Note: Descriptions are shown in the official language in which they were submitted.
CA 02230896 1998-03-02
W O 97/09325 PCT~US96/14089
Description
PYRIMIDINE CARBOXYLATES AND RELATED COMPOUNDS AND
METHODS FOR TREATING INFLAMMATORY CONDITIONS
Technical Field
The present invention relates generally to compounds that block
intracellular signal transduction and activation of transcription factors, and to methods
for preventing or treating immunoinfl~mm~tory and autoimmune diseases.
Back~round of the Invention
Signals necessary for cell growth, differentiation, response to
bioregulatory molecules, infectious agents and physiological stress involve changes in
the rates of gene expression. The ability to respond a~ o~l;ately to such .signz~ling
15 events challenge the survival of the cell and ultimately the org~ni~m Perturbations in
the normal regulation of these specific genetic responses can result in pathogenic events
which lead to acute and chronic disease.
In certain autoimmune diseases or chronic infl~mm~tory states,
continuous activation of T-cells eventually leads to a self perpetuating destruction of
20 normal tissues or organs. This is caused by the induction of adhesion molecules,
chemotaxis of leukocytes, activation of leukocytes and the production of me~ tors of
infl~mm~tion. All of these events are regulated at the level of transcription for the
production of new proteins, including cytokines. The production of cytokines, as well
as a number of other cellular regulators, is controlled by a family of proteins known as
25 transcription factors (TFs). These transcription factors, when activated, bind to specific
regions on the DNA and act as molecular switches or messengers to induce or
upregulate gene expression. The activation of these TFs is caused by a variety of
external signals including physiological stress, infectious agents and other bioregulatory
~ molecules. Once the plasma membrane receptors are activated. a cascade of protein
30 kinases and second messengers are induced which, in turn, result in the production of
-
WO 97/09325 CA 0 2 2 3 0 8 9 6 l 9 9 8 - O 3 - O 2 PCT/US96/14089
RNA ~ SC~ S. The end result is the production of proinfl~ ry proteins via
- translation and proces~ing of the RNA transcripts.
This activation system can, at times, be very robust. For example, a
specific set of ~xtt-rnal signals could result in a single transcription factor to induce
5 many proteins responsible for a given disease. Therefore, regulating this process by
disrupting the production of activated TF(s) has the potential to att~n-l~te the production
of the associated pathological proteins, thereby halting or ~ g the course of the
disease.
Two transcription factors, NFKB and AP-l, have been shown to regulate
10 the production of many proinflamm~tory cytokines and related proteins that are
elevated in immunoinfl~mmatory diseases. These TFs regulate interleukin-l (IL-l),
interleukin-2 (IL-2), tumor necrosis factor-oc (TNFa), interleukin-6 (IL-6) and
interleukin-8 (IL-8) levels in a variety of cell types. For exarnple, NFKB and other
related complexes are involved in the rapid induction of genes whose products function
15 in protective and proliferative responses upon exposure of cells to ex~ l stimuli.
Similarly, AP-l has a significant role in the regulation of interleukin-2 (IL-2) and tumor
necrosis factor-a (TNF-a) transcription during T-cell activation. In addition, TNF-a
and IL-I are strong activators of collagenase, gel~tin~e and stromelysin gene
e~lession, which require a single AP-l binding site in the promoter region of these
20 genes. Therefore, an inhibitor of NFKB and/or AP-l activation would coordinately
repress the activities of a series of protein~ec In addition, cell adhesion molecules are
also controlled by these TFs. All of these proteins have been shown to play a role in
~ esl~e~ including osteoarthritis, transplant rejection, ischemis- reperfusion injury,
trauma, certain cancers and viral disorders, and autoi."~ c diseases such as
25 rheumatoid arthritis, multiple sclerosis, psoriasis, inflammatory bowel cli~e~ce,
glomerulonephritis, lupus and juvenile diabetes. In summary, the role of these TFs is to
act as a tr~n~lncer for certain stimuli that lead to immllne, inflammatcrv, and acute
phase responses.
Since many ~ e~es are caused by the inappropriate production of
30 proteins, conventional ~hcld~t;ulic approaches have focused on inhibiting function or
-
CA 02230896 1998-03-02
W O 97/09325 PCT~US96/14089
activity of individual effector proteins. These treatments have not always proved to be
effective and, at times, are associated with many undesirable side effects. Therefore,
there is a need for new therapies for the prevention and/or tre~tment of
immunoinfl~mm~tory and autoimmune diseases. More specifically, there is a need for
S compounds that prevent, preferably by inhibiting transcription at an early stage, the
production of proteins associated with immunoinfl~mm~tory and autoimml-ne diseases.
Furthermore, these compounds should inhibit the kinase(s) that regulate the activation
of TFs such as NFKB and AP-l . The present invention fulfills these needs and provides
further related advantages.
Summary of the Invention
In brief, this invention is directed to compounds that block the activation
of transcription factors (TFs), particularly NFKB and AP- 1, and are believed to function
through inhibition of a family of specific kinases. This results in a decrease in a number
15 of proinfl~mm~tory proteins, including IL-l, IL-2, IL-8 and/or TNFa, which are
responsible for tissue and organ damage associated with diseases such as rheumatoid
arthritis, osteoarthritis, related autoimmnne disorders and tissue rejection. Accordingly,
compounds of the present invention are useful in, for example, the prevention of organ
and tissue rejection associated with transplantation. Furthermore, the compounds of
20 this invention also have utility in the prevention and/or treatment of
immllnr)infl~mm~tory and autoimmlme diseases, as well as having general activity as
anti-infl~mm~tory agents.
In one embodiment of this invention, compounds are disclosed having
the following general structure (I):
N ~N
R2
(I)
-
CA 02230896 l998-03-02
WO 97/09325 PCTAUS96/14089
wherein R2, R4, R5 and R6 are as defined in the following detailed description.
In another embodiment, a ph~rm~elltical composition is disclosed
co~ i"g one or more compounds of this invention in combination with
pharm~el-tically or prophylactically acceptable carrier or diluent.
In a further embo~1im~nt methods are disclosed for ~lcv~llLillg and/or
treating infl~mm~tQry conditions by ~-1mini~t(~ring to a warm-blooded animal in need
thereof an effective amount of a compound of this invention. Such infl~mm~t )ry
conditions include both immlln-)infl~mm~tory conditions and alllo;-",~,lln~ e~ In
the practice of the disclosed methods, the compounds are preferably ~-lmini~tered to the
warm-blooded animal in the form or a ph~rm~ce~ltical composition.
These and other aspects of this invention will become evident upon
reference to the ~tt~ehed figures and the following detailed description.
Brief Description of the Drawin~s
Figures 1 and 2 illustrate reaction schemes for the synthesis of
e~ se~ ive compounds ofthis invention.
Figure 3 illustrates the ability of a representative compound of this
invention to inhibit the activation of NFKB and AP- 1.
Figure 4 illustrates the ability of a representative compound of this
invention to inhibit IL-2 and IL-8.
Detailed Description of the Invention
As mentioned above, the compounds of this invention block activation of
Ll~lscl.~Lion factors (TFs), and thus have utility as anti-infl~mm~tory agents in general,
and in the ~lcv~llLion and/or tre~tment of a variety of conditions, including (but not
limited to) immunoinfl~mm~tory and autoimmlme ~ e~e~ The compounds are
believed to function by inhibiting, at an early stage, transcription of deleterious proteins
associated with such conditions or ~ e~es It is believed that this is achieved by
inhibiting the kinase(s) that regulate the activation of TFs, such as NFKB and/or AP-l.
By disrupting the production of these activated TFs, synthesis of pathological proteins,
including proinfl~mm~tory cytokines, associated with a series of immlln- infl~mm~tory
,
CA 02230896 1998-03-02
W O 97/09325 PCT~US9~/14089
and autoimmlme diseases are effectively blocked at a transcriptional level.
Accordingly, the compounds of this invention have activity in both the prevention and
trP~tment of immllnoinfl~mm~tory (li~e~e~ such as rheumatoid arthritis, osteoarthritis
and transplant rejection (tissue and organ), as well as autoimmune diseases such as
5 multiple sclerosis.
The compounds of this invention are generally represented by the
following structure (I):
Rs
R4 ~N~R6
R2
(I)
10 wherein R2, R4, R5 and R6 are as defined below.
In structure (I) above, R5 is selected from the following chemical
moieties (i), (ii) and (iii):
O OR7 O R8~J~o- 7
'0,1
(i) (ii) (ia
1 5 wherein
R7 is selected from hydrogen and an unsubstituted or substituted
Cl 8alkyl, C6 l2aryl or C7 l2aralkyl; and
R8 is an unsubstituted or substituted C, 8alkyl, C6 ,2aryl or C7 l,aralkyl. In
a ~ r~ d embodiment, R7 is a C, 8 alkyl, and in a more preferred embodiment is
20 selected from methyl and ethyl. Further, in a ~l~r~ ;d embot1iment R8 is selected from
methyl and phenyl.
WO 97/09325 CA 0 2 2 3 0 8 9 6 19 9 8 - O 3 - O 2 PCT/US96/14089
The compounds of this invention further include rhzrm~celltically and
- prophylactically acceptable salts of compounds of structure (I). Compounds of
structure (I) may contain proton clor~ting groups (e.g., a carboxylic acid group) and/or
proton accepting groups (e.g., a group with a nitrogen atom having a free lone pair of
5 electrons, such as an amine group), and the salts of compounds of structure (I) may be
formed and utilized in the practice of the invention. Thus, compounds of the invention
may be in t-h-e form of a base addition salt (i.e., a salt of a proton donating group) or in
the form of an acid addition salt (i. e., a salt of a proton accepting group), as well as the
free acid or free base forms thereof.
10Acid addition salts of a free base amino compound of the invention may
be prepared by methods well known in the art, and may be formed from organic andinorganic acids. Suitable organic acids include acetic, ascorbic, ben7~nesll1fonic,
benzoic, fumaric, maleic, meth~neclllfonic~ and succinic acids. Suitable inorganic acids
include hydrochloric, hydrobromic, sulfuric, phosphoric and nitric acids. Base addition
15 salts of a free acid carboxylic acid compound of the invention may also be prepared by
methods well known in the art, and may be formed from organic and inorganic bases.
Thus, the compounds of this invention also include those salts derived from inorganic
bases such as the hydroxide or other salt of sodium, potassium, lithium, ammonium,
calcium, magnesium, iron, zinc, copper, m~ngslnese, aluminum, and the like, and
20 organic bases such as substituted ammonium salts.
As used herein, the above terms have the following me~ning:
A "Cl 8alkyl" is a straight chain or brzlncherl, cyclic or non-cyclic,
~tllr~te~l or unsaturated carbon chain co..~ g from 1 to 8 carbon atoms. In one
embodiment, the C,.8alkyl is a fully saturated, straight chain alkyl selected from methyl,
25 ethyl, n-propyl, n-butyl, n-pentyl and n-hexyl. In another embodiment, the Cl 8alkyl is a
fully saturated cyclic alkyl selected from (but not limited to) cyclopropyl, cyclobutyl,
cyclopentyl, cyclohexyl, methylenecyclopropyl and methylenecyclohexyl. In still a
further embodiment, the C, 8alkyl is a fully saturated, branched alkyl selected from (but
not limited to) isopropyl, sec-butyl, isobutyl, tert-butyl, isopentyl and isohexyl. In yet a
CA 02230896 1998-03-02
W O 97/09325 PCT~US96/14089
further embodiment, the Cl 8alkyl is an wlsdluldLed straight chain alkyl selected from
(but not limited to) ethylenyl, propylenyl, l-butenyl, l-pentenyl and l-hexenyl.A "C6 l2aryl'l is an aromatic moiety co~ from 6 to 12 carbon
atoms. In one embodiment, the C6 l2aryl is selected from (but not limited to) phenyl,
5 tetralinyl, and napthalenyl. In a preferred embodiment, the C6 l2aryl is phenyl.
A "C, ,2aralkyl" is an arene cont~inin~ from 7 to 12 carbon atoms, and
has both ~ h~tic and aromatic units. In one embodiment, the C7 lzaralkyl is selected
from (but not limited to) benzyl, ethylbenzyl, propylbenzyl and isobutylbenzyl.
A "substituted" C, 8alkyl, C6 ,2aryl or C7 l2aralkyl is a C, 8alkyl, C6 ,2aryl
10 or C, ,2aralkyl having one or more hydrogens replaced with a substituent selected from
halogen (including -F, -Cl, -Br and -I), -OH, -R, -OR, -COOH, -COOR, -COR,
-CONH2, -NH2, -NHR, -NRR, -NO2, -SH, -SR, -SOOR, -SO3R and -SOR, where each
occurrence of R is independently selected from an unsubstituted or substituted C, 8alkyl,
C6 ,2aryl or C, ,2aralkyl as defined above. In one embodiment, the substituted C, 8alkyl
15 is a C, 8haloalkyl including (but not limited to) -CF3 and -C2F5.
In one embodiment of structure (I) above, R2 is R2~ and R4 is R4a~ In this
embodiment, R4a is selected from hydrogen, halogen and an unsubstituted or substituted
C, 8alkyl, C6 ,2aryl, C, ,2aralkyl, C3 ,2heterocycle or C4 ,6heterocyclealkyl, and R2a is
selected from the following chemical moieties (iv) through (vii):
R9 ~ A. ~ 'N ~ H , N~N ~ Rll
(iv) (v) (vi) (vii)
wherein Rg is selected from hydrogen, -C(=O)-D-R, and an unsubstituted C, 8alkyl or
25 C"2aralkyl; and Rlo and R" are the same or different and independently selected from
hydrogen and an unsubstituted or substituted C, 8alkyl or C6 ,2aryl; n is an integer from 0
to 4 and represents the number of substitllent~ on the benzene ring of chemical moiety
(iv); D represents a direct bond, -O- or -NH-; and each occurrence of A is independently
WO 97/09325 CA 0 2 2 3 0 8 9 6 l 9 9 8 - O 3 - O 2 PCT/US96t,14~89
selected from a sllbstih~nt as identified above. In a preferred embodiment, D is a direct
- bond; Rg is selected from hydrogen, -CH3, -CH2CH3 and -CH2C6H5; Rlo and Rl~ are the
same or dirr~ and independently selected from hydrogen, -CH3, -CF3, -(CH2)l 5CH3,
-C6H5, -CH2C6H5, and a substituted phenyl or benzyl moiety; and n is 0.
In another embodiment of structure (I) above, R2 is R2b and R4 is R4b. In
this embodiment, R2b is selected from hydrogen, halogen and an unsul,~LiluLGd orsubstituted Cl 8alkyl, C6 ,2aryl, C, l2aralkyl, C3 l2heterocycle or C4 l6heterocyclealkyl; and
R4b is selected from chemical moieties (iv) through (vii) identified above.
As used herein, a "C3 l2heterocycle" is a moiety that contains a ring made
up of more than one kind of atom, and which contains 3 to 12 carbon atoms. In one
embodiment, the C3 l2heterocycle is selected from (but not limited to) pyrrolyl, furanyl,
thienyl, imi~ 7.olyl, oxazolyl, thiazolyl, pyrazolyl, pyrrolidinyl, pyridinyl, pyrimidinyl,
purinyl, and thianaphthyl.
A "C4 l6heterocyclealkyl" is a moiety that contains a C3 ,2heterocycle
linked to a C, 8alkyl, and which contains 4 to 16 carbon atoms. In one embodiment, the
C4 ,6heterocyclealkyl is a methylene furan having the following structure:
A "substituted" C3 ,2heterocycle or C4 l6heterocyclealkyl is a
C3 ~2heterocycle or C4 l6heterocyclealkyl having one or more hydrogens replaced with a
substituent selected from halogen (including -F, -Cl, -Br and -I), -OH, -R, -OR, -COOH,
-COOR, -COR, -CONH2, -NH2, -NHR, -NRR, -NO2, -SH, -SR, -SOOR, -SO3R and
-SOR, where each occurrence of R is independently selected from an unsl-bstih~ or
substituted Cl 8alkyl, C6 l2aryl, C, l2aralkyl, C3 ,2heterocycle or C4 ,6heterocyclealkyl as
defined above.
In structure (I) above, R6 is selected from hydrogen, -CH3, -CH2C6H5, -F
and -CF3.
,
CA 02230896 1998-03-02
W O 97/09325 PCT~US96/14089
In one embodiment, the compounds of this invention have structure (I)
above wherein Rs is the chemical moiety (i). In this embo~iment, the compounds
disclosed herein have the following structures (II) and (III):
o;~OR7 O~OR7
R4a ~R6 R4b ~R6
N ~ N N ~ N
R2a R2b
S (II) (III)
wherein R2a~ R2b, R4a~ R4b, R6 and R7 are as defined above.
In a prefened embodiment, the compounds of this invention have
structure (II) above, wherein R2a, R4a~ R6 and R, are selected from the moieties identified
in Table 1 below.
CA 02230896 1998-03-02
W O 97/09325 PCTAUS96/14089
Table 1
- Co,.. poullds of Structure (II)
R2a R4a R6 R,
-Cl -H -CH3
H ~~ CH3
,N~N ~ -CF3 -CF3 -CH2CH3
o -CH3 -CH3 -H
H ~ C~5 -phenyl
'N ~ -(cH2)l-2cH3
~~ -C~F3
~X
o CH3
/
H3
o
wherein X, Y and ~ are the same or different, and indepPn~lently selected from
hydrogen, -OH, -R, -OR, -COOH, -COOR, -COR -CONH2, -NH2, -NHR, -NRR,
-NO2, -SH, -SR, -SOOR, -SO3R and -SOR, where each occurrence of R is
independently selected from an unsubstituted or substituted C, 8alkyl, C6 l2aryl,
C, l2aralkyl, C3 l2heterocycle or C4 l6heterocyclealkyl.
S In a further preferred embodiment, the compounds of this invention have
structure (III) above, wherein R2b, R4b, R6 and R, are selected from the moieties
identified in Table 2 below.
