Note: Descriptions are shown in the official language in which they were submitted.
CA 022320~9 1998-03-13
W O 97/10240 PCT~B96/00668
PHENOL DERIVATIVES WITH PHARMACEUTICAL ACTIVITY
Backaround of the Invention
The preser,l invention relates to prodrugs for the neuroprotective agent (3R,4S)-
3-[~(~fluorophenyl)-4-hydroxy-piperidin-1-yl]~l.rc,r.,~4,7-diol which is a N-methyl-~
asp~ lic acid (NMDA) anta~oni~.l. The preser,l invention further relates to methods of
using, and pl,ar",&ceutical comrosilions containing, the prodrugs described herein.
Prodrugs are compounds that have little or no i, Ill i"sic biological activity until converted
Tnto another, biologically active che",i--' Sp~-8S in the body of a recipient (e.g. a
human). Upon delivery to a ,e~ nl by any of several routes (such as orally,
p&re~ rally, or rectally), the prodrug is l~ .rur--,ed into a new cGI"pound (parent drug)
that possesses desirable biological activity. Conversion of the prodrug into the parent
drug can occur in the body at a variety of locations (e.g. gut wall, liver, kidney, blood,
etc.) and by any of a number of mechani~l"s (e.g. enzymatic hydrolysis, oxidative
mt:l~bclisn" etc.).
Prodrugs can be useful to overcome a variety of ' nil~lions of the parent drug.
For e~ F 'e, the parent dnug may not have an ~ ~¢ eFt '~ le bioavailability when delivered
by an otherwise desi,atls route of administration. The parent drug may suffer from
e)~ler sive first pass metabolism that eflectively removes the drug from the body at an
excessively rapid rate that inhibHs effective therapy. The parent drug may have
undesirable physical properties, such as poor solubility or stability, or H may have other
properties that make formulation of the parent drug difficult or expensive to ad" ,inisler.
In some i"~ ces, the prodrug may be easier to synthesize.
Once administered to an appropli~le re ~ nt, the prodrugs of the present
invention are converted into (3R,4S)-3-[4-(~fluorophenyl)4-hydroxy-piperidin-1-yl]-
chroman~,7-diol (parent chrol"anol) by one or more of the mechani_n,s desaiLed
above. The prodrugs of the pr~ser,l invention overcome the limHations of the parent
chromanol including one or more of the Ijl~jlal;GnS ACsoci~lpd with parent drugs as
described above. In particular, the prodrugs of the present invention are more stable
in solution than the parent chromanol and, as a result, are better suited for intravenous
admini~l~aliGIl.
The parent chlumanol is an NMDA antagonist and, as such, is useful in the
treatment of head trauma, stroke and CNS degenerative ~I;r6~~es such as Alzheimer's
CA 02232059 1998-03-13
W O 97/10240 PCT~B9~
AC~, Hu~ ylon~s d;s~ , rL~ hil,so"'s ~ise~se and other CG nJitiGns alleviated byblocking the NMDA receptor.
NMDA ~ ,lagon;sls are compounds that block the NMDA r eceptor by i, ller~cti"y
with the glutarnat3 binding site or other sites on the raceptor "~o~ 'e The ability of
5 a particular cGmpound to competitively bind to the NMDA glutarnate rdceptor can be
evaluated using a radialig~-.d binding assay. See Murphy et al., British J. Pl.ar-.,acol.
95, 932-938 (1988). The &~t~o.nists can be distinguished from the a~oni~l~ using a
rat cortical wedge assay. See l I&,i~on and Si",monds, British J. Pl-L..nacol., 84, 381-
391 (1984). Ex..n-Flss of competitive NMDA anlagGnisls include D-2-amino-5-
10 phosphonopel,l~.oi~ acid (D-AP5), and D-2-amino-7-phosphonoheptanoic acid,
Schoepp et al., J. Neur. Transm., 85, 131-143 (1991).
Antagonists of neu,ut,ansl..is~ion at NMDA receptor~ are useful ll-erapeutic
agents forthe l.t,~l-"enl of neu-~lag ~ ' disorders. U.S. Pat. No. 4,902,695 is directed
to a series of competitive NMDA ~ll~onbl~ useful for the l,~al")e"l of neurological
15 disGrder:,, including, ~ , 5y, stroke, anxiety, c~raLI~l ischer,~ia, musc~'--spasms, and
neurodegenerative d;_Grde,~ such as Alzheimer's ~lise~se and Hu,lti,lylon's .l ~U.S. Pat. No. 4,968,878 is directed to a second series of co"".etili~e NMDA l~ceptor
antagonic.ls useful for the treatment of similar neurological disorders and
neurodegenerative d is-rder:.. U.S. Pat. No. 5,192,751 provides a method of l,a&li--g
20 urinary inconli- ~ence through use of a competitive NMDA &r.l~.gonist.
NMDA antagoni~.t~ are ~Iso useful therapeutic agents with anticonvulsant,
anxiolytic, muscle ~elax~rll, and antipsychotic activity. J. Lehm~r), The NMDA
neceptor. Druqs of the Future, 14(11),1059 (1989). NMDA antagonists have also been
repolled to be effective for l.et-ling migraine (Can. J. Neurol. Sci., 19(4), 487 (1992));
25 drug addiction (Sc-once. 251, 85 (1991)); and neuro-psychi~l,ic disorders related to
AIDS (PIPS, 11, 1, (1990)).
