Note: Descriptions are shown in the official language in which they were submitted.
CA 02233173 2004-05-25
DOPAMINE AND SEROTONIN TRANSPORTER
LIGANDS AND IMAGING AGENTS
FIELD OF THE INVENTION
This invention relates to novel tropane-based ligands that display selective
binding to central nervous system receptors, such as dopamine and serotonin
transporters (reuptake sites), and have utility, inter alia, as imaging agents
for the
central nervous system. Also within the scope of this invention are methods
for
utilizing these ligands as diagnostic agents. Methods for preparing the novel
ligands of
the invention and intermediates useful in their preparation are also
presented.
GOVERNMENT SUPPORT
The work reported herein was supported in part by National Institute of Health
NS-18509 and NS-24538.
BACKGROUND
Neural transmitters are chemicals in the brain that are used to send messages
from one brain cell to another. Neurotransmitters bind to special receptor
proteins in
the membranes of nerve cells, like a lock in a key, triggering a chemical
reaction
within the cell. Dopamine is an example of a central nervous system (CNS)
neurotransmitter.
Dopamine is a catecholamine belonging to a class of biogenic amine
neurotransmitters, along with norepinephrine, serotonin, and histamine. The
CA 02233173 1998-04-16
WO 97/14445 1'CT//1TS96/16908
-2-
catecholomines (particularly dopamine and serotonin) are involved in the
control of
movement; mood; attention; and possibly, certain endocrine, cardiovascular,
and stress
responses. Imbalances in neurotransmitter production have been implicated in a
variety
of mental and physical disorders, such as Parkinson's disease (PD). It is thus
desirable
to diagnose and monitor such imbalances and to monitor the effectiveness of
drugs and
substances that affect brain chemistry.
New and powerful imaging methods that enable one to assess the living brain
in vivo and thereby monitor brain chemistry and the effectiveness of drugs and
substances that affect brain chemistry have been developed. Methods such as
positron
emission tomography (PET) and single photon emission computed tomography
(SPELT) involve administering to a patient a radioactive tracer substance
comprising a
ligand that binds to the presynaptic or postsynaptic neuroreceptors in the
patient's
brain. Emissions (primarily gamma rays are emitted from the positrons or
photons
from the radioactive tracer) are measured. These emissions are indicative of
the
number and degree of occupancy of blocking of the neuroreceptors. The number
of
neuroreceptors and the degree of occupancy or blocking is calculated utilizing
a
mathematical model, and compared with an infra-person or inter-person control
to
determine the degree of drug response. Further treatment of the patient with
drugs is
based on the comparisons made. For these methods to be useful, however, a
ligand
that has a high specificity and affinity for the desired receptor is required.
It is believed that certain radioactive ligands may be selective for dopamine
transporters and are thus potentially useful in evaluating changes in
dopamine. function
in vivo and in vitro, especially for patients with Parkinson's disease (PD),
which is
characterized by a selective loss of dopamine neurons in the basal ganglia and
substantia nigra. Recently, a large number of dopamine transporter imaging
agents
based on cocaine or its closely related congeners, tropane derivatives, have
been
reported. (Carroll, F. L, et al, Med. Res. Rev. I99S, 15, 419-444; Carroll, F.
L, et
al., J. Med. Chem. 1994, 37, 2865-2873; Carroll, F. L, et al., J. Med. Chem.
1995,
38, 379-388). The regional brain distribution of cocaine is largely
concentrated in the
basal ganglia, where the dopamine neurons are located. [1'C]-N-methyl labeled
cocaine
(Yu, D.-W., et al., J. Med. Chem. 1992, 35, 2178-2183; Fowler, J. S., et al.,
Synapse 1992, 12, 220-227) is a very useful PET (positron emission computed
CA 02233173 2004-05-25
-3-
tomography) ligand for studying the pharmacology and drug effects of cocaine
itself;
however, additional modifications on the cocaine molecule have led to
development of
positron emission tomography (PET) imaging agent, CFT (WIN35,428) (Clarke,
R.L.
et al. J.' Med. Chem. 1973, 16, 1260-1267; Clarke, R.L. et al. J. Med. Chem.
1978,
21, 1235-1242; Frost, J. J., et al., Ann. Neurol. 1993, 34, 423-431; Wong, D.
F., et
al., Synapse 1993, 15, 130-142), and single photon emission computed
tomography
(SPELT) imaging agents ~B-LIT (Innis, R. B., et al., Proc. Natl. Acad. Sci.
U.S.A.
1993, 90, 11965-11969; Seibyl, J. P., et al., J. Nucl. Med. 1996, 37, 222-228;
Kuikka, J. T., et al., Eur. J. Nucl. Med. 1995, 22, 682-686; Neumeyer, J. L.,
et al.,
J. Med. Chem. 1994, 37, 1558-1561.), IPT (Goodman, M. M. et al. J. Med. Chem.
1994, 37, 1535-1542; Mozley, P. D., et al., J. Nucl. Med. 1996, 37, 151-159),
and
other related derivatives that display much higher binding affipity and
selectivity to
dopamine reuptake sites. Both of the agents for PET and SPELT imaging
displayed
excellent specific uptake in the striatum (basal ganglia) area and are more
suitable than
_ GBR12,935 in imaging dopamine reuptake sites (dopamine transporters).
(Kilbourn, M.
R., Life Sci. 1988, 42, 1347-1353). The dopamine reuptake site ligands are
useful in
evaluating changes in dopamine reuptake sites in vivo and in vitro, especially
for
patients with PD. Recent publications describing :he use of ["C]-CFT
(WIN35,428)
(Frost, J. J., et al., Ann. Neurol. 1993, 34, 423-431) and ['~I]-~-LIT (Innis,
R. B., et
at., Proc. Natl. Acad. Sci. U.S.A. 1993, 90, 11965-11969; Innis, R. B., Eur.
J. Nucl.
Med. 1994, 21, 1-5) suggest a strong correlation between the decrease in
localization
of dopamine transporters in the anterior putamen area and PD symptoms. -
Currently, PET and SPELT imaging studies of dopamine transporters are
under investigation. Recent publications using ["C]-CFT and [1~I]-~B-LIT
suggest a
strong correlation between the decrease in localization in the anterior
putamen area and
PD symptoms. See Innis, R. B. et al. Proc. Natl. Acad. Sci. U.S.A. 1993, 90,
11965-
11969; Innis, R. B. Eur. J. Nucl. Med. 1994, 2l , 1-5; Frost, J. J. et al.
Ann.
Neurol. 1993, 34, 423-431.
Central nervous system (CNS) receptor function-has also been successfully
evaluated in vivo using C" (T,n = 20 minutes, j3+) or Fl$ (Tln = 120 minutes,
a+)
labeled agents for positron emission tomography (PET) imaging and '~I (Tl,~ =
13
CA 02233173 2004-05-25
-4-
hours, 159 KeV) labeled agents for single photo, emission computed tomography
(SPELT) imaging. See Eckehnan, W. C. Nucl. Med. Biol. 1992, 18, iii-v; Fowler,
J.
S. et al. Ann. Rep. Med. Chem. 1989, 24, 277-286; Fowler, J. S. et al. Ann.
Rep.
Med. Chem. 1990, 25, 261-268.
A ligand that is -being widely investigated as an agent for diagnosing and
treating PD patients is ['~I]-LIT. However, one of the drawbacks of ['~I]-~-
LIT is
the length of time ( > 18 hours) required for reaching optimal uptake ratio in
the target
area (semi-equilibrium state) (the basal ganglia (BG)) versus the nontarget
area (the
frontal cortex (CTX)). Because of the need for agents with faster equilibrium
times,
new ligands such as ['23I]-IPT (N-(3-iodopropen-2-yl)-2(3-carbomethoxy-3~3-(4-
chlorophenyl)tropane) and ["~I]-S-LIT-FP are under investigation, both of
which reach
equilibrium in less than one hour. See Kung, M.-P. et al. Synapse 1995, 20,
316-324;
Malison, R. T. et al. J. Nucl. Med. 1995, in Press; Mozley, P. D. et al. J.
Med.
Chem. 1994, 37, 1558-1561.
Despite the success in developing such new techniques using PET and SPELT
for imaging CNS receptors, their use in routine procedures is hampered by the
cost
([123p costs about $30/mCi) and the limited supply of the three isotopes
mentioned
above, 1231, 1'C, and 'sF, all of which are produced by cyclotron.
A radionuclide that is widely used in diagnostic nuclear medicine is
technetium
[~"'Tc] (T;,~ = 6 hours, 140 KeV). It is well established that when Tc-99m
pertechnetate (Tc04-), the most commonly available starting material, is
reduced in the
presence of a reducing agent, such as stannous chloride, and a "soft"
chelating ligand,
including NZSZ and NS3, a [Tc"OJ3+N~S2 er [Tc"O]3 +NS3 center core is formed.
Technetium [~"'Tc] is readily produced by a [~"'Tc]/Mo-99 generator, and its
medium
gamma-ray energy emission (140 KeV) is suitable for gamma camera detection
with a
far less cost ($l/mCi). In the past ten years, significant progress has been
made in
defining technetium chemistry using the chemical level of [~Tc] (Tln = 2.1 x
105. yr),
and non-radioactive rhenium as a surrogate substitute, that will potentially
benefit
millions of patients who receive [~''"Tc] agents for routine nuclear medicine
~tHagnosis.
Over 85 % of the routine nuclear medicine procedures currently performed use
CA 02233173 2004-05-25
radiopharmaceutical methodologies based on ['~"'Tc] . In addition, comparable
rhenium
complexes labeled with ['~Re] (T'"~ = 90 hours) or ['~Re] (TIn = 17 hours) may
also
be potentially useful for in vivo imaging of dopamine transporters.
The potential of developing [~'"Tc] labeled agents for CNS receptor imaging is
well recognized. Several recent reports demonstrate that it is possible to
incorporate
[TcvO]3+N2S2 (bisaminoethanethiol, BAT) into potential receptor selective
imaging
agents for muscarinic receptors, vesamicol sites, and steroid hormone
receptors. See
Del Rosario, R. B. et al. Nucl. Med. Biol. 1994, 21, 197-203; Chi, D. Y. et
al. J.
Med. Chem. 1994, 37, 928-935; DiZio, J. P. et al. Bioconj. Chem. 1991, 2, 353-
366;
DiZio, J. P. et al. J. Nucl. Med. 1992, 33, 558-569; O'Neil, J. P. et al.
Inorg. Chem.
1994, 33, 319-323; O'Neil, J. P. et al. Bioconj. Chem. 1994, S, 182-193;
Jurisson, S.
et al. Chem. Rev. 1993, 93, 1137-1156; 3urisson, S. S. et al. Nucl. Med. Biol.
1995,
22, 269-281; 3urisson, S. et al. Chem. Rev. 1993, 93, 1137-1156; Steigman, J,
et al.
National Academy Press: Washington, D.C. 1992; Lever, S. Z. et al. Nucl. Med.
Biol. 1994, 21, 157-164, Chi, D.Y. et al. Am. Chem. Soc. 1993, 115, 7045-7046.
However, these [99mTc] imaging agents have demonstrated limited success in in
vivo
studies, this is believed to be attributable to the low initial brain uptake
and poor
selective binding to the receptor after attaching the molecules with [99"'Tc].
Recently, a series of neutral and lipophilic conjugated complexes, containing
N-alkylthiolatotropane, aminobisethylthiolato and a [~'"'Tc]Tc03+ center core,
were
prepared and evaluated as CNS dopamine transporter imaging agents in rats_
(Meegalla,
S. K., et al., J. Am. Chem. Soc. 1995, 117, 11037-11038). One of the
compounds,
[~"'Tc] technetium, [methyl 3-(4-chlorophenyl)-8-(2-mercaptoethyl)-8-
azabicyclo
[3.2.1]octane-2-carboxylato-S][(2,2'-(methylimino) bis[ethanethiolato]](2-)-
N,S,S']oxo,
displayed low initial uptake in rat brain (0.1 ~~ at 2 minutes post
intravenous injection),
but the striatal/cerebellar (ST/CB) ratio reached 3.50 at 60 minutes after an
intravenous
injection. The Rhenium complexes were also discussed. These neutral [~'"Tc]
labeled
three plus one complexes designed for brain imaging, as well as mixed-ligand,
aminothiol plus aminothiol complexes (two plus two complexes) designed as
[~''"'Tc]
steroid analogs are quite stable in .vivo and in vitro. However,. since these
agents are
CA 02233173 2004-05-25
-6-
not designed to cross the blood-brain barrier, they are inferior for imaging
CNS
receptors, such as dopamine and serotonin.
Technepine compounds containing technetium and rhenium have been
investigated as potential dopamine transporter agents. Madras, B. K., et al.,
Synapse
1996, 22, 239-246. These compounds showed poor biodistribution, which is
believed
to be a result of the amide moiety present on the N2S2 ligand.
Despite its attractive physical properties, technetium is a difficult element
for
designing suitable SPECT ligands. Technetium is a transition metal and
requires a
complexing agent to stabilize it at different valence states. Steigman, 1992,
supra.
Valance states can vary from plus 7 (as pertechnetate) to zero (0), depending
on the
reaction conditions and chelating agents used during preparation. After
complexation,
the molecules invariably become big and bulky, which is the limiting factor in
designing a molecule targeted to a specific biological process(es). Additional
requirements for Tc-99m labeled complexes as CNS receptor imaging agents are:
i)
small (molecular weight < 750), with good lipophilicity (partition coefficient
-50-1,000); ii) high binding affinity (Kd < 10 nM) and high selectivity; iii)
minimum
brain uptake in rats should be 0.5 % dose/organ at 2 minutes post intravenous
injection.
The [~'''"Tc] brain imaging agents that have been developed thus far have been
aimed at measuring perfusion or its changes due to a particular disease state,
and not as
diagnostics directed to evaluating neuronal functions, such as the
biochemistry of
dopamine or serotonin receptors. See Mastrostamatis, S. G. et al. , J. Med.
Chem.
1994, 37, 3212-3218; Spies, H. et al. , Technetium and Rhenium in Chemistry
and
Nuclear Medicine, 1995, 4, 243-246; S. G. Editoriali, Ed.: Padova, Italy 1995,
4,
243-246; Spies, H. et al. Angew. Chem. Int. Ed. Engl. 1994, 33, 1354-1356;
Chi, D.
Y. et al. J. Amer. Chem. Soc. 1993, 115, 7045-7046; Chi, D. Y. et al. J. Med.
Chem.
1994, 37, 928-935.
Thus, there remains a need for new imaging agents, such as CNS receptor-
based imaging agents, for evaluating neuronal functions that do not present
the
problems associated with prior agents, as discussed above. These agents should
have
good selectivity, affinity, and specific activity for the target. Several
additional factors
are also of importance such as radiochemistry (preparation time for short-
lived labeled
CA 02233173 1998-04-16
WO 97/14445 PCT/CTS96/16908
_7_
agents), suitable modeling for kinetics of receptor uptake and retention, and
metabolism. The imaging agents should also be economical and readily
available.
The present invention addresses these, as well as other needs, by providing
novel tropane-based technetium- or rhenium- labeled imaging agents useful,
inter alia,
for imaging the CNS, in particular dopamine and serotonin receptors, to
diagnose CNS
abnormalities. It is also expected that the novel compounds of the invention
possess
pharmacological activity. It is believed that the compounds of the invention
are the
first imaging agents of their kind displaying specific regional uptake
directly
proportional to dopamine neuronal distribution in the brain.
SUMMARY OF THE INVENTION
This invention presents, inter alia, a novel class of technetium- or rhenium-
labeled dopamine or serotonin transporters imaging agents based on a tropane
core.
The compounds of the invention transfer through the blood brain barner, which
makes
them good candidates for diagnostic and therapeutic agents for the central
nervous
system.
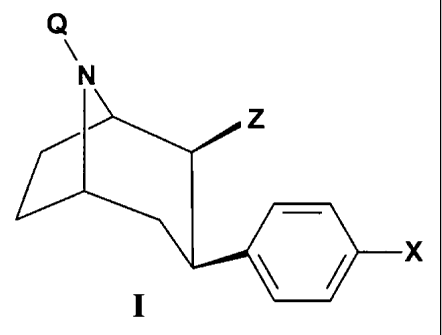
In one aspect of this invention, the compounds of the invention have the
following general formula (I):
Q~~ N
'- Z
X
I
wherein X is selected from the group consisting of H, C1-C4 alkyl, F, Cl, Br,
and I; Q
is selected from the group consisting of H, Cl-CS alkyl, Al, A2, A3, A4, A5,
A6, A.,,
and A8; Z is selected from the group consisting of R, CONRR,, CORI, C02Ri,
COZRa,
COaAI, CO2A2, COZA3, COZA4, COZAS, CO2A6, C02A~, COZAg, COA i . COA2, COA3,
COA4, COAS, COA6, COA,, COAB, C 002, CH20A3, CH20A~, CH20A5, CH20A6,
CHZOA.,, CH20A8, CH2NHA1, CHZNHA2, CHZNHA3, CHZNHA4, CHZNHAS,
CH2NHA,6, CH2NHA.,, CHZNHAB, Al, A2, A3, A4, As, As, A.,, and A8; Y is -
(CHZ)n; R
is H, C~-CS alkyl; Rl is H, C1-CS alkyl; R2, is a heterocyclic amine;
CA 02233173 1998-04-16
WO 97/14445 PCT/fIS96/16908
_g_
CHiNHAs, CHZNHA6, CH2NHA,, CHZNHAB, A~, Ax, A3, A4, As, As, A,, and As; Y
is -(CH2)"; R is H, C,-Cs alkyl; Ri is H, Cl-Cs alkyl; RZ is a heterocyclic
amine;
A1 is
( SRS RCS.
- ~,.~ NV
A2 is
>_ ~-~~
A3 is
sR3 k3~
NH'"
~._ Y _~
O
n
A4 is ~ Sv I I ~
~_._. Y, ~.. ~ ,;~
0
As is ~ ~3 R3~
~t _ _ Y.J
i
- Y
A, is S\ N' ~
M
~-r s' 's'
A8 S SR 3
~- Y"
CA 02233173 1998-04-16
WO 97/14445 PCT/US96/16908
-9-
E is C,-CZ alkyl; n is 0, 1, 2, 3, 4, 5; M is selected from the group
consisting of Tc
and Re; R3 is selected from the group consisting of H, CR,, substituted or
unsubstituted C1-CS alkoxy, substituted or unsubstituted C6-C24 aryl, and
substituted or
unsubstituted phenylallcoxy; and R4 is selected from the group consisting of H
and CI-
Cs alkyl, optionally substituted with a substituted or unsubstituted phenyl
group;
' provided that, one of Q and Z, and only one of Q and Z, includes a moiety
selected
from the group consisting of At, A2, A3, A, As, A6, A,, and As.
The compounds of the invention include compounds that comprise an NZSZ
ligand (bis-aminoethanethiol) in which the ligand complexes with either the
technetium
or rhenium, or "three plus one complexes" (NS3) where a tropane thiolate and
an
aminobisethanethiolate ligaad are involved in complexation. In one series of
the NiS2
complexes, the N2S2 ligand is attached to the 2(3 position of the tropane
core, at the Z
position of Formula I. In another series of the NZS2 complexes, the NZS2
ligand is
attached to the bridge head nitrogen of the tropane core. The "three plus one
complexes" may have the ligand attached to either the 2(3 position of the
tropane core
or the bridge head nitrogen of the tropane core.
This invention further relates to compounds that are useful, inter alia, as
intermediates for preparing the compounds of the invention. For example,
compounds
having the following general Formula B, are useful, inter alia, as
intermediates for
preparing certain three plus one complexes of the invention. A compound of
Formula
II has the following general formula:
i
-r'r Nw.
~SH H ~S
11
wherein E is C1-C2 alkyl.
A compound of Formula II can be used in combination with a compound of
Formula I wherein X is C1-Cd alkyl, F, Cl, Br, or I; Q is As, wherein Y is -
(CH~a
and n is 0, 1, 2, 3, 4, or 5; and Z is C02R1 or COZR2, wherein R, is Ci-Cs
alkyl and
t R2 is a heterocyclic amine.
The compounds of Formula I, where A" is A8, and the compounds of Formula
II are useful, inter alia, as intermediates for preparing certain three Plus
one complexes
CA 02233173 1998-04-16
WO 97/14445 PCT/US96/16908
-10-
of the invention, and further useful as kit components. Kits comprising these
compounds and kits comprising other Formula I compounds are also within the
scope
of this invention.
In other aspects of the invention, methods of utilizing the novel Iigands of
the
invention in diagnostic imaging techniques are presented.
Test results indicate that the novel Iigands of the invention are highly
selective
for CNS receptors, particularly dopamine receptors, which make these compounds
useful, when appropriately labelled, as imaging agents for the evaluation of
such
receptors. The compounds of the invention should also have pharmacological
activity
associated with such CNS receptors as a result of their high selectivity.
BRIEF DESCRIPTION OF THE DRAWINGS
Figure 1 depicts a general reaction scheme that may be used for synthesizing
compounds of Formula I comprising a two plus one complex and where the N~S~
ligand is attached to the 2~3 position of the tropane ring via an amide
linkage.
Figure 2 depicts a general reaction scheme that may be used for synthesizing
compounds of Formula I comprising two plus one complexes and having the NZS2
ligand in the 2(3 position of the tropane ring and two plus one complexes
having an
amide on the NzS2 Iigand.
Figure 3 depicts general reaction schemes that may be used for synthesizing
two plus one complexes containing either rhenium or technetium and two plus
one
complexes having an amide moiety on the NZSZ ligand.
Figure 4 depicts an HPLC chromatogram of a racemic mixture of a
technetium containing compound of the invention.
Figure 5 depicts the ratios of regional brain uptakes at 60 minutes post
intravenous injection of [~''"'Tc]TRODAT-1 (1.19a) in control rats and rats
pretreated
with haldol (1 mg/kg, iv) or (3-CIT (1 mg/kg, iv) 5 minutes prior to
radiotracer
injection. Values shown are means ~ SD (n=3-4, p < 0.05, student t-test). ST:
striatum; HP: hippocampus; CX: cortex; CB: cerebellum. The ST area, where
dopamine transporters are Located, displayed selective regional brain uptake,
with the
highest concentration ratio (ST/CB = 2.6). Pretreatment with ~3-CIT, which
competes
CA 02233173 1998-04-16
WO 97/14445 PCT/IJS96/16908
-lI-
with dopamine transporter binding, showed blocking of the specific uptake of
j99mTc]TRODAT-1 (1.19a) in the ST area.
Figure 6 depicts a transaxial, coronal and sagital SPECT images (1.34 mm
thick) of monkey brain at 60-90 post-intravenous injection of 9 mCi of
[99mTc]TRODAT-1 (1.19a). SPECT images were acquired by a Picker T3000
scanner. The SPECT images were co-registered with the same sections of MRI of
the
same baboon. A high accumulation of [99mTc]TRODAT-1 (1.19a) was observed in
caudate and putamen, areas of the brain where dopamine transporters are
concentrated.
Figure 7 depicts a time course of regional brain uptakes as measured by
SPECT imaging following intravenous injection of [99mTc]TRODAT-1 (1.19a) in a
baboon. Regions of right and left caudat and putamen, cerebellum (CB) is
expressed
as counts/pixel/minute.
Figure 8 depicts a general reaction scheme that may be used for synthesizing
two plus one complexes where the NaS2 ligand is connected to the bridge head
nitrogen
. of the tropane core.
Figure 9 depicts a general reaction scheme that may be used for radiolabelling
an N2S2 ligand.
Figure 10 depicts a reaction scheme that may be used for preparing a rhenium-
containing compound of the invention.
Figure 11 depicts HPLC chromatograms of a racemic mixture of an NZSZ
compound of the invention where the N2S2 ligand is attached to the bridge head
nitrogen of the tropane _core and an HPLC chromatogram of the resolved
isomers.
Figure 12 depicts a reaction scheme that may be used for synthesizing a
tridentate ligand that is useful as an intermediate for preparing three plus
one
complexes of the invention.
