Note: Descriptions are shown in the official language in which they were submitted.
CA 02229036 1998-02-06
W O 97/09335 PCT/GB96/02113
Substituted PhosPhinic Compounds and their Use as Pharmaceuticals
This invention relates to chemical compounds which are substituted phosphinic acids or
salts or esters thereof, their prepaLration and their use as pharmaceuticals.
In WO 94122843 there are described phosphinic acids of forrnula
o
~ N OH
R2 H
wherc Rl and R~ are both H, Rl and R2 are both methyl or Rl and R2 together with the
attached carbon atom are cyclopentyl. These col~lpoullds are said to act as GABAB
antagonists.
It has now been found that compounds having remarkably high GABAB receptor binding
affinity can be provided by preparing novel substituted phosphinic acids containing a
morpholine ring.
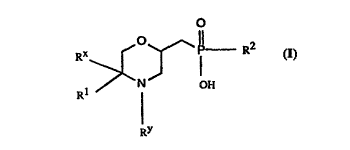
Accordingly, the present invention provides compounds which are substituted phosphinic
acids of formula
RX>~ ~--P-- R2
7 OH
RY
or salts or esters thereof,
where R1 is a monovalent aromatic or araliphatic group connected through a carbon atom
thereof to the in~ir~t~-,cl carbon atom, R2 is an unsubstituted or substituted hydrocarbyl
group, Rx is hydrogen or an unsubstituted or substituted hydrocarbyl group, RY is
hydrogen, RYa or a NH- protecting group, and RYa is an unsubstituted or substituted
CA 02229036 1998-02-06
W O 97/09335 PCT/GB96/02113
hydrocarbyl group.
Rl as an aromatic group may have up to 40 carbon atoms a ld may be an aryl group such
as a phenyl, tolyl, xylyl or naphthyl group or a heterocyclic aromatic group such as a
thienyl, furyl, indolyl or pyridyl group, which groups may be unsubstituted or substituted
by one or more substituents such as halogen, hydroxy, C1 to C4 alkoxy, carboxyl,functionally modified carboxyl inrl~ ng esterified carboxyl, ~mi-l~tP~l carboxyl and
cyano, carboxy-C1-C8 alkyl, fllnrtion~lly modified carboxy-Cl-C8 alkyl or nitro.
Preferably, Rl as an aromatic group is an aryl group of 6 to 15 carbon atoms which may be
uns--hstitllt~fl or substituted in one or more positions by halogen, carboxyl, functionally
m~lifie~ carboxyl, carboxy-Cl-C8 alkyl, functionally modified carboxy-Cl-C8 alkyl or
nitro, or Rl as an aromatic group is a 5 to 10-membered heterocyclic aromatic group
having one or two nitrogen atoms in the ring system. More preferably, Rl as unsubstituted
or substituted aryl is phenyl or phenyl substituted in one or more of the meta and para
positions, with respect to the carbon atom thereof linked to the in~lic~t~ll morpholine ring,
by halogen, carboxyl, functionally modified carboxyl, or nitro. Examples of suchsubstituted phenyl groups include phenyl mono-or di-substituted by chloro; bromo; iodo;
carboxyl; -CooR3 where R3 is Cl to C8 alkyl such as methyl, ethyl, propyl, butyl, pentyl,
hexyl, heptyl or octyl optionally substituted by halogen, hydroxy or Cl to C4 alkoxy;
carbarnoyl; N-C1-C4 alkyl carbamoyl, such as methyl- or ethyl-carbamoyl, N, N-di(Cl-C4
alkyl) carbamoyl such as dimethyl- or diethyl-carbamoyl; cyano; carboxy-Cl-C4 alkyl
such as carboxymethyl; Cl to C8 alkoxy-carbonyl-Cl-C4 alkyl such as methoxy- or
ethoxy- carbonylmethyl; carbamoyl-Cl-C4 alkyl such as carbamoylmethyl; N-CI-C4
alkylcarbamoyl-C1-C4 alkyl such as methyl- or ethyl-carba;noylmethyl, N,N-di(Cl-C4
alkyl)carbamoyl-Cl-C4 alkyl such as dimethyl- or diethyl-carbamoylmethyl; cyano-Cl-C4
alkyl such as cyanomethyl; or nitro. More preferably, R1 as a heterocyclic aromatic group
is a S to 10-membered heterocyclic group having a nitrogen atom as the only ring hetero
atom, e.g. pyridyl or indolyl.
Rl as an araliphatic group may have 7 to 40 carbon atoms and may be phenyl-lower alkyl,
for example benzyl or 2-phenylethyl, a, a-diphenyl-lower alkyl such as diphenylmethyl,
or a-naphthyl-lower alkyl such as naphthylmethyl, any of which ~roups may be
unsubstituted or substituted in one or more positions, which may be ortho, meta or para
positions, by a substituent chosen from those hereinbefore specified for Rl as an aromatic
group. Preferably, Rl as an araliphatic group is a-phenyl-Cl-C4 alkyl, which is
unsubstituted or substituted in one or more positions by halogen, carboxyl, functionally
modi~ed carboxyl or nitro.
CA 02229036 1998-02-06
W O 97/09335 PCT/GB96/02113
In especi~lly preferred cc,~ ounds of the invention, Rl is phenyl, 3-iodophenyl, 3,
4-dichlorophenyl, 3-carboxyphenyl, 3-cyanophenyl, 3-(methoxycarbonyl)phenyl,
3-nitrophenyl, benzyl, 4-iodobenzyl, 4-carboxybenzyl, 4-ethoxycarbonylbenzyl or
indol-3-yl.
R2 as an unsubstituted or substituted hydrocarbyl group may, in general, have 1 to 40
carbon a~oms. It may be for example an alkyl, cycloalkyl, aL~enyl or alkynyl group or an
alkyl, cycloalkyl or alkenyl group substituted by one or more substituents such as halogen,
hydroxy, Cl to C~3 alkoxy, thio, Cl to C8 alkylthio, cyano, acylamino, C3 to C8 cycloaL~cyl,
C3 to C8 cycloalkyl substituted for example by one or more substit-l~nt~ such as hydroxy,
Cl to C8 alkoxy, thio or Cl to C8 alkylthio, C3 to Cg cyclo~lk~nyl,C6 to C15 aryl, C6 to Cls
aryl substituted for example by one or more substituents such as hydroxy, Cl to C8 aLkoxy,
halogen or trifluoromethyl, het~ a~yl or heteroaryl substituted by one or more
substituents such as halogen.
Aliphatic radicals R2 are, for example, lower alkyl, lower alkenyl, lower alkynyl,
oxo-lower alkyl, 'hydroxy- or dihydroxy-lower alkyl, hydroxy-lower alkenyl, mono-, di- or
poly-halo-lower alkyl, mono-, di- or poly-halo-lower alkenyl, mono-, di- or poly-halo-
(hydroxy)-lower alkyl, mono-, di- or poly-halo(hydroxy)-lower alkenyl, lower alkoxy-
lower alkyl, di-lower alkoxy-lower alkyl, lower alkoxy(hydroxy)-lower alkyl, lower
alkoxy(halo)-lower alkyl, lower alkylthio-lower alkyl and di-lower alkylthio-lower aL~cyl.
Cycloaliphatic radicals R2 are, for example, cycloalkyl, hydroxycycloalkyl, oxa, dioxa-,
thia- and dithia-cycloalkyl.
Cycloaliphatic-aliphatic radicals R2 are, for example, cycloalkyl-lower alkyl, cyclo-
alkenyl-lower aL~cyl, cycloalkyl(hydroxy)-lower alkyl and (lower aLkylthio)cycloalkyl-
(hydroxy)-lower alkyl.
Araliphatic radicals R2 are, for example, phenyl-lower alkyl radicals that are unsubstituted
or mono-, di- or tri-substituted by lower alkyl, lower alkoxy, hydroxy, halogen and/or by
trifluoromethyl, preferably a-phenyl-lower alkyl substituted as in~lir~tefl or unsubstituted
a,a-diphenyl- or a-naphthyl-lower alkyl.
Heteroarylaliphatic radicals R2 are, for example, thienyl-, furyl- or pyridyl-lower alkyl
radicals that are unsubstituted or substituted, especially mono- or di-substituted, by
halogen, preferably unsubstituted a-thienyl-, a-furyl- or a-pyridyl-lower alkyl.
CA 02229036 1998-02-06
W O 97/09335 PCT/GB96/02113
-4-
Hereinbefore and hereinafter, lower radicals and compounds are to be lln~lerstood, for
example, as those containing up to and including 7, preferably up to and in~ Aing 4,
carbon atoms.
Lower aLIcyl is, for example, Cl-C7alkyl, preferably Cl-C4alkyl, such as methyl, ethyl,
propyl, isopropyl or butyl, but may also be isobutyl, sec-butyl, tert-butyl or a Cs-C7aLIcyl
group, such as a pentyl, hexyl or heptyl group.
Lower alkenyl is, for example, C2-C4alkenyl, such as vinyl, allyl or but-2-enyl, but may
also be a C5-C7alkenyl group, such as a pentenyl, hexenyl or heptenyl group.
Lower alkynyl is, for exarnple, C2-C7aL~cynyl, preferably C3-CsaL~cynyl, that carries the
double bond in a position higher than the a,~-position, for example 2-propynyl
(propargyl), but-3-yn- 1 -yl, but-2-yn- 1 -yl or pent-3-yn- 1 -yl.
Oxo-lower alkyl carries the oxo group preferably in a position higher than the a-position
and is, for example, oxo-C2-C7alkyl, especially oxo-C3-C6alkyl, such as 2-oxopropyl, 2-
or 3-oxobutyl or 3-oxopentyl.
Phenyl-lower aL~yl is, for example, benzyl, l-phenylethyl, 2-phenylprop-2-yl or, in the
second place, 2-phenylethyl, 2-phenylprop-1-yl or 3-phenylprop-1-yl.
Thienyl-, furyl- or pyridyl-lower alkyl is, for exarnple, thienyl-, furyl- or pyridyl-methyl,
1-thienyl-, l-furyl- or 1-pyridyl-ethyl, 2-thienyl-, 2-furyl- or 2-pyridyl-prop-2-yl, or, in the
second place, 2-thienyl-, 2-furyl- or 2-pyridyl-ethyl, 2-thienyl-, 2-furyl- or 2-pyridyl-
prop-1-yl or 3-thienyl-, 3-furyl- or 3-pyridyl-prop-1-yl.
Hydroxy-lower alkyl carries the hydroxy group preferably in the a- or ~-position and is,
for example, corresponding hydroxy-C2-C7alkyl, such as 1-hydroxyethyl, 1- or 2-hydroxy-
propyl, 2-hydroxyprop-2-yl, l- or 2-hydroxybutyl, l-hydroxyisobutyl or 2-hydroxy-3-
methylbutyl.
Dihydroxy-lower alkyl carries the hydroxy groups especially in the a,~-position and is, for
example, a"B-dihydroxy-C3-C7alkyl, such as 1,2-dihydroxyprop-2-yl.
Hydroxy-lower alkenyl carries the hydroxy groups preferably in the a-position and the
double bond preferably in a position higher than the a,~-position and is, for example,
CA 02229036 1998-02-06
W O 97/09335 PCT/GB96/02113
c~ yonding a-hydroxy-C3-Csalkenyl, for example l-hydroxybut-2-enyl.
Mono-, di- or poly-halo-lower alkenyl is, for exarnple, mono- di- or t~i-fluoro-C2-Cs-
alkenyl, such as l-fllloro~ut-2-enyl.
Mono-, di- or polyhalo(hydroxy)-lower alkyl carries the hydroxy group preferably in the
a-position and the halogen atoms preferably in a position higher than the a-position and
is, for example, corresponding mono- di- or tri-fluoro-cx-hydroxy-C2-C7alkyl, such as
4,4,4-~ifluoro- 1-hydroxybutyl.
Mono-, di- or poly-halo-lower alkyl is, for example, mono- di- or tri-fluoro-c2-c5aL~yl~
such as 3,3,3-trifluoLupiupyl, 4,4,4-trifluorobutyl, 1- or 2-fluorobutyl or l,l-difluorobutyl.
Lower alkoxy is, for exarnple, C1-C7alkoxy, preferably Cl-C4aLt~oxy, such as methoxy,
ethoxy, propoxy, isopropoxy or butoxy, but may also be isobutoxy, sec-butoxy, tert-
butoxy or a Cs-C7alkoxy group, such as a pentyloxy, hexyloxy or heptyloxy group.
Acylamino-lower aLkyl is, for exarnple Cl-C4 alkylcarbonylamino-CI-C4 alkyl such as
acetylarninopropyl or C6-C10 arylcarbonylamino-CI-C4 aL~cyl such as
benzoylaminomethyl.
Cyano-lower aL~cyl is, for exarnple cyano-CI-C4 alkyl, such as cyanomethyl or
2-cyanoethyl.
Mono-, di- or poLy-halo(hydroxy)-lower aLkenyl ca~ies the hydroxy group preferably in
the ~ positlon and the halogen atoms preferably in a position higher than the a-position
and is, for example, corresponding mono-, di- or tri-fluoro-oc-hydroxy-C2-CsaLkerlyl, such
as 2-fluoro-1-hydroxybu~en-2-yl.
Lower aLkoxy-lower aLkyl is, for example, Cl-C4aLkoxy-C~-C4alkyl, such as methoxy- or
ethoxy-methyl, 2-methoxyelhyl, 2-ethoxyethyl, 3-methoxy- or 3-ethoxy-propyl or 1- or
2-methoxybutyl.
Di-lower alkoxy-lower alkyl is, for example, di-CI-~-alkoxy-CI-C4alkyl, for example
dimethoxymethyl, dipropoxymethyl, 1,1- or 2,2-diethoxyethyl. diisopropoxymethyl,dibutoxymethyl or 3,3-dimethoxypropyl.
Lower alkoxy~hydroxy)-lower alkyl is, for exarnple, Cl-c4aLlcoxy-c2-cr(hydroxy)aLk
CA 02229036 1998-02-06
W O 97/09335 PCT/GB96/02113
such as 2-hydroxy-3-methoxyprop-2-yl.
Lower alkoxy(halo)-lower alkyl is, for example, Cl-C4alkoxy-C2-Cs-(halo)aLlcyl, such as
2-fluoro-3-methoxybutyl.
Lower alkylthio-lower alkyl is, for example, Cl-C4alkylthio-CI-C4alkyl, such as methyl-
thio- or ethylthio-methyl, 2-methylthioethyl, 2-ethylthioethyl, 3-methylthio- or3-ethylthio-propyl or 1- or 2-methylthiobutyl.
Di-lower alkylthio-lower alkyl is, for example, di-CI-C4alkylthio-Cl-C4alkyl, for example
dimethylthiomethyl, dipropylthiomethyl, 1,1- or 2,2-diethylthioethyl, diisopropylthio-
methyl, dibutylthiomethyl or 3,3-dimethylthiopropyl.
Halogen is halogen having an atomic number of up to and including 53, i.e. fluorine,
chlorine, bromine or iodine.
Cycloalkyl is, for example, C3-C8cycloalkyl, especially C3-C6cycloalkyl, such as cyclo-
propyl, cyclobutyl, cyclopentyl or cyclohexyl.
Hydroxycycloalkyl is, for example, a-hydroxy-C3-C6cycloaL~yl, such as 1-hydroxycyclo-
propyl, 1-llyd~o~ycyclobutyl or 1-hydroxycyclohexyl.
Oxa- or thia-cycloalkyl is, for example, oxa- or thia-C3-C8cycloalkyl, especially oxa- or
thia-C3-C6cycloalkyl, such as 2-oxacyclopropyl (oxiranyl), 2- or 3-oxacyclobutyl (oxetan-
yl), 2- or 3-thiacyclobutyl (thietanyl), 2- or 3-oxacyclopentyl (tetrahy~lluful~nyl), 2- or
3-thiacyclopentyl (thiolanyl) or 2-oxacyclohexyl (tetrahydropyra~nyl).
Dioxacycloalkyl is, for example, 1,3-dioxa-C3-C8cycloaL~cyl, such as 1,3-dioxolan-2-yl or
1,3-dioxan-2-yl.
Dithiacycloalkyl is, for example, 1,3-dithia-C3-C8cycloalkyl, such as 1,3-dithiolan-2-yl or
1,3-dithian-2-yl.
Cycloalkyl-lower alkyl is, for example, C3-C8-cycloalkyl-Cl-C4alkyl, especially C3-C6-
cycloalkyl-Cl-C4alkyl, such as a-(C3-C6cycloalkyl)-Cl-C4alkyl, for example cyclopropyl-
methyl, cyclobutylmethyl, cyclopentylmethyl or cyclohexylmethyl.
Cycloalkenyl-lower alkyl is, for example, C3-C8cycloalkenyl-Cl-C4alkyl, especially
CA 02229036 1998-02-06
W O 97/09335 PCT/GB96/02113
-7-
C3-C6cycloalkenyl-Cl-C4a'lkyl1 such as a-(C3-C6cyclo~lk~1lyl)-cl-c4alkyL for example
cyclopent-1-enylmethyl, cyclopent-2-enylmethyl, cyclopent-3-enylmethyl, cyclohex-1-
enyl rmethyl, cyclohex-2-enylmethyl or cyclohex-3-enylmethyl.
..
CycloaLkyl(hydroxy)-lower alkyl is, for example, C3-C6cycloalkyl-C~-C4(hydroxy)alkyl,
such as ~c-(C3-C6cycloalkyl)-a-hydroxy-CI-C4a'lkyl, for example cyclopropyl(hydroxy)
methyl, cyclobutyl(hydroxy)methyl, or cyclohexyl(hydroxy)methyl.
(Lower alkylthiocycloalkyl)(hydroxy)-lower alkyl is, for example, 1-(Cl-C4alkylthio-
C3-C6cycloalkyll-l-hydroxy-CI-C4alkyl, such as (2-methylthiocycloprop-1-yl)hydroxy-
methyl.