CA 02232059 1998-03-13
W O 97/10240 PCT~B96/00668
Summary of the Invention
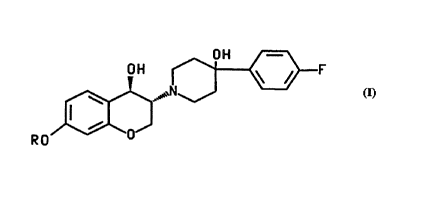
The invention relates to cGn-pounds of the formula
O H ~F
~ N~
RO O
and ph&.nAce-nically ~ t-~'s salts thereof,
wherein R is C,-C~ alkyl, C4-C8 cycloalkyl, RlC(O)-, or RlOC(O)-; and,
R1 is Cl-C~ alkyl, C~-C8 cycloalkyl, benzyl, C~-C10 aryl, or C3-C,~ heteroaryl
wherein said aryl, h~ter~aryl and the phenyl moiety of said benzyl are optionally
s~hstit~ted with from one to three s~hstitl~ents sele ' from the group consi~li..y of
hydroxy, chloro, bromo, fluoro, and -NR2R3, w:-el. . R2 and R3 are independe"lly15 se'e;l ' from the group cons;~ g of hyd~uyen~ Cl-C~ alkyl, (Cl-C" alkyl)C(O)-, (Cl-C~s
alkyl)OC(O)-, (C~s-Clo aryl)C(O)-, (benzyl)OC(O)-, and (C"-C10 aryl)OC(O)-.
VVth respect to the cor..pounds of formula 1, and their phar--,P~re-ltically
Arc~Ft-'le salts, as used in accord with the preser.l invention, it is to be underalood
that there are sl~r~isG,..eric forrns such as optical and geGr..~t,ic isomers due to
20 asymmetric carbon atoms and that the use of such isomers is also included within the
scope of the invention.
The term ~alkyl~, as used hsrein, unless otherwise indicated, includes saturatedmonovalent hydrocarbon radicals having straight or branched moieties.
The term ~cycloalkyl~, as used herein, includes saturated monovalent cyclic
25 hyd,ocE; L,on radicals including cyclobutyl, cyclopenlyl and cyclohaptyl.
Theterm ~aryl~, as used herein, unless oli.erv~,.e indicated, includes an organic
radical derived from an nr~l--alic hyd~ocarL,on by removal of one hydrogen, including
phenyl and naphthyl.
The term ~h~ler~,&ryl~, as used herein, unless otherwise indicated, includes an
30 org~n-c radical derived from an arom&lic heterocyclic cornpound by removal of one
hydrogen, such as pyridyl, furyl, pyrryl, thienyl, isoli.i&zolyl, imidazolyl, benzi",ii~olyl,
t~ olyl, pyrazinyl, pyrimidyl, quinolyl, isoquinolyl, benzofuryl, isobenzofuryl,ben~vlhie~yl, pyrazolyl, indolyl, isoindolyl, purinyl, c&rL.a~olyl, isoxS~olyl, thiazolyl,
CA 02232059 1998-03-13
W O 97/10240 PCT~B96/00668
oxazolyl, benzU.i~ ~olyl or ben~qY~olyl. E~..F'~s of C3-C8 heteroaryl m~ ;es include
thiazolyl (C3 hetero~rl), furyl (C~ hetero~ryl), and quinolyl (Cg h~teloar~
The term ..~_I...ent~ as used herein, unless otherwise indicated, includes (i)
methods to cure a condition or ~ s that is actively occurring in a ...~..mal, such as
5 a human, or to relieve the s~---ptoms Acsc- - ~' with such condition or ~iisç--c, (ii)
methods to prevent said condition or ' ~ from occurring in a mammal, and (iii)
methods to slow the onset of said cGr.dition or . I;~ Fe in a .namr.,al.
The term ~ther~suticAlly effective amount~ as used herein, unless otherwise
indicated, means an amount effective to block NMDA sites in a mammal, such as a
10 human, or an arnount that is effective in treating or preventing the specifiG conditions
for which the m&r.~r..al is being treated.
R,~rer.ed cG,npounds of forrnula I include those in which R is R'C(O)- or
Rl OC(O) .
Other pr~, . ad compounds of formula I include those in which R is Rl C(O)- and
15 Rl is benzyl, C,s-C10 aryl, or C3-C~ taro&ir~/l.
Other pr~l-ed cor..pounds of formula I include those in which R is RlC(O)- and
R1 is Cl-C~, alkyl or C4-C8 cycloalkyl, and more ,c.r~r~r~bly those in which R' is Cl-C~
alkyl.
Other ,~l t,r~, . ad compounds of formula I include those in which R is R1 OC(O)-
20 and Rl is benzyl, C~-C10 aryl, or Ca-C~ h~tero~yl.
Other pr~ d compounds of formula I include those in which R is R10C(O)-
and Rl is Cl-C~, alkyl or C~-C8 cycloalkyl, and more praferably those in which Rl is Cl-
C" alkyl, and still more pr~r~r~bly those in which Rl is ethyl.
Srec ~;c compounds of formula I include those sslçcted from the group
25 consi~ y of:
(3R ,4S)-7-Ethoxy~4-(4-fluorophenyl)~hydroxyl-piperidin-1 -yl] -chroman-4-ol,
(3R,4S)-7-Propoxy-3-[4-(4-fluorophenyl)~-hydroxyl-piperidin-1 -yl] -chroman-4-ol,
(3R,4S)-7-lsopropoxy-3-[4-(4-fluorophenyl)-4-hydroxyl-piperidin-1 -yl] -chroman-4-ol,
(3R,4S)-7-Cyclopentyloxy-3-[4-(4-fluorophenyl)4-hydroxyl-piperidin-1 -yl] -chroman~ol,
30 and pharmaceutically accept '18 salts of said compounds.
Other specHic compounds of formula I include those sqlQcted from the group
col-si~li..g of:
(3R,4S)-7-Acetoxy-3-[4-(4-fluorophenyl)~hydroxyl-piperidin-1 -yl]-chroman-4-ol,
CA 02232059 1998-03-13
W O 97/10240 PCT~B96/00668
(3R,4S)-7-Pivaloxy-3-[4-(4-fluorophenyl)-4-hydroxyl-piperidin-1 -yl] -chroman-4-ol,
(3R,4S)-7-Benzoyloxy-3 [1 (q fiuGrvphel)yl)~hydroxyl-piperidin-1-yl]-chroman-4-ol~
and pheu.,~nce~tically acceptable salts of said compounds.
