Note: Descriptions are shown in the official language in which they were submitted.
CA 02235942 1998-04-23
WO~ 97/17356 PCT/US96116585
TRICYCLIC ERYTHROMYCIN DERIVATIVES
- Technical Field
The present invention relates to novel semisynthetic macrolides having
antibacterial
activity and useful in the treatment and prevention of bacterial infections.
More particularly,
the invention relates to tricyclic erythromycin derivatives, compositions
containing such
compounds and methods for using the same, as well as processes for making such
compounds.
Background of the Invention
Erythromycins A through D, represented by formula (E),
CH3
NMez
O
HO,,~
g 2'
CH3.,~ OH ~
CH
~
~ Erythromycin Ra Rb
3
Ho,""
s
,",.
o
o"
cH3
A -OH -CH3
, H B -H -CH3
HsC H
CH3
CH3
'
~,"~s~"
C -OH -H
CH3
4"
H
cH3
, D -H -H
~
OH
O
''~'
CH3
ORb
are well-known and potent antibacterial agents, used widely to treat and
prevent bacterial
infection. As with other antibacterials, however, bacterial strains having
resistance or
insufficient susceptibility to erythromycin have been identified. Also,
erythromycin A has
only weak activity against Gram-negative bacteria. Therefore, there is a
continuing need to
identify new erythromycin derivative compounds which possess improved
antibacterial
activity, which have less potential for developing resistance, which possess
the desired
Gram-negative activity, or which possess unexpected selectivity against target
microorganisms. Consequently, numerous investigators have prepared chemical
derivatives
of erythromycin in an attempt to obtain analogs having modified or improved
profiles of
antibiotic activity.
Ka.shimura et al. have disclosed 6-O-methylerythromycin derivatives having a
tricyclic basic nuclear structure in European Application 559896, published
November 11,
1991. Also, Asaka et al. have disclosed 5-O-desoaminylerythronolide
derivatives containing
a tricyclic carbamate structure in PCT Application WO 93/21200, published
April 22, 1992.
-1-
CA 02235942 1998-04-23
WO 97/17356 PCT/LTS96/16585
~ummarv of the Invention
The present invention provides a novel class of antibacterial tricyclic
erythromycin
compounds which possess antibacterial activity.
In one aspect of the present invention are disclosed novel tricyclic
erythromycin
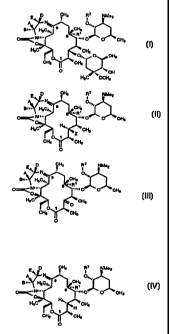
compounds selected from the group having the formulas:
CH3 R2 NMe2
'a
A ~ 0.,
H3C''~. CH3
..
,~~ R~
O~ Nn.. s
....a O CH3
O
HsCv.,, CHa
~0,,,. O CH3
O
CH3 CH3
O .~~~ OH
(I) H3C '~OCH3
CH3 RZ NMe2
~, 1
A N
H 9 . CH3 O''~.
gm..
6 .~eRi ~
O~ Nm.. ~~~n O O_ ' CH3
O
HsC~~'. H~~~ CH
3
O 1
CH3 CH3
(B) O
p CH3 R2 NMe2
.:
N
A
gi ~.. ' ~ O'',
H3C~~,. 9 CH3
.~.R~
O~Nn..
""~ O O CH3
O
H3Cv.,. CHs
_O
O
CH3 CH3
C > and
-2-
CA 02235942 1998-04-23
WO 97/17356 PCT/US96/16585
p CH3 RZ NMe2
E,
0...
gm.. H3C~~, 9 CH3
~~~ R~
O~ Nm. s ...n O Cl-13
O
HaCv,. H~.. CHs
'H
O
CH3 ~ CH3
(n') ° ;
as well as the pharmaceutically acceptable salts and esters thereof. 1n
formulas (1) - (IV)
above,
A, B, D and E are independently selected from the group consisting of:
(a) hydrogen;
(b) C1-C6-alkyl, as defined below, optionally substituted with one or
more substituents selected from the group consisting of:
(i) aryl, as defined below;
(ii) substituted-aryl, as defined below;
(iii) heteroaryl, as defined below;
(iv) substituted-heteroaryl, as defined below;
(v) heterocycloalkyl, as defined below;
(vi) hydroxy;
(vii) C1-C6-alkoxy, as defined below;
(viii) halogen consisting of Br, Cl, F or I; and
(ix) NR3R4, where R3 and R4 are independently selected from
hydrogen and C1-C6-alkyl, or R3 and R4 are taken with the nitrogen atom to
which they are
connected to form a 3- to 7-membered ring optionally containing a hetero
function consisting
of -O-, -NH-, -N(C1-C6-alkyl-)-, -N(aryl-C1-Cs-alkyl-)-, -N(substituted-aryl-
C1-C6-alkyl-
)-, -N(heteroaryl-C1-C6-alkyl-)-, -N(substituted-heteroaryl-C1-C6-alkyl-)-, -S-
or -S(O)S-,
wherein n is 1 or 2;
(c) C3-C7-cycloalkyl;
(d) aryl;
(e) substituted-aryl;
(fj heteroaryl;
(g) substituted-heteroaryl;
(h) heterocycloalkyl;
~ and
(i) a group selected from option (b) above further substituted with -M-
R5, wherein M is selected from the group consisting of
-3-
CA 02235942 1998-04-23
WO 97/17356 PCT/US96/16585
(aa) -C(O)-NH-;
(bb) W-C(O)-;
(cc) -NH-
(dd) -N(CH3)-
(ee) -O-
(ff) -S(O)S-, wherein n is 0, 1 or 2; and
(gg) -C(=NH)-NH-;
and RS is selected from the group consisting of:
(aaa) Ci-C6-alkyl, optionally substituted with a substituent selected
i0 from the group consisting of:
(i) aryl;
(ii) substituted-aryl;
(iii) heteroaryl; and
(iv) substituted-heteroaryl;
(bbb) aryl;
(ccc) substituted-aryl;
(ddd) heteroaryl;
(eee) substituted-heteroaryl; and
(fff) heterocycloalkyl;
or
any one pair of substituents, consisting of AB, AD, AE, BD, BE or DE, is taken
together with the atom or atoms to which they are attached to form a 3- to 7-
membered ring
optionally containing a hetero function consisting of:
-O-,
-NH-,
-N(C i-C6-alkyl-)-,
_N(aryl_C i _C6_~Yl-)_~
-N(substituted-aryl-C i-C6-allcyl-)-,
-N(heteroaryl-Ci-C6-alkyl-)-,
-N(substituted-heteroar 1-C
Y i-C6-aikYl-)-,
-S- or -S(O)S-, wherein n is 1 or 2;
-C(~)-NH-;
-C(O)-NR5-, wherein R5 is as described above;
W-C(O)-;
-NR5-C(O)-, wherein R5 is as described above; and ,
-C(=NH)-NH-;
Ri is selected from the group consisting of:
(a) hydrogen;
-4-
CA 02235942 1998-04-23
WO 97/17356 PCT/US96/16585
(b) hydroxy;
(c) _O_Cl_C3_alkyl;
(d) -O-C3-C5-cycloalkyl;
(e) -O-Cl-C3-alkyl-C3-CS-cycloalkyl;
(f) -O-C(O)-Cl-C3-alkyl;
(g) -O-C(O)-O-Cl-C3-alkyl;
and
(h) _O_C(O)_NH_C1_C3_~'l;
R2 is hydrogen or a hydroxy-protecting group, as defined below; and
Z is hydroxy or protected-hydroxy;
with the provisos that when the compound is of Formulas (I) or (IIII) then A,
B, D, and E
may not all be hydrogen, D and E may not be C1-C3-alkyl when A and B are
hydrogen, nor
may one of D and E be hydrogen and the other be C1-C3-alkyl when A and B are
hydrogen.
In another aspect of the present invention are disclosed pharmaceutical
compositions
comprising a therapeutically effective amount of a compound of the invention
in combination
with a pharmaceutically acceptable carrier and treatment of antibacterial
infections with such
compositions. Suitable carriers and methods of formulation are also disclosed.
The
compounds and compositions of the present invention have antibacterial
activity.
In a further aspect of the present invention are provided processes for the
preparation
of tricyclic macrolide derivatives of Formulas (I), (II), (IIn and (IV) above.
~et~iled Description of the Invention
In one embodiment of the present invention are compounds selected from the
group
having formula (1) above, wherein A, B, D, E, and R1-RS are as described
above.
In a second embodiment of the present invention are compounds selected from
the
group having formula (II) above, wherein A, B, D, E, R1-RS and Z are as
described above.
In another embodiment of the present invention are compounds selected from the
group having formula (III) above, wherein A, B, D, E, and R 1-RS are as
described above.
In yet another embodiment of the present invention are compounds selected from
the
group having formula (IV) above, wherein A, B, D, E, and Ri-RS are as
described above.
In one preferred embodiment of the present invention are compounds of formula
(III)
above wherein R1 is hydrogen, hydroxy or methoxy, R2 is hydrogen, and A, B, D,
E and
R1-RS are as described above.
In another preferred embodiment of the present invention are compounds of
formula
(III) above wherein R1 is hydrogen or methoxy, R2 is hydrogen, and any three
of the A, B,
D and E groups are hydrogen and the other group is selected from a singly
substituted Ci-
C6-alkyl group comprised of -(CH2)mR6 where m=l, 2, 3 or 4 and R6 is:
(a) aryl;
(b) substituted-aryl;
-5-
CA 02235942 1998-04-23
WO 97/17356 PCT/US96/16585
(c) heteroaryl;
(d) substituted-heteroaryl;
(e) heterocycloalkyl;
(f) hydroxy;
(g) C1-C6-alkoxy
(h) NR3R4, where R3 and R4 are independently selected from hydrogen and Cl-
C6-alkyl, or R3 and R4 are taken with the nitrogen atom to which they are
connected to form
a 3- to 7-membered ring optionally containing a hetero function consisting of -
O-, -NH-,
-N(Cl-C6-alkyl-)-, -N(aryl-C1-C6-alkyl-)-, -N(substituted-aryl-C1-C6-alkyl-)-,
l0 -N(heteroaryl-C1-C6-alkyl-)-, -N(substituted-heteroaryl-Ci-C6-alkyl-)-, -S-
or -S(O)S-,
wherein n is 1 or 2;
(i) halogen consisting of Br, Cl, F or I;
Ci-C3 alkyl; or
(k) -(CH2)r-M-(CH2)S-R~ wherein r=0,1 or 2; s=0,1 or 2 and M is
(aa) -C(O)-NH-;
(bb)
(cc) -NH-
(dd) -N(CH3)-
(ee) _O_
(ff) -S(O)S-, wherein n is 0, 1 or 2; and
(gg) -C(=NH)-NH-;
and R~ is selected from the group consisting of:
(aaa) Ci-C3-alkyl,
(bbb) aryl;
(ccc) substituted-aryl;
(ddd) heteroaryl; and
(eee) substituted-heteroaryl.
In yet another preferred embodiment of the present invention are compounds of
formula (IIn above wherein R1 is hydrogen or methoxy, R2 is hydrogen, B=E=H,
and A
and D taken together is selected from the group consisting of:
(a) -CH2-Z-CH2-, wherein Z is
(aa) -C(O)-NH_;
(bb) -C(O)-NR5-, wherein R5 is selected from the group consisting
of:
(aaa) Ci-C6-alkyl, optionally substituted with a substituent
selected from the group consisting of:
(i) aryl;
(ll) substituted-aryl;
-6-
CA 02235942 1998-04-23
WO 97/17356 PCT/US96/16585
(iii) heteroaryl; and
(iv) substituted-heteroaryl;
(bbb) aryl;
(ccc) substituted-aryl;
(ddd) heteroaryl;
(eee) substituted-heteroaryl; and
(fff) heterocycloalkyl;
(cc) -NH-C(O)-;
(dd) -NR5-C(O)-, wherein R5 is as defined .above;
(ee) -NH-
(ff) -N(CH3}-
(gg) -
(hh) -S(O)n-, wherein n is 0, 1 or 2; and
(ii) -C(=NH)-NH-;
(b) -CH2-N(-(CH2)S-R')-CH2-, wherein s=0,1 or 2, and R~ is selected from the
group consisting of:
(aaa) C1_C3_alkYl,
(bbb) aryl;
(ccc) substituted-aryl;
(ddd) heteroaryl; and
(eee) substituted-heteroaryl;
(c) -CH2-N(-(CH2)r-M-(CH2)S-R')-CH2-, wherein r=0,1 or 2; s=0,1 or 2, and
M and R~ are as defined above; and
(d) -CH2-(CH2)r-CH2-, wherein r=0,1 or 2.
Representative of the compounds of the invention include:
Compound of Formula (IV); Ri=methoxy; R2=hydrogen; A=B=D=E=hydrogen;
Compound of Formula (III), A=B=E=H, D=benzyl, R1=methoxy, R2=hydrogen;
Compound of Formula (III), A=B=D=H, E=benzyl, R 1=methoxy, R2=hydrogen;
Compound of Formula (III); A=benzyl, B=D=E=H, Rl=metlloxy;
Compound of Formula (III); B=benzyl, A=D=E=H, R1=methoxy, R2=hydrogen;
Compound of Formula (III); A=E=phenyl, B=D=H, R1=methoxy, R2=hydrogen;
Compound of Formula (III); A=methyl, B=D=E=H, Rl=methoxy, R2=hydrogen;
Compound of Formula (III); B=methyl, A=D=E=H, R1=methoxy, R2=hydrogen;
Compound of Formula (III); A=D=methyl; B=E=H, R i=methoxy, R2=hydrogen;
Compound of Formula (III); A=E=methyl; B=D=H, R1=methoxy, R2=hydrogen ;
Compound of Formula (III); B=D=H; A and E taken together is -CH2CH2CH2-,
Ri=methoxy, R2=hydrogen;
CA 02235942 1998-04-23
WO 97/17356 PCT/US96/16585
Compound of Formula (III); B=E=H; A and D taken together is -CH2CH2CH2CH2-,
Ri=methoxy, R2=hydrogen ;
Compound of Formula (I); A=B=D=E=hydrogen; R i=hydrogen, R2=hydrogen;
Compound of Formula (III): R'=OCH3, RZ=H; A=B=D=H; E=-CH2NH2;
Compound of Formula (III): R1=OCH3, RZ=H; A=B=E=H; D=-CH2NH2;
Compound of Formula (III): RI=OCH3, RZ=H; B=E=H; A and D taken together is
-CH2CH2CH2-;
Compound of Formula (III): R1=OCH3, R2=H; B=E=H; A and D taken together is
-CH20CHz-;
Compound of Formula (III): R1=OCH3, RZ=H; B=E=H; A and D taken together is
-CH2-NH-CH2-;
Compound of Formula (DI): R1=OCH3, R2=H; B=E=H; A and D taken together is
-CH2-N(Cbz)-CH2-;
Compound of Formula (III): R I=OCH3, Rz=H; B=E=H; A and D taken together is
-CH2-N(benzyl)-CH2-;
Compound of Formula (III): R1=OCH3, R2=H; B=E=H; A and D taken together is
-CH2-N(benzoyl)-CH2-;
Compound of Formula (llI): R1=OCH3, RZ=H; B=E=H; A and D taken together is
-CH2-N(phenyl-CH2-CH2-)-CHZ-;
Compound of Formula (III): R1=OCH3, RZ=H; B=E=H; A and D taken together is
-CH2-N(4-Cl-phenyl-CH2-)-CH2_;
Compound of Formula (la): R1=OCH3, R2=H; B=E=H; A and D taken together is
-CH2-N(4-pyridyl-CH2-)-CH2-;
Compound of Formula (DI): R1=OCH3, R2=H; B=E=H; A and D taken together is
-CH2-N(2-pyridyl-CH2-)-CH2-;
Compound of Formula (III): RI=OCH3, RZ=H; B=E=H; A and D taken together is
-CH2-N(3-pyridyl-CH2-)-CH2-;
Compound of Formula (DI): RI=OCH3, R'=H; B=E=H; A and D taken together is
-CH2-N(4-quinolyl-CH2-)-CH2-;
Compound of Formula (III): R1=OCH3, R2=H; B=E=H; A=D=-CH2-O-CH2-
phenyl;
Compound of Formula (III): R 1=OCH3, R2=H; B=E=H; A=D=-CH2-OH;
Compound of Formula (III): R'=OCH3, R2=H; B=E=H; A=D=-CH2-O-phenyl;
Compound of Formula (lII): RI=OCH3, RZ=H; A=B=H; D and E taken together is
-CH2-CH2-CH2-CH2-;
Compound of Formula (III): R1=OCH3, RZ=H; A and B taken together is
-CH2-CH2-CH2-CH2-; D=E=H;
_g_
CA 02235942 1998-04-23
W~ 97/17356 PCT/US96/16585
Compound of Formula (III): R1=OCH3, R2=H; A=B=H; D and E taken together is
-CH2-O-CH2-;
Compound of Formula (III): R1=OCH~, R2=H; A=D=E=H; B=-CH2-CH2-phenyl ;
Compound of Formula (III): R1=OCH3, R2=H; A=D=E=H; B=-
CH2-CH2-CH2-phenyl ;
Compound of Formula (III): R1=OCH3, R2=H; A=D=E=H; B=-
CH2-O-CH2-phenyl ;
Compound of Formula (III): R'=OCH3, R~=H; A=D=E=H; B=-
CH2-CH2-(4-OCH3-phenyl );
Compound of Formula (III): R'=OCH3, R2=H; A=-CH2-CH2-phenyl; B=D=E=H ;
Compound of Formula (III): R1=OCH3, R2=H; A=-CH2-CH2-CH2-phenyl;
B=D=E=H ;
Compound of Formula (III): R1=OCH3, RZ=H; A=-CH2-O-CH2-phenyl ;
B=D=E=H ;
i5 Compound of Formula (III): Rl=OCH3, R2=H; B=D=E=H; A=-
CH2-CH2-(4-OCH3-phenyl);
Compound of Formula (III): R'=OCH3, RZ=H; A=B=D=H; E=-CH2-CH2-Ph;
Compound of Formula (III): RI=OCH3, R2=H; A=B=E=H; D=-CH2-CH2-Ph;
Compound of Formula (III): RI=OCH3, R2=H; A=B=D=H; E=-
2o CH2-CH2-CH2-Ph;
Compound of Formula (III): Ri=OCH3, R2=H; A=B=E=H; D=-
CH2-CH2-CH2-Ph;
Compound of Formula (III): R1=OCH3, RZ=H; A=- CH2-CH2-OPh; B=D=E=H;
Compound of Formula (III): R1=OCH3, R2=H; A=- CH2-CH2-NHCbz;
25 B=D=E=H;
Compound of Formula (III) R1=OCH3, R2=H; A=-CH2-CH2-NH2; B=D=E=H;
Compound of Formula (III): R~=OCH3, R2=H; A=- CH2-CO2Benzyl; B=D=E=H;
Compound of Formula (III) Rl=OCH3, R2=H; A=-CH2-COOH; B=D=E=H;
Compound of Formula (III) R1=OCH3, R2=H; A=-CH2-CHz-OH; B=D=E=H;
30 Compound of Formula (III): R1=OCH3, R2=H; A=- CH2-CH2-NH(4-Pyridyl-);
B=D=E=H
Compound of Formula (III): R'=OCH3, RZ=H; A=B=D=H; E=-CH2-OH;
Compound of Formula (III): R1=OCH~, R2=H; A=B=E=H; D=-CH2-OH;
Compound of Formula (III): RI=OCH3, R2=H; A=B=E=H; D=-CH2-NHBenzoyl;
35 Compound of Formula (III): R'=OCH3, R2=H; A=B=E=H; D=-CH2-NHBenzyl;
Compound of Formula (III): Rl=OCH~, R2=H; A=B=D=H; E=-CH2-NHBenzoyl;
Compound of Formula (III): R'=OCH3, RZ=H; A=B=D=H; E=-CH2-NHBenzyl;
-9-
CA 02235942 1998-04-23
WO 97/17356 PCT/US96/16585
Compound of Formula (IIT): R1=OCH3, RZ=H; B=D=H; A=E=-CH20CH2(4-Cl-
phenyl);
Compound of Formula (III): R1=OCH3, R'=H; A=B=E=H; D=-CH2-N(CH3)-
Benzyl;
Compound of Formula (III): R1=OCH3, R2=H; A=B=D=H; E=-CH2-N(CH3)-
Benzyl;
Compound of Formula (III): Ri=OCH3, R2=H; A=B=D=H; E=-CH2-NH-phenyl;
and
Compound of Formula (III): R1=OCH3, R'=H; A=B=E=H; D=-CH2-NH-phenyl.
A selected group of representative compounds includes:
Compound of Formula (IV); Ri=methoxy; R2=hydrogen; A=B=D=E=hydrogen;
Compound of Formula (lII), A=B=E=H, D=benzyl, R i=methoxy, R2=hydrogen;
Compound of Formula (III), A=B=D=H, E=benzyl, R i=methoxy, R2=hydrogen;
i5 Compound of Formula (III); A=benzyl, B=D=E=H, Ri=methoxy, R2=hydrogen;
Compound of Formula (III); B=benzyl, A=D=E=H, Ri=methoxy, R2=hydrogen;
Compound of Formula (III); A=E=phenyl, B=D=H, R 1=methoxy, R2=hydrogen;
Compound of Formula (BI); A=methyl, B=D=E=H, R1=methoxy, R2=hydrogen;
Compound of Formula (III); B=methyl, A=D=E=H, Ri=methoxy, R2=hydrogen;
Compound of Formula (lII); A=D=methyl; B=E=H, R i=methoxy, R2=hydrogen;
Compound of Formula (III); A=E=methyl; B=D=H, R i=methoxy, R2=hydrogen ;
Compound of Formula (III); B=D=H; A and E taken together is -CH2CH2CH2-,
R i=methoxy, RZ=hydrogen;
Compound of Formula (III); B=E=H; A and D taken together is -CH2CH2CH2CH2-,
R i=methoxy, R2=hydrogen ; and
Compound of Formula (I); A=B=D=E=hydrogen; R i=hydrogen, R2=hydrogen.
One object of the present invention is to provide a process for the
preparation of
tricyclic macrolide derivatives having the Formulas:
CH3 R2 NMe2
.:
0.,
gn.. H3C~~, 9
CH3
.~~ Ri
N~~..
.. O CH3
O CH3
O .~~~ OH
H3C .'~OCH3 ~ (I),
CH3
CH3 CH3.
- 10-
CA 02235942 1998-04-23
W(3 97/17356 PCT/CTS96/16585
CHs R2 NMe2
p ~' ~ 0...
CHs
gn.. H3C,~,.
s ... R ~
O~ Nm ..
CHs
O
HaC~~,. H~.. CHs
'Z
O
CHs CHs
O , (~),
p CHs R2 NMe2
E~. I
A N~ 0...
H3C,~,. 9 CHs
~ ... R
O~Nm.. ...u O O CHs
O
H3C~~~~ ~ CHs
'O
O 1
CHs CHs
(III) or
CHs R2 NMe2
p ~' ~ 0.,.
gm.. H3C,~, 9 CHs .
~~~R
O~ N~ ~.. s ...» O CHs
O
H3Cv,. H~.. CHs
'H
O
CHs CHs
O ~ (IV)~
wherein
A, B, D and E are independently selected from the group consisting of:
(a) hydrogen;
(b) C1-C6-alkyl, as defined below, optionally substituted with one or
more substituents selected from the group consisting of:
(i) aryl, as defined below;
' (ii) substituted-aryl, as defined below;
(iii) heteroaryl, as defined below;
' (iv) substituted-heteroaryl, as defined below;
(v) heterocycloalkyl, as defined below;
(vi) hydroxy;
-il-
CA 02235942 1998-04-23
WO 97/17356 PCT/US96/16585
(vii) C1-C6-alkoxy, as defined below;
(viii) halogen consisting of Br, Cl, F or I; and
(ix) NR3R4, where R3 and R4 are independently selected
from
hydrogen and C1-C6-alkyl,
or R3 and R4 are
taken with the
nitrogen atom
to which they
are
connected to 3- to 7-membered ring optionally containing a hetero
form a function consisting
of -O-, -NH-, -N(C1-C6-alkyl-)-, -N(aryl-Ci-C6-alkyl-)-, -N(substituted-aryl-
C1-C6-alkyl-)-,
-N(heteroaryl-Ci-C6-alkyl-)-,
-N(substituted-heteroaryl-C1-C6-alkyl-)-,
-S- or -S(O)n-,
wherein n is 1
or 2;
(c) C3-C~-cycloalkyl;
l0 (d) aryl;
(e) substituted-aryl;
(f7 heteroaryl;
(g) substituted-heteroaryl;
(h) heterocycloalkyl;
~d
(i) a group selected from option (b) above further substituted
with -M-
R5, wherein M is
selected from
the group consisting
of:
(aa) -C(O)-NH-;
(bb) -~-C(O)-;
(cc) -NH-
(dd) -N(CH3)-
(ee) -O-
(rr) -S(O)S-, wherein n is 0, 1 or 2; and
(gg) -C(=NH)-NH-;
and R5 is selected
from the group
consisting of:
C1-C6-~yl, optionally substituted with a substituent
selected from
the group consisting
of:
(i) aryl;
(ii) substituted-aryl;
(iii) heteroaryl; and
(iv) substituted-heteroaryl;
(bbb) aryl;
(ccc) substituted-aryl; ,
(ddd) heteroaryl;
(eee) substituted-heteroaryl; and ,
(fffj heterocycloalkyl;
or
-12-
CA 02235942 1998-04-23
WO 97/17356 PCT/US96/16585
any one pair of substituents, consisting of AB, AD, AE, BD, BE or DE, is
taken together with the atom or atoms to which they are attached to form a 3-
to 7-membered
ring optionally containing a hetero function consisting of
-O-,
-NH-,
-N(C 1-C6-a~Yl-)->
-N(~.3'1-C1-C6-a~Yl-)->
-N(substituted-aryl-C1-C6-alkyl-)-,
-N(heteroaryl-C1-C6-alkyl-)-,
-N(substituted-heteroaryl-C 1-C6-alkyl-)-,
-S- or -S(O)S-, wherein n is 1 or 2;
-C(O)-NH-;
-C(O)-NRS-, wherein RS is as described above;
-NH-C(O)-;
-NRS-C(O)-, wherein RS is as described above; and
-C(-NH)_NH_;
Rl is selected from the group consisting of:
(a) hydrogen;
(b) hydroxy;
(c) -O-C1-C3-alkyl;
(d) -O-C3-CS-cycloalkyl;
(e) -O-C1-C3-alkyl-C3-Cg-cycloallcyl;
(f) -O-C(O)-C1-C3-alkyl;
(g) -O-C(O)-O-Ci-C3-alkyl; and
(h) -O-C(O)-NH-C1-C3-alkyl;
R2 is hydrogen or a hydroxy-protecting group, as defined below; and
Z is hydroxy or protected-hydroxy;
except when the compound is of Formulas (I) or (III) then A, B, D,
and E may not all be hydrogen, D and E may not be C1-C3-alkyl when A and B are
hydrogen, nor may one of D and E be hydrogen and the other be Cl-C3-alkyl when
A and B
are hydrogen;
the method comprising:
(a) treating a compound having the formula
-13-
CA 02235942 1998-04-23
WO 97/17356 PCT/US96/16585
CHa NMez
O zRO,,
HaCi.. RC Ha ~
HOm.. ....r O' _CHa
HO
H3C~~ CHa
Or... CHa
CHa CHa
O .rOR2
Hac ~''ocHa , (compound (2) from Scheme 1),
CHa RZ NMe2
O
O,,
H°C~~~ 'RCHa
HO r...