CA 02230896 1998-03-02
W O 97/09325 PCTAUS96/14089
Table 2
ComPounds of Structure (III)
R2b R4b R6 1~,
-Cl H CH3 -H -CH3
-CF3 'N'N~ -CF3 -CH2CH3
-CH3 o -CH3 -H
-phenyl O~ c6H5
-(cH2)'-2cH3 ~N~N~
-C2F3 ~
~¢CH~
~X H ~3
CIH3 o
N
o
wherein X, Y and ,_ are the same or diL elellt, and independently selected from
hydrogen, -OH, -R, -OR, -COOH, -COOR, -COR -CONH2, -NH2, -NHR, -NRR,
-NO2, -SH, -SR, -SOOR, -SO3R and -SOR, where each occurrence of R is
independently selected from an unsubstituted or substituted Cl 8alkyl, C6 12aryl,
C7 12aralkyl, C3 12heterocycle or C4 16heterocyclealkyl.
In another embodiment, the compounds of this invention have structure
(I) above wherein R5 is the chemical moiety (ii). In this embodiment, the compounds
disclosed herein have the following structures (IV) and (V):
W O 97/0932~ CA 02230896 1998-03-02 PCTrUS96/14089
O~Rg O~R~
R4a~R~ R4b~f R6
N~N N~N
R2a ~2b
(IV) (V)
wherein R2a, R2b, R4a~ R4b, R6 and R8 are as defined above.
In another embodiment, the compounds of this invention have structure
5 (I) above wherein R5 is the chemical moiety (iii). In this embodiment, the compounds
disclosed herein have the following structures (VI) and (VII):
O 'O
oJ~o, R7 OJ~O, R7
R4a~R6 0,1 NrN
R2a R2b
(VI) (VII)
wherein R2z, R2b, R4a~ R4b, R6 and R, are as defined above.
In one embodiment, the compounds of this invention have structure (II)
or (III) above and include (but are not limited to) the following: ethyl 2-(N-(1'-
15 aminocitraconamido))-4-trifluoromelhyl~ylilllidine-5-carboxylate; ethyl 2-(N-(1'-
aminophth~limi~ ))-4-trifluoromethylpyrimidine-5-carboxylate; 5-acetyl-2-(N-(l'-aminocikraconamido))-4-kifluorom~;Lhylpy .; " .i(line; ethyl 2-(N-( I '-amino-3'-
phenylmaleimido))-4-trifluoromethylpyrimidine-5-carboxylate, ethyl 2-(N-( 1 '-amino-
3',4'-dimethylmaleimido))-4-kifluoromt;lhyll~ylilllidine-5-carboxylate; ethyl 2-(N-(l'-
20 aminocikraconamido)-N-methyl)-4-trifluoromethylpyrimi~line-5-carboxylate; ethyl 4-
(N-(l'-amino-3'-phenylmaleimido))-2-kifluoromethylpyrimidine-5-carboxylate; ethyl 4-
CA 02230896 1998-03-02
W O 97/09325 PCTAJS96/14089
13
(N-( 1 '-amino-3 ',4'-dimethylmaleimido))-2-trifluoromethylpyrimidine-S-carboxylate;
ethyl 2-(N-( 1 '-aminocitraconamido))-4-m~lhyl~ylhllidine-5-carboxylate; ethyl 2-(N-( 1'-
aminocitraconamido))-4-pentafluoroethylpyrimidine-5-carboxylate; ethyl 2-(N-( 1'-
aminocitraconamido))-4-phenylpyrimidine-5-carboxylate; methyl 2-(N-( 1'-
5 aminocitraconamido))-4-(3'-pyridyl)pyrimidine-5-carboxylate; diethyl 2-(N-(l'-aminocitraconamido))pyrirnidine-4,5-dicarboxylate; ethyl 2-[N-( 1 '-amino-3'-
methylsuccinimido)]-4-trifluoromethyl-pyrimidine-5-carboxylate; methyl 2-[N-(l'-amino-3'-methylsuccinimido)]-4-trifluoromethyl-pyrimidine-5-carboxylate; ethyl 2-[N-
( 1 '-amino-4'-methylphth~l imido)]-4-trifluoromethyl-pyrimidine-5-carboxylate; ethyl 2-
10 [N-( 1 '-amino-3 ,4-dichlorophth~limido)]-4-trifluoromethyl-pyrimidine-5-carboxylate;
ethyl 2-[N-(l'-aminocitraconamido)]-pyrimidine-5-carboxylate; ethyl 2-[ N-(l'-
aminocitraconamido)]-4-ethyl-pyrimidine-5-carboxylate; ethyl 2-[N-(l'-aminocitraconamido)-N-methyl]-4-ethyl-pyrimidine-5-carboxylate; ethyl 2-[N-acetyl-
N-( 1 '-aminocitraconamido)]-4-propyl-pyrimidine-5-carboxylate; ethyl 2-[N-acetyl-N-
15 (1'-aminocitraconamido)]-4-trifluoromethyl-pyrimidine-5-carboxylate; methyl 2-[N-(l'-
aminocitraconamido)]-4-pentafluoroethylpyrimidine-5-carboxylate; methyl 2-[N-
(methyl)-N-( 1 '-amino-3'-methylmaleimido)]-4-pentafluoroethylpyrimidine-5-
carboxylate; t-butyl-2-[N-(l'-aminocitraconamido)]-4-trifluoromethyl-pyrimidine-5-
carboxylate; methyl-2-LN-( 1 '-aminocitraconamido)]-4-trifluoromethyl-pyrimidine-5-
20 carboxylate; methyl-2-[N-(l'-aminocitraconamido)-N-methyl]-4-trifluoromethyl-
pyrimidine-5-carboxylate; methyl 2-[N-( 1 '-aminocitraconarnido)]-4-(2'-
thienyl)pyrimidine-5-carboxylate; ethyl 2-[N-( 1 '-aminocitraconamido)-N-benzyl]-4-
trifluoromethyl-pyrimidine-5-carboxylate; ethyl 2-[N-(l'-aminocitraconamido)-N-
methyl]-4-(2'-thienyl)pyrimidine-5-carboxylate; 2-[N-( 1 '-aminocitraconamido)]-4-
25 trifluoromethyl-pyrimidine-5-carboxylic acid; ethyl 2-[N-(l'-aminocitraconamido)]-4-
(3'-thienyl)pyrimidine-5-carboxylate; ethyl 2-[N-(l'-aminocitraconamido)-N-methyl]-4-
- (3'thienyl)pyrimidine-5-carboxylate; ethyl 2-[N-(l'-aminocitraconamido)]-4-(5'-methyl-
2'-thienyl)pyrimidine-5-carboxylate; ethyl 2-[N-(l'-aminocitraconamido)]-4-(2'-
furanyl)pyrimidine-5-carboxylate; ethyl 2-[N-(l'-aminocitraconamido)-N-methyl]-4-(5'-
30 methyl-2'-thienyl)pyrimidine-5-carboxylate; ethyl 2-[(1'-amino-3'-methylmaleimido)]-
WO 97/09325 CA 0 2 2 3 0 8 9 6 19 9 8 - O 3 - O 2 PCT/US96/14089
14
4-(2'-thi~n~phthyl)pyrimi~1ine-5-carboxylate; methyl-2-~N-(l'-aminocitracnn~mi-lo)-N-
- ethyl]-4-trifluoromethyl-pyrimidine-5-c~b~xylate; ethyl 2-[N-(1'-aminocitraconamido)-
N-butanoyl]-4-trifluoromethyl-pyrimidine-5-carboxylate; ethyl 2-[N-( 1'-
aminocitraconamido)-N-(N-methylcarboxamidyl)-]-4-trifluorome~lylL~y .; . "itlin~-5-
S carboxylate; ethyl 2-[N-( 1 '-aminocitraconamido)-N-( 1 -oxo-2-phenylethyl)]-4-
trifluoromethyll~y,; " ~ i~1ine-5-carboxylate; ethyl 2-[N-( 1 '-aminociL,dcollaL,lido)3-4-
methoxymel~,yl~yfllllidine-5-carboxylate; ethyl 2-[N-(l'-aminocitraconamido)-N-
(ethoxyc~ubolly~1)]-4-trifluoromel~lyl~ylilnidine-5-carboxylate; ethyl 2-[N-(1~-aminocitraconamido)-N-benzoyl]-4-trifluoromethylpyrimidine-5-carboxylate; ethyl-2-
10 [N-(1'-aminocitraconamido)-N-methyl]-4-pentafluoroethyl-pyrimidine-5-carboxylate;
ethyl 2-[N-(1 '-aminocitraconamido)]-4- (2'-thianaphthyl)pyrimidine-5-carboxylate;
ethyl 2-[N-(1'-aminocitraconamido)]-4-(2'-thiazolyl)pyrimi~1ine-5-carboxylate; ethyl 2-
[N-( 1 '-aminocitraconamido)-N-methyl]-4-(2'-thiazolyl)pyrimidine-5-carboxylate; ethyl
2-[N-(1'-aminocitraconamido)]-4-cyclo~lopylpylilllidine-5-carboxylate; and ethyl 2-[N-
15 ( 1 '-aminocitraconamido)-N-methyl]-4-cyclopropylpyrimidine-5 -carboxylate.
In another embodiment, the compounds of this invention have structure
(IV) or (V) above and include (but are not limited to) the following: 5-benzoyl-2-
chloro-4-trifluoromethyl~yfllllidine; 5-acetyl-2-[N-(1 '-aminocitraconamide)]-4-
trifluoromeLllyl~!ylilllidine; 5-benzoyl-2-[N-(1 '-aminocitracon~mit1~)] 4
20 trifluoromethyl~ylilllidine; 5-benzoyl-2-[N-(l'-aminocitracnn~micl--)] 4-ethylpyrimidine;5-benzoyl-2-[N-(l'-aminocitracon~mi~lo)-N-methyl]-4-
e~lhyl~ylilllidine; and5-butanoyl-2-[N-(1'-aminocitraconamido)-N-methyl]-4-
ethylpyrimicline.
In yet another embodiment, the compounds of this invention have
25 structures (VI) or (VII) above, and include (but are not limited to) the following: 5-
methylol-2-[N-(1'-aminocitraconamido)]-4-ethylpyrimidine; 5-methylol-2-[N-(1'-
aminocitraconamido)-N-methyl]-4-ethylpyrimi~line; 5-methoxymethane-2-rN-(1'-aminocitraconamido)-N-methyl]-4-ethylpyrimicline; 5-methoxymethane-2-[N-(1'-aminocitracon~midc-)]-4-trifluoromelhyl~yl ;",icline; and 5-ethoxymethylcarbonate-2-
3 0 [N-( 1 '-aminocitraconamido)]-4-trifluoromethylpyrimidine.
-
CA 02230896 1998-03-02
W O 97/09325 PCTnJS96/14089
Preferred compounds of the invention are ethyl 2-(N-(l'-
aminocitraconamido))-4-trifluoromethylpyrimidine-5-carboxylate; ethyl 2-(N-(l '-
- aminophth~limiclo))-4-trifluoromelhyl~-y~ lidine-5-carboxylate; 5-acetyl-2-(N-(l'-
aminocitraconamido))-4-trifluoromethylpyrimidine-5-carboxylate; ethyl 2-(N-(1'-5 amino-3'-phenylm~leimido))-4-trifluoromethylpyrimidine-5-carboxylate; ethyl 2-(N-(l'-
amino-3',4'-dimethylmaleimido))-4-trifluoromethylpyrimidine-5-carboxylate; ethyl 2-
(N-( 1 '-aminocitraconamido)-N-methyl)-4-trifluoromethylpyrimi~lin~-5-carboxylate;
ethyl 4-(N-(l'-amino-3'-phenylmaleimido))-2-trifluoromethylpyrimidine-5-carboxylate;
ethyl 4-(N-(l'-amino-3', 4'-dimethylmaleimido))-2-trifluoromethylpyrimidine-5-
10 carboxylate; ethyl 2-(N-( I '-aminocitraconamido))-4-methylpyrimidine-5-carboxylate;
ethyl 2-(N-(l'-aminocitraconarnido))-4-pentafluoroethylpyrimidine-5-carboxylate; ethyl
2-(N-( 1 '-aminocitraconamido))-4-phenylpyrimidine-5-carboxylate; methyl 2-(N-( 1'-
aminocitraconamido))-4-(3'-pyridyl)pyrimi~line-5-carboxylate; diethyl 2-(N-(l'-
aminocitraconamido))pyrirnidine-4,5-dicarboxylate; ethyl 2-(N-( I '-
15 aminocitraconamido))-4-(2'-thienyl)pyrimidine-5-carboxylate; ethyl 2-(N-( 1'-
aminocitraconamido)-N-methyl)-4-ethylpyrimidine-5-carboxylate; methyl 2-(N-( 1'-aminocitraconamido))-4-(2'-thienyl)pyrimidine-5-carboxylate; ethyl 2-(N-( 1'-
aminocitraconamido)-N-methyl)-4-(2'-thienyl)pyrimidine-5-carboxylate; ethyl 2-(N-(l'-
aminocitraconamido))-4-(5'-methyl-2'-thienyl)pyrimidine-5-carboxylate; ethyl 4-(N-(1'-
20 aminocitraconamido))-2-phenylpyrimidine-5-carboxylate; and ethyl 4-(N-( 1'-
aminocitraconamido))-2-(2'-thienyl)pyrimi~line-5-carboxylate.
The compounds of this invention may be made by one skilled in organic
synthesis by known techniques, as well as by the synthetic routes disclosed herein.
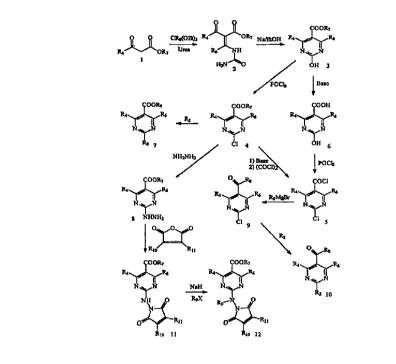
Referring to Figure 1, the compounds of this invention may be made from
25 commercially available ,B-keto esters 1 by heating at elevated temperatures (75-110~C)
with a mixture of urea and triethylorthoformate (or a substituted orthoformate) to
- provide ureido derivatives 2. Tre~tment of these intermediates with sodium alkoxides,
such as sodium ethoxide in an alcoholic solvent at 3 5-1 00~C, gives 2-
hydroxypyrimidine esters 3 which, upon treatment with a chlorinating agent such as
W O 97/09325 CA 02230896 1998-03-02 PCT~US96/14089
16
phosphorous oxychloride at elevated temperatures (75-120~C), yields 2-
- chlo~ y,h,lidine esters 4.
Compound 4 may be reacted with various nucleophiles in an aprotic
solvent at ambient temperature to provide derivatives 7. Compound 4 may also be
S converted to the carbonyl chloride 5 by tre~tmPnt with base, such as hydroxide in water,
followed by a chlorinating agent, such as oxalyl chloride in methylene chloride.Compound 5 can be treated with an organometallic, such as methyl ma~ .lesiu"l bromide
in a solvent such as THF or ether at -35~C to -65~C, to give ketone 9. This ketone may
be treated with various nucleophiles in an aprotic solvent and at ambient te;m~e.dlul~; to
10 provide compound 10.
~ It~rn:~tively, compound 3 may be converted to the hydroxy carboxylic
acid 6 by tre~tment with a strong base, such as sodium hydroxide, or strong acid, such
as HCl, at elevated temperature (70-110~C). The hydroxy carboxylic acids may be
converted to the chloro carbonyl chloride with thionyl chloride and/or phosphorous
1 5 oxychloride.
Compound 4 can also be treated with hydrazine at ambient temperature
in a solvent, such as THF, with pyridine as a catalyst to provide the interme~ tPS of
structure 8. These hydrazino derivatives can be reacted with cyclic anhydrides, such as
citraconic anhydride, in a solvent, such as chloroforrn, at elevated lell,p~ res20 (35-65~C) to provide compounds of structure 11. Subsequent tre~trnent of 11 with a
strong base, such as sodium hydride, in an aprotic solvent, such as THF, at ambient
te~ c~ re followed by an alkyl iodide, such as methyl iodide, provides the alkylated
derivatives of structure 12.
Compounds of structures (VI) and (VII) may be prepared by reducing
25 any of the compounds in Figure 1 so as to convert a carboxylate group to a methylol
(-CH2OH~ group. Lithium al--minnm hydride is a suitable re~lncin~ agent. In any event,
the methylol group may, if desired, be converted to -CH2OR7 by standard alkylation
chemistry (e.g, using a strong base and a nucleophile).
Referring to Figure 2, an alternative synthetic procedure is disclosed. In
30 this procedure, commercially available diethyl ethoxymethylenemalonate 13 is treated
:
CA 02230896 1998-03-02
W O 97/0932S PCTAUS96/14089
17
with an amidine, such as trifluoromethylamidine, in a protic solvent, such as ethanol, in
the presence of an alkoxide, such as NaOEt, at elevated len.~c.dLures (75-11 0~C) to give
the hydroxy pyrimidine ester 14. Chlorination with a chlorinating agent, such as POCl3
or thionyl chloride, yields the chloroester derivative 15 which can be treated with
5 various arnines at ambient teu~ ,ldlule in an aprotic solvent, such as THF, to provide
the substituted pyrimidines 16. Derivative 15 may also be treated with hydrazine in a
solvent such as THF in the presence of pyridine to give the hydrazino intermediates 17.
Tre~tment of 17 with various cyclic anhydrides, such as citraconic anhydride, in a
solvent, such as chloroform, at elevated temperatures (34-65~C) provide compounds of
10 structure 18. Alkylation of these compounds with a hydride, such as sodium hydride,
followed by an alkyl halide in an aprotic solvent, such as THF, gives the alkylated
derivatives of structure 19.
Again, compounds of structures (VI) and (VII) may be prepared from the
corresponding carboxylate compounds as discussed above in connection with Figure 1.
15 Compounds of the invention wherein Rg is -C(=O)R7 may be plep~d from the
corresponding compound wherein Rg is hydrogen (as ~re~ d according to either of
Figure l or 2) using standard acylation chemistry. For example, a compound having R9
equal to hydrogen may be treated with a strong base (e.g, NaH), followed by an
acylating agent (e.g, Cl-C(=O)R7).