Figure 13 depicts a reaction scheme that may be used for synthesizing a
monothiol compound that is useful as an intermediate for preparing three plus
one
complexes of the invention.
Figure 14 depicts a reaction scheme for the synthesis of a three plus one
complex from Formula II and Formula III intermediates.
Figure 15 depicts an X-ray crystallographic structure of rhenium complex
containing a tropane moiety, 3.23. Meegalla, 1995, supra.
CA 02233173 2004-05-25
-
Figure I6 depicts ratios of regional brain uptakes at 60 minutes post-
intravenous injection of 3.25 in contml rats and rats pretreated with haldol
(1 mg/kg,
iv) or ~i-CIT (1 mg/kg, iv} at 5 minutes prior to radiotracer injection.
Values shown
are means ~ SD (n=3-4, p < 0.05, student t-test). ST: striatum; HP:
hippocampus;
CX: cortex; CB: cerebellum. The ST area, where dopamine transporters are
located,
displayed selective regional brain uptake, with the highest concentration
ratio (ST/CB
= 2.6). Pretreatment with /3-CIT, which competes with dopamine transporter
binding,
showed blocking of the specific uptake of 3.25 in the ST area, where dopamine
transporters are located. '
Figure 17 depicts an in vitro autoradiogram of 3.25 binding to dopamine
transporters in a rat brain section demonstrated that it localized in caudate
putamen and
olfactory tubercle, areas known to have a high dopamine transporter density. A
comparable section labeled with [1~I]IPT, a known iodinated dopamine
transporter
ligand, Kung, 1995, supra, showed an almost identical regional labeling
pattern. The
eoronal section of rat brain shown corresponds to Plate 17 in the atlas by
Paxinos and
Watson. Paxinos, G. et al. The Rat Brain In Stereotaxic Coordinates (New York,
Academic Press 1986).
Figure 18 is a comparison of HPLC traces of 3.23 (UV) and 3.25 (radio trace)
on a C-18 column (Partisil 10-ODS-3, 250 x 4.6 mm) with MeOH/NH4HC03 (O.1M,
pH 7, ratio 8:2, flow rate 1 mllmin).
Figure 19 depicts a reaction scheme where the Z moiety of Formula I is an
ester moiety.
Figure 20 depicts a reaction scheme where the Z moiety of Formula I is an
ether moiety.
Figure 21 depicts a reaction scheme where the A moiety of Formula I is in the
Z position and the Z moiety is COA2.
Figure 22 depicts a reaction scheme where the A moiety is in the Z position
and the Z moiety is CO2A2.
Figure 23 depicts a reaction scheme where the A moiety is in the Z position
and is connected to the tropane core by an amide linkage.
CA 02233173 1998-04-16
WO 97/14445 PCT/US96/16908
-13-
Figure 24 depicts a reaction scheme where a compound of Formula I is
prepared from a direct linkage reaction by eliminating an ester moiety at the
Z
position.
Figure 25 depicts a reaction scheme for the synthesis of a compound of
S Formula I using cocaine as a starting material.
DETAILED DESCRIPTION OF THE INVENTION
The term "ligand," as used herein, refers to compounds chelating with a
central atom in a complex compound, and may be used generally to refer to the
compounds of the invention. It is contemplated that certain of the compounds
of the
invention may be chiral or have chiral centers and form R and S isomers
(enantiomers), which may be resolved, for example, using a chiral HPLC column.
The racemic mixtures as well as the resolved isomers of the ligands are within
the
scope of this invention.
_ A "complex compound, " as used herein, refers to any group of chemical
compounds in which part of the molecular bonding is of the coordination type.
In the
context of this invention, "coordination compound" refers to compounds having
a
central atom or ion and a group of ions or molecules surrounding it.
In the context of this invention, "tridentate ligand" refers to a chelating
agent
having three groups capable of attachment to a metal ion. As used herein, the
terminology "chelating agent" refers to orgaruc compounds in which atoms form
more
than one coordinate bond with metals in solvent. _
As used herein, the term "alkyl" refers to radicals that are formed from the
loss of a hydrogen from an alkane (CnH2n+2)- The alkyl compounds may be
straight or
branched chain compounds, cyclic or acyclic, and further substituted with
hetero atoms
(i. e. , O, S, N) . Also included within this definition are structural
isomers (i. e. , chain
isomers, position isomers, and functional isomers) and stereoisomers (i. e. ,
enantiomers
and diasteriomers).
In the context of this invention, the term "aliphatic" refers to any carbon-
hydrogen containing compounds having either saturated or unsaturated bonds
(alkanes,
alkenes, alkynes). Such compounds may be cyclic or acyclic, straight or
branched
chains, and may further be substituted with hetero atoms (i. e. , O, S, N).
The term
CA 02233173 1998-04-16
WO 97/14445 PCT/US96/16908
-14-
"aromatic," as used herein, refers to any compound that contains at least one
benzene
ring. The term "aryl," as used herein refers to a compound having at least one
benzene radical or benzene ring, and may be substituted with other aliphatic
compounds.
The terminology, "heterocyclic amines, " refers to aliphatic compounds having
a ring structure that incorporates other atoms in addition to carbon atoms.
Heterocyclic amines include, without limitation, pyrrole and its derivatives,
imidazole
and its derivatives, pyridine and its derivatives, pyrimidine and its
derivatives,
qunioline and its derivatives, piperidine and its derivatives, pyrrolidone and
its
derivatives, purine and its derivatives, pyrrolidine and its derivatives, and
morpholine
and its derivatives. Derivatives refers to compounds having the core
heterocyclic
structure (e.g., pyrrole) substituted with one or more of an aliphatic moiety,
aromatic
moiety, alkoxy moiety, phenoxy moiety, amine moiety, vitro moiety, nitrite
moiety,
halogens, or hetero atoms.
As used herein, the term "phenoxy" is used in its conventional sense and
refers to, generally, phenyl radicals attached to a molecule through an
oxygen. It is
contemplated that the phenoxy compounds of the invention may be further
substituted.
Also included within this definition are structural isomers (e. g. , chain
isomers, position
isomers, and functional isomers) and stereoisomers (e. g. , enantiomers and
diasteriomers).
The term "alkoxy," as used herein, is used in its conventional sense and
refers
to, generally, alkyl radicals attached to a molecule through an oxygen. Alkyl
radicals
are generally derived from an aliphatic hydrocarbon by removal of one hydrogen
atom.
It is contemplated that the alkoxy compounds of the invention may be further
substituted with, for example, other aliphatic compounds. Also included within
this
definition are structural isomers (i. e. , chain isomers, position isomers,
and functional
isomers) and stereoisomers (i. e. , enantiomers and diasteriomers).
The term "substituted," as used herein refers to single or multiple
substitutions '
of a molecule with a moiety or moieties distinct from the core molecule.
Substituents
include, without limitation, halogens, hetero atoms, vitro moieties, amine
moieties,
nitrite moieties, hydroxy moieties, alkoxy moieties, phenoxy moieties, other
aliphatic
CA 02233173 1998-04-16
W~ 97/14445 PCT/US96/16908
-15-
or aromatic moieties. Substituted compounds may be referred to as derivatives
of the
core structure.
As used herein, the terminology "radionuclide-containing" compound refers to
any compound that contains at least one atom of a compound capable of being
used in
radiopharmaceutical techniques, such as technetium or rhenium. As used herein,
"technetium-containing compound" refers to any compound that contains at least
one
atom of technetium (Tc). In the context of this invention, the term "rhenium-
containing compound" refers to compounds having at least one atom of rhenium
(Re).
The terminology "protecting group," as used herein, is used in its
conventional
sense and refers to compounds that are used for blocking reactive sites on a
multifunctional molecule to prevent such reactive sites from taking part in a
chemical
reaction.
In one series of compounds the N2S2 ligand is in the 2(3 position of the
tropane
core. These compounds may be synthesized by the general reaction schemes shown
in
Figures 1-3. For example, a ligand, such as 1.16a, 1.16b or 1.17, is dissolved
in
ethanol and hydrochloric acid. To this is added hydrochloric acid and a
reducing
agents, such as Sn-glucoheptonate (containing 136 micrograms SnCl2 and 200
micrograms Na-glucoheptonate, pH 6.67) and 50 microliters EDTA solution.
[99mTc]Pertechnetate in saline is then added to the reaction mixture, which is
then
heated for about 30 minutes at 100°C. After cooling to room
temperature, the reaction
mixture is neutralized with a saturated sodium bicarbonate or phosphate buffer
solution. It is possible that tropane derivatives containing a bis-
aminoethanethiol group
will form stereoisomers.
Other methods for preparing these kinds of compounds are depicted in Figures
21-24, but any such methods known to those skilled in the art may be used
without
departing from the spirit of the invention.
Another series of the two plus one complexes has the N2Sz ligand attached to
' the bridge head nitrogen of the tropane core. These compounds may be
prepared
according to the general reaction schemes depicted in Figures 8-10.
Radiolabeling was
achieved by a method similar to that disclosed above in connection with the
compounds
having the N2S2 ligand in the 2(3-position of the tropane core. The tropane
derivatives
CA 02233173 2004-05-25
-16-
(2.6a) containing a bis-aminoethanethiol group are believed to form
stereoisomers.
(Table 2.1 a, b, c).
Other methods for preparing these kinds of compounds are depicted in Figures
19, 20, and 25, but any such methods known to those skilled in the art may be
used
without departing from the spirit of the invention.
The three plus one complexes (NS3) (those containing, for example, an A,
moiety) may generally be made from two key intermediates -- a tridentate
ligand
aminodithiol (Formula II) and a monothiol ligand (Formula I, where Z is A~,
which is
particularly applicable when the A" moiety is in the Q position of Formula I.
These
intermediates may be prepared by using methods analogous to those in the
reaction
schemes set forth in Figures 12 and 13. The monothiol intermediate compound
can be
prepared according to the reaction scheme of Figure 13 or reactions analogous
thereto.
Generally, a demethylated tropane derivative is prepared from cocaine in
4 steps, as previously reported. See Meltzer, P.C. et al. J. Med. Chem. 1993,
36, 855-
862. N-alkylation of the tropane derivative is achieved by reacting it with a
triphenylmethyl thioether (S-trityl) protected 2-bromoethanethiol and 3-
bromopropanethiol. Dhar, T.G.M. et al. J. Med. Chem. 1994, 37, 2334-2342. The
protecting group (trityl) may then be cleaved by acid hydrolysis or with heavy
metal
ions to yield the monothiol ligand of Formula II. See Ohmomo, Y. et al. J.
Med. Chem.
1992, 35, 157-162; Kolb, U. et al. Inorg. Chem. 1994, 33, 4522-4530;
Dhar,1994,
supra. Any protecting groups known to those skilled in the art may be used
without
departing from the spirit of the invention.
For example, the particular tropane derivatives of formulas 3.19-3.22 may be
prepared as follows: a halogenated nortropane derivative is refluxed with a
triphenylalkyl mercapto (trityl) compound to form a protected mercapto
halogenated
nortropane intermediate. The tritylated nortropane derivative is then
dissolved in a
suitable solvent system, such as trifluoromethyl carboxylic acid and anisole,
followed
by the addition of a mercury oxide, such as Hg(OAc)2,.to cleave the protecting
group.
The resultant mixture is stirred at 0°C for 30 minutes. The reaction
mixture is then
concentrated in vacuo to obtain a brownish red oil, which is then dried undei
high
vacuum for 1 hour. Anhydrous ether is then added to the above oil and the
mixture is
CA 02233173 2004-05-25
-17-
then sonicated for 15 minutes, followed by magnetic stirring for an additional
30
minutes. This results in the formation of a colorless precipitate, which is
then
collected by suction filtration. The collected precipitate is then dried under
a high
vacuum for 15 minutes and then redissolved in ethanol. Hydrogen sulfide gas is
then
bubbled throughout the ethanol solution for 15 minutes, forming a black
precipitate.
The resultant black precipitate is then filtered through a thick pad of celite
The
resultant filtrate is then concentrated in vacuo to obtain a colorless oil,
which is then
dried in a high vacuum for 30 minutes. An aqueous solution of hydrochloric
acid and
an aqueous solution of ether is then added to the dried oil and the resulting
mixture is
vigorously stirred for 15 minutes. The mixture is then transferred to a
separating
funnel where the aqueous layer is separated and basified with concentrated
ammonium
hydroxide and the resulting colorless product is ex~racted with methylene
chloride (20
x 2 mL). The methylene chloride layer is then dried with sodium sulfate and
concentrated in vacuo to yield the desired tropane derivative. Other tropane
derivatives of Formula II may be made by analogous methods that would be known
to
those skilled in the art.
This invention presents a novel series of compounds based on a tropane core,
which compounds comprise NZSZ Iigands and three plus one complexes that
comprise
an NS3 ligand.
Tridentate ligand aminodithiol intermediates, Formula II, can be synthesized
according to the reaction scheme of Figure 12 or reactions analogous thereto.
In certain preferred embodiments the aminobisethanethiol ligand has gem-
dimethyl groups, 3.6, and can be synthesized according to methods known in the
art, as
shown in Figure 12. See, e.g., Ohmomo,1992, supra. The aminobisethanethiol
ligand
~ without gem-dimethyl groups, 3.7, can be synthesized according to methods
known in
the art. Kolb,1994, supra. The N-ethyl substituted aminobisethylthiols, 3.8,
may be
prepared according to the procedure employed for the synthesis of 3.7, using N-
ethyl
bischloroethyl amine as the starting material. The other N-substituted
bisethanethiols,
3.9 to 3.12, may be prepared by bis-alkylation of benzyl-, iso-butyl-,
morpholinoethyl-
~d ~~N-bisethylamino)ethyl-amine with ethylene sulfide, respectively, as
disclosed in
CA 02233173 2004-05-25
-18-
Corbin, 1984, supra.
In certain preferred embodiments the tridentate ligand aminodithiol (Formula
II) will be N,N -di[(2-(4'-methoxybenzylthio)-2-methylpropyl)] amine (3.4).
This can
be prepared by adding a solution of BH3~THF to a solution of N-[(2-(4'-
methoxybenzylthio)-2-methylpropyl)] 2-(4'-methoxybenzylthio)-2-methyl-
propionamide
(3.3), and heating the resultant mixture at reflux under nitrogen gas for 12
hours. The
reaction mixture is then cooled in an ice bath, and water is carefully added.
The
resulting solution is then concentrated in vacuo to obtain a viscous oil that
is suspended
in hydrochloric acid. This mixture is then heated at reflux for 1 hour. The
reaction
mixture is then cooled in an ice bath and then basified with concentrated
ammonium
hydroxide. The product (3.4) is then extracted with methylene chloride and
purified
on silica.
In other preferred embodiments the tridentate ligand aminodithiol (Formula II)
, is N,N-di[(2-(4'-methoxybenzylthio)-2-methylpropyl))-methylamine (3.5). This
can be
prepared by adding NaBH3CN to a soluiicn of coripound 3.4 and methanol in
acetonitrile. The resulting mixture is then stirred for 15 minutes and glacial
acetic acid
is added dropwise until the solution tests neutral. The mixture is then
stirred for 45
minutes and the solvents are removed. Potassium hydroxide is then added to the
residue, and the resulting mixture is extracted with ether (3 x 10 mL). The
ether
extracts are combined and washed with potassium hydroxide and then extracted
with
hydrochloric acid. The acid extracts are combined and neutralized with solid
potassium
hydroxide and then re-extracted with ether (3 x 10 mL). The ether layers are
combined
and concentrated in vacuo to obtain a viscous oil (3.5), which is purified on
silica.
In certain other preferred embodiments, the tridentate ligand aminodithiol
(Formula II) is N,N-di[(2-mercapto)-2-methylpropyl)j-methylamine (3.6). This
compound can be prepared by mixing the amine compound 3.5 and anisole in
trifluoracetic acid and cooling to 0°C. Then Hg(OAc)2 is added and the
mixture
stirred at 0°C for 15 minutes and concentrated in vacuo at room
temperature. The
residue is then dried under a high vacuum for 30 minutes and dry ether is
added. The
resulting solid is then collected by suction filtration and redissolved in
ethanol:
Hydrogen sulfide gas is then bubbled through the ethanol solution for 15
minutes and
CA 02233173 1998-04-16
WO 97/14445 PCT/US96/16908
-19-
the black precipitate that forms .is then filtered through a thick pad of
celite. The
filtrate is concentrated in vacuo to obtain a colorless oil that is then dried
under a high
vacuum for 30 minutes. Hydrochloric acid and ether are added to the oil, and
the
resulting mixture is vigorously stirred for 15 minutes and then transferred to
a
separating funnel. The aqueous layer is separated and is then basified with
concentrated ammonium hydroxide, and ti'~e resulting colorless product is then
extracted with methylene chloride (20 x 2 mL). The methylene chloride layer is
then
dried in sodium sulfate and then concentrated in vacuo to obtain a viscous oil
(3.6)
which is then stored under a blanket of argon.
Other tridentate ligand aminodithiols of Formula II for use as intermediates
in
preparing other compounds of Formula I, may be prepared by analogous methods
that
would be known to those skilled in the art.
The dithiols seem to have only moderate stability and tend to produce a white
solid, presumably disulfides, over time. However, the monothiols, 3.15-3.18,
seem to
_have fairly good stability when stored at a low temperature under NZ. The X-
ray
crystallographic structure of the Re-complex 3.23 (Figure 15), shows that none
of the
reaction conditions employed in preparation of the monothiols 3.15-3.18 and
the Re
complex altered the stereochemistry at the C-2 position of tropane ring.
The compounds of Formula I comprising a three plus one complex may be
synthesized by using the monothiol intermediate and tridentate aminodithiol
intermediate (Formula II) in methods analogous to those illustrated in the
general
reaction scheme of Figure 14.
Other compounds ef Formula I where Z is an ester moiety and the A" moiety
is in the Q position may be synthesized according to the general reaction
scheme
depicted in Figure 19 or ones analogous thereto.
Other compounds of Formula I where Z is an ether moiety and the A°
moiety
is in the Q position may be synthesized according to the general reaction
scheme
depicted in Figure 20 or ones analogous thereto.
In other embodiments of the invention, Z is COA2 and may be prepared
according to the general reaction scheme depicted in Figure 21 or methods
analogous
thereto.
CA 02233173 2004-05-25
-20-
Generally, methods of.making compounds of the invention having an ether
linkage couple an alcohol and a halide using NaH.
In certain other embodiments, Z is C02A2 and may be prepared according to
the general reaction scheme depicted in Figure 22 or ones analogous thereto.
In
addition to the ester linkage described above and in Figure 22, amide linkages
are also
contemplated and may be synthesized according to the general reaction scheme
depicted in Figure IZ or ones analogous thereto.
Other illustrative synthetic methods include those using a direct linkage and
elimination of the ester functional group as depicted in Figure 23. See, e. g.
, Kung, H.
F. et al. , Nucl. Med. Biol. , 1991, 18, 215-226 (Reference A); and
Kozikowski, A. P.
et al. , J. Med. Chem. , 1995, 38, 3086-3093 (Reference B)..
Persons skilled in the art, once armed with the present disclosure, will be
able
to select the appropriate starting materials and prepare any of the claimed
compounds
of the invention.
The compounds of this invention lend themselves easily to the formation from
materials that may be assembled in kit form and provided to users. When the
novel
compounds of the invention are to be used as diagnostic tools, such as imaging
agents,
the compounds are labeled with a radionuclide, such as technetium. Kits for
forming
the three plus one complex imaging agents of the invention may contain, for
example,
a lyophilized composition comprising the tridentate ligand aminodithiol
(Formula II)
and the monothiol ligand (Formula I, where Z is Ag), a reducing agent,
preferably the
lyophilized composition is a powder. A user would dissolve the lyophilized
composition comprising the tridentate Iigand aminodithiol and the monothiol
ligand in
ZS saline and hydrochloric acid. A reducing agent, such as stannous
glucoheptonate, and
sodium [~'"Tc] pertechnetate, in.saline solution is then added to the mixture.
The
mixture is then kept at room temperature for about 30 minutes. This is
followed by
neutralizing the reaction mixture with sodium carbonate. The resulting
compounds are
extracted into ethyl acetate and the compounds are purified by HPLC. ,
Preferred compounds of Formula II for inclusion in the kits of the invention
are those where, independently or in combination, A is Cl or Fl; B is A6; and
D is
C02R5, wherein RS is C,-CS alkyl, or C02R2, wherein RZ is a heterocyclic
amine.
CA 02233173 1998-04-16
WO 97/14445 PCT/US96/16908
-21-
Preferred compounds of Formula II for inclusion in the kits are those where E
is Cl-CZ
alkyl.
Reducing agents suitable for practicing this invention include, but are not
limited to, stannous glucoheptonate, stannous chloride, sodium bisulfate, or
~ 5 combinations thereof, with stannous glucoheptonate being preferred. Any
other
reducing agents known to those skilled in the art may be used without
detracting from
the spirit of the invention.
Radionuclide-containing compounds suitable for practicing this invention
include, but are not limited to, sodium [~'"Tc] pertechnetate (Na~'''"Tc04),
and sodium
pertechnetate (NaRe04), with sodium [99"'Tc] pertechnetate (Na~''"TcO,~ being
preferred. Other radionuclide-containing compounds known to those skilled in
the art
may be used without detracting from the spirit of the invention.
Kits containing N2S2 (bas-aminoethanethiol) complexes are also within the
scope of the invention. These kits would generally comprise a lyophilized
powder of a
- the N2S2 compound, stannous chloride, sodium glucoheptonate, EDTA,
hydrochloric
acid, and ethanol. A user would dissolve the lyophilized compound ethanol and
hydrochloric acid. A reducing agent, such as stannous glucoheptonate, and
sodium
[99mTc] pertechnetate, and EDTA is added to the mixture. The mixture is then
autoclaved for about 30 minutes. After the autoclaving, the mixture is allowed
to cool
to room temperature. The amount of the solution necessary for analysis is
mixed with
sodium phosphate before use. The information discussed above in connection
with the
three plus one complexes is applicable here.
For dopamine transporter imaging agents, the target area of the brain is the
striatum, where dopamine transporters are highly concentrated. The cerebellum
region
is suitable for use as the background region, because it has no dopamine
transporters. .
The specific uptake is measured by the ratio of % dose/gram of striatum
divided by
dose/gram of cerebellum (ST/CB ratio)--the higher the value, the better the
specific
uptake, and the more promising as a dopamine transporter imaging agent.
When using the compounds of the invention as diagnostic agents (such as in
imaging techniques) or as therapeutic agents, it is preferable to keep the Tc
or Re
complex core of the compounds of the iiavention small. There are stringent
structural
requirements for achieving both neutral-lipophilicity and dopamine transporter
binding
CA 02233173 1998-04-16
WO 97/14445 PCT/LTS96/16908
-22-
affinity for this series of tropane derivatives. The addition of gem-dimethyl
groups to
a compound of Formula I, dramatically enhances lipophilicity and significantly
reduces
specific binding to the striatal regions of the brain, where dopamine reuptake
sites are
located. Fine tuning of size and lipophilicity of technetium or rhenium
complexes
appears to be important for brain uptake and receptor binding. It is also
important that
the compounds of the invention have a neutral charge for them to be able to
cross into
the brain, and therefore, it is preferable that only one nitrogen on the
tropane core be
substituted. It is also preferable that the COZR moiety (Z) and the
halogenated phenyl
moiety (X) be in the beta position. The examples show that substitution of the
NZS2
moiety (An moiety) with gem-dimethyl groups destroys the specificity of the
compound
(Table 3.1), same is true when the N of the A" moiety is substituted with
groups larger
than C3 or with additional amino groups (Table 3.1). Both of these
observations are
not readily predictable by simple estimation of partition coefficient or
molecular weight
or size. The partition coefficient value is also very important for the
evaluation of
potential brain imaging agents, because they must be neutral and lipophilic in
order to
penetrate the intact blood-brain barrier. Generally, the optimal range of
partition
coefficient values for good brain uptake are between 100-1000. Apparently, the
in viva
biodistribution and receptor binding site for dopamine transporters place
unique and
highly selectively requirements on this novel groups of agents.
