Note: Descriptions are shown in the official language in which they were submitted.
CA 02235133 1998-04-17
WO 97730031 PCT/EP97/00584
-1-
ESTERS OF 3-HYDROXY-PIPERIDINEMETHANOL DERIVATIVES
The present invention is concerned with novel compounds of formula (I) having
superior gastrokinetic properties. The invention further relates to methods
for preparing
such novel compounds, pharmaceutical compositions comprising said novel
compounds as well as the use as a medicine of said compounds.
Journal of Medicinal Chemistry, 1993, 36, pp 4121-4123 discloses ( 1-butyl-4-
piperidin-
yl)methyl-8-amino-7-chloro-1,4-benzodioxane-5-carboxylate hydrochloride, i.e.
SB
204070, as a highly potent and selective 5-HT4 receptor antagonist.
Naunyn-Schmiedeberg's .Archives of Pharmacology (1994) 349, pp 546-548
discloses
(I-butyl-4-piperidinyl)methyl 8-amino-7-iodo-1,4-benzodioxan-5-carboxylate as
being
a selective and high affinity 5-HT4 -receptor antagonist, in particular for
human atrial
5-HT4 -receptors.
WO 93/05038, published on March 18, 1993 {SmithKline Beecham PLC) discloses a
number of substituted 4-piperidinylmethyl 8-amino-7-chloro-1,4-benzodioxan-
5-carboxylates having 5 HT4 receptor antagonistic activity.
WO 94/10174, published on May I l, 1994 (SmithKline Beecham PLC) discloses
5-( 1-{3-pyridylmethyl)-4.-piperidinyl)methyl-8-amino-7-chloro-1,4-benzodioxan-
carboxylate, [I-(2-carboethoxyethyl)-4-piperidinyl]methyl-8-amino-7-chloro-
I,4-benzodioxan-5-carboxylate, [I-(3-hydroxybutyl)-4-piperidinyl]methyl-8-
amino-
7-chloro-1,4-benzodioxan-5-carboxylate having 5 HT4 receptor antagonistic
activity.
The above prior art documents all disclose substituted 4-piperidinylmethyI 8-
amino-
7-chloro-I,4-benzodioxan-5-carboxylates and the analogues thereof having 5
HTq.
receptor antagonistic activity. Compounds showing SHTq. antagonism are taught
to
have potential interest in the treatment of, for example, irritable bowel
syndrome, in
particular the diarrhoea aspects of irritable bowel syndrome, i.e. these
compounds block
the ability of SHT {which stands for 5-hydroxytryptamine, i.e. serotonin) to
stimulate
gut motility (see WO 93/05038, page 8, lines 12 to 17). The present
gastroprokinetic
compounds differ in structure mainly by the presence of a hydroxy- or an
alkyloxy
group on the central piperidine ring.
CA 02235133 1998-04-17
WO 97730031 PCT/EP97/00584
-2-
Bioorganic & Medicinal Chemistry Letters ( 1994) vol 4, No 5, pp 667-668
discloses
_ oxazolo, oxazino and oxazepino[3,2-a]indole derivatives as being 5 HT4
receptor
antagonists.
The present gastroprokinetic compounds differ in structure mainly by the
presence of a
2,3,4,5-tetrasubstituted phenylmoiety in stead of the oxazoio, oxazino and
oxazepino[3,2-a]indole moiety.
WO 95/25100, published on 21 September 1995, discloses the use of substituted
piperidinyl ethyl or propyl 4-amino-5-chloro-2-methoxybenzoic esters as 5 HT4
agonists. The present compounds differ in structure by the different
orientation of the
central piperidine ring and the different substitution pattern on said
piperidine ring.
EP 0 299 566, published on I8 January 1989, discloses N-(3-hydroxy-4-piperidin-
I5 yl)benzamides having gastrointestinal motility stimulating activity.
EP 0 309 043, published on 29 March 1989, discloses substituted N-(1-alkyl-
3-hydroxy-4-piperidinyl)benzamides having gastrointestinal motility
stimulating
activity.
EP 0 389 037 discloses N-(3-hydroxy-4-piperidinyl)(dihydrobenzofuran, dihydro-
2H-
benzopyran or dihydrobenzodioxin)carboxamide derivatives as having
gastrointestinal
motility stimulating activity.
The latter three prior art documents all disclose carboxamide derivatives,
while the
compounds of the present invention all have an ester function and there is a
methylene
between the ester oxygen and the piperidine ring.
The problem that this invention sets out to solve is to provide compounds
having
gastrointestinal motility stimulating properties, particularly having superior
gastric
emptying activity. Said compounds are also shown to be orally active.
The solution to this problem is provided by the novel compounds of formula
(I), that
differ structurally from the prior art, inter alia, by the presence of a
hydroxy or a
C1_6aikyloxygroup on the 3 position of the central piperidine ring.
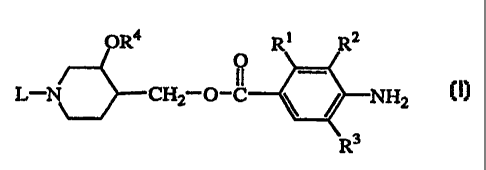
The present invention concerns compounds of formula (I)
CA 02235133 1998-04-17
WO 97130031 PCT/~P97/00584
-3-
OR4 R f RZ
O _
L-N~CHZ O-C \ / NHZ (I)
R3
a stereochemically isomeric form thereof, an N-oxide form thereof or a
pharmaceutically accegtabie acid or base addition salt thereof,
wherein
RI is CI_6alkyloxy, C2_6aIkenyloxy or C2_6alkynyloxy;
R2 is hydrogen or C1_6alkyloxy,
or taken together R1 and R2 may form a bivalent radical of formula
-O_CH2_O_ (a-1)~
-O-CH2-CH2- (a-2),
-O-CH2-CH2-~- (a-3)~
-O-CH2-CH2-CH2- (a-4),
-O-CH2-CH2-CH2-O- (a-5),
-O-CH2-CH2-CH2-CH2- (a-6),
wherein in said bivalent radicals one or two hydrogen atoms may be substituted
with
C I _6alkyl,
R3 is hydrogen or halo;
R'~ is hydrogen or CI_6alkyl;
L is C3_6cycIoaIkyl, C5_6cycloalkanone, C2_6alkenyl optionally substituted
with aryl,
or L is a radical of formula
-Alk-RS (b-1 ),
-Alk-X-R6 (b-2),
-Alk-Y-C(=O)-R8 (b-3}, or
-AIk-Y-C(=O)-NR I oR I I (b-4),
wherein each Alk is CI_l2alkanediyl; and
RS is hydrogen, cyano, CI_6aIkylsulfonylamino, C3_6cycloalkyi,
Cg_gcycloalkanone,
aryl, di(aryl)methyI or Hetl;
R6 is hydrogen, CI_6alkyl, hydroxyCl_6alkyl, C3_6cycloalkyl, aryl or Het2;
X is O, S, S02 or NR7; said R~ being hydrogen, C~_6alkyl or aryl;
R8 is hydrogen, Ci_6alkyl, C3_6cycloalkyl, aryl, arylCl_6alkyl,
di(aryl)methyl,
C1_galkyloxy or hydroxy;
Y is NRg or a direct bond; said R9 being hydrogen, CI_galkyl or aryl;
RIO and RI I each independently are hydrogen, CI_6alkyl, C3_6cycloalkyl, aryl
or
arylCl_6alkyl, or RIO and RI I combined with the nitrogen atom bearing RIO and
CA 02235133 1998-04-17
WO 97730031 PCT/EP97/00584
Rt 1 may form a pyrrolidinyl or piperidinyl ring both being optionally
substituted
with C~_6alkyl, amino or mono or di(C1_6alkyl)amino, or said RIO and Ri 1
combined with the nitrogen bearing R I ~ and R 11 may form a piperazinyl or
4-morpholinyl radical both being optionally substituted with C1_6alkyl;
each aryl being unsubstituted phenyl or phenyl substituted with 1, 2 or 3
substituents
each independently selected from halo, hydroxy, Ci_galkyl, Cr_6aIkyloxy, amino-
sulfonyl, C1_6alkylcarbonyl, nitro, trifluoromethyl, amino or aminocarbonyl;
and
Heti and Het2 each independently are selected from furan; furan substituted
with
C1_6alkyl or halo; tetrahydrofuran; a tetrahydrofuran substituted with
Ci_6alkyl; a
dioxolane; a dioxolane substituted with CI_6alkyl, a dioxane; a dioxane
substituted
with C1_6alkyl; tetrahydropyran; a tetrahydropyran substituted with C1_6alkyl;
pyrrolidinyl; pyrrolidinyl substituted with one or two substituents each
independently
selected from halo, hydroxy, cyano, or C1_6alkyl; pyridinyi; pyridinyl
substituted with
one or two substituents each independently selected from halo, hydroxy, cyano,
25 Ct_6alkyl; pyrimidinyl; pyrimidinyl substituted with one or two
substituents each
independently selected from halo, hydroxy, cyano, C~_6alkyl, C1_$alkyloxy,
amino and
mono and di(C;_6alkyl)amino; pyridazinyl; pyridazinyl substituted with one or
two
substituents each independently selected from hydroxy, C1_6alkyloxy, C1_6alkyl
or
halo; pyrazinyl; pyrazinyl substituted with one ore two substituents each
independently
selected from halo, hydroxy, cyano, C1_6alkyl, C1_6alkyloxy, amino, mono- and
di(C1_6alkyl)amino and C1_6alkyloxycarbonyl;
Hetl can also be a radical of formula
O
R1~-N N- R12-N. 'N- RIZ-N' \N- / N-.
\ ~ ,N
(c-1) (c-2) (c-3)
Hetl and Het2 each independently can also be selected from the radicals of
formula
0 0 0
N / s ~Rlz
N I
N
13 s~ ~ 13 \ N
S N R N R N
(d_1) (d_2) (d_3) (d_4)
CA 02235133 1998-04-17
WO 97/30031 PCT/EP97/00584
-5-
R12 and R13 each independently are hydrogen or Cl_q.alkyl.
As used in the foregoing definitions halo is generic to fluoro, chloro, bromo
and iodo;
C1_4aIkyl defines straight and branched chain saturated hydrocarbon radicals
having
from I to 4 carbon atoms such as, for example, methyl, ethyl, propyl, butyl, 1-
methyl-
ethyl, 2-methylpropyl and the like; C1_6alkyl is meant to include Cl~alkyl and
the
higher homologues thereof having 5 or 6 carbon atoms, such as, for example, 2-
methyl-
butyl, pentyl, hexyl and the like; C3_6cycloalkyl is generic to cyclopropyl,
cyclobutyl,
cyclopentyl and cyclohexyi; C2_6alkenyl defines straight and branched chain
IO unsaturated hydrocarbon radicals having from 2 to 6 carbon atoms, such as
ethenyl,
propenyl, butenyl, pentenyl or hexenyl; C2_dalkynyl defines straight and
branched chain
hydrocarbon radicals having 2 to 6 atoms containing a triple bond, such as
ethynyl,
propynyI, butynyl, pentynyl or hexynyl; C1-l2alkanediyl defines bivalent
straight or
branched chain hydrocarbon radicals containing from i to 12 carbon atoms such
as, for
I5 example, 1,2-ethanediyl, 1,3-propanediyl, 1,4-butanediyl, 1,5-pentanediyl,
1,6-hexanediyl, 1,7-heptanediyl, 1,8-octanediyl, 1,9-nonanediyl, I,IO-
decanediyl,
I, I 1-undecanediyl, I, I2-dodecanediyl and the branched isomers thereof.
C1_6alkanediyl is definedin an analogous way as C1_l2alkanediyl
20 The term "stereochemically isomeric forms" as used hereinbefore defines all
the
possible isomeric forms which the compounds of formula (I) may possess. Unless
otherwise mentioned or indicated, the chemical designation of compounds
denotes the
mixture of all possible stereochemically isomeric forms, said mixtures
containing all
diastereomers and enantiomers of the basic molecular structure. More in
particular,
25 stereogenic centers may have the R- or S-configuration; substituents on
bivalent cyclic
(partially) saturated radicals may have either the cis- or trans-
configuration.
Compounds encompassing double bonds can have an E or Z-stereochemistry at said
double bond. Stereochemically isomeric forms of the compounds of formula (I)
are
obviously intended to be embraced within the scope of this invention.
The pharmaceutically acceptable acid addition salts as mentioned hereinabove
are
meant to comprise the therapeutically active non-toxic acid addition salt
forms which
the compounds of formula (I) are able to form. The latter can conveniently be
obtained
by treating the base form with such appropriate acid. Appropriate acids
comprise, for
example, inorganic acids such as hydrohalic acids, e.g. hydrochloric or
hydrobromic
acid, sulfuric, nitric, phosphoric and the like acids; or organic acids such
as, for
example, acetic, propanoic, hydroxyacetic, lactic, pyruvic, oxalic (i.e,
ethanedioic),
CA 02235133 1998-04-17
WO 97/30031 PCT/EP97/00584
-6-
malonic, succinic (i.e. butanedioic acid), malefic, fumaric, malic, taxtaric,
citric,
methanesuIfonic, ethanesulfonic, benzenesulfonic, p-toluenesulfonic, cyclamic,
salicylic, p-aminosalicylic, pamoic and the like acids.
