Note: Descriptions are shown in the official language in which they were submitted.
CA 02234484 l998-04-08
W O 97/13776 PCT~R96/01588
Nouveaux dérives aminés de 2",3" didésoxyglycosides d'épipodophyllotoxine,
leur procedé de prép~r~tion, leur utilis~tion comme médicament
et leur utilisation destinée aux traitements anticancéreux.
La presente invention concerne des nouveaux dérivés d'arnino 2",3 "
S didésoxyglycosides d'épipodophyllotoxine, leur procédé de préparation, leur utilisation
comrne mél1ic~ment et leur utilisation destinée aux traitementc anticancéreux.
Parmi les dérivés issus de lign~n~s naturels, il existe la classe des
épipodophylloïdes, ayant le squelette de base de la podophyllotoxine. Y figurent les
dérivés hémisynthétiques tels que l'Etoposide ou le Téniposide lesquels sont utilisés de
10 façon usuelle dans la préparation de mé~ mentc pour le tr~it~m~?nt du cancer. Ils sont
considérés cornrne des produits majeurs dans ce domaine.
L'Etoposide possède des propriétés ~nfitllm~rales et perrnet de traiter en
particulier le cancer du poumon à petites cellules et le cancer des testicules.
L'inconvénient de ces produits est leur manque d'hydrosolubilité et rencontrent
15 par conséquent des rliffic--ltes au niveau de la formulation et de l'~minictration.
L'objet de la présente invention est de montrer que des dérivés de la 4'-déméthyl
épipodophyllotoxine, possédant en position 4, une substitution de structure 2"-
désoxyglycoside, permet, par l'incorporation d'un ou plusieurs azotes, de former des
composés dont les sels d'addition possèdent une solubilité aqueuse permettant de20 répondre au problème et montrent l'activité anticancéreuse recherchée.
Le brevet EP-0 196 6 l 8 relate des dérivés hydrosolubles de 4'-déméthyl
épipodophyllotoxine de formule:
O ~
<~~o
.. ~ o
~ MeO ~ OM-
OEI
25 où R1 = Me, Xl = NH2, NMe2, X2 = OH
CA 02234484 1998-04-08
W O 97/13776 PCT~FR96/01588
Le brevet EP-0 415 453 mentionne des dérivés ,B-D-altrosides de 4'-deméthyl
épipodophyllotoxine de formule
I ~ O ~2
0~ 0
~ O
<~~0
,~
ll
MeO ~ O Me
OH
où Rl = Me R2, R3 = OH et NH2 ou F et NH2 de même que JP 0161, 423.
D'autres publications mentionnent des dérivés analogues (Carbohydr. Res. 1990,
206, 219; Chem. Pharm. Bull. 1986, 34, 3733; Chem. Pharm. Bull 1986, 34, 3741;
Chem. Lett. 1987, 799).
Le fait d'utiliser un glycoside ayant une position 2"-désoxy est tout à fait
particulier. Il permet d'obtenir des composés plus lipophiles que les analogues
hydroxylés et, par conséquent, avoir un spectre d'activite ~ntitnmoral elargi. Cela leur
permet d'avoir une meilleure pénétration membranaire et de pouvoir atteindre plus
facilement la cible biologique comme, par exemple, les tumeurs solides peu irriguées
15 En outre, l'avantage de posséder une fonction amino par exemple en position 3", confere
une possibilité de salification donc d'hydrosolubilité s~ffic~nte pour une meilleure
formulation et une meilleure ~lminictration.
CA 02234484 1998-04-08
W O 97/13776 PCT~R96/01588
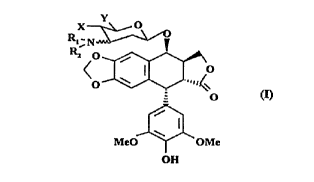
La p~ésel,le invention concerne donc un composé de formule générale I
~,N
<O~o
MeO ~ O Me
OH
5 dans laquelle le groupement en 3" N(Rl R2) est en position ~ (serie 2-désoxy DArabino) ou c~ (série 2-désoxy D ribo) par rapport au cycle, Rl et R2 identiques ou
dirr~ reprrsrntent un atome d'hydrogène, un groupe alkyle de Cl à C6, pouvant
former un cycle, ce cycle pouvant comporter un hétéroatome comme un oxygène ou un
azote, un groupe ~mino~lkyle en Cl à C6 ou cyanométhyle
X et Y peuvent être identiques ou différents et représent.ont un OH, CH3, CH2-
NH~, X et Y peuvent également être liés et constituer un cycle, comme par exemple, un
2-méthyl 1,3 dioxane, formant ainsi un squelette bicyclique osidique de type 4,6-
éthylidène 3-amino-2,3-didésoxy-,B-D arabino ou ribo-hexo pyranoside.
Elle concerne également leurs sels d'addition avec des acides minéraux ou
organiques s~lifi~nt le ou les atomes d'~ote, en particulier les chlorhydrates
D'une manière avantageuse, le groupe NRlR2 est un groupement NH2 ou
N(CH3)2
Le groupe NRlR2 peut eg~lement être un groupe amino substitué une ou deux
fois par Ull méthyl, CH2CN, CH2-CH2-NH2, former un cycle comme la morpholine
De manière avantageuse, les composés de formule générale I seront choisis avec
un glycoside pour lequel X et Y forment un cycle avec un enrh~înrment
OCH(CH3)0CH2, tel qu'un 4,6 éthylidène-3-amino-2,3-didésoxy-,B-D arabino-hexo
pyranoside ou bien 4,6 éthylidène-3-amino-2,3-dldesoxy-~-D-ribo-hexopyranoside.
CA 02234484 1998-04-08
W O 97/13776 PCT~FR96/01588
En particulier, les composés selon l'invention sont sélectionnés parmi les
composes suivants:
~ 4'-Déméthyl-4-0(3-amino-4,6-éthylidène-2,3-didésoxy-13-D-arabinohexopyranosyl)
épipodophyllotoxine,
~ 4'-Déméthyl-4-0(3-amino-4,6-éthylidène-2,3-didésoxy-~-D-ribohexopyranosyl)
épipodophyllotoxine,
4'-Déméthyl-4-0(3-diméthylamino-4,6-éthylidène-2,3-didésoxy-,B-D-arabinoh~co-
pyranosyl) épipodophyllotoxine,
~ 4'-Déméthyl-4-0(3-diméthylamino-4,6-éthylidène-2,3-didésoxy-~-D-ribohexo-
pyranosyl) épipodophyllotoxine,
4'-Déméthyl-4-0(3-cyanométhylarnino-4,6-éthylidène-2,3-didésoxy-~-D-ribohexo-
pyranosyl) épipodophyllotoxine,
4'-Déméthyl-4-0(3-(N-morpholino)-4,6-éthylidène-2,3-didésoxy-~-D-ribohexo-
pyranosyl) épipodophyllotoxine,
~ 4'-Déméthyl-4-0[3(2-aminoéthylamino)-4,6-éthylidène-2,3-didésoxy-,B-D-ribohexo-
pyranosyl)] épipodophyllotoxine,
4'-Déméthyl-4-0(3-amino-2,3,6-tridesoxy-~-D-ribohexopyranosyl)
épipodophyllotoxine,
~ 4'-Déméthyl-4-0(3,6-diamino-2,3,6-tridésoxy-,B-D-ribohexopyranosyl)
épipodophyllotoxine.
La ~.éselIte invention concerne aussi les compositions pharmaceutiques
comprenant au moins un composé de formule générale I selon l'invention et un excipient
a~p.oylié.
Les compositions pharmaceutiques peuvent être présentées de façon adaptée par
25 l'~tlmini~tration par voie injectable ou par voie orale sous forme de capsule, de gélules,
de co~ hllés à la posologie de I à 200 mg/m2 par voie inJectable et de 5 à 500 mg/m2
par voie orale par période de 24 h.
Ces dérivés peuvent ainsi être ~lmini~trés en clinique hum~ine pour traiter
différentes formes de cancer tels le cancer du poumon à petites cellules, le cancer des
30 testicules, les tumeurs embryonnaires, les neuroblastomes, le cancer du rein, les
CA 02234484 l998-04-08
W O 97/13776 PCT~R96/01588
Iyphomes Hodgkiniens et non Hodgkiniens, les leucemies aiguës, les cancers
colorectau.~;, les mélanomes, les choriocarcinomes placentaires et les adénocarcinomes
.~ rn m~ires.
La présente invention est également relative aux procedcs de préparation des
S composés de formule I ainsi que de leurs sels d'addition avec les acides minérau:; ou
organiques pharmaceutiquement acceptables.
La présente invention concerne donc les procédés de préparation des composés
de forrmule générale I selon l'invention, dans lesquels on fait réagir un composé de
forrnule III ou IV ou V
~''o~~,~- ji + ~~~~~Fi + P o~oFi
0 lll IV V
avec la 4'-Déméthyl 4'-benzyloxycarbonyl épipodophylloto:~ine avec l'éthérate de ~3,
ou le triméthyl silyl trifluorométh~neslllforate dans un solvant inerte a basse
température;
dans la formule III et IV, le substituant en position 3 peut etre ~ ou ~, NRIR~
peut être un amino protégé par un groupe Z,
dans la forrnule V, P represente un groupe protecteur d'alcool et les produits
resultants de cette condensation sont déprotéges et hydrogénes pour fournir les
composés de forrnule 1,
les amines primaires en position 3 du glycosyl sont methylées par le formol et le
cyanoborohydrure de sodium.