Certain preferred Formula I compounds useful as imaging agents are those
comprising a three plus one complex as the ligand and where, independently or
in
combination, Q or Z includes a compound selected from the group consisting. of
an A,
or Ag moiety or groups including an A ~ or A $ moiety; X is Cl or Br; Z is CO
~ for
COZRa; and E is C1-CZ alkyl. A preferred compound is one where X is Cl; Q is A
.nor
A$ and n is 2; M is Tc; and Z is CO~CH3. Other preferred compounds are those
where
X is Cl; 'Q is A~ or A$ and n is 2; M is Tc; and Z is CO zR a wherein R 2 is
piperidine,
pyrolidine, or morpholine. Still other preferred compounds are those where X
is Br; Q
is A~ or A8 and n is 2; M is Tc; and Z is COZCH3. Other preferred compounds
are
those where X is Br; Q is A~ or Ag and n is 2; M is Tc; and Z is COZR~,
wherein RZ is
piperidine, pyrolidine, or morpholine.
In other preferred embodiments .the Formula I compounds useful as imaging
agents are those comprising an N2S2 ligand in which the ligand is attached to
the
CA 02233173 1998-04-16
WO 97/14445 PCT/US96/16908
-23-
bridgehead nitrogen of the tropane core, and where X is C1-C4 alkyl, F, Cl,
Br, or I;
Q is Al, Aa, A3, or A4; Z is CO~RI, or C02R~ In other preferred embodiments X
is
Cl or Br; Q is A2 and n is 2; Z is C02CH3; and M is Tc. Other preferred
embodiments are those where X is Cl or Br; Q is AI and n is 2; Z is COZCH3;
and M
S is Tc. In still other preferred embodiments, X is Cl or Br; Q is AZ and n is
2; Z is
4
COZRZ, wherein R2 is piperidine, pyrolidine, or morpholine; and M is Tc. In
still other
preferred embodiments, X is Cl or Br; Q is A1 and n is 2; Z is C02R2, wherein
RZ is
piperidine, pyrolidine, or morpholine; and M is Tc. The Tc-99m labeled tropane
derivative, 2.6a, displayed a significant brain uptake and specific uptake to
the striatum
area of the rat brain. This compound showed a distinctive difference from both
the
prior. art, technepine, and 3.25.
Particularly preferred Formula I compounds are those comprising an NZSZ
ligand (bis-aminoethanethiol) where the ligand is attached to the 2~i position
of the
tropane core, and where X is C~-C4 alkyl, F, Cl, Br, or I; Z is Al, A2, A3, or
A4; Y is
-(CH~ri ; R3 is H; and Q is H or Ci-C4 alkyl; and M is Tc. A particularly
preferred
compound is one where Q is CH3, X is Cl; Z is A3 or A4; and Y is CHZ. A
particularly preferred compound is one where Q is CH3, X is Br; Z is A3 or A4;
and Y
is CH2. Another preferred compound is one where Q is CH3, X is Br, Z is A, or
A2,
and Y is CH2. Another preferred compound is one where Q is CH3, X is Cl, Z is
Al
or A~, and Y is CH2. Experimental data, as discussed in Example 12,
demonstrates
that chemical compounds 1.19a, and 1.20a having an A2 moiety are particularly
useful
dopamine transporter imaging agents. -
This novel series of NaS~ compounds differ from those previously reported in
that the substitution of the bis-aminethanethiol ligazd is attached at the 2~3-
position of
the tropane core structure. The corresponding Tc-99m labeled agent displayed a
three-
to fourfold increase in initial brain uptake (0.1 % for N-substituted compound
(Meegalla, supra) vs. 0.3-0.4% in brain at 2 minutes post-injection) and
concomitantly
' retained the specific uptake in the striatum area of the brain. This
observation suggests
that, in this series of compounds, the 3(3-p-fluoro- is slightly less
favorable then the
' 30 corresponding 3(3-p-chlorophenyl derivative. As previously reported for
the same series
of tropane derivatives, the 3(3-p-fluoro- derivative displayed a lower brain
uptake,
CA 02233173 1998-04-16
WO 97/14445 PCT/ITS96/16908
_2ø_
which may be due to its lower binding affinity to dopamine transporters.
Carroll,
1995, supra.
Pharmaceutical formulations of the novel ligands of the invention are also
within the scope of this invention. Pharmaceutically acceptable diluents
suitable for
practicing this invention include, but are not limited to, non-pyrogenic
physiological
saline, and water, with saline being preferred. Any other suitable diluents
known to
those skilled in the art may be used without departing from the spirit of the
invention.
Pharmaceutically accepted salts of the novel compounds of the invention are
also within the contemplated scope of the invention. Pharmaceutically
acceptable salts
suitable for practicing this invention include, but are not limited to,
hydrochloride and
tartrates.
Another aspect of this invention relates to methods for utilizing the
compounds
of the invention as CNS imaging agents. Imaging techniques are non-invasive
diagnostic techniques that generally involve administering a compound with
marker
_atoms that can be detected externally to the mammal. Generally, these methods
comprise administering to a mammal a compound of the invention, dissolved or
dispersed in a suitable pharmaceutical carrier or diluent. The compound of the
invention selectively binds to CNS receptors, such as dopamine, thus
permitting the
imaging of CNS receptors and the ability to, inter alia, evaluate brain
chemistry, the
effectiveness of drugs, and neuronal functions. Imaging techniques suitable
for
practicing the present invention include, but are not limited to, single
photon emission
computed tomography (SPELT) and positron emission tomography (PET).
The compounds of the invention are the first receptor specific imaging agents,
in particular, the first technetium [~''mTc] labeled CNS and are believed to
provide a
convenient source of short-lived imaging agents for routine diagnosis of CNS
abnormality, in conjunction with, for example, single photon emission computed
tomography (SPELT). The technetium [9~'"Tc] isotope has better physical
properties
than those that are currently available in the art. Por example, [~mTc] has a
half life
of 6 hours versus a half-life of 13 hours for iodine [lzsl]. Also, [~''mTc] is
more readily
available than ['~I]. The [1~I] is a cyclotron produced isotope and is
believed to be
available from only two commercial sources -- Nordion, located in Canada, and
Emencia, located in Great Britain. By contrast, [~''"'Tc] is generated using
[99'"Tc] /Mo-
CA 02233173 1998-04-16
WO 97/14445 PCT/LTS96/16908
-25-
99, which is a generator that is used in nuclear medicine techniques and is
virtually in
all hospitals. Another advantage to the use of [~'"Tc] is its cost. Using the
generator
described above, [~''mTc] can be made at a cost of $1 per millicurie while the
cost of
preparing the same amount of iodine would be 30 to 50 times greater. It is
also
possible to follow the same radiochemistry reactions using radionuclide-
containing
compounds containing rhenium, which has similar advantages.
The following tables (A-R) provide examples of compounds of Formula I and
are intended only for illustrative purposes and in noway limit the scope of
the
invention. The examples are also illustrative of the invention and are not
intended to
limit the scope of the invention. The examples illustrate the preparation of
compounds
within the scope of this invention as well as compounds used for comparison.
CA 02233173 1998-04-16
WO 97/14445 PCT/US96/16908
- 26 -
TABLE A
Q n X Z R, E' M
Az, A,, 2 F C02R, CHz CHz Tc"''
As, or
A,
Az, A" As, 2 CI COIR, CH, CHz Tc"'
or A~
S Az, A~, 2 Br CO=R, CH3 CHz Tc'g'"
A.s, or
A~
Az, A" As, I F C02R, CHz CHz Tc"'
or A~
Az, A,, I CI CO=R, CH3 CHz Tc9''
As, or
A,
Az, A" As, i Br COzR, CH3 CHz Tc~
or A.,
Az, A,, 3 F COZR, ~CHz CHz Tc"'
As, or
A~
1 Az, A" As, 3 Cl CO=R, CHz CHz Tc'v"'
~ or A~
Az, A" As, 3 Br C02R, CH, CHz Tc"'
or A~
Az, A" As, 4 F CO=R, CFiz CHz Tc'9"'
or A.,
Az, A,, 4 CI COzR, CHz CHz Tc""'
As, or
A,
Az, A" As, 4 Br COzR, CH, CHz Tc""'
or A.,
IS Az, A,, 2 F COzR, CH2CH, CHz Tcs'"'
As, er
A,
Az, A" As, 2 Cl COzR, CH=CH; CHz Tc"'
or A,
Az, A,, 2 Br COzR, CH2CHz CHz Tc""'
As, or
A,
Az, A" As, 1 F COZR, CHzCH; CHz Tc"'"
or A,
Az. A" As, I Cl COzR, CH2CHz CHz Tc'9'
or A7
Az, A" As, I Br CO2R, CH=CHz CHz Tc""'
or A,
Az, A" As, 3 F CO,R, CH2CH, CHz Tc""'
or A,
Az, A" As, 3 CI C02R, CHZCHz CHz Tc9''"
or A~
Az, A" As, 3 Br C02R, CH,CH, CHz Tc"'"
or A,
Az, A" As, 4 F C02R, CH2CH, CHz Tc""'
or A,
ZS Az, .Aq, 4 Cl CO=R, CHZCHz CHz Tc"'
As, or
A,
Az, A" .4s,4 Br COzR, CH2CHz CHz Tc"'
or A.,
Applies when A is A,
CA 02233173 1998-04-16
WO 97/14445 PCT/US96/16908
-27-
TART.F R
Q n X Z R, E' M
Az, A" 2 F COxR, CH, CHZCH, Tc9''
Afi, or
A,
A~, A" 2 CI COZR, CH, CHzCH, Tc""'
.A6, or
A,
S A=, A" 2 Br COiR, CH, CH2CH, Tc~'""
Afi, or
A,
A=, A" i F C02R, CH, CHiCH~ Tc"'"
A6, or
A,
A,, A" i Cl COIR, CH, CH,CH, Tc'~'"
.46, or
A,
A,, A" 1 Br C02R, CH, CH2CH, Tc'~"'
.A6, or
A,
Az, A" 3 F COzR, CHI CH2CH, Tc"'
A~, or
A,
IO A2, A" 3 CI C02R, CH, CH,CH, Tc""'
A6, or
A,
A=, A" 3 Br CO=R, CH3 CH2CH, Tc"'"
A", or
A,
AZ, A" 4 F C02R, CHI CH2CH, Tc~'"
A", or
A,
A2, A" 4 Cl CO=R, CH3 CH2CH, Tc""'
A", or
A,
A" A" A6, 4 Br C02R, CH, CH2CH~ Tc9''
or A,
ZS A=, A" 2 F COzR, CH2CH, CH2CH, Tc"'"
.46, or
A,
Az, A" 2 CI COiR, CH=CH~ CH2CH3 Tc'~
A", or
A,
A" A" A6, 2 Br C02R, CH,CH, CH=CH, Tc""'
or A,
A2, A" i F COzR, CH2CH, CH=CH3 Tc~
A", or
A,
A" A,. i CI CO,R, CHZCH, CH2CH, Tc'9'
A", or
A,
ZO AZ, A" 1 Br C02R, CH,CH~ CH~CH, Tc9""
A~, or
A,
A2, A" 3 F CO,R, CHZCH, CH,CH, Tc""'
A6, or
A,
A=, A" 3 Cl COTR, CH2CH3 CHZCH~ Tc"m
Afi, or
A,
A2, A" 3 Br CO,R, CH,CH~ CH2CH, Tc""'
A", or
A,
AZ, A" 4 F C02R, CH2CH, CH2CH, Tc"'
Afi, or
A,
ZS A=, A" 4 Ci C02R, CH=CH3 CH2CH, Tc9'"'
A6, or
A,
A=, A" 4 Br C02R, CH=CH, CH=CH3 Tc'~'"
A", or
A,
Applies when A is A,
CA 02233173 1998-04-16
WO 97/14445 PCT/US96/16908
- 28 -
TABr.E c
Q n X Z R= E' M
A" A" A,~,2 F C02R= piperidine, pyrrolidine,CHi Tc"'"
or A, or
morpholine
A=, A" 2 CI C02Rz piperidine, pyrrolidine,CHi Tc"'"
A,~, or or
A,
morpholine
Jr Az, A" 2 Br COiRZ piperidine, pyrrolidine,CH= Tc"'"
A" or or
A,
moipholu..:
A=, A" I F CO=R= piperidine, pyrrolidine,CH= Tc"'
A," or or
A,
morpholine
A=, A" I CI C02R= piperidine, pyrrolidine,CHI Tc"'"
A.6, or or
A,
morpholine
Ai, A" l Br C02R2 piperidine, pyrrolidinc,CH2 Tc~"'
A6, or or
A,
morpholine
Ai, A" 3 F CO,R= piperidine, pyrtnlidine,CH2 Tc"'"
A,~, or or
A,
moipholine
1~ A=, A" 3 Cl C02R= piperidine, pyrrolidine,CH2 Tc~'''"
A", or or
A,
morpholine
AZ, A," 3 Br COxRx piperidine, pyrrolidine,CH2 Tc'9'"
.A6, or or
A,
morphoiine
A=, A" 0 F COzR2 piperidine, pyrrolidinc,CH= Tc'~"'
A6, or or
A,
morpholine
A2, A" 0 CI C02Rz piperidine, pyrrolidine,CHZ Tc""'
.A6, or or
A,
morpholine
A=, A" 0 Br CO=R2 piperidine, pyrrolidine,CHi Tc""'
A6, or or
A,
morpholine
IJ~
Applies
when
A
~s
A,
CA 02233173 1998-04-16
WO 97/14445 PCT/US96/16908
-29-
TABLE D
Q n X Z E' R, M
Az, A" 2 F COR, CHZ CH, Tc'9"
A6, or
A,
A=, A" 2 Cl COR, CH= CH, Tc""'
Ab, or
A,
S AZ, A" 2 Br COR, CH2 CH, Tc""'
A" or
A,
A=, A" 1 F COR, CH2 CH, Tc'm
A,, or
A,
A2, A" 1 Cl COR, CH= CH, Tc"
A", or
A.,
A=, A" 1 Br COR, CH= CH, Tc"'"
Aa, or
A,
A=, A" 3 F COR, CH, CH, Tc"'
A6, or
A.,
1~ A2, A" 3 C( COR, CH2 CH, Tc"m
A6, or
A,
A=, A" 3 Br COR, CHZ CH, Tc"'
A" or
A,
A" A" A" 0 F COR, CH. CH, Tc""'
or A,
A=, A" 0 CI COR, CHz CH, Tc""
A" or
A,
A" A" A" 0 Br COR, CH, CH, Tc"'"
or A,
IS A" A" A" 2 F COR, CHi CH,CH, Tc"'
or A~
A" A" .4" 2 CI COR, CHI CH=CH, Tc"'
or A.,
A=, A" 2 Br COR, CHT CH=CH, Tc""'
A" or
A,
Ai, A" I F COR, CHr CH,CH, Tc"'
A6, or
A,
A" A" A" I CI COR, CH2 CH=CH, Tc""
or A,
ZO A" A" A,, I Br COR, CHT CH=CH, Tc"'
or A,
A=, A" 3 F COR, CH= CHiCH, Tc"
A," or
A,
A,. A" 3 CI COR, CHI CH=CH, Tc"'
A", or
A,
A=, A" 3 Br COR, CHZ CH=CH, Tc''"
A" or
A,
A" A" A" 0 F COR, CHz CH=CH, Tc""'
or Ar
ZS A:, A" 0 CI COR, CH= CH=CH, Tc''
A" or
A.,
A=, A" 0 Br COR, CH2 CHiCH, Tc''
A" or
A.,
Applies when A is A7
CA 02233173 1998-04-16
WO 97/14445 PC'd'/US96/16908
-30-
TABLE E
Q n X Z E' R, M
A=, A" 2 F COR, CH,CH, CH, Tc"'
A" or
A,
A,. A" 2 CI COR, CHZCH, CH, Tc""'
A6, or
A,
S A=, A" 2 Br COR, CHZCH, CH, Tc"
A" or
A,
A" A" A6, 1 F COR, CH=CH, CH, Tc"
or A,
A=, A" 1 CI COR, CH,CH, CH, Tc""
A" or
A,
A" A" P." ( Br COR, CHZCH, CH, Tc"
or A,
A" A" A" 3 F COR, CH,CH, CH, Tc"
or A,
IO A=, A" 3 CI COR, CH,CH, CH, Tc""
A" or
A,
A" A" A,. 3 Br COR, CHZCH, CH, Tc"'
or A,
A=, A" 0 F COR, CH,CH, CH, Tc'""
A" or
A,
AE, A" 0 CI COR, CH,CH, CH, Tc'"
A" or
A,
A" A" A" 0 Br COR, CHrCH, CH, Tc"'
or A,
IS A" A" A" 2 F COR, CH,CH, CH,CH, Tc'""
or A,
A" A" A" 2 C( COR, CH=CH, CHiCH, Tc"'"
or A,
A" A" A" 2 Br COR, CH CH, CH=CH, Tc'*'
or A,
Ar, A" I F COR, CH,CH, CH,CH, Tc"'
A" or
A,
A" A" Aa, 1 CI COR, CH,CH, CH=CH, Tc'"'
or A,
ZO A" A" A" I Br COR, CH,CH, CH,CH, Tc"'
or A,
A,, A" 3 F COR, CH=CH, CH,CH, Tc'~
A" or
A,
A" A" A" 3 CI COR, CHZCH, CH,CH, Tc"'"
or A,
A" A" A" 3 Br COR, CH,CH, CH=CH, Tc'""
or A,
A" A" Aa, 0 F COR, CHrCH, CH,CH, Tc"'
or A,
ZS AZ, A,, 0 C( COR, CH,CH, CH=CH, Tc'm
A" or
A,
A,, A" 0 Br COR, CHZCH, CH,CH, Tc's'
As, or
A,
Applies when A is A,
CA 02233173 1998-04-16
WO 97/14445 PCT/US96/16908
-31-
TABLE F
Q n X Z E' M
CH, 2 F COA" COA" COAa,CHz Tc'""
or COA,
CH, 2 CI COA=, COA" C0.46,CH, Tc""
or COA,
S CH, 2 CI COA=, COA" COA6,CH= Tc'~'
or COA,
CH, 1 F COA=. COA" COA"CHI Tc"'
o: COA,
CH, I CI COA=, COA" COA"CHi Tc'"
or COA,
CH, 1 F COA=, COA" COA"CHi Tc"'
or COA,
CH, 3 F COA=, COA" COA"CH= Tc"'
or COA,
IO CH, 3 CI COA=, COA" COA"CH= Tc"''
or COA,
CH, 3 CI COA2, COA" CO.A"CHx Tc"'"
or COA,
CH, 0 F COAT, COA" COA"CH= Tc'"'
or COA,
CH, 0 CI COAZ, COA" COA"CH= Tc'""
or COA,
CH, 0 CI COAT, COA" COA"CH, Tc"'"
or COA,
ZS CH, 2 F COA=, COA" COA"CH, Tc''
or COA,
CH, 2 CI COAT, COA" COA6,CH= Tc"'
or COA,
CH, 2 F COAZ, COA" COA"CH= Tc"'"
or COA,
CH, t F COAx, COA" COA"CHr Tc"'
or COA,
CH3 1 CI COA" COA" COA6,CHi Tc""'
or COA,
ZO CH, 1 F COAT, COA" COA6,CH, Tc''
or COA,
CH, 3 F COA2, COA" COA"CH, Tc"'"
or COA,
CH, 3 CI COAL, COA" COA,,CHZ Tc""
or COA,
CH, 3 F COAT, COA" COA6,CHr Tc'""
or COA,
CH, 0 F COAT, COA" COA6,CHr Tc"'"
or COA,
ZS CHI 0 C1 COA2, COA,. CH= Tc"'"
COAa, or COA,
CH, 0 F COAT, COA" COAe,CH2 Tc'""
or COA,
Applies when A is A,
CA 02233173 1998-04-16
WO 97/14445 PCT/CTS96/16908
-32-
TABLE G
Q n X Z E' M
CH, 2 F COA" COA" COAa,CH,CH, Tc"'
or COA,
CH, 2 Cl COA" COA" COAs,CH,CH, Tc"
or COA,
S CH, 2 CI COAT, COA" COA"CH=CH, Tc"'"
or COA,
CH, I F COA" COA,, COA"CH,CH, Tc"
or COA,
CH, 1 CI COA=, COA" COA"CH,CH, Tc"
or COA,
CH, 1 F COA,, COA" COA,,CH,CH, Tc""'
or COA,
CH, 3 F COA" COA" COAa,CH,CH, Tc'
or COA,
IO CH, 3 CI COA" COA" COA" CH,CH, Tc""
or COA,
CH, 3 CI COA,, COA" COAa,CH,CH, Tc""'
or COA,
CH, 0 F COA" COA" COAs,CH=CH, Tc""
or COA,
CH, 0 Cl COA,, COA" COAL,CH,CH, Tc"'
or COA,
CH, 0 CI COA=, COA" COA6,CH,CH, Tc'~
or COA,
IS CH, 2 F COA" COA" COAs,CH,CH, Tc"'"
or COA,
CH, 2 CI COA" COA" COA",CH=CH, Tc"m
or COA,
CH, 2 F COA" COA" COA6,CH,CH, Tc""'
or COA,
CH, t F COA" COA" COAa,CH,CH, Tc""'
or COA,
CH, t C( COAT, COA" COA"CH,CH, Tc"'"
or COA,
LO CH, 1 F COA" COA" COA" CH,CH, Tc"'
or COA,
CH, 3 F COA" COA" COA" CH,CH, Tc"'"
or COA,
CH, 3 Ct COA" COA" C0.4a,CH,CH, Tc"'
or COA,
CH, 3 F COAZ. COA" COAa,CH2CH, Tc'"'
or COA,
CH, 0 F COA" COA" COA" CH=CH, Tc"'
or COA,