Once synthesi7~1, the compounds of this invention may be f~rmlll~ted
for ~1mini~tration to a warm-blooded animal by a variety of techniques known to those
skilled in the art. In one embodiment, the compound is in the form of a ph~rm~eutical
composition for prophylactic or therapeutic use, and which contains at least onecompound of this invention in combination with a ph~rms~ceutically acceptable carrier
25 or diluent. The compound is present in the composition in an amount which, upon
~t1mini~tration to the animal, is effective in preventing or treating the condition of
~ interest. Preferably, the composition includes a compound of this invention in an
amount ranging from 0.01 mg to 250 mg per dosage, depending upon the route of
z~lmini~tration~ and more preferably from l mg to 60 mg. Appropriate con(~entr~tions,
CA 02230896 1998-03-02
W O 97/09325 PCT~US96/14089
dosages and modes of ~lmini~tr~tion may be readily cletPrmined by one skilled in the
art.
Suitable carriers or ~liluent~ are fz~mili~r to those skilled in the
formulation field. For compositions formnl~ted as liquid solutions, acceptable carrier or
S ~lihlpnt~ include saline and sterile water, and may optionally include ~ntio~ nt~,
buffers, bacteriostats and other common additives. The compositions of this invention
may also be formlll~tecl as pills, ç~pslllf c, gr~nllle,c or tablets which contain, in addition
to the compound of this invention, ~lih~ent~, dispersing and surface active agents,
binders and lubricants. One skilled in the art may further form~ te the compounds of
10 this invention in any ~plo~liate manner, and in accordance with accepted practices,
such as those disclosed in Remington's Pharmaceufical Sciences, Gennaro, Ed., Mack
Publishing Co., Easton, PA, 1990 (incorporated herein by reference).
In another embo~liment the present invention provides methods for
preventing or treating a variety of conditions. Such methods include ~lmini~tpring a
15 compound of this invention to a warm-blooded animal in need thereof in an amount
sufficient to prevent or treat the condition. Such methods include systemic
mini~tration of a compound of this invention, preferably in the form of a composition
as disclosed above. As used herein, systemic ~imini~tration includes oral and parental
methods of ~s~lministration. For oral ~flmini~tration, suitable ph~rm~celltical
20 compositions include powders, granules, pills, tablets and capsules, as well as liquids,
syrups, suspensions and emulsions. These compositions may also include flavorants,
preservatives, suspen~ling~ thickening and emulsifying agents, and other
rh~rm~celltically acceptable additives. For parental ~imini~tration, the compounds of
the present invention may be prepared in aqueous injectable solutions which may
25 contain, in addition to the compound of this invention, buffers, antio~ nt~,
bacteriostats and other additives commonly employed in such solutions.
As mentioned above, compounds of the present invention can be used to
prevent or treat a wide variety of disorders, diseases and/or illnes~es In particular, the
compounds may be ~tlmini~t~red to a warm-blooded animal for prevention or trç~tment
30 of rheumatoid arthritis, osteoarthritis, tissue and/or organ transplant rejection, sepsis,
CA 02230896 1998-03-02
W O 97/09325 PCTAUS96/14089
ARDS, ~ethm~, trauma, oxidative stress, cell death, irradiation damage, ischemi~,
reperfusion, cancer, viral infection, and autoimmllne diseases such as psoriasis,
infl~mm~tory bowel ~li.ee~ee, glomerulonephritis, lupus, uveitis and chronic hepatitis.
Compounds of this invention may be screened by known and accepted
S techniques for their ability to function as prophylactically and/or thc.d~ lically active
agents. For example, the compounds may be evaluated in in vitro and/or in vivo assays
indicative ofthe compound's anti-infl~mm~tQry and immlmo~u~lcs~ive p~ clLies. Tothis end, such compounds may first be evaluated in a number of cell-based assays which
determine the ability of a compound to prevent activation of NFKB and AP-l (see
10 Example 126). Next, the compound's ability to ~ttPnll~tP cytokine levels (such as IL-2
and IL-8), which are known to be elevated in certain disease states, may be ~letprmined
(see Example 127). The compounds may then be evaluated in an a~lol)liate animal
model, including rodent models of infl~mm~tion and immuno~u~ ,s~ion (see Exarnple
128).
It should be recognized that, for example, in the case of
immuno~u~.le;,sive drugs and other agents which have utility for the trez~tment of
rheumatoid arthritis (RA), numerous studies have been performed directed to the
activity of such drugs. To this end, cyclosporin A has been used in clinical trials since
the late 1970's as a second-line drug, and is recommen-1ed to be used only in p~tiente
20 with active RA. Thus, Experiment 128 may be performed ntili7in~ cyclosporin A as a
positive control. A recent review of such immlmo~u~lcs~ive drugs, including relevant
assays for the same, is presented by R.P. Carlson in Exp. Opin. Invest. Drugs 4(9):853-
859, 1995 (incorporated herein by reference in its entirety, including cited references).
The following examples are presented for purpose of illustration, not
25 limitation.
EXAMPLES
To summarize the examples that follow, Examples 1-124 disclose the
synthesis of representative compounds of this invention, as well as intermediates
30 thereto; Example 125 discloses the synthesis of representative compounds by
CA 02230896 1998-03-02
W O 97/0932~ PCT~US9U14089
combinational ch~mi~try techniques; Examples 126-127 disclose the ability of
- lG~l~se.lLdli~e compounds of this invention to inhibit NFKB, ~P-1 and cytokines; and
Example 128 discloses assays for evaluating activity of lc~r~ç~ re compounds of
this invention in both graft versus host disease and contact sensitivity models.
s
Exarn~le 1
2-CHLORO-5-[3',5'-BIS(TRIFLUOROMETHYL)PHENACYL]-
4-TRIFLUOROMETHYLPYRIMIDINE
To m~gn~.sillm t-lrnings (0.026 g, 1.06 mrnol) in Et20 (15 mL) was
10 added a solution of 3,5-bistrifluoromethyl iodobenzene (0.300 g; 0.882 mrnol) in Et2O
(5 mL). The reaction was refluxed under an atmosphere of N2 for 2 h and then cooled
to 0~C. A solution of 2-chloro-4-trifluoromethylpyrimidine-5-carbonyl chloride (0.205
g, 0.838 mmol) in Et2O (5 mL) was added dropwise via syringe. After stirring 1 hour
at 0~C, water (15 mL) was added and the mixLul~ extracted with Et20 (2 X 20 mL).15 The organic layer was washed with brine (15 mL), dried over MgSO4, filtered and
concelllld~ed. The residue was chromatographed (SiO2, h~ n.os/EtOAc 8:1) to give the
title compound (0.078 g, 21% yield) as an oil; 'H NMR (CDCl3) ~ 8.90 (s, lH), 8.21 (s,
lH), 8.19 (s, 2H).
Example 2
5 -BENZOYL-2-CHLORO-4-TRIFLUOROMETHYLPYRIMIDINE
The title compound was prepared as described in Example 1, but
employing phenyl m~gne~iurn bromide (0.23 mL, 0.69 mmol) and the acid chloride
(0.17 g, 0.69 mrnol), res-llting in a yield of 30%; lH NMR (CDCl3) ~ 8.81 (s, lH), 7.4-
7.8 (m, SH).
CA 02230896 l998-03-02
PCTAUS96/14089
W O 97/0932S
Example 3
ETHYL UREIDOMETHYLENE ACETOACETATE
A Ilfi~ c of ethyl acetoacetate (200 g, 1.54 mole), urea (105 g, 1.54
mole) and triethyl orthoformate (228 g, 1.54 mole) was heated at 140~C under N2 for 22
5 h The reaction mixture was cooled and filtered to provide the title compound in a 51 %
yield (156 g), m.p. 173-174~C.
Example 4
ETHYL UREIDOMETHYLENE BENZOYLACETATE
The title compound was prepared as described in Example 3, but
employing ethyl benzoylacetate (30.0 g, 156 mmol), resulting in a yield of 21% (12g);
m.p. 124-126~C.
Example S
ETHYL 2-HYDROXY-4-METHYLPYRIMIDINE-5-CARBOXYLATE
A solution of ethyl ureidomethylene acetoacetate (50 g, 250 mmol)
NaOEt (22.1 g, 325 mmol) in EtOH (500 mL) was stirred at room t~m~ d~ lre under N2
for 3 days. The resulting solid was filtered and dried to yield the title compound as a
sodium salt in a yield of 88% (45 g); m.p. >220~C (dec.).
Example 6
ETHYL 2-HYDRoxy-4-pHENyLpyRlMlDlNE-5-cARBoxyLATE
The title compound was prepared as described in Example 5, but
employing ethyl ureidometl1ylene benzoyl acetate (12 g, 45 mmol), resulting in a yield
25 of 15% (6 g); m.p. >260~C, (dec.).
CA 02230896 1998-03-02
W O 97/09325 PCT~US96/14089
F~nnpl~ 7
ETHYL 2-cHLoRo-4-METHyLpyRlMIDrNE-s-cARBoxyLATE
A solution of ethyl 2-hydroxy-4-m~ yl~y-il--idine-S-carboxylate (S g,
27.5 mmol) and POCl3 (84 g, 550 mmol) was heated at reflux under N2 for 1 h The
5 reaction was cooled and concentrated. The residue was partitioned between CHCl3 and
H20 and the organic layer was dried (Na2SO4), filtered, and concenlldl~d to yield the
title compound in a yield of 27% (1.5 g); ~H NMR (CDCl3) ~ 9.04 (s, lH), 4.42 (q,
2H), 2.85 (s, 3H), 1.43 (t, 3H).
Example 8
ETHYL 2-CHLORO-4-PHENYLPYRIMIDINE-S-CARBOXYLATE
The title compound was prepared as described in Example 7, but
employing 2-hydroxy-4-phenylpyrimi~1ine-5-carboxylate (6 g, 25 mmol) to give the title
compound (5.5 g, 86%), m.p. 45-47~C.
Example 9
2-CHLORO-4-METHYLPYRlMlDlNE-5-CARBOXYLlC ACID
A solution of ethyl 2-chloro-4-methylpyrimidine-5-carboxylate (1.0 g, 5
mmol), NaOH (0.24 g, 6 m~nol) in H2O (30 mL) was stirred at room temperature for 3
20 h The solution was acidified with 6N HCI and the resulting solid was filtered and dried
to give the title compound (0.67 g 78%), ~H NMR (DMSO-d6) ~ 9.01 (s, lH), 2.75 (s,
3H).
Exam~le 10
2-CHLORO-4-PHENYLPYRlMlDlNE-5-CARBOXYLlC ACID
The title compound was prepared as described in Example 9, but
employing 2-chloro-4-phenyl~ylinlidine-5-carboxylate (4.5 g, 17 mmol), resllltin~ in a
yield of 87% (3.9 g), m.p. 105-110~C.
CA 02230896 1998-03-02
W O 97/09325 PCTAUS96/14089
Example 11
2-CHLORO-4-METHYLPYRIMIDINE-5-CARBONYL CHLORIDE
A solution of 2-chloro-4-methylpyrimidine-5-carboxylic acid (0.81 g,
4.70 mmol), oxalyl chloride (0.89 g, 7.05 mmol), DMF (2 drops) in CH2Cl2 (23 mL)
5 was stirred at room t~ e under N2 fior 4 h The solution was conc~lll.dLed and
distilled to give the title compound (0.55 g, 61%), b.p. 90-100~C, 1.3 mm/Hg; 'H NMR
(CDCl3) ~ d 9.02 (s, lH), 2.74 (s, 3H).
Example 12
102-CHLORO-4-PHENYLPYRIMlDlNE-5-CARBONYL CHLORIDE
The compound was pl~ .ed as described above in Example 11, but
employing 2-chloro-4-phenyl~yli-I-idine-5-carboxylic acid (3.8 g, 14 mmol), resulting
in a yield of 53%; m.p. 42~C.
Example 13
2-CHLOROPYRIMIDINE-5-CARBONYLCHLORIDE
The compound was prepared as described in the literature (see Arukwe,
J. Und*eim, K Acta ChemicaScand. 1~40:764, 1986).
Example 14
ETHYL ETHOXYMETHYLENE-4,4,4-TRIFLUOROACETOACETATE
A solution of ethyl 4,4,4-trifluoroacetoacetate (46 g, 0.25 mol) triethyl
orthoformate (74 g, 0.50 mol) and Ac2O (77 g, 0.75 mol) was heated at 120-140~C for
7 h The mixture was concentrated and distilled to give the title compound in a 98%
25 yield (58.6 g); b.p. 80-90~C, 1.5 mm/Hg.
WO 97/09325 CA 0 2 2 3 0 8 9 6 19 9 8 - O 3 - O 2 PCT/US96/14089
24
Example lS
- ETHYL 2-TRIFLuoRoMETHYL-4-HYDRoxYPyRlMlDlNE-s-cARsoxyLATE
A solution of diethyl ethoxymethylenemalonate (35.0 g, 162 mmol),
trifluoroacetamidine (18 g, 162 mmol) and NaOEt (11.0 g, 162 mmol) in EtOH (200
S mL) was heated at reflux for 6 h. The reaction mixture was concenlldl~d and HzO (48
mL) was added. The resulting solid was filtered, washed with Et2O (300 mL) and H2O
(200 rnL), and dried to give the title compound (21 g, 50% yield); m.p. >220~C (dec.);
'H NMR (DMSO-d6) ~ 8.38, 4.16 (q, 2H), 1.25 (q, 3H).
Example 16
2-TRIFLUROROMETHYL-4-CHLOROPYRIMIDINE-5-CARBONYL CHLORIDE
A solution of ethyl 2-trifluoromethyl-4-hydroxypyrimic1in~-5-
carboxylate (5.00 g, 19.4 mmol) and NaOH (0.93 g, 23.3 mmol) in H2O (20 mL) was
stirred at 60~C for 15 h. The reaction was acidified (concentrated HCl) and
15 concentrated until a solid began to form. The solid was filtered and dried to give 2-
trifluoromethyl-4-hydro~y~,ylhllidine-S-carboxylic acid (2.1 g, 53% yield); 'H NMR
(DMSO-d6) ~ 8.83 (s, lH).
A solution of 2-trifluoromethyl-4-hydroxypyrimidine-5-carboxylic acid
(2.0 g, 10.4 mmol), POC13 (32 g, 212 mmol) and SOCl2 (25 g, 212 mmol) was heated at
20 reflux for 4 days. The reaction was concentr~tt?~l and distilled (b.p. 90-95~C, 1.5
mm/Hg) to provide the title compound (2.1 g, 81% yield), 'H NMR (CDCI3) ~ 9.45 (s,
lH).
Example 17
2-CHLORO-4-PENTAFLUOROETHYLPYRIMIDINE-5-CARBONYL CHLORIDE
A solution of ethyl 2-hydroxy-4-pentafluoroethylpyrimi~line-5-
carboxylate (4.0 g, 13 mmol) and NaOH (1.60 g, 39 mmol) in EtOH (20 mL) and H2O
(45 mL) was heated at reflux for 1 h. The solution was cooled and acidified
(concentrated HCl). The resllltin~ solid was filtered and dried to provide 2-hydroxy-4-
.-
WO 97/09325 CA 02230896 l 998 - 03 - 02 PCT/US96/14089
pentafluoroethylpyrimidine-5-carboxylic acid (3.3 g, 98% yield), 'H NMR (DMSO-d6)
9.90 (bs, lH), 8.43 (s, lH).
A solution of 2-hydroxy-4-pentafluoroethylpyrimidine-5-carboxylic acid
(3.33 g, 12.9 mmol) in SOCl2 (27.7 g, 233 mmol) was heated at reflux for 0.5 h Then
5 POCl3 (35.6 g, 233 mmol) was added to the reaction ~ lulc; and heating c~ntinIled for
36 h. The reaction llli~L~ was then concelll.a~d and distilled (b.p. 80-85~C, 1
mm/Hg) to give the title compound (1.2 g, 35% yield); 'H NMR (DMSO-d6) o 9.18 (s,
lH).
Example 18
ETHYL 4-HYDRAZINO-2-TRIFLUOROMETHYLPYRIMIDINE-5-CARBOXYLATE
A solution of ethyl 4-chloro-2-trifluoromethylpyrimidine-5-carboxylate
(0.20 g, 0.79 mmol), hydrazine (0.18 g, 6.0 mmol) and THF was stirred for 1 h at room
temperature. The solution was filtered and dried to give the title compound in a 96%
15 yield; 'H NMR (CDC13) o 9.26 (bs, lH), 8.90 (s, lH), 4.40 (q, 2H), 4.24 (bs, 2H), 1.41
(t, 3H).
Example l9
ETHYL 2-HYDRAZINO-4-TRIFLUOROMETHYLPYRIMIDINE-5-CARBOXYLATE
The title compound was prepared as described in Example 18, but
employing ethyl-2-chloro-4-trifluorom~lhyl~ylilllidine-5-carboxylate (0.20 g, 0.79
mmol), rçsnltin~ in a yield of 91% (0.18 g); m.p. 89-90~C.
Example 20
ETHYL 2-[N-(1~AMINOCITRACONAMIDO)]-4-TRIFLUOROMETHYL
PYRIMIDINE-5 -CARBOXYLATE
A solution of ethyl 2-hydrazino-4-trifluoromethylpyrimidine-5-
carboxylate (0.18 g, 0.72 mmol) and citraconic anhydride (0.08 ml, 0.94 mmol) in
CHCl3 (10ml) was refluxed for 0.5 h. The solution was cooled, concentrated and
WO 97/09325 CA 02230896 1998 - 03 - 02 PCT/US96~14089
chromatographed (SiO2, he~n~s/EtOAc) to give the title compound (0.10 g, 39%
- yield); IH NMR (CDCl3) ~ 9.94 (s, IH), 7.72 (s, lH), 6.53 (s, lH), 4.41 (q, 2H), 2.19 (s,
3H), 1.36 (t, 3H).
ExamPle 21
ETHYL 4- [N-(1 '-AMINOCITRACONAMIDO)]-2-TRIFLUOROMETHYL-
PYRIMIDINE-5-CARBOXYLATE
The title compound was prepared as described in Example 20, but
employing ethyl 4-hy~ lo-2-trifluorom~lllylpylilllidine-5-carboxylate (0.19 g, 0.76
mmol), resulting in an 80% yield (0.21 g); m.p. 85-86~C.
Example 22
ETHYL 2-[N-( 1 '-AMINOPHTHALIMIDO)]-4-TRIFLUOROMETHYL-
PYRIMIDINE-5 -CARBOXYLATE
A solution of ethyl 2-chloro-4-trifluoromethyl-5-pyrimi-line ester (0.25
g, 1.0 mmol), N-aminophth~limi~le (0.17 g, 1.0 mmol) and pyridine (0.09 ml, 1.0 mmol)
in THF was heated at 60~C for 5 h and then conc~ l The residue was
chromatographed (SiO2, hexanes/EtOAc, 1:1) to give the title compound (0.07 g, 17%
yield); m.p. 46-48 ~C.