""r O O CHa
HO
HsC.: CHa
p O-Cbz
CHa CHa
(compound (6) from Scheme 2),
CHa RZ NMe2
O
O,,
i
HaC''~ 'RCHa
HO r...
-"'r O O CHa
HO
H3C'~ O CHa
O
CHa CHa
O
(compound (7) from Scheme 3), or
CHa RZ NMe2
O
0,,,
HaCa .~RCHa
HO r...
".v O O CHa
HO
HaC.: CHa
O
CHa CHa
O
respectively,
wherein R1 is as described above and R2 is a hydroxy-protecting group,
with a base, such as sodium hydride, lithium hydride, potassium carbonate, for
example,
and followed by reaction with carbonyldiimidazole, in an aprotic solvent, as
defined below,
which does not adversely affect the reaction, preferably dichloromethane,
chloroform, DMF,
tetrahydrofuran (THF), N-methyl pyrrolidinone or a mixture thereof, with
cooling or
heating, depending on the conditions used, at a reaction temperature from -
20°C to 70°C,
-14-
CA 02235942 1998-04-23
WO~ 97/17356 PCTNS96/16585
preferably from 0°C to reaction temperature, for a period from 0.5
haurs to 10 days,
preferably 1-5 days, to prepare first intermediate compounds having the
formulas:
CH3 NMez
O zRO,,
HaC ~'RCH
3
/ "'m O O CH3
N~ N~ O
II HaC.: CHs
O ~0,,,. O CH3
CH3 CHI
O ..,~ ORz
H3c ~~'ocH, , (compound (3) from Scheme 1),
CH3 Rz NMez
O I
a,,
H3C ~ RCHa ~
N N O / ...u O~ CH3
U
HsCr:
OCb H'
CH3~ CH3
(compound (6) from Scheme 2),
CHs Rz NMez
O I
H3C RCH
~---~ 3 ~
N~N O / ....r O' _ CHy
U
HsC~, CHI
'O
CH~~ CH3
(compound (8) from Scheme 3), or
CH3 Rz NMez
I
0,,,
N N 1 O~ CH3
O ~Ha
,(compound ( tt7) from scheme 4),
respectively; wherein Ri and R2 are as described above;
(b) reacting said first intermediate compounds (3), (6), (8), or (10) with a
compound having the formula:
D
NHz
E r...
A
g' NHz
-15-
CA 02235942 1998-04-23
WO 97/17356 PCT/US96/16585
wherein A, B, D, and E are as described above, preferably in a solvent such as
aqueous
acetonitrile, DMF or aqueous DMF, to give the bicyclic second intermediate
compounds
having the formulas:
NHZ CH3 NMe2
E rr..
O ZRO,,
A
H3C ' CH
3
O~ Nn..' ~...r p O CH3
O
H3C~~ CH3
O -O,~ O CHs
CH3 CH3
.~° OFD
H3c ~~'ocH, (compound (11), from Scheme 5),
p NHZ CH3 NMei
E r''Y O
ZRO
H C RCH ~~~.
O Nr...
O ~"" O O CH3
HsCo.~ CHs
O OCbz
CH3 CH3
(compound ( 15), from Scheme 7),
p NHZ CH3 NMe2
E ,
A O RO',,.
B,. HaC 'RCH3
N a..
O O ...n O O CH3
H3C~, O CH3
O
CH3 CH3
O
(compound ( 19) from Scheme 9),
p NHZ CH3 NMe2
E r'Y O
'' IAA
B'~~ H3C ~RCH3
O~ Nn.. ~.." O O CH3
O
HsC.: CHa
O
CH3 CH3
O
(compound (23), from Scheme 11 ), respectively; ,
wherein R1 and RZ are as described above;
(c) deprotecting said second intermediate compounds ( 11 ), ( 15), ( 19) or
(23) by ,
treatment with methanol or ethanol when OR2 is an ester or with fluoride in
THF or
acetonitrile when R2 is a trialkylsilyl group, for from 1 to 24 hours, to give
the third
intermediate compounds
-16-
CA 02235942 1998-04-23
Wd~ 97/17356 PCT/US96/16585
CHa NMeZ
NHZ
E i... O
p R~ HOn..
' g~.,. H3C . CHa
O? N i...
"'m0 O CHa
O
HaC~~ ~CHa
O ' O,, O CHa
CHa CHa
O ~~ ~ OH
Hac ~~'ocHa (compound ( 12), from Scheme 5),
D NH2 CHa NMe2
Ey p W..
A
HaC ~'RCHa
O~Nn.. ...np O CHa
\O
HaC~ ~CHa
O OCbz
CHa CHa
(compound ( 16), from Scheme 7),
D NHZ CHs NMe2
E n. O
q
R'
Ha ~~ CHa
O~N a..
""' O O CHa
O
HaC~ 'CHa
O
O
CHa CHa
(compound (20) from Scheme 9),
D NHZ CHa NMe2
E,~ O HO
A~ ,
B" HaC .RCHa
O~N~... .....0 O CHa
O
1-IsG~ ~CHa
O
CHa CHa
° (compound (24) from Scheme 11), respectively; and
(d) cyclizing said third intermediate compounds ( 12), ( 16), (20) or (24) by
treatment with dilute acid, such as acetic acid or HCl, for example, in an
organic solvent,
preferably ethanol or propanol, for a period of from 4 hours to 10 days to
give the desired
compounds (~, (II), (III) or (IV) above.
Another object of the present invention is to provide an alternate process for
the
preparation of tricyclic macrolide derivatives having the Formulas:
-17-
CA 02235942 1998-04-23
WO 97/17356 PCT/US96/16585
D CHs R2 NMe2
E
A ~ 0.,
gn.. H3C~~.. s CHs
... R i
O~ Nn.. s
O CHs
O
HsC~.,. CHs
~0,,~. O CHs
O
CHs CHs
O .~~~ OH
H3C ~~~OCH3
> (I)>
E p CHs R2 NMe2
':
A ~ 0.,
g~... H3C~~, s
CHs
O~ Ni ~..
CHs
O
H3C~~,. H~~~' CH
3
O 1
CHs CHs
O
CHs R2 NMe2
.:
A NW 0....
gi... s CH3
HsC'~~.
O~N~...
O CHs
O
HaCv.,. CHs
'O
O 1
CHs CHs
O
> (III) or
CHs R2 NMep
.:
A ~ 0.,.
gn.. H3C~~I s CHs
..e R 1
O~ Ni... s ....~ O
O CHs
. . H~..
H3C~ . CHs
'H
O 1
CHs CHs
O
wherein ' ~N)>
A, B, D and E are independently selected from the group consisting of:
- i8 -
CA 02235942 1998-04-23
WO 97/17356 PCT/US96/16585
(a) hydrogen;
(b) C1-C6-alkyl, as defined below, optionally substituted with one or
more substituents selected from the group consisting of:
(i) aryl, as defined below;
(ii) substituted-aryl, as defined below;
(iii) heteroaryl, as defined below;
(iv) substituted-heteroaryl, as defined below;
(v) heterocycloalkyl, as defined below;
(vi) hydroxy;
(vii) C1-Cf,-alkoxy, as defined below;
(viii) halogen consisting of Br, Cl, F or I; and
(ix) NR3R4, where R3 and R4 are independently selected from
hydrogen and C1-C6-alkyl, or R3 and R4 are taken with the nitrogen atom to
which they are
connected to form a 3- to 7-membered ring optionally containing a hetero
function consisting
of -O-, -NH-, -N(C1-C6-alkyl-)-, -N(aryl-C1-C6-alkyl-)-, -N(substituted-aryl-
C1-C6-alkyl-
)-, -N(heteroaryl-C1-C6-alkyl-)-, -N(substituted-heteroaryl-C1-C6-allcyl-)-, -
S- or -S(O)S-,
wherein n is 1 or 2;
(c) C3-C~-cycloallcyl;
(d) aryl;
(e) substituted-aryl;
(f) heteroaryl;
(g) substituted-heteroaryl;
(h) heterocycloalkyl;
and
(i) a group selected from option (b) above further substituted with -M-
R5, wherein M is selected from the group consisting of:
(aa) -C(O)-NH_;
(bb) -NH-C(O)-;
(cc) -NH-
(dd) -N(CH3)-
(ee) -O-
(rr) -S(O)n-, wherein n is 0, 1 or 2; and
(gg) -C(=NH)-~-;
and R5 is selected from the group consisting of:
(aaa) C1-Ct,-alkyl, optionally substituted with a substituent selected
from the group consisting of:
(i) aryl;
(ii) substituted-aryl;
-19-
CA 02235942 1998-04-23
WO 97/17356 PCT/US96/16585
(iii) heteroaryl; and
(iv) substituted-heteroaryl;
(bbb) aryl;
(ccc) substituted-aryl;
(ddd) heteroaryl;
(eee) substituted-heteroaryl; and
(fff) heterocycloalkyl;
or
any one pair of substituents, consisting of AB, AD, AE, BD, BE or DE, is taken
l0 together with the atom or atoms to which they are attached to form a 3- to
7-membered ring
optionally containing a hetero function consisting of:
-O-,
-NH-,
-N(C1-C6-a~Yl-)->
15 -N(aryl-C1-C6-alkyl-)-
-N(substituted-aryl-C1-C6-alkyl-)-,
-N(heteroaryl-C1-C6-alkyl-)-,
-N(substituted-heteroaryl-C 1-C6-alkyl-)-,
-S- or -S(O)n-, wherein n is 1 or 2;
20 -C(O)-NH-;
-C(O)-NR$-, wherein RS is as described above;
-C(O)-;
-NRS-C(O)-, wherein RS is as described above; and
-C(=NH)-NH-;
25 R1 is selected from the group consisting of:
(a) hydrogen;
(b) hydroxy;
(c) -O-C1-C3-alkyl;
(d) -O-C3-Cg-cycloalkyl;
30 (e) -O-C1-C3-alkyl-C3-Cg-cycloalkyl;
-O-C(O)-Cl-C3-alkyl;
(g) -O-C(O)-O-Cl-C3-alkyl; a.nd
(h) -O-C(O)-NH-C1-C3-alkyl;
R2 is hydrogen or a hydroxy-protecting group, as defined below; and
35 Z is hydroxy or protected-hydroxy;
except when the compound is of Formulas (I) or (1Tl) then A, B, D, and E may
not all be
hydrogen, D and E may not be C1-C3-alkyl when A and B are hydrogen, nor may
one of D
and E be hydrogen and the other be Cl-C3-alkyl when A and B are hydrogen;
-20-
CA 02235942 1998-04-23
W~ 97/17356 PCT/US96/16585
the method comprising:
(a) treating a compound having the formula:
' CH3 NMe2
O 2R0~~
HaCi,,, .RCH3
HO n.~ ..." O CH3
HO
HaCr ~ CHa
O,, CH3
CH3 CH3 ~~
O ~~~ ORZ
H,c ~~°ocH, , (compound (2) from Scheme 1),
CHs RZ NMe2
O
O,,
HaC~. ~'RCHa
HO i...
....,0 O CH3
HO
H ~; CH
O O-Cbz
CHI CH3
(compound (6) from scheme 2),
CHs RZ NMe2
O
0,,,
HsCi.. ~'RCH~
F~ a..
""~O O CH3
HO
HaCr O CHa
O
CHs CH3
O
(compound (7) from Scheme 3), or
CHs RZ NMez
O
O,,
i
H3C~~ ~RCH3
HO,...
""~O O CHI
HO
HaCo; CHa
O
CH3 CHI
O
(compound (9) from Scheme 4),
respectively,
wherein R1 is as described above and RZ is a hydroxy-protecting group,
with a base, such as sodium hydride, lithium hydride, potassium carbonate, for
example,
and followed by reaction with carbonyldiimidazole, in an aprotic solvent, as
defined above,
which does not adversely affect the reaction, preferably dichloromethane,
chloroform, DMF,
tetrahydrofuran (THF), N-methyl pyrrolidinone or a mixture thereof, with
cooling or
-21-
CA 02235942 1998-04-23
WO 97/17356 PCT/iJS96/16585
heating, depending on the conditions used, at a reaction temperature from -
20°C to 70°C,
preferably from 0°C to reaction temperature, for a period from 0.5 to
24 hours, preferably 1-
8 hours, to prepare first intermediate compounds having the formulas:
CH3 NMaz
O zRO,,
H3C Ri
'~ C H3
/ "~~~O O CH3
N~ N O
H3C~ CH3
p ' O,, O CH3
CHa CH3
O ~~ ~ ORz
H3c ~~~'ocH3 , (compound (3) from Scheme 1),
CH3 RZ NMep
0,,.
R'
H3C ~ CHs
N~ N O / .~.n O CH3
HsC~' OC ZHa
CHs x ' CH3
(compound (6) from Scheme 2)
CH3 RZ NMe2
' I
O,,
N~ N\ _ O CH3
'OIL =Hs
(compound (8) from Scheme 3), or
CHa RZ NMe2
N~ N\ _ O CH3
'O'~ %Ha
(compound ( 10) from Scheme 4),
respectively; wherein R 1 and R2 are as described above;
(b) reacting said first intermediate compounds (3), (6), (8), or ( 10) with a
compound having the formula:
-22-
CA 02235942 1998-04-23
WO 97/17356 PCT/US96/16585
D
OH
E ~...
A
g NHz
wherein A, B, D, and E are as described above, preferably in a solvent such as
aqueous
acetonitrile, DMF or aqueous DMF, to give the bicyclic second intermediate
compounds:
D OH CHs NMez
E,..
A O zRO,,.
Bv H3C ~ RCH3
O? N ,.... ....~ p O CH3
\O
HsC. ~CH3
' 0,,,_ O CH3
CHa CH3
O ...~ ORz
H3c '''ocH3 , (compound ( 14), from Scheme 6, Y=OH),
D ~ CHs NMez
E,...
A~ O ~ 2R0...
H3C RCH3
O~ Ni... ...n O O CHa
O
HsC~ CHs
O OCbz
CHI CH3
(compound ( 18), from Scheme 8, Y=OH),
CHa NMez
Ei~ R
O zRO,,
A
B,. Hs '' CH5
O?Nn.~ ~~.uO O CHa
\O _
HaC~ ~CHa
O
O
CH3 CHs
(compound (22) from Scheme 10, Y=OH), or
D OH CH3 NMez
Ey
O zRO,,,
~A
B» H3C ~RCH~
O~ N~... °.u O O CH3
O~
H3C
(compound (26), from Scheme 12, Y=OH),
respectively;
(c) reacting the hydroxy group of the said bicyclic second intermediate
compounds ( 14), ( 18), (22) and (26) by treatment triphenylphosphine and
diphenyl-
- 23 -
CA 02235942 1998-04-23
WO 97/17356 PCT/US96/16585
phosphoryl azide-DEAD in tetrahydrofuran, under Mitsunobu reaction conditions,
to prepare
the third intermediate compounds:
N3 CHs NMe2
E r~.. O
A
g~"~ H3C ' CH
a
O~ N~.... ~~.~r p O CH3
O
HsC~~ CH3
O o,,..~oYcH,
CH3 CH3
~~ ~ OH
(compound ( 14), from Scheme 6, Y=N3),
p N3 CHa NMe2
E r~.. O
A
H3C
.~RCHs
O Nn..
O ~...r O O CH3
H3C~. CHa
OCbz
CHa CH3
(compound ( 18), from Scheme 8, Y=N3),
p Na CHa NMe2
Ey O
A
g" HaC 'RCH
3
O? Nr...
\O ""'O O CH3
HaCS: CHa
O
O
CHy CH3
, (compound (22) from Scheme 10, Y=N3), or
p Na CH3 NMe2
E r~.. O
A
gy HaC 'RCH3
O N~...
""'O O CHs
HaC.: CHa
O
CHs CH3
(compound (26), from Scheme 12, Y=N3),
respectively;
(d) reducing the third intermediate compounds having an azido group, with
suitable reducing reagents such as triphenylphosphine-water, hydrogen with a
catalyst,
sodium borohydride, or dialkylaluminum hydride, to prepare the fourth
intermediate
compounds having the formulas:
-24-
CA 02235942 1998-04-23
Way 97/17356 PCT/US96/16585
p NHZ CHa NMez
E r.. O
A ~~..
gy HaC ~~ CHa
o~Nr...
"'~ O O CHa
O
H3C'~ ~CHa
p -0,,,. O CHa
CHa CHa
~~ ~ OH
Hac ~~'ocHa , (compound (12), from Scheme 6),
NHZ CHa NMe2
ErY O
p
B" HaC JRCHa
OaNr... ...'~O O CHa
\O
HaC.: CHa
O OCbz
CHa CHa
(compound (16), from Scheme 8),
NHZ CHa NMe2
Ey O
A
B" HaC RCH
3
p NI~..
"'rr O O CHa
H3C' ~CHa
O
O
CHa CHa
(compound (20) from Scheme 10), or
NHZ CHa NMe2
E r., O
A HO...
~ R'
B"'-\ HsC , CHa
O~N ' O O CH
O .~ n a
HaC' ~CHa
O
CHa CHa
, (compound (24), from Scheme 12),
respectively; and
(e) cyclizing said fourth intermediate compounds ( 12), ( 16), (20) or (24),
wherein Y is an amino group, by treatment with dilute acid, such as acetic
acid or HCI, for
example, in an organic solvent, preferably ethanol or propanol, for a period
of from 4 hours
to 10 days to give the desired compounds of Formulas (I), (II), (III) or (IV).
In an alternate embodiment of the alternate process above, the third
intermediate
compounds may be prepared by a two-step sequence (replacing step (c) thereof)
which
comprises ( 1 ) reacting the hydroxy group of the bicyclic second intermediate
compounds
- 25 -
CA 02235942 1998-04-23
WO 97/17356 PCT/US96/16585
with an alkyl or aryl sulfonyl chloride, an alkyl or aryl sulfonic anhydride
or
trifluoromethanesulfonic anhydride in an aprotic solvent at -78°C to
reaction temperature to
give the corresponding sulfonate, and (2) reacting the said sulfonate with
lithium azide or
sodium azide in an aprotic solvent at 0°C to 100°C to give the
third intermediate compound.
peflnitions
The terms "C1-C3-alkyl" or "C1-C6-alkyl" as used herein refer to saturated,
straight-
or branched-chain hydrocarbon radicals containing between one and three or one
and six
carbon atoms, respectively. Examples of C1-C3 alkyl radicals include methyl,
ethyl, propyl
and isopropyl, and examples of C1-C6-alkyl radicals include, but are not
limited to, methyl,
ethyl, propyl, isopropyl, n-butyl, tert-butyl, neopentyl and n-hexyl.
The term "Cl-C6-alkoxy" as used herein refers to an C1-C6-alkyl group, as
previously defined, attached to the parent molecular moiety through an oxygen
atom.
Examples of Ci-C6-alkoxy, but are not limited to, methoxy, ethoxy, propoxy,
isopropoxy,
~a-butoxy, tert-butoxy, neopentoxy and n-hexoxy.
The term "C1-C3-alkyl-amino" as used herein refers to one or two Cl-C3-alkyl
groups, as previously defined, attached to the parent molecular moiety through
a nitrogen
atom. Examples of C1-C3-alkyl-amino include, but are not limited to
methylamino,
dimethylamino, ethylamino, diethylamino, and propylamino.
The term "aprotic solvent" as used herein refers to a solvent that is
relatively inert to
proton activity, i.e., not acting as a proton-donor. Examples include, but are
not limited to,
hydrocarbons, such as hexane and toluene, for example, halogenated
hydrocarbons, such
as, for example, methylene chloride, ethylene chloride, chloroform, and the
like,
heterocyclic compounds, such as, for example, tetrahydrofuran and N-
methylpyrrolidinone,
and ethers such as diethyl ether, bis-methoxymethyl ether. Such compounds are
well known
to those skilled in the art, and it will be obvious to those skilled in the
art that individual
solvents or mixtures thereof may be preferred for specific compounds and
reaction
conditions, depending upon such factors as the solubility of reagents,
reactivity of reagents
and preferred temperature ranges, for example. Further discussions of aprotic
solvents may
be found in organic chemistry textbooks or in specialized monographs, for
example:
(.~reanic Solvents Physical Properties and Methods of Purification, 4th ed.,
edited by John
A. Riddick et al., Vol. II, in the Techniques of Chemistry Series, John Wiley
& Sons, NY,
1986.
The term "aryl" as used herein refers to unsubstituted carbocyclic aromatic
groups
including, but not limited to, phenyl, 1- or 2-naphthyl and the like.
The term "C3-CS-cycloalkyl- and C3-C7-cycloalkyl" as used herein refers to
carbocyclic groups of 3 to 5 or 3 to 7 carbons, respectively, for example,
cyclopropyl,
cyclobutyl, cyclopentyl, cyclohexyl, and cycloheptyl.
-26-
CA 02235942 1998-04-23
WO 97/17356 PCT/US96116585
The term "C1-C3-alkyl-C3-C5-cycloalkyl", as used herein refers to a C3-C5-
cycloallcyl radical, as defined above, attached to a Cl-C3-alkyl radical by
replacement of a
hydrogen atom on the Latter.
The terms "halo" and "halogen" as used herein refer to an atom selected from
fluorine, chlorine, bromine and iodine.
The term "heteroaryl", as used herein, refers to a cyclic aromatic radical
having from
five to ten ring atoms of which one ring atom is selected from S, O and N;
zero, one or two
ring atoms are additional heteroatoms independently selected from S, O and N;
and the
remaining ring atoms are carbon, the radical being joined to the rest of the
molecule via any
of the ring atoms, such as, for example, pyridinyl, pyrazinyl, pyrimidinyl,
pyrrolyl,
pyrazolyl, imidazolyl, thiazolyl, oxazolyl, isooxazolyl, thiadiazolyl,
oxadiazolyl, thiophenyl,
furanyl, quinolinyl, isoquinolinyl, and the like.
The term "heterocycloallcyl" as used herein, refers to a non-aromatic 5-, 6-
or 7-
membered ring or a bi- or tri-cyclic group comprising fused six-membered rings
having
between one and three heteroatoms independently selected from oxygen, sulfur
and nitrogen,
wherein (i) each 5-membered ring has 0 to 1 double bonds and each 6-membered
ring has 0
to 2 double bonds, (ii) the nitrogen and sulfur heteroatoms may optionally be
oxidized, (iii)
the nitrogen heteroatom may optionally be quaternized, and (iv) any of the
above heterocyclic
rings may be fused to a benzene ring. Representative heterocycles include, but
are not limited
to, pyrrolidinyl, pyrazolinyl, pyrazolidinyl, imidazolinyl, imidazolidinyl,
piperidinyl,
piperazinyl, oxazolidinyl, isoxazolidinyl, morpholinyl, thiazolidinyl,
isothiazolidinyl, and
tetrahydrofuryl.
"Hydroxy-protecting group", as used herein, refers to an easily removable
group to
which are known in the art to protect a hydroxyl group against undesirable
reaction during
synthetic procedures and to be selectively removable. The use of hydroxy-
protecting groups
is well known in the art for protecting groups against undesirable reactions
during a synthetic
procedure and many such protecting groups are known, cf, for example, T.H.
Greene and
P. G. M. Wuts, Protective Group in Or anic Synthesis 2nd edition, John Wiley &
Sons,
New York ( 1991 ). Examples of hydroxy-protecting groups include, but are not
limited to,
methylthiomethyl, tert-dimethylsilyl, tert-butyLdiphenylsilyl, acyl
substituted with an
aromatic group and the Like.
A the term "protected-hydroxy" refers to a hydroxy group protected with a
hydroxy
protecting group, as defined above, including benzoyl, acetyl, trimethylsilyl,
triethylsilyl,
methoxymethyl groups, for example.
The term "protogenic organic solvent" as used herein refers to a solvent that
tends to
provide protons, such as an alcohol, for example, methanol, ethanol, propanol,
isopropanol,
butanol, t-butanol, and the Like. Such solvents are well known to those
skilled in the art, and
it will be obvious to those skilled in the art that individual solvents or
mixtures thereof may
-27-
CA 02235942 2004-08-19
WO 97/17356 PCTNS96/16585
be preferred for specific compounds and reaction conditions, depending upon
such factors as
the solubility of reagents, reactivity of reagents and preferred temperature
ranges, for
example. Further discussions of protogenic solvents may be found in organic
chemistry
textbooks or in specialized monographs, for example: Organic Solvents P~~sical
Pro erties
aid Methods of Purification, 4th ed., edited by John A. Riddick et al., Vol.