In plcfG~lGd compounds of the invention, R2 is lower alkyl, lower alkenyl, lower alkynyl,
oxo-lower alkyl, hydroxy- or dihydroxy-lower alkyl, hydroxy-lower alkenyl, mono-, di- or
poly-halo-lower a~kyl, mono-, di- or poly-halo-lower alkenyl, mono-, di- or poly-halo-
(hydroxy)-lower alkyl, mono-, di- or poly-halo(hydroxy)-lower alkenyl, lower aLlkoxy-
lower alkyl, di-lower alkoxy-lower alkyl, lower alkoxy(hydroxy)-lower alkyl, lower
aLkoxy(halo)-lower alkyl, lower alkylthio-lower alkyl, di-lower alkylthio-lower alkyl,
cyano-lower alkyl, acylamino-lower alkyl, cycloalkyl, hydroxycycloalkyl, oxa-7 dioxa-,
thia- and dithia-cycloalkyl, cycloalkyl-lower alkyl, cycloalkenyl-lower alkyl, cycloalkyl-
(hydr~xy)-lower alkyl, (lower alkylthio)cycloalkyl(hydroxy)-lower alkyl, or mono- or di-
phenyl-lower aLkyl that is unsubstituted or mono-, di- or tri-substituted by lower alkyl,
lower alkoxy, halogen, hydroxy and/or by trifluoromethyl, naphthyl-lower alkyl or unsub-
stituted or halo-substituted thienyl-, furyl- or pyridyl-lower alkyl.
In more preferrel1 compounds of formula I, R2 is C1-C7alkyl, such as methyl, ethyl, propyl,
isopropyl, butyl isobutyl or pentyl, a,a-di-C1-C4alkoxy-Cl-C4alkyl, especially a,a-di-
Cl-C4aLcoxy-methyl or ethyl, such as dimethoxy- or diethoxy-methyl or 1,
l-dieehoxyethyl, cyano-C1-C4 alkyl such as cyanomethyl or 2-cyanoethyl,
acylamino-Cl-C5 aL~yl such as acetyl:lminoethyl, acetylaminopropyl, acetylaminopentyl
or benzoylaminomethyl, C3-C6cycloalkyl-C1-C4alkyl, such as cyclopropyl- or cyclo-
hexyl-methyl, C.,-C6cyclo~1kt-nyl-C1-C4aLcyl, such as cyclohex-3-enylmethyl, or is
phenyl-C1-C4alkyl, such as benzyl, that is unsubstituted or mono-, di- or tri-substituted by
C1-C4alkyl, such as methyl, C1-C4alkoxy, such as methoxy, hydroxy and/or by halogen,
such as fluorine, chlorine or iodine.
In more preferred compounds of the invention, R2 is C1-C5 alkyl such as methyl, ethyl or
butyl, a,o~-di-(CI-C4 alkoxy)methyl such as diethoxymethyl, a,a-di-(Cl-C4 aL~coxy)ethyl
CA 02229036 1998-02-06
W O 97/09335 PCT/GB96/02113
such as 1, 1-diethoxyethyl, C3-C6 cycloalkyl-Cl-C4 aLkyl such as cyclopropylmethyl or
cyclohexylmethyl, benzyl or 4-methoxybenzyl. In especially preferred compounds, R2 is
cyclohexylmethyl or 4-methoxybenzyl.
Rx as an unsubstituted or substituted hydrocarbyl group may have up to 40 carbon atoms
and may be a Cl to C10 alkyl, C2 to C10 alkenyl, C3 to C8 cycloaL~cyl, C4 to C13cycloalkylalkyl, C6 to C1O aryl or C7 to C13 aralkyl group, any of which groups may be
substituted by one or more subsliLuc--ls chosen from those hereinbefore specified for Rl.
Preferably R~ is hydrogen, lower alkyl, C3 to C6 cycloalkyl, C6 to C8 aryl or C7 to Cg
aralkyl, especially hydrogen or isopropyl.
RY as an unsubstituted or substituted hydrocarbyl group RYa may have up to 40 carbon
atoms and may be, for exarnple, a Cl to C10 alkyl, C3to C8 cycloaLkyl or C7to C~3 araL~cyl
group, any of which groups may be unsubstituted or substituted by hydroxy or Cl to C4
aL~oxy. RY as a NH-protecting group may be, for example, an acyl group such as acetyl,
trifluoroacetyl, benzoyl or p-nitrobenzoyl or an aL~oxycarbonyl or aralkoxycarbonyl group
such as tert-butoxycarbonyl or benzyloxyc~l,onyl. Preferably RY is hydrogen, lower
alkyl, C7 to Cg aralkyl, acetyl, benzoyl, tert-butoxycarbonyl or benzyloxycarbonyl,
especially hydrogen, methyl, ethyl, benzyl, acetyl, benzoyl, tert-butoxycarbonyl or
benzyloxycarbonyl.
Specific especially ~l~re..~,d compounds of the invention are those of formula I in which
Rl is phenyl, 3-iodophenyl, 3, 4-dichlorophenyl, 3-cyanophenyl,
3-(methoxycarbonyl)phenyl, 3-carboxyphenyl, 3-nitrophenyl, benzyl, 4-iodobenzyl,4-carboxybenzyl, 4-ethoxycarbonylbenzyl or indol-3-yl, R2 is cyclohexyimethyl or4-methoxybenzyl, Rx is hydrogen or isopropyl and RY is hydrogen, methyl or
benzyloxycarbonyl, and salts and esters thereof.
The compounds of formula I may be in the form of internal salts and can form both acid
addition salts and salts with bases by conventional salt-forming reactions.
Acid addition salts of compounds of formula I are, for example, their pharmaceutically
acceptable salts with suitable mineral acids, such as hydrohalic acids, sulfuric acid or
phosphoric acid, for example hydrochlorides, hydrobromides, sulfates, hydrogen sulfates
or phosphates, or salts with suitable aliphatic or aromatic sulfonic acids or N-substituted
sulfamic acids, for example meth~neslllfonates, benzenesulfonates, p-toluenesulfonates or
N-cyclohexylslllf~ tes (cyclamates).
CA 02229036 1998-02-06
W O 97/09335 PCT/GB96/02113
Salts of compounds of formula I with bases are, for example, their salts with ph~Tn~reut-
ically acceptable bases, such as non-toxic metal salts derived from metals of groups Ia, Ib,
IIa and IIb, for example aL~ali metal salts, especially sodium or potassium salts. ~lk~linf~.
earth metal salts, especially c~lri-lm or m~En~sil~m salts, and also ammonium salts with
ammonia or organic amines or quaternary ammonium bases, such as unsubstituted orC-hydroxylated aliiphatic ~minÇs7 especially mono-, di- or tri-lower alkyl~minPs, for
example methyl-, ethyl- or diethyl-amine, mono-, di- or tri-(hydroxy-lower alkyl)~min~s,
such as ethanol-, diethanol- or trieth~nQl-amine, tris(hydroxymethyl)methylamine or 2-
hydroxy-tert-butylamine, or N-(hydroxy-lower alkyl)-N,N-di-lower alkyl~minPs or
N-(polyhydroxy-lower aL~cyl)-lower aL~cyl~m;nes, such as 2-(dimethylamino)ethainol or
D-gluc~mine7 or quaternary aliphatic ammonium hydroxides, for example tetrabutyl-
ammonium hydroxide.
As well as forming salts with bases, the hydroxy group ~tt~rhefl to phosphorus in formula
I may also be esterified. Thus, the invention infl~ s compounds of formula I in the form
of their esters with an alcohol, which may be a Cl to C10 aLkanol in which the a~kyl radical
is unsubstituted or substituted, for example by halogen, cyano or Cl to C4 alkoxy, such as
methanol, ethanol, isopropanol, isobutanol, 2-ethylhexanol, 2-chloroethanol,
2-cyanoethanol, 2;-ethoxyethanol or 2-n-butoxyethanol, a C3 to C8 cycloaliphatic alcohol
such as cyclopropanol, cyclobutanol, cyclopentanol, cyclohexanol, cycloheptanol,methylcyclohexanol or cyclooctanol, or a C7 to Cl3 araliphatic alcohol such as benzyl
alcohol.
Provided asymmetric carbon atoms are present, the compounds according to the invention
may be in the fonn of isomeric mixtures, especially in the form of racemates, or in the
form of pure isomers, especially optical antipodes.
I~crc~l~d isomers of compounds of formula I are those in which Rl and the group ~tt~ch~-l
to the 2- position of the in-1ir~te-1 morpholine ring are trans with respect to each other,i.e.
those of formula
O
R~ R2
J I ~A)
~ - N OH
Rl
RY
or of fonnula
CA 02229036 1998-02-06
W O 97/09335 PCT/GB96/02113
- 10-
RX ~ ~ P R2
~ N J OH (E-)
R 1 RY
where R1, R2, Rx and RY are as hereinbefore defined.
Other preferred isomers of formula I are those in which Rl and the group ~t~rhell to the
2-position of the indicated morpholine ring are cis with respect to each other, i.e. those of
formula
~ ...""'~11
~ N ) P R2 (IC)
Rl RY OH
or of formula
RX j~ ~--P R2
Rl Nl ¦ (ID)
RY OH
where Rl, R2, Rx and RY are as hereinbefore clefinecl
Examples of specific compounds of formula I are
3- { (3R*,6R*)-6-[(5-acetylaminopentyl)hydroxyphosphinoylmethyl]
morpholin-3-yl } benzoic acid,
3- { (3R*,6R*)-6- [(cyclohexylmethyl)hydroxyphosphinoylmethyl]
morpholin-3-yl } benzoic acid,
3- { (3R*,6R*)-6- [(4-methoxyphenylmethyl)hydroxyphosphinoyl-
methyl]morpholin-3-yl } benzoic acid,
3-[(3R*,6R*)-6-(butylhydroxyphosphinoylmethyl)-
CA 02229036 1998-02-06
W O 97/0~335 PCT/G1396/02113
morpholin-3-yl]'benzoic acid,
3- { (3R*,6R*)-6 -[(diethoxymethyl)hydroxyphosphinoylmethyl]-
morpholin-3-yl } benzoic acid,
3-[(3R*,6R*)-6-(benzylhydroxyphosphinoylmethyl)-
morpholin-3-yl]'benzoic acid,
diethoxymethyl- { (2R*,SR*)-5-[(3-methoxycarbonyl)phenyl]-
morpholin-2-ylmethyl } phosphinic acid,
cyclohexylmeth yl-[(2R*,SR*)-5-phenylmorpholin-2-ylmethyl]-
phosphinic acid,
diethoxymethyl [(2R*,5R*)-5-(3-nitrophenyl)-
morpholin-2-ylnnethyl]phosphinic acid,
butyl-[(2R*,SRY )-5-(3-iodophenyl)morpholin-2-ylmethyl]-
phosphinic acid,
[(2R*,5R*)-5-(3-cyanophenyl)morpholin-2-ylmethyl]-
phenylmethylphosphinic acid,
5-acetylaminopentyl-[(2R*, 5R*)-5-(3,4-dichlorophenyl)morpholin-2-ylmethyl]-
phosphinic acid,
cyclohexylmeth yl-[(2R*,5R*)-5-(3,4-dichlorophenyl)-
morpholin-2-ylnnethyl]phosphinic acid,
butyl-[(2R*,5R*)-5-(3,4-dichlorophenyl)morpholin-2-ylmethyl]-
phosphinic acid,
benzyl-L(2R*,5R*)-5-(3,4-dichlorophenyl)morpholin-2-ylmethyl]-phosphinic acid,
[(2R*,5R*)-5-(3 ,4-dichlorophenyl)morpholin-2-ylmethyl] -
pyridin-2-ylmethylphosphinic acid,
[(2R*,5R*)-5-(3 ,4-dichlorophenyl)morpholin-2-ylmethyl]-
diethoxymethylphosphinic acid,
[(2R*,5R*)-5-(3 ,4-dichlorophenyl)morpholin-2-ylmethyl] -
4-methoxyphenylmethylphosphinic acid,
[(2R*,5R*)-5-benzylmorpholin-2-ylmethyl]-4-methoxyphenyl-
methylphosphinic acid,
4- { (3R~',6R*)-6-[(cyclohexylmethyl)hydroxyphosphinoylmethyl]-
morpholin-3-ylrneehyl } benzoic acid,
4- { (3R*,6R*)-6-[(4-methoxyphenylmethyl)hydroxyphospinoyl-
methyl]morpholin-3-ylmethyl } benzoic acid,
' 4-[(3R*,6R*)-6- (benzylhydroxyphosphinoylmethyl)-
morpholin-3-ylrnethyl]benzoic acid,
4-[(3R*,6R*)-6- (bul:ylhydroxyphosphinoylmethyl)morpholin-
3-ylmethyl]benzoic acid,
CA 02229036 1998-02-06
W O 97/09335 PCT/GB96/02113
- 12-
4- { (3R*,6R*)-6-[(diethoxymethyl)hydroxyphosphinoylmethyl]-
morpholin-3-ylmethyl } benzoic acid,
cyclohexylmethyl- [(2R* ,SR*)-5-(4-iodobenzyl)morpholin-2-
ylmethyl]phosphinic acid,
[(2R*,SR*)-5-(4-iodobenzyl3morpholin-2-ylmethyl] -4-
methoxyphenylmethylphosphinic acid,
cyclohexylmethyl- { (2R*,5R*)-5-[4-ethoxycarbonyl)-
phenylmethyl]mo;pholin-2-ylmethyl] phosphinic acid,
4-[(3R*,6R*)-6-(butylhydroxyphosphinoylmethyl)-N-methyl-
morpholin-3-ylmethyl]benzoic acid,
4- { (3R*,6R*)-6-[(cyclohexylmethyl)hyd;oxyphosphinoylmethyl]-
N-benzyloxycarbonylmorpholin-3-ylmethyl } benzoic acid,
3- { (3R*,6R*)-[(5-acetylaminopentyl)hydroxyphosphinoyl-
methyl]-3-methylmorpholin-3-yl ~ benzoic acid,
3- ~ (3R*,6R*)-6-[(diethoxymethyl)hydroxyphosphinoylmethyl]
-3-methylmorpholin-3-yl ) benzoic acid,
3-[(3R*,6R*)-6-(butylhydroxyphosphinoylmethyl)
-3-methylmorpholin-3-yl]benzoic acid,
3-[(3R* ,6R*)-6-(benzylhyd;oxyphosphinoylmethyl)
-3-methylmorpholin-3-yl]benzoic acid,
3- { (3R*,6R*)-6-[(4-methoxyphenylmethyl)hydroxyphosphinoyl-
methyl]-3-methylmorpholin-3-yl } benzoic acid,
3- { (3R*,6R*)-6-[(cyclohexylmethyl)hydroxyphosphinoylmethyl]-
3-methylmorpholin-3-yl } benzoic acid,
S-acetylaminopentyl-[(2R*,SR*)-5-(6-oxo- 1 ,6-dihydropyridin-3-yl)-
morpholin-2-ylmethyl]phosphinic acid,
cyclohexylmethyl-[(2R*,5R*)-5-(6-oxo- 1 ,6-dihydropy;idin-3-yl)-
morpholin-2-ylmethyl]-phosphinic acid,
butyl-~(2R*,5R*)-5-(6-oxo- 1 ,6-dihydropyridin-3-yl)moipholin-
2-ylmethyl]phosphinic acid,
benzyl-[(2R*,5R*)-5-(6-oxo- 1 ,6-dihydropyridin-3-yl)mo~pholin-
2-ylmethyl]phosphinic acid,
4-methoxyphenylmethyl-[(2R*,SR*)-5-(6-oxo- 1 ,6-dihydropyridin-3-yl)-
morpholin-2-ylmethyl]phosphinic acid,
diethoxymethyl-[(2R*,5R*)-5-(6-oxo- 1 ,6-dihydropyridin-3-yl)-
morpholin-2-ylmethyl]phosphinic acid,
CA 02229036 1998-02-06
W O 97/0933S PCT/G]B96/02113
cyclohexylmethyl-[(2R*,SR*)-5-(2-oxo- 1 ,2-dihydropyridin-4-yl)-
morpholin-2-ylmethyl]phosphinic acid,
S-acetylaminopentyl-[(2R*,5R*)-5-(2-oxo-1,2-dihydropy;idin-4-yl)-
morpholin-2-ylmethyl]phosphinic acid,
butyl-[(2R*,5R*)-5-~2-oxo- 1 ,2-dihydropyridin-4-yl)morpholin-
2-ylmethyl]phosphinic acid,
diethoxymethyl- [(2R*,SR*)-5-(2-oxo- 1 ,2-dihydropyridin-4-yl)-
morpholin-2-ylmethyl]phosphinic acid,
benzyl-[(2R*,5R~*)-5-(2-oxo- 1,2-dihydropy;idin-~yl)morpholin-
2-ylmethyl]phosphinic acid,
4-methoxyphenylmethyl-[(2R*,5R*)-5-(2-oxo- 1 ,2-dihyd;opyridin-4-yl)-
morpholin-2-ylmethyl]phosphinic acid,
2- { (3R*,6R*)-6--[(cyclohexylmethyl)hydroxyphosphinoylmethyl]-
morpholin-3-ylmethyl } benzoic acid,
2- { (3R*,6R*)-6 [(4-methoxyphenylmethyl)hydroxyphosphinoylmethyl] -
morpholin-3-ylmethyl } benzoic acid,
2- { (3R*,6R*)-6 [(diethoxymethyl)hydroxyphosphinoylmethyl]-
morpholin-3-ylmethyl}benzoic acid,
2- { (3R*,6R*)-6 [(5-acetylaminopentyl)hydroxyphosphinoylmethyl]-
morpholin-3-ylmethyl } benzoic acid,
2-[(3R*,6R*)-6-(butylhydroxyphosphinoylmethyl)morpholin-
3-ylmethyl]benzoic acid,
