Note: Descriptions are shown in the official language in which they were submitted.
CA 02223989 2008-11-27
PRODRUGS OF PHARMACEUTICALS
WITH IMPROVED BIOAVAILABILITY
The present invention relates to lipid derivatives of pharmaceutical agents.
It
relates particularly to lipid prodrugs of pharmaceutical agents, methods for
improving
the oral and/or tissue bioavailability of pharmaceutical agents, and the use
of the lipid
prodrugs in the treatment of cancer, viral infections, inflammatory disease,
and
proliferative disease.
Drugs administered orally can be absorbed through the oral mucosa, through the
lining of the stomach and primarily through the small and large intestines;
however, the
rate of absorption depends on the ability of the drug to pass through the
lipid barrier
of epithelial membranes. For example, alcohol, -a lipid soluble, non-ionic
compound,
is rapidly absorbed into the bloodstream by diffusion across the gastric
mucosa. Weak
acids are also well absorbed through the lining of the stomach, while weak
bases are
absorbed mainly in the small intestine. Drugs that are ionized, or lipid
insoluble, for
example, quaternary ammonium compounds and streptomycin, are poorly absorbed
in
the digestive tract, and must be administered by injection. Although injected
drugs are
not subject to the gastrointestinal 'barriers to bioavailability which are
imposed on drugs
administered orally, nonetheless, minimal tissue uptake or lack of tissue
retention often
interfere with the injected drug's bioavailability.
Under normal circumstances, intact dietary lipids, mostly triglycerides and
phospholipids, are not readily absorbed through the intestinal mucosa.
Phospholipids
are present physiologically in the gut as phosphatidylcholine,
phosphatidylethanolamine,
phosphatidylinositol, phosphatidylserine, phosphatidylglycerol and
phosphatidic acid.
The norrial physiological mechanism for lipid absorption requires conversion
of the
phospholipid to lysophospholipids by removal of the sn-2 acyl group by the
hydrolytic
action of the pancreatic enzyme phospholipase A2 on the sn-2 acyl ester bond.
Conversion of phospholipids to lysophospholipids provides the normal mechanism
for
absorption and transport of this class of lipids from the gut and accounts for
the uptake
of several grams of phospholipid per day.
While the need continues for less toxic, more selective, and more effective
prodrugs of all types, bioavailability of pharmaceutical agents.remains an
important
1
CA 02223989 2008-11-27
problem. Many oral drug candidates fail because of difficulty in oral
absorption or in
penetration of the cellular membranes of target tissues in the body after
injection.
Many intravenous, intraperitoneai, or other injectable drug candidates fail
because of
difficulty in penetrating cellular membranes of target tissues and/or failure
to be
retained in the tissue.
For example, the antiviral compounds phosphonoacetate and phosphonoformate,
which were first synthesized in 1924 (Nyl6n, Chem. Berichte 57:1023), have the
ability
to inhibit viral enzymes selectively. This ability was not, immediately
demonstrated.
Helgstrand, et at, Science 201:819-821 (September. 1,_1978) disclosed that
both
phosphonoacetic acid and phosphonoformic acid inhibit several, DNA.polymerases
and
preferentially inhibit several viral DNA polymerases. Phosphonoformate and
phosphonoacetate are presently known to selectively inhibit the DNA polymerise
of
many viruses, including human cytomegalovirus (HCMV), herpes simplex virus
(HSV)
and the reverse irunscriptase of human immunodeficiency virus (HIV). Chrisp =
and
Clissold ((1991) Drugs 41:104) review the pharmacology of these agents.
Phosphonoacetate is too toxic for use in humans, but phosphonoformate
(Foscavir,
Astra) is approved for human use in HCMV-infected AIDS patients. However, it
is not
highly potent, requires prolonged -intravenous administration and has
substantial toxicity
to the kidney and other organs. Ericksson, et at.. U.S. Patent Nos. 4,215,113;
4,339,445;
4,665,062; 4,771,041 teach the use of phosphonoformic acid as, the selective
agent in
treating viral infections, including herpes virus type land II and
cytomegalovirus, in
treating cancer caused by virus, and also opposing transformation of cells
caused by
oncogenic viruses.
Derivatized ` forms of phosphonoacids and pharmaceutical formulations
comprising these compounds are known. U.S. Patent, No. 5,072,032 to McKenna )
discloses thiophosphonoacids; Nos. 4,386,081 and 4,591,583 to Helgstrand et
al.
disclose phosphonoformic acid esters of alkyl, alkylene, alkoxy and related
cyclic and
aromatic groups and. some of these are shown to inhibit herpes virus and the
functions
and intracellular multiplications of influenza virus. U.S. , Patent No.
5,194,654 to
Hostetler et al., discloses phospholipid derivatives of phosphonoacids, their
incorporation into liposomes and their use as selective antiviral and
antiretroviral agents.
2
CA 02223989 2008-11-27
It would be useful to identify chemical structures for pharmaceutical prodrugs
which
enhance oral bioavailability and/or cellular uptake and retention regardless
of the route
of administration. The optimized prodrugs would be metabolized in target
tissues to
release the pharmaceutical agent, which agent would persist in the target
tissue to exert
its intended action either directly or after metabolic conversion to the
active form.
The present invention provides a series of improved prodrugs and their analogs
having substantial increases in -desired . activity over the parent compounds
against
various cancers, viral . diseases, autoimmune diseases, and other -
inflainmatory- and
proliferative diseases.
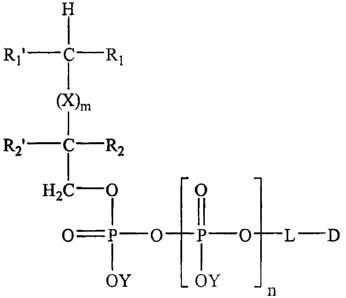
In one aspect, the present invention provides compounds of the formula:
(R1 1k -f-RI
(X)m
(R21k R2
n
O= P -_O_(P -Oj --(L)-D
I
OY OY
[I)
wherein R1 and Rl' are each independently an O-alkyl or S-alkyl group, wherein
the
alkyl group comprises a linear or branched, substituted or unsubstituted C, to
C24 group,
having from 1 to 6 double bonds; or an 0-acyl or S-acyl moiety, wherein the
acyl
group comprises a linear or branched, substituted or unsubstituted C, to C24
group,
having from 1 to 6 double bonds;
X is CH-R2;
m=0to6;
each k is independently = 0 or 1;
each R2 or R2' are' independently selected from the group consisting of H, =0,
the
halogen group consisting of fluorine, chlorine, iodine and bromine; O(CH2)pCH,
where
p is 0 to 7; NH2; O-alkyl or S-alkyl group, wherein the alkyl group comprises
a linear
or branched, substituted or unsubstituted C, to C8 group having from 0 to 3
double
3
CA 02223989 2008-11-27
bonds; O-acyl or S-acyl group, wherein the acyl group comprises a linear or
branched,
substituted or unsubstituted C, to C1 group having from 0 to 3 double bonds; N-
acyl
group wherein the acyl group comprises a linear - or branched, substituted or
unsubstituted C, to C, group having from 0 to 3 double bonds; N -alkyl or N-
dialkyl
group wherein the alkyl group comprises a linear or branched, substituted or
unsubstituted C, to C24 group having from 0 to 6 double bonds;
wherein Y is AA when said compound is in the form4of a salt or combination
thereof,
each AA is independently selected f om the group pousisting of W, Na, Lt1s, r,
NHS+;
amines selected from the group consisting _Qf mono-, di-, - trialkylamines,
and other
610 physiologically acceptable cations;
n=0, 1,or2;
wherein L is a linking molecule of the formula J-(CH2),-G wherein J and G are
functional groups independently selected from the group consisting of
hydroxyl,
sulfhydryl, carboxyl, and amine groups, wherein t = 1 to 24; or L is absent;
and
wherein D is a drug having a functional group selected from the group
consisting of
hydroxyl, sulfhydryl, carboxyl and amino groups.
Figure 1 shows spleen weights of mice treated with 1-O-octadecyl-sn-glycero-3-
phospho-AZT.
Figure 2 shows tissue distribution after 24 hours following oral uptake of
["C] PFA
analogs.
It is an object of the invention to provide lipid prodrugs of pharmaceutical
agents
that retain the phadnacological effects of the parent compounds and which
provide
improved oral bioavailability and/or tissue bioavailability. Unexpectedly, it
has been
found that the compounds of the present invention have advantageous
pharmacological
effects either over previously known- prodrugs of this type or over non-
derivatized
parent drugs. After oral, intravenous, intraperitoneal, intramuscular,
subcutaneous or
topical administration and upon uptake in the target tissues, the claimed
prodrugs are
converted to the active pharmaceuticals which persist intracellularly to exert
their
expected action. The claimed structures are advantageous in terms of target
tissue
4
CA 02223989 2008-11-27
uptake, conversion to active form, persistence in target tissue, and exertion
of expected
action. Accordingly, the compounds of the invention may be administered to
subjects
orally, intravenously, intraperitoneally, topically, subcutaneously,
intramuscularly or by
inhalation to treat diseases of mammals according to the use of the free;-
i.e. non.
derivatized drug. In comparison to lipid-conjugate derivative prodrugs, 'the
claimed
lipid-conjugates of the same drugs demonstrate superior oral absorption and
tissue
uptake by virtue of the lack of a free hydroxyl group at the R2 position, as
described
below.
The invention accordingly provides a series of improved prodrugs and their
analogs having substantial increases in desired activity over--be parent
compounds
against various cancers, viral diseases, autoimmune diseases, and other
inflammatory
and proliferative diseases. These enhanced activities can be demonstrated in
cell
culture, for example, by means of the in vitro susceptibility assays described
in
Examples 35 to 37 and in pharmacokinetic experiments described in Example 38
and
)15 Figure 2.
The invention provides compounds which are prodrugs of pharmaceutical agents
and which have the advantage of improved bioavailability after oral,
intravenous,
intraperitoneal, intramuscular, subcutaneous or topical administration. A
number of
drugs that have poor bioavailability, whether by oral, intravenous,
intraperitoneal,
intramuscular, subcutaneous or topical route of administration, can be made
suitable for
a route of administration by conversion to the lipid derivatives of the
invention,
particularly to substituted or unsubstituted 1-0-alkyl-propanediol-phosphate
derivatives,
wherein an C, to C24 alkyl group is attached to the 1-position of the
propanediol moiety
by an ether linkage.:.` It has been determined that the improved oral
bioavailability
and/or tissue uptake of the compounds of the present invention relies on the
lack of a
free hydroxyl at the R2 position in the claimed compounds. The inventors do
not fully
understand the reasons for the improved bioavailability but hypothesize that
without a
free hydroxyl group at the R2 position, the claimed compounds cannot be
metabolized
to less favorable alkyl/acyl species in vivo, which species are less effective
in oral
absorption and in tissue uptake and retention in vivo. Furthermore, the
propanediol
5
CA 02223989 1997-12-05
WO 96/39831 PCTIUS96/10054
lipid compounds lacking a hydroxyl at R2 are more hydrophobic and may cross
cell
membranes more effectively and may also be less cytotoxic.
The claimed method or strategy for making the claimed compounds is applicable
to any drug which has a chemical group capable of covalently binding to a
phosphate
group or capable of covalently binding to a linking group that can covalently
bind to
a phosphate group. As disclosed herein, drugs or pharmaceutical agents having
an
available hydroxyl, sulfhydryl, carboxyl or amine group can be covalently
linked by
either strategy to a phosphate group of a 1-O-alkyl-propanediol-3-phosphate or
to the
corresponding 1-0-acyl, 1-S-alkyl, and 1-S-aryl analogs to promote improved
bioavailability and/or tissue bioavailability of the drug. The linking group
is a
multifunctional molecule having the required covalent binding properties; for
example,
an hydroxylated carboxylic acid or an amino acid or a polypeptide. The alkyl
group
of the alkylpropanediols of the invention can be a straight, branched or
cyclic
hydrocarbon chain, having from 2 to 24 carbons, and can be saturated or
unsaturated
with up to six double bonds. Preferably the alkyl group has 8 to 24 carbon
atoms.
Alkyl groups having from 16 to 20 carbon atoms are most preferred. Taking note
of
formula I above, particularly preferred are compounds wherein Rl is an 0-
octadecyl
group. The alkyl group is attached to the propanediol moiety by an ether or
vinyl ether
bond. Also preferred are compounds wherein R2 is an O-benzyl or an OCH3 group.
In other embodiments, RI may be attached to the sn-3 position of propanediol
or
glycerol while the phosphate, linker and drug moieties are attached at the sn-
1 position.
Alternatively, the lipid moieties may also be racemic.
According to yet another aspect of the invention, there are provided 2-carbon
analogs of the compounds of the invention, having the general structure
(Formula [II]).
O 0
E~R_jj II
P-O-(PO)-(L)-D
OY OY
[ I]
6
CA 02223989 1997-12-05
WO 96/39831 PCT/US96110054
A preferred compound according to this embodiment is
1-0-octadecyl-1,2-ethanediol-2-phosphate adduct of pharmaceutical agents.
The- preferred lipid derivatives of the invention are of the formula:
(R1 )k-r-R1
(X)m
(R2)k L R2
O
0
II
0= i -0-(I -0)n -(L) -D
OY OY
wherein R1 and Rl' are each independently an O-alkyl or S-alkyl group, wherein
the
alkyl group comprises a linear or branched, substituted or unsubstituted C, to
C24 group,
having from 1 to 6 double bonds; or an O-acyl or S-acyl moiety, wherein the
acyl
group comprises a linear or branched, substituted or unsubstituted C, to C24
group,
having from 1 to 6 double bonds;
X is CH-R2;
m = 0 to 6;
each k is independently = 0 or 1;
each R2 or R2' is independently selected from the group consisting of H, =0,
the
halogen group consisting of fluorine, chlorine, iodine and bromine; O(CH2)PCH3
where
p is 0 to 7; NH2; O-alkyl or S-alkyl group, wherein the alkyl group comprises
a linear
or branched, substituted or unsubstituted C, to C. group having from 0 to 3
double
bonds; O-acyl or S-acyl group, wherein the acyl group comprises a linear or
branched,
substituted or unsubstituted C, to C. group having from 0 to 3 double bonds; N-
acyl
group wherein the acyl group comprises a linear or branched, substituted or
unsubstituted C, to C. group having from 0 to 3 double bonds; N-alkyl or N-
dialkyl
group wherein the alkyl group comprises a linear or branched, substituted or
unsubstituted C, to C24 group having from 0 to 6 double bonds;
7
CA 02223989 2008-11-27
wherein Y is A when said compound is in the form of a salt or combination
thereof,
each A' is independently selected from. the group consisting of H', Na', Li',
1', NHS';
amines selected from the group consisting of mono-, di-, trialicylamines, and
other
physiologically acceptable cations;
n=0, 1, or 2;
wherein L is a linking molecule of the formula J-(CH21-G wherein j and G are
functional groups independently selected from the group consisting of
hydroxyl,
sulfhydryl, carboxyl, and amine groups, wherein t s I to 24; or L is absent;
and
wherein D is a drug having a functional group selected from the group
consisting of
hydroxyl, sulfhydryl, carboxyl and amino grows. Linking groups can be any of
several molecules having multifunctional groups
comprising hydroxyl, sulfhydryl, carboxyl, and amino groups. Particularly
suitable for
use as linkers. are:
(1) 7 the amino alcohols, having the general structure HO-(CH?)IINH2, where n
= 1 to 24,
preferably where n = 2 or 3, and suitable for insertion at the carboxyl group
of a
candidate drug which is an active drug moiety or a chemically modified drug. A
1-0-
alkylpropanediol-3-phosphoetbanobamine is a naturally occurring phospholipid
that
incorporates a linker of the amino alcohol type, and a l-O-alcyl-propanediol-3-
phosphoethanolamine can be conveniently coupled to drugs having an available
carboxyl group to prepare a lipid prodrug of the invention..