Other speçific cv,npounds of formula I include those slls e ~' from the group
5 consisting of:
(3R,4S)-7-Methoxy~ Lonyloxy-3 [q (q ~ lorv,.~h~ .~1) 4hydroxyl-piperidin-1-yl]~hlv" ,~
ol,
(3R,4S)-7-Ethoxycarbonyloxy~[q (4 . ~orvphenyl)~hydroxyl-piperidin-1-yl]-chlvl,,&n 1-
ol,
10 (3R,4S)-7-Propoxycarbonyloxy~[q (~ .~ Ioropher yl)~hydroxyl-piperidin-1 -yl] cl .r
01,
(3R,4S)-7-lsopropoxycarbonyloxy-3-[4-(4-fluorophenyl)4-hydroxyl-piperidin-1 -yl]-
chrv,..an~ol, and pl1&"~P~ceuticAlly accep; bl8 SJtS of said cvr"pounds.
Other sre~;r;C colnpounds of formula I include those S8'~ C' from the group
15 consi~li, .y of:
(3R,4S)-7-ButyloxycLrLvr,~loxy~4-(4-fluvrvph~"~l)~hydroxyl-piperidin-1-yl] duvman 4
ol,
(3R,4S)-7-tert-Butyloxycarbonyloxy-3-14-(4-fluorophenyl)4-hydroxyl-piperidin-1 -yl]-
chrv, "en~ol,
20 (3R,4S)-7-Pentyloxycarbonyloxy-3-[4-(~fluon~vph~,~I)~hydroxyl-piperidin-1-yl] chlvr"~-
4-ol,
(3R,4S)-7-Phenoxy~r,yloxy~[~ (4 . uorophenyl)~hydroxyl-piperidin-1-yl]-cl,romA~-~ol, and ph~,--~-ceutiç~"y A~c~Ft-~'e salts of said col..pounds.
The preser,l invention further relates to a ph&. " .aceutical composition for ll ~lil ,y
25 a ~ '-S~--E or condition, the l._t...ent of which can be facilitated by blocking NMDA
sites in a m_mmal, such as a human, co~"p,isi"g a II-er~eutically effective amount of
a compound of formula 1, or a ph&.",r~ceutiç-"y accep~ salt thereof, and a
ph~"l"nceutic~"y accept~ble carrier.
The presenl invention further relates to a ph&. " ~Aceutic~l composition for treating
30 a di E--E or condition sQ'Qcted from degenerative CNS disorders such as stroke,
Alzheimer's ~ ence~ Parkinson's ~I;seAce, and Hu"li"ylon's ~ nce; eFil~6y, anxiety,
muscl ~ spasr n5, multiinfarct dementia, head trauma, traumatic brain injury, pain, AID
related der..er,lia, hypoglycemia, migraine, amyol.uphic lateral sclerosis, drug and
CA 02232059 1998-03-13
W O 97/10240 PCT~B96/00668
alcohol addiction, drug and alcohol v- ' ~d~ .~N~I SY. nptoms, psychotic COnLi;l;OnS, tinnitus
and urinary illcor,li. ~ence in a mc;ri.,nal, such as a human, col..,Gri~ y a therapeuticaily
effective amount of a cG:"pound of formula 1, or a ph&."~nrel~ticaily a~c~apl~ble salt
thereof, and a pl .~ . . ,~-cel~ic ~ "y - cc~F l ' 1 8 carrier.
The pr~~ent invention further relates to a, .h~ " ~AreuticAI composition for l- ~lli. ~g
an ischemic event arising from CNS surgery, open heart surgery or any procedure
during which the function of the cardiovascular system is COmpl't.lllisf_i in a ".u~...ai,
such as a human, co,.,p,iai.,g a ther~euticnlly effective amount of a compound of
formula 1, or a phar",~-ceutic~lly r~c~t~~ls sait thereof, and a pharm~eutic~lly10 accept r ~ le carrier.
The presellt invention further relates to a method of l,~li"g a ~ Gn~G or
condition, the l,.,~t",er,l of which can be facilitated by blocking NMDA sites in a
mr-r...--ai, such as ahuman, cori.~,,iai"y ~ .-;ni~l~li..g to said ...u~.l,.ai atherapeuticaily
effective amount of a cGI"pound of formula 1, or a ph~",s-ceuticaily ArceFt-~le sait
1 5 thereof.
The pr~se,lt invention further relates to a m ~thod of l~ y a ~'iSf~S6 or
condition s~'scto :' from degenerative CNS disorder:, such as stroke, Alzheimer's
~ e, P~hi~son~s~ 3AcG~ ~md Hu~ y1on~s ,li e~; a, leFsy, anxiety, musc~
sp~r"s, muitiinfarct dementia, head trauma, traumatic brain injury, pain, AIDS related
20 dementia, hypoglycemia, migraine, amyot,opl-ic laterai s~'~rosis, drug and aicohol
A~;ction, drug and ~cohol wilhd~ vai Sy""~t~l"s, psychotic condilions, tinnitus and
urinary incontinence in a ,nar"mal, such as a human, Col"p,isi..~ administering to said
Ill&mmal a ther~peutically effective amount of a compound of formula 1, or a
ph&r.... ....)rceutic~lly n~cept~'- le salt thereof.
The pr~5el 1l invention further relates to a method of treating an ischemic event
arising from CNS surgery, open heart surgery or any pluce.Jure during which the
function of the cardiovascular system is col"prom;sed in a m~-.rnal, such as a human,
comprising ~td~. .isteri..g to said mammal a therapeutically effective amount of a
compound of formula 1, or a pharmaceutically acceplable salt thereof.
Ex~ Fl~s of ~ 3S or conditions suscepl;l ~IQ to treatment by a compound of
formula 1, or a pharm~ceuticnlly accept bl~ salt thereof, include degenerative CNS
~i_order:, such as stroke, Al l,~ er's d;6e--e, Parkinson's ~ eP~S2~ and Hu,lli,lglon's
n~; sFil~Fsy, anxiety, muscu'~- spasr"s, multiinfarct dementia, head trauma,
CA 02232059 1998-03-13
W O 97/10240 PCT~B96/00668
traumatic brain injury, p in, AIDS related der, .e"lia, hypoglycemia, migraine,
amyol,ophic lateral sclerosis, drug and alcohol addiction, drug and alcohol wilhdl ~ '
sy..,plvr ,s, psychotic cor,clilions, tinnitus and urinary incGr,li"ence.