2-[(3R*,6R*)-6-(benzylhyd;oxyphosphinoylmethyl)morpholin-
3-ylmethyl]benzoic acid,
3- { (3R*,6R*)-6 -[(cyclohexylmethyl)hydroxyphosphinoylmethyl]-
morpholin-3-ylmethyl } benzoic acid,
3- { (3R*,6R*)-6 -[(4-methoxyphenylmethyl)hydroxyphosphinoyl-
methyl]morpholin-3-ylmethyl } benzoic acid,
~ 3- { (3R*,6R*)-6 -[(diethoxymethyl)hydroxyphosphinoylmethyl]-
mo~pholin-3-yln:lethyl } benzoic acid,
3- { (3R*,6R*)-6 -[(S-acetylaminopentyl)hydroxyphosphinoylmethyl]-
morpholin-3-yln;lethyl } benzoic acid,
3-[(3R*,6R*)-6- (butylhydroxyphosphinoylmethyl)morpholin-
3-ylmethyl]benz:oic acid,
3-[(3R*,6R*)-6-(benzylhydroxyphosphinoylmethyl)morpholin-
3-ylmethyl]benzoic acid,
CA 02229036 1998-02-06
W O 97/09335 PCT/GB96102113
- 14-
benzyl- ( (2R*,5R*)-5-[4-([ 1 ,3,4]o7c~ 7r 1 -2-yl)phenyl]morpholin-
2-ylmethyl}phosphinic acid,
butyl- { (2R*,5R*)-5-[4-(5-trifluoromethyl-[ 1 ,2,4]ox~ 7--1-3-yl)-
phenyl] morpholin-2-ylmethyl } phosphinic acid,
1-(4- { (3R*,6R*)-6-[(4-methoxybenzyl)hydroxyphosphinoylmethyl]-
morpholin-3-yl}phenyl)-lH-rl,2,4]triazole-3-carboxylic acid,
{ (2R*,SR*)-5-[4-(3-amino-[ 1 ,2,4]oxadiazol-5-yl)phenyl]morpholin-
2-ylmethyl}cyclohexylmethylphosphinic acid,
{(2R*,5R*)-5-[3-(3-amino-[1,2,4]o~ 7nl-5-yl)phenyl]morpholin-
2-ylmethyl } cyclohexylmethylphosphinic acid,
diethoxymethyl- { (2R*,5R*)-5-[3-(lH-tetrazol-5-yl)phenyl]morpholin-
2-ylmethyl}phosphinic acid,
3- { (3R*,6S*)-6-[(5-acetylaminopentyl)hydroxyphosphinoyl-
methyl]morpholin-3-yl}benzoic acid,
3- { (3R* ,6S *) -6- [(cyclohexylmethyl)hydroxyphosphinoylmethyl] -
morpholin-3-yl } benzoic acid,
3-~(3R*,6S*)-6-[(4-methoxyphenylmethyl)hydroxyphosphinoyl-
methyl]morpholin-3-yl } benzoic acid,
3-[(3R*,6S *)-6-(butylhydroxyphosphinoylmethyl)-
morpholin-3-yl]benzoic acid,
3- { (3R*,6S *) -6- [(diethoxymethyl)hydroxyphosphinoylmethyl] -
morpholin-3-yl } benzoic acid,
3-[(3R*,6S *)-6-(benzylhydroxyphosphinoylmethyl)morpholin-
3-yl]benzoic acid,
diethoxymethyl- { ~2R*,5S*)-5-[(3-methoxycarbonyl)phenyl]-
morpholin-2-ylmethyl}phosphinic acid,
cyclohexylmethyl-[(2R*,SS*)-S-phenylmorpholin-2-ylmethyl]-
phosphinic acid,
diethoxymethyl-[(2R*,5S*)-5-(3-nitrophenyl)morpholin-
2-ylmethyl]phosphinic acid,
butyl-[(2R*,5S*)-5-(3-iodophenyl)morpholin-2-ylmethyl]-
phosphinic acid,
[(2R*,SS*)-5-(3-cyanophenyl)morpholin-2-ylmethyl]phenyl-
methylphosphinic acid,
S-acetylaminopentyl-[(2R*, 5S*)-5-(3,4-dichlorophenyl)-
morpholin-2-ylmethyl]phosphinic acid,
:
CA 02229036 1998-02-06
W O 97/0933~ PCT/GB96/02113
cyclohexylmethyl-[(2R*,SS*)-5-(3,4-dichlorophenyl)-
morpholin-2-ylmethyl]phosphinic acid,
butyl-[(7R*,SS * )-5-(3,4-dichlorophenyl)-
morpholin-2-ylmethyl]phosphinic acid,
benzyl-[(2R*,5S *)-5-(3,4-dichlorophenyl)-
morpholin-2-ylmethyl]phosphinic acid,
[(2R*, SS*)-5-(3,4-dichlorophenyl)morpholin-
2-ylmethyl]pyridin-2-ylmethylphosphinic acid,
[(2R*,5S *)-5-(3 ,4-dichlorophenyl)morpholin-2-ylmethyl]-
diethoxymethylphosphinic acid,
[(2R*,5S *)-5-(3 ,4-dichlorophenyl)morpholin-2-ylmethyl] -
4-methoxy-phen.ylmethylphosphinic acid,
[(2R*,SS *)-5-benzylmorpholin-2-ylmethyl]-4-methoxyphenyl-
methylphosphinic acid,
4- { (3R*,6S *)-6- [(cyclohexylmethyl)hydroxyphosphinoylmethyl] -
morpholin-3-ylmethyl}benzoic acid,
4- { (3R*,6S *)-6- [(4-methoxyphenylmethyl)hydroxyphospinoyl-
methyl]morpholin-3-ylmethyl } benzoic acid,
4-[(3R*,6S *)-6-(benzylhydroxyphosphinoylmethyl)morpholin-
3-ylmethyl]benz:oic acid,
4-[(3R*,6S*)-6-(butylhydroxyphosphinoylmethyl)morpholin-
3-ylrnethyl]benzoic acid,
4- { (3R*,6S *)-6- [(diethoxymethyl)hydroxyphosphinoylmethyl]-
morpholin-3-ylmethyl } benzoic acid,
cyclohexylmethyl-[(2R*,5S*)-5-(4-iodobenzyl)morpholin-
2-ylmethyl]phosphinic acid,
[(2R*,SS*)-5-(4-iodobenzyl)morpholin-2-ylmethyl]-
4-methoxyphenylmethylphosphinic acid,
cyclohexylmethyl- 1 ~2R*,5S*)-5-[4-ethoxycarbonyl)phenyl-
methyl]morpho].in-2-ylmethyl]phosphinic acid,
4-[(3R*,6S*)-6-(butylhydroxyphosphinoylmethyl)-
N-methylmorpholin-3-ylmethyl]benzoic acid,
4- { (3R*,6S*)-6 -[(cyclohexylmethyl)hydroxyphosphinoylmethyl]-
N-benzyloxycarbonylmorpholin-3-ylmethyl } benzoic acid,
3- { (3R*, 6S*)-6-[(5-acetylaminopentyl)hydroxyphosphinoyl-
methyl] -3-meth ylmorpholin-3-yl } benzoic acid,
3-{(3R*,6S*)-6-[(diethoxymethyl)hydroxyphosphinoylmethyl]-
3-methylmorpholin-3-yl } benzoic acid,
CA 02229036 1998-02-06
W O 97/09335 PCT/GB96/02113
- 16-
3-[(3R*,6S*)-6-(butylhydroxyphosphinoylmethyl)-3-methyl-
morpholin-3-yl]benzoic acid,
3-[(3R*,6S *)-6-(benzylhydroxyphosphinoylmethyl)-3-methyl-
morpholin-3-yl]benzoic acid,
3- ~ (3R*,6S *)-6-[(4-methoxyphenylmethyl)hydroxyphosphinoyl-
methyl]-3-methylmorpholin-3-yl } benzoic acid,
3- { (3R*,6S*)-6-[(cyclohexylmethyl)hydroxyphosphinoylmethyl]-
3-methylmorpholin-3-yl ~ benzoic acid,
5-acetylaminopentyl-[(2R*,SS*)-5-(6-oxo- 1 ,6-dihydropyridin-3-yl)-
morpholin-2-ylmethyl]phosphinic acid,
cyclohexylmethyl-[(2R*,SS *)-5-(6-oxo- 1 ,6-dihydropy;idin-3-yl)-
morpholin-2-ylmethyl]phosphinic acid,
butyl-[(2R*,5S*)-5-(6-oxo-1,6-dihydropyridin-3-yl)morpholin-
2-ylmethyl]phosphinic acid,
benzyl-[(2R*,5S*)-5-(6-oxo- 1 ,6-dihydropy;idin-3-yl)mo;pholin-
2-ylmethyl]phosphinic acid,
4-methoxyphenylmethyl-[(2R*,5S*)-5-(6-oxo- 1 ,6-dihydropy;idin-
3-yl)morpholin-2-ylmethyl]phosphinic acid,
diethoxymethyl-[(2R*,SS *)-5-(6-oxo- 1 ,6-dihydropyridin-3-yl)-
morpholin-2-ylmethyl]phosphinic acid,
cyclohexylmethyl-[(2R*,SS*)-5-(2-oxo- 1,2-dihydropyridin-4-yl)-
morpholin-2-ylmethyl]phosphinic acid,
5-acetylaminopentyl-[(2R*,SS *)-5-(2-oxo- 1 ,2-dihydropy;idin-4-yl)-
morpholin-2-ylmethyl]phosphinic acid,
butyl-[(2R*,SS *)-5-(2-oxo- 1 ,2-dihydropyridin~-yl)morpholin-
2-ylmethyl]phosphinic acid,
diethoxymethyl-[(2R*,SS*)-5-(2-oxo- 1,2-dihyd;opy;idin-4-yl)-
mo;pholin-2-ylmethyl]phosphinic acid,
benzyl-[(2R*,SS *)-5-(2-oxo- 1 ,2-dihydropy;idin-4-yl)morpholin-
2-ylmethyl]phosphinic acid,
4-methoxyphenylmethyl-[(2R*,5S*)-5-(2-oxo-1,2-dihydropyridin-
4-yl)morpholin-2-ylmethyl]phosphinic acid,
2- { (3R*,6S*)-6-[(cyclohexylmethyl)hydroxyphosphinoylmethyl]-
morpholin-3-ylmethyl } benzoic acid,
2- { (3R*,6S*)-6-[(4-methoxyphenylmethyl)hydroxyphosphinoylmethyl]-
CA 02229036 1998-02-06
W O 97/0~335 PCT/G1396/02113
morpholin-3-ylrnethyl } benzoic acid,
2- { (3R*,6S *)-6- [(diethoxymethyl)hydroxyphosphinoylmethyl] -
morpholin-3-ylrnethyl } benzoic acid,
2- { (3R It,6S*)-6--t(5-acetylaminopentyl)hydroxyphosphinoylmethyl]-
morpholin-3-ylrnet,hyl } benzoic acid,
2-[(3R*,6S*)-6-(butylhydroxyphosphinoylmethyl)morpholin-
3-ylmethyl]benzoic acid,
2-[(3R*,6S*)-6-(benzylhydroxyphosphinoylmethyl)morpholin-
3-ylmethyl]benzoic acid,
3- { (3R*,6S*)-6 -[(cyclohexylmethyl)hydroxyphosphinoylmethyl]-
morphc,lin-3-ylmethyl ~ benzoic acid,
3-{(3R*,6S*)-6-[(4-methoxyphenylmethyl)hydroxyphosphinoylmethyl]-
morpholin-3-ylmethyl } benzoic acid,
3- { (3R*,6S*)-6-[(diethoxymethyl)hydroxyphosphinoylmethyl]-
morpholin-3-ylmethyl } benzoic acid,
3- { (3R*,6S *)-6-[(5-acetylaminopentyl)hydroxyphosphinoylmethyl] -
morpholin-3-ylmethyl } benzoic acid,
3-[(3R*,6S*)-6- (butylhydroxyphosphinoylmethyl)morpholin-
3-ylmethyl]benzoic acid,
3-[(3R*,6S*)-6- (benzylhydroxyphosphinoylmethyl)morpholin-
3-ylmethyl]benzoic acid,
benzyl-{ (2R*,SS*)-5-[4-([1,3,4]oxadiazol-2-yl)phenyl]morpholin-
2-ylmethyl}phosphinic acid,
butyl- { (2R*,5S ~)-5-[4-(5-trifluoromethyl-[ 1 ,2,4]o~ 7nl -3-yl)-
phenyl}morpholin-2-ylmethyl } phosphinic acid,
1-(4- { (3R*,6S*)-6-t(4-methoxybenzyl)hydroxyphosphinoylmethyl]-
morpholin-3-yl ~ phenyl)- lH-[ 1 ,2,4]triazole-3-carboxylic acid,
{ (2R*,5S*)-5-[4-(3-amino-[1,2,4]oxadiazol-5-yl)phenyl]morpholin-
- 2-ylmethyl } cyclohexylmethylphosphinic acid,
{ (2R*,5S*)-5-[3-(3-arnino-[1 ,2,4]oxadiazol-5-yl)phenyl]morpholin-
~ 2-ylmethyl } cyclohexylmethylphosphinic acid,
diethoxymethyl-{(2R*,SS*)-5-[3-(lH-tetrazol-5-yl)phenyl]morpholin-
2-ylmethyl } phosphinic acid.
It has been found that the compounds of formula I and their pharm~re~ltic~lly acceptable
salts have valuable pharmacological p,o~ ies. They exhibit an effective binding to the
CA 02229036 1998-02-06
W O 97/09335 PCT/GB96/02113
-18-
GABAB receptor and have been found to be antagonists of GABA (~-aminobutyric acid)
at that receptor. With regard to the mççh~nicm, antagonism at GABAg ,~cepl~ . can
increase the release of rapid stimulant amino acid tr~ncmittprs~ that is to say, ~lnt~m~te
and aspartate, and thus improve information processing in the brain. This is in keeping
with the finding that the late post-synaptic inhibition potential in the hippocampus, which
is attributed to a GABAB mech~nicm, is broken down by the ~nt~gonictC and thus permits
a faster nerve impulse tr~ncmicsiQn sequence.
It has also been found that chronic tre~tm~nt with anti-depressants and repeated electric
shocks increase the number of GABAB receptors in the cerebral cortex of rats. In accord-
ance with receptor theories, chronic treatment with GABAB antagonists should have the
sarne effect. For this and other reasons, GABAB antagonists can accordingly act as anti-
depresc~ntc
The GABAB antagonists according to the invention interact at the GABAB ~~,ce~lul with
IC50 values from 10-7 to 10-1~M (mole/litre) on cerebral cortex membranes of rats. In
contrast to GABAB agonists, such as baclofen, they do not potentiate the stimulation by
noradrenalin of adenylate cyclase on sections of the cerebral cortex of rats but act as
antagonists of the baclofen action. The antagonists not only exhibit ~nt~gonicm tow~ds
baclofen but also have an independent action as antagonists of endogenous GABA.
In view of their excellent GABAB antagonistic lJ~u~e- Lies, the compounds of the invention
are suitable for use in the treatment or prevention of conditions characterised by
stimulation of GABAB receptors. Thus they are suitable for use as nootropics,
antidepressants and anxiolytics, for example in the treatment of central nervous system
disorders such as anxiety, depression, cerebral insufficiency, epilepsy of the "petit mal"
type, i.e. absence epilepsy in children and adolescçntc, atypical absences such as the
Lennox-Gastant syndrome, in the treatment of conditions requiring enh~nrernent of
cognitive l clro,l--ance and as an antidote to baclofen.
Compounds of formula I where RY is hydrogen may be prepared by reacting a compound
of formula
CA 02229036 1998-02-06
W O 97/09335 PCT/GB96/02113
- 19 -
OH
CH2
R
R4 / \MH2
where R4 is R1 as hereinbefore defined except that R4 may not be substituted by carboxyl,
and Rx is as hereinbefore defined except that it may not be substituted by carboxyl with a
compound of folmula
X /\ ll R2 m
IR5
where R2 is as hereinbefore f1efin~-1 X is halogen, e.g. chlorine or bromine, and Rs is Cl
to C8 alkyl, e.g. n-hexyl, n-octyl, preferably Clto C4 alkyl such as methyl, ethyl, isopropyl
or isobutyl, especially ethyl, in the presence of a base, to give a compound of forrnula
RX~ ~ l l R2
~ IV
/ N 5
R4 H OR
where ~4 and R~ are as defined in formula II, followed, where required, by one or more
substitution reactions to change the nature of a substituent in R4 and/or Rx andlor by
hydrolysis of an ester substituent in R4 and/or RX to carboxyl and/or by conversion of the
~ ester group -oR5 to -OH.
- By alJ~ iate selection of the base and reaction conditions, the reaction of compounds of
formulae II and rII, which proceeds by monoalkylation of the amino group followed by
cycli~tion, may be effected in a one-step procedure. Preferably, to avoid complit~tions
resul~ing from dialkylation of the amino group, the reaction is carried out in two stages. In
the first stage, a weak base, for example a hindered amine such as
1, 8-diazabicyclo~5.4.0]undec-7-ene (DBU) is added slowly to a mixture of the
CA 02229036 1998-02-06
W O 97/0933~ PCT/GB96/02113
-20-
compounds of formulae II and III in a solvent, preferably a hydrocarbon such as benzene,
toluene or xylene, at a tel.lp~,lature of 70 to 110~C, to give a novel intermeAi~tP product of
formula
R
R4 H R2
where R2, R4, R5 and Rx are as hereinbefore defined. This interme~ t~ is then treated
with a base under harsher conditions than those employed in its formation, for example
with a similar base at a higher temperature or, preferably, with a stronger base such as an
a~cali metal hydride at a temperature from 10 to 50~C. The treatment of the intermediate
with base may be carried out in a solvent, preferably a hydrocarbon such as toluene,
benzene or xylene.