II, in the
Techniques of Chemistry Series, John Wiley & Sons, NY, 1986.
The term "substituted aryl" as used herein refers to an aryl group as defined
herein
substituted by independent replacement of one, two or three of the hydrogen
atoms thereon
with Cl, Br, l~, I, OH, Ct-C3-alkyl, Ct-C6-alkoxy, methoxymethoxy, amino, or
C1-C3-
alkyl-amino.
The teml "substituted heteroaryl" as used herein refers to a heteroaryl group
as
defined herein substituted by independent_replacement of one, two or three of
the hydrogen
atoms thereon with Cl, Br, F, I, OH, Ct-C3-alkyl, Ct-C6-alkoxy,
methoxymethoxy, amino,
or Ci-C3-alkyl-amino.
The term "substituted heterocycloalkyl'' as used herein, refers to a
heterocycloallcyl
group, as defined above, substituted by independent replacement of one, two or
three of the
hydrogen atoms thereon with Cl, Br, F, I, OH, Ct-C3-alkyl, Ct-C6-alkoxy,
methoxymethoxy, amino, or C1-C3-alkyl-amino.
Numerous asymmetric centers may exist in the compounds of the present
invention.
Exceptwhere otherwise noted, the present invention contemplates the various
stereoisomers
and mixtures thereof. Accordingly, whenever a bond is represented by a wavy
line, it is
intended that a mixture of stereo-orientations or an individual isomer of
assigned or
unassigned orientation may be present.
As used herein, the term "pharmaceutically acceptable salt" refers to those
salts which
are, within the scope of sound medical judgment, suitable for use in contact
with the tissues
of humans and lower animals without undue toxicity, irritation, allergic
response and the
like, and are commensurate with a reasonable benefit/risk ratio.
Pharmaceutically acceptable
salts are well known in the art. For example, S. M. Berge, et al. describe
pharmaceutically
acceptable salts in detail in J_. Pharmaceutical Sciences. 66: 1-19 (1977),
The salts can be prepared in situ during the final isolation and purification
of
the compounds of the invention, or separately by reacting the free base
function with a
suitable organic acid. Examples of pharmaceutically acceptable, nontoxic acid
addition salts
are salts of an amino group formed with inorganic acids such as hydrochloric
acid,
hydrobromic acid, phosphoric acid, sulfuric acid and perchloric acid or with
organic acids
such as acetic acid, oxalic acid, malefic acid, tartaric acid, citric acid,
succinic acid or maloruc
acid or by using other methods used in the art such as ion exchange. Other
pham~aeeutically
acceptable salts include adipate, alginate, ascorbate, aspartate,
benzenesulfonate, benzoate,
bisulfate, borate, butyrate, camphorate, camphorsulfonate, citrate,
cyelopentanepropionate,
-28-
CA 02235942 1998-04-23
W~ 97/17356 PCT/US96/16585
digluconate. dodecylsulfate, ethanesulfonate, formate, fumarate,
glucoheptonate,
glycerophosphate, gluconate, hemisulfate, heptanoate, hexanoate, hydroiodide,
2-hydroxy-
ethanesulfonate, lactobionate, lactate, laurate, lauryl sulfate, malate,
maleate, malonate,
methanesulfonate, 2-naphthalenesulfonate, nicotinate, nitrate, oleate,
oxalate, palmitate,
pamoate, pectinate, persulfate, 3-phenylpropionate, phosphate, picrate,
pivalate, propionate,
stearate, succinate, sulfate, tartrate, thiocyanate, p-toluenesulfonate,
undecanoate, valerate
salts, and the like. Representative alkali or alkaline earth metal salts
include sodium, lithium,
potassium, calcium, magnesium, and the like. p'urther pharmaceutically
acceptable salts
include, when appropriate, nontoxic ammonium, quaternary ammonium, and amine
cations
formed using counterions such as halide, hydroxide, carboxylate, sulfate,
phosphate, nitrate,
loweralkyl sulfonate and aryl sulfonate.
As used herein, the term "pharmaceutically acceptable ester" refers to esters
which
hydrolyze in vivo and include those that break down readily in the human body
to leave the
parent compound or a salt thereof. Suitable ester groups include, for example,
those derived
from pharmaceutically acceptable aliphatic carboxylic acids, particularly
alkanoic, alkenoic,
cycloalkanoic and a.lkanedioic acids, in which each alkyl or alkenyl moiety
advantageously
has not more than 6 carbon atoms. Examples of particular esters includes
formates, acetates,
propionates, butyates, acrylates and ethylsuccinates.
The pharmaceutical compositions of the present invention comprise a
therapeutically
effective amount of a compound of the present invention formulated together
with one or
more pharmaceutically acceptable Garners. As used herein, the term
"pharmaceutically
acceptable carrier" means a non-toxic, inert solid, semi-solid or liquid
filler, diluent,
encapsulating material or formulation auxiliary of any type. Some examples of
materials
which can serve as pharmaceutically acceptable carriers are sugars such as
lactose, glucose
and sucrose; starches such as corn starch and potato starch; cellulose and its
derivatives such
as sodium carboxymethyl cellulose, ethyl cellulose and cellulose acetate;
powdered
tragacanth; malt; gelatin; talc; excipients such as cocoa butter and
suppository waxes; oils
such as peanut oil, cottonseed oil; safflower oil; sesame oil; olive oil; corn
oil and soybean
oil; glycols; such a propylene glycol; esters such as ethyl oleate and ethyl
laurate; agar;
buffering agents such as magnesium hydroxide and aluminum hydroxide; alginic
acid;
pyrogen-free water; isotonic saline; Ringer's solution; ethyl alcohol, and
phosphate buffer
solutions, as well as other non-toxic compatible lubricants such as sodium
lauryl sulfate and
magnesium stearate, as well as coloring agents, releasing agents, coating
agents,
sweetening, flavoring and perfuming agents, preservatives and antioxidants can
also be
present in the composition, according to the judgment of the formulator. The
pharmaceutical
compositions of this invention can be administered to humans and other animals
orally,
rectally, parenterally, intracisternally, intravaginally, intraperitoneally,
topically (as by
powders, ointments, or drops), bucally, or as an oral or nasal spray.
-29-
CA 02235942 1998-04-23
WO 97/17356 PCT/US96/16585
Liquid dosage forms for oral administration include pharmaceutically
acceptable
emulsions, microemulsions, solutions, suspensions, syrups and elixirs. In
addition to the
active compounds, the liquid dosage forms may contain inert diluents commonly
used in the
art such as, for example, water or other solvents, solubilizing agents and
emulsifiers such as
ethyl alcohol, isopropyl alcohol, ethyl carbonate, ethyl acetate, benzyl
alcohol, benzyl
benzoate, propylene glycol, 1,3-butylene glycol, dimethylformamide, oils (in
particular,
cottonseed, groundnut, corn, germ, olive, castor, and sesame oils), glycerol,
tetrahydrofiufuryl alcohol, polyethylene glycols and fatty acid esters of
sorbitan, and
mixtures thereof. Besides inert diluents, the oral compositions can also
include adjuvants
0 such as wetting agents, emulsifying and suspending agents, sweetening,
flavoring, and
perfuming agents.
Injectable preparations, for example, sterile injectable aqueous or oleaginous
suspensions may be formulated according to the known art using suitable
dispersing or
wetting agents and suspending agents. The sterile injectable preparation may
also be a sterile
5 injectable solution, suspension or emulsion in a nontoxic parenterally
acceptable diluent or
solvent, for example, as a solution in 1,3-butanediol. Among the acceptable
vehicles and
solvents that may be employed are water, Ringer's solution, U.S.P. and
isotonic sodium
chloride solution. In addition, sterile, fixed oils are conventionally
employed as a solvent or
1
1
suspending medium. For this purpose any bland fixed oil can be employed
including
20 synthetic mono- or diglycerides. In addition, fatty acids such as oleic
acid are used in the
preparation of injectables.
The injectable formulations can be sterilized, for example, by filtration
through a
bacterial-retaining filter, or by incorporating sterilizing agents in the form
of sterile solid
compositions which can be dissolved or dispersed in sterile water or other
sterile injectable
25 medium prior to use.
In order to prolong the effect of a drug, it is often desirable to slow the
absorption of
the drug from subcutaneous or intramuscular injection. This may be
accomplished by the
use of a liquid suspension of crystalline or amorphous material with poor
water solubility.
The rate of absorption of the drug then depends upon its rate of dissolution
which, in turn,
30 may depend upon crystal size and crystalline form. Alternatively, delayed
absorption of a
parenterally administered drug form is accomplished by dissolving or
suspending the drug in
an oil vehicle. Injecta.ble depot forms are made by forming microencapsule
matrices of the
drug in biodegradable polymers such as polylactide-polyglycolide. Depending
upon the ratio
of drug to polymer and the nature of the particular polymer employed, the rate
of drug
35 release can be controlled. Examples of other biodegradable polymers include
poly(orthoesters) and poly(anhydrides). Depot injectable formulations are also
prepared by
entrapping the drug in liposomes or microemulsions which are compatible with
body tissues.
-30-
CA 02235942 1998-04-23
WO 97/17356 PCT/US9C/16585
Compositions for rectal or vaginal administration are preferably suppositories
which
can be prepared by mixing the compounds of this invention with suitable non-
irritating
excipients or carriers such as cocoa butter, polyethylene glycol or a
suppository wax which
are solid at ambient temperature but liquid at body temperature and therefore
melt in the
rectum or vaginal cavity and release the active compound.
Solid dosage forms for oral administration include capsules, tablets, pills,
powders,
and granules. In such solid dosage forms, the active compound is mused with at
least one
inert, pharmaceutically acceptable excipient or earner such as sodium citrate
or dicalcium
phosphate and/or a) fillers or extenders such as starches, lactose, sucrose,
glucose, mannitol,
i0 and silicic acid, b) binders such as, for example, carboxymethylcellulose,
alginates, gelatin,
polyvinylpyrrolidinone, sucrose, and acacia, c) humectants such as glycerol,
d)
disintegrating agents such as agar-agar, calcium carbonate, potato or tapioca
starch, alginic
acid, certain silicates, and sodium carbonate, e) solution retarding agents
such as paraffin, f)
absorption accelerators such as quaternary ammonium compounds, g) wetting
agents such
15 as, for example, cetyl alcohol and glycerol monostearate, h) absorbents
such as kaolin and
bentonite clay, and i) lubricants such as talc, calcium stearate, magnesium
stearate, solid
polyethylene glycols, sodium lauryl sulfate, and mixtures thereof. In the case
of capsules,
tablets and pills, the dosage form may also comprise buffering agents.
Solid compositions of a similar type may also be employed as fillers in soft
and hard-
20 filled gelatin capsules using such excipients as lactose or milk sugar as
well as high
molecular weight polyethylene glycols and the like.
The solid dosage forms of tablets, dragees, capsules, pills, and granules can
be
prepared with coatings and shells such as enteric coatings and other coatings
well known in
the pharmaceutical formulating art. They may optionally contain opacifying
agents and can
25 also be of a composition that they release the active ingredients) only, or
preferentially, in a
certain part of the intestinal tract, optionally, in a delayed manner.
Examples of embedding
compositions which can be used include polymeric substances and waxes.
Solid compositions of a similar type may also be employed as fillers in soft
and hard-
filled gelatin capsules using such excipients as lactose or milk sugar as well
as high
30 molecular weight polethylene glycols and the like.
The active compounds can also be in micro-encapsulated form with one or more
excipients as noted above. The solid dosage forms of tablets, dragees,
capsules, pills, and
granules can be prepared with coatings and shells such as enteric coatings,
release
controlling coatings and other coatings well known in the pharmaceutical
formulating a.rt. In
35 such solid dosage forms the active compound may be admixed with at least
one inert diluent
such as sucrose, lactose or starch. Such dosage forms may also comprise, as is
normal
practice, additional substances other than inert diluents, e.g., tableting
lubricants and other
tableting aids such a magnesium stearate and microcrystalline cellulose. In
the case of
-31-
CA 02235942 1998-04-23
WO 97/17356 PC'T/US96/16585
capsules, tablets and pills, the dosage forms may also comprise buffering
agents. They may
optionally contain opacifying agents and can also be of a composition that
they release the
active ingredients) only, or preferentially, in a certain part of the
intestinal tract, optionally,
in a delayed manner. Examples of embedding compositions which can be used
include
polymeric substances and waxes.
Dosage forms for topical or transdermal administration of a compound of this
invention include ointments, pastes, creams, lotions, gels, powders,
solutions, sprays,
inhalants or patches. The active component is admixed under sterile conditions
with a
pharmaceutically acceptable carrier and any needed preservatives or buffers as
may be
required. Ophthalmic formulation, ear drops, eye ointments, powders and
solutions are also
contemplated as being within the scope of this invention.
The ointments, pastes, creams and gels may contain, in addition to an active
compound of this invention, excipients such as animal and vegetable fats,
oils, waxes,
paraffins, starch, tragacanth, cellulose derivatives, polyethylene glycols,
silicones,
bentonites, silicic acid, talc and zinc oxide, or mixtures thereof.
Powders and sprays can contain, in addition to the compounds of this
invention,
excipients such as lactose, talc, silicic acid, aluminum hydroxide, calcium
silicates and
polyamide powder, or mixtures of these substances. Sprays can additionally
contain
customary propellants such as chlorofluorohydrocarbons.
Transdermal patches have the added advantage of providing controlled delivery
of a
compound to the body. Such dosage forms can be made by dissolving or
dispensing the
compound in the proper medium. Absorption enhancers can also be used to
increase the flux
of the compound across the skin. The rate can be controlled by either
providing a rate
controlling membrane or by dispersing the compound in a polymer matrix or gel.
According to the methods of treatment of the present invention, bacterial
infections
are treated or prevented in a patient such as a human or lower mammal by
administering to
the patient a therapeutically effective amount of a compound of the invention,
in such
amounts and for such time as is necessary to achieve the desired result. By a
"therapeutically
effective amount" of a compound of the invention is meant a sufficient amount
of the
compound to treat bacterial infections, at a reasonable benefit/risk ratio
applicable to any
medical treatment. It will be understood, however, that the total daily usage
of the
compounds and compositions of the present invention will be decided by the
attending
physician within the scope of sound medical judgment. The specific
therapeutically effective
dose level for any particular patient will depend upon a variety of factors
including the
disorder being treated and the severity of the disorder; the activity of the
specific compound
employed; the specific composition employed; the age, body weight, general
health, sex and
diet of the patient; the time of administration, route of administration, and
rate of excretion of
the specific compound employed: the duration of the treatment; drugs used in
combination or
-32-
CA 02235942 1998-04-23
WO 97/17356 PCT/US96/16585
coincidental with the speck compound employed; and like factors well known in
the
medical arts.
The total daily dose of the compounds of this invention administered to a
human or
other mammal in single or in divided doses can be in amounts, for example,
from 0.01 to 50
mg/kg body weight or more usually from 0.1 to 25 mg/kg body weight. Single
dose
compositions may contain such amounts or submultiples thereof to make up the
daily dose.
In general, treatment regimens according to the present invention comprise
administration to
a patient in need of such treatment from about 10 mg to about 1000 mg of the
compounds)
of this invention per day in single or multiple doses.
Abbreviations
Abbreviations which have been used in the descriptions of the scheme and the
examples that follow are: AIBN for azobisisobutyronitrile; Bu3SnH for
tributyltin hydride;
CDI for carbonyldiimidazole; DBU for 1,8-diazabicyclo[5.4.0]under-7-ene; DEAD
for
diethylazodicarboxylate; DMF for dimethyl formamide; DPPA for
diphenylphosphoryl azide;
EtOAc for ethyl acetate; MeOH for methanol; NaN(TMS)2 for sodium
bis(trimethylsilyl)amide; NMMO for N-methylmorpholine N-oxide; TEA for
triethylamine;
THF for tetrahydrofuran; TPP for triphenylphosphine.
Synthetic Methods
The compounds and processes of the present invention will be better understood
in
connection with the following synthetic schemes which illustrate the methods
by which the
compounds of the invention may be prepared. The groups A, B, D, E, R1 and R2
are as
defined above unless otherwise noted below.
-33-
CA 02235942 1998-04-23
WO 97/17356 PCT/US96/16585
Scheme 1
Preparation of Starting Materials, 12-imidazolylcarbonyloxy
macrolide (3) for Compounds of Formula (I)
CH3 NMe2
CH3 NMe2
O ~ ~~.. O 2R0
HaCi. R ~ i..
CH3 2 ~"~3C~~ CH3
HOd.. ~ ...., O O CH HOn..
HO 3 protect HO ""'O O CH3
HaC.; CHa -----~ HaC'.. CHa
O O,, O CHa
CHs CH3 - CH O C03 ~. O CH3
O 4" . O
OH .~' ORZ
HsC ~~OCHa HaC ~~OCH3
('1 ) (2)
R' is as described for formula I R2 is a hydroxy-protecting group
NaH-
CDI
CH3 NMe2
O ZROi.
H3C RCH3
""~O O CH3
N~ N O
H3C~ CH3
O O,, O CHI
CH' CH3..
O ~~ ~ ORZ
H3C ~OCH3
(3)
R ~ is as described for formula I
R2 is a hydroxy-protecting group
Scheme 1 illustrates a general procedure for preparing the starting material,
imidazolylcarbonyloxy macrolide (3), for compounds of Formula (I). The 2'- and
4"-
hydroxyl groups of compound ( 1 ) are protected by reacting ( 1 ) with
suitable hydroxy group
protecting reagents (cf. T. W. Greene and P. G. M. Wuts, protective nouns in
Org nic
Synth, 2nd ed., John Wiley & Son, Inc., 1991) such as acetic anhydride,
benzoic
anhydride, benzyl chloroformate or a trialkylsilyl chloride in an aprotic
solvent, as defined
above, which does not adversely affect the reaction, preferably
dichloromethane,
chloroform, DMF, tetrahydrofuran (THF), N-methyl pyrrolidinone or a mixture
thereof.
-34-
CA 02235942 1998-04-23
W~ 97/17356 PCT/LTS96/16585
The protected macrolide (2) is reacted under anhydrous conditions with base
such as sodium
hydride, lithium hydride, potassium carbonate and followed by
carbonyldiimidazole to form
compound (3) in an aprotic solvent, as defined above, which does not adversely
affect the
reaction, preferably dichloromethane, chloroform, DMF, tetrahydrofuran (THF),
N-methyl
pyrrolidinone or a mixture thereof. The reaction may require cooling or
heating, depending
on the conditions used. The reaction temperature may be from -20°C to
70°C, and preferably
from 0°C to reaction temperature. The reaction may require 0.5 hours to
10 days, and
preferably 1-5 days, to complete.
Scherrle 2
Preparation of Starting Materials, 12-imidazolylcarbonyloxy
macrolide (6). for Compounds of Formula (II)
CH3 NMe2 ~Ha RZ NMe2
O HO~.
~R~
iaCi.. . CHa
r.. ~..rr0 O CH3 H+ CHI
CH3
0 0,,..~oYCH3
CH3 CH3
r 'OH
HaC~~OCH~ (4a) R2 = li
R1 is as described for formula II (4) R2 = hydroxy- protecting group
CH3 RZ NMe2 CHI RZ NMe2
O ~ O
0,,, O,r
H3C 'RCH Ri
H3C,,. v CH3
3
/ ...rr0 O CHI O~Om.. ~rn0 O CH3
N~N~H3C~' CHs NaH-CDI ~.' cH,
O ~O-Cbz O 'O-Cbz
CH3 CHI CHa CHs
O
(g) (
R1 is as described for formula II R2 is a hydroxy-protecting group
R2 is a hydroxy-protecting group
Scheme 2 illustrates a general procedure for preparing the starting materials,
12-
imidazolylcarbonyloxy macrolide (6), for the preparations of compounds of
formula (II).
The cladinose moiety of macrolides of formula (1) is removed either by mild
aqueous acid
hydrolysis of by enzymatic hydrolysis. In accordance with Scheme 2, a
suspension of (1) in
-35-
CA 02235942 1998-04-23
WO 97/17356 PCT/fJS96/16585
a solution of protogenic organic solvent (for example methanol, ethanol,
isopropanol or
butanol) and water is treated with dilute hydrochloric acid, chloroacetic
acid, dichloroacetic
acid or trifluoroacetic acid for 0.5 to 24 hours. The reaction temperature is
preferably -10 to
35 °C to give the des-cladinose compounds (4a). The 2'-hydroxy group of
(4a) is protected
and converted to (4a) by treatment with a suitable hydroxy protecting reagent
(cf. T. W.
Greene and P. G. M. Wuts, Protective Groups in Organic Synthesis, 2nd ed.,
John Wiley &
Son, Inc., 1991 ) such as acetic anhydride, benzoyl anhydride, benzyl
chloroformate or
trialkylsilyl chloride in an aprotic solvent, as defined above, preferably
dichloromethane,
chloroform, DMF, tetrahydrofuran (THF), N-methyl pyrrolidinone or a mixture
thereof.
The protected macrolide (4) is reacted under anhydrous conditions with base
such as sodium
hydride, lithium hydride, potassium carbonate or dimethylaminopyridine and
followed by
phosgene, diphosgene, triphosgene or benzyl chloroformate in an aprotic
solvent, as defined
above. The reactive intermediate is then trapped with benzyl alcohol to give
compound (5).
Compound (5) is reacted under anhydrous conditions with NaH and CDI in an
aprotic
solvent, preferably THF, DMF or a mixture thereof. The reaction may require
cooling of
heating, depending upon the conditions used. The reaction temperature may be
from -20°C
to 70°C, and preferably from 0°C to reaction temperature. The
reaction may require 0.5
hours to 10 days, and preferably 1-5 days, to complete.
-36-
CA 02235942 1998-04-23
WO 97/17356 PCT/US96/16585
Scheme 3
Preparation of Starting Materials, 12-imidazolylcarbonyloxy
xnacrolides, for Compounds of Formula (III)
CHa R2 NMeZ CHs Rz NMez
o I
O~~ O
i
H3C~~~ RCH3 H3Cir~ ~CH3
HOr... .~~~r0 O CHs ~m~. ...rr0 O CH3
HO Hp
H3C~'~ 3 CHs ~~ H3Cv' 3 CHI
O OOH O O
CHs CH3 CH3 CH3
O O
(4)
NaH-CDI
CHs RZ NMep CH3 RZ NMea
I I
O 0,,, O Oi~.
iC .R~ HaC RCHs
CH3
/ ....r p ~ O CHs O~ / ...rr O O CH3
NaH-CDI
CHa ---w. HaC~: O CHs
O O
O CH3 CH3
CH3 CH3
O O
~a)
R' is as described for formula III
R2 is a hydroxy-protecting group
Scheme 3 illustrates two general procedures for the synthesis of 12-
imidazolylcarbonyloxy macrolide (8), starting materials for the preparation of
compounds of
formula (III). In accordance with scheme 3, the 3-hydroxy group of a 2'-
protected des-
cladinose macrolide (4) is oxidized to the corresponding 3-oxo compound (7)
using a
modified Swern oxidation procedure. In scheme 3, suitable oxidizing agents are
N-
chlorosuccinimide-dimethyl sulfide or carbodiimide-dimethylsulfoxide. In a
typical
example, (4) is added into a pre-formed N-chlorosuccinimide and dimethyl
sulfide complex
in a chlorinated solvent such as methylene chloride at -10 to 25 °C.
After being stirred for
0.5-4 hours, a tertiary amine such as triethylamine or Hunig's base is added
to produce the
oxidized compound (7).
In the first approach in scheme 3, (7) is reacted with sodium hydride or
lithium
hydride and CDI under anhydrous conditions in an aprotic solvent, as defined
above, which
does not adversely affect the reaction, preferably dichloromethane,
chloroform, DMF,
_ -37-
CA 02235942 1998-04-23
WO 97/17356 PCT/US96/16585
tetrahydrofuran (THF), N-methyl pyrrolidinone or a mixture thereof. The
resulting allcoxide
is then reacted with excess carbonyldiimidazole for 0.5 hours to 10 days in
the same reaction
mixture to produce (8). The preferred temperature is from -10 °C to
reaction temperature.
In the second approach in Scheme 3, (7) is converted to (7a) with sodium
hydride or
lithium hydride and phosgene, diphosgene or triphosgene under anhydrous
conditions
followed by aqueous work up. Alternatively, (7) is converted to its
corresponding 11-
mesylate by reacting (7) with methanesulfonic anhydride in pyridine. The 11-
mesylate is
then converted to (7a) with an amino base such as DBU or dimethylaminopyridine
in acetone
or acetonitrile. (7a) is then reacted with sodium hydride or lithium hydride
under anhydrous
1o conditions in an aprotic solvent, as defined above, which does not
adversely affect the
reaction, preferably dichloromethane, chloroform, DMF, tetrahydrofuran (THF),
N-methyl
pyrrolidinone or a mixture thereof. The reactive alkoxide is then reacted with
carbonyldiimidazole for 0.5 hours to 10 days in the same reaction mixture to
produce (8).
The preferred temperature is from -10 °C to reaction temperature.
-38-
CA 02235942 1998-04-23
W~ 97/17356 PCT/US96/16585
Scheme 4
Preparation of 12-imidazolylcarbonyloxy
macrolides (10). for Compounds of Formula IIVI
CH3 Rz NMe2 CH3 R2 NMe2
O R~ O'.,. O R ~ O'.,.
H3C~'~ ° CH3 H3C~~.~ ~ CH3
Ho",. '...,o o cH3 1. NaH CS eo~,..