Conversely said salt forms can be converted by treatment with an appropriate
base into
the free base form.
The compounds of formula (I) containing an acidic proton rnay also be
converted into
their non-toxic metal or amine addition salt forms by treatment with
appropriate
organic and inorganic bases. Appropriate base salt forms comprise, for
example, the
ammonium salts, the alkali and earth alkaline metal salts, e.g. the lithium,
sodium,
potassium, magnesium, calcium salts and the Like, salts with organic bases,
e.g. the
benzathine, N-methyl-D-glucamine, hydrabamine salts, and salts with amino
acids such
as, for example, arginine, lysine and the like.
I5
The term addition salt as used hereinabove also comprises the solvates which
the
compounds of formula (I) as well as the salts thereof, are able to form. Such
solvates
are for example hydrates, alcoholates and the like.
Some of the compounds of formula (1) may also exist in their tautomeric form.
Such
forms although not explicitly indicated in the above formula are intended to
be included
within the scope of the present invention. For instance, when an aromatic
heterocyclic
ring is substituted with hydroxy the keto-form may be the mainly populated
tautomer.
The N-oxide forms of the compounds of formula (I), which may be prepared in
art-
known manners, are meant to comprise those compounds of formula (I) wherein
one or
several nitrogen atoms are oxidized to the so-called N-oxide. Particularly
those N-
oxides are envisaged wherein the piperidine-nitrogen is N-oxidized.
R1 is suitably methoxy and R2 is hydrogen;
when taken together R1 and R2 suitably form a radical of formula (a-I), (a-2),
(a-3),
(a-4) or (a-5), wherein optionally one or two hydrogen atoms are substituted
with
methyl;
R3 is suitably fluoro, chloro or bromo;
R4 is suitably hydrogen or methyl.
CA 02235133 1998-04-17
WO 97130031 PCT/EP97/00584
-7-
Interesting compounds are those compounds of formula (I) wherein R 1 is
methoxy, R2
is hydrogen and R3 is chloro.
Also interesting compounds are those compounds of formula {I) wherein R1 and
R2
taken together form a radical of formula (a-2), (a-3) or (a-4).
More interesting compounds are those interesting compounds of formula (I)
wherein R4
is hydrogen or methyl.
Particular compounds are those more interesting compounds with the trans
configuration, i.e. the hydroxy or methoxy is in the trans position in
relation to the
methylene on the central piperidine moiety.
Very particular compounds are those compounds wherein L is
C2_6alkenyl, especially butenyl;
a radical of formula (b-1), wherein each Alk is Cl_6alkanediyl, and
R5 is hydrogen, cyano, Cl_6alkylsulfonylamino, C3_6cycloalkyl or Hetl,
Hetl being tetrahydrofuran, dioxolane substituted with C1_6alkyl, or a radical
of
formula (c-3) or a radical of formula (d-1);
a radical of formula (b-2), wherein each Alk is C 1 _6alkanediyl, X being O or
NR~
wherein R~ is hydrogen and
R6 is hydrogen, Ci_6alkyl, hydroxyCl_6alkyl, C3_6cycloalkyl, aryl or Het2,
aryl being phenyl or phenyl substituted with halo,
Het2 being pyridinyI; pyridinyl substituted with cyano; pyridazinyl
substituted with
one or more substituents selected from hydroxy, halo and CZ_6alkyl; pyrazinyl
substituted with C1_6alkyl;
a radical of formula (b-3) wherein Y is a direct bond, and
R8 is Cl_6aIkyl or C1_6alkyloxy;
a radical of formula (b-4) wherein Y is a direct bond, and
R1~ and R11 combined with the nitrogen atom bearing R1~ and R11 form a
pyrrolidinyl.
Particularly interesting compounds are those compounds wherein L is butyl,
methoxypropyl, methylcarbonylpropyl, hydroxyethoxyethyl, 2-[2-methyl-1,3-
33 dioxolane]propyl, ethyl substituted with 4-methyl-2-pyridazinon, ethyl
substituted with
4-chloro-2-pyridazinon, propyl with 4-methyl-2-pyridazinon, or propyl with 4-
chloro-2-
pyridazinon.
CA 02235133 1998-04-17
WO 97/30031 PCT/EP97/00584
-g_
Preferred compounds are
traps-( I -butyl-3-hydroxy-4-piperidinyl]methyl 4-amino-5-chloro-2,3-dihydro-
7-benzofurancarboxylate;
traps-[3-hydroxy-I-[2-(2-hydroxyethoxy)ethyl]-4-piperidinyl]methyl 8-alnino-
7-chloro-2,3-dihydro-1,4-benzodioxin-5-carboxylate;
traps-[3-hydroxy-1-[3-(2-methyl-1,3-dioxolan-2-yl)propyl]-4-piperidinyl]methyl
8-amino-7-chloro-2,3-dihydro- I ,4-benzodioxin-5-carboxylate;
traps-[3-hydroxy- I -(3-methoxypropyl)-4-piperidinyl]methyl 8-amino-7-chloro-
2,3-dihydro-1,4-benzodioxin-5-carboxylate ethanedioate( 1:1 );
traps-[3-hydroxy-1-(3-methoxypropyl)-4-piperidinyl]methyl 4-amino-5-chloro-
2,3-dihydro-7-benzofurancarboxylate;
traps-[3-hydroxy-1-[3-(2-methyl-1,3-dioxolan-2-yl)propyl)-4-piperidinyl]methyl
4-amino-5-chloro-2,3-dihydro-7-benzofurancarboxylate;
traps-[3-hydroxy-1-(4-oxopentyl)-4-piperidinyl)methyl 4-amino-5-chloro-2,3-
dihydro-
7-benzofurancarboxylate; and
traps-[ 1-[2-( 1,6-dihydro-3-methyl-6-oxo- I -pyridazinyl)ethyl]-3-hydroxy-4-
piperidinyl]methyl 4-amino-5-chloro-2,3-dihydro-2,2-dimethyl-7-bertzofuran-
carboxylate; the stereochemically isomeric forms thereof and the
pharmaceutically
acceptable acid addition salts thereof.
Most preferred is (t)-traps-(I-butyl-3-hydroxy-4-piperidinyl)methyl 8-amino-7-
chloro-2,3-dihydro-1,4-benzodioxin-5-carboxylate, a pharmaceutically
acceptable acid
addition salt thereof or a stereoisomeric form thereof. Especially, the Iaevo-
rotatory
isomer of traps-(1-butyl-3-hydroxy-4-piperidinyl)methyl 8-amino-7-chloro-2,3-
dihydro-I,4-benzodioxin-5-carboxylate is most preferred.
The compounds of formula (I) may be prepared by reacting an intermediate of
formula
(II) with an carboxylic acid derivative of formula {III) or a reactive
functional derivative
thereof, such as for example carbonyl imidazole derivatives. Said esterbond
formation
may be performed by stirring the reactants in an appropriate solvent in the
presence of a
base, such as sodium imidazolide.
OR4 R! R2
L-N CH2 OH ~. HO-O
(I)
(m (~ R3
CA 02235133 1998-04-17
WO 97/30031 PCT/EP97/00584
-9-
Another way of preparing compounds of formula (I) is by N-alkylating an
intermediate
of formula (IV), wherein W represents an appropriate leaving group such as
halo, with
a reagent of formula (V). Said N-alkylation reaction may be performed by
stirring the
reactants in an appropriate solvent. Optionally a base may be present.
OR4 R~ R2
O
L-W + HN CHz O-C ~ ~ NHZ --~. (I)
(N) (V) R3
Alternatively, an intermediate of formula (V) is reductively N-alkylated with
an
appropriate ketone or aldehyde intermediate of formula L'=O (VI), said L'=O
being a
compound of formula L-H, wherein two geminal hydrogen atoms in the Ct_l2alkane-
diyl moiety are replaced by =O, with a piperidine of formula (V).
OR4 R ~ RZ
O _
L'=O + H-N CH2 O-C ~ / NHZ ~ (I)
(VI) (V) R3
Further, compounds of formula (I) may be prepared by carbonylation of an
intermediate
of formula (XIII), wherein X is bromo or iodo, in the presence of an
intermediate of
formula (II).
R4 Ri R2
CO
L-N CHZ OH + X \ / iVH2 ---~ (I)
catalyst
~) (XIII) Rs
Said carbonylation reaction is carried out in a reaction-inert solvent such
as, e.g.
acetonitrile or tetrahydrofuran, in the presence of a suitable catalyst and a
tertiary amine
such as, e.g. triethylamine, and at a temperature ranging between room
temperature and
the reflux temperature of the reaction mixture. Suitable catalysts are, for
instance,
palladium(triphenylphosphine) complexes. Carbon monoxide is administered at
atmospheric pressure or at an increased pressure. Analogous carbonylation
reactions
are described in Chapter 8 of "Palladium reagents in organic syntheses",
Academic
CA 02235133 2004-09-23
WO 97/30031 " ~ PCT/EP97I00584
-10-
Press Ltd., Benchtop Edition 1990, by Richard F. Heck; and the references
cited
therein.
Said ester formation reaction is known from the above mentioned reference with
metal
catalysts which are soluble such as palladium(triphenylphosphine) complexes.
Unexpectedly, we have found that these reactions can also be performed on
metal
catalysts which are insoluble or immobilized on a solid carrier. Suitable
catalysts are for
example palladium-on-carbon, Raney nickel or Cu20. These insoluble catalysts
or
catalysts on a solid phase are much less expensive than the metal complexes
and are
often much easier to handle when synthesis is done on an industrial scale.
In other words, we deem to have found a novel and inventive way to prepare
esters in
the following way
[Rd]n catalyst O /[Rd]n
haIid ----~ R'O \
\ l
co
R'OH
In the above formulas Rd represent any substituent possible on a phenyl, n is
an integer
from 1 to S, and R'O is the alcohol-rest of any alcohol. In other words, R'OH
can be
any alcohol. The term halide suitably refers to chloro, bromo, iodo. Preferred
halides
are bromo and iodo.
The preferred catalyst is palladium-on-carbon.
The pressure of CO, i.e. carbon monoxide, may vary widely and a person skilled
in the
art will certainly be able to find the suitable range after straightforward
experimentation. The preferred pressure of CO, i.e. carbon monoxide, is 50
kg/cm2
(about 4.9 x 106 Pa). It may suitably range between about 1 kg/cm2 (about lx
105 Pa)
and about 100 kglcm2 (about 10 x 106 Pa).
The reaction temperature may range from room temperature and the reflux
temperature
of the reaction mixture.
This reaction is preferably performed in a solvent, which can be in the
alcohol R'OH,
itself or in acetonitrile or in tetrahydrofuran. Preferred solvent is
acetonitrile.
Depending upon the solvent side-reactions may occur. In acetonitrile, few side-
products
were formed.
* Trade-mark
CA 02235133 1998-04-17
WO 97/30031 PCT/EP97/00584
-11-
Suitably a base is also present. An interesting base is for instance
triethyIamine.
Preferably said alcohol is a primary alcohol, more preferably R'OH is a
primary
alcohol.
The preparation of esters of formula (I) as shown hereinabove is a preferred
embodiment of said novel and inventive way of preparing esters.
An intermediate of formula (V) may be prepared by reacting an intermediate of
formula
(VII), wherein PG represents an appropriate protective group, such as for
example a
t-butoxycarbonyl or a benzyl group or a photoremovable group, with an acid of
formula
(III) or an appropriate reactive functional derivative thereof, and subsequent
deprotection of the thus formed intermediate, i.e, removal of PG by art-known
methods.
OR4 R1 R2
O _
PG-N CHz-OH -H HO-C NH -----
~ (V)
R3
{VII)
(III)
An intermediate of formula (VII), wherein R~ is a hydrogen and PG is a benzyl
group,
having the traps configuration, is known from J. Med. Chem. 1973, I56.
An intermediate of formula (II) may be prepared by reacting an intermediate of
formula
(VIII), with an intermediate of formula (IV). Said intermediate of formula
(VIII) may
be prepared by deprotection of an intermediate of formula (VII). Some of the
intermediates of formula (VII) are art-known. For instance cis-3-hydroxy-4.-
hydroxy-
methylpiperidine is known from J.Org. Chem, 1969, 34( I I ), 3674.
OR4 OR4
L-W
PG-N~CHz-OH --.-~.- H-N~CHZ-OH -----
~5 (VII) (VIII)
In some cases, it may be appropriate to protect the primary alcohol
functionality during
the reactionsequence starting from intermediate (VII) to intermediate (II).