Interrne~ te compounds of formula V may also themselves be used as pharmaceuticals,
for example in the treatment or prevention of a condition characterised by stimulation of a
GABAB receptor, particularly in de-esterified form, i.e. where Rs as aL~yl has been
replaced by hydrogen and any carboxylic ester group in R4 and/or Rx has been converted
into a carboxyl group. Accordingly, the invention includes novel compounds of formula
OH
RX ~ ¦ VA
Rl N ~ P R2
H
OH
where Rl, R2 and Rx are as hereinbefore defined, or salts or esters thereof.
Compounds of formula I or IV in which Rx and/or Rl or R4 respectively con~ins a cyano
substituent on an aryl or heteroaryl ring may be prepared by reacting an aL~cali metal
cyanide with a compound of formula I or IV where Rx and/or Rl or R4 respectivelycontains a halogen substituent on an aryl or heteroaryl ring, which compound may be
pre~ed by diazotisation, followed by reaction with an aL~ali metal halide, of a co~ oul~d
of formula I or IV where Rx and/or Rl or R4 respectively contains an amino group on an
aryl or heteroaryl ring, which compound may be prepared by re~ ction of a compound of
formula I or IV in which Rx and/or R1 or R4 respectively contains a ni~o group on an aryl
CA 02229036 1998-02-06
W O 97/0~335 PCT/G1396/02113
-21-
or he~eroaryl ring. All of these reactions can be effected using known procedures.
Compounds of f~rmula I or IV in which Rx and/or R1 or R4 respectively cQnt:~inc an
esteriifled carboxyl substituent can also be prepared from other compounds of formula I or
IV respectively. For example, they may be prepared by reacting a compound of forrnula I
or IV in which Rx and/or Rl or R4 respectively contains a halogen substituent on an aryl or
hete~u~yl ring vvith carbon monoxide and an alcohol in the presence of a p~ m
complex as cata]lyst, using known procedures.
Compounds of formula I in which Rx and/or Rl cont~inc a carboxyl substituent may be
d by hydrolysis of a compound of forrnula I or IV in which Rx and/or Rl or R4
respectively contains an esterified carboxyl substituent using conventional hydrolysis
procedures.
Where, in a compound of formula IV or V, R4 cont~inc an esterified carboxyl group, this
may be hydrolysed to a free carboxyl group using conventinn~l methods. Where R4 in the
compound of fo:rmula IV contains a nitro group on an aryl or hetclua-yl ring, this group
may be converted in turn to amino by reduction, to halo by diazotisation of arniino
followed by reaction with an alkali metal halide, to cyano by reaction of halo ~ith an
alkali metal cyanide and thence to carboxyl by hydrolysis of cyano, these reactions
conveniently be ing carried out using known procedures.
The conversion of the ester group -ORs in a compound of formula IV or V into -OH can
be effected by tre~tment with a suitable basic or acidic agent, such as an alkali metal
hydroxide, for example sodium hydroxide or lithium hydroxide, an alkali metal halide,
especia]ly an aLkali metal bromide or iodide, such as lithium bromide or sodium iodide,
thiourea, an alkali metal thiophenolate, such as sodium thiophenQl~t~. or a protonic acid or
a Lewis acid, such as a mineral acid, for example hydrochloric acid, or a tri-lower
alkyl-halosilane, for example trimethylchlorosilane. rhe replacement reaction can be
effected in the absence or presence of a solvent and, if necessary, with heating or with
cooling in a closed vessel and/or under an inert gas atmosphere.
The conversion of -oR5 in a compound of formula IV or V into -OH can also be carried
out by treatmenlt with an acid under hydrolytic conditions, especially with a mineral acid,
such as a hydrohalic acid, for example hydrochloric acid, which is used in dilute or
concentrated aqueous form, or by treatment with an organic silyl halide, such astrimethylsilyl iodide or bromide, and, if necessary, by subsequent hydrolysis. The reaction
is preferably calTied out at elevated temperature, for example by m~inr~ining the reaction
CA 02229036 1998-02-06
W O 97/09335 PCT/GB9~/02113
mixture at reflux te~llpc.dtul~, and, where appropriate, using an organic diluent in a closed
vessel and/or under an inert gas atmosphere.
Compounds of formula II are in some in~t~nres commçrcially available, e.g. (R) - and (S)
- phenyl glycinols. Compounds of formula II may be prepared by re~ ction of an
~minoc~rboxylic acid of formula R4C(RX)(NH2)CooH, where R4 and R~ are as
hereinbefore defined in formula II, by reaction with borane dimethyl sulphide in the
presence of a boron trifluoride complex such as boron trifluoride diethyl etherate. This
reaction may be carried out using known procedures. Novel colll~oullds of formula II,
where (i) R4 is iodobenzyl, particularly 4-iodobenzyl, and R~ is hydrogen and (ii) R4 is
phenyl and Rx is isopropyl, may be prepared by this method.
The compounds of formula II where R4 is substituted by nitro may be ~Icp~,d from an
aminocarboxylic acid of formula R4C(RX)(NH2)CooH where R4 is otherwise
unsubstituted by nitration to introduce a nitro group into R4, converting the amino group in
the product into a protected amino group, for example by reaction with di-tert-butyl
dicarbonate to form a tert-butylcarbamate group, esterifying the carboxyl group in the
proL~ Gd product for example by conversion into a methyl ester, then reducing the ester
group to -CH20H by treatment with an ~ u~liate reducing agent such as an alkali metal
borohydride and finally removing the amino-protecting group by tre~tment with acid to
re-form a free amino group. These reactions may be carried out using known procedures
or minor mo lific~tions thereof. The known compound of forrnula II where R4 is
nitrophenyl may be prepared by such a reaction sequence.
Compounds of formula II may also be prepared by a Strecker synthesis in which analdehyde or ketone of fo;mula R4C(=o)RX, where R4 and Rx are as hereinbefore ~lefin~l
is reacted with a compound of formula R6NH2, where R6 is hydrogen or an alkyl group of
1 to 8 carbon atoms optionally substituted by a C6 to C10 aryl group which is unsubstituted
or substituted, for example by hydroxy or Cl to C4 alkoxy, and an alkali metal cyanide to
give a compound of formula
CN
VI
R4 1-- NHR6
where R4, R6 and Rx are as hereinbefore clefin~-l, reacting the compound of formula VI
CA 02229036 1998-02-06
W O 97/0~33~ PCT/GB96/02113
with an alcohol of forrnula R70H, where R7 is an alkyl group of 1 to 10 carbon atoms, e.g.
n-hexyl, 2-ethylhexyl, n-octyl or decyl, preferably Clto C4 alkyl such as methyl, ethyl,
isopropyl or n-butyl, especially methyl or ethyl, in the presence of an acid to form a
compound of folmula
CooR7
R4 ¦ - NHR6 V~
where R4, R6, R 7 and Rx are as hereinbefore de~med, removing R6, when this is other than
hydrogen, from ~he compound of formula VII using, for exarnple, known procedures to
give a compound of formula
CoOR7
V~
R4 C NH2
I X
where R4, R7 and Rx are as hereinbefore fl~fin~-l, for exarnple, where R6 is an optionally
substituted benzyl group, by catalytic hydrogen~tion in the presence of an organic acid,
e.g. acetic acid, l:o give a compound of formula VIII in the form of a salt the;eof with the
organic acid, reacting the compound of formula VIII with an amino-protecting agent such
as tert-butyl dicarbonate to convert the amino group into a protected amino group,
reducing the ester group -CooR7 in the ~l~.t~lt;d compound to-CH2OH by reaction with
an ap~ liate re~ cing agent such as an aLkali metal borohydride, and finally removing
the protecting group to form a free amino group. This sequence of reactions may be
carried out using known procedures, or minor moriific~tion~ thereof. Where R4 issubstituted by a carboxylic ester group, the protected amino group formed should be a
group such as a eert-butyl carbamate group which will permit the ester group -CooR7 to
be reduced to -CH20H while leaving the ester group in R4 and then be removable by a
re~rtion, for exarnple in a non-aqueous medium, which leaves the ester group in R4.
In a mo-lifir~tion of the Strecker synthesis hereinbefore described, the compound of
formula VI may be subjected to acid hydrolysis, for example using convention:~l
procedures, to convert the in(lic~te~l cyano group to carboxyl and the resulting
CA 02229036 1998-02-06
W O 97/09335 PCT/GB96/02113
-24-
aminocarboxylic acid may be reduced to a compound of formula II by reaction withborane dimethyl sulphide in the presence of a boron trifluoride complex such as boron
trifluoride diethyl etherate, for in~t~nre using known procedures.
Compounds of formula II, VI, VII or VIII in which R4 is 3-methoxycarbonylphenyl, which
may be prepared from an aldehyde or ketone of forrnula R4C(=o)RX by the reactionsequence as hereinbefore described, are believed to be novel per se. Compounds of
formula II or VIII where R4 is 3,4-dichlorophenyl and R~ is Cl-C1o alkyl are also believed
to be novel.
Compounds of formula II in which R4 is a monovalent aromatic group as hereinbefore
defined and Rx is an unsubstituted or substituted hydrocarbyl group are believed to be
novel, with the exception of compounds of formula II in which R4 is phenyl and Rx is
methyl, chloromethyl, ethyl, -(CH2)3 S CH3, allyl or methylol, compounds of forrnula II in
which Rx is aminomethyl and R4 is phenyl, p-hydroxyphenyl or p-methoxyphenyl,
compounds of formula II in which Rx is methylol and R4 is 4-decylphenyl or
5-[(7-chloro-4-quinolinyl)amino]-2-hydroxyphenyl, a compound of forrnula II in which R4
is 4-methoxyphenyl and Rx is ethyl,and a compound of forrnula II in which R4 is
2,4-dichlorophenyl and Rx is N-triazolylmethyl.
Compounds of formula II in which R4 is a monovalent araliphatic group as hereinbefore
defined and Rx is an unsubstituted or substituted hydrocarbyl group as hereinbefore
defined other than methylol are believed to be novel, with the exception of compounds of
formula II where Rx is methyl and R4 is benzyl, 4-chlorobenzyl, 3,4-dichlorobenzyl,
3,4-dimethoxybenzyl, 2-phenylethyl, 1,3-benzodioxol-5-methyl, 3-phenyl-1-aminopropyl,
a-bydroxybenzyl, a-hydroxy-a-methylbenzyl, or a-hydroxy-a-methyl-4-ni~-)be,-~yl, and
with the exception of compounds of formula II where R4 is benzyl and Rx is allyl or
-CH2CH2SCH3.
Compounds of formula II in which R4 is iodobenzyl, particularly 4-iodobenzyl, and Rx is
hydrogen or an unsubstituted or substituted hydrocarbyl group, as hereinbefore defined are
also believed to be novel.
Compounds of formula III which, with the exception of the compound where R2 is methyl
and Rs is ethyl, are believed to be novel, particularly those where R2 is cycloalkylalkyl
such as cyclohexylmethyl, may be prepared by reacting a compound of formula
CA 02229036 1998-02-06
W O 97/0933S PCT/G]B96/02113
-25-
H P R2
oR5
with a compound of formula
X - CH - CH CH2 X
where R~2, Rs and X are as hereinbefore defimed, in the presence of a silylating ;agent such
as a bis(trialkylsilyl) derivative of an arnide, which agent undergoes reaction with the
compound of formula IX to form a P(III) silyl compound which then reacts with the
compound of formula X. The reaction may be carried out at a ~e~pe-dture frorn O to
50~C; it is preferably carried out in a solvent, for exarnple a hydrocarbon such as toluene
or a halohydrocarbon such as dichloromethane.
Esters of forrnula ~ may be p.~ d by reacting a protected phosphinate ester of forrnula
Il
Q P H Xl
IR5
where R5iS as hereinbefore defined and Q is a P-H-protecting group, with a compound of
forrnula
R2Z XII
where R2 is as hereinbefore defined and Z is a leaving moiety, to give a compound of
formula
~ Il
Q p R XI~
I
ORS
and then replacing the protecting group Q in the compound of formula XIII by hydrogen.
CA 02229036 1998-02-06
W O 97/09335 PCT/G B96/02113
- 26 -
The leaving moiety Z may be, for example, a halogen atom or an organic sulphon~te
group. Preferably Z is chlorine, bromine, iodine, or a meth~nPs~llphonate~
trifluorometh~nesulphonate or p-toluenesulphonate group. The reaction between the
compounds of formulae XI and XII and the depl~)te-;Lion reaction on the co~ ou.,d of
formula XIII may be carried out using known procedures, for example as described in EP
0569333.
Protected phosphinate esters of formula XI may be prepared by known methofl~, for
example as described in US 4 933 478. Compounds of formula XII are either
comm~rcially available or may be prepared by known procedures.
Compounds of formula X are rlih~loalk~-nes which are either commercially available or
may be ~-c~ared using known methods.
Compounds of formula I where RY is RYa may be Illcl ~1d by reacting a compound of
formula I where RY is hydrogen with a co~lpoulld of formula RYaZ, where RYa and Z are as
hereinbefore defined, or by reductive alkylation using an aldehyde of formula RYb CHO,
where RYb is hydrogen or RYa as hereinbefore ~l~fin~l, and a reducing agent which reduces
imines to amines, for example sodium cyanoborohydride. Such reactions may be carried
out using conventional procedures.
Compounds of formula I where RY is a NH-protecting group may be plcpa cd by reacting
a compound of formula I where RY is hydrogen with a reagent known to introduce the
desired protecting group. For exarnple, where the protecting group is an acyl group, the
compound of forrnula I where RY is hydrogen may be reacted with an acyl halide or
carboxylic acid anhydride such as acetyl chloride, acetic anhydride or benzoyl chlnri~
for inst~nce using known procedures. Where the protecting group is an alkoxycarbonyl or
aralkoxycarbonyl group, the compound of formula I where RY is hydrogen may be reacted
with an alkoxycarbonyl or aralkoxycarbonyl halide or an alkyl or aralkyl dicarbonate such
as benzyl chloroforrnate or di-tert-butyl dicarbonate, for example using known procedures.
In general, compounds of formula I where RY is RYa or a NH-~ tccLi.-g group may also be
e~ d by the method hereinbefore described for the preparation of compounds of
formula I where RY is hydrogen, in which method the compound of formula II is replaced
by a compound of formula
CA 02229036 1998-02-06
W O 97/09335 PCT/GE~96/02113
OH
CH2
RX
\ ~A
R4 NHRY
where R4 and RX are as hereinbefore defined in formula II and RY is RYa or a
NH-protecting group, the reaction of the compound of formula IIA with the cornpound of
formula III in the presence of a base giving directly a compound of formula
O ~ 1I R2
N I rVA
oR5
R4 RY
where R4 and Rx are as defined in formula II, R2 and R5 are as hereinbefore defined and
RY is RYa as hereinbefore defined or a NH-protecting group. This reaction may be carried
out in a solvent, usually a hydrocarbon such as benzene, toluene or xylene, and is
generally carried. out under harsher conditions than those used for the reaction of
compounds of formulae II and III, for exarnple using sodium hydride as the base and at a
temperature of 10~C to 70~C. This reaction may be followed, where required, by one or
more substitl-tiol- reactions to change the nature of a substituent in R4 and/or R' and/or by
hydrolysis of an ester substituent in R4 and/or Rx to carboxyl and/or by conversion of the
ester group -ORs to -OH.
Compounds of forrnula I may also be ~ a,ed by reacting a compound of formula
~' R2
N~ ¦
R4 ¦ ORS
RY
to convert the in~ic~tefl primary hydroxyl group into a leaving moiety Z as hereinbefore
CA 02229036 1998-02-06
W O 97/09335 PCT/GB96/02113
-28-
defin~od, thereby effecting cyclisation to give a compound of formula
~~--R2
4~N xv
R I oR5
RY
where R2, R4, Rs, Rx and RY are as hereinbefore defined followed, where required, by
replacement of RY as an NH-protecting group by hydrogen andlor by one or more
subs~ -SiQn re~c tion~ to change the nature of a substituent in R4 and/or Rx and/or by
hydrolysis of an ester substituent in R4 and/or Rx to carboxyl and/or by conversion of the
ester group -oR5 to -OH.
The conversion of the prima;y hydroxyl group in the compound of formula XIV into Z
may be carried out using known procedu.~s. For exarnple, where Z is an iodine atom, the
conversion may be effected by reacting the compound of formula XIV with
triphenylphosphine, im~ 7ole and iodine in a solvent such as acetonitrile or
tetrahydrofuran at 0~C to 50~C, and where Z is a trifluoromethanesulphonate group, the
conversion may be effected by reacting the compound of formula XIV with
trifluoromethanesulphonic anhydride in pyridine at -100~C to 50~C.
The replacement of RY as an NH-protecting group by hydrogen may be carried out using
known procedures for removal of the NH-protecting g,roup. For example, where RY is an
acyl group such as acetyl or benzoyl, replacement by hydrogen may be effected byreaction with aqueous hydrochloric acid, while where RY is trifluoroacetyl, replacement by
hydrogen may be effected by reaction with aqueous po~,.c~itlm carbonate.
The other optional subsequent reactions of compounds of formula XV may be carried out
as hereinbefore described for corresponding reactions of compounds of formula IV.
Compounds of formula XIV, which are themselves believed to be novel, may be l,lcpal~d
by reacting a compound of formula II with a compound of formula
CA 02229036 1998-02-06
W O 97/0933~ PCT/GB96/02113
-29-
HO
\~ ' R2 XVI
oR5
z
where R2, R5 and Z are as hereinbefore rlefint~.r1, in the presence of a hindered base, to give
a compound of formula
RX~ R2 XVII
R4 lRS
where R2, R4, Rs and Rx are as hereinbefore nP,fin~-~l, and replacing the in-lir~t~cl hydrogen
att~<~h~l to nitrogen by a NH-protecting group RY as hereinbefore defined, for example
using known procedures such as those hereinbefore described. The reaction between the
compounds of formula II and XVI may be carried out, for example, at a temperature of 20
to 10û~C, preferably in an organic solvent such as an alcohol, especially ethanol. The
hindered base may be, for example, a diazabicyclo compound such as 1,5-diazabicyclo
t4.3.0]non-5-ene or 1,8-diazabicyclo t5.4.0]undec-7-ene or preferably, a tertiary amine
such as dicyclohexyl(ethyl)amine or, especially, diisopropylethylamine.