Another 'i~ or eondition suseeplil)lQ to tl ac.l"~er,l by a compound of formula
I, or a ph&"!AceutiQ~lly ~rc~rt 'Ic salt thereof, is an ischel"ic event arising from CNS
surgery, open heart surgery or any proeedure during whieh the function of the
cardiovascular system is cGr"~,,or"-;e ~.
Detailed DescLri~tiGn of the Invention
The prodrugs of formula I are readily prepared using (3R,4S)-3-[4-(4-
fluorophenyl)-4-hydroxy-piperidin-1-yl]~hr~r"~l-4,7-diol (the parent chro",&nol) as a
starting l"al~,ial. This chrom&~ol can be preparad as .Jes~,iLed in U.S. Patent No.
5,356,905 (issued October 18, 1994), U.S. patent aFp' ~oflon serial no. 08/189,479 ffiled
January 31, 1994), and U.S. provis -nal patent aFF'i -tlon of M. Meltz et al., which is
entitled Tl.acas~ ForThe nEs~lutiQn Of Cis nacernic 7-Benzyloxy~ [1 (4 rluGrophenyl)-
4-Hydroxy-Piperidin-1-yl]-Cl,rol"an 4 ol Dibenzoyl-D-Tartrate~ (filed July 20, 1995), all
three of which are herein i~CGI~ orc.lad by l~t,rt,r,ce in their entirety. The starting
"~ ri&l~ and leagenl:. required for the sy"ll,esis of the parent chlon,&)ol are readily
av ' - ' ' 8, either commercially, according to synthetic methods ~ c losefJ in the
literature, or by synthetic ,netl,ods exer"~lifi6 1 in the clescri~liGn provided below.
The parent chromanol can be pr~p red by f~ctional crystallization of the L-
proline ester of ,~cer"ic cis-7-benzyloxy-3 [1 (1 fluoluphenyl)4-hydroxy-piperidin-1-yl]-
chro" ,an4-ol, as described in U.S. patent F-FFllr -~ion serial no. 08/189,479, r~fer,ad to
above. In a pr~f~:r,ad method, the l~solution method desc-liLed in U.S. provisional
patent aF~r' c-~ion entitled ~ C655 ForThe Resol ~tion OfCisr~acernic 7-Benzyloxy-3-
t4-(4-Fluorophenyl)-4-Hydroxy-Piperidin-1-yl]-Chroman4-ol Dibenzoyl-D-Tartrate~,r~"ad to above, is f_llovI~d. In this method, the parent chrol"&nol is prepared by
d;~solv;.lg r~cenlic cis-7-benzyloxy-3-[4-(4-fluorophenyl)-4-hydroxy-piperidin-1-yl]-
chroman-4-ol with an equal molar arnount of dibenzoyl-~tartaric acid in boiling
Aq-lQous ethanol. I acer,.ic cis-7-benzyloxy-3-[4-(4-fluorophenyl)-4-hydroxy-piperidin-1-
yl~-cl~ron)&n~ol is prepared as described in U.S. patent arpli- tion serial no.
08/189,479, le~r,ad to above. The concer,l~lion of Aqueous ethanol is not critical and
may be varied hQ~.een 75% and 95% ethanol (ETOH). A concenl,~lion of
9:1/ETOH:H20 has been found to be effective and is pl~r~r~ad. A sufficient arnount Of
CA 02232059 1998-03-13
W O 97/10240 PCT~B96/00668
the A~lueous ethanol solvent to di~solve the ,c.cer..ic compound is required. This
amount has been found to be about 17ml psr gram of lacen, c compound.
Upon stirring while heating under reflux, the ~acen.ic cG,npound d;~solves to
form a hazy solution which is -"DW6~ to cool with stirring whereupon the (+) isomer,
5 (3R,4S)-7~zyloxy~[~4 (q ~ ~o~uph~ )~hydroxy-piperidin-yl]~h~uman~ol dibenzoyl-~tartrate, prec;l i'-~es and may be COIIE l~ ' by f;l~ ;Gn and w~-hed with aqueous
ethanol. This initial product is of about 90% optical purity. If a higher purity is desired,
the product may be heated again with Aqueous ethanol, cooled and the product
~0115~t~:' and washed. Two such t.aal..)er.l~ were found to yield the (+) isomer of
10 99.4% optical purity in an overall yield of 74%. This procedure is ,urere..ed over the
procedure descriLed in U.S. patent ~lic 'ic n serial no. 08/189,479, lu~u~d to above,
in that it avoids a reduction step with lithium aluminum hydride and is therefore more
suitable for bulk operdions. This pruce. ure also produces a siyl .ifics~nlly higher yield
of the desired product.
The above descriLed (+) isomer can be converted to the parent chromanol by
standard procedures. For example, treatment with dilute base can be used to free the
piperidinyl base and s~hsequent hyd~ùyeneration removes the 7-benzyl group to yield
the parent chlol"anol.
In general, the parent chror. .&nol is converted to the prodrugs of formula I using
20 simple alkylation and acylation metl.G.I~ well known to those skilled in the art and
desc.il~ed in the literature. See, for ex~..rle, J. March, Advanced Orqanic Cl.er.,i~l,y,
4th edition, J. Wiley and Sons, New York, chapter 10 (pages 293-500)(1992).
To pr~p~e the compounds of formula I wherein R is C,-C~, alkyl or C4-C8
cycloalkyl, the parent chro,..P-nol is reacted with 1 molar equivalent (preferably a slight
25 excess) of RX where X is an iqppro,G,i~le leaving group, such as halogen, tosylate,
triflate, or mesylate. The reaction is pe.h.r...ed in a reaction inert solvent, such as
dimethylfc,,-..~-~'s (DMF), dimethylsulfoxide (DMSO), ac6.l0ne, methyl ethyl ketone, or
tetrahydrofuran (THF) at a ter"per~lure from about 0~C up to the reflux temperature of
the solvent. To f~ t~ this reaction, it is preferable to include 1-2 molar equivalents
30 of a base, such as an alkali metal carbonate (e.g. pot~ ci~n carbonate), trialkylamine
(e.g. triethylamine), or sodium hydride.