Protecting
groups for primary alcohol functionalities are art-known. These protecting
groups may
then be removed at the appropriate time during the further synthesis.
CA 02235133 2004-09-23
WO 97130031 ' ' PCT/EP97100584
-12-
The intermediates of formula (VIII) wherein R4 is C1_6alkyl and having the cis
configuration can be prepared by hydrogenating an intermediate of formula (IX)
following art-known methods.
In intermediate (IX), PG is as defined hereinabove while PG' is a protective
group
which is preferably removable by hydrogenation, such as, for example, a benzyl
group.
The intermediate (IX) can be prepared by reacting a protected piperidone of
formula
(X) with a phosphonium reagent of formula [(aryl)3P-CH2-O-PG']+ -halide , in
appropriate conditions for carrying out a Wittig-type reaction.
OC 1~~,1 ~C 1-6alkyl OC1_6alkyl
PG-N~O ---~ PG°N --~ H-NCH.,-OH
O-PG'
(X) (~) cis-(VIII)
The intermediates of formula traps-(VIII) having R4 being C1_6alkyl may be
prepared
4
by alkylating the intermediates of formula traps-(VIII) wherein R is hydrogen,
which
may be prepared as shown hereinunder.
A novel way of preparing an intermediate of formula (VII) having the trans-
configuration was found. Said novel preparation starts from an intermediate of
formula
(XI) having the cis-configuration or from an intermediate of formula (XII)
having the
cis-configuration. In said intermediates of formula (XI) and (XII) PG is as
defined
above, R4a is hydrogen, C1_6alkyl or a protective group such as for example,
benzyl, t~
butoxycarbonyl and the like.
OR4a OR4a
H2
PG-N~CH20H pG-N~ ~ ~ ~CH20H
CuO.Cr203
cis-(XI) traps-(VII)
OR4a OR4a
H2
PG-N COOC~.~alkyl PG-N~~~~CH20H
CuO.Cr20 ~/3
cis-(XII) traps-(VII)
CA 02235133 1998-04-17
WO 97/30031 PCT/EP9%/00584
-13-
Said inversion-reaction is carried out in an appropriate solvent, such as, for
example an
ether, e.g. tetrahydrofuran in the presence of CuO.Cr203 under a hydrogen
atmosphere
and in the presence of an appropriate base, such as, for example calciumoxide.
The preferred hydrogen pressure and reaction temperature is dependent upon the
starting material. Starting from cis-(XI) the hydrogen pressure preferably
ranges from
900 to 2000 kPa (measured at room temperature) and the reaction temperature
ranges
from room temperature up to 200°C, preferably the reaction temperature
is about
120°C.
When starting from cis-(XII), the preferred hydrogen pressure range is from 1
S00 kPa
to 2200 kPa, preferably between 1800 kPa to 2000 kPa. The reaction temperature
is
between 100°C and 200°C preferably at about 125°C.
Apparently an equilibrium is
reached, typically with a diastereomeric ratio of about 65:35 (traps : cis )
as determined
by gas chromatography. However via recrystalIization it is possible to purify
the
desired traps-isomer. A suitable solvent for recrystallization is an ether,
e.g.
diisopropyl ether.
The pure intermediate of formula (VIII) having the traps configuration can
also be
obtained by chromatographic techniques, such as, for example gravitation
chromatography or (H)PLC.
Still another novel way of preparing intermediates of formula traps-(VIII) is
to react an
intermediate of formula (XIII) with borane or a borane derivative. Borane
itself is
commercially available as a borane-tetrahydrofuran complex. Borane
derivatives,
especially chiral borane derivatives are also commercially available. The
reaction with
borane is performed in a reaction inert solvent, preferable an ether, e.g.
tetrahydrofuran.
While adding the borane or the borane derivative the reaction mixture is kept
at
temperatures below 0°C, interestingly at a temperature below -50
°C and preferably at a
temperature of about - 70 °C. After adding the borane or the borane
derivative to the
reaction mixture the mixture is allowed to heat up while stirring is
continued. The
mixture is stirred for several hours. Subsequently, a hydroxide, e.g. sodium
hydroxide
. is added as well as a peroxide, e.g. hydrogen peroxide and the reaction
mixture is
stirred at elevated temperatures for several hours. After this treatment the
reaction
product was isolated in art-known manner.
CA 02235133 1998-04-17
WO 9'7130031 PCT/EP97/00584
-I4-
'~ BH3 OH
PG-N\~CH20H PG-N ~nuCH20H
NaOH, H20.,
(XIII) trans-(VII)
The compounds of formula {I), the N-oxide forms, the pharmaceutically
acceptable
salts and stereoisomeric forms thereof possess favourable intestinal motility
stimulating
properties. In particular the present compounds show significant gastric
emptying
activity as is evidenced in pharmacological example P-2, the "Gastric emptying
of an
acaloric liquid meal delayed by administration of lidamidine in conscious
dogs"-test.
Moreover, the compounds of the present invention also show an improvement of
the
gastrointestinal motility as is evidenced in pharmacological example P-I :
"Telemetxic
recording of motility of the antrum, pylorus and duodenum in the conscious
dog"-test.
The compounds of formula (i) also are shown to have a beneficial effect such
as
increase of basal pressure of the LES, i.e. Lower Esophageal Sphincter.
Most of the intermediates of formula (V) have shown to have analogous activity
as the
final compounds of formula (I).
In view of the capability of the compounds of the present invention to enhance
the
gastrointestinal motility, and in particular to activate gastric emptying, the
subject
compounds are useful to treat conditions related to a hampered or impaired
gastric
emptying and more generally to treat conditions related to a hampered or
impaired
gastrointestinal transit.
In view of the utility of the compounds of formula (I), it follows that the
present
invention also provides a method of treating warm-blooded animals, including
humans,
(generally called herein patients) suffering from conditions related to a
hampered or
impaired gastric emptying or more generally suffering from conditions related
to a
hampered or impaired gastrointestinal transit. Consequently a method of
treatment is
provided for relieving patients suffering from conditions, such as, for
example, gastro-
oesophageal reflux, dyspepsia, gastroparesis, constipation, post-operative
ileus, and
intestinal pseudo-obstruction. Gastroparesis can be brought about by an
abnormality in
the stomach or as a complication of diseases such as diabetes, progressive
systemic
sclerosis, anorexia nervosa and myotonic dystrophy. Constipation can result
from
CA 02235133 1998-04-17
WO 97/30031 PCT/EP97/OOS84
-I5-
conditions such as lack of intestinal muscle tone or intestinal spasticity.
Post-operative
ileus is an obstruction or a kinetic impairment in the intestine due to a
disruption in
muscle tone following surgery. Intestinal pseudo-obstruction is a condition
characterized by constipation, colicky pain, and vomiting, but without
evidence of
physical obstruction. The compounds of the present invention can thus be used
either
to take away the actual cause of the condition or to relief the patients from
symptoms of
the conditions. Dyspepsia is an impairment of the function of digestion, that
can arise as
a symptom of a primary gastrointestinal dysfunction, especially a
gastrointestinal
dysfunction related to an increased muscle tone or as a complication due to
other
disorders such as appendicitis, galbladder disturbances, or malnutrition.
The symptoms of dyspepsia may also arise due to the intake of chemical
substances,
e.g. SS1ZI's.
Some of the compounds also additionally show stimulating kinetic activity on
the
colon.
The compounds of formula (I) and of formula (V) are cardio haemodynamically
and
cardio electrophysiologically safe.
Hence, the use of a compound of formula (I) as medicine is provided, and in
particular
the use of a compound of formula (I) for the manufacture of a medicine for
treating
conditions involving a decreased motility of the stomach. Both prophylactic
and
therapeutic treatment are envisaged.
To prepare the pharmaceutical compositions of this invention, an effective
amount of
the particular compound, in base or acid addition salt form, as the active
ingredient is
combined in intimate admixture with a pharmaceutically acceptable carrier,
which
carrier may take a wide variety of forms depending on the form of preparation
desired
for administration. These pharmaceutical compositions are desirably in unitary
dosage
form suitable, preferably, for administration orally, rectaIly or by
parenteral injection.
For example, in preparing the compositions in oral dosage form, any of the
usual
pharmaceutical media may be employed, such as, for example, water, glycols,
oils,
alcohols and the like in the case of oral liquid preparations such as
suspensions, syrups,
elixirs and solutions; or solid carriers such as starches, sugars, kaolin,
lubricants,
binders, disintegrating agents and the like in the case of powders, pills,
capsules and
tablets. Because of their ease in administration, tablets and capsules
represent the most
advantageous oral dosage unit form, in which case solid pharmaceutical
carriers are
CA 02235133 1998-04-17
WO 97!30031 PCT/EP97/00584
-16-
obviously employed. For parenteral compositions, the carrier will usually
comprise
sterile water, at least in large part, though other ingredients, for example,
to aid
solubility, may be included. Injectable solutions, for example, may be
prepared in
which the carrier comprises saline solution, glucose solution or a mixture of
saline and
glucose solution. Injectable suspensions may also be prepared in which case
appropriate
liquid carriers, suspending agents and the like may be employed. In the
compositions
suitable for percutaneous administration, the carrier optionally comprises a
penetration
enhancing agent and/or a suitable wetting agent, optionally combined with
suitable
additives of any nature in minor proportions, which additives do not cause a
significant
deleterious effect to the skin. Said additives may facilitate the
administration to the skin
and/or may be helpful for preparing the desired compositions. These
compositions may
be administered in various ways, e.g., as a transdermal patch, as a spot-on,
as an
ointment. Acid addition salts of (I) due to their increased water solubility
over the
corresponding base form, are obviously more suitable in the preparation of
aqueous
compositions.
It is especially advantageous to formulate the aforementioned pharmaceutical
compositions in dosage unit form for ease of administration and uniformity of
dosage.
Dosage unit form as used in the specification and claims herein refers to
physically
discrete units suitable as unitary dosages, each unit containing a
predetermined quantity
of active ingredient calculated to produce the desired therapeutic effect in
association
with the required pharmaceutical carrier. Examples of such dosage unit forms
are
tablets {including scored or coated tablets), capsules, pills, powder packets,
wafers,
injectable solutions or suspensions, teaspoonfuls, tablespoonfuls and the
Like, and
segregated multiples thereof.
In general it is contemplated that a therapeutically effective amount would be
from
about 0.001 mg/kg to about 10 mg/kg body weight, preferably from about 0.02
rng/kg
to about 5 mg/kg body weight. A method of treatment may also include
administering
the active ingredient on a regimen of between two or four intakes per day.
Experimental part
In the procedures described hereinafter the following abbreviations were used
: "THF",
which stands for tetrahydrofuran; "DIPE" stands for diisopropylether; "EtOAc"
stands
for ethyl acetate; "NHq.Oac" stands for ammonium acetate; "HOAc" stands for
acetic
acid.
CA 02235133 1998-04-17
WO 97/30031 PCT/EP97/00584
-17-
For some chemicals the chemical formula was used, e.g. NaOH for sodium
hydroxide,
K2C03 for potassium carbonate, H2 for hydrogen gas, CH3CN for acetonitrile,
MgS04
for magnesium sulfate, CuO.Cr203 for copper chromite, N2 for nitrogen gas,
CH2CI2
for dichloroanethane~ CH30H for methanol, NH3 for ammonia, HCl for
hydrochloric
acid, NaH for sodium hydride, CaC03 for calcium carbonate, CO for carbon
monoxide,
KOH for potassium hydroxide.
Preuaration of the intermediates
Example I-I
a) A mixture 1,2,3,6-tetrahydro-I-(phenylmethyl)-4-pyridinemethanol {0.367
mol) and
THF ( 1000 ml) was stirred at - 70 °C and a solution of borane in THF (
1 M) was added
dropwise thereto . After the addition, the reaction mixture was allowed to
warm up to
room temperature and stirred at room temperature for 18 h. The reaction
mixture was
cooled to - 10 °C and water (23 ml) was added dropwise, NaOH (3M in
water, 18 ml)
was added dropwise and hydrogen peroxide (30 % solution in water, 28 mI} was
added
dropwise at the same time. Then; simultaneously, NaOH (3M in water, 36 ml) and
the
hydrogen peroxide (30 % solution in water, 29 mI) was added dropwise. Again
NaOH
(50 % in water, 80 mI) was added. The reaction mixture was stirred at reflux
for 4
hours. The reaction mixture was cooled and filtered. The filtrate was
evaporated. The
resulting precipitate was dissolved in water (500 ml) and saturated with
K2C03. The
product was extracted with CH2Cl2. The resulting solution was dried over MgS04
and
evaporated. The residue was crystallized from DIPE/CH3CN. After several
crystallizations a total yield of 40, 8 g of (~)-traps-I-(phenylmethyl)-3-
hydroxy-4-
piperidinemethanol was obtained (Yield : 50.1 %)
h) A mixture of (~)-traps-I-(phenylmethyl)-3-hydroxy-4-piperidinemethanol
(17.8 g,
0.085 mol) (already described in J. Med. Chem., 1973, 156) in methanol (250
mI) was
hydrogenated, at 50 °C, with palladium on activated carbon ( 10%) (2 g)
as a catalyst.