L'intermédiaire de formule IV est prépare en faisant réagir un mélange de
diacétoxy azido glycoside VI
AcO
AcO
N~
~,~ Vl
CA 02234484 l998-04-08
W O 97/1~776 PCT~FR96/OlS88
avec le chlorure de tertbutyl dimethylsilyl en présence d'imidazole, en ce que l'on sépare
les produits résultant de cette réaction, en ce que chacun de ces produits sont deacétyles,
cyclises en 4,6-ethylidène avec l'acétal de l'acétaldehyde en milieu acide catalytique
D'autres caractéristiques du procedé selon l'invention apparaîtront à la lumierede la description qui suit, not~mment du procédé de synthèse reporté sur le schéma I.
SCEEMA 1
Ac~
Ac(~L I ) H20 80~C AcO~
AcO~ g 2) N~N3 / AcOH AcO~ OH
N~ ;~
TsDMSiCI / Imid;~olc/
CH2C12
AcO
AcO ~ ~ OSi+ AcO ~ j_ OS~
~, 4 3
oll
s ~-- ~ osl ~
011
~IcO
~T.
C~13CIl(OE.)2
OU ,~r rs'
Cl 13C~i
R,T,
~12 CJrd ~ 0-~. ~ ~~ OS~
A~OEt.~.E~3 ~ 6
zcl / NEt3 ~/
ZN~OS~ c1l2cl2 ~0~= o1s~f
1) N~u~ F,TIIF
C112C12
2) BF3.E~20
DMErT ~ oz
CA 02234484 1998-04-08
W O 97113776 PCTM~R96/01588
-
li2 CtPd 10'~.
~~0 ~" O ~cOEt, NEt3 ~~ 0,~_
D.~1ErT 10 D.~1 IrT
I ) llCI1O
Nai3113CN'
Cl ItC12 ~~ 0
2 ) I ICI ~lrOH 1 1 D ~r~loE
011
l~leONn 011
4 ~~ osl ~ ~~ ~
~J 12 Clt3CN N~ 13
A~ , ' ' ,
T.'~tSOTt
C112C12
T~mis ~1~
D:~IEPT ~'-0~0
U F3-E120
Cli2C12 J 14 DhlEPT
D ~l EPT ~~ '-OZ
112 Urd 10~/.
EtOH-AcOEI
~f ~o~~o c~c ~1 f
~N ~ D.~IEPT IMI NE13 / D.~lF I D~1EPT
o 17 16 OH
O ( C}12 C}12 1 ) 2 /
C}12CI'
RT.
I C~12Ci-i2NHZ
D;~lF Na8H3CN
C}13CN
R.T.
'- H2 C/Pd ~f
~~0 ~ 0 ~cOEl-EtOll ~O~
o ~ ~f o~o r~ N 20 4'-OH
~-~ ~ 4'-OH Z~. ~ }iN' D4h~oE~PT
CA 02234484 l998-04-08
W O 97/13776 PCT~FR96/01588
Elle se fait selon une méthodologie décrite (J. C. Florent et C. Monneret, J.
Chem. Soc. Chem. Comm. 1987, 1171 et B. Abbaci, J. C. Florent et C. Monneret, Bull.
Soc. Chim. Fr. 1989, 667) à partir du glucal 1. L'ion azoture est condensé pour fournir
un intermédiaire glycosidique 2 dont le OH anomérique est protégé par un groupement
silyl exclusivement en position ,B selon la technique décrite (C. Kolar et G. Kneissl
Angew. Chem. Int. Ed 29, 809 (1990)) pour fournir le mélange des 2 azides épimères: 3
et 4 séparables chromatographiquement à ce stade. Par déacétylation basique en
présence de méthylate de sodium, les diols en position 4, 6 sont obtenus: composés 5 et
12. Le composé diol azide ~ 5 est cyclisé en éthylidène classiquement à l'aide de l'acétal
de l'acétaldehyde en catalyse acide pour conduire au composé 6 dont l'azide se réduit en
amine 7 pour être protégée par un groupement benzyloxycarbonyle (Z) en composé 8,
nécessaire pour effectuer le couplage avec la déméthyl épipodophyllotoxine, elle-même
protégée sur son phénol en 4' par un groupe benzyloxycarbonyle, cet intermédiaire sera
appelé DMEPT4'-OZ. Ce couplage s'effectue dans un premier temps par clivage du
silyle protecteur par les ions F- suivi du traitement par l'éthérate de BF3 à basse
température dans le même milieu. La déprotection des groupements Z par
hydrogénolyse fournit le composé 10 de formule générale I (NRlR2 = ~NH2; XY =
OCH(CH3)OCH2). Le composé 11 de formule générale I (NRIR2 = ~NMe2; XY =
OCH(CH3)0CH7) s'obtient par méthylation de l'amine primaire précedente par action
du formol et du cyanoborohydrure de sodium.
L'intermédiaire glycosidique ~ide a 12 suit la même suite de réaction que son
épimère et fournit de façon identique le dérivé de couplage avec la DMEPT4'0Z à partir
de l'azide 3" a 13, dans ce cas le couplage est effectué plus facilement selon deux
techniques.
La première technique consiste à traiter le dérivé ribohexopyranoside 13 par le
triméthyl silyl trifluorométhane sulfonate (TMSOTf) à - 40~C dans du CH2C12. La
deuxième technique consiste à utiliser l'éthérate de BF3 dans le CH2C12 à - 15~C.
L'interrné~ ire obtenu 14 est alors réduit catalytiquement pour conduire à 15
correspondant à la formule générale I où NRlR2 = aNH2 et XY = OCH(CH3)OCH2.
La même méthylation que pour le composé ,~NH2 10, par le formol et le
CA 02234484 l998-04-08
W O 97/13776 PCT~R96/01588
cyanoborohydlul~ conduit au dérivé diméthylamino correspondant: formule générale I
(NR1R2 = aNMe2; XY = OCH(CH3)0CH2). A partir de l'amine primaire lS, il est
possible d'alkyler l'azote avec un dérivé halogéné comme l'iodoacétonitrile dans des
conditions faiblement basiques avec la triéthylamine dans la DMF et conduire au dérivé
S 16 de forrnule générale I où NRlR2 = aNHCH2CN; XY = OCH(CH3)0CH2 De la
même façon, le dérivé 17 s'obtient en formant le cycle morpholine par cyclisation de
l'éther diiodé sur le même interrnédiaire amine primaire 15 La même al~ylation avec la
iodoéthylamine protégée par un groupement Z sur le composé 15 fournit le dérivé 18
qui est réduit catalytiquement en 19 dérivé ~i~rnino correspondant à la formule générale
10 I avec NRlR2 = a-NHCH2CH2NH2; XY = OCH(CH3)0CH2.
Les composés de formule I où XY ne forme pas un cycle s'obtiennent de la façon
suivante ,~.esentés sur le schéma 2:
CA 02234484 l998-04-08
W O 97/13776 PCT/FR96/01588
SCHEMA 2
Ol~ OT~
Ho~~5~ ¦ T Cl / P idi ~ ~ osl
N~ 12 CH2C12 N3 21 N 22
CICH2COCI
Pvridine
CH2C12
N~AcO~O RMTNF3 ClAc~ DMEPT4'-OZ os!
N~ 27 DMEPT
4 OZ ~ 24 4'-OZ ~ 23
Annberlite410 \H2C12/l~-eOH
N3 \~ \
110~ 0 I~O--~O AcOÉt HO--~O
112 C/Pd NH26 ~'-OH 25 ~-oz
NEt3
H2N
HO ~,~_ O
J''1}2 DMEPT
OH
l'azide a silylé 12 est tosylé sélectivement avec le chlorure de tosyle dans la pyridine en
21, le tosylate de l'alcool primaire est échangé en dérivé iodo 22. A ce stade, I'alcool
5 secondaire en position 4 est protegé par un chloroacétate pour fournir le 2-desoxysucre
fonctionnalise 23 prêt à être condensé avec la DMEPT 4'-OZ dans les conditions
usuelles avec l'éthérate de BF3 dans le chlorure de méthylène à basse température.
L'intermediaire 24 obtenu, permet par passage sur la résine amberlite IRA410 traitée en
milieu basique de deprotéger le groupe chloroacetate en composé 25 et l'hydrogénation
10 catalytique finale permet en une étate de reduire la fonction azide en arnino 3"a, de
déprotéger la fonction Z en position 4' et de réduire le carbone 6" en méthyle pour
fournir le dérive 26 correspondant à la formule générale I où NR1R2 = 3"aNH2, X =
CA 02234484 l998-04-08
W 0 97/13776 PCT~R96/01588
- 11
OH; Y = CH3. L'intermédiaire 24 obtenu précéclemment peut réagir avec l'ion azoture
dans la DMF à telllLJel~L~ ambiante pour fournir le 3"aN3, 6"-N3 co~ olL~lt un
azidoacétate sur la position 4" 27, lequel peut se couper en alcool 4" par un traitement
similaire au précédent: le passage sur résine échangeuse Amberlite IRA 410 pour
S fournir le rli~7.i(1o~lcool 28. L'étape de réduction catalytique finale permet d'obtenir le
dérivé di~miné 29 correspondant à la formule générale I (où NR1R2 = 3"aNH2; X =
OH, Y = CH2NH2). Les sels formés à partir des composés azotés sont, par exemple,des chlorhydrates et se forment de façon classique en traitant une solution méthanolique
du composé azoté par une solution stoechiométrique par rapport aux sites à salifier
de m~th~n-l chlorhydrique prealablement dosé. Le chlorhydrate cristallisé peut
éventuellement s'obtenir par précipitation dans le milieu réactionnel par addition d'éther
éthylique.