(2) the hydroxyalkyl carboxylic acids, having the general structure HO-(CH2),
COON,
where n = 1 to 12, and' suitable for insertion at the amino group. of an
active candidate
drug. Naturally occurring molecules such as hydroxy fatty acids, beta-
hydroxybutyric
acid, and hydroxyamino acids such as serine and hydroxyproline may also be
conveniently used.
Finally, the phosphate-linker-drug moiety of the invention may be replaced by
phosphonoformate, phosphonoacetate, thiophosphonoformate and
thiophosphonoacetate
or their respective carboxymethyl or carboxyethyl esters.
The present invention provides claimed structures of -pharmaceutical agents
and
methods of use which provide advantages compared to the free, non-derivatized
forms
of the pharmaceutical agents in terms of target tissue uptake, conversion to
active form,
8
CA 02223989 2008-11-27
persistence in target tissue, and exertion of expected action. Advantages of
the claimed
structures and methods are manifest as improved usefulness, efficacy,
biological half
life;. transport across cellular membrane, i.e. bioavailability after oral,
intravenous,
intraperitoneal, intramuscular, subcutaneous or topical administration of any
drug having
a chemical structure. suitable for binding as described herein. The method of
the
invention is advantageously applicable to drugs that are poorly bioavailable,
regardless
of route of administration 'Examples of the variety of therapeutic classes of
drugs that
can. be.. effectively administered by the oral route comprise 1-0-alky 1-0-
acyl,
__ _L-S-alkyl (thioether), or 1-S-acyl (thioester) pr+opanediol derivatives,
of
(a) anticancer agents, comprising nucleoside, analogs, for example,
I -P-D-arabinofuranosylcytosine (hereinafter, cytosine " arabinoside or ara-
G'), 9-13-)-
arabinofmanosyladenine (hereinafter, adenine. arabinoside or araTA), 5-
fluorouridine,
6-mercaptopurine riboside, or 2'-ara-fluoro-2-clilorodeoxyadenosine;
(b) antiviral nucleosides, particularly the I-O-alkyl-propanediol-3-phosphate
derivatives
1S of acyclovir, ganciclovir and the antiviral nucleosides disclosed in U.S.
Patent no. 5,223,263
(c) therapeutic peptides. or peptidomimetics, or peptides that are enzyme
inhibitors,
comprising D-amino acids, L-amino acids, or amino-acid analogs, and having up
to
about 35 amino. acids, preferably less than 6 amine acids, or ' analogs
thereof
particularly the lipid derivatives disclosed in U.S. Patent no. 5,554,728
In a preferred embodiment of this species,
a l -O-alkyl-propanediol-3-phosphate derivative of desmopressin, n- muramyl
tripeptide,
or cnalkiren is synthesized and administered orally.
(d) antibiotics, particularly those of the penicillin and cephalosporin class,
including
penicillin G, cefazolin,' ceftazidime, ceftriaxone, or piperacillim
(e) phosphonoacid compounds, particularly the 1-0-alkyl propanediol
derivatives of
phosphonofgrmic acid and_phosphonoacetic acid, and nucleoside phosphonates
disclosed
in U.S. patent no. 5,194,694
(f) 5-amino(I-beta-D-ribofuranosyl) imidazole carboxamide or 1-beta-D-
ribofuranosyl
1,2,4-triazole-3-carboxamide, which are used for the treatment of allergy,
including
9
CA 02223989 2008-11-27
asthma and urticaria eczema; autoimmune disease, including Lesch Nyhan
disease,
cardiac disorders related to restricted blood flow or viral diseases.
(g) non-sterroidal 'anti-inflammatory compounds,particularly the 1-0-
alkylphospholipid
derivatives of these compounds
Table 1 lists preferred drug candidates for the method of the invention
according to
therapeutic class.
TABLE I
Candidate UM .for Preparation of Orally Bioavailable
Lipid Prodrugs -r- , _
THERAPEUTIC CLASS: MERCK INDEX
Antuciolas~c .agents
1.
actinomycin D:
bleomycin 1324-
cisplatu- anti- Pt analogs:
carboplatin, iproplatin 2319,1828
cytosine arabinoside 2790:
daunorubicin 2825
doxofluoridine 3426
doxorubicin 3428
etoposide 3842
floxuridine 4045
mithramycin
mitomycin= C 6133
mitoxanthrone 6135
pentostatin (deoxycoformycin) 7091
phosphonoacids
streptozotocin 8794
taxol and taxotere 9049
ulna alkaloids:
vincristine 9291
CA 02223989 2008-11-27
vinblastine 9887
vindesine 9892
II. Anti-Infectives
aminoglycerides:
netilmycin 6389
amikacin 416
gentamycin 4284
streptomycin 8786
kanamycin A 5161
tobramycin 9413
neomycin B 6369
pliocarmycin 7510
amphotericin B 620
vancomycin 9869
)15 III. Antivirals
(AZT; anti-HIV) 139
acyclovir (herpes simplex, anti-HSV) 4166
foscarnet 4166
ganciclovir (anti-CMV) 4262
idoxuridine (anti-HSV keratitis) 4262
ribavirin 8199
5-fluoro-3'-thia-2',3'-dideoxcytidine 9599
(anti-HBV, HIV)
trifluridine` (herpes group, eye)
vidarabine (HSV encephalitis)' 9881
IV. Short Peptides or p,eptidomimetics
corticotropin (ACTH) 127
calcitonin 1640
desmopressin (DDAVP) 2904
gonadotropin RH (LH-RH) 5354
ry1
CA 02223989 2008-11-27
goserelin (LHRF) 4433
insulin 4887
lypressin 5503
beta-melanotropin (fl-MSH) 6206
alpha melanotropin (a-MSH) 6206
muramyl dipeptide 6214
oxytocin 6934
vasopressin 9843
FK-506
octreotide 6682
enalkiren renin inhibitor
aspartyl protease inhibitors (anti-HIV)
serine protease inhibitors
V. Miscellaneous Agents
morphine (narcotic analgesic) 6186
prostaglandin 7891
leukotrienes 5339
cyclosporins (immunosuppressive) 2759
A significant aspect of the compounds of the invention and related methods for
oral administration of drugs is that 1-0-alkyl-, 1-0-acyl-, I -S-alkyl-, and
1-S-acylpropanediol-3-phosphate derivatives require no metabolic conversions
for oral
absorption. These lipid prodrugs are in this way distinct from phosphatidyl
derivatives,
for which metabolic processing requires preliminary conversion to a
lysophospholipid.
Furthermore, the alkyl group at the 1-position of the. propanediol moiety of
the
1-0-alkyl derivative cannot be degraded by intestinal lysophospholipases
because of the
ether bond linking the alkyl group to the glycerol structure. This metabolic
feature
prevents digestive degradation and facilitates the intestinal uptake of the
intact
1-S-alkyl- and 1-O-alkyl-propanediol-3-phosphate drug conjugate together with
other
lysophospholipids that are undergoing membrane transport in the small
intestine. The
12
CA 02223989 2008-11-27
1-O-acyl and the corresponding thioester analogs may also be absorbed
substantially but
are less preferred in applications wherein this property is required.
= In contrast to the prior 1-0-alkyl-glycero-phospho-drug compounds, an
important
design feature of the lipids of this invention is the absence of a free
hydroxyl at the
2-position of glycerol. This prevents the formation of unfavorable 1-alkyl,
2-acyl-glycero-phosphate-drug metabolites which may not be subject to prompt
passage
through the small intestine and, further, may not be metabolized readily
intracellularly
to yield the desired active drug moiety. In addition, the compounds of to
invention
are more hydrophobic and may cross cell. membranes more readily and may
exhibit less
cytotoxicity.
Coupling of Lipid Moiety to a Candidate Drug
The compounds of the invention are formed according to synthetic procedures
which couple a substituted or unsubstituted I-O-alkyl-propanediol-3-phosphate,
or
1-0-acyl, I-S-alkyl, or 1-S-acyl analogs thereof to a drug or which couple a
substituted
)15 or unsubstituted I -0-alkyl propanediol or 1-0-acyl, 1-S-alkyl, or 1-0-
acyl analogs
thereof, to a phosphorylated functional group of a drug.
The 1-0-alkyl propanediol moiety, or any other analog described above, and the
drug can be covalently linked through mono-, di-, or triphosphate groups at
the 3
position of the propanediol structure. When the 1-0-alkyl propanediol and the
drug are
joined through a linking group, the linker molecule is conveniently attached
to the
terminal phosphate of, for example, 1-0-alkyl-propanediol-3-phosphate. In
either case,
the candidate drug has an available functional group.
A reaction is typically carried out at a temperature of 25 to 60 C,
preferably
to 50 C for a period of from 2 to 25h, preferably 8 to l0h. N,N'-Dicyclohexyl-
25 carbodiimide (DCC) is added in measured portions generally over a period of
0.5 to 3h,
preferably 0.75 to 1.5h. The reaction mixture is worked up by addition of
water and
azeotroped by successive additions of toluene and ethanol. The resulting crude
product
is purified by ion exchange and silica chromatography to afford the desired
compound
with the desired purity.
30 The process of the invention is preferably conducted in the liquid phase.
Upon
addition of either triisopropylbenzenesulfonyl chloride (TIPS) or
13
CA 02223989 2008-11-27
N,N'-dicyclohexylcarbodiimide (DCC), the reaction mixture is heated to a
temperature
of 30 to 60 C. It is noted that the presence of equivalent (or more than
stoichiometric), amounts of either TIPS or DCC does not impede the course of
the
reaction.
The temperature of the reaction mixture can rise up to its boiling point. The
heat of the reaction can be removed by external cooling of the reaction vessel
or by
means of a cooled reflux condenser.
Suitable solvents for the reaction are amines or derivatives thereof.
Preferred
solvents include tertiary amines such as diisopropylethylamine, triethylamine,
tributylamine, or heterocyclic amines such as pyridine or picolines. 1-0-alkyl
analogs
of the invention, for example, 1-O-octadecyl-propanediol-3-phosphate
derivatives, or
any of the other 1-0-acyl or 1-S-acyl or 1-S-alkyl analogs, can be produced by
any of
the synthetic organic procedures known to those in the art, for example,
condensation
of 1-0-alkyl-propanediol and the monophosphate of the drug candidate such as
ara-C
monophosphate as described in Example 2 (compound IIa). An alternative
approach
links 1-O-alkylpropanediol-3-phosphate to the hydroxyl of a candidate drug in
the
presence of a condensing agent such as DCC or TIPS (Example 5).
In another variation of the method, 1-O-octadecyl-2-O-benzyl-sn-glycero-3-
phosphate was condensed with ara-C while the hydroxy group in 2-position, of
the batyl
alcohol was protected as the benzyl ether.
Lipid Prodrug Derivatives of Taxol-Related Compounds
Lipid derivatives of.taxol are prepared according to a procedure wherein the
amino alcohol and hydroxy carboxylic acid units of the taxol side chain are
covalently
attached to a phosphatidic acid, preferably a 1-O-alkylpropanediol-3 phosphate
as set
forth in Examples 13 through 16. The side chain can be derivatized by the
insertion
of an aliphatic group (CH2)p to increase lipophilicity.
According to the general procedure, a substituted f3-amino-a-hydroxy-benzene
propanoate is covalently linked to a phosphatidylglycerol or a 1-0-alkyl- or 1-
0-acyl-2
benzyl-sn-glycero-3-phosphatidic acid in the presence of a condensing agent,
such as
DCC, to provide compounds of the formula:
14
CA 02223989 2008-11-27
(Qm
~t 0 H
n
F-o-C-(CH2n--CO2R3
0 c H3- -phenyI
NHR4
wherein
R3 is any hydrolyzable ester group, for example, methyl, ethyl, or pivaloyl;
,R4 is benzoyl, pivaloyl, acetate, peptides, or amino acids; and
n is 0-10.
In alternative embodiments, R, and R2 are attached' to the glycerol group by
thioester or thioether bonds. Ina preferred embodiment, R, is an ether-linked
1-0-
octadecyl propanediol group, and R4 is benzyl, and an l-O-alkyl-propanediol-3-
phosphate is condensed with a a-(benzoylamino)-ac-hydrobenzene propanoate
ester to
form a lipid derivative of the taxol side chain. The propanoate ester is then
hydrolyzed
to yield the propanoic acid which is ready for coupling with baccatin III, or
10-deacetyl
baccatin, having the formula:
0
N tOH
40
CA 02223989 1997-12-05
WO 96/39831 PCT/US96/10054
to form an orally bioavailable taxol compound.
Lipids comprising fatty acids, fatty alcohols, glycerides, and phospholipids
for
use in preparing the lipid prodrugs of the invention may be purchased from
commercial
suppliers (Avanti Polar Lipids, Inc., Pelham, Ala.; or Genzyme Corp.,
Cambridge,
Mass.) or may be synthesized according to known methods. 1-O-Octadecyl-sn-
glycerol
(batyl alcohol) is available from Sigma, St. Louis, MO, and a 2-O-benzyl
derivative of
batyl alcohol is available from Bachem, Inc., Basel, Switzerland. Other
lysophosphatidic acids useful in preparing the prodrugs of the invention are
available
from Genzyme, Cambridge, Mass. The drugs to which these lipids are covalently
linked can be purchased from the pharmaceutical manufacturers.
It is important that all traces of water be removed from the reactants in
order for
the coupling reactions to proceed. Therefore, the lipids are first either
freeze-dried by
solvent evaporation under vacuum, or in a vacuum oven over P205. The reactions
are
also carried out under an inert gas, such as, for example, argon.
The synthetic reactions are followed using thin layer chromatography (TLC)
with
appropriate solvents. When the reaction is complete as determined by TLC, the
product
is extracted with an organic solvent and purified by chromatography on a
support
suitable for lipid separation, for example, silicic acid.
Efficacy and Potency of 1-O-alkvlpropanediol-3-nhosnhate Prodrugs
The lipid derivative prodrugs of the invention, preferably l-O-
alkylpropanediol-
3-phosphate prodrugs, have advantageous pharmacological properties in
comparison to
the non-derivatized drugs.
The efficacy of the lipid prodrugs of the invention was demonstrated in tests
carried out both in vitro and in vivo. 1-O-Octadecyl-sn-glycero-3-phospho-3'-
azido-3'-
deoxythymidine (AZT) was used in oral absorption studies. This compound has an
18-
carbon alkyl ether at position 1 of glycerol; the hydroxyl at position 2 of
glycerol is
open, and position 3 is linked by a phosphodiester bond to 3'-azido-3'-
deoxythymidine
(AZT)-5'-monophosphate. 1-O-Octadecyl-sn-glycero-3-phospho-AZT does not
require
any metabolic conversions for absorption and appears to be absorbed directly
from the
gastrointestinal tract. It is not subject to deacylation by lysophospholipases
in the gut
16
CA 02223989 1997-12-05
WO 96139831 PCT/US96/10054
because of the ether bond at position 1 of glycerol. Its metabolism is not
known but
it is hypothesized that the compound is metabolized by cellular enzymes and
phosphodiesterases releasing 3'-azido-3'-deoxythymidine (AZT) or AZT-MP inside
the
cell.