After uptake of H2 (1 equiv.), the catalyst was filtered off and the filtrate
was
evaporated, yielding I2 g of (~)-traps-3-hydroxy-4-piperidinemethanol (interm.
1-a)
(quantitative yield; used in next reaction step, without further
purification). The
corresponding cis-isomer is known from J. Org. Chem., 1969, 34 (I I).
A mixture of intermediate ( I-a) (0.022 mol) and butanal (0.025 mol) in
methanol ( 150
ml) was hydrogenated with palladium on activated carbon (IO%) (1 g) as a
catalyst in
the presence of a solution of thiophene (4% in THF) (1 ml). After uptake of H2
(1
equiv.), the catalyst was filtered off and the filtrate was evaporated,
yielding 4.1 g of
CA 02235133 1998-04-17
WO 97/30031 PCT/EP97100584
-18-
(t)-trans-1-butyl-3-hydroxy-4-piperidinemethanol (interrn. I-b) (99.5%, used
in next
reaction step without further purification).
The corresponding cis-isomer was prepared in an analogous manner
Table I-1
OR4
L-N CHZOH
Interm. L OR4 h sical data
No.
i-c CH3(CH2)3- OH cis
1-d CH3CH2- OCH3 cis
Example I-2
a) A mixture of (~)-cis-1-(phenylmethyl)-3-hydroxy-4-piperidinemethanol (0.11
moI)
and calcium oxide (S g) in tetrahydrofuran (250 ml) was hydrogenated with
copper
chromite (CuO.Cr203) (4 g) as a catalyst at 120°C. The catalyst was
filtered off and
the filtrate was evaporated. The residue was purified by column chromatography
(CH2C1~,/CH30H/CH30H(NHg) : 93/5/2). The fractions were evaporated, yielding
8.S g of (~)-cis-1-{phenylmethyl)-3-hydroxy-4-piperidinemethanol and 6.32 g of
{t)-trans- I -(phenylmethyl)-3-hydroxy-4-piperidinemethanol .
b) A mixture of ethyl (~)-cis-I-(phenylmethyl)-3-hydroxy-4-
piperidinecarboxylic acid
(40g) (90% cis; 10% trans) (0.15 mol) and calcium oxide ( 10 g) in
tetrahydrofuran
(500 ml) was hydrogenated with copper chromite {CuO.Cr203) (10g) as a catalyst
at
12S°C. After uptake of hydrogen, the catalyst was filtered off and the
filtrate was
evaporated. The residue was solidified and crystallized twice in DIPE. The
residue
was filtered off and dried in vacuo at 40°C, yielding 16 g of (~)-trans-
1-{phenylmethyl)-
3-hydroxy-4-piperidinemethanol (96% trans and 3% cis).
Example I-3
a) Reaction under N2 flow. A suspension of triphenyl [(phenylmethoxy)methyl]
phosphonium chloride (0.18 mol) in THF (600 ml) was stirred anti cooled to -
75°C. A
solution of n-butyl lithium in hexanes 2.5 M (0.18 mol) was added dropwise at -
75°C.
The mixture was stirred for 90 minutes at -75°C. A suspension of 3-
methoxy-1-
(phenylmethyl)-4-piperidone (0.12 mol) in THF (180 ml) was added at -
75°C and the
resulting reaction mixture was allowed to warm to room temperature. The
reaction
CA 02235133 1998-04-17
WO 97)30031 ~ PCTJEP97/00584
-19-
mixture was stirred for one hour at room temperature. The mixture was filtered
and the
filtrate was evaporated. The residue was dissolved in CH2Cl~, and purified by
column
chromatography over silica gel (eluent: EtOAc/hexane 30/70). Two desired
fractions
were collected and the solvent was evaporated, yielding 4 g of fraction I
(10.3%) and
17 g of fraction 2. Fraction 2 was purified by column chromatography over
silica gel
' (eluent: hexanelEtOAc 80/20). The desired fractions were collected and the
solvent
was evaporated, yielding 13 g (33.5%) of (~)-3-methoxy-4--[(phenylmethoxy)-
methylene]-1-(phenylmethyl}piperidine (interm. 3a).
b) A mixture of intermediate (3a} (0.043 mol) in THF (250 ml) was hydrogenated
with
platinum on activated carbon (5%) (3 g) as a catalyst. After uptake of H2 ( 1
equiv.),
the catalyst was filtered off and the filtrate was evaporated. The residue was
purified by
high-performance liquid chromatography over LiChroprep RP-i8~ (750 g; 8 cm;
DAC
column; eluent A: (0.5% NH40Ac in H20)/CH30H 75/25; eluent B: CH3CN; step
gradient). The pure fractions were collected and the solvent was evaporated,
yielding
8.34 g (59.6%) of (~)-3-methoxy-4-[(phenylmethoxy)methyl]-1-(phenylmethyl)-
piperidine (interm. 3b).
c) A mixture of intermediate (3b) (0.0256 mot) in THF (250 ml) was
hydrogenated
with palladium on activated carbon ( 10%) (2 g) as a catalyst. After uptake of
H2 ( 1
equiv.), bis (1,1-dimethylethyl) dicarbonate (0.0256 mol) was added and
hydrogenation
was continued. After uptake of H2 (1 equiv.), the catalyst was filtered off
and the
filtrate was evaporated. The residue was purified by column chromatography
over
silica gel (eluent: CH2C12/CH30H 95/5). The desired fractions were collected
and the
solvent was evaporated. The residue was solidified and crystallized from
petroleum
ether (2 x). The precipitate was filtered off and dried (vacuum; 30
°C}, yielding 2.6 g
(40%) of (t)-I,1-dimethylethyl cis-4-(hydroxymethyl)-3-methoxy-I-piperidine-
carboxylate (interm. 3c).
d) A mixture of intermediate (3c) (0.04 mol) in a mixture of HCl in diethyl
ether (25
ml) and methanol (250 ml} was stirred and refluxed for 30 minutes. The
reaction
mixture was cooled and the solvent was evaporated. Toluene was added and
azeotroped on the rotary evaporator, yielding 8 g (90%) of (~}-cis-methoxy-4-
piperidinemethanol hydrochloride (interm. 3d).
e) A mixture of intermediate (3d) (0.04 mol), potassium acetate (5 g) and
butanal (0.04
moI} in methanol (150 ml) was hydrogenated with palladium on activated carbon
(IO%)
(2 g) as a catalyst in the presence of thiophene (4%o in THF) ( 1 ml). After
uptake of H2
( 1 equiv.), the catalyst was filtered off and the filtrate was evaporated.
The residue was
dissolved in CH2C12/(CH30H/NH3) and purified by short column chromatography
over
silica gel (eluent: CH2C12/(CH30H/NH3) 95/5). The desired fractions were
collected
CA 02235133 1998-04-17
WO 97130031 PCT/EP97/00584
-20-
and the solvent was evaporated, yielding 7.2 g (> 90%) of (~)-cis-1-butyl-3-
methoxy-
4-piperidinemethanol (interm. 3e).
Example I-4 '
4a) A mixture of (tetrahydro-2-furanyl)methyl methanesuIfonate (0.05 mol),
intermediate (3d) (0.046 mol) and N,N-diethylethanamine (0.12 mol) in N,N-
dimethyl-
formamide { 150 ml) was stirred for 20 hours at 60 °C. More (tetrahydro-
2-furanyl)-
methyl methanesulfonate (0.01 mol) was added and the resulting reaction
mixture was
stirred for 4 hours at 60 °C. The reaction mixture was cooled and the
solvent was
evaporated. The residue was purified by column chromatography over silica gel
(eluent: CH2Cl2/ (CH30H/ NHg) 95/5). The pure fractions were collected and the
solvent was evaporated, yielding 3.1 g (32010) of (t)-cis-1-(2-alninoethyl)-3-
hydroxy-4-
piperidinemethanol {interrn. 4a).
25 The following intermediates, as depicted in table I-2, were prepare
according to this
method.
Table I-2
OR4
L-N\ r--CHZOH
Interm. OR4 L physical data
No.
4-a OCH3 cH
cis
4-b OCH3 (cHz)3-o ~ ~ F cis
4-c pH -(CH2)3-CN trans
4-d
OH ~(CH2)g-C-OCH3 trans
4-a
OH CH2~ C1S
~
4-f OH o~ o cis
--(CHZ)g ~CHg
4'g OH -(CH2)3-O-CH3 trans
4-h OH -(CHy)3-O-( j cis
4-i OH -(CH2)3-CN
cis
CA 02235133 1998-04-17
WO 97/30031 ' PCT/EP9'7/005~4
-21-
Interm. OR4 L physical data
' No.
o
4 J OH -(CHZ)3'C-N~ cis
4-k OH ~Jo
~CH3 C1S
~
-(CHy)2-N
N-CH
~3
4-1 OH /~
o trans
-(CH
)j ~CH
Z
3
OH vCH2)2-NH-S02-CH3
trans
4-n
OH cH2~ trans
V
4-o OH o
-(cH2)2-~ ' 103-104C , trans
Ny
CH3
4-P OH -{CH2)3-O-CH3 i
c
s
OH
-(cH2)z-~ I 131-132C, cis
N~.
CHq
'fir OH (cH2)3-o ~ ~ F trans
4-s OH o
~rHZ-cH3 trans
-(CHZ)z-
4_t OH o
i
c
s
-(CHi)2 ~ ~IV~
CHg Lj~ 'S
4-a OH (CH2)g-CH(4-fluorophenyl)2traris
4_v OH o trans
-
c
(CHZ)3-
_~
4_w o
OH
~Hz traps
--(CHa)3-NN~N ~CH3
CA 02235133 1998-04-17
WO 97/30031 PCT/EP97/00584
-22-
Interm. OR4 L physical data
No.
4-x OH
cH2 > 260C, trans.HCl
~
-(CHZ)3-NN~N ~CHg
4"Y OH
~2 CIS
--(CH
-NN~
)
/ ~
Z
3
N
CHg
4-Z
OH ~ cHZ > 260C, cis.HCI
-(CHZ)3-
/ \~3
~ /
4-as OH
~cH3 trans
~
-(CHZ)2-N
N-CH
1 cH
L
3
.
4-bb pH
trans
(cHZ)Z ~
Ny
cl
4-cc pH ,-.~2_cH:cH ~ ~ E-trans
F
4-dd OH
cis
'-_(CHq)2~' i
N~
CI
4-ee
OH
~(CH ) -C-OCH C1S
Example I-5 -
A mixture of (~)-cis-1-(2-aminoethyl)-3-hydroxy-4.-piperidinemethanol (0.064
mol),
2-chloro-3-methylpyrazine (0.068 mol) and calcium oxide (0.145 mol) was
stirred for 5
hours at 120 °C. The reaction mixture was cooled. The mixture was
dissolved in
CH2C12/CHgOH and filtered over dicalite. The filtrate was evaporated and the
residue
was purified by column chromatography over silica geI (eluent:
CH2C12/(CH30H/NH3)
90/10). The desired fractions were collected and the solvent was evaporated.
The oily
residue was triturated in CH3CN: The precipitate was filtered off and dried,
yielding
CA 02235133 1998-04-17
WO 97130031 PCT/EP97/00584
-23-
4 g {23.5%) of (~)-cis-1-[2-[(3-methyl-2-pyrazinyl)amino]ethyl]-3-hydroxy-4.-
piperi-
dinemethanol (interm. Sa).
In a similar manner were also prepared
Table I-3
OR4
L ; CHZ-OH
Interm. OR4 L physical data
No.
5-a OH N
_-(CH2)2--NH~ ~ C1S
N
CH3
5-b OCH3 ~-/
-(CHZ)2--NH~ C1S
/~ N
CH3
S-C N~
OH -(CH2)z-NH~~ traIlS
/\~ 'N
CHI
5-d OH cN
-(CHy)3-NH ~ tran5
N
5-a OH cN
-(CHZ)4-NH ~~ tranS
N
5- f OH cN
-(CHz)3-NH ~~ C1S
N
5 g OH
--(CHZ)4-NH ~ C1S
N
5 h OH ~N
N trallS
CH3
5-i OH
~(CH2)3-NH-'C
trans
.~xampie t-a
A mixture of (t)-I,1-dimethylethyl cis-4-[[{4-amino-5-chloro-2-
methoxybenzoyl)oxy]
methyl]-3-hydroxy-1-piperidinecarboxylate (0.053 moI) in THF (250 ml) and a
mixture
of HCl and 2-propanol (25 ml) was stirred for 2 hours at room temperature,
then for 10
minutes at 50 °C, then cooled again to room temperature. The mixture
was alkalinized
CA 02235133 1998-04-17
WO 97130031 PCT/EP97/005$4
-24-
with NHg. The organic layer was separated, dried (MgSOq}, filtered and the
solvent
was evaporated. The residue was purified by column chromatography over silica
gel
(eluent: CH2Cl2/ {CH30H/NH3) 90110). The pure fractions were collected and the
solvent was evaporated, yielding 4.6 g {29%) (~) cis-(3-hydroxy-4-
piperidinyl)methyl
4-amino-5-chloro-2-methoxybenzoate (interm. 6-a}.