A titre indicatif et pour illustrer les différentes étapes de la synthèse mais, de
façon non limitative, les exemples suivants sont donnés.
F~FMPr.F 1
Formule générale I: NRlR2 = ,~NH2; XY = OCH(CH3)OCH2
4'-Déméthyl-4-0(3 -amino-2,3-didésoxy-4,6-éthylidène-~-D-arabinohexopyranosyl)
épipodophyllotoxine (composé 10)
Stade I
Une solution de tri-O-acétyl-D-glucal I (50 g; 183 mmol) dans de l'eau (400 ml)
est chauffée durant 3 h à 80~C. Le milieu réactionnel est alors refroidi à 20~C avant
addition d'azoture de sodium (17,9 g; 275 mmol) et d'acide acétique (38 ml;
600 mrnol) Après agitation à température ambiante durant 24 h, le milieu est neutralisé
par du NaHCO3 (sel). La phase est extraite avec de l'acétate d'éthyle (3 x 500 ml). Les
phases organiques sont réunies, séchées sur MgSO4 puis concentrées sous pressionréduite. Ceci fournit 51 g de produit brut 2 immédiatement traité comme suit.
Caractéristiques: CCM: cyclohexane/AcOEt: 1/1; Rf = 0,43
CloH1sN3O6 M = 273
CA 02234484 l998-04-08
W O 97/13776 PCT~FR96/01588
- 12
St~
Tertbutyldiméthylsilyl-3 -azido-2,3 -didésoxy-4,6-di-O-acétyl-,B-D-arabino-
hexopyranoside 3 et terbutyldiméthylsilyl-3-azido-2,3-didéso~{y-4,6-di-O-acétyl-,B-D-
ribohexopyranoside 4
Aunesolutiondumélangebrut2(16,1 g;58mmol)obtenuaustade I dansdu
dichlorométhane anhydre (200 ml) préalablement refroidi à 0~C, sont ajoutés
successivement, sous argon, 6 g d'imidazole (87,8 mmol) et 13,24 g de chlorure de
tertbutyldiméthylsilyle (87,8 mmol). Après agitation durant 15 min à 0~C et 19 h à
20~C, le milieu réactionnel est versé dans 500 ml H20. La phase aqueuse est ex~aite
par du CH2C12 (200 ml) puis après séchage sur MgSO4, la phase organique est
concentrée sous pression réduite et le résidu (18,7 g) est chromatographié sur gel de
silice (cyclohexane/AcOEt: 9/1). On isole ainsi 10,7 g de 3 (sirop; 48 %) et 4,7 g de 4
(sirop; 21 %); cependant que des fractions intermediaires renferment un mélange de 3
et 4 (3,3 g; 15 %).
Caracteristiques: 3 CCM: cyclohexane/AcOEt: 4/1
Rf= 0,45
[a]D20 = - 10~ (c = 1,4; CHC13)
SM: m/z 405 (M +NH4)
C16H29N3O6Si M= 387
4 Rf= 0,52
[a]D20 = + 10~ (c = l; CHC13)
SM: mJz 405 (M + NH4)+
C16H2gN3O6Si M= 387
lH RMN 300MHz CDC13 ~
Dérivé 3: 0,13 (3H,s, SiCH3); 0,14 (3H, s SiCH3);
0,92 (9H, s, tBu); 1,72 (lH, m, J2a-1 = 9~5 Hz~
J2a-2e = 12,5 Hz, J2a-3 = 12,5 Hz,H2a)
2,05 (3H, s, COCH3); 2,15 (3H, s, COCH3); 2,25
(lH, ddd, J2e-1 = 1,5 Hz, J2e-2a= 12~5 Hz~ J2e-3
= 4,5 Hz, H2e); 3,55 - 3,62 (2 H, m, H3 et Hs);
CA 02234484 1998-04-08
4,10 (IH, dd, J6-s = 2,5 Hz, H6);
4, 20 (IH, dd, J6-s = 6 Hz, J6-6~ = 12 Hz, H6');
4,86 (IH, t, J = 9,5 Hz, H4);
4, 86 (lH, dd, Jl-2a = 9,5 Hz, Jl-2e = 1,5 Hz~ Hl)
s Dérivé 4: 0,1 (6H, s, Si(CH3)2); 0,88 (9H, s, tBu);
1,64 (IH, ddd, J2a-2e = 14 Hz, J2a-l = 8,5 Hz,
J2a-3 = 3,5 Hz, H2a);
2,03 (lH, ddd, J2e-2a = 14 Hz, J2e-1 = 2 Hz, J2e-3
= 4 Hz, H2e);
2,05 (3H, s, COCH3); 2,13 (3H, s, COCH3);
4,05 -4,12 (lH, m, Hs);
4,17 - 4,22 (3H, m, H3, H6 et H6 ); 4,89 (1 H, dd,
J4 5 = 9,5 Hz, J4 3 = 3,5 Hz, H4);
5,02 (lH, dd, Jl-2a = 8,5 Hz, Jl-2e = 2 Hz, Hl).
Stade 3
Tertbutyldiméthylsilyl-3-azido-2,3-didésoxy-,~-D-arabino-hexopyranoside S
A une solution du dérivé 3 (3 g; 7,7 mmol) obtenu au stade 2 dans du
méthanol anhydre (40 ml) est ajoutée, sous argon, une solution de méthanolate desodium lM (1,9 ml). Après réaction durant 1 h 30 à 20~C, le milieu réactiormel est
ramené à pH = 7 par addition de réside H+ (AmberliteTM IRC 50 S). Le mélange
réactionnel est filtré et le filtrat concentré sous pression réduite conduisant à 2,27 g de 5
(97 %).
Caractéristiques: CCM: cyclohexane/AcOEt: 2/1; Rf = 0,36
[(x]D20 = - 26~ (c = 1; CHC13)
SM: m/z 304 (M + H)+ 321 (M + NH4)+
Pf = 70 - 72~C
C12H2sN304Si M = 303
Sta~e 4
Tertbutyldiméthylsilyl-3-azido-2,3-didésoxy-4,6-O-éthylidène-,B-D-arabino-
hexopyranoside (6)
A une solution de 5 (0,20 g; 0,6 mmol) obtenu au stade 3 dans 5 ml
~U~ ~~IF~~E
CA 02234484 l998-04-08
W O 97/13776 PCT~R96/01588 14
d'acétonitrile, on ajoute 0,94 ml (6,6 mmol) du diéthylacétal de l'acétaldéhyde puis
15 mg d'acide paratoluènesulfonique (0,08 mmol). Le milieu réactionnel est agité à
température ambiante durant 1 h puis dilué par de l'acétate d'éthyle (~0 ml) avant lavage
par une solution d'hydrogénocarbonate de sodium (pH = 9) (20 ml) puis par de l'eau
5 (20 ml). La phase organique est séchée sur MgS04 puis concentrée sous pressionréduite pour fournir 0,25 g de produit brut. Une purification sur gel de silice
(cyclohexane/AcOEt: 9515) perrnet d'isoler 0,19 g de 6 pur (B6 %).
Caractéristiques: CCM: cyclohexane/AcOEt: 7/3; Rf= 0,89
r~]D20 =- 19~ (C I,1, CHC13)
SM: m/z 347 (M + NH4)+
C14H27N304Si M = 329
C H N
Calculé 5 1,04 8,26 12,75
Trouvé 51,64 8,43 12,51
15 Stade ~
Tertbutyldiméthylsilyl-3 -amino-2,3-didésoxy-4,6-0-éthylidène-~-D-arabino-
hexopyranoside (7)
A une solution de 6 (2 g; 6 mmol) obtenu au stade 4 dans 30 ml d'acétate
d'éthyle sont ajoutés 50 ~11 de triéthylarnine puis 0,5 g de charbon palladié à 10 %. Le
~0 milieu réactionnel est mis sous atmosphère d'hydrogène (pression atmosphérique).
