterion for statistical significance was P c 0.05.
RESU~TS
Antinociceptive efficacy of histogranin and related peptides
Intracerebroventricular administration of HN and
related peptides in mice induced dose- and structure-dependent
analgesic activitie~ as assessed by their ability to inhibit
writhing in response to acetic acid (i.p.; fig. 1). Histone
H4-(86-100) was 5.4 times more potent than HN with an EDso of
4.1 nmol/mouse as compared with 22.3 nmol/mouse for HN (table
1). The chemically stable HN analogue, [Serl]HN, displayed an
analgesic potency similar to that of HN. The unmodified C-
terminal fragment of histone H4, osteogenic growth peptide
(OGP; 32), was 2 times less potent than HN. The analgesic
activity of HN was shown to reside in the C-te~m;nAl portion
of the peptide, since HN-(7-15) was 3.0 times more potent than
HN itself with an EDso of 7.5 nmol/mouse, while the N-te~m;
fragment, HN-(1-10), was inactive at 50 nmol/mouse. The
m;n;m~l core peptide for analgesic activity was HN-(7-10) (SL-
99), with a potency ratio of 2 as compared with HN. [Ala9]HN-
(7-10) (S~-100) was 5.7 times as potent as HN, whereas the
cyclic form of this peptide (SL-102) was 7.7 times as potent
as HN (EDso of 2.9 nmol/mouse; fig.1). [Arg7, Ala9~HN-(7-10)
- 16 -
72591-10
CA 02219437 1997-10-24
(SL-101 or [Ala9s]H4-(92-95)) was 4.5 times as potent a~ HN,
whereas the pentadecapeptide [Serl, Ala9]HN (SL-104) was 5.4
times as potent as HN (table 1).
[Serl]HN (50 nmol/mouse) produced an analge~ic effect
that lasted approximately 45 min (fig. 2). Its m~;m~l
antinociceptive effect lasted approximately 15 min. The
tetrapeptide SL-100 (10 nmol/mouse) produced an effect that
lasted only 15 min. However, cyclization of this tetrapeptide
(SL-102) greatly enhanced its length of action (an effect that
lasted more than 45 min after i.c.v. injection of the
compound, Fig 2).
NMDA receptor-mediated analgesic activity
In order to verify which receptor was involved in the
antinociceptive activity of [Ser1]HN, the peptide was
coadministered i.c.v. with the opioid antagonist naloxone or
the competitive or non-competitive NMDA antagonists, CPP and
MK-801, respectively (fig. 3). Naloxone alone (1 nmol/mouse)
did not show significant analgeaic activity in the mouse
writhing test, and in combination with [Serl]HN (50nmol/mouse),
it did not significantly modify the antinociceptive activity
of the peptide (fig. 3). Both NMDA receptor antagonists, CPP
(0.1 nmol/mouse) and MK-801 (0.3 nmol/mouse) injected alone,
did not display significant analge8ic activity; but they
significantly antagonized the analgesic activity of [Ser1]HN
(50 nmol/mouse; fig. 3).
Peripheral Activity
Peripheral (i.p., i.v. and s.c.) administration of
dynorphin A-(1-13) was recently shown to cause analgesia in
- 17 -
72591-10
CA 02219437 1997-10-24
the mouse writhing assay (33). In order to verify if the
naloxone-insensitive antinociceptive effects of HN related
peptides could be ob~erved after such type of administration,
histone H4-(86-100), [Serl]HN, HN-(7- 15), SL-100 and cyclic
SL-100 (or SL-102) were administered i.p. at 5 ~mol/kg, and
the percentage of mice showing analge~ia was mea~ured 10 min
after the injection of the peptides and compared with that
obtained with morphine (5 ~mol/kg, i.p.; fig. 4). The five
peptides produced significant antinociceptive activity, the
incidence of positive re~pon~es being 50%, 63%, 62% and 65%,
respectively, as compared with 90% for morphine.
- 18 -
72591-10
CA 02219437 1997-10-24
Table 1: Relative potency of histogranin (HN) and related peptides (i.c.v.) in producing
analgesia in the r~ouse writhing pain assay.
Peptide EDso (nmol/mouse) Potency
(95% CL)a ratiob (95% CL)
HN 22.3 (12.141.1 ) 1.0
H4-(86-100) 4.1 (0.9-17 9) 5.4 (0.7~0.1)[Ser']HN 17.4 (7.043.0) 1.3 (0.4-3.8)
H4-(89-102) (OGP)C 40.9 (25.8-65) 0.5 (0.25-1.17)
HN-(7-15) 7.5 (2.3-24.4) 3.0 (0.8-11.2)
HN-( 1-10) NA
H N-(7-10) (S L-99) 11.3 (4.2-30.4) 2.0 (0.6-6.3)
Ala~ H N-(7-10) (S L-100) 3.9 (1.7-9.1) 5.7 (2.0-15.9)*
lArg', Ala9]H N-(7-1 0) (SL-1 01) 4.9 (1.8-1 3.Z) 4.5 (1.4-14.3)*
cyclo-(S L-1 01) (SL-1 0 2) 2.9 (0.8-9.8) 7.7 (1.3-46.6)~
[Ser', Ala~ H N (S L-104) 4.1 (1.5-11.5) 5.4 (1.6-18.0)~
~95% Confidence limit. bAs compared with HN. COGP: osteoblastic growth peptide. NA:
not active at 50 nmollmouse (i.c.v.). ~ P < 0.05 as compared with HN.
- 19 -
72591-10