In a similar manner were also prepared
Table I-4
OR4 C1
O
H-N CHZ O~C~ ~ ~ NH2
Rt R2
Tnterm. OR4 R1 R2 physical
No. data
6-b OCH3 -CH30 I -H cis
6-c OCHg -O-C(CHg)2-CH2- ciS
6-d OH -O-(CH2)2-O- traps
6-a OH -O-C(CH2)2-CH2- cis
6-f OH -O- (CH2)3-O- traps
6-g OH -O-CH2-O- traps
6-h OH -O-(CHZ)3-O- cis
6-i OH -O-(CH2)2-O- cis
6-j OH -OCHg -OCH3 169-170C
traps
OH -O-CH2-O- 185-186C
cis
6-1 OH -O-(CH2)2- >280C
traps
6-m OH -O-C(CH3)2-CH2- 197-198C
trans.HCl
6-n OH -O-C(CH3)2-CH2- ~.~s
CA 02235133 1998-04-17
WO 97/30031 PCT/EP97/00584
-25-
Example I-7
NaH (60 % oily dispersion) (0.05 mol) was added under NZ atmosphere to a
mixture of
(t)-trans-3-hydroxy-4-piperidinemethanol (0.053 mol) and I H-imidazole (0.12
mol) in
THF (150 ml) . The mixture was stirred for 5 min at room temperature. I-(4-
amino-5-
chloro-2-methoxybenzoyl)-IH-imidazole (0.023 mol) was added at room
temperature
' and the resulting reaction mixture was stirred for 5 min at room
temperature. The
solvent was evaporated. The residue was diluted with water and extracted twice
with
CHZCIa. The organic layer was separated, dried (MgS04), filtered and the
solvent was
evaporated. The residue was crystallized from CH3CN. The precipitate was
filtered off
and dried, yielding I.69 g of intermediate 7-a (see table). This fraction was
recrystallized from methanol. The precipitate was filtered off and dried
yielding 0.83 of
intermediate 7-a ( 11.5 % ) (mp 160 °C)
OH Cl
O
H-N CHZ-O-C ~ ~ NH2
R1 Rz
In this manner and in a similar manner were prepared
Table I-5
Interm. OR4 R I R2 physical
No. data
7-a OH -O-(CH2)2- cis
7-b OH -OCH3 -H I59-I60C
tranS
7-c OH -OCH3 -OCH3 cis, I/z
C~H2~4(*)
227-228C
7-d OH -OCH3 -OCH3 cis.
7-a OH -O-C(CH3)2-CH2- cis
7-f OH -O-CH2-O- tTans
(*)1/2
C2H~04
indicates
a
ethanedioic
acid
salt
(2:1)
CA 02235133 1998-04-17
WO 97/30031 PCT/EP97J00584
-26-
Example I-8
a) CaC03 (32.5 g) was added to a mixture of 3,4-dihydro-2H-1,5-benzodioxepin-6-
amine (J. Med. Chem. (1988}, 31 (10)), 1934} (0.25 mol) in CH2Cl~ (400 ml) and
CH30H (200 rnl). This mixture was stirred at room temperature. N,N,N-
trimethylbenzenemethanaminium dichloroiodate (0.25 mot) was added portionwise
at
room temperature. The resulting reaction mixture was stirred for 30 min at
room
temperature. The mixture was diluted with water. The layers were separated.
The
aqueous phase was extracted with CHZCI2. The combined organic layers were
washed
with water, dried (MgS04), filtered and the solvent evaporated. The residue
was
purified by column chromatography over silica gel (eluent: CH2Cl~). Two pure
fractions were collected and the solvent was evaporated. The first residue (7
g) was
crystallized from petroleum benzin. The precipitate was filtered off and
dried, yielding:
5.26 g ( 1 ). The second residue (45 g) was crystallized from petroleum
benzin. The
precipitate was filtered off and dried, yielding: 40.5 g (2). Total yield:
45.76 g or 62.9%
I5 of 3,4-dihydro-9-iodo-2H-1,5-benzodioxepin-6-amine.{intermed 8-a)
b} Acetic acid anhydride {0.13 mol) was added dropwise to a mixture of
intermediate
8a (0.127 mol) in HOAc (450 mI), stirred at room temperature. The reaction
mixture
was stirred for 30 min at room temperature. The reaction mixture was poured
out into
water (500 ml). The precipitate was filtered, washed with water, then dried,
yielding:
39.2 g or 92.7% of N-[3,4-dihydro-9-iodo-2H-1,5-benzdiazepin-6-yl]acetamide
(intermed. 8-b)
c) A mixture of intermediate 8-b (0.1 i 6 mol), KOAc (20 g) and palladium on
carbon
(10 %) (2 g, as a catalyst) in CH30H {500 ml) was stirred at 150 °C
under 4.9 106 Pa
pressure of CO, during 16 hours. The reaction mixture was cooled, filtered
over
dicalite, and the filtrate was evaporated. The residue was diluted with water,
then
extracted twice with CH2CI~,. The combined organic layers were dried (MgS04),
filtered and the solvent evaporated. The residue was dissolved in acetic acid
and acetic
acid anhydride (5 ml) was added. The mixture was stirred for 10 min at room
temperature, then diluted with water and extracted twice with CH2CIa. The
separated
organic phase was washed. with water, with NaOH ( 10 % aqueous solution),
again
with water, then dried (MgS04), filtered and the filtrate was evaporated. The
residue
was suspended in DIPE, filtered off and dried, yielding: 24 g or
78.1°!0 of methyl 3,4-
dihydro-9-acetylamino-2H-1,5-benzodiazepine-6-carboxylate. (intermed 8-c)
d) A mixture of intermediate 8-c (0.10 mol) and N-bromosuccinimide (0.11 moI)
in
CH3CN (250 ml) was stirred and refiuxed for one hour. The reaction mixture was
cooled, and poured out into water. This mixture was extracted twice with
CH2CI2. The
separated organic layer was washed with water, dried (MgS04), filtered and the
solvent
CA 02235133 1998-04-17
WO 97730031 PCT/EP97/00584
-27-
evaporated. The residue was suspended in DIPE, filtered off and dried,
yielding 27.38
g (91.3%) of methyl 8-chloro-3,4-dihydro-9-acetylamino-2H-1,S-benzodiazepine-6-
carboxylate (intermed. 8-d).
e) Intermediate 8-d (0.091 moI) was added to KOH (0.91 mol) in H20 (S00 ml}
and the
resulting reaction mixture was stirred and refluxed for 3 hours. The reaction
mixture
was cooled and acidified with HCl {36% aqueous solution) {pH = ~ 4). The
precipitate
was filtered off, washed with water, then dried, yielding: 21.53 g (97.1 %) of
8-chloro-
3,4-dihydro-9-acetylamino-2H-1,5-benzodiazepine-6-carboxylic acid (intermed 8-
e).
Example I-9
a) A mixture of N-(2,3-dimethoxyphenyl)-acetamide (Eur.J.Med.Chem. (1988), 23,
6,
pp SOl-S10) (0.91 mol) and N-bromosuccinimide (0.91 rnol) in CH3CN (2000 ml)
was
stirred and refluxed for 1 hour. The mixture was cooled, poured out into H20
(2000
mI) and extracted twice with CHZC12. The combined organic layers were washed
with
H20, dried (MgS04), filtered and the solvent was evaporated. The residue was
crystallized from DIPE. The precipitate was filtered off and dried. Yielding:
93.8 g N-
{6-chloro-2,3-dimethoxyphenyl)-acetamide (44.9%) (intermed 9-a).
b) A solution of intermediate 9-a (O.S9 mot) in HCI (20 % aqueous solution)
(IS00 ml)
was stirred and refluxed for 2 hours. The mixture was cooled, alkalinized with
NaOH
(SO% aqueous solution) and extracted twice with CH2CIz. The combined organic
layers
were washed with water, dried (MgS04), filtered, and the solvent was
evaporated,
yielding 1 l Og ( 100%) of 6-chloro-2,3-dimethoxybenzenamine (intermed 9-b).
c) A mixture of intermediate 9-b (O.S9 mol) and CaC03 (75 g) in CHZCIZ (600
ml) and
CH30H {300 ml) was stirred at room temperature. N,N,N-trimethylbenzene-
metanaminium dichloroiodate {0.6 moI) was added portionwise and the mixture
was
stirred and refluxed for I hour. The mixture was cooled and diluted with H2O (
1.SI).
The organic layer was separated and the aqueous layer was extracted again with
CH2CI2. The combined organic layers were washed with H20, dried (MgS04),
filtered,
and the solvent was evaporated. The residue (170 g) was purified by column
chromatography over silica gel (eluent: CH30H1H20 80/20). The desired
fractions
were collected and the solvent was evaporated, yielding: 114.16g (61.7%) of 6-
chloro-
4-iodo-2,3-dimethoxybenzenamine(intermed 8-c)
d) A mixture of intermed 8-c (0.36 mol), potassium acetate (4S g) and
palladium on
carbon (10%) (2 g) in CH30H (4S0 ml) was stirred at 125°C under CO (4.9
I06 Pa CO
CA 02235133 1998-04-17
WO 97130031 PCTlEP97/00584
-28_
pressure) for 18 hours. The mixture was cooled and filtered over hyflow. The
filtrate
was evaporated. The residue was diluted with HZO and extracted three times
with
CHaCl2. The combined organic layers were dried (MgS04), filtered, and the
solvent
was evaporated. The residue was purified by column chromatography over silica
gel
(eluent: CH2Cl~). The. desired fractions were collected and the solvent was
evaporated,
yielding: 67.1 g (75.9%) of 4-amino-5-chloro-2,3-dimethoxy-benzoate methyl
ester
(intermediate 8-d).
e) A mixture of intermed 8-d (0.27 moI) and KOH (2.7 moI) in H20 ( 1000 mI)
was
stirred and refluxed for 2 hours. The reaction mixture was cooled and
acidified with
HCI (36%), and the resulting precipitate was filtered off, washed with water
and dried,
yielding: 53 g (84.8%) 4-amino-5-chloro-2,3-dirnethoxy-benzoic acid (intermed
8-e).
Example I-IO
A mixture of 4-amino-5-chloro-2,3-dihydro-2,2-dimethyl-7-benzofurancarboxylic
acid
(0.3 mol) and 1,1'-carbonyIdiimidazole (0.3 mol) in CH3CN (1000 ml) was
stirred for
1.5 hours at room temperature. The solvent was evaporated. The residue was
partitioned between water and CHZCl2. The organic layer was separated, dried
{MgS04), filtered and the solvent evaporated. The residue was suspended in
DIPE,
filtered off and dried (vacuum, 50 °C), yielding 50 g (58%) of N-[4-
amino-5-chloro-
2,3-dihydro-2,2-dimethyI-7-benzofurancarboyl] IH-imidazole (intermediate 9-a).
In a similar manner were also prepared
N-[4-amino-5-chloro-2,3-dihydro-7-benzofuranoyI]-1H-imidazole (intermed 9-b)
N-[8-chloro-3,4-dihydro-9-acetylamino-2H-I,5-benzdiazepine-6-oyI]-1H-imidazole
(intermed 9-c)
N-[4-amino-5-chloro-2,3-dimethoxybenzoyl]-1H-imidazole (intermed 9-d).
Preparation of the final compounds
Example F-1
Reaction under N2 flow. Sodium hydride (60% oily dispersion) {0.013 mol) was
added
to a solution of (t)-traps-1-butyl-3-hydroxy-4.-piperidinemethanol (0.013 mol)
in THF
(80 m1). The mixture was stirred and refluxed for 3 hours, then cooled
(solution I).
8-amino-7-chloro-2,3-dihydro-1,4-benzodioxin-5-carboxylic acid (0.013 mol) was
dissolved in acetonitrile (80 ml). 1,1'-Carbonylbis-IH-imidazole (0.015 mol)
was
CA 02235133 1998-04-17
WO 97/30031 PCT/EP97100584
-29-
added. The resulting mixture was stirred for 2 hours at room temperature. The
solvent
was evaporated. The residue was dissolved in THF (80 ml) (= solution II).