The in vivo study as described in Example 16 demonstrates that a 1-O-alkyl-sn-
glycero-3-phosphate drug derivative has the same pharmacological efficacy as
that of
the non-derivatized agent. It further demonstrates that oral dosing with the 1-
0-
octadecyl-glycero-3-phosphate derivative can allow more convenient and
effective
administration of AZT. 1-O-Octadecyl-sn-glycero-3-phospho-AZT was compared to
free AZT in treating mice infected with Rauscher murine leukemia virus (RLV).
RLV
is a murine retrovirus, and RLV-infected mice are useful as a model system for
evaluating therapeutic effectiveness of candidate anti-AIDS drugs against
retrovirus
induced disease in vivo. RLV infects splenocytes and the infected animals
exhibit
massive splenomegaly. Effective antiviral agents inhibit the splenomegaly, and
a
reduction in organ weight correlates with the elimination of virus (Ruprecht,
R_, et al.,
Nature 323:467-469 (1986)). Because AZT has a short physiological half-life,
the most
effective mode of AZT therapy should be continuous oral administration. The
closest
practical approach to optimum administration is the intake of AZT in drinking
water.
Oral administration of batylphosphate-AZT on a once a day regimen of gavage
proved
to be as effective, in comparable doses, as virtually continuous free AZT
administration,
as determined by inhibition of splenomegaly in the infected mice (FIG 1).
It is anticipated that the lipid compositions of the invention, when linked to
AZT, will have equivalent or superior activity to 1-O-octadecyl-sn-glycero-3-
phosphate-
AZT.
Lipid Prodrugs of the Phosphonoacids
It is an object of the invention to provide lipid prodrugs of the
phosphonoacids
that retain the pharmacological effects of the parent compounds. Unexpectedly,
it has
been found that the compounds of the present invention have advantageous
pharmacological effects over previously known prodrugs of this type. The
invention
accordingly provides a series of improved prodrugs of phosphonoformate and
17
CA 02223989 2008-11-27
phosphonoacetate and their analogs having substantial increases in antiviral
activity over
the parent compounds against human cytomegalovirus (HCMV), herpes simplex
virus
(HSV), and human immunodeficiency virus (HIV-1). This enhanced antiviral
activity
can be demonstrated in cell culture, for example, by means of the in vide
susceptibility
assays described in Examples 35-37.
Synthesis of improved prodrugs of the phosphonoacids
(R1)--,---R1
(gym
(R2) R2
O !Z!
P (CH4--COY
OY
25 The compounds that are synthesized are outlined in Scheme I with the
various
substitutes on Cl, C2 and C3 of the glycerol, and similarly in Schemes II and
III for
compounds wherein (X). is (CH-R2), and inal.
Identification of Starting Materials- and Products
The phosphonoacids to which various lipid moieties are coupled in the
preparation of the lipid prodrugs of the invention are designated by acronyms,
as
follows:
n = 0, Z = O'(PFA) Phosphonoformic acid
n = 0, Z = S (PFSA) Thiophosphonofoninic acid
18
CA 02223989 2008-11-27
n = 0, Z = Sc (PFSeA) Selenophosphonoformic acid
n = 1, Z =O'(PAA) Phosphonoacetic acid
n - 4, Z = S (PASA) Thiophosphonoacetic acid
n - 1, Z = Se (PASeA) Selenophosphonoacetic acid
The various lipid prodrug derivatives are designated herein by acronyms
derived
from those above, and defined in the legends of Tables I-III.
19
CA 02223989 2008-11-27
SCHEME I
--OH --ORI -OR,
a b, c
` ~O --30- HO-
0- 0 Trityl
d
RI R, ORS
Z f Z e Z
H OTrityl
O=P-COOR3
0'
4
R 1 = -(CH2)nCH3, n = 7, 9, 11, 13, 15, 17
Z = Cl, Br, I, -OCH3
R3 = -CH3, -CH2CH3 and -CH2CH2CH3
a: NaH, DMF, R1-OSO2Me; b: Acetic acid; c: Trityl chloride, pyridine;
d: NaH, DMF, R2-Br e: TFA, CH2CI2; f: C12P000OR3; H2O
CA 02223989 1997-12-05
WO 96/39831 PCT/US96/10054
SCHEME II
Example: Synthesis of phosphonoformic acid analog =CHOH, m=1, R2=OH,
R1=Octadecyl
HO O HO
OH i OH n p~ '
OH OH
CH2OH CH2OBn CH2OBn
1 2
D-Erythrose
4CH2OR H2OR CHOOH
t ~>< v --~< iv
CH2OH CH2OBn CH2OBn
4
vi
CH2OR CH2OR
vii, viii ON OH
~ OH
CH2O- i -COOEt CH2O- i COONa
OH OH
Z $
i. BnBr, NaH; ii. DMP, H+; iii. NaBH4; iv. ROSO2Me (R=Octadecyl), NaH;
v. H21 Pd/C; vi. DCC, ethyl phosphonoformate; vii. TFA, CH2C12;
21
CA 02223989 1997-12-05
WO 96/39831 PCTIUS96/10054
SCHEME III
Example: Synthesis of phosphonoformic acid analog =CHOH, m=2, R2=0H,
R1=Octadecyl
SMe SMe
0 CH-SMe CHSMe
OH OH ii OH
OH OH OH
OH OH OH
CH2OH CH2OH CH2OBn
Q (D-Ribose) 14 11
SMe
I
CH2OH CHO CH SMe
OMOM OMOM iv OMOM
OMOM OMOM .0 OMOM
ONION! OMOM OMOM
CH2OBn CH2OBn CH2OBn
14 12 12
vi
CH2OR CH2OR CH2OR
OMOM vii. viii OMOM ix, x 4_OH
OMOM OMOM 30 OH
OMOM OMOM OH
CH2OBn 0i 1
CH2O-P - COOEt CH2O- P - COONa
OH OH
L~ 11
i. McSSLMe3, Zn12; ii. BnBr, NaH; iii. CH3OCH2C1 (MOM chloride), NaH, THF;
iv. AgNO3, Ag20; v. NaBH4; vi. ROSO2Me, NaH; vii. H2, Pd/C; viii. DCC, ethyl
phosphonoformate; ix. Acetic acid; x. NaOH, ethanol
22
CA 02223989 2008-11-27
Synthesis Procedures
- Lipid prodrugs of the phosphonoacids listed above are prepared according to
the
procedures described in Examples 28, 29 and 33. A synthesis flow diagram
particularly
relevant to the synthesis of compounds wherein m = 0 and Y is absent is set
forth in
Scheme 1; flow diagrams particularly relevant to the chemical synthesis of
compounds
wherein m > 0 and Y is present are set forth in Sc mes II and M. v
Antiviral Activity
The antiviral activity of various lipid derivatives of phosphonoacids
according
to the invention was determined in cultures of human cell lines infected with
HCMV,
HSV, or MV -1 as described in Examples 35-37. The results are shown in Tables
I-III.
The predictive value of in vitro susceptibility testing for virus infections
is discussed
by Kern,. E.R. (1990) Preclinical evaluation of antiviral agents: In vitro and
animal
model testing. Galasso, G. et al. eds., Antiviral Agents and Viral Disease of
Man 3rd
Edition, Raven Press, NY, pp. 87-123.
The most preferred compounds which exhibit greatly increased antiviral
activity
(Tables I-ITI) have 1-0-alkyl'groups at RI, and 0-methyl or O-benzyl or 2,2-
dimethoxy
groups at R2.
Antiviral 'activity of the improved phosphonoacid prodrugs against human
cytomegalovirus-infected MRCS human lung fibroblast cells is shown in Table
II. The
most preferred prodrugs of phosphonoformate have remarkable increases in
antiviral
activity. Previous attempts to produce prodrugs of phosphonofonmate with
increased
activity have identified a few compounds which have very small increases in
activity,
but no compound having increases in activity over PFA greater than 1.9 fold
have been
shown previously (Nor6n, J.O., et al., J. Med Chem. 2664-270, 1983). The most
active PFA prodrugs, B-PFA, BB-PFA, MB-PFA, and ODDMOP-PFA exhibit 107-,
72-, 38- and 209- fold increases in activity and represent the most active PFA-
containing compounds yet reported. These compounds have a 1-0-alkyl group at
the
Rl position of glycerol and either a hydroxyl, -0-benzyl or -0-methyl or 2,2
dimethoxy
function as the R2 of glycerol or propane. Prodrugs having H, halogen or amino
as R2
23
CA 02223989 1997-12-05
WO 96/39831 PCTIUS96/10054
will also be highly active and substitution at X of S or Se for 0 will provide
similar
results.
- The improved PFA prodrugs also exhibit greatly increased activity versus PFA
in herpes simplex virus-1 infected human lung fibroblasts (Table III). MB-PFA,
B-PFA
and BB-PFA are 72-, 43- and 34-times more active than PFA and represent the
most
active PFA derivatives yet reported. The order of activity is slightly
different from that
observed with human cytomegalovirus; MB-PFA is the most active compound
followed
by B-PFA and BB-PFA. Similar results were obtained with human immunodeficiency
virus-1 infected cells in vitro (Table IV). With HIV-1, MB-PFA was the most
active
compound followed by B-PFA and BB-PFA; the compounds were 104-, 37- and 9-fold
more active than PFA in HIV-infected HT4-6C cells and represent the most
active
anti-HIV derivatives of PFA reported. MB-PFA was more active than B-PFA to a
statistically significant degree.
Summaries of the antiviral activity of selected compounds according to the
invention are provided in Tables V and VI.
Therapy of Viral Diseases
The lipid derivatives of antiviral nucleoside analogs and phosphonoacids
disclosed herein are useful in treating diseases caused by viruses such as
influenza,
herpes simplex virus (HSV), human herpes virus 6, cytomegalovirus (CMV),
hepatitis
B virus, Epstein-Barr virus EBV), and varicella zoster virus (VZV). They are
useful
in the treatment of AIDS and other retroviral diseases, as well.
Lipid derivatives of antiviral drugs may be applied topically to the skin,
eyes or mucus
membranes or into the interior of the body, for treating susceptible virus
infections in
man and animals. They can be introduced internally, for example orally,
intratracheally
or otherwise by the pulmonary route, enternally, rectally, nasally, vaginally,
lingually,
intravenously, intra-arterially, intramuscularly, intraperitoneally,
intradermally, or
subcutaneously. The present pharmaceutical preparations can contain the active
agent
alone, or can contain further pharmaceutically valuable substances. For
example,
formulations comprising lipid phosphonoacid prodrugs of the invention can
additionally
comprise another antiviral agent, such as for example, a viral protease
inhibitor, or an
24
CA 02223989 2008-11-27
antiviral nucleoside analog. They can further comprise a pharmaceutically
acceptable
carrier.
- Lipid derivatives of antiviral agents may have a prolonged antiviral effect
as
compared to the lipid free agents; therefore they provide therapeutic
advantages as
medicaments even when not incorporated into liposomes. These phosphonoacid
prodrug antiviral agents may be used alone or in combination with antiviral
nucleosides
as given conventionally: The use of combination therapy may greatly reduce A
he
tendency for drug resistant HIV mutant strains to appear and would therefore
increase
the likelihood of stopping_the progression of HIV infection. The some argument
would
hold equally well in treating' cy omegalovirus or herpes virus infections with
regard to
the likelihood of developing resistant strains.
Formulations
? Pharmaceutical preparations containing lipid derivatives of antiviral
nucleoside
analogs or phosphonoacids are produced by conventional dissolving and
lyophilizing
processes to contain from approximately 0. Wo to 100%, preferably from
approximately
I% to 90% of the active ingredient. They can be prepared as ointments, salves,
tablets,
capsules, powders or sprays, together with effective excipients, vehicles,
diluents,
fragrances or flavor to make the formulations palatable or pleasing to use.
Formulations fore oral ingestion are in the form of tablets, capsules, pills,
ampules of
powdered active agent, or oily or aqueous suspensions or solutions. Tablets or
other
non-liquid oral compositions - may contain acceptable excipients, known to the
art for
the manufacture of pharmaceutical compositions, comprising diluents, such as
lactose
or calcium carbonate; binding agents=such as gelatin~or starch; and one or
more agents
selected from the group consisting of sweetening agents, flavoring agents,
coloring or
preserving agents to provide a palatable preparation. Moreover, such oral
preparations
may be coated by known techniques to further delay disintegration and
absorption in
the intestinal tract. The preparations can also comprise bile salts and
detergents.
Aqueous suspensions may contain the active ingredient in admixture with
pharmacologically acceptable excipients, comprising suspending agents, such as
methyl
cellulose; and wetting agents, such as lecithin, lysolethicin or long-chain
fatty alcohols.
CA 02223989 1997-12-05
WO 96/39831 PCTIUS96/10054
The said aqueous suspensions may also contain preservatives, coloring agents,
flavoring
agents and sweetening agents in accordance with industry standards.
Preparations for topical and local application comprise aerosol sprays,
lotions, gels and
ointments in pharmaceutically appropriate vehicles which may comprise lower
aliphatic
alcohols, polyglycols such as glycerol, polyethylene glycol, esters of fatty
acids, oils
and fats, and silicones. The preparations may further comprise antioxidants,
such as
ascorbic acid or tocopherol, and preservatives, such as p-hydroxybenzoic acid
esters.
Parenteral preparations comprise particularly sterile or sterilized products.
Injectable
compositions may be provided containing the active compound and any of the
well
known injectable carriers. These may contain salts for regulating the osmotic
pressure.
Liposomal Formulations
If desired, the claimed compounds can be incorporated into liposomes by any
of the reported methods of preparing liposomes for use in treating viral
diseases such
as, but not limited to HCMV, HSV, and HIV-1. The present invention may utilize
the
antiviral derivatives noted above incorporated in liposomes in order to direct
these
compounds to macrophages, monocytes, other cells and tissues and organs which
take
up the liposomal composition. The liposome-incorporated antiviral derivatives
of the
invention can be used to treat HCMV, HSV or AIDS patients by parenteral
administration, enhancing delivery of the antiviral compound to macrophages
and
monocytes, an important reservoir of viral infections. This will allow for the
efficacious use of lower doses of the modified phosphonoacids, reducing
toxicity of the
compound. Ligands may also be incorporated to further focus the specificity of
the
liposomes.