Après agitation durant 6 h à température ambiante, le catalyseur est éliminé par filtration
et la phase organique concentrée sous pression réduite pour fournir 1,82 g de 7 pur
(98 %)~
Caractéristiques: CCM: cyclohexane/AcOEt: 1/1; Rf= 0,23
[a~D20 = - 28~ (C = 1,3; CHCI3)
SM: ml'z 304 (M + H)+
C14H2gN04Si M = 303
St~-le 6
Tertbutyldiméthylsilyl-3 -aminobenzyloxycarbonyl-2,3 -didésoxy-4,6-0-éthylidène-~-D-
30 arabino-hexopyranoside (8)
-
CA 02234484 1998-04-08
W O 97/13776 PCT~FR96/01588
Le chlorure de benzyloxycarbonyle (1,17 ml; 7,88 mmol) est ajouté, sous argon,
à une solution préalablement refroidie à 0~C de l'acétal 7 (1,82 g; 6 mmol) obtenu au
stade ~ dans un mélange de dichlorométhane (30 ml) et de triéthylamine (1,27 ml; 9,1
mmol) anhydres. Après agitation durant 8 h, le milieu réactionnel est versé dans 100 ml
- 5 H2O et la phase aqueuse est extraite par du CH2C12 (100 ml). La phase organique est
séchée sur MgSO4, concentrée sous pression réduite et le résidu est purifié par
chromatographie sur gel de silice (cyclohexane/AcOEt: 6/1 et 4/1) afin d'isoler 1,9 g de
8 (72 %).
Caractéristiques: CCM: cyclohexane/AcOEt: 1/1; Rf= 0,64
[a]D20 = - 27~ (c = 1,14; CHC13)
SM: m/z 438 (M + H)+
Pf= 102~C
C22H35No6si M = 437
C H N
Calculé 60,38 8,06 3,20
Trouvé 60,27 8,10 3,29
Stade 7
3-aminobenzyloxycarbonyl-2,3-didésoxy-4,6-O-éthylidène-~-D-arabino-
hexopyranoside de la 4'-benzyloxycarbonyl-épipodophylloto~cine (9)
Au sucre 8 (2,0 g; 4,57 mrnol) obtenu au stade 6 en solution dans du
dichlorométhane anhydre (100 ml) sont ajoutés 5,06 ml de fluorure de tétra-
butylammoniurn (solution 1,1 M dans le THF; 5,5 mrnol). Lorsque la disparition totale
de 8 est constatée par CCM (2 h d'agitation), le milieu réactionnel est refroidi à - 20~C.
On additionne alors successivement la DMEPT 4'-OZ (2,57 g; 4,8 mmol) puis 8,44 ml
de BF3.Et2O (68,6 mmol). Apres réaction durant 1 h à - 20~C, le milieu réactionnel est
verse dans 200 ml d'une solution de NaHCO3 saturée (addition de sels de NaHCO3)
(pH = 9). La phase organique est séchée sur MgSO4, concentrée sous pression réduite,
puis le résidu brut (6,2 g) est chromatographié sur gel de silice (CH2Cl2/Acétone: 98/2
puis 97/3) pour fournir 9 (2,1 g; 54 %).
-
CA 02234484 1998-04-08
- W O 97/13776 PCT~FR96/OlS88
16
Caractéristiques: CCM: CH2CI~/Acétone: 92/8; Rf= 0,61
[(x]D20 =- 74~ (c = 1,1; CHC13)
SM:m/z857(M+NH4)+
Pf= 175~C
C4sH4sNO 15 M = 839
C H N
Calculé 64,36 5,40 1,67
Trouvé 64,21 5,30 1,58
StZlllt? 8
10A une solution de 9 (0,28 g; 0,33 mmol) dans 20 ml d'acétate d'éthyle sont
ajoutés 30 !11 de triéthylamine puis 150 mg de charbon palladié a 10 %. Le milieu
réactionnel est mis sous atmosphère d'hydrogène (pression atmosphérique). Après
agitation durant 1 h 30 à température ambiante, le catalyseur est éliminé par filtration et
la phase organique concentrée sous pression réduite puis chromatographiée sur gel de
15silice (CH2C12/MeOH: 97/3 puis 95/5) pour fou~ir 172 mg du composé 10 pur (90 %).
(Recri~t~llic~tion dans CH2C12/pentane).
Caractéristiques: CCM: CH2C12/MeOH: 95/5; Rf= 0,31
[a]D20=- 120~ (c= 1,05; CHC13)
SM: ITI/Z 594 (M + Na)+ 610 (M+ K)+
Pf= 21g~ C
C2gH33NO1 1 M = 571
C H N
Calculé 60,94 5,82 2,45
Trouvé 60,45 5,78 2,58
lH RMN 300 MHz CDC13 ~ 1,36 (3H, d, J = SHz, CH3-CH); 1,51 (lH, m, H2~a);
2,05 (lH, m, H2~e); 2,88 (lH, m, H3); 3,02 (2H, m, H3~ et H4~); 3,28 (lH, m,
J2-1 = 5,2 Hz, H2); 3,30 (lH, m, Hs~); 3,57 (lH, t, J = 10 Hz, H6~a), 3,75 (6H, s,
OCH3); 4,15 (lH, dd, J = S Hz, J = 10 Hz, H6~e); 4,41 (lH, dd, J = 9 Hz, Hl la); 4,21
(lH,t,J=9Hz,Hllb);4,59(1H,d,J=5,2Hz,Hl);4,75(1H,q,J=SHz,H7~);4,85
(lH, dd, J = 9 Hz, J = 2 Hz, Hl~); 4,94 (lH, d, J = 3,3 Hz, H4); 5,98 (lH, d,
CA 02234484 1998-04-08
W O 97/13776 PCT~FR96/01588
17
OCHAO); 6,00 (lH, d, OCHgO); 6,24 (2H, s, H~ et H6~); 6,55 (lH, s, Hg); 6,75 (lH,
s, H5).
- Préparation du chlorl~ydrate
A l'amine 10 (61 mg; 0,10 mmol) en solution dans du dichlorométhane anhydre
(6 ml) est ajoutée une solution de méthanol chlorhydrique 0,098 M (1,09 ml;
0,106 mmol). Le milieu réactionnel est agité durant 10 minutt-s Le produit attendu est
précipité après addition d'éther (20 ml). On recueille 56 mg (86 %) du chlorhydrate de
10.
Car~ctérictiques: Pf= 230~ C
C29H32N~1 lCl M = 606
Essai de solubilité: 2,56 mg dans 0,3 ml d'eau
C = 0,014 M
F~FMPT F 2
Formule générale I (NR1R2 = ~BNMe2; XY = OCH(CH3)OCH2)
4'-Déméthyl-4-0(3-diméthylarnino-2,3-didesoxy-4,6-éthylidène-,B-D-
arabinohexopyranosyl) épipodophyllotoxine (Composé 11)
A une solution de 10 (0,19 g; 0,33 mmol) dans du dichlorométhane anhydre
(15 ml) sont ajoutés successivement du formaldéhyde (13,5 ~11) et du cyanoborohydrure
de sodium (85 mg). Après agitation durant 45 min à température arnbiante, ces mêmes
20 reactifs sont additionnés et la réaction est poursuivie durant 45 min. Le milieu
réactionnel est dilué par du CH2C12 (30 ml) et lavé par de l'eau (40 ml). La phase
organique est séchée sur MgSO4 et concentrée sous pression réduite. Le résidu est
chromatographié sur gel de silice (CH2C12/MeOH: 97/3). Ceci fournit 101 mg de 11 (51 %).
Caractéristiques: CCM: CH2C12/MeOH: 95/5; Rf = 0,4
[a]D20 =-121~ (c = 1; CHC13)
SM: m/z 600 (M + H)+
Pf= 270~C
C31H37NOll M 599
1HRMN 300 MHz CDC13 ~ 1,38 (3H, d, J = 5 Hz, CH3 - CH); 1,55 (lH, m, H2~a);
CA 02234484 l998-04-08
W O 97/13776 PCT~FR96/01588
- 18
1,96 (lH, m, H2~a); 1,96 (lH, m, H2~e); 2,33 (3H, s, CH3 - N); 2,91 - 2,82 (lH, m,
H3); 2,91 - 2,82 (lH, m, H3~); 3,35 - 3,25 (lH, m, Hs~); 3,38 (lH, t, J4~ 5~ = 9 Hz,
J4~ 3~ = 9 Hz, H4 ); 3,58 (lH, t, J6~a-s~ = 10 Hz, J6~a-6~e = 10 Hz, H6"a); 3,75 (3H~
s, CH3 O); 4,16 (lH, dd, J6~e-5~ = 5 Hz, J6~e-6~a = 10 Hz, H6~e); 4,21 (lH, t, Jgb ga
= 9 Hz, Jgb 3 = 8 Hz, Hgb) ; 4,42 (1H, dd, Jga gb = 9 Hz,
Jga 3 = 10,5 Hz, Hga); 4,59 (lH, d, J1-2 = 5,2 Hz, H1); 4,74 (lH, q, J = 5 Hz, CH -
CH3); 4,82 (lH, dd, Jl~-2~a= 9,5 Hz, Jl~-2~e = 2 Hz, Hl~); 4,95 (lH, d, J4 3 = 3,2
Hz, H4); 5,97 (lH, d, OCHAO); 6,00 (lH, d, OCHgO); 6,25 (2H, s, H2, et H6~); 6,55
(lH, s, Hg); 6,76 (lH, s, Hs).
10 Pré~r~tion du chlorhvdrate
A l'amine 11 (101 mg; 0,17 mmol) en solution dans du dichlorométhane
anhydre (7 ml) est ajoutée une solution de méthanol chlorhydrique 0,098 M (1,72 ml;
0,17 mrnol). Le milieu réactionnel est agité durant 10 min~ltP~. Le produit attendu est
précipité après addition d'éther (20 ml). On recueille 86 mg (81 %) du chlorhydrate de
15 11.