' O O CH3
H° 2. Bu3SnH, AIBN Ho
H3C~ ~CH3 H3C~ CH3
p 'OH O
CHa CH3 CH3 CH3
O O
(5) (9)
NaH-CDI
N CHI RZ NMe2
I
o,,,
HaC RCH3
O~ / ~~~u O O CH3
O
H..C~' CH3
CH3 ~ ~ CH3
R' is as described for formula III
R2 is a hydroxy-protecting group
In accordance with Scheme 4, a protected descladinose compound of formula (5)
is
dissolved in an aprotic solvent such as THF, then reacted with an excess of
NaH at from 0°C
to -30°C under an inert atmosphere, followed by reaction of the
intermediate anion with CS2,
then CH3I at -5 to 10°C, to form a 3-O-xanthyl compound. This xanthate
intermediate is
then reacted with 1.1-1.3 equivalents of Bu3SnI-I under an inert atmosphere in
the presence
of a catalytic amount of AIBN in a solvent suitable for a free radical
reaction, such as
benzene or toluene, for example, at reflux conditions to afford the desired
compound (9).
Compounds (9) are then reacted with carbonyldiimidazole and NaH under
anhydrous
conditions in an aprotic solvent, as defined above, which does not adversely
affect the
reaction, preferably dichloromethane, chloroform, DMF, tetrahydrofuran (THF),
N-methyl
pyrrolidinone or a mixture thereof, at a temperature from 0°C to
reaction temperature for 0.5
hours to 10 days to provide the compounds of formula ( 10).
-39-
CA 02235942 1998-04-23
WO 97/17356 PCT/US96/16585
Scheme 5
Preparation of Compound of Formula (I)
CHs NMez
O Z p NH2 CEb NMei
FiOr.. p Err..
Ri O x
H3C ~ CH3 E rr.. NHZ A~ R~ ROr'~~
/ A B~,~.~/ I-6C ~~ CHs
\ N O ~~~np O CI~ '~ O~Nr... ...rr O CH
g' NHz a
w
H3C~ C~ It,C~~ CH
O ~0~, O CH3
CHa CHs .
CH3
CH3 CH3 ~~
O
I~rORz O
(11) .,.oRz
(3) HsC .'FOCI-~
R 1 is as described for formula I
R2 is a hydroxy-protecting group
deprotect
D CH3
E~~~ NMex D NHz ~3 NMe2
Err.. p HO
''/ ..
B~ r'' A I-bC R
CHa O
CH3 ~
O
~Nn.. ~~,rr
O CHa O
g O' - CH3
O
Nrr~. ~~,rr CyCIIZe
H3C~~ CH3 w
_ F~,C~~ _ CH3
p ~1,,. CH3
Or., CH3
CHa CH3 CI-h .
O ~~~ ~ O .,~ OH
(13) H,c ~~'ocH, (12) Et,c ~°ocH,
Compound of Formula (I).
In accordance with Scheme 5, a starting material compound of formula (3) is
reacted
with a diamine compound having substituents A, B, D and E as defined above but
with C2
or Cs symmetry or A=B=H, in a suitable solvent, such as for example, aqueous
acetonitrile,
DMF or aqueous DMF, to give the bicyclic compound of formula ( 11 ). The 2'
and 4"
hydroxy protecting group of compound ( 11 ) are then removed by standard
methods (cf. T.
W. Greene and P. G. M. Wuts, Protective C~roun~ in rganic Svnthe~ie, 2nd ed.,
John
Wiley & Son, Inc., 1991 ) to give ( 12). When OR2 is an ester, for example,
such as acetate
or benzoate, the compound may be deprotected by treatment with methanol or
ethanol.
When R2 is a trialkylsilyl group, the compound may be deprotected by treatment
with
fluoride in THF or acetonitrile, for example. Compound ( 12) is then cyclized
by treatment
with dilute acid, such as acetic acid or HCI, for example, in a suitable
organic solvent, such
as methanol, ethanol or propanol, for example, for a period of from 4 hours to
10 days,
from reaction temperature to reflux, in order to prepare the compound of
formula (13).
-40-
a~t~ ~ f ~~~ ~}
CA 02235942 1998-04-23
WO 97/17356 PCT/CTS96/16585
Scheme 6
Alternate Preparation of Compounds of Formula (n
NMe~ p y C!-la NMez
O ~p~y D ~ O 'RCL..
EI,C ~~ E~... ~ A, H~ R.
. A 8
"'~ O O C rY... ,
Fi, ~ .. "O O CF[,
NHx O
':
H~ CH,
O O i'.. O ~' Y=~H O ~.. O CH,
OR O ..' OR:
H,c ~ocH, (14
H,c ocEh
R' is as described for formula I
RZ is a hydroxy-protecting group
En. D Oli, NMei p'' NHi ~' NMe,
E ~..
A ~ HCy.. p~ O H~~.
R'
I-hCi, ~ ~ g~. HOC ~ CH
' p Nr...
p Nr.... - ~~ ? ~ O O
O O CHI Cy0111e
H,Oa ~ ~ H,O' OH,
O, ~ CFL,
D On,, O CH, O
O O ~~~~ OH
OH
(13~ 1-t~C OCH, (12~ H,C ~~OCH~
Compound of Formula (I).
Scheme 6 illustrates an alternative preparation of compounds of formula (I).
Starting
material (3) is reacted with a beta-aminoalcohol (Y=OH) having substituents A,
B, D and E,
as defined above, in a suitable solvent system such as aqueous acetonitrile,
DMF or aqueous
DMF at 0 - 70 °C to give compound (14) where Y=OH. The azido
intermediate, compound
( 14) Y=N3, is prepared by Mitsunobu reaction by reacting compound ( 14) Y=OH
with
triphenylphosphine and diphenylphosphoryl azide-DEAD in tetrahydrofuran.
Compound
(14) is then deprotected by standard methods (cf. T. W. Greene and P. G. M.
Wuts,
Protective Groups in OrfJanic Synthesis, 2nd ed., John Wiley & Son, Inc.,
1991). When
OR2 is an ester, for example, such as acetate or benzoate, the compound may be
deprotected
by treatment with methanol or ethanol. When R2 is a trialkylsilyl group, the
compound may
be deprotected by treatment with fluoride in THF or acetonitrile, for example.
The azido
intermediate, compound (14) Y=N3, is then reduced to the amino compound (12).
Suitable
reducing reagents are triphenylphosphine-water, hydrogen with a catalyst,
sodium
-41 -
CA 02235942 1998-04-23
WO 97/17356 PCT/LTS96/16585
borohydride, or dialkylaluminum hydride. Compound (12) is then cyclized by
treatment with
dilute acid, such as acetic acid or HCl, for example, in a suitable organic
solvent, such as
methanol, ethanol or propanol, for example, for a period of from 4 hours to 10
days, in
order to prepare the compound of formula ( 13).
Alternatively in scheme 6, the hydroxy group (Y=OH) in ( 14) is activated by
treatment with sulfonyl chloride, alkyl or aryl sulfonic anhydride or
trifluoromethanesufonic
anhydride in an aprotic solvent (e.g., diethyl ether, dichloromethane,
tetrahydrofuran,
chloroform, pyridine or a mixture thereof). The reaction requires cooling or
heating,
depending on the conditions used. The reaction temperature is preferably -100
to 10 °C.
The reaction may require 20 minutes to 24 hours to complete. The activated
hydroxy group
in (14) (e.g., Y=-OS02CF3) is then converted to the corresponding azide (Y=N3,
14) by
reacting with lithium azide or sodium azide in the same solvent described
above. The
reaction temperature is preferably 0-100 °C. The azido compound is then
converted to (I3)
according to the procedures described above.
- 42 -
CA 02235942 1998-04-23
WO 97/17356 PCT/US96/16585
Scheme 7
Preparation of Compounds of Formula (II)
aa,
NMe, p N~ G-la NMe.
'RO, p Eu... zR~,
R' tdii A ~ Rn
a ~ En...
/ ..." A
O O CHI p? N....
~N B' ~.. \O ...np
~/ O H'O ~ ~'bC'° ci-L.
~ O~x
C
o (6)
O (iCJ)
R~ is as described for formula I
Rz is a hydroxy-protecting group
deprotect
E~ p ~' . NMe1 p NHi ~' NMei
En..
A \ HCy.. A HO~.
B~ ~cm. 'Rq.b g,. H~
Nr... , Q Nr...
.. o o ~ cyclize ~ ...,.o a~,
~c'~~ ~ °H~ H,c''
oceZ ocbz
cr6 cH, a-6 cH,
0 0
(17) (16)
Compound of Formula (11)
In accordance with Scheme 7, a starting material compound (6) is reacted with
a
diamine compound having substituents A, B, D and E as defined above but with
C2 or Cs
symmetry or A=B=H, in a suitable solvent, such as for example, aqueous
acetonitrile, DMF
S or aqueous DMF, to give the bicyclic compound of formula (15). Compound (15)
is then
deprotected to prepare the compound of formula ( 16). The deprotection is
accomplished by
standard methods described (cf. T. W. Greene and P. G. M. Wuts, Protective
Groups in
Glr a~ic Synthesis, 2nd ed., John Wiley & Son, Inc., 1991). When OR2 is an
ester, for
example, such as acetate or benzoate, the compound may be deprotected by
treatment with
methanol or ethanol. When R2 is a trialkylsilyl group, the compound may be
deprotected by
treatment with fluoride in THF or acetonitrile, for example. Compound (16) is
then cyclized
by treatment with dilute acid, such as acetic acid or HCl, for example, in a
suitable organic
solvent, such as ethanol or propanol, for example, for a period of from 4
hours to 10 days,
from reaction temperature to reflux, in order to prepare the compound of
formula ( 17).
- 43 -
SUBSTITUTE SHEET (RULE 26)
CA 02235942 1998-04-23
WO 97/17356 PC"T/US96/16585
Scheme 8
Alternate Preparation of Compounds of Formula (IIl
H NMh ~~NH; CH, HMS
O ,per E n.~~'.
O
' 'AO,,
Ha A
'' CH, Em. NH' H R.
B'
"'NO O CH, A~ O W,.. n
H~N~O B~' NH. ~ ~~~ O O GH,
C H, O
O H'C~~ CH,
O OCbx H,C
CH, CH, O OCbx
CH, CH,
O
(6)
o (15)
R~ is as described for formula I
R2 is a hydroxy-protecting group
deprotect
CH, NM~,
NHr CH, NM~,
A N HO,I Em O
A H O,,
Bn~G... ' CH B H' '' CH
O~ N,... . O? N".. ....,0 ~ C H
° ~~"o o cH, cycllze o '
HaCV: CH, H,C4: CH,
O OCbx O OCbx
CH, CH, CN, CH,
O O
(17) (16)
Compound of Formula (II)
Scheme 8 illustrates an alternative preparation of compounds of formula (II).
Starting material (6) is reacted with a beta-aminoalcohol (Y=OH) having
substituents A, B,
D and E, as defined above, in a suitable solvent system such as aqueous
acetonitrile, DMF
or aqueous DMF at 0 - 70 °C to give compound ( 18) where Y=OH. The
azido intermediate,
compound (18) Y=N3, is prepared by Mitsunobu reaction by reacting compound
(18)
Y=OH with triphenylphosphine and diphenylphosphoryl azide-DEAD in
tetrahydrofuran.
Compound ( 18) is then deprotected to prepare the compound of formula ( 16)
wherein R2 is
H. The deprotection is accomplished by standard methods (cf. T. W. Greene and
P. G. M.
Wuts, Protective Groups in Or. anic Synthesis, 2nd ed., John Wiley & Son,
Inc., 1991).
When OR2 is an ester, for example, such as acetate or benzoate, the compound
may be
deprotected by treatment with methanol or ethanol. When R2 is a trialkylsilyl
group, the
compound may be deprotected by treatment with fluoride in THF or acetonitrile,
for
example. The azido intermediate, compound ( 18) Y=N3, is then reduced to the
amino
compound (16). Suitable reducing reagents are triphenylphosphine-water,
hydrogen with a
SUBSTITUTE SHEET (RULE 26)
CA 02235942 1998-04-23
WO~ 97/17356 PCT/iJS96/16585
catalyst, sodium borohydride, or dialkylaluminum hydride. Compound ( 16) is
then cyclized
by treatment with dilute acid, such as acetic acid or HCI, for example, in a
suitable organic
solvent, such as methanol, ethanol or propanol, for example, for a period of
from 4 hours to
days, from reaction temperature to reflux, in order to prepare the compound of
formula
. s (17).
Alternatively in scheme 8, the hydroxy group (Y=OH) in (18) is activated by
treatment with sulfonyl chloride, alkyl or aryl sulfonic anhydride or
trifluoromethanesufonic
anhydride in an aprotic solvent (e.g., diethyl ether, dichloromethane,
tetrahydrofuran,
chloroform, pyridine or a mixture thereof. The reaction requires cooling or
heating,
1o depending on the conditions used. The reaction temperature is preferably -
100 to 10 °C.
The reaction may require 20 minutes to 24 hours to complete. The activated
hydroxy group
in (18) (e.g., Y=-OS02CF3) is then converted to the corresponding azide (Y=N3,
18) by
reacting with lithium azide or sodium azide in the same solvent described
above. The
reaction temperature is preferably 0-100 °C.- The azido compound is
then converted to (13)
according to the procedures described above.
- 45 -
CA 02235942 1998-04-23
WO 97/17356 PCT/US96/16585
Scheme 9
Preparation of Comt~ounds of Formula IIII)
C_H, NMv,
C ~RO,, p Em. NH CH NMV,
H~ R' NHr ~ ~O rROrr
'' CH Er.., R.
...., A 6~~ H' '' CHI
O O CH, O Nr",
O B' NH, ~ .~'~r0 O CH,
CH, O
O HrC: O O HrCa; O CHr
CH, CH, O
CH, CH,
O
(8) o (19)
R~ is as described for formula I
RZ is a hydroxy-protecting group deprotect ~pro~~
cyclize
CHr NMv, NH CH'
NM~,
A HOp Eh~. r0 HO,.
aC R~ A~_ [/ R.
r'' ' CHr 9"' \ Ha '' CH,
o w.... ~ w....
"'~~O O CH, O~ "'.r0 O CH,
O~ CYCIIZA p
H C~ I ~ o CHa ~ H~~' CH,
O O
CH, CH, CH, CH,
O O
(21 ) (2~)
Compound of Forrnula (III)
In accordance with Scheme 9, a starting material compound(8) is reacted with a
diamine compound having substituents A, B, D and E as defined above but with
C2 or Cs
S symmetry or A=B=H, in a suitable solvent, such as for example, aqueous
acetonitrile, DMF
or aqueous DMF, to give the bicyclic compound of formula ( 19). Compound ( 19)
is then
deprotected prepare the compound of formula (20). The deprotection is
accomplished by
standard methods (cf. T. W. Greene and P. G. M. Wuts, Protective Groups in Or
anic
nthe is, 2nd ed., John Wiley & Son, Inc., 1991). When OR2 is an ester, for
example,
such as acetate or benzoate, the compound may be deprotected by treatment with
methanol or
ethanol. When R2 is a trialkylsilyl group, the compound may be deprotected by
treatment
with fluoride in THF or acetonitrile, for example. Compound (20) is then
cyclized by
treatment with dilute acid, such as acetic acid or HCI, for example, in a
suitable organic
solvent, such as ethanol or propanol, for example, for a period of from 4
hours to 10 days,
from reaction temperature to reflux, in order to prepare the compound of
formula (21 ).
-46-
SUBSTITUTE SHEET (RULE 26)
CA 02235942 1998-04-23
WQ~ 97/17356 PCT/US96/16585
Alternately, as is readily apparent to those skilled in the art it is possible
to cychze compound
(19) first, then deprotect, to obtain the compound (21).
Scheme 10
Alternate Preparation of Compounds of Formula (l~l
NMei p ,, CI-Lr NMer
O r ZRO~. p E u. O TROT
E6C E rr.. Y A R.
~'r ~ A g ~ ~p ° CH,
" O O CH, O N ... ..nr
~ N~ ~ O O CH,
O H~ O CHr H~C~. CHI
~O
Y=OH ~o
G-I, CFh
o ($)
o (22)
R' is as described for formula I
RZ is a hydroxy-protecting group
p ~ NMei p NHz ~' NMar
E...LL Err.
A- / ~ Rr. HO, A O Rr. HQ~
B' ~ FhC~.. ~ CHa g H,C '~ CH
a
~Nr... ~ ..rrO O ~ ~N.o' ~.nO O CH
p CYCItZe
H,C O CH' ~ HoC~~' ~ CE6
O O
CJ-la CH, CH, Ch6
O O
(21 ) (2~)
Compound of Formula (III)
Scheme 10 illustrates an alternative preparation of compounds of formula
(III).
Starting material (8) is reacted with a beta-aminoalcohol (Y=OH) having
substituents A, B,
D and E in a suitable solvent system such as aqueous acetonitrile, DMF or
aqueous DMF at
0 - 70 °C to give compound (22) where Y=OH. the azido intermediate,
compound (22)
Y=N3, is prepared by Mitsunobu reaction by reacting compound (22) Y=OH with
triphenyl-
phosphine and diphenylphosphoryl azide-DEAD in tetrahydrofuran. Compound (22)
is then
deprotected by standard methods (cf. T. W. Greene and P. G. M. Wuts,
Protective Groups
in Organic Synthesis, 2nd ed., John Wiley & Son, Inc., 1991 ). When OR2 is an
ester, for
example, such as acetate or benzoate, the compound may be deprotected by
treatment with
methanol or ethanol. When R2 is a trialkylsilyl group, the compound may be
deprotected by
treatment with fluoride in THF or acetonitrile, for example. The deprotected
azido
- 47
SUBSTITUTE SHEET (RULE 2fi)
CA 02235942 1998-04-23
WO 97/17356 PCT/US96/16585
intermediate, compound (22) Y=N3, is then reduced to the amino compound (20).
Suitable
reducing reagents are triphenylphosphine-water, hydrogen with a catalyst,
sodium
borohydride, or dialkylaluminum hydride. Compound (20) is then cyclized by
treatment with
dilute acid, such as acetic acid or HCl, for example, in a suitable organic
solvent, such as
methanol, ethanol or propanol, for example, for a period of from 4 hours to 10
days, from
reaction temperature to reflux, in order to prepare the compound of formula
(21 ).
Alternatively in scheme 10, the hydroxy group (Y=OH) in (22) is activated by
treatment with sulfonyl chloride, alkyl or aryl sulfonic anhydride or
trifluoromethanesufonic
anhydride in an aprotic solvent (e.g., diethyl ether, dichloromethane,
tetrahydrofuran,
chloroform, pyridine or a mixture thereof. The reaction requires cooling or
heating,
depending on the conditions used. The reaction temperature is preferably -100
to 10 °C.
The reaction may require 20 minutes to 24 hours to complete. The activated
hydroxy group
in (22) (e.g., Y=-OS02CF3) is then converted to the corresponding azide (Y=N3,
14) by
reacting with lithium azide or sodium azide in the same solvent described
above. The
reaction temperature is preferably 0-100 °C. The azido compound is then
converted to (13)
according to the procedures described above.
- 48 -
CA 02235942 1998-04-23
WO 97/17356 PCT/US96/16585
Scheme 11
Preparation of Compounds of Formula (N7
NMei D ~s CHa NMei
O 'ROy p Er.. O a
RO.,,
F[,C .R~~ Er... N~ A C Rn
a ~ H~ ' ~b
.....0 O O~~ A O Nr... ....,
O g~ N~ ~ O O CH,
O H~ ~ wCH~ H~C~,. CH,
O O
CI-l, CH,
p (10)
p (23)
R~ is as described for formula I
RZ is a hydroxy-protecting group d~rotect
D CHI NMe, D NHs U~~ NMer
W R. A R.
A~N HC1.. Er "\ O H4~~
B E6Cr... ' CHI ~ g.r HOC ~ CH
O~t~l... ....e0 O Cfi~ p~Nr... ..rr0 O CFir
p CYCIIZe
~Cv: C~ ~-~C~: wCH,
O
CHI CH, C~"6 CF6
O O
(24)
Compound of Formula (IV)
In accordance with Scheme 11, a starting material compound ( 10) is reacted
with a
diamine compound having substituents A, B, D and E as defined above but with
C2 or Cs
symmetry or A=B=H, in a suitable solvent, such as for example, aqueous
acetonitrile, DMF
or aqueous DMF, to give the bicyclic compound of formula (23). Compound (23)
is then
deprotected to prepare the compound of formula (24) by standard methods (cf.
T. W.
Greene and P. G. M. Wuts, Protective Groups in Organic Synthesis, 2nd ed.,
John Wiley
& Son, Inc., 1991 ). When OR2 is an ester, for example, such as acetate or
benzoate, the
compound may be deprotected by treatment with methanol or ethanol. When R2 is
a
trialkylsilyl group, the compound may be deprotected by treatment with
fluoride in THF or
acetonitrile, for example. Compound (24) is then cyclized by treatment with
dilute acid,
such as acetic acid or HCI, for example, in a suitable organic solvent, such
as ethanol or
propanol, for example, for a period of from 4 hours to 10 days, from reaction
temperature to
reflux, in order to prepare the compound of formula (25).
-49-
SUBSTtTUTE SKEET (RULE 26)
CA 02235942 1998-04-23
WO 97/17356 PCT/US96/16585
Scheme 12
Alternate Prena_ration of Compounds of Formula fIV)
NMei
D E D~~ y'Y ~' NMei
E r~ Y q~ O R~' Z RO,,,
B HOC
r0 O CI-h g ~2 O~Nr... '~~~.r0 O Cfir
O
H~O~~' OHM
Y=OH
~h G-6
(10)
~ (26)
R~ is as described for formula I
RZ is a hydroxy-protecting group
c
E r.. H, ~°~ D NHx ~' NMs,
q HO~, E O
q HO.,
B"". Hero.. ~ CFir g HOC
Nr... ~
"' O O q.h Nr... ...r p~
CyCIiZB
E60 Clip H~'f CH,
O
CHI ~ CF{, CI-[,
D O
(25) (24)
Compound of Formula (IV)
Scheme 12 illustrates an alternative preparation of compounds of formula (IV).
Starting material ( 10) is reacted with an beta-aminoalcohol (Y=OH) having
substituents A,
B, D and E, as defined above, in a suitable solvent system such as aqueous
acetonitrile,
S DMF or aqueous DMF at 0 - 70 °C to give compound (26) where Y=OH.
The azido
intermediate, compound (26) Y=N3, is prepared by Mitsunobu reaction by
reacting
compound (26) Y=OH with diphenylphosphoryl azide-DEAD in tetrahydrofuran.
Compound (26) is then deprotected by standard methods (cf. T. W. Greene and P.
G. M.
Wuts, Protective Groups in Oreanic Synthesis, 2nd ed., John Wiley & Son, Inc.,
1991).
When OR2 is an ester, for example, such as acetate or benzoate, the compound
may be
deprotected by treatment with methanol or ethanol. When R2 is a trialkylsilyl
group, the
compound may be deprotected by treatment with fluoride in THF or acetonitrile,
for
example. The deprotected azido intermediate, compound (26) Y=N3, is then
reduced to the
amino compound (24). Suitable reducing reagents are triphenylphosphine-water,
hydrogen
with a catalyst, sodium borohydride, or dialkylaluminum hydride. Compound (24)
is then
-50-
SUBSTITUTE SHEET (RULE 26)
CA 02235942 1998-04-23
W~ 97/17356 PCT/US96/16585
cyclized by treatment with dilute acid, such as acetic acid or HCl, for
example, in a suitable
organic solvent, such as methanol, ethanol or propanol, for example, for a
period of from 4
hours to 10 days, in order to prepare the compound of formula (25).
Alternatively in Scheme 12, the hydroxy group (Y=OH) in (26) is activated by
treatment with sulfonyl chloride, alkyl or aryl sulfonic anhydride or
trifluoromethanesufonic
anhydride in a solvent does not adversely affect the reaction (e.g., diethyl
ether,
dichloromethane, tetrahydrofuran, chloroform, pyridine or a mixture thereof.
The reaction
requires cooling or heating, depending on the conditions used. The reaction
temperature is
preferably -100 to 10 °C. The reaction may require 20 minutes to 24
hours to complete. The
activated hydroxy group in (26) (e.g., Y=-OS02CF3) is then converted to the
corresponding
azide (Y=N3, 26) by reacting with lithium azide in the same solvent described
above. The
reaction temperature is preferably 0-70 °C. The azido compound may be
deprotected and
converted to (25) according to the procedures described above.
The compounds and processes of the present invention will be better understood
in
connection with the following examples, which are intended as an illustration
of and not a
limitation upon the scope of the invention.
Exaunple 1
Compound (IV): R~--methoxy: R~hydro~en: A=B=D=E=hydrogen
la. 5-O-desosaminyl-6-O-methylerythronolide A
A sample of clarithromycin (3-O-cladinosyl-5-O-desosaminyl-6-O-methyl-
erythronolide A, Abbott Labs> 142.38 g, 190.35 mmol) was suspended in an
ethanol-water
solution (1700/600 mL), and 341 mL of 1N HCl was added. The reaction mixture
was
stirred for 24 hours, and 2 M NaOH (170 mL) and an additional 250 mL of water
were
added with vigorous stirring. The precipitate was collected by filtration,
washed with water,
and dried to afford the title compound (95.00 g, 84%). MS m/z: 590 (M+H)+.
lb. 3-O-xanth~ 1-5-O-desosaminyl-6-O-methylerythronolideA
To a solution of 5-O-desosaminyl-6-O-methylerythronolide A (11.79 g, 20 mmoL,
from step 1 a above) in THF (100 mL) at -20°C under an inert atmosphere
NaH ( 1.80 g, 60
mmoL, 60% dispersion) was added slowly over a 5 minute period. Several minutes
later
CS2 (1.2 mL, 20 mmoL) was added. After 5 minutes of stirring CH3I (1.24 mL, 20
mmol)
was added, the reaction mixture was allowed to gradually warm to -5 -
0°C, and the mixture
was stirred for 1 hour. The reaction mixture was diluted with EtOAc (400 mL),
and the
mixture was washed with saturated aqueous NaHC03 and brine, dried (MgS04), and
concentrated to afford the crude product. The residue was purified by
chromatography on
silica gel, eluting with CHCl3 and CHC13-MeOH (95:5), to afford the title
compound (7.68
-51-
CA 02235942 1998-04-23
WO 97/17356 PCT/US96/16585
g; 56%). MS m/z: 680 (M+H)+. Anal. Calcd. for C32H5~NO1pS2: C, 56.52; H, 8.45;
N,
2.06; Found: C, 56.95; H, 8.65; N, 1.92.
