Note: Descriptions are shown in the official language in which they were submitted.
CA 02233560 1998-03-31
WO 97/19068 PCT/US96/16761
-1-
SULFONAMIDE 2NHIBITORS OF MATRIX METALLOPROTEINASES
~ 5
FIELD OF THE INVENTION
The present invention relates to sulfonamide
compounds that inhibit matrix metalloproteinases,
pharmaceutical compositions that include these
compounds, and pharmaceutical methods of treatment
using these compounds.
BACKGROUND OF THE INVENTION
The novel compounds of the present invention are
inhibitors of matrix metalloproteinases, e.g.,
stromelysin-1 and gelatinase A (72 kDa gelatinase).
More particularly, the compounds of the present
invention are useful in the treatment of
atherosclerotic plaque rupture, aortic aneurism, heart
failure, restenosis, periodontal disease, corneal
ulceration, burns, decubital ulcers, chronic ulcers or
wounds, cancer metastasis, tumor angiogenesis,
arthritis, multiple sclerosis, and other autoimmune or
inflammatory disorders dependent on the tissue invasion
of leukocytes or other activated migrating cells.
Stromelysin-1 and gelatinase A are members of the
matrix metalloproteinase (MMP) family (Woessner J.F.,
FASEB J. 1991;5:2145-2154). Other members include
fibroblast collagenase, neutrophil collagenase,
geTatinase B (92 kDa gelatinase), stromelysin-2,
stromelysin-3, matrilysin, collagenase 3 (Freije J.M.,
Diez-Itza I., Balbin M., Sanchez L.M., Blasco R.,
Tolivia J., and Lopez-Otin C., J. Biol. Chem.,
1994;269:16766-16773), and the newly discovered
membrane-associated matrix metalloproteinases (Sato H.,
CA 02233560 1998-03-31
WO 97/19068 PCT/US96/16761
-2-
Takino T., Okada Y., Cao J., Shinagawa A., Yamamoto E.,
and Seiki M., Nature, 1994;370:61-65).
The catalytic zinc in matrix metalloproteinases is
the focal point for inhibitor design. The modification
of substrates by introducing chelating groups has
generated potent inhibitors such as peptide
hydroxamates and thiol-containing peptides. Peptide
hydroxamates and the natural endogenous inhibitors of
MMPs (TIMPs) have been used successfully to treat
animal models of cancer and inflammation.
The ability of the matrix metalloproteinases to
degrade various components of connective tissue makes
them potential targets for controlling pathological
processes. For example, the rupture of atherosclerotic
plaques is the most common event initiating coronary
thrombosis. Destabilization and degradation of the
extracellular matrix surrounding these plaques by MMPs
has been proposed as a cause of plaque fissuring. The
shoulders and regions of foam cell accumulation in
human atherosclerotic plaques show locally increased
expression of gelatinase B, stromelysin-1, and
interstitial collagenase. In situ zymography of this
tissue revealed increased gelatinolytic and
caseinolytic activity (Galla Z.S., Sukhova G.K.,
Lark M.W., and Libby P., "Increased expression of
matrix metalloproteinases and matrix degrading activity
in vulnerable regions of human atherosclerotic
plaques", J. Clin. Invest., 1994;94:2494-2503). In
addition, high levels of stromelysin RNA message have
been found to be localized to individual cells in
atherosclerotic plaques removed from heart transplant
patients at the time of surgery (Henney A.M.,
Wakeley P.R., Davies M.J., Foster K., Hembry R., ,
Murphy G., and Humphries S., "Localization of
stromelysin gene expression in atherosclerotic plaques
CA 02233560 1998-03-31
WO 97/19068 PCT/US96/16761
-3 -
by in situ hybridization", Proc. Nat'1. Acad. Sci.,
1991;88:8154-8158).
Inhibitors of matrix metalloproteinases will have
utility in treating degenerative aortic disease
associated with thinning of the medial aortic wall.
Increased levels of the proteolytic activities of MMPs
have been identified in patients with aortic aneurisms
and aortic stenosis (Vine N. and Powell J.T.,
"Metalloproteinases in degenerative aortic diseases",
Cliri. Sci., 1991;81:233-239).
Heart failure arises from a variety of diverse
etiologies, but a common characteristic is cardiac
dilation which has been identified as an independent
risk factor for mortality (Lee T.H., Hamilton M.A.,
Stevenson L.TnT., Moriguchi J.D., Fonarow G.C.,
Child J.S., Laks H., and Walden J.A., "2mpact of left
ventricular size on the survival in advanced heart
failure", Am. J. Cardiol., 1993;72:672-676). This
remodeling of the failing heart appears to involve the
breakdown of extracellular matrix. Matrix
metalloproteinases are increased in patients With both
idiopathic and ischemic heart failure (Reddy H.K.,
Tyagi S.C., Tjaha I.E., Voelker D.J., Campbell S.E.,
and Weber K.T., "Activated myocardial collagenase in
idiopathic dilated cardiomyopathy", Clin. Res.,
1993;41:660A; Tyagi S.C., Reddy H.K., Voelker D.,
Tjara I.E., and Weber K.T., "Myocardial collagenase in
failing human heart", Clin. Res., 1993;41:681A).
Animal models of heart failure have shown that the
induction of gelatinase is important in cardiac
dilation (Armstrong P.W., Moe G.W., Howard R.J.,
Grima E.A., and Cruz T.F., "Structural remodeling in
heart failure: gelatinase induction", Can. J.
Cardiol., 1994;10:214-220), and cardiac dilation
precedes profound deficits in cardiac function
(Sabbah H.N., Kono T., Steir_ P.D., Mancini G.B.; and
CA 02233560 1998-03-31
WO 97/19068 PCT/LTS96/16761
-4-
Goldstein S., "Left ventricular shape changes during
the course of evolving heart failure", Am. J. Phvsiol.,
1992;263:H266-H270).
Neointimal proliferation, leading to restenosis,
frequently develops after coronary angioplasty. The
migration of vascular smooth muscle cells (VSMCs) from
the tunics media to the neointima is a key event in the
development and progression of many vascular diseases
and a highly predictable consequence of mechanical
injury to the blood vessel (Bendeck M.P., Zempo N.,
Clowes A.W., Galardy R.E., and Reidy M., "Smooth muscle
cell migration and matrix metalloproteinase expression
after arterial injury in the rat", Circulation
Research., 1994;75:539-545). Northern blotting and
zymographic analyses indicated that gelatinase A was
the principal MMP expressed and excreted by these
cells. Further, antisera capable of selectively
neutralizing gelatinase A activity also inhibited VSMC
migration across basement membrane barrier. After
injury to the vessel, gelatinase A activity increased
more than 20-fold as VSCMs underwent the transition
from a quiescent state to a proliferating, motile
phenotype (Pauly R.R., Passaniti A., Bilato C.,
Monticone R., Cheng L., Papadopoulos N., Gluzband Y.A.,
Smith L., Weinstein C., Lakatta E., and Crow M.T.,
"Migration of cultured vascular smooth muscle cells
through a basement membrane barrier requires type IV
collagenase activity and is inhibited by cellular
differentiation", Circulation Research, 1994;75:41-54).
Collagenase and stromelysin activities have been
demonstrated in fibroblasts isolated from inflamed
gingiva (Uitto V.J., Applegren R., and Robinson P.J.,
"Collagenase and neutral metalloproteinase activity in
extracts from inflamed human gingiva", sT. Periodontal
Rtes., 1981;16:417-424), and enzyme levels have been
correlated to the severity of gum disease
CA 02233560 1998-03-31
WO 97/19068 PCT/US96/16761
-5-
(Overall C.M., Wiebkin O.W., and Thonard J.C.,
"Demonstrations of tissue collagenase activity in vivo
L
and its relationship to inflammation severity in human
gingiva", J. Periodontal Res., 1987;22:81-88).
Proteolytic degradation of extracellular matrix has
been observed in corneal ulceration following alkali
burns (Brown 5.2., Weller C.A., and Wasserman H.E.,
"Collagenolytic activity of alkali burned corneas",
Arch. Obthalmol., 19&9;81:370-373). Thiol-containing
peptides inhibit the collagenase isolated from alkali-
burned rabbit corneas (Burns F.R., Stack M.S.,
Gray R.D., and Paterson C.A., Invest. O~ththamol.,
1989;30:1569-1575).
Stromelysin is produced by basal keratinocytes in
a variety of chronic ulcers (Saarialho-Kere U.K.,
Ulpu K., Pentland A.P., Birkedal-Hansen H., Parks W.C.,
Welgus H.G., "Distinct populations of basal
keratinocytes express stromelysin-1 and stromelysin-2
in chronic wounds", J. Clin. Invest., 1994;94:79-88).
Stromelysin-1 mRNA and protein were detected in
basal keratinocytes adjacent to but distal from the
wound edge in what probably represents the sites of
proliferating epidermis. Stromelysin-1 may thus
prevent the epidermis from healing.
Davies, et al., (Cancer Res., 1993;53:2087-2091)
reported that a peptide hydroxamate, BB-94, decreased
the tumor burden and prolonged the survival of mice
bearing human ovarian carcinoma xenografts. A peptide
of the conserved MMP propeptide sequence was a weak
inhibitor of gelatinase A and inhibited human tumor
cell invasion through a layer of reconstituted basement
membrane (Melchiori A., Albili A., Ray J.M., and
- Stetler-Stevenson W.G., Cancer Res., 1992;52:2353-
2356), and the natural tissue inhibitor of
metalloproteinase-2 (TIMP-2) also showed blockage of
tumor cell invasion in in vitro models (DeClerck Y.A.,
CA 02233560 1998-03-31
WO 97/19068 PCT/US96/16761
-6-
Perez N., Shimada H., Boone T.C., Langley K.E., and
Taylor S.M., Cancer Res., 1992;52:701-708). Studies of
J
human cancers have shown that gelatinase A is activated
on the invasive tumor cell surface (Strongin A.Y.,
Mariner B.L., Grant G.A., and Goldberg G.I., J. Biol
hue, 1993;268:14033-14039) and is retained there
through interaction with a receptor-like molecule
(Monsky W.L., Kelly T., Lin C.-Y., Yeh Y.,
Stetler-Stevenson W.G., Mueller S.C., and Chen W.-T.,
Cancer Res., 1993;53:3159-3164).
Inhibitors of MMPs have shown activity in models
of tumor angiogenesis (Taraboletti G., Garofalo A.,
Belotti D., Drudis T., Borsotti P., Scanziani E.,
Brown P.D., and Giavazzi R., Journal of the National
Cancer Institute, 1995;87:293; and Benelli R.,
Adatia R., Ensoli B., Stetler-Stevenson W.G., Santi L.,
and Albini A., Oncoloav Research, 1994;6:251-257).
Several investigators have demonstrated consistent
elevation of stromelysin and collagenase in synovial
fluids from rheumatoid and osteoarthritis patients as
compared to controls (Walakovits L.A., Moore V.L.,
Bhardwaj N., Gallick G.S., and Lark M.W., "Detection of
stromelysin and collagenase in synovial fluid from
patients with rheumatoid arthritis and post-traumatic
knee injury", Arthritis Rheum., 1992;35:35-42;
Zafarullah M., Pelletier J.P., Cloutier J.M., and
Marcel-Pelletier J., "Elevated metalloproteinases and
tissue inhibitor of metalloproteinase mRNA in human
osteoarthritic synovia", J. Rheumatol., 1993;20:693-
697). TIMP-1 and TIMP-2 prevented the formation of
collagen fragments, but not proteoglycan fragments, .
from the degradation of both the bovine nasal and pig
articular cartilage models for arthritis, while a .
synthetic peptide hydroxamate could prevent the
formation of both fragments (Andrews H.J.,
Plumpton T.A., Harper G.P., and Cawston T.E., Aaents
CA 02233560 1998-03-31
WO 97/19068 PCT/CTS96/16761
-7-
Actions, 1992;37:147-154; Ellis A.J., Curry V.A.,
Powell E.K., and Cawston T.E., Biochem. Biophvs. Res.
Commun., 1994;201:94-101).