Compounds of formula XVI may be ~ ~.,d using the procedures described in J. Med.Chem, 1995, 38, 3313.
Compounds of fc,rmula XIV or XVII may them~çlves be used as pharmaceuticals, forexample in the treatment or prevention of a co~ tiQn characterised by stimulation of a
GABAB receptor, particularly in de-esterified form, i.e. where Rs as aL~cyl has been
replaced by hydrogen and any carboxylic ester group in R4 and/or Rx has been converted
into a carboxyl g3roup, for example using known procedures. Accordingly, the invention
inclllcles novel compounds of formula
CA 02229036 1998-02-06
W O 97/09335 PCT/GB96/02113
-30-
~1 xvm
R4 OH
~Y
where Rl, R2, Rx and RY are as hereinbefore f~efin~ , or salts or esters thereof.
Compounds of the invention obtained as salts can be converted into the free compounds in
a manner known ~ se, for example by tre~tment with a base, such as an alkali metal
hydroxide, a metal carbonate or metal hydrogen carbonate, or ammonia, or another of the
salt-forming bases mentioned hereinbefore, or with an acid, such as a mineral acid, for
example with hydrochloric acid, or another of the salt-forming acids mentiQn~
hereinbefore.
Salts of the invention can be converted into different salts of the invention in a manner
known per se; for example, acid addition salts can be converted by treatment with a
suitable metal salt, such as a sodium, barium or silver salt, of another acid in a suitable
solvent in which an inorganic salt being forrned is insoluble and is thus excluded from the
reaction equilibrium, and base salts can be converted by freeing the free acid and
converting into a salt again.
The compounds of formula I, inl~hl-ling their salts, may also be obtained in the form of
hydrates or may include the solvent used for cryst~ tiQn
Owing to the close rel~tion~hip between the novel compounds in free form and in the form
of their salts, hereinbefore and hereinafter the free compounds and their salts are also
optionally to be understood as being the corresponding salts and free compounds, respec-
tively, where a~ op~iate and where the context so allows.
For compounds of forrnula I, and interme~ tto,s in the preparation thereof,
diastereoisomeric mixtures and mixtures of r~cem~t~os can be separated in known manner
into the pure diastereoisomers and racemates, respectively, on the basis of the physico-
chernic~l differences between their constituents, for example by chromatography and/or
fractional crystallisation.
Resulting racemates can also be resolved into the optical antipodes by known methods, for
example by recrystallisation from an optically active solvent, with the aid of micro-
CA 02229036 l998-02-06
W O 97/0~335 PCT/GB96/02113
- 31 -
or~nicm~ or, by reaction of the resulting diastereoi~Qm~ric ,.,i~Lu~ or r~rPm~e with an
optically active auxiliary compound, for example according to the acidic, basic or
fimrtiQn~lly morlifi~ble groups cont~inrfl in compounds of formula I, with an optically
~ active acid, base or an optically active alcohol, into ,.,i~Lules of diastereoisomeric salts or
functional derivatives, such as esters, separation of the same into the diastereoisomers
from which the desired enantiomer can be freed in c~ tnm~ry manner. Suitable bases,
acids and alcohols for the purpose are, for example, optically active ~lk~loicl bases, such as
strychnine, cinchonine or brucine, or D- or L-(l-phenyl)ethylamine, 3-pipecr~line,
ephedrine, amphet~minP and similar bases that can be obtained by synthesis, optically
active carboxylic or sulfonic acids, such as quinic acid or D- or L-tartaric acid, D- or
L-di-o-toluoyltartaric acid, D- or L-malic acid, D- or L-m~ndelic acid, or D- or L-camphor-
sulfonic acid, or optically active alcohols, such as borneol or D- or L-(1-phenyI)eth~nol
Compounds of formula I, VA or XVIII may be isotopically labelled, particularly with llC,
l4C, 2H, 3H or ~25I, for use in ~ gnostics.
The compounds of formula I, VA or XVIII may be used, for exarnple, in the form of
ph~l~n~re,utical compositions that comprise a therapeutically effective amount of the
active ingredient, where a~ u~,iate together with pharm~reu~ic~lly acceptable carriers
that are suitable for enteral, for example oral, or parenteral ~lmini~tration, which carriers
may be solid or liquid and organic or inorganic. For example, tablets or gelatin capsules
are used that contain the active ingredient together with diluents, for example lactose, dex-
trose, saccharose, m~nnitol, sorbitol, cellulose and/or lubricants, for example silica, talc,
stearic acid or salts thereof, such as m~gnesillm or c~lcinm stearate, and/or polyethylene
glycol. Tablets may also contain binders, for example m~gnesillm ~lllmininm silicate,
star~hes, such as corn, wheat, rice or arrowroot starch, gelatin, tr~g~r~nth, methyl-
cellulose, sodium carboxymethylcellulose and/or polyvinylpyrrolidone, and, if desired,
disintegrators, for example starches, agar, alginic acid or a salt thereof, for example
sodium alginate, and/or effervescent mixtures, or absorbents, colourings, flavourings an
sweeteners. The compounds of formula I can also be used in the form of parenterally
~ aflmini~trable compositions or in the form of infusion solutions. Such solutions are prefer-
ably isotonic aqueous solutions or suspensions which, for example in the case of- lyophilised cornpositions that comprise the active ingredient on its own or together with a
carlier, for example m~nnitol, can be prepared before use. The pharmaceutical
compositions rnay be sterilised and/or may comprise excipients, for example preserv-
atives, stabilisers, wetting agents and/or emulsifiers, solubilisers, salts for regulating the
osmotic ~rtS'.U~, and/or buffers. The present pharmzlreutiral compositions which, if
desired, may comprise other pharmacologically active substances, may be prepared in a
CA 02229036 1998-02-06
W O 97/09335 PCT/G B96/02113
manner known ~er se, for example by conventional mixing, gr~n~ ting, confectioning,
dissolving or lyophilising processes, and may co~ lise a~lu~illlately from 0.1 % to
100 %, especially from approxim~t~ly 1 % to ap~,u~ ately S0 %, and, in the case of
lyophilic~tes, up to a~lù~d",ately 100 %, active ingredient.
The invention relates also to the use of the compounds of formula I, VA or XVIII, or salts
or esters thereof, preferably in the form of pharm~ceuri~ ~l compositions.
The dose may depend on various factors, such as the mode of ~flminictration, specieC, age
and/or individual condition. The doses to be ~-lminictered daily may, in the case of oral
~-lminictration~ be from approximately 1 to ap~io~ -ately 50 mg/kg, especially from 5 to
approximately 25 mg/kg, and, in the case of warm-blooded ~nim~lc having a body weight
of approximately 70 kg, preferably from a~p,u~d~-~ately 70 mg to a~,u,~i".ately 3500 mg,
especially from approximately 350 to a~ul"u~il"ately 1750 mg, expediently divided into
from 2 to 6, for example 3 or 4, single doses.
The invention accordingly includes a method of treating or preventing a conflition in
warm-blooded m~mm~lc, particularly humans, characterised by stimulation of a GABAB
~cce~tor which comprises ~-lminictering to the warm-blooded m~mm~l a compound offormula I, VA or XVIII, or a pharm~centic~lly acceptable salt or ester thereof.
The invention is illustrated by the following Examples.
Compound D used in the Examples is prepared as follows. In subsequent formulae, Boc
denotes tert-butoxycarbonyl.
Phenyl glycine (42g) is dissolved in concentrated sulphuric acid (210ml) and the solution
is cooled to 0~C. Fuming nitric acid (15.5ml) is added dropwise to the cooled solution
over 30 minutes and the mixture is stirred for a further 30 minutes at 0~C, then 18 hours at
room temperature. The solution obtained is poured over l litre of ice and carefully adjusted
to pH7 by adding approximately 875ml of lOM aqueous sodium hydroxide whilst keeping
the temperature of the solution below 20~C. The resulting mixture is stirred for 3 hours at
room temperature and the precipitate obtained is filtered off. The precipitate is washed
three times with water and recrystalized from l litre of water to afford
3-nitrophenylglycine, mpt 165-6~C.
3-Nitrophenyl glycine (Sg) is added to a mixture of methanol (200ml) and triethylamine
(20ml) and the mixture is stirred vigorously for lO minutes at room tem~,aLulc.
CA 02229036 1998-02-06
wo 97/09335 PCT/GB96/02113
- 33 -
Di-tert-butyl dicarbonate (11.13g) is added and the reaction mixture is heated under reflux
for 2 hours. The solution obtained is cooled to room te,l,~ dture and then conc~n~ t~ to
dryness under reduced pressure to afford a dark orange residue. The residue is purified by
flash chromatography [silica gel, CH2Cl2 (95%), CH30H (2.5%), CH3COOH (2.5%)] toafford Compound A as an orange foam.
COOH
NHBoc ~ mr~ A
N02
13Cnmr (lOOMH.z; CDC13): ~ (ppm)27.9 (q), 5B.2(d), 82.6(s), 122.4(d), 123.0(d), 129.5(d),
133.0(d), 140.4(s), 148.2(s~, 156.8(s), 172.1(s).
A solutic-n of Compound A (14.54g) and p-tolllent-sl-lrhonic acid (1.87g) in meth~nol
(200ml) is stirred at room temperature for 24 hours. Ethyl acetate and saturated aqueous
NaHCO3 are added, the organic phase is separated, dried over MgSO4 and filtered, and the
filtrate is evaporated under reduced pressure. The product obtained is purified by flash
chromatography [silica gel, hexane:ethyl acetate (2:1)] to yield Compound B as a yellow
solid, mpt = 86~C,
COOCH3
mrlmfi B
NHBoc
N02
Analysis: calculated for Cl4Hl8N2O6; C, 54.19; H, 5.85; N, 9.03%.
Found, C, 54.32; H, 6.00; N, 8.93%.
A solution of sodium borohydride (2.33g) in absolute ethanol (30ml) is added dropwise to
a stirred solution of Compound B (9.57g) in absolute ethanol (120ml) at room tem~.d~ure~
The reaction mixture is stirred for 18 hours at room temperature, after which excess
sodium borohydride is destroyed by adding glacial acetic acid. The solvents are removed
under reduced pressure and the residue is triturated with ethyl acetate (3 x 50ml). The
combined ethyl acetate phases are evaporated to dryness and the residue is then
CA 02229036 1998-02-06
W O 97/09335 PCT/GB96/02113
- 34-
co-evaporated with toluene (3 x 50ml). The res-llt~nt residue is purified by flash
chromatography [silica gel, ethyl acetate: hexane (1:1)] to afford Compound C, m.p. 100~C,
OH
C-, 'C
N02
Analysis: calculated for C13H,8N2Os; C, 55.31; H, 6.43; N, 9.92%. Found, C, 55.25; H,
6.63; N, 9.76%.
Trifluo;oacetic acid (60ml) is added to a flask cont~ining Compound C (5g) cooled to 0~C.
The resulting solution is stirred for 20 minutes at 0~C, then allowed to warm to room
temperature and stirred for a further 3 hours. The solvent is ;emoved under reduced
~ ,S~.ulc and the residue is purified by ion e~rh~nge chromatography (Dowex 50WX2-200 (H+ form) resin, elutant 3% aqueous ammonium hydroxide solution) to affordCompound D as a pale brown foam.
OH
Nh2 ~nm7~ D
N02
Analysis: c~lcnl~te~l for C8HloN203; C, 52.74; H, 5.53; N, 15.38%.
Found, C, 53.12; H, 5.75; N, 15.07%.
13C nmr (100MHz; MeOH): ~ (ppm) 58.0(d), 68.2(t), 122.8(d), 123.1(d), 130.4(d), 134.5(d),
146.3(s), 149.6(s).
Compound G used in the Exa~mples is ~le~al~,d as follows:
Sodium cyanide (4.9g, 0.1M) and ammonium chloride (5.88g, 0.11M)are stirred in water
(20ml) at room temperature. A solution of 3,4-dichlorobenzaldehyde (17.5g, 0.1M) in
meth~nol (30ml) is added dropwise over one minute. Aqueous ammonia soluhon (lOml,
CA 02229036 1998-02-06
W O 9'7/0~335 PCT/GB96/02113
specific gravity 0.88) is added and the reaction is sti;red for 3 hours at room t~ alu~G.
Etnyl acetate is added and the organic phase s~dLGd, dried over m~gn~Sillm s;ulphate,
filteTed and evaplorated. The residue is dissolved in ethyl acetate and repeatedly çY~ceA
with 2N hydrochloric acid. The combined aqueous layers are adjusted to pH9 usingaqueous ammonia solution and re-extracted repeatedly with ethyl acetate. The combined
organic layers a~ dried over m~gn~Sillm s~lph~tP, filtered and evaporated under reduced
.SUlc: to afford an orange Oil which is purified by flash chromatography on silica using
h~y~.ne:ethyl acetate (1:1) as eluant to afford Compound G. m.p. 64-65~C
CN
~ (~nm~~ l C
J~ NH2
Cl
Cl
Found: C, 47.81; H, 2.99; N, 13.92%.
C8H6Cl2N2 requires C, 47.79; H, 3.01; N, 13.93%.
Compound J used in the Examples is ~l~pa,ed as follows:
Using subst,.nh~lly the same procedure as described for the preparation of compound G, a
mixture of 3-bromo-ben7~ ehyde (18.5g, O.lM), sodium cyanide (4.9g, O. lM),
~mmon;nm chloride (5.9g, 0.1 lM) and aqueous ammonia solution ( lOml, specific gravity
0.88) in methanoVwater (30ml/20ml) is reacted for 24 hours at room temperature to afford
Compound H as a redlbrown waxy solid.
CN
NH2 r~m~..~H
Br
3C nmr (100 MHz; CDc13):~ (ppm) 46.6 (d), 120.4 (S), 122.9 (S), 125.2 (d), 129.7 (d),
130.5 (d), 132.1(d), 138.3 (S).
A mixture of Compound H (10.5g, 49.8mM) in 6M hydrochloric acid (200ml) is heated
under r,flux for 68 hours. The supernatent is ~lec~nt~(l off, cooled to room temperature
CA 02229036 1998-02-06
W O 97/0933S PCT/GB96/02113
- 36-
and adjusted to pH7 using aqueous ammonia soluhon The ~ te~l product is
collected by filtration, washed with water and dried. Trituration with ethyl acetate
followed by drying affords Compound J as a brown solid. m.p. 201-204~C (dec).
CO2H
NH2 Cnm~.._~J
Br
~3C nmr (lOOMHz; CD30D): ~ (ppm) 56.9 (d), 124.0 (S), 128.1 (d), 132.2 (2 x d), 134.0
(d), 136.0 (S), 170.1 (S).
Example 1
CN
~~nmr~lm~l 1
H ~
OCH3
COCC~3
3-methoxycarbonylben7~1dlohyde (1.6g, lO.OmM) in methanol (lOml) is added to a solution
of ~methoxybenzylamine hydrochloride (1.7g, lO.OmM) and sodium cyanide (0.490g,
lO.OmM) in water (lOml) and the mixture is stirred for 3 hours at room temperature. Water
(20ml) is added and the mixture extracted with dichloromethane. The organic phase is
washed with brine, dried (MgS04) and evaporated to dryness to afford an oil which is
purified by silica gel chromatography using 20% ethyl acetate in hexane as eluant to
afford Compound 1.
Analysis: calculated for C18 Hl8 N2 ~3; C, 69.66; H, 5.85; N, 9.03%.
Found, C, 69.40; H, 5.94; N, 8.73%.
13C nmr (lOOMHz; CDC13):~(ppm) 50.6(t), 52.2(q), 52.8(d), 55.2(q), 114.0(d), 118.3(s),
128.3(d), 129.0(d), 129.6(d), 129.8(s), 130.1(d), 130.9(s), 131.6(d), 135.3(s), 159.1(s), 166.2(s).
-
CA 02229036 1998-02-06
W O 97/09335 PCT/G]B96/02113
Example 2
COOCH3
H ~ Cn~ 2
OCH3
COOCH3
A solution of Compound 1 (7g, 22.56mM) in methanol (75ml) is cooled to 0~C and
saturated with hydrogen chloride gas. Once saturated, the reaction ~ Lul~;iS stored at
-20~C for 4 days and then concentrated under reduced ~ S~ to a quarter of its original
volume. Ethyl acetate and saturated aqueous sodium bicarbonate solution are added and
the organic phase is separated, washed with water and brine, d~ied over mAgnes~ n
sulphate, filtered and evaporated to give an oil. Purific~*on by flash chromatography on
silica using hexane: ethyl acetate (1:1) as eluant affords Compound 2 as an oil.
31C nmr (100MHz; CD C13):~5 (ppm) 50.7 (t), 52.1(q), 52.3(q), 55.2(q), 63.8(d), 113.7(d),
128.69(d), 128.72(d), 129.2(d), 129.4(d), 130.5(s), 131.2(s), 132.0(d), 138.5(s), 158.7(s),
166.7(s~, 172.9(s).
Example 3
COOCH3
NH2-HOCOCH3
Cn~..~A3
COOCH3
A mixture of Compound 2 (8.0g, 23.3mM) and p~ m black (2.0g) in glacial acetic
acid (SOml) and meth~nol (SOml) is hydrogen~t~d for 4 hours at room ~ ture. After
cht-~'cing completion of the reaction by thin layer chromatography (tlc), the mixture is
filtered and the filtrate evaporated under reduced p-es ,ure. The residue is co-evapol~ted
three times with toluene (3 x 20ml) and then purified by flash chromatography on silica
using ethyl acetate as eluant to afford Compound 3 as a pale yellow oil.