Example 23
5-ACETYL-2-[N-(1 '-AMINOCITRACONAMIDO)]-
4-TRIFLUOROMETHYLPYRIMIDINE
To a solution of 2-chloro-4-trifluoromethylpyrimidine-5-carbonyl
chloride (0.50 g, 2.0 mmol) in THF at -78~C under N2 was added MeMgBr (0.75 ml,
2.3 mmol). The reaction was stirred 0.75 h, quenched with H2O (lml) and diluted with
EtOAc (30 ml). The organic layer was washed with H2O, brine and then dried over
MgSO4. The residue was chromatographed (SiO2, he~n~/EtOAc, 2:1) to provide the
5-acetyl-2-chloro-4-trifluorom~ ylpy~ idine (0.20 g) in 43% yield. The title
CA 02230896 1998-03-02
W O 97/0932S PCTAUS96/14089
compound was then prepared as described for ethyl 2-[N-(l'-aminocitraconamide)]-4-
trifluoromethyl~y~ idine-S-carboxylate of Example 20, resulting in a 29% yield (0.08
g); m.p. 61-62~C.
Example 24
ETHYL UREIDOMETHYLENE PROPIONOYLACETATE
The title compound was ~ ,p~,d as described in Example 3, but
employing ethyl propionylacetate (5.15 g, 35.7 mmol), resulting in a yield 43%
(3.29 g); m.p. 148-150~C.
Example 25
ETHYL UREIDOMETHYLENE BUTYRYLACETATE
The title compound was prepared as described in Example 3, but
employing ethyl butyrylacetate (25 g, 158 mmol), resllltin~ in a yield of 47% (17g);
15 m.p. 145- 147~C.
Example 26
ETHYL UREIDOMETHYLENE 2-THIENYLACETATE
The title compound was ~Icl~ed as described in Example 3, but
20 employing ethyl 2-thienoylacetate (7.22 g, 36.4 mrnol), resulting in a yield of 41% (4.0
g); m.p. 149-150~C.
Example 27
ETHYL 2-HYDROXY-4-ETHYLPYRIMIDINE-5-CARBOXYLATE
The title compound was prepared as described in Example 5, but
employing ethyl ureidomethylene ethanoylacetate (2.5 g, 11.7 mmol), resulting in a
- yield of 52% (1.2 g); ~H NMR (DMSO-d6) ~ 8.05 (d, lH), 4.24 (q, 2H), 2.87 (m, 2H),
1.29 (t, 3H).
WO 97/09325 CA 02230896 1998 - 03 - 02 PCT/US96/14089
28
Example 28
ETHYL 2-HYDRoxy 4-PROPYLPYRIMIDINE-5-CARBOXYLATE
The title compound was prepared as described in Example 5, but
employing ethyl ureidomethylene propionoylacetate (10.96 g, 48 mmol), resulting in a
5 yield of 73% (7.3 g); ~H NMR (CDCl3) ~ 8.2 (s, lH), 4.35 (q, 2H), 3.0 (t, 2H), 1.75
(m, 2H), 1.37 (t, 3H), 1.0 (t, 3H).
Example 29
ETHYL 2-HYDROXY-4-(2'-THIENYL)PYRIMIDINE-5-CARBOXYLATE
The title compound was prepared as described in Example 5, but
employing ethyl ureidomethylene 2-thiophenoylacetate (4.0 g, 14.9 mmol), resulting in
a yield of 79% (2.9 g); m.p. 144-146~C.
Example 30
ETHYL 2-CHLORO-4-ETHYLPYRIMIDINE-5-CARBOXYLATE
The title compound was pl~a.ed as described in Example 7, but
employing ethyl 2-hydroxy-4-~Lhyl~yIi-l-idine-5-carboxylate (1.2 g, 6.1 mmol),
rtoslIltin~ in a yield of 77% (1.0 g); GC/MS calcd for C9HIIN202Cl (M+) 214, found 214.
Example 31
ETHYL 2-CHLORO-4-PROPYLPYRIMIDINE-S-CARBOXYLATE
The title compound was prepared as described in Example 7, but
employing ethyl 2-hydroxy-4-propylpyrimidine-5-carboxylate (1.05 g, 5 mmol),
resulting in a yield of 79% (0.9 g); GC/MS calcd for C,oH,3N202CI (M+) 228, found
25 228.
CA 02230896 1998-03-02
W O 97/09325 PCT~US96/14089
29
Example 32
ETHYL 2-CHLORO-4-(2'-THIENYL)PYRIMIDINE-S-CARBOXYLATE
The title compound was ~l~paled as described in Example 7, but
employing ethyl 2-hydroxy-4-(2'-thienyl)pyrimidine-5-carboxylate (1.0 g, 4.0 mmol),
S resulting in a yield of 19% (0.2g); GC/MS calcd for C"H9N202SCl (M+) 268, found
268.
Example 33
ETHYL 2-HYDRAZINO-4-ETHYLPYRIMIDINE-S-CARBOXYLATE
The title compound was prepared as described in Example 18, but
employing ethyl 2-chloro-4-~;thyl~ylilllidine-5-carboxylate (1 g, 4.7 mmol), resulting in
a yield of 41 % (0.4 g); GC/MS calcd for CgHI4N402 (M+) 210, found 210.
Example 34
ETHYL 2-HYDRAZINO-4-PROPYLPYRIMIDINE-S-CARBOXYLATE
The title compound was prepared as described in Example 18, but
employing ethyl 2-chloro-4-propylpyrimi~line-5-carboxylate (0.9 g, 3.9 mmol),
resulting in a yield of 97% (0.85 g); GC/MS calcd for C~oHl6N4o2 (M+) 224, found 224.
Example 35
ETHYL 2-HYDRAZINO-4-(2'-THIENYL)PYRIMIDINE-5-CARBOXYLATE
The title compound was prepared as described in Example 18, but
employing ethyl 2-chloro-4-(2'-thienyl)pyrimidine-5-carboxylate (0.2 g, 0.7 mmol),
resulting in a yield of 92% (0.17 g); GC/MS calcd for CI,H,2N402S (M+) 264, found
25 264.
CA 02230896 1998-03-02 PCT~US96/14089
W O 97/09325
Example 36
ETHYL 2-[N-(1 '-AMINOCITRACONAMIDO)]-4-ETHYLPYRIMIDINE-5-CARBOXYLATE
The title compound was p~ ucd as described in Example 20, but
employing ethyl 2-hydrazino-4-~ ylpy,ill~idine-5-carboxylate (0.4 g, 1.9 mmol),
5 resllltin~ in a yield of 32% (0.13 g), 'H NMR (CDCl3) ~ 8.84 (s, lH), 6.51 (s, lH), 5.94
(s, lH), 4.32 (m, 2H),3.05 (q, 2H), 2.18 (s,3H), 1.35 (m, 6H).
Example 37
ETHYL 2-[N-(1~-AMINOCITRACONAMIDO)]-4-PROPYLPYRIMIDINE-5-CARBOXYLATE
The title compound was pl~J~ed as described in Example 20, but
employing ethyl 2-hydrazino-4-propylpy~ ine-5-carboxylate (0.77 g, 3.4 mmol),
rçslllting in a yield of 73% (0.7 g); m.p. 103-105~C.
Example 38
ETHYL 2-[N-(1 '-AMINOCITRACONAMIDO)]-4-(2'-THIENYL)PYRIMIDINE-5-CARBOXYLATE
The title compound was prepared as described in Example 20, but
employing ethyl 2-hydrazino-4-(2'-thienyl)pynmiclin~-5-carboxylate (0.17 g, 0.64mmol), resulting in a yield of 55% (0.127 g), m.p. 123-126~C
Exam~le 39
5-AMIDO-2-[N-(l '-AMINOCITRACONAMIDO)]-
4-TRIFLUOROMETHYLPYRIMIDINE
To a solution of 2-chloro-4-trifluoromethylpyrimi(1ine-5-carbonyl
chloride (0.50 g, 2.0 mmol) in THF at 0~C under N2 was added 2M NH3/MeOH (1.0 ml,
25 2.0 mrnol). The reaction was stirred 5 minlltçs, diluted with EtOAc (10 ml), filtered and
concentrated. Chromatography (SiO2, hexanes/EtOAc, 4:1) provided the 5-amido-2-
chloro-4-trifluoromethyl~yl;",i~lin~ (0.20 g) in 44% yield. The title compound was then
~,G~ed as described for ethyl 2-[N-(l'-aminocitraconamide)]-4-
trifluoromethylpyrimidine-5-carboxylate, resulting in a 15% yield (0.04 g), 'H NMR
CA 02230896 1998-03-02
W O 97/09325 PCT~US9~14089
(CDCI3) ~ 8.69 (s, lH), 8.30 (s, 1H), 6.52 (s, lH), 6.51 (s, lH), 6.28 (s, lH), 2.19 (s,
3H).
ExamPle 40
ETHYL 2-[N-(1 '-AMINO-3'-PHENYLMALEIMIDO)]-4-TRIFLUOROMETHYL-
PYRIMIDINE-5-CARBOXYLATE
The title compound was ~ ,d as described for ethyl 2-[N-(l'-
aminocitraconamide)]-4-trifluoromethylpyrimi~line-5-carboxylate but employing 3-phenylmaleic anhydride (0.21 g, 1.2 mmol), res-llting in a 74% yield (0.18 g), m.p.
10 52-53~C.
Example 41
ETHYL 2-[N-(1 '-AMINO-3 ',4'-DIMETHYLMALEIMIDO)]-4-TRIFLUOROMETHYL-
PYRIMIDINE-5-CARBOXYLATE
The title compound was prepared as described for ethyl 2-tN-(l'-
aminocitraconamido)]-4-trifluoromethylpyrimi~line-5-carboxylate but employing 3,4-
dimethylmaleic anhydride (0.08 g, 0.63 mmol), resulting in a 47% yield (0.10 g), m.p.
110-111~C.
Example 42
ETHYL 2- [N-( l '-AMINOCITRACONAMIDO)-N-METHYL]-4-TRIFLUOROMETHYL-
PYRIMIDINE-5 -CARBOXYLATE
To a solution of ethyl 2-[N-(l'-aminocitraconamido)]-4-trifluoromethyl-
pyrimidine-5-carboxylate (0.10 g, 0.29 mmol) in THF at 0~C under nitrogen was added
25 NaH (0.01 g, 0.44 mmol). After 5 minlltes, MeI (0.10 ml, 1.6 mmol) was added and the
reaction was allowed to warm to room te~ ldLu~e. After stirring 0.5 h at room
temperature, the reaction was diluted with EtOAc (15 mL), washed with H2O and brine.
The organic layer was dried over MgSO4, filtered and concentrated. The residue was
chromatographed (SiO2, he~ne~/EtOAc 8:1) to give the title compound (0.05 g, 60%
CA 02230896 1998-03-02 PCT~US96/14089
W O 97/09325
yield); 'H NMR (CDCl3) ~ 9.05 (s, IH), 6.48 (s, lH), 4.37 (q, 2H), 3.61 (s, 3H), 2.17 (s,
- 3H), 1.35 (t, 3H).
ExamPle 43
ETHYL 4-[N-(1 '-AMIN0-3'-PHENYLMALEIMIDO)]-2-TRIFLUOROMETHYL-
PYRIMIDINE-S -CARBOXYLATE
The title compound was prepared as described in Example 20, but
employing a solution of ethyl 4-hydrazino-2-trifluorom~ yl~y~il,lidine-5-carboxylate
(0.09 g, 0.36 mmol) and 3-phenylmaleic anhydride (0.13 g, 0.72 mlnol) r~sllltin~ in a
69% yield (0.10 g); m.p. 179-180~C.
ExamPle 44
ETHYL 4-[N-(1 '-AMINO-3 ', 4'-DIMETHYLMALEIMIDO)]-2-TRIFLUOROMETHYL-
PYRIMIDINE-5 -CARBOXYLATE
The title compound was prepared as described in Exarnple 20, but
employing a solution of ethyl 4-hydrazino-2-trifluoromethylpyrimidine-5-carboxylate
(0.09 g, 0.36 mmol) and 3,4-dimethylmaleic anhydride (0.09 g, 0.72 mmol) rçsnlting in
a 89% yield (0.12 g); m.p. 116-117~C.
ExamPle 45
ETHYL 2-HYDRAZINO-4-METHYLPYRIMIDINE-5-CARBOXYLATE
The title compound was prepared as described in Example 18, but
employing a solution of ethyl 2-chloro-4-methylpyrimidine-5-carboxylate (0.08 g, 0.39
mmol) and hydrazine (0.06 g, 2.0 rnmol) in THF (7.8 mL) resulting in a 93% yield(0.07 g); ~H NMR (CDC13) ~ 8.86 (s, lH), 6.64 (bs, lH), 4.33 (q, 2H), 2.70 (s, 3H), 1.38
(t, 3H).
CA 02230896 1998-03-02
W O 97/09325 PCT~US96/14089
Example 46
ETHYL 2- [N-(1 '-AMINOCITRACONAMIDO)] -4-METHYLPYRIMIDINE-
S-CARBOXYLATE
The title compound was ~rcp~ed as described in Example 20, but
5 employing a solution of ethyl 2-hyd~ o-4-methylpyrimidine-5-carboxylate (0.07 g,
0.36 mmol) and citraconic anhydride (0.08 g, 0.72 mmol) resulting in a 52% yield
(0.06 g ), m.p. 49-50~C.
Example 47
10ETHYL 2-HYDRAZlNO-4-PENTAFLUOROETHYLPYRlMlDlNE-
S-CARBOXYLATE
The title compound was ~r~ed as described in Example 18, but
employing a solution of ethyl 2-chloro-4-pentafluoroethylpyrimidine-5-carboxylate
(0.30 g, 0.99 Inmol) and hydrazine (0.16 g, 5.0 mmol) in THF (20 mL) resulting in a
15 95% yield (0.28 g); IH NMR (CDCI3) ~ 8.86 (bs, lH), 7.12 (bs, lH), 4.37 (q, 2H), 1.72
(bs, 2H), 1.38 (t, 3H).
ExaInple 48
ETHYL 2-rN-(1'-AMINOClTRACONAMlDO)]-4-PENTAFLUOROETHYLPYRlMIDlNE-
S-CARBOXYLATE
The title compound was ~lcp~ed as described in Exarnple 20, but
employing a solution of et~1yl 2-hydrazino-4-pent~fllIoroethylpyrimidine-S-carboxylate
(0.28 g, 0.93 mmol) and citraconic anhydride (0.13 g, 1.1 mmol) resulting in a 68%
yield (0.25 g), m.p. 73-74~C.
Example 49
ETHYL 2-HYDRAZINO-4-PHENYLPYRIMIDINE-S-CARBOXYLATE
The title compound was ~lc~aled as described in Example 18, but
employing a solution of ethyl 2-chloro-4-phenylpyrimicline-5-carboxylate (0.09 g, 0.34
CA 02230896 1998-03-02
W O 97/09325 PCTAUS96/14089
mmol) and hyd.d~ e (0.06 g, 1.7 mmol) in THF resulting in a 91% yield (0.08 g); m.p.
74-75~C.
Example 50
ETHYL 2-[N-(1~-AMINOCITRACONAMIDO)]-4-PHENYLPYRIMIDINE-5-CARBOXYLATE
The title compound was ~rG~al~d as described in Example 20, but
employing a solution of ethyl 2-hydrazino-4-phGnyl~ylilllidine-S-carboxylate (0.08 g,
0.31 mmol) and citraconic anhydride (0.04 g, 0.37 mmol) resulting in a 64% yield (0.07
g); m.p. 165- 167~C.
Examl~le 51
ETHYL 2-HYDRAZINO-4-BENZYLPYRIMIDINE-S-CARBOXYLATE
The title compound was plGpal~d as described in Example 18, but
employing a solution of ethyl 2-chloro-4-benzyll~ylilllidine-5-carboxylate (0.34 g, 1.2
15 mmol) and hydla~lle (0.2 g, 6.1 mmol) in THF r~s-lltin~ in a 99% yield (0.33 g, oil);
GC/MS, 272(M+).
Examl~le 52
ETHYL 2-[N-(1 '-AMINOCITRACONAMIDO)]-4-BENZYLPYRIMIDINE-5-CARBOXYLATE
The title compound was prepared as described in Example 20, but
employing a solution of ethyl 2-hy~llo-4-ben;~yl~y~ icline-5-carboxylate (0.34 g,
1.2 mmol) and citraconic anhydride (0.22 g, 2.0 mmol) resulting in a 37% yield (0.15 g)
of the title compound; m.p.34-36~C.
Exarnple 53
METHYL 2-HYDROXY-4-(3'-PYRIDYL)PYRIMIDINE-5-CARBOXYLATE
The title compound was plGpa ~d as described in Example 5, but
employing a solution of methyl ureidomethylene nicotinoyl acetate (8.4 g, 34 mmol) Na
CA 02230896 1998-03-02
W O 97/09325 PCTAUS96/14D89
(1.0 g, 44 mmol) in EtOH (200 mL) resulting in a 52% yield (4.4 g), lH NMR (DMSO-
d6) ~ 8.59 (s, lH), 8.51 (m, 2H), 7.72 (m, lH), 7.36 (m, lH), 3.51 (s, 3H).
Example 54
METHYL 2-CHLORO-4-(3'-PYRIDYL)PYRIMIDINE-5-CARBOXYLATE
The title compound was prepared as described in Example 7, but
employing a solution of methyl 2-hydroxy-4-(3'-pyridyl)pyrimi-linç-5-carboxylate (4.42
g, 18 mmol) and POC13 (53.6 g, 350 mmol) resulting in a 25% yield (1.1 g); lH NMR
(DMSO-d6) ~ 9.11 (s, lH), 8.77 (m, 2H), 8.03 (m, lH), 7.45 (m, lH), 3.85 (s, 3H).