~1 c. 3-deoxv-5-O-desosaminyl-6-O-methyler5rthronolide A
A solution of 3-O-xanthyl-5-O-desosaminyl-6-O-methyl erythronolide A (20.00 g;
29.41 mmoL, from step lb), Bu3SnH (9.49 mL, 35.29 mmol) and AIBN (~50 mg,
catalytic) in benzene (200 mL) was heated at reflux (adding 25 mg portions of
AIBN
periodically) for 8 hours. The organic layer was separated and washed with 10%
aqueous
KF and brine, dried (MgS04), and concentrated to afford the crude product as
an oil. The
residue was purified by chromatography on silica gel, eluting with CHCl3 and
CHC13-
MeOH (97.5:2.5). The material was recrystallizated from hexane to afford the
title
compound (5.48 g; 32%). MS m/z: 574 (M+H)+. Anal. Calcd. for C3pH55NO9: C,
62.80; H, 9.66; N, 2.44; Found: C, 63.02; H, 9.74; N, 2.30.
ld 2'-O-acetyl-3-deox~-5-O desosaminyl 6 O methylerythronolide A
Samples of 3-deoxy-5-O-desosaminyl-6-O-methylerythronolide A (573 mg, 1.0
mmol, from Example lc above), acetic anhydride (0.188 mL) and TEA (0.278 mL)
were
dissolved in 10 mL of methylene chloride, and the reaction mixture was stirred
at reaction
temperature for 20 hours. The reaction mixture was diluted with 40 mL of
methylene
chloride, and the organic solution was washed with saturated aqueous NaHC03
and brine,
dried and concentrated to obtain the title compound (600 mg). MS m/z: 616
(M+H)+.
le. Compound 1101 from Scheme 4- R.1--methoxy: R2=ace 1
A sample of 2'-O-acetyl-3-deoxy-5-O-desosaminyl-6-O-methylerythronolide A
(0.63
g, 1.023 mmol, from Example ld above) was dissolved in 10 mL of THF, and the
solution
was cooled to -60°C. To this stirred solution was added sodium
bis(trimethylsilyl) amide
(1.22 mL, 1.0 M in THF). After 4 hours l, l-carbonyldiimidazole (0.66 g, 4.09
mmol) was
added as a solution in 6 mL of 2:3 DMF:THF, and the reaction mixture was
slowly warmed
to reaction temperature and stirred for 16 hours. The reaction was quenched by
addition of
5% aqueous NaH2P04, and the resulting mixture was extracted with chloroform.
The
organic layer was washed with water, dried over MgS04 and concentrated to give
the title
compound. MS m/z: 692 (M+H)+.
lf. Compound 1231 from Scheme 11' Ri-methoxv: R2=acetyl: A=B=D=E=hvdro en
A sample of the compound from step le above (0.25 g, 0.36 mmol) was dissolved
in
3 mL of acetonitrile, ethylenediamine (0.24 mL, 3.6 mmol) was added, and the
reaction
mixture was stirred at reaction temperature for 3 hours. The solvent was
removed under
vacuum, and the residue was dissolved in ethyl acetate. This solution was
washed with
saturated aqueous NaHC03 solution, dried over Na2S04 and concentrated. The
residue was
-52-
CA 02235942 1998-04-23
WO 97/17356 PCT/US96/16585
twice purified by flash chromatography on silica gel, eluting with 2-20'%
ethanol in
chloroform containing 0.5-2% NHq.OH to give the title compound. MS m/z: 684
(M+H)+.
1~,;- Compound 1241 from Scheme 11: R.1=methoxv: A=B=D=E=h5rdrog_en
A sample of the compound from step if was dissolved in methanol and stirred at
reaction temperature for 64 hours. The title compound (170 mg) was obtained
after filtration
and removal of the solvent.
lh. Compound (IVl : R1-methoxy: R2=hydr~en: A=B=D=E=hydrogen
A sample of the compound from step 6b ( 170 mg, 0.265 mmol) was dissolved in 2
mL of ethanol to which was added 0.03 mL of acetic acid, and the reaction
mixture was
stirred at reaction temperature for 16 hours. The solvent was removed, and the
residue was
suspended in water. The solution was adjusted to approximately pH 10-11 with
2M NaOH,
and the mixture was extracted with ethyl acetate. The organic extract was
washed with
brine, dried over Na2S04 and concentrated. The residue was purified by flash
chromatography on silica gel, eluting with 10% ethanol in chloroform
containing 0.1 %
NH40H. The product was re-chromatographed, eluting with 0-15% ethanol in
chloroform
to give the title compound (55 mg). MS m/z: 624 (M+H)+, 646 (M+NHq.)+. Anal.
Calcd.
for C33Hg7N3Og: C, 63.53; H, 9.20; N, 6.73; Found: C, 64.68; H, 9.27; N, 7.01.
13CNMR: C=O (16) 156.3, NCH2 (17) 42.4, NCH2 (18) 49.1
ExaBnple 2
Preparation of Intermediate Starting Material:
Compound (8) from Scheme 3: R~--methoxv-R~benzovl
2a. 5-O-desosaminvl-6-O-meth~crvthronolide A
A sample of clarithromycin (3-O-cladinosyl-5-O-desosaminyl-6-O-
methylerythronolide A, Abbott Labs, 900 g, 1.2 mole) was suspended in water (
10.8 L) and
ethanol (4.0 L), and the resulting slurry was stirred at reaction temperature
until
homogeneous (about 20 minutes). HCl (1.00 M, 2.16 L) was added over 15
minutes, and
the reaction mixture was stirred for 20 hours. NaOH solution (2.00 M, 1.20 L)
was added
over 30 minutes until pH 10.5-11.0 was reached, and the reaction mixture was
stirred for 2
hours. The precipitate was collected and washed with cold water, which was
dried under
vacuum at 50°C to afford 601 g of the title compound. MS m/z (M+H)+:
590.
2b. 2'-O-benzovl-5-O-desosaminyl-6-O-methylerythronolide A
To a solution of 5-O-desosaminyl-6-O-methylerythronolide A, (600 g, 1.01 mol
from step 2a above) in methylene chloride (2.0 L) was added 90% technical
grade benzoic
anhydride (380 g, 1.59 mol). Triethylamine (222 mL, 1.59 mol) was added over
10
minutes, and the thick solution was stirred for 48 hours. Sodium bicarbonate
solution
-53-
CA 02235942 1998-04-23
WO 97/17356 PCT/US96/16585
(10%, 1.5 L) was added, and the mixture was stirred for 30 minutes. The layers
were
separated, and the organic fraction was washed with water (3 x 600 mL) and
brine (600
mL). The organic layer was dried (Na2S04) and filtered, and the volatiles were
removed on
a rotary evaporator to leave a syrup. Trituration with a warm solution of
hexane (2.0 L) and
ethyl acetate (100 mL) converted the product to white crystals. The product
was filtered,
washed with hexane and dried in a vacuum oven overnight at ambient temperature
to give the
title compound (691 g). MS m/z (M+H)+: 694.
2c. 2'-O-benzovl-5-O-desosaminvl-3-deoxv 3 oxo 6 O methylervthronolide A
A sample of N-chlorosuccinimide (57.0 g, 0.42 mol) was slurried in anhydrous
methylene chloride (600 mL), and dimethyl sulfide (36.0 mL, 0.49 mol) wa.s
added
dropwise over 30 minutes. A sample of the compound from step 2b (200.0 g, 0.29
mol)
was dissolved in methylene chloride ( 1.20 L), and this solution was added to
the reaction
mixture over 45 minutes. After stirring for 30 minutes a solution of
triethylamine (40.0 mL)
in methylene chloride (200 mL) was added dropwise over 30 minutes at
0°C under nitrogen.
The resulting solution was washed with sodium bicarbonate ( 10%, 3 x 600 mL)
and brine
(600 mL). The organic fraction was dried (Na2S04) and filtered, and the
volatiles were
removed on a rotary evaporator to give a thick syrup, which became a solid
upon standing.
The solid was crushed and dried overnight at ambient temperature in a vacuum
oven to give
2o the title compound ( 196 g). MS m/z (M+H)+: 692.
2d. 2'-O-benzoyl-5-O-desosaminyl-3-deoxy-3-oxo-6-O-
methvl-11-O-methanesulfonyl-6-O-methylervthronolide A
To a solution of 2'-O-benzoyl-5-O-desosaminyl-3-deoxy-3-oxo-6-O-methyl-
erythronolide A from step 2c above (20.00 g, 28.9 mmole) in pyridine (40 mL)
cooled to 0°
and held under N2 was added methanesulfonic anhydride ( 14.6 g, 83.81 mmole),
and the
reaction was allowed to stir at reaction temperature for 17 hours. The
pyridine was removed
under vacuum, and the residue was dissolved in EtOAc (400 mL). This solution
was
washed with saturated aqueous NaHC03, H20 and brine, dried (MgS04),
decolorized with
charcoal, and filtered through a diatomaceous earth filter aid. The solvent
was removed
under vacuum to afford the crude product (24.46 g). This material was taken
directly to the
next step without further purification.
2e. 10,11-anhydro-2'-O-benzoyl-5-O-desosaminyl-
3-deoxy-3-oxo-6-O-methylerythronolid~A
The mesylate from step 2d above was dissolved in acetone (70 mL), and DBU
(5.22
mL, 34.9 mmole) added. After stirring at reaction temperature for 22 hours the
acetone was
removed under vacuum, EtOAc (250 mL) was added, and the organic layer was
washed
with 100-mL portions of sat. aq. NaHC03 (2x), H20 (lx), and brine (lx). The
solution
was dried (MgS04), decolorized with charcoal and filtered through a
diatomaceous earth
-54-
CA 02235942 1998-04-23
WO 97/17356 PCT/US96/16585
falter aid. The solvent was removed under vacuum to afford the crude product (
18.54 g).
This material was purified by chromatography on silica gel, eluting wittu 40%
ethyl
acetate/hexanes containing 0.25 % concentrated NH40H. The appropriate
fractions were
combined and concentrated to give the product. MS m/z (M+H)+: 674.
~f. Compound (8) from Scheme 3: R1-methoxy: R~=benzovl
A 500 mL flask was charged with 60% NaH (1.05 g, 26.3 mmole). The NaH was
ransed with 3 portions of hexanes, and dried under a N2 stream. Freshly
distilled THF (90
mL) was added, and the solution was cooled to 0 °C under N2. The 10,11-
anhydro
compound from step 2d above (8.40 g, 12.5 mmol) was then added over a one
minute
period. After stirring for 15 minutes, a solution of carbonyl diimidazole
(5.98 g, 36.9
mmole) in 60 mL of THF was added to the reaction mixture via cannula over a
period of 15
minutes. After stirring for 5 hours, the reaction mixture was quenched with 5%
KH2PO4
solution, and the mixture was stirred at 0 °C for 20 minutes. The
mixture was extracted with
ethyl acetate. The combined organic extracts were washed with brine, dried
over Na2SOq.
and concentrated under reduced pressure. The residue was purified by
chromatography on
silica gel, eluting with 25% -40% acetone/hexanes. The appropriate fractions
were
combined and concentrated to give the product. MS m/z (M+H)+: 768. 1H NMR
(CDC13):
0.90(t, 3H), 0.95(d, 3H), 1.21(d, 3H), 1.27(d, 3H), 1.32(s, 3H), 2.25(s, 6H),
2.78(s,
3H), 2.97(m, 1H), 3.58(m, 1H), 2.63(q, 1H), 4.14(d, 1H), 4.50(d, 1H), 5.00(dd,
1H),
5.65(dd, 1H), 6.75(s, 1H), 7.05(m, 1H), 7.35(m, 1H), 7.43(dd, 2H), 7.54(t,
1H),
8.02(d, 2H), 8.07(s, 1H); 13C NMR (CDC13): 204.8, 168.8, 165.0, 145.9, 138.4,
138.1,
137.0, 132.7, 130.8, 130.5, 129.7, 128.2, 117.0, 102.1, 84.5, 81.0, 78.5,
76.9, 72.0,
69.2, 63.7, 50.9, 50.2, 47.2, 40.7, 40.3, 38.8, 31.1, 30.8, 22.5, 20.9, 20.7,
20.0, 18.8,
14.8, 14.2, 13.2, 10.4.
Example 3
Compound of Formula IIII). A. B & E=hydrogen, D=benzvl-R~=methoxy: R.~-
hydr~o.~en
3a. 2-lR)-BOC-amino)-3-phenyl-1-propanol
To a 5.2 g (23.8 mmole) sample of di-t-butyl dicarbonate in 20 mL of methylene
chloride held at 0°C was added (R)-2-amino-3-phenyl-1-propanol (3.0 g,
19.8 mmole,
Aldrich), and the reaction mixture was stirred 1.5 hours at reaction
temperature. The solvent
was removed, and the residue was dried under high vacuum and taken directly to
the next
Step.
3b. 2-(R)-(BOC-amino)-1-O-methanesulfon~lox~phen ly_nropane
The material from step 3a was dissolved in 20 mL of methylene chloride and 5
mL of
THF, and the solution was cooled to 0°C. Triethylamine (4.1 mL, 29.4
mmole) was added,
-55-
CA 02235942 1998-04-23
WO 97/17356 PCT/US96/16585
then methanesulfonyl chloride (1.9 mL, 24.5 mmole) was added slowly. The
mixture was
stirred 45 minutes at reaction temperature, then the solvent was removed under
vacuum. The
residue was dissolved in ethyl acetate, and the solution was washed with water
and brine,
dried (Na2SOq.) and filtered. The solvent was removed under vacuum to afford
6.38 g of the
title compound. MS m/z (M+H)+: 330, MS m/z (M+NH4)+: 347.
3c. 1-azido-2-(Rl-(BOC-aminol-3-phenylpropane
The compound from step 3b above (6.36 g, 193 mmole) was dissolved in 25 mL of
DMF, and 2.5 g (38 mmole) of NaN3 was added. The reaction mixture was stirred
for 24
l0 hours at 62°C. The solution was cooled to reaction temperature, then
extracted with ethyl
acetate. The organic extract was washed with water and brine, dried (Na2S04)
and filtered.
The solvent was removed under vacuum to afford 4.34 g of the title compound.
MS m/z
(M+H)+: 277, MS m/z (M+NH4)+: 294.
3d. 1-azido-2-(Rl-amino-3-phenylpropane
The compound from step 3c (4.3 g,15.6 mmole) was dissolved in 30 mL of 4 N HCl
in ethanol, and the reaction mixture was stirred for 1.5 hours at reaction
temperature. The
solvent was stripped and chased with ether. The residue was dissolved in
water, NaCI was
added, and the mixture was extracted with ethyl ether, which was discarded.
The aqueous
layer was adjusted to pH 12 with K2C03, saturated with NaCI, then extracted
with CHCl3.
The organic extract was washed with brine, dried (Na2SOq.) and filtered. The
solvent was
removed under vacuum to afford 2.17 g of the title compound. MS m/z (M+H)+:
177, MS
m/z (M+NH4)+: 194.
3e. 1 2-fR)-diamino-3- henylpro ane
A sample of the compound from step 3d (1.2 g, 6.8 mmole) was hydrogenated (4
atm) in ethanol over 1.2 g of 10% Pd/C for 21.5 hours at reaction temperature.
The mixture
was filtered to remove the catalyst, and the solvent was removed to afford the
title compound
(1.055 g). MS m/z (M+H)+: 151, MS m/z (M+NH4)+: 168.
mnound ( 19) from Scheme 9 A=B=E=H D-benzyl R~--methoxy-R.~= enz 1
Samples of the compound of formula (8) (Scheme 3; R1=methoxy; R2=benzoyl;
from Example 2, 750 mg, 0.98 mmole) and l, 2-(R)-diamino-3-phenylpropane (from
step
3e above, 1.04 g, 6.92 mmole) were dissolved in 4 mL of acetonitrile and 0.5
mL of water.
The reaction mixture was stirred for 24 hours at reaction temperature. The
solvent was
removed, and the residue was dissolved in ethyl acetate. The solution was
washed with
water and brine, dried (Na2SOq.), filtered, and the solvent was removed. The
residue was
purified by chromatography on silica gel, eluting with hexane to 30% acetone
in hexane to
give 236 mg of the title compound. MS m/z (M+H)+: 850.
-56-
CA 02235942 1998-04-23
WO 97/17356 PCT/US96/16585
3g. Compound (20) from Scheme 9, A=B=E=H,
D=bend. R.1=methoxy, R2=hydrogen
A sample (217 mg, 0.26 mmole) of the compound from step 3f above in 6 mL of
methanol was stirred at reflux for 4 hours. The solvent was removed, and the
residue was
purified by column chromatography on silica gei, eluting with 1.5% methanol in
chloroform
containing 1 % NH40H to afford 176 mg of the title compound. MS rn/z (M+H)+:
746.
3h. Compound of Formula lIlP, A=B=E=H, D=benzyl. RI=methoxy. R~h,~gen
A sample of the compound from step 3g (148 mg, 0.20 mmole) was dissolved in
2.6
l0 mL of ethanol, and of acetic acid (0.026 mL, 0.45 mmole) was added. The
reaction mixture
was stirred for 24 hours at reflux, then the solvent was removed. The residue
was dissolved
in ethyl acetate, which was washed with aqueous K2C03, water and brine. The
solution
was dried (Na2SO4), filtered, and the solvent was removed under vacuum to
afford 150 mg
of product. This material was purified by column chromatography on silica gel,
eluting with
0.5% to 0.6% methanol in chloroform containing 0.5% NH40H to afford 134 mg of
the title
compound. MS m/z (M+H)+: 728. 1H NMR (CDC13): 0.82(t, 3H), 1.02(d, 3H),
1.18(d,
3H), 1.32(s, 3H), 1.35(d> 3H), 1.43(s, 3H), 1.50(m, 1H)> 1.87(m, 1H), 2.27(s,
6H)>
2.32(s, 3H), 2.56(dd, 1H), 2.66(m, 2H), 3.03(m, 1H), 3.19(dd, 1H), 3.63(s,
1H),
3.75(q, 1H), 3.94(dd, 1H), 4.04(m, 1H), 4.13(d, 1H), 4.28(d, 1H), 4.85(dd,
1H), 7.10-
7.35(m, 5H); 13C NMR (CDC13): 204.0, 177.8, 169.4, 156.0, 138.3, 130.1, 127.9,
126.1, 103.9, 81.4, 79.4, 78.5, 76.4, 70.3, 69.5, 65.9, 59.5, 57.9, 51.2,
48.7, 48.3,
46.5, 42.8, 40.2, 38.5, 36.0, 31.6, 28.2, 22.0, 21.2, 19.6, 19.0, 16.7, 14.4,
14.1, 12.7,
11.0, 10.4;
Example 4
Compound of Formula (III). A=B=D=H, E=benzyl, R~--methoxy~R2=hydrogen
4a. 1-azido-2-fS)-amino-3-phenyl-propane
Following the procedure of Example 3a, except substituting (S)-2-amino-3-
phenyl-1-
propanol (Aldrich) for the (R)-2-amino-3-phenyl-1-propanol thereof, and
carrying the
product forward as in Example 3, steps a-d, the title compound was prepared
(1.74 g). MS
m/z (M+H)+: 177, MS m/z (M+NHq.)+: 194.
4b 12-(S)-diamino-3-phen~~luropane
A sample of the compound from step 4a (790 mg, 4.48 mmole) was dissolved in 30
mL of THF and 6 mL of water, triphenylphosphine (5.0 g, 19.1 mmole) was added,
and the
reaction mixture was stirred for 24 hours at reflux. The solvent was removed,
and the
residue was dissolved in 2 N HCl. NaCI was added, and the solution was
extracted with
ethyl ether. The aqueous layer was adjusted to pH 12 with K2C03, saturated
with NaCI,
-57-
CA 02235942 1998-04-23
WO 97/17356 PCT/US96/16585
then extracted with CHCl3 and CHC13 containing 15% isopropanol. The solvent
was
removed under vacuum to afford 439 mg of the title compound. MS m/z (M+H)+:
151, MS
m/z (M+NH4)+: 168.
4c. Comuound (191 Scheme 9. A=B=D=H E=benzyl R1-methoxy. R~benzovl
Samples of the compound of formula (8) (Scheme 3; R1=methoxy; R2=benzoyl;
from Example 2, 450 mg, 0.59 mmole) and 1,2-(S)-diamino-3-phenylpropane (from
step 4b
above, 435 mg, 2.90 mmole) were dissolved in 2 mL of acetonitrile and 0.25 mL
of water.
The reaction mixture was stirred for 24 hours at reaction temperature. The
solvent was
removed, and the residue was dissolved in ethyl acetate. The solution was
washed with
brine, dried (Na2S04), filtered, and the solvent was removed. The residue was
purified by
chromatography on silica gel, eluting with hexane to 25% acetone in hexane to
give 405 mg
of the title compound. MS m/z (M+H)+: 850.
4d. Compound (20). A=B=D=H E=benzyl R.1=methoxy. R~hydrogen
A sample (386 mg, 0.45 mmole) of the compound from step 4c above in 7 mL of
methanol was stirred at reflux for 4 hours. The solvent was removed, and the
residue was
purified by column chromatography on silica gel, eluting with 1.5 % methanol
in chloroform
containing 1 % NH40H to afford 315 mg of the title compound. MS m/z (M+H)+:
746.
4e. Compound of Formula IIIII A=B=D=H E=phenyl R.~methox3r: R~hvdro en
Following the procedure of Example 3h, except substituting the (S)-compound
(150
mg, 0.20 mmole) from step 4d for the (R)-isomer of 3h, the title compound (136
mg) was
prepared. MS m/z (M+H)+: 728. 1H NMR (CDCl3): 0.86(t, 3H), 1.08(d, 3H),
1.19(d,
3H), 1.33(x, 3H), 1.39(d, 3H), 1.47(s, 3H), I.54(m, 1H), 1.92(m, 1H), 2.28(s,
6H),
2.58(s, 3H), 3.19(dd, 1H), 3.52(m, IH), 3.67(s, 1H), 3.77(q, 1H), 4.12(d, 1H),
4.27(d,
1H), 4.92(dd, 1H), 7.10-7.40(m, 5H); 13C NMR (CDC13): 203.7, 176.0, 169.4,
156.5,
139.2, 130.0, 129.7, 128.3, 127.9, 126.3, 126.1, 104.1, 80.9, 80.5, 78.6,
78.1, 70.3,
69.5, 65.9, 62.0, 61.1, 51.2, 49.5, 48.8, 48.6, 45.9, 43.3, 42.9, 41.4, 40.2,
40.0, 35.2,
28.2, 22.2, 21.1, 19.7, 19.4, 17.1, 16.8, 15.3, 14.5, 14.1, 10.4;
Example 5
Compound of Formula lIlI1 A=benzyl B=D=E=H R1-methoxy. R~hvdro~en
5a. Compound (22) (Scheme 10), A=benzyl;
B=D=E=H. R1-methoxy-R~=benzoyl: Y=OH
Compound (8) (Scheme 3; R1=methoxy; R2=benzoyl; from Example 2, 450 mg,
0.59 mmole) and (S)-2-amino-3-phenyl-1-propanol (1.97 g, 13.0 mmole, Aldrich)
were
dissolved in 4.5 mL of acetonitrile and 0.5 mL of water. The reaction mixture
was stirred
for 7 days at reaction temperature. The solvent was removed, and the residue
was dissolved
- 58 -
CA 02235942 1998-04-23
WO 97/17356 PCT/US96/16585
in ethyl acetate. The solution was washed with 20% aqueous KH2P04, water and
brine,
then dried (Na2S04) and filtered. The solvent was removed, and the residue
dried under
high vacuum to afford I.09 g of product. This material was purified by
chromatography on
silica gel, eluting with hexane to 20% acetone in hexane to give 644 mg of the
tide
compound. MS m/z (M+H)+: 851.
Sb. Compound (22) (Scheme 10), A=benzyl;
B=D=E=H. RI=methoxy. R~=benzoyl: Y=azido
A sample of the compound from step Sa above (617 mg, 0.72 mmole) was dissolved
i0 in l2mL of THF, and triphenylphosphine (610 mg, 2.33 mmole) was added. This
solution
was cooled to 0°C, DEAD (0.375 mL, 2.38 mmole) was added dropwise over
3 minutes,
and the mixture was stirred for 10 minutes. Next was added DPPA (0.515 mL,
2.37
rnmole) dropwise over 3 minutes, and the mixture was stirred for 2 hours at
0°C and 48
hours at reaction temperature. The volatiles were removed under vacuum to
leave an oily
residue. The residue was purified by chromatography on silica gel, eluting
with hexane to
30% ethyl acetate in hexane followed by 20% acetone in hexane to afford 321 mg
of the title
compound. MS m/z (M+H)+: 876.
Sc. Compound (22) (Scheme 10), A=benzyl;
B=D=E=H. R.1=methoxyiR~=hydrogen: Y=azido
A sample of the compound from step Sb above (317 mg, 0.36 mmole) was dissolved
in 5 mL of methanol, and the mixture was stirred at reflex for 4.5 hour. s.
The solvent was
removed, the residue was purified by chromatography on silica gel, eluting
with 1:1 acetone
in hexane , and the residue was dried under high vacuum to afford 218 mg of
the title
compound. MS m/z (M+H)+: 772.