ZS CH, 0 CI COA" COA" COA,,CHZCH, Tc"'
or COA,
CH, 0 F COA=, COA" COA"CH,CH, Tc""'
or COA,
nppnes wnen n is n,
CA 02233173 1998-04-16
WO 97114445 PCT/US96/16908
-33-
TABLE H
Q Z n E' X M
CH, CH,NHA, I CH, C1, F, Tc""'"
or Br
CH,CH, CH,PTHA,1 CH, CI, F, Tc''"
or Br
S CH_CFt,CH,CHrTIHA,1 CtL_ CI, F, Tc""
or Br
CH_CHrCH,CH,CFI_NHA,1 CH: CI, F, Tc'~"
or Br
CH, CH,NHA, 2 CH, CI, F, Tc'""
or Br
CH,CH, CH_NFIA,2 CH, CI, F, Tc'"'
or Br
CH.CH,CH,CH,NHA, 2 CHr Ci, F, Tc~'
or Br
I CH:CH,CH,CH,CH,NHA, 2 CH, CI. F, Tc"-
O or Br
CH, CHrNHA, 3 CH, CI, F, Tc"""
or Br
CHiCH, CH,NHA, 3 CH, CI, F, Tc"'"
or Br
CH,CH,CH,CH,NHA, 3 CH, CI, F, Tc'"
or Br
CH,CH,CH,CH,CH.NHA, 3 CH, CI, F, Tc""'
or Br
IS CH, CH,NF3A,0 CH, CI, F, Tc""
or Br
CH,CH, CH,NHA, 0 CH, CI, F, Tc""'
or Br
CH,CH,CH,CH:NHA, 0 CH= CI, F, Tc"""
or Br
CH,CH,CH,CH,CH,NHA, 0 CHr CI, F, Tc"'"
or Br
App nes when A a A,
CA 02233173 1998-04-16
WO 97/14445 PCT/US96/16908
-34-
TABLE I
Q 2 n E' X M
CH, CH_NHA, I CH,CH, CI, F, Tc'"
or Br
CH,CH, CH,NHA, 1 CH,CH, CI, F, Tc~~"
or Br
S CH_CH_CH,CFiNHA, i CFI,CH, CI, F, Tc'""
or Br
CH,CFL_CH,CH,CH,NHA, 1 CH,CH, CI, F, Tc"'"
or Br
CH, CH,NFIA,2 CH,CH, CI, F, Tc''
or Hr
CH,CH. CH,NtIA,2 CH,CH, Ci, F, Tc'~"
or Br
CH,CH,CH,Cft_NFtA,2 CH,CH, CI, F, Tc""
or Br
IO CH,CH CIi_NFIA,2 CH,CH, Cf, F, Te"
CH,CH, or Br
CH, CH,NHA, 3 CH,CH, CI, F, Tc""'
or Br
CH,CH, CHrNHA, 3 CH,CH, CI. F, Tc"'"
or Br
CH,CH.CH,CH,NHA, 3 CIi,CH, CI, F, Tc'"~
or Br
CH,CH,CH,CH,CH,NHA, 3 CFi_CH, CI, F, Tc"'"
or Br
15 CH, CH.NHA, 0 CH,CH, CI, F, Tc"'"'
or Br '~
CH,CH, CH,NEN, 0 CH,CH, CI, F, Tc""
or Br
CH,CH,CH,CH_NFtA,0 CH,CH, CI, F, Tc"'
or Br
CH,CH,CH,CH,CH,NHA, 0 CFIrCH, CI, F, Tc""
or Br
,ppncs wnrn n ,s ,
CA 02233173 1998-04-16
WO 97/14445 PCT/US96/16908
-35-
TABLE J
Q Z n X E' M
CH, CO,A,. CO,A" I CI, F, CH, Tc"'"
COsA" or CO,A, or Br
CH,CH, CO,A" CO,A" t CI, F, CH, Tc""
COrA" or CO,A or Br
S CH,CH,CH,C0.A~ CO,A" 1 CI, F, CH, Tc'~"
CO,A" or CO,A or Br
CH,CH.CH,CH,CO,A" CO,A" 1 CI, F, CH, Tc""'
CO,A" or CO,A or Br
CH, CO._A" CO,A" 2 CI, F, CH, Tc""'
CO,A" or CO.A or Br
CH,CH, CO,Ar, CO,A" 2 CI, F, CH. Tc"'"
CO,A" or COiA or Br
CH,CH.CH,CO:A" CO,A" 2 CI, F, CH, Tc"'"
CO,A" or COrA or Br
I CH,CH,CFI:CH,CO_A_, CO_A" 2 CI, F, CH, Tc""'
Q C0,.1", or or Br
CO,A
CH, CO,A" CO:A" 3 CI, F, CH. Tc""'
CO,A" or CO.A cr Br
CH,CH, CO,A~ CO,P." 3 C4 F, or CH, Tc'""
CO:A", or CO,A Br
CH,CH,CH,CO_A.:, CO_.4" 3 Cl, F, CH, Tc'""
CO,A", or CO,A or Br
CH,CH,CH,CH,f CO,A" CO,A" 3 CI, F, CH, Tc~""
CO,A" or CO,A or Br
IS CH, CO:A" CO,A" 0 CI, F, CH. Tc"'"
CO,r~, or CO:A, or Br
CH_CH, CO,A" CO;A" 0 CI, F, CH. Tc"~
CO,A", or CO.A or Br
C3i_CH_CH,CO,A_, CO=P." 0 C1, F, CH: Tc'"
CO_A" or CO;A or Br
CH,CH,CH,CH,CO,Ar, CO,A" 0 CI, F, CH, Tc'"'
CO,A", or CO,A or Br
- APPlies when A is A,
CA 02233173 1998-04-16
WO 97/14445 PCT/US96/16908
-36-
TABLE IC
Q Z o X E' M
CH, CO,A" CO,A" I CI, F, CH,CH,Tc'"'"
CO,(~, ar CO or Br
~
CH,CH, CO,A;, CO,A" I CI, F, CH,CH,Tc~'"
CO,A" or CO.A or Br
S CFLCH,CH,CO,A,. CO,A" 1 CI, F, CH,CH,Tc'""
COrA" or CO,A or Br
CFGCH,CH,CH,CO,A~ CO,A" I C4 F, or CF!_CH,Tc'"'"
CO,A" or CO,A Br
CH, CO;A" CO:A" 2 CI, F, CH,CH,Tc""'
CO_A" or CO,A or Hr
CH,CH, CO,A" CO,A" 2 Ci. F, CH,CH,Tc""'
CO,A" or CO,A or Br
CFl_CH,CH,C0.A" CO,A" 2 CI, F, CH,CH,Tc"
CO,A" or CO,A or Br
I CFI_CH:CH,CH,CO:A=, CO,A" 2 C1, F, CH.CH,Tc""'
O CO A", or CO.A or Br
CH, CO,A" CO,A" 3 CI, F, CH,CH,Tc'~"
CO,A" ar CO,A or Br
CH,CH, CO:A" CO,A" 3 CI. F, CH.CH,Tc""'
CO,A" or CO,A or Br
CH,CH,CH,CO,A., CO,A" 3 CI, F, CFi:CH,Tc'""
CO,A" or CO,A or Br
CH,CH,CH,CH,CO;A,, CO,A" 3 CI, F, CH,CH,Tc""
COiA", or CO_A or Br
1 CH, CO,A" CO,A" 0 CI, F, CH,CH,Tc'""
S COiA", or CO.A or Br
CH,CH, CO,A" CO_A" 0 CI, F, CH,CH,Tc""'
CO,A", or CO.A or Br
CH,CH.CH,CO,A" CO,A" 0 I, F, or CH;CH,Tc""'
CO,A" or COA Br
C
CH_CH,CH,CH,CO,A" CO_A" 0 CI. F, CH.CH,Tc"""
CO,A" or C0.A or Br
~APPm worn n ~s w,
CA 02233173 1998-04-16
WO 97/14445 PCT/US96/16908
-37-
~r~sm.F r
Z n X E' M
CH, COA" COA" COA" 1 CI, F, CH, Tc'""
or COA, or Br
CH,CH, COA" COA" COA" 1 CI, F, CH, Tc~"
or COA, or Br
S CH,CH,CH,COh, COA" COA" 1 Cl, F. CH, Tc""'
or COA, or Hr
CH:CH,CH,CH,COA" COA" COA" I CI, F, CH, Tc""
or COA, or Br
CH, COA,, COA" COA"2 CI, F, CH, Tc"""
or COA, or Br
CH,CH, COA" COA,. COA"2 CI, F, CH, Tc"'
or COA, or Br
CH,CH,CH,COAL COA" COA" 2 CI, F, CFi_ Tc""'
or COA, or Br
1 CH,CH,CH,CH,COA;, COA" COA",2 CI, F, CH; Tc'""
O or COA, or Br
CH, COA" COA" COA" 3 Ci, F, CH, Tc''"
or COA, or Br
CH,CH, COA. COA" COA" 3 CI. F, CH, Tc'"'
or COA, or Br
CH_CH,CH,COA" COA" COA".3 CI, F, CH, Tc"'
or COA, or Br
CH,CH,CH,CH,COA" COA" COA" 3 CI, F, CH, Tc""'
or COA, or Br
15 CH, COA" COA" COA" 0 CI, F, CH; Tc"""
or COA, or Br
CH,CH, COA" COA" COA" 0 CI, F, CH, Tc'"'
or COA, or Br
CH,CFI,CH,COAL COA" COA" 0 CI, F, CH, Tc'~"
or COA, or Br
CH;CFGCH,CH,COP." COA" COAM0 CI, F, CH, Tc"""
or COA, or Br
-Apphu when A ,s A,
CA 02233173 1998-04-16
WO 97/14445 PCT/US96/16908
- 38 -
TABLE M
Q Z s X E' M
CH, COA" COA" COA" 1 CI, F, CH,CH,Tc"
or COA, or Hr
CH,CH, COA" COA" COA" t CI, F, CH.CH,Tc""'
or COA, or Br
S CH,CH,CH,COA,. COA" COA"1 CI, F, CH,CH,Tc'""
or COA, or Hr
CH,CH,CH,CH,COA" COA" COA" t CI, F, CH,CHZTc~
or COA, or Br 3
CH, COA" COA" COA" 2 CI, F, CH,CH,Tc"'"
or COA, or Br
CH,CH, COAL COA" COA" 2 CI, F, CH,CH,Tc'""
or COA, or Br
CH,CH,CH,COA,, COA" COA"2 CI, F, CH.CH,Tc'~"
or COA, or Bc
IO CH,CH,CH,CH,COA,. COA" COA"2 ~I, F, CH,CH,Tc'~"
or COA, or Br
CH, COA" COA" COA" 3 CI, F, CH,CH,Tc""'
or COA, or Br
CH,CH, COA" COA" C0.4"3 CI, F, CH,CH,Tc"'"
or COA, or Br
CH,CH,CH,COA,, COA" COA"3 Cl, F, CH,CH,Tc'""
or COA, or Br
CH,CH,CH,CH,COAL COA" COA" 3 CI. F, CH,CH,Tc'"""
or COA, or Br
I CH, COA,, COA" COA"0 CI, F, CH,CH,Tc"'"
S or COA, or Br
CH,CH, COA,, COA" COA"0 Cl, F, CH,CH,Tc""
or COA, or Br
CH,CH,CH,COAT COA" COA,.0 Ci, F, CH,CH,Tc'""
or COA, or Br
CH,CH,CH,CH,COA,, COA" COALO CI, F, CH,CH,Tc'""
or COA, or Br
r,pp"es wnca ms ,
CA 02233173 1998-04-16
WO 97/14445 PCT/US96/16908
-39-
TABLE N
Q Z n X E' M
CH, CH,OA,, CH,OA" 1 CI, F, CH, Tc'~"
CH,OA" or CHtOA, or Br
CH,CH, CH,OA,, CH,OA" I CI, F, CH, Tc~
CH,OA" ar CH,OA, or Br
S CH,CH,CH,CH,OA" CH,OA" CH,OA"1 CI, F, CH, Tc"'
or CH,OA, or Br
CH,CH,CH,CH,CH,OA,, CH,OA" I Cl, F, CH, Tc'""
CFI_OA" or CH,OA, or Br
CH, CH,OA" CH,OA" CH,OA,"2 CI, F, CH, Tc"'"
or CH,OA, or Hr
CH,CH, CH,OA,. CH,OA" 2 CI, F, CH, Tc'"'"
CH,OA" or CH,OA, or Br
CH,CH,CH,CH,OA,, CH,OA" 2 CI, F, CH, Tc""'
CH,OA" or CH,OA, or Br
IQ CH,CH,CH,CH,CH,OA,, CH,OA" 2 CI, F, CH, Tc""'
CH,OA" or CH,OA, or Br
CH, CH,OA" CH,OA" CH,OA"3 CI, F, CH, Tc'""
or CH,OA, or Br
CH,CH, CH,OA_. CH,OA" 3 CI, F, CH, Tc"'"
CH,OA,, or CH,OA, or Br
CH,CH,CH,CH,OM, CH,OA,. 3 CI, F, CH, Tc'""
CH,OA" or CH,OA, or Br
CH,CH,CH,CH,CH,OA,, CH,OA" 3 Cl, F, CH, Tc""'
CH,OA" or CH,OA, or Br
IS CH, CH,OA,, CH,OA,. 0 CI, F, CH, Tc"'""
CH,OA,, or CH,OA, cr B.
CH,CH, CH,OA,, CH,OA" 0 CI. F, CH, Tc'"'
CH,OA" or CH,OA, or Br
CH_CH_CH,CH,OA,, CH,OA" O CI, F, CH: Tc'"'
CH,OA" or CH,OA, or Br
CH,CH,CH,CH,CH,OA:, CH,OA" 0 CI, F, CH, Tc~'
CH,OA" or CH,OA, or Br
Applies when A ,5 A,.
CA 02233173 1998-04-16
WO 97/14445 PCTlCTS96/16908
-40-
TABLE O
Q Z n X E' M
CH, CH,OA" CH,OA,. CH,OA,"1 C1, F, CH,CH,Tc""'
or CH,OA, or Br
CH,CH, CH,OA" CH,OA" CH,OA"1 CI, F, CH,CH,Tc'"
or CH.OA, or Br
S CH,Cfi,CH,CFI,OA~ CHrOA" CH._OA"t CI, F, CH,CH,Tc"~
or CH,OA, or Br
CH,CH_CFi,CH,CFI,OA~ CH,OA" CHiOA"I Ci, F, CH,CH,Tc""
or CH,OA, or Br
CH, CH,OA,. CH,O A" 2 Ci, F, CH,CH,Tc"'"
CH,OA" or CH,OA, or Br
CH,CH, CH,OA" CH,OA" CH,OA,"2 CI, F, CH,CH,Tc""'
or CH,OA, or Br
CH,CH,CH,CH_OA" CF40A" CH,OA"2 CI, F, CH,CH,c""'
or CH,OA, T or Br
I CH,CH_CH.CH,CFL_OA:, CH,OA" 2 Ci, F, CH,CH,Tc'""
O CH,OA" or CHrOA, or Br
CH, CH;OA_, CH:OA" CH,OA"3 CI, F, CH,CH,Tc'""
or CH,OA, or Br
CH,CH, CH,OA" CH,OA" CH,OA",3 CI. F, CH,CH,Tc"'
or CHrOA, or Br
CH,CH,CH,CFL,OA., CH,OA" 3 CI, F, CH,CH,Tc'""
Cfi,OA" or CH,OA, or Br
CFLCH,CH,CH,CH,OA_, CH,OA" CHrOAa,3 i, F, or CH.CH,Tc'""
or CH,OA, Br
C
I CH, CH:OA" CH,OA" CH,OA",0 CI, F, CH,CH,Tc'""
S or CH:OA, or Hr
CH,CH, CH~OA" CH,OA,. CH,OA"0 Ci, F, CH,CH,Tc'""
or CH,OA, or Br
CH,CH,CH,CH,OA" CH,OA" CH,OA"0 CI, F, CH,CH,Tc'~"
or CH,OA, ar Br
CH,CH,CH,CH,CH:OA=, CH=OA" CH:OA"0 Cl, F, CI-GCH,Tc'"'
or CH_OA, or Br
CA 02233173 1998-04-16
WO 97/14445 PCT/US96/16908
-41-
TABLE P
Q Z R, n R, X M
A,. A,. CO,R, CH, 1 H CI Tc"'"
A,. or or
A, F
A" A" A" CO,R, CH, 1 CH,CH,CH,CI Tc"
or A, or
F
S A" A" A" CO,R, CH, 1 methoxybenrylCI Tc""'
or A, or
F
A" A" A" CO,R, CH, 1 uityl CI Tc'""
or A, or
F
A" A" A,, CO,R, CH,CH, 1 H CI Tc''"
or A, or
F
A" A" A" CO,R, CH,CH, I CH,CH,CH,CI Tc""'
or A, or
F
A" A" A" CO,R, CH,CH, 1 methoxyhrnrylCI Tc~"'
or A, or
F
ZO A" A,. CO,R, CH,CH, I irityi CL Tc"'"
A" or or
A, F
A" A" A" CO,R, CH,CH,CH,1 H CI Tc'-
ar A, or
F
A" A" A" CO,R, CH,CH,CH,1 CH,CH,CH,CI Tc~"
or A, or
F
A" A" A" CO:R, CH,CHrCH,1 uityl CI Tc""
or A, or
F
A" A" A" CO:R, Cf4CF4CH,I methoxybenrylCI Tc'""
or A, or
F
IS A" A" A" CO,R, CH,CH,CH,CH,1 methoxybenrylCI Tc'"'
or A" ar
F
A" A" A" CO,R, CH,CH,CH,CH,1 H CI Tc""
or A, or
F
A" A" A" CO,R, CH,CH,CH,CH,I CH_CH,CH,CI Tc'"'
or A" or
F
A" A" A,. CO,R, CFI,CFI,CFL,CH,1 uityl CI Tc'"
or A, or
F
A" A" A,, CO,R, CH, 2 H CI Tc"'"
or A" or
F
ZO A" A" A" CO,R, CH, 2 CH,CH,CH,CI Tc"'"
or A, or
F
A" A" A" CO,R, CH, 2 methoxybmrylCI Tc""'
or A, or
F
A" A" A" C0.R, CH, 2 trityl CI Tc"'"
or A, or
F
A" A" A" CO=R, CH,CH, 2 methoxybenrylCI Tc""
or A, or
F
A" A" A" CO,R, CH,CH, 2 uityl Cl Tc""
or A, or
F
ZS A" A" A" CO,R, CF4CH, 2 H CI Tc""
or A, or
F
A" A" A" CO,R, CFi,CH, 2 CH,CH,CH,CI Tc'"'
or A, or
F
A" A" A" CO,R, CH,CH,CH,2 H CI Tc""
or A, or
F
A" A" A" CO,R, CH,CH,CH,2 CH,CH,CH,C( Tc'""
or A, or
F
A" A" A,, CO,R, CH,CH,CH,2 mahoxybenrylCI Tc'""
ar A, or
F
3O A" A" A" CO,A, CH,CFI_CH,2 triTyl CI Tc~'"
or A, or
F
A" A" A" CO,R, CH,CH,CH,CH,2 methoxybenrylCI Tc""'
or A" or
F
A" A" A" CO,R, CH,CH,CH,CH,2 CH,CH,CH,CI Tc~-
or A, or
F
A" A" A" CO,R, CH,CH,CH,CH,2 H CI TP"
or A, or
F
A" A" A" CO:R, CH,CH,CEI_CH,2 trityl CI Tc"-
or A, or
F
CA 02233173 1998-04-16
WO 97/14445 PCT/(JS96/16908
-42-
TABLE Q
Q Z R, o R, X M
A" A" As. COR, CH, i H CI Tc'"'
or A, or
F
A" A" As, COR, CH, I CH,CH,CH,Cl Tc'""
or A" ~ or
F
S A" A" As, COR, CH, 1 methoxybmrylCI Tc'~"
or A, or
F
A" A" As, COR, CH, 1 aityl CI Tc'""
or A, or
F
A" A" A" COR, CH,CH, I methoxybmrytCI Tc""
or A, or
F
A,. A" COR, CH,CH, i trityl CI Tc~'"
Aa, or or
A, F
A" A" Aa. COR, CH,CH, I H CI Tc"'"
or A, or
F
I A" A" A" COR, CH,CH, 1 CH_CFI,CH,CI Tc"'"
Q or A, or
F
A" A" As, COR, CH,CH,CH,i H CI Tc"'"
or A, or
F
A" A" Aa, COR, CH,CH,CH,1 CH.CH,CH,CI Tc""'
or A" or
F
A" A" Aa, COR, CH,CHsCH,1 methoxybmrylCI Tc~"
or A" or
F
A" A" A" COR, CH,CH,CH,1 trityi CI Tc'""
or A, or
F
I A" A" As, COR, CH,CH,CH,CH,I methoxybmzyiCI Tc"'"
S or A, or
F
A" A" As, COA, CH,CH,CH,CH,I CH,CH,CH,CI
or A, or
F
A" A" As, COR, CH.CH,CH,CH,t H CI
or A, or
F
A" A" p" COR, CH.CFisCH_CH,1 oityl CI Tc"'"
or A, or
F
A" A" Aa. COR, CH, 2 H CI Tc""
or A, or
F
GO A" A" Aa. COR, CH, 2 CH,CH,CH,CI Tc""
or A, or
F
A" A" A" COR, CH, 2 methoxybmrylCf Tc"'
or A, or
F
A" A" Aa, COR, CH, 2 trityl Cl Tc"'"
or A, or
F
A" A" A" COR, CH,CH, 2 methoxybmrylCt Tc'""
or A, or
F
A" A" Aa, COR, CH,CH, 2 trityl CI Tc'""
or A, or
F
ZS A" A" As, COR, CH,CH, 2 CFI,CF1_CH,CI Tc"
or A" or
F
A" A,. COR, CH,CH, 2 H CI Tc'"'
Aa. or or
A, F
A" A" As, COR, CH,CH,CH,2 H C! Tc""'
or A, or
F
A" A" As, COR, CH,CH,CH,2 CH,CH,CH,CI Tc""'
or A, or
F
A" A" Aa, COR, CH,CHsCH,2 vityl Cl Te'""
or A, or
F
3~ A" A" As, COR, CH,CH,CH,2 methoxybenrylCI Tc'~"
or A, or
F
A" A" As, COR, CH,CH,CH,CH,2 mnhoxybmrylCI Tc""'
or A, or
F
A" A" As, COR, CH,CH,CH,CH,2 CH,CH,CH,Cl Te""
or A, or
F
A" A" As, COR, CF~,CIi_CH,CH,2 H CI Tc"'
or A, or
F
A" A" As, COR, CH,CH,CH,CH,2 trityi C! Tc"'
or A, or
F
CA 02233173 1998-04-16
WO 97/14445 PCT/LTS96/16908
-43-
TABLE R
Q Z R, n IL X M
A" A" A" COR, CH, 3 H CI Tc'""
or A" or
F
t
A" A" A" COR, CH, 3 CH'CH'CH,CI Tc""
or A, or
F
S A" A" A" COR, CH, 3 mahoxybrnrylCI Tc''"
or A, or
F
A" A" A" COR, CH, 3 trityi CI Tc""'
or A, or
F
A" A" A" COR, CH,CK, 3 methoxybenrylCI Tc""
or A, or
F
A" A" A" COR, CH,CH, 3 uityl CI Tc'~"
or A, or
F
A" A" A" COR, CH,CH, 3 CH,CH,CH,CI Tc'""
ar A, or
F
I O A" A" A" COR, CH,CH, 7 H CI Tc~"'
or A" or
F
A,. A" COR, CH,CH'CH,3 H CI Tc'""
A" or or
A, F
A" A" A,. COR, CH,CH,CH,3 CH_CfI,CH,CI Tc"'"
or A, or
F
A" A" A" COR, CH,CH'CH,3 methoxybrnrylCI Tc'~"
or A, or
F
A" A" A" COR, CH,CH,CH,3 trityl CI Tc'""
cr A, or
F
IS A" A,. COR, CH,CH,CH,CH,3 methoxybmrylCI Tc'""
A,. or or
A. F
A" A" A" COR, CH_CH,CFI_CH'3 H CI Tc"'
or A, or
F
A" A" A" COR, CH,CH'CHrCH,3 CH,CH,CH,CI Tc~'
or A, or
F
A" A" A" COA, CH,CH,CH,CH'3 uityi CI Te"'
or A, or
F
A" A" A" COR, CH, 0 H CI Tc'"'
or A, or
F
ZO A,. A" COR, CH, 0 CH,CH,t:H,CI Tc~'"
A" or or
A" F
A" A" A" COR, CH, 0 methoxybenrylCI Tc'""
or A" or
F
A" A" A" COR, CH, 0 trityi CI Tc""
or A, or
F
A" A" A" COR, CH,CH, 0 methoxybenrylCI Tc''"
or A, or
F
A" A" A" COR, CH,CH, 0 uityl CI Tc'""
or A" or
F
GS A" A" A" COR, CHzCH, 0 CH,CH,CH,CI Tc""'
or A, or
F
A" A" A" COR, CH,CH, 0 H CI Tc'""
or A, or
F
A" A" A" COR, CH,CH'CH,0 H CI Tc'~'
or A, or
F
A" A" A" COR, CH'CH,CH,0 CH,CH,CH,CI Tc""'
or A, or
F
A" A" A" COR, CHrCH,CH,0 methoxybenrylCI Tc"'"
or A, or
F
3O A" A" A" COR, CH,CH'CH,0 trityl CI Tc"'"
or A. or
F
A" A" A" COR, CH,CH'CH'CH,0 methoxybenzylCI Tc'""
or A, or
F
A" A" A,. COR, CFL,CH_CFI,CH,0 H CI Tc'
or A, or
F
A" A" A" COR, CH,CH,CH,CH,0 CH,CH,CH,CI Tc"'
or A, or
F
~S A" A" A" COR, CH,CH,CH'CH'0 trityl CI Tc"
or A, or
F
CA 02233173 1998-04-16
WO 97/14445 PCT/US96/16908
EXAMPLES
General Experimental For Examples 1 to 13
Reagents used in the syntheses were purchased from Aldrich (Milwaukee, WI)
or Fluka (Ronkonkoma, NY), and were used without further purification unless
otherwise indicated. Anhydrous Na2S04 was used as a drying agent. Reaction
yields
are reported without attempts at optimization. Thin layer chromatography was
performed on EM Science (Gibbstown, NJ) precoated (0.2 mm) silica gel 60
plates,
and the spots were detected with iodine vapor and/or UV light. Silica gel 60
(70-230
mesh), obtained from EM Science (Gibbstown, NJ), was used for column
chromatography. 1H NMR spectra were obtained on a Bruker spectrometer (Bruker
AC 300). All samples prepared for NMR analysis were dissolved in CDCl3,
purchased
from Aldrich. Chemical shifts are reported as d values with chloroform or TMS
as the
internal reference. Coupling constants are reported in Hz. The multiplicity is
defined
by s (singlet), d (doublet), t (triplet), brs (broad signal), dt (doublet of
triplet) and m
IS .(multiplet). IR spectra were recorded with a Mattson Polaris FT-IR
spectrometer and
are reported in cm-1. Melting points were determined on a Meltemp apparatus
(Cambridge, MA), and are uncorrected. Elemental analyses were performed by
Atlantic Microlabs (Norcross, GA). High resolution mass spectrometry was
performed
by the Nebraska Center for Mass Spectroscopy, University of Nebraska (Lincoln,
NE).