Gijbels, et al., (J. Clin. Invest., 1994;94:2177-
2182) recently described a peptide hydroxamate, GM6001,
that suppressed the development or reversed the
clinical expression of experimental allergic
encephalomyelitis (EAE) in a dose dependent manner,
suggesting the use of MMP inhibitors in the treatment
of autoimmune inflammatory disorders such as multiple
sclerosis.
A recent study by Madri has elucidated the role of
gelatinase A in the extravasation of T-cells from the
blood stream during inflammation (Ramanic A.M. and
Madri J.A., "The Induction of 72-kD Gelatinase in
T Cells upon Adhesion to Endothelial Cells is VCAM-1
Dependent", J. Cell Biolocrv, 1994;125:1165-1178). This
transmigration past the endothelial cell layer is
coordinated with the induction of gelatinase A and is
mediated by binding to the vascular cell adhesion
molecule-1 (VCAM-1). Once the barrier is compromised,
edema and inflammation are produced in the CNS.
Leukocytic migration across the blood-brain barrier is
known to be associated with the inflammatory response
in EAE. Inhibition of the metalloproteinase
gelatinase A would block the degradation of
extracellular matrix by activated T-cells that is
necessary for CNS penetration.
These studies provided the basis for the belief
that an inhibitor of stromelysin-1 and/or gelatinase A
~ will treat diseases involving disruption of
extracellular matrix resulting in inflammation due to
. lymphocytic infiltration, inappropriate migration of
metastatic or activated cells, or loss of structural
integrity necessary for organ function.
CA 02233560 1998-03-31
WO 97/19068 PCT/US96/I6761
_g_
We have identified a series of sulfonamide
compounds that are inhibitors of matrix
metalloproteinases, particularly stromelysin-1 and
gelatinase A, and thus useful as agents for the
treatment of multiple sclerosis, atherosclerotic plaque
rupture, restenosis, aortic aneurism, heart failure,
periodontal disease, corneal ulceration, burns,
decubital ulcers, chronic ulcers or wounds, cancer
metastasis, tumor angiogenesis, arthritis, or other
autoimmune or inflammatory diseases dependent upon
tissue invasion by leukocytes.
SUMMARY OF THE INVENTION
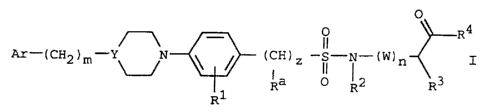
The present invention provides compounds of the
Formula I
O
4
- O R
Ar-(CH2 )m- ~ ~ ~ (CH) Z -Vii- ~ - (W) n I
Ra O R2 R3
R1
wherein:
Ar is selected from phenyl;
phenyl substituted with alkyl, -N02, halogen, -ORS,
-CN, -C02R5, -S03R5, -CHO, -CORS, -CONHRS, -NHRS,
or -NHCORS;
heteroaryl; or
2-naphthyl;
R1 is hydrogen, methyl, -N02, -C1, -NH2, -NHC02CH3,
-OH, or -C02H;
R2, R3, and Ra are the same or different and are
independently selected from hydrogen, alkyl,
-(CH2)v-aryl, -(CH2)V-heteroaryl,
-(CH2)v-cycloalkyl, -(CH2)p-X-(CH2)q-aryl,
CA 02233560 2004-03-05
-g_
-(CH2)p-X-(CH2)q-heteroazyl, -(CH2)tNR6R6a
-(CH2)vR~, -(CH2)VC02R5, -(CH2)~CONR6R6a, or
-(CH2)vSRS;
m is zero or 1;
Y is CH or N; provided that when m = 1, Y does not = N;
z is zero or 1;
w is -CHR8;
n is zero or 1;
R4 is -OH, -NR6R6a, or -NHOR9;
R5 is hydrogen or alkyl;
v is 1 to 5 ;
X is O or S;
p and q are independently 1 to 5, provided that p+q is
not greater than 5;
t is 1 to 9 ;
R6 and R6a are each the same or different and are
hydrogen or alkyl;
R~ is 1,3-dihydro-1,3-dioxo-2H-isoindol-2-yl, or
1,3-dihydro-1,3-dioxo-benzo[f]isoindol-2-yl;
R8 is hydrogen or alkyl; and
R9 is hydrogen, alkyl, or benzyl; or
a pharmaceutically acceptable salt thereof.
In a preferred embodiment, the present invention
provides compounds of Formula I wherein:
Ar is phenyl;
m is 0 or 1;
Y is CH or N;
R1 is hydrogen;
z is zero;
R2 is hydrogen or alkyl;
R3 is hydrogen, alkyl, - (CHZ) o-aryl, or
- (CHZ) "-heteroaryl;
R4 is -OH or -NHOH;
v is 1 to 5;
n is 0 or 1; and
W is -CHZ-; or
a pharmaceutically acceptable salt thereof.
CA 02233560 1998-03-31
WO 97/19068 PCT/CTS96/16761
-10-
In other preferred embodiments of the present
invention relating to the compounds of Formula I, Z is _
zero, or Ar is phenyl, or Y is C, or m is zero, or R2
is hydrogen, or R1 is hydrogen, or n is zero, or R4 is
-OH, and pharmaceutically acceptable salts of these
compounds provided that when m = 1, Y does not = N.
In a most preferred embodiment, the compounds of
Formula I are:
[4-(4-Phenyl-piperidin-1-yl)-benzenesulfonyl-
amino]-acetic acid;
N-Hydroxy-2-[4-(4-phenyl-piperidin-1-yl)-
benzenesulfonylamino]-acetamide;
3-[4-(4-Phenyl-piperidin-1-yl)-benzenesulfonyl-
amino]-propionic acid;
(R)-4-Methyl-2-[4-(4-phenyl-piperidin-1-yl)-
benzenesulfonylamino]-pentanoic acid;
(S)-4-Methyl-2-[4-(4-phenyl-piperidin-1-yl)-
benzenesulfonylamino]-pentanoic acid;
(S)-3-Phenyl-2-[4-(4-phenyl-piperidin-1-yl)-
benzenesulfonylamino]-propionic acid;
(R)-3-Phenyl-2-[4-(4-phenyl-piperidin-1-yl)-
benzenesulfonylamino]-propionic acid;
(S)-3-(1H-Indol-3-yl)-2-[4-(4-phenyl-piperidin-1-
yl)-benzenesulfonylamino]-propionic acid;
(t)-5-Phenyl-2-[4-(4-phenyl-piperidin-1-
yl)-benzenesulfonylamino]-pentanoic acid;
[4-(4-Phenyl-piperazin-1-yl)-benzenesulfonyl-
amino]-acetic acid;
{Isobutyl-[4-(4-phenyl-piperidin-1-yl)-benzene-
sulfonyl]amino}-acetic acid;
(S)-4-Phenyl-2-[4-(4-phenyl-piperidin-1-yl)- -
benzenesulfonylamino]-butyric acid;
(R)-2-[4-(4-Phenyl-piperidin-1-yl)-benzene-
sulfonylamino]-3-tritylsulfanyl-propionic acid sodium
salt;
CA 02233560 1998-03-31
WO 97/19068 PCT/US96/16761
-11-
(R)-3-(1H-Indol-3-yl)-2-[4-(4-phenyl-piperidin-
1-yl)-benzenesulfonylamino]-propionic acid, disodium
salt, monohydrate;
(S)-2-{4-[-4-(4-Hydroxy-phenyl)-piperazin-1-yl]-
benzenesulfonylamino}-3-phenyl-propionic acid;
(S)-2-{4-[-4-(4-Chloro-phenyl)-piperazin-1-yl]-
benzenesulfonylamino}-3-phenyl-propionic acid,
hydrochloride;
(R)-3-Mercapto-2-[4-(4-phenyl-piperidin-1-yl)-
benzenesulfonylamino]-propionic acid, trifluoracetic
acid salt;
(S)-2-[4-(4-Benzyl-piperidin-1-yl)-benzene-
sulfonylamino]-3-phenyl-propionic acid;
(S)-3-(4-Benzyloxy-phenyl)-2-[4-(4-phenyl-
piperidin-1-yl)-benzenesulfonylamino]-propionic acid;
(S)-3-(4-Hydroxy-phenyl)-2-[4-(4-phenyl-piperidin-
1-yl)-benzenesulfonylamino]-propionic acid;
(S)-3-Phenyl-2-[4-(4-phenyl-piperazin-1-yl)-
benzenesulfonylamino]-propionic acid;
(S)-2-{4-[-4-(3-Methoxy-phenyl)-piperazin-1-yl]-
benzenesulfonylamino}-3-phenyl-propionic acid;
(S)-2-{4-[-4-(3-Hydroxy-phenyl)-piperazin-1-yl]-
benzenesulfonylamino}-3-phenyl-propionic acid hydro-
bromide; and
(S)-2-{4-[-4-(4-Methoxy-phenyl)-piperazin-1-yl]
benzenesulfonylamino}-3-phenyl-propionic acid.
The present invention also provides a method of
inhibiting a matrix metalloproteinase, the method
comprising administering to a patient in need of matrix
metalloproteinase inhibition a matrix metallo-
proteinase inhibiting amount of a compound of
Formula I.
In a preferred embodiment, the matrix metallo-
proteinase is stromelysin-1 or gelatinase-A.
CA 02233560 1998-03-31
WO 97/19068 PCTlUS96/16761
-12-
In another embodiment, the present invention
provides a method of preventing atherosclerotic plaque
rupture comprising administering to a patient suffering
from an atherosclerotic plaque a therapeutically
effective amount of a compound of Formula I.
In another embodiment, the present invention
provides a method of inhibiting aortic aneurism
comprising administering to a patient having an aortic
aneurism a therapeutically effective amount of a
compound of Formula I.
In another embodiment, the present invention
provides a method of preventing restenosis comprising
administering to a patient following balloon
angioplasty, graft or shunt implantation, or
atherectomy, a therapeutically effective amount of a
compound of Formula I.
In another embodiment, the present invention
provides a method of treating periodontal disease
comprising administering to a patient suffering
therefrom a therapeutically effective amount of a
compound of Formula I.
In another embodiment, the present invention
provides a method of treating burns comprising
administering to a patient suffering therefrom a
therapeutically effective amount of a compound of
Formula I.
In another embodiment, the present invention
provides a method of treating decubital ulcers
comprising administering to a patient suffering
therefrom a therapeutically effective amount of a
compound of Formula I.
In another embodiment, the present invention
provides a method of treating chronic ulcers or wounds -
comprising administering to a patient suffering
therefrom a therapeutically effective amount of a
compound of Formula I.
CA 02233560 1998-03-31
WO 97/19068 PCT/US96/16761
-13-
In another embodiment, the present invention
provides a method of treating cancer comprising
administering to a patient suffering therefrom a
therapeutically effective amount of a compound of
Formula I.
In another embodiment, the present invention
provides a method of treating multiple sclerosis
comprising administering to a patient suffering
therefrom a therapeutically effective amount of a
compound of Formula I.
In another embodiment, the present invention
provides a method of treating arthritis comprising
administering to a patient suffering therefrom a
therapeutically effective amount of a compound of
Formula I.
In another embodiment, the present invention
provides a method of treating autoimmune or
inflammatory disorder dependent upon tissue.invasion by
leukocytes comprising administering to a patient
suffering from an autoimmune or inflammatory disorder
dependent upon tissue invasion by leukocytes a
therapeutically effective amount of a compound of
Formula I.
In another embodiment, the present invention
provides a pharmaceutical composition comprising a
compound of Formula I and a pharmaceutically acceptable
carrier.