The derivatives described have several unique and novel advantages over the
liposomal water soluble antiviral drugs. First, they can be formulated in
liposomes to
much higher ratios of drug to lipid because they are incorporated into the
wall of the
liposome instead of being located in the aqueous core compartment. Secondly,
the
liposomes containing the lipophilic antiviral derivatives noted above do not
leak during
storage, providing improved product stability. Furthermore, these compositions
may be
26
CA 02223989 1997-12-05
WO 96/39831 PCT/US96/10054
lyophilized, stored dry at room temperature, and reconstituted for use,
providing
improved shelf life. They also permit efficient incorporation of antiviral
compounds
into liposomal formulations without significant waste of active compound. A
further
advantage is that the compositions used in in vivo treatment cause a larger
percentage
of the administered antiviral prodrug to reach the intended target. For
example, the use
of the compositions reduces the amount being taken up by the kidney and bone,
thereby
decreasing the toxic side effects of the phosphonoacid drugs. The toxic side
effects of
the phosphonoformates may be further reduced by targeting the liposomes in
which they
are contained to actual or potential sites of infection by incorporating
ligands into the
liposomes. The liposome-incorporated lipid-antiviral conjugate is administered
to
patients by any of the known procedures utilized for administering liposomes.
The
liposomes can be administered intravenously, intraperitoneally,
intramuscularly,
intravitreally or subcutaneously as a buffered aqueous solution. Any
pharmaceutically
acceptable aqueous buffer or other vehicle may be utilized so long as it does
not destroy
the liposome structure or the activity of the lipid phosphonoacid analog. One
suitable
aqueous buffer is isotonic sorbitol containing 5mM sodium phosphate with a pH
of
about 7.4, or other physiological buffered salt solutions.
The therapeutically effective amount of the lipid derivatives is determined by
reference to the recommended dosages of the active antiviral drug, bearing in
mind that,
in selecting the appropriate dosage in any specific case, consideration must
be given to
the patient's weight, general health, metabolism, age and other factors which
influence
response to the drug. The dosage for a mammal, including a human, may vary
depending upon the extent and severity of the infection and the activity of
the
administered compound. Dosage levels of liposomal lipid analogs of antivirals
will be
about the same as for the antiviral itself. Dosage levels for antiviral
nucleosides and
phosphonoformate through conventional administration by intravenous infusion
are well
established (Lambert, R., et al. (1989) J. Med. Chem. 32:367-374; Szoka, F.
and Chu,
C-J., Antimicrobial Agents and Chemotherapy 32(6):858-864 (1988); Ericksson et
al.
U.S. Patent No. 4,771,041). Foscarnet is administered by i.v. infusion at
200mg/kg/day
for treatment of HCMV in humans.
27
CA 02223989 2008-11-27
The phosphonoacid prodrugs of the invention are administered to the patient on
.a daily basis in an oral dose of about 0.1mg/icilogram to 1000mg/kilogram and
more
preferably from about 1 mg/kilogram to about 200mg/kilogram. The parenteral
dosage
will be appropriately 20 to 100% of the oral dose.
i iposome - ion
After synthesis and purification, the lipid derivative -mof the antiviral is
incorpor$ted into liposomes, or other suitable carrier. The incorporation, c
6n be carried
out, according to well known liposome preparation pwoodums, such as sonication
and
extrusion. Suitable conventional methods of liposome preparation include, but
are not
limited to, those disclosed by Bangham, et al. (Bangham, A.D.,9Standish, M.M.
and
Watkins, J.C. (1965) J. Mol. Biol.. 23:238-252.) Olson, et al. (Olson, F.,
Hunt, C.A.,
Szoka, F.C., Vail, W:J. - . and Papahadjopoulos, D. (1979) Biochim. Biophys.
Acts
557:9-23), Szoka, F. and Papahadjopoulos, D. (1978) Proc. Nat. Acad ScL
75:4194-4198, Mayhew, E.= et al. (1984) Biochim. Biophys. Acta 775:169175),
Kim, S.
et al. (1983) B ochim:-Biophy& Acts 728:339:348, and Mayer, et al. (1986)
Biochim.
Biophys. Acta 858:161-168.
The liposomes may be made from the lipid derivatives of antivirals in
combination with any of the conventional. synthetic or natural liposome
materials including phospholipids from natural sources such as egg, plant or
animal
sources such as phosphatidylcholine, phosphatidylethanolamine,
phosphatidylglycerol,
sphingomyelin, phosphatidylserine, or phosphatidylinositol. Synthetic
phospholipids
that may also be used, include, but are not limited to:
dimyristoylphosphatidylcholine,
dioleoylphosphatidylcholine, dipalmitoylphosphatidylcholine and
distearoylphosphatidyeholine, and the.. corresponding synthetic
phosphatidylethanolamines and phosphatidylglycerols. Cholesterol or other
sterols,
cholesterol hemisuccinate, glycolipids, cerebrosides, fatty acids,
gangliosides,
sphingolipids, 1,2-bis(oleoyloxy)-3-(trimethyl ammonio) propane (DOTAP), N-[1-
(2,3-
dioleoyl) propyl-N,N,N-trimethylammonium chloride (DOTMA), and other cationic
lipids may be incorporated into the liposomes, as is known to those skilled in
the art.
The relative amounts of phospholipid and additives used in the liposomes may
be varied
28
CA 02223989 2008-11-27
if desired. The preferred ranges are from about 60 to 90 mole percent of the
phospholipid; cholesterol, cholesterol hemisuccinate, fatty acids or cationic
lipids may
be used in amounts ranging from 0 to 50 mole percent. The amounts of
antivirals
incorporated into the lipid layer of liposomes can be varied with the
concentration of
their lipids ranging from about 0.01 to about 50 mole percent.
Using conventional methods, approximately 20 to 30% of the free
phosphonoacid present in solution can be entrapped in liposomes; thus,
appwximate1
70 to 80% of the active compound is wasted. In contrast, when the lipid
--~ phasphonoacid is incorporated into liposomes, virtually all of the
antiveo~mpound
0 is incorporated into the liposome, and essentially none of the active
compound is
wasted.
The liposomes with the above formulations may be made still more specific for
their intended targets with the incorporation of monoclonal antibodies or
other ligands
specific for a target. For examplemonoclonal antibodies to the CD4 (T4)
receptor may
be incorporated into the liposome by linkage to phosphatidylethanolamine (PE)
incorporated into the liposome by the method of Leserman, L. et al. (1980)
Nature
288:602-604.
Therapeutic Use of the Lipid Derivatives
The dosage of 1-0-allrylpropenediol-3-phosphate prodrugs for a mammal,
including a human, may vary depending upon the extent and severity of the
condition
that is treated and the activity of the administered compound. The dosage of
the lipid
prodrug is determined by reference to the recommended dosages of the active
agent,
bearing in mind that, in selecting the appropriate dosage in any specific
case,
consideration must be given to the patient's weight, general health,
metabolism, age and
other factors which influence response to the, drug. Dosage levels for most
commercially available therapeutic agents, as well. as many agents that are
being
clinically investigated, are well established For. example, the dosage of AZT
is
reported to be from about 7 to about 21mg/kg/day. The dosage of 1-0-octadecyl
propanediol-3-phosphate-AZT, for example, can be from about 1 to 25mg/kg/day,
preferably about 4-8mg/kg/day.
29
CA 02223989 2008-11-27
Formulations for oral ingestion are in the form of tablets, capsules, pills,
ampoules of powdered active agent, - or oily or aqueous suspensions or
solutions.
Tablets or other non-liquid oral compositions may contain acceptable
excipients,
vehicles, diluents, fragrances, or flavors known to the art for the
manufacture of
pha maceutical compositions, to make the medication palatable or pleasing to
use. The
formulation can therefore include diluents, such as lactose or calcium
carbonate; binding
agents such as gelatin or stanch; and one or more agents selected from the
group
consisting of sweetening -agents,. flavoring agents, coloring or preserving
agents to
provide -a. palatable-preparation. Moreover, such oral preparations may be
coated by
known techniques - to furdrer delay disintegration and absorption in the
intestinal tract.
Aqueous suspensions may contain the active ingredient in admixture with
pharmacologically acceptable excipients, comprising suspending agents, such as
methyl
cellulose; and wetting agents, such as lecithin or long-chain fatty alcohols.
The aqueous
suspensions may also contain preservatives, coloring agents, flavoring agents
and )
sweetening. agents in accordance with industry standards. The preparations may
further
comprise antioxidants, such as ascorbic acid or tocopherol, and preservatives,
such as
a-hydroxybenzoic acid esters.
The present invention is described below in detail using the following
examples,
but the chemical reactions described are disclosed in terms of their general
application
to the preparation of the lipid prodrugs of the invention. Occasionally, the
reaction may
not be applicable as described to each compound included within the disclosed
scope
of the invention. The compounds for which this occurs will be readily
recognized by
those skilled in the art. In all such cases, either the reactions can be
successfully
performed by conventional modifications known to those skilled in the art,
e.g., by
appropriate protection of interfering groups, by changing to alternative
conventional
reagents, or by routine modification of reaction conditions. Alternatively,
other
reactions disclosed herein or otherwise conventional will be applicable to the
preparation of the -corresponding compounds of the invention. In all
preparative
methods, all starting materials are known or readily preparable from known
starting
materials; all temperatures are set forth uncorrected in degrees Celsius; and,
unless
otherwise indicated, all parts and percentages are by weight.
38
CA 02223989 2008-11-27
It is believed that one skilled in the art can, using the preceding
description,
utilize the invention to its fullest extent. The following preferred
embodiments are,
therefore, to be construed as merely illustrative and not limitative for the
remainder of
the disclosure in any way whatsoever.
~1
CA 02223989 2008-11-27
EXPERIMENTAL
In the experimental disclosure which follows, the following abbreviations
apply:
eq (equivalents); M (Molar); mM (millimolar); M (mic romolar); N (Normal);
mol (moles); mmol (millimoles); mol (micromoles); nmol (nanomoles);
kg (kilograms); gm (grams); mg (milligrams); ug (micrograms); ng (nanograms);
L (liters); mL (milliliters); Al (microliters); vol (volumes); and C (degrees
Centigrade).
EXAMPLE 1
Preparation--Lipid Moieties Used in the. Coupling Procedures:
(a) Synthesis of 1 O-alkyl-2-O-benzyl-sn-glycero-3-phosphatidic acid:
To a vigorouly stirred solution of 1-octadecyl-2-O-benzyl glycerol (Sachem,
Inc., Basel, Switzerland), hereinafter referred to as OBG, a mixture of
pyridine,
triethylamine and tetrahydrofuran (THF) was added. Neat phosphorous
oxychioride,
POC13, was added dropwise while maintaining the temperature between -5 to 5
C. The
reaction mixture was stirred for 90 minutes at a temperature of 4 C. The
precipitated
triethylamine hydrochloride was filtered and the residue treated with toluene
at least
twice (2 x IOmL) and the solvent removed under reduced pressure. The resulting
oil
was converted to the ammonium salt upon careful addition of methanolic
ammonium
hydroxide. The yield was 55%, and the target compound was a white to pale
yellow
solid.
(b) Preparation of I-O-octadecyl-1,3-propanediol:
1-O-trityl-1, 3-Propanediol, synthesized according to the procedure of Sela,
et
al. (1987) Nucleic Acids Research 15:3124, was treated with octadecyl
methanesulfonate (NuChek Prep, Inc.) in the presence of sodium hydride in
dimethylformamide. The product, l -O-octadecyl-3-O-tritylpropanediol, was
isolated and
purified by flash chromatography. The trityl protecting group was removed by
treatment with trifluoroacetic acid in dichloromethane to yield 1-O-octadecyl-
1,3-
propanediol. The compound was converted to the corresponding 3-phosphate by a
procedure identical to the one in example (a).
(c) Synthesis of 1-O-octadecyl-2,2-dimethoxypropanediol-3-phosphate:
32
CA 02223989 2008-11-27
2,2-Dimethoxypropanediol was synthesized according to the procedure of
Cesarotti et
al. (Hely Chim. Acta 1993, 76, 2344).
To a solution of 2,2-dimethoxypropanediol (2.0gm, 14.7mmol) in
dimethylformamide (100mL) was added sodium hydride (0.7gm, 17.6mmol) and the
mixture stirred at room temperature for 30 min. Octadecyl methanesulfonate
(5.63gm,
16.2mmol) was added as a solid in one portion, and the mixture was stirred at
room
temperatuure under an atmosphere of nitrogen gas overnight. The mixture; was
poured
into ice-water (100mL), upon' which a solid separated. The solid was filtered
off and
dried. The solid was then dissolved- in-ethyl ~ux~te and flash cbromatographed
over
silica gel with 10% ethyl acetate in hexane ,s eluting solvent to yield pure
product.
This product was converted to the 3 phosphataaby the procedure used in example
(a).
(d) N-ti itylethanolamine:
A mixture of ethanolamine, trityl chloride and pyridine was refluxed for 15h.
Water
was added slowly to the cooled reaction and the precipitate collected by
filtration The
crude product recrystallized from a 1:1 mixture of ethanol and water.
A mixture of N-tiityl-O-(1-O-octadecyl-2-O-benzyl-sn-glycero-3-
phosphoryl)-ethanolamine: N-tritylethanolamine, 1-O-octadecyl-O-benzyl-sn-
glycerol
and triisopropylbenzenesulfonyl chloride in pyridine was stirred at a
temperature of
C for a period of 24h. The desired compound was extracted from the reaction
20 mixture and detritylation was carried out by methods familiar to those
skilled in the art.
EXAMPLE 2
Coupling of 1-O-octadecyl-1,3-propanediol to a Phosphorylated Drug Derivative
I. Synthesis of 1-0-octadecyl-1,3-propanediol-3: phospbate-Acyclovir:
25 Preparation from ACV monophosphate and 1-O-octadecyl-1,3-propanediol:
Acyclovir was phosphorylated by addition of phosphorous oxychloride (POC13).
After 1-2h at 0 C, acyclovir was extracted with ether as a phosphoryl
dichloride. A 2N
NaOH solution was added to an aqueous solution of the dichloride to bring the
pH to
about 9 to 10, converting the compound to the disodium form. Chromatography on
Dowex 50 converted the disodium salt to acyclovir monophosphate. A solution of
acyclovir monophosphate as its salt, such as tributylamine or trioctylamine,
in pyridine
33
CA 02223989 2008-11-27
was treated with batyl alcohol followed by triisopropylbenzenesulfonyl
chloride (TIPS)
.at a temperature of 45 C, for a period of 28h. The dark-colored solution was
treated
with water, followed by toluene, and the resulting solution was concentrated
under
reduced pressure. The crude product was purified by ion exchange
chromatography
followed by silica column chromatography to obtain the desired compound as a
white
chloroform- soluble powder in a yield of 50% with a purity > 95%. In. a
similar
manner, l-0-octadecyl-2 3-dimethoxy-13-pr+opanedioI was coupled with acyclovir
I monophosphate to yield : the corresponding acyclovir derivative.
II. Synthesis of 1-O-gctadecy1-W,2-ethanediol-2 phosphate-ara C:
as A. solution :of
cytosine arabuu side(ara C)-5'-monophosphate (Sigma, St Lours,
MO), 1-O-octadecyl-1,2-ethanediol, and tr iisopropylbenzenesulfonyl chloride
(TIPS) in
pyridine was allowed to stir,;at a temperature of 45 C over a period of 25h.
Water was
added to the reaction mixture followed by toluene and the solvents removed
under
reduced pressure.: The-crude =product,was chromatographed on silica gel to
afford the
desired compound.
III. Preparation of 1-O-octadecyl-2-0,-benzyl-sn-glycero-3-phosphate-ara-C:
The title compound can .be prepared starting from 1-O-octadecyl-2-O-benzyl-sn-
glycerol (OBG) as delineated in the preparation of II in which OBG can be used
to
couple with ara-C-monophosphate.