The compound reference numbers used in the examples and Tables correspond to
the
compounds depicted in the reaction schemes.
EXAMPLE 1
Preparation Of Compounds 1.5a And 1.5b
Oxalyl chloride (2 mmol, 1 mL from 2M solution in CHZC12) was added to
tropane acid 1.3 (1 mmol) in CH2Cl2 (10 mL) at room temperature under N2. The
resulting mixture was stirred for 1.5 hours and concentrated in vacuo at
30°C to obtain
a viscous oil which was dried in a vacuum for 15 minutes. The acid chloride
obtained '
was dissolved in CHZC12 (10 mL) and cooled to -10°C and amine 1.4 (1
mmol, 470
mg) in CHZCl2 (10 mL) was added, followed by Et3N (3 mmol, 0.42 mL), under N2.
The resulting solution was stirred at room temperature for 6 hours. Water (20
mL) was
then added to the reaction mixture and the product was extracted into CHZCl2
(3 x 20
CA 02233173 1998-04-16
WO 97/14445 PCT/US96/16908
-45-
mL). CH2C12 layers were combined, dried (Na2S04) and concentrated in vacuo to
obtain an oil that was chromatographed on silica (EtOAc:MeOH:NH40H 8.5:1:0.5)
to
obtain the title compound as a viscous oil.
l.Sa: yield 59%; IR 1640 cm-l; 1H NMR (CDCl3) d 1.59 (m, 3H), 2.05-2.5
_ 5 (m, 9H), 2.7-2.9 (m, 4H), 3.1-3.2 (m, 4H), 3.3-3.5 (m, 3H), 3.5 and 3.6
(s, 2H
each), 3.72 and 3.74 (s, 3H each), 6.36 (brs, 1H), 6.8 (d, 2H, J = 6 Hz), 6.84
(d,
2H, J = 6 Hz), 7.02-7.21 (m, 8H).
1.5b: yield 66%; IR 1636 cm-l; 1H NMR (CDC13) d 1.5-1.7 (m, 3H),
2.05-2.5 (m, 9H), 2.7-2.9 (m, 4H), 3.1-3.2 (m, 4H), 3.3-3.5 (m, 3H), 3.5 and
3.6 (s,
2H each), 3.72 and 3.74 (s, 3H each), 6.4 (brs, 1H), 6.8 -6.9 (m, 6H), 7.02-
7.25 (m,
6H).
EXAMPLE 2
Preparation Of Compounds 1.6a And 1.6b
_ BH3~THF (5 mmol, 5 mL of 1M solution in THF) was added to compound
1.5 (1 mmol) in THF (10 mL) under N2 and the resulting mixture was heated at
reflux
for 12 hours. The reaction mixture was cooled to 0°C and 1N HCl was
added
dropwise until there was no more gas evolution, then the mixture was
concentrated in
vacuo. 1N HCI (10 mL) was added to the viscous oil obtained and the resulting
mixture was stirred at 50°C for 30 minutes. The reaction mixture was
cooled to 0°C
and basified with concentrated NH40H. The product was extracted into CHZCIZ
and
purified by chromatography on silica (EtOAc:MeOH:NH40H 8.5:1:0.5) to give the
title compounds.
1.6a: yield 19 % ; IR 1661 cm-1; 1H NMR (CDCl3) d 1.4-1.9 (m, SH),
2.05-3.1 (m, 18H), 3.0-3.15 (m, 1H), 3.26-3.28 (m, 1H), 3.48-3.51 (m, 1H),
3.67
and 3.681 (s, 2H each), 3.72 and 3.73 (s, 3H each), 6.8 (m, 4H), 7.02-7.21 (m,
8H).
1.6b: yield 23 % ; IR 1659 cm-1; 1H NMR (CDC13) d 1.4-1.9 (m, SH),
2.05-3.1 (m, 18H), 3.0-3.15 (m, 1H), 3.26-3.28 (m, 1H), 3.48-3.51 (m, 1H),
3.57
and 3.61 (s, 2H each), 3.72 and 3.73 (s, 3H each), 6.75 -6.8 (m, 4H), 6.87-
6.90 and
6.99-7.04 (m, 2H each), 7.12-7.18 (m, 4H).
CA 02233173 1998-04-16
WO 97/14445 PCT/US96/16908
-46-
EXAMPLE 3
Preparation Of Compounds l.lla and l.llb
Oxalyl chloride (2 mmol, 1 mL from 2M solution in CH2C1~ was added to
tropane acid 1.3 (1 mmol) in CHZCIz (10 mL) at room temperature under N2. The
resulting mixture was stirred for 1.5 h and concentrated in vacuo at
30°C to obtain a
viscous oil, which was dried in a vacuum for 15 minutes. The acid chloride
obtained
was dissolved in CH2C12 (10 mL) and cooled to -10°C and amine 1.9 (1
mmol, 197
mg) in CH2C12 (10 mL) was added, followed by Et3N (2 mmol, 0.28 mL) under N~.
The resulting solution was stirred at room temperature for 6 hours. Water (20
mL) was
then added to the reaction mixture and the product was extracted into CH~C12
(3 x 20
mL). The CHZCl2 layers were combined, dried (NazSO,~ and concentrated in vacuo
to
produce an oil that was chromatographed on silica (EtOAc:MeOH:NH~OH 8.5:1:0.5)
to obtain the title compound as a viscous oil.
l.lla: yield 63 % ; IR 1656 cm-1; 1H NMR (CDCl3) d 1.55-1.78 (m, 3H),
_2.05-2.6 (m, 9H), 3.1-3.4 (m, SH), 3.69 (s, 2H), 3.79 (s, 3H), 6.84 (d, 2H,
J=6 Hz),
7.09 (d, 2H, J=6 Hz), 7.18 (d, 2H, J=6 Hz),7.24 (d, 2H, J=6 Hz), 9.8 (brs,
1H).
l.llb: yield 59%; IR 1653 cm-1; 1H NMR (CDCl3) d 1.59-1.76 (m, 3H),
1.97-2.4 (m, 6H), 2.4 -2.5 (m, 3H), 3.09 (m, 1H), 3.2-3.38 (m, 4H), 3.68 (s,
2H),
3.78 (s, 3H), 6.8-6.9 (m, 4H), 7.08-7.13 (m, 2H), 7.2 (d, 2H, J=10 Hz),9.8
(brs,
1H).
EXAMPLE 4
Preparation Of Compounds 1.13a and 1.13b
BH3~THF (5 mmol, 5 mL of 1M solution in THF) was added to compound
1.11 ( 1 mmol) in THF ( 10 mL) under Na and the resulting mixture was heated
at
reflux for 12 hours. The reaction mixture was cooled to 0°C and 1N HCl
was added
dropwise until there was no more gas evolution, then the mixture was
concentrated in
vacuo. 1N HCl (10 mL) was added to the viscous oil obtained and the resulting
mixture was stirred at 90°C for 30 minutes. The solution was then
cooled to 0 (C and
basified with conc. NHQOH. The product was extracted into CH2Cl2 and purified
by
chromatography on silica (EtOAc:MeOH:NH40H 8.5:1:0.5) to give the title
compound.
CA 02233173 1998-04-16
WO 97/14445 PCT/US96/16908
-47-
1.13a: yield 79%; IR 1608 cm-1; 1H NMR (CDC13) d 1.5-1.8 (m, SH),
2.05-2.2 (m, 4H), 2.23 (s, 3H), 2.34 -2.39 (m, 2H), 2.47-2.52 (m, 2H), 2.65
(dd,
1H, J1 = 5.3, J2 = 7.8), 3.03 (td, 1H, J1 = 4, J2 = 8.8), 3.25 (m, 2H), 3.5
(s,
2H), 3.78 (s, 3H), 6.84 (d, 2H, J = 6 Hz), 7.09 (d, 2H, J = 6 Hz), 7.18 (d,
2H, J =
- 5 6 Hz),7.24 (d, 2H, J = 6 Hz).
1.13b: yield 83 % ; IR 1600 cm-l; 1H NMR (CDCl3) d 1.59-1.76 (m, 3H),
1.97-2.4 (m, 6H), 2.4 -2.5 (m, 3H), 3.09 (m, 1H), 3.2-3.38 (m, 4H), 3.68 (s,
2H),
3.78 (s, 3H), 6.8-6.9 (m, 4H), 7.08-7.13 (m, 2H), 7.2 (d, 2H, J = 10 Hz).
EXAMPLE 5
Preparation Of Compounds 1.14a and 1.14b
Amine 1.9 (12.5 mmol, 2.46 g) in CH2C12 (15 mL) was added dropwise to a
stirred solution of chloroacetyl chloride (12.5 mmol, 1 mL) in CHZCl2 (15 mL),
followed by Et3N (12.5 mmol, 1.7 mL) at -78°C under N~. The reaction
mixture was
-allowed to warm to room temperature and stirred for 1 hours. Water (20 mL)
was then
added and the organic layer was separated and washed with 1N HCl (20 mL),
brine
(20 mL) and water (20 mL). The organic layer was concentrated in vacuo and
dried in
vacuo for 30 minutes. The oil was dissolved in EtOAc (50 mL) and hexane (100
mL).
The turbid solution obtained was concentrated to half the volume and stored in
a
freezer for 4 hours. The solid formed was collected by suction filtration to
give
compound 1.12, which was used for further reactions without additional
purification.
Amine 1.13 (1 mmol), chloro compound 1.12 (2 mmol, 548 mg) and Et3N (2
mmol, 0.28 mL) in acetonitrile (10 mL) were heated at reflux for 24 hours. The
reaction mixture was concentrated in vacuo and the product was partitioned
between
water (20 mL) and CHZCl2 (20 mL). The oil obtained by concentrating the
organic
phase was chromatographed on silica (2 % MeOH: CHZC12 and 10 % MeOH: CH2C12)
to
produce the title compound as a yellow oil.
1.14a: yield 53 % ; IR 1654 cm-1; 1H NMR (CDC13) d 1.4-1.8 (m, SH),
2.05-2.9 (m, 14H), 2.92-3.5 (m, 7H), 3.52 (s, 2H), 3.95 (s, 2H), 3.76 (s, 6H),
6.8-6.85 (m, 4H), 6.9 (d, 2H, J = 6 Hz), 7.15-7.25 (m, 6H), 8.4 (brs, 1H).
CA 02233173 1998-04-16
WO 97/14445 PCT/LTS96/16908
-48-
1.14b: yield 66%; IR 1650 cm-1; 1H NMR (CDCl3) d 1.38-1.76 (m, SH),
2.0-2.81 (m, 14H), 2.88-3.46 (m, 7H), 3.58 (s, 2H), 3.68 (s, 2H), 3.77 (s,
6H),
6.79-6.85 (m, 4H), 6:9-7.03 (m, 2H), 7.18-7.25 (m, 4H), 8.4 (brs, 1H).
EXAMPLE 6
Preparation Of Compounds 1.15a and 1.15b
LAH (1.5 eq) was added to a solution of compound 1.14 (1 mmol) in THF
(10 mL) under N2 and the resulting mixture was heated at reflux for 12 hours.
Usual
work up, followed by chromatography of the resulting product on a
chromatotrone
(EtOAc:MeOH:NH40H 8.5:1:0.5), gave the title compound.
1.15a: yield 63 % . IR 1609 cm-1; 1H NMR (CDCl3) d 1.5-1.8 (m, SH),
2.05-2.3 (m, SH), 2.3 (s, 3H), 2.3-2.6 (m, 7H), 2.7-2.9 (m, SH), 3.1 (td, IH,
Jl =
4, J2 = 8.8), 3.35-3.45 (m, 2H), 3.59 (s, 2H), 3.65-3.7 (m, 2H), 3.76 (s, 6H),
6.8-6.85 (m, 4H), 7.15-7.25 (m, 8H).
_ 1.15b: yield 53 % . IR 1603 cm-1; 1H NMR (CDC13) d 1.4-1.7 (m, SH),
1.95-2.29 (m, SH), 2.25-2 (m, lOH), 2.7-2.9 (m, SH), 3.0 (td, 1H, JI = 4, J2 =
8.8), 3.2 (m, 1H), 3.35 (m, 1H), 3.6 and 3.65 (s, 2H each), 3.7 and 3.77 (s,
3H
each), 6.7-6.8 (m, 4H), 6.9-6.96 and 7.01-7.06 (m, 2H each), 7.16-7.22 (m,
4H).
EXAMPLE 7
General procedure for compounds 1.7, 1.16 and 1.17
Substrate 1.6, 1.14 or 1.15 (1 mmol) was dissolved in TFA (7.5 mL) and
anisole (0.25 mL) at 0°C and Hg(OAc)a (636 mg, 2 mmol) was added. The
resulting
mixture was stirred for 30 min and concentrated in vacuo to obtain a viscous
oil which
was dried in vacuo for 30 minutes. Dry ether (10 mL) was then added to the
above oil
and the resulting suspension was sonicated for 5 minutes. The colorless solid
that
formed was collected by suction filtration, dried in vacuo for 20 minutes and
dissolved
in absolute EtOH (10 mL). H2S gas was passed through the solution for 20
minutes
and the reaction mixture was filtered through a pad of celite. The filtrate
was
concentrated in vacuo to obtain the trifluoroacetate salt of the title
compound, which
was used for further reactions without additional purification.
CA 02233173 1998-04-16
WO 97/14445 PCT/US96/16908
-49-
EXAMPLE 8
Preparation of 2b-{oxo[N,N'-bis-(2'-mercaptoethyl)-ethylenediaminato)] rhenium
(~-methylamino}-3b-(4-chlorophenyl)tropane (1.22)
A solution of compound 1.16a (428 mg, 1 mmol) in MeOH (IO mL) was
added to a solution of Bu4NReOCl4 (588 mg, 1 mmol) in MeOH (2 mL) under NZ at
0°C. Et3N (0.5 mL, 4 mmol) was then added, and the resulting mixture
was stirred at
room temperature for 12 hours. The reaction mixture was concentrated in vacuo.
The
residue obtained was chromatographed on silica (EtOAc:MeOH:NH40H 9.5:0.4:0.1)
to
obtain a purple solid, which was recrystallized (MeOH: CHZCl2) to give pinkish
needles. Yield 43 % ; mp 158 (C; IR (Kbr) 967 cm-1.
EXAMPLE 9
Radiolabeling with Tc-99m
_ A sample of ligand (1.7a, 1.7b, 1.16a, 1.16b or 1.17; 0.2-0.4 ~mol) was
dissolved in 100 ~.L EtOH and 100 ~,L HCl (1 N). 500 ~.L HCl (1 N) and 1 mL
Sn-glucoheptonate solution (containing 136 ~cg SnCl2 and 200 ~,g Na-
glucoheptonate,
pH 6.67) and 50 ~.L EDTA solution (0.1 N) were successively added.
[99mTc]Pertechnetate (100-200 p,L; ranging from 1-20 mCi) saline solution was
then
added. The reaction was heated for 30 minutes at 100°C, cooled to room
temperature
and neutralized with a sat. NaHC03 solution. After extracting the complex from
the
aqueous reaction medium with ethyl acetate (1 x 3 mL, 2 x 1.5 mL) and passing
it
through a small column of Na2S04, ethyl acetate was removed under a flow of
N2. The
residue was dissolved in 200 ~,L EtOH and purified by HPLC (PRP-1 column, 250
x
4.1 mm, CH3CN/3,3-dimethylglutarate buffer, 5 mM, pH 7, volume ratio 8:2, flow
rate 1 mL/min). The retention times for the mixtures of 1.19a were 10.5 to
11.5
minutes (radiochemical yield 88%, radiochemical purity >98%). All the
complexes
displayed stability at 4 and 24 hours after preparation. Little change in
radiochemical
purity was observed. Identical labeling and HPLC conditions were used for the
preparation of mixtures of 1.19b, 1.20a, and 1.20b (retention times of 10.6
and 12,
9.4 and 10.1, 8.0 and 8.2 and 7.8 and 8.9 minutes respectively) with
radiochemical
yields of 70, 88 and 82 % , respectively, and radiochemical purities of > 98 %
. For 21,
CA 02233173 1998-04-16
WO 97/14445 PCT/LTS96/16908
-50-
the radiochemical yield was 80 % , with a purity of 97 % (PRP-1 column,
CH3CN/3,3-dimethylglutarate buffer, 5 mM, pH 7, vol. ratio 6:4).
EXAMPLE 10
Evaluation .
10a. Partition coefficients
Partition coefficients were measured by mixing each Tc-99m compound with 3
g each of 1-octanol and buffer (pH 7.0 or 7.4, 0.1 M phosphate) in a test
tube. The
test tube was vortexed for 3 minutes at room temperature, then centrifuged for
5
minutes. Two weighed samples (0.5 g each) from the 1-octanol and buffer layers
were
counted in a well counter. The partition coefficient was determined by
calculating the
ratio of cpm/g of octanol to that of buffer. Samples from the octanol layer
were
repartitioned until consistent partition coefficient values were obtained. The
measurement was repeated 3 times.
_ lOb. Biodastribution in rats
IS Male Sprague-Dawley rats (225-300 g) allowed free access to food and water
were used for in vivo biodistribution studies. Kung, 1984, supra; Kung, 1985,
supra.
While under ether anesthesia, 0.2 mL of a saline solution containing 1.19a,
1.20a or
1.21 (50-100 ~cCi) was injected directly into the femoral vein of rats, and
the rats were
sacrificed by cardiac excision at various time points post-injection. The
organs of
interest were removed and weighed, and the radioactivity was counted with an
automatic gamma counter (Packard 5000). The percentage dose per organ was
calculated by a comparison of the tissue counts to suitably diluted aliquots
of the
injected material. Total activities of blood and muscle were calculated under
the
assumption that they were 7 % and 40 % of the total body weight, respectively.
Regional brain distribution in rats was obtained after an injection of 1.19a,
1.20a or 1.21. Samples from different brain regions (cortex, striatum,
hippocampus,
and cerebellum) were dissected, weighed and counted, and the percentage dose
per
gram of sample was calculated by comparing the sample counts with the count of
the
diluted initial dose. The uptake ratio of each region was obtained by dividing
the
percentage dose per gram of that region by that of the cerebellum. For
blocking
studies, rats were injected with either (3-CIT or haloperidol (iv, 1 mg/Kg) 5
min prior
CA 02233173 2004-05-25
51 -
to injection of 1.19a. The rats were dissected and.brain tissue samples were
counted as
described above. The specific uptake of the compound was expressed as ratio of
ST/CB (% dose/g of striatum divided by the same of cerebellum).
lOc. SPECT and MRI imaging in a baboon
A baboon ( -- Z S kg) was the subject of a SPECT imaging study. Prior to
imaging, the animal was fasted, immobilized with ketamine (10-20 mg/kg, i.m.)
and
xylazine (2-3 mg/kg, i.m. ), intubated and maintained on a 1.5-2.0 %
isofluorane/98.5
oxygen mixture (flow rate of 200-500 cc/min). The animal was injected with
glycopyrrolate (10 ~,g/kg, s.c.), an anticholinergic drug that does not cross
the
blood-brain barrier, in order to decrease digestive and respiratory
secretions. Body
temperature was maintained using a hot water circulating heating pad and was
monitored with a rectal thermometer. The animal's head was immobilized using a
. vacuum-packed bean-bag device that hardens upon evacuation when molded
around the
head. No-carrier-added [~"'Tc]TRODAT-1 (1.19a; 9 mCi) was administered as an
- intravenous bolus in the saphenous vein of the baboon. Immediately after
injection,
sequential 5 minute per frame dynamic SPECT scans were acquired on a triple-
headed
Picker Prism 3000 camera (FVVHM: 7 mm) equipped with fan beam collimators for
2
hours. The acquisition parameters were a 20% energy window at 140 KeV, 120
projection angles over 360 degrees, a 128 x 128 matrix, and a zoom factor of
1.78 in a
slice thickness of 2 mm. The projection data was reconstructed with a count
dependent
3-D Wiener filter. Chang's first order correction method was used to
compensate for
attenuation. Magnetic resonance images (MRI) of the baboon brain were acquired
with
a 1.5 Tesla machine (GE Medical Systems, Milwaukee, WI). The spoiled GRASS
acquisition parameters included a repetition time (TR) of 5 msec and a flip
angle of 35
degrees, which produced 1 mm thick slices. The data were reinterpolated in 256
x 256
matrices with cubic voxels of 2 mm per side to match the SPECT images. The
SPECT
images of [99mTc]TRODAT-1 (1.19a) at 60-90 minutes post-injection were summed
and reformatted to match the MRI scans. Both data sets were imported into a
locally
developed software package for co-registration. The program simultaneously
displayed
three orthogonal views of either the MRI, the SPECT scans (coronal, transaxial
and
sagital views), or the fused images of both sets.
CA 02233173 1998-04-16
WO 97/14445 PCT/US96/16908
-52-
EXAMPLE 11
Experimental For Reaction Schemes Depicted In Figures 1 and 2.
Syntheses of the 3(3-p-chloro and p-fluorophenyl tropane ester, 1.2a and 1.2b,
respectively, were carried out according to the procedure described previously
S (Meltzer, 1993, supra) and the corresponding carboxylic acids 1.3a and 1.3b
were
made following a literature procedure (Carroll, 1992, supra) and are shown in
Figure
1. The corresponding acyl chlorides of these carboxylic acids were prepared by
the
action of oxalyl chloride at room temperature intercepted with amine 1.4 to
obtain
amide 1.5. Diborane reduction of these amides in THF furnished corresponding
tertiary
amide 1.6. The removal of the 4-methoxybenzyl groups of protected dithiols
compound
1.6 was achieved by treating the substrate with Hg(OAc)a in TFA followed by
the
removal of mercury as its sulfide.
The synthetic strategy employed for the preparation of compound 1.16 is shown
in
Figure 2. Acyl halide 1.3 was converted to secondary amide 1.11 by reacting it
with
_ amine 1.9 in the presence of triethyl amine. Secondary amine 1.13 was
prepared by
diborane reduction of amide 1.11, and was converted to amide 1.14 by
alkylations with
alkyl chloride 1.12. The amide functions of compound 1.14 were reduced with
LAH to
yield compound 1.15. Amine 1.15 and amide 1.14 were deprotected with Hg(OAc)~
to
obtain trifluoroacetate salts of dithiols 1.16 and 1.I7. Since the instability
of the
disulfides 1.7, 1.16 and 1.17 prevented the purification of these dithiols,
they were
obtained as their triflate salts, and used directly without further
characterization for the
preparation of rhenium (22) and technetium (1.19a, 1.19b, 1.20a, 1.20b and
1.21)
complexes.
The radiolabeling of 1.7, 1.16 and 1.17 with sodium [99'"Tc]pertechnetate was
successfully achieved by using stannous(II) glucoheptonate as the reducing
agent to
produce 1.19a, 1.19b, 1.20a, 1.20b and 1.21 in good yield (80% ) and
radiochemical
purity ( > 95 % ) .