DETAILED DESCRIPTION OF THE INVENTION
The present invention provides compounds having
the Formula I
CA 02233560 2004-03-05
-14-
O
4
/~ - II R
Ar-(CH2)m- \ /N \ / (iH)Z -li-N-(W)n I
Ra O ~ 2 R3
R1
wherein:
Ar is selected from phenyl;
phenyl substituted with alkyl, -N02, halogen, -ORS,
-CN, -C02R5, -S03R5, -CHO, -CORS, -CONHRS, -NHRS,
or -NHCORS;
heteroaryl; or
2-naphthyl;
R1 is hydrogen, methyl, -N02, -C1, -NH2, -NHC02CH3,
-OH, or -C02H;
R2, R3 and R' are the same or different and are
independently selected from hydrogen, alkyl,
-(CHZ)v-aryl, -(CH2)v-heteroaryl,
-(CH2)v-cycloalkyl, -(CH2)p-X-(CH2)q-aryl,
-(CH2)p-X-(CH2)q-heteroaryl, -(CH2)tNR6R6a~
-(CH2)vR~, -(CH2)VC02R5, -(CH2)vCONRSR6a, or
-(CH2)vSRS;
m is zero or 1;
Y is CH or N; provided that when m = 1, Y does not = N;
z is zero or 1;
W is -CHR8;
n is zero or 1;
R4 is -OH, -NR6R6a, or -NHOR9;
RS is hydrogen or alkyl;
v is 1 to 5 ;
X is O or S;
p and q are independently 1 to 5, provided that p+q is
not greater than 5;
t is 1 to 9;
R6 and R6a are each the same or different and are
hydrogen or alkyl;
CA 02233560 2004-03-05
-15-
R' is 1,3-dihydro-1,3-dioxo-2H-isoindol-2-yl, or
1,3-dihydro-1,3-dioxo-benzo[f)isoindol-2-yl;
R8 is hydrogen or alkyl; and
R9 is hydrogen, alkyl, or benzyl; or
a pharmaceutically acceptable salt thereof.
In a preferred embodiment, the present invention
provides compounds of Formula I wherein:
Ar is phenyl;
m is 0 or 1;
Y is CH or N;
R1 is hydrogen;
z is zero;
R2 is hydrogen or alkyl;
R3 is hydrogen, alkyl, - (CH2) ~-aryl, or
- (CHZ) "-heteroaryl;
R° is -OH or -NHOH;
v is 1 to 5;
n is 0 or 1; and
W is -CHZ-; or
a pharmaceutically acceptable salt thereof, provided that
when m = l, Y does not = N.
In a further preferred embodiment Y is N and m is 0.
The term "alkyl" means a straight or branched chain
hydrocarbon radical having from 1 to 8 carbon atoms.
Representative examples of alkyl groups include, but are not
limited to, methyl, ethyl, n-propyl, isopropyl, n-butyl, sec-
butyl, tert-butyl, isobutyl, n-pentyl, n-hexyl, n-heptyl and
n-octyl.
The term "alkoxy" and "thioalkoxy" mean 0-alkyl or S-
alkyl having from 1 to 6 carbon atoms. Examples of alkoxy
groups include, but are not limited to, methoxy, ethoxy,
propoxy, isopropoxy, and butoxy.
The term "cycloalkyl" means a saturated hydrocarbon
ring having 3 to 8 carbon atoms. Examples of cycloalkyl
groups include, but are not limited to, cyclopropyl,
cyclobutyl, cyclopentyl, cyclohexyl, cycioheptyl, and
cyclooctyl.
CA 02233560 1998-03-31
WO 97/19068 PCT/US96/1676I
-16-
The term "aryl" means an aromatic radical. For
example, the aryl group can be a phenyl group, or a
phenyl group substituted with 1 to 4 substituents
(phenyl is abbreviated "Ph"). The substituents can be
the same or different and can be selected from alkyl,
alkoxy, thioalkoxy, hydroxy, halogen, trifluoromethyl,
amino, alkylamino, dialkylamino, -N02, -CN, -C02H,
-C02alkyl, -S03H, -CHO, -COalkyl, -CONH2, -CONH-alkyl,
-CONHRS, -CON(alkyl)2, -(CHZ)n-NH2, where n is 1 to 5,
-(CH2)n-NH-alkyl, -NHRS, or -NHCORS.
The term "heteroaryl" means an aromatic compound
that includes one or more heteroatom. Examples of
heteroatoms include O, S, and N. Examples of
heteroaryl groups are 2- or 3-thienyl, 2- or 3-furanyl,
2- or 3-pyrrolyl, 2-, 3- or 4-pyridinyl, 2-pyrazinyl,
1H-indol-6-yl, 1H-indol-5-yl, 1H-benzimidazol-5-yl, or
1H-benzimidazol-6-yl.
The term "halogen" means fluorine, chlorine,
bromine, or iodine.
Some of the compounds of Formula I are capable of
further forming both pharmaceutically acceptable acid
addition and/or base salts. All of these forms are
within the scope of the present invention.
Pharmaceutically acceptable acid addition salts of
the compounds of Formula I include salts derived from
nontoxic inorganic acids such as hydrochloric, nitric,
phosphoric, sulfuric, hydrobromic, hydriodic,
hydrofluoric, phosphorous, and the like, as well as the
salts derived from nontoxic organic acids, such as
aliphatic mono- and dicarboxylic acids, phenyl-
substituted alkanoic acids, hydroxy alkanoic acids, .
alkanedioic acids, aromatic acids, aliphatic and
aromatic sulfonic acids, etc. Such salts thus include .
sulfate, pyrosulfate, bisulfate,. sulfite, bisulfate,
nitrate, phosphate, monohydrogenphosphate,
dihydrogenphosphate, metaphosphate, pyrophosphate,
CA 02233560 1998-03-31
WO 97/19068 PCT/LTS96/16761
-17-
chloride, bromide, iodide, acetate, trifluoroacetate,
propionate, caprylate, isobutyrate, oxalate, malonate,
succinate, suberate, sebacate, fumarate, maleate,
mandelate, benzoate, chlorobenzoate, methylbenzoate,
dinitrobenzoate, phthalate, benzenesulfonate,
toluenesulfonate, phenylacetate, citrate, lactate,
maleate, tartrate, methanesulfonate, and the like.
Also contemplated are salts of amino acids such as
arginate and the like and gluconate, galacturonate
(see, for example, Berge S.M., et al., "Pharmaceutical
Salts," J. of Pharma Sci., 1977;66:1).
The acid addition salts of said basic compounds
are prepared by contacting the free base form with a
sufficient amount of the desired acid to produce the
salt in the conventional manner. The free base form
may be regenerated by contacting the salt form with a
base and isolating the free base in the conventional
manner. The free base forms differ from their
respective salt forms somewhat in certain physical
properties such as solubility in polar solvents, but
otherwise the salts are equivalent to their respective
free base for purposes of the present invention.
Pharmaceutically acceptable base addition salts
are formed with metals or amines, such as alkali and
alkaline earth metals or organic amines. Examples of
metals used as cations are sodium, potassium,
magnesium, calcium, and the like. Examples of suitable
amines are N,N'-dibenzylethylenediamine, chloro-
procaine, choline, diethanolamine, dicyclohexylamine,
ethylenediamine, N-methylglucamine, and procaine (see,
for example, Berge S.M., et al., "Pharmaceutical
Salts," J. of Pharma Sci., 1977;66:1).
The base addition salts of said acidic compounds
are prepared by contacting the free acid form with a
sufficient amount of the desired base to produce the
salt in the conventional manner. The free acid form
CA 02233560 1998-03-31
WO 97/19068 PCT/US96/16761
-1$-
may be regenerated by contacting the salt form with an
acid and isolating the free acid in the conventional
manner. The free acid forms differ from their
respective salt forms somewhat in certain physical
properties such as solubility in polar solvents, but
otherwise the salts are equivalent to their respective
free acid for purposes of the present invention.
Certain of the compounds of the present invention
can exist in unsolvated forms as well as solvated
forms, including hydrated forms. In general, the
solvated forms, including hydrated forms, are
equivalent to unsolvated forms and are intended to be
encompassed within the scope of the present invention.
Certain of the compounds of the present invention
possess one or more chiral centers and each center may
exist in the R or S configuration. The present
invention includes all diastereomeric, enantiomeric,
and epimeric forms as well as the appropriate mixtures
thereof. Additionally, the compounds of the present
invention may exist as geometric isomers. The present
invention includes all cis, trans, syn, anti, entgegen
(E), and zusammen (Z) isomers as well as the
appropriate mixtures thereof.
Also provided by the present invention is a method
of inhibiting a matrix metalloproteinase, the method
comprising administering to a patient in need of matrix
metalloproteinase inhibition a matrix metallo-
proteinase inhibiting amount of a compound of
Formula I. In a preferred embodiment, the matrix
metalloproteinase is stromelysin-1 or gelatinase-A.
The term "patient" means humans and other animals. .
A patient in need of matrix metalloproteinase
inhibition is a patient who may suffer from
atherosclerotic plaque rupture or restenosis, or a
patient who suffers from aortic aneurism, periodontal
disease, burns, decubital ulcers, chronic ulcers or
CA 02233560 1998-03-31
WO 97/19068 PCT/US96/1676I
-19-
wounds, cancer, arthritis, multiple sclerosis, or other
_ autoimmune or inflammatory disorders dependent upon
tissue invasion by leukocytes.
Also provided by the present invention is a method
of preventing atherosclerotic plaque rupture comprising
administering to a patient suffering from an
atherosclerotic plaque a therapeutically effective
amount of a compound of Formula I.
Also provided by the present invention is a method
of inhibiting aortic aneurism comprising administering
to a patient having an aortic aneurism a
therapeutically effective amount of a compound of
Formula I.
Also provided by the present invention is a method
of preventing restenosis comprising administering to a
patient following balloon angioplasty, graft or shunt
implantation or atherectomy, a therapeutically
effective amount of a compound of Formula.
Also provided by the present invention is a method
of treating periodontal disease comprising
administering to a patient suffering therefrom a
therapeutically effective amount of a compound of
Formula I.
Also provided by the present invention is a method
of treating burns comprising administering to a patient
suffering therefrom a therapeutically effective amount
of a compound of Formula I.
Also provided by the present invention is a method
of treating decubital ulcers comprising administering
to a patient suffering therefrom a therapeutically
effective amount of a compound of Formula I.
Also provided by the present invention is a method
of treating chronic ulcers or wounds comprising
administering to a patient suffering therefrom a
therapeutically effective amount of a compound of
Formula I.
CA 02233560 1998-03-31
WO 97/19068 PCT/US96/16761
-20-
Also provided by the present invention is a method
of treating cancer metastasis comprising administering
to a patient suffering therefrom a therapeutically
effective amount of a compound of Formula I.
Also provided by the present invention is a method
of treating arthritis comprising administering to a
patient suffering therefrom a therapeutically effective
amount of a compound of Formula I.
Also provided by the present invention is a method
of treating multiple sclerosis comprising administering
to a patient suffering therefrom a therapeutically
effective amount of a compound of Formula I.
Also provided by the present invention is a method
of treating an autoimmune or inflammatory disorder
dependent upon tissue invasion by leukocytes comprising
administering to a patient suffering from an autoimmune
or inflammatory disorder dependent upon tissue invasion
by leukocytes a therapeutically effective amount of a
compound of Formula I.
The compounds of the present invention can be
prepared and administered in a wide variety of oral and
parenteral dosage forms. Thus, the compounds of the
present invention can be administered by injection,
that is, intravenously, intramuscularly,
intracutaneously, subcutaneously, intraduodenally, or
intraperitoneally. Also, the compounds of the present
invention can be administered by inhalation, for
example, intranasally. Additionally, the compounds of
the present invention can be administered
transdermally. It will be obvious to those skilled in
the art that the following dosage forms may comprise as .
the active component, either a compound of Formula I or
a corresponding pharmaceutically acceptable salt of a
compound of Formula I.
For preparing pharmaceutical compositions from the
compounds of the present invention, pharmaceutically
CA 02233560 1998-03-31
WO 97/19068 PCT/US96/16761
-21-
acceptable carriers can be either solid or liquid.
Solid form preparations include powders, tablets,
pills, capsules, cachets, suppositories, and
dispersible granules. A solid carrier can be one or
more substances which may also act as diluents,
flavoring agents, solubilizers, lubricants, suspending
agents, binders, preservatives, tablet disintegrating
agents, or an encapsulating material.
In powders, the carrier is a finely divided solid
which is in a mixture with the finely divided active
component.
In tablets, the active component is mixed with the
carrier having the necessary binding properties in
suitable proportions and compacted in the shape and
size desired.