Caracteristiques: Pf= 199~C
C3 1 H33NO 1 1 Cl M = 635
Essai de solubilité: 2,5 mg dans 0,1 ml d'eau
C = 0,03~ M
F.XFMPT.F 3
Formule générale I: NRlR2 = a-NH2; XY = OCH(CH3)OCH2
4'-Déméthyl-4-0-(3 -amino-2,3 -didésoxy-4,6-éthylidène~-D-ribohexopyranosyl)
épipodophyllotoxine (composé 19)
St~e 1
Tertbutyldiméthylsilyl-3-azido-2,3-didesoxy-,B-D ribo hexopyranoside (composé 12)
De façon similaire, au stade 3 de l'exemple l, mais en tltili~nt le composé 4, on
obtient le composé 12 qui est engagé dile.,~e,llent dans le stade 2.
st~e 2
Tertbutyldiméthylsilyl-3-azido-2,3-didésoxy-4,6-O-éthylidène-,B-D-ribo hexopyranoside
(13)
CA 02234484 l998-04-08
W O 97/13776 PCT~FR96/01588 19
A une solution de 12 (0,25 g; 0,8 mmol) obtenu au stade 1 dans 10 ml
d'acétonitrile, on ajoute 1,1 ml (8 mmol) du diéthylacétal de l'acétaldéhyde puis 52 mg
- d'acide c~mphorsulfonique (0,24 mmol~. Le milieu réactionnel est agite à température
ambiante durant 9 h puis dilué par de l'acétate d'éthyle (30 ml) avant lavage par une
5 solution d'hydrogénocarbonate de sodium (pH = 9) puis par de l'eau (30 ml). La phase
organique est séchée sur MgS04 puis concentrée sous pression réduite pour fournir
0,3 g de produit brut. Une purification sur gel de silice (cyclohexane/AcOEt: 15ll)
permet d'isoler 0,15 g de 13 pur (55 %).
Caractéristiques: CCM: cyclohe~cane/AcOEt: 4/1; Rf= 0,77
[a]D20 =- 35~ (c = I,1 CHC13)
C14H27N3O4Si M = 379
Stade 3
3-azido-2,3-didésoxy-4,6-O-éthylidène-~-D-ribo-hexopyranoside de la 4'-
ben2yloxycarbonyl-épipodophyllotixne (composé 14)
15 lère voie de synthèse:
A un mélange de DMEPT 4'-OZ (438 mg; 0,82 mmol), de 13 (270 mg;
0,82 mmol) obtenu au stade 2 et de tamis moleculaire 4 A (1,5 g) dans du
dichlorométhane anhydre (30 ml) refroidi à - 40~C, est ajouté le triméthylsilyl-trifluorométh~n~sl~lfonate (TMSOTf) (446 !11; 2,46 mmol). Après réaction durant I h
15 à - 40~C, le milieu réactionnel est neutralisé par de la triéthylamine (347 ~1), filtré
puis lavé par une solution saturée de NaCl (20 ml). La phase organique est séchée sur
MgSO4, concentrée sous pression réduite puis, le résidu brut est chromatographié sur
gel de silice (cyclohexane/AcOEt: 65/35) pour fournir 14 (260 mg; 45 %).
2ème voie de synthèse:
A un mélange de DMEPT 4'0Z (1,85 g; 3,46 mmol), de 13 (1,20 g; 3,64 mmol)
obtenu au stade 2 dans du dichlorométh~ne anhydre (100 ml) refroidi à - 15~C, est
a~outé l'éthérate de trifluorure de bore (BF3.Et2O) (425 ~Ll; 3,46 mmol). Après réaction
durant 2 h à - 15~C, le milieu réactionnel est dilué par 100 ml de C~I2C12 puis, versé
dans 200 ml dlune solution de NaHCO3 saturée. La phase organique est séchée sur
MgSO4, concentrée sous pression réduite puis, le résidu brut (2,9 g) est
CA 02234484 1998-04-08
W O 97/13776 PCT~FR96/01588
chromatographié sur gel de silice (cyclohe~cane/AcOEt: 65/35) pour foumir 14 (1,19 g;
47 %) (recristallisation Et20/hexane).
Caractéristiques: CCM: cyclohe.Yane/AcOEt: 6/4; Rf= 0,41
cyclohe~ane/AcOEt: 65/3; Rf= 0,77
[a]D20=- 105~ (c = 1,05; CHC13)
SM: m/z 749 (M + NH4)+
Pf = 139~C
C37H37N3O 1 3 M = 731
St~ 4
3-amino-2,3-didésoxy-4,6-O-éthylidène-~-D-~Libo-hexopyranoside
d'épipodophyllotixine 15
A une solution de 14 (110 mg; 0,15 mmol) obtenu au stade 3 dans un mélange
~ de 10 ml d'éthanol et de 5 ml d'acétate d'éthyle sont ajoutés 20 ~LI de triéthylamine puis
20 mg de charbon palladié à 10 %. Le milieu réactiolmel est mis sous atmosphère
15 d'hydrogène (pression atmospherique). Après agitation durant 2 h à température
ambiante, le catalyseur est éliminé par filtration et la phase organique concentrée sous
pression réduite puis chromatographiée sur gel de silice (CH2C12/MeOH: 97/3 puis
95/5) pour fo~3rnir 63 mg de 15 pur (7 2 %).
Caracteristiques: CCM: CH~CI~/MeOH: 95/5; Rf= 0,39
[a]D~0=- 100~ (c= 1,05; CHC13)
SM: m/z 572 (M + 1) 589 (M + NH4)+
Pf= 217~C
C2gH33NO1 1 M = 571
lHRMN 300 MHz CDC13 o: 1,35 (3H, d, J = 5 Hz, CH3-CH); 1,73 (lH, m, H2~a);
1,90 (lH, m, H2~e); 2,83 (lH, m, H3); 3,22 (lH, dd, J2-1 = 5,2 Hz, J2-3 = 14 Hz, H2);
3,42 (lH, dd, J4~ 3~ = 9,5 Hz, J4~ 5~ = 3,5 Hz, H4~); 3,74 (6H, s, OCH3); 3,49 - 3,60
(2H, m, H3~ et H6~); 3,94 - 4,02 (lH, m, Hs~); 4,10 - 4,20 (2H, m, H6~ et Hl lb); 4,42
(lH~ dd~ Jlla-3 = 9Hz, Jlla-llb = 9,5 Hz, Hlla); 4,57 (lH, d, Jl-2 = 5,2 Hz, Hl);
4,78(1H,q,J=5Hz,H7~);4,91(1H,d,J43=3,4Hz,H4);5,38(1H,dd,J1~2~e=2
Hz, Jl~-2~a= 9 Hz, Hl~); 5,93 l~lH, s, OCHAO); 5,97 (lH, s, OCHgO); 6,25 (2Ht s,
CA 02234484 l998-04-08
W O 97/13776 ~CT~R96/01588
21
H2~etH6~);6,51 (lH,s,Hg);6,86(1H,s,Hs).
Préparation du chlorhydrate
- A l'amine lS (50 mg; 0,087 mmol) obtenue au stade 4 sont a~outés 890 ,ul d'une
solution de mçth~nl l chlorhydrique 0,098 M (0,087 mmol). Après agitation durant 10
min~ltec et addition d'éther (10 ml), les cristaux ou chlorhydrate (52 mg, 98 %) sont
obtenus par filtration.
Caractéristiques: Pf = 1 75~C
C29H34N~l 1 Cl M = 607
Essai de solubilité: 2,2 mg dans 0,2 ml d'eau
C = 0,02 M
F~.MPr F. 4
Formule générale I: NR1R2 = a-NMe2; XY = OCH(CH3)OCH2
4'-Déméthyl-4-0(3 -N,N-diméthylamino-2,3 -didésoxy-4,6-O-éthylidène-~-D-ribo-
hexopyranosyl) épipodophyllotoxine 20
A une solution de lS (29 mg: 0,05 mmol) obtenu au stade 4 de l'exemple 3 dans
ml d'acétonitrile sont ajoutés successivement du formaldéhyde ( 10,3 ~ll) et du
cyanoborohydrure de sodium ( 12 mg). Après agitation durant 2 h à température
ambiante, le milieu réactionnel est dilué par du CH2C12 (20 ml) et lavé par de l'eau
(20 ml). La phase organique est séchée sur MgSO4 et concentrée sous pression réduite.
20 Le résidu est remis en réaction dans les mêmes conditions et apres un traitement
identique chromatographié sur gel de silice (CH ~Cl2/MeOH : 97/3). Ceci fournit
29 mg de 20 (95 %).