IV., Preparation of 1-0-octadecy12,2-dimethoxy :1,3-propanediol-3 .phosphate-
ara-C j
A' solution of ara-C-monophosphate, 1-O-octadecyl-2,2-dimethoxy-1,3-
propanediol and triisopropylbenzenesulfonyl. chloride (TIPS) in pyridine was
allowed
to stir at a temperature of 45 C, over a period of 25h. Water was added to the
reaction
mixture followed by toluene and the solvents removed under reduced pressure.
The
crude product was purified by silica chromatography to afford the desired
compound
with the desired purity.
34
CA 02223989 2008-11-27
EXAMPLE 3
Coupling of Drugs Having a Free Carboxyl Group to the Amino Group of a
Monoglyceride 'Phosphorylethanolamine:
Preparation of the 1-O-Octadecyl-1,3-propanediol Derivative of Cefazolin:
1-O-octadecyl-1,3-propanediol-3-phosphoethanolamine (lnunol) and cefazolin
(1.2mmol, 3 [(5-methyl-l.3.-thiadiazol-2-yl)thio]-8-oxo-7](1H-tetrazol-1
yl)acetyl]
amino]5thia--l-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid) were dissolved
inpyridine
followed by N,N dicyclohexylcarbodiimide (3nimol, DCC). The reaction mixture
was
stirred for 24h at 10 C. The reaction- vaa-stopped by the addition of cold
water and the
solvents were evaporated and the product was purified by preparative thin
layer
chromatography. The following compounds w&e similarly coupled to
1-O-octadecyl-1,3-propanediol-3-phosphoethanolamine by using the above
procedure:
3a: ceftazidime 1-[[7-[[(2-amino-4-thiazolyl)[(1-carboxy-l-
methylethyloxy)imino]-
acetyl]amino]-2-carboxy-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-en-3-yl]methyl]-
pyridinium hydroxide);
3b: ceftriaxone {7-[[(2-amino-4-thiazolyl)(methoxyimino)acetyl]amino]-
8-oxo-3-[[(1,2,5,6-tetrahydro-2-methyl-5,6-(Hoxo-1,2,4-triazin-3-
yl)thio]methyl]-
5-this-l-azabicyclo[4.2.0]oct 2-ene-2-carboxylic acid); and
3c: piperacillin (6-[[[[(4-ethyl-2,3-dioxo-l-piperazinyl)carbonyl]amino]phenyl
acetyl]amino]-3,3-dimethyl-7-oxo-4-thia-l-azabicyclo[3.2.0]heptane-2-
carboxylic acid).
EXAMPLE 4
Coupling Drugs Containing a Free Amino Group to a 1-0-Octadecyl-1,3-
propanediol-3-
Phosphate Through an Aliphatic Chain Linker:
Preparation of 1-O-Octadecyl-l,3-propanediol-3-phosphate derivatives of
cefazolin
4a: Hydroxycarboxylic acid linker
Hydroxybutyric acid sodium salt (0.5 mol, Aldrich) was dissolved in methanol
and dry HCl was passed to convert the acid to its methyl ester. Methanol was
evaporated and the dry methyl ester linking compound was coupled to 1-0-
octadecyl-
1,3-propanediol-3-phosphate by using N,N'-dicyclohexylcarbodiimide (DCC) as a
CA 02223989 2008-11-27
coupling agent. The resulting compound was subjected to a base-catalyzed
methanolysis by using 0.5N methanolic. sodium hydroxide and the free acid
derivative
was again coupled to various drugs containing free amino groups, such as, for
example,
the methyl ester of ceftazidime, or sulfamethazine as described above. The
protective
ester group was removed from the drug by treatment with base.
4b: Dihydroxyl linker
In another embed me nt, the carboxylic acid group of the linker was. aced to
an alcohol group (after coupling to 1-0-octadecy1-1,3: propanediol-3
pliospfiate) to
couple to flee drugs having a free acid moiety. ---- - -
EXAMPLE 5
1-O-Octadecyl-2-O-benzyl-sn-glycem-3-phospho-Ara-C:
5a A solution of 1-O-octadecyl-2-O-benzyl-sn-glycxm-3-phosphatidic acid (1)
and
ara-C in pyridine was treated with TIPS at a temperature of 40 C, over a
period of 24h.
The reaction was stopped by addition of water and the solvent evaporated under
reduced pressure. The crude product purified by chromatography to afford the
title
compound.
5b: Alternative preparation of this compound involved the coupling of OBG and
ara-C
monophosphate using pyridine as the solvent and TIPS as the coupling agent.
Purif ication was effected using the standard procedures.
Using the above methods, the corresponding lipid derivatives of the following
nucleoside analogs can be obtained:
5c: 2'-ara-fluoro-2-chlorodeoxyadenosine
5d: 5-fluorouridine
5e: 6-mercaptopurine riboside
5f: 3'-thia-dideoxycytidine
Sg: 3 -this-S-fluoro-dideoxycytidine
5h: Ganciclovir
Si: Acyclovir
36
CA 02223989 1997-12-05
WO 96139831 PCTIUS96/I0054
EXAMPLE 6
Synthesis of 1-O-octadecyl-rac-glycero-3-phospho-5'-(3'-azido-3'-
deoxy)thymidine:
Dry 1-O-octadecyl-rac-3-glycerol (batyl alcohol, 250mg), 3'-azido-3'-
deoxythymidine monophosphate sodium salt (0.725gm) and
2,4,6,-triisopropylbenzenesulfonyl chloride (TIPS, 1.219gm) were mixed in dry
pyridine
and stirred overnight under nitrogen. Chloroform (5OmL) was added and the
reaction
mixture was washed twice with cold 0.2N HCl and 0.2N sodium bicarbonate. The
organic phase was removed in vacuo with a rotary evaporator and the product
was
crystallized at -20 C from 20mL of chloroform/acetone (12:8 by volume). The
final
purification of the compound was done by preparative thin layer chromatography
using
500 micron layers of silica gel G developed with
chloroform/methanol/concentrated
ammonia/water (70/30/1/1 by volume).
EXAMPLE 7
Synthesis of 1-O-Octadecyl-sn-glycero-3-phosphonoformate:
1-O-Octadecyl-2-O-benzyl-sn-glycerol (9.9gm, 23mmol) in dry pyridine (25mL)
was added dropwise to an ice-cold solution of ethoxycarbonyl
phosphodichloridate
(7.0gm, 36mmol) in dry chloroform (50mL). The mixture was warmed to room
temperature and stirred for 24 hours. The reaction was stopped by the addition
of cold
water (5mL) and stirring for two hours. The reaction mixture was poured in
water
(100mL) and the organic phase was separated. The aqueous phase was extracted
chloroform (3 x 25mL) and the organic extracts were combined. The combined
organic
phase was washed with aqueous saturated sodium chloride (5OmL), dried over
anhydrous magnesium sulfate and concentrated in-vacuo to yield an oil. The oil
was
purified by flash chromatography with 10% methanol in chloroform as eluent to
yield
1-O-octadecyl-2-O-benzyl-sn-glycero-3-ethylphosphonoformate as a colorless oil
(6.2gm, 46%).
A portion of the above oil (1.67gm) was dissolved in absolute ethanol (IOOmL),
Pd/C (300mg) were added and the mixture hydrogenated at 60psi for 24 hours.
The
catalyst was filtered off to yield 1-O-octadecyl-sn-glycero-3-
ethylphosphonoformate
(1.0gm, 71%)
37
CA 02223989 1997-12-05
WO 96/39831 PCTIUS96/10054
To the ethylphosphonoformate (1.0gm) in absolute ethanol (50mL) was added
.an aqueous solution of sodium hydroxide (8mL, iN) and the mixture stirred at
room
temperature for 30 minutes. The mixture was centrifuged and the solid
isolated. The
solid was washed with absolute ethanol (3x25mL) and dried to give batyl
phosphonoformate (0.7gm).
EXAMPLE 8
Coupling 1-O-octadecyl-1,3-propanediol-3-phosphatidic Acid (and compounds of
example 1) to the Amino Group of a Peptide:
1-O-octadecyl-l,3-propanediol-3-phosphatidic acid, (or any of the lipid-
phosphate moieties) prepared as in Example 1 above, was partitioned between
chloroform/methanol (2:1 (v/v); 200mL) and cold IN HCl (50mL). The aqueous
layer
was re-extracted with chloroform methanol (2:1) (v/v); I OOmL). The combined
organic
phase was evaporated and dried under vacuum over P205. The resulting free
phosphatidic acid was dissolved in a mixture of DMF (2mL) and pyridine (2mL)
and
to the solution was added the appropriate peptide having a free amino group (i
mmol)
followed by NN'- dicyclohexylcarbodiimide (DCC; Aldrich Chemical Co.,
Milwaukee,
WI, MW:206, 620mg, 3mmol). The reaction mixture was stirred for 24 hours at
room
temperature. The solvents were evaporated and the product was purified by
flash
chromatography over silica gel column (2.5 x 50cm) using a linear gradient of
0 to 50%
methanol in chloroform. Fractions containing the desired product as indicated
by TLC
and HPLC were pooled and evaporated. The product was further purified, if
necessary,
by preparative HPLC or by crystallization yielding the 1-O-octadecyl-l,3-
propanediol-3-
phospho-(NH)-peptide. Any therapeutic peptide may be coupled in a like manner.
EXAMPLE 9
Coupling 1-O-octadecyl-2-O-benzyl-3-phosphoethanolamine to the Amino Group of
a
Therapeutic Peptide Using Succinate as a Linking Group:
A solution of 1-O-octadecyl-2-O-benzyl-3-phosphatidic acid and ethanolamine
in pyridine was treated with N,N'-dicyclohexylcarbodiimide and the mixture was
allowed to stir at room temperature for a period of 24h. The solvents were
evaporated
38
CA 02223989 2008-11-27
1 .
and the product purified by chromatography. Fractions containing the desired
product
were pooled and evaporated. The 1-O-octadecyl-2-O-benzyl-3-phosphoethanolamine
was - next treated with succinic anhydride to afford the hemisuoeinate of
1-0-octadecy1-2-0-benzyl-3-phosphoethanolamine. The free carboxyl group of the
hemisuccinate was coupled to the N-terminal amino group of a HIV protease
inhibitor
[D-Phe]-D-a-napthylalanine]-pipecolic acid-[a-OH Leu]-Val amide (VST 7140) or
a
peptide-suc as VST 7194 or a renin inhibitor, enalkiren (A64662). Any
therapeutically
useful peptide may be coupled in a like manna.
EXAMPLE 10
Coupling 1 4)-alkyl-2-O-benzyl-sn-glycero-3-phosphatidic Acid to the Hydroxy
Group
of a Peptide:
1-O-alkyl-2-O-benzyl-sn-glycero-3-phosphatidic acid (immol) prepared as above
was dissolved in a mixture of DMF (2mL) and pyridine (2mL) and to the solution
was
added the appropriate peptide having a free hydroxyl group (lmmol). The
reaction was
carried out and the product was isolated as described in Example 9.
The condensation of the phosphatidic acid and the hydroxyl group of a peptide
was also
conveniently carried out by using 2,4,6-triisopropylbenaenesulfonyl chloride
(TOS-Cl;
Aldrich Chemical Co., Milwaukee, WI; MW:302.86; 758mg, 2.Smmol) as a coupling
agent in place of DCC.
EXAMPLE 11
Coupling a Peptide Containing a Free Carboxyl Group 1 O-Octadecyl-1,3-
propanediol
3-phosphoethanolamine:
A mixture of the appropriate peptide (lmmol), and 1-O-octadecyl-1,3-
propanediol-3-phosphoethanolamine (l mmol) was dissolved in pyridine (5mL) and
DCC
(3mmol) followed by 1-hydroxybenzotriazole (HOBt; Aldrich Chemical Co.,
MW:153;
450mg, 3mmol) were added. The reaction mixture was stirred for 24 hours at
room
temperature and the product was purified by silica gel chromatography as
described in
Example I followed by debenzylation as in Example 10. Any therapeutically
useful
peptide may be coupled in a like manner.
39
CA 02223989 1997-12-05
WO 96/39831 PCT/US96/10054
EXAMPLE 12
Synthesis of Lipid Derivative of a Taxol Side Chain:
Synthesis of (3-(Benzoylamino)-a-(1-O-octadecyl-1,3-propanediol-
3-phospho)-benzenepropanoate, ester (1).
To a solution of 1-O-octadecyl-1,3-propanediol-3-phosphate (0.5 mol) and
(3-(benzoylamino)-a-hydroxybenzene-propanoate ester either in an ethereal
solvent like
diethyl ether, tetrahydrofuran or a halogenated solvent like dichloromethane
or
chloroform was added DCC either neat or as a solution and allowed to stir for
2-25h
at a temperature of 4 C. Water was added to the reaction mixture and the
solvents
removed under reduced pressure. The crude product was chromatographed on
silica gel
to afford the desired compound.
Synthesis of P-(Benzoylamino)-a-(1-O-octadecyl-2-benzyl-sn-glycero-3-phospho)-
benzenepropanoate ester (2).
A solution of 1-O-octadecyl-2-O-benzyl-sn-glycero-3-phosphatidic acid (0.1mol)
and p-(benzoylamino)-a-hydroxybenzenepropanoate ester in pyridine or
chloroform was
stirred in the presence of DCC (0.4mol) at a temperature of 4 C for a period
of 6h.
Water was added to the reaction mixture and the contents extracted with
chloroform.
The solvent was removed under reduced pressure and the crude product purified
by
chromatography to afford the benzenepropanoate ester.
EXAMPLE 13
Synthesis of (3-amino Substituted Taxol Side Chain:
Synthesis of (3-Amino -a-(1-O-octadecyl-2-O-benzyl-sn-glycero-3-phospho)-
benzenepropanoate ester.
To a solution of 1-O-octadecyl-2-O-benzyl-sn-glycero-3-phosphatidic acid
(0.lmol) and (3-Amino-a-hydroxybenzene propanoate ester (0.lmol) in chloroform
or
pyridine was added DCC (0.4mol) and allowed to stir at a temperature of 4 C
for a
period of 5h. Water was added to the reaction mixture and the contents
extracted with
chloroform or other halogenated solvent. The solvent was removed under reduced
CA 02223989 2008-11-27
pressure and the crude product purified by chromatography to afford the
substituted
ethanolamine of 1-O-octadecyl-2-O-benzyl-sn-glycero-3-ph~osphatidic acid.
EXAMPLE 14
Hydrolysis of Propanoate Esters of Lipid Derivatized Taxol Side Chain:
Synthesis of P-(Benzoylamino)-a-(1-O-alkyl-1,3-propanediol-3-phospho)-
benzeneprapanoic acid (3). 4
T propanoate ester (0.1mol) flan (1)_was- hydrolyzed using sodium methoxide
in methanol or sodium carbonate in methanol at=a-temperature of 5 C for a
period of
4h to affordthe desired compound which is ready for coupling with baccatin M.
Synthesis of p-( ylamiao)-a-(I-O-octadecyl-2-0-benzyl sn-glycero-3-phospho)-
benzenepropano' acid.