The powders and tablets preferably contain from
five or ten to about seventy percent of the active
compound. Suitable carriers are magnesium carbonate,
magnesium stearate, talc, sugar, lactose, pectin,
dextrin, starch, gelatin, tragacanth, methylcellulose,
sodium carboxymethylcellulose, a low melting wax, cocoa
butter, and the like. The term "preparation" is
intended to include the formulation of the active
compound with encapsulating material as a carrier
providing a capsule in which the active component, with
or without other carriers, is surrounded by a carrier,
which is thus in association with it. Similarly,
cachets and lozenges are included. Tablets, powders,
capsules, pills, cachets, and lozenges can be used as
solid dosage forms suitable for oral administration.
For preparing suppositories, a low melting wax,
such as a mixture of fatty acid glycerides or cocoa
. butter, is first melted and the active component is
dispersed homogeneously therein, as by stirring. The
molten homogenous mixture is then poured into
CA 02233560 1998-03-31
WO 97/19068 PCTICTS96/16761
-22-
convenient sized molds, allowed to cool, and thereby to
solidify.
Liquid form preparations include solutions,
suspensions, and emulsions, for example, water or water
propylene glycol solutions. For parenteral injection,
liquid preparations can be formulated in solution in
aqueous polyethylene glycol solution.
Aqueous solutions suitable for oral use can be
prepared by dissolving the active component in water
and adding suitable colorants, flavors, stabilizing,
and thickening agents as desired.
Aqueous suspensions suitable for oral use can be
made by dispersing the finely divided active component
in water with viscous material, such as natural or
synthetic gums, resins, methylcellulose, sodium
carboxymethylcellulose, and other well-known suspending
agents.
Also included are solid form preparations which
are intended to be converted, shortly before use, to
liquid form preparations for oral administration. Such
liquid forms include solutions, suspensions, and
emulsions. These preparations may contain, in addition
to the active component, colorants, flavors,
stabilizers, buffers, artificial and natural
sweeteners, dispersants, thickeners, solubilizing
agents, and the like.
The pharmaceutical preparation is preferably in
unit dosage form. In such form, the preparation is
subdivided into unit doses containing appropriate
quantities of the active component. The unit dosage
form can be a packaged preparation, the package
containing discrete quantities of preparation, such as
packeted tablets, capsules, and powders in vials or
ampoules. Also, the unit dosage form can be a capsule,
tablet, cachet, or lozenge itself, or it can be the
appropriate number of any of these in packaged form.
CA 02233560 2004-03-05
-23 -
The quantity of active component in a unit dose
preparation may be varied or adjusted from 1 mg to
1000 mg, preferably 10 mg to 100 mg according to the
particular application and the potency of the active
component. The composition can, if desired, also
contain other compatible therapeutic agents.
In therapeutic use as agents for the treatment of
multiple sclerosis, atherosclerotic plaque rupture,
aortic aneurism, heart failure, restenosis, periodontal
disease, corneal ulceration, burns, decubital ulcers,
chronic ulcers or wounds, cancer, multiple sclerosis,
arthritis, or other autoimmune or inflammatory
disorders dependent upon tissue invasion by leukocytes,
the compounds utilized in the pharmaceutical method of
this invention are administered at the initial dosage
of about 1 mg to about 100 mg per kilogram daily. A
daily dose range of about 25 mg to about 75 mg per
kilogram is preferred. The dosages, however, may be
varied depending upon the requirements of the patient,
the severity of the condition being treated, and the
compound being employed. Determination of the proper
dosage for a particular situation is within the skill
of the art. Generally, treatment is initiated with
smaller dosages which are less than the optimum dose of
the compound. Thereafter, the dosage is increased by
small increments until the optimum effect under the
circumstance is reached. For convenience, the total
daily dosage may be divided and administered in
portions during the day if desired.
CA 02233560 2004-03-05
-23a-
In a preferred embodiment, the invention comprises a
commercial package comprising a container containing therein
a compound according to claim 1 and written matter which
states that the compound is for use for one or more of the
following purposes:
a. inhibiting stromelysin-1,
b. inhibiting gelatinase-A,
c. preventing atherosclerotic plaque rupture,
d. inhibiting aortic aneurism,
e. preventing restenosis following balloon
angioplasty, graft or shunt implantation or
atherectomy,
f. treating periodontal disease,
g. treating decubital ulcers,
h. treating chronic ulcers or wounds,
i. treating cancer,
j. treating arthritis,
k. treating autoimmune or inflammatory disorders
dependent upon tissue invasion by leukocytes, and
1. treating multiple sclerosis.
In a further preferred embodiment, the invention
comprises a commercial package comprising a container
containing therein a composition according to claim 27 and
written matter which states that the composition is for use
for one or more of the following purposes:
a. inhibiting stromelysin-l,
b. inhibiting gelatinase-A,
c. preventing atherosclerotic plaque rupture,
d. inhibiting aortic aneurism,
e. preventing restenosis following balloon
angioplasty, graft or shunt implantation or
atherectomy,
f. treating periodontal disease,
g. treating decubital ulcers,
h. treating chronic ulcers or wounds,
i. treating cancer,
CA 02233560 2004-03-05
-23b-
j. treating arthritis,
k. treating autoimmune or inflammatory disorders
dependent upon tissue invasion by leukocytes, and
1. treating multiple sclerosis.
The following examples illustrate particular
embodiments of the invention and are not intended to limit
the specification, including the claims, in any way.
CA 02233560 1998-03-31
WO 97/19068 PCT/US96/16761
-24-
EXAMPLES
A compound of Formula I can be made by the general
route, as set forth in Scheme I below.
With reference to Scheme I, a compound of
Formula II is reacted with a compound of Formula III
(commercially available from Sigma Chemical Company,
St. Louis, Missouri, or can be synthesized according to
Schemes V and VI) in the presence of a suitable base
such as triethylamine, sodium carbonate or potassium
carbonate in a suitable solvent such as water,
methanol, tetrahydrofuran, or some combination thereof,
at temperatures between 0°C and 50°C to obtain a
compound of Formula IV. The compound of Formula IV is
then reacted with a compound of Formula V in the
presence of an excess of a suitable base such as sodium
carbonate or potassium carbonate in a suitable solvent
such as dimethylsulfoxide (DMSO) or dimethylformamide
(DMF) at temperatures between 25°C and 180°C to obtain
a compound of Formula Ia, wherein the variables are
defined as above, except that z = 0 and R4 = OH.
Specific compounds of the present invention can be
prepared by various routes, all of which are well known
in the art.
CA 02233560 1998-03-31
WO 97/19068 PCT/LTS96/16761
-25
SCHEME 2
C02H
base
S02C1 + ~-(W)n R3
R2
R1
II III
R3
Ar-( CH2 ) m Y\
~JV
F S02N - ( W ) n C02H
base DMSO
Rl
IV
R3
R
Ar-( CH2 ) m -Y' N S02N - ( H1 ) n C02H
~/ R1
Ia
[Z = O, R4 = OH]
35
CA 02233560 1998-03-31
WO 97/19068 PCT/LJS96/16761
-26-
Compounds of Formula I wherein z = n = 0, R1 and
R2 are hydrogen, Y = CH, R4 - OH, and Ar, m and R3 are
as defined in Formula I, can be prepared according to
the sequence described in Scheme II below.
With regard to Scheme II, the halide (1), wherein -
halo is defined as iodine, bromine or chlorine, is
reacted with a suitable metallating agent (M), such as
an alkyl lithium, for example, n-butyl lithium, sec-
butyl lithium, or tert-butyl lithium, or magnesium
metal, in a suitable solvent such as tetrahydrofuran
(THF) or diethyl ether (Et20) at temperatures between
-80°C and 60°C, followed by 1-(phenylmethyl)-4-
piperidinone at temperatures between -80°C and 25°C to
obtain the 4-piperidinol (2). The 4-piperidinol (2) is
dehydrated by stirring in a suitable solvent such as
acetic acid (AcOH) with a strong acid catalyst such as
concentrated hydrochloric acid (HC1) at temperatures
between 0°C and reflux to obtain the 1,2,5,6-
tetrahydropyridine (3) as an acid salt. The 1,2,5,6-
tetrahydropyridine (3) is reduced by catalytic
reduction using a suitable catalyst such as. 10~
palladium on carbon (Pd/C) and hydrogen gas (H2) at
pressures between 10 p.s.i. and 100 p.s.i. in a
suitable solvent such as absolute ethanol, acetic acid,
or tetrahydxofuran to yield the piperidine
hydrochloride (4).
The sulfonamide (6) wherein R3 is as defined in
Formula I, may be prepared by reacting the amino acid
(5) which is commercially available from a variety of
vendors, e.g., Sigma Chemical Company, St. Louis,
Missouri, or synthesized by standard methods well known
in the art, (set forth in Schemes V & VI below) with
4-fluoro-benzenesulfonyl chloride in the presence of a
suitable base such as triethylamine, sodium carbonate
(Na2C03) or potassium carbonate in a suitable solvent
CA 02233560 1998-03-31
WO 97/19068 PCT/US96/16761
-27-
such as water, methanol, tetrahydrofuran, at
temperatures between 0°C and 50°C.
The piperidine hydrochloride (4) is reacted with
the sulfonamide (6) in the presence of an excess of a
suitable base such as sodium carbonate or potassium
carbonate in a suitable solvent such as dimethyl-
sulfoxide or dimethylformamide at temperatures between
25°C and 180°C to obtain the (sulfonylamino)-carboxylic
acid (7) .
CA 02233560 1998-03-31
WO 97/19068 PCT/US96/16761
-28
SCHEME II
Ar(CH)mhalo (1)
1. M THF or Et20
2. O N~
Ph
HO
Ar-(CH2)m N~Ph ~2~
3
HC1 R2 R
ACOH
~ (5)
HN-(W)n C02H
Ar-(CH2)m ~ N~Ph (3)
~HC1 F ~ S02C1
2 0 H2 Pd/C Na2C03
R2 R3
Ar-(CH ) NH ~HC1 (4) F SO N-(W) ~CO H
2 m O 2 n 2 ( )
a
R2 R3
Ar- CH N SO N-( W ) ~CO H 7
( 2)m O 2 n 2 ( )
CA 02233560 1998-03-31
WO 97/19068 PCT/US96/16761
-29-
Compounds of Formula I wherein z = n = 0, R1 and
R2 are hydrogen, Y = CH, R4 = NHOR9 or NR6R6a, and Ar,
m and R3 are as defined in Formula I, can be prepared
according to the sequence described in Scheme III.
With regard to Scheme III, the (sulfonylamino)-
carboxylic acid (7) can be reacted with a suitable
O-substituted hydroxylamine hydrochloride of the
formula H2NOR9 HCl in the presence of a suitable base
such as triethylamine (Et3N) or N,N-diisopropyl-N-
ethylamine and a suitable coupling agent such as 1,1'-
carbonyldi-imidazole (CDI) or N,N'-dicyclohexylcarbodi-
imide (DCC) and 1-hydroxybenzo-triazole (HOBT) in a
suitable solvent such as tetrahydrofuran (THF),
dichloromethane, or N,N-dimethyl-formamide (DMF) at
temperatures between 0°C and 100°C to yield the
O-substituted hydroxamic acid (8). When R9 is defined
as benzyl (R9 = CH2Ph), the O-substituted-hydroxamic
acid (8) can be reduced to yield the hydroxamic acid
(9) by catalytic reduction using hydrogen gas at
pressures between 10 p.s.i. and 100 p.s.i. and a
suitable catalyst such as 5~ or 10~ palladium on barium
sulfate in a suitable solvent such as THF or ethanol.
Alternatively, the (sulfonylamino)-carboxylic acid (7)
can be reacted with various amines of the formula
R6R6aNH in the presence of a suitable coupling agent
such as 1,1'-carbonyldiimidazole (CDI) or N,N'-
dicyclohexylcarbodiimide (DCC) and 1-hydroxy-
benzotriazole (HOBT) in a suitable solvent such as
tetrahydrofuran, dichloromethane, or N,N-dimethyl-
formamide at temperatures between 0°C and 100°C to
yield the (sulfonylamino)-carboxamides (10).