Caractéristiques: CCM: CH2Cl2/MeOH: 95/5; Rf= 0,5
[a]D20=- 85~ (c= 1,06; CHCl3)
SM: m/z 600 (M + H)+
Pf= 140~C
C31H37NOl 1 M = 599
lH RMN 300 MHz CDC13 o: 1,35 (3H, d, J = 5 Hz); 1,55 (lH, m, H2~a); 2,20 (lH,
m, H2~e); 2,36 (3H, s, CH3 N); 2,62 (lH, m, 2,62, H3~); 2,85 (lH, m, H3); 3,23 (lH,
dd, J2-1 = 5,2 Hz, J2-3 = 14 Hz, H2); 3,52 (lH, dd, J4~ 3 = 3 Hz, J4~ 3~ = 9 Hz, H4~
CA 02234484 l998-04-08
W O 97/13776 PCT~R96/01588
22
3t61 (lH, t, J6~a-6~e = 10 Hz, J6~a-5~ = 10 Hz, H6~a); 3,75 (3H,s, CH3 O); 4,22 - 4,02
(lH, m, Hs~); 4,22 - 4,02 (lH, m, H6~e); 4,22 - 4,02 (lH, m, Hgb); 4,43 (IH, dd, Jga
gb = 9 Hz, Jgb 3 = 10,5 Hz, Hgb); 4,62 - 4,57 (lH, m~Jl-2 = 5,2 Hz, Hl); 4,62 - 4,57
(3H, m, J = 5 Hz, CH-CH3), 4,88 (lH, d, J4 3 = 3,4 Hz, H4); 5,96 (lH, d, OCHA ~);
5,98 (lH, d, OCHg ~); 6,25 (2H, s, H2 et H6~); 6,52 (lH, s, Hg); 6,80 (lH, s, Hs).
Préparation du chlor~ydrate
A l'amine 20 (59 mg; 0,1 mmol) en solution dans du méthanol anhydre (2 ml)
est ajoutée une solution de methanol chlorhydrique 0,098 M (1 ml; 0,1 mmol). Le
milieu réactionnel est agité durant 10 minl-t~s Le produit attendu est précipité après
addition d'éther (20 ml). On recueille 33 mg (53 %) de cristaux.
Caractéristiques: Pf = 1 50~C
C31H38N~l lCl M = 635
Essai de solubilité: 2,6 mg dans 0,1 ml d'eau
C = 0,04 M
F~FrvIpr F. S
Formule générale ~: NR1R2 = ~-NHCH2CN; XY = OCH(CH3)OCH2
4'-Deméthyl-4-0-(3 -cyanométhylamino-2,3 -didésoxy-4,6-éthylidène-,B-D-ribo-
héxopyranosyl) épipodophyllotoxine (Composé 16)
A une solution de 15 (120 mg; 0,21 mmol) obtenu au stade 4 de l'exemple 3
dans 4 ml de diméthylformamide sont ajoutés 200 ~LI de triéthylamine (1,47 mmol) puis
100 ~Ll d'iodoacétonitrile (1,47 mmol). Le milieu réactionnel est agité durant 20 h à
température ambiante puis dilué par de l'acétate d'éthyle (30 ml) avant lavage par de
l'eau (4 x 30 ml). La phase organique est séchée sur MgSO4, concentrée sous pression
réduite puis chromatographiée sur gel de silice (CH2C12/acétone: 92/8) pour fournir
71 mg de 16 pur (55 %).
Caractéristiques: CCM: CH2C12/MeOH: 95/5; Rf= 0,25
[a]D20 =- 86~ (c = 0,80; CHCl3)
SM: m/z 611 (M + H)+ 628 (M + NH4)+
C31H34N2~ll M=610
CA 02234484 l998-04-08
W O 97/13776 PCT~R96/OlS88
23
lH RMN 300 MHz CDC13 ~: 1,35 (3H, d, J = SHz, CH3CH); 1,69 (lH, m, J2~a-1 =
9,5 Hz, J2~a-2~e = 13 Hz, J2~a-3 = 3 Hz, H2"a~; 1,93 (lH, m, H2~e); 2,86 (lH, m,H3); 3,25 (lH, dd, J2-1 = 5,2 Hz, J2-3 = 14 Hz, H2); 3,51 - 3,56 (2H, m, H4~ et
H6~a); 3,56 (2H, m, CH2CN); 3,76 (6H, s, OCH3); 3,88 (lH, m, Hs~); 4,17 (2H,
t, Jlla-3 = 8 Hz, Jlla-llb = 10 Hz, H3~ et Hllb); 4,14 (lH, dd, J6a~-6~b = 10 Hz,
J6"b-5" = 5 Hz, H6~b); 4,41 (lH, dd, Jl la-3 = 9 Hz, Jl la-l lb = 10 Hz, Hl la); 4~60
(lH, d, Jl-2 = 5,3 Hz, Hl); 4,75 (lH, q, J = 5 Hz, H7~); 4,90 (lH, d, J4 3 = 3,3 Hz,
H4); 5,17 (lH, dd, Jl"-2"c = 2 Hz, Jl~-2~a = 9,5 Hz, Hl~); 5,99 (lH, s, OCHAO);
6,00 (lH, s, OCHgO); 6,25 (2H, s, H2 et H6~); 6,54 (lH, s, Hg); 6,79 (lH, s, Hs).
10 F~F.MPLF 6
Formule générale I: NRlR2 = a-morpholino; XY = OCH(CH3)0CH2
4'-Déméthyl-4-0-(3 -N-morpholino-2,3-didesoxy-4,6-éthylidène-,~-D-ribo-
hexopyranosyl) épipodophyllotoxine (Composé 17)
A une solution de 15 (60 mg; 0,10 mmol) obtenu au stade 4 de l'exemple 3 dans
2 ml de diméthylformamide sont ajoutés 58 ~11 de triéthylarnine (0,42 mmol) puis
512 mg de di-iodoéthyléther (1,57 mmol). Le milieu réactionnel est agité durant 96 h à
température ambiante et à l'obscurité puis, dilué par de l'acetate d'éthyle (30 ml) avant
lavage par de l'eau (4 x 30 ml). La phase organique est séchée sur MgS04, concentrée
sous pression réduite puis chromatographiée sur gel de silice (CH2Cl2/acétone: 92/8)
pour fournir 46 mg de 17 pur (68 %).
Caractéristiques: CCM: CH2C12/acetone: 92/8; Rf= 0,31
[a]D20 = - 98~ (c = 1,04; CHC13)
C33H3gN012 M = 641
lH RMN 300 MHz CDC13 ~: 1,33 (3H, d, J = SHz, CH3CH); 1,55 (lH, m, J2~a-1~ =
9,5 Hz, J2~a-2~e= 13 Hz, J2~a-3~ = 3 Hz, H2"a); 2,15 (lH, m, H2~e); 2,84-2,90 (5H,
m, CH2N et H3); 2,80 (lH, dd, J3~ 4~ = 3Hz, J3~-2~a = 3 Hz, J3~-2~e = 3 Hz, H3~
3,23 (lH, dd, J2-1 = 5,2 Hz, J2-3 = 14 Hz, H2); 3,48 (lH, t, J6~a-6~b = 12 Hz, J6"a-s"
= 12 Hz, H6~a); 3,57 (lH, dd, J3~ 4~ = 3Hz, J4~ 5~ = 9 Hz, H4~); 3,66-3,76 (lOH, m,
OCH3 et OCH2); 4,08-4,16 (2H, m, Hs~ et H6"b); 4,20 (lH, t, Jl la-3 = 8 Hz, Jl la-
1lb = 9 Hz, Hlla); 4,43 (lH, dd, Jllb-3 = 9 Hz, Jllb-lla= 9 Hz, H1lb); 4,57-4,62
CA 02234484 l998-04-08
W O 97/13776 PCT~FR96/01588
24
(2H, m, H1 et H7~); 4,89 (lH, d, J4 3 = 3,4 Hz, H4); 5,20 (lH, dd, Jl~-2~e = 2 Hz,
Jl~-2~a= 9,5 Hz, Hl~); 5,97 (lH, s, OCHAO); 6,00 (lH, s, OCHgO); 6,25 (2H, s, H2et H6~); 6,54 (lH, s, Hg); 6,73 (lH, s, Hs).
F~FMPT E 7
Formule générale I: NRlR2 = a-NH2(CH2)2NH2; XY = OCH(CH3)OCH2
4'-Déméthyl-4-0[3-(2-Aminoéthylamino) 2,3-didésoxy-4,6-éthylidène-~-D-ribo-
hexopyranosyl] épipodophyllotoxine 18
A une solution de 15 (173 mg; 0,30 mmol) obtenu au stade 4 de l'exemple 3
dans 10 ml de diméthylfom~ le sont ajoutés de la triéthylamine (127 ~Ll; 0,91 mmol)
et du N-benzyloxycarbonyl-2-iodoéthyl-amine (0,28 g; 0,91 mmol). Le milieu
réactionnel est agité durant 5 jours à température ambiante puis, dilué par de l'eau
(30 ml). Après extraction de l'acétate d'éthyle (30 ml) lavage à l'eau (5 ~c 20 ml), la phase
organique est séchée sur MgSO4, concentrée sous pression réduite, puis
chromatographiée sur gel de silice (CH2C12/MeOH: 97/3) pour fournir 155 mg de 18pur (68 %).
Caractéristiques: CCM: CH2C12/MeOH: 95/5; Rf = 0,70
[a]D20 = - 74~ (c = 1,17; CHC13)
SM: }n/z 749 (M + H)+
C3gH44N2O 1 3 M = 748
qui est engagé directement dans l'étape suivante de débenzylation:
A une solution de 18 (0,15 g; 0,20 mmol) dans un mélange d'acétate d'éthyle et
d'éthanol (10 ml, 1/1) sont ajoutés de la triéthylamine (30 ,ul) puis du charbon palladié a
10 % (0,1 g). Le milieu réactionnel est mis sous atmosphère d'hydrogène (pression
atmosphérique) .