ExamPle 55
METHYL 2-HYDRAZINO-4-(3'-PYRIDYL)PYRIMIDINE-5-CARBOXYLATE
The title compound was prepared as described in Example 18, but
employing a solution of methyl 2-chloro-4-(3'-pyridyl)pyrimiciin~-5-carboxylate (0.10
15 g, 0.42 mmol) and l,ydld~lle (67 mg, 2.1 mmol) rçsnl~ing in a 97% yield (0.10 g); IH
NMR (DMSO-d6) ~ 9.09 (s, lH), 8.76 (d, lH), 8.69 (d, lH), 8.35 (bs, lH), 7.89 (d,
lH), 7.42 (m, lH), 3.76 (s, 2H), 2.19(bs, 2H).
Exarnple 56
METHYL 2-[N-(1 '-AMINOCITRACONAMIDO)]-4-(3 '-PYRIDYL)-
PYRIMIDINE-5 -CARBOXYLATE
The title compound was prepared as described in Example 20, but
employing a solution of methyl 2-hydrazino-4-(3'-pyridyl)pyrimidine-5-carboxylate
(0.1 g, 0.39 mmol) and citraconic anhydride (0.04 g, 0.39 mmol) r~snltin~ in a 42%
25 yield (0.06 g); m.p. 93-94~C.
WO 97/09325 CA 02230896 1998 - 03 - 02 PCT/US96/14089
36
Exam~le 57
ETHYL 2-HYDROXY-4-BENZYLPYRIMIDINE-5-CARBOXYLATE
A solution of benz~ cet- ~cetate (7.0 g, 34 mmol) and N,N-
dimethylforrn~nni~le dimethyl acetal (4.0 g, 34 mmol) was stirred at room temperature
5 for 0.25 h. The solution was concçntr~te~l and treated with urea (2.2 g, 37 mmol) and
Na (1.2 g, 51 mmol) in EtOH (100 mL). The ~ LulG was heated at reflux for 18 h,
concentrated, acidified and purified by chromatography (SiO2) to yield the desired
product (2.6 g, 30%); m.p. 57-61~C.
Example 58
ETHYL 2-CHLORO-4-BENZYLPYRIMIDINE-5-CARBOXYLATE
The title compound was I~lGlJ~Gd as described in Example 7, but
employing a solution of ethyl 2-hydroxy-4-bG-~yl~y~ ne-s-carboxylate (1.1 g, 4.3
mmol) and POC13 (16 g, 107 mmol) re~ ng in a 37% yield (0.44 g); ~H NMR
15 (DMSO-d6) 8 9.00 (S, lH), 7.6-7.4 (m, SH), 4.55 (s, 2H), 4.25 (q, 2H), 1.14 (t, 3H).
Example 59
DIETHYL UREIDOMETHYLENE OXALACETATE
A solution of diethyl ox~l~cet~te (5.7 g, 30.4 mmol), triethyl
20 orthoformate (8.8 g, 30 rnmol) and urea (2.0 g, 30 rnmol) was heated at reflux for 1.5 h
The product was filtered, washed with water and ether to give (3.5 g, 45%) of the title
compound; ~H NMR (DMSO-d6) ~ 11.29 and 10.81 (dd, lH), 8.6-7.4 (m, 3H), 4.2 (m,
4H), 1.24 (m, 6H).
25Exarnple 60
METHYL UREIDOMETHYLENE NICOTINOYL ACETATE
The title compound was prepared as described in Example 3, but
employing a solution of methyl nicotinoyl acetate (10 g, 56 mmol), triethyl
CA 02230896 1998-03-02
W O 97/09325 PCT~US96/14089
orthoformate (8.3 g, 56 mmol) and urea (3.4 g, 56 mmol) resIlItin~ in a 60% yield (8.4
g); 'H NMR (DMSO-d6) ~ 10.85 and 10.36 (dd, lH), 8.8-7.2 (m, 7H), 3.58 (d, 3H).
Example 61
5DIETHYL 2-HYDROXYPYRIMIDINE-4,5-DICARBOXYLATE
A solution of diethyl ureidomethylene oxzlI~ret~t~ (18.3 g, 71 mmol) in
xylene (95 mL) was heated at reflux for 18 h. The reaction was cooled and the resulting
solid was filtered. The solid was recryst~lli7~1 from EtOAc to give the desired
compound (8.2 g, 48%); IH NMR (DMSO-d6) ~ 8.61 (s, lH), 4.25 (m, 4H), 1.27 (m,
1 0 6H).
ExamPle 62
DIETHYL 2-CHLOROPYRIMIDINE-4,5-DICARBOXYLATE
The title compound was ple~ed as described in Example 7, but
15 employing a solution of diethyl 2-hydroxypyrimidine-4,5-dicarboxylate (1.0 g, 4.2
mmol) and POCl3 (7.7 g, 50 rnmol) resulting in a 30% yield (0.32 g); 'H NMR (CDCI3)
9.09 (d, lH), 4.35 (m, 4H~, 1.30 (m, 6H).
Example 63
20DIETHYL 2-HYDRAZINOPYRIMIDINE-4,5-DICARBOXYLATE
The title compound was L)~ aled as described in Example 18, but
employing a solution of diethyl 2-chloroypyrimidine-4,5-dicarboxylate (0.21 g, 0.83
mmol) and hydrazine (0.13 g, 4.2 mmol) in THF (10 mL) resulting in 96% yield (0.20
g); 'H NMR (CDC13) ~ 8.93 (bs, lH), 8.34 (bs, lH), 4.40 (m, 4H), 4.18 (bs, 2H), 1.35
25 (m, 6H).
CA 02230896 1998-03-02
W O 97/09325 PCTAUS96~1~089
38
Exam~le 64
DIETHYL 2-[N-(1I-AMINOCITRACONAMIDO)]
PYRIMIDINE-4,5-DICARBOXYLATE
The title compound was ~l~pd,~d as described in Example 20, but
5 employing a solution diethyl 2-hydrazinopyrimidine-4,5-dicarboxylate (0.20 g, 0.79
mmol) and citraconic anhydride (0.11 g, 0.95 mmol) r~sllltinp~ in a 77% yield (0.21 g);
NMR (CDCl3) ~ 8.94 (s, lH), 8.88 (bs, lH), 6.53 (s, lH), 4.35 (m, 4H), 2.16 (s, 3H),
1.33 (m, 6H).
Example 65
E~THYL 2-[N-(1 '-AMINO-3'-METHYLSUCCINIMIDO)]-4-
TRIFLUOROMETHYLPYRIMIDINE-5 -CARBOXYLATE
To a solution of ethyl 2-[N-(l'-aminocitraconamido)]-4-
15 trifluoromeLhylpy.;~--iciine-5-carboxylate (0.05 g, 0.15 mmol) in EtOH (20 ml) was
added Pd/C (0.1 g). The reaction was stirred under an atmosphere of H2 overni~ht
The mixture was filtered over celite and concentrated to give the title compound (0.047
g, 94% yield); ~H NMR (CDCl3) ~ 9.00 (s, lH), 8.39 (s, lH), 4.40 (q, 2H), 3.10 (m, 2H),
2.44 (m, lH), 1.38 (m, 3H).
Example 66
METHYL 2- rN-( l '-AMINO-3 '-METHYLSUCCINIMIDO)] -4-
TRIFLUOROMETHYLPYRIMIDINE-5 -CARBOXYLATE
The title compound was prepared as described in Example 65, but
employing methyl 2-[N-(l'-aminocitracon~midQ)]-4-trifluoromethylpyrimi~line-S-
carboxylate (0.02 g, 0.06 mmol), resulting in a yield of 94% (0.019 g), m.p. 66-68~C.
CA 02230896 1998-03-02
W O 97t0932~ PCTAUS96/14089
Example 67
ETHYL 2-[N-(1 '-AMINOCITRACONAMIDO)]-4-
[2'-(5"-cHLoRoTHIENYL]PYRIMIDINE-5-cARsoxyLATE
A solution of 5-chlorothiophene-2-carboxylic acid (16.3 g, 0.1 mole) and
5 carbonyl lliimi~l~7c)le (27.8 g, 0.17 mol) in THF (200 mL) was stirred 30 mimltec.
Bis(ethyl malonate)m~gne~illm salt (14.4 g, 0.051 mol) was added and the mixture was
refluxed for 7 hours, cooled to RT and conct;llLldled EtOAc (200 mL) and 6N HCl (50
mL) were added and the organic layer was separated, dried (MgS04), and concentrated.
The ethyl 5-chloro-2-thiophenoylacetate (23 g, 0.1 mol) and N,N-dimethylf~lrmzlmicle
10 dimethyl acetal (12 g, 0.1 mol) were stirred for 1 hour and concentrated. To this was
added, a solution of NaOEt previously prep~ed using sodiurn in EtOH [Na (2.3 g, 0.1
mol) dissolved in 500 mL EtOH] and S-methyl isothiouronium sulfate (13.9 g, 0.05mol) that was allowed to stir for 30 min-~t~s. The mixture was refluxed for 4 hours,
cooled to RT, coneçntr~tPtl, dissolved in EtOAC (500 mL), washed with 4N HCl (2 x
15 50 mL), dried (MgS04) and the solvent replaced with CH2Cl2 (300 mL). The solution
was cooled to 0~C, mCPBA (51 g, 0.3 mol) was slowly added, and the solution was
allowed to reach RT. After stirring for 2 hours, the solution was quenched by slowly
pouring into a cold satd. NaHSO3 solution (100 mL). The organic layer was then
washed with satd. NaHCO3 (100 mL), dried (MgSO4), and concentrated. From this, the
20 title compound was prepared as described in Example 20, but employing the ethyl 2-
hydrazino-4-[2'-(5'-chlorothiophene)]pyrimi(line-5-carboxylate (3.0 g, 0.017 mol)
~prepared according to the procedure of Example 19, but employing ethyl 2-mesyl-4-
[2'-(5'-chlorothiophene)]pyrimidine-5-carboxylate described above}, resulting in a yield
of 38% (1.3 g); m.p. 137-138~C.
WO 97/09325 CA 02230896 1998 - 03 - 02 PCT/US96/14089
ExamPle 68
ETHYL 2-[N-(1 '-AMINOCITRACONAMIDO)-N-METHYL]-4-
[2'-(5"-CHLOROTHIENYL]PYRIMIDINE-5-CARBOXYLATE
The title compound was ~ ed as described in Example 67 from 4-[2'-
5 (5'-chlorothiophene)-2-hydrazino-5-pyrimidine carboxylate (0.5 g, 1.7 mmol), but
employing methyl hydrazine (0.35 g, 7.5 mmol) where hydrazine was used, resulting in
a yield of 48% (0.32 g); GC/:MS calcd for Cl,HI5N404SCl (M+) 406, found 406.
Example 69
ETHYL 2-[N-(1'-AMINO-4~-METHYLPHTHALIMIDO)]-4
TRIFLUOROMETHYLPYRIMIDINE-5 -CARBOXYLATE
The title compound was prepared as described in Example 20,
employing ethyl 2-hy&~illo-4-trifluoromethylpyrimidine-5-carboxylate (0.20 g, 0.8
15 mmol) and 4-methylrh~h~lic anhydride (0.26 g, 1.6 mmol) where citraconic anhydride
was used, resulting in a yield of 29% (0.09 g); m.p. 50-51 ~C.
Example 70
20ETHYL 2-[N-(1 '-AMINO-4',5'-DICHLOROPHTHALIMIDO)]-4-
TRIFLUOROMETHYLPYRIMIDINE-5 -CARBOXYLATE
The title compound was prepared as described in Example 20,
employing ethyl 2-hydrazino-4-trifluoromethylpyrimidine-5-carboxylate (0.20 g, 0.8
mmol) and 4,5-dichlorophthalic anhydride (0.35 g, 1.6 mmol) where citraconic
25 anhydride was used, resulting in a yield of 61 % (0.22 g); m.p. 142-144~C.
CA 02230896 1998-03-02
W O 97/09325 PCT~US96/14089
41
Example 71
ETHYL 2-[N-(1'-CITRACONAMIDO)]-PYRIMIDINE-5-CARBOXYLATE
The title compound was ~lepaled by (a) treating 2-chl~,ropylllllidine-5-
carbonylchloride (Example 13, 110 mg, 0.62 mmol) with ethanol (200 mg, 4.4 rnmol in
5 1 mL of ethyl acetate) to provide 60% of ethyl 2-chloropyrimidine-S-carboxylate, (b)
reaction of ethyl 2-chloropyrimidine-5-carboxylate (70 mg, 0.37 mmol) with hydld~ e
(100 mg, 3 mmol) to afford 44% of ethyl 4-hydrazinopyrimidine-S-carboxylate as in
Example 19, (c) reaction of ethyl 4-hydrazinopyrimidine-S-carboxylate (0.36 g, 2.0
mmol) with citraconic anhydride (0.34 g, 3.0 mmol) in analogy to Example 21 to afford
10% (0.05 g) of the title compound; m.p. 95-98~C
Example 72
5-BENZOYL-4-ETHYL-2-METHYLTHIOPYRIMIDINE
l S The title compound was prepared by (a) reaction of ethyl
propionylacetate (19.4 g, 0.13 mol) and N,N-dimethylformamide dimethyl acetal (16.0
g, 0.13 mol) in analogy to Example 67, (b) reaction with NaOEt (0.18 mol) and S-methyl isothoulolliulll sulfate (18.7 g, 0.067 mol) also described for Example 67, (c)
hydrolysis of the ethyl 4-ethyl-2-methylthiopyrimidine-S-carboxylate (18.0 g, 0.08
20 mmol) with NaOH (12.7 g, 0.32 mol) in H2O (200 mL) as described in Example 67, (d)
reaction of the 4-ethyl-2-methylthiopyrimidine-S-carboxylic acid (0.50 g, 2.5 mmol)
and oxalyl chloride (0.32 g, 2.5 mmol) as described in Example 11, and (e) reaction of
the 4-ethyl-2-methylthiopylimidine-5-carbonyl chloride (0.50 g, 2.3 mmol) and phenyl
m~gn~ium bromide as described for Example 23, resulting in a yield of 22% (0.14 g)
25 from the 4-ethyl-2-methylthiopyrimidine-S-carboxylic acid; ); 'H NMR (CDCl3) ~ 9.18
(s, lH), 7.80 (d, 2H), 7.62 (t, lH), 7.50 (t, 2H), 2.81 (q, 2H), 2.61 (s, 3H), 1.22 (t, 3H).
CA 02230896 1998-03-02
W O 97/09325 PCT~US96/14089
42
Exam~le 73
ETHYL 2-[N-(1 -AMINOCITRACONAMIDO)-N-METHYL]-4-
ETHYLPYRIMIDINE-5 -CARBOXYLATE
The title compound was prepared as described in Example 42, but
S employing ethyl 2-[N-(l'-aminocitraconamido)]-4-ethylpyrimi~line-5-carboxylate (0.05
g, 0.16 mmol), rçslllting in a yield of 76% (0.04g); 'H NMR (CDCl3) ~ 8.80 (d, lH),
6.46(s, lH),4.30(q,2H),3.58(s,3H),3.18(m,2H),2.16(s,3H), 1.33 (m, 6H).
Example 74
ETHYL 2-[N-ACETYL-N-(l'-AMINOCITRACONAMIDO)]4-
PROPYLPYRIMIDINE-5-CARBOXYLATE
Ethyl 2-[N-(l'-aminocitraconamido)]-4-propylpyrimi-line-5-carboxylate
(0.060 g, 0.19 mmole) and 1.5 mL of an acetic anhydride and pyridine (1:1 molar)
15 solution were stirred overnight. Concentration and chromatography (SiO2,
he~s3n~?s/EtOAc, 3 :2) provided the title compound (0.03 g, 44% yield); 'H NMR
(CDCl3) ~ 9.0 (s, lH), 6.5 (s, lH), 4.4 (q, 2H), 3.1 (t, 2H), 2.82 (s, 3H), 2.19 (s, 3H), 1.7
(m, 2H), 1.39 (t, 3H), 0.99 (t, 3H).
Example 75
ETHYL 2-[N-ACETYL-N-(1 -AMINOCITRACONAMIDO)]-4-
TRIFLUOROMETHYLPYRIMIDINE-5 -CARBOXYLATE
The title compound was prepared as described in Example 74, but
25 employing ethyl 2-[N-(1 '-aminocitraconamido)]-4-trifluoromethylpyrimidine-5-
carboxylate (0.14 g, 0.4 mmol), resulting in a yield of 67% (0.11 g); ' H NMR (CDC13)
9.14 (s, lH), 6.55 (s, lH), 4.45 (q, 2H), 2.83 (s, 3H), 2.20 ( s, 3H), 1.39 (t, 3H).
CA 02230896 1998-03-02
W O 97/09325 PCTAUS96/14089
43
Exarnple 76
METHYL 2-HYDRAZINO-4-PENTAFLUOROETHYLPYRIMIDINE-
5-CARBoXYLATE
The title compound was prepared according to the procedure of Example
5 47 but employing a solution of methyl 2-chloro-4-pentafluoroethylpyrimidine-5-
carboxylate (0.55 g, 1.9 mmol; itself prepared by a reaction of methanol with 2-chloro-
4-pentafluoro~ yl~ylilllidine-5-carbonyl chloride (see Fx~nple 17)) and hydrazine (0.3
g, 9.4 mmol), resnltin~ in a yield of 99% (0.54 g); ~HNMR (CDCl3) ~, 8.87 (s, lH), 7.09
(s, lH), 3.92 (s, 3H), 1.7 (s, 2H).
Example 77
METHYL 2-[N-(1 '-AMINOCITRACONAMIDO)]-4-
PENTAFLUOROETHYLPYRIMIDINE-5-CARBOXYLATE
The title compound was prepared according to the procedure of Exarnple
20 but employing 2-hydrazino-4-pentafluoroethylpyrimidine-5-carboxylate (0.54 g, 1.9
mrnol) and citraconic anhydride (0.25 g, 2.3 mmol), resulting in a yield of 98%
(0-71 g); m.p. 94-95~C.
Example 78
METHYL 2-[N-(1 '-AMINOCITRACONAMIDO)]-N-METHYL]-4-
PENTAFLUOROETHYLPYRIMIDINE-5 -CARBOXYLATE
The title compound was prepared according to the procedure of Example
25 73 but employing methyl 2-[N-(1 '-aminocitraconamido)]-4-pentafluoroethylpyrimidine-
5-carboxylate (0.05 g, 0.13 mmol), a base and methyl iodide (0.04 g, 0.26 mrnol);
~HNMR (CDC13) ~ 9.02 (s, lH), 8.79 (s, lH), 6.49 (m, lH), 3.92 (s, 3H), 3.59 (d, 3H),
2.16 (d, 3H).