CA 02233560 1998-03-31
WO 97/19068 PCT/LTS96/16761
-30-
SCHEME III
R3
Ar-(CH2)m N ~ S02NH C02H (7)
~R6R6a
~20R DCC, HOBT
DCC, HOBT
R3
NHR6R6a
Ar -(CH2)m N ~ S02NH
O (10)
20
R3
NHOR9
Ar-( CH2 ) m N ~ S02NH
(8)
O
H2N, 5~ Pd/BaS04
Methanol
R3
2 5 Ar- ( CH2 ) m N ~ SO2 NH OOH
(9)
O
CA 02233560 1998-03-31
WO 97/19068 PCT/US96/16761
-31-
Compounds of Formula I wherein z = m = n = 0, R1
and R2 are hydrogen, Y = N, and Ar, R3, and R4, are as
defined in Formula I, can be synthesized according to
the sequence described in Scheme IV below.
With regard to Scheme IV, the amine (11} is
reacted with bis(2-chloroethyl)amine hydrochloride of
the formula HN(CH2CH2C1)2 HCl, in a suitable solvent
such as chlorobenzene, at temperatures between 25°C and
180°C to yield the piperazine hydrochloride (12). The
piperazine hydrochloride (12) is reacted with the
sulfonamide (6) in a manner similar to that previously
described for compound (7) to obtain the corresponding
piperazine-carboxylic acid (13). The piperazine-
carboxylic acid (13) can be reacted with a suitable
O-substituted hydroxylamine hydrochloride of the
formula H2NOR9 IiCl in the presence of a suitable base
such as triethylamine (Et3N} or N,N-diisopropyl-N-
ethylamine and a suitable coupling agent such as 1,1'-
carbonyldi-imidazole (CDI) or N,N'-dicyclohexyl-
carbodiimide (DCC) and 1-hydroxybenzotriazole (HOBT) in
a suitable solvent such as tetrahydrofuran (THF),
dichloromethane, or N,N-dimethylformamide (DMF) at
temperatures between 0°C and 100°C to yield the
O-substituted-hydroxamic acid (14). Alternatively, the
piperazine-carboxylic acid (13) can be converted to the
free hydroxamic acid (15) by first reacting with a
suitable activating agent such as isobutyl
chloroformate of formula (CH3)2CHCH2COC1 in the
presence of a suitable base such as triethylamine or
N,N-diisopropyl-N-ethylamine in a suitable solvent such
- as dichloromethane or tetrahydrofuran at temperatures
between -78°C and +25°C followed by a suitable
O-substituted-hydroxylamine such as O-(tri-
methylsilyl)-hydroxylamine of formula H2NOSi(CH3)3
(TMSONH2) or O-(tert-butyldimethylsilyl)-hydroxylamine
of formula H2NOSi(CH3)2C(CH3)3 and then quenching the
CA 02233560 1998-03-31
WO 97/19068 PCT/US96/16761
-32-
reaction with aqueous acid. Alternatively, the
piperazine-carboxylic acid (13) can be reacted with
various amines of the formula HNR~R6a in the presence
of a suitable coupling agent such as 1,1'-
carbonyldiimidazole (CDI) or N,N'-dicyclohexyl-
carbodiimide (DCC) and 1-hydroxy-benzotriazole (HOBT)
in a suitable solvent such as tetrahydrofuran,
dichloro-methane, or N,N-dimethyl-formamide at
temperatures between 0°C and 100°C to yield the
piperazine-carboxamides (16).
CA 02233560 1998-03-31
WO 97/19068 PCT/US96/16761
-33-
x
0
x
z
' o
N
x
N
M O
3~
r-I
o_ o
N
z
x ~ ~ ° 2 v ~o
~ v H x
x r; c~ ni
o ( ~ o
z
0
I
c~ O
~x
~~w o
0
x
U O
z
v ~ z
v ~ o z
. z x
z
O N
_U
v N
_U U I
1
x xw
U
x
o ti r.~
N fx ~ H
z
=., o 0
x '~' q ~.
x
00
z
N
- x
_U
CA 02233560 1998-03-31
WO 97/19068 PCT/CTS96/16761
-34-
Compounds of Formula I wherein z = 0, n = 1, R1 is
hydrogen, and Ar, Y, R2, R3, R4, and R8 are as defined
in Formula I can be prepared as set forth in Scheme V
below.
With regard to Scheme V, an aldehyde (17) is
reacted with trimethylphosphono-acetate of formula
(CH30)2P(O)CH2C02CH3 in the presence of a suitable base
such as sodium hydride or lithium diisopropylamide
(LDA) in a suitable solvent such as tetrahydrofuran at
temperatures between -78°C and +60°C to yield the
unsaturated ester (18). The unsaturated ester (18) is
reacted with lithium (R)-(+)-N-benzyl-N-a-
methylbenzylamine, prepared in situ by a slow addition
of n-butyl lithium to (R)-(+)-N-benzyl-N-oc-
methylbenzylamine, in a suitable solvent such as
tetrahydrofuran at -78°C followed by the addition of
R3-halo wherein halo is defined as chlorine, bromine,
or iodine, and R3 is as defined in Formula I, and
allowing the temperature to slowly warm from -78°C to
+25°C overnight to yield the amino ester (19). (The
designates a chiral carbon.) The diastereomers of the
amino ester (19) can be separated by column
chromatography. The complimentary diastereomer of the
amino ester (19) can be prepared by following the
procedure described previously and substituting
(S)-(-)-N-benzyl-N-a-methylbenzylamine for (R)-(+)-
N-benzyl-N-oc-methylbenzylamine. The single
stereoisomers of amino ester (19) can be reduced
separately by reacting with hydrogen gas in the
presence of a suitable catalyst such as 5~ to 30~
palladium on carbon in a suitable solvent such as
tetrahydrofuran, acetic acid, methanol, or mixtures
thereof at pressures between atmospheric and 100 p.s.i.
and temperatures between 25°C and 100°C to yield the
amino ester (20). The amino ester (20) is then reacted
in a suitable aqueous acid mixture such as 6 M
CA 02233560 1998-03-31
WO 97/19068 PCT/US96/1676I
-3 5-
hydrochloric acid at temperatures between 25°C and
_ reflux to yield the amino acid hydrochloride (21).
Alternatively, the amino ester (20) can be reacted with
an alkyl halide of formula R2-halo, wherein R2 is as
defined in Formula I and halo is defined as chlorine,
bromine, or iodine, in the presence of a suitable base
such as triethylamine or N,N-diisopropyl-N-ethylamine
in a solvent such as diethyl ether or tetrahydrofuran
at temperatures between 0°C and 50°C followed by
conversion of the free amino ester to the amino ester
hydrochloride (22). The amino ester hydrochloride (22)
is then reacted in a suitable aqueous acid mixture such
as 6 M hydrochloric acid following the procedure
described above to yield the amino acid hydrochloride
(23). When in the procedure described by Scheme II,
the amino acid hydrochlorides (21) or (23) are
substituted for the amino acid (5) and reacted with
4-fluoro-benzenesulfonyl chloride, the sulfonamides
(24) and (25) can be prepared, respectively. When in
the procedures described for Schemes II and IV the
sulfonamides (24) or (25) are substituted for the
sulfonamide (6) and reacted with either the piperidine
hydrochloride (4) or the piperazine hydrochloride (12)
(generically represented by V), respectively, the
(sulfonylamino)-carboxylic acids (26) and (27) can be
prepared, respectively. When in the procedures
described for Schemes III and IV the (sulfonylamino)-
carboxylic acids (26) and (27) are substituted for the
(sulfonylamino)-carboxylic acids (7) or (13) and the
appropriate methodology for either the piperidines
- (Scheme III) or piperazines (Scheme IV) is followed,
the compounds (28) and (29) can be prepared,
- respectively.
CA 02233560 1998-03-31
WO 97/19068 PCT/US96/16761
-36-
M
x N
x
N
O
N M
E
~ ~'
O x
w a~ x
0
-~, ~ o ~ a~ U
a ~ o .~ x
M
x
.~ m ~ ~ ~
p., ~ ~ O r-I N N
N M U ~ ~- P4
m
c ~ o
N
v z
U x
U
N n7 x
v ~ ~ x .~o
H
_ . o x
N cn U O
fps ~ ~ U
Q
o'
., z /
o ~ - ~w z
w x
U
x
CA 02233560 1998-03-31
WO 97/19068 PCT/US96/16761
-37-
x ~ d, °'
~ N
U
0
* ~ *
x ~
N
v 00
I N-~ z _ ~
o ~~ ~ o
N
* U
R',
O r~C',
1 ~ -i
z z
0 00
00
w '~ !
x x
U _U
.,'' I I
O.
U
x ~ ~, o~
ON
U
r, z o
o ~ O * ~
x
o I x x
N
U ~ x O O
_U
R' * ~ 7.,
x
O --1
z z1
O
00
.
w
N N
~ x
U
CA 02233560 1998-03-31
WO 97/19068 PCT/US96/1676I
-38-
Compounds of Formula I wherein z = n = 0, R1 is
hydrogen, and Ar, m, Y, R2, R3, and R4 are as defined
in Formula I, can be synthesized according to the
sequence described in Scheme VI below.
With regard to Scheme VI, an amine of formula
R2NH2, wherein R2 is as defined in Formula I, is
reacted with a bromo-ester (30), wherein R3 is as
defined in Formula I, in the presence of a suitable
base such as triethylamine (Et3N) or N,N-diisopropyl-
N-ethylamine in a solvent such as diethyl ether or
tetrahydrofuran at temperatures between -10°C and 50°C
to afford the free amino ester which is converted to
the amino ester hydrochloride (31). Alternatively, the
amino ester hydrochloride (31) can be prepared by
reacting an alkyl halide of the formula R2-halo,
wherein R2 is as defined in Formula I and halo is
defined as chlorine, bromine, or iodine, with an amino-
ester hydrochloride (30a), wherein R3 is as defined in
Formula I, following the procedure described for (31).
The amino-ester hydrochloride (31) is reacted in a
suitable aqueous acid mixture such as 6 ~t hydrochloric
acid following the procedure described previously for
Scheme V to yield the amino-acid hydrochloride (32).
When in the procedure described for Scheme II the
amino-acid hydrochloride (32) is substituted for the
amino acid (5) and reacted with 4-fluoro-
benzenesulfonyl chloride, the sulfonamide (33) is
obtained. When in the procedures described for
Schemes II and IV the sulfonamide (33) is substituted
for the sulfonamide (6) and reacted with either the
piperidine hydrochloride (4) or the piperazine
hydrochloride (12), respectively, the (sulfonylamino)-
carboxylic acid (34) can be prepared. When in the
procedures described for Schemes III and IV the
(sulfonylamino)-carboxylic acid (34) is substituted for
either the (sulfonylamino)-carboxylic acid (7) or (13)
CA 02233560 1998-03-31
WO 97/19068 PCT/CTS96/16761
-39-
and the appropriate methodology for either the
piperidines (Scheme III) or piperazines (Scheme IV) is
followed, the compound (35) can be prepared, where R4
is defined as NHOR9 or NR6R6a.
CA 02233560 1998-03-31
WO 97/19068 PCTlUS96/16761
-40-
SCHEME VI
R3 R3
2 + ~ 2 +
R ~2 Br C02Et R -Halo H2N C02Et.HCl
(30) (30a)
Et3N Et3N, Et20
R3
H2N
(31)
R2 N C02Et~HCl
H
6M HC1
(32)
R2 H C02H~HC1
F ~ S02C1, Na2C03
~2 R3
S02N
C02H (33 )
Ar-(CH2)m ~ , K2C03, DMSO
R3
Ar-(CH2 ) m-Y' N S02N' 'CO H ( 34 )
2
1 2
R
R3
R4
Ar-(CH2)m- ~ ~ S02N (35)
R2 0
3
N
R4 = HNORg; NR6R6a
CA 02233560 1998-03-31
WO 97/19068 PCT/1JS96/16761
-41-
Compounds of Formula I wherein z = 0, Ar, Y, m, n,
R1, R2, and R3 are as defined in Formula I, and R4 is
OH, can be synthesized according to the sequence
described in Scheme VII below.