~25 Après agitation durant 1 h 30 à température ambiante en présence d'hydrogene à
pression atrnosphérique, le catalyseur est éliminé par filtration et la phase organique
concentrée sous pression réduite et chromatographiée sur gel de silice
(CH2C12/MeOH(NH3): 97/3) pour fournir 107 mg (84 %) de 19.
Caractéristiques: CCM: CH2C12/MeOH(NH3): 95/5; Rf= 0,22
[a]D20=- 77~ (c= 1; CHC13)
CA 02234484 l998-04-08
W 0 97/13776 PCT/FR96/01588
Pf= 130~ C
C31H38NOl 1 M = 614
lH Rl~ 300 M[Hz CDC13~: 1,33 (3H, d, J = 5 Hz); 1,5~ (lH, m, H2~a); 2,15 (lH, m,H2~e); 2,33 (3H, m, NH2 et NH échangeables); 2,84-2,90 (SH, m, CH2-N et H3); 3,20
(lH~ dd~ J3~ 4~ = 3 Hz, J3~-2~a= 3 Hz, J3~-2~e = 3 Hz, H3~); 3,23 (lH, dd, H2), 3,48
(lH~ t, Jb~a-6~b = 12 Hz, J6"a-s" = 12 Hz, H6~a); 3,57 (lH, dd, J4~ 3~ = 3 Hz, J4~ 5~ =
9 Hz, H4~); 3,75 (6H, s, OCH3); 4,08-4,16 (2H, m, Hs~ et H6~b); 4,20 (lH, t, J1 lb-3
= 8, Jl lb-l la = 9 Hz, Hl lb); 4,43 (lH, dd, Jl la-3 = 9 Hz, Jl la-l lb = 9 Hz, Hl la~;
4,57-4,62 (2H, m, H1 et H7~); 4,89 (lH, d, J4 3 = 3,4 Hz, H4); 5,20 (lH, dd, J1~-2~e =
2Hz, J1~-2~a = 9,5 Hz, H1~); 5,97 et 6,00 (2H, d, OCH2O); 6,25 (2H, s, H2~ et H6~);
6,54(1H,s,Hg);6,73(1H,s,Hs).
Plc:p~.dLion du chlorhydrate
A la di-amine 19 (64 mg; 0,10 mmol) en solution dans du méthanol anhydre
(3 ml) est ajoutée une solution de méthanol chlorhydrique 0,098 M (2,13 ml;
0,21 mmol). Le milieu réactionnel est agité durant 10 minl-tes Le produit attendu est
précipité après addition d'éther (20 ml). On recueille 50 mg (73 %) du chlorhydrate.
Caractéristiques: Pf = 1 70~C
C31H39N2~l 1Cl~ M = 649
Essai de solubilité: 2,0 mg dans 0,05 ml d'eau
C = 0,06 M
FXF.MPT F 8
Formule générale I: NRlR2 = a-NH2; X = OH; Y = CH3
4'-Déméthyl-4-0(3-amino-2,3,6-tridésoxy~-D-ribo-hexopyranosyl) épipodophyllotoxine
(composé 26)
25 Stade 1
Tertbutyldiméthylsilyl-3-~ido-2,3-didésoxy-6-0-tosyl-~-D-ribo-hexopyranoside 2 1
Une solution de chlorure de tosyle (1,55 g; 8,15 mmol) dans la pyridine (10 ml)
est ajoutée, goutte à goutte, à une solution du diol 12 (2,06 g; 6,79 mmol) obtenu au
stade 1 de l'exemple 3 préalablement refroidie à 0~C. Après agitation à la même
le~ dlLlre durant 1 h, puis durant 18 h à 20~C, le milieu réactionnel est dilué par du
CA 02234484 l998-04-08
W O 97/13776 PCT~FR96/01588
26
dichlorométhane (100 ml). La phase organique est lavée par de l'eau (2 x 100 ml),
séchée sur MgSO4 et concentrée sous pression réduite. Le résidu est purifié sur gel de
silice (cyclohexane/EtOAc: 8/2). Ceci fournit ~,0 ~ g de 21 (65 %).
Caractéristiques: CCM: cyclohexane/AcOEt: 2/1; Rf= 0,46
SM: m/z 475 (M + H)+
C 1 gH31N3O6SSi M = 457
St~le 2
Tertbutyldiméthylsilyl-3-azido-6-iodo-2,3,6-tridésoxy-~B-D-ribo-hexopyranoside 22
Le composé 21 (2,02 g; 4,42 rnmol) obtenu au stade 1 en solution dans 120 ml
d'acétone est chauffé au reflux durant 72 h en presence d'iodure de sodium (2,65 g;
17,68 mmol). Après refroicliccement, le milieu réactionnel est concentré sous pression
réduite (30 ml) puis dilué par du dichlorométhane (100 ml). Après lavage par unesolution aqueuse de thiosulfate de sodium à 10 %, puis séchage sur MgSO4 et
évaporation sous pression réduite, on obtient un residu qui est purifie sur silice
(cyclohexane/EtOAc: 8/2) foll~ic~nt 1,5 g (82 %) de 22.
Caractéristiques: [a]D20 =- 30~ (c = 1,06; CHCl3)
SM: m/z 431 (M + NH4)+
C12H24N3O3ISi M = 413
C H N
Calculé 34,87 5,81 10,17
Trouvé 35,07 5,76 10,25
Stade 3
Tertbutyldiméthylsilyl-3 -azido-6-iodo-4-O-chloroacétyl-2,3,6-tridésoxy-,B-D-ribo-
hexopyranoside (composé 23).
Du chlorure de chloroacétyle (396 ml; 5 mmol) est ajouté à une solution de 22
(1,03 g; 2,5 mmol) obtenu au stade précédent dans un mélange de dichlorométhane
(20 ml) et de pyridine (404 ~11; 5 mmol). Après 1 h d'agitation à - 10~C, le milieu
réactionnel est dilué par du CH2Cl2 (30 ml) et lavé par de l'eau (3 x 20 ml). Untraitement habituel, suivi d'une chromatographie sur silice (cyclohexane/EtOAc: 10/1)
livre 1,1 g (90 %) de composé 23.
) CA 02234484 1998-04-08
27
Caractéristiques: [a]D20 = - 14~ (c = 1,03; CHC13)
SM: m/z 507 (M + NH4)+
Cl~H2sN3O~CHSi M = 489
C H N
s Calculé 34,35 5,11 8,59
Trouvé 34,69 5,16 8,22
Stade 4
4'-Déméthyl-4-0(3-azido-6-iodo-2,3,6-tridésoxy-~-D ribohexopyranosyl) épipodo-
phyllotoxine (composé 24)
A un mélange de DMEPT 4'-OZ (1 g; 1,85 mmol), de 23 (1 g; 2,04 mmol)
du stade précédent dans du dichlorométhane anhydre (100 ml) refroidi à - 15~C, est
ajouté l'éthérate de trifluorure de bore (BF3.Et20) (455 1ll; 3,7 mmol). Après réaction
durant 5 h (- 15~C ~ 0~C), le milieu réactionnel est dilué par du CH2C12 (100 ml) puis,
versé dans une solution de NaHCO3 saturée (200 ml). La phase organique est séchée sur
MgSO4, concentrée sous pression réduite. Le résidu brut est chromatographié sur gel de
silice (cyclohexane/AcOEt: 7/3) pour fournir 24 (0,8 g; 48 ~/O).
Caractéristiques: CCM: cyclohexane/AcOEt: 1/1; Rf = 0,58
[a]D20 =- 85~ (c= 1,26; CHC13)
SM: m/z 909 (M + NH4)+
Pf = 143~C
C37H3sN3O13ClI M = 891
Stade S
4'-Déméthyl-4-0(3-Azido-6-iodo-2,3 ,6-tridesoxy-,~-D-ribo-hexopyranosyl)4'-benzyloxy
carbonyl épipodophyllotoxine (composé 25)
A une solution de l'azido-glycoside 24 (257 mg; 0,29 mrnol) dans un
mélange CH2Cl2/MeOH (15 ml, 2/1) est ajoutée de la résine OH- (AmberliteTM IRA
410). Après réaction durant 3 h à 20~C, le milieu réactionnel est filtré puis concentré
sous pression réduite pour fournir 0,22 g de 25 pur (94%).
Caractéristiques: CCM: cyclohexane/AcOEt: 1/1; Rf= 0,46
[a]D20 =- 57~ (c = 1,02; CHCl3)
,0~IFI~E
CA 02234484 l998-04-08
W O 97/13776 PCT~FR96/01588
28
SM: m/z 833 (M + NH4)+
Pf= 115~C
C3sH34N3O12I M = 815
St~ 6
4'-Déméthyl-4-0(3-amino-2,3,6-tridesoxy-,B-D ribohexopyranosyl) épipodophyllotoxine
(composé 26)
A une solution de 25 (0,20 g; 0,25 mmol) du stade précédent dans 10 ml
d'acétate d'éthyle sont ajoutés de la triéthylamine (100 ,ul) puis du charbon palladié a
10 % (0,2 g). Après agitation durant 30 h à température arnbiante en présence
d'hydrogène à pression atmosphérique, le catalyseur est éliminé par filtration et la phase
organique concentrée sous pression réduite puis chromatographiée sur gel de silice
(CH2C12/MeOH: 92/8) pour fournir 50 mg (38 %) de 26.