WO 97/09325 CA 02230896 1998 - 03 - 02 PCT/US96/14089
Example 79
t-BUTYL 2-[N-(1 -AMINOCITRACONAMIDO)]-4-
TRIFLUOROMETHYLPYRIMIDINE-5 -CARBOXYLATE
The title compound was pl~,p~u~,d firom t-butyl 2-hydrazino-4-
trifluorometh~/lpylilli~dine-5-carboxylate (0.88 g, 3.2 mmol) (prepared as described for
Example 19, but employing t-butyl 2-chloro-4-trifluoromt;lhyl~y,illlidine-5-
carboxylate) as described in Example 20, resulting in a yield of 10% (0.12 g); IH NMR
(CDCl3) ~ 8.84 (s, lH), 7. 50 (s, lH), 6.53 (s, lH), 2.20 (s, 3H), 1.55 (s, 9H).
Example 80
METHYL 2-[N-(1~-AMINOCITRACONAMIDO)]-4-
TRIFLUOROMETHYLPYRIMIDINE-5-CARBOXYLATE
The title compound was prepared firom methyl 2-hydr~ino-4-
trifluoromethyl~ylilllidine-5-carboxylate (0.01 g, 0.42 mmol) (prepared as described for
Example 19, but employing methyl 2-chloro-4-trifluoromethylpyrimit1ine-5-
carboxylate) as described in Example 20, r~slllting in a yield of 69% (0.096 g); m.p.
118-120~C.
Example 81
METHYL 2- [N-(1 '-AMINOCITRACONAMIDO)-N-METHYL] -4-
TRIFLUOROMETHYLPYRIMIDINE-5 -CARBOXYLATE
The title compound was prepared firom methyl 2-[N-(l'-
aminocitraconamido)]-4-trifluoromethylpyrimidine-5-carboxylate (0.20 g, 6.1 mmol) as
described in Example 42, resulting in a yield of 10% (0.02 g); 'H NMR (CDCI3) ~ 9.05
(s, lH), 6.49 (s, lH), 3.91 (s, 3H), 3.62 (s, 3H), 2.18 (s, 3H).
CA 02230896 1998-03-02
W O 97/09325 PCT~US96/14089
Example 82
METHYL 2-HYDRAZINO-4-(2 -THIENYL)PYRIMIDINE-5-CARBOXYLATE
A solution of ethyl 2-hydroxy-4-(2'-thienyl)pyrimidine-5-carboxylate
(2.7 g, 10.8 mmol) and NaOH (1.3 g, 32.4 mmol) in H2O (25 mL) was stirred overnight
then acidified to pH<2 using concentrated HCl. The precipitate was collected, dried
under vacuum, and POCl3 (16.5 g, 108 mmol) was added. To this was added
diethylaniline (1.6 g, 10.8 rnmol), and the ~ e was refluxed for 30 mimlt~ The
solution was cooled, concentrated, poured over ice and extracted with ether (200 mL).
The 2-chloro-4-(2'-thienyl)pyrimidine-5-carboxylic acid was then dissolved in CH2Cl2
(30 mL), treated with oxalyl chloride (5.4 g, 42.7 mmol) and DMF (2 drops), and
allowed to stir overnight. The solution was then concentrated, dissolved in MeOH (10
mL) and concentrated after stirring 10 minutes. The title compound was prepared from
the crude methyl 2-chloro-4-(2'-thienyl)pyrimidine-5-carboxylate as described inExample 19, resulting in a overall yield of 6% (0.16 g); GC/MS calcd for CloHIoN4O2S
(M+) 250, found 250.
Exarn~le 83
METHYL 2-[N-(1'-AMINOCITRACONAMIDO)]-4-
(2'-THIENYL)pyRlMIDlNE-s-cARsoxyLATE
The title compound was ~le~ed from methyl 2-hydrazino-4-(2'-
thienyl)pyrimidine-5-carboxylate (0.16 g, 0.64 mmol) according to the procedure of
Example 20, resulting in a yield of 90% (0.20 g); m.p. 112-113~C.
WO 97/09325 CA 0 2 2 3 0 8 9 6 19 9 8 - O 3 - O 2 PCT/US96/14089
46
Exarn~le 84
ETHYL 2-[N-(1 -AMINocITRAcoNAMlDo-N-sENzYL)]-4-
TRIFLUOROMETHYLPYRIMIDINE-5 -cARsoxYLATE
The title compound was ~ ~ed from ethyl 2-(hydrazino-1'-benzyl)-4-
5 kifluoromethylpyrimidine-s-carboxylate (0.2 g, 0.78 mmol) (prepared according to the
procedure of E~xample 19 but employing benzylhydrazine) as described in Example 20,
resulting in a yield of 12% (0.05 g); 'H NMR (CDCl3) ~ 9.08 (d, lH), 7.3 (m, SH), 6.39
(s, lH), 5.21 (m, 2H), 4.37 (m, 2H), 2.09 (s, 3H), 1.37 (m, 3H).
Example 85
ETHYL 2-[N-(1'-AMINOCITRACONAMIDO-N-METHYL)]-4-
(2'-THIENYL)PYRIMIDINE-S -cARsoxYLATE
The title compound was ~lc~l,d from ethyl 2-[N-(l'-
lS aminocikaconamido)]-4-(2l-thienyl)pyrimidine-s-carboxylate (0.05 g, 0.14 mmol)
according to the procedure of Example 42, resulting in a yield of 19% (0.01 g); 'H NMR
(CDCl3) ~ 8.83 (d, lH), 7.78 (m, lH), 7.41 (m, lH), 7.10 (m, lH), 6.47 (d, lH), 4.34
(m, 2H), 3.61 (s, 3H), 2.20 (d, 3H), 1.33 (m, 3H).
ExamPle 86
2- [N-(1 '-AMINOCITRACONAMIDO)] -4-
TRIFLuoRoMETHYLPyRlMlDlNE-s-cARsoxyLlc AclD
A solution of t-butyl 2-[N-(1 '-aminocitraconamido)]-4-
25 trifluoromethyl~y. ;I~i(line-s-carboxylate (0.20 g, 0.5 mmol) in concenkated formic acid
(8 mL) was refluxed for one hour, cooled, diluted with H20 (10 mL), exkacted with
EtOAc (2 x 20 mL), dried (MgSO4) and concenkated The solid was washed with
CH2Cl2 (2 x 20 mL) and dried under vacuum to give the title compound in 60% yield
(0.10 g); m.p. 107-109~C.
CA 02230896 1998-03-02
W O 97/09325 PCT~US96/14089
Example 87
5_BENZOYL 2-[N-(1 '-AMINOCITRACONAMIDO)]-4-
5TRIFLUOROMETHYLPYRIMIDINE
The title compound was prepared from 5-benzoyl-2-hydrazino-4-
trifluoromethylpyrimidine (0.15 g, 0.53 mmol) (~ ,d as described for Example 19,
but employing 5-benzoyl-2-mesyl-4-trifluoromethylpyrimidine) as described in
Example 20, resulting in a yield of 6% (0.015 g); 'H NMR (CDCl3) ~ 8.5 (s,lH), 7.8
10 (m,2H), 7.65 (m,lH), 7.5 (m,2H), 6.5 (s,lH), 3.45 (s,lH), 2.2 (s,3H).
Example 88
ETHYL 2-rN-(l'-AMlNoclTRAcoNAMlDo)]-4
15(3 '-THIENYL)PYRIMIDINE-5-CARBOXYLATE
The title compound was prepared from ethyl 2-hydraz~no-4-(3'-
thienyl)pyrimidine-5-carboxylate (0.22 g, 0.88 mmol) (prepared in the same manner as
Example 35) as described in Example 20, resulting in a yield of 40% (0.11 g); m.p. 47-
48~C.
Example 89
ETHYL 2-[N-(1 '-AMlNoclTRAcoNAMlDo-N-METHyL)]-4
(3 ' -THIENYL)PYRIMIDINE-5 -CARBOXYLATE
The title compound was pI~ep~u~d from ethyl 2-[N-( l '-
aminocitraconamido)]-4-(3'-thienyl)pyrimidine-5-carboxylate (0.05 g, 0.14 mmol) as
described in Example 42, resulting in a yield of 19% (0.01 g); 39-41~C.
WO 97/09325 CA 02230896 1998 - 03 - 02 PCT/US96/14089
48
Examl~le 90
ETHYL 2-rN-(1 -AMINOCITRACONAMIDO)]-4-
[2'-(5'-METHYLTHIENYL)]PYRIMIDINE-5-CARBOXYLATE
The title compound was l~lepdlcd from ethyl 2-hydrazino-4-[2'-(5'-
5 methylthienyl)]pyrimidine-5-carboxylate (0.22 g, 0.88 mmol) (prepared in the same
manner as Example 67) as described for Example 20, rçsIllting in a yield of 40% (0.11
g); m.p. 138-139~C.
ExamPle 91
ETHYL 2- [N-( 1 '-AMINOCITRACONAMIDO)]-4-
(2'-FURANYL) PYRIMIDINE-5-CARBOXYLATE
The title compound was prepared by (a) reaction of ethyl 2'-
fiuranoylacetate (10 g, 0.055 mol) with N,N-dimethylformamide dimethyl acetal (7.3 g,
15 0.055 mol) as described for Example 67, (b) subsequent reaction with a solution NaOEt
(0.058 mol) and S-methyl isothouroniurn sulfate (7.7 g, 0.028 mol) also described for
Example 67, (c) oxidation with mCPBA (9.8 g, 0.057 mol) also described for Example
67, (d) reaction with hydrazine (1.5 mL, 0.05 mol) as described for Example 19, and (e)
reaction with citraconic annydride (4 mL, 0.05 mol) as described for Example 20,
20 resulting in an overall yield of 1% (0.18 g); m.p. 38-40~C.
Example 92
ETHYL 2-rN-(l -AMlNoclTRAcoNAMlDo-N-METHyL)l-4
[2'-(5'-METHYLTHIENYL)]PYRIMIDINE-5-CARBOXYLATE
The title compound was prepared from ethyl 2-(1'-methylhy&~ino)-4-
[2'-(5'-methylthienyl)]pyrimidine-5-carboxylate (3.0 g, 10.0 mmol) (prepared in the
same marmer as Example 67 employing methyl hydrazine) as described in Example 20,
CA 02230896 1998-03-02
W O 97/09325 PCTAUS96J14089
49
rçs-llting in a yield of 40% (1.6 g); 'H NMR (CDCl3) ~ 8.71 (d,lH), 7.7 (d,lH), 6.74
(d,lH), 6.5 (d,lH), 4.33 (q,2H), 3.6 (s,3H), 2.5 (d,3H), 2.2 (d,3H), 1.32 (t,3H).
Example 93
ETHYL 2- [N-( l '-AMINOCITRACONAMIDO)]-4-
(2'-THlANAPHTHYL)PYRlMlDlNE-S-cARsoxYLATE
The title compound was ~Icpa~cd by (a) reaction of ethyl 2'-
benzothienoylacetate (24.8 g, 0.1 mol) with N,N-dimethylforrn~micle dimethyl acetal
(7.3 g, 0.055 mol) as described for Example 67, (b) subsequent reaction with a solution
of NaOEt (0.12 mol) and S-methyl isothouronium sulfate (13.9 g, 0.05 mol) also
described for Example 67, (c) oxidation with mCPBA (8.5 g, 0.05 mol) also described
for Example 67, (d) reaction with hydrazine (1.5 mL, 0.05 mol) as described for
Example 19, and (e) reaction with citraconic anhydride (4 mL, 0.05 mol) as described
for Example 20, resulting in an overall yield of 0.1 % (0.05 g); m.p. 165- 166~C .
Example 94
METHYL 2-[N-(1 -AMINoclTRAcoNAMlDo-N-ETHYL)~-4-
TRIFLuoRoMETHYLPyRlMlDlNE-s-cARsoxyLATE
The title compound was pIe~alcd from methyl 2-[N-(l'-
aminocitraconamido)]-4-tri~luoromethylpyrimidine-5-carboxylate (0.20 g, 0.61 mmol)
as described for Example 42, but employing ethyliodide, rçslllting in a yield of 18%
(0.04 g); 'H NMR (CDCl3) ~ 9.01 (d, lH), 6.48 (s, lH), 4.05 (q, 2H), 3.87 (s, 3H), 2.14
(s, 3H), 1.27 (t, 3H).
WO 97/09325 CA 0 2 2 3 0 8 9 6 19 9 8 - O 3 - O 2 PCT/US96/14089
Exarnple 95
ETHYL 2- [N-( l '-AMlNocITRAcoNAMIDo-N-suTANoyL)]-4
TRlFLuoRoMETHyLpyRlMlDINE-s-cARsoxyLATE
The title compound was prepared from ethyl 2-[N-( l '-
5aminocikacl~n~mi~lo)]-4-trifluoromet-h-ylpyrimidine-5-carboxylate (0.10 g, 0.29 mmol)
as described for Example 74, but employing butyric anhydride, rç~-llting in a yield of
23% (0.03 g); 'H NMR (CDCl3) ~ 9.13 (s, lH), 6.54 (s, lH), 4.42 (q, 2H), 3.17 (t, 2H),
2.18 (s, 3H), 1.77 (q, 2H), 1.36 (t, 3H), 1.01 (t, 3H).
Example 96
ETHYL 2-[N-(l'-AMlNoClTRACoNAMlDo)-N-(N'-METHYLcARsoxAMIDYL)]-4-
TRlFLUoROMETHYLPYRlMlDlNE-5-CARBOXYLATE
Ethyl 2 - [N-( l '-aminocitraconamido)] -4-trifluoromethylpyrimi-dine-5 -
15carboxylate (0.05 g, 0.15 mmol) and methylisocyanate (0.3 mL) were heated to 60~C
for 3 minlltes and allowed to cool to RT. Af~ter 30 minllte~, the residual isocyanate was
removed under vacuum, and the solid was washed with hexanes to provide the titlecompound in a 70% yield (0.03 g); m.p. 132-134~C.
Example 97
ETHYL 2-[N-( l '-AMINocITRAcoNAMlDo)-N-( l ''-oxo-2~-PHENYLETHYL]-4
TRIFLUOROMETHYLPYRIMIDINE-5 -cARsoxYLATE
A solution of ethyl 2-[N-(1 '-aminocitraconamido)]-4-
25trifluoromethylpyrimidine-5-carboxylate (0.04 g, 0.12 mmol), phenylacetylchloride
(0.08 mL, 0.6 mmol) and pyridine (0.05 mL, 0.6 mmol), in ~H2C12 (15 mL) was stirred
1.5 hours, washed with H2O, dried (MgSO4), concentrated and chromatographed (SiO2,
he~ne~/EtOAc~ 2:1) to give the title compound in 42% yield (0.021 g); IH NMR
CA 02230896 1998-03-02
WO 97/0932S PCT~US96/14089
(CDCI3) ~ 9.11 (s, lH), 7.27 (m, 5H), 6.53 (s, lH), 4.46 (s, 2H), 4.42 (q, 2H), 2.17 (s,
3H), 1.37 (t, 3H).
S Example 98
ETHYL 2-[N-(1 '-AMINOCITRACONAMIDO)]-4-METHOXYMETHYL
PYRIMlDlNE-5-eARsoxYLATE
'rhe title compound was prepared by (a) reaction of ethyl
methoxyketo~cet~t~ (10.5 g, 0.07 mol) with kiethyl orthoformate (9.7 g, 0.07 mol) and
urea (3.9 g, 0.07 mol) as described for Example 3, (b) reaction with NaOEt (0.02 mol)
as described for Example 5, (c) reaction with POCl3 (6.5 mL, 0.07 mol) as described for
Example 7, (d) reaction with hydrazine (2 mL, 0.07 mol) as described for Example 19,
and (e) reaction with cikaconic anhydride (6 mL, 0.07 mol) as described for Example
20, resulting in an overall yield of 8% (1.8 g); 'H NMR (CDCI3) ~ 8.87 (s, lH), 8.2 (s,
lH), 6.5 (s, lH), 4.87 (s, 2H), 4.35 (q, 2H), 3.47 (s, 3H), 2.18 (s, 3H), 1.35 (t, 3H).
Example 99
ETHYL 2-[N~ -AMINoelTRAeoNAMlDo)-N-(ETHoxYeARsoNYL)]-4
TRlFLuOROMETHYLPYRlMlDlNE-5-CARBOXYLATE
The title compound was prepared from ethyl 2-[N-(1'-
aminocikaconamido)]-4-trifluoromethylpyrimidine-5-carboxylate (0.06 g, 0.19 mmol)
as described for Example 97, but employing ethyl chloroformate, resulting in a yield of
71% (0.055 g); 'H NMR (CDCl3) ~ 9.13 (s, lH), 6.54 (s, lH), 4.39 (m, 4H), 2.18 (s,
25 3H), 1.31 (m, 6H).
CA 02230896 1998-03-02
W O 97/09325 PCT~US96/14089
ExamPle 100
ETHYL 2-[N-(l'-AMINoelTRAeoNAMlDo)-N-sENzoYL]-4-
TRI~LUOROMETHYLPYRIMIDINE-5 -eARso~YLATE
The title compound was prepared from ethyl 2-[N-( l '-
S aminoeitrac~n~mitlc-)]-4-trifluoromt;L}lyl~y.;...i~lin~-5-carboxylate (0.09 g, 0.26 mmol)
as deseribed for Example 97, but employing benzoyl chloride, resulting in a yield of
69% (0.08 g); IH NMR (CDCl3) ~ 8.99 (s, lH), 7.72 (d, 2H), 7.52 (dd, lH), 7.41 (dd,
2H), 6.55 (s, lH), 4.39 (q, 2H), 2.19 (s, 3H), 1.35 (t, 3H).
Example 101
S -BENZOYL 2- [N-(1 '-AMINOCITRACONAMIDO)] -4-ETHYLPYRIMIDINE
The title compound was prepared by (a) reaction of 5-benzoyl-4-ethyl-2-
methylthiopyrimidine (0.14 g, 0.54 mmol) and mCPBA (0.28 g, 1.6 mmol) as deseribed
15 for Example 67, (b) reaction with hydrazine (0.09 g, 2.7 mmol) as deseribed for
Example 19, and (c) reaetion with eitraeonie anhydride as described for Example 20,
resulting in a overall yield of 25% (0.046 g); m.p. 49-50~C.