The commercially available fluorosulfonic acids
(36) as their sodium salts are reacted with a suitable
halogenating agent such as a mixture of phosphorus
pentachloride (PC15) in phosphorus oxychloride (POC13)
at temperatures between -20°C and 50°C to yield the
sulfonyl chloride (37). The sulfonyl chloride (37) is
reacted with either the amino acid (5) from Scheme II,
the amino acid hydrochloride (21) from Scheme V, the
amino acid hydrochloride (23) from Scheme V, or the
amino acid hydrochloride (32) from Scheme VI, all of
which may be represented by the general structure
designated by Formula III of Scheme I, in the presence
of a suitable base such as triethylamine, sodium
carbonate or potassium carbonate in a suitable solvent
such as water, methanol, tetrahydrofuran or some
combination thereof, at temperatures between 0°C and
50°C to give the (sulfonylamino)-carboxylic acid (38).
When in the procedures described for Schemes II and IV
the sulfonamide (38) is substituted for the sulfonamide
(6) and reacted with either the piperidine
hydrochloride (4) or the piperazine hydrochloride (12),
respectively, represented by the general structure V,
the (sulfonylamino)-carboxylic acid (40) can be
prepared. ln~h.en in the procedures described for
Schemes III and IV the (sulfonylamino)-carboxylic acid
(40) is substituted for either the (sulfonylamino)-
carboxylic acid (7) or (13) and the appropriate
methodology for either the piperidines (Scheme III) or
piperazines (Scheme IV) is followed, the compound (40a)
can be prepared, where R4 is defined as NHOR9 or
NR:6R6a.
CA 02233560 1998-03-31
WO 97/19068 PCT/US96/16761
-42-
SCHEME VII
Co2H
* ~HC1
HN- ( W ) ~ 3 I I I
F S03Na --1 F S02C1 n R
R1 R1
(36) (37)
Co H
2 Ar-(CH2)m- ~ H
F S02N- (W) n 3
base
R2
R1 (38)
C02H
Ar-(CH2 ) m-Y/ N S02N- (W) n 3
R
R2
R
(40)
O R4
Ar-(CH2)m- ~ S02N-(W)n R3
12
R1
(40a)
CA 02233560 1998-03-31
WO 97/19068 PCT/US96/16761
-43-
Compounds of Formula I wherein z = 1, R1 is
hydrogen, and Ar, Y, R2, R3, R4, W, m, and n are as
defined in Formula I, can be prepared according to the
sequence described in Scheme VIII.
With regard to Scheme VIII, the compound of
Formula V is reacted with 4-fluorobenzoic acid, ethyl
ester in the presence of an excess of a suitable base
such as sodium carbonate (Na2C03) or potassium
carbonate in a suitable solvent such as dimethyl-
sulfoxide (DMSO) or dimethylformamide at temperatures
between 25°C and 180°C to obtain the ester (41). The
ester (41) is reduced with a suitable reducing agent
such as lithium aluminum hydride (LiAlH4) in a suitable
solvent such as tetrahydrofuran at temperatures between
0°C and 60°C to yield the alcohol (42). The alcohol
(42) is reacted with a suitable halogenating agent such
as phosphorous tribromide (PBr3) in dichloromethane at
temperatures between -40°C and 40°C to yield the halide
(43). The halide (43) is reacted with sodium
thiosulfate (Na2S203) in water with or without a phase
transfer agent such as N-methyl-N,N,N-tri(n-octyl)-
ammonium chloride at temperatures between 0°C and 100°C
in the presence of chlorine gas (C12) to yield the
sulfonyl chloride (44). Alternatively, the halide (43)
can be reacted with sodium thiosulfate (Na2S203) in
water with or without a phase transfer agent such as
N-methyl-N,N,N-tri(n-octyl)ammonium chloride at
temperatures between 25°C and 100°C to yield the
sulfonate (45). The sulfonate (45) is then reacted
with a suitable halogenating agent such as a mixture of
- phosphorus pentachloride (PC15) in phosphorus
oxychloride (POC13) at temperatures between -20°C and
150°C to yield the sulfonyl chloride (44). The
sulfonyl chloride (44) is reacted with either the amino
acid (5) from Scheme II, the amino acid hydrochloride
(21) from Scheme V, the amino acid hydrochloride (23)
CA 02233560 1998-03-31
WO 97/19068 PCT/US96/16761
-44-
from Scheme V, or the amino acid hydrochloride (32)
from Scheme VI, all of which may be represented by the
general structure designated by Formula III of
Scheme I, in the presence of a suitable base such as
triethylamine, sodium carbonate or potassium carbonate
in a suitable solvent such as water, methanol,
tetrahydrofuran or some combination thereof, at
temperatures between 0°C and 50°C to give the
(sulfonylamino)-carboxylic acid (46). Alternatively,
the sulfonyl chloride (44) can be reacted with
tert-butylamine in a suitable solvent such as
diethylether or tetrahydrofuran in the presence of
excess base such as tert-butylamine or triethylamine to
yield the sulfonamide (48). The sulfonamide (48) can
be reacted with two equivalents of a strong base such
as n-butyl lithium, sec-butyl lithium, or tert-butyl
lithium in a suitable solvent such as tetrahydrofuran
at temperatures between -78°C to +25°C, followed by the
addition of an alkyl halide of the formula R2-halo,
wherein R2 is as defined in Formula I, and halo is
defined as chlorine, bromine, or iodine, to yield the
sulfonamide (49). The sulfonamide (49) can be reacted
with a strong acid such as trifluoroacetic acid (TFA)
either neat or in a suitable solvent such as
dichloromethane to yield the sulfonamide (50). The
sulfonamide (50) can be reacted with a suitable base
such as sodium hydride (NaH) in tetrahydrofuran as
solvent or sodium ethoxide in ethanol as solvent,
followed by the addition of the bromoester (30),
wherein R3 is as defined in Formula I, to yield the
(sulfonylamino)-ester (51). The (sulfonylamino)-ester
(51) can be reacted with either lithium, sodium, or
potassium hydroxide in a suitable solvent such as
ethanol followed by acidification to yield the
(sulfonylamino)-carboxylic acid (52).
CA 02233560 1998-03-31
WO 97/19068 PCT/US96/16761
-45-
When in the procedures described for Schemes III
and IV, the (sulfonylamino)-carboxylic acids (46) or
!52) is substituted for either the (sulfonylamino)-
carboxylic acid (7) or (13) and the appropriate
methodology for either the piperidines (Scheme III) or
piperazines (Scheme IV) is followed, the compounds (47)
and (53) can be prepared, where R4 is defined as NHOR9
or NR6R6a.
CA 02233560 1998-03-31
WO 97/19068 PCT/US96/16761
-46-
.-~
U
N
O
O W O U1
N '
N x
r-I
N
x O U
o ~ o ~ o
z
w
z ~ ~ z ~ ~ z ~ --v
o ~
o ~
z ~ N x x
x U U
U
H
H O
U
d~ c~7
U07 ch
o °w w 2
N
N
N
f~ U
z
x
z
z r, I
N
_ x
N
_U I I
N '
_U
I
CA 02233560 1998-03-31
WO 97/19068 PCT/CTS96/16761
-47-
x
N
O
U O
_C
3
x x
N N
iI1 Lx
fl1
t0
t0
z
z
0
x --~ z
C~ C~
N N
x x
U U_
I I N x
a v
0
MC'a M
N N N N N
O
y, Ll~ pc,' tl~ 0.~' UI W' UI p',, iJ~
00 O~ \ ~ O ~ 1-1 \ ~ N ~ M
z z z z z z
C ~ --~C ~--C ~ ~C ~--C ~ --C ~
a coo ~ H ~ v I o
a a ~M ~ w a
N ~ ~ N N ~--- a x O N N
x
U_ _ _ U_ U_ cx0 p~q ~ U x x U U
I r-1 N ~i N I
~N
CA 02233560 1998-03-31
WO 97/19068 PCT/US96/16761
-48-
The O-substituted-hydroxylamine hydrochlorides of
the formula H2NOR9 HCl can be purchased from commercial
sources or prepared as set forth in Scheme IX.
CA 02233560 1998-03-31
WO 97/19068 PCT/CTS96/16761
-49
SCHEME IX
O 1. R3-Halo, THF, Et3N
2. H2NNHCH3, THF, 0-25°C
N-OH H2NOR9 ~ HC1
3. HC1(g), Et20
O
15
25
35
CA 02233560 1998-03-31
WO 97/19068 PCT/US96/16761
-50-
EXAMPLE 1
f4-(4-Phenyl-pi~eridin-1-vl)-benzenesulfonvlaminol-
acetic acid
(a) (4-Fluoro-benzenesulfonvlamino)-acetic acid
A mixture of 4-fluoro-benzenesulfonyl chloride
(9.68 g, 0.497 mol), glycine (4.48 g, 0.0598 mol), and
sodium carbonate (16.99 g, 0.160 mol) in water (60 mL)
was stirred at room temperature for 42 hours. The
mixture was carefully acidified to pH 8 to 9 with
concentrated hydrochloric acid, and washed 2 times with
dichloromethane. The aqueous layer was acidified
further to pH 2, and the resulting white suspension was
extracted two times with ethyl acetate. The extracts
were combined, washed with saturated sodium chloride
solution, and dried over magnesium sulfate. The dried
solution was rotary evaporated to give a white solid,
which was dried in vacuo; yield 4.7 g (41~),
mp = 154.0-155.5°C.
(b) f4-(4-Phenyl-t~iperidin-1-vl?benzenesulfonvl-aminol
acetic acid
A stirred mixture of (4-fluoro-benzenesulfonyl-
amino)-acetic acid (0.0895 g, 0.000384 mol), 4-phenyl-
piperidine (0.618 g, 0.000383 mol), and potassium
carbonate (0.109 g, 0.000789 mol) in dry dimethyl
sulfoxide (0.10 mL) in a tightly capped vial was placed
in a hot sand bath (115°C). After 21 hours, the
reaction mixture was cooled and partitioned between
ethyl acetate and water. The mixture was acidified
with 1M hydrochloric acid (3.2 mL, 0.0032 mol) and the
layers were separated. The aqueous layer was washed
with additional ethyl acetate. The organics were
combined, dried (MgS04), and rotary evaporated to give -
a glass. The glass was dissolved in methanol, silica
gel was added (4.2 g, 230-400 mesh), and the mixture
was rotary evaporated to dryness. The resulting powder
CA 02233560 1998-03-31
WO 97/19068 PCT/US96/16761
-51-
was poured onto a column of silica gel (14 g,
230-400 mesh), and eluted with a mixture of hexanes-
ethyl acetate-acetic acid (15:15:1, 11 x 15 mL).
Fractions containing product were combined and rotary
evaporated. The residue was crystallized from
methanol-water (1:1) after a hot filtration to give the
title compound as a pale yellow solid; yield 0.070 g
(49~), mp = 154.5-155.5°C.
EXAMPLE 2
N-Hvdroxv-2-f4-(4-phenvl-piperidin-1-vl)-benzene-
sulfonvlaminol-acetamide
(a) N-f(Phenvlmethvl?oxvl-2-f4-(4-phenvl-piperidin-1-
yl)-benzenesulfonvlaminol-acetamide
A suspension of O-benzylhydroxylamine hydro-
chloride (0.110 g, 0.000689 mol) in a mixture of
triethylamine (0.096 mL, 0.00069 mol) in anhydrous
tetrahydrofuran (7 mL) was heated on a steam bath, and
dimethylformamide (~5 mL) was added until all solids
had dissolved. The mixture was cooled to room
temperature. The solids which precipitated were
filtered off and set aside.