Analysis: e~le~ tel1 for Cl3HI7NO6; C, 55.12; H, 6.05; N, 4.94%.
CA 02229036 1998-02-06
W O 97/09335 PCT/GB96/02113
Found; C, 55.46; H, 5.99; N, 5.05%
13C nmr (lOOMHz; cDcl3):~(ppm) 20.8 (q), 52.1(q), 52.5(q), 57.9(d), 127.9(d), 128.9(d),
129.3(d), 130.7(s), 131.4(d), 139.9(s), 166.6(s), 173.5(s), 175.9(s).
Example 4
OH
NH2 rnmro~A4
COOCH3
Step 1
A solnti~ n of di-tert-butyl-dicarbonate (4.5g, 20.83mM) in methanol (lOml) is added to a
vigorously stirred solution of Compound 3 (3.1g, lO.9mM) and triethylamine (lOrnl,
71.75mM) in methanol (40ml). The mixture is then heated at 60~C for 30 minute-~, cooled
to room ~ .dture and evaporated under reduced pressure. The residue is purified by
flash chromatography on silica using hexane: ethyl acetate (4:1) as eluant to afford
Compound E m.p. 88-90~C.
COOCH3
e3~NHBoc
Cnmro~E
COOCH3
Analysis: calculated for C16H21NO6; C, 59.43; H, 6.55; N, 4.33%.
Found, C, 59.54; H, 6.72; N, 4.32%.
Step 2
Sodium borohydride (800mg, 21.2mM) is added in eight equal portions, at 30 minute
intervals, to a stirred solution of Compound E (3.2g, 9.9mM) in methanol (50ml). On
completion of the reaction (tlc), the rem~ining sodium borohydride is destroyed with
glacial acetic acid and the reaction mixture evaporated under reduced pressure to afford an
CA 02229036 1998-02-06
W O 97/09335 PCT/GB96/02113 - 39 -
oily solid. This ~esidue is co-evaporated with toluene (2 x 20ml) and tritll~te~ with e~hyl
acetate. Evaporation of the ethyl acetate extracts under reduced ~)le;~:iUlc affords a
colourless oil which is purified by flash chromatography on silica using h~Y~ne ethyl
acetate (1:1) as eluant to afford Compound F, m.p. 102-104~C.
CH20H
~3--NHBoc
rnm~n~F
COOCH3
Analysis: c~ t~.l for C15H2lNO5; C, 61.01; H, 7.17; N, 4.74%.
Found; C, 61.11; H, 7.23; N, 4.70%.
Step 3
Trifluoroacetic acid (3.0ml, 39.17mM) is added to a stured solution of Co-nl,.au-ld F (2.2g,
7.45mM) in dry dichloromethane (25ml) under argon at room te---~c-~ture. The ~ is
stirred for 5 hours at room temperature. On completion of the reaction (tlc), the mixture is
evaporated under reduced ~l-,S~ul~ without heating and the residue is co-evaporated with
chloroform (2 x 20ml). After drying under high vacuum, the residue is purified by ion
exch~nge chromatography on Amberlyst A21 resin using water as eluant to afford
Compound 4 as a colourless oil.
Found: C, 58.98; H, 6.73; N, 6.53%.
Cl0 Hl3 NO3 . lH20 requires C, 58.8; H, 6.91; N, 6.86%.
13C nrnr (100 MHz; CD30D): ~ (ppm) 52.6(q), 58.1(d), 67.4(t), 129.1(d), 129.8(2 x d),
131.6(s), 132.9(d)V 142.4(s), 168.2(s).
CA 02229036 1998-02-06
W O 97/09335 PCT/GB96/02113
- 40 -
Example S
COOCH3
~ NH2
Cl
Cl Cnmr~ S
A solution of Compound G (1.4g, 5.98mM) in meth~nol (20ml) is cooled to 0~C and
s~hl~te~l with hydrogen chloride gas. Once saturated, the reaction mixture is stored at
-20~C for 2 days. The solvent is removed under reduced pressure and the residue
co-evaporated with methanol (3 x 20ml). The residue is suspended in ethyl acetate and
the organic phase is washed successively with saturated aqueous sodium bicarbonate
solution, water and brine. The organic layers are dried over m~nesillm sulphate, filtered
and evaporated under reduced pressure to afford an oil which is purified by flash
chromatography on silica using hexane: ethyl acetate (1:1) as eluant to afford Compound 5.
Found: C, 46.12; H, 3.85; N, 6.09%. CgHgCI2NO requires C, 46.18; H, 3.88; N, 5.98%.
13C nmr (100MHz; CD C13):~ (ppm) 52.5(q), 57.5(d), 126.2(d), 128.9(d), 130.8(d), 131.9(s),
132.6(s), 140.2(s), 173.4(s).
ExamDle 6
COOCH3
~NHBoc
Cl C~ , '6
Cl
Compound 5 (6.0g, 25.63mM), di-tert-butyl dicarbonate (11.19g, 51.26mM) and
CA 02229036 1998-02-06
W O 97/09335 PCT/GB96/02113
-41-
triethylamine (20ml, 143.50mM) are reacted in meth~nol (200m1) using s~lbst~nti~lly the
same procedure as ~les~he~l for the prep~tion of Compound E in Example 4. The crude
product is purified by chromatography on silica using 2()% ethyl acetate in hexane as
eluant to afford Compound 6 as a yellow solid. m.p. 90-92~C.
Found: C, SO.55; H, 5.16; N, 4.08%. Cl4Hl7C12NO4 requires C, 50.32; H, 5.13; N, 4.19%.
Exa~mple 7
_,,OH
~NHBoc
Cl / T
Cl Cnm7l~lm~7
To a solution of Compound 6 (6.78g, 20.28mM) in absolute ethanol (lOOml) is dropwise
added a solution of sodium borohydride (1.15g, 30.43mM) in absolute ethanol (30ml). The
reaction is stirred for 6 hours at room temperature and then left to stand for 48 hours at
room temperature. The solvent is removed under reduced ~,-es~ur~ and the residue is
purified by flash chromatography on silica using hexane: ethyl acetate (1:1) as eluant to
afford Compound 7 as a white solid. m.p. 113-114~C.
Found: C, 51.21; H, 5.69; N, 4.43%. Cl3HI7C12NO3 requires C, 51.00; H, 5.60; N, 4.57%.
Example 8
_" OH
J ~ NH2
Cl Cl Cn~ m~s
Compound 7 (4.7, 15.35mM) is reacted with trifluo~acetic acid (75ml) using subst~ntis~.lly
CA 02229036 1998-02-06
W O 97/09335 PCT/GB96/02113
-42-
the same procedure as described for the preparation of Compound D from Compound C.
The crude product is purified by ion ~yrh~n ~ chromatography on Dowex SOWX 2-200~H~ form) resin using methanol: water: aqueous ammonia solution (50%: 47%: 3%) as
eluant to afford Compound 8 as a cream coloured solid. m.p. 65-67~C.
Found: C, 46.61; H, 4.37; N, 6.59%. C8HgCl2NO requires C, 46.63; H, 4.40; N, 6.80%.
Example 9
Br
~1--0
OC2H5 (~. ~ 9
Bis(trimethylsilyl)~et:lmi-le (28.5 lml) is added d~.~pwise to a sohltion of 18.22g of ethyl
cyclohexylmethylphosphinate, p.~ d as described in EP 0569333, in lOOml of dryCH2Cl2 under argon. The solution is stirred at room tem~cl~Lu~c for 1 hour, then trimethyl
phosphate (13.42ml) is added, followed by 1,3-dibromopropene (mixture of cis/trans
isomers) (9.57ml). After stirring the solution at room temperature for 18 hours, it is poured
into saturated aqueous NaHCO3 solution (lOOml) and stirred for lQ mimltes The product
is extracted with CH2C12 (3 x 50 ml) and the combined organic extracts are washed with
brine, then dried with MgSO4 and filtered. The filtrate is evaporated under reduced
l,.cs~.u.e, then excess trimethyl phosphate is removed by evaporation at 80~C at 0.45mm
Hg. The residue is purified by flash chromatography (silica gel, ethyl acetate) to yield
Compound 9 as a mixture of cis and trans isomers.
31p nmr (162MHz, CDC13): ~ (ppm) 51.1 and 52.2.
Example 10
~OH
~HN !P ~
~C2H5 Cnmro~ln~ 10
A mixture of (R)-2-amino-2-phenylethanol (0.88g, 6.47mM) and Compound 9 (1.Og,
3.23mM) in toluene (lOml) is heated underreflux. 1, 8-Diazabicyclo[5.4.0]undec-7-ene
CA 02229036 1998-02-06
W O 97/0933S PCT/GB96/02113
- 43 -
(0.48ml, 3.23mM) is added in ten aliquots at 30 minute intervals. The reaction is heated
under reflux for a further hour and allowed to stand overnight at room ~ )e.dtul~. The
C; is filtered and the filtrate evaporated under reduced ~ ul., to afford a yellow oil
which is purified by flash chromatography on silica using 5% methanol in
dichlort)m~th~ne as eluant to afford Cu...~ound 10 as a 1: 1 mixture of diasL.,~,olllers at
phosphorus.
31P nmr (162Hz; CDCl3): ~ (ppm) 43.49 and 43.55.
Example 1 1
OH
O
0--'H ~P ~ ~1
~. . Il
(S)-2-arnino-2-phenyl ethanol (2.66g,19.4mM), Compound 9 (3.0g, 9.7mM) and
1,8-diazabicyclorS.4.0]undec-7-ene (1.40g, 9.7mM) are reacted in toluene (30ml) using the
procedure described for the preparation of Compound 10. The crude product is purified by
flash chromatography on silica using 5% methanol in dichloromethane as eluant to afford
Compound 11 as a 1: 1 mixture of diastereomers at phosphorus.
31p nmr (162MHz; CDC13): ~(ppm) 43.37 and 43.43.
Example 12
OH
/ O
~J~N ~P ~
OC2H5
COOCH3 ~ mr~ n~ 12
A mixture of Compound 4 (1.3g, 6.66mM) and Compound 9 (2.06g, 6.66mM) in
tohlene~rI~ (25ml, 1: 1 mixture) is heated to 80~C under argon. A solution of 1,8-diazabicyclo [5.4.0]undec-7-ene (1.52g, 9.95mM) in toluene/l~ (lSml, 1:1 mixture) is
CA 02229036 1998-02-06
W O 97/09335 PCT/GB96/02113
-44-
added over 5 hours. The mixture is cooled to room temperature and allowed to stand for
18 hours. The mixture is filtered and the filtrate evaporated under reduced ~S~ , to
afford a yellow oil which is purified by flash chromatography on silica using 5% meth~n
in dichlorometh~ne as eluant to give Compound 12 as a 1:1 mixture of dia~Lw~ol.lers at
phosphorus.
31p (162MHz; CDCl3): ~ (ppm) 43.53 and 43.58.
ExamPle 13
~ OH
OC2H5
Cl
Cl
cn nr~n~ 13
Using substantially the same procedure as described for the ~l~aldlion of Compound 12,
a mixture of Compound 8 (2.76g, 13.39mM) and Compound 9 (4.14g, 13.39mM) in
toluene/THF (SOmU4ml) is reacted with a solution of 1,8-diazabicyclo[5.4.0~undec-7-ene
(2.03g, 13.39mM) in THF (6ml) at 110~C to afford Compound 13 as a 1:1 mixture ofdiastereomers at phosphorus.
31P nmr (162MHz; CDC13): ~ (ppm) 43.73 and 43.82.
Example 14
OH
OC2H5
NO2 rnm~~ 14
Using substantially the same procedure as described for the preparation of Compound 12,
a mixture of Compound D ~3.19g, 17.50mM) and Compound 9 (5.41g, 17.~0mM) in
toluenel I~ (5QmU4ml) is reacted with a solution of 1 ,8-diazabicyclo[5.4.0]undec-7-ene
CA 02229036 l998-02-06
W O 97/09335 PCT/G]B96/02113
-4~-
(2.66g, 17.5mM) in THF (6ml) at 110~C to afford Compound 14 as a 1~ U;~-t of
diastereomers at phosphorus.
31P nmr (162MHz; CDCl3): ~ (ppm) 43.60 and 43.66.
Exarnple lS
nA 15
A suspension of sodium hydride (0.079g, 3.31mM) in dry toluene (lOml) is stirred at 0~C.
A solution of Compound 10 (l.lg, 3.01mM) in dry toluene (20ml) is added dropwise.
The reaction mixture is allowed to warm to room temperature and stirred for 20 hours.
Sa~u~dLed aqueous arnmonium chloride solution (Sml) is added, then the reaction llli~lu~G
is partitioned between ethyl acetate and water. The aqueous layer is extracted with ethyl
acetate and the combined organic phases are dried over ma~neSillm sulphate, filtered and
evaporated. The residue is purified by flash chromatography on silica using 5% merh~n
in dichloromethane as eluant to afford Compound lS as a mixture of dia.LGl~,o-llers at
phosphorus.
Mass spec. (FAB): (m +1)+ m/z = 366.
31p nmr (162MHz; CDC13): ~ (ppm) 54.0~ and 54.63.
ExamPle 16
o
N~
C~ n-i 16
Using subst~nti~lly the same procedure as described for the preparation of Compound lS,
Compound 11 (1 20g, 3.28mM) and sodium hydride (0.086g, 3.61mM) are reacted in
toluene (40ml) to afford Compound 16 as a mixture of diastereomers at phosphorus.
-
CA 02229036 1998-02-06
W O 97/09335 PCT/GB96/02113
- 46 -
Found: C, 63.39; H, 8.75; N, 3.70%. C2oH32NO3P . 0.75 H20 l~,~uil~,s C, 63.39; H, 8.91;
N, 3.70%.
31p (162MHz; CD C13):~ (ppm) 54.08 and 54.65.
Example 17
H ~
~nm~ln~17
COOCH3
A solution of Compound 12 (50mg, 0.12mM) in dry toluene (O.Sml) is stirred at room
Le.l-p~,ldture. A suspension of sodium hydride (6.2mg, 0.26mM) in toluene (0.5ml) is
added in one portion and the reaction mixture is stirred for 3 hours at room tempeldtulG.
The reaction is then quenched with glacial acetic acid and the product is extracted with
ethyl acetate. The combined organic phases are washed with water and brine, then dried
over m~gne~ m sulphate, filtered and evaporated. The residue is purified by flash
chromatography on silica using 5% meth~nol in dichloromethane as eluant to afford
trans-2,5-disubstituted morpholine racemic Compound 17 as a mixture of diastereomers at
phosphorus.
31p nmr (162MHz; CDC13): ~ (ppm) 54.06 and 54.64.
CA 02229036 1998-02-06
W O 97/09335 PCT~GB96/02113
EXa'inPle 18
O
,,~,J~ N ~o ~u
Cl
Cl Cnm~''~18
Using subst~nti~lly the same procedure as described for the ple~a.dLion of Compound 15,
Compound 13 (2.24g, 5.15mM) and sodium hydride (0.136g, 5.67mM) are ;eacted in
toluene ~80ml) to afford trans-2,5-disubstituted morpholine T~ mi~ Compound 18 as a
mixture of diastereomers at phosphorus.
Analysis: Found, C, 54.21; H, 7.08; N, 3.11%. C20H30Cl2 NO3P . 0.5H2O requin_s C, 54.18;
H, 7.05; N, 3.16%.
31p nmr (162MHz; CDCl3): ~ (ppm) 53.80 and 54.40.
Example 19
N ~
N02 ~ A 19
Using subst~nti~lly the same procedure as descTibed for the preparation of Compound 15,
Compound 14 (5 38g, 13.1mM) and sodium hydride ~0.346g, 14.41mM) are reacted in
toluene (150ml) to afford trans-2,5-disubstituted morpholine racemic Compound 19 as a
mixture of dia~le;l~,GIl~ers at phosphorus.
CA 02229036 l998-02-06
WO 97/09335 PCT/GB96/02113
- 48 -
31p nmr (162MHz, CDCl3): ~ (ppm) 53.73 and 54.33.
Example 20
N~t~
('nmro~71A 20
To a stirred solution of Compound 15 (650mg, 1.78mM) in dichlorometh~ne (25ml) under
argon is added dropwise bromotrimethylsilane (0.939ml, 7.12mM). The reaction mixture
is stirred for 24 hours at room temperature. The reaction is then quenched by the ~Mitir~n
of a mixture of meth~nol: water (95:5). T~le solvent is removed under reduced ~l~,'7~Ult to
afford an oily residue which is purified by ion exchange chromatography on DowexSOVVX 2-200 resin (H~ form) using methanol: water: aqueous ammonia solution (50%:
47%: 3%) as eluant. The resulting product is dried under high vacuum (< O.O5mm Hg) to
affo;d Compound 20 as a white solid. m.p. > 250~C. [a]D = +10.8~ (C=l, CH30H).
Found: C, 63.72; H, 8.44; N, 4.02%. Cl8H28NO3P requires C, 64.08; H, 8.36; N, 4.15%.
31p nmr (162MHz, D2O): ~ (ppm) 55.22.
Example 21
N~ t'H--[~
C~ . '21
Using the same procedure as described for the preparation of Compound 20, Compound
16 (670mg, 1.83mM) and bromotrimethylsilane (l.lg, 7.3mM) are reacted in
dichloromtoth~n~o (20ml) to afford Compound 21. m.p. > 250~C. [a]D = -10.5~ (C = 1,
CA 02229036 1998-02-06
W O 97/09335 PCT/GB96/02113
-49-
CH3~H)-
Found: C, 63.62; H, 8.50; N, 4.05%. C18H2gNO3P requires C, 64.08; H, 8.36; N, 4.15%.