Example 102
5 -BENZOYL 2- [N-( l '-AMINoelTRAeoNAMlDo)-N-METHYL] -4-ETHYLPYRIMIDINE
The title eompound was ~ ~ed as described for Exarnple 42, but
employing 2-[N-(l'-aminoeitraeonamido)]-5-benzoyl-4-ethylpyrimidine (0.06 g, 0.18
mmol), resulting in a yield of 40% (0.025 g); 'H NMR (CDC13) ~ 8.41 (d, lH), 7.78 (d,
2H), 7.60 (dd, lH), 7.44 (dd, 2H), 6.43 (s, lH), 3.61 (s, 3H), 2.68 (m 2H), 2.19 (s, 3H),
1.2 (m 3H).
CA 02230896 1998-03-02
W O 97/09325 PCT~US96/14089
Exarnple 103
ETHYL 2 [N (1'-AMINOCITRACONAMIDO)-N-METHYL]-~-
PENTAFLUOROETHYLPYRIMIDINE-S -CARBOXYLATE
The title compound was prepared according to the procedure of Example
5 78 but employing ethyl-2-[N-(l'-aminocitraconamide)~-4-pell~luoroe~lyl~.,y, ;-"ic1in~-S-
carboxylate (0.1 g, 0.3 mmol) and methyl iodide (0.07 g, 0.5 mmol) under basic
conditions, resulting in a yield of 47% (0.05 g); ~HNMR (CDCl3) ~i 9.01 (s, 1 H), 8.80 (s,
lH),6.49(m, lH),4.39(q,2H),3.62(s,3H),2.18(d,3H), 1.36(t,3H).
Example 104
ETHYL 2-[N-(1 '-AMINOCITRACONAMIDO)]-4-
(2"-THIANAPHTHYL)PYRIMIDINE-S -CARBOXYLATE
The title compound was prepared from ethyl 2-mesyl-4-(2'-
5 thianaphthyl)pyrimidine-S-carboxylate (2.0 g, 5.5 mmol) as described in Example 93,
but employing methyl hydrazine (0.9 mL, 16.5 mmol) where hydrazine was used, this
followed by reaction with citraconic anhydride (1.5 mL, 16.5 mmol) also in anology to
Example 93; reslllting in a yield of 69% (1.6 g); 'H NMR (CDCl3) ~ 8.8 (d, lH), 8.15
(d, lH), 7.8 (m, 2H), 7.3 (m, 2H), 6.6 (d, lH), 4.37 (q, 2H), 3.65 (d, 3H), 2.17 (s, 3H),
20 1.34 (t, 3H).
ExamT~le 105
ETHYL 2-[N-(1 '-AMINOCITRACONAMIDO)]-4-
25 (2'-THIAZOLYL)PYRIMIDINE-S-CARBOXYLATE
2-Bromothiazole (8.25 g, 0.05 moles) in anhydrous ether (60 mL) is
added dropwise to a solution of nBuLi (34 mL, l.SM solution, 0.051 mmol) in
anhydrous ether (60 mL) cooled to -78~C and stirred for 30 minIlfes Carbon dioxide is
bubbled into the solution and after saturation is achieved, the reaction mixture is poured
CA 02230896 1998-03-02
WO 97/09325 PCT~US96/14089
over dry-ice. H2O (10 mL) is added and the mixture basified to pH=9 with NaOH (1- N). The aqueous layer is acidified with concentrated HCl to pH <3 then extracted into
ether, dried (MgSO4) and concentrated to provide the thi~ole-2-carboxylic acid in 57%
yield (3.2 g); 'H NMR (MeOD) ~ 8.00 (d, lH); 7.94 (d, lH). The title compound was
then prepared by (a) reaction of thiazole-2-carboxylic acid (3.7 g, 29.0 mmol) and
bis(ethyl malonate)m~gne~ium salt (4.3 g, 15.0 mmol) as described in Exarnple 67, (b)
reaction of 2-thiazolylacetate (2.5 g, 12.8 mmol) and N,N-dimethylformamide dimethyl
acetal (2.42 g, 12.8 mmol) as described for Example 67, (c) reaction with NaOEt (13.8
mol) and S-methyl isothouronium sulfate (1.78 g, 6.4 mol) also described for Exarnple
67, (d) oxidation with mCPBA (6.1 g, 35.0 mmol) also described for Example 67, (e)
reaction of ethyl 2-mesyl-4-(2'-thiazolyl)pyrimidine-5-carboxylate (1.08 g, 3.45 mmol)
with hydrazine (0.33 mL, 10.3 mmol) as described for Example 19, and (f) reaction
with cikaconic anhydride (0.31 mL, 3.5 mmol) described for Example 20, rçs-IItin~ in
an overall yield of 8% (0.10 g) from ethyl 2-mesyl-4-(2'-thiazolyl)pyrimidine-5-carboxylate; m.p 42-44~C.
Example 106
ETHYL 2-[N-(1'-AMINoclTRAcoNAMlDo)-N-METHYL]-4-
(2'-THlAzOLYL)pyRlMlDlNE-5-cARsoxyLATE
The title compound was prepared as described in Example 20, but
employing ethyl 2-(1 '-methylhydrazino)-4-(2'-thiazolyl)pyrimidine-5-carboxylate(0.96 g, 3.4 mmol) (prepared according to the procedure of Example 19 but employing
ethyl 2-mesyl-4-(2'-thiazolyl)pyrimidine-5-carboxylate and methyl hydrazine where
hydrazine was used). resulting in a yield of 50% (0.64 g); 'H NMR ~ 8.69 (bs, lH); 7.92
(d, lH); 7.50 (d, lH); 6.50 (d, lH); 4.35 (q, 2H); 3.62 (s, 3H); 2.2 (s, 3H); 1.27 (t, 3H).
.
CA 02230896 l998-03-02
W O 97/09325 PCTAJS96/14089
Example 107
S-BUTANOYL 2-[N-(1 -AMINOCITRACONAMIDO-N-METHYL)]-4-ETHYLPYRIMIDINE
The title compound was prepared as described for Example 101, but
employing 5-butanoyl-4-ethyl-2-methylthiopyrimidine (1.16 g, 5.2 mmol) (prepared as
5 described for Example 72) where 5-benzoyl-4-ethyl-2-methylthiopyrimidine was used,
resulting in an overall yield of 15% (0.24 g); iH NMR ~ 8.80 (d, lH), 6.42 (d, lH), 3.60
(s, 3H), 2.65 (m ,4H), 2.20 (s, 3H), 1.65 (m,3H), 1.23 (m, 2H), 0.99 (m, 3H).
10Example 108
ETHYL 2-[N-(1 '-AMINOCITRACONAMIDO)]-4-
CYCLOPROPYLPYRIMIDINE-5-CARBOXYLATE
The title compound was prepared as described for Example 67, but
employing cyclopIvpallecarboxylic acid (5.5 g, 63.9 mmol) where 5-chlorothiophene-2-
5carboxylic acid was used, resulting in an overall yield of 0.8% (0.09 g); m.p. 76-78~C.
Example 109
ETHYL 2-~-(1 -AMlNoclTRAcoNAMlDo-N-METHyL)]-4
20CYCLOPROPYLPYRIMIDINE-S-CARBOXYLATE
The title compound was prepared as described for Example 108 from
cyclo~Io~ ecarboxylic acid (5.5 g, 63.9 mmol), but employing methyl hydrazine
where hydrazine was used, resulting in an overall yield of 0.3% (0.062 g); m.p. 53-
55~C.
CA 02230896 1998-03-02 PCTnUS96/14089
W O 97/09325
Example 110
4-ETHYL-5-(HYDROXYMETHYL)-2-METHYLTHIOPYRIMIDINE
To a solution of ethyl 4-ethyl-2-methylthiopyrimidine-5-carboxylic acid
(2.0 g, 10.1 mrnol) (prepared as described in steps a-c of Example 72) and N-methyl
5 morpholine (1.16 m~, 10.6 mmol) in dimethyl glycol (50 mL) at 0~C was added
isobutylchloroformate (1.38 mL, 10.6 mmol). The mixture was stirred 10 minntt?s,filtered, and cooled to 0~C. A solution of NaBH4 (0.40 g, 10.6 mmol) in H2O (10 mL)
was added to the filtrate and the mixture was stirred for 10 minutes H2O (10 mL) was
added and the ~ Lul~; was extracted with EtOAc (100 mL),washed with brine (20 mL),
10 and ct-nl~entrAtç-l to provide the title compound in 56% yield (1.04 g); GC/MS calcd for
C8H,2N2OS (M+) 184, found 184.
Example 111
2-[N-(1 '-AMINocITRAcoNAMlDo)-N-METHYL]-4-
(HYDROXYMETHYL)PYRIMIDINE
A solution of 5-hydroxymethyl-4-ethyl-2-methylthiopyrimi~1in~ (0.50 g,
2.72 mmol), t-butyldimethylsilylchloride (0.49 g, 3.26 mmol), and imifl~7~1e (0.20 g,
2.99 mmol) in DMF (S mL) was stirred overnight and concentrated to give of S-t-
20 butyldimethylsiloxymethyl-4-ethyl-2-methylthiopyrimidine in a 94% yield (0.76 g);
GC/MS calcd for Cl4H26N20SSi (M+) 299, found 299. The title compound was then
p~ ,d by (a) OX~ tion of 5-t-butyldimethylsiloxymethyl-4-ethyl-2-
methylthiopyrimidine (0.30 g, 1.0 mmol) with mCPBA (0.35 g, 2.0 mmol) as described
for Example 67, (b) reaction with methyl hydrazine (0.16 mL, 3.0 mmol) followed by
25 citraconic anhydride (0.11 g, 1.0 mmol) also described in Example 67; (c) reaction of 2-
[N-(l'-aminocitraconamido)]-S-t-butyldimethylsiloxymethyl-4-ethyl~ylhl~idine (0.08 g,
0.21 mmol) and concentrated HCI (0.5 mL) in EtOH (20 mL) for 1 hour, concentration
and chromatography (Hexanes/EtOAc, 2:1) to provide the title compound in 76% yield
(0.043 g) from 2-rN-(1'-aminocitraconamido)]-5-t-butyldimethylsiloxymethyl-4-
30 ethylpyrimidine; m.p. 108-110~C.
CA 02230896 l998-03-02
W O 97/09325 PCT~US96/14089
ExamPle 112
2-[N-(1 '-AMINOCITRACONAMIDO)-N-METHYL]-~-ETHYL-S-
S (METHOXYMETHYL)PYRIMIDINE
A solution of 4-ethyl-5-hydroxymethyl-2-methylthiopyrimi~ine (0.63 g,
3.42 mmol), Ag2O (1.6 g, 6.9 mmol) and MeI (1.1 mL, 17.1 mmol) in CH3CN (10 mL)
was stirred overnight, filtered and concc~nlldl~d to give 4-ethyl-5-methoxymethyl-2-
methylthiopyrimidine in 98% yield (0.66 g); GC/MS calcd. for CgHI4N2OS (M+) 198,found 198. The title compound was then prepared by (a) oxidation of 4-ethyl-5-
methoxymethyl-2-methylthiopyrimidine (0.66 g, 3.35 mmol) with mCPBA (1.18 g,
6.85 mmol) as described for Example 67, and (b) reaction with methyl hydrazine (0.36
mL, 6.85 mmol) followed by citraconic anhydride (0.77 g, 6.85 mmol) also described in
Example 67; resulting in a yield of 41% (0.40 g) from 4-ethyl-5-methoxymethyl-2-methylthiopyrimidine; 'H NMR (CDC13) o 8.14 (s, lH), 6.43 (s, lH), 4.29 (s, 2H), 3.53
(s, 3H), 3.36 (s, 3H), 2.68 (n1, 2H), 2.14 (s, 3H), 1.13 (m, 3H).
Example 113
2-[N-(l'-AMINOCITR~CONAMIDO)]-S-(METHOXYMETHYL)-4-
TRIFLUOROMETHYLPYRIMIDINE
The title compound was prepared as described for Example 112, but
employing of 4-trifluoromethyl-5-hydroxymethyl-2-methylthiopyrimidine (0.50 g, 2.23
mmol) (plt;p~ed in analogy to Example l lO) where 4-ethyl-5-hydroxymethyl-2-
25 methylthiopyrimidine was used, and employing hydrazine (0.09 mL, 2.87 mmol) wheremethyl hydrazine was used, resulting in an overall yield of 35% (0.25 g); 'H NMR
(CDC13) â 8.63 (s, lH), 8.20 (s, lH), 6.48 (s, lH), 4.46 (s, 2H), 3.75 (s, 3H), 2.12 (s,
3H).
-
W O 97/09325 CA 02230896 1998-03-02 PCT~US96/14089
Exarnple 114
2- [N-( l '-AMINOCITRACONAMIDO)]-S -(ETHOXYMETHYLcARsoNATE)-4-
TRIFLUOROMETHYLPYRIMIDINE
S To a solution of 2-[N-(1 '-aminocitraconamido)]-4-
trifluoromethylpyrimi~1ine-5-carboxylic acid (0.10 g, 0.317 mrnol) and N-
methylmorpholine (0.035 mL, 0.317 mmol) in THF (10 mL) at 0~C was added
ethylchloroformate (0.030 mL, 0.317 mrnol). After stirring S min~ s, NaBH4 (0.040g,
1.05 mmol) was added, MeOH (7 mL) was added, and the mixture was stirred 10
10 minlltes then concentrated. The oil was dissolved in EtOAc (30 mL) and washed with
1 N HCl (10 mL), 1 N NaHCO3 (10 mL), and brine ~10 mL), dried (MgSO4),
concentrated and chromatographed (SiO2, hexanes/EtOAc, 2:1) to provide the titlecompound in 19% yield (0.022 g), IH NMR (CDCl3) o 9.03 (s, lH), 6.53 (s, lH), 4.88
(s, 2H), 4.36 (q, 2H), 2.19 (s, 3H), 1.32 (t, 3H).
Example 115
ETHYL 4- [N-(1 '-AMlNocITRACoNAMlDo)]-2-PHENYLPYRIMlDlNE-s-cARsoxyLATE
The title compound was prepared by (a) reaction of diethylethoxy-
20 methylenemalonate (31 g, 144 mmol) with benzamidine (22 g, 144 mmol) under basic
conditions to afford ethyl 2-phenyl-4-hydroxypyrimidine-5-carboxylate in 64% yield in
analogy to F.x~mple 15, (b) reaction of ethyl 2-phenyl-4-hydro~y~y.;.~ lin~-5-
carboxylate (1.5 g, 6 mmol) and POC13 (9.4 g, 62 mmol) to afford 81% of ethyl 2-phenyl-4-chlo~ ylhllidine-5-carboxylate in analogy to Exarnple 7, (c) reaction of ethyl
25 2-phenyl-4-chlolv~ylilllidine-5-carboxylate (12 g, 46 mmol) with hydrazine (4.4 g, 137
mmol) to afford 99% of ethyl 4-hydrazino-2-phenylpyrimidine-5-carboxylate in
analogy to Example 18, (d) reaction of ethyl 4-hydrazino-2-phenylpyrimidine-5-
carboxylate (12 g, 46 mrnol) with citraconic anhydride (10 g, 92 mmol) to afford 75%
(12 g) ofthe title compound (m.p. 155-156~C) in analogy to Example 21.
CA 02230896 1998-03-02
WO 97/09325 PCT~US96/14089
59
Example 116
ETHYL 4 [N (1 -AMINOCITRACONAMIDO)-N-METHYL]-2-
PHENYLPYRIMIDINE-5-CARBOXYLATE
The title compound was ~lG~ed according to the procedure of Example
68 but employing ethyl 2-[N-(1 '-aminocitraconamide)]-4-phenylpyrimidine-5-
carboxylate (0.1 g, 0.3 mmol) and methyl iodide (0.09 g, 0.7 mmol) under basic
conditions, resulting in a yield of 45% (0.05 g); m.p. 118- 119~C.
Example 117
ETHYL 4-[N-ACETYL-N-(1 '-AMINOCITRACONAMIDO)]-2-
PHENYLPYRIMIDINE-5-CARBOXYLATE
The title compound was ~.e~aled according to tlle procedure of Example
75 but employing ethyl 2-[N-(l'-aminocitraconamide)]-4-phenylpyrimidine-5-
carboxylate (0.5 g, 1.3 mmol, ~ ~ed in Example 115) and acetic anhydride (5 mL)
under basic conditions, rt?sl7ltinp~ in a yield of 10% (0.05 g), IHNMR (CDCl3) ~ 9.15 (d,
lH), 8.19 (d, 2H), 7.47 (m, 3H), 6.70 (d, lH), 4.42 (q, 2H), 2.28 (d, 6H), 1.43 (t, 3H).
Example 118
ETHYL 4-[N-(1 '-AMINOCITRACONAMIDO)]-2-
METHYLPYRIMIDINE-5 -CARBOXYLATE
The title compound was prepared by (a) reaction of
diethylethoxymethylenemalonate (6 g, 28 mmol) with acetamidine hydrochloride (3 g,
31 mmol) to afford 67% of ethyl 2-methyl-4-hydroxypyrimidine-5-carboxylate in
analogy to Example 15, (b) reaction of ethyl 2-methyl-4-hydroxypyrimidine-5-
carboxylate (2.8 g, 15 mmol) with POCl3 (46 g, 300 mmol) to afford 28% of ethyl 4-
WO 97/09325 CA 0 2 2 3 0 8 9 6 19 9 8 - O 3 - O 2 PCT/US96/14089
chloro-2-m~lhyl~y. ;...ir~in~-S-carboxylate, (c) reaction of ethyl 4-chloro-2-
- methylpyrimidine-S-carboxylate (0.25 g, 1.25 mrnol) with hydrazine (0.2 g, 6.3 mmol)
to afford 96% of 4-hydrazino-2-m~ yl~y~ idine-5-carboxylate in ~nP1ogy to Example
18, (d) reaction of 4-hydrazino-2-methylpyrimidine-5-carboxylate (0.24 g, 1.2 mmol)
S with citraconic anhydride (0.16 g, 1.44 mmol) to afford 70% (0.24 g) of the title
compound (m.p. 98-99~C) in analogy to Example 21.