In a separate flask containing a cool (5°C),
stirred solution of [4-(4-phenyl-piperidin-1-yl)
benzenesulfonylamino]-acetic acid (0.2307 g,
0.0006161 mol) and 1-hydroxy-benzo-1,2,3-triazole
(0.0842 g, 0.000623 mol) in anhydrous tetrahydrofuran
(10 mL) was added in one portion 1,3-dicyclohexyl-
carbodiimide (0.1449 g, 0.000702 mol). The mixture was
stirred for 30 minutes at 5°C then allowed to warm to
room temperature. After 3 hours at room temperature,
the mixture was added in one portion to the filtrate
containing O-benzylhydroxylamine. The mixture was
stirred at room temperature for 16 hours, then refluxed
(74°C) for 1 hour. The volatiles were rotary
evaporated off, and the residue was partitioned between
CA 02233560 2004-03-05
-52-
ethyl acetate and water. The aqueous layer was
extracted with ethyl acetate. The organics were
combined, washed with 0.1 M NaOH, water, 0.1 M HC1,
water, and saturated sodium chloride. The organic
layer was dried (MgS04) and rotary evaporated. The
residue was dissolved in chloroform, and
chromatographed on silica gel (34 g, 230-400 mesh)
eluting with dichloromethane-acetone (9:1, 10 x 30 mL).
Fractions containing product were rotary evaporated to
give a white solid. The solid was dried in vacuo;
yield 0.1448 g (49g), mp = 163-165°C.
(b) N-Hvdroxy-2-f4-(4-t~henyl-t~igeridin-1-vl)-benzene-
sulfonvlaminol-acetamide
A room temperature mixture of N-[phenyl-methyl)-
oxy)-2-[4-(4-phenyl-piperidin-1-yl)-benzenesulfonyl-
amino]-acetamide (0.1115 g, 0.0002325 mol) in methanol-
tetrahydrofuran (1:1, 25 mL) was hydrogenated at
50 p.s.i. over 5~ palladium on barium sulfate (0.018 g)
for approximately 10 hours. Additional catalyst
(0.020 g) was added, and the mixture hydrogenated again
for approximately 10 hours. The mixture was filtered
through celite, and the filtrate was rotary evaporated
to give a glaze. The glaze was dissolved in
chloroform-methanol, silica gel (1.6 g, 230-400 mesh)
was added, and the mixture was rotary evaporated to
dryness. The powder was poured onto a column of silica
gel (10 g, 230-400 mesh) and eluted with hexanes-ethyl
acetate-acetic acid (10:20:1, 16 x 10 mL and 10:20:2,
16X10 mL). Fractions containing product were rotary
evaporated, and the residue triturated with chloroform.
The chloroform suspension was filtered, and the
filtercake was dried in vacuo; yield 0.0054 g (6.0~),
mp = 164-166°C.
3 5 *Trade-mark
CA 02233560 1998-03-31
WO 97/19068 PCT/US96/16761
-53-
EXAMPLE 3
3-f4-(4-Phenvl-~i~eridin-1-vl)-benzenesulfonvlaminol-
~ropiox~.ic acid
3-f4-Fluoro-(benzenesulfonvlamino)1-~ropionic
acid. sodium salt
A mixture of 4-fluoro-benzenesulfonyl chloride
(1.920 g, 0.009866 mol), 3-amino-propionic acid
(0.980 g, 0.0110 mol), and sodium carbonate (2.33 g,
0.0220 mol) in water (15 mL) was stirred at room
temperature for 28 hours, then briefly heated on a
steam bath. The mixture was allowed to cool, then
stirred at room temperature overnight. The mixture was
reheated on a steam bath, gravity filtered hot, and
allowed to cool. The filtrate was acidified to
approximately pH 5 with concentrated hydrochloric acid.
A white precipitate was filtered off and dried
in vacuo; yield 1.907 g (78~).
1H-NMR (DMSO-d6): 8 7.85 (m, 2H), 7.80 (br s, 1H),
7.45 (m, 2H), 2.93 (t, 2H), 2.35 (t, 2H).
(b) 3-f4-(4-Phenvl-piperidin-1-vl)-benzenesulfonyl-
aminol-pronionic acid
A stirred mixture of 3-(4-fluoro-(benzenesulfonyl
amino)]-propionic acid, sodium salt (0.248 g,
0.00100 mol), 4-phenyl-piperidine hydrochloride
(0.218 g, 0.00110 mol), and sodium carbonate (0.317 g,
0.00299 mol) in dry dimethyl sulfoxide (3 mL) was
heated in a sand bath (130°C) under nitrogen for
22 hours. The mixture was cooled and partitioned
between ethyl acetate and 1 M hydrochloric acid. The
- aqueous layer was extracted with additional ethyl
acetate. The organics were combined, washed with
. saturated sodium chloride, dried (MgS04), and rotary
evaporated. The residue was dissolved in dichloro-
methane and chromatographed on silica gel (14 g,
230-400 mesh) eluting with dichloromethane-methanol
CA 02233560 1998-03-31
WO 97/19068 PCT/L1S96/16761
-54-
(15:1, 10X15 mL). Fractions containing product were
combined, rotary evaporated and rechromatographed to
give the title compound as a peach-colored solid; yield
0.082 g (21~), mp = 145-147°C.
EXAMPLE 4
(R)-4-Methyl-2-f4-(4-t~henvl-pi~eridin-1-vl)-benzene-
sulfonvlaminol-~entanoic acid
(a) (R)-2-l4-Fluoro-benzenesulfonvlamino)-4-methvl-
~entanoic acid
A mixture of 4-fluoro-benzenesulfonyl chloride
(1.65 g, 0.00848 mol), (R)-2-amino-4-methyl-pentanoic
acid (1.233 g, 0.009398 mol), and sodium carbonate
(1.91 g, 0.0180 mol) in water (15 mL) was stirred at
room temperature for 5 days. The solution was
filtered, and the filtrate was acidified with
concentrated hydrochloric acid to pH = 4. The mixture
was extracted with ethyl acetate. The extract was
washed with saturated sodium chloride, dried (MgS04),
and rotary evaporated to a yellow oil. The oil was
chromatographed on silica gel (320 g, 230-400 mesh)
eluting with dichloromethane-methanol (10:1,
10X300 mL). Fractions containing product were combined
and rotary evaporated to give a pale yellow oil. The
oil was dried in vacuo; yield 1.44 g (59~).
1H-NMR (DMSO-d6): 8 8.1 (br s, 1H), 7.80 (m, 2H), 7.38
(m, 2H), 3.55 (t, 1H), 3.33 (br s, H20),1.56 (m, 1H),
1.36 (dd, 2H), 0.75 (dd, 6H).
(b) ~R)-4-Methyl-2-f4-(4-phenyl-piperidin-1-y~ -
benzenesulfonvlaminol-pentanoic acid .
In a manner similar to Example 3(b), 4-phenyl-
piperidine hydrochloride was condensed with (R)-2- .
(4-fluoro-benzenesulfonylamino)-4-methyl-pentanoic acid
to give the title compound, mp = 163-165°C.
CA 02233560 1998-03-31
WO 97/19068 PCT/US96/16761
- -55-
EXAMPLE 5
(S)-4-Methyl-2-f4-(4-phenyl-~i~eridin-1-yl)-benzene-
sulfonvlaminol-pentanoic acid
_ (a) (S)-2-l4-Fluoro-benzenesulfonvlamino)-4-methvl-
nentanoic acid
In a manner similar to Example 4(a) (S)-2-amino-4-
methyl-pentanoic acid was substituted for (R)-2-amino-
4-methyl-pentanoic acid; yield 14.0 g (55~).
300 MHz 1H-NMR (DMSO-d6): 8 8.17 (br s, 1H), 7.83
(m, 2H), 7.40 (m, 2H), 3.64 (t, 1H), 1.57 (m, 1H), 1.38
(m, 2H), 0.76 (dd, 6H).
(b) (S)-4-Methyl-2-f4-l4-uhenvl-pineridin-1-vl
benzenesulfonvlamina~entanoicacid
In a manner similar to Example 3(b), 4-phenyl-
piperidine hydrochloride was condensed with (S)-2-(4-
fluoro-benzenesulfonylamino)-4-methyl-pentanoic acid to
give the title compound, $C,H,N found: 63.96, 6.96,
6.44.
EXAMPLE 6
(S)-3-Phenyl-2-f4-(4-t~henvl-pit~eridin-1-vl)-benzene-
sulfonvlaminol-~ropionic acid
(a) (S)-2-(4-Fluoro-benzenesulfonvlamino)-3-phenvl-
t~ro~ionic acid, sodium salt
In a manner similar to Example 4(a), 4-fluoro-
benzenesulfonyl chloride and (S)-2-amino-3-phenyl-
propionic acid were condensed to give the title
compound as a white solid, mp = 108-111°C.
- (b) lS)-3-Phenyl-2-f4-(4-phenyl-nineridin-1-yl)-
benzenesulfonylaminol-propionic acid
In a manner similar to Example 3(b), (S)-2-(4-
fluoro-benzenesulfonylamino)-3-phenyl-propionic acid,
sodium salt and 4-phenyl-piperidine hydrochloride were
condensed to give the title compound, mp = 167-169°C.
CA 02233560 1998-03-31
WO 97/19068 PCT/US96/16761
-56-
EXAMPLE 7
(R)-3-Phenvl-2-f4-(4-~henvl-biberidin-1-vl)-benzene-
sulfonvlaminol-nronionic acid
(a) (R)-2-(4-Fluoro-benzenesulfonvlamino)-3-phenvl-
r~ro~ionic acid, disodium salt
In a manner to Example 4(a), (R)-2-amino-3-phenyl-
propionic acid was condensed with 4-fluoro-
benzenesulfonyl chloride to give the title compound,
mp = 246-248°C.
(b) lR)-3-Phenvl-2-f4-(4-nhenvl-~iperidin-1-vl)-
benzenesulfonvlaminol-t~ronionic acid
In a manner similar to Example 3(b), (R)-2-(4-
fluoro-benzenesulfonylamino)-3-phenyl-propionic acid,
disodium salt was condensed with 4-phenyl-piperidine
hydrochloride to give the title compound,
mp = 168-170°C.
EXAMPLE 8
lS)-3-(1H-Indol-3-vl)-2-f4-(4-bhenvl-pi~eridin-1-vl)-
benzenesulfonylaminol-propionic acid
(a) (S)-2-(4-Fluoro-benzenesulfonylamino)-3-l1H-Indol-
3-vl)-pro~ionic acid
In a manner to Example 4 (a), 4-fluoro-benzene-
sulfonyl chloride was condensed with (S)-2-amino-3-(1H-
Indol-3-yl)-propionic acid to give the title compound,
mp = 57-60°C.
(b) (S)-3-(1H-Indol-3-vl)-2-f4-(4-phenyl-~iperidin-1-
yl)-benzenesulfonvlaminol-propionic acid
In a manner similar to Example 3(b), (S)-2-(4- -
fluoro-benzenesulfonylamino)-3-(1H-Indol-3-yl)-
propionic acid was condensed with 4-phenyl-piperidine -
hydrochloride to give the title compound,
mp = 103-107°C.
CA 02233560 1998-03-31
WO 97/19068 PCT/LTS96/16761
_57-
EXAMPLE 9
(t)-5-Phenvl-2-f4-(4-nhenvl-pit~eridin-1-vl)-benzene
~»~ fonvlaminol -t~entanoic acid
(a) (t)-2-Amino-5-~henvl-bentanoic acid
A stirred suspensiori of (t)-2-(acetylamino)-5-
phenyl-pentanoic acid (0.5003 g, 0.002126 mol) in 2.8 M
hydrochloric acid was refluxed for 2 hours, and the
resulting brown solution was allowed to cool. A tan
precipitate formed upon cooling. The solids were
filtered off, and the filtrate was rotary evaporated to
give a yellow gum. The gum was dissolved in hot water,
gravity filtered, and allowed to cool. The mixture was
made basic with 1 M sodium hydroxide to pH = 5. The
resulting precipitate was filtered off, washed with
water, and dried in vacuo to give a yellow solid; yield
0.205 g (50~), mp = 213-215°C.