CA 02233173 1998-04-16
WO 97/14445 PCT/US96/16908
-53-
EXAMPLE 12
Evaluation And Results Comparing Compounds Of The Invention Having the NZS2
Ligand In The 2~-Position Of The Tropane Core With Other Compounds And
Prior Art Compounds
The novel series of compounds differ from those previously reported in that
the substitution of the bis-aminethanethiol ligand is attached at the 2a-
position of the
tropane core structure. The corresponding Tc-99m labeled agent displayed a
three- to
fourfold increase in initial brain uptake (0.1 % for N-substituted compound
(Meegalla,
1995, supra) vs. 0.3-0.4 % in brain at 2 min post injection) and concomitantly
retained
the specific uptake in the striatum area of the brain. This observation
suggests that, in
this series of compounds, the 3a-p-fluoro- is slightly less favorable then the
corresponding 3~i--p-chlorophenyl derivative. As previously reported for the
same
series of tropane derivatives, the 3(3-p-fluoro- derivative displayed a lower
brain
uptake, which may be due to its lower binding affinity to dopamine
transporters.
' Carroll, 1995, supra.
12a. Biodistribution comparison
The major criteria for determining the potential usefulness of in vivo
dopamine
transporter imaging agents are based on an animal biodistribution study. In
this case,
rats were used as an animal model. After an intravenous injection of the Tc-
99m
labeled compounds, the values of initial brain uptake ( % dose/whole organ) at
2
minutes post intravenous injection are used to measure the ability to
penetrate the intact
blood-brain barrier. Compounds displaying higher brain uptake are the better
agents
(minimum requirement > 0.1 % dose in brain). The second criteria is the
specific
uptake in the striatum region of the brain, where dopamine transporters are
located.
The uptake value (% dose/gram) of the striatum (ST) is divided by the
background
area, cerebellum (CB; essentially devoid of dopamine transporter); the ratio
of ST/CB
is an indicator for specific uptake (minimum requirement for ST/CB > 1.5).
Five
structurally similar tropane derivatives were examined in this biological
study. (Tables
1.1 to 1.4). Compounds 1.19a and 1.24c displayed high initial uptake and high
specific retention in brain.
CA 02233173 1998-04-16
WO 97/14445 PCT/CTS96/16908
-54-
12b. Partition coefficients evaluation
One of the key properties for these potential dopamine transporter imaging
agents is their neutrality and lipophilicity. The [~''"Tc] labeled complexes,
1.19a,
1.20a, 1.20c and 1.21, displayed excellent medium range lipophilicity
(partition
coefficients between 1-octanol and pH 7.0 buffer of 99-1818). It is noteworthy
that,
contrary to the lowered lipophilicity commonly observed with the addition of
an amide
group, complex 1.21, containing an amide functional group, exhibited an almost
tenfold higher partition coefficient compared to the corresponding reduced
compound,
1.19a.
The lipophilicity, as measured by the partition coefficient (1-octanol/pH 7.0
buffer), displayed very unpredictable values. Most striking is the value for
1.21, which
contains an amide inside the chelating ring (partition coefficient = 1818).
The partition
coefficient value is very important for the evaluation of potential brain
imaging agents,
because they must be neutral and lipophilic in order to penetrate the intact
blood-brain
_ barner. Generally, the optimal range of partition coefficient values for
good brain
uptake are between 100-1000.
12c. Brain uptake
Biodistribution studies of 1.19a, 1.20a, 1.20c and 1.21 in rats were performed
after an intravenous injection of a tracer dose. The results showed a
distribution pattern
reflecting regional perfusion (i. e. , high uptake in muscle, kidney, liver,
brain and skin;
Tables 1. l and 1.2). However, the brain uptake is moderate, ranging from 0.43-
0.11
dose/organ at two minutes for the five complexes. The complex containing an
amide
group, 1.2I, showed the lowest brain uptake (Table 1.1) despite its high
lipophilicity
(P.C. = 1818). Increasing the lipophilicity of these compounds does not appear
to
improve brain uptake in rats. The most significant fording of this initial
biodistribution
study is that the brain uptake of these complexes, except 1.21, was highly
concentrated
in the striatal area, where the dopamine transporters are located, compared to
a region
with no dopamine neurons (i.e., the cerebellar region); therefore, at 60
minutes after
an intravenous injection, the ratios of striatum to cerebellum (ST/CB) were
found to be
2.66, 2.18, and 2.82 for 1.19a, 1.20a and 1.20c, respectively. However,
complex 21
showed little specific uptake (ST/CB = -1.17) at 60 minutes post intravenous
injection.
The initial biodistribution study suggested that [~'mTc]TRODAT-I (1.19a) is a
good
CA 02233173 1998-04-16
WO 97/14445 PCT/US96/16908
-55-
imaging agent candidate in this series of complexes. It displayed the highest
initial
brain uptake, and high retention in the target area (i. e. , striatum) at
later time points;
therefore, a more detailed biodistribution study was carried out. Specific
uptake of
s
[~'mTc]TRODAT-1 (1.19a) at different time points displayed a prolonged
retention, and
the ST/CB ratio reached a maximum value of 4.07 at 4 hours after injection
(Table
1.3).
Blocking studies in rats were performed at 60 minutes post-injection to
characterize further the uptake and binding, and [~'"Tc]TRODAT-1 (1.19a) did
indeed
show binding to dopamine transporter sites. In this series of regional brain
uptake
studies in rats, the ST/CB ratio at 60 minutes post-injection was 2.66 (Fig.
1.1). The
specific uptake of [99mTc]TRODAT-1 11.19a.) in rat striatum could be blocked
by
pretreating rats with a dose of ~3-CIT (1 mg/kg, iv), a known dopamine
transporter
ligand. The specific binding, as indicated by ST/CB, was reduced to 1.0 after
pretreatment with (3-CIT. The specific binding was not reduced by pretreatment
with
_haldol (1 mg/kg, iv), an agent with a mixed pharmacological profile (binding
to
various CNS receptors but not to the dopamine transporter); no blocking effect
was
observed. The ST/CB ratio (2.6) was identical to that in control rats (Fig.
5). The
results suggested that uptake in the rat striatum was specifically related to
the dopamine
transporter in rat brain.
To evaluate further the potential of [99"'Tc]TRODAT-1 (1.19a) as a dopamine
transporter imaging agent, a SPECT imaging study was carried out in a female
baboon.
To facilitate the identification of anatomical localization, the coronal,
transaxial and
sagital SPECT images (1.34 mm thick) obtained 60-90 minutes post injection
were
coregistered with the MRI images of the same baboon. The fused images
displayed
excellent consistence with the expected localization of this agent in caudate
and
putamen areas, where the dopamine transporters are known to be located. The
images
also showed good correlation with PET imaging agent ['1C]CFT and SPECT imaging
agent [1231]/3-CIT, reported previously. In vitro binding studies of Re
complex 1.22 in
rat striatal homogenates displayed good binding affinity (Ki = 14 nM, using
['~I]IPT
(Kung, 1995, supra) as the ligand; Kd = 0.2 nM, data not shown); while the
bis-ethanethiol, 1.16a, displayed a comparable affinity (Ki = 7 nM). However,
upon
intravenous co-injection of the bis-ethanethiol ligand into rats (200 mg/dose,
which is
CA 02233173 1998-04-16
WO 97/14445 PCT/US96/16908
-56-
equivalent to I mg/Kg), the specific uptake (Table 1.2) was not blocked by the
competing agent, which suggests that the free t<hiol compound may not be able
to
penetrate the intact blood-brain barrier and compete with the binding of
[99mTc]TRODAT-1 (1.19a) as a dopamine transporter in the brain. Although the
binding affinity of the corresponding Re complex, 1.22, is not as potent as
other
iodinated tropane derivatives, the brain uptake and retention of [~mTc]TRODAT-
1
(1.19a) appears to be sufficient for in vivo SPECT imaging in nonhuman
primates.
The initial brain uptake at 2 minutes after intravenous injection in rat brain
is
the key indicator for evaluating the compound's penetration of the blood-brain
barrier.
For compounds with high first pass extraction, the brain uptake values at 2
minutes are
in the range of 2-3 % dose/whole brain after iv injection in rats. Comparing
the four
structurally similar compounds (Table 1.4), only 1.19a displayed significant
uptake; the
other three compounds, 1.21, 1.24 and 1.25, showed at least 300 % lower
uptake.
This observation is very surprising and not consistent with what one would
_predict by their partition coefficient. Based on this novel finding,
compounds
containing an amide in the N2S2 chelating ring system are specifically removed
from
further development. The prior art, technepine (Madras, 1996, supra), used a
N2S~
chelating ring system containing an amide group (technepine), which provides
less
favorable biological properties.
For dopamine transporter imaging agents, the target area of the brain is the
striatum, where dopamine transporters are highly concentrated. The cerebellum
region
is suitable for use as the background region, because it has no dopamine
transporters.
The specific uptake is measured by the ratio of % dose/gram of striatum
divided by
dose/gram of cerebellum (ST/CB ratio}--the higher the value, the better the
specific
uptake, and the more promising as a dopamine transporter imaging agent. Again,
the
data in Table 1.1 shows that ST/CB ratio for 1.19a is the best among this
group of
compounds. The specific uptake of these agents in rat brain clearly displays
the novel
finding that, among the structurally similar compounds containing a N~S2
chelating
group, only 1.19a displays the selective localization in the striatum, where
dopamine
transporters are located. Single photon emission computed tomography (SPELT)
images of the selected and claimed agent displayed a high contrast in the
target area
(striatum) of a baboon (nonhuman primate) brain. The data clearly demonstrate
that the
CA 02233173 1998-04-16
WO 97/14445 PCT/CTS96/16908
-57-
new technology can be reduced to practice and is potentially useful for human
imaging
study of dopamine transporter in brain.
Based on the novel findings regarding biodistribution discussed above, the
specific chemical compounds (1.19a and 1.20a) not containing amide groups and
with
an AZ moiety are particularly useful dopamine transporter imaging agents.
EXAMPLE 13
Evaluation Of Stereoisomeric Compounds
It is possible that tropane derivatives containing a bis-aminoethanethiol
group
will form stereoisomers, which requires the separation and characterization of
these
isomers (Table 1.5 a, b, c). Initial evaluation of the stereoisomers suggests
that Peak A
and Peak B displayed different brain uptake and specific localization in
striatum area:
Peak A showed higher initial brain uptake (0.5 vs. 0.28% dose/organ, at 2
minutes
post-injection for Peak A and Peak B, respectively), but Peak B had a higher
ratio
_ (ST/CB 2.72 vs. 3.79 at 60 min post-injection for Peak A and Peak B,
respectively).
Correlation of dopamine transporter uptake as measured by SPECT imaging and
the
actual neuronal integrity requires extensive kinetic modeling studies to
validate and
estimate the potential clinical utility. Optimized imaging protocol and
reproducibility of
SPELT imaging of this new agent remain to be investigated.
CA 02233173 1998-04-16
WO 97/14445 PCT/LTS96/16908
-58-
Table 1.1. Brain uptake of six Tc-99m labeled tropane derivatives (%
dose/organ)
H~C~Y CSTc.~ S'O
N ~ Y Qi s.Y C Tc '
Y Y
x ( 1.11 )~ a
TRODAT-1 (a.a9.) n
( LZOs) F
( lZOc) 1~
Compound 2 min 60 min ST/CB Ratio P.C
at 60 min
1.19x, TRODAT-1 0.43 f 0.16 0.12 t 0.001 2.66 t 0.01 227
1.20a 0.37 t 0.09 0.098 t 0.0142.18 t 0.32 99
1.20c 0.41 t 0.03 0.18 t 0.01 2.82 t 0.19 262
1.21 0.11 t 0.02 0.07 t 0.014'1.17 t O.OIO 1818
'Stria_tal/Cerebellum (ST/CB) ratio: % dose per dram of striatum/ % does per
gram of
cerebellum
b P.C.: measured between 1-octanol/pH 7.0 phosphate buffer
~ Data were from rats sacrificed at 30 minutes
CA 02233173 1998-04-16
WO 97/14445 PCT/US96/16908
-59-
Table 1.2. Biodistribution in rats post intravenous injection of
[99mTc]TRODAT-1, 1.19a'
Organ 2 minutes 60 minutes 60 minutes'
Blood 4.94 t 0.46 2.14 t 0.17 6.29 t 1.12
S Heart 1.47 t 0.16 0.19 t 0.02 0.25 t 0.05
Muscle 10.84 t 1.95 10.28 t 1.36 10.37 t 0.48
Lung 6.82 t 0.37 1.86 t 0.36 1.76 t 0.12
Kidney 5.13 t 0.75 3.06 t 0.14 1.76 t 0.32
Spleen 0.42 t 0.16 0.45 t 0.05 0.51 t 0.06
Liver 16.62 t 2.11 17.67 t 3.42 20.35 t 0.99
Skin 2.66 t 0.35 3.96 t 0.22 4.11 t 0.29
Brain 0.43 t 0.16 0.12 t 0.00 0.12 t 0.01
Regional Brain Distribution (°lo does/g)
Region 2 min (ST/CB) 60 min (ST/CB) 60 mini (ST/CB)
Cerebellum 0.284 t 0.066 0.057 t 0.003 (1.00)0.059 t 0.007 (1.00)
Striatum 0.266 t 0.066 O.I51 t 0.002 (2.66)0.156 t 0.018 (2.65)
Hippocampus 0.268 t 0.090 0.096 t 0.015 (1.69)0.104 t 0.011 (1.77)
Cortex 0.347 t 0.140 0.094 t 0.012 (1.65)0.090 t 0.009 (1.53)
Remainder 0.269 t 0.076 0.076 t 0.005 (1.34)0.079 t 0.006 (1.34)
' % does/organ, average of 3 rats t SD
b Biodistribution in rats post iv injection of [~'''"Tc]TRODAT-1 with excess
ligand
(100 mg/does; % does/organ, average of 3 rats t SD)
CA 02233173 1998-04-16
WO 97/14445 PCT/US96/16908
-60-
Table 1.3. Biodistribution in rats post intravenous injection of
[99mTc]TRODAT-1, 1.19a'
2 min 30 min 60 min 120 min 240 min 360 min
ST/CB 2.6610.01 3.9011.334.0710.54 2.97 t
0.9310.23 2.0410.18 0.14
-
Ratio
~ Striatal/Cerebellum (ST/CB) ratio: % dose per gram of striatum/ % dose per
gram of
cerebellum
CA 02233173 1998-04-16
WO 97/14445 PCT/US96/16908
-61-
Table 1.4 Biodistribution in rats (brain uptake, % dose/organ) after an
intravenous injection of ["'°'Tc]TRODAT-1 and related compounds (%
dose/organ,
average of 3 rats t SD)
s. o, o~
Y
Q-t ~~Y C Tc\
O
Q
( 1.21 )
TRODA'T-i (1.19x)
C~..~.~ Cso~
--~ ~ H. ~...~
a
( 1.Z4)
( 1.Z5)
ST/CB ratioPartition
Organ 2 ~ 30 min (30 min) coefficient
S TRODAT-1(1.19x) 0.42510.1200.23210.0212.0 22?
(1.21) O.iIt0.02 0.07010.0141.17 1818
(1.24) 0.02810.0070.02210.002- 1.28
(1.25) 0.04010.0070.01310.002- 1-07
CA 02233173 1998-04-16
WO 97/14445 PCT/US96/16908
-62-
Table 1.5. Biodistribution in rats (brain uptake, % dose/organ) after an
intravenous injection of ['9"'Tc]TRODAT-1 (1.19a) (racemic) and stereoisomers
(Peak A and Peak B) (% dose/organ, average of 3 rats t SD)
Table 1.5a. Biodistribution in rats after an intravenous injection of peak A
(1.19a)
(% dose/organ, average of 3 rats t SD)
Organ 2 min 60 min
Blood 1.98 t 0.19 1.00 ~ 0.15
Heart 1.76 t 0.09 0.21 t 0.00
Muscle 33.61 t 4.31 14.98 t 0.49
i
Lung 9.68 t 0.51 3.06 0.?.8
Kidney 5.47 t 1.04 2.04 t 0.21
Spleen 0.27 t 0.04 0.46 t 0.18
Liver 9.55 t 2.19 17.75 t 1.81
Skin 2.73 t 0.39 3.56 t 0.65
IS Brain 0.50 t 0.06 0.21 t 0.01
I
Regional brain distribution (% dose/g)
Region 2 min 60 min Ratio ~ 60
min
Cerebellum 0.289 t 0.017 0.086 t 0.007 1.00
Striatum 0.318 t 0.063 0.235 t 0.009 2.72
2,0 Hippocampus 0.287 t 0.082 0.138 t 0.003 1.61
Cortex 0.387 t 0.047 0.139 t 0.007 1.61
Remainder 0.283 t 0.047 0.119 t 0.007 1.38
CA 02233173 1998-04-16
WO 97/14445 PCT/ITS96/16908
-63-
Table l.Sb. Biodistribution in rats after an intravenous injection of peak B
(1.19a)
(% dose/organ, avg. 3 rats t SD)
Organ 2 min 60 min
Blood 4.02 t 0.20 1.89 t 0.21
Heart 1.28 t 0.15 0.19 t 0.06
Muscle 41.46 f 11.33 8.60 t 2.51
Lung 6.00 t 1.11 1.23 t 0.89
Kidney 4.47 t 0.69 1.87 f 0.80
Spleen 0.27 t 0.03 0.41 t 0.24
1~ Liver 9.07 t 1.66 18.71 t 2.54
Skin 3.43 t 0.62 4.11 t 0.62
Brain 0.28 t 0.01 0.12 f 0.02
Regional Brain Distribution (% dose/g)
Region 2 min 60 min Ratio (~ 60
min
15 Cerebellum 0.167 t 0.019 0.047 t 0.010 1.00
Striatum 0.155 f 0.019 0.177 t 0.008 3.79
Hippocampus 0.137 t 0.003 0.083 t 0.012 1.77
Cortex 0.199 t 0.012 0.083 t 0.016 1.78
Remainder 0.149 t 0.007 0.069 t 0.009 1.48
CA 02233173 1998-04-16
WO 97/14445 PCT/US96/16908
_(,ø_
Fable l.Sc. Brain uptake in rats after an intravenous injection of the
Racemate
(1.19a) (% dose/organ, average of 3 rats t SD)
Organ 2 min 60 min
Brain 0.43 ~ 0.16 0.12 t 0.00
Regional brain distribution (% dose/g)
Region 2 min 60 min Ratio ~ 60 min
Cerebellum 0.284 t 0.066 0.057 t 0.003 1.00
Striatum 0.266 t 0.066 0.151 t 0.002 2.66
Fiippocampus 0.268 t 0.090 0.096 t 0.015 1.69
1~ Cortex 0.347 t 0.140 0.094 t 0.012 1.65
Remainder 0.269 t 0.076 0.076 t 0.005 1.34
CA 02233173 1998-04-16
WO 97/14445 PCT/US96/16908
-65-
General Experimental For Examples 14 To 17
Reagents used in the syntheses were purchased from Aldrich (Milwaukee, WI)
or Fluka (Ronkonkoma, NY), and were used without further purification unless
otherwise indicated. Anhydrous Na2S04 was used as a drying agent. Reaction
yields
are reported without attempts at optimization. Thin layer chromatography was
performed on EM Science (Gibbstown, NJ) precoated (0.2 mm) silica gel 60
plates,
and the spots were detected with iodine vapor and/or UV light. Silica gel 60
(70-230
mesh), obtained from EM Science (Gibbstown, NJ), was used for column
chromatography. 1H NMR spectra were obtained on a Broker spectrometer (Broker
AC 300). All samples prepared for NMR analysis were dissolved in CDCl3,
purchased
from Aldrich. Chemical shifts are reported as d values with TMS as the
internal
reference. Coupling constants are reported in Hz. The multiplicity is defined
by s
(singlet), t (triplet), td (triplet of doublet), dt (doublet of triplet) and m
(multiplet). IR
spectra were recorded with a Mattson Polaris FT-IR spectrometer and are
reported in
cm-1. Melting points were determined on a Meltemp apparatus (Cambridge, MA)
and
are uncorrected. The compound reference numbers used in the Examples and
Tables
correspond to the compounds depicted in Figures 8-11.
EXAMPLE 14
Preparation of 2.2
A mixture of nortropane derivative 2.1 (1 g, 3.9 mmol), KI (664 mg, 4
mmol), K2C03 (1.4 g, 10 mmol) and bromopropanol (0.37 mL, 4 mmol) in dioxane
(25 mL) was heated at reflux under N2 for 12 hours. The reaction mixture was
allowed to cool to room temperature and then filtered. The filtrate was
partitioned
between water and CH2Cl2. The CH2C12 layer was concentrated in vacuo to obtain
a
viscous oil, which was purified on silica (2 % MeOH: CH2C12 drop of NH3) to
obtain
the title compound as a colorless oil. Yield 46%; IR (CHC13) 3330, 1739; 1H
NMR
1.45 -1.8 (SH, m), 1.95-2.2 (m, 2H), 2.37-2.5 (m, 3H), 2.85-2.8 9 (m, 1H),
2.98 (td,
J1= 5.1, J2= 12.8, 1H), 3.45 (s, 3H), 3.57-3.63 (m, 2H), 3.69-3.78 (m, 2H),
7.11
and 7.19 (d, J = 8.55, 2H each).
CA 02233173 1998-04-16
WO 97/14445 PCT/US96/16908
-66-
EXAMPLE 15
Preparation of 2.3
A solution of alcohol, 2.1 (337 mg, 1 mmol) and Et3N (0.14 mL, 1 mmol) in
CH2Ci2 was cooled to -10°C under N2 and MsCI 9 0.07 mL, 1 mmol) was
added over
5 minutes. The resulting solution was stirred for 20 minutes. and water (10
mL) was
added and allowed to warm to room temperature. The CHZCI~ layer was separated
and
concentrated in vacuo at 25°C to obtain a viscous oil which was dried
in vacuo for 30
minutes. The mesylate obtained was dissolved in dry acetone (10 ml) and
anhydrous
Liar (108 mg, 1.2 mmol) was added. The resulting mixture was heated at reflux
for 5
hours under N2. The reaction mixture was allowed to cool to room temperature
and
concentrated in vacuo to obtain a viscous oil which was purified on silica (1
% MeOH:
CH2Cl~ to obtain the title compound as a viscous oil. 62 % ; IR (CHC13) 1740;
1H
NMR 1.58-2.15 (m, 7H), 2.38 (t, J=6.18, 2H), 2.55 (dt, Jl =2.88, J1= 12.36,
1H),
2.7-3 9 (m, 2H), 3.36-3.37 (m, 1H), 3.5-3.68 (m, 6H), 7.16 and 7.23 (d, J=8.7,
2H
each).
EXAMPLE 16
Preparation of 2.4
A solution of bromo compound B.3 (1.4 g, 3.6 mmol), N,N'-
bis-(2-S-4-methoxybenzyl-2-mercapto)ethyl-ethylenediamine (3.3 g, 7.9 mmol),
KI
(770 mg, 3.6 mmol), KZC03 (1.4 g, 10.1 mmol) in dioxane (50 mL) was heated at
reflux for 15 hours. The reaction mixture was allowed to cool to room
temperature and
filtered. The filtrate was concentrated in vacuo to obtain a viscous oil,
which was
partitioned in between water and CH2Cl2. The CH2C12 layer was concentrated in
vacuo
to obtain an oiI which was purified on silica (EtOAc: MeOH: NH40H; 9: 0.9:
0.1).