F.~r~mple 119
ETHYL 4-[N-(l'-AMINoclTRAcoNAMlDo)]-2-
sENzYLPYRIMlDlNE-5 -cARsoxYLATE
The title compound was ~ d by (a) reaction of diethyl
ethoxymethylenemalonate (11 g, 52 mmol) with phenyl~ce~midine (8 g, 60 mmol) to
afford 40% of ethyl 2-benzyl-4-hydroxypyrimidine-S-carboxylate in analogy to
15 Example lS, (b) reaction of 2-benzyl-4-hydroxylJylilllidine-S-carboxylate (3 g, 12
mmol) with POC13 (18 g, 116 mmol) to afford 45% of 2-benzyl-4-chloropyrimi~lin~-5-
carboxylate in analogy to Example 7, (c) reaction of 2-benzyl-4-chloropyrimic1ine-s-
carboxylate (l.S g, 5.5 mmol) with hydld~ille (0.5 g, 16 mmol) to afford 75% of 2-
benzyl-4-hydrazinopyrimidine-S-carboxylate in analogy to Example 18, (d) reaction of
20 ethyl 2-benzyl-4-hydrazinopyrimidine-S-carboxylate (1 g, 3.5 mmol) with citraconic
anhydride (lg, 11 mmol), in analogy to Example 21, to afford 70% (0.9 g) of the title
compound, m.p. 117-118~C.
Example 120
ETHYL 4-[N-(1 '-AMINOCITRACONAMIDO)]-2-
(3 '-NITROPHENYL)-PYRIMIDINE-S -cARsoxYLATE
The title compound was prepared by (a) reaction of diethyl
ethoxymethylenemalonate ( S g, 24 rnmol) with 3-nitrober~LLllidine (S g, 25 mmol) to
30 afford 86% of ethyl 4-hydroxy-2-(3'-nitrophenyl)pyrimidine-S-carboxylate in analogy
-
CA 02230896 1998-03-02
W O 97/09325 PCT~US9~/14089
to Example lS, (b) reaction of ethyl 4-hydroxy-2-(3'-nitrophenyl)pyrimidine-5-
carboxylate (6 g, 21 mmol) with (chloromethylene)dimethylammonium chloride (4 g,- 31 mmol) concentration of reaction llliX~ , followed by hexane and ether wash afford
45% of ethyl 4-chloro-2-(3'-nitrophenyl)pyrimidine-S-carboxylate, (c) reaction of ethyl
5 4-chloro-2-(3'-nitrophenyl)pyrimidine-5-carboxylate (1.9 g, 6 mmol) with hydrazine
(0.6 g, 18 mmol) to afford 33% of ethyl 4-hydrazino-2-(3'-nitrophenyl)pyrimidine-5-
carboxylate in analogy to F~:~mple 18, (d) reaction of ethyl 4-hydrazino-2-(3'-
nitrophenyl)pyrimidine-S-carboxylate (1.8 g, 6 mmol) with citraconic anhydride in
analogy to Example 21, to afford 80% (0.5 g) of the title compound; m.p. 118-120~C.
Example 121
ETHYL 4-[N-(1'-AMINoClTRACoNAMlDo)]-2-
(4~-TRIFLuoRoMETHYLPHENyL)pyRlMlDlNE-5-cARsoxyLATE
15 The title compound was prepared by (a) of diethyl
ethoxymethylenemalonate (6.3 g, 29 mmol) with 4-trifluoromethylphenylben7~mi~1ine
(5.5 g, 29 mmol) to afford 50% of ethyl 4-hydroxy-2-(4'-
trifluoromethylphenyl)pyrimidine-5-carboxylate in analogy to Example 15, (b) reaction
of ethyl 4-hydroxy-2-(4'-trifluoromethylphenyl)pyrimidine-5-carboxylate (3 g, 9.6
20 mmol) with POC13 (49 g, 322 mmol) to afford 97% of ethyl 4-chloro-2-(4'-
trifluoromethylphenyl)pyrimidine-5-carboxylate in analogy to Example 7, (c) reaction
of ethyl 4-chloro-2-(4'-trifluoromethylphenyl)pyrimidine-5-carboxylate (2 g, 6 mmol)
with hydrazine (0.6 g, 18 mmol) to afford 60% of ethyl 4-hydrazino-2-(4'-
trifluoromethylphenyl)pyrimidine-5-carboxylate in analogy to Example 18, (d) reaction
25 of ethyl 4-hydrazino-2-(4'-trifluoromethylphenyl)pyrimidine-5-carboxylate (1 g, 3
mmol) with citraconic anhydride (1 g, 9 mmol) to afford 40% (0.5 g) of the titlecompound (m.p. 148-150~C) in analogy to Example 21.
CA 02230896 1998-03-02
WO 97/09325 . PCT~US96/14089
62
Exam~le 122
ETHYL 4- [N-(1 ~-AMINOCITRACONAMIDO)]-2-
(2'~THIENYL)-PYRIMIDINE-5-eARsoxyLATE
The title compound was prepared by (a) reaetion of diethyl
ethoxymethyl~nem~lonate (8 g, 35 rnmol) with thienyl-2-font~nnic1in~ hydroehloride
(5.7 g, 35 mmol) to afford 74% of ethyl 4-hydroxy-2-(2'-thienyl)pyrimidine-5-
carboxylate in analogy to Example 15, (b) reaetion of ethyl 4-hydroxy-2-(2'-
thienyl)pyrimic1ine-5-earboxylate (2 g, 8 mmol) with POCI3 (24 g, 160 mmol) to afford
96% of ethyl 4-ehloro-2-(2'-thienyl)pyrimidine-5-earboxylate in analogy to Example 7,
(e) reaction of ethyl 4-chloro-2-(2'-thienyl pyrimidine-S-carboxylate (0.57 g, 2.2 mmol)
with hydrazine (0.34 g, 11 mmol) to afford 99% of ethyl 4-hydrazino-2-(2'-thienyl)-
pyrimidine-S-earboxylate in analogy to Example 18, (d) reaetion of ethyl 4-hydrazino-
2-(2'-thienyl)pyrimidine-5-earboxylate (0.55 g, 2.1 mmol) with citraconic anhydride
(0.71 g, 6.3 mmol) in analogy to Example 21 to afford 80% (0.61 g) of the title
eompound, m.p. 164- 164~C .
Example 123
ETHYL 4- [N-(1 '-AMINOCITRACONAMIDO)-N-METHYL] -2-
(2'-THlENYL)-pyRlMlDlNE-s-cARsoxyLATE
~ he title eompound was prepared by (a) reaetion of ethyl 4-chloro-2-(2'-
thienylpyrimidine-S-carboxylate (0.3 g, 1.1 mmol)) with methylhydrazine (0.25 g, 5.6
mmol)) to afford 99% of ethyl 4-(1'-methylhydrazino)-2-(2'-thienyl)pyrimidine-5-earboxylate in analogy to Example 18, (b) reaetion of ethyl 4-(1'-methylhydrazino)-2-
25 (2'-thienyl)pyrimidine-5-carboxylate (0.3 g, 1 mmol)) with citraconic anhydride (0.4 g,
3 mmol) in analogy to F~mple 21 to afford 52% (0.21 g) of the title compound; m.p.
126-127~C.
CA 02230896 l998-03-02
W O 97/09325 PCTAUS96/14089
ExamDle 124
ETHYL 4-[N-(1~-AMINOCITRACONAMIDO)]-2-(3~4~-DICHLOROBENZYL)-
PYRIMlDlNE-5-cARsoxYLATE
The title compound was prepared by (a) of diethyl
ethoxymethylenemalonate (6 g, 27 mmol) with 3',4'- dichlorophenylacet~mi~1ine (5.5 g,
27 mmol) to afford 37% of ethyl 4-hydroxy-2-(3',4'-dichlorobenzyl)pyrimidine-5-
carboxylate in analogy to Example 15, (b) reaction of ethyl 4-hydroxy-2-(3',4'-
dichlorobenzyl)pyrimidine~5-carboxylate (2 g, 6 mmol) with POCl3 (14 g, 92 mmol) to
afford 62% of ethyl 4-chloro-2-(3',4'-dichlorobenzyl)pyrimidine-5-carboxylate inanalogy to Example 7, (c) reaction of ethyl 4-chloro-2-(3',4'-dichlorobenzyl)pyrimidine-
5-carboxylate (1 g, 2.8 mmol) with hydld~ille (0.27 g, 8.3 mmol) to afford 99% of ethyl
4-hydrazino-2-(3',4'-dichlorophenyl)pyrimidine-5-carboxylate in analogy to Example
18, (d) reaction of ethyl 4-hydrazino-2-(3',4'-dichlorobenzyl)pyrimidine-5-carboxylate
(0.9 g, 2.7 mmol) with citraconic anhydride (0.49 g, 4.4 mmol) in analogy to Example
21 to afford 47% (0.3 g) of the title compound; m.p. 119- 121 ~C.
Example 125
SYNTHESIS OF REPRESENTATIVE COMPOUNDS
BY COMBINATORIAL CHEMISTRY TECHNIQUES
This example illustrates the synthesis of a representative class of
compounds of this invention, 2-~ub~Liluled-4-trifluoromethyl-5-pyrimidine carboxylic
acid methyl esters, by combinatorial chemi~try. It should be understood that, while a
specific class of compounds are illustrated in this example, the following procedure
25 may be employed to synthesize other compounds of this invention.
A mixture of TentaGel resin (TentaGel S PHB, Advanced Chem Tec
Louisville, Kentucky, 17 g, 0.2 mmol/g reactive sites) con1~ininp; a free hydroxy as the
reactive site and DMF (75 mL) was stored for 0.25 h. Then a solution of 2-chloro-4-
trifluoromethyl pyrimidine 5-carbonyl chloride (2.20 g, 10.2 mmol) in DMF (15 mL)
30 was added. The llliXLul~ was gently shaken for 3 h and filtered. The resin was washed
CA 02230896 1998-03-02
WO 97/0932~ PCTAUS96/14089
64
thoroughly with DMF (3 X100 mL) and CH2Cl2 (3 X100 mL) and then dried under
- vacuum. The resin was divided into 80 equal portions and placed into 80 separate
reaction vessels (dispersion tubes) The dispersion tubes were placed into separate test
tubes each cnnt~ininp a di~,le-lL amine (2.5 mmol, Appendix I) and 2 mL of a 0.1M
S solution of pyridine/DMF (5 molar equivalents of each arnine in each test tube). The
entire set of 80 reaction vessels was gently shaken for 5 hours to ensure complete
reaction. Then each of the dispersion tubes was removed from the test tube co~ i..g
the amine and rinsed se~dL~ly to remove any unreacted m~t~-ri~l~ The ~lieper.eion
tubes were then dried and submersed into 80 new test tubes (previously tared), each
0 contztining a solution of NaOMe (2.5 mL, 0.012 M solution, 0.03 mmol). The reaction
was allowed to proceed overnight at room te~ t;ldLule under N2. Then the dispersion
tubes were removed from the solution and individually rinsed with MeOH. The
individual MeOH solutions were concelllld~ed in the tared test tubes to provide known
amounts of the desired 80 individual methyl esters substituted with different groups at
15 the 2-position. Each compound was >85% pure by HPLC and had the correct
molecular weight as fl~tertnin~(l by GC/MS.
ExamPle 126
INHIBITION OF THE ACTIVATION OF NFKB AND AP-1
20 A. NFKB ASSAY
Stable human Jurkat T-cells cont~ining an NFKB binding site (from the
MHC promoter) fused to a minim~l Sv~O promoter driving luciferase ex~l~s~ion were
used in this ~ .;lllent. Cells were split to 3 x 105 cells/mL every 2-3 days (cell
concentration should not exceed 1 x 106 cells/mL to keep the cells proliferating in log
25 phase). These cells were counted, resuspended in fresh medium c0~ l;tl~ 10%
Serum-Plus at a density of 1 x 106 cells/mL and plated in 96 well round bottom plates
(200 IlA per well) 18 hours prior to starting the experiment.
Compounds of this invention, dissolved in dimethyl sulfoxide (3.3, 0.33
and 0.03 ~g/mL), were then added to the 96 well plates co~ n;-lg the cells and the
CA 02230896 1998-03-02
W O 97/09325 PCTnUS96/14089
plates were incubated for 0.5 h at 37~C. Then 50 ng/mL of phorbol 12-myristate-13-
acetate (PMA) and 1 ~lg/mL of phytohemagglulinin (PHA) were added to each well and
the cells were inGllb~ted for an additional 5 h at 37~C. The plates were c~ iruged at
2200 RPM for 3 minutes at room temperature and then the medium was removed. To
5 each well was added 60 ~lL of cell lysis buffer and the plates were left at room
telll~ d~ule for 0.25 h. Then 40 ~L of each cell extract was transferred to a black 96
well plate and 50 IlL of luciferase subskate buffer was added. Lllmin~scence wasimmediately measured using a Packard TopCount.
B. AP-l ASSAY
For AP-l, the assay was run as described above for NFKB except stable
Jurkat T-cells were used that contained a collagenase promoter driving luciferase
expression. In addition, the concentration of PMA used was 5 ng/mL.
C. RESULTS
The results of the above assays for a representative compound of this
15 invention, ethyl 2-[N-(l'-aminocitraconamido)]-4-pentafluoroethylpyrimidine-5-
carboxylate (see Example 48), as percent inhibition versus control are presented in
Figure 3. This figure also indicates activity of ~-actin which was employed in these
assays as a control cell line indicating effects on transcription. The lack of ,B-actin
activity evidences selectivity of the test compounds for the transcription factors AP-1
20 and NFKB.
Expressed as IC50's, the results of these assays on additional test
compounds are sllmm~ri7f d in Table 3 below. The IC50 values reported in Table 3 are
the average of the measurements obtained from the NFKB and AP-1 assays describedabove.
WO 97/0932~ CA 02230896 1998 - 03 - 02 PCT~US96/14089
66
Table 3
- Abilitv of Representative Compounds of Structure (I) to Inhibit NFKB and AP- I
Test IC5~ Test IC~o Test IC50
Compound (I LM)Compound (~M)Compound (~M)
(Example No.) (Example No.) (Example No.)
0.7 79 0.2 103 0.4
21 0.5 81 0.1 104 0.2
23 5-10 83 0.08 105 2.4
36 0.15 84 0.45 106 1.7
37 0.15 85 0.02 107 0.5
38 0.04 87 0.55 108 1.5
1.0 88 0.5 109 0.2
41 3.9 89 0.2 111 4.9
42 5 90 0.06 112 0.6
44 10-30 91 1.1 113 1.0
46 4.0 92 0.3 114 1.1
48 0.09 93 0.8 115 0.03
1.0 94 1.1 116 1.4
52 2.0 95 0.3 117 4
64 1.0 96 0.7 118 17
68 0.4 97 0.9 119 7
73 0.05 98 6.0 120 8
74 0.45 99 0.4 121 0.4
0.4 100 0.8 122 0.02
77 1.3 101 0.6 123 0.8
78 0.15 102 0.4 124 4
Based on the above results, representative compounds of this invention
were found to be effective at inhibiting the activation of transcription factors (i.e.,
NFKB and AP-l) involved in gene transcription, and therefore have utility as, for
exarnple, immnnnsuppressive agents.
F~mr1e 127
INHIBITION OF CYTOKINES
To determine the effects of compounds on PMA/PHA-incl~ce~l cytokine
production, ~llpern~t~nt~ from either the NFKB (for IL-8) and AP-l (for IL-2) reporter
CA 02230896 1998-03-02
W O 97/09325 PCTAUS96/14089
67
gene assays of Example 55 were collected and saved. Cytokine levels in the
sup~ (25-50 ~uL aliquots) were ~let~rmined by ELISA. The results of this
~x~e~ ent for a representative compound of this invention, ethyl 2-rN-(l'-
aminocitraconamido)]-4-pentafluoroethylpyrimidine-5-carboxylate (see Example 48), is
5 presented in Figure 4 (expressed as percent inhibition versus control).
Example 128
ACTIVITY OF REPRESENTATIVE COMPOUND
IN GRAFT VS. HOST AND CONTACT SENSITIVITY MODELS
The murine popliteal lymph node (PLN) assay is a graft vs. host model
that predicts activity of compounds in blocking human transplant rejection. The
delayed-type hypersensitivity response to oxazolone is a standard contact sensitivity
model. Both of these models are used routinely to evaluate compounds that are used
clinically. For example, cyclosporin and cyclophospharnide are active in these models
15 and are used clinically (Morris et al., Transplantation Proceedings 22(Suppl. 1):110-
112, 1990).
A. POPLITEAL LYMPH NODE MODEL
Spleens are removed from donor BALBlc mice and splenocytes are
isolated then irradiated (3,000 rads) to prevent donor cell proliferation. After washing
20 and adjusting cell density, 2.5x106 cells are injected subcutaneously into the left hind
footpad of C3H mice. On day 4, the mice are sacrificed and left popliteal lymph nodes
(PLNs) weighed.
A repres~ntzlfive compound of this invention is ~fimini~tt?red once daily
by intraperitoneal injection beginning one day before footpad injection (day 0) through
25 day 4. The compound is suspended, immediately prior to use, at a concentration of ~
mg/mL in 0.25% methyl cellulose (Sigma) using a glass-Teflon homogenizer. For
doses of 10, 20 and 30 mg/kg, ~p~ iate dilutions of the stock solution are made so
that 0.1 mL/10 g body weight is sl-imini~tered by intraperitoneal injection.
-
CA 02230896 1998-03-02
W O 97/09325 PCTAUS96/14089
68
B. DELAYED TYPE HYPERSENSITIVITY STUDY
On day 0, oxazolone (100 mL of a 3% solution) is applied to the shaved
abdomen of mice. On day 7, a ch~llenge application of oxazolone is applied (10 mL)
around the right ear. A representative compound of this invention is ~-iminictered from
5 days -2 to 7 by intraperitoneal injection. The injectable solution is preparedimme~ tely prior to use by suspending the compound in 0.25% methyl cellulose
(Sigma) using a glass-Teflon homogenizer. For each dose, 0.1 mL/10 g body weight of
the suspension is ~lmini~t~red. The compound is prepared at the highest concentration
for this study and a~lupliate dilutions of the stock solution are made so that 0.1 mL/10
10 g body weight is :~lmini~tered. Twenty four hours later, the difference in right vs. left
ear thickness is measured.
It will be appreciated that, although specific embodiments of this
invention have been described herein for purpose of illustration, various modifications
15 may be made without departing from the spirit and scope of the invention.
-