In another method, compounds of formula I wherein R is C,-C,~ alkyl or C4-C8
cycloalkyl are prepared by reacting the parent chron,anol with a Cl-C~, alcohol (e.g.
CA 02232059 1998-03-13
W O 97/10240 PCT~B96/00668
ethanol) or a C~-C8 cyclic alcohol (e.g. cyclope~ ol) in the presence of
l,i~Jhenylphosphine and a dialkyl azodicarboxylate (e.g. diethyl azodicarboxylate). This
is the so called Mitsunobu re&_tion which is well known in the literature (Synthesis 1
1981).
To prepare the compounds of formula I wherein R is R1C(O)- or RlOC(O)-, the
parent chrol~,anol is leactecl with an applo,bii~lQ acylating agent such as an anhydride
(e.g. acetic anhydride) or an acid halide (e.g. benzoyl chlo.ida or ethyl chlor~,fui",ala)
in a r &ction inert solvent (e.g. THF or methylene chl~ ide) at a ter,.per~lure ranging
from about 0 ~ C to the reflux te. ~ .~,erc.1~lre of the solvent. This, eaction can be facilitated
by the ~dditiol, of a base such as sodium hydride or a trialkylamine (e.g. triethylamine).
Another ...~ll.od to prep&re compounds of formula I wherein R is RlC(O)- is to
react the parent cl ,--,,nanol with an acid cG.,~sponding to the group to be added (e.g.
pivalic acid or phenylacetic acid) employing a ~ cagenl, such as
dicy. IohexylcarLc ~ 9 or CL. bor,~l dii~ ,18, to activate the acid prior to coupling
15 with the chr~m~ol. In this re&_tion the acylating agent is pl~prred in situ. This
process is described in J. March, Advanced Or~anic Cl,er,.i~tl-l 4th Ed., ~;I,~ter 10,
re~u~d to above, and is well known to those skilled in the art. The pr~fe..ed
conliliGI)s for the leactions in this method are the same as those desc,iLed in the
preceding par~ ph. DMF can be added as a cosolvent to I ~ e dissolution of the
20 .-~&ger.l~ if necesss-ry.
The prodrugs of the ~reser,l invention are converted in vivo into the parent
chr~ ol, a ssle c~ c NMDA s~r,lagon;~1. They are 11 ,erefure useful in the treatment of
J;_orde,~ and con- ilions, the 1,~a1",er,1 of which can be facilitated by blocking NMDA
sites in a mamms~l. Ex~mr ~s of such d is-~~es and condi1ions include degenerative
25 CNS disorders such as stroke, Akl~ e~s ~;~eA~Se, P&rki,.rorls ~ e and
Hu..1i..y1Or.'s .li~,&asG; eFi ~Fsy anxiety, musa~ spa~--,s multiinfarct dementia head
trauma, traùmatic brain injury, pain AIDS related dementia hypoglycemia migraineamyc.1,ophic lateral sc ~rosis drug snd alcohol addiction drug and alcohol withdrawal
sy.... ...p1u",s, psychotic conditions, tinnitus and urinary i"col,1i"ence.
The prodrugs of the presen1 invention can be r~ F ,i~lered as pharl-.&ceuticallyaccep~ hl~ salts of the compounds of formula 1. Such salts include conventional acid
addilion salts and cation salts. Tha cGr.,pounds of formula I contain an amine group
which is basic, and so are cArA~le of forming such salts. Said salts include, but are
CA 02232059 1998-03-13
W O 97/10240 PCTnB96/00668
-10-
not limited to, those with HCI, HBr, HNO3, H2SO", H3PO~, CH3SO3H, e-CH3C~sH4SO3H,
CH3CO2H, gluconic acid, tartaric acid, lactic acid, maleic acid and succinic acid. The
salts can be pr~pared by conver,liGnal methods, e.g., by combining a compound offormula I with at least one molar equivalent of the acid in a su4~'18 solvent.
The prodrugs of formula I convert to the parent chlcmanol through metAhol ~
proces~es within the body of a m-ri,l"al. The parent chrc.",anol has sel~o~ c
neu, oprot~li~/a ~ ,li:_cher, ,io and -~ccil~o, ~ amino acid l~ ki, ,y activity that reflects its
valuable utility in the treatment of neurological ~ rd6rs such w eFilEFsy and stroke,
and degenerc~ e CNS Ji~orJer:, such as Al~l ,e "er's ~Ise~s2, r~ hinson's -~;s ~ - - e and
Hulllillyl~n~s ~iseAse. In the systemic Ir~&l~eriL of such ~iseA es einFloy;.,g a
therapeutically effective amount of a compound of formula 1, the dosA~e is typically
from about 0.02 to 20 mg/kg/day (0.001-1 g/day for a typical human weighing about 50
kg) in single or divided doses, regardless of the route of admini~.l,alion. Of course,
depending upon lthe exact cGmpound and the exact nature of the individual illness,
doses outside this range may be prt.sc,ibed by the attending physician.
The compounds of the pr.~,ser,l invention are generally admini~.lered in the form
of phar" ,aceutical comrQsitions con ,p, i ~;"y at least one of the compounds of the
formula 1, together with a pl1~..,~ceutically Acceptr'-le vehicle or diluent. Such
CGi nr osilions are generally formulated in a conver,lional manner utilizing solid or liquid
20 vehicles or diluents as ~ppn~p,i~l,a to the mode of desired LJ~n~ lion: for oral
admini~ lion, in the form of tablets, hard or soft gelatin cAps~'es, suspensions,
granules, powders and the like; snl.po~ J,ies, including rectal; for parenle,~l
administration (intravenous, s~hcut~neous, intramuscu'--), in the form of inje~ tle
solutions or suspensions, and the like; and for topical admin;~l,c.lion, in the form of
25 solutions,lotions,~oi.,l,.,e,.l~,salvesandthelike.