To , ; Solution of 2 (0.1 mol) in methanol was added a solution of sodium
methoxid in methanol and the resulting solution was stirred at a temperature
of 50C
fore a'period of 4h. The reaction mixture was neutralized and the resulting
solution
concentrated under reduced pressure to afford the crude product. Purification
by
cold chromatography gave the desired compound which is suitable for coupling
with
b atin III or 10-deacetyl baccatin.
EXAMPLE IN
Coupling of Lipid Derivative of Taxol Side chain to Baccatin:
A. Coupling of lipid, derivative of phosophoethanolamine side chain to 10-
deacetyl
baccatin II.
To a solution of f 3-(benzoylamino)-a-(1-0-alkyl-1,3-propanediol-3-phospho)-
benzenepropanoic acid (example 14) (0.1 mol) and 10-deacetyl baccatin III (0.1
mol) in
chloroform was added DCC (0.4mol) and allowed to stir at a temperature of 25 C
for
a period of 7h. Water was added to the reaction mixture and the contents
extracted
with chloroform. The organic layer was separated and the aqueous phase was
extracted
with chloroform. The combined organic layer was concentrated under reduced
pressure
4,1
CA 02223989 2008-11-27
- to
and the crude product purified by chromatography to afford the 1-O-alkyl-l,3-
propanediol-3-phosphoethanolamine derivative of taxol.
B. Coupling of 1-O-octadecyl-2-O-benzyl-3-phospho ethanolamine side chain to
10-deacetyl baccatin III.
To a solution of a-(benzoylamino)-a-(1-0-octadecyl-2-0-benzyl-
sn-glyaero-3-phospho)-benzenepropanoic acid (0.1mol), 10-deacetyl baccatin III
(0.Insh1) in chloroform was added DCC (0.4mol) and allowed to stir at room
-~ - --temperature for a period of iOh. _ Water was added to the reaction..
mixture and.-the---
contents extracted with chloroform. The organic layer was Separated. and the
aqueous
layer was extracted with chloroform. The combined organic layer was
concentrated
under reduced pressure and the- crude .product -purified by chromatography to
afford the
batylbenzylphosphoethanolamine derivate of taxol.
In the preceding, synth .proton NMR spectra were obtained with a General
Electric QE-300 spectrometer, using tetramethylsilane as internal standard
(key:
s = singlet, d = doublet, t triplet,, q = quartet, dd = doublet of doublets, b
= broad).
UV. spectra were... recorded on Shimadzu UV-160, spectrophotometer. Fast atom
bombardment mass. spectra were determined by Mass Spectrometry Service
Laboratory,
University of Minnesota. Elemental analyses were determined by Galbraith
Laboratories, Knoxville, TN and Schwarzkopf Microanalytical Laboratory, NY.
Melting points were obtained with a Fischer-Johns melting apparatus. Column
chromatography was carried out on Merck silica gel 60 (70-230 mesh). Rf values
were
obtained with HPTLC Merck, Kieselgel 60 pre-coated plates, 10 x 10cm.
Anhydrous
pyridine, 2,4,6-Triisopropylbenzenesulfonyl chloride (TIPS), 3'-azido-3'-
deoxythymidine
(AZT) and 1,3-propanediol were purchased from Aldrich Chemical Co., Milwaukee,
WI
and 1-0-octadecyl-2-benzylglycerol was purchased from Bachem Bioscience Inc.,
Philadelphia, PA.
EXAMPLE 16
Single-Dose 1-O-octadecyl-sn-glycero-3-phospho-AZT Oral Administration
Compared
to Continuous Oral AZT Administration:
42
CA 02223989 2008-11-27
Treatment of Rauscher Leukemia Virus-Infected Mice:
Female BALB/C mice were infected with 1X10' plaque-forming units (PFU) of
Rauscher leukemia virus complex (RLV) on day 0. Control animals were injected
with
saline. Beginning on day 2, groups of the infected mice as indicated in FIG. 1
were
tceatcd with AZT at doses from about 1.0mg/kg/day to 15.Omg/kg/day for 21 days
either by- offering AZT in drinking water or by gavaging with 1-06octadecyl-sn-
glycero-
e-AZT 'once a day. On day 23 post inoculaddf, the mice in both treatment
protocols wet+e Sacrificed, and the spleen weights of the haimals were
determined. The
mean spleen weights, indicating relative Ievel_bfvmisL ` 'Y 'for each dose
level in
the two protocols, are rested in the bar gra* -v1 "II.' 1. The effective doses
(ED50) of daily 1"-O-oetadecyl-sn-glycero-3-phosphafe-ALT given by asingle
oral
administration and AZT given by oral administration in the drinking water were
comparable.
It is apparent from the foregoing that otter 1-0-alkyl-1,3-propanediol-3-
phosphate derivatives and the other lipid adducts in ale 1. of therapeutic
drugs can
be substituted to obtain similar results of delivering a drug, otherwise
poorly orally
bioavailable, more effectively through the oral route. It should be further
emphasized
that the present invention is notiimited'to the use of any particular drug or
c
agent in the compounds of the invention; rather the beneficial results of the
invention
now from the synthesis of the lipid moieties of Example I linked via a
substituted or
unsubstituted propanediol-3-phosphate, with or without a linker to these drugs
and
agents. ~ Thus, regardless of whether a specific drug or agent is presently
known, or
whether it becomes. known - in the future, the methods of forming the
presently
contemplated lipid prodrugs therefrom are based on established chemical
techniques,
as will 'be apparent to those of skill in the art, and therefore these
compounds are
broadly enabled by the preceding disclosure. It should be emphasized again
that the
present. syntheses are broadly applicable to formation of compounds from
essentially
all drugs' having an appropriate structure, and the effectiveness of which can
be
improved - by preparing a lipid prodrug form for use in the practice of the
invention.
43
CA 02223989 2008-11-27
EXAMPLE 17
Synthesis of 1-0-Octadecyl-2-O-methyl-sn-glycero-3-phospho-acyclovir.
Acyclovir monophosphate (1 mmol) was converted to the tri-n-butylammonium
salt (TBA) by treatment with td-n-butylamine in methanol followed by
lyophilization
of the product. The TBA salt was dried by coevaporation with 1OmL pyridine
twice.
To a mixture of the dry acyclovir TBA salt and 1-O-octadecyl 2-O-methyl-sn-
glycerol
(lmmol) in dry. pyridine (2OmL) was added 2,4,6-triisopropylbee sul onyl
chloride
(3mmol), upon which the mixture turned bright yellow. The mixture was
stirred'at-i
room temperature for 48h, and quenched by addition of methanol (2OmL). fixture
. was, pur fied.'by flash chromatography over silica gel with an increasing
gradient.of
-:methanol in dichloromethane as the eluting solvent. The appropriate
fractions were
pooled and concentrated in vacuo to yield the title compound as a colorless
amorphous
solid. Any pharmaceutically useful nucleoside analog monophosphate may be
utilized.
as.noted - in - the example to obtain the corresponding 1-0-allcyl, 2-0-methyl-
sn-glycero-3-phosphate analog, or other analogs thereof.
EXAMPLE 1S
Synthesis of 1-O-Octadecyl-'i,3-propanediol-3-phospho-acyclovir:
animonium
Acyclovir- monophosphate - (lmmol) was converted to the td-n-butyl
salt (TBA) by treatment with tri- n-butylamine in methanol followed by
lyophilization
of the product The TBA salt was .dried by coevaporation with I OmL pyridine
twice.
To a mixture of the dry acyclovir TBA salt and 1-0-octadecyl-1,3-propanediol
(lmmol)
in dry pyridine (2OmL) was added 2,4,6-triisopropylbenzene sulfonyl chloride
(3mmol),
upon which the mixture turned bright- yellow. The mixture was stirred at room
temperature for 48h, and quenched by addition. of.methanol (20mL). The mixture
was
purified by flash chromatography over silica gel with an increasing gradient
of methanol
in dichloromethane as the eluting solvent. The appropriate fractions were
pooled and
concentrated in vacuo to yield the title compound as an amorphous solid. Any
pharmaceutically useful nucleoside analog monophosphate may be utilized as
noted in
the example to obtain the corresponding 1-0-alkyl, 2-O-methyl-sn-glycero-3-
phosphate-analog.
44,
CA 02223989 2008-11-27
f@ `t TrrS$, ra
EXAMPLE 19
.1-0-Octadecyl-2-0-methyl-sn-glycero-3-phospho-ethanolamine-Peptide 7194 (HIV
Protease inhibitor) conjugated through the C-terminus:
Pentapeptide 7194, with -a tert-butyloxycarbonyl protecting group at the
N-terminus (t-BOC-L-Phe-[B-D-NAL]-PIP-[a-OH-Leu]-Val-000H) (2mmo1) was
mixed with 1-0-octadecyl-2-0-methyl-sn glycem-3-phaspho-ethanolamine (2mmol)
in
dry pyridine (SOmL)'and' cooled in an ice-salt bath. A solution of
dicyclohexyl-
carbodiimide (6mmol) in dry dichioromethane was added to the mixture dropwise
with
stirring. The-resulting mixture was allowed to stir at room t due overnight.
The
reaction mixture was filtered and the filtrate was concentrated to dryness in
vacu. The
residue was purified by silica gel flash chromatography with an increasing
gradient of
methanol ` in chloroform as the eluent to yield the title compound, with the t-
BOC
protecting group at the N-terminus of the peptidyl moiety. The protecting
group was
removed by treatment with 10% trifluoroacetic acid in dichloromethane to yield
title
compound. Any pharmaceutical peptide drug having a tert- butyloxycarbonyl
protecting
group at the N-terminus may be converted to the corresponding 1-O-octadecyl-2-
O-
methyl-sn glycero-3-phosphoethanolamine analog using the method described
above.
EXAMPLE 20,
t O Dctadecyl-2-O-methyl-sn-glycero-3-phospho-L-Peptide 7194 (HIV Protease
inhibitor) conjugated through the N-terminus: (L-O-CH2CH2-COO-):
'Pentapeptide 7194, as its methyl ester at C-terminus (2mmol) was mixed with
3-hydroxy, propanoiq acid (2mmol) in dry dichioromethane (SOmL). The mixture
was
cooled in an ice-salt bath and N,N-dicyclohexylcarbodiimide (2.4mmol) was
added.
The mixture was stirred at 0 C for 3h and at room temperature overnight. The
mixture
was filtered and the filtrate was concentrated to dryness in vacuo. The
residue was
flash chromatographed over silica gel with an increasing gradient of methanol
in
dichloromethane as the eluting solvent to obtain pure product as a foam.
To a mixture of the above foam (lmmol) and 1-O-octadecyl-2-O-methyl-
sn-glycero-3-phosphatidic acid (l mmol) in dry pyridine (50mL), cooled in an
ice-salt
bath, was added a solution of dicyclohexylcarbodiimide (3mmol) in dry
CA 02223989 1997-12-05
WO 96/39831 PCTIUS96/10054
dichloromethane dropwise with stirring. The mixture was allowed to stir at
room
temperature overnight. The resulting reaction mixture was filtered and the
filtrate was
concentrated to dryness in vacuo. The residue was purified by silica gel flash
chromatography with an increasing gradient of methanol in chloroform as the
eluent to
yield the title compound as the methyl ester at the C-terminus. The ester was
treated
with ethanolic sodium hydroxide to yield title compound. Similarly, any
pharmaceutical
peptide drug having a methyl ester at the C-terminus may be converted to the
corresponding 1-O-octadecyl-2-O-methyl-sn-glycero-3-phosphate-analog using the
method of this example.
EXAMPLE 21
General method of synthesis of 1-O-Octadecyl-2-O- methyl-sn-glycero-3-phospho-
5'-
oligonucleotides:
To the fully protected oligonucleotide attached to the solid support on a DNA
synthesis column, and having a free 5'-hydroxyl group (l mol) was added a
mixture
of 1-O-octadecyl-2-O-methyl-sn-glycero-3-phosphatidic acid (5 mol) and
dicyclohexylcarbodiimide (5pmol) in pyridine (2mL). The reaction was allowed
to
proceed at room temperature overnight.
The column with the derivatized oligonucleotide was washed with pyridine
(2mL) and acetonitrile (2mL). Iodine in tetrahydrofuran (0.1 M, lmL) was added
to the
column over 5 mins. The lipid-oligonucleotide was freed from the solid support
by the
addition of ammonia, and deblocked completely by treatment with ammonia at 55
C
overnight. The resulting 1-O-octadecyl-2-O-methyl-sn-glycero-3-phospho-5'-
oligonucleotide was purified by HPLC to yield pure compound. Oligonucleotides
having from 2-24 bases may be derivatized in this manner, using any of the
lipid-
phosphate moieties in Example 1.
EXAMPLE 22
Synthesis of 1-O-Octadecyl-2-O-methyl-sn-glycero-3-phospho-ganciclovir:
Ganciclovir monophosphate (lmmol) was converted to the tri-n-butylammonium
salt (TBA) by treatment with tri-n-butylamine in methanol followed by
lyophilization
46
CA 02223989 1997-12-05
WO 96/39831 PCTIUS96/10054
of the product. The TBA salt was dried by coevaporation with lOmL pyridine
twice.
To a mixture of the dry ganciclovir TBA salt and 1-O-octadecyl-2-O-methyl-sn-
glycerol (lmmol) in dry pyridine (20mL) was added 2,4,6-triisopropylbenzene
sulfonyl
chloridg (3mmol), upon which the mixture turned bright yellow. The mixture was
stirred at room temperature for 48h, and quenched by addition of methanol
(2OmL).
The mixture was purified by flash chromatography over silica gel with an
increasing
gradient of methanol in dichloromethane as the eluting solvent. The
appropriate
fractions were pooled and concentrated in vacuo to yield the title compound as
a
colorless amorphous solid.
EXAMPLE 23
Synthesis of 1-0-Octadecyl-1,3-propanediol-3-phospho-ganciclovir:
Ganciclovir monophosphate (1 mmol) was converted to the tri-n-butylammonium
salt (TBA) by treatment with tri- n-butylamine in methanol followed by
lyophilization
of the product. The TBA salt was dried by coevaporation with 10mL pyridine
twice.
To a mixture of the dry ganciclovir TBA salt and 1-O-octadecyl-1,3-propanediol
(lmmol) in dry pyridine (20mL) was added 2,4,6-triisopropylbenzene sulfonyl
chloride
(3mmol), upon which the mixture turned bright yellow. The mixture was stirred
at
room temperature for 48h, and quenched by addition of methanol (20mL). The
mixture
was purified by flash chromatography over silica gel with an increasing
gradient of
methanol in dichloromethane as the eluting solvent. The appropriate fractions
were
pooled and concentrated in vacuo to yield the title compound as an amorphous
solid.
EXAMPLE 24
1-O-Octadecyl-2,2-dimethoxy-1,3-propanediol-3-phosphonoformate, disodium salt:
To a solution of 1-O-octadecyl-2,2-dimethoxy-l,3-propanediol (1.92gm, 5mmol)
and ethyl phosphonoformate (1.19 g, 5mmol) in dry pyridine (5OmL), cooled in
an ice-
salt bath, was added a solution of N,N-dicyclohexylcarbodiimide (3.1 g,
15mmol) in dry
dichloromethane (20mL). The resulting mixture was stirred at room temperature
over.