~d. Compound 120) (Scheme 10), A=benzvl: B=D=E=H, R-1=methoxy, R.~~,1 ydro~en
A sample of the compound from step Sc above (208 mg, 0.27 mmole) was dissolved
in 3 mL of THF and 0.5 mL of water, triphenylphosphine (425 mg, 1.62 mmole)
was
added, and the reaction was stirred for 24 hours at reflex. The solvent was
removed under
vacuum, chased with toluene, and the residue dried under high vacuum. The
residue was
purified by chromatography on silica gel, eluting with chloroform containing
1.5% methanol
and 1 % NH40H to afford I 96 mg of the rifle compound. MS m/z (M+H)~: 746.
Se. Compound of Formula (IIII: A=benzvl, B=D=E=H. R1-methoxy-R~hvdroEen
A sample of the compound from step Sd above ( 128 mg, 0.17 mmole) was
dissolved
in 2.3 mL of ethanol and 0.023 mL (0.40 mmole) of acetic acid was added. The
reaction
mixture was stirred at reflex for 48 hours, and the solvent was removed. The
residue was
dissolved in ethyl acetate, and the solution was washed with aqueous K2C03,
water and
brine. The solution was dried (Na2S04), filtered, and the solvent was removed
under
-59-
CA 02235942 1998-04-23
WO 97/17356 PCT/US96/16585
vacuum to afford 100 mg of a tan compound. The residue was purified by
chromatography
on silica gel, eluting with chloroform containing 0.6°lo methanol and
0.5% NHq.OH
progressing to chloroform containing 0.7% methanol and 0.5% NH40H to afford 52
mg of
the title compound. MS m/z (M+H)+: 728. 1H NMR (CDCl3): 0.86(t, 3H), 1.08(d,
3I-~,
1.34(d, 3H), 1.37(d, 3H), 1.44(s, 3H), 1.52(s, 3H), 1.93(m, 1H), 2.29(s, 6H),
2.48(m,
1H), 2.67(dd, 1H), 2.86(s, 3H), 3.06(dd, 1H), 3.57(m, 1H), 3.74(q, 1H),
3.86(s, 1H),
4.33(d, 1H), 4.39(d, 1H), 4.95(dd, 1H), 7.18-7.40(m, 5H); 13C NMR (CDCl3):
204.4,
169.6, 155.8, 138.0, 129.5, 129.3, 128.6, 128.5, 126.5, 103.8, 81.4, 78.8,
77.3, 70.3,
69.6, 65.9, 57.3, 53.7, 51.2, 50.8, 49.8, 47.4, 42.7, 40.3, 38.5, 35.6, 31.6,
28.2, 22.6,
22.2, 21.2, 20.6, 20.3, 15.4, 14.9, 14.1, 13.4, 11.1, 10.4;
Example 6
Compound of Formula fIII): B=benzvl A=D=E=H R.l--methoxy, R~hydrogen
Following the procedures of Example 5, except replacing the (S)-2-amino-3-
phenyl-
1-propanol of step 5a with (R)-2-amino-3-phenyl-1-propanol (1.97 g, 13.0
mmole,
Aldrich), and carrying the product forward according to the procedures of
steps 5b-5e, the
title compound (83 mg) was prepared. MS m/z (M+H)+: 728. iH NMR (CDC13):
0.73(t,
3H), 1.05(d, 3H), 1.24(d, 3H), 1.29(d, 3H), 1.33(s, 3H), 1.37(d, 3H), 1.41(s,
3H),
2.26(s, 6H), 2.44(m, 1H), 2.68(m, 1H), 2.79(s, 3H), 3.18(dd, 1H), 3.46(m, 1H),
3.60(s,
1H), 3.71(dd, 1H), 3.81(q, 1H), 3.94(dd, 1H), 4.30(dd, 1H), 4.72(dd, 1H), 7.10-
7.38(m, 5H); 13C NMR (CDCl3): 204.4, 180.7, 169.3, 154.5, 138.4, 128.9, 128.8,
128.6, 128.3, 126.8, 126.2, 125.1, 104.2, 103.7, 81.1, 78.5, 78.3, 78.1, 77.2,
76.2,
70.3, 69.5, 65.8, 65.7, 62.4, 58.1, 53.6, 51.2, 51.0, 49.4, 47.6, 42.9, 40.2,
38.5, 36.7,
36.0, 31.5, 28.2, 25.2, 22.6, 21.6, 21.1, 19.7, 19.3, 16.2, 15.6, 14.6, 14.3,
14.0, 12.6,
11.1, 10.4, 10.2;
Example 7
Compound of Formula (lII)~ A=E=phenyl B-D-H R.~-methoxy, R~=hydro en
Comuound (19) from Scheme 9~ A=E=phenyl B-D-H R.1--=methoxy-R~benzovl
Compound (8) (Scheme 3; R1=methoxy; R2=benzoyl; from Example 2, 400 mg,
0.52 mmole) and (1S,2S)-1,2-diphenyl-1,2-ethylenediamine (500 mg, 2.36 mmole,
Aldrich) were dissolved in 2 mL of acetonitrile and 0.25 mL of water. The
reaction mixture
was stirred for 10 days at reaction temperature. The mixture was diluted with
methylene
chloride, the solution was dried over NaCl and Na2S04, filtered, and the
solvent was
removed. The residue (958 mg) was taken directly to the next step . MS (m/z) :
666
(M+H)+.
-60-
CA 02235942 1998-04-23
WO 97/17356 PCT/US96/16585
7b. Compound (19) from Scheme 9;
A=E=phenyl. B=D=H. R1=methoxy. R~h~g_en
The compound from step 7a (958 mg) was dissolved in 15 mL of methanol, and the
solution was heated at reflux for 24 hours. The solvent was removed, and the
residue was
purified by column chromatography on silica gel, eluting with 0 to S% methanol
in
methylene chloride to afford 340 mg of the title product as the C10-epi-
isomer. MS (m/z)
808 (M+H)+. 1H NMR (300 MHz, CDCl3) : 8 6.90-7.20 (m, lOH), 5.48 (dd, 1H),
5.04
{d, 1 H), 4.27 (d, 1H), 4.23 (d, 2H), 3.93 (q, 1 T-i), 3.76 (d, 1 H), 3.62 (d,
1 H, H 11 ), 3.52
{m, ZH)3.12-3.22 (m, 4H), 3.07 (s, 3H, OMe), 2.75 (dd, 1H), 2.75 (m, 1H), 2.2-
2.5 (m),
l0 2.24 (s, 6H, NMe2), 1.94 (dq, 2H), 1.59 (s, 3H), 1.46 (d, 3H), 1.29 (d,
3H), 1.22 (d,
3H), 0.90 (t, 3H), 0.84 (d, 3H), 0.73 (d, 3H). 13C NMR (75 MHz, CDC13) : 8
214.2,
205.5, 171.0, 156.3, 143.5, 140.5, 128.0, 128.0, 127.9, 127.2, 126.9, 126.8,
104.1,
83.5, 78.8, 78.7, 78.2, 77.4, 77.0, 76.6, 70.2, 69.9, 69.3, 67.6, 65.6, 57.9,
51.7, 50.7,
49.0,48.5, 41.1, 41.0, 40.1, 28.2, 21.2, 21.1, 21.0, 19.3, 18.0, 16.0, 14.4,
10.5, 9.9.
7c. Compound of Formula IIII): A=E=phenyl. B=D=H. Ri-methoxy-R~hydrogen
Following the procedure of Example Se, except replacing the compound from Sd
with the compound from 7b, the title compound was prepared. MS (m/z) : 790
(M+H)+.
13C NMR {125 MHz, CDC13): selected signals: 8 203.3, 175.7, 169.5, 156.5,
104.1.
Example 8
Compound of Formula (nI): A=methyl. B=D=E=hydro eg n R.1--methoxy. R~hydrogen
8a. Compound (22) from Scheme 10;
A=methyl. B=D=E=H. R~methoxy. R~benzoyl. Y=OH
Compound (8) (Scheme 3; R1=methoxy; R2=benzoyl; from Example 2, 400 mg,
0.52 mmole) and (S)-2-amino-1-propanol (0.200 mL, 2.6 mmole, Aldrich) were
dissolved
im 0.9 mL of acetonitrile, 0.1 mL of water was added, and the reaction was
stirred for 40
hours at reaction temperature. The solvent was removed, and the residue was
dissolved in
ethyl acetate. The solution was washed with water and brine, dried (Na2SOq.),
filtered, and
the solvent was removed. The residue was purified by chromatography on silica
gel, eluting
with 20% to 30% acetone in hexane to give 135 mg of the title compound. MS m/z
(M+H)+:
'775.
8b_ Compound (22) from Scheme 10; A=methyl,
B=D=E=H. R-1--methoxy. R~benzoyl. Y=azido
A sample of the compound from step 8a above (277 mg, 0.362 mmole) was
dissolved in 6 mL of THF, and triphenylphosphine (302 mg, 1.15 mmole) was
added. This
solution was cooled to 0°C, DEAD (0.190 mL, 1.21 mmole) was added
dropwise over 3
minutes, and the mixture was stirred for 10 minutes. Next was added DPPA
(0.260 mL,
-61 -
CA 02235942 1998-04-23
WO 97/17356 PCTNS96/16585
1.21 mmole) dropwise over 3 minutes, and the mixture was stirred for 3 hours
at 0°C and 24
hours at reaction temperature. The volatiles were removed under vacuum. The
residue was
purified by chromatography on silica gel, eluting with 15% to 20% acetone in
hexane, then
rechromatographed on silica gel> eluting with 40% ethyl acetate in hexane to
25% acetone in
hexane, to afford 140 mg of the title compound. MS m/z (M+K)~-: 838.
8c. Compound (22) from Scheme 10; A=methyl,
B=D=E=H. R 1=methoxv, R2=hydrogen. Y=azido
A sample of the compound from step 8b ( 133mg, 0.17 mmole) was dissolved in 2
mL of methanol, and the solution was stirred for 4 hours at reflux. The
solvent was
removed under vacuum, and the residue was dried under high vacuum to give a
glassy
residue. The residue was purified by chromatography on silica gel, eluting
with 1:1 acetone
in hexane to 100% acetone to afford the title compound (98 mg). MS m/z (M+H)+:
696.
8d. Compound (20) from Scheme 10;
A=methyl. B=D=E=H R 1=methoxv-R~hydro~en
A sample of the compound from step 8c above (185 mg, 0.27 mmole) was dissolved
in 4 mL of THF, and 2 mL of H20 and triphenylphosphine (425 mg, 1.62 mmole)
was
added. This solution was stirred at reaction temperature for 40 hours and at
reflux for 24
hours. The volatiles were removed under vacuum. The residue was purified by
chromatography on silica gel, eluting with 3% methanol and 1% NH40H in
chloroform to
afford the title compound ( 128 mg). MS m/z (M+H)+: 670.
Vie. Comr~nd of Formula fIIIO A=methyl B-D-E-H Ri=methoxy-R~hydrogen
A sample of the compound from step 8d above ( 123 mg, 0.18 mmole) was
dissolved
in 2.6 mL of ethanol and 0.24 mL of acetic acid, the reaction was heated at
reflux for 24
hours, then stirred at reaction temperature for 48 hours. The solvent was
removed under
vacuum, and the residue was dissolved in ethyl acetate. The solution was
washed with 10%
aqueous K2C03, water and brine, then dried (Na2S04) and filtered. The solvent
was
removed, and residue was dried under high vacuum. The residue was purified by
chromatography on silica gel, eluting with 0.5% NH40H and 2% to 3% methanol in
chloroform to afford the title compound (38 mg). MS m/z (M+H)+: 652. 1H NMR
(CDC13): 0.86 (t, 3H), 1.04 (d, 3H), 1.21 (d, 3H), 1.24 (d, 3H), 1.50 (s, 3H),
1.87-1.98
(m, 2H), 2.26 (s, 6H), 2.41-2.50 (m, 1 H), 2.78 (s, 3H), 3.11-3.23 (m, 3H),
3.51-3.59
(m, 1 H), 3.76 (dd, 1 H), 3.76 (s, 1 H), 3.85 (q, 1 H), 3.92 (dd, 1 H), 4.31
(d, 1 H), 4.37 (d,
1H), 4.90 (dd, 1H); 13C NMR (CDCl3): 204.3, 181.5, 169.6, 155.6, 103.7, 81.3,
78.6,
77.9, 76.6, 70.3, 69.5, 65.8, 56.5, 53.4, 51.1, 50.6, 47.4, 47.2, 42.6, 40.2,
38.4, 35.5,
28.1, 22.0, 21.2, 20.5, 20.1, 15.8, 15.2, 14.8, 13.2, 10.6, 10.3; High
resolution mass
-62-
CA 02235942 1998-04-23
WO 97/17356 PCT/US96/16585
spectrum: calculated (M+H)+ m/z for C34HSgN3O9=652.4173; observed (M+H)+
m/z=652.4175.
Example 9
C~~ound of Formula fIIIh B=methyl A=D=E=hydrogen Rl.--methox~, R~hydro~en
Following the procedures of Example 8, except with a larger amount of the
starting
compound from Example 2 (1.23 g, 1.6 mmole) and replacing the (S)-2-amino-1-
propanol
of step 8a with an appropriately larger amount of (R)-2-amino-1-propanol (1.23
mL, 16.2
l0 mmole, Aldrich), carrying the product forward as in steps 8b-8e, the tide
compound was
prepared (71 mg). 1H NMR (CDCl3): 0.87 (t, 3H), 1.45 (s, 3H), 1.82-2.01 (dd,
2H),
2.26 (s, 6H), 2.37-2.51 (m, 1H), 2.72 (s, 3H), 3.07 (dd, 1H), 3.18 (dd, 1H),
3.22-3.36
(m, 1H), 3.47-3.59 (m, 1H), 3.61 (s> 1H), 3.77-3.91 (m, 2H), 4.25 (d, 1H),
4.29 (d,
1H), 5.01 (dd, 1H); 13C NMR '(CDC13): 204.1, 180.7, 169.7, 154.7, 103.8, 80.6,
78.9,
78.5, 76.4, 70.3, 69.6, 65.9, 62.3, 56.6, 53.1, 51.1, 49.3, 47.8, 42.8, 40.2,
38.6, 36.5,
34.6, 31.5, 28.2, 25.3> 22.6, 21.9, 21.2, 20.7, 19.6, 19.3, 17.1, 15.8> 14.4,
14.1, 12.9,
11.0, 10.3; MS (M+H)+ m/z=652. High resolution mass spectrum: calculated
(M+H)+ m1z
for C34HggN3O9=652.4173; observed (M+H)+ m/z=652.4188.
2o Example 10
Compound of Formula (lm: A=D=methyl: B=E=H, Ri-methoxy, R2=hydrogen
lsla. meso-2.3-bis(methanesulfon~ylbutane
Samples of meso-2,3-butanediol (10 g, 111 mmole, Aldrich) and triethylamine
(92.8
mL, 666 mmole) were dissolved in methylene chloride. The solution was cooled
to -78°C,
and methanesulfonyl chloride (25.8 mL, 333 mmole) was added dropwise. A
precipitate
formed. The mixture was diluted with additional methylene chloride, and the
mixture was
stirred for 20 minutes at -78°C and at 0°C for 2 hours. The
reaction mixture was warmed to
reaction temperature, diluted with additional solvent, and washed with H20,
aqueous
3o NaHC03 and aqueous NaCI. The organic solution was dried over MgS04, and the
solvent
was removed to afford the title compound (25.01 g). IH NMR (300 MHz, CDCl3) :
8 4.91
(q, 2H), 3.10 (s, 6H), 1.45 (d, 6H).
lOb. meso-2.3-diazidobutane
A sample of the compound from step l0a (25 g) was dissolved in 250 mL of DMF,
and NaN3 (40 g) was added. The mixture was stirred vigorously at 85°C
for 24 hours, then
cooled to reaction temperature. The mixture was diluted with 800 mL of ether,
washed with
H20, aqueous NaHC03 and aqueous NaCI, then dried over MgS04. The solution was
filtered and concentrated to afford the title compound ( 13.00 g). 1H NMR (300
MHz,
CDC13) : 8 3.50 (m, 2H), 1.30 (d, 6H).
- 63 -
CA 02235942 1998-04-23
WO 97/17356 PCT/US96/16585
lOc. meso-2 3-butanedia_minP
A sample of the compound from step l Ob ( 13.0 g, 125 mmole) was dissolved in
ethanol and hydrogenated at 4 atm over 10% Pd/C for 20 hours at reaction
temperature. The
catalyst was removed by filtration, and the solvent was removed under vacuum
to afford the
title compound. 1H NMR (300 MHz, CDC13) : 8 2.70 (m, 2H), 1.45 (br, 4H), 1.05
(d,
6H). MS (m/z) : 89 (M+H)+.
l Od. Compound ( 19) from Scheme 9' A-D-methyl B-E-H R.1--methoxy. R~benzoyl
to A sample of 10,11-anhydro-2'-O-benzoyl-5-O-desosaminyl-6-O-methyl-3-oxo-
erythronolide A 12-O-imidazolyl carbamate (from Example 2, 500 mg, 0.651
mmole) and
meso-2,3-butanediamine (500 mg, from step lOd above) were dissolved in 3 mL of
acetonitrile and 0.3 mL of water, and the reaction was stirred for 72 hours at
reaction
temperature and 17 hours at reflux. The solution was diluted with methylene
chloride, dried
IS over NaCI and MgS04, and filtered. The solvent was removed, and the residue
was taken
directly to the next step.
~Oe. Compound (201 from Scheme 9~ A-D-methvl~ B-E-H R1=methoxv
The compound from step lOd above was dissolved in methanol, and the solution
was
20 heated at reflux for 12 hours. The solvent was removed, and the residue was
taken directly
to the next step. MS (m/z) : 684 (M+H)+.
l Of. Comvound of Formula (IIW A-D-methyl- B-E-H R 1-methoxv, R~hydro en
To the material from the previous step, dissolved in methanol, acetic acid was
added,
25 and the reaction mixture was heated at reflux for 24 hours. A solution of
NH3 in methanol
was added, and the solution was concentrated. The residue was dissolved in
ethyl acetate,
and the solution was washed with 1 N NaOH, H20 and brine, then dried over
Na2S04. The
solvent was removed, and the residue was purified on silica gel, eluting with
10% methanol
in methylene chloride to 10% methanol (containing NH3) in methylene chloride
to afford the
3o title compound. This material was rechromatographed on pH 8 silica gel,
eluting with 4:1
ethyl acetate:hexane to 100% ethyl acetate to afford 116 mg of the title
compound. The
NMR analysis confirmed the product to be the A=D=methyl isomer. MS (m/z) : 666
(M+H)+. HRMS Calc. for C35H~N309: 666.4330; Observed: 666.4326. 'H NMR (300
MHz, CDCl3) : 8 4.81 (dd, 1H), 4.27 (d, 1H), 4.22 (d, 2H), 4.05 (m, 1H), 3.96
(m, 1H),
35 3.78 (q, 1H), 3.65 (s, 1H, H11), 3.48 (m, 1H), 3.10 (dd, 1H), 3.06 (m, 1H),
2.75 (q,
1H), 2.67 (s, 3H), 2.65 (m, 1H), 2.37 (m, 2H), 2.19 (s, 6H), 1.85, 1.49 (m,
2H), 1.59,
1.15 (m, 2H), 1.55 (m, 2H), 1.42 (s, 3H), 1.27 (d, 3H), 1.27 (s, 3H), 1.25 (d,
2H), 1.24
(d, 3H), 1.17 (d, 3H), 1.16 (d, 3H), 1.04 (d, 3H), 0.96 (d, 3H), 0.78 (t, 3H).
'3C NMR
(75 MHz, CDC13) : 8 204.2, 179.0, 169.5, 155.6, 118.8, 103.7, 81.3, 78.6,
77.7, 76.8,
CA 02235942 1998-04-23
WO~ 97/17356 PCT/US96/16585
70.2, 69.4, 65.8, 56.1, 55.5, 51.9, 51.0, 50.4, 47.0, 42.5, 40.1, 38.4, 35.2,
28.1, 22.3,
22.0, 21.1, 20.7, 20.1, 15.0, 14.8, 13.1, 11.5, 10.6, 12_2. IR (film) : 3460
(w), 2972,
1.750, 1718 (w), 1647, 1456, 1423, 1372, 1305, 1246, 1163, 1107, 1051, 989,
757 cm 1.
Example 11
ompound of Formula IIIII: A=E=me~yl: B=D=H. R1=methoxv-R~hvdro~en
Following the procedures of Example 10 steps a-c, except substituting (R,R)-
2,3-
butanediol (1 g, 11.1 mmole, Aldrich) for the meso-isomer thereof of step 10a,
and carrying
the product forward as in steps lOb and l Oc, the title compound (494 mg) was
prepared.
MS (m/z) : 89 (M+H)+. 1H NMR (300 MHz, CDC13) : 8 2.62 (m, 2Id], 1.45 (br s,
4H),
1.08 (d, 6H).
11 b. Compound ( 191 from Scheme 9: A=E=methyl: B=D=H. RI-methox3r, R~benzovl
A sample of 10,11-anhydro-2'-O-benzoyl-5-O-desosaminyl-6-O-methyl-3-oxo-
erythronolide A 12-O-imidazolyl carbamate (from Example 2, 1.56 g, 2.03 mmole)
and 2S,
3S-butanediamine (400 mg, 4.54 mmole, from step 11 a above) were dissolved in
16 mL of
20% aqueous acetonitrile, and the reaction mixture was stirred at reaction
temperature for 7
days. The solution was diluted with methylene chloride, dried over NaCI and
MgS04, and
filtered. The solvent was removed, and the residue was taken directly to the
next step.
l~jc. Compound f201 from Scheme 9: A=E=methyl: B=D=H. R1-methoxv
The compound from step 1 lb above was dissolved in methanol, and the solution
was
heated at reflux for 12 hours. The solvent was removed, and the residue was
taken directly
to the next step.
l ld. Compound of Formula 1I11): A=E=methyl: B=D=H. R1-methoxy-R~h~rdrogen
The material from the previous step was dissolved in 20 mL of ethanol, acetic
acid
(0.80 mL) was added, and the reaction mixture was heated at reflux for 3 days.
NH3/
methanol was added, and the solvent was removed. The residue was dissolved in
ethyl
acetate, and the solution was washed with 1 N NaOH, H20 and brine, then dried
over
IVa2S04. The solvent was removed, and the residue was purled on neutral silica
gel,
eluting with 4:1 ethyl acetate:hexane to 5% methanol in ethyl acetate to
afford 682 mg of the
title compound. MS (m/z) : 666 (M+H)+. HRMS Calc. for C35H~N3O9: 666.4330;
Observed: 666.4333. 1H NMR (300 MHz, CDC13): 8 4.85 (dd, 1H), 4.35 (d, 1H),
4.30
(d, 1H), 4.12 (q, 2H), 3.65 (s to d, 1H, J=1.2 Hz), 3.56 (m, 1H), 3.46 (br s,
1H), 3.21
(dd, 1H), 3.13 (t, 1H), 2.86 (q, 1H), 2.78 (s, 3H), 2.68 (m, 2H), 2.46 (dt,
1H), 2.28 (s,
6H), 1.95 (m, 1H), 1.68 (m, 3H), 1.52 (s, 3H), 1.38 (d, 3H), 1.36 (d, 3H0,
1.33 (s, 3H),
4.0 1.32 (d, 3H), 1.31 (d, 3H), 1.26 (d, 3H), 1.21 (d, 3H), 1.05 (d, 3H); 0.87
(t, 3H). 13C
- 65 -
CA 02235942 1998-04-23
WO 97/17356 PCT/US96/16585
NMR (75 MHz, CDCl3) : 8 203.9, 177.9, 169.5, 156.4, 103.8, 81.1, 78.5, 78.4,
77.6,
70.3, 69.5, 65.8, 60.4, 56.9, 52.3, 50.9, 50.6, 47.6, 43.9, 40.2, 39.5, 35.0,
28.1, 21.7,
21.1, 20.8, 20.2, 19.9, 18.3, 16.3, 15.6, 14.7, 14.1, 13.6, 10.1.
IR (KBr) : 3441, 2972, 2938, 1755, 1117 (w), 1653, 1457, 1425, 1376, 1306,
1248,
1167, 1109, 1053 crri 1.
Example 12
Compound of Formula (III); B=D=H;
A and E taken together is -CH~CH~CH2- Rl--methoxy. R~hy ro~en
to
12a. racemic-tra r-1 2-cYclonen anP~iiamine
Following the procedures of Example 10 steps a-c, except substituting (DL)-1,2-
cyclopentanediol (5.0 g, 49.0 mmole, Aldrich) for the diol of step 10a, and
carrying the
product forward as in steps lOb and lOc, the title compound (2.76 g) was
prepared. MS
(m/z) : 101 (M+H)+. 1H NMR (300 MHz, CDCl3) : 8 2.75 (m, 2H), 2.00 (m, 2H),
1.65
(m, 2H), 1.50 (s, 4H), 1.30 (m, 2H).
12b. Compound (19) from Scheme 9; B=D=H;
A and E taken together is -CH~CH~~=, R1-methoxv-R~=benzovl
Samples of 10,11-anhydro-2'-O-benzoyl-5-O-desosaminyl-6-O-methyl-3-oxo-
erythronolide A 12-O-imidazolyl carbamate (from Example 2, 200 mg, 0.26 mmole)
and
racemic-tram-1,2-cyclopentanediamine (104 mg, 1.04 mmole, from step 12a) were
dissolved in 20% aqueous acetonitrile, and the reaction mixture was stirred at
reaction
temperature for 4 days and at reflux for 8 hours. The solution was diluted
with 100 mL of
ethyl acetate, dried over NaCl and MgS04, and filtered. The solvent was
removed, and the
residue was purified by chromatography on silica gel, eluting with 5% to 10%
methanol in
methylene chloride to afford the title compound (88 mg). MS (m/z) : 800
(M+H)+.