Caractéristiques: CCM: CH2C12/MeOH: 90/10; Rf= 0,33
[a]D20 = - 90~ (c = 0,5; CHC13)
SM:m/zS30(M+H)+
Pf= 140~C
C27H31NOlo M = 529
P~paldtion du chlorhydrate
A l'amine 26 (70 mg; 0,14 mmol) précé~lente en solution dans du methanol
anhydre (6 ml) est ajoutée une solution de méthanol chlorhydrique 0,098 M (1,44 ml;
0,14 mmol). Le milieu réactiormel est agité durant 10 minutes. Le produit attendu est
précipité après addition d'éther (20 ml). On recueille 40 mg (53 %) de chlorhydrate
cri~t~ é
Caractéristiques: Pf = 164~C
C27EI32NO l oCl M = 565
Essai de solubilité: 2,6 mg dans 0,2 ml d'eau
C = 0,02 M
FXFMPT F.9
Formule générale I: NR1R2 = a-NH2; X = OH; Y - CH2NH2
4'-Démethyl-4-0-(3,6-diamino-2,3,6-tridésoxy-~-D ribohexopyranosyl) epipodo-
CA 02234484 1998-04-08
29
phyllotixine (composé 29)
Stade 1
4 ' -Déméthyl-4 ' -benzyloxycarbonyl-4-0(3,6-diazido-4-azidoacétyl-2,3,6-tndesoxy-,~ -D
ribo-hexopyranosyl) épipodophyllotoxine (composé 27)
A une solution de 24 (0,45 g; 0,51 mmol) obtenu au stade 4 de l'exemple 8 dans
10 ml de dimethylformamide est ajouté de l'azoture de sodium (0,1 g; 1,5 mmol). Le
milieu réactionnel est agité durant 64 h à température ambiante, dilué par de l'eau (30
ml) et de l'acétate d'éthyle (30 ml). La phase organique est lavée par de l'eau (4 x 20
ml), séchée sur MgSO4, concentrée sous pression réduite et chromatographiée sur gel
0 de silice (cyclohexane/AcOEt: 7/3) pour fournir 0,36 g de 27 (90 %).
Caractéristiques: CCM: cyclohexane/AcOEt: 6/4; Rf= 0,44
[a]D20 = - 64~ (c = l; CHC13)
SM: m/z 831 (M + NH4)+
Pf= 120~C
C37H35NgO 13 M = 813
Stade 2
4' -Déméthyl-4' -benzyloxycarbonyl-4-0(3,6-diazido-2,3,6-tridésoxy-,~-D-ribo-
hexopyranosyl) épipodophyllotoxine (composé 28)
A une solution de l'azido-glycoside 27 (70 mg; 0,08 mmol) dans 3 ml d'un
mélange CH2C12/MeOH (2/1, v/v) est ajoutée de la résine OH: (AmberliteTM IRA 410).
Après réaction durant 5 h à 20~C, le milieu réactionnel est filtré puis concentré sous
pression réduite pour fournir 59 mg de 28 pur (94 %).
Caractéristiques: CCM: cyclohexane/AcOEt: 6/4; Rf = 0,25
[a]D20 =- 53~ (c= 1,04; CHC13)
Pf = 125~C
C3sH34N6ol2I M= 730
Stade 3
4'-Déméthyl-4-0(3,6-diamino-2,3,6-tridesoxy-,~-D-ribo-hexopyranosyl)
épipodophyllotoxine (composé 29)
A une solution de 28 (0,11 g; 0,15 mmol) du stade précédent, dans un mélange
~ ~','' L' ~ irlE~
CA 02234484 l998-04-08
- W O 97/13776 PCT~FR96/01588
d'acétate d'éthyle et d'éthanol (10 ml, 1/1) sont ajoutes de la triéthylamine (20 ~11) puis
du charbon palladié à 10 % (70 mg). Après agitation durant 16 h à température arnbiante
en présence d'hydrogène à pression atmosphérique, le catalyseur est éliminé par
filtration et la phase organique concentrée sous pression réduite pour fournir 78 mg
5 (9S %) de 29~
Caractéristiques: CCM: CH2Cl2/MeOH(NH3): 90/10; Rf= 0,06
C27H32N2Olo M = 544
Préparation du Oichlorlurdrate
A la di-arnine 29 (78 mg; 0,14 mmol) précéderlt~ en solution dans du méthanol
10anhydre (2 ml) est ajoutée une solution de méthanol chlorhydrique 0,098 M (2,92 ml;
0,78 mmol). Le milieu réactionnel est agité durant 10 minutes. Le produit attendu est
précipité après addition d'éther (20 ml). On recueille î0 mg (79 %) de dichlorhydrate
cristallisé.
Caractéristiques: Pf= 9~~C
C27H34N2~ I oCl2 M = 616
Essai de solubilité: ~,1 mg dans 0,05 ml d'eau
C = 0,07 M
F~cpérimentation biologique
20Les molécules ont été testées en experimentation biologique et ont montré leurintérêt en tant qu'agent anticancéreux dans les tests de la leucémie P 388 in vivo chez la
souris. Ce test est cour~mment utilisé dans le domaine de la recherche de substances
anticancéreuses (Protocols for screening chemical agents and natural products against
animal tumors and other biological systems, R. Geran, N.H. Greenberg, M.M.
25MacDonald, A.M. Sch-lm~h~r and B.J. Abbott, Cancer Chemotherapy reports 1972, 3,
N~ 2).
Cependant, ce modèle e~cpérimental est extrêmement chimiosensible et de très
nombreux composés manifestent une bonne activité ce qui rend ce test peu discrimin~nt
Nous avons modifié le protocole du test pour le rendre plus sélectif.
30L'~-lmini~tration des cellules tumorales se fait par voie intraveineuse et non par voie
CA 02234484 l998-04-08
W O 97/13776 PCTIFR96/01588 31
intrapéritonéale. Elles se disséminent ainsi par la circulation dans tout l'organisme
rapidement. L'~Aminictration du produit à tester est faite ensuite par voie
intrapéritonéale. Deux paramètres sont définis pour mettre en évidence l'activité des
composés:
5 ~ détermination de la dose efficace 50 (DEso) qui représente la dose minimum unique
du composé à ~minictrer pour obtenir une survie des ~nim~l-x significative par
rapport aux ~nim~lly témoins non traités,
~ détennin~tion du temps de survie maximum des ~nim~lx quelque soit la dose
~lminictrée par injection unique. Le fait de pouvoir ~minictrer une dose importante
du composé et d'observer une survie importante, permet d'obtenir une mesure de
l'efficacité thérapeutique maximale du produit que l'on peut atteindre.
Orioine de la tumeur
La leucémie P 388 a été chimiquement induite en 1955 par le 3-
méthylcholanthrène sur une souris DBA/2 (Am. J. Pathol. 33, 603, 1957).
Procédure pharrnacolorique
Les tumeurs sont m~int~?n~les par passages hebdomadaires sous forme d'ascite
dans le péritoine de souris DBA/2 (lignée d'origine) et les expérimentations sont
effectuées sur les souris femelles hybrides CDFl (bal b/c femelles XDBA/2 mâles) de
20 + 2 g (Cancer chemother, Rep. 3, 9, 1972). Les cellules tumorales sont implantées
par voie intraveineuse (106 cellules par souris) au jour 0. Les ~nim~l~x sont randomisés
et répartis par groupe de 2 pour chaque série.
Les sllbst~nces ~ntitllmorale5 sont ~-lminictrées par voie intrapéritonéale (ip) un
2~ jour après l'inoculation des cellules leucémiques (traitement aigu). Les solutions sont
injectées à raison de 10 ml/lcg de souris. Le critère d'évaluation de l'activité antitumorale
est la prolongation de la survie des Zlnim~llx traités. 86 % des souris meurent le 7ème
jour après la greffe tumorale, Une substance sera considérée comme active si elle induit
une survie supérieure à 8 jours.
Le tableau suivant permet de mettre en évidence la solubilité aqueuse des
CA 02234484 1998-04-08
- W O 97/13776 PCT~FR96/01588
32
produits de l'invention, e~;primée en mg/ml, l'activité de ces composés en terme de
DEso, de survie exprimee en jours ou en T/C %, qui represente le rapport entre la survie
moyenne du groupe d'~nim~n~ traités et la survie moyenne du groupe des ~nim~n~c
contrôle.
Solubilite P 388
Survie
aqueusemg/ml DE50 (mg/kg) ma~;imum (J) T/C %
Animaux contrôle avec 6-8
a-lmini~tration iv des
(mediane-7)
cellules tumorales
Composes
Etoposide 0,01 10 19 271
Teniposide < 0,01 20 15 214
13 10 21 300
1 1 25 1 0 1 7 2~
7 10 14 ~00
1 8 257
Il apparaît ainsi que les composes de l'invention ont conservé le niveau d'activite
des composés de référence comme l'Etoposide et ont en plus l'avantage d'avoir une
solubilité aqueuse avantageuse pour la formulation et l'administration.