The mixture was filtered, and the filtrate was concentrated to dryness. The
residue was
flash chromatographed with a gradient of 0-10% methanol in dichloromethane to
give
47
CA 02223989 1997-12-05
WO 96/39831 PCTIUS96/10054
the target compound. In a similar manner, use of ethyl phosphono- acetate
resulted in
the corresponding 1-O-octadecyl-2,2-dimethoxy-1,3-propane diol-3-
ethylphosphonoacetate.
A suspension of 1-O-octadecyl-2,2-dimethoxy-1,3-propanediol-3-
phosphonoformate (0.91gm, 1.7mmol) in absolute ethanol (20mL) was treated with
IN
aqueous sodium hydroxide (3.8mL), and the resulting mixture stirred at room
temperature for 1.5h. The mixture was centrifuged, the resulting solid
suspended in
absolute ethanol (20mL). The suspension was vortexed and centrifuged. The
solid was
dried to yield product as an amorphous powder (0.9 gm). In a similar manner,
the
corresponding phosphonoacetate derivative was synthesized.
EXAMPLE 25
1-O-Octadecyl-2-O-benzyl-sn-glycero-3-phosphonoformate, disodium salt:
1-O-Octadecyl-2-O-benzyl-sn-glycerol (lmmol) and ethyl phosphonoformate
(lmmol) were dissolved in pyridine (20mL). The solution was cooled in an ice-
salt
bath and N,N-dicyclohexylcarbodiimide (2.4mmol) in dichloromethane was added.
The
mixture was stirred at O C for 3h and at room temperature overnight. The
mixture was
filtered and the filtrate was concentrated to dryness in vacuo. The residue
was flash
chromatographed over silica gel with an increasing gradient of methanol in
dichloromethane as the eluting solvent to obtain pure product as a foam.
A suspension of the above foam (2mmol) in absolute ethanol (20mL) was treated
with
IN aqueous sodium hydroxide (4.1mL), and the resulting mixture stirred at room
temperature for 1.5h. The mixture was centrifuged, the resulting solid
suspended in
absolute ethanol (20mL). The suspension was vortexed and centrifuged. The
solid was
dried to yield product as an amorphous powder.
EXAMPLE 26
1-0-Octadecyl-1,3-propanediol-3-phosphonoformate, disodium salt:
1-O-Octadecyl-1,3-propanediol (1.41gm, 4mmol) and ethyl phosphonoformate
(1.19gm, 5mmol) were dissolved in pyridine (20mL). The solution was cooled in
an
ice-salt bath and N,N-dicyclohexylcarbodiimide (2.4mmol) in dichloromethane
was
48
CA 02223989 2008-11-27
added. The mixture was stirred at O C for 3h and at room temperature overnight
The
mixture was filtered and the filtrate was concentrated to dryness in vacuo.
The residue
was flash chromatographed over silica gel with an increasing gradient of
methanol in
dichloromethane as the eluting solvent to obtain 1-0-octadecyl-1,3 propane
diol 3-ethyl
phosphonoformate (1.15gm, 62%) as an amorphous powder.
A suspension of the ethyl phosphonoformate ester (0.8gm, 1.72mmol) in
absolute ethanol (2OmL) was treated with IN aqueous sodium hydro,dde i(4.3mL),
and
the resulting mixture stirred at room temperature for 1.5h. The mixture was
centrifuged, the resulting solid suspended. in absolute ethanol - (2OmL)..:
The suspension
was vortexed and centrifuged. The solid was dried to yield title compound 11
(0.64gm,
77%) as an amorphous powder.
} EXAMPLE 27
1-0-Octadecyl-2,2-dimethoxy-l,3-propanediol-3=phospho=Acyclovir.
Toa-solution of 1-0-octadecyl-2,2-dintetio)y-1,3propanediol (1.92gm, Smmol)
and ethyl phosphonoformate (1.5gm, Smmol) in dry pyridine-(5OmL), cooled in an
ice-
salt bath, was added-4k solution of NN-dicyclohexylcarbodiimide (3.lgm,
15mmol) in
dry dichloromethane (2OmLL). The resulting mixture was stirred at room
temperature
overnight The, mixture wai filtered, and the filtrate was concentrated to
dryness. The
residue ' was flash chromatographed with a gradient of 0-10% methanol in
dichloromethane to give the target compound. In a similar manner, use of
ganciclovir
mono phosphate resulted in the corresponding 1-0-octadecyl-2,2-dimethoxy-l,3-
propanediol-3-phospho-ganciclovir.
EXAMPLE 28
Synthesis of 1-0-alkyl-2-halo-propane-3-phospho-Acyclovir and
1-O-alkyl-2-amino-sn-glycero-3-phospho-Ganciclovir:
The stereocontrolled synthesis of 1-0-alkyl-2-halo-propane-3-phospho-Acyclovir
is outlined in Scheme I. 2,3-Isopropylidene-sn-glycerol, upon treatment with
the
appropriate alkylmethane sulfonate, leads to the intermediate 2. Removal of
the
isopropylidene group by treatment with acetic acid followed by tritylation
with trityl
CA 02223989 2008-11-27
chloride and pyridine results in compound 3, with a free 2-hydroxyl group.
Treatment
of 3 with n-halosuccimide and triphenyl phosphine according to the procedure
of Bose
and Lal (1973, Tetrahedron Len. 40:3937) will lead to intermediate 4. The
replacement
of the hydroxyl group with halogen proceeds with complete inversion (SN2
displacement) and in yields from 65-95%. Removal of the trityl-group with
trifluoroacetic acid in dichloromethane leads to the halo compound 5. Reaction
of 5
with acyclovir monophosphate and dicyclohexylcarbodiimide in pyridine will
lead to
the target compound.
While this procedure works when X is Cl, Br and I, a slightly different
approach
is needed for the !analog when_ X is F. Treatment of the intermediate 3 with
diphenyltrifluorophosphorane according to a procedure reported by Kobayashi
and
coworkers (1968, Chem. Pharm. Bull. 16(9):1784) leads to the conversion of
alcohol
3 to the fluorinated compound 4 in good yields. Subsequent steps to the fluoro
analogs )
are identical to those described- earlier.
Treatment of bromo intermediate 4 described in Scheme I with liquid ammonia
in a steel bomb will result in.amination at the 2-position. Treatment of the
resulting
2-amino compound with benzyl bromide will protect. the 2-amino group.
Subsequent
steps will be identical to those. described in Scheme I up to intermediate
1-O-alkyl-2-benzylamino-sn-glycero-3-phospho-ganciclovir. The benzyl
protecting
group can b removed at this point by hydrogenolysis with Pd/C to give the
target
compound.
Treatment of the amino intermediate with acyl chloride will result in the N-
acyl
compound. Alternatjvely, treatment of the bromo intermediate 4 with mono or
dialkyl )
amine will result in the monoalkyl amino and dialkylamino derivatives.
The procedures described above can be carried out with readily available
starting
materials, the methodology is well documented and those skilled in the art can
recognize the modifications needed in starting materials and methods to
synthesize the
2-amino analogs that have been listed.
fio.
CA 02223989 2008-11-27
EXAMPLE 29
Synthesis of thiophosphonoacids and their lipid prodrugs:
Thiophosphonoformic acid is synthesized according to the procedure of
McKenna (U.S. Patent 5,072,032) by the reaction of trimethyl phosphonoformic
acid
with Lawesson's reagent [2,4-bis(4-metlwxyphenyl)-1,3-dithia-2,4-diphosphetane-
2,4 -
sulfide]. Thiophosphonoacetic acid is also synthesized in a similar manner.
Selenophosphonoformic acid can be synthesized= by treatment of
trimethylphosphonoformic acid with elemental selenium by adaptation of
procedures
reported by Stec and co-workers (1976, Stec, WJ., Okcruszek, A and bfichalski,
J., J.
Org. Chem. 41, 233), and by Buina et al. (1979, Buina, N.A-;. Sibgatuffins,
F.G.;
Neureldinor, I.A., Izv. Akad Nauks ., Ser. Khim. 10,2362).
Seldiophosphonoacetic
acid can also be synthesized in a similar manner. The selenoacids can than be
j converted to the corresponding lipid prodcugs by procedures identical to
those for the
oxo- and thio- analogs.
' The synthesis of 1-O-octadecy1-1,3-propanediol=3-thiophosphooofonmic acid
was
carried out in a manner similar to the synthesis of 1-O-octadecyl-
1,3propaanediol-
3 phosphonofor mic acid, with some modifications. 1-O-Octadecyl-1,3
propanediol was
coupled to thiophosphonoformic acid ethyl ester using
dicyclohexylcarbodiimide. After
purification by silica gel chromatography, the target compound was obtained by
base
hydrolysis of the ester. 1-O-Octadecyl-13-propanediol-3-thiophosphonoacetic
acid was
synthesized in a similar manner, as were 1-O-octadecyl-2,2-dimethoxy-1,3-
propanediol-
3-thiophosphonoformate and the corresponding thiophosphonoacetate. -
1
0
EXAMPLE 30
Synthesis of 1-O-octadecyl-1,2-ethanediol-2-phosphonoformate:
To a solution of carbethoxyphosphodichloridate (1.6mmol) in chloroform
(25mL), cooled to 0 C in an ice salt bath, was _ added a solution of
1-0-octadecylethanediol (Immol) in pyridine (15mL), dropwise with stirring.
The
mixture was allowed to warm to room temperature and stirred at room
temperature
overnight. The mixture was cooled, and 1mL water was added. After stirring at
0 C
for 2h, the mixture was concentrated in vacuo. The residual oil was flash
5.1
CA 02223989 2008-11-27
chromatographed with chloroform:methanol 95:5 as eluent to yield product:
1-O-octadecylethanediol-2-ethylphosphonoformate.
The ethyl ester was dissolved in 1:1 mixture of ethanol and 0.1N NaOlI (5OmL)
and sonicated for 15 minutes. The resulting mixture was stirred at 60 C in an
oil bath
for 2h. The mixture was filtered and the filtrate was concentrated to dryness
in vacuo.
The resulting solid was resuspended in water, cooled and lyophilized to yield
target
compound.
EXAMPLE 31
Synthesis of 1-O-octadecyl-2-0=methyl-sn-glycero-3 phosphonoformate:
To a solution of carbethoxyphosphodichloridate (1.6mmol) in chloroform
(2OmL), cooled . to O C in an ice - salt bath was added a solution of 1-0-
octadecyl-
2-0-methyl-sn-glycerol (1..Ommol) in pyridine (15mL) dropwise. The mixture was
stirred at room temperature overnight. The mixture was cooled, 2mL of. water
added
and the resulting -mixture stirred at room temperature for 2h. The reaction
mixture was
then concentrated in vacuo, and the residue was flash chromatographed over
silica gel
with chloroform:methanol as eluent.
The ethyl ester was dissolved in a 1:1 mixture of ethanol and O.1N NaOH and
sonicated for 15 minutes. The; mixture was heated with stirring at 60 C for
2h, filtered
and the filtrate evaporated to dryness. The residue was recrystallized from
25%
aqueous ethanol .to yield pure product.,
EXAMPLE 32
Synthesis of nucleoside analog species wherein X = CH-OH, m = 1, RI =
octadecyl
(Compound #8 on Scheme II):
D=Erythrose, 1 (purchased from Aldrich Chemical Company), upon treatment
with NaH and benzyl bromide in DMF at -70 C will give the selectively
protected
species 4-0-benzyl-erythrose, 2. Treatment of this intermediate with
dimethoxypropane
and acetone with trace amounts of perchloric acid will lead to
2,3-di-0-isopropylidene-4- .0-benzyl erythrose 3. Reduction of compound 3 with
sodium borohydride leads to the protected erythritol 4.. Treatment of 4 with
2
5
CA 02223989 2008-11-27
octadecylmethanesulfonate yields the l-O-octadecyl-2,3-di-O-isopropylidene-
.4-O-benzyletythritol 5. Debenzylation with Pd/C and hydrogen followed by DCC
coupling with a nucleoside analog monophosphate will yield intermediate 7.
Deblocking of 1 with 10% TFA in CH2C12 followed by base hydrolysis yields
target
compound 8.
EXAMPLE 33
Synthesis of nucleoside analog derivatives wherein
R2 = H, X = CH-OH, R = octadecyl, m = 2 {compound 17 on Scheme III):
Commercially available D-ribose 9 (Sigma Chemical Co.) upon treatment with
trimethylsilyl methylmercaptan by adaptation ofa procedure by Evans and Co-
workers
(Evans, D.A., Truesdale, L.K., Giamm, K.G., Nesbitt, S.L., J. Am' Chem. Soc.
99, 5009,
1977) will result in the dimethyldithioacetal 10,. This locks the ribose in
the open chain
conformation. Treatment of the protected ribose with benzyl bromide in DMF at -
70 C
will result in selective blocking of the 5-primary hydroxyl group of ribose
leading to
compound 11. Such selective blocking has been reported in the literature
(1987,
Yukuzawa, A., Sato, H., Masamune, T., Tetrahedron Lett. 28, 4303). The 2, 3,
and 4
hydroxyl groups on the 1,5 derivatived ribose can be protected by treatment
with
methoxymethyl chloride (1972, Stark, G., Takashi, T., J. Am. Chem. Soc. 94,
7827) to
yield the fully protected ribose intermediate 12.
The aldehyde at the C position is regenerated by treatment of intermediate 12
with AgNO2/Ag20 (1977, Corey, EJ., Shibasaki, M., Knolle, J., Sugahara, T.,
} Tetrahedron Lett. 985) to yield compound 13. Reduction with sodium
borohydride
followed by allcylation with octadecylmethanesulfonate yields the 1-O-
octadeeyl-2,3,4-
tri-O-methoxymethyl-5-O-benzyl ribose 15. Removal of the benzyl group by
hydrogenation with Pd/C followed by coupling with nucleoside analog phosphate
using
dicyclohexylcarbodiimide yields the intermediate 16. Treatment with acetic
acid
removes the methoxymethyl protecting groups and leads to the target compound,
17.
53
CA 02223989 2009-02-05
EXAMPLE 34
1-S-Octedecyi- l -thiopropane-3-phospho-Acyclovir:
To a stirred mixture of 3-mercapto-1; propanol. (10.0gm, 0.l.lmol) and dry DMF
(400mL) was added sodium hydride (3.6gm, 0.lSmol). When hydrogen evolution
ceased, octadecylmethanesulfonate (35.9gm, 0.1 Imol) was added. Stirring was
c onntinued 2 hours then the mixture was poured into crushed ice .(5008m). The
pI product was colec ted by vaawm filtration, washed with n anol (100dL)
s
and dried to give 1-S octadecyl-1 io-3 +opanol (24.2gm, 64%).
1-S-Octadecyl-1-t1uo-3-propanol =(0.3gm,0.9mmoi)aud
.14 0.30gm, l mol) were dissolved in pyridine (20mL) and A M& The solutiaai
was
cooled in an ice-salt bath and N, N-dicydohexylcarbodumide (0.56gm,
2.7mmol))in
didiloromethaae (IOmL) was added Tice mixture was stored at 0 C for 3 hours
and
at room temperature overnight Tice mixture was Stand and do filtrate vjas
concentrated to dryness In vacuo. The residue was flash clr~omatographhed over
silica
get with an increasing gradient of methanol in dlcbloromede as the eluting
solvent
to obtain 1-S-oetadocyl-i-tbiopropane-3- o. ho-acyclov r as a white solid.