31p nrnr ~162MHz; D2O/DCl): ~ (ppm) 55.36.
Example 22
Cl ~ ~ OH
Cl Cnmro~ 22
Using the same procedure as described for the preparation of Compound 20, Compound
18 (0.979g, 2.25mM) and bromotrime~hylsilane (0.89ml, 6.76mM) are reacted in
dichloromethane (40ml) to afford trans-2,5-disubstituted morpholine racemic Compound
22. m.p. > 200~C (dec).
Found: C, 52.45; H, 6.53; N, 3.29%. Cl8H26C12NO3P . 0.25H20 requires C, 52.63; H,
6.50; N, 3.41%.
31P nmr (202.5MHz; d4-acetic acid): ~ (ppm) 45.45.
Sodium salt, 31P nmr (162MHz; D2O/DCl): ~ (ppm) 41.89.
CA 02229036 1998-02-06
W O 97/09335 PCT/GB96/02113
- 50 -
Example 23
~/~ N ~ 0;~
N02 ~nm~lln~l 23
Using the same procedure as described for the preparation of Compound 20, Compound
19 (0.50g, 1.20mM) and bromotrimethylsilane (0.48ml, 3.65mM) are reacted in
dichloromethane (25ml) to afford trans-2,5-disubstituted morpholine the racemic
Compound23. m.p. 128-130~C.
Found: C, 54.23; H, 7.25; N, 6.93%. Cl8H27N2OsP . H2O requires C, 53.99; H, 7.30; N,
7.00%.
31p nmr (162MHz; CD30D): ~ (ppm) 37.64.
Example 24
~)~ N U~
~-nmrllntl 24
~H2
A mixture of Compound 19 (2.98g, 7.26mM) and 10% p~ m on activated charcoal
(0.5g) in absolute ethanol (150ml) is hydrogenated for 18 hours. The mixture is filtered
and the filtrate is evaporated. The residue is purified by flash chromatography on silica
using 10% methanol in dichloromethane as eluant to afford trans-2,5-disubstituted
morpholine racemic Compound 24 as a mixture of diastereomers at phosphorus. m.p.115-118~C.
CA 02229036 1998-02-06
W O 97/09335 PCT/GB96/02113
- 51 -
~ Found: C, 62.52; H, 8.88; N, 7.18%. C2oH33N203P . 0.25 H20 requires C, 62.40; H, 8.77;
N, 7.31%.
31p (162MHz; CDCl3): ~ (ppm) 54.16 and 54.72.
Exa~mple 25
~ N ~0~ ~
C-~m7l~o-lnf1 25
I
Crushed ice (l5g) is added to a stirred solntion of Compound 24 (2.43g, 6.38mM) in
con~en~rated hydrochloric acid (50ml) and the resulting mixture is cooled to OIDC. A
solution of sodium nitrite (0.48g, 7.02mM) in water (25ml) is added dropwise and the
r~s--lting mixture is stirred for 10 minu~t~.s at 0~C. The resulting solution is then added
dropwise to a solution of potassium iodide (11.13g, 67.01mM) in water (200ml). The
reaction mixture is stirred for a further 2~ hours at room Le~ ,.dture and then allowed to
stand overnight at room temperature. Ethyl acetate is added and the two phases sep~ n-.cl
The aqueous phase is neutralized by addition of solid sodium bicarbona~e and extracted
witn ethyl acetate. These ethyl acetate extracts are combined with the original organic
phase and the combined organic phases are washed with aqueous 10% sodium hydroxide
solution followed by aqueous 5% sodium bisulphite solution then water. The organic
phase is dried over m~gnesillm sulphate, filtered and evaporated under reduced ~res~.ulc~
The residue is purified by flash chromatography on silica using 10% meth~nQl in ethyl
acetate as eluar t to afford trans-2,5-disubstituted morpholine racemic Compound 25 as a
mixture of diasLt~Go."ers at phosphorus. Mass spec. (CI/NH3): (m+l)+ m/z = 492.
31p nmr (162MEIz; CDC13): ~ (ppm) 54.21 and 54.77.
CA 02229036 1998-02-06
W O 97/09335 PCT/GB96/02113
Example 26
N gH
~m~ol-71~26
Using snbst~nti~lly the same procedure as described for the ~l~,p~Lion of Compound 20,
Co~ .ound 25 (0.lg, 0.20mM) and bromo~imethylsilane (0.427ml, 3.20mM) are reacted
in dichloromethane (lOml) for 78 hours at room temperature. The crude product is purified
by ion exch~n~e chromatography on Dowex 50WX 2-200 resin (H+ forrn) using meth~n-)l
water: aqueous ammonia solution (50%: 47%: 3%) as eluant and the product is dried
under high vacuum (~ 0.05 mm Hg) to afford trans-2,5-~ ubstitllte~ morpholine racemic
Compound 26. m.p. > 230~C (dec).
Found: C, 44.48; H, 5.70; N, 2.95%. Cl8H27INO3P . 1.2H20 requires C, 44.59; H, 6.11; N,
2.89%.
31p nmr (162MHz; CD30D/DCI): ~ (ppm) 53.08.
Example 27
N~oc~
mr~l~n~l 27
CN
Sodium cyanide impregnated alumina (SmM NaCN per gram of ~ min~) is plG~a.~d by
the p~cedure of S. L. Regen, S. Quici and S. J. Liaw described in the Journal Organic
Chemistry,1979, 44(12), 2029. To a mixture of tris (dibenzylideneacetone) ~ip~ rlillm
CA 02229036 1998-02-06
W O 97/0933~ PCT/G B96/02113
(O) [0.17g, 0.18lmM], sodium cyanide impregnated ~ min~ (4.7g) and
tri(2-furyl)phosphine (0.34g, 1.45mM) under argon is added a solution of Compound 25
(0.89g, 1.8mM) in dry degassed toluene (SOml). The reaction is heated at 80~C for 12
hours. The reaction is monitored by tlc and if required further tris(dibenzyli~lenes~cetc?nf~)
rlir~ m (O) ~0.17g, 0.18mM] and tri(2-furyl)phosphine (0.34g, 1.45mM) are added and
the reaction heated for 8 hours at 80~C. On completion of the reaction, the ~ LulciS
filtered and the solids washed with ether. The combined filtrate is evaporated under
reduced l11GS~I11C: and the residue is purified by flash chromatography on silica using 10%
methanol in ethyl acetate as eluant to afford trans-2,5-disubstituted morpho~ine racemic
Compound 27 as a mixture of diastereomers at phosphorus.
Mass spec. (CVNH3): (M+l)+ m/z = 391.
31p nmr (162MHz; CDCl3): ~ (ppm) 53.95 and 54.57.
Example 28
N ~ OH
CN ~n~r~n~28
Using substantially the same procedure as described for the preparation of Compound 20,
Compolmd 27 (0.16g, 0.41mM) and bromotrimethylsilane (0.81ml, 6.14mM) are reacted in
dichlorometh~ne to afford trans-2,5-disubstituted morpholine racemic Compound 28.
Mass spec. (CI, NH3): M+ m/z = 362.
31p nmr (162~;z; D20tDCl): ~ (ppm) 55.31.
CA 02229036 1998-02-06
W O 97/09335 PCT/GB96/02113
-54-
Example 29
N ~ OH
C~m~ 29
COOH
A mixture of Compound 27 (0.09g, 0.23mM), 6M hydrochloric acid solution (lOml) and
ethanol (lrnl) is heated under reflux for 72 hours. On completion of the reaction (31p nmr),
the solvent is removed under reduced ~l~,S~ulc. The residue is purified by ion exchange
chromatography on Dowex 50WX 2-200 resin (H+ form) using methanol: water: aqueous
~mmoni~ solution (50%: 47%: 3%) as eluant and the product is dried under high vacuum
(<0.05mm Hg) to afford trans-2,5-disubstituted morpholine racemic Compound 29. m.p.
> 260~C.
31p nmr (162MHz; D2O): ~ (ppm) 40.67.
Example 30
A mixture of Compound 17 (0.50g, 1.18mM), 6M hydrochloric acid solution (25ml) and
glacial acetic acid (5ml) is heated at 100~C for 16 hours. On completion of the reaction
(31p nmr), the solvent is removed under reduced pressure. The residue is purified by ion
exch~nge chromatography on Dowex 50WX 2-200 resin (H+ form) using methanol: water:
aqueous ammonia solution (50%: 47%: 3%) as eluant and the product is dried under high
vacuum (S0.05mm Hg) to afford trans-2,5-disubstituted morpholine racemic Compound
29.
Found: C, 58.68; H, 7.35; N, 3.71%. ClgH28N OsP . 0.5H2O requires C, 58.45; H, 7.49; N,
3.59%.
31p nmr (162MHz; D20): ~ (ppm) 40.71.
CA 02229036 1998-02-06
W O 97/09335 PCT/G~96/02113
-55-
Exam~le 31
.
,~/~ N OH--O
H
('nm~l~nri 30
COOCH3
A solution of Compound 29 (317mg, 0.83mM) in methzanol (20ml) is saturated with
gaseous hydrogen chloride and the reaction ~ Lul~ stirred for 16 hours at room
temp~ ldLu~G. The solvent is removed under reduced plGS~ulc and the residue is purified by
ion exchange chromatography on Dowex 50WX 2-200 resin (H+ form) using methanol:
water: aqueous ammonia solution (50%: 47%: 3%) as eluant. The product is d~ied under
high vacuum (S0.05 mm Hg) to afford trans-2,5-disubstituted morpholine racemic
Compound 30.
Found: C, 60.26; H. 7.57; N, 3.45%. C20H30N OsP . 0.25 H2O requires C, 60.01; H, 7.68;
N, 3.~0%.
31P nmr (162MH:z; CD30D): ~ (ppm) 37.21.
Example 32
OH
O
o;;
cnn.~n.l 31
Using substzln*z~lly the same procedure as described for the preparation of Compound 20,
Compound 10 (90mg, 0.246mM) and bromotrimethylsilane (200~1,1.57mM) are reacted in
CA 02229036 1998-02-06
W O 97/09335 PCT/GB96/02113
-56-
dichloromçth~ne (2ml) to afford Compound 31.
[a]D -38.5~ (C = 0.6, CH30H)
31p nmr (162MHz; CD30D): ~ (ppm) 29.00.
Example 33
OH
r O
IH~
OIm~ 32
Using substantially the same procedure as described for the preparation of Cu~ oul~d 20,
Compound 11 (9Omg, 0.246mM) and bromotrimethylsilane (200,u1,1.57mM) are reacted in
dichloromethane to afford Compound 32.
[a]D +41.3~ (C = 0.6, CH30H)
31P nmr (162MHz; CD30D): ~ (ppm) 28.84.
Example 34
~OH
~N ~P--O
COOH (~nm~l ~n~ 33
To a stirred solution of Compound 12 (lOOmg, 0.24mM) in dichloromethane (2ml) under
argon is added bromotrimethylsilane (300~L1, 2.4mM) and the reaction is stirred for 24
CA 02229036 1998-02-06
W O 97/09335 PCT/G B96/02113
hours at room te.npeldture. The solvent is removed under reduced ~l-,SSulc and the
residue is co-evaporated with a 1:1 mixture of meth~nnl: water. The resulting residue is
then dissolved in a mixture of 6M hydrochloric acid (5ml) and methanol (0.3ml) and the
mixture is heated under reflux for 4 hours. The solvent is removed under reduced pl~,S ,
and the residue is co-e~dpoldted three times with water. The resulting residue ,;s purified
by ion exch~nge chromatography on Dowex SOWX 2-200 resin using methanol: water:
aqueous ammonia sohltion (50%: 47%: 3%) as eluant to afford Compound 33.
31P nmr (162MHz; CD30D): ~ (ppm) 29.28.
Example 35
N ~~P~O
Cl Cl
('n~-~nri 34
Using subst~nti~lly the same procedure as described for the ~repdlduon of Compound 20,
Compolmd 13 (O.Sg, 1.15mM) and bromotrimethylsilane (0.91ml, 6.9mM) are reacted in
dichloromethane (lSml) to afford Compound 34.
31p nmr (162MHz; CD30D): ~ (ppm) 29.06.
CA 02229036 1998-02-06
W O 97/09335 PCT/GB96/02113
Exam~,~le 36
OH
0
N02 Cnm~"n" 35
Using subst~nri~lly the same procedure as described for the preparation of Compound 20,
Compound 14 (9Omg, 0.219mM) and bromotrimethylsilane (200~11, 1.57mM) are reacted in
dichloromethane (2ml) to afford Compound 35.
31p nmr (162MHz; CD30D): ~ (ppm) 29.57.
ExamPle 37
~ OH
I NH2
('nm~-m-l 36
Boron trifluoTi~le ethyl etherate ( 1.25ml, lOmM) is added dropwise to a su~pen~ion of
2-amino-3-(4-iodophenyl)propionic acid (2.9g, lOmM) in THF (lOml) over 20 minut~os
The mixture is heated under reflux for 2 hours, then borane dimethyl sulphide complex
(l.lml, 1 lmMol) is added dropwise over 1 hour whilst m~int:~ining the mixture at reflux.
The mixture is heated under reflux for a further S hours and then stirred for lS hours at
room temperature.
A 1:1 mixture of water and THF (20ml) is added followed by SN sodium hydroxide
solution (7.5ml). The reaction mixture is heated under reflux for 7 hours, cooled to room
temperature and filtered. The filter cake is washed with THF (2 x lOml) and the filtrate is
evaporated to 25% of its original volume and then extracted with dichloromethane. The
combin~o-1 organic layers are dried over magnesium sulphate, filtered and evaporated under
reduced ~lGS:il.llG to afford a pale yellow solid which is recrystallised from hexane: ethyl
acetate (1:2) to afford Compound 36, m.p. 105-107~C.
Found: C, 38.69; H, 4.40; N, 4.92~Yo. C9H,2INO requires C, 39.01; H, 4.37; N, 5.06%.
CA 02229036 1998-02-06
W O 97/09335 PCT/GB96/02113
59 _
Example 38
~ oc~
I OH
~ ' . '37
Using substantially the same procedure as described for the preparation of Comlpound 12,
a mixt~-e of Compound 36 (2.40g, 8.66mM) and Compound 9 (2.7g, 8.66mM) in toluene
(20ml) is reactecl with a sohltion of 1,8-diazabicyclo~5.4.0]undec-7-ene (1.30g, 8.66mM)
in TH~ (lOml) at 75~C to afford Compound 37 as a 1:1 mixtuIe of diastereomers atphosphorus.
31p nmr (202MHz; CDCL3) ~(ppm) 43.69 and 43.72.
Example 39
~~~ P~
38 l~ans]
C~.,Y.~n.i 39 [c~s]
A suspension of sodium hydride (O.105g, 4.4mM) in dry toluene (5ml) is added
portionwise over 30 seconds to a stirred solution of Compound 37 (2.0g, 4.0m~) in dry
toluene (25ml). The mixture is stirred for 30 minUt~s at 0~C and then for 3 hours at room
lenlp~.~ture. Glacial acetic acid (lml) is added, then the reaction mixture is diluted with
ethyl acetate (75ml). The organic phase is washed with saturated sodium bicarbonate
solution, water and tnen brine. The combined organic phases are dried over m~ne~ m
sulphate, filtered and evaporated. The residue is purified by flash chromatography on
silica using 20% methanol in ethyl acetate as eluant to afford trans-2, S-disubstituted
morpholine racemic Compound 38 and cis-2, 5- disubstituted morpholine racemic
CA 02229036 l998-02-06
W O 97/09335 PCT/GB96/02113
- 60 -
Compound 39 each as a mixture of diasL~.~o",ers at phosphorus.
Compound 38: 31p nmr (162MHz; CDCl3) ~ (ppm) 53.93 and 54.60
Compound 39: 31p nmr (162MHz; CDCl3) ~ (ppm) 54.25 and 54.82 ppm.
Example 40
\\~ ~N O N~
('nmr~l~nfJ 40
To a stirred solution of Compound 38 (180 mg, 0.356 mM) in dichloromethane (5ml)under argon is added dropwise brc.motrimethylsilane (0.20ml, 1.52mM). The reaction
mixture is stirred for 20 hours at room temperature. The solvent is removed under reduced
pl~ C and the residue coevaporated with 1: 1 water:methanol (2 x 0.5ml). The residue is
purified by ion exchange chromatography on Dowex SOWX 2-200 resin (H+ form) using
meth~noh 2% sodium hydroxide solution (1:1) to elute the product. The resulting product
is further purified by gel filtration on a Bio-Gel P2 column using water as eluant to afford
trans-2, 5-disubstituted morpholine racemic Compound 40, m.p. >250~C (dec).
Found: C, 41.42; H, 5.40; N, 2.42%. ClgH28I NO3 P.Na. 3H2O requires C, 41.20; H, 6.10;
N, 2.53%.
31p nmr (161MHz; D2O): ~ ~ppm) 41.~1.
CA 02229036 l998-02-06
W O 97/09335 PCT/GB96/02113
-61-
Example 41
o
CH3CH2~
N OcH2cH
C.~ 4 1
A mixture of Compound 38 (0.180g, 0.36mM) and bis triphenylphosphine p~ m (II)
chloride (0.200g, 0.28mM) in absolute ethanol (2ml) and triethylamine (lml) is rleg~
by sparging with argon for 5min. The mixture is heated under reflux and vigorously
stirred under an atmosphere of carbon monoxide for 3 hours. The mixture is cooled to
room temperature and the solvent is removed under reduced pressure to afford an oily
solid which is triturated with ethyl acetate. The combined ethyl acetate washings are
evaporated under reduced ~ u~c and the residue is purified by flash chromatography on
silica using 10% methanol in chloroform as eluant to afford trans-2, S-disubstituted
morpholine racemic Compound 41 as a mixture of diastereomers at phosphorus.
31p nmr (161 M Hz; CDC13): ~ (ppm) 53.93 and 54.60.