21 % ; IR (neat) 1746; IH NMR 1.4-1.7 (m, 4H), 2-2.4 (m, 6H), 2.41-2.7 (m,
I2H), '
2.75 (t, J = 6.8, 2H), 2.85-2.93 (m, 2II), 3.34-3.36 (m, 2H), 3.45 (s, 3H),
3.63-3.69
(m, SH), 3.77 (s, 6H), 6.82 (d, J = 8.7, 4H), 7.14-7.26 (m, 8H). '
CA 02233173 1998-04-16
WO 97/14445 PCT/US96/16908
-67-
EXAMPLE 17
Preparation of 2.7a
Substrates 2.Sa (1 mmol) was dissolved in TFA (7.5 mL) and anisole (0.25
mL) at 0°C and Hg(OAc)2 (636 mg, 2 mmol) was added. The resulting
mixture was
stirred for 30 minutes. and concentrated in vacuo to obtain a viscous oil
which was
dried in vacuo for 30 minutes. Dry ether (10 mL) was then added to the above
oil and
the resulting suspension was sonicated for 5 minutes. The colorless solid
formed was
collected by suction filtration and dried in vacuo for 20 minutes and
dissolved in
absolute EtOH (10 mL). H2S gas was passed through the solution for 20 minutes
and
the reaction mixture was filtered through a pad of celite. Filtrate was
concentrated in
vacuo and redissolved in CH2Cla (IO mL) and washed with saturated Na2C03 (10
mL).
The CH2Cl2 layer was concentrated in vacuo to obtain 2.Sa as a viscous oil.
Bu4NReOCI4 (588 mg, I mmol) was dissolved in MeOH (5 mL) under Ar and cooled
to 0°C. Dithiol 2.Sa (485 mg, 1 mmol) in MeOH (5 mL) was then added
followed by
Et3N (0.56 mL, 4 mmol). The resulting solution was stirred at room temperature
for
12 hours and concentrated in vacuo. The residue obtained was subjected to
preparative
TLC (CHZCI2:MeOH:NH40H 9:0.9:0.1) to obtain stereoisomeric mixture of 2.Sa.
23 % ; mp mixture 95-110°C (sub.). IR (KBr) 1743, 941; 1H NMR 2.8-3.2 9
(m),
3.2-3.45 (m), 3.49 and 3.52 (s each), 3.52-3.7 (m), 4-4.25 (m),7.14 (d,
J=8.7), 7.15
(d, J=8.7), 7.34 (m). Radiochemistry was achieved by Figure 9. The
corresponding
rhenium complex, 2.6, was prepared as surrogate for chemical analysis. Figure
10.
CA 02233173 1998-04-16
WO 97/14445 PCT/US96/16908
-68-
Table 2.1a. Biodistribution in- rats after intravenous injection of peak A of
(2.6a)
(% dose/organ, avg. of 3 rats t SD)
Organ 2 min 30 min
Blood 2.41 t 0.33 1.16 t 0.06
S Heart 0.79 t 0.03 0.22 t 0.02
Muscle 7.78 t 0.32 10.41 t 1.26
Lung 15.63 f 2.31 9.65 t 1.37
Kidney 2.97 t 0.09 1.25 f 0.11
Spleen 0.42 t 0.10 0.42 t 0.05
1~ Liver 14.29 t 2.56 20.87 t 2.23
Skin 1.59 t 0.68 5.69 t 0.73
Brain 0.25 t 0.04 0.13 t 0.02
CA 02233173 1998-04-16
WO 97/14445 PCT/US96/16908
-69-
Table 2.1b. Biodistribution in rats after intravenous injection of peak B of
(2.6a)
(% dose/organ, avg. of 3 rats t SD) * n = 1
Organ 2 min 30 min
Blood 3.96 t 0.70 1.82 t 0.43
S Heart 0.70 t 0.21 0.24 t 0.08
Muscle 9.92 t 1.04 8.09 t 1.686
Lung 19.16 t 2.06 12.51 t 1.46
Kidney 2.03 t 0.45 1.24 t 0.18
Spleen 0.42 t 0.10 0.67 t 0.17
Liver 19.56 t 3.21 28.57 t 2.94
Skin 1.25 t 0.17 3.25 t 0.76
Brain 0.19 t 0.041 0.094 t 0.026
Regional brain distribution (% dose/g)
Region 2 min 30 min Ratio v. CB
IS Cerebelium 0.13 t 0.01 0.041 t 0.009 -
Striatum 0.10 t 0.03 0.057 t 0.014 1.4
Hippocampus 0.090 t 0.01 0.054 t 0.012 1.3
Cortex 0.14 t 0.03 0.064 t 0.016 1.6
Remainder 0.10 t 0.02 0.052 t 0.015 1.3
CA 02233173 1998-04-16
WO 97/14445 PCT/US96/16908
-70-
Table Z.lc. Brain uptake in rats after intravenous injection of the Racemate
of
(2.6a) (2.5; % dose/organ, avg. of 3 rats t SD)
Organ 2 min 30 anin
Brain 0.27 t 0.02 0.11 t 0.02
Regional brain distribution (% dose/g)
Region 2 min 30 min Ratio v. CB
Cerebellum 0.161 t O.OIS 0.048 t 0.004 -
Striatum 0.159 t 0.025 0.086 t 0.010 1.8
Hippocampus 0.139 t 0.016 0.063 t 0.008 1.4
1~ Cortex O.I83 t 0.014 0.072 t 0.003 1.5
Remainder 0.146 t 0.017 0.062 t 0.007 1.4
CA 02233173 1998-04-16
WO 97/14445 PCT/US96/16908
-71-
General Experimental For Examples 18 To 28
Reagents used in the syntheses were purchased from Aldrich (Milwaukee, WI)
or Fluka (Ronkonkoma, NY), and were used without further purification unless
otherwise indicated. Anhydrous Na2S04 was used as a drying agent. Reaction
yields
., 5 are reported without attempts at optimization. Thin layer chromatography
was
performed on EM Science (Gibbstown, NJ) precoated (0.2 mm) silica gel 60
plates,
and the spots were detected with iodine vapor and/or UV light. Column
chromatography was performed on silica gel 60 (70-230 mesh) obtained from EM
Science (Gibbstown, NJ). The spectra of 'H and 13C were obtained using a
Bruker
spectrometer. All samples prepared for NMR analysis were dissolved in CDCl3
purchased from Aldrich. Chemical shifts are reported as d values with
chloroform or
TMS as the internal reference. Coupling constants are reported in Hz. The
multiplicity
is defined by s (singlet), d (doublet), t (triplet), brs (broad signal), dt
(doublet of
triplet) and m (multiplet). IR spectra were recorded with a Mattson Polaris FT-
IR
_ spectrometer and are reported in cm 1. Melting points were determined on a
Meltemp
apparatus (Cambridge, MA), and are uncorrected. Elemental analyses were
performed
by Atlantic Microlabs (Norcross, GA). HRMS was performed by the Nebraska
Center
for Mass Spectroscopy, University of Nebraska (Lincoln, NE).
Compounds 3.7, 3.8, 3.10 and 3.12 were prepared according to the literature
methods. Satisfactory elemental analyses (withi_n 0 4 % ) could not be
obtained for the
thiols and dithiols and hence HRMS data for those compounds are reported. The
compound reference numbers used in the examples and Tables correspond to the
compounds depicted in the reaction schemes.
EXAMPLE 18
Preparation of N-[(2-(4'-methoxybenzylthio)-2-methylpropyl)] 2-(4'-
methoxybenzylthio)-2-methyl-propionamide (3.3)
2-(4'-methoxybenzylthio)-2-methylpropionic acid (2.4 g, 10 mmol) and SOC12
(7 mL) in CHC13 (50 mL) were heated at reflux for 3 hours. The reaction
mixture was
allowed to cool to room temperature and concentrated in vacuo. The resulting
oil, 3.1,
was dried under a high vacuum, redissolved in CHzCl2 (20 mL) and cooled to -
20°C.
A solution of 2-(4'-methoxybenzylthio)-2-methylpropanamine, 3.2, and Et3N (1.7
mL)
CA 02233173 1998-04-16
WO 97/14445 PCT/US96/16908
-72-
in CH~CIa (20 mL) was then added, and the resulting mixture was stirred at
room
temperature for 12 hours. Water (50 mL) was added and the product, 3.3, was
extracted with CH2Cl2 (2 x 25 mL). The CH~C121ayer was dried (Na2S0,~ and
concentrated in vacuo to obtain a viscous oil which was chromatographed on
silica
S (50 % EtOAc: hexane) to obtain the title compound, 3, as a waxy solid. Yield
63 % ; IR
(CHC13) 1664 cm 1; 1H NMR (300 Mhz) D 1.33 (s, 12H), 2.56 (s, 2H), 3.69 (s,
4H),
3.76 (s, 6H), 6.80 and 7.25 (D, J = 8.67 Hz, 4H each); HRMS (FAB) m/z 447.1902
calculated for C~H3gNOgS2, M+ +1, 448.1994.
EXAMPLE 19
Preparation of N,N-di[(2-(4'-methoxy'~enzylthio)-2-methylpropyl)] amine (3.4)
BH3~THF (100 mL, 1M solution in THF) was added to a solution of
compound 3.3 (Example 18 compound) (4.4 g, 10 mmol) in THF (100 mL), and the
resulting mixture was heated at reflux under NZ for 12 hours. The reaction
mixture
was cooled in an ice bath, and water (20 mL) was carefully added. The
resulting
solution was concentrated in vacuo to obtain a viscous oil that was suspended
in 6N
HCl (50 mL). This mixture was heated at reflux for 1 hour. After the reaction
mixture was cooled in an ice bath, it was basified with concentrated NH40H,
and the
product, 3.4, was extracted with CH2C12 and purified on silica (EtOAc). Yield
63 % ;
m.p. 58-60'C; IR (CHCl3) 1609 cm-'; 1H NMR (300 MHz) D 1.35 (s, 12H), 2.59 (s,
4H), 3.71 (s, 4H), 3.78 (s, 6H), 6.81, and 6.24 (D, J = 9.92 Hz, 4H each);
HRMS
(FAB) m/z 433.2109 calculated for C24H3502SaN, M+ +1, 434.2198. Anal.
calculated
for C24H35~2S2N: C 66.51; H 8.06; Found: C 66.12; H 8.62.
EXAMPLE 20
Preparation of N,N-di[(2-(4'-methoxybenzylthio)-2-methylpropyl)]-methylamine
(3.5)
NaBH3CN (500 mg, 8 mmol) was added to a solution of 3.4 (Example 19
compound) (2 g, 5 mmol) and methanol (2 mL, 37% aq.) in CH3CN. The resulting
mixture was stirred for 15 minutes, and glacial acetic acid was added dropwise
until
the solution tested neutral on wet pH papers. The mixture was stirred for 45
minutes
and the solvents were removed. 2N KOH (10 mL) was added to the residue, and
the
CA 02233173 1998-04-16
WO 97/14445 PCT/US96/16908
-73-
resulting mixture was extracted with ether (3 x 10 mL). The ether extracts
were
combined, washed with 0.2N KOH (10 mL) then extracted with 1N HCl (2 x 20 mL).
The acid extracts were combined, neutralized with solid KOH and re-extracted
with
ether (3 x 10 mL). Ether layers were combined and concentrated in vacuo to
obtain a
viscous oil, 3.5, which was purified on silica (EtOAc). Yield 73 % ; oil; IR
(CHCl3)
1604 cm 1; 1H NMR (300 MHz) D 1.38 (s, 12H), 2.56 (s, 3H), 2.66 (s, 4H), 3.74
(s,
4H), 3.77 (s, 6H), 6.83, and 7.25 (D, J = 8.67 Hz, 4H each); HRMS (FAB) m/z
447.2265 calculated for C~H3~OZS2N, M+ * l, 448.2361.
EXAMPLE 21
Preparation of N,N-di[(2-mer capto)-2-methylpropyl)]-methylamine (3.6)
Amine 3.5 (Example 20 compound) (2.6 g, 6 mmol) and anisole (1.5 mL) in
TFA (45 mL) were cooled to 0'C, and Hg(OAc)a (1.91 g, 6 mmol) was added. This
mixture was stirred at 0' C for 15 minutes and concentrated in vacuo at room
tertiperature. The residue was dried under a high vacuum for 30 minutes and
dry ether
(50 mL) was added. The resulting solid was collected by suction filtration and
redissolved in ethanol (100 ml). Hydrogen sulfide was then bubbled through the
ethanol solution for 15 minutes, and the black precipitate that formed was
filtered
through a thick pad of celite. The filtrate was concentrated in vacuo to
obtain a
colorless oil that was dried under a high vacuum for 30 minutes. 1N HCl (20
mL) and
ether (20 mL) were added to the above oil, and the resulting mixture was
vigorously
stirred for 15 minutes and transferred to a separating funnel. The aqueous
layer was
separated and basified with concentrated NH40H, and the resulting colorless
product
was extracted with CH2C12 (20 x 2 mL). The CH~Cl2 layer was dried (Na2S04)
concentrated in vacuo to obtain a viscous oil, 3.6, which was stored under a
blanket of
argon. Yield 66 % ; oil; IR (CHCl3) 1604 cmw; 1H NMR (300 MHz) D 1.3 (s, 12H),
2.17 (brs, 2H), 2.5 (s, 3H), 2.63 (s, 4H); HRMS (EI) m/z 207.1115 calculated
for
CgIi2INS, M+ -2, 205.0955.
CA 02233173 1998-04-16
WO 97/14445 PCT/LTS96/16908
-74-
EXAMPLE 22
Preparation of N,N-di[(2'-mercaptoethyl)]-2-methylpropylamine (3.9)
Ethylene sulfide (2 g, 22 mmol), isobutyl amine (1.4 mL, 14 mmol) and
toluene (5 mL) were heated at 110°C in a sealed tube for 8 hours. Upon
cooling, the
solution was filtered to remove colorless solid formed during the reaction and
the
filtrate obtained was concentrated in vacuo. The product, 3.9, was obtained by
fractional distillation. Yield 23 % ; by 78-80°C (4 Hgmm); IR (neat)
2964, 2799, 2551
cm-1; 1H NMR (300 MHz) d 0.82 (d, J = 9 Hz, 6H), 1.64 (brs, 3H), 2.03 (d, J =
9
Hz, 2H), 2.54 (m, 8H); HRMS (EI) m/z calculated for C8H19NS2, M+ -.
EXAMPLE 23
Preparation of N,N-di[(2-mercaptoethyl)] -2-aminoethyl-4-morphoIine) (3.11)
Ethylene sulfide (5 g, 38 mmol), 4-(2-aminoethyl)morpholine (7.6 mL, 76
mmol) and 'toluene (30 mL) were heated at 110 iC in a sealed tube overnight.
Upon
cooling, the solution was filtered to remove colorless solid formed during the
reaction
and the filtrate obtained was concentrated in vacuo. The product was purified
by flash
column chromatography on silica (5 % ethanol/ethyl acetate) to obtain the
titled
compound, 3.11. 1H NMR (300 MHz) d 1.95 (s, 2H), 2.43-2.45 (m, 6H), 2.65-2.75
(m, lOH), 3.68 (m, 4H). Anal. calculated for CloIi~OSZN2: C 37.15; H 7.48; N
8.66
Found: C 37.42; H 7.33; N 7.29.
EXAMPLE 24
General procedure for the preparation of trityl compounds (3.15-3.18)
Tritylthiol (Ph3CSH) (I g, 3.6 mmol) in THF (5 mL) was added over 5
minutes to a pentane washed NaH (60% dispersion, 145 mg, 3.6 mmol) in THF (5
mL) at 0' C, and the resulting mixture was stirred at room temperature for 10
minutes.
It was then cooled to 0'C and treated with 1,2-dibromoethane (0.31 rnL, 3.6
mmol).
After stirring at room temperature for 30 minutes, the reaction mixture was
concentrated and partitioned between EtOAc (20 mL) and water (10 mL). The
EtOAc
layer was dried over NaZS04 and concentrated in vacuo to obtain a yellow oil
that
solidified on standing.
CA 02233173 1998-04-16
WO 97/14445 PCT/US96/16908
-75-
1,4-dioxane (25 mL), Na2C03 (1.15g, 10.8 mmol), NaI (543 mg, 3.6 mmol),
and nortropane (3.13 or 3.14) (671 mg, 2.64 mmol) were sequentially added to
the
bromo compound obtained above (1 g, 3.6 mmol) and the contents were heated at
reflux for 18 hours. The reaction mixture was concentrated and partitioned
between
.> 5 CHZC12 (50 mL) and water (20 mL). The CHZCl2 layer was separated, dried
over
Na2S04 and concentrated in vacuo to obtain a brownish yellow oil that was
purified by
flash chromatography on silica (10% EtOAc:hexane).
a. N-[2'-(triphenylmethylmercapto)ethyl]-3b-(4"-fluorophenyl) tropane-
2b-carboxylic acid methyl ester 13.15)
Yield 51 % ; mp 49-51 ' C; IR (CHC13) 1743 cm 1; iH NMR (300 MHz)
d 1.43 (m, 3H), 1.92 (m, 2H), 2.17 (m, 1H), 2.28 (m, 2H), 2.45 and 2.62 (m,
each
1H), 2.93 (m, 2H), 3.3 and 3.5 (m, each 1H), 3.43 (s, 3H), 7.3, and 7.5 (m, 19
H);
HRMS (FAB) m/z 565.2450 calculated for C36Hs60zNSF, M+ + 1, 566.2263. Anal.
calculated for C36H360aNSF: C 76.46; H 6.37. Found: C 76.27; H 6.07.
- b. N-[2'-(triphenylmethyhnercapto)ethyl]-3b-(4"-chlorophenyl)
tropane-2b-carboxylic acid methyl ester (3.16)
Yield (1.2 g) 57.4 % ; mp 120-122' C; IR (CHC13) 1739 cmu; 1H NMR
(300 MHz) D 1.47 (m, 3H), 1.92 (m, 2H), 2.17 (m, 1H), 2.28 (m, 2H), 2.45 and
2.62 (m, each 1H), 2.93 (m, 2H), 3.3 and 3.5 (m, each 1H), 3.43 (s, 3H), 7.3,
and
7.5 (m, 19H); HRMS (EI) m/z 581.2155 calculated for C36FIseOzNSCI, M+ -Tr,
338.0983; FAB 581.2155 calculated for C36Hs60aNSCl, M+ +1, 582.2233. Anal.
calculated for C36Hs602NSCl: C 74.35; H 6.19. Found: C 74.01; H 6.53.
c. N-[2'-(Triphenylmethyhnercapto)propyl]-3b-(4"-fluorophenyl)
tropane-2b-carboxylic acid methyl ester (3.17)
Yield 45.9% ; mp 72-74 ~C; IR (CHCl3) 1743 cm-l; IH NMR (300 MHz) d
1.5 (m, SH), 2.12 (m, 6H), 2.55 (dt, J1 = 2.88, J2 = 12.27, 1H), 2.88 (t, J =
4.2,
1H), 2.95 (td, J1 = 4.83, J2 = 12.27, 1H) 3.32 and 3.59 (m, each 1H), 3.43 (s,
3H),
6.9-7.4 (m, 19 H); HRMS (FAB) m/z 579.2607 calculated for C3~H3g02NSF, M++ 1,
579.2693. Anal. calculated for C3~H3802NSF: C 76.55, H 6_55. Found: C 76.10; H
6.74.
CA 02233173 1998-04-16
WO 97/14445 PCT/US96/16908
-76-
d. N-[2'-(Triphenylmethylanercapto)propyl]-3b-(4"-chlorophenyl)
tropane-2b-carboxylic acid methyl ester (3.18)
Yield 41 % ; a foam; IR (CHC13) 1743 cm-1; 1H NMR (300 MHz) d 1.5 (m,
SH), 2.12 (m, 6H), 2.55 (dt, J1 = 2.88, J2 = 12.27, 1H), 2.88 (t, J = 4.2,
1H),
2.95 (td, Jl = 4.83, J2 = 12.27, 1H) 3.32 and 3.59 (m, each 1H), 3.43 (s, 3H),
6.9
-7.4 (m, 19 H); HRMS (FAB) m/z 595.2311 calculated for C3~H3gOZNSC1, M++ 1,
596.2316. Anal. calculated for C3~H3g02NSC1: C 74.62, H 6.38. Found: C 74.23;
H
6.74.
EXAMPLE 25
General procedure for the preparation of mercapto compounds (3.19-3.22)
A trityl compound (3.15-3.18) (1 mmol) was dissolved in CF3COOH (6.5
mL), and anisole (0.2 mL) at 0'C and Hg(OAc)2 1382 mg, 1.2 mmol) were added.
The resulting mixture was stirred at 0' C for 30 minutes. The reaction mixture
was
_ then concentrated in vacuo to obtain a brownish red oil that was dried under
high
vacuum for 1 hour. Anhydrous ether (50 mL) was added to the above oil, and the
mixture was kept under sonication for 15 minutes. The resulting mixture was
magnetically stirred for an additional 30 minutes. The colorless precipitate
that formed
was collected by suction filtration, dried under a high vacuum for 15 minutes
and
redissolved in ethanol (50 mL). Hydrogen sulfide gas was bubbled through the
ethanol
solution for 15 minutes, and the black precipitate that formed was filtered
through a
thick pad of celite. The filtrate was concentrated in vacuo to obtain a
colorless oil
which was dried in a high vacuum for 30 minutes. 1N HCl (20 mL) and ether (20
mL) were added to this oil, and the resulting mixture was vigorously stirred
for 15
minutes and transferred to a separating funnel. The aqueous layer was
separated and
basified with concentrated NH40Ii, and the resulting colorless product was
extracted
with CHZC12 (20 x 2 mL). The CH~Cl2 layer was dried (Na2S04) concentrated in
vacuo.
a. N-[2'-(mercapto)ethyl]-3b-(4"-fluorophenyl)tropane-2b-carboxylic
acid methyl ester (3.19)
Yield 68 % ; MP 46-48' C; IR (CHC13) 1743 cm I; 'H NMR (300 MHz) d (m,
3H), 1.92 (m, 2H), 2.17 (m, 1H), 2.28 (m, 2H), 2.45 and 2.62 (m, each 1H),
2.93
CA 02233173 1998-04-16
WO 97/14445 PCT/US96/16908
-77-
(m, 2H), 3.3 and 3.5 (m, each 1H), 3.43 (s, 3H), 7.3 and 7.5 (m, 19 H); HRMS
(FAB) m/z 323.1355 calculated for CI~H~OZNSF, M+ +1, 324.1596. Anal.
calculated
for C"H~02NSF: C 63.15; H 6.88. Found: C 62.55; H 6.38.
b. N-[2'-(mercapto)ethyl]~~3b-(=t"-chlflrophenyl)tropane-2b-carboxylic
S acid methyl ester (3.20)
Yield (263 mg) 77.5 % ; mp 69-71 ' C; IR (CHCl3) 1743 cm-1; 'H NMR (300
MHz) d 1.7 (m, 4H), 2.05 (m, 3H), 2.17 (m, 3H), 2.60 (dt, Jl=2.97, JZ=12.39,
1H),
2.94 (m, 2H), 3.39 and 3.64 (m, each 1H), 3.52 (s, 3H), 7.22 (m, 4 H); HRMS
(FAB) m/z 339.1060 calculated for Ci~H~O~NSCI, M+ +l, 340.1141. Anal.
calculated for C1~HZZOZNSCI: C 60.17; H 6.48. Found: C 56.72; H 5.83.
c. N-[2'-(mercapto)propyl]-3b-(4"-fluorophenyl)tropane-2b-carboxylic
acid methyl ester (3.21)
Yield 71 % ; waxy solid; IR (CHC13) 1741 cm-1; 1H NMR (300 MHz) d 1.49
and 2.09 (m, each 6H), 2.53 (dt, Jl = 2.88, J2 = 12.27, 1H), 2.86 (t, J = 4.2,
1H),
_ 2.95 (td, Jl = 4.83, J2 = 12.27, 1H), 3.32 and 3.59 (m, each 1H), 3.43 (s,
3H), 6.9
and 7.1 (m, each 2H); HRMS (FAB) m/z 337.1512 calculated for C18H240zNSF,
M++1, 338.1579.
d. N-[2'-(mercapto)propyl]-3b-(4"-chlorophenyl)tropane-2b-carboxylic
acid methyl ester (3.22)
Yield 66% ; waxy solid; IR (CHCl3) 1'741 cm-1; d 1.7 (m, 6H), 2.05 (m, 3H),
2.17 (m, 3H), 2.60 (dt, J1 = 2.97, J2 = 12.39, 1H), 2.94 (m, 2H), 3.39 and
3.64
(m, each 1H), 3.52 (s, 3H) and 7.22 (m, 4H); HRMS (FAB) m/z 353.1216-
calculated
for C1gH24O2NSCl, M++1, 354.1211.