EXAMPLE 35
Human Cytomegalovinus (HCMV) AntivI a1 bil ty Assay:
Subconthreit MRC-5 cells in -24-well culture dishes were pretreated for 24
hours
with various concentrations of drug in ISM medium eomaining 2% FBS and
antibiotics. The media was removed and virus added at a dilution that .will
result in a
3-4+ cytopathic effect (CPE) in the no-drug vas in five :days. This was
absorbed for
1 hour at 37 C, aspinaed and replaced with the drug dilutions. After five days
of
incubation HCMV DNA was quantified in triplicate by nucleic acid hybridization
using
a CMV Antiviral Susceptibility Test Kit from Diagnostic Hybrids, Inc. (Athens,
OH).
The media was removed and cells lysed according to the man's instructions.
After absorption of the lysate, the Hybriwix i filters were hybridized
overnight at 60 C.
The Hybriwixx were washed for'30 minutes at 73 C and counted in a gamma
counter.
The results are expressed as HCMV DNA as a percentage of the untreated
HCMV-infected control cells.
54
CA 02223989 2008-11-27
TABLE H.
ANTI-HUMAN CYTOMEGALOVIRUS ACTIVITY OF
PHOSPHONOFORMIC ACID AND VARIOUS PRODRUGS
COMPOUND ICs., M M SD FOLD p VALUE
INCREASE IN
ACTIVITY
PFA 46 19 (4)
B-PFA 0.43 0.27 (9) 107 '0.0001
BB-PFA' 0.54 0.65 (8) -- - 72 '0.0001
MB-PFA 0.64 0.14. (3) 72 0.0050
ODDMOP-PFA 0.24 0.06 (3) 192 0.0096
ABBREVIATIONS: PFA, phosphonoformate; B-PFA, 1-O-octadecyl-sn-
glycero-3-phosphonoformate; BB-PFA, 1-0-octadecyl-2-0-benzyl-sn-glycero-3-
phosphonoformate; and MB-PFA, 1-O-octadecyl-2-O-methyl-sn-glycero-3-
phosphonoformate; ODDMOP-PFA, 1-O-octadecyl-2,2-dimethoxypropane-3-
phosphonoformate:
The compounds of this invention are all substantially more active than the
free drug,
PFA. MB-PFA and BB-PFA are equivalent in antiviral activity to B-PFA while
ODDMOP-PFA is substantially more active.
EXAMPLE 36
HSV Antiviral Susceptibility Assay
Subconfluent MRC-5 cells in 24-well culture dishes were inoculated by
removing the media and adding HSV-1 virus at a dilution that will result in a
3-4+ CPE
in the no-drug well in 20-24 hours. This was absorbed for 1 hour at 37 C,
aspirated
and replaced with various concentrations of drugs in MEM medium containing 2%
FBS
and antibiotics. After approximately 24 hours of incubation, HSV DNA was
quantified
in triplicate by nucleic acid hybridization using a HSV Antiviral
Susceptibility Test Kit
from Diagnostic Hybrids, Inc. (Athens, OH). The media was removed and cells
lysed
according to the manufacturer's instructions. After absorption of the lysate,
the
Hybriwix filters were hybridized overnight at 60 C. The Hybriwix were
washed for
CA 02223989 2008-11-27
30 minutes at 73 C and counted in a gamma counter. The results are expressed
as a
-percentage of the untreated HSV-infected control cells.
TABLE III
INHIBITION OF HUMAN. SIMPLEX
VIRUS 1 REPLICATION BY PHOSPHONOFORMATE
AND PHOSPHONOFORMATE PRODRUGS
COMPOUND ICs, pM (n) FOLD p_ VALUE
INCREASE IN
ACTIVITY
PFA 47, 20 (6) --
B-PFA 0.63 -0.22 (6) 75 0.0003
BB-PFA 1.4 0.4 (4) 34 0.0003
MB-PFA 0 33,, 0,17 (3) 64 0.0009
is ODDMOP-PFA 16'' 1.9 (4) 29 0.0022
-7
ABBREVIATIONS: PFA, phosphonoformate; B-PFA, 1-O-octadecyl-sn-glycero-
3-phosphonoformate BB-PFD, 1-0-actadecyl:2-O-henzyl-sn-giycero-3-
phosphonoformate; MB-PFA, I-O-octadecyl-2-O-methyl-sn-glycero-3-
phosphonofotmate; ODDMOP.PFA, 1-O-octadecyl-2,2-dimethoxypropane-3-
phosphonofoimate.
All compounds of the invention are more active than free PFA. _
EXAMPLE 37
Plaque Reduction Assay for HIV-1 Replication in HT4-6C Cells
HT4-6C cells and plaque reduction assay, CD4-expressing HeLa cells, HT4-6C
cells (Chesebro, B. and K. Wehrly (1988) J. ViroL 62:3779-3788), were obtained
from
Bruce Chesebro, Hamilton, Mont. The effect of antiviral compounds. on HIV
replication was measured by a plaque reduction assay. Briefly, monolayers of
HT4-6C
cells were infected with 100 to 300 PFU of virus per well in 24-well
microdilution
plates. Various concentrations of drug were added to the culture medium,
Dulbecco's
Modified Eagle Medium containing 5% fetal bovine serum and antibiotics, as
noted
56
CA 02223989 1997-12-05
WO 96/39831 PCT/US96/10054
above. After 3 days at 37 C, the monolayers were fixed with 10% formaldehyde
solution in phosphate-buffered saline and stained with 0.25% crystal violet to
visualize
virus plaques (Larder, B. et al. (1989) Science 243:1731-1734). Antiviral
activity was
assessed as the percentage of control Plaques measured in drug-treated
samples.
TABLE IV
ANTIVIRAL EFFECT OF PHOSPHONOFORMATE AND
PHOSPHONOFORMATE PRODRUGS ON HUMAN
IMMUNODEFICIENCY VIRUS-1
REPLICATION IN HT4-6C CELLS
COMPOUND IC50, M (n) FOLD p VALUE
INCREASE IN
ACTIVITY
PFA 133 t 54 (8) -- --
B-PFA 3.60 f 1.51 (3) 37 0.0015
BB-PFA 14.8 t 7.3 (2) 9.0 0.0091
MB-PFA 1.28 0.73 (3) 104 0.00141
OODMOP-PFA 0.58 0.16 (3) 229 0.00262
All compounds of this invention are more active than PFA. MB-PFA and ODDMOP-
PFA are significantly more active than B-PFA and BB-PFA.
EXAMPLE 38
Synthesis of Radiolabeled Compounds and Pharmacokinetic Studies in Mice
A. Synthesis of 1-O-octadecyl-2-O-methyl-sn-glycero-3-[14 C)-PFA
2.5mCi (.083mmol) of ethoxy[ 14C]carbonylphosphonic dichloride (3011Ci/ mol)
in 0.5mL chloroform was cooled to 0 C. 23mg (0.064mmol) of
' p = 0.0373 versus B-PFA
2 p = 0.0262 versus B-PFA
57
CA 02223989 2008-11-27
a` f A" . ~ y S4
1-O-octadecyl-2-O-methyl-sn-glycerol in 1.5mL of chloroform/pyridine (2:1) at
0 C was
.added while stirring. After 18 hours at room temperature the reaction was
stopped with
0.5niL of water and the mixture dried under nitrogen. The sample was dissolved
in
80:20:1:1 (C/M/NH4/VV) and loaded onto a I gm. silica gel column (70-230 mesh)
which
was eluted with the same solvent. Fractions containing the ethylester of
1-O-octadecyl-2-O-methyl-sn glycero-3-PFA were pooled and dried under
nitrogen.
Half of the ethyl ester was deblocked by dissolving in 1mL of 50% ethanol and
adding.
24 mol of NaO}L The mixture was dried and redissolved in lmL of 50% ethanol
and
again dried under nitrogen. The deblocked - product was dissolved in 80:20:1:1
and
loaded onto a 1 g silica column, washed with 5mL of 80:20:1:1 and eluted with
15mL
of 70:58:8:8. The fractions containing the purified product were pooled, dried
under
nitrogen and redissolved in C/M/W(23:1). The final yield was 7%. The specific
activity is assumed to have remained at 301ACi/ imol.
B. Synthesis of 1-O-Octadecyl-sn-glycem-3-("C}-PFA
SmCi (.167mmol) of ethoxy["C]carbonylphosphonic dichloride (3014Ci/ mol)
in 1mL chloroform was added to 87mg (0.2.mmol) of
1-O-octadecyl-2-O-benzyl-sn-glycerol in 1.5mL of chloroform/pyridine (2:1)
stirring at
0 C. After 18 hours at room temperature the reaction was stopped with 0.5mL of
water
and the mixture dried under nitrogen. The sample was dissolved in 3.5mL
ethanol and
placed in a hydrogenation vessel overnight at 60psi hydrogen with 10%
palladium on
carbon. The process was repeated with the addition of 10% palladium hydroxide
on
carbon. The catalyst was removed and the sample dried under nitrogen. The
sample j
was redissolved in 80:20:1:1 (C/M/NH4/W) and loaded onto a 5gm silica gel
column
(70-230 mesh) which was eluted with the same solvent. The purest fractions
containing
the ethyl ester of Batyl-PFA were pooled and dried under nitrogen. Half of the
ethyl
ester was deblocked by dissolving in 2mL of 50% ethanol and adding 50pmol
(2.Seq)
of NaOH. The mixture was dried and redissolved in 2mL of 50% ethanol and again
dried under nitrogen. The deblocked product was dissolved in 80:20:1:1 and
eluted
with 20mL of 70:58:8:8. The fractions containing the purest product were
pooled, dried
under nitrogen and redissolved in 2mL of C/M/W(2:3:1). The final yield was
2.5%.
58
CA 02223989 1997-12-05
WO 96/39831 PCTIUS96110054
C. Oral Administration of 14C-PFA Lipid Prodrugs to Mice:
1-O-Octadecyl-sn-glycero-3-PFA[14C] and]-O-octadecyl-2-O-methyl-sn-glycero-
3-PFA[14C] (30 microcuries/micromole) were dried with
dioleoylphosphatidylcholine/
cholesterol/drug in a molar ratio of 60/30/10 in a nitrogen stream,
lyophilized overnight
and isotonic sorbitol with 50 mM sodium acetate (pH 5.4) was added. After
vortexing
and warming, the vessel was sonicated at maximum output in a Heat Systems cup
horn
sonicator for one hour. The resulting liposome preparation was filtered
through a 0.2
micron filter and administered to mice by oral gavage in a dose of 20mg/kg of
the
respective PFA lipid prodrugs. After 24 hours the animals were sacrificed and
tissues
rinsed, removed, blotted dry, weighed and homogenized. Aliquots were counted
and
the amount of radioactivity determined. The results are expressed as nanomoles
of lipid
prodrug PFA[14C] per gm tissue.
As can be appreciated from Figure 2, liposomes containing Batyl-PFA (1-0-
octadecyl-sn-glycero-3-PFA) provided lower levels of drug in most tissues
studied
including liver, spleen, jejunum and ileum. However, with the compound of the
invention, methylbatyl-PFA(1-O-octadecyl-2-O-methyl-sn-glycero-3-PFA), it was
surprisingly observed that drug levels were lower in the small intestine and
higher tissue
levels of drug were observed in liver, spleen, lung, brain, kidney and lymph
node. This
suggests that batyl-PFA is not fully delivered to the circulation after its
uptake into the
small intestinal cells, resulting in lower tissue levels of drug. Conversely,
a compound
of the invention, methyl-batyl-[ 14C]-PFA, is more readily cleared from the
small
intestine providing higher levels of drug in liver, lung, kidney, lymph node,
spleen and
brain. It is anticipated that the other lipid moieties claimed in this
invention will also
provide similar results. Furthermore, the benefits arise not from the drug
moiety
attached to the lipid, but from the lipid moiety itself which enhances small
intestinal
uptake and tissue penetration as shown in Figure 2.
59
CA 02223989 1997-12-05
WO 96/39831 PCT/US96/10054
EXAMPLE 39
ANTI-HEPATITIS B VIRUS ACTIVITY AND CYTOTOXICITY OF ACV AND
ITS 1-O-ALKYLGLYCEROL PHOSPHATE OR -PROPANEDIOL PHOSPHATE
CONJUGATES IN 2.2.1 CELLS.
Using the cell culture test system as described by Korba and Gerin (Antiviral
Research 19:55-70, 1992) in the example above, nucleoside derivatives were
also tested
for anti-HBV activity and the data is presented in Table V below.
TABLE V
Selectivity
Compound cc,5O ECG" ELM Index
ACV 684 >100 >100 n.m.
ODGP-ACV 843 6.8 30 124
HDPDP-ACV >300 3.0 10 >100
Data are expressed as M concentration of drug required to reduce hepatitis B
virus
replication by 50% (EC50) or 90% (EC90) or to reduce cell viability by 50%
(CC50).
Selectivity index is the CCSO/EC90. Abbreviation: n.m. - not meaningful; ACV -
acyclovir; ODGP-ACV-1-O-octadecyl-sn-glycero-3-phospho-acyclovir; HDPDP-ACV,
1-O-hexadecyl-propanediol-3-phospho-acyclovir.
1-O-hexadecylpropanediol-3-phospho-acyclovir has an EC50 of 3.0 as compared
to 6.8 for ODGP-ACV, and is significantly more active than acyclovir itself in
inhibiting HBV replication in vitro. The selectivity index of this compound is
also
>100. Thus, these data indicate HDPDP-ACV to be considerably more active and
selective than ODGP-ACV.
EXAMPLE 40
CA 02223989 1997-12-05
WO 96/39831 PCT/US96110054
ANTI-HSV ACTIVITY AND CYTOTOXICITY OF ACV AND ITS 1-0-
ALKYLGLYCEROL PHOSPHATE OR PROPANEDIOL PHOSPHATE
CONJUGATES IN HSV-INFECTED MRC-5 CELLS.
Using the cell culture system described in Example 36 above, acyclovir
derivatives were also tested for anti-HSV activity and the data is presented
in Table V
below in Table VI.
TABLE VI
HSV-1 (wild type)
Compound EC50
ACV 0.03 0.01 (5)
ODGP-ACV 0.07 0.03 (5)
HDPDP-ACV 0.33 0.25 (5)
DM21 HSV-1 (TK-)
Compound EC50
ACV 33 12 (4)
ODGP-ACV 36 19 (3)
HDPDP-ACV 6.8 2.0 (3)
Data are expressed as M concentration of drug required to reduce herpes
simplex virus
replication by 50% (EC50). Abbreviation: ACV - acyclovir; ODGP-ACV - 1-0-
octadecyl-sn-glycero-3-phospho-acyclovir; HDPDP-ACV,1-O-hexadecyl-propanediol-
3-
phospho-acyclovir; HSV-1 (wt) herpes simplex virus-1 (wild type); DM21 HSV-1
(TK-), DM21 strain of HSV-1 deficient in viral thymidine kinase.
Against HSV-1, HDPDP-ACV is less active than ODGP-ACV. However, in the
DM21 HSV-1, which lacks thymidine kinase, the propanediol compound is four
fold
more active than ODGP-ACV and acyclovir itself.
61