Example 42
N-+ O2C N ~ ¦
c. . 42
A mixture of Compound 41 (0.095g, 0.21mM) and 6M hydrochloric acid solution (4ml) is
heated under reflux ~or 20 hours. On completion of the reaction (31p nmr) the solvent is
removed under reduced ~ s~ure and the residue is purified by ion exchange
CA 02229036 l998-02-06
W O 97/09335 PCT/G B96/02113
- 62 -
chromatography on Dowex 50WX 2-200 resin (H+ form) using meth~nol- 2% sodium
hydroxide solution (1: 1) to elute the product. The resulting product is further purified by
gel filtration on a BIO-GEL P2 column using water as eluant to afford trans-2,
5-disubstituted morpholine racemic Compound 42, m.p. >250~C (dec).
Found: C, 50.53; H, 7.40; N, 2.96%. C20H2gNOsP. Na. 2H20 ~ u-l~s C, 50.52; H, 6.79;
N, 2.95%.
31p nmr (202MHz, D20): ~ (ppm) 42.38.
Example 43
~ 1~
C02~Na ~n~ro~ln~43
A buffered stock solution of formaldehyde is ~lGpal~,d by dissolving sodium acetate (1.8g,
21.4mM), acetic acid (1.3ml, 22.7mM) and40% aqueous formaldehyde sohlticn (7.0ml;
lOlmM) in water (Sml). An aliquot (lOml) of the above stock solution is added to a
mixture of Compound 29 (O.lOg, 0.262mM) in methanol (2ml) and the mixture is stirred
for 10 mimlt~s at room temperature. Sodium cyanoborohydride (0.165g, 2.62mM) is
added portionwise over 2 minutes. The ~ cLule iS stirred for 24 hours at room
lc~ ,.dture. The solvent is removed under reduced p~es~u~c; and the residue purified by
ion e~ch~nge chromatography on Dowex 50WX 2-200 resin (H+ form) using meth~nol
2% sodium hydroxide solution (1:1) to elute the product. The resulting product is further
purified by gel filtration on a BIO-GEL P2 column using water as eluant to afford the
trans-2, 5- disubstituted morpholine racemic Compound 43, m.p. >250~C (dec).
Found: C, 52.60; H, 6.82; N, 3.10%. C20H28NOsP. 2Na. H20 requires C, 52.52; H, 6.61;
N, 3.06%.
31p nmr (202 MHz; D20) ~ (ppm) 41.93.
CA 02229036 1998-02-06
W O 97/09335 PCT/GB96/02113
-63-
Example 44
N ~ j/~
o~a+
C02~a+ ~3
I'nmr~
Compound 29 (0.25g, 0.66mM) is dissolved in a 1: 1 mixture of dioxan: water ~4mlj and
the pH of the resulting solution is adjusted to pH 9 by the addition of 0. lM sodium
hydroxide solution. The mixture is vigorously stirred and benzyl chloroformate (0.188ml,
1.32mM) is added dropwise over lS minnttos The pH of the Illi~lUlC iS adjusted to pH9 by
further addition of 0. lM sodium hydroxide solution and the mixture is stirred for 20 hours
at room temperature. The Illi~Lulcisconcentrated under reduced p,es~.u,~ to one half of its
~rigin~l volume and ?~ci-lified with concentrated hydrochloric acid. The mixture is
extracted with ethyl acetate and the combined organic phases are washed with water then
brine, dried over m~gnç~illm sulphate, fi1tered and evaporated. The residue is purified by
flash chromatography on silica using acetic acid: meth~nol: chlolv~l-n (2% :10%: 88%)
as eluant. The product is further purified by ion exchange chromatography on Dowex 50
WX 2-2!00 resin (H~- form) using THF: water (3:1) as eluant. The reslllting product is
dissolved in 1% sodium hydroxide solution (2.5ml) and purified by gel filtration on a
BIO-GEL P2 column using water as eluant to afford trans-2, 5-disubstituted morpholine
~ racemic Compound 44, m.p. > 250~C (dec).
Found: C, 52.27; H, 6.00; N, 2.24%. C27 H32 N O7 P. 2 Na. 3H2O requires C, 52.85; H,
6.24; N, 2.28%.
31p nmr (161 MHz; D2O) ~ (ppm) 41.75.
CA 02229036 1998-02-06
W O 97/09335 PCT/GB96/02113
- 64 -
Example 45
OH
NH2
~ nm7r-171~45
Boron t;iflu~-ri~le ethyl etherate (75.0ml, 0.61M) is added dropwise to a suspension of
compound J (70.2g, 0.3 lM) in THF (350ml) over 20 minutes. The mixture is heatedunder reflux for 2 hours, then borane dimethyl sulphide complex (57.9ml, 0.61M) is added
dropwise over 1.5 hours whilst m~int~ining the mixture at reflux. The mixture is heated
under reflux for a further 3 hours and then stood for 18 hours at room tem~.,,dLulc.
A 1: 1 mixture of water and THF (350ml) is added followed by SM sodium hydroxidesolution (350ml). The reaction mixture is heated under rcflux for 5 hours then cooled to
room L~ dture. The two layers are separated and the aqueous layer extracted withethyl acetate. The combined organic phases are washed with brine, d;ied over m~nesinm
sulphate, filtered and evaporated under reduced plCSSUlC to afford a brown oil.
This residue is triturated with diethyl ether/hexane then recrystallised fro;n ethyl acetate to
afford compound 45, m.p. 74-76~C.
Found C, 44.40; H, 4.67; N, 6.35%.
C8HlOBr NO requires C, 44.46; H, 4.67; N, 6.48%.
CA 02229036 1998-02-06
W O 97/09335 PCT/GB96/02113
-65-
Example 46
.
OH
C02CH3
On..~ ri 4
A mixture of compound 45 (15.0g, 69.4mM) and bis(triphenylphosphine) p~ rn (II)
chloride (4.0g, 5.70mM) in methanol (lOOml) and triethylamine (25ml) is degassed by
sparging with argon for 5 minuteS The mixture is saturated with carbon monoxide and
then pressurised ~o 30 psi in a IJlCS~UlC vessel. The mixture is slowly heated to 100~C
whilst m~int~ining the ~l~,..'.Ult; below 50psi for 5 hours. The mixture is cooled to room
.ature, filtered and evaporated. The residue is triturated with ethyl acetate and the
filtrate evaporated. The residue is purified by flash chromatography on silica using a
gradient from 10% to 20% methanol in chloroform as eluant to afford compound 4.
13C mnr (lOOMHz; CD30D): ~(ppm) 52.6 (q), 58.3 (d), 68.1 (t), 129.0 (d), 129.5 (d), 129.7
(d), 131.5 (s), 132.9 (d), 143.6 (s), 168.4 (s).
Example 47
OH O
¦ H ¦
H OCH2CH3
f'~m~-~n-1 46
(2R/S)-2-amino-2- (lH-indol-3-yl)ethanol is prepared by the procedure of A.H. Katz et.al
described in the Journal ~lic inz~l Chemistry, 1988, 31, 1244.
Using subst~nti~lly the same procedure as described for the preparation of Compound 12,
a uli~Lulc of (2R,/S)-2-amino-2-(lH-indol-3-yl) ethanol (0.39g, 2.21mM) and Compound 9
CA 02229036 1998-02-06
W O 97/09335 PCT/GB96/02113
- 66 -
(0.68g, 2.20mM) in toluene/l~ (lOmU15ml) is reacted with a sollltion of1,8-diazabicyclo ~5.4.0~undec-7-ene (0.33g, 2.17mM) in toluene (Sml) at 75~C to afford
Compound 46 as a 1 ~ cLul~, of diasL~.~ul--ers at phosphorus.
31p nmr (162 MHz; CDCl3): â(ppm) 43.25 and 43.43.
Example 48
0\ N ~ OCII~C lll
47
A sncp~oncion of sodium hydride (0.014g, 0.59mM) in dry toluene (lOml) is stirred at 0~C.
A solution of Compound 46 (0.200g, 0.49mM) in dry toluene (7ml) is added dropwise.
The reaction mixture is allowed to warm slowly to room temperature and stirred for 4
hours. Glacial acetic acid is added to quench the reaction, then the mixture is filtered and
evaporated. The residue is purified by flash chromatography on silica using 20%
methanol in ethyl acetate as eluant to afford trans-2,5-disubstituted morpholine racemic
Compound 47 as a mixture of diastereomers at phosphorus.
Mass spec. (CI/NH3): (M+1)+ m/z = 405.
31 p nmr (162 MHz; CDC13): ~ (ppm) 54.44 and 55.32.
CA 02229036 1998-02-06
W O 97/09335 PCT/GB96/02113
- 67 -
Example 49
H o N.~
Cn~.ln.1 4g
Using subst~nh~lly the same procedure as ~1~scrihe~l for the preparation of Compound 40,
Compound47 (O.llSg,0.28mM) andbromotrimethylsilane (0.15ml, 1.13mM) arereacted
in dichloromethane (5ml) for 3 days at room ~ eldture to afford trans-2,5-disub~ ut~d
morpholine race:mic Compound 48.
13C nrrLr (lOOMHz; D20) ~ (ppm) 28.6 (t), 28.7 (t), 35.1 (d), 37.6 (t), 37.7 (t), 37.9 (t), 38.3
(t), 41.6 (t), 53.8 (d), 54.2 (t), 74.7 (t), 75.6 (d), 114.8 (d), 115.6 (s), 121.3 (d), 122.3 (d),
125.0 (d), 125.4 (d), 128.3 (s), 138.8 (s).
31p nmr (202 MHz; D20) ~ (ppm) 42.6.
Example 50
CH3 OH
CH3 ~
0/ NH2
Cnm~n~49
Using subs~n*~lly the same procedure as described in Example 37,
DL-2-amino-3-methyl-2-penylbutyric acid (10.Og, 51.8mM), boron trifluoride ethyletherate (6.4ml, S 1.8mM) and borane dimethyl sulphide complex (4.9ml, S l.~mM) are
reacted in dry THF (SOml) to afford Compound 49.
CA 02229036 1998-02-06
W O 97/0933~ PCT/G B96/02113
- 68 -
Mass spec. (CI, NH3): (m+l)+ m/z = 180.
13C nmr (lOOMHz; CDCl3) 16.8 (q), 17.4 (q) 34.8 (d), 61.7 (s), 69.2 (t), 126.2 (d), 126.4
(d), 127.8 (d), 144.2 (s).
Example S 1
CH ~ N
H OCH2CH3
Cnn~ nA 50
Using substantially the same procedure as described in Example 12, a mixture of
Compound 49 (1.07g, 6.0mM) and Compound 9 (1.86g, 6.0mM) in toluene (20ml) is
reacted with a solution of 1, 8 - diazabicyclo [5.4.0]undec-7-ene (l.lg, 7.2mM) in toluene
(Sml) at 75~C to afford Compound 50 as a 1: 1 mixture of diastereomers at phosphorus.
r~ass spec. (CI, NH3): (m + 1)+ m/z = 408.
31p (202.~ MHz; CDC13) ~ (ppm) 44.22 and 44.25.
Example 52
OCU2CH3
(2R~, 5R*) Cnmrl~nA 51
(2R~. SS~ Imr~llnr1 52
Using subst~nti~lly the same procedure as described in Example 39, Compound 50
(200mg, 0.49mM) and sodium hydride (12mg, 0.49mM) and sodium hydride (12mg,
0.49mM) are reated in dry toluene (3ml). The crude product is purified by flash
chromatography on silica using 10% methanol in ethyl acetate as eluant to afford (2R*,
5R*) morpholine racemic Compound 51 and (2R*, 5S*) morpholine racemic Compound
CA 02229036 1998-02-06
W O 97/09335 PCT/GB96/02113
-69-
52 each as a mixlure of diastereomers at phosphorus.
Compound 51: 3 I P nmr (162MHz; CDCl3) ~ (ppm) 54.3 and 55Ø
Compound 52: 31p nmr (162MHz; CDCl3) ~ (ppm) 54.2 and 55.5.
Example 53
CH~ ~N ~o ~O
~~ nrr~ n~1 53
Using subst~nri~lly the same procedure as described in Example 40, Compound 51 (40mg,
O.lmM) and bromotrimethylsilane (0.066ml, 0.5mM) are reacted in d~ry dichloromethane
(1 ml) to afford (2R*, 3R*) morpholine racemic Compound 53.
31p nmr (202.5MHz; D20) ~ (ppm) 42.5.
Example 54
CH3 ~ N
1 O~Na+
CH3
Cnm~ n~S4
Using subst~nti~lly the same procedure as described in Exarnple 40, Compound 52
(180mg, 0.2mM)I and bromotrimethylsilane (0.132ml, l.OmM) are reacted in dry
dichloromethane (2 ml) to affo;d (2R*, 5S*) morpholine racemic Compound 54.
31P nmr (202.5M:Hz; D20) ~ (ppm) 42.2.
CA 02229036 1998-02-06
W O 97/09335 PCT/GB96/02113
-70-
Example 55
o CH3 OcH2cH3
~ ~ OCH2CH3
CH30 OCH2CH3
t'nmrolm-i 55
A 0.5M solution of potassium bis(trimethylsilyl)amide in toluene (50ml, 25mM) is added
dropwise to a cooled (-70~C) solution of ethyl l,l-diethoxyethylphosphinate (5.25g,
25mM) in dry THF (30ml). The mixture is stirred for 0 5 hours at -70~C. The rçslliting
solution is added dropwise over ten min~t~s to a cooled solution of 4-methoxybenzyl
chloride (3.9g, 25mM) in THF (30ml). The resulting mixture is stirred for 1 hour at -70~C
and then allowed to warm to room lc-..peldture. Reaction is stirred for 18 hours at room
temperature. Glacial acetic acid is added and the reaction is evaporated. The residue is
partitioned between ethyl acetate and aqueous sodium bicarbonate solution. The organic
phase is separated and washed with water then brine, dried over magnt~sillm sulphate,
filtered and evaporated. The residue is purified by flash chromatography on silica using
ethyl acetate: hexane (2:1) as eluant to afford Compound 55.
31P nmr (162MHz; CDC13) ~ (ppm) 44.6.
Example 56
~- H
~1
.
CH30 OCH2CH3
Comrol~nA56
Chlorotrimethylsilane (3.8ml, 30.3mM) is added to a solution of Compound 55 (l.Og,
3.03mM) in a 9:1 mixture of chl~oful.,l: ethanol (lOml) and the mixture is stirred for 18
hours at room le,--~G~ture. The mixture is evaporated under reduced pressure and the
residue is co-evaporated with chloroform. After drying under high vacuum, the residue is
CA 02229036 l998-02-06
W O 97/09335 PCT/GB96/02113
purified by flash chromatography on silica using ethyl acetate as eluant to afford
Compound 56 as a colourless oil.
Mass spec. (CI, N H3): (m + N H4)+ m/z = 232.
31p mnr (162MH[z; CDCl3): ~ (ppm) 37.3.
Example 57
~ i'~~--6~'Br
CH30 OCH2CH3
Cnm~nAS7
Using substantially the same procedure as described in Example 9, Compound 56 (O.Sg,
2.33mM), bis(trimethylsilyl)acetamide tO.69ml, 2.80mM), trimethyl phosphate (0.33ml,
2.80mM) and 1,3-dibromo~lvpene (mixture of cis/trans isomers) (0.23ml, 2.33mM) are
reacted in dry dichloromethane (lOml) to afford Compound 57 as a mixture of cis and
trans isomers.
31p nmr (162MHz; ~L)C13) ~ (ppm) 47.2 and 48.1.
Example 58
OH O
N ~ ~y6~ ~
OCH2CH3 OCH3
02CH3
rn~..nA58
Using subst~nti~llly the same procedure as described in Example 12, a mixture ofCompound 4 (O.193g, 0.99mM) and Compound 57 (0.330g, 0.99mM) in toluene/l~IF
(lOml, 4:1 mixhlre) is reacted with a solution of 1,8-diazabicyclo [5.4.0]undec-7-ene
(0.181g7 1.19mM) in toluene (2ml) at 80~C to afford Compound 58 as a 1:1 mixture of
diastereomers at phosphorus.
Mass spec. (CI, NH3): (m+1)+ m/z = 448.
CA 02229036 1998-02-06
W O 97/09335 PCT/G B96/02113
31p nmr (162MHz; CDCl3) ~ (ppm) 39.40 and 39.49.
Example S9
OCnt~l~ 3
C02CH3
~nm~lm-l 59
Using substantially the same procedure as described in Example 39, Compound 58
(200mg, 0.45mM) and sodium hydride (10.8mg, 0.45mM) are reacted in toluene (2ml) to
afford trans-2,5-disubstituted morpholine racemic Compound 59 as a mixture of
diastereomers at phosphorus.
31p nmr (162MHz; CDC13) ~ (ppm) S0.0 and 50.8.
ExamPle 60
~ OCH3
CO~2Na+
c~ 60
Bromotrimethylsilane (0.074ml, O.SSmM) is added to a solution of Compound 59 (50mg,
0.1 lmM) in dichloromethane (1 ml) and the reaction is stirred for 24 hours at room
Jcldture. The solvent is removed under reduced pressure and the residue is
co-evaporated with a 1: 1 mixture of methanol: water. The resulting residue is dissolved in
6M hydrochloric acid (2ml) and the mixture is heated under reflux for 4 hours. The
solvent is removed under reduced pressure and the residue is co-evaporated three times
with water. The resulting residue is purified by ion exchange chromatography on Dowex
CA 02229036 1998-02-06
W O 97/09335 PCT/GB96/02113
50 WX 2-200 resin (H+ form) using meth~n~ 2% sodium hydroxide solution (1:1) to
elute the product:. The resulting product is further purified by gel filtration on a. BIO-GEL
P2 column using water as eluant to afford the trans-2,5-disubstituted morpholine racemic
Compound 60.
31p nmr (2Q2.5MHz; D20) ~ (ppm) 37 4