Solution
(II) was poured out into solution (I) and the resulting reaction mixture was
stirred for 20
hours at room temperature. The solvent was evaporated. The residue was
partitioned
between water and CH2Cl2. The organic layer was separated, dried (MgS04),
filtered
and the solvent was evaporated. The residue was purified by column
chromatography
over silica gel (eluent: CH2Cl2/CH30H 93/7). The desired fractions were
collected
and the solvent was evaporated. The residue was dissolved in 2-propanol and
converted
into the ethanedioic acid salt (1:1). The mixture was cooled to 0 °C
and the resulting
precipitate was filtered off and dried (vacuum, 60 °C}, yielding 1.5 g
(24%) of (~)-
trans-{ 1-butyl-3-hydroxy-4-piperidinyl)methyl 8-amino-7-chloro-2,3-dihydro- I
,4-
benzodioxin-5-carboxylate ethanedioate (1:1); mp. 201.6°C (comp. 30}.
Example F-2
A mixture of (~)-cis-(3-hydroxy-4-piperidinyl)methyl 4-amino-5-chloro-2-
methoxy
benzoate (0.OI6 moI), bromobutane (0.02 mot) and N,N-diethylethanamine (0.03
mol)
in N,N-dimethylformamide (80m1) was stirred for 20 hours at 60 °C. The
reaction
mixture was cooled and the solvent was evaporated. The residue was purified by
column chromatography over silica gel (eluent: CH2C12/CH30H 90/IO). The pure
fractions were collected and the solvent was evaporated. The residue was
repurified by
column chromatography over silica gel (eluent: CH2CI2/(CH30HlNH3) 95/5). The
pure fractions were collected and the solvent was evaporated. The residue was
crystallized from CH3CN. The resulting precipitate was filtered off and dried
(vacuum;
50 °C), yielding I .4 g (24%) of (~)-cis-( 1-butyl-3-hydroxy-4-
piperidiliyI}methyl 4-
amino-5-chloro-2-methoxybenzoate; mp. I2I.4°C (comp. 3).
Example F-3
(~)-traps-( 1-butyl-3-hydroxy-4-piperidinyl)methyl 8-amino-7-chloro-2,3-
dihydro-1,4-
benzodioxin-5-carboxylate ethanedioate(I:1) (0.0055 mot) was,prepurified by
reversed-phase column chromatography over RP-18 (eluent: (0.5% NH40Ac in
H20}/CH30H 35/65), then separated into its optical isomers (resolution) by
chiral
column chromatography over Chiralcel OD (eluent: hexane/ethanol 85/15). Two
desired fraction groups were collected and their solvent was evaporated. Each_
residue
was (separately) purified by column chromatography over silica gel (eIuent:
CH2C12/CH30H 94/6). The pure fractions of each enantiomer were collected and
the
solvent was evaporated, giving 0.540 g of the (-)-enantiomer and 0.330 g of
the (-r)-
enantiomer. The oil comprising the (-)enantiomer was suspended in DIPE,
filtered off,
CA 02235133 1998-04-17
WO 97/30031 PCTJEP97/00584
-30-
then dried (vacuum, 40 °C), yielding 0.49 g of ((-)-trans)-{1-butyl-3-
hydroxy-4-
piperidinyl)methyl 8-amino-7-chloro-2,3-dihydro-1,4-benzodioxin-5-carboxylate
(comp. 32); [a]D - -31.12 (conc. = 4.89 mg / 5ml in CH30H). The oil comprising
the
{-i-)-enantiomer was suspended in DIPE, filtered off, then dried (vacuum, 40
°C),
5 yielding 0.3 g ((+}-trans}-(I-butyl-3-hydroxy-4-piperidinyl)methyl 8-amino-7-
chloro-
2,3-dihydro-1,4-benzodioxin-5-carbox /ate com .33 20
Y ( p )~ [a]D = + 31.31 (cone = 5.27
mg / 5 ml in CH30H).
Example F-4
10 A mixture of compound 37 (see table 1)(0.0052 mot) and HCI (5 ml) in THF
(52 ml)
was stirred and refluxed for one hour. The reaction mixture was cooled,
alkalinized
with NH3, and the solvent was evaporated. The residue was partitioned between
NH3/water and CH2Cl~. The organic layer was separated, dried (MgS04), filtered
and
the solvent was evaporated. The residue was triturated in DIPE, filtered off,
then
15 crystallized from CH3CN. The precipitate was filtered off and dried,
yielding 0.75 g
(37.5%) of compound 38 (see table I).
Example F-5
Intermediate 6-d (0.007 mol) was dissolved in CH30H (70 ml). Oxirane (gas) was
20 allowed to bubble through the solution during 2 hours. The solvent was
evaporated.
The residue was purified by column chromatography over silica gel (eluent:
CH2C12/CH30H 90/10). The pure fractions were collected and the solvent was
evaporated. The residue was crystallized from CH3CN (0 °C). The
precipitate was
filtered off and dried {vacuum; 50 °C), yielding 0.85 g (32%) of
compound 78 (see
table 5).
Example F-6
A mixture of intermediate 6-d (0.01 mol) and ethanal (0.01 moi; solution in
THF) in
THF (150 ml) was hydrogenated with platinum on carbon (1 g) as a catalyst in
the
presence of thiophene (4% solution, 1 ml). After uptake of HZ (1 equiv.), the
catalyst
was filtered off and the filtrate was evaporated. The residue was purified by
column
chromatography over silica gel (eluent: CH~Cl2/(CH30H/NH3) 95/5). The pure
fractions were collected and the solvent was evaporated. The residue was
crystallized
from DIPE with a small amount of CH3CN. The precipitate was filtered off and
dried
(vacuum, 40 °C), yielding I g (27%) of compound 79 (see table 5).
CA 02235133 1998-04-17
WO 97!30031 PCT/EP97l00584
-31-
Example F-7
In an autoclave (CO-pressure = 50 kg}, a mixture of intermediate 4-m (0.019
mo1), 6-
chlor-2,3-dihydro-8-iodo-1,4-benzodioxin-5-amine (O.OI3 moI), palladium on
carbon
( 1 g) and N,N-diethylethanamine (4 g) in CH3CN { 100 mI} was heated for 20
hours at
I00 °C. The reaction mixture was cooled, filtered and the filtrate was
evaporated. The
residue was partitioned between water and CH2CI2. The organic layer was
separated,
dried (MgS04), filtered and the solvent was evaporated. The residue was
purified by
column chromatography over silica gel {eluent: CH2CI2/{CH3OH/NH3) 95/5). The
desired fractions were collected and the solvent was evaporated. The residue
was
dissolved in 2-propanol and converted into the ethanedioic acid salt (1:1).
The
precipitate was filtered off and dried (vacuum, 40 °C), yielding 2.5 g
(33%) of
compound i08 {see table 5). This fraction was repurified by column
chromatography
over silica gel. (eluent: CH2C12/CH30H 95/5). The pure fractions were
collected and
the solvent was evaporated. The residue was solidified and crystallized as an
ethanedioic acid salt in 2-propanol. The precipitate was filtered off and
dried. Yielding:
1 g compound 108 (see table 5).
Example F-8
Compound 32 (0.0025 mol) was stirred in 2-propanol (20 mI) for 1 S min at
room.
temperature. The resulting suspension was heated until complete dissolution
(nearly at
refIux temperature). The mixture was cooled to room temperature. HCl in 2-
propanol
( I8 drops) was added dropwise over a 15-min period and slowly precipitation
resulted.
The reaction mixture was stirred for one hour at room temperature. The
precipitate was
filtered off, washed with 2-propanol (5 ml), then dried (vacuum, 40°C).
The filtrate
was treated with HCl in 2-propanol (21 drops} and 2-propanol (30 ml), and the
above
reaction procedure was repeated. A total yielding of 1.5 g compound 109 (see
table 5).
Table F-1
OR4 OCHg
O _
L N~CH2-O-C ~ ~ NH.,
C1
CA 02235133 1998-04-17
WO 97!30031 PCT/EP97/00584
-32-
Co. Ex. -L OR4 physical data
No. No. m . in C
1 F-2 -(CH2)3-CH3 OCH3 107.9C; cis
2 F-2 -(CH2)2-O-(CH2)2-OH -- OCH3 130.4C; cis
3 F-2 -(CH2)3-CH3 OH 121.4C; cis
4 F-2 -CH2-CH3 OCH3 9g.9C; cis
S F-I -(CH2)3-O-(4-fluorophenyl) OCH3 72.5C; cis
6 F-1 -(CH2)2-~-(3-methyl-2-pyrazinyl)OH 158.4C; cis
7 F-1 -(CH2)2-NH-(3-methyl-2-pyrazinyl)OCH3 172.2C; cis
8 F-1 -(CH2)3-CH3 OH 95.3C; trans
9 F-1 -(CH2)2-NH-(3-methyl-2-pyrazinyl)OH 80-90C; trans
34 F-1 -(CH2)3-CN OH 129-130C; trans
35 F-1 -(CH2)3-O-CH3 OH l I7-118C;
cis
36 F-1 -(CH2)3-O-(4-fluorophenyl) OH 164-165C; traps
37 F-1 0 o OH 1 i9-120C;
cis
'CH
-(CH2)3
3
38 F-4 -~cH _~_cH OH 118-i 19C,
7~3 3 cis
39 F-1 -CH2-2-tetrahydrofuranyyl OH 168-169C; traps
40 F-1 OH I40C; traps
(CH )3 N
Nw
CH3
Table F-2
OR4 O
O _
L N~CH2-O-~C ~ ~ NHZ
m
Co. Ex. -L OR4 physical data
No. No. m . C
F-1 -(CH2)3-~3 OH 152.6C; cis
11 F-1 -(CH2)3-CH3 OCH3 112.4C; cis
I2 F-I -CH2-CH3 OCH3 145.1C; cis
I3 F-I -(CH2)3-O-(4-fluorophenyl) OCH3 168.4C; cis
CA 02235133 1998-04-17
WO 97/30031 PCTlEP99/00584
-33-
Co. Ex. -L OR4 physical data
No. No. m . C
14 F-1 -CH2-(2-tetrahydrofuranyl) OCH3 144.6C;.(COOH)2;
cis
I5 F-1 -(CH2)2-NH-(3-methyl-2-pyrazinyl)OCH3 195.0C; cis
I6 F-1 -(CH2)3-CH3 OH 82.I C; trans
41 F-I -CH2-(2-tetrahydrofuranyI) OH i
c
s
o~
o
42 F-1 ~ OH cis
'
/ ~cH
(cH2)
,
cN
43 F- (CHZ)3-NH
I OH i
N c
s
0
44 F-4 -(cHZ)3-c~-cH3 OH 124C; cis
45 F-1 ~(cH2)3- OH 117C; cis
46 F-1 '~' OH 148C; cis
.--(CHz)3-NH
N
47 F-I O OH 152C; cis
-(CHZ)3-C-N~
48 F-I
OH i
cH3 c
~ s
-(CHZ)z-N
N-CH
~C
H3
49 F-i -(CH2)3-O-CH3 OH 98C; trans
50 F-I -CH2-(2-tetrahydrofuranyl) OH 136-137C; trans
51 F-1 o OH 110C; trans
~
CHg
'--(CI-I
O
52 F-4 ~(CHz)3-C~-CH3 OH 103C; trans
53 F-1 -(CH2)3-O-CH3 OH 139C, cis
54 F-i OH 139-I40C,cis
w-(CHZ)z- ~
N~
CH;
55 F-1 OH 109-1IOC,cis
-(CHz)z- ~ ~ \
N~
56 F-2 -(CH2)2-O-(CH2)2-OH OH 123-124C,trans
57 F-1 -(CH2)3-~ OH I35-136C,cis
58 F-I -(CH2)3-CN OH 114-115C,trans
CA 02235133 1998-04-17
WO 9T130031 PCTIEP97/00584
-34-
Co. Ex. -L OR4 physical data
No. No. m . C
59 F-2 -(CH2)2-O-(CH2)2-OH OH
99-100
C,cis
0
60 F-1 ~(oH~3 ~~~ OH I90C; traps
61 F-1 cH2 OH
160-165 C
trans
CH3 ,
-(CHZ)3_.. ;
~
62 F-I
~ OH
~cH3 traps
-(CHy)2-N
N-CH
V ~H3
0
63 F-1 ~(CH2)3-ICS-~H3 OH 75-78C,cis
64 F-1 OH decomposition; traps
~(CH~Z- i
N~
C1
Table F-3
CH3 CH3
OR4 O
O _
i~
L-N CH?-O-C , ~ NH,
CI
Co. Ex. -L OR4 physical data
No. No. m . C
I7 F-2 -CH2-CH3 OCH3 61.0C; cis; 1!2
H20
I F-1 -(CH2)3-CH3 OH 140.1 C; cis
8
I9 F-I -(CH2)3-CH3 OCH3 102.1C; cis
20 F-1 -(CH2)3-O-(4-fluorophenyl)OCH3 123.0C; cis
2I F-I -(CH2)3-CH3 OH 77.2C; traps
22 F-1 -(CH2)2-NH-(3-methyl-2-pyrazinyi)OH 194.4C; cis
.