12c. Compound (20) from Scheme 9; B=D=H;
A and E taken together is -C''H2~H~~H2- RL=methoxY
The compound from step 12b above (88 mg) was dissolved in methanol, and the
solution was heated at reflux for 10 hours. The solvent was removed, and the
residue was
purified by chromatography on silica gel, eluting with 5% methanol in
methylene chloride to
afford the title compound. MS (m/z) : 696 (M+H)+.
12d. Compound of Formula (ITI); B=D=H;
A and E taken to~~ether is -CH~CH~~H2 RL=methoxv. R~hvdro, en
Following the procedure of Example 1 ld the title compound was prepared. MS
(m/z) : 678 (M+H)+.
CA 02235942 1998-04-23
WO 97/17356 PCT/US96/16585
Example 13
Compound of Formula (IQ); B=E=H;
A and D taken together is -CHZCH2CH~CH2-. R~=methoxv. R.~h~droaen
13a. Compound (19) from Scheme 9; B=E=H;
A and D taken together is -CH2CH2CH2CH~yR.I--methoxy. R2= nz
Following the procedure of Example 1 l, step l lb, except substituting cis-1,2-
cyclohexanediamine (800 mg, 1.04 mmole, Aldrich) for the diamine thereof, the
title
compound was prepared.
13b. Compound of Formula (Iln; B=E=H;
A and D taken together is -CH~CH~CH~CH~-. RI=methox~. R~=hvdro~en
Following the procedures of Example 11 steps c and d, substituting the
compound of
step 13a for the compound of l lb thereof, the title compound was prepared.
'H NMR (300 MHz, CDCl3): 8 4.87 (dd, 1H), 4.34 (d, 1H), 4.31 (d, 1H), 4.10 (br
s,
1 H), 4.07 (br m, 1 H), 3.84 (q, 1 H), 3.70 (s, 1 H, H 11 ), 3.56 (m, 1 H),
3.22 (dd, 1H),
3.14 (pent, 1H), 2.78 (s, 3H, OMe), 2.49 (m, 1H), 2.30 (s, 6H), 1.30-2.00 (m),
1.49 (s,
1H), 1.39 (s, 3H), 1.37 (d, 3H), 1.32 (d, 3H), 1.26 (d, 3H), 1.22 (d, 3H),
1.06 (d, 3H),
0.85 (t, 3H).
13C NMR (75 MHz, CDCl3) : 8 203.8, 178.1, 169.3, 155.6, 103.7, 81.1, 78.6,
78.1,
76.64, 70.2, 69.3, 65.7, 56.3, 55.9, 55.8, 51.0, 50.4, 47.4, 42.6, 40.1, 38.2,
35.4, 34.4,
28.1, 25.2, 25.1, 25.0, 21.9, 21.0, 20.2, 20.1, 19.2, 15.6, 14.7, 12_9, 10.6,
10.2.
MS (m/z) : 692 (M+H)+.
Example 14
Compound of Formula (IIII: A=B=D=E=H. Rl.=h,~g_en. R~hydrogen
14a. 2'-O-acetyl-6-deoxy-erythromycin A
A sample of 6-deoxyerythromycin A (4.34 g, 6.04 mmole, Abbott Labs) was
dissolved in 250 mL of methylene chloride, and acetic anhydride ( 1.54 g, 15
mmole) and
triethylamine ( 1.22 g, 12 mmole) were added. The reaction mixture was stirred
at reaction
temperature for 16 hours, then poured into 5% aqueous NaHC03. The mixture was
extracted with methylene chloride, and the organic layer was dried, filtered
and concentrated.
The residue was chased with toluene, ethylene dichloride, chloroform and
methylene
chloride. The residue then dried under high vacuum to afford 4.46 g of the
title product,
which was taken to the next step without further purification. MS m/z (M+H)+:
760.
14b. 2'-O-acetyl-6-deoxy-4"-triethylsilyl-erythromycin A
A sample of the compound from step 14.a (4.44 g, 5.84 mmole) was dissolved in
100 mL of dry methylene chloride, and imidazole ( 1.59 g, 23.3 mmole) was
added. The
-67-
CA 02235942 1998-04-23
WO 97/17356 PCT/US96/16585
mixture was cooled to 0°C, and a solution of tziethylsilyl chloride (
1.76 g, 117 mmole) in 25
mL of methylene chloride dropwise. The reaction mixture was stirred for 1 hour
at 0°C and
at reaction temperature for 16 hours. The reaction mixture was poured into 5%
aqueous
NaHC03, and the mixture was extracted with CHC13. The organic extract was
washed with
saturated brine, dried over MgSOq, filtered and concentrated. The residue was
dried under
high vacuum for 48 hours to afford 5.38 g of the title product, which was
taken to the next
step without further purification. . MS m/z (M+H)+: 874.
14c. 2'-O-acetyl-10,11-anhydro-6-deoxy-4"-triethylsilyl-erythromycin A
12-O-imdazolyl carbamate (Compound (3) from Scheme 5,
RI=hydI'Ogen. 2'-R~acetyl: 4"-R2=trieth~yl)
A sample of the compound from step 14b (5.36 g, 6.13 mmole) was dissolved in
45
mL of dry DMF, and carbonyldiimidazole (4.98 g, 30 mmole) was added. After the
reagent
had dissolved, the solution was diluted with 15 mL of THF, and the solution
was cooled to
0°C. To this solution was added NaH (807 mg, 60% dispersion, 20.2
mmole) in portions.
The reaction mixture was stirred for 30 minutes, and 10 mL of H20 was added.
The
reaction mixture was poured into saturated brine, and the resulting mixture
was extracted
with ethyl acetate. The ethyl acetate solution was washed with brine, dried
over MgS04,
filtered and concentrated. The residue was co-distilled with ethylene
dichloride, chloroform
and methylene chloride. The residue was dried under high vacuum for 16 hours
to afford
6.95 g of the title product, which was taken to the next step without further
purification. MS
m/z (M+H)+: 986.
14d. Compound (11) from Scheme 5;
A=B=D=E=H: R1-_hydrogen. 2'-R~acetvl: 4"- R~triet yl ilvl
A sample of the compound from step 14c (6.93 g, 6.13 mmole) was dissolved in
50
rnL of acetonitrile, ethylene diamine was added (4.31 g, 7.19 mmole), and the
solution was
stirred for 17 hours at reaction temperature. The reaction mixture was poured
into saturated
brine containing NHq.OH. This mixture was extracted with chloroform, and the
organic
extracts were combined, filtered and concentrated. The residue was co-
distilled with
ethylene dichloride, chloroform and methylene chloride. The residue was dried
under high
vacuum for 72 hours to afford 5.77 g of the title product which was taken to
the next step
without further purification. MS m/z (M+H)+: 924.
14e. Compound (12) from Scheme 5;
A=B=D=E=H' R~hydrogen. 2'-R~4"-R~=hvdro en
A sample of the compound from step 14d (5.75 g) was dissolved in THF ( 120 mL)
and 14.3 mL of tetrabutylammonium fluoride ( 1 M in THF) was added in one
portion. The
reaction mixture was stirred at reaction temperature for 4 hours, diluted with
400 mL of
methanol, and stirred at reaction temperature for 64 hours. The solution was
concentrated
- 68 -
CA 02235942 1998-04-23
W~ 97/17356 PCT/US96/16585
and then poured into 5% aqueous NaHC03 containing 0.5% NH40H. The mixture was
extracted with CHCl3, the extract was washed with brine, and dried over
MgSOq.. The
solvent was removed, and the residue was co-distilled with ethylene chloride
and methylene
chloride. The residue was further dried under vacuum for 16 hours to afford
the title
compound (5.59 g).
14f. Compound of Formula CI): A=B=D=E=H: R1=h~ e~ n-R~hydroaen
A sample of the compound from step 14e (5.56 g, 7.07 mmole) was dissolved in
60
mL of ethanol, and acetic acid (637 mg, 106 mmole) was added. The solution was
heated at
reflux for 3.5 hours, cooled to reaction temperature, then poured into 5%
aqueous NaHC03.
This mixture was extracted with CHCl3, and the organic extracts were combined,
dried over
MgS04, filtered and concentrated. The residue was co-distilled with ethylene
dichloride,
chloroform and methylene chloride, then dried under high vacuum for 72 hours
to afford
4.12 g of the product. This material was purified by chromatography on silica
gel, eluting
with 1:6 TEA:ethyl acetate, to afford the title compound (1_29 g). MS m/z
(M+H)+: 768.
1H NMR (CDCl3) 8: 0.88 (m), 2.28 (s), 3.28 (s), 4.23 (m), 4.85 (m), 4.92 (m).
13C
NMR (CDCI3) b: 40.3 (N(CH3)2), 42.8 (NCH2), 49.4 (OCH~), 49.9 (NCH2), 155.8 (O-
CO-N). IR (CDC13): 1755 cm-1.
Example 15
Compound of Formula 1I111: Rl=OCH~ R~=H: A=B=D=H: E~CH2NH2
15a. 1.2-diazido-3-IBOC-aminolpropane
A sample of 3-(BOC-amino)propene (Aldrich, 15.72 g, 0.1 mole), manganese
triacetate (80.43 g, 0.3 mole) and NaN3 (65.01 g, 1.0 mmole) were suspended in
400 mL of
acetic acid, and the mixture was heated at 100°C for 10 minutes. The
mixture was cooled in
an ice bath and diluted with 400 mL of 1N NaOH. The mixture was extracted with
ethyl
acetate, which was washed wrath 1 N cold NaOH, H20, NaHC03, brine and dried
over
Na2S04. The solvent was removed, and the residue was chromatographed on silica
gel,
eluting with 4:1 hexane:ether to give 7.18 g of the title compound. MS m/z
(M+NH4)+:
259.
15b. 1.2-diamino-3-BOC-aminolpropane
The compound of the previous step was dissolved in ethanol and hydrogenated (4
atm H2) over Pd/C for 22 hours at reaction temperature. The solvent was
removed, and the
product was taken to the next step. MS m/z (M+H)+: 190.
15c. Compound of Formula (III): Rl-= OCH~ R~=H: A=B=D=H: E=-CH2NH2
Following the procedures of Example 3 steps f h, samples of the compound of
formula (8) (Scheme 3; R1=methoxy; R2=benzoyl; from Example 2, 400 mg, 0.52
mmole)
-69-
CA 02235942 1998-04-23
WO 97/17356 PCT/LTS96/16585
and the compound from step 15b ( 1.478 g, 7.81 mmol) were reacted and the
title product
was obtained. Chromatographic separation of the isomers of the N-BOC compound,
followed by deprotection with HCl in ethanol at reaction temperature gives the
title
compound. MS m/z (M+H)+: 889.
Example 16
compound of Formula (IIT1~ Rl--~ R~=H: A=B=E=H: D=-CH2NH2
The compound is obtained by deprotection with HCl in ethanol at reaction
temperature of the isomer separated by chromatography from the mixture
obtained in
Example 15c.
Example 17
Compound of Formula (III): R1=OCH3,
R~=H; B=E=H: A and D taken together is -CH2CH2CH2
17a. cvclopentane cis-1 2-diamine
Following the procedures of Example 10 steps a-c, except substituting cis-1,2-
cyclopentanediol for the diol of step 10a, and carrying the product forward as
in steps lOb
and l Oc, the title compound is prepared.
17b. Compound of Formula (III): R1=OCH3,
R~=H~ B=E=H~ A and D taken togeth r;s -CH2CH2CH2-
Following the procedures of Example 10 steps d-f, except substituting cis-1,2-
cyclopentanediamine, from the previous step, for the meso-2,3-butanediamine
thereof, the
title compound is prepared.
Example 18
Compound of Formula (III): RI=OCH3,
R~=H: B=E=H~ A and D taken together is -CH20CH2
18a. tetrahydrofuran cis-3 4-diamine
Following the procedures of Example 10 steps a-c, except substituting 1,4-
anhydroerythritol for the diol of step 10a, and carrying the product forward
as in steps lOb
and lOc, the title compound is prepared.
18b. Compound of Formula (III):
Rl--OCH~ R~=H: B=E=H: A and D taken to ether is -CH OCH2
Following the procedures of Example 10 steps d-f, except substituting
tetrahydrofuran cis-3,4-diamine, from the previous step, for the meso-2,3-
butanediamine of
step lOd, the title compound is prepared.
-70-
CA 02235942 1998-04-23
WAD 97/17356 PCT/US96/16585
Example 19
Compound of Formula (Iln: R1=OCH3,
RAH: B=E=H: A and D taken together is -CH2-NH-CH2-
19a cis-3 4-diamino-pyrrolidine
Pyrolline (Aldrich) is N-protected by treatment with
benzyloxycarbonyloxysuccinimide in ethyl acetate at reaction temperature. The
N-Cbz-
pyrroline is oxidized with catalytic Os04 and excess N-methylinorpholine N-
oxide. (NNMO)
in THF and t-butanol according to the procedure of Tetrahedron Lett., 1976:
1973 to give
to the N-Cbz-pyrroline-3,4-diol. Following the procedures of Example 10 steps
a-c, except
substituting N-Cbz-pyrroline-3,4-diol for the diol of step 10a, and carrying
the product
forward as in steps lOb and lOc, the title compound is prepared.
19b. Compound of Formula (III):
i5 Rl=OCH,~ R~=H: B=E=H: A and D taken to~~ether is -CH2-NH-CH2-
Following the procedures of Example 10 steps d-f, except substituting cis-3,4-
diamino-( 1-Cbz-pyrrolidine), from the previous step, for the meso-2,3-
butanediamine of
step lOd, the title compound is prepared.
20 Example 20
Compound of Formula (111): RI=OCH3,
,~~=H: B=E=H' A and D taken together is -CH2-NlCbz)-CH2-
Starting with the Compound of Formula (III): Rl=QCH~, R2=H; B=E=H; A and D
taken together is -CH2-NH-CH2-, from Example 19 above, adding the Cbz group by
25 treatment with benzyloxycarbonyloxysuccinimide in ethyl acetate at reaction
temperature, the
title compound is prepared.
Example 21
Compound of Formula (lII): RI=OCH3,
30 $~=H: B=E=H' A and D taken together is -CH2-N(benzyl)-CH2-
Starting with the Compound of Formula (IQ): Rl=OCH3, R2=H; B=E=H; A and D
taken together is -CH2-NH-CH2-, from Example 19 above, reacting the amine of
the -CH2-
NH-CH2- group with benzyl chloride in the presence of triethylamine, the title
compound is
prepared.
Example 22
Compound of Formula (III): Rl=OCH3,
$~_--H: B=E=H: A a_n_d D taken together is -CH2-Nlben~o~n -CH2-
Starting with the Compound of Formula (111): R'=OCH3, R2=H; B=E=H; A and D
taken together is -CH2-NH-CH2-, from Example 19 above, reacting the amine of
the -CH2-
-71-
CA 02235942 1998-04-23
WO 97/17356 PCT/US96/16585
NH-CH2- group with benzoyl chloride in the presence of triethylamine, the
title compound is
prepared.
Example 23
Compound of Formula (III): R'=OCH3,
$~=H: B=E=H; A and D taken tether is -CH2-N(nhenyl-CH2-CH2-1-CH2-
Starting with the Compound of Formula (III): R'=OCH3, R~=H; B=E=H; A and D
taken together is -CH2-NH-CH2-, from Example 19 above, reacting the amine of
the -CH2-
NH-CH2- group with 2-phenylethyl bromide in the presence of triethylamine, the
title
compound is prepared.
Example 24
Compound of Formula (III): Rl=OCH3,
$2=H: B=E=H: A and D taken toeether i - H2-N(4-Cl-yahenvl-CH2-1-CH2-
Starting with the Compound of Formula (III): R'=OCH3, RZ=H; B=E=H; A and D
taken together is -CH2-NH-CH2-, from Example 19 above, reacting the amine of
the -CH2-
NH-CH2- group with 4-chlorobenzyl chloride in the presence of triethylamine,
the title
compound is prepared.
Example 25
Compound of Formula (III): R'=OCH3,
R~=H. B=E=H: A and D taken together is -CH2-Nl4-pyridyl-CH2-)-CH2-
Starting with the Compound of Formula (III): R1=OCH3, R2=H; B=E=H; A and D
taken together is -CH2-NH-CH2-, from Example 19 above, reacting the amine of
the -CH2-
NH-CH2- group with 4-picolyl chloride in the presence of triethylamine, the
title compound
is prepared.
Example 26
Compound of Formula (111): R'=OCH3,
$~=H: B=E=H~ A and D taken together is -CH2-Ni2-p~ridyl-CH2-)-CH2-
Starting with the Compound of Formula (III): RI=OCH3, R2=H; B=E=H; A and D
taken together is -CH2-NH-CH2-, from Example 19 above, reacting the amine of
the -CH2-
NH-CH2- group with 2-picolyl chloride in the presence of triethylamine, the
title compound
is prepared.
Example 27
Compound of Formula (IIn: R'=OCH3,
$~=H: B=E=H: A and D taken togethPT;s -CH2-NH(3-wridyl-CH2-)-CH2-
Starring with the Compound of Formula (III): R'=OCH3, RZ=H; B=E=H; A and D
taken together is -CH2-NH-CH2-, from Example 19 above, reacting the amine of
the -CH2-
-72-
CA 02235942 1998-04-23
WO 97/17356 PCT/US9b/16585
NH-CH2- group with 3-picolyl chloride in the presence of triethylamine, the
title compound
is prepared.
Example 28
Compound of Formula (111): Rl=OCH3,
$~=H: B=E=H: A and D taken together is -CH2-N(4-quinolxl-CH2-)-CH2-
Stardng with the Compound of Formula (III): R1=OCH3, R2=I-i; B=E=H; A and D
taken together is -CH2-NH-CH2-, from Example 19 above, reacting the amine of
the -CH2-
NH-GH2- group with 4-chloromethylquinoline in the presence of triethylamine,
the title
compound is prepared.
Example 29
Compound of Formula (III): Rl-~ R~=H: B=E=H: A=D=-CH2-O-CH2=phenyl
29a. PhCH20CH2CH(NH2) H(NH2)CH20CH2Ph
Meso-erythritol (Aldrich) is 1,4-dibenzylated by reaction with NaH and benzyl
bromide in DMF according to the procedure of El Amin et al., J. Org. Chem.
44:3442,
(1979). Following the procedures of Example 10 steps a-c, except substituting
1,4-
dibenzyl-meso-erythritol for the diol of step 10a, and carrying the product
forward as in
steps lOb and lOc, the title compound is prepared.
29b. Compound of Formula (III):
Rl-OCH~,,~~=H: B=E=H: A=D=-CH2-O- H2-~n3r1
Following the procedures of Example 10 steps d-f, except substituting
IPhCH20CH2CH(NH2)CH(NH2)CH20CH2Ph, from the previous step, for the meso-2,3-
butanediamine of step lOd, the title compound is prepared.
Example 30
Compound of Formula /TTT1~ R1R~=H: B=E=H: A=D=-CH2-OH
Starting with the Compound of Formula (III): RI=OCH3, R2=H; B=E=H; A=D=-
CH2-O-CH2-phenyl, from Example 29 above, removing the benzyl groups by
hydrogenation over Pd/C, the title compound is prepared.
Example 31
Compound of Formula fIIII- Rl-- .~H R~=H~ B=E=H~ A=D=-CH2-O-p_h~~~l
Starting with the Compound of Formula (III): R1=OCH3, R2=I-i; B=E=H; A=D=-
CH2-OH, from Example 30 above, reacting the -CH2-OH groups (of substituents A
and D)
with phenol, triphenylphosphine and DEAD under Mitsunobu conditions the title
compound
is prepared.
- 73 -
CA 02235942 1998-04-23
WO 97/17356 PCT/US96/16585
Example 32
Compound of Formula (III): Ri=OCH3,
R~=H: A=B=H: D and E taken together is -CH2-CH2-CH2-CH2-
Following the procedures of Example 3 except substituting 1-amino-1-cyclo-
pentanemethanol (Aldrich) for the 2-(R)-amino-3-phenyl-1-propanol thereof, the
title
compound is obtained.
Example 33
Compound of Formula (111): R1=OCH3,
~~=H: A and B taken together is -CH2-CH2-CH2-CHI-~ D-E-H
Following the procedures of Example 5, except replacing the (S)-2-amino-3-
phenyl-
1-propanol of step Sa with 1-amino-1-cyclopentanemethanol (Aldrich), the title
compound is
obtained.
Example 34
Compound of Formula (III): RI=OCH3,
Rz'=H; A=B=H: D and E taken together is -CH2-O-CH2-
34a. 3-amino-3-aminomethyloxetane
A sample of tris(hydroxymethyl)methylamine (Aldrich) is reacted with di-t-
butyldicarbonate and triethylamine to give N-BOC-
tris(hydroxymethyl)methylamine. This
compound is reacted with 1 equivalent of methanesulfonyl chloride and
triethylamine to give
3-(BOC-amino)-3-hydroxymethyloxetane. This compound is converted to the title
compound according to the procedures of Example 3 steps b-e.
34b. Compound of Formula (III): R1=OCH3,
R~=H: A=B=H: D and E taken together is -CH2-O-CH2-
Following the procedures of Example 3 steps f h except substituting the
compound
from step 34a for the (R)-3-phenyl-1,2-propanediamine thereof, the title
compound is
obtained.
Example 35
Compound of Formula IIIII: Rl--OCH~ RAH: A=D=E=H~ B=-CH2-CH2-phen~
Following the procedures of Example 5, except replacing the (S)-2-amino-3-
phenyl-
1-propanol of step Sa with D-homophenylalaninol (prepared by LiAlH4 reduction
of the D-
homophenylalanine available from Aldrich), the title compound is obtained.
-74-
CA 02235942 1998-04-23
WO 97/17356 PCT/US96/16585
Example 36
Coa~r~nound of Formula (IIII: Rl-~ R2=I-I: A=D=E=H: B=-CH2-CH2-CH2=phenyl-
36a. 2-(R)-amino-5-phen~nentanoic acid
(t)-2-Amino-5-phenylpentanoic acid (35 g, from Example 40a) was suspended in
water (3 L) and solubilized by adjusting the pH to 12 with 7 N NaOH solution.
The pH was
readjusted to pH 8 with 1 M phosphoric acid while stirring at 45°C. The
solution was cooled
to 40°C, and L-amino acid oxidase (Sigma, 0.7 unit/mg) was added. The
reaction was
stirred with good aeration for 2 weeks. The reaction mixture was concentrated
to 500 mL
under vacuum, the pH was adjusted to 5, and the precipitate was collected. The
material was
recrystallized from ethanol-water to afford 17.32 g of the title compound).
36b. 2-(R)-amino-5-phenylpentanol
The compound from step 36a is reduced with LiAIHq. under standard conditions
to
give the title compound.
36c. Compound of Formula (BI):
RI=O H R' =H: A=D=E=H' B=-CH2-CH2-CH2=phenyl
Following the procedures of Example 5, except replacing the (S)-2-amino-3-
phenyl-
1-propanol of step Sa with the 2-(R)-amino-5-phenylpentanol from step 36b, the
title
compound is obtained.
Example 37
Compound of Formula (III): Rl--OCHx R~=H: A=D=E=H: B=-CH2-O-CH2=phenyl
37a. 1-N-(CBZ)-2-IS)-diamino-3-O-ben~lpropane
N-BOC-O-benzyl-D-serine (Bachem) is reduced under the reaction conditions
described by Kotokos, Synthesis, 299-301 (1990) to give the N-BOC-O-benzyl-D-
serinol.
This compound is treated according to the procedures of Example l0a-c to give
the
corresponding amine. The amine is converted to the benzyloxycarbonyl (CBZ)
derivative,
and the BOC group is removed to give the title compound.
37b. Compound of Formula (III):
~1--~H R~=Ii: A=D=E=H: B=- H2-O- H2~heB,y1
Following the procedures of Example 10 steps d-f, except substituting the
material
from the previous step, for the meso-2,3-butanediamine of step lOd, the title
compound is
prepared.
- 75 -
CA 02235942 1998-04-23
WO 97/17356 PCT/US96/16585
Example 38
Compound of Formula (IIII): R'=OGH3,
R"=H. A=D=E=H~ B=-CH2-CH2-(4-OCH~- hen 1
38a D-homo-O-methvl osinnl
D-Homo-O-methyltyrosine (prepared according to the procedure of Melillo et
al., J.
Org. Chem., x:5149-5150 (1987)) is reduced with LiAIHq under standard
conditions to
give the title compound.
38b. Cimpound o~Formula (III):
~ .,~3. =H: A=D=E=H' B=- H2- -H~-tq.-OCH~- n 1
Following the procedures of Example 5, except replacing the (S)-2-amino-3-
phenyl-
1-propanol of step Sa with the compound of step 38a, the title compound is
obtained.
Example 39
Compound of Formula (IIII: Rl---I R~=H' A=-CH2-CH2-ph~nvl- B=D=E=H
Following the procedures of Example S, except substituting L-
homophenylalaninol
(prepared by LiAIHq. reduction of the L-homophenylalanine (Aldrich) compound)
for the
(S)-2-amino-3-phenyl-1-propanol of step Sa , the title compound is obtained.
Example 40
Compound of Formula ,(IIII: Rl---I ,~~=H: A=-CH2-CH2-CH2-~henvl~ B=D=E=H
40a. (~)-2-Amino-5-~rhenvlnentann;c a~-;rt
Diethyl acetamidomalonate (220 g) in 1 L of absolute ethanol was added to a
stirred
solution of sodium ethoxide in ethanol, prepared by dissolving sodium (24 g)
in absolute
ethanol (500 mL), under nitrogen. The reaction mixture was refluxed under
nitrogen for 30
minutes, then 1-bromo-3-phenylpropane (200g) was added. The reaction mixture
was
refluxed overnight, cooled to ambient temperature, filtered, and the solvent
was removed
under vacuum. Concentrated HCl (800 mL) was added to the residue, and the
reaction
mixture was refluxed for 14 hours. The cooled aqueous solution was washed with
ether,
and the residual ether in the aqueous phase was removed by bubbling nitrogen
through the
solution. The pH of the aqueous phase was adjusted to pH 7-8 by the addition
of
ammonium hydroxide. The title compound was collected by filtration, air dried
and
recrystallized from ethanol-water (150 g) m.p. 255-257°C. MS m/z: 194
(M+H)+.