Since the prodrugs of the present invention are more stable in solution than theparent chromanol, the prodrugs are better suited than the parent chromanol for
intravenous admini~ lioll. Intravenous administration is the pr~l~r.ed parenteral
method of administration. The preparation of solutions for intravenous administration
30 is known to those skilled in the art. The intravenous solution should be osmotically
b&l~ced and have a neutral pH. Applop,iate solutions include 5% dextrose solution,
isotonic saline, and phosph&le-buffered salina.
CA 02232059 1998-03-13
W O 97/10240 PCT~B96/00668
In the f~ v; ng Pl.apar~lion and E)-~mFles, unless otherewise in~
I~&_tiGils and other pu ceJures were run at ambient ter"per~ re (20-25~C), and all
non-n1ueo~s reactions were run under nil,o~en for conveni~:)ce and generally to
~ ,.,~cilni~e yields. All solvents/diluents were dried according to standard published
"rocedures or pu, ~:hased in a ~r~l~ ied form. All reactions ware stirred eithermF~nelically or mech~li~ally. NMR spectra are recolded at 2S0 or 300 MHz and arereported in ppm. The NMR solvent was CDCI3 unless otherwisa s,~e~ d. IR spectra
are lepGilecl in cm l, generally specifying only strong signals. The f_llov~i.,gabbreviations are used: DMF for dimethylfoi"~ar,li !e, THF for tetrahydrofuran, HRMS
for high r~solution mass spectrum.
PREPARATION
(3R 4S)-7-Benzyloxy-3-r4-(4-fluoroPhenvl)-4-hydroxv-piperidin-yl1-chroman-
4-ol dibenzoyl-D-tartrate
A. I t~cerl .ic cis-7-banzyloxy-3-[4-(4-fluorophenyl)~hydroxy-piperidin-1 -yl]-
chro".&n~ol (2.079, 4.6 mmol) and dibenzoyl-D-tartaric acid (1.65g, 4.6 mmol) were
suspended in 30 ml, 90% ethanol/water. The resulting mixture was stirred and heated
to reflux; an addilional 5 ml, 90% ethanol/water was added and a hazy solution was
obtained. The resulting solution was stirred overnight at room temperature. The solid
which formed was colle~: ~ by filtration and washed twice with 3ml, 95% ethanol to
yield 1.559 (83.4%) of the title product which was shown to be of 87% purity by HPLC.
B. The above product (1.29) was suspended in 21.4 ml of 90% EtOH:H2O,
stirred and heated under reflux for 1.5 hours and then cooled to room temperature.
The solid product was ccllQcted by filtration and washed with two 3 ml portions of 90%
ETOH:H2O. The yield was 1.19 of 98.0% optical purity.
C. The p,ocedure of step B was repe~tçd with the product of step B yielding
97% of a product which had 99.4% optical purity.
Optical purity was determined by HPLC using a 250 x 4.6 mm Chiralpak0 AD
column (Chiral Technologies, Exton, PA) with the mobile phase comprising 600 ml
hexane, 400 ml isopropanol,1 ml trifluoroacetic acid and 0.5 ml diethylamine. The flow
rate was 0.7 ml/min with an injaction volume of 20 ~I containing 0.1 to 0.4 mg
sample/ml. Detection was set for 220 nm.
CA 022320~9 1998-03-13
W O 97/10240 PCT~B96/00668
-12-
EXAMPLE 1
(3R .4S)-7-Acetoxv-3-r4-(4-fluorophenvl)~hydroxyl-piperidin-1 -yll -chroman-4-olA mixture of (3R,4S)-3-[4 (4-fluorophenyl) 1 hydroxy]-piperidin-1-yl-chroman4,
7-diol (0.50 9, 1.39 mmol), 2-ac~toxyL,er,z ~ ic acid (0.26 g, 1.44 mmol). 1-(3-
5 dimethylaminopropyl)~ethylcarbodiimide hydrochloride (0.28 9. 1.46 mmol) and 4-
dimethylaminopyridine (0.17 g, 1.39 mmol) in methylene chloride (2S mL) was stirred
at a m~ c- nl te, . .~erc.t.lre ovemight. The . eF-ction was conce~ tad and the residue was
partitioned b~tween ether/ethyl acetate (2:1) and water. The phases were separated
and the organic layer was washed with brine, dried over calcium sulfate, and
10 concel.l.~led to afford a white solid. The solid was triturated with ether/ethyl acetate
(2:1) and then recrystallized from metl.anol/ethyl acetate to give 0.062 g (12%) of
(3R,4S)~acetoxy-3-[4-(4-fluorophenyl)4-hydroxy]piperidin-1 -yl-chru, . .arl-4-ol which
had the f ll~w:.,g characteristics: melting point 181.~182~C: [a]D = +86.13~ (c = 0.31
in methano!). Analysis c~ 'sd forC22H24FNO5; C, 65.82; H, 6.03; N, 3.49. Found:
15 C, 65.33; H, 6.12; N, 3.41.
EXAMPLE 2
(3R,4S)-7-PivaloxY-3-r4-(4-fluorophenY1)-4-hydroxy-Piperidin-1 -vll-chroman4-ol
A mixture of (3R,4S)-3-[4-(~fluorophenyl)~hydroxy]-piperidin-1-yl-chroman-4,
7-diol (0.30 g, 0.83 mmol), pivalic acid (0.195 mL, 1.70 mmol), 1 -(3-
20 dimethylaminopropyl)-3-ethylc& Lodiimide hydroch'o ;de (0.33 9, 1.72 mmol) and 4-
dimethylaminopyridine (0.20 g. 1.64 mmol) in methylene chloride (15 mL) was stirred
at ambient temperature ovemight. The reaction was concer.l-dted and the residue was
partitioned bet~ecn ~ther and water. The phases were separated and the organic layer
was washed with brine, dried over calcium sulfate, and concenl.~ted to afford an oily
25 white solid. The solid was triturated with ~thyl acetate and then flash chromatographed
on silica gel (1 x 4 inches, packed with 25% ethyl AcetAts/hexane). Elution proceeded
as follows: 25% ethyl P~retAt~/hexane (200 mL), nil; 50% ethyl ~o-cet~ot~/hexane (200 mL),
nil; 75% ethyl ~r,et~ts/hexane (200 mL) and ethyl acetate (200 mL), white solid product.