23 F-I -(CH2)2-~-(3-methyl-2-pyrazinyl)OH
traps
65 F-2 -CH2-(2-tetrahydrofuranyl)OH
cis
CA 02235133 1998-04-17
WO 97!30031 PCT/EP97/00584
-35-
Co. Ex. -L OR4 physical data
No. No. m . C
66 F-2 o OH i
c
s
'CH
-(CH2)g
3
0
184 F-4 ---~CH~3-c-CH3 OH
cis
67 F-1 ~N OH
139
C; cis
---(CHp)g-NH
N
68 F-1 ~fCH2)3-~~ OH
cis
69 F-1 cN OH I38C
ci
;
s
-(CHZ)y NH
N
70 F-I -(cH -~~ OH 158C; cis
2)3
71 F-1 -(CH2)3-CN OH 162-163C, traps
72 F-1 -(CH2)3-~-CH3 OH 117-118C, traps
73 F-1 OH 199-200C, traps
(CHp)2- f
N~
CH3
74 F-2 -(CH2)2-O-(CH2)2-OH OH 99-100C, traps
75 F-1 OH i56C
~ cis
~
3 ,
-(CHZ)Z-N
N-CH
'cH,
Table F-4
oR4
L-N CHZ
Co. No. Ex. -L OR4 physical data
No.
m .C
24 F-1 -(CH2)3-CH3 OCH3 101.1C; cis
25 F-1 -(CH2)3-CH3 OH 185.1C; cis
CA 02235133 1998-04-17
WO 97/30031 PCT/EP97/00584
-36-
10
Table F-4-bis
OR4 C1
O
L-N~CH~-O-C,-~ ~ ~NH,
Co. No. Ex. -L OR4 physical data
No.
m .C
76 F-1 ~ OH 169-170C
traps
0, ,
,0
x
-(CHZ?3 ~CH3
77 F-4 ~ OH 199-200C, traps
-(CHZ)g--C-CHg
Table F-5
OR4 O O
O _
L-N CHI-O-C ~ ~ NH=
CI
Co. Ex. -L OR4 physical data
No. No. m . C
26 F-1 -(CH2)3-CH3 OCH 197.8C; cis
3
27 F-I -(CH2)3-CH3 OH I00C; cis
28 F-1 -(CH2)2-NH-(3-methyl-2-pyrazinyl)OH 176.6C; cis
29 F-1 -(CH2)2-NH-(3-methyl-2-pyrazinyl)OCH 184.7C; cis
3
30 F-1 -(CH2)3-CH3 OH 201.6C; traps;
.(COOH)2
31 F-I -(CH2)2-NH-(3-methyl-2-pyrazinyl)OH
traps
3? F-3 -(CH2)3-CH3 OH 106C; A-traps
33 F-3 -(CH2)3-CH3 OH 106C; B-traps
78 F-5 -(CH2)2-OH OH
traps
CA 02235133 1998-04-17
WO 97/30031 PCT/EP97/00584
-37-
Co. Ex. -L OR4
_ ~ No. physical data
No. m . C
79 F-6 -CH2-CH3 OH 130C; traps
80 F-2 -(CH2)3-CN OH 168C; txans
81 F-6 -(CH2)5-CH3 OH 148C; traps
82 F-2 -(CH2)3-O-(4-fluorophenyl)OH 158C; traps
83 F-1
cN OH
135
C; traps
~(CHZ)g-NH
N
84 F-2 -CH2-(2-tetrahydrofuranyl)OH
traps
85 F-2
OH -!-142C; traps
-(cH2)-
HaC Is~ S
86 F-2 -(CH2)2-O-(CH2 j2-OH OH 120C; traps
87 F-I -(CH2)3-O-CH(CH3)2 OH 180C; traps
88 F-2 0~ OH 170C; traps
c
~
-(CHZ) ~
CH~
89 F-2 -(CH2)2-(2-tetrahydrofuranyl)OH i30C; traps
90 F-5 ~ OH 200C; traps
(CHz )~-C-CHg
9 I F- I c~ OH
traps
--(CH2Ja-N'H
N
92 F-2 OH 138C; traps
~(CH,)i i
M\
CI
O
93 F-1 ~cH2~i-~-o-~3 OH 138C; traps
94 F-2 -CH2-CH=CH-CH3 OH 98C; E-traps _
95 F-I -CH2-(2-tetrahydrofuranyl)OH 119C; cis
96 F-2
OH 170C; traps
-(CHZ)2 N
N\
CHI
97 F-2 OH 140C; traps
-(cH2n- ~
y
h\
a
' CA 02235133 1998-04-17
WO 97/30031 PCTIEP97/00584
-38-
Co. Ex. -L OR4 physical data
No. No. m . C
98 F_1 cN
OH i
-(cH2)3-NH ~ ~ c
N s
99 F-1 -(CH2)3-O-CH3 OH
traps
100 F-2
OH I60C; traps
-(CHZ)3 N
N~
CH3
101 F-1 CH2cyclopropyI OH
traps
102 F-
I
,-(cHZ)3- OH cis
103 F-1 ~ OH 1 I2C; cis
c
--(CHZ) ~ ~CH
3
104 F-2
OH
116
C; cis
-(CHz).t'~
N
105 F-5 OH 75C; cis
-...(cH2),-c-cH,
106 F-2 OH I76C; traps
--(CHz)~-C-N
I 07 F-
I OH
~cH3 cis
~
-(CH2),-N
N-CH
~CH~
108 F-7 -(CH2)2-NH-S02-CH3 OH
traps
109 F-8 -(CH2)3-CH3 OH A-traps, HCI
110 F-8 -(CH2)3-CH3 OH A-traps, HBr
11 I F-8 -(CH2)3-CH3 OH A-traps, fumarate
112 F-8 -(CH2)3-CH3 OH A-traps, nitrate
l I3 F-g -(CH2)3-CH3 OH A-traps, malate
(S)
1 I4 F-8 -(CH2)3-CH3 OH A-traps, stearate
115 F-g -(CH2)3-CH3 OH A-traps, phosphate
I16 F-1 -(CH2)3-O-CH3 OH I 17-118C, cis
117 F-1
OH 169-I70C
t
,
rans
--(CHZ)z- i'
N\ //
118 F-1
OH 189-190C, traps
-(CHy)2- ~N-CH~CH;
CA 02235133 1998-04-17
WO 97/30031 PCT/EP97/00584
-39-
Co. Ex. -L OR4 physical data
No. No. m . C
119 F-1 _
OH 169-I70C
i
, c
-(CHz) s
N S
H3C
120 F-I -(CH2)3-CN OH 179-180C
i
, c
I21 F-I -(CH2)3-CH(4 fluorophenyl)OH s
2
149-150
C, traps
I 22 F-1
~ OH 147-148C, traps
(CHZ)3-N
N..CHZ_CHg
I23 F-1 cH~~N~ OH traps
N
CH;
124 F-1
~ OH 165-167C, cis
N_CHZ_CH~
-(CH2)9-N
125 F'- ~ OH 200-201 C
I traps
iCH3 ,
-(CHZ)2- ~N CH\
CH
3
126 F-2 -(CH2)2-O-(CH2)2-OH OH I80C; cis
127 F- I -cH,-cH=cH ~ ~ F OH 151 C, E-traps
128 F-1 ~ OH 180C, cis
-(CHZ7;- i'
N~
CH3
O
129 F-7 ~(CHZ)t~NH-C ~ ~ OH 141 C, traps
130 F-I OH I63-164, cis
(CH~)3-C-OCH3
Table F-6
OH C~
O
L.-IV~CH,-O-C ~ ~ r(H~
/ O ~O
I f
CHI CH;
CA 02235133 1998-04-17
WO 97/30031 PCT/EP97/00584
-40-
Co. Ex. -L OR4 physical data
No. No. m , in C
131 F-1 -(CH2)3-CH3 OH I 10C; trans
132 F-8 0~ OH I48C;
~(CH2)3 ~cH lhCqH
O
C
H
0(
3 4
4.
3
8
*), trans
133 F-1 0~ OH trans
-<CH
)g ~CH
2
;
OH 190 C
134 F-4 ~(CH2)3_,C-CH3 ; trans
135 F-1 -(CH2)3-CH3 OH I84C; cis
136 F-I o~ OH 158C; cis
-(CH
\CH
)
2
3
3
OH 170 C
137 F-4 ~(cH2)3~~-cH3 ; cis
185 F-1 _CH2-(2-tetrahydrofuranyl) OH cis
138 F-1 -CH2-(2-tetrahydrofuranyl) OH 193-194C, trans
i39 F-I -(cH2)3-.a OH 160-161C, cis
140 F-1 OH 150-ISIC, trans
-(CH2)2-N I
N~
H3
141 F-I
OH 127-128C, cis
~(CHy)2- t
N~
~3
142 F-1
cN OH 153-154C, cis
--(CH2)4-NH
N
143 F-I -(CH2)3-O-CH3 OH 179-I80C, trans
I44 F-2 -(CH2)2-O-(C~I2)2-OH OH I36-137C, cis
145 F-2 -(CH2)2-O-(CH2)2-OH OH I59-160C, trans
146 F-I
~ OH 130C, cis
N CH~CH3
-(CH2)2-N
~J ~cH
,
CA 02235133 1998-04-17
WO 97/30031 PCTIEP97l00584
-41-
Co. Ex. -L OR4 physical data
No. No. m . in C
147 F-I cN
OH 105C, cis
-(CHy)g-NH
N
148 F-2 -(cHZ)2-NH---rN, OH
traps
3 N
(*)I~ZCaHaO4.C3Hs0 means (E)-2-butenedioic acid salt (2:1). 2-propanol
Table F-7
OH CI
O
L-N_ ,-CHZ-O-C ~ ~ NHZ
O~O
Co. Ex. -L OR4 physical data
No. No. m . in C
149 F-1 -(CH2)3-CH3 OH 102C; traps
150 F-1 0 o OH 118C;trans
c
~
-(CHy)g
CH3
O
I51 F-4 -(CH~)3-C-CH; OH 148C; traps
152 F-1 -(CH2)3-CH3 OH 100C; cis
153 F-1 0~ OH 1 I9-I20C ,cis
c
-(CHZ)g ~CH3
OH 97-98 C
I54 F-4 -(CH2)3-CI-CHg ,C1S
155 F-I -CH2-(2-tetrahydrofuranyl) OH 119-120C .cis
156 F-I -CH2-(2-tetrahydrofuranyl) OH 205C; traps
157 F-1 (cH2),-o~ OH 89-90C, cis
~
158 F-1 -(CH2)3-O-CH3 OH I52C, cis
159 F-1 -(CH2)3-CN OH 158C, cis
CA 02235133 1998-04-17
WO 97J30031 PCT/EP97/00584
-42-
- Co. Ex. -L OR4 physical data
No. No. m . in C
160 F-1 ~ OH 164C
cis
'--(CH2)Z~-N~N CHI H3 ,
CH
3
161 F-1 ~
OH 190C,trans
-(cHz)z- ~
N~
C1
162 F-1
OH 200C
ci
,
-(CH~2- i s
N~
C1
I63 F-I -(CH2)3-O-CH3
OH I76C, traps
164 F-2 -(CH2)2-O-(CH2)2-OH OH 126C, traps
Table F-8
OH CI
O
L-N CH2-O._C ~ ~ NH
O O
Co. Ex. -L OR4 physical data
No. No. m . in C
165 F-1 OCH2)3-CH3 OH 153C; traps
166 F-1 0~ OH traps
c
-(CHZ)3 'CH
3
167 F-8 0~ OH 1/z C4H4O4
(*),
c
-(cH2), ~cH3 traps
168 F-4 ~ OH 182C; traps
(CH2)g-C-CHg
169 F-1 -(CH2)3-CH3 OH 98C; cis
CA 02235133 1998-04-17
WO 97130031 PCTJEP97/00584
-43-
Co. Ex. -L OR4 physical data
No. No. m . in C
170 F-I -CH2-(2-tetrahydrofuranyl) OH 166C; trans
171 F-1 -CH2-(2-tetrahydrofuranyl) OH 137C; cis
172 F-8 a~ OH C2H204 (**},
cis
-(CH
)
CH
y
g
g
I73 F-4 ~ OH I04C; cis
(CHZ)3-C-CH3
174 F-1 0 o OH 166C; cis
c
-(CH
)~ ~CH
i
3
175 F-1 -(cH~3-o~ OH 158C; cis
176 F-1
cN OH 134C; cis
-(CHZ)4-~
N
177 F-1 OH 115-116C
- cis
cH
-
-
( ,
z)3
c
N
cN
I78 F-1 ~ ~ OH 138C, cis
-(CHZ)3-NH
N
179 F-1 OH 124-I25C,trans
-(CHz)2- I
N~
CHg
180 F-1 -(CH2)3-O-CH3 OH 118-119C ,trans
181 F-1 OH 191C, cis
~
N cH''cH3
-(cH2)x-N
~CH
g
182 F-6 -(CH2)2-OH OH 150C, trans
183 F-1 -(CH2)~-NH~N~ OH trans
CHg N
(~')1/z C:4H404 means (E)-2-butenedioic acid salt (2:1)
(**)CZH~O4means ethanedioic acid salt (I:I)
Pharmacological example P-1
. Gastrointestinal motility in conscious dog
Strain gauge force transducers were calibrated before implantation (Schuurkes
et al.,
1978). Female beagle dogs, weighing 7-17 kg, were implanted with isometric
force
CA 02235133 1998-04-17
WO 97130031 PCT/EP97/00584
transducers, under general anesthesia and aseptic precautions. After a median
Iaparotomy, 4 transducers were sutured on the serosal side of either stomach
and
duodenum (to measure antroduodenal motility). To study motility of the
stomach, the
small intestines and the antroduodenal-coordination, transducers were placed
on the
antrurn (4 cm distance of the pylorus), on the pylorus and the duodenum (4 and
8 cm
distance of the pylorus). The wires were led via a subcutaneous tunnel on the
left costal
flank through a stab-wound between the scapulae. The connector was soldered to
the
lead wires and protected by a canvas jacket. Dogs were allowed a recovery
period of at
least two weeks. Experiments were started after a fasting period of -~- 20
hours, during
which water was available ad Iibitum. During the experiments, dogs were free
to move
in their cages. The cages were built in a special room, provided with glass
pervious to
light in one direction, i.e. the observator can see the dogs while the dogs
can not see the
observator. Via this system it was possible to observe the dogs for behavioral
changes
and to determine defecation events. The information from the transducers was
transmitted in digitized form {sampled at 5 Hz) by a small transmitter box.