(b) (f)-2-(4-Fluoro-benzenesulfonvlamino)-5-t~henvl-
pentanoic acid
A mixture of (t)-2-amino-5-phenyl-pentanoic acid
(0.188 g, 0.000973 mol), 4-fluoro-benzenesulfonyl
chloride (0.189 g, 0.000971 mol), and sodium carbonate
(0.208 g, 0.00196 mol) in water (4 mL) was stirred at
room temperature for 4 days. The mixture was heated
briefly on a steam bath to give a cloudy solution. The
solution was gravity filtered hot, and the filtrate
allowed to cool. The resulting solid that crystallized
was filtered off, washed with water, and dried
in vacuo; yield 0.131 g, (38~). 1H-NMR (DMSO-d6):
8 7.81 (m, 2H), 7.37 (t, 2H), 7.25 (t, 2H), 7.14
- (m, 4H), 3.34 (br s, H2), 3.04 (t, 1H), 2.46 (m, 2H),
1.6-1.4 (m, 4H).
CA 02233560 1998-03-31
WO 97/19068 PCT/LTS96/16761
-58-
(c) lt)-5-Phenyl-2-f4-t4-phenyl-piperidin-1-vl)-
benzenesulfonvlaminol-nentanoic acid
In a manner similar to Example 3(b), (t)-2-(4-
fluoro-benzenesulfonylamino)-5-phenyl-pentanoic acid
was condensed with 4-phenyl-piperidine hydrochloride to
give the title compound, mp = 59-62°C.
EXAMPLE 10
f4-(4-Phenyl-piperazin-1-vl)-benzenesulfonvlaminol-
acetic acid
In a manner similar to Example 3(b), (4-fluoro-
benzenesulfonylamino)-acetic acid was condensed with
4-phenyl-piperazine to give the title compound,
mp = 120-124°C.
EXAMPLE 11
~Isobutvl-f4-(4-phenyl-piberidin-1-vl)-benzene-
sulfonvllamino~-acetic acid
(a) Isobutvlamino-acetic acid, ethyl ester
hydrochloride
A mixture of isobutylamine (0.90 mL, 0.0091 mol),
bromoacetic acid, ethyl ester (1.0 mL, 0.0090 mol), and
triethylamine (1.28 mL, 0.00918 mol) in diethylether
(15 mL) was stirred at room temperature for 24 hours.
The resulting suspension was filtered off and washed
with diethylether. The filtrate and washings were
combined and rotary evaporated to an oil. The oil was
chromatographed on silica gel (150 g, 230-400 mesh)
eluting with dichloromethane-diethylether (19:1,
8 x 125 mL; 15:1, 7 x 125 mL; 10:1, 15 x 125 mL).
Fractions containing product were combined and rotary
evaporated to give an oil. The oil was dissolved in
diethylether, concentrated hydrochloric acid (0.52 mL,
0.0063 mol HC1) was added, and the volatiles were
rotary evaporated to give a white solid. The solid was
dried in vacuo; yield 1.0 g (59~).
CA 02233560 1998-03-31
WO 97/19068 PCT/US96/16761
-59-
1H-NMR (DMSO-d6): b 9.23 (br s, 2H), 4.22 (q, 2H),
_ 3.96 (m, 2H), 3.41 (br s, H20), 2.78 (m, 2H), 2.00
(m, 1H) , 1.25 (t, 3H) , 0.94 (d, 6H) .
(b) f(4-Fluoro-benzenesulfonvl)-isobutvl-aminol-acetic
acid
A mixture of isobutylamino-acetic acid, ethyl
ester hydrochloride (0.359 g, 0.00183 mol) in 6 M
hydrochloric acid (10 mL) was refluxed for 20 hours and
allowed to cool. The mixture was made basic with 50~
wt/wt sodium hydroxide and 1 M sodium hydroxide to
pH = 5, and the volatiles were rotary evaporated. The
residue was triturated 3 times with boiling methanol,
and the triturates were combined and rotary evaporated.
The residue was triturated 3 times with hot acetic
acid, and the triturates were combined and rotary
evaporated. The residue was dissolved in water and
freeze-dried to give a white solid. This solid was
combined with 4-fluoro-benzenesulfonyl chloride
(0.3334 g, 0.001713 mol) and sodium carbonate (0.547 g,
0.00516 mol) in water, and the mixture was stirred at
room temperature for 3 days. The mixture was acidified
with concentrated hydrochloric acid and extracted with
ethyl acetate. The extract was dried (MgS04) and
rotary evaporated to a white solid. The solid was
chromatographed on silica gel (15 g, 230-400 mesh)
eluting with dichloromethane-methanol (10:1, 10 x 15
mL). Fractions containing product were combined and
rotary evaporated to give a white solid. The solid was
dried in vacuo; yield 0.32 g (64~ overall).
1H-NMR (DMSO-d6): 8 7.85 (m, 2H), 7.39 (m, 2H), 3.91
(s, 2H), 3.32 (br s, H20), 2.94 (d, 2H), 1.77 (m, 1H),
- 0.79 (d, 6H) .
CA 02233560 1998-03-31
WO 97/19068 PCT/US96/16761
-60-
(c) {Isobutvl-f4-(4-phenvl-x~i~eridin-1-vl)-benzene-
sulfonvllamino~-acetic acid
In a manner similar to Example 3(b), [(4-fluoro-
benzenesulfonyl)-isobutyl-amino -acetic acid was
condensed with 4-phenyl-piperidine hydrochloride to
give the title compound, mp = 140-143°C.
EXAMPLE 12
(S)-2-f4-(4-Benzvl-nineridin-1-vl)-benzenesulfonyl-
aminol-3-xahenvl-propionic acid
In a manner similar to Example 3(b), (S)-2-
(4-fluoro-benzenesulfonylamino)-3-phenyl-propionic
acid, sodium salt, and 4-benzyl-piperidine were
condensed to give the title compound, mp = 164-165°C.
EXAMPLE 13
(S)-3-(4-Benzvloxv-~henvl)-2-f4-(4-phenyl-bineridin-
1-vl)-benzenesulfonvlaminol-pro~ionic acid
(a) (S)-3-(4-Benzvloxv-phenvl)-2-(4-fluoro-benzene-
sulfonvlamino)-propionic acid
A mixture of (S)-2-amino-3-(4-benzyloxy-phenyl)-
propionic acid (2.7 g, 0.010 mol), 4-fluoro-benzene-
sulfonyl chloride (2.0 g, 0.010 mol), and sodium
carbonate (2.2 g, 0.020 mol) in a mixture of tetra-
hydrofuran (20 mL) and water (20 mL) was stirred at
room temperature for 3 days. The reaction mixture was
partitioned between ethyl acetate and 1 M hydrochloric
acid. The organic layer was washed with saturated
sodium chloride solution, dried (MgS04), and rotary-
evaporated under reduced pressure to give an oil. The
oil was chromatographed on silica gel (445 g,
230-400 mesh) eluting with dichloromethane-methanol
(20:1), and the fractions containing product were
rotary-evaporated to give a solid. The solid was
recrystallized from toluene to give the title compound
CA 02233560 1998-03-31
WO 97/19068 PCT/US96/16761
-s1-
as a light yellow solid; yield 0.22 g (5~),
mp = 138-139°C.
(b) lS)-3-(4-Benzyloxv-phenyl)-2-f4-(4-phenvl-
~ineridin-1-vl)-benzenesulfonvlaminol-~ropionic
a i
In a manner similar to Example 3(b), (S)-3-
(4-benzyloxy-phenyl)-2-(4-fluoro-benzenesulfonylamino)-
propionic acid was condensed with 4-phenyl-piperidine
hydrochloride to give the title compound, mp = 75-78°C.
EXAMPLE 14
(S)-3-(4-Hvdroxv-phenyl)-2-f4-(4-phenyl-~iberidin-
1-vl)-benzenesulfonvlaminol-~roQionic acid
To a room temperature, stirred mixture of
(S)-3-(4-benzyloxy-phenyl)-2-[4-(4-phenyl-piperidin-
1-yl)-benzenesulfonylamino~-propionic acid (0.033 g,
0.000058 mol) in thioanisole (0.34 mL) was added
trifluoroacetic acid (1 mL), and the mixture was
stirred for 18 hours. The reaction mixture was poured
into water and extracted with ethyl acetate. The
organic layer was dried (Na2S04) and rotary-evaporated
under reduced pressure to remove volatiles. The
resulting yellow solution was chromatographed on silica
gel (5.5 g) eluting with dichloromethane (10 x 5 mL)
followed by dichloromethane-methanol (14:1). Fractions
containing product were rotary-evaporated. The residue
was suspended in water and stirred to give the title
compound as an off-white solid; yield 0.0079 g (28~),
mp = 108-110°C.
EXAMPLE 15
lS)-3-Phenyl-2-f4-(4-phenyl-biperazin-1 yl)-benzene-
sulfonvlaminol-bro~ionic acid
In a manner similar to Example 3(b), (S)-2-
(4-fluoro-benzenesulfonylamino)-propionic acid, sodium
CA 02233560 1998-03-31
WO 97/19068 PCT/US96/16761
-62-
salt and 4-phenyl-piperazine were condensed to give the
title compound as a beige solid, mp = 192-193°C.
EXAMPLE 16
lS)-2-~4-f-4-(3-Methoxv-x~henvl)-~i~erazin-1-vll-
benzenesulfonvlamino}-3-phenvl-propionic acid
In a manner similar to Example 3(b), (S)-2-
(4-fluoro-benzenesulfonylamino)-propionic acid, sodium
salt and 4-(3-methoxy-phenyl)-piperazine were condensed
to give the title compound as a pale red-brown solid,
mp = 237-139°C.
EXAMPLE 17
-2-f4-f-4-(3-Hvdrox~phenvl)-piperazin-1-vl
benzenesulfonvlamino~-3-nhenvl-~ro~ionic acid hvdro-
bromide
To a stirred suspension of (S)-2-{4-[4-(3-methoxy-
phenyl)-piperazin-1-yl]-benzenesulfonylamino}-3-phenyl-
propionic acid (0.103 g, 0.000208 mol) in dichloro-
methane (2 mL) at -~78°C under nitrogen was added
dropwise a 1.0 M solution of boron tribromide in
dichloromethane (1.0 mL, 0.0010 mol). The mixture was
stirred for 15 minutes at -78°C and then allowed to
warm to +3°C. After 6 hours, the reaction mixture was
diluted with water. The resulting suspension was
stirred overnight. The solids were filtered off,
washed with additional water, and dried in vacuo to
give the title compound as an off-white solid; yield
0.069 g (69~), mp = 229-230°C.
EXAMPLE 18 -
(S)-2-14-f-4-(4-Methoxv-nhenvl)-piperazin-1-vll-
benzenesulfonvlamino}-3-~henvl pronionic acid
In a manner similar to Example 3(b), (S)-2-
(4-fluoro-benzenesulfonylamino)-propionic acid, sodium
salt and 4-(4-methoxy-phenyl)-piperazine dihydro-
CA 02233560 1998-03-31
WO 97/19068 PCT/US96/16761
-63-
chloride were condensed to give the title compound as a
brown solid, mp = 203-205°C.
INHIBITION STUDIES
Experiments were carried out which demonstrate the
efficacy of compounds of Formula I as potent inhibitors
of stromelysin-1 and gelatinise A. Experiments were
carried out with the catalytic domains. Table 1 shows
the activity of the Examples 1-12 versus GCD
(recombinant gelatinise A catalytic domain); SCD
(stromelysin-1 catalytic domain). IC50 values were
determined using a thiopeptolide substrate, Ac-Pro-Leu-
Gly-thioester-Leu-Leu-Gly-OEt (Ye Q.-Z., Johnson L.L.,
Hupe D.J. and Baragi V., "Purification and
Characterization of the Human Stromelysin Catalytic
Domain Expressed in Escherichia coli", Biochemistry,
1992;31:11231-11235).
CA 02233560 1998-03-31
WO 97/19068 PCT/US96/16761
-64-
TABLE 1
Compound
IC50 ~M/SCD IC50 uM/GCD
Number
1 0.02 0.21
2 0.019 0.81
3 1.24 4.8
4 0.036 0.93
5 0.011 0.084
6 0.014 0.22
7 0.012 0.12
8 0.01 0.32
9 0.30 0.40
10 0.05 0.50
11 0.17 3.3
12 0.60 3.2
13 0.19 5.3
14 0.015 0.13
15 0.021 0.088
16 0.062 0.33
17 0.077 0.18
18 0.014 0.033