This solid was further triturated with ether/hexane (1 :1) and air dried to afford 0.10 9
30 (22%)of(3R,4S)-7-pivaloxy-3-[4-(4-fluorophenyl)~hydroxy-piperidin-1-yl]-chroman4-ol
which had the following characteristics: melting point 219-220~C; [a']D = +83.9~ (c =
0.31 in methanol). Analysis e-' u'o~ed for C2sH28FN05; C, 67.70; H, 6.82; N, 3.16.
Found: C, 67.61: H, 6.88: N, 3.24.
CA 02232059 1998-03-13
W O 97/10240 PCTnB~ 6~8
EXAMPLE 3
(3R,4S)-7-r~ u~,oxv~r4-(4-fluGroPhQnyl)~hvdroxv-piDeridin-l -vll-chrolnan-4-ol
In a dry three neck flask, sodium hydride (60% .Ji..pe,;,ion in oil, 0.043 9, 1.07
- mmol) was washed with hexane to remove the oil. DMF (3 mL) was added and the
mixture was chilled to 0~C. (3R,4S)-3 [~1 (1 rluorophenyl)-4-hydroxy- piperidin-1-yl]-
chroman-4,7-diol (0.350 g, 0.974 mmol) was added and the mixture was stirred for 30
min. Propyl iodide (0.104 mL, 1.07 mmol) was added and the reaction was allowed to
stir at Ln.'- e ll lempeiall~re for 1 hour. The mixture was diluted with ethyl acetate and
extracted with 1 N P~queous lithium chloride. The aqueous lithium chloride was back
extracted with ethyl acetate (twice) and the combined org&n c phase was washed with
lN ~queous lithium chloride and brine. The organic layer was dried over calcium
sulfate and concer,l-dtad to a white solid. The solid was recrystallized from
isopropanol/methanol to afford 0.130 g (33%) of (3R,4S)-7-propoxy-3-[4-(4-
fluorophenyl)4-hydroxy-piperidin-1-yl~-cl"~."&n~ol as a white solid which had the
following oh&r~cteri~tics: melting point 148.5-149.5~C; [O]D = + 83.33~ (c = 0.27 in
methanol). Analysis ~ Q~'9~ for C25H28FNO~: C, 68.81; H, 7.û3; N, 3.49. Found:
C, 68.65; H, 7.05; N, 3.38.
EXAMPLE 4
(3R.4S)-7 A~ ',oxv-3-r4-(4-fluorol~heriyl)~hydroxv-piperidin-1-yl1-chroman~-ol
In a dry three neck flask, sodium hydride (6û% dispersion in oil, 0.0275 g, 0.689
mmol) was w--hed with haxane to remove the oil. DMF (2 mL) was added and the
mixture was chilled to 0~C. (3R,4S)-3 [~ (q Fluorophenyl)4-hydroxy-piperidin-1-yl]-
chrc,r"~4,7~iol (0.225 g, 0.626 mmol) was added and the mixture was stirred for 30
minutes. Methyl iodide (0.043 mL, 0.689 mmol) was added and the reaction was
allowed to stir at an~-~nt ter"peral,~re for 1 hour. The mixture was diluted with ethyl
acetate and extracted with 1N ~gueous lithium chlo.ide. The aqueous lithium chloride
was back exl,~.cted with ethyl acetate (twice) and the couJhi.~ed organic phase was
washed with 1 N ~lueous lithium chlaride and brine. The organic layer was dried over
calcium sulfate and cGncerll,alecl to a white solid. The solid was recrystA"i~ed from
I. ,ell ,~ ,ol to afford 0.105 g (44%) of (3R,4S)-7-methoxy~[4-(4-fluorophenyl)4-hydroxy-
piperidin-1-yl]-chr~")&n4-ol as a white solid which had a melting point of 181-1 82~C.
Analysis calculated for C21H24FN05: C, 67.55; H, 6.48; N, 3.75. Found: C, 67.41; H,
6.47; N, 3.60.
CA 02232059 1998-03-13
W O 97/10240 PCT~B96/00668
EXAMPLE 5
(3R,4S)-7-Ethoxv~r4-(4-fluoroPhenvl) 1 hydroxy-piPeridin-1-yll-chrul"&n-4-ol
In a dry three-neck flask, sodium hydride (60% .li~per:.ion in oil, 0.037 g, 0.918
mmol) was washed with hexane to remove the oil. DMF (3 mL) was added and the
mixture was chilled to 0~C. (3R,4S)~[4-(~Fluorophenyl)4-hydroxy-~ ri " 1-1-yl]-
chrom~n4,7-diol (0.300 g, 0.835 mmol) was added and the mixture was stirred for 30
min. Ethyl iodide (0.073 mL, 0.918 mmol) was added and the r~a~,1ion was allowed to
stir at ~h Ih;Q~ ,t temperature for 1 hour. The mixture was diluted with ethyl acetate and
e~t,~_ted with 1N A~lueous lithium chla.ide. The A~lueous lithium chloride was back
~AII~.cted with ethyl acetate (twice) and the co,.,'-i ,ed or~ , phase was washed with
1N a~ueous lithium chloride and brine. The oryanic layer was dried over calcium
sulfate and concer,l,~ted to a white solid. The solid was recrystallized from methanol
to afford 0.175 9 ~54%) of (3R,4S)-7-ethoxy-3-[~(4-fluorophenyl)~hydroxy-pipQridin-1-
yl]-chf~,i "an~ol as a white solid which had the f~ . .g char~ct~ri~lics: melting point
191-192~C; la]D = + 87.47~ (c = 0.375 in rneU~ol). Analysis c~ 'ot~d for
C22H26FNO4: C, 68.20; H, 6.76; N, 3.62. Found: C, 68.02; H, 6.76; N, 3.40.