This box
was placed in a jacket worn by the dog. The signals were received via a
microphone
above each cage and were transmitted to a central computer system {5 Hz rate).
It is
possible to monitor the motility of 8 dogs (4 channels per dog) 24 hours a
day.
Dogs with strain gauge force transducers, were fed in the quiescent phase of
the
interdigestive state, 30 min after the passage of a migrating motor complex
(MMC}
(phase III). The meal consisted of 75 g dog feed.
Two hours after feeding, solvent or test compound was administered p.o. (in a
volume
5 m1, preceded and followed by 2.5 mI water, via an orogastric tube).
Antroduodenal
motility was followed for at least 10 hours after drug administration.
The mean amplitude of the contractions of antzum, pylorus and duodenum during
a
period of 30 min was calculated as % of the mean maximal contractions during a
migrating motor complex (MMC). Drug effects on the amplitude of contractions
one
hour after administration were expressed in percentage of the amplitude 30 min
before
administration of the drugs. These effects were compared with the effects
after solvent
administration on the same dogs in the same period.
Improvement of coordination was also evaluated.
Table P-1
Effect of compound 30 on antrum, pylorus, duodenum and coordination
CA 02235133 1998-04-17
WO 97130031 PCT/EP97IOOS84
-45-
DA means the mean amplitude increase of the contractions of the antrum
(expressed in
MMC).
' DP means the mean amplitude increase of the contractions of the pylorus
(expressed in
% MMC).
' dD means the mean amplitude increase of the contractions of the duodenum
(expressed in % MMC).
Dose Changes in Improvement
(mg/kg P.O.) amplitude of
(expressed coordination
in %
MMC)
DA DP pD
0.00031 5 2I 9 3/4
0.00125 9 29 13 3/4
0.005 i 3 35 I3 4/6
0.02 12 34 12 5/6
0.08 24 37 I 3 4/6
0.3 I I 1 30 11 4/6
Phanmacolo~ical example P-2
Gastric emptying of an acaloric liquid test meal delayed by administration of
Iidamidine, in conscious dogs.
Female beagle dogs, weighing 7-14 kg, were trained to stand quietly in Pavlov
frames.
They were implanted with a gastric cannula under general anesthesia and
aseptic
precautions. After a median laparatomy, an incision was made through the
gastric wall
in the longitudinal direction between the greater and the lesser curve, 2 cm
above the
nerves of Latarjet. The cannula was secured to the gastric wall by means of a
double
purse string suture and brought out via a stab wound at the left quadrant of
the
hypochondrium. Dogs were allowed a recovery period of at least two weeks.
Experiments were started after a fasting period of 24 hours, during which
water was
available ad libitum. At the beginning of the experiment, the cannula was
opened in
order to remove any gastric juice or food remnants.
The stomach was cleansed with 40 to 50 ml lukewarm water. The test compound
was
administered LV. (in a volume <_ 3 ml via the vena cephalica), S.C. {in a
volume
<_ 3 ml) or P.O. (in a volume of 1 ml/kg body weight, applied intragastrically
via the
cannula with a device that filled the lumen of the cannula; after injection of
the test
CA 02235133 1998-04-17
WO 97/30031 PCT/EP97/00584
-46-
compound, 5 ml NaCl 0.9 % was injected in order to correct for the dead space
in the
injection system). Immediately after administration of the test compound or
its solvent,
lidamidine 0.63 mg/kg was administered subcutaneously. 30 min later, the
cannula was
opened to determine the amount of fluid present in the stomach, promptly
followed by
reintroduction of the fluid. Then the test meal was administered via the
cannuIa. This
test meal consisted of 250 ml distilled water containing glucose (5 g/I) as a
marker.
The cannula remained closed for 30 min, whereafter the gastric contents were
drained
from the stomach to measure total volume (t = 30 min). For later analysis 1 ml
of the
gastric contents was taken, promptly followed by reintroduction of the rest
volume into
the stomach. This sequence was repeated 4 times with 30 min intervals (t = 60,
90,
120, 150 min).
In the 1 ml samples of the gastric contents, the glucose concentrations were
measured
on a Hitachi 717 automatic analyzer by the hexokinase method (Schmidt, 1961 ).
These
data were used to determine the absolute amount of glucose that remained in
the
I5 stomach after each 30 min period, as a measure for the rest volume of the
meal itself,
independent of acid secretion.
Curves were fitted to the measurement points (glucose versus time) using
weighed non-
linear regression analysis. Gastric emptying was quantified as the time needed
to
empty 70% of the meal {t 70010). The control emptying time was calculated as
the mean
t 7p% of the last 5 solvent experiments of the same dog. Acceleration of
delayed
gastric emptying (fit) was calculated as the time difference between t 70%
compound
and t 70% solvent. To correct for variations in emptying rate between dogs, Ot
was
expressed as % of t 70% solvent {Schuurkes et a1, 1992)).
Table P-2
Acceleration of gastric emptying of a liquid meal delayed by Iidamidine in
conscious
dog by compound 30.
Dose(m /k Acceleration ex ressed in
) OTIT
0.00016 + 0.16
0.00063 - 0.26
0.0025 - 0.46
0.01 - 0.60
0.04 - 0.67
CA 02235133. 1998-04-17
WO 97130031 PCT/EP97/00584
-4~-
Table P-3
The acceleration emptying liquid ayed ne
in
of gastric of meal by
a del lidamidi
conscious ds
dog was measured at
for the following a
compoun dose
of
0
0025
. mg/lcg.
Co. No. TIT 77 -0 1
43
. 08 -0.49
3 -0.01 78 -0.25 126 -0.15
- 0.22 79 -0.40 127 -0.25
16 -0.54 80 -0.39 132 -0.06
21 -0.25 81 -0.41 134 -0.45
27 -0.12 83 -0.22 135 -0.I7
30 -0.55 84 -0.38 138 -0.35
31 -0.31 85 -O.lI 139 -0.25
32 -0.54 86 -0.51 140 -0.39
34 -0.77 87 -0.38 141 -0.12
35 -0.28 88 -0.53 143 -0.37
41 -0.45 89 -0.25 15I -0.52
42 -0.25 90 -0.46 I52 -0.15
43 -0.17 91 -0.34 153 -0.40
44 -0.43 92 -0.44 154 -0.2I
46 -0.04 93 -0.41 156 -0.36
47 -0.26 94 -0.55 158 -0.34
48 -0.39 95 -0.13 159 -0,09
49 -0.58 96 -0.48 165 -0.14
SO -0.73 97 -0.35 167 -0.15
51 -0.49 98 -0.30 169 -0,1
g
52 -0.49 99 -0.71 I70 -0.25
54 -0.05 100 -0.50 172 -O.I7 _
56 -0.31 101 -0.45 181 -0.08
65 -0.23 102 -0.01 182 -0.40
66 -O.I4 103 -O.I3 184 -0.20
68 -0.32 104 -0.29 I85 -0.23
71 -0.20 105 -0.23
73 -0.72 106 -0.39
76 -0.59 I07 0.24
CA 02235133 1998-04-17
WO 97730031 . PCTJEP97/00584
_48_
Table P-4
The acceleration of gastric emptying of a liquid meal delayed by Iidamidine in
conscious dog was measured for the following intermediates at a dose of 0.0025
mg/kg.
Interm. OT/T
No
6-j -0.18
6-k -0.49
6-I -0.75
6-m -0.30
7-a -0.32
Composition examples
The following formulations exemplify typical pharmaceutical compositions in
dosage
unit form suitable for systemic or topical administration to warm-blooded
animals in
accordance with the present invention.
"Active ingredient" (A.L) as used throughout these examples relates to a
compound of
formula (I), a N-oxide form, a pharmaceutically acceptable acid or base
addition salt or
a stereochemically isomeric form thereof.
Examt~Ie C-1 : Oral solutions
9 g of methyl 4-hydroxybenzoate and 1 g of propyl 4-hydroxybenzoate are
dissolved in
4 I of boiling purified water. In 3 I of this solution are dissolved first 10
g of
2,3-dihydroxybutanedioic acid and thereafter 20 g of the A.I. The latter
solution is
combined with the remaining part of the former solution and 121 of 1,2,3-
propanetriol
and 3 I of sorbitol 70% solution are added thereto. 40 g of sodium saccharin
are
dissolved in 0.5 I of water and 2 m1 of raspberry and 2 ml of gooseberry
essence are
added. The latter solution is combined with the former, water is added q.s. to
a volume
of 201 providing an oral solution comprising 5 mg of the A.i. per teaspoonful
(5 m1).
The resulting solution is filled in suitable containers.
Exarnpie C-2 : Capsules
20 g of the A.L, 6 g sodium Iauryl sulfate, 56 g starch, 56 g lactose, ~0.8 g
colloidal
silicon dioxide, and 1.2 g magnesium stearate are vigorously stirred together.
The
resulting mixture is subsequently filled into 1000 suitable hardened gelatin
capsules,
each comprising 20 mg of the A.L.
CA 02235133 1998-04-17
WO 97130031 PCT/EP97/00584
-49-
Example C-3 : Film-coated tablets
_ . Preparation of tablet core
A mixture of 100 g of the A.L, 570 g lactose and 200 g starch is mixed well
and
thereafter humidified with a solution of 5 g sodium dodecyl sulfate and IO g
polyvinyl-
pyrrolidone in about 200 ml of water. The wet powder mixture is sieved, dried
and
' sieved again. Then there are added 100 g microcrystanline cellulose and 15 g
hydrogenated vegetable oil. The whole is mixed well and compressed into
tablets,
giving 10.000 tablets, each comprising 10 mg of the active ingredient.
Coating
To a solution of IO g methyl cellulose in 75 rnl of denaturated ethanol there
is added a
solution of 5 g of ethyl cellulose in ISO ml of dichiorornethane. Then there
are added 75
ml of dichloromethane and 2.5 ml 1,2,3-propanetriol. IO g of polyethylene
glycol is
molten and dissolved in 75 ml of dichloromethane. The latter solution is added
to the
former and then there are added 2.5 g of magnesium octadecanoate, 5 g of
I5 polyvinylpyrrolidone and 30 ml of concentrated color suspension and the
whole is
homogenated. The tablet cores are coated with the thus obtained mixture in a
coating
apparatus.
Examine C-4 : Iniectable solution
I .8 g methyl 4-hydroxybenzoate and 0.2 g propyl 4-hydroxybenzoate were
dissolved in
about 0.5 I of boiling water for injection. After cooling to about 50°C
there were added
while stirring 4 g lactic acid, 0.05 g propylene glycol and 4 g of the A.I.
The solution
was cooled to room temperature and supplemented with water for injection q.s.
ad 1 1
volume, giving a solution of 4 mg/ml of A.I. The solution was sterilized by
filtration
and filled in sterile containers.
Example C-5 : Suppositories
3 Grams A.I. was dissolved in a sonution of 3 grams 2,3-dihydroxybutanedioic
acid in
25 ml ponyethylene glycol 400. 12 Grams surfactant and 300 grams triglycerides
were
molten together. The latter mixture was mixed well with the former solution.
The thus
obtained mixture was poured into molds at a temperature of 37-38°C to
form I00
suppositories each containing 30 mglml of the A.I.