49b l+1- 2-amino-5-phenvlpen annl
The compound from step 40a is reduced with LiAlH4 under standard conditions to
give the title compound.
-76-
CA 02235942 1998-04-23
WO 97/17356 PC'1'/US96/16585
40c. Compound of Formula (af):
Rl-,-~ R~=H: A=-CHz-CHz-CHz-phenyl: B=D=E=H
Following the procedures of Example 5, except replacing the (S)-2-amino-3-
phenyl
1-propanol of step Sa with the (~)- 2-amino-5-phenylpentanol from step 40b,
and separating
the isomers by chromatography the title compound is obtained.
Example 41
Compound of Formula IIIi): Rl-=OCH~ R~=H: A=-CH2-O-CH2-phenyl ~ B=D=E=H
Following the procedures of Example 37, except substituting the N-BOC-O-benzyl-
L-serine (Sigma) for the N-BOC-O-benzyl-D-serine thereof the title compound is
prepared.
Example 42
Compound of Formula (111): R1=OCH3,
$~-_-H: B=D=E=H: A=- H2-CH2-l4-OCH~-_ henvll
42a. L-homo-O-methyltvros~n_ol
L-Homo-O-methyltyrosine (prepared according to the procedure of Melillo et
al., J.
drg. Chem., ,x:5149-5150 (1987)) is reduced with LiAlHø under standard
conditions to
give the title compound.
42b. Compound of Formula (III):
Rl=OCH~ R~=H: B=D=E=H: A=-CH2-CH2-(4-OCH~- hen 1
Following the procedures of Example 5, except replacing the (S)-2-amino-3-
phenyl-
1-propanol of step Sa with the compound of step 42a, the title compound is
obtained.
Example 43
Compound of Formula lIln: Rl=OCH~. R~=H: A=B=D=H: E=-CH2CH2Ph
Following the procedures of Example 3 except replacing the 2-(R)-amino-3-
phenyl-
1-propanol of step 3a thereof with L-homo-phenylalaninol (prepared by LiAIHq.
reduction
under standard conditions of the L-homo-phenylalanine compound available from
Aldrich)
the title compound is prepared.
Example 44
Compound of Formula (IIIO Rl-OCH~ R~=H. A=B=E=H: D=-CH2~H2Ph
Following the procedures of Example 3 except replacing the 2-(R)-amino-3-
phenyl-
1-propanol of step 3a thereof with D-homo-phenylalaninol (prepared by LiAIHq.
reduction
under standard conditions of the D-homo-phenylalanine compound available from
Aldrich)
the title compound is prepared.
_77_
CA 02235942 1998-04-23
WO 97/17356 PCT/LJS96/16585
Example 45
Compound of Formula (IIII: Rl----OCH~ R2=H: A=B=D=H: E=~HzCH2CHzPh
Following the procedures of Example 3 except replacing the 2-(R)-amino-3-
phenyl-
1-propanol of step 3a thereof with (~)- 2-amino-5-phenylpentanol (prepared in
Example
40b) and separating the desired isomer by chromatography the title compound is
prepared.
Example 46
Compound of Formula (III): Rte--OCH~ R~=H: A=B=E=H: D=-CH2CH2CH2Ph
Following the procedures of Example 3 except replacing the 2-(R)-amino-3-
phenyl-
l0 1-propanol of step 3a thereof with 2-(R)-amino-5-phenylpentanol (prepared
in Example
36b) the title compound is prepared.
Example 47
Compound of Formula (IIII R 1=OCH~,R2=H: A=-CH2CH20Ph~ B=D=E=H
47a. N-a-Boc-L-homo-serine benz~rl ester
The title compound is prepared from N-a-Boc-L-aspartic acid a-benzyl ester,
ethyl
chloroformate, N-methylinorpholine and sodium borohydride in tetrahydrofuran
and
methanol according to the procedures described by Kokotos, Synthesis, (1990):
299-301.
47b. 4-Phenoxy-2-(S)-Boc-aminobutyric acid benzyl ester
A solution of N-a-Boc-L-homo-serine benzyl ester, phenol and
triphenylphosphine
in THF is treated with diethylazodicarboxylate (DEAD) at 0 °C. After
being stirred at
reaction temperature for 2 hours, the reaction is worked up and product purled
by silica gel
chromatography. (c~ Organic Reactions, Vol. 42, John Wiley & Son, Inc, 1992).
47c. 4-phenoxy-2-(S 1-Boc-aminobutane-1-0l
Lithium aluminum hydride is added into a stirred solution of 4-phenoxy-2-(S)-
Boc-
aminobutyric acid benzyl ester, from step 47b, in THF at 0 °C. The
reaction mixture is
heated to reflux for 0.5 hour and cooled to ice-cold temperature. Water, equal
weight to the
lithium aluminum hydride used, is added and stirred at reaction temperature
overnight. The
reaction is diluted with ethyl acetate and dried over sodium sulfate. After
filtration and
removal of solvent in vacuo, the product is purified by silica gel
chromatography.
47d. 4-nhenoxv-2-(S)-aminobutane-1-of
The amino protecting group of 4-phenoxy-2-(S)-Boc-aminobutane-1-of from step
47c is removed by treatment of the protected compound with hydrogen chloride
in dioxane.
(cf. T. W. Greene and P. G. M. Wuts: Protective Groups in Organic Synthesis.
2nd ed.,
John Wiley and Son, 1991, pp 309-315.)
_7g_
CA 02235942 1998-04-23
WO 97/17356 PCT/US96116585
47e. Compound (22) Scheme 10:
R~-OCH~~ R~'=Bz: A~CH2CH2-OPh: B=D=E=H: Y=OH
The title compound is prepared from 4-phenoxy-2-(S)-aminobutane-1-of and
compound (8) in aqueous acetonitrile according to the procedures described in
Example 5.
47f. Compound l207 Scheme 10: Rl=_ OCHY: A=-CH2CH2-OPh: B=D=E=H
The title compound is prepared from the compound of step 47d. according to the
procedures described in Example 5.
47 g. Compound of Formula (III) R 1=OCH~, R2=H: A=-CH2CH20Ph: B=D=E=H
The title compound of Example 47 is prepared from the compound of 47f
according
to the procedures described in Example 5.
Example 48
i5 Compound of Formula~III) R1=OCH~, R2=H: A=-CH2CH2NHCbz: B=D=E=H
48a. N-a-!S)-Boc -N-y Cbz-2.4-diaminobutvric acid benzyl ester
Diphenylphosphoryl azide is added into a solution of N-a-(S)-Boc-L-glutamic
acid a
-benzyl ester in THF and the resulting solution is refluxed for 2 hours.
Benzyl alcohol is
added and the reaction mixture is refluxed for an additional hour. The product
is purified by
silica gel chromatography.
4$b. N-a-!S)-Boc -N-'y Cbz-2.4-diamino-butane-1-of
Lithium aluminum hydride is added into a stirred solution of N-oc-(S)-Boc -N-y
Cbz-
diaminobutyric acid benzyl ester, from step 48a, in THF at 0 °C. The
reaction mixture is
heated to reflux for 0.5 hour and cooled to ice-cold temperature. Water, equal
weight to the
lithium aluminum hydride used, is added and stirred at reaction temperature
overnight. The
reaction is diluted with ethyl acetate and dried over sodium sulfate. After
filtration and
removal of solvent in vacuo, the product is purified by silica gel
chromatography.
48c. 2-!S 1-4-N-Cbz-diamino-1-butanol
The amino protecting group of N-a-(S)-Boc -N-'y Cbz-2,4-diamino-butane-1-of
from step 48b is removed by treatment of the protected compound with hydrogen
chloride in
dioxane according to literature method. (cf. T. W. Greene and P. G. IVI. Wuts:
Protective
Groups in Organic Synthesis. 2nd ed., John Wiley and Son, 1991, pp 309-315.)
48d. Compound (22) Scheme 10:
$1--OC ~H ;~~=Bz~ A=-CH2CH2-NHCbz~ B=D-E-H~ Y-OH
The title compound is prepared from 2-(S)-amino-4-N-y Cbz-diamino-butane-1-ol,
from step 48c, and compound (8) from Scheme 3 in aqueous acetonitrile
according to the
procedures described in Example 5.
-79-
CA 02235942 1998-04-23
WO 97/17356 PCT/US96/16585
48e. Compound (201 Scheme 10' Rl=~; A=-CH2CH2-NHCbz; B=D=E=H
The title compound is prepared from the compound of step 48d according to the
procedures described in Example 5.
48f. Compound of Formula (IIII R1=OCH~ R2=H: A=-CH2CH2NHCbz~ B=D=E=H
The title compound is prepared from the compound of step 48e according to the
procedures described in Example 5.
1o Example 48-A
Compound of Formula (Im Rl=OCH~, R2=H: A=-CH2CH2NH2~ B=D=E=H
The title compound is prepared from the compound of Example 48 and hydrogen in
the presence of Pd-C in ethanol.
Example 49
Compound of Formula (III1 R 1=OCH~ R2=H: A=-CH2C02Bzl: B=D=E=H
49a. 4-hvdroxy-3-N-(S)-Boc-aminobutvric acid benzyl ester
The title compound is prepared from N-a-Boc-L-aspartic acid y benzyl ester,
ethyl
chloroformate, N-methylinorpholine and sodium borohydride in tetrahydrofuran
and
methanol according to the procedures described by Kokotos, Synthesis, (1990):
299-301.
49b. 4-hydroxy-3-(S)-aminobutyric acid Benz ly ester
The amino protecting group of 4-hydroxy-3-N-(S)-Boc-aminobutyric acid benzyl
ester, from step 49a, is removed by treatment of the protected compound with
hydrogen
chloride in dioxane according to literature method.(cf. T. W. Greene and P. G.
M. Wuts:
Protective Groups in Organic Synthesis. 2nd ed., John Wiley and Son, 1991, pp
309-315.)
49c. Compound (22) Scheme 10:
R~-OCH~- R~=Bz: A~CH2C02Bz1~ B=D=E=H' Y=OH
The title compound is prepared from 4-hydroxy-3-(S)-aminobutyric acid benzyl
ester, from step 49b and compound (8) of Scheme 3 in aqueous acetonitrile
according to the
procedures described in Example 5.
49d. Compound (20) Scheme 10- Rl=y A=-CH2C02Bzl: B=D=E=H
The title compound is prepared from the compound of step 49c according to the
procedures described in Example 5.
-80-
CA 02235942 1998-04-23
WO 97/17356 PCT/US96/16585
49e. Compound of Formula (III) R1=OCH~, R2=H; A=-CH2C02Bzl: B=D=E=H
The title compound is prepared from compound from the compound of step 49d
according to the procedures described in Example 5.
Example 49-A
Compound of Formula (IIP Rl=OCH~, R2=H, A=-CH2COOl~i~ B=D=E=H
The title compound is prepared from the compound of Example 49 and hydrogen in
the presence of Pd-C in ethanol.
to Example 49-B
Formula (IIII R1=OCH~ R2=H: A=-CH2CH20H~ B=D=E=H
The title compound is prepared from the title compound of Example 49-A, ethyl
chloroformate, N-methyhnorpholine and sodium borohydride in tetrahydrofuran
and
methanol according to the procedures described by Kokotos, Synthesis, (1990):
299-301.
Example 50
Compound of Formula (IIII R1=OCHY R2=H; A=-CH2CH2NH(4'-fyridyll B=D=E=H
The title compound is prepared from the compound of Example 48-A, 4-
chloropyridine, and Cu0 or CuBr-K2C03 heated at 70-90 °C overnight. The
reaction
mixture is partitioned between ethyl acetate and water. The organic phase is
washed once
with dilute hydrochloric acid followed by saturated sodium bicarbonate. The
product is
purified by silica gel chromatography.
Example 51
Compound of Formula (IIIO Rl--OCH~ R~1~; A=B=D=H; E=-CH20H
~ 1 a. 1 2-diamino-3-nropanol
Allyl acetate (Aldrich) is reacted with manganese triacetate, sodium azide and
acetic
acid. The resulting compound is treated with NaHC03 and methanol followed by
hydrogenation to give the title compound.
~ 1 b. Compound of Formula IIII~ ~ Rl=O~ R~=H: A=B=D=H: E=-CH20H
Following the procedures of Example 3 steps f h, except substituting 1,2-
diamino-3
propanol from the previous step, for the l, 2-(R)-diamino-3-phenylpropane
thereof, the title
compound is prepared, and the diastereomers are separated by chromatography.
Example 52
Compound of Formula (III)' Rl-~ RAH; A=B=E=H: D=-CH20H
The compound is separated from the diastereomeric mixture of Example 51b by
chromatography.
-81-
CA 02235942 1998-04-23
WO 97/17356 PCT/US96/16585
Example 53
Compound of Formula (III): R~-~ R~=H: A=B=E=H: D=-CH2NHBenzo~l
The compound is prepared from the compound of Example 16 by treatment with
benzoyl chloride and TEA.
Example 54
Compound of Formula (III): Rl=OCHY R~=H: A=B=E=H: D=-CH2NHBenzvl
The compound is prepared from the compound of Example 16 by treatment with
benzaidehyde, NaCNBH3 and acetic acid in methanol at reaction temperature.
Example 55
Compound of Formula IIII): Rl-~ R~=H: A=B=D=H: E=-CH2NHBenzovl
The compound is prepared from the compound of Example 15 by treatment with
benzoyl chloride and TEA.
Example 56
Compound of Formula (III): Rl-~ R~=H: A=B=D=H: E=-CH2NHBenzyl_
The compound is prepared from the compound of Example 15 by treatment with
benzaldehyde, NaCNBH3 and acetic acid in methanol at reaction temperature.
Example 57
Compound of Formula (III): Rl-~ R~=H: B=D=H: A=E=-CH20CH2(4-Cl-phenyl-)
~7a. (R.R)-(+)-1.4- bis-O-(4-chlorobenzvl)-2.3-butanediamine
Following the procedure of Example 10 steps a-c, replacing the meso-2,3-
butanediol
thereof with (R,R)-(+)-1,4- bis-O-(4-chlorobenzyl)-D-threitol (Aldrich) the
title compound is
obtained.
57b. Compound of Formula (III):
Rl--I R2=H: B=D=H: A=E=-CH20CH2(4-Cl-phenyl-1
Following the procedures of Example 10 steps d-f, replacing the meso-2,3-
butanediamine of step d thereof with the diamine from step 57a, the title
compound is
prepared.
Example 58
Compound of Formula (III): Rl=OCH3. R~=H: A=B=E=H: D=-CH2-N(CH~)-Benzx_1
The title compound is prepared by treating the compound of Example 54 with
HCHO, NaCNBH3 and acetic acid in methanol at reaction temperature.
-82-
CA 02235942 1998-04-23
WO 97/17356 PCT/US96/16585
Example 59
Compound of Formula (IIIZ: Rl=OCH3. R~=H; A=B=D=H: E=-CH2-N(CH~ -Benz 1
The title compound is prepared by treating the compound of Example 56 with
I~CHO, NaCNBH3 and acetic acid in methanol at reaction temperature.
Example 60
Compound of Formula (III): Rl=OCH3. R~=H; A=B=D=H: E=-CH2-NH-phenvl
The title compound is prepared by treating the compound of Example 51 with
l0
triphenyl phosphine, DEAD and aniline under Mitsunobu conditions.
Example 61
Compound of Formula fIII): Rl=OCH3. R~=H: A=B=E=H: D=-CH2-NH- hn envl
The title compound is prepared by treating the compound of Example 52 with
triphenyl phosphine, DEAD and aniline under Mitsunobu conditions.
Example 62
In Vitro Assay of Antibacterial Activity
Representative compounds of the present invention were assayed in vitro for
antibacterial activity as follows: Twelve petri dishes containing successive
aqueous dilutions
of the test compound mixed with 10 mL of sterilized Brain Heart Infusion (BHn
agar (Difco
0418-O1-5) were prepared. Each plate was inoculated with 1:100 (or I:10 for
slow-growing
strains, such as Micrococcus and Streptococcus) dilutions of up to 32
different
microorganisms, using a Steers replicator block. The inoculated plates were
incubated at 35-
37 °C for 20 to 24 hours. In addition, a control plate, using BHI agar
containing no test
compound, was prepared and incubated at the beginning and end of each test.
An additional plate containing a compound having known susceptibility patterns
for
the organisms being tested and belonging to the same antibiotic class as the
test compound
was also prepared and incubated as a further control, as well as to provide
test-to-test
comparability. Erythromycin A was used for this purpose.
After incubation, each plate was visually inspected. The minimum inhibitory
concentration (MIC) was defined as the lowest concentration of drug yielding
no growth, a
slight haze, or sparsely isolated colonies on the inoculum spot as compared to
the growth
control. The results of this assay, shown below in Table 2 demonstrate the
antibacterial
activity of the compounds of the invention.
-83-
CA 02235942 1998-04-23
WO 97/17356 PCT/US96/16585
Table 2
Atltibacterial Activity (MIC's) of Selected Compounds
Ery A Cmpd of Cmpd
of
MICROORGANISM (Ref. Ex 1 Ex 3
std)
STAPHYLOCOCCUS AUREUS ATCC 0.2 6.2 0.39
6538P
STAPHYLOCOCCUS AUREUS A5177 6.2 6.2 0.39
STAPHYLOCOCCUS AUREUS A-5278 > 100 > 100 > 100
STAPHYLOCOCCUS AUREUS CMX 642A0.39 6.2 0.39
STAPHYLOCOCCUS AUREUS NCTC10649M0.2 6.2 0.39
STAPHYLOCOCCUS AUREUS CMX 553 0.2 6.2 0.39
STAPHYLOCOCCUS AUREUS 1775 > 100 > 100 > 100
STAPHYLOCOCCUS EPIDERMIDIS 0.39 12.5 0.39
3519
ENTEROCOCCUS FAECIC1M ATCC 0.05 0.78 0.05
8043
STREPTOCOCCUS BOVIS A-5169 0.05 0.39 0.02
STREPTOCOCCUS AGALACTIAE CMX 0.05 0.39 0.01
508
STREPTOCOCCUS PYOGENES EES61 0.05 0.39 0.01
STREPTOCOCCUS PYOGENES 930 >100 >100 100
STREPTOCOCCUS PYOGENES PIU 6.2 0.78 na
2548
MICROCOCCUS LUTEUS ATCC 9341 0.02 0.39 0.1
MICROCOCCUS LUTEUS ATCC 4698 0.2 0.39 0.39
ESCHERICHIA COLI JUHI. 100 na >100
ESCHERICHIA COLI SS 0.2 na na
ESCHERICHIA COLI DC-2 >100 0.78 >100
ESCHERICHIA COLI H560 25 50 >100
ESCHERICHIA COLI KNK 437 50 25 >100
ENTEROBACTER AEROGENES ATCC 100 >100 >100
13048
KLEBSIELLA PNEUMOMAE ATCC 8045100 >100 >100
PROVIDENCIA STUARTII CMX 640 >100 100 >100
PSEUDOMONAS AERUGINOSA BMH10 >100 >100 >100
PSEUDOMONAS AERUGINOSA 5007 > 100 > 100 > 100
PSEUDOMONAS AERUGINOSA K799/WT50 >100 >100
PSEUDOMONAS AERUGINOSA K799/610.78 >100 3.1
PSEUDOMONAS CEPACIA 296I > 100 12.5 > 100
ACINETOBACTERCALCOACETICUSCMX6696.2 >100 >100
PSEUDOMONAS AERUGINOSA DPHD-5263>100 50 >100
PSEUDOMONAS AERUGINOSA DPHD-2862>100 >100 >100
CANDll~A ALBICANS CCH442 >100 >100 >100
MYCOBACTERIUM SMEGMATIS ATCC 0.02 3.1 >100
114
-84-
CA 02235942 1998-04-23
W(y 97/17356 PCT/US96/16585
Table 2
Antibacterial Activit3r (MIC's) of Selected Compounds
Cmpd of Cmpd of Cmpd
of
MICROORGANISM Ex 4 Ex 6 Ex 8
STAPHYLOCOCCUS AUREUS ATCC 6538P 0.2 0.1 0.78
STAPHYLOCOCCUS AUREUS A5177 0.2 0.05 0.78
STAPHYLOCOCCUS AUREUS A-5278 >100 >100 >100
STAPHYLOCOCCUS ALTREUS CMX 642A 0.78 0.1 0.78
STAPHYLOCOCCUS AUREUS NCTC10649M 0.78 0.1 0.78
STAPHYLOCOCCUS AUREUS CMX 553 0.78 0.2 0.78
STAPHYLOCOCCUS AUREUS 1775 > 100 > 100 > 100
STAPHYLOCOCCUS EPIDERMIDIS 3519 0.78 . 0.1 0.78
LNTEROCOCCUS FAECIUM ATCC 8043 0.1 0.1 0.78
STREPTOCOCCUS BOVIS A-5169 0.02 0.01 0.1
STREPTOCOCCUS AGALACTTAF CMX 508 0.005 0.02 0.1
STREPTOCOCCUS PYOGENES EES61 0.05 0.02 0.1
STREPTOCOCCUS PYOGENES 930 > 100 > 100 > 100
STREPTOCOCCUS PYOGENES PICT 2548 na 0.39 0.78
1VIICROCOCCUS LUTEUS ATCC 9341 0.1 0.02 1.56
MICROCOCCUS LUTEUS ATCC 4698 0.39 0.1 1.56
ESCHERICHIA COLI JCTHI. >100 12.5 100
ESCHERICHIA COLI SS na 0.02 0.78
LSCHERICHIA COLI DC-2 100 12.5 50
LSCHERICHIA COLI H560 25 3.1 25
ESCHERICHIA COLI KNK 437 >100 6.2 100
ENTEROBACTER AEROGENES ATCC 13048 100 12.5 100
KLEBSIELLA PNEUMONIAE ATCC 8045 >100 12.5 100
PROVIDENCIA STUARTII CMX 640 >100 100 >100
PSEUDOMONAS AERUGINOSA BMH10 >100 50 100
PSEUDOMONAS AERUGINOSA 5007 > 100 100 > 100
1'SEUDOMONAS AERUGINOSA K799/WT >100 25 100
PSEUDOMONAS AERUGINOSA K799/61 1.56 0.39 na.
1'SEUDOMONAS CEPACIA 296I >100 50 >100
ACINETOBACTER CALCOACETICUS CMX 669 25 12.5 25
1'SEUDOMONAS AERUGINOSA DPHD-5263 >100 12.5 >100
1'SEUDOMONAS AERUGINOSA DPHD-2862 >100 100 >100
CANDIDA ALBICANS CCH 442 100 > 100 > 100
MYCOBACTERIUM SMEGMATIS ATCC 114 na na 6.2
-85-
CA 02235942 1998-04-23
WO 97/17356 PCT/(TS96/16585
Tabte 2
Antibacterial Activity (MIC'sl of Selected Com -hounds
Cmpd of Cmpd of Cmpd
of
MICROORGANISM Ex 9 Ex 10 Ex 11
STAPHYLOCOCCUS AUREUS ATCC 0.39 0.39 0.39
6538P
STAPHYLOCOCCUS AUREUS A5177 0.39 na 0.39
STAPHYLOCOCCUS AUREUS A-5278 > 100 > 100 > 100
STAPHYLOCOCCUS AUREUS CMX 642A0.39 0.39 0.39
STAPHYLOCOCCUS AUREUS NCTC10649M0.2 0.39 0.39
STAPHYLOCOCCUS AUREUS CMX 553 0.39 0.39 0.39
STAPHYLOCOCCUS AUREUS 1775 >100 >100 >100
STAPHYLOCOCCUS EPIDERMIDIS 0.39 0.39 0.39
3519
ENTEROCOCCUS FAECIiJM ATCC 0.05 na 0.1
8043
STREPTOCOCCUS BOVIS A-5169 na 0.05 0.01
STREPTOCOCCUS AGALACTIAE CMX 0.01 na 0.05
508
STREPTOCOCCUS PYOGENES EES61 0.01 na 0.005
STREPTOCOCCUS PYOGENES 930 > 100 > 100 > 100
STREPTOCOCCUS PYOGENES PIU 0.39 0.39 0.39
2548
MICROCOCCUS LUTEUS ATCC 9341 0.1 0.1 0.2
MICROCOCCUS LUTEUS ATCC 4698 0.39 0.2 0.39
ESCHERICHIA COLI JUHI. na 25 25
ESCHERICHIA COLI SS na 0.2 0.39
ESCHERICHIA COLI DC-2 25 ~5 ~5
ESCHERICHIA COLI H560 25 25 6.2
ESCHERICHIA COLI KNK 437 100 na 25
ENTEROBACTER AEROGENES ATCC 25 50 25
13048
KLEBSIELLA PNEUMONIAE ATCC 25 na 12.5
8045
PROVIDENCIA STUARTII CMX 640 > 100 > 100 > 100
PSEUDOMONAS AERUGINOSA BMH10 100 >100 >100
PSEUDOMONAS AERUGINOSA 5007 > 100 > 100 > 100
PSEUDOMONAS AERUGINOSA K799/WT100 na >100
PSEUDOMONAS AERUGINOSA K799/616.2 na na
PSEUDOMONAS CEPACIA 296I >100 100 >100
ACINETOBACTER CALCOACETICUS 12.5 25 6.2
CMX 669
PSEUDOMONAS AERUGINOSA DPHD-5263>100 >100 >100
PSEUDOMONAS AERUGINOSA DPHD-2862> 100 > 100 > 100
CANDIDA ALBICANS CCH 442 >100 >100 >100
MYCOBACTERIUM SMEGMATIS ATCC 3.1 6.2 6.2
114
-86-