EXAMPLE 6
(3R,4S)-7-Ethoxvc&rLonyloxy-3-r~(4-fluoroPhenvl)~hydroxv-piperidin-1 -vll-chrul "nn-
4-ol
A mixture of (3R,4S)-3 [q (1 fluorophQnyl)4-hydroxy-piperidin-1-yl]-chroman-4,
7-diol (0.378 9,1.05 mmol), l~ol--~si~m carbonate (0.345 9. 2.5 mmol) and 1-chloroethyl
ethyl carbonate (0.155 mL, 1.15 mmol) in acetone (10 mL) was gently refluxed
overnight. The reaction mixture was diluted with ethyl acetate, filtered and
concenl~tffd. The residual solid was flash chn,",aloy,~phcd on silica gel (1 x 3inches) with elution proceeding as follows: 25% ethyl Acet~h3/hexane (500 mL), nil;
50% ethyl ncet~tQ/hex_ne (50 mL), unweighed unidentified impurity; 50% ethyl
Acet~te/hexane (600 mL), 0.355 9 (78%) of (3R,4S)-7-ethoxycarbonyloxy-3-[~(4-
fluorophenyl)4-hydroxy-piperidin-1-yl]-chroman-4-ol which had the following
characte,i-lics: melting point 153-154~C; [a]D = + 83.0~ (c = 0.49 in methanol).Analysis c~'c~'o~ed for C231126FNO6: C, 64.03; H, 6.07; N, 3.25. Found C, 63.75; H,
6.10; N, 2.95.
CA 02232059 1998-03-13
W O 97/10240 PCT~B96/00668
- ~ - EXAMPLE 7
(3R 4S)-7-lso~rolJoxy~r4-(4-fluorophenvl)4-hydroxy-,. ~ -1 -vl1-chror"an-4-olIn a dry three-neck flask, sodium hydride (60% ~JiSp~l~iGn in oil, 0.037 9 0.918mmol) was w--hed with hexane to remove the oil. DMF (3 mL) was added and the
5 mixture was chilled to 0~C. (3R,4S)-3-l4-(4-Fluorophenyl)-4-hydroxy- piperidin-1-yl]-
chroman-4 7-diol (0.300 g 0.835 mmol) was added and the mixture was stirred for 30
minutes. Isopropyl Lrc,i"ic ~ (0.086 mL, 0.918 mmol) was added and the reaction was
~"~v.ed to stir at Lr"' ~nl ten,pe,~lure ovemight. Addilional sodium hydride (0.01 9)
and isop,opyl br~,~"i 8 (0.016 mL) were added and the l~t tion was heated to 60~C
10 ovemight. The mixture was diluted wKh ethyl acetate and eA~ ted with 1N aqueous
lithium chloride. The A~lueous lithium chla.ide was back ~xll..cted with ethyl acetate
(twice) and the COrll . ,ed organic phase was washed with 1 N aqueous lithium chloride
and brine. The or~lic layer was dried ovar calcium sulfate and concer,l,c.led to a
white solid. The solid was flash chlorn~luy,..plleJ on silica gel (1 x 43 inches, packed
15 in methylene chloride) with elution proceeding as follows: 3% m~th&nol / methylene
chlcride (250 mL), 0.191 9 of white solid product. This product was recrystpll 7ed from
isopropDnol to afford 0.154 g (46%) of (3R4S)-7-isoplopoxy-3-[4-(4-fluorophenyl)~-
hydroxy-piperidin-1-yl~-cl ,rom~-n~ol as a white solid which had the following
characteristics: melting point 151-152~C; [O]D = + 88.15~ (c c 0.27 in methanol).
20 Analysis ~- u'oted for C23H28FN04: C, 68.81; H, 7.03; N, 3.49. Found: C 68.80; H
7.07; N 3.38.
EXAMPLE 8
(3R,4S)-7-CvcloPentvloxy-3-r4-(4-fluoroPhenyl)4 hvdroxv-Piperidin-l-yll-chro,nan4-ol
In a dry three neck flask, sodium hydride (60% dispersion in oil, 0.037 g, 0.91825 mmol) was washed with hexane to remove the oil. DMF (3 mL) was added and the
mixture was chilled to 0~C. (3R,4S)-3-[4-(4-Fluorophenyl)-4-hydroxy- piperidin-1-yl]-
cl ,ro",an4 7-diol (0.300 g 0.835 mmol) was added and the mixture was stirred for 30
min. Cyclopentyl br~.i"i~e (0.098 mL 0.918 mmol) was added and the reaction was
"ov.~d to stir at ambient le""~erature over"i_ ,l. Additional sodium hydride (0.014 9)
30 and cyclopentyl bromide (0.027 mL) were added and the reaction was heated to 60~C
ovemight. The mixture was diluted with ethyl acetate and extracted with 1 N aqueous
lithium chlc.ide. The aqueous lithium chloride was back extracted with ethyl acetate
(twice) and the combined organic phase was washed with 1 N aqueous lithium chloride
CA 02232059 1998-03-13
W O 97/10240 PCT~B96/00668
and brine. The organic layer was dried over calcium sulfate and concer.l,c~ll3d to a
white solid. The solid was flash chr~ l ~hed on silica gel (1 x 43 inches, packed
in methylene chlo.ide) with elution proceeding as follows: 3% methanol/methylenechloride (200 mL), 0.220 g of white solid product. A sample of the product was
5 recrystallized from isoprop&nol/ethyl acetate to afford white crystals of (3R,4S)-7-
cyclopentyloxy-3-~4-(4-fluorophenyl)~hydroxy-piperidin-1-yl]-chroman4-ol which had
the f~ w:..g cha;~clt~ .lics: melting point 180-181 ~C; [O]D = + 80.73~ (c = 0.275 in
methanol). Analysis c~'c~ ted for C25H30FNO~: C, 70.24; H, 7.07; N, 3.28. Found:C, 69.98; H, 7.07; N, 3.13.