EXAMPLE 26
Radiolabeling with [~"'Tc] Described For Ligand Mixture 3.20 And 3.7 (3.25)
A lyophilized sample of a mixture of 3.20 (3 ~cmol) and 3.7 (2 ~,mol) was
dissolved in 200 ~cL CH3CN, and 100 ~L 1N-HCl and 1 mL Sn-glucoheptonate were
successively added. [~''"'Tc]Pertechnetate (1 mL; ranging from 1 to 90 mCi)
saline
solution was then added. The reaction was allowed to stand at room temperature
for 30
minutes. After extracting the complex from the aqueous reaction medium with
ethyl
acetate (2 x 1.5 mL) and drying the organic solution over NaaS04, ethyl
acetate was
CA 02233173 1998-04-16
WO 97/14445 PCT/US96/16908
_78_
removed with a flow of N2. The residue was dissolved in 200 microlitre ethyl
acetate
and purified by HPLC on a PRP-1 column (250 x 4.1 mm) with CH3CN/DMGA buffer
(5 mM, pH 7; 8:2) as eluent and a flow rate of 1 mL/minute. The retention time
of
3 .25 was 16. 6 min (radiochemical yield 40 % , radiochemical purity > 95 % )
. All the
S complexes displayed stability at 4 and 24 h after preparation; little change
in
radiochemical purity was observed. Identical labeling procedures were used for
the
preparation of the labeled complexes 3.24 to 3.34, with radiochemical yields
of 46, 40,
36, 46, 20, 10, 33, 22, 10, 5 and 21 % , respectively (radiochemical purities
were all
> 95 %).
EXAMPLE 27
Evaluation
27a. Partition coefficients
The partition coefficient was measured by mixing the Tc-99m compound with
3 g. each of 1-octanol and buffer (pH 7.0 or 7.4, 0. I M phosphate) in a test
tube. The
test tube was vortexed for 3 min at room temperature, then centrifuged for 5
minutes.
Two weighed samples (0.5 g each) from the 1-octanol and buffer layers were
counted
in a well counter. The partition coefficient was determined by calculating the
ratio of
cpm/g of octanol to that of buffer. Samples from the octanol layer were
repartitioned
until consistent partition coefficient values were obtained. The measurement
was
repeated 3 times.
The complexes with gem-dimethyl groups, [~''"'Tc] 3.26 and 3.27, displayed a
much higher partition coefficient than those without, [~''"'Tc] 3.24 and 3.25.
In
addition, the partition coefficient of the complex containing the fluorine
atom, [~''mTc]
3.24 and 3.26, is always lower than the corresponding complex with the
chlorine atom.
It is apparent that the partition coefficient of [~'"Tc] 3.25 appears to be
the best among
this series of three plus one Tc complexes.
The lipophilicities of [99mTc] 10-13 (described in Example 26) were measured
by partition between n-octanol and buffer (Table 3.1).
27b. In vitro autoradiography
Male Sprague-Dawley rats (200-250 mg) were sacrificed by decapitation, and
the brains were removed, placed in OTC embedding medium (Miles Laboratory,
CA 02233173 2004-05-25
-79-
Elkhart, IN) frozen in powdered dry ice. After equilibration to -15' C, 20 ~cm
coronal
sections were sliced on a cryostat (Hacker Instruments, Fairfield, Nn, thaw
mounted
onto gelatin-coated slides, desiccated at 4' C for 3 hours and kept at -70' C
until use.
Prior to the experiment, the slides were dried at room temperature and
preincubated
for 30 minutes in buffer containing 50 mM Tris-HCl (pH 7.4, 120 mM NaCI). The
slides were then incubated for 2 hours in preincubation buffer and the
[~'"'Tc]
TM
compound 3.25 (385,000 cpm/200 p.L) in a coplan jar, washed for 1 hour in cold
Tris
buffer, with one change of buffer, dipped in ice-Cold water to remove buffer
salts and
allowed to dry at room temperature. Nonspecific binding was determined in the
TM
presence of 1 ~.M IPT. These slides were simultaneously exposed to DuPont x-
ray
film in an autoradiographic cassette for 18 hours. The exposed film was
developed
TM
with a Kodak automatic film processor.
27c. In vivo biodistribution
Male Sprague-Dawley rats (225-300 g) that were allowed free access to food
and water were used for in vivo biodistribution studies. (Kung, 1984, supra;
Kung,
1985, supra). While under halothane anesthesia, 0.2 mL of a saline solution
containing
3.24 to 3.34 (SO-100 MCi) was injected directly into the femoral vein of rats,
and they
were sacrificed by cardiac excision at various time points post-injection. The
organs of
interest were removed and weighed, and the radioactivity was counted with an
TM
automatic gamma counter (Packard 5000). The percentage dose per organ was
calculated by a comparison of the tissue counts to suitably diluted aliquots
of the
injected material. Total activities of blood and muscle were calculated under
the
assumption that they were 7% and 40% of the total body weight, respectively.
Regional brain distribution in rats was obtained after an injection of 3.24 to
3.34. Samples from different brain regions (cortex, striatum, hippocampus, and
cerebellum) were dissected, weighed and counted, and the percentage dose per
gram of
sample was calculated by comparing the sample counts with the count of the
diluted
initial dose. The uptake ratio of each region was obtained by dividing the
percentage
dose per gram of that region by that of the cerebellum. For blocking studies,
rats vtcere
injected with either (3-CIT or haloperidol (iv, 1 mg/Kg) 5 min prior to
injection of
3.25. The rats were dissected and brain tissue samples were counted as
described
above.
CA 02233173 1998-04-16
WO 97/14445 PCT/US96/16908
_80_
The brain uptakes of these complexes appeared to be similar and were all
relatively low (0.1 % dose/organ or less at 2 minutes post-injection) despite
the
different lipophilicities measured by partition coefficient between n-octanol
and
phosphate buffer (Tables 3.I). Slow washout from the brain was observed for
all these
complexes. Interestingly, only complexes 3.24 and 3.25, without any gem-
dimethyl
groups, displayed specific uptake in striatum, where dopamine transporters are
highly
concentrated; striatum to cerebellum ratio (ST/CB) was 1.93 and 2.2 at 30
minutes
post-injection for [~'mTc] 3.24 and 3.25, respectively. The cerebellum area,
which
does not contain dopamine transporters, was used as the background region for
comparison.
Biodistribution of a [9smTc] compound 3.25 at 2, 30, 60, and 120 minutes post
intravenous injection indicated that the complex followed the initial blood
flow and
localized in organs with high blood flow, such as muscle, Liver, and kidney.
The
[99mTC] complex 3.25 cleared from muscle, while liver accumulation increased
with
time (Table 2). Accumulation of radioactivity was highest in all brain regions
at 2
minutes post intravenous injection and declined gradually over the 120 minute
period.
The washout was slowest from the striatal region, followed by cortex, and
hippocarnpus. Maximum regional contrast ratio observed for
striatum(ST)/cerebellum(CB) was 2.2 and 3.5 at 30 and 60 minutes post-
injection,
respectively (Table 3). Therefore, additional blocking studies in rats were
performed
at this time point to further characterize the uptake of this [~'''"Tc]
compound. In this
series of regional brain uptake studies in rats, the ST/CB ratio at 60 minutes
after
injection was 2.6 (Fig. 1). The specific uptake of the [~9'"Tc] compound 3.25
in
striatum could be blocked by pretreating rats with a dose of RTI-55 (N-methyl-
2b-
carbomethoxy-3b-(4-iodophenyl)tropane) (1 mg/kg, iv), a dopamine transporter
ligand
(ST/CB =1.0); but not with haloperidol (1 mg/kg, iv), an agent with a mixed
pharmacological profile (binding to various CNS receptors but not to the
dopamine
transporter); no blocking effect was observed (ST/CB =2.6; Fig. 1). The
results
suggested that the uptake in the striatum of rat brain was specifically
related to the
dopamine transporter.
In vitro autoradiography of rat brain sections incubated with a [~'''"Tc]
compound 3.25 exhibited elevated labeling in striatum, major islands of
Calleja and
CA 02233173 2004-05-25
-81-
olfactory tubercle regions, where dopamine neurons are known to be
concentrated. See
Malison, R.T. et al. J. Nucl. Med. 1995, 36(12), 2290-2297; Motley, P.D. et
al. J. Nucl.
Med. 1995, 36(7), 1322-1331; Neumeyer, J.L. et al. J. Med. Chem. 1994, 37,
1558-1561.
EXAMPLE 28
Experimental For Reaction Schemes Depicted in Figures 12-14
The aminobisethanethiol ligand with gem-dimethyl groups, 3.6, was
synthesized according to a previously reported procedure (Ohmomo, 1992,
supra), as
shown in Figure 12. The aminobisethanethiol ligand without gem-dimethyl
groups, 3.7,
was synthesized according to the literature (Kolb, 1994, supra). The N-ethyl
substituted aminobisethylthiols, 3.8, were prepared according to the procedure
employed for the synthesis of 7, using N-ethyl bischloroethyl amine as the
starting
material. The other N-substituted bisethanethiols, 3.9 to 3.12, were prepared
by
bis-alkyladon of benzyl-, iso-butyl-, morpholinoethyl- and
(AI,N-bisethylamino)ethyl-amine with ethylene sulfide, respectively. (Corbin,
1984,
supra).
The syntheses of thiol tropane . ligands 3.19 to 3.22 are shown in Scheme 13.
Demethylated tropane derivatives 3.13 and 3.14 were prepared from cocaine in 4
steps,
as previously reported (Meltzer, 1993, supra). N-alkylation was achieved by
reacting
them with S-trityl protected 2-bromoethanethiol and 3-bromopropanethiol (Dhar,
1994,
supra), and the resulting alkylated products, 3.15-3.18, were successfully
deprotected
with Hg(OAc)2 to produce free thiols '3.19-3.22. Although the bis-alkylation
of amines
with ethylene sulfide generally produced the expected bis-ethanethiols in poor
yields, it
provided a very short route for obtaining the required dithiols quickly for
the initial
studies. No attempts were made to improve the yields of these reactions. The
vacuum
distillation yielded fairly pure dithiols but further reduced the yields. The
dithiols seem
to have only moderate stability and tend to produce a white solid, presumably
disulfides, over time. However, the monothiols, 3.15-3.18, seem to have fairly
good
stability when stored at a low temperature under N2. As is evident from the X-
ray
crystallographic structure of Re-complex 3.23, none of the reaction conditions
employed in preparation of monothiols 3.15-3.18 and the Re complex altered the
stereochemistry at the C-2 position of tropane ring.
CA 02233173 2004-05-25
Labeling with [~"'Tc] was carried out by reacting the appropriate ligands in a
molar ratio of 1:1.5 (aminobisethanethiolaropanethiol ligand) with sodium
[99mTc)pertechnetate, in the presence of tin(In glucoheptonate as a reducing
agent at
room temperature, with yields of about 40% (Figure 14).
Byproducts were labeled complexes in which either the tridentate ligand
aminobisethylthiol or the monothiol is complexed to a Tc0-core to form neutral
binuclear complex Tc,Z02[RN(CH2CHZS),~3 (Revert, 1983, supra) and ionic
complexes
(Spies, 1990, supra).
The labeled compounds were purified by HPLC and final radiochemical purity
of > 95 % was obtained. All of the [99mTc] complexes 3.24-3.34 displayed good
in
vitro stability. The lipophilicities of 3.24-3.34 were measured by partition
between
n-octanol and buffer (Table 3.1). The complexes with gem-dimethyl groups, 3.26
and
3.27, displayed a much higher partition coefficient than the analogous
complexes, 3.24
and 3.25, without gem-dimethyl groups. In addition, the partition coefficients
of the
complexes containing the fluorine atom, 3.24, 3.26 and 3.28, are always lower
than
those of the corresponding complexes with the chlorine atom. In the series of
alkyl
substituted NS2-ligand complexes, 3.25, 3.30 and 3.31, the partition
coefficient follows
an expected trend; the PC increases with the size of alkyl substituent (the
PC's for
N-methyl, N-ethyl and N-i-butyl are 307, 369 and 881, respectively). Having a
hetero
atom in the substitution group at the NS2-part (3.33, 3.34) did not follow any
general
trend in partition coefficient.
Coinjections of Tc-99m and Re-3.23 into HPLC under various elution
conditions appeared to confirm that they have a similar retention time. It is
likely that
the Tc-99m complexes have the same chemical structures as those of the
corresponding
Re-complexes. Compounds 3.25 and 3.23 behaved similarly under identical HPLC
conditions (coinjection), suggesting that 3.23, is indeed a good surrogate for
3.25. The
retention time of 3.25 is similar to that of 3.23; on a C-18 column (Partisil
10-ODS-3,
250 x 4.6 mm) with MeOH/NHaHC03 (O.1M; pH 7, ratio 8:2, flow rate 1 mL/min) as
the eluent, the retention times were 14.9 and 15.3 min for 3.25 and 3.23,
respectively
TM
(Fig. 17). On a Chiralpak AD column (250 x 4.6 mm) with hexane/EtOH (1:1, flow
rate 1 ml/min) as the eluent, the retention times were 10.4 and 10.9 min for
x.25 and
3.23, respectively. On a PRP-1 column (Hamilton 250 x 4.1 mm) with
CA 02233173 1998-04-16
WO 97/14445 PCT/US96/16908
- 83 -
CH3CN/dimethylglutarate buffer (5 mM, pH 7, ratio 9:1, flow rate 1 ml/min) as
the
eluent, the retention times were 10.2 and 9.8 minutes for 3.25 and 3.23,
respectively.
Whether the 99mTc complex has indeed the same chemical structure as Re complex
3.23 (methyl anti to the Re=O) cannot be determined definitively at this
point. Under
the applied conditions, only one of the two possible isomers (anti or syn) can
be
detected. If the other isomer is indeed present, the ratio must be > 98:2 and
the
compounds must behave very similarly.
In vivo biodistribution studies of various Tc-99m labeled complexes,
3.24-3.34, were evaluated in male Sprague-Dawley rats. The brain uptakes of
these
IO complexes appeared to be similar and were all relatively low (0.1 %
dose/organ or less
at 2 min post-injection) despite the different lipophilicities measured by
partition
coefficient between n-octanol and phosphate buffer (Table 3.1). Slow washout
from the
brain was observed for all these complexes. Interestingly, only complexes
without any
gem-~dimethyl groups displayed specific uptake in striatum, where dopamine
tr~nsporters are highly concentrated. Furthermore, only complexes with smaller
R
groups on the NS2-ligand showed any specific brain uptake; striatum to
cerebellum
ratio (ST/CB) was 1.93, 2.2 and 1.97 at 30 minutes post-injection for 3.24,
3.25 and
3.30, respectively. Increasing the chain length in the tropane thiol ligand
from two to
three carbons (compounds 3.28 and 3.29) did not change the brain uptake
dramatically,
but the ST/CB ratio was decreased (1.71 and 1.72, respectively). The
cerebellum area,
which does not contain dopamine transporters, was used as the background
region for
comparison. -
Biodistribution showed that 3.25 had the highest ST/CB ratio (2.2) after 30
minutes post intravenous injection. Therefore 3.25 was studied further in
rats. The
study at 2, 30, 60 and 120 min post intravenous injection indicated that the
complex
followed the initial blood flow and localized in organs with high blood flow,
such as
muscle, liver and kidney. The Tc-99m i:omplex cleared from muscle, while liver
accumulation increased with time (Table 3.2). Accumulation of radioactivity
was
highest in all brain regions at 2 minutes post intravenous injection and
declined
gradually over the 120 minute period. The washout was slowest from the
striatal
region, followed by cortex and hippocampus. Maximum regional contrast ratio
observed for striatum/cerebellum (ST/CB) was 3.5 at 60 minutes post-injection
(Table
CA 02233173 1998-04-16
WO 97/14445 PCT/LTS96/16908
-84-
3.3). Therefore, additional blocking studies in rats were performed at 60
minutes post-
injection to further characterize the uptake of this Tc-99m compound. In this
series of
regional brain uptake studies in rats, the ST/CB ratio at 60 min post-
injection was 2.6
(Fig. 15). The specific uptake of 3.25 in striatum could be blocked by
pretreating rats
with a dose of ~i-CIT (1 mg/kg, iv), a dopamine transporter ligand (ST/CB =
1.0); but
not with haldol (1 mg/kg, iv), an agent with a mixed pharmacological profile
(binding
to various CNS receptors but not to the dopamine transporter); no blocking
effect was
observed. The ST/CB ratio (2.6) was identical to that in control rats (Fig.
15). The
results suggested that uptake in the rat striatum was specifically related to
the dopamine
transporter. In addition, in vitro autoradiography of a rat brain coronal
section
incubated with 3.25 clearly demonstrated a regional distribution pattern of
intense
labeling in caudate putamen and olfactory tubercle, areas known to have a high
density
of dopamine transporters (Fig. 16).
F,XAMPLE 29
Comparison Study Of Technetrin And Rhenium Complexes
To further characterize the chemical structure of the technetium complexes,
the corresponding Re0(III) complex, Ra-3.25, was prepared and characterized
(Kung,
J. Amer. Chem. Soc., in submission; Meegalla, 1995, supra). The non-
radioactive
rhenium is chemically close to technetium-99, but without the usual hazards
associated
with the handling of radioactive material. X-ray crystallography of the
heterodimeric
complex, Re-3.25, displayed an expected structure, with a pyramidal Re=O core
and a
N-methyl group at the anti position to the Re=O functionality. In vitro
binding studies
with rat striatal homogenates displayed excellent binding affinity (Ki = 0.3
nM, using
[luI]-IPT as the ligand; Kd = 0.2 nM).
In vitro binding studies of Re complex 3.23 with rat striatal homogenates
displayed excellent binding affinity (Ki = 0.3 nM, using [125I]-IPT as the
ligand; Kd
= 0.2 nM). Kung, 1995, supra. The major drawback of this series of agents is
that
the brain uptake (0.1 % dose/organ in rat brain at 2 min post intravenous
injection) is
too low to be useful for human imaging study. New complexes that produce at
least
0.5 % dose/organ in rat brain must be obtained before any are further
developed as
dopamine transporter imaging agents. Nevertheless, this series of [~'''"Tc]
mixed ligand
CA 02233173 1998-04-16
WO 97/14445 PCT/LJS96/16908
-85-
complexes (aminobisethanethiol and monothiol complexed to a Tc03+ center core)
is
the first example of technetium labeled agents displaying specific regional
uptake in rat
brain that reflects receptor distribution.
CA 02233173 1998-04-16
WO 97/14445 PCT/LJS96/16908
-86-
Table 3.1. Partition coefficient, brain uptake and striatum/cerebeUum ratios
of
various Tc-99m complexes
Retention
Time(min)/ Brain Uptake Striatum/Cerebellum
Complex Partition (1o dose) ,
Coefficient Ratios (30
2 min 30 min)
min
3.26 17.6'/1800 0.08 t 0.030.05 t 0.0021.0 (n=3)
3.24 11.6'/97 0.09 t 0.010.063 t 0.0031.93 (n=3)
3.27 27.4'/2000 0.051 t 0.046 t 0.0021.0 (n=3)
0.009
3.25 16.6'/307 0.100 t 0.07 t 0.0032.2 (n=12)
0.068
3.28 14.5'/99 0.082 t 0.045 t 0.0081.71 (n=3)
0.014
3.29 12.5"/125 0.12 t 0.030.063 t 0.0131.72 (n=3)
3.30 10.8"/369 0.097 t 0.058 t 0.0061.97 (n=3)
0.003
3.31 14.8"/881 0.080 t 0.051 t 0.0060.97 (n=3)
0.009
3.32 23.6"/146 0.048 t 0.046 t 0.200.72 (n=3)
0.021
3.33 9.8"/86 0.062 t 0.046 t 0.0090.47 (n=3)
0.008
5.34 14.5"/92 0.11 t 0.020.061 t 0.0110.53 (n=6)
* Retention times of the complexes were measured by HPLC on a PRP-1 column
(250x4.1 mm) eluted with acetonitrile-5 mM dimethylglutaric acid buffer, pH 7
(80:20), flow rate: 1 mL/min.
# Retention times of the complexes were measured by HPLC on a PRP-1 column
(250x4.1 mm) eluted with acetonitrile-5 mM dimethylglutaric acid buffer, pH 7
(90:10), flow rate: 1 mL/min.
i
CA 02233173 1998-04-16
WO 97/14445 PCT/US96/16908
_87_
Table 3.2. Biodistribution of 3.25 in rats (intravenous injection; °lo
dose/organ
standard deviation, n=3-~
Organ 2 min 30 min 60 min 120 min
Blood 4.72 t 1.37 2.75 f 0.62 2.83 t 0.42 2.31 t 0.27
$ Heart 1.46 t 0.37 0.30 t 0.04 0.22 t 0.23 0.12 t 0.013
Muscle 23.30 t 3.82 16.70 t 3.62 13.63 t 0.99 4.33 t 1.20
Lung 6.41 t 2.04 2.09 t 0.47 1.26 t 0.06 0.69 f 0.061
Kidney 5.67 t 0.94 3.10 t 0.68 2.87 t 0.43 2.74 t 0.096
Spleen 0.36 t 0.07 0.61 t 0.10 0.50 t 0.09 0.58 t 0.009
1~ Liver 13.51 f 0.36 29.61 t 2.22 32.15 t 3.00 35.37 t 1.52
Skin 2.94 t 0.36 4.71 t 1.27 4.57 t 0.65 2.30 t 0.082
Brain 0.10 t 0.02 0.06 t 0.02 0.046 t 0.0020.025 t 0.002
CA 02233173 1998-04-16
WO 97/14445 PCT/LTS96/16908
_88_
Table 3.3. Regional brain distribution of 3.25 in rat brain (% dose/g)
Region 2 min 30 min 60 min 120 min
Cerebellum 0.066 t 0.0170.027 t 0.0060.018 t 0.0010.011 t 0.002
Striatum 0.054 t 0.0100.060 t 0.0110.063 t 0.0010.031 t 0.003
Hippocampus 0.046 t 0.0050.028 t 0.0080.025 t 0.0030.011 t 0.000
Cortex 0.060 t 0.0090.038 t 0.0070.026 t 0.0020.015 ~ 0.003
ST/CB ratio 0.82 2.2 3.5 2.8
CA 02233173 1998-04-16
WO 97114445 PCT/CTS96/16908
-89-
EXAMPLE 30
Kit Formulation For Compound 1.19a
' A kit would contain: compound 1.19a (180-250 mg); stannous chloride (100-
200 mg); sodium glucoheptonate (200-4.00 mg); disodium EDTA (300-350 mg); 2.0
N
hydrochloric acid (200-250 mL); and ethanol (100-200 mL).
Preparation of compound 1.19a using kit formulation:
Vial A (reaction vial): 200 micrograms 1.19a
Vial B: 2N HCl solution
Vial C: SnCl2/glucoheptonate, pH 6.7
Stannous chloride anhydrous (100 mg/mL)
Sodium glucoheptonate (200 mg/mL)
Vial D: 0.05 mL EDTA
0.1 M Sodium EDTA
Vial E: Sterile sodium phosphates, injection, USP (Abbott, lot 10-288-
DK)
One sterilized empty vial and one ethanol (absolute) vial.
Mix 1.95 mL of ethanol with 0.05 mL of 2N HCl in the sterilized empty vial.
Add 0.1 mL of the mixture to Vial A and shake well. Add 0.2 mL of the solution
from Vial B to Vial A and 1 mL of the solution from Vial C to Vial A. Add 0.05
mL
of the solution for Vial D to Vial A. Add Tc-99m solution (0.1-0.5 mL) to Vial
A.
Place Vial A in the autoclave at 112°C for about 30 minutes. Following
autoclaving,
cool the reaction vial (A) to room temperature. When the compound is to be
used for
analysis, draw as much as the solution from vial A with a 5 mL syringe which
is
prefilled with 0.4 mL of sodium phosphate (Vial E) and mix well. Check the
radiochemical purity of the residual aliquot in